Изобретение относится к способу нитрования толуола до динитротолуола (ДНТ) нитрующей кислотой (нитрующей смесью - азотная кислота + серная кислота) в двухстадийном процессе, причем первую стадию осуществляют адиабатически, а вторую стадию - изотермически.
Динитротолуол (ДНТ) является промежуточным продуктом получения толуилендиизоцианата, используемого в качестве исходного вещества для получения полиуретанов. Обычно употребляемым в технике способом для получения динитротолуола является изотермическое двухстадийное взаимодействие толуола с нитрующей кислотой из азотной и серной кислот (Ullmanns Encyklopädie der technischen Chemie, 4 Auflage, Band 17, Seite 392, Verlag Chemie, Weinheim 1979). При этом сначала получают смесь изомеров мононитротолуола (МНТ), которая превращается в динитротолуол на второй отдельной стадии способа. Однако способ имеет тот недостаток, что получаемая на обеих стадиях кислотная фаза (главным образом, серная кислота) должна быть освобождена от поглощенной воды с энергетическими затратами.
Известно также одностадийное превращение толуола в динитротолуол в адиабатическом процессе (европейская заявка на патент ЕР-А-597361). При этом толуол взаимодействует, по меньшей мере, с двумя эквивалентами нитрирующей кислотой определенного состава в адиабатическом процессе, причем конечная температура достигает свыше 120°С. После разделения фаз при этой температуре кислотную фазу подвергают концентрированию (процесс мгновенного упаривания в вакууме), причем для концентрирования требуется сохранение кислотой тепла. Сконцентрированную кислоту подкрепляют азотной кислотой и возвращают в процесс.
Однако при таком способе возникают трудности, заключающиеся в том, что при мгновенном упаривании в газовую фазу вместе с отогнанной водой переходит определенная растворившаяся в кислоте часть динитротолуола (ДНТ), затвердевающая затем при конденсации вторичного пара в условиях конденсации воды (точка затвердевания смеси изомеров 52-58°С) и отлагающаяся в теплообменнике. Кроме того, при проведении процесса температура повышается до такой температуры, при которой динитротолуол (ДНТ) не является долго стабильным в присутствии побочных продуктов. Для гарантии безопасности процесса допустимое время пребывания потока вещества, содержащего динитротолуол, при высоких температурах не должно быть значительно превышено. Это требует существенных затрат на оборудование, обеспечивающее технологическую безопасность.
Для устранения проблемы нарушения конденсации, связанной с содержанием во вторичном паре динитротолуола, предлагались различные решения, такие как попеременно работающие теплообменники с шаговым перемещением, конденсаторы смешения или эжекторные конденсаторы (R.F.Vauck, Н.А.Müller, Grundoperationen chemischer Verfahrenstechnik, 5, Auflage, VEB Leipzig 1962, s.447). Кроме того, в европейской заявке на патент А-0696569 описан способ адиабатического получения динитротолуола (ДНТ), причем для снижения отложений в теплообменнике в продукте реакции адиабатического нитрования целенаправленно оставляют, по меньшей мере, 5% мононитротолуола.
Все эти способы либо являются технически или энергетически затратными, либо они не позволяют получить динитротолуол с очень незначительным содержанием мононитротолуола. Помимо этого, при адиабатическом проведении реакции сохраняется проблема жесткого воздействия высокой температуры на динитротолуол и среду, содержащую значительное количество динитротолуола.
Поэтому задачей настоящего изобретения явилось создание технически простого способа получения динитротолуола нитрованием толуола, при котором, по меньшей мере, часть выделяющегося при реакции тепла может быть использована для концентрирования отработанной кислоты и одновременно при котором динитротолуол (ДНТ) и среды, содержащие динитротолуол (ДНТ), не подвергаются воздействию каких-либо технически небезопасных температур.
Изобретение относится к способу получения динитротолуола двухстадийным нитрованием толуола, в котором:
а. На первой стадии толуол подвергают адиабатическому взаимодействию с нитрующей кислотой, причем, по меньшей мере, 90%, предпочтительно, по меньшей мере, 98% толуола вступает в реакцию и максимум 70%, предпочтительно, до 50% используемого толуола превращается в динитротолуол, непосредственно после чего органическую фазу, содержащую мононитротолуол, и водную кислотную фазу, содержащую серную кислоту, разделяют, водную содержащую серную кислоту кислотную фазу концентрируют мгновенным упариванием со сбросом давления, а полученную при этом сконцентрированную серную кислоту возвращают в реакцию на первой стадии и/или в реакцию на второй стадии и/или на концентрирование на второй стадии.
