Настоящее изобретение касается нового способа каталитического окисления соединений, содержащих аллильную группу, в котором используют катализаторы на основе рутения. Способ является, как правило, пригодным для окисления соединений, содержащих аллильные водороды или спирты, и в частности для окисления Δ-5-стероидных соединений.
Предпосылки создания изобретения
Основным посредником андрогенной активности у некоторых органов-мишеней, например, простаты, является 5-α- дигидpoтecтocтeрон ("DHT"), локально образованный в органе-мишени посредством действия 5-α- редуктазы, которая превращает тестостерон в DHT. Результатом гиперандрогенной стимуляции, вызванной чрезмерной аккумуляцией тестостерона ("Т") или подобных андрогенных гормонов в метаболической системе, являются определенные нежелательные физиологические проявления, такие как обыкновенные юношеские угри, себорея, гирсутизм женщин, андрогенная алопеция, которая включает алопецию у женщин и мужчин, и доброкачественная гиперплазия простаты. Ингибиторы 5-α- редуктазы будут служить для предотвращения или уменьшения симптомов гиперандрогенной стимуляции в этих органах. Смотри, в особенности, патент США N 4377584, опубликованный 22 марта 1983, и N 4760071, опубликованный 26 июля 1988, правопреемниками которых является Мерк энд Ко., Инк. Кроме того, в настоящее время известно, что существует второй изофермент 5α- редуктазы, который взаимодействует с кожными тканями, главным образом в скальпированных тканях. Смотри, например, g.Horris, et al. Proc. Natl. Acad. Sci. USA, Vol. 89, pp. 10787-10791 (Nov. 1992). Изофермент, который преимущественно взаимодействует в кожных тканях, обозначают обычно как 5α- редуктаза 1 (или 5α-редуктаза типа 1), в то время как изофермент, который преимущественно взаимодействует в тканях простаты, обозначают обычно как 5α-редуктаза 2 (или 5α- редуктаза типа 2).
Окисление Δ-5-стероидных алкенов в соответствующие еноны является важной стадией в синтезе стероидных конечных продуктов, пригодных в качестве ингибиторов 5α-редуктазы. Для окисления аллильных групп ранее использовали окисления, основанные на применении хрома, но они являются неприемлемыми с точки зрения окружающей среды, и при этом необходима силикагельная хроматография. Настоящее изобретение обеспечивает усовершенствованный альтернативный способ окисления Δ-5- стероидных алкенов, который является удобным для реализации и приемлемым для окружающей среды. Кроме того, выход и чистота окисленных промежуточных соединений, полученных настоящим способом, соответствуют выходу и чистоте, которые были получены с использованием ранее известных способов окисления, или превосходят их.
Сущность изобретения
Новый способ этого изобретения включает окисление соединений, содержащих аллильную спиртовую группу или аллильные водороды, в соответствующие еноны с использованием катализатора на основе рутения в присутствии гидропероксида. Это изобретение, в частности, включает превращение Δ-5-стероидных алкенов в Δ-5-7-кетостероидные алкены с использованием катализатора на основе рутения в присутствии гидропероксида. В качестве примера нового способа может служить его следующий вариант:
Соединения формулы II являются пригодными в качестве промежуточных соединений 7β- замещенных 3-кето-4-азастероидных соединений, пример таких, которые являются ингибиторами 5α- редуктазы. Ингибиторы 5α- редуктазы являются пригодными при лечении гиперандрогенных расстройств, таких как доброкачественная гиперплазия простазы, обыкновенные юношеские угри, себорея, гирсутизм у женщин, андрогенная алопеция, и при предотвращении и лечении рака предстательной железы.
Подробное описание изобретения
Новый способ этого изобретения включает обнаружение того, что стероидные соединения, содержащие C5-C6 двойную связь (т.е. Δ-5- стероидные алкены), могут быть окислены до соответствующих 7-кетосоединений путем обработки гидропероксидом в присутствии катализатора на основе рутения.
С использованием такого же способа соединения, содержащие аллильную спиртовую группу, могут быть подобно окислены в их соответствующие котоны. Для справки: стандартная нумерация стероидной структуры с незамещенным ядром и буквенное обозначение колец является следующим: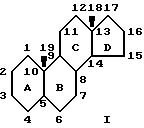
К удивлению было обнаружено, что настоящий способ окисления можно реализовать с использованием любого катализатора на основании рутения. В данной области известно много катализаторов, основанных на рутении, и такие катализаторы на основе рутения могут быть использованы в этом способе. Примеры катализаторов на основе рутения, которые могут быть использованы в этом способе, включают: RuW11O39SiNa5, RuCl3, RuCl2(PPh3)3, Ru(ацац)3, Ru(диметилглиоксимато)2 (PPh3)2, RuO2, Ru(TPP) (CO)(THF), Ru(бипи)2Cl2, Ru(TPP)(CO)(THF), Ru/C и K5SiRu(H2O)W11O39, однако, при этом не ограничиваются только ими. "TPP" представляет тетрафенилпорфин; "ацац" является ацетилацетонатом; "бипи" является бипиридином. Катализаторы на основе рутения описаны, например, в источнике J.Am. Chem., Soc., Vol. 112, 6025 (1990); S-I.morahashi, Tetrahedron Letters, Vol. 34, 1299 (1993).
Используют, в частности, катализатор, основанный на вольфрамате натрий-рутения и более конкретно RuW11O39SiNa5. В этой реакции применяют каталитическое количество рутениевого соединения. Специалисты в данной области знают об использовании каталитических количеств реакционных катализаторов, которое может быть использовано, может изменяться в зависимости от масштаба реакции и особенностей применяемого катализатора на основе рутения. Примерное количество катализатора на основе рутения находится в диапазоне от около 0,05 до 5 мол. % и в особенности от около 0,5 мол.% катализатора на мол.% исходного материала, но при этом также являются приемлемыми варианты, находящиеся за пределами этого диапазона.
Исходный алкен обрабатывают гидроперекисью в присутствии катализатора на основе рутения для превращения в соответствующий енон. В данной области известно много гидроперекисей, и любая такая гидроперекись может быть использована в этом способе. Примеры гидроперекисей, которые могут быть использованы в этом способе, включают гидроперекись трет-бутила (трет-BuOOH), гидроперекись кумена, перекись водорода и перекись бензоила, но при этом не ограничиваются только ими, предпочтительным является трет-BuOOH. В способе используют количество гидроперекиси, достаточное для полного окисления, например, по крайней мере, около 2 молей и предпочтительно от около 8 до 10 молей на моль исходного материала. На этой стадии способа может быть использован любой коммерчески пригодный растворитель или его комбинации, например алканы, простые эфиры, спирты, галогенированные растворители, вода и т. д. Примеры множества растворителей, которые могут быть использованы, включают толуол, этилацетат, гексан, хлорбензол, гептан, трет-бутилметиловый эфир (МТВЕ), бензол, ацетонитрил, циклогексан, метиленхлорид, 1,2-дихлорэтан и трет-бутиловый спирт (трет-BuOH), или их комбинации, но при этом не ограничиваются только ими. Когда в качестве катализатора применяют RuW11O39SiNa5, предпочтительным растворителем является гептан. При использовании в качестве катализатора RuCI2(PPh3)3 предпочтительными растворителями являются хлорбензол или бензол.
Способ окисления можно осуществлять при температуре между от около -20oC до температуры перегонки используемого растворителя, например около 100oC, и в особенности между от около 5oC и до примерно 50oC, и наиболее предпочтительно при около 15oC. Реакцию можно осуществлять при любом pH, и в частности при кислых значениях pH, и в особенности при pH около 1. Перед добавлением трет-BuOOH путем добавления водного раствора кислоты, такой как серная кислота, можно отрегулировать pH реакционной смеси. Реакцию предпочтительно проводят в инертной атмосфере, например азоте или аргоне, хотя это и не является необходимым.
Δ-5- стероидные алкены, которые могут быть использованы в этом способе, являются известными в данной области. Например, смотри такие, которые перечислены и доступны от Сигма Кэмикал Ко.
