Предпосылки изобретения
Финастерид, выпущенный в продажу под торговым наименованием PROSCAR Компанией Merck & Co., Inc. представляет собой  и является ингибитором 5α- редуктазы для использования в лечении угрей, женского гирсутизма и особенно доброкачественной простатической гиперплазии. См. патент США 4760071 (1988), включенный сюда в качестве ссылки.
и является ингибитором 5α- редуктазы для использования в лечении угрей, женского гирсутизма и особенно доброкачественной простатической гиперплазии. См. патент США 4760071 (1988), включенный сюда в качестве ссылки.
Синтез финастерида, раскрытый в патенте США 4760071, включает реакцию 
с т-бутиламином. Другой синтез финастерида, описанный в Synthetic Communications 30 (17), стр. 2683-90 (1990), включенном сюда в качестве ссылки, включает реакцию 17-ацилимидазола 4-аза-5α-андрост-I-ен-3-она с т-бутиламином.
Однако обе эти реакции требуют использования гетероциклических ароматических аминов, которые являются дорогостоящими, а их применение проблематичным, если учитывать их токсичность и опасность для окружающей среды. Оба эти промежуточных соединения получают из 17β - карбоновой кислоты.
Реакция Bodroux, описанная F.Bodroux в ссылках, 33, 831 (1905), 35, 519 (1906), 1, 912 (1907), Bull. Soc. Chim. 138, 1427 (1904), 140, 1108 (1905), 142, 401 (1906), раскрывает реакцию галоидмагниевых солей аминов со сложными эфирами. Однако там нет ни описания, ни изучения возможности применения реакции в случае стерически блокированного амина, например т-бутиламина со стерически блокированным сложным эфиром, таким, как I.
В данной области техники нужен способ синтеза финастерида, который был бы безопасен для окружающей среды, нетоксичен и не использовал ароматический гетероциклический амин. Предпочтительно исходным соединением может быть 17-бета сложный эфир (I), который избавит способ от одной стадии при получении вышеупомянутых гетероциклических промежуточных соединений.
В настоящем изобретении предложен способ получения финастерида II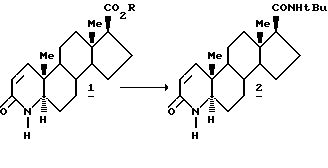
где
R - C1-C10 линейный, разветвленный или циклический алкил, незамещенный или замещенный одним или несколькими фенилами, включающий стадии:
(1) контактирования сложного эфира I с т-бутиламино магний галидом, в котором молярное соотношение т-бутиламино магний галида и сложного эфира составляет по меньшей мере около 2:1, в инертном органическом растворителе в атмосфере инертного газа, (2) выдерживание реакционной смеси при температуре по меньшей мере 10oC и (3) выделение продукта финастерида II.
Предлагаются также промежуточные соединения, используемые для синтеза финастерида. Кроме того, предлагается способ синтеза, включающий выделение и кристаллизацию определенных полиформных кристаллических форм финастерида, а также сами полиморфные формы.
Подобное описание изобретения
Мы выявили, что  может реагировать с т-бутил амином вместе с реагентом алифатическим/арил магний галидом, например этил бромидом магния, где реагент магний галид и т-бутил амин присутствуют в молярном соотношении по меньшей мере около 2:1 к сложному эфиру (I), чтобы получить финастерид II с хорошим выходом. Реакция между алифатическим/арил магний галидом и т-бутиламином дает т-бутиламино магний галид. Один моль т-бутиламино магний галид можно использовать для депротонирования сложного эфира A-кольца лактама, растворяя таким образом стероид, второй моль требуется для реакции амидирования и третий моль можно использовать для депротонирования вновь образованного амида. В другом случае сложный эфир (I) можно депротонировать реагентом Гриньяра отдельно, а затем подвергнуть реакции с двумя молями т-бутиламино магний галид для проведения амидирования.