б. На второй стадии органическую фазу, содержащую мононитротолуол с первой стадии, подвергают изотермическому полному взаимодействию с нитрующей кислотой, непосредственно после чего органическую фазу и водную кислотную фазу, содержащую серную кислоту, разделяют и водную содержащую серную кислоту кислотную фазу концентрируют упариванием в вакууме, а полученную при этом сконцентрированную серную кислоту возвращают в реакцию на первой стадии и/или на второй стадии реакции.
В способе по изобретению на первой стадии может вступить в реакцию, предпочтительно, 98% толуола, причем, предпочтительно, до 50%, особенно предпочтительно, до 30% используемого толуола превращается в нитротолуол.
Первая адиабатическая стадия может проводиться в любом соответствующем реакторе. Однако, предпочтительно, используют трубчатые реакторы, длина которых может составлять от 2 до 20 м, а диаметр - от 25 до 2000 мм. При этом длину и диаметр выбирают предпочтительно из того расчета, что среднее время пребывания в нем составляет от 10 до 300 секунд. Может быть использован как трубчатый реактор со встроенным приспособлением для диспергирования двухфазной смеси (предпочтительный вариант), так и без него. Предпочтительно используемыми встроенными приспособлениями являются, например, ситчатые тарелки или статичные перемешивающие элементы, например, такие как описаны в европейских заявках на патент ЕР-А-0708076 и ЕР-А-0489211.
Используемая нитрующая кислота, предпочтительно, содержит от 80 до 100мас.% неорганических компонентов, состоящих, главным образом, из 50-90мас.% серной кислоты, 1-20мас.% азотной кислоты и, по меньшей мере, 5мас.% воды. Молярное соотношение азотной кислоты к толуолу, предпочтительно, составляет, по меньшей мере, 0,7:1 и максимум 1,8:1, особенно предпочтительно, максимум 1,5:1. Используемая азотная кислота может иметь концентрацию 50-100мас.% Предпочтительно, используют азотную кислоту с концентрацией 62-70мас.%
Нитрующую кислоту на входе в реактор смешивают с толуолом, что может осуществляться посредством простого питающего трубопровода или посредством специального элемента смешения, такого как сопло или специальный статический смеситель. Предпочтительно, толуол диспергируют в нитрующей кислоте посредством диспергирующего устройства, например, струйного диспергатора.
На входе в реактор смесь, предпочтительно, имеет температуру от 80 до 120°С, а на выходе из реактора температура повышается до 120-170°С за счет выделяющегося при реакции тепла. Повышение температуры составляет при этом, предпочтительно, от 20 до 80°С.
После выхода из реактора двухфазную смесь разделяют на водную кислотную фазу, содержащую серную кислоту, и органическую фазу, содержащую мононитротолуол, что происходит обычно в статическом фазовом сепараторе.
Органическая фаза, полностью или частично, может быть использована затем для экстрагирования отработанной кислоты со второй стадии и затем направлена на вторую стадию нитрования. Предпочтительно, она содержит максимум 10мас.%, особенно предпочтительно, менее 2мас.% толуола и, предпочтительно, максимум 70мас.%, особенно предпочтительно, до 50мас.% и, наиболее предпочтительно, до 30мас.% динитротолуола (ДНТ).
Водную кислотную фазу (отработанную кислоту) освобождают, предпочтительно, от 60-110% образовавшейся при реакции и привнесенной азотной кислотой воды посредством мгновенного упаривания со сбросом давления, преимущественно, при давлении от 10 до 400 мбар, в случае необходимости, при дополнительном подводе тепла посредством теплообменника, а затем возвращают обратно на вход в реактор.
Предпочтительно, выпаривают количество воды, равное количеству воды, возникшему при реакции и привнесенному с азотной кислотой. Смещение от этого, предпочтительно, равновесного состояния может быть, однако, выровнено посредством обмена количества серной кислоты из циркуляционного контура первой стадии на отработанную кислоту из циркуляционного контура второй стадии и/или сконцентрированную серную кислоту после ее концентрирования на второй ступени.
В другом предпочтительном варианте осуществления способа стадии мгновенного выпаривания со сбросом давления и разделения фаз осуществляют в обратной последовательности, то есть двухфазную смесь подвергают мгновенному упариванию со сбросом давления при вышеназванных условиях, после чего охлажденную смесь разделяют на кислотную фазу и органическую фазу.
