Изобретение относится к способам осуществления экологического мониторинга, а именно сорбционного контроля состояния и загрязнения водных объектов, и может быть использовано для целей экологического контроля природных и техногенных вод, включая речные водоемы, шахтные и карьерные воды, а также промышленные стоки различной природы. Изобретение, в частности, может быть использовано для постоянного контроля состава промышленных сбросных вод, а также для выявления скрытых залповых сбросов, приводящих к превышению предельно допустимых норм по концентрациям токсичных металлов в промстоках в относительно короткие промежутки времени, за пределами которых происходит их естественное разбавление.
Известен способ сорбционного контроля загрязнения природных водных объектов кадмием и другими токсичными металлами, включающий сорбционное концентрирование ионов тяжелых металлов контактированием известного объема образца контролируемой воды с волокнистым хелатообразующим сорбентом ПОЛИСОРГ VII M, десорбцию катионов металлов азотной кислотой и последующее определение их концентраций в полученных растворах методом атомной адсорбции [1]. При этом стадия концентрирования может быть осуществлена непосредственно после отбора пробы в месте осуществления контроля, что обеспечивает соответствие ее состава составу контролируемого объекта.
Недостатками метода являются необходимость обеспечения определенного, постоянного pH контролируемого раствора, необходимость выбора индивидуальных условий для каждого контролируемого иона, сложность осуществления непрерывного контроля объекта.
Наиболее близким техническим решением к предложенному является способ сорбционного загрязнения водных объектов, в частности промстоков, включающий пропускание контролируемой воды через слой сорбционного материала и определение содержания загрязняющих ионов [2].
В известном способе проводят групповое сорбционное концентрирование ионов тяжелых металлов с помощью волокнистых сорбентов на основе полиакрилонитрила путем разового отбора из контролируемого водного раствора (отстойника, контрольного колодца и т.д.) проб воды известного объема, которые подвергают сначала механической фильтрации, а потом пропускают через колонки с волокнистым сорбентом в OH--форме, после чего сорбент сушат на воздухе и проводят его химанализ.
Недостатками указанного способа являются ограниченные возможности сорбента по ассортименту определяемых ионов металлов, а также сложность в организации непрерывного контроля качества сбросной воды.
Задачей, решаемой в настоящем изобретении, является упрощение и повышение эффективности сорбционного способа контроля загрязняющих микрокомпонентов за счет обеспечения возможности использования в качестве внутренних стандартов объективно-правильной оценки скорости фильтрации через колонну ионообменных характеристик сопутствующих макрокомпонентов, образующих постоянный солевой фон сбросных вод.
Поставленная задача решается тем, что в способе сорбционного контроля загрязнения водных объектов, включающем пропускание контролируемой воды через слой сорбционного материала и определение содержания загрязняющих ионов, в качестве сорбционного материала используют ионообменный материал в исходной форме иона, слабее сорбируемого этим материалом по сравнению с ионами макрокомпонентов контролируемой воды, пропускание контролируемой воды проводят непосредственно в стоке контролируемой воды и непрерывно, но не дольше, чем до обнаружения на выходе из слоя сорбционного материала наиболее слабо сорбируемого данным материалом иона из числа макрокомпонентов указанной контролируемой воды, содержание загрязняющего иона в контролируемой воде определяют по формуле
Cv=Mi/V,
где
Mi - общее количество иона, накопленного в сорбционном материале за время пропускания контролируемой воды;
V - объем контролируемой воды, пропущенной через материал, определяемый как функция скорости перемещения в слое материала концентрационного профиля слабее сорбируемого иона из числа макрокомпонентов контролируемой воды.
Кроме того, в качестве сорбционного материала используют ионообменный материал в форме иона, слабее сорбируемого, чем ионы макрокомпонентов, содержащихся в контролируемой воде в концентрациях, не менее чем на порядок превышающих концентрацию в ней каждого из загрязняющих ионов.
А также в случае наличия в контролируемой воде газообразующих анионов и бикарбонат-ионов пропускание контролируемой воды проводят последовательно через слой сорбционного материала в форме слабее сорбируемого иона, не образующего с компонентами контролируемой воды газообразных компонентов, и вспомогательный слой такого же или другого сорбционного материала в форме иона, образующего с компонентами контролируемой воды газообразные компоненты.