Один вариант настоящего изобретения включает стадию обработки соединения формулы 1:
гидроперекисью в присутствии катализатора на основе рутения в растворителе для образования соединения формулы II: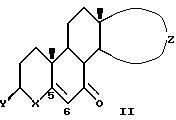
в которой Y является гидроксигруппой, этерифицированной гидроксигруппой, кето или этиленкеталем, X является -CH2-, -NH- или -N(CH3) - или -N-2,4-диметоксибензилом, и Z является
На реакцию окисления не оказывает влияние заместитель в 16- или 17-положении стероида и поэтому "A" может быть любым синтетически возможным заместителем. Гибкость и широкая применимость этого способа иллюстрируется тем фактом, что в нем не ограничен выбор заместителя в 16 и 17-положениях стероида. Характерные примеры "A" включают: -H; кето(=O); защищенную гидрокси, например, диметил-трет-бутилсилилокси, триметилсилилокси, триэтилсилилокси, три-изопропилсилилокси, трифенилсилилокси; ацетат, гидрокси; защищенную амино, например, ацетиламино; амино; C1-10 алкил, например, метил, этил, 1,5-диметилгексил, 6-метилгепт-2-ил-холестанил с 17-боковой цепью, прегнан или стигмастерол с 17-боковой цепью; арилзамещенный C1-10-алкил, например омега-фенилпропил, 1-(хлорфенокси)этил; арилкарбамоилзамещенный C1-10алкил, например 2-(4-пиридинилкарбамоил)этил; C1-10алкилкарбонил, например, изобутилкарбонил; арилкарбонил, например, фенилкарбонил; эфирозамещенный C1-10-алкил, например, 1-метоксиэтил, 1-этоксиэтил; кетозамещенный C1-10-алкил, например, 1-кетоэтил; гетерозамещенный C1-10-алкил, например омега-(4-пиридил) бутил; карбокси; сложные эфиры карбоновых кислот, например, сложные эфиры C1-10 -алкилкарбоновых кислот, такие как карбометокси; карбоксамиды, например C1-10-алкилкарбоксамиды или аралкилкарбоксамиды, такие как: N, N-диизо-пропилкарбоксамид, н-трет-бутилкарбоксамид или N-(дифенилметил) карбоксамид; карбаматы, такие как C1-10 -алкилкарбаматы, особенно трет-бутилкарбамат; замещенные или незамещенные производные анилида, в которых фенил может быть замещен 1-2-заместителями, выбранными из этила, метила, трифторметила или галогена (F, Cl, B, I); мочевины, например, C1-10-алкилкарбониламиномочевины, такие как трет-бутилкарбониламиномочевина;
C1-10-алкилкарбониламино, например, трет-бутилкарбониламино; простые эфиры, например, н-бутилокси, этиленкеталь; замещенные и незамещенные ариловые эфиры, такие как: хлорфенокси, метилфенилокси, фенилокси, метилсульфонилфенилокси, пиримидинилокси и т.п.
Термин "алкил" включает алкильные группы как с прямой, так и с разветвленной цепью, и "арил" включает фенил, пиридинил и пиримидинил.
Специалистам в данной области известны защитные группы для гидрокси и амино, при этом могут быть использованы любые такие защитные группы. Подходящими защитными группами для гидрокси являются, например, ацетатные, бензоатные, эфирные и силильные защитные группы. Стандартные силильные защитные группы имеют общую формулу: -Si(Xa)3, где каждая Xa-группа независимо является алкильной или арильной группой, и включают, например, триметилсилил, триэтилсилил, три-изо-пропилсилил, трифенилаллил, а также трет-бутил-ди (Xв)-силил, где Xв является метилом, этилом, изопропилом или фенилом (Ph). Стандартные защитные группы для амино имеют общую формулу: -C(O)-Xc, где Xc является алкилом, арилом, O-алкилом или O-арилом, и включают, например N-трет-бутоксикарбонил. Смотри также описание защитных групп в источнике Protective groups in Organic Synthesis, T.W. green et.al. (John Wiley and Sons, 1991).
Специалистам в данной области понятно, что когда Y является этерифицированной гидроксигруппой, имеются в виду заместители, например, такие, которые представлены формулой III.
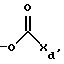 (III)
(III)
где Xd может образовывать любую синтетически возможную группу сложного эфира. Способ не ограничивается выбором конкретной группы сложного эфира для Y. Характерные примеры Xa включают алкил с прямой или разветвленной цепью, например, C1-18-алкил, фенил, моно или двузамещенный фенил, где заместители включают, например, галоген, алокси и амино.
Для получения 7β- замещенных, 4-азастероидных соединений и в частности таких, которые являются ингибиторами 5α-редуктазы, является пригодным промежуточное соединение II. Примеры таких соединений включают соединения, представленные в патентах США N 4377584 и 4760071; в WO 93/23419 и W0 93/23420. Более конкретно, соединения, которые могут быть получены из промежуточного соединения II, включают соединения общей формулы IV:
в которой R является H или метилом, Z является
и Alk выбирают из C1-25 линейного или разветвленного алкила, например, метила (Me), этила (Et), пропила (Pr), изопропила (i-Pr), н-бутила (H-Bu), втор-бутила, изо-бутила, трет-бутила (трет-Bu) и т.п.; C3-6-циклоалкила, например, циклопропила, циклобутила, циклопентила и циклогексила; и аллила. Способы получения таких соединений представлены, например в патентах США N 5237064, WO 93/23419 и WO 93/23420 и заявке PCT, имеющей порядковый номер US 94/12071.
Далее следует пример схемы синтеза, показывающей, как можно получить соединения формулы IV.



Исходными материалами для способа являются обычно 3-ацетоксиандрост-5-ены, которые являются известными и доступными в данной области.
Z является
Термин "A" описан выше и может быть любым заместителем, предпочтительно инертным и не оказывающим влияния на конкретные условия реакции каждой стадии, представленной в вышеприведенной реакционной схеме.
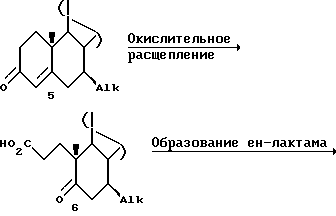
Группа A может быть защищенной гидроксигруппой или защищенной аминогруппой, которая подвергается указанной реакционной последовательности и затем ее удаляют, или она может быть удалена во время протекания конкретной стадии для того, чтобы она не служила препятствием указанной реакции. Когда A является, например, -O-TBDMS, т.е. трет-бутилдиметилсилилокси, силильная защитная группа может быть удалена, например, во время стадии замыкания цикла секокислоты 6 в 4-азастероид 7, с тем, чтобы последующие стадии осуществляли с 16- или 17-OH-соединением.
Исходная группа A может быть также предшественником конечной желательной группы A и может быть, кроме того, одновременно превращена на одной стадии. Когда, например, группа A содержит двойную связь, например, аналог стигма-стерола, двойная связь в 16- или 17-боковой цепи может быть также окислена во время образования секокислоты при переходе из 5 в 6.
Как показано на вышеприведенной реакционной схеме "Alk", заместитель может быть обычно введен в кольцо B 4-азастероида путем использования реакции присоединения металлоорганического карбонила, например, реакции Гриньяра, в которой 7-карбонильная группа может взаимодействовать с реактивом Гриньяра, содержащим "Alk" в качестве R радикала в RMgX. Условия реакции Гриньяра являются обычными и включают, например, применение метил-, аллил- или циклоалкилмагнийхлорида, этилмагнийбромида, циклопропилмагнийбромида и т.п. Реактив Гриньяра используют предпочтительно с CeCl3. Используемые сухие растворители включают, например, тетрагидрофуран (THF), диэтиловый эфир, диметоксиэтан и ди-н-бутиловый эфир. Реакцию проводят в безводной среде, обычно в диапазоне температуры от 0oC до 40oC. Для завершения реакции обычно необходимо от 6 до 24 часов. На этой стадии могут быть использованы другие реакции присоединения металлоорганического карбонила, например такие, в которых используют литиевые и цинковые металлоорганические реагенты, которые являются известными в данной области.