Переведенные во вторичный пар вследствие летучести водяного пара органические компоненты конденсируют вместе с водой. Благодаря высокому содержанию мононитротолуола (МНТ) не возникает какой-либо опасности, что в конденсаторе вторичного пара образуются твердые отложения динитротолуола (ДНТ) или смеси динитротолуол/мононитротолуол. Двухфазный конденсат вторичного пара разделяют на органическую и водную фазы, что можно осуществить, предпочтительно, в статическом сепараторе. Органическую фазу, полностью или частично, можно использовать для экстрагирования отработанной кислоты со второй стадии, а затем направлять на вторую стадию нитрования.
Вторую изотермическую стадию проводят в соответствующем реакторе или серии реакторов с устройством для охлаждения. Предпочтительно, используют реактор в виде котла с перемешиванием или петлевые реакторы с циркуляционным контуром и теплообменниками, такие как описаны в Ullmanns Encyklopädie der technischen Chemie, 4 Auflage, Band 17, Seite 392, Verlag Chemie, Weinheim 1979. При этом температуру реакции регулируют как обычно отводом тепла охлаждающей водой. Предпочтительно, она находится в пределах от 60 до 95°С, особенно предпочтительно, от 60 до 80°С. Время выдержки составляет, предпочтительно, от 1 до 10 минут.
В реактор или в каскад реакторов подают органическую фазу с первой стадии, содержащую мононитротолуол, которая ранее, полностью или частично, могла быть использована для экстракции водной кислотной фазы со второй ступени, и смешивают ее с нитрующей кислотой. Нитрующую кислоту, предпочтительно, получают смешением сконцентрированной серной кислоты с концентрацией от 83 до 98мас.%, особенно предпочтительно, 85 до 98мас.% и азотной кислоты. При этом количество нитрующей кислоты на второй стадии выбирают из расчета, что сумма использованной на первой и второй стадиях азотной кислоты составляет 1,9-2,2 моль, предпочтительно, 2,0-2,05 моль на один моль использованного толуола. Концентрация использованной азотной кислоты, предпочтительно, равна ее концентрации на первой стадии.
Поступающую со второй стадии реакции двухфазную смесь разделяют на органическую фазу и водную фазу, содержащую серную кислоту, что может осуществляться посредством центрифуг или, предпочтительно, в статических сепараторах. Органическую фазу соответствующим образом освобождают от следов кислоты, например, экстракцией водой и/или раствором соды, и в результате получают конечный продукт - динитротолуол.
Полученную кислотную фазу (отработанная кислота), предпочтительно, сначала экстрагируют органической фазой с первой стадии, содержащей мононитротолуол (МНТ) для снижения содержания растворенного динитротолуола (ДНТ). Для этого может использоваться или часть органического конденсата вторичного пара с первой стадии и/или вся либо часть органической фазы с первой стадии. Это может быть осуществлено одностадийно или многостадийно, причем могут быть использованы секции смеситель-сепаратор или также перемешивающие-экстракционные колонны. Часть отработанной кислоты перед экстракцией или после экстракции может отбираться и добавляться серная кислота с первой стадии нитрования перед ее концентрированием или после концентрирования.
Отработанную кислоту со стадии динитрования после экстракции освобождают от воды, образовавшейся при реакции и привнесенной с азотной кислотой, в соответствующей дистилляционной установке. Пригодный способ дистилляции описан, например, в немецкой заявке на патент DE-A-19636191. Поступающая в дистилляционную установку отработанная кислота, предпочтительно, имеет концентрацию серной кислоты от 75 до 90мас.% и подвергается концентрированию, предпочтительно, до концентрации серной кислоты от 83 до 98мас.%, особенно предпочтительно, до 85-98мас.%
Для выравнивания баланса серной кислоты на первой стадии на это концентрирование кислоты может быть направлено дополнительно до 30% полученной по адиабатическому режиму (на первой стадии) части отработанной кислоты, предпочтительно серной кислоты, после мгновенного упаривания со сбросом давления. Для выравнивания баланса серной кислоты на первой стадии с изотермическим режимом работы, соответствующее количество серной кислоты, отходящей со стадии концентрирования кислоты, должно затем добавляться в циркуляционный контур кислоты первой ступени.
Потери серной кислоты на обеих стадиях реакции могут быть возмещены коммерчески доступной 80-100 мас.%-ной, предпочтительно, 90-100 мас.%-ной свежей серной кислотой.
Способ по изобретению в нижеследующем поясняется более подробно посредством фигур.