может реагировать с т-бутил амином вместе с реагентом алифатическим/арил магний галидом, например этил бромидом магния, где реагент магний галид и т-бутил амин присутствуют в молярном соотношении по меньшей мере около 2:1 к сложному эфиру (I), чтобы получить финастерид II с хорошим выходом. Реакция между алифатическим/арил магний галидом и т-бутиламином дает т-бутиламино магний галид. Один моль т-бутиламино магний галид можно использовать для депротонирования сложного эфира A-кольца лактама, растворяя таким образом стероид, второй моль требуется для реакции амидирования и третий моль можно использовать для депротонирования вновь образованного амида. В другом случае сложный эфир (I) можно депротонировать реагентом Гриньяра отдельно, а затем подвергнуть реакции с двумя молями т-бутиламино магний галид для проведения амидирования.
В другом альтернативном варианте можно вначале подготовить заранее в том же или отдельном реакторе т-бутиламино магний галид при температуре окружающей среды и затем подвергнуть взаимодействию с 4-аза-стероидным сложным эфиром I в молярном соотношении по меньшей мере 3:1 реагента галида к сложному эфиру, предпочтительно с последующим нагреванием до около 100oC. Еще в одном варианте т-бутил магний галид можно получить в том же или отдельном реакторе в молярном соотношении к сложному эфиру I 2:1 и затем подвергнуть взаимодействию со сложным эфиром I, который предварительно был введен во взаимодействие с тем же или другим реагентом Гриньяра в молярном соотношении 1:1 для депротонирования и растворения сложного эфира.
В одном конкретном варианте этого изобретения способ получения финастерида II включает стадии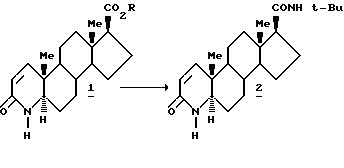
(1) взаимодействия в одном реакторе 4-аза-стероидного сложного эфира I с т-бутил амином и алифатическим/арил магний галидом в инертном органическим растворителе в атмосфере инертного газа при температуре в диапазоне от -20o до 10oC с перемешиванием реакционной смеси для получения т-бутил магний галида in situ по меньшей мере в молярном соотношении к сложному эфиру I 3: 1, без взаимодействия сложного эфира с алифатическим/арил магний галидом, чтобы избежать получения нежелательных продуктов кетона и спирта; (2) нагревания реакционной массы до 15 - 100oC для реакции сложного эфира с т-бутил амино магний галидом и (3) выделения указанного продукта финастерида II (где т-Bu означает третичный бутил).
Промежуточная галидная магний соль 4-аза-стероида I имеет следующую формулу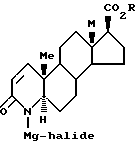
в которой
R - C1-C10 линейный, разветвленный или циклический алкил, незамещенный или замещенный одним или несколькими фенилами.
Исходный сложный эфир I и его синтез описаны в патенте США 4760071. Соединение, используемое для получения I, представляет собой известный стероидный сложный эфир, который насыщен в 1, 2 положении, который дегидрогенируется дегидрогенирующим агентом, таким, как бензолселеновый ангидрид в хлорбензоле при температуре кипения.
Исходным сложным эфиром может быть соединение, в котором R - C1-C10 линейный, разветвленный или циклический алкил, в котором алкильная цепь необязательно замещена одним или несколькими фенилами. Эфирная часть включает, например, метил (Me), этил (Et), пропил, бутил, пентил, гексил, гептил, октил, нонил, децил, изо-пропил, циклогексил и др., и бензил, -CH2CH2-фенил, -CH2CH2CH2-фенил и пр. R - алкил с прямой цепью, который незамещен или монозамещен фенилом и лучше - метилом. Можно также использовать алкильные группы с более длинной цепью, но этого не требуется.