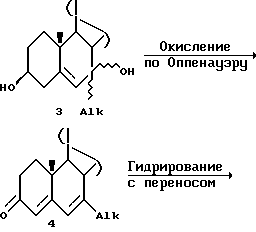
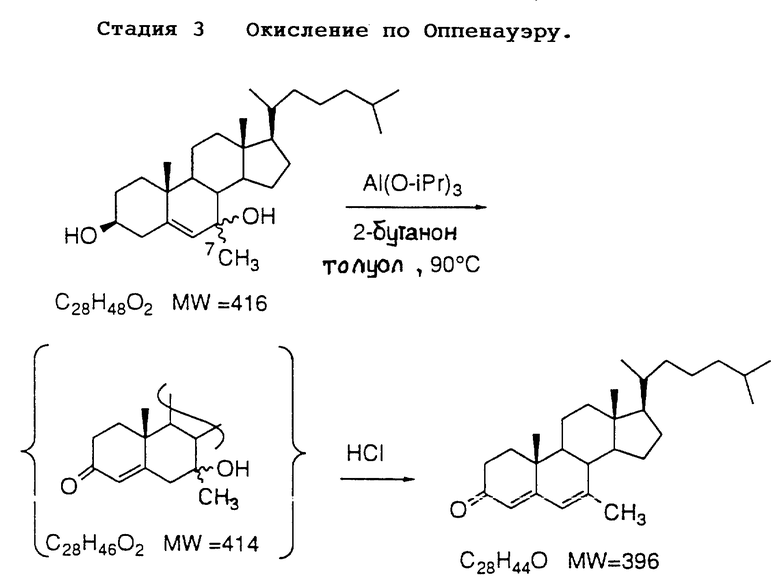
Затем аддукт  с помощью, например, алюминийизопропоксида и циклогексанона (условия окисления по Оппенауэру) окисляют при нагревании в колбе с обратным холодильником в толуоловом растворителе для получения 7-алкил-4,6-диен-3-она
с помощью, например, алюминийизопропоксида и циклогексанона (условия окисления по Оппенауэру) окисляют при нагревании в колбе с обратным холодильником в толуоловом растворителе для получения 7-алкил-4,6-диен-3-она  . Другими реагентами, которые могут быть использованы, являются, например, этилат алюминия или трет-бутилат алюминия. Другие растворители, которые могут быть использованы, включают, например, метилэтилкетон (МЕК) и ксилол. Температура находится обычно в диапазоне от около 60oC до около 120oC, реакцию осуществляют в безводных условиях и обычно для ее завершения необходимо от около 2 до около 24 часов.
. Другими реагентами, которые могут быть использованы, являются, например, этилат алюминия или трет-бутилат алюминия. Другие растворители, которые могут быть использованы, включают, например, метилэтилкетон (МЕК) и ксилол. Температура находится обычно в диапазоне от около 60oC до около 120oC, реакцию осуществляют в безводных условиях и обычно для ее завершения необходимо от около 2 до около 24 часов.
Затем, диен-3-он- превращают в 4-ен
превращают в 4-ен  путем обработки палладием на углероде, DBU и циклогексеном в растворителе, таком как этанол.
путем обработки палладием на углероде, DBU и циклогексеном в растворителе, таком как этанол.
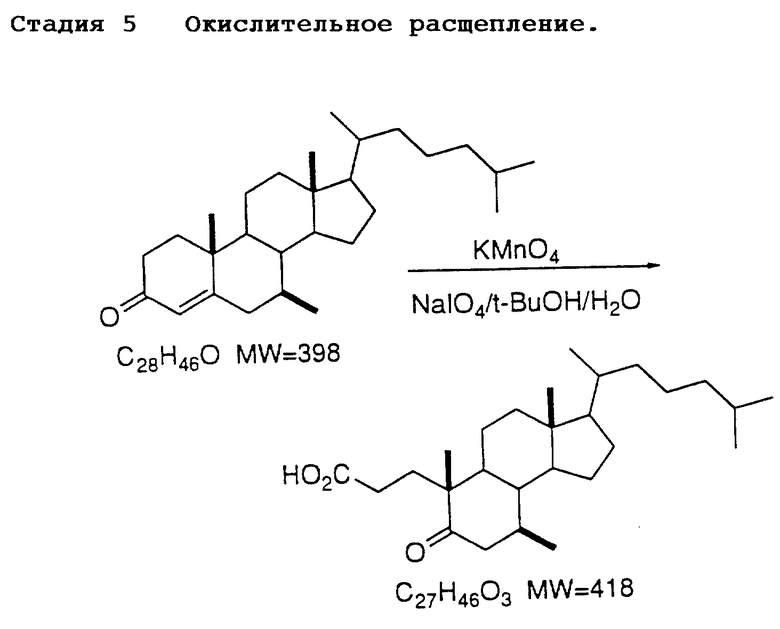
После этого раскрывают цикл A, например, путем обработки перманганатом калия, периодатом натрия, например, в трет-бутиловом спирте при температуре 80oC для получения соответствующей секо-кислоты  . Другие окислительные реагенты, которые могут быть использованы, включают тетраоксид рутения и озон. Другими используемыми растворителями являются: CH3CN, CCl4, метанол (MeOH) и CH2Cl2. Для полного осуществления реакции обычно необходимо от 2 до 4 часов.
. Другие окислительные реагенты, которые могут быть использованы, включают тетраоксид рутения и озон. Другими используемыми растворителями являются: CH3CN, CCl4, метанол (MeOH) и CH2Cl2. Для полного осуществления реакции обычно необходимо от 2 до 4 часов.
Секо-кислоту в C2-4-алкановой кислоте, например уксусной кислоте (HOAc), обрабатывают ацетатом аммония при температуре около 15-30oC, после чего нагревают в колбе с обратным холодильником в течение времени от 2 до 4 часов. После охлаждения до примерно 50-70oC добавляют воду и в смесь вносят затравку, чтобы вызвать кристаллизацию ен-лактама  .
.
Гидрирование ен-лактама осуществляют с использованием катализатора, которым является благородный металл; например, Pd(OH)2, PtO2, Pd на углероде, Ph на углероде или Ph/Al2O3, и предпочтительно с использованием Ph на углероде или Ph/Al2O3, в C2-4-алкановой кислоте, например уксусной кислоте, или спирте, таком как этанол, или этилацетате при давлении водорода 50-70 psi (3,5155-4,9217 кг/см2). Реакцию проводят при температуре 15-25oC в течение времени от 8 до 12 часов, и затем температуру поднимают, например, до 50-70oC, и проводят до тех пор, пока реакция завершится полностью. Фильтрацией удаляют катализатор и фильтрат концентрируют досуха. Затем продукт  можно очистить, например, путем перекристаллизации.
можно очистить, например, путем перекристаллизации.
Последнюю стадию N-метилирование осуществляют путем обработки раствора лактама  в ароматическом растворителе, таком как бензол или толуол, в присутствии кислого сульфата тетрабутиламмония и водного раствора щелочи, например гидроксида калия или гидроксида натрия, газом хлористым метилом при температуре 40-60oC и быстром перемешивании до тех пор, пока реакция завершится полностью, обычно в течение времени около 20-30 часов.
в ароматическом растворителе, таком как бензол или толуол, в присутствии кислого сульфата тетрабутиламмония и водного раствора щелочи, например гидроксида калия или гидроксида натрия, газом хлористым метилом при температуре 40-60oC и быстром перемешивании до тех пор, пока реакция завершится полностью, обычно в течение времени около 20-30 часов.
Характерные методики экспериментов с использованием нового способа подробно описаны ниже. Эти методики служат только в качестве примера и их не следует истолковывать как ограничения для нового способа этого изобретения.
Пример 1
Получение 4,7β- диметил-4-аза- -5α- холестан-3-она
Стадия 1 (см. схему и табл. в конце описания).
В трехгорлую колбу объемом 2000 мл добавили дигидрат вольфрамата натрия (3,3 г), метасиликат натрия (0,315 г) и 70 мл воды и перемешивали до получения гомогенного раствора. Раствор нейтрализовали (pH 6-7) концентрированной серной кислотой (0,45 мл). При добавлении кислоты при 4oC отметили выделение тепла. Добавили гидрат трихлорида рутения (240 мг) и смесь перемешали в течение 10 минут. К каталитической смеси добавили холестерилацетат (78,1 г) и гептан (300 мл). Скорость перемешивания составляла 225-275 оборотов в минуту при перемешивании лопастной мешалкой.
В течение 10 минут добавили 70% трет-BuOOH (189 г). Внутреннюю температуру поддерживали равной 15-20oC путем охлаждения водяной баней. После индукционного периода, равного 5-15 мин, температура загрузки начала медленно подниматься. Реакционную смесь перемешивали до тех пор, пока осталось менее чем 1,5 вес.% s.m. (исходного материала) и менее чем 2% промежуточного соединения 7-гидроксихолестерилацетата, в течение примерно 20-24 часов.
Реакцию контролировали с помощью УМС основоной колонки, при соотношении ацетонитрила к воде 90:10, скорость потока = 1,5 мл/мин, ультрафиолетовое обнаружение при 200 нм. Время удержания : tR (время удержания) холестерилацетата = 17,0 мин, tR 7-кетохолестерилацетата = 7,8 мин, tR промежуточных соединений 7-гидроксипероксидов, 7-олов = 6,8, 6,9, 7,0, 8,2 мин. Примесями, элюируемыми через 18 и 19 минут, являются 7-трет-BuOO-холестерилацетаты.