Показано:
Фиг.1 - схематическое изображение способа, в котором сначала разделяют органическую фазу и водную кислотную фазу с первой стадии, а затем водную кислотную фазу подвергают концентрированию мгновенным упариванием со сбросом давления.
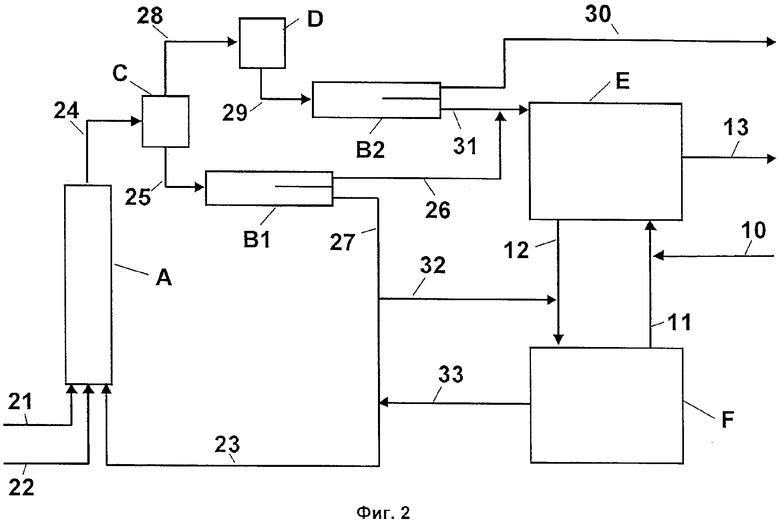
Фиг.2 - схематическое изображение способа, при котором органическую фазу и водную кислотную фазу совместно сначала подвергают мгновенному упариванию со сбросом давления, а затем разделяют органическую фазу и сконцентрированную водную кислотную фазу.
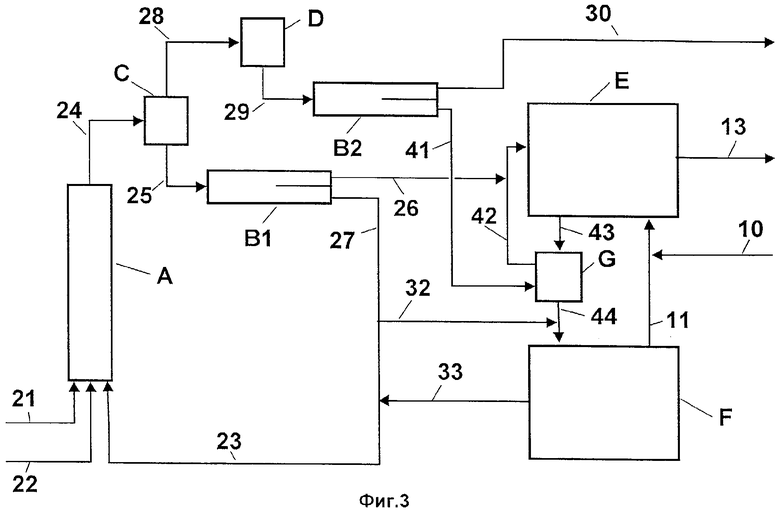
Фиг.3 - схематическое изображение способа, при котором органическую фазу и водную кислотную фазу совместно сначала подвергают мгновенному упариванию со сбросом давления, а затем разделяют органическую фазу и сконцентрированную водную кислотную фазу и при котором водную кислотную фазу со второй стадии экстрагируют органическим конденсатом вторичного пара с первой стадии.
На Фиг.1 схематично изображен один из вариантов осуществления способа по изобретению. При этом А обозначает реактор первой стадии, В - фазовый сепаратор реакционной смеси, С - испаритель мгновенного действия (выпарной аппарат с мгновенным испарением и с теплообменником в нижней части) водной кислотной фазы первой стадии, D - конденсатор испарения мгновенного действия, Е - реактор и фазовый сепаратор второй стадии и F - вакуумный испаритель для концентрирования водной кислотной фазы со второй стадии. В реактор А подают и смешивают толуол (поток 1), азотную кислоту (поток 2) и возвращенную после концентрирования серную кислоту (поток 3). Непосредственно после этого полученную двухфазную реакционную смесь (поток 4) разделяют в фазовом сепараторе В на органическую фазу (поток 5) и водную кислотную фазу (поток 6). Водную кислотную фазу (поток 6) концентрируют в испарителе мгновенного действия со сбросом давления. Образованный в испарителе мгновенного действия С вторичный пар (поток 8) конденсируют в конденсаторе D, и он может быть выведен из процесса. Сконцентрированную водную кислотную фазу (главным образом, серная кислота) (поток 3) подают в реактор А первой стадии. Органическую фазу (поток 5) направляют в реактор Е второй стадии для взаимодействия в нем с нитрующей кислотой с образованием динитротолуола. Разделение органической фазы, содержащей динитротолуол, и водной кислотной фазы не изображено на Фиг.1 в виде самостоятельной стадии. Органическую фазу выводят из процесса в виде потока 13 и подвергают дальнейшей переработке. Водную кислотную фазу 12 (отработанная кислота) направляют в вакуумный испаритель F второй стадии, где происходит концентрирование отработанной кислоты. Сконцентрированную водную кислотную фазу затем вновь возвращают на вход в реактор Е в виде потока 11. Для компенсации потерь серной кислоты добавляют свежую серную кислоту (поток 10). Добавление осуществляют в потоки сконцентрированной серной кислоты 3 и 11.