Определение содержания общего количества иона, накопленного в сорбционном материале за время пропускания контролируемой воды, проводят посредством регенерации сорбционного материала и последующего анализа регенерата или посредством химического анализа известного количества усредненной пробы материала.
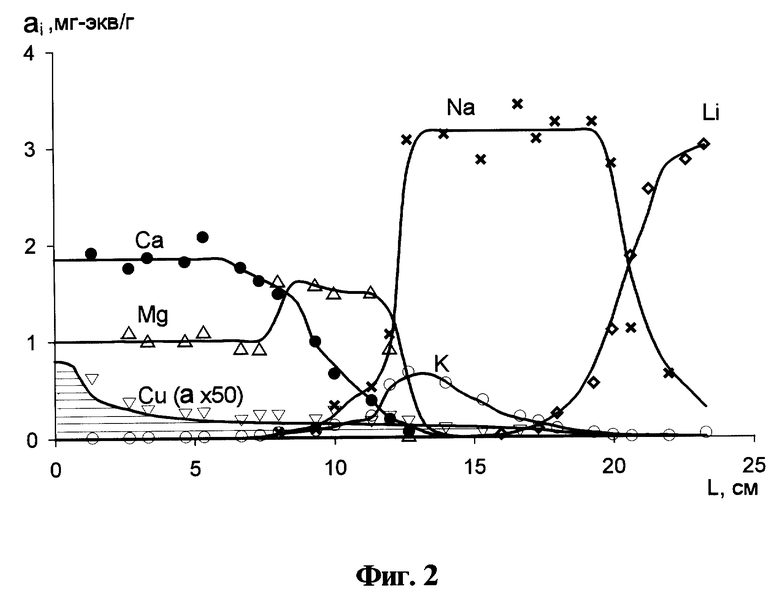
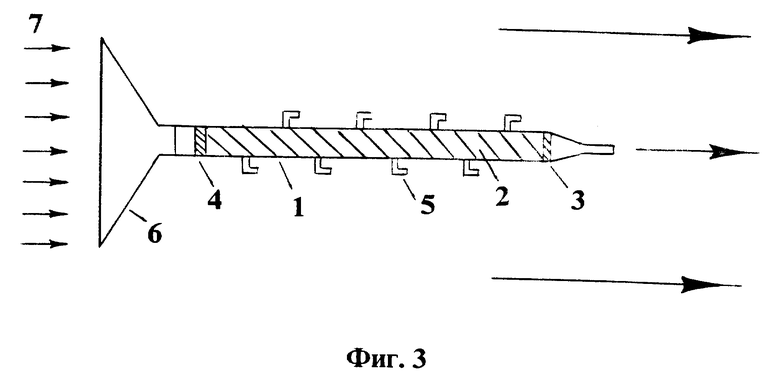
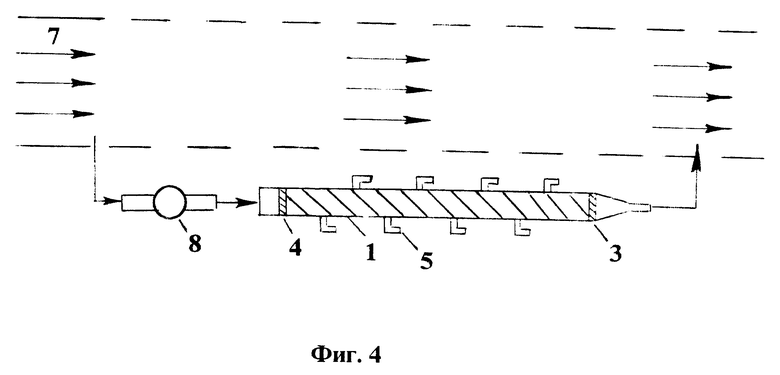
На фиг. 1 представлены типичные кривые распределения анализируемых компонентов в слое сорбционного материала в виде концентрации в материале в зависимости от координаты вдоль его слоя: A - исходная ионная форма, B и C - макрокомпоненты контролируемой воды и D - загрязняющий компонент; на фиг. 2 - экспериментальные кривые распределения, полученные при анализе загрязнения водного объекта; на фиг. 3 - схема установки без принудительной прокачки воды для осуществления сорбционного способа контроля загрязнения водных объектов; на фиг. 4 - схема установки с принудительной прокачкой воды для осуществления способа контроля загрязнения водных объектов.