К реакционной смеси добавили 550 мл МЕК, 390 мл воды и 39 г сульфата натрия. Смесь нагревали до 70oC до тех пор, пока исчезли примеси ендиона, примерно в течение 3 часов. Реакционную смесь охладили, затем перенесли в делительную воронку, и фракцию водного слоя и затем органический слой промыли 100 мл 1% рассола. Затем МЕК и трет-BuOH удаляли посредством азеотропной дистилляции гептаном (после доведения исходной концентрации до 300 мл добавили 800 мл гептана) до тех пор, пока осталось менее чем 0,7% смеси, состоящей из МЕК и трет-BuOH, что было установлено с помощью анализа GC (газовой хроматографией).
С помощью газовой хроматографии и с использованием колонки HP-5 при 35oC и скорости потока 0,5 мл проконтролировали гептан на содержание МЕК и трет-BuOH. tR МЕК=4,9 мин, tR трет-BuOH=5,3 мин, tR гептана=7,7 мин. Объем установили равным 350 мл, охладили до -5oC и фильтровали, дважды промыли 150 мл гептана при 0oC. После сушки получили продукт, выход 62% (в целом 51,5 г, 94 вес.%, 97 A%) в виде не совсем белого твердого соединения. "A%" является % площади.
Температура плавления (т.р): 155-157oC; ЯМР (1H, 300МН, CDCl3): 5,70 (c, 1H), 4,7 (м, 1H), 2,5-0,8 (м, 43H), 0,6 (с, 3H).
(Далее см. стадию 2 в конце описания).
Безводный хлорид церия (16,6 г) перемешивали в виде суспензии в THF (150 мл) при 20oC под азотом в течение 2 часов.
Хлорид церия получили в виде гептагидрата и сушили в вакууме при температуре печи 170oC в течение периода времени от 3 до 4 дней. Высушенный хлорид церия, как показал анализ TG (термогравиметрией), имел потерю веса 0,7%. Через 2 часа удалили пробу суспензии и под микроскопом наблюдали мелкие игольчатые кристаллы. К суспензии добавили реактив Гриньяра (160 мл) и образованную светло-пурпурную смесь подвергли выдержке в течение 30 минут.
К охлажденной смеси (20oC) добавили кеон (60 г при степени чистоты 95%, как показал количественный анализ 57 г) в THF (150 мл) в течение 50 мин. При добавлении кетона к реактиву Гриньяра наблюдали выделение тепла, экзотерму регулировали посредством скорости добавления. Чтобы обеспечить полное растворение, перед добавлением раствора кетона к реактиву Гриньяра раствор кетона в THF следовало нагреть до 30oC.
Ход реакции контролировали посредством HPLC (жидкостной хроматографией под высоким давлением). К 10 мл 0,1N HOAc добавили 0,5 мл пробу и затем с помощью CH3CN разбавили до 50 мл. Условия HPLC: (Фенильная колонка Зорбакса®, CH3CN, вода, фосфорная кислота; градиент элюирования от 75:25:0,1 до 90:10:0,1 в течение 18 мин. Скорость потока=1,5 мл/мин, ультрафиолетовое обнаружение при 200 нм). Время удержания: tR 3,7-диола=5,6 и 5,9 мин, tR исходного кетона=10,9 мин, tR промежуточного соединения 7-OH, 3-OAc=9,8 мин и 10,8 мин 95% площади соответствовало 3,7-диолу (приблизительно 85 мг/мл). (Примечание: любой оставшийся исходный материал или промежуточные соединения можно превратить в продукт с использованием дополнительного количества реактива Гриньяра).
После завершения реакции реакционную смесь быстро охладили путем добавления ее к смеси раствора лимонной кислоты (115 г в 300 мл воды) и толуола (300 мл), находящейся при 0oC. Резкое охлаждение было экзотермическим. (Примечание: скорость добавления следует регулировать осторожно, чтобы сохранить внутреннюю температуру ниже 10oC).
Двухфазную смесь перемешали в течение 30 минут и выдержали в течение 10-15 минут для соответствующего разделения фаз. рH водного слоя составил около 2. Органическую фазу отделили, промыли водой (200 мл, pH 3 после промывки) и насыщенным раствором NaHCO3 (240 мл, pH 8 после промывки). Указанное дало возможность получить 750 мл органического слоя, который содержал 66 мг/мл диола, выход 49,5 г (93%). Водный слой содержал менее чем 1% продукта.
Загрузку концентрировали в вакууме до 300 мл (100-200 мм), разбавили толуолом до 600 мл и повторно концентрировали до 360 мл. Изменение растворителя на толуол сочли завершенным, когда, как показала газовая хроматография, % площади THF составил < 2% относительно % площади толуола (Примечание: первые 200 мл дистиллята обладали склонностью к образованию пены при низких давлениях. После того, как образование такой фазы было завершено, вакуум следовало снизить до 100 мм. Температура перегонки медленно возросла от 20oC до примерно 45oC, когда изменение растворителя на толуол подошло к завершению).
Пробы дистиллята с использованием газовой хроматографии анализировали на достаточное содержание THF. Пробу в количестве около 0,1 мл разбавили до 1 мл метанолом. Условия для газовой хроматографии: (HP-5 колонка (25 м, 1D 0,32 мкм) при использовании инжектора с нагревательным блоком, изотерма при 35oC, скорость потока = 0,5 мл/мин), * tR THF = 6,2 мин, tR тоуола = 10,1 мин. Конечный анализ провели, используя пробу из загрузки. *MeOH tR = 5,5 мин.
Органический слой содержал 134,4 мг/мл диолов, общий выход составил 48,4 г (90%). (примечание: перед протеканием следующей стадии KF загрузки должен быть ниже 100 мкг/мл). (Далее см. Стадию 3 в конце описания)
К толуоловому раствору диола (256 мл, 118 мг/мл) добавили 2-бутанон (126 мл) и изопроксид алюминия (18,9 г). Раствор нагрели в колбе с обратным холодильником (92oC) под азотом. Ход реакции контролировали посредством HPLC.
Перед добавлением изопроксида алюминия посредством газовой хроматографии загрузку анализировали на содержание 2-бутанона. Пробу в количестве примерно 0,1 мл разбавили MeOH до 1 мл. Условия для газовой хроматографии:
HP-5 колонка (25 м, 1D 0,32 мкм) при использовании инжектора с нагревательным блоком при 250oC, изотерма при 35oC, скорость потока = 0,5 мл/мин, tR 2-бутанона = 6,1 мин, MeOH = 5,5 мин, tR толуола = 10,1 мин. KF исходной смеси 70 мкг/мл.
0,1 мл пробу реакционной смеси резко охладили 0,1N раствором HOAc (2-3 мл) и затем разбавили до 10 мл CH3CN в мерной колбе. Условия HPLC [25 см фенильная колонка Зорбакса®; CH3CN:H2O с 0,1% фосфорной кислоты, градиент элюирования от 75:25 до 90:10 в течение 18 мин, при выдержке до 22 мин 90: 10, скорость потока = 1,5 мл/мин, ультрафиолетовое обнаружение при 210 нм]; tR исходных диолов = 5,4 мин, 5,8 мин; tR промежуточного Δ- 4-енона = 6,4 мин, tR диенона = 12,1 мин.
Реакцию посчитали завершенной, когда исходный диол был менее чем на 3% площади (8 часов). После завершения реакции загрузку охладили до 15-20oC и затем резко охладили 3N HCl (120 мл). Двухфазную смесь перемешали в течение 20 минут и затем обеспечили ее отстаивание. Удалили нижний водный слой и органический слой промыли 5% NaCl (120 мл). Загрузку концентрировали в вакууме до половины объема (40-60oC при 150 мм). Перегонкой из загрузки удалили избыток 2-бутанона. Уровень 2-бутанона в конечной загрузке составил < 2% относительно толуола (при использовании газовой хроматографии) и KF 60 г/мл.
Раствор толуола обработали концентрированной соляной кислотой (3,5 мл) при 25oC под азотом. Реакционную смесь анализировали посредством HPLC до тех пор, пока промежуточный третичный спирт полностью превратился в диенон (приблизительно через 1 час). Раствор промыли деионизированной водой (60 мл) и насыщенным NaHCO3 (60 мл). pH бикарбонатной промывки составил 8,5. (Примечание: реакция разложения будет протекать в обратном направлении, если опыт проводить в течение времени более чем 8 часов). Полученный красный раствор (128 мл) содержал 202 мг/мл диенона, выход - 25,9 г (90%). (Далее см. Стадию 4 в конце описания).