На Фиг.2 показан альтернативный вариант осуществления способа по изобретению. В реакторе А подвергают взаимодействию толуол (поток 21), азотную кислоту (поток 22) и сконцентрированную серную кислоту (поток 23) с образованием смеси мононитротолуола и динитротолуола. Полученную двухфазную реакционную смесь (поток 24) направляют затем в испаритель мгновенного действия С. Полученный при этом вторичный пар (поток 28) конденсируют в конденсаторе D, а конденсат (поток 29) направляют в фазовый сепаратор В2 и там разделяют на органическую фазу (поток 31) и водную фазу (поток 30). Водную фазу (поток 30) выводят из процесса. Полученную в испарителе мгновенного действия С жидкую фазу (сконцентрированная серная кислота и органическая фаза) (поток 25) разделяют в фазовом сепараторе В1 разделяют на органическую фазу (поток 26) и водную кислотную фазу (поток 27). Часть сконцентрированной серной кислоты (поток 32) направляют вместе с потоком 12 в вакуумный испаритель F второй стадии. Остальное количество потока 27 сводят вместе со сконцентрированной серной кислотой (поток 33) второй стадии и вновь направляют в виде потока 23 в реактор А первой стадии. Органические фазы из фазовых сепараторов В1 (поток 26) и В2 (поток 31) объединяют и направляют в реактор Е второй стадии. Органические фазы, содержащие мононитротолуол, (поток 26+31) взаимодействуют там с нитрующей кислотой с образованием динитротолуола. Разделение органической фазы, содержащей динитротолуол, и водной кислотной фазы на Фиг.2 не изображено в виде самостоятельной стадии. Органическую фазу выводят из процесса в виде потока 13 и подвергают дальнейшей обработке. Водную кислотную фазу направляют в вакуумный испаритель F второй стадии, где происходит концентрирование отработанной кислоты. Сконцентрированную серную кислоту вновь возвращают на вход в реактор Е в виде потока 11. Для компенсации потерь серной кислоты добавляют свежую серную кислоту (поток 10). При этом добавление осуществляют в поток кислоты 11.
На Фиг.3 показано особое осуществление варианта способа, представленного на Фиг.2, дополненного стадией экстракции. В экстракционном аппарате G экстрагируют водную кислотную фазу (поток 43) со второй стадии органическим конденсатом вторичного пара (поток 41) с первой стадии. В результате этого снижается содержание динитротолуола (ДНТ) водной кислотной фазы (поток 44). Органическую фазу из фазового сепаратора В1 (поток 26) и экстрактора G (поток 42) объединяют и направляют в реактор Е второй стадии. В остальном обозначения на Фиг.3 имеют то же значение, что и на Фиг.2.
Примеры
Пример 1
Использовали аппарат согласно Фиг.1. Температуры и составы потоков вещества приведены в Таблице 1. На входе в реактор А (с размерами: длина (L)=5 м, диаметр (D)=25/80 мм) на первой стадии с адиабатическим режимом интенсивно перемешивали 50,6 кг/ч толуола (поток 1), 63,0 кг/ч 68 мас.%-ной азотной кислоты (поток 2) и 1066,6 кг/ч 76,8 мас.%-ной сконцентрированной отработанной кислоты (поток 3). В снабженном перфорированными дисками для редиспергирования трубчатом реакторе температура поднималась до 131°С. При среднем времени выдержки 15 минут реакционную смесь (поток 4) разделяли в фазовом сепараторе на 78,1 кг/ч органической фазы (поток 5) и 1102,1 кг/ч водной кислотной фазы (поток 6). 74,5 мас.%-ную отработанную кислоту концентрировали в испарителе мгновенного действия С, работающем при давлении 40 мбар, до концентрации серной кислоты 76,8 мас%. Кроме того, в нижней части испарителя мгновенного действия она затем незначительно нагревалась через теплообменник. Сконцентрированную отработанную кислоту (поток 3) вновь направляли в реактор.