В основе предложенного способа лежит возможность создания специальных условий проведения ионообменных процессов в сорбционной колонне, при которых, используя закономерности движения вдоль колонны концентрационных профилей различных макрокомпонентов по мере прохождения процесса, можно из картины распределения этих профилей в колонне в конце процесса восстановить объективно правильную среднюю скорость фильтрации раствора через колонну и точный объем анализируемого раствора, которому соответствует все количество поступивших в колонну загрязняющих ионов за известное время анализа. Для этого осуществляют определенные процедуры и расчеты в приводимой здесь последовательности.
Сорбционный материал и его исходная ионная форма A выбираются таким образом, чтобы все ионы-макрокомпоненты контролируемого водного объекта более селективно сорбировались этим материалом, чем ион A. Например, если ставится проблема загрязнения катионами цветных и тяжелых металлов природной пресной воды, содержащей всегда такие макрокомпоненты, как Na+, Ca2+, K+, Mg2+, то в качестве ионообменного материала могут быть взяты катиониты. В случае, если взяты сильнокислотные катиониты типа КУ-2 (Dowex-50), в качестве A-формы, в которую их следует предварительно переводить, можно выбрать H+- или Li+-форму, поскольку ряд селективности для катионитов типа КУ-2 имеет вид Ca2+>Mg2+>K+>Na+>Li+>H+.
Если ставится проблема загрязнения анионами (сурьма, мышьяк, бор, селениды, сульфиды, анионные формы переходных металлов) на фоне пресной воды, содержащей такие макрокомпоненты, как SO4 2-, HCO3 - и Cl-, в качестве сорбционного материала могут быть взяты аниониты. В случае, если взяты сильноосновные аниониты типа AB-17 (Dowex-1), в качестве исходной A-формы, в которую их следует предварительно переводить, можно выбрать F-- или OH-- форму, поскольку ряд селективности подобных анионитов имеет вид: SO
Выбор других типов катионитов или анионитов и соответствующих ионных форм A проводится с учетом содержания фоновых маркокомпонентов в контролируемой воде и загрязняющих ионов на основании литературных сведений, результатов исследований или других доступных данных по рядам селективности ионитов или значениям коэффициентов селективности типа K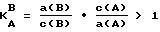 , где a и c - равновесные концентрации компонентов в ионите и в растворе соответственно в ряду селективности B>A).
, где a и c - равновесные концентрации компонентов в ионите и в растворе соответственно в ряду селективности B>A).
При соблюдении указанного правила выбора сорбционного материала и его исходной A-формы по мере прохождения процесса, т.е. пропускания контролируемой воды через слой ионообменного материала, ион A вытесняется из слоя макрокомпонентами воды, например, B и С в режиме вытеснительной хроматографии. При этом формируется передняя (по движению концентрационных профилей) чистая зона наиболее слабо сорбируемого иона их числа ионов-макрокомпонентов, например иона B, как показано на фиг. 1. Тогда скорость движения концентрационного профиля иона B ω (t) по слою ионообменного материала в колонке связана с линейной скоростью пропускания контролируемой воды через колонку ν(t) (скоростью расхода воды, отнесенной к площади сечения колонки) следующим соотношением: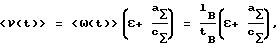
где
aΣ и cΣ- полная обменная емкость ионообменного материала (в экв/единицу объема слоя) и суммарная концентрация солей в контролируемой воде (в экв/единицу объема раствора) соответственно;
ε - порозность слоя сорбционного материала;
lВ - расстояние от входа в слой сорбционного материала до середины концентрационного профиля иона B, как показано на фиг. 1;
tВ - продолжительность пропускания контролируемой воды через слой сорбционного материала.
Как показано в приведенной формуле, физическая скорость пропускания воды ν(t) может быть непостоянной во времени, в соответствии с этим и скорость движения концентрационного профиля ω(t) может являться функцией времени. Поэтому в приведенной формуле через время tВ и расстояние lВ определяются усредненные по времени испытаний значения скоростей <ω(t)> и <ν(t)>.
После определения "физической" линейной скорости пропускания раствора через колонну точный объем пропущенного раствора V можно определить следующим образом:
V = f(<ω(t)>) = f′(<ν(t)>) = <ν(t)>•S•tB,
где
S - площадь сечения колонки.