Толуоловый раствор диенона (150 мл, 214,6 мг/мл) разбавили этанолом (120 мл) и циклогексеном (120 мл) и DBU (0,62 мл). К смеси добавили 5% палладий на углероде (9,0 г увлажненного 50% воды). Смесь дегазировали с использованием вакуума/продувки азотом (3х). Затем суспензию нагрели в колбе с обратным холодильником (температура перегонки 72oC). Реакцию контролировали посредством HPLC. 2 мл пробу реакционной смеси фильтровали через Solka Flok. Фильтрат (0,1 мл) разбавили CH3CN до 10 мл и анализировали HPLC: [25 см фенильная колонка Зорбакса®;ацетонитрил/вода, содержащая 0,1% фосфорной кислоты, градиент элюирования от 75:25 до 90:10 CH3CN/H2O в течение 18 минут, при выдержке до 22 минут. 90:10; скорость потока 1,5 мл/мин; ультрафиолетовое обнаружение при 200 нм].
tR диенона = 12,1 мин, tR Δ-4-енона = 13,2 мин, Δ-5-енона 14,1 мин, tR избыточно восстановленного кетона = 14,4 мин; tR этиленового эфира = 20,9 мин. Избыточно восстановленный кетон следует анализировать при 192 нм.
Реакцию посчитали завершенной, когда диенон был менее чем на 2% площади и уровень Δ-5-енона составил 5% (примерно через 10 часов). Когда реакция завершилась, смесь охладили до окружающей температуры. Фильтрацией через Solka Flok удалили палладий и фильтровальный осадок промыли этанолом (150 мл).
Загрузка содержала 51 мг/мл енона. (Примечание: продолжительного протекания реакции следует избегать, т.к. может произойти избыточное восстановление. Если исходный материал израсходовался и уровень Δ-5- енона составляет более 5% через 10 часов, тогда палладий следует отфильтровать, и реакция изомеризации завершится без присутствия катализатора).
Раствор концентрировали под пониженным давлением (75 мм) приблизительно до объема 150 мл. Загрузку разбавили этанолом (225 мл) и повторно концентрировали до 150 мл.
Изменение растворителя на этанол посчитали завершенным, когда уровень толуола, определенный газовой хроматографией, составил менее 2% относительно этанола, обнаруживаемого циклогексена не было. (Примечание: удаление циклогексена является важным, т. к. он реагирует на последующей стадии окислительного расщепления, что приводит к непродуктивному расходу периодата). 0,1 мл пробу разбавили этанолом до 1 мл и анализировали на содержание циклогексена (и 1,1,1-трихлорэтан - на содержание толуола). Условия газовой хроматографии: [HP-5 (25М х 0,32 мкм 1D ), с использованием инжектора с нагревательным блоком при 250oC, температура колонки 35oC, скорость потока = 0,5 мл/мин] , tR этанола = 5,6 мин, tR циклогексена = 7,7 мин, tR трихлорэтана = 7,7 мин, tR толуола = 10,2 мин. Присутствие циклогексена также обнаруживается посредством 1H ЯМР (CDCl3) раствора: протоны циклогексенвинила при δ = 5,64 част. на миллион, протон енонвинила при δ = 5,69 част. на миллион.
Концентрат разбавили гексаном (250 мл) и 3N HCl (150 мл). Двухфазную смесь нагревали до 40oC до тех пор, пока завершился гидролиз енолового эфира. Слои разделили и органический слой промыли полунасыщенным раствором бикарбоната натрия (100 мл). Гексановую фазу, которая имела объем 291 мл и содержала менее чем 5% этанола по объему, анализировали и установили, что она содержит 92 мг/мл енона.
Раствор концентрировали под пониженным давлением до 100 мл (100 мм/15oC). Загрузку разбавили трет-бутанолом (175 мл) и повторно концентрировали до 100 мл (100 мм/40oC). Загрузка содержала 260 мг желательного 7-β- метиленона/мл, выход составил 26,8 г (85%).
(Примечание: эти соединения могут быть также обнаружены посредством газовой хроматографии. Применения газовой хроматографии для протекания этой реакции следует избегать, так как енон диспропорционируется в колонке. Условия газовой хроматографии: [HP-5 (25М) колонка, инжекция в колонке при 285oC]; tR избыточно восстановленного енона = 12,8 мин, tR 7-альфа-эпимера = 15,7 мин, продукта = 17,3 мин, tR (исходного материала) = 21,3 мин.
(Далее см. Стадию 5 в конце описания).
В 5 л круглодонную колбу загрузили деионизированную воду (4,93 л), периодат натрия (1,55 кг) и перманганат калия (11,1 г). Для получения раствора суспензию перемешали при 65oC в течение 30 минут.
К раствору енона (300 г) в трет-бутаноле (4,60 л) добавили раствор карбоната натрия (159 г) в воде (2,3 л). Двухфазную смесь нагрели до 65oC. Енон не должен содержать толуол, этанол и циклогексен. (Примечание: Концентрация енона в органическом слое составляет около 56 мг.мл-1). К раствору енона при быстром перемешивании в течение трех часов добавили раствор периодата натрия при поддержании температуры реакции 65oC. Суспензию выдержали в течение 2 часов при температуре 65oC. Через нагретую капельную воронку добавили раствор периодата.
Во время реакции выделился газ диоксид углерода. Медленное добавление обеспечивает регулируемое выделение газа. Во время добавления выделения тепла не обнаружили. При этом во время добавления образовалась суспензия пурпурно-коричневого цвета.
Ход реакции контролировали посредством HPLC, 2 мл пробу реакционной смеси охладили до 15oC и фильтровали. Фильтрат (0,1 мл) разбавили до 10 мл смесью воды и CH3CN (1:3). Условия HPLC [Основная колонка УМС 25 см х 4,6 мм, CH3CN, 0,01М H3PO4 90:10, изократный поток = 1,5 мл/мин, ультрафиолетовое обнаружение при 200 нм]; tR енона = 11,5 мин, tR секокислоты = 5,5 мин.
Реакцию посчитали завершенной, когда содержание исходного енона составило < 0,5 мг/мл. Добавили воду (3,0 л) и для разложения остатка KMnO4 (цвет изменился от пурпурного до коричневого) и растворения большей части твердых частиц, осажденных на стенках сосуда, суспензию нагрели в колбе с обратным холодильником в течение 2 часов. Полученную суспензию охладили до 15oC и фильтровали через дикалит (50 г). Сосуд и фильтровальный осадок промыли трет-бутанолом/водой (1:2; 6,0 л).
Фильтровальный осадок анализировали на содержание секокислоты путем растворения 200-400 мг фильтровального осадка в 50 мл воды и 50 мл ацетонитрила и последующего фильтрования в пробирку через диатомит для удаления незначительного количества твердых оранжевых частиц марганца. Фильтраты (pH 9,0-10,5) экстрагировали гептаном (5,0 л).
К водной смеси добавили этилацетат (2,6 л) и путем добавления концентрированной HCl (250 мл) установили pH равным 2,5±0,3. Водный слой удалили.
Органический слой промыли 5% водным рассолом (2х1,2 л). Раствор этилацетата концентрировали (150 мм Hg, 30oC) до примерно 10% объема. Добавили уксусную кислоту (7,4 л) и остаток этилацетата удалили концентрированием (100 мм Hg, 60oC) до содержания 1% по объему. (Как показала HPLC, < 0,5% площади). Путем добавления уксусной кислоты конечный объем установили равным 5 л. Удаление этилацетата проконтролировали с помощью HPLC при использовании вышеприведенных условий, за исключением того, что скорость потока составила 0,5 мл/мин и ультрафиолетовое обнаружение осуществили при 210 нм. tR этилацетата = 7,4 мин, tR уксусной кислоты = 6,9 мин. Выход анализируемого вещества составил 275 г, что соответствует 88%. Раствор уксусной кислоты непосредственно использовали на следующей стадии (образование енлактама). (Далее см. Стадию 6 в конце описания).
К раствору секокислоты в уксусной кислоте (265 г в 5,3 л), полученному на предыдущей стадии, добавили ВНТ (5,3 г) и ацетат аммония (488 г) при 20oC. Суспензию медленно нагревали в колбе с обратным холодильником в течение 3 часов под атмосферой азота. При 30oC получили раствор. Внутренняя температура при нагревании в колбе с обратным холодильником составила 120oC. Цвет изменялся от желтого до красного и затем до красно-коричневого. Применение незначительных количеств уксусной кислоты привело к замасливанию продукта на стадии кристаллизации.