т-Бутил амин и алифатический/арил магний галид каждый используется в молярном соотношении к сложному эфиру (I) по меньшей мере 3:1, чтобы получить молярное соотношение 3:1, а предпочтительно 3,5:1 - 5,5:1 т-бутил амин магний галида к сложному эфиру (I), с тем, чтобы обеспечить хорошее и полное превращение (I) в (II) и свести к минимуму примеси. Реакции можно наглядно изобразить как реакцию трех молей т-бутиламино магний галида, образованного реакцией между алифатическим/арил магний галидом и т-бутиламином, с одним молем сложного эфира I. В другом случае реакцию можно изобразить как реакцию двух молей т-бутил магний галида с одним молем галид магниевой соли сложного эфира (I).
Алифатический/арил магний галид доступен и может быть выбран следующим образом:
(1) алифатическая/арильная часть представляет собой C1-C18 линейный, разветвленный или циклический алкил, например метил, этил, пропил, изопропил, н-бутил, втор-бутил, т-бутил, гексил, циклогексил, октил, децил, додецил, тетрадецил, октадецил, бензил, аллил, винил, этинил и др. и (2) арильная часть представляет собой фенил, или моно-, ди- или три-замещенный фенил, в котором заместители могут включать C1-C4 алкил, C1-C4 алкокси, например, метил, метокси фторо и др.
Галид является хлоридом, бромидом, фторидом или йодидом, предпочтительно бромидом или хлоридом, еще более предпочтительно бромидом. Предпочтителен этил магний бромид. Термин "алифатический/арил магний галид" охватывает алифатический магний галид и арил магний галид.
Используемым инертным растворителем является обычный растворитель Гриньяра и может быть C4-C8 линейным или циклическим эфиром, включающим диэтиловый эфир, ди-н-бутиловый эфир, диметоксиэтан, тетрагидрофуран, диоксан и другие. Растворитель должен быть сухим в условиях реакции, которая обычно проводится в атмосфере инертного газа, например, сухого азота, с перемешиванием.
Первоначально реакция проводится при температуре, достаточной, чтобы достичь образования продукта, и может проводиться, например, между около -40o и 40oC, лучше при пониженной температуре, например от около -20o до 10oC в течение
(1) реакции т-бутиламина и алифатического/арил магний галида с образованием т-бутиламино магний галида и
(2) реакции между сложным эфиром I и т-бутил амино магний галидом или реагентом Гриньяра с образованием магниевой соли галида сложного эфира I.
После этого реакционная смесь перемешивается и выдерживается при температуре, достаточной, чтобы могла пройти реакция амидирования. Реакционной смеси обычно дают дойти до температуры, например, около 10oC или около комнатной температуры и в дальнейшем нагревают до 100oC или до температуры кипения растворителя. Обычно время нагревания составляет от 2 до 12 часов.
В другом случае т-бутиламино магний галид можно получить заранее, например, при температуре окружающей среды и затем провести реакцию с 4-аза- сложным эфиром стероида (I) при комнатной температуре.
Для обработки неочищенного финастерида используются традиционные способы, также как устройство для его проведения. В основном, хроматография на силикагеле и/или кристаллизация из подходящего растворителя, например метиленхлорида/этилацетата или уксусной кислоты/воды, используются для очистки финастерида.
Порядок добавления сложного эфира, т-бутиламина и алифатического арил магний галида можно изменить и сделать обратным с хорошими результатами. В частности т-бутил амин может вначале прореагировать с алифатически/арил магний галидом, чтобы предварительно образовать т-бутиламино магний галид до взаимодействия со сложным эфиром I.
Следующие примеры иллюстрируют способ, заявленный здесь, и их не следует рассматривать как ограничивающие объем раскрытого здесь изобретения.
Пример 1.