Отделенный в испарителе мгновенного действия С вторичный пар (35,7 кг/ч, поток 8) затем конденсировали в теплообменнике D. Вследствие высокого содержания мононитротолуола (78мас.% от содержания органики в потоке вторичного пара) в конденсаторе вторичного пара никаких отложений не образовывалось.
Отделенную в фазовом сепараторе В органику (78,1 кг/ч, поток 5) направляли на вторую стадию (реактор Е). Там в охлажденном примерно до 70°С петлевом реакторе с циркуляционным контуром Е посредством добавления нитрующей кислотной смеси получили 96,0 кг/ч динитротолуола (ДНТ) (поток 13). Образованную при этом отработанную кислоту освобождали от избытка воды на стадии концентрирования кислоты F.
Благодаря адиабатическому режиму проведения реакции стало возможным полезное использование реакционного тепла для концентрирования отработанной кислоты на первой стадии. В сравнении с изотермическим режимом проведения мононитрования вследствие экономии греющего пара примерно в 40% исследований технологической безопасности реакционной смеси (поток 4) при температуре адиабатической реакции 131°С для всего процесса не получено каких-либо признаков начавшегося разложения. При продолжительности воздействия свыше 90 минут также не обнаружилось никаких рискованных с точки зрения технологической безопасности экзотермических реакций.
Пример 2
Использовали аппарат согласно Фиг.2. Температуры и составы потоков вещества приведены в Таблице 2.
На входе в реактор А (размер: длина L=8 м, диаметр D=80 мм) на первой стадии с адиабатическим режимом интенсивно перемешивали 50,6 кг/ч толуола (поток 21), 62,9 кг/ч 68 мас.%-ной азотной кислоты (поток 22) и 736,0 кг/ч 78,6 мас.%-ной сконцентрированной отработанной кислоты (поток 23). В снабженном перфорированными дисками для редиспергирования трубчатом реакторе температура поднималась до 138°С. Реакционную смесь (поток 24) подвергали мгновенному расширению со сбросом давления посредством сопла в работающем при давлении 40 мбар испарителе мгновенного действия С, где испарялись часть воды и органические компоненты. Вторичный пар затем конденсировали в теплообменнике D. Несмотря на охлаждение холодной водой с температурой 18°С в конденсаторе вторичного пара D никаких отложений не образовывалось. Конденсат вторичного пара (поток 29) разделяли в фазовом сепараторе В2 на водную фазу (21,3 кг/ч, поток 30) и органическую фазу (57,6 кг/ч, поток 31). Органическую фазу, состоящую, главным образом, из мононитротолуола, направляли на вторую стадию (реактор Е).
Охлажденную до 94°С посредством мгновенного испарения со сбросом давления смесь кислота/динитротолуол/мононитротолуол (поток 25) освобождали от нерастворенных динитротолуола/мононитротолуола в фазовом сепараторе В1. Разделенную смесь (20,2 кг/ч, поток 26) вместе с потоком 31 направляли на динитрацию Е. Часть потока (83,8 кг/ч, поток 32) сконцентрированной до концентрации 77,1мас.% отработанной кислоты (750,4 кг/ч, поток 27) со стадии концентрирования кислоты F направляли на вторую стадию, где ее концентрировали до концентрации 93мас.%, а затем возвращали в циркуляционный контур кислоты на первой стадии (поток 33).
В процессе динитрования Е в охлажденном примерно до 70°С петлевом реакторе с циркуляционным контуром из обоих притоков 31 и 26 посредством добавления нитрующей кислотой получали 96,1 кг/ч динитротолуола (ДНТ) (поток 13). Образованную при этом отработанную кислоту освобождали от избыточного количества воды на стадии концентрирования кислоты F.
Благодаря адиабатическому режиму проведения реакции на первой стадии возможно было отделить значительную часть привнесенной воды без подвода энергии из вне.