Ход реакции контролировали HPLC. Условия HPLC [SB фенил, CH3CN, 0,01М H3PO4, при соотношении 80:20 в течение 30 мин, скорость потока = 1,5 мл/мин, ультрафиолетовое обнаружение при 190/200 нм]. Время удержания: tR енлактама = 9,4 мин, tR секoкислоты = 5, 3 мин. Ультрафиолетовое обнаружение в ходе реакции при 190 нм и для исходного материала, и анализируемого продукта - при 200 нм. Реакцию посчитали завершенной, когда секокислоты осталось менее чем на 0,05% площади, т.е. примерно через 3-4 часа.
Реакционную смесь охладили до 60oC и через 15 мин добавили воду (398 мл). (Примечание: является важным добавление к раствору уксусной кислоты точного количества воды 7,5% по объему).
Раствор охладили до 50oC и внесли затравку енлактама (1,0 г). При 50oC произошла кристаллизация. Полученную суспензию выдержали при 50oC в течение 1 часа и затем охладили в течение 2 часов до 0-2oC.
Суспензию фильтровали и светлое рыжевато-коричневое твердое промыли смесью уксусной кислоты и воды при соотношении компонентов в смеси 5:1 (1,0 л). Твердое в течение ночи сушили в вакууме при 30oC, получив при этом, как показал количественный анализ, 255 г, 87 вес.% (остаток - уксусная кислота), что соответствовало выходу 88%. Профиль HPLC, UV при 200 нм, вещество было на 99,4% площади, температура плавления (m. p.) сольвата = 112 - 115oC. Температура плавления чистого вещества = 175 - 178С, размягчения - 162oC.
(Далее см. Стадию 7 в конце описания).
К 20 г 83 вес. % енлактама добавили 100 мл уксусной кислоты, которая содержала 200 мг ВНТ. Для растворения суспензию нагрели до 60oC под атмосферой азота, затем охладили до 50oC. После этого добавили 10 мл воды. Затем смесь охладили в течение 1,5 часов до 5oC, выдержали в течение 1 часа и затем отфильтровали твердое вещество. (Примечание: перед охлаждением до 5oC раствор при 50oC начинал кристаллизоваться). После добавления ВНТ кГ раствора составила примерно 0,2 - 0,4. в/в.
Маточный раствор контролировали посредством HPLC. Условия HPLC: [SB фенил, CH3CN, 0,01М H3PO4, при соотношении 80:20, в течение 30 минут, скорость потока 1,5 мл/мин, ультрафиолетовое обнаружение при 200 нм]. Время удержания: tR енлактама = 9,4 мин. Отобрали пробу объемом 100 мкл и разбавили до 10 мл ацетонитрилом.
Суспензию отфильтровали и светлое рыжевато-коричневое твердое промыли смесью уксусной кислоты и воды при соотношении компонентов в смеси 5:1 (40 мл) при 5oC. Твердое в течение ночи сушили в вакууме при 30oC, получив при этом, как показал количественный анализ, 18,5 г, 84 вес.% (остаток - уксусная кислота), степень извлечения - 94%.
Профиль HPLC, ультрафиолетовое обнаружение при 200 нм, вещество было на 99,4% площади.
Температура плавления сольвата = 112 - 115oC. Температура плавления чистого вещества 175 - 178oC, размягчения - 162oC. (Далее см. Стадию 8 в конце описания).
В уксусной кислоте (1,7 л) при 20oC растворили BHT (3,8 г). Раствор дегазировали с помощью продувки азотом в течение 30 мин и одной порцией добавили енлактам (218 г, 87 вес.%). Полученный раствор в течение 15 мин продули азотом. Добавили 10% Pd/C (50% влажн.) (38 г) и суспензию перенесли в автоклав с мешалкой объемом 1 галлон (3,785 л). Для промывки суспензии, подаваемой в автоклав, использовали дегазированную уксусную кислоту (190 мм). (Примечание: Перед добавлением енлактама к уксусной кислоте следует добавлять ВНТ. Применение уксусной кислоты, стабилизированной ВНТ, является необходимым вследствие неустойчивости енлактама к окислению).
После вакуумной продувки азотом смесь поместили в сосуд под давлением 60 psi (фунтов/дюйм2) (4,2186 г/см2) H2 и перемешивали при 20oC. Через 10 часов выдержки при температуре 20oC температуру реакции увеличивали до 60oC до тех пор, пока реакция завершилась на > 99,9%.
Ход реакции контролировали HPLC [25 см фенильная колонка Зорбакса® SB, CH3CN : 0,01% H3PO4 90:10, 1,5 мл/мин, двойное UV обнаружение при 210 и 240 нм] . Время удержания: tR енлактама - 8,50 мин, tR транс-лактама - 12,4 мин, tR цислактама - 18,4 мин. Отобрали пробу объемом 20 мкл и разбавили ацетонитрилом до 2 мл.
По завершении реакции (т.е. степень превращения >99,9%) смесь охладили до 20oC и фильтровали через Solka-Flok (20 г). Фильтровальный осадок промыли уксусной кислотой (1,9 л). Фильтраты соединили и концентрировали при 30oC/10 мм Hg до объема 570 мл. Добавили гептан (в целом 3,8 л) и концентрирование продолжили при атмосферном давлении (точка кипения азеотропа = 91-92oC) для удаления уксусной кислоты. (Примечание: Удаление уксусной кислоты до <2% по объему является важным вследствие очень высокой растворимости продукта в уксусной кислоте). Конечная температура кипения составила 98-99oC. Уксусную кислоту контролировали при 200 нм посредством HPLC с использованием 25 см фенильной SB колонки Зорбакса® при использовании в качестве элюента смеси CH3CN и воды при соотношении компонентов в смеси 90:10, скорость потока 0,5 мл/мин. Отобрали пробу объемом 100 мкл и разбавили ацетонитрилом до 10 мл.
Раствор концентрировали до 570 мл и добавили МЕК (в общем 2,5 л). Азеотропной перегонкой при атмосферном давлении удалили гептан до содержания, как было определено газовой хроматографией, дистиллятов и загрузки <5% по объему. Условия газовой хроматографии: DB - 520 м, 0,5 мл/мин гелия, изотерма при 35oC; tR МЕК = 6,4; tR гептана = 8,0 мин. Во время удаления гептана произошла кристаллизация.
Установили объем раствора равным 600 мл и раствор в течение 3 часов охладили до 20oC. Образованную суспензию выдержали в течение 2 часов при -10oC. На фильтре собрали твердое вещество и промыли холодным МЕК (150 мл). Твердое вещество сушили в вакууме при 20oC. Выход - 170 г, 99%; при 210 нм продукт находился более чем на 99,2% площади. Выход стадии - 89%.
(Далее см. Стадию 9 в конце описания).
Автоклав объемом 5 галлонов (18,927 л) загрузили при комнатной температуре суспензией лактама (3,0 кг), BnMe3NCl (150 г) и гидроксидом калия на оксиде алюминия (1:1, 3,0 кг) в толуоле (12 л). При 20oC и медленном перемешивании ввели метилхлорид (453 г). Суспензию нагрели при медленном перемешивании до 65oC и выдержали в течение 1 часа. На датчике температуры отметили экзотерму при температуре 52-53oC в виде острого выступа.
Ход реакции контролировали HPLC. Условия HPLC [Фенильная SB колонка Зорбакса®, CH3CN: 0,01М H3PO4 90:10, скорость потока = 1,5 мл/мин, UV обнаружение при 200 нм], tR лактама = 12,4 мин, tR IV-a = 15,0 мин. 25 мкл пробу толуолового слоя разбавили ацетонитрилом до объема 2 мл. Реакцию контролировали до получения полного превращения (>99,95%). Реакция завершилась при 60oC в течение времени <60 мин.
Реакционную смесь охладили до 20oC и продули азотом (4х) для удаления избытка MeCl. Раствор толуола фильтровали через Solka Flok (100 г), и сосуд и фильтровальный осадок промыли толуолом (2 л). Смешанные фильтраты концентрировали при давлении 100 мм Hg и 20-30oC до остаточного масла. Масло должно быть однородно распределено в гептане (10 мл/г) без образования мути.
Масло подвергли анализу газовой хроматографией на содержание толуола при температуре 35oC. Продукт (100 мл) растворили в метаноле (0,5 мл) и инжектировали 1 мкл tR толуола = 4,4 мин, tR метанола = 2,7 мин.
Масло сохраняли под вакуумом до тех пор, пока количество растворителя стало < 2%. Масло влили в стеклянный поток, ввели затравку IV-a (1,25 г) и выдерживали в вакууме (20 мм Hg) всю ночь.