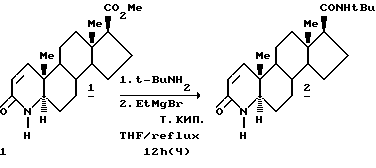
В колбу, снабженную верхней мешалкой, входом для азота и обратным холодильником, помещалось 840 мл сухого ТГФ и 20.0 г ΔI- метилового эфира (I). Полученная суспензия охлаждалась до -5 - -10oC и добавлялось 27.6 мл т-бутиламина. При поддержании температуры реакционной смеси ниже 10oC добавлялся раствор этилмагний бромида в ТГФ (122 мл, 2 M). Реакционная смесь нагревалась при температуре кипения 12 часов и добавлялась к холодному (10oC) раствору 25% хлорида аммония в воде. Смесь нагревалась до 25oC и оставлялась для осаждения. Раствор ТГФ отделялся, концентрация проводилась атмосферной перегонкой до 200 мл и продукт кристаллизовался добавлением приблизительно 600 мл водного раствора HCl. Полученное твердое вещество белого цвета выделялось фильтрацией и высушивалось при 70oC в вакууме с получением 21.7 г (выход 97%) финастерида. Продукт финастерида можно очищать традиционными способами, например перекристаллизацией из метиленхлорида/этилацетата или уксусной кислоты/воды, т.п. 261oC.
Пример 2.
В колбу, снабженную верхней мешалкой, входом для азота и обратным холодильником помещалось 516 мл сухого ТГФ и 27.6 мл т-бутиламина. Раствор охлаждался до 10oC и при поддержании температуры реакции ниже 30oC добавлялось 244 мл 1 M бромида этилмагния в ТГФ. Добавлялась суспензия, содержащая 10.0 г ΔI- метилового эфира I в 100 мл сухого ТГФ. Реакционная смесь нагревалась при температуре кипения 4-6 часов и добавлялась к холодному (10oC) раствору 25% хлорида аммония в воде. Смесь нагревалась до 25oC и оставлялась для осаждения. Раствор ТГФ отделялся и концентрировался атмосферной перегонкой до 200 мл и продукт кристаллизовался добавлением 200 мл водного раствора HCl. Полученное твердое вещество белого цвета выделялось и высушивалось при 70oC в вакууме с получением 21.6 г (выход 97%) финастерида.
Полиморфизм можно определить как способность одного и того же химического вещества существовать в разных кристаллических структурах. Их называют полиморфы, полиморфные модификации или формы. Финастерид, как было определено, существует по меньшей мере в двух полиморфных несольватированных формах, форме I и форме II, каждую из которых можно получать при тщательном соблюдении условий кристаллизации.
Полиморфную форму I можно получить:
(1) кристаллизацией из смеси финастерида в органическом растворителе и 0% или больше по весу воды, так, чтобы количество органического растворителя и воды в смеси было достаточным, чтобы был превышен предел растворимости несольватированной формы финастерида формы I в смеси и несольватированная форма финастерида была менее растворимой, чем любая другая форма финастерида в смеси: (2) выделением полученной твердой фазы и (3) удалением из нее растворителя.
Органические растворители, используемые в этом способе, включают любые растворители, в которых можно растворить финастерид. Некоторые примеры органических растворителей включают, например, тетрагидрофуран (ТГФ), органические кислоты, этилацетат (Et OAc), толуол, изо-пропилацетат и другие. Кроме того, органический растворитель может быть тем, который известен в области техники как водосмешивающийся. Термин "водосмешивающиеся растворители" здесь включает растворители, которые не образуют двухфазную систему с водой в условиях, достаточных для кристаллизации указанных полиморфов. Например, водосмешивающиеся растворители включают, но не ограничены ТГФ и органическими кислотам, такими, как муравьиная кислота, уксусная кислота, пропионовая кислота и другие. Кроме того, органическим растворителем может быть растворитель, который известен в области как водонесмешивающийся. Термин "водонесмешивающиеся растворители" здесь включает растворители, которые образуют двухфазную систему с водой в условиях, достаточных для кристаллизации данных полиморфов. Например, водонесмешивающиеся растворители включают, но не ограничиваются толуолом, этилацетатом, изо-пропилацетатом и другими.
Если используются водосмешивающиеся растворители в вышеописанном способе, полиморфную форму I финастерида можно получить использованием относительно влажной смеси растворителей, например, если использовать ледяную уксусную кислоту для получения формы I при температуре окружающей среды около 25oC, можно использовать около 83% или больше по весу воды.