Пример 3
Использовали аппарат согласно Фиг.3. Температуры и составы потоков веществ представлены в Таблице 3. На входе в реактор А (с размерами: длина L=5 м, диаметр D=80 мм) на первой стадии с адиабатическим режимом интенсивно перемешивали 50,6 кг/ч толуола (поток 21), 63,0 кг/ч 68 мас.%-ной азотной кислоты (поток 22) и 1306,6 кг/ч 82,4 мас.%-ной сконцентрированной отработанной кислоты (поток 23). В снабженном перфорированными дисками для редиспергирования трубчатом реакторе температура поднималась до 132°С. Реакционную смесь (поток 24) подвергали мгновенному расширению со сбросом давления посредством сопла в работающем при давлении 40 мбар испарителе мгновенного действия С, где испарялись часть воды и органические компоненты. Вторичный пар затем конденсировали в теплообменнике D. Несмотря на охлаждение холодной водой с температурой 18°С в конденсаторе вторичного пара D никаких отложений не образовывалось. Конденсат вторичного пара (поток 29) разделяли в фазовом сепараторе В2 на водную фазу (18,3 кг/ч, поток 30) и органическую фазу (56,4 кг/ч, поток 41).
Охлажденную до 109°С посредством мгновенного испарения со сбросом давления смесь кислота/динитротолуол/мононитротолуол (поток 25) освобождали от нерастворенных динитротолуола/мононитротолуола в фазовом сепараторе В1. Разделенную смесь (21,4 кг/ч, поток 26) направляли на вторую стадию (в реактор Е). Часть потока (137,5 кг/ч, поток 32) от сконцентрированной до концентрации 81,3 мас.% отработанной кислоты (1324,1 кг/ч, поток 27) со стадии концентрирования кислоты F направляли на вторую стадию, где ее концентрировали до концентрации 93мас.%, а затем возвращали в циркуляционный контур кислоты на первой стадии (поток 33).
На второй стадии в охлажденном примерно до 70°С петлевом реакторе с циркуляционным контуром Е из обоих притоков 42 и 26 посредством добавления нитрующей кислотой смеси получали 96,2 кг/ч динитротолуола (поток 13). Из образованной при этом отработанной кислоты (поток 43) удаляли часть растворенного динитротолуола (ДНТ) в экстракторе G посредством органического конденсата вторичного пара с первой стадии (поток 41). Затем отработанную кислоту (поток 44) освобождали от избыточного количества воды в процессе концентрирования кислоты F. Органическую фазу после экстракции G (59,4 кг/ч, поток 42) вместе с потоком 26 направляли на динитрование.
Благодаря адиабатическому режиму проведения реакции на первой стадии возможно было отделить значительную часть привнесенной воды без подвода энергии из вне.
Температуры и составы описанных в примерах 1-3 потоков представлены в Таблицах 1-3.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ УДАЛЕНИЯ И РЕГЕНЕРАЦИИ СМЕСЕЙ НИТРУЮЩИХ КИСЛОТ ИЗ ПРОЦЕССОВ НИТРОВАНИЯ И ПРОМЫШЛЕННАЯ УСТАНОВКА ДЛЯ НИТРОВАНИЯ | 2006 |
|
RU2356885C2 |
| СПОСОБ И УСТАНОВКА ДЛЯ РЕГЕНЕРАЦИИ ОТРАБОТАННОЙ СЕРНОЙ КИСЛОТЫ ИЗ ПРОЦЕССОВ НИТРОВАНИЯ | 2010 |
|
RU2511380C2 |
| СПОСОБ И УСТАНОВКА ДЛЯ КОНЦЕНТРИРОВАНИЯ ОТРАБОТАННОЙ СЕРНОЙ КИСЛОТЫ ИЗ ПРОЦЕССОВ НИТРОВАНИЯ | 2007 |
|
RU2404917C2 |
| СПОСОБ ИЗОТЕРМИЧЕСКОГО НИТРОВАНИЯ ОРГАНИЧЕСКИХ АРОМАТИЧЕСКИХ СОЕДИНЕНИЙ | 2006 |