Полученное твердое вещество разрезали на блоки и измельчили в смесителе Варинга (Waring), содержащем воду (10 л), находящуюся при температуре 2oC, до крупности частиц <50 мкм. Суспензию отфильтровали, промыли водой (5,0 л) и сушили в потоке азота всю ночь. Выход продукта = 3,0 кг, 97%.
Пример 2.
Получение 7-кетохолестерилацетата
Катализатор хлорид трис(трифенилфосфин) рутения (II) (96 мг, 0,1 ммоля) растворили в хлорбензоле (10 мл), затем в холестерилацетате (4,3 г, 10,0 ммолей). Смесь дегазировали продувкой азота в вакууме (3х), затем охладили до +5oC. К смеси в течение 15 часов под азотом добавили 90% трет-бутилпероксид водорода (4,4 мл, 40 ммолей). Анализ HPLC показал 285 г (65%) 7-кетохолестерилацетата.
Загрузку фильтровали через Solka-Flok и в вакууме удалили растворитель. Остаток растворили в метаноле и затем загрузку охладили до +5oC и выдержали в течение 30 минут. Загрузку фильтровали и промыли холодным метанолом. Твердое вещество сушили на воздухе до получения 2,26 г (51%) 7-кетохолестерилацетата.
Пример 3.
Получение 7-кетохолестерилацетата (см. схему и таблицу в конце описания)
В трехгорлую колбу объемом 2000 мл при воздушном перемешивании добавили гидрат трихлорида рутения (240 мг), 55 мл воды, холестерилацетат (78,1 г) и гептан (310 мл). Скорость воздушного перемешивания лопастной мешалкой составила 225-275 оборотов в минуту.
В течение 4 часов медленно добавили 70% трет-BuOOH (229 г). При охлаждении водяной баней внутреннюю температуру поддерживали равной 15-20oC. После периода индукции, равного 5-15 мин, температура загрузки начала медленно подниматься.
Реакционную смесь перемешивали до тех пор, пока осталось менее чем 1,5 вес.% исходного материала и менее чем 2% промежуточного соединения 7-гидроксихолестерилацетата, т.е. примерно в течение 20-24 часов.
Ход реакции контролировали с помощью основной колонки УМС, смесь ацетонитрила и воды, при соотношении компонентов в смеси 90:10, скорость потока = 1,5 мл/мин, UV обнаружение при 200 нм. Время удержания: tR холестерилацетата = 17,0 мин, tR 7-кетохолестерилацетата = 7,8 мин, tR ендиона = 4,5 мин, tR промежуточных соединений 7-гидропероксидов, 7-олов = 6,8, 6,9, 7,0, 8,2 мин. Позднее элюируемыми примесями (через 18 и 19 минут) были 7-трет-BuOO-холестерилацетаты.
К реакционной смеси добавили 550 мл МЕК, 390 мл воды и 39 г сульфита натрия. Смесь нагревали до 70oC до тех пор, пока пропала примесь ендиона, примерно в течение 3 часов. Реакционную смесь охладили, затем для удаления солей рутения фильтровали через прокладку Solka-Flok. Прозрачный раствор перенесли в делительную воронку, отделили водный слой и затем органический слой промыли 100 мл 1% рассола. Затем азеотропной перегонкой с гептаном (после начального концентрирования до 300 мл добавили 800 мл гептана) удаляли МЕК и трет-BuOH до тех пор, пока осталось менее чем 0,7% смеси МЕК и трет-BuOH, как показал анализ газовой хроматографией.
Гептан проверили на содержание МЕК и трет-BuOH газовой хроматографией с использованием HP-5 колонки при 35oC и скорости потока 0,5 мл, tR МЕК = 4,9 мин, tR трет-ВuOH = 5,3 мин, tR гептана = 7,7 мин.
Установили объем 350 мл, охладили до -5oC и фильтровали, дважды промыли 150 мл гептана при 0oC. После сушки получили продукт, выход 62% (51,5 г, 94 вес.%, 97% площади) в виде не совсем белого твердого вещества.
Пример 4.
Следуя по существу той же самой методике, которая описана в примере 1, стадия 1, из соответствующих исходных материалов формулы 1, в которой является
и A, X и Y определены следующим образом:
(a) A = 6-метилгепт-2-ил, X=-CH2- и Y=-OH;
(b) A = этиленкеталь, X=-CH2- и Y=этиленгликоль;
(c) A = трет-бутил-ди-метилсилилокси (TBDMS-O-), X=-CH2- и Y=-OC(O)CH3 и
(d) A = 6-метилгепт-2-ил, X=-N(CH3)- и Y=кето(=O),
получили соединения формулы (II).
Кроме того, циклогексенол окислили в циклогексенон с использованием по существу той же самой методики, которая описана в примере 1, стадия 1.
Исходный материал для (b) получили путем обработки коммерчески пригодного 4-андростен-3,17-диона этиленгликолем и HCl при использовании стандартных условий реакции. Исходный материал для (с) получили путем обработки коммерчески пригодного 5-андростен-3,17-диол-3-ацетата TBDMS-Cl и имидазолом с применением стандартных условий реакции. Исходный материал для (d) получили с использованием стандартных методик синтеза; т.е. окислительного расщепления коммерчески пригодного холестанона (методика описана в примере 1, стадия 5) и последующей обработки NH2CH3.
Пример 5. (схему и таблицу см. в конце описания).
К раствору 16-ТВS эфира (100 г, 16-трет-бутил-силилового эфира) в гексане (500 мл) при 20oC добавили воду (300 мл) и гидрат трихлорида рутения (0,46 г). Двухфазную смесь перемешали и охладили до 10oC. В течение 5 часов при поддержании температуры реакции 10-15oC добавили трет-BuOOH (70 вес.%) (432 г).
Реакция была слегка экзотермической. Для поддержания температуры между 10 и 15oC использовали смесь воды со льдом.
После этого осуществляли HPLC с применением фенильной SB колонки Зорбакса®, 25 см, смесь ацетонитрила и воды при соотношении компонентов в смеси от 30:70 до 80:20 в течение 25 мин, затем осуществили выдержку в течение 15 мин. UV обнаружение при 200 нм, скорость потока 1,5 мл/мин.
Время удержания: tR - Мин.
OTBS эфир - 29,4
7-кетон - 23,8
TBHP - 3,25
Реакцию посчитали завершенной, когда осталось <2% исходного материала (<1,5 мг/мл). Типичное время протекания реакции = 10 часам. После завершения реакции добавили древесный уголь (10 г), затем сульфит натрия (20 г) и суспензию перемешали в течение 30 минут.
Сульфит натрия разлагает остаточный трет-BuOOH и другие гидропероксиды. При добавлении сульфита натрия происходило выделение небольшого количества тепла и количество добавки зависело от концентрации остаточного трет-BuOOH. С помощью HPLC зарегистрировали полное удаление трет-BuOOH. Двухфазную смесь фильтровали через Дикалит (в спеченной воронке (7,62 см) и фильтровальный осадок промыли гексаном (300 мл). Отделили водный слой и слой гексана промыли водой (100 мл х2).
Добавлением ацетонитрила (20 мл) удалили небольшое количество твердого слоя на поверхности раздела.
Слой гексана концентрировали до небольшого объема и промыли большим количеством гексана (400 мл). Раствор концентрировали до конечного объема 150 мл (отношение гексан : подложка составляет 2:1) и использовали как таковой на стадии очистки.
Перед обработкой диоксидом кремния раствор гексана сушили.
Обработка диоксидом кремния.
Указанный выше раствор гексана загрузили на силикагельную колонку (470 г, предварительно суспендировали в гексане, 60-230 меш). Для удаления непрореагировавшего исходного материала колонку элюировали гексаном (800 мл). Затем колонку элюировали 10% этилацетатом в гексане (1000 мл), обеспечив при этом 7-кетон. Подробности, касающиеся колонки: 100 мл фракции, после колонки следовала TLC (тонкослойная хроматография) (20% этилацетат/гексан) или альтернативно HPLC (как указано выше).
Фракции 14-17 смешали и концентрировали до общего объема 100,0 мл.