Если использовать органические растворители, считающиеся водонесмешивающимися, например толуол, этилацетат, изо-пропилацетат и другие, данный способ получения формы I проводится в относительно сухом растворителе. Например, чтобы получить форму I финанстерида из этилацетата/воды, количество используемой воды самое большее должно быть около 3,5 мг/мл, а из смеси изо-пропилацетата/воды не должно быть больше около 1.6 мг/мл, при температуре окружающей среды около 25oC для обоих растворителей.
Примеры кристаллизации, приведенные выше, даны для методик, проводимых при температуре окружающей среды. Специалистам данной области известно, что количество воды, необходимое для получения формы I в любой данной смеси органического растворителя, будет меняться с температурой, так как изменения температуры изменят растворимость вещества. Например, если использовать изо-пропилацетат для получения формы I, при указанных температурах количества воды будут следующими:
Температура - Кол-во воды
1,4oC - 0,8 мг/мл или меньше
6oC - 0,9 мг/мл или меньше
12oC - 1,0 мг/мл или меньше
18oC - 1,3 мг/мл или меньше
Полиморфную форму I можно также получить нагреванием полиморфной формы II финастерида по меньшей мере до около 25oC в воде или органическом растворителе в течение времени, достаточном для полного преобразования формы II в форму I, и выделением полученной твердой фазы, например, фильтрацией.
Полиморфную форму II можно получить: (1) кристаллизацией из смеси финастерида в органическом растворителе и воде, так, чтобы количество органического растворителя и воды в смеси было достаточным, чтобы был превышен предел растворимости сольватированной формы финастерида в смеси, и сольватированная форма финастерида была менее растворимой, чем любая другая форма финастерида в смеси; (2) выделением полученной твердой фазы, и (3) удалением из нее растворителя.
Органические растворители, используемые в этом способе, те же, что и выше, и также включают водосмешивающиеся и водонесмешивающиеся растворители. Однако при получении формы II из водосмешиваемого растворителя вес.% воды, используемой в растворительной смеси, будет меньше, чем при получении формы I из того же водосмешивающегося растворителя. Например, чтобы получить форму II финастерида из смеси ледяной уксусной кислоты/воды, вес.% воды в растворительной смеси будет менее 83% при температуре окружающей среды около 25oC.
Кроме того, если для получения формы II использовать водонесмешивающийся растворитель, такой, как этилацетат или изо-пропилацетат, количество воды в растворительной смеси будет больше используемой в получении формы I из того же органического растворителя. Например, чтобы получить форму II финастерида из смеси этилацетата/воды, количество используемой воды будет больше 3,5 мг/мл, и из смеси изо-пропилацетат/вода, это количество будет больше 1,6 мг/мл, в обоих случаях при температуре окружающей среды около 25oC. Как объяснялось выше, специалистам понятно, что изменения температуры могут повлиять на количество воды, необходимое для получения формы II из любой данной растворительной смеси.
Полиморфную форму II можно также получить нагреванием полиморфной формы I финастерида до температуры по меньшей мере около 150oC в течение времени, достаточного для полного превращения формы I в форму II, например около часа, и выделением полученной твердой фазы.
Следующие примеры иллюстрируют способы получения полиморфных форм I и II финастерида (Proscar® ,MK 906) и некоторые характеристики. Примеры даются для дальнейшей подробной иллюстрации способа получения соединений настоящего изобретения. Примеры никоим образом не ограничивают объем данного изобретения и их не следует рассматривать как ограничивающие его.
Пример 3.