|
RU2418783C2 |
| СПОСОБ ПОЛУЧЕНИЯ МОНОНИТРОТОЛУОЛОВ | 1995 |
|
RU2119909C1 |
| СПОСОБ НЕПРЕРЫВНОГО ПОЛУЧЕНИЯ МОНОНИТРОТОЛУОЛОВ | 2001 |
|
RU2274634C2 |
| СПОСОБ ПРОМЫВКИ ДИНИТРОТОЛУОЛА | 2013 |
|
RU2627308C2 |
| СПОСОБ ПОЛУЧЕНИЯ МОНОНИТРОТОЛУОЛА С ПОВЫШЕННЫМ СОДЕРЖАНИЕМ П-ИЗОМЕРА | 2007 |
|
RU2346930C1 |
| СПОСОБ НЕПРЕРЫВНОГО ИЗОТЕРМИЧЕСКОГО ПОЛУЧЕНИЯ МОНОНИТРОТОЛУОЛОВ В ПРИСУТСТВИИ ФОСФОРНОЙ КИСЛОТЫ | 2002 |
|
RU2293722C2 |
| СПОСОБ СОВМЕСТНОГО ПОЛУЧЕНИЯ ИЗОЦИАНАТОВ И ХЛОРА | 2007 |
|
RU2443682C2 |
Изобретение относится к способу получения динитротолуола двухстадийным нитрованием толуола, в котором: а) на первой стадии толуол подвергают адиабатическому взаимодействию с нитрирующей кислотой, причем, по меньшей мере, 90% вступает в реакцию и максимум 70% используемого толуола превращается в динитротолуол, непосредственно после чего органическую фазу, содержащую мононитротолуол, и водную кислотную фазу, содержащую серную кислоту, разделяют, водную содержащую серную кислоту кислотную фазу концентрируют мгновенным упариванием со сбросом давления, а полученную при этом сконцентрированную серную кислоту возвращают в реакцию на первой стадии и/или в реакцию на второй стадии и/или на концентрирование на второй стадии; б) на второй стадии органическую фазу, содержащую мононитротолуол с первой стадии, подвергают изотермическому полному взаимодействию с нитрующей кислотой смесью, непосредственно после чего органическую фазу и водную кислотную фазу, содержащую серную кислоту, разделяют и водную содержащую серную кислоту кислотную фазу концентрируют упариванием в вакууме, а полученную при этом сконцентрированную серную кислоту возвращают в реакцию на первой стадии и/или на второй стадии реакции. 2 з.п. ф-лы, 3 ил., 3 табл.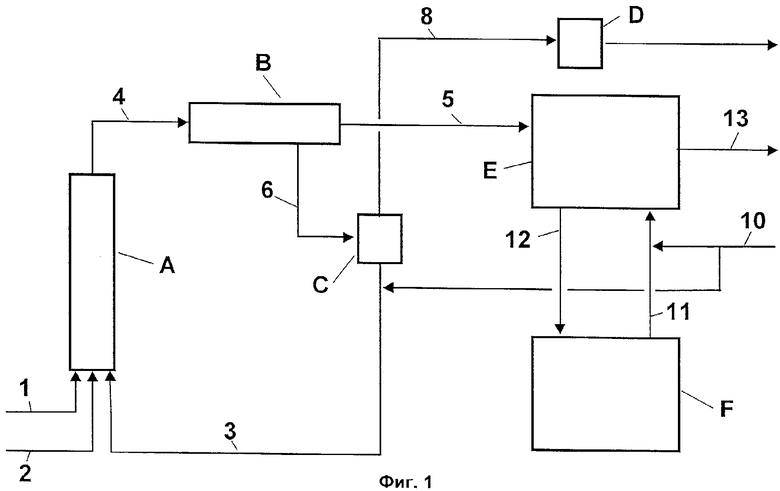
а) на первой стадии толуол подвергают адиабатическому взаимодействию с нитрующей кислотой, причем, по меньшей мере, 90% толуола вступает в реакцию и максимум 70% используемого толуола превращается в динитротолуол, непосредственно после чего органическую фазу, содержащую мононитротолуол, и водную кислотную фазу, содержащую серную кислоту, разделяют, водную содержащую серную кислоту кислотную фазу концентрируют мгновенным упариванием со сбросом давления, а полученную при этом сконцентрированную серную кислоту возвращают в реакцию на первой стадии, и/или в реакцию на второй стадии, и/или на концентрирование на второй стадии; и
б) на второй стадии органическую фазу, содержащую мононитротолуол с первой стадии, подвергают изотермическому полному взаимодействию с нитрующей кислотой, непосредственно после чего органическую фазу и водную кислотную фазу, содержащую серную кислоту, разделяют и водную содержащую серную кислоту кислотную фазу концентрируют упариванием в вакууме, а полученную при этом сконцентрированную серную кислоту возвращают в реакцию на первой стадии и/или на второй стадии.
| Вяжущее | 1980 |
|
SU903336A1 |
| DE 4309140 A1, 29.09.1994 | |||
| СПОСОБ ПОЛУЧЕНИЯ ДИНИТРОТОЛУОЛА | 1993 |
|
RU2106338C1 |