Хотя в предшествующем описании представлена сущность изобретения и с целью иллюстрации обеспечены примеры, следует понимать, что на практике изобретение охватывает все из возможных вариантов, переделок и модификаций, которые входят в объем последующей формулы изобретения и ее эквивалентов.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ЭПОКСИДА | 1995 |
|
RU2137769C1 |
| ПРОИЗВОДНЫЕ ФЕНОКСИФЕНИЛУКСУСНОЙ КИСЛОТЫ | 1994 |
|
RU2139273C1 |
| СПОСОБ ПОЛУЧЕНИЯ 7β-ЗАМЕЩЕННЫХ 4-АЗАαАНДРОСТАН-3-ОНОВ И СПОСОБ ПОЛУЧЕНИЯ ЗАМЕЩЕННЫХ 7β-АЛКИЛ-АНДРОСТ-5-ЕН-3-ОНОВ | 1993 |
|
RU2114117C1 |
| ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ ДЛЯ СИНТЕЗА ИНГИБИТОРОВ ВИЧ-ПРОТЕАЗ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 1994 |
|
RU2125561C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ИНДОЛА | 1993 |
|
RU2126003C1 |
| СПОСОБ ПОЛУЧЕНИЯ 5-(1,2,4-ТРИАЗОЛ-1-ИЛ)ТРИПТАМИНОВЫХ СОЕДИНЕНИЙ И 2-[5-(1,2,4-ТРИАЗОЛ-1-ИЛ- МЕТИЛ)-1Н-ИНДОЛ-3-ИЛ/ЭТИЛОВЫЙ СПИРТ | 1995 |
|
RU2138496C1 |
| СПОСОБЫ ПОЛУЧЕНИЯ ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ БЕТА-МЕТИЛКАРБАПЕНЕМОВ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ | 1993 |
|
RU2130927C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ, ИСПОЛЬЗУЕМЫХ ДЛЯ СИНТЕЗА ИНГИБИТОРОВ ВИЧ-ПРОТЕАЗЫ | 1994 |
|
RU2134263C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ТИРОЗИНА | 1994 |
|
RU2097377C1 |
| ИНГИБИТОРЫ HIV ПРОТЕАЗЫ | 1994 |
|
RU2137768C1 |












Описывается новый способ окисления Δ5-стероидного алкена общей формулы I, где Y, X, Z указаны в п.1 формулы, отличающийся тем, что Δ5-стероидный алкен общей формулы I обрабатывают гидроперекисью в растворителе в присутствии катализатора на основе рутения. Описывается также способ получения соединения. Технический результат - упрощение процесса получения целевых продуктов. 3 с. и 25 з.п. ф-лы.


где Z является
или
А является -Н; кето (=O); защищенная гидрокси например, диметил-трет-бутилсилилокси, триметилсилилокси, триэтилсилилокси, триизопропилсилилокси, трифенилсилилокси; ацетат, гидрокси; защищенная амино, например, ацетиламино; амино; С1 - С10-алкил, например, метил, этил, 1,5-диметилгексил, 6-метилгепт-2-ил, арилзамещенный С1 - С10-алкил, например, омегафенилпропил, 1-(хлорфенокси)этил; арилкарбамоилзамещенный С1 - С10-алкил, например, 2-(4-пиридинилкарбамоил)этил; С1 - С10-алкилкарбонил, например, изобутилкарбонил; арилкарбонил, например, фенилкарбонил; эфирозамещенный С1 - С10-алкил, например, 1-метоксиэтил, 1-этоксиэтил; кетозамещенный С1 - С10-алкил, например, 1-кетоэтил; гетерозамещенный С1 - С10-алкил, например, омега-(4-пиридил)бутил; карбокси; сложные эфиры карбоновых кислот, например, сложные эфиры С1 - С10-алкилкарбоновых кислот, такие как карбометокси; карбоксамиды, например, С1 - С10-алкилкарбоксамиды или аралкилкарбоксамиды, такие, как N, N-диизопропилкарбоксамид, н-трет-бутилкарбоксамид или N-(дифенилметил)карбоксамид; карбаматы, такие, как С1 - С10-алкилкарбаматы, особенно трет-бутилкарбамат; замещенные или незамещенные производные анилида, в которых фенил может быть замещен 1 - 2 заместителями, выбранными из этила, метила, трифторметила или галогена (F, Cl, Br, I); мочевины, например, С1 - С10-алкилкарбониламиномочевины, такие, как трет-бутилкарбониламиномочевины; С1 - C10-алкилкарбониламино, например, трет-бутилкарбониламино; простые эфиры, например, н-бутилокси, этиленкеталь; замещенные и незамещенные ариловые эфиры, такие, как хлорфенокси, метилфенокси, фенилокси, метилсульфонилфенилокси, пиримидинилокси;
Y - гидроксигруппа, этерифицированная гидроксигруппа;
Х - -СН2-, NH-группа,
в Δ5-7-кетостероидный алкен общей формулы II
где Х, Y и А имеют вышеуказанные значения,
отличающийся тем, что Δ5-стероидный алкен общей формулы I обрабатывают гидроперекисью в растворителе в присутствии катализатора на основе рутения.

где Z является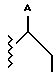
или
Y выбирают из гидрокси, этерифицированной гидроксигруппы, кето и этиленкеталя;
Х выбирают из -CH2-, -NH- или N(CH3)-, или -N-2,4-диметоксибензила;
А является любым синтетически возможным заместителем.
(а) защищенную гидроксигруппу выбирают из диметил-трет-бутилсилилокси, триметилсилилокси, триэтилсилилокси, триизопропилсилилокси и трифенилсилилокси;
(b) защищенной аминогруппой является ацетиламино;
(с) С1 - С10-алкил выбирают из метила, этила, 1,5-диметилгексила, 6-метилгепт-2-ила и 1-метил-4-изопропилгексила;
(d) арилзамещенный С1 - С10-алкил выбирают из омега-фенилпропила и 1-(хлорфенокси)этила;
(с) арилкарбамоилзамещенным С1 - С10-алкилом является 2-(4-пиридинилкарбамоил)этил;
(f) С1 - С10-алкилкарбонилом является изобутилкарбонил;
(g) арилкарбонилом является фенилкарбонил;
(h) эфирозамещенный С1 - С10-алкил выбирают из 1-метоксиэтила и 1-этоксиэтила;
(i) кетозамещенным С1 - С10-алкилом является 1-кетоэтил;
(j) гетероарилзамещенным С1 - С10-алкилом является омега(4-пиридил)бутил;
(k) сложными эфирами карбоновых кислот являются сложные эфиры С1 - С10-алкилкарбоновых кислот, выбранные из карбометокси и карбоэтокси;
(l) карбоксамиды выбирают из N, N-диизопропилкарбоксамида, N-трет-бутилкарбоксамида и N-(дифенилметил)карбоксамида;
(m) карбаматы выбирают из трет-бутилкарбамата и изопропилкарбамата;
(n) замещенные или незамещенные производные анилида выбирают из таких производных, где фенил может быть замещен заместителями в количестве от одного до двух, выбранными из этила, метила, трифторметила или галогена (F, Cl, Br, I);
(о) мочевиной является трет-бутилкарбониламиномочевина;
(р) С1 - С10-алкилкарбониламино является трет-бутилкарбониламино;
(q) простой эфир выбирают из н-бутилокси и этиленкеталя;
(r) замещенный и незамещенный ариловый эфир выбирают из хлорфенилокси, метилфенилокси, фенокси, метилсульфонилфенилокси и пиримидинилокси.
20. Способ по п.19, отличающийся тем, что А выбирают из 6-метилгепт-2-ила, трет-бутилкарбамоила, фенилкарбамоила, 2,5-дитрифторметилфенилкарбамоила, 4-метилсульфонилфенокси, изобутилкарбонила, фенилкарбонила, 1-метоксиэтила, 2-(4-пиридинилкарбамоил)этила, хлорфеноксиэтила и 1-кетоэтила.
23. Способ по п.22, отличающийся тем, что А является фенокси, хлорфенокси, метилфенокси, 2-пиримидинилокси и трет-бутилсилилокси.
отличающийся тем, что осуществляют стадии
(а) Δ-5-7-кетостероидный алкен превращают в соединение 3'
(b) соединение 3' подвергают окислению для образования соединения 4'
(c) соединение 4' подвергают гидрированию для образования соединения 5'
(d) соединение 5' подвергают окислительному расщеплению для образования соединения 6'
(e) соединение 6' лактанизируют для образования NH-енлактама 7'
(f) NH-енлактам 7' восстанавливают для образования соединения 8'
(g) соединение 8' метилируют для образования соединения 9'
| US 4377584, 22.03.1983 | |||
| ФИЗЕР Л., ФИЗЕР М | |||
| Стероиды | |||
| - М.: Мир, 1964, с.234-242 | |||
| J | |||
| of Chem | |||
| Soc | |||
| Приспособление для установки двигателя в топках с получающими возвратно-поступательное перемещение колосниками | 1917 |
|
SU1985A1 |