Форму I финастерида можно получить растворением финастерида в ледяной уксусной кислоте (около 100 мг/мл) и добавлением воды с перемешиванием, до тех пор, пока вес.% воды не будет равным или превысит 84%. Полученная твердая фаза собирается фильтрацией и высушивается в вакууме при температуре около 50oC. Полученная форма I характеризуется кривой дифференциальной сканирующей калориметрии (DSC) при скорости нагревания 20oC/мин и в закрытом резервуаре, демонстрируя минимальную эндотерму с пиковой температурой около 232oC, температурой начала экстраполяции около 223oC и теплом около 11 Дж/г и максимальную эндотерму расплава с пиковой температурой около 261oC, температурой начала экстраполяции около 258oC с соответствующим теплом около 89 Дж/г. Рентгеновская картина дифракции порошка характеризуется d-интервалами в 6.44, 5.69, 5.36, 4.89, 4.55, 4.31, 3.85, 3.59 и 3.14. FT-ИК спектр (в KBr) показывает полосы на 3431, 3237, 1692, 1666, 1602 и 688 см-1. Растворимость в воде и циклогексане при 25oC составляет 0.05+ 0.02 и 0.27 + 0.05 мг/г соответственно.
Кроме того, форму I финастероида можно получить перекристаллизацией из сухого (H2O < 1 мг/мл) этилацетата и изопропилацетата при температуре окружающей среды (25oC). Выделенное твердое вещество высушиваются в вакууме при 50oC и имеет те же самые физические характеристики, что указаны выше.
Кроме того, форма I была получена перемешиванием формы II в течение ночи в сухом толуоле при комнатной температуре и выделением полученной твердой фазы. Форма I была также получена перемешиванием формы II в течение ночи в сухом ацетонитриле при окружающей температуре и выделением полученной твердой фазы.
Пример 4.
Форму II финастерида можно получить растворением финастерида в ледяной уксусной кислоте (около 100 мг/мл) и добавлением воды с перемешиванием до тех пор, пока вес.% воды не будет равным 75%, но не будет превышать 80%. Полученная твердая фаза собирается фильтрацией и высушивается в вакууме около 100oC. Полученная форма II характеризуется DSC кривой при скорости нагревания в 20oC/мин и в закрытом резервуаре, демонстрируя одну эндотерму расплава с пиковой температурой в около 261oC, температурой начала экстраполяции около 258oC с соответствующим теплом около 89 Дж/г. Рентгеновская картина дифракции порошка характеризуется d-интервалами в 14,09; 10,36; 7,92; 7,18; 6,40; 5,93; 5,66; 5,31; 4,68; 3,90; 3,60 и 3,25. FT-ИК спектр (в KBr) показывает полосы на 3441, 3215, 1678, 1654, 1597, 1476 и 752 см-1. Растворимость в воде и циклогексане при 25oC составила 0,16+0,02 и 0,42+0,05 мг/г соответственно.
Кроме того, форму II финастерида можно получить перекристаллизацией из этилацетата, содержащего от 3,5 до 30 мг/мл воды, или из изопропилацетата, содержащего от около 1,6 до 15 мг/мл воды при температуре окружающей среды (25oC). Выделенные твердые вещества высушиваются в вакууме при около 80oC и обладают теми же физическими характеристиками, что указаны выше.
Форму II можно также получить нагреванием формы I до 150oC, выдерживанием в течение часа и охлаждением опять до комнатной температуры. Такая форма II имеет те же физические характеристики как представлены выше.
В изобретении раскрывается новый способ получения финастерида, 17β -[N-трет. бутилкарбамоил] -4-аза- 5α -андрост-1-ен-3-он (2), который включает реагирование сложного эфира 1], 17β -карбоалкокси-4-аза- 5α -андрост-1-ен-3-она с т-бутиламино магний галидом, присутствующим в молярном соотношении к сложному эфиру по меньшей мере 2:1, при этом продукт образуется из т-бутил амина и алифатического/арил магний галида при температуре окружающей среды в инертном органическом растворителе в атмосфере инертного газа с последующим нагреванием и выделением продукта финастерида. Раскрываются также две полиморфные кристаллические формы I и II финастерида и способы их получения. Финастерид является ингибитором 5α- редуктазы. Способ синтеза финастерида безопасен для окружающей среды и нетоксичен.

6 с. и 29 з.п.ф-лы.
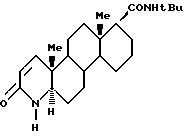
отличающийся тем, что включает 1) контактирование сложного эфира формулы I
где R представляет собой линейный, разветвленный (C1-C10) алкил,
с т-бутиламиномагний галидом, где молярное соотношение т-бутиламиномагний галида и сложного эфира составляет по меньшей мере около 2:1, в инертном органическом растворителе в атмосфере инертного газа; 2) выдерживание реакционной смеси при температуре по меньшей мере 10oC и 3) выделение продукта финастерида формулы II.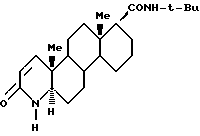
который существует в полиморфной форме I, имеющий характеристики полос абсорбции, полученный от рентгеновской дифракции порошка со спектральными d-интервалами в 6,44; 5,69; 5,36; 4,89; 4,55; 4,31; 3,85; 3,59 и 3,14, кривой дифференциальной сканирующей калориметрии, со скоростью нагревания 20oC/мин, которая демонстрирует минимальную эндотерму с пиковой температурой около 232oC и температуру начала экстраполяции около 223oC с соответствующим теплом около 11 Дж/г и которая демонстрирует максимальную эндотерму расплава с пиковой температурой около 261oC и температуру начала экстраполяции около 258oC с соответствующим теплом около 89 Дж/г FT-ИК-спектр (вKBr), с полосами на 3431, 3237, 1692, 1666, 1602 и 688 см-1 и растворимость в воде и циклогексане при 25oC 0,05+0,02 и 0,27+0,05 мг/г соответственно.
которое существует в полиморфной форме I, подученное нагреванием формы II финастерида в органическом растворителе или в воде до температуры по меньшей мере около 25oC и выделением полученной твердой фазы.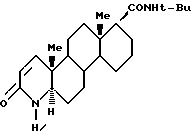
который существует в полиморфной форме II, имеющий характеристики полос абсорбции, полученные от рентгеновской дифракции порошка со спектральными d-интервалами в 14,09; 10,36, 7,92; 7,18; 6,40; 5,93; 5,66; 5,31; 4,68; 3,90; 3,60 и 3,25, кривой дифференциальной сканирующей калориметрии, со скоростью нагревания 20oC/мин, которая демонстрирует одну эндотерму расплава с пиковой температурой около 261oC и температурой начала экстраполяции около 258oC с соответствующим теплом около 89 Дж/г, FT-ИК спектром (в KBr) с полосами на 3441, 3215, 1678, 1654, 1597, 1476 и 752 см-1, и растворимостью в воде и циклогексане при 25oC 0,16+0,02 и 0,42+0,05 мг/г соответственно.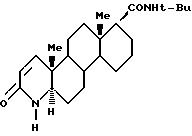
которое существует в полиморфной форме II, полученное способом, включающим: 1) кристаллизацию из смеси финастерида в органическом растворителе и воде так, что количество органического растворителя и воды в смеси достаточное, чтобы был превышен предел растворимости сольватированной формы финастерида в смеси, и сольватированная форма финастерида была менее растворима, чем любая другая форма финастерида в смеси; 2) выделение полученной твердой фазы; 3) удаление из нее растворителя.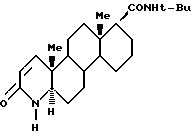
которое существует в полиморфной форме II, полученное нагреванием формы I финастерида до температуры по меньшей мере около 150oC.
Приоритет по пунктам:
19.11.92 - по пп.1-12;
29.01.93 - по пп.13, 14, 19, 20, 24-27, 31, 32;
05.11.93 - по пп. 15-18, 21-23, 28-30, 33-35.
| US, A, 4760071, (Merck & Co Inc.), 1988 | |||
| A | |||
| Bhattacharja et al | |||
| Practical Synthesis of Proscar (R), Synthetic Comm | |||
| Способ обработки медных солей нафтеновых кислот | 1923 |
|
SU30A1 |
| EP, A, 0200855, (Merck & Co Inc), 1986 | |||
| Физер Л., Физер М | |||
| Стероиды, М.: Мир, 1964, с.257-258. | |||