Если анализ накопления какого-либо загрязняющего иона в сорбционной колонне показал, что вся масса этого иона составила Mi, то содержание этого иона в контролируемом водном объекте равно значению Ci=Mi/V.
Отметим, что все указанные закономерности не будут выполняться при неправильном выборе сорбционного материала и его исходной формы. Например, если для проведения сорбционного контроля тяжелых металлов на фоне макрокомпонентов пресной воды выбрать неорганический сорбент с поверхностными OH-группами [2], то в связи с более высокой селективностью таких сорбентов к собственным H+ - группам по сравнению с макрокомпонентами контролируемой воды, в слое сорбционного материала не будет образовываться чистая зона одного из макрокомпонентов, переносимая по слою со скоростью, определяемой приведенной формулой.
В отличие от формы профиля макрокомпонента B в слое ионообменного материала, показанной на фиг. 1, для контролируемых загрязняющих ионов, сбрасываемых в фоновый водный поток периодически, концентрационный профиль формируется в виде сформированного, как это показано на фиг. 1, или еще несформированного, как показано на фиг. 2, хроматографического пика для компонента D.
Целесообразно использовать сорбционный материал в форме иона, слабее сорбционного, чем ионы макрокомпонентов, содержащихся в контролируемой воде в концентрациях, не менее чем на порядок превышающих концентрацию в ней каждого из загрязняющих ионов. Указанное уточнение позволяет дать определение понятиям "макрокомпоненты" и "микрокомпоненты" контролируемой воды, а также компонента В, в форме которого используется сорбционный материал и к которому последний должен проявлять селективность меньшую, чем к любому из макрокомпонентов. В природных водах макрокомпонентами-катионами практически всегда являются ионы Na+, Ca2+, K+ и Mg2+, а анионами - Cl-, SO
Отличительной особенностью предлагаемого способа является то, что в нем не требуется изначально знать или каким-либо независимым образом измерять объем контролируемой воды, пропущенной через слой ионообменного материала в процессе анализа, также как не требуется поддерживать строго постоянную скорость пропускания или измерять ее по мере проведения сорбционного процесса. Все необходимые параметры, как было описано выше, косвенным образом, но с большой точностью "восстанавливаются" из кривой распределения вдоль колонны концентрации одного из макрокомпонентов. Этим компонентом является наиболее слабо сорбируемый макрокомпонент, который, тем не менее, сильнее сорбируется, чем ионы исходной формы и вытесняет их в виде неразмывающегося концентрационного фронта. Поэтому для осуществления способа необходимо, чтобы указанный концентрационный фронт "не покидал" колонну, а оставался в пределах слоя ионообменного материала.
Целесообразно поэтому пропускание контролируемой воды через слой сорбционного материала проводить не дольше, чем до обнаружения в выходящей из слоя воде указанного наиболее слабо сорбируемого макрокомпонента.
Во всех природных и многих техногенных водах содержатся бикарбонат-ионы, а также могут содержаться другие газообразующие анионы, которые в кислой среде могут привести к газовыделению в колонне в процессе ее работы и тем самым к нарушению гидродинамических режимов в слое сорбента. В связи с этим возникают дополнительные ограничения на выбор исходной ионной формы используемого ионообменного материала. В частности, возникают проблемы с использованием водородных форм катионита, хотя для сильнокислотных катионов именно эта форма предпочтительна, исходя из приведенных выше рядов селективности. При этом не всегда имеется возможность напрямую использовать какую-либо другую ионную форму. Например, в случае сильнокислотных катионитов можно было бы использовать литиевую форму, но в некоторые контролируемые водные среды нельзя вводить литий (выходящий из колонны в процессе ионного обмена) даже в малых количествах. В этом случае вслед за литиевой колонной можно поставить т.н. подавительную колонну в водородной форме. К подавительной колонне не предъявляется никаких требований с точки зрения гидродинамики кроме требования достаточности ионообменного материала (достаточности емкости).
Целесообразно, таким образом, пропускание контролируемой воды проводить последовательно через слой сорбционного материала в форме слабее сорбируемого иона, не образующего с компонентом контролируемой воды газообразных компонентов, и вспомогательный слой такого же или другого сорбционного материала в форме иона, образующего с компонентом контролируемой воды газообразные компоненты.
Процесс анализа распределения различных компонентов в слое сорбционного материала может быть проведен различным образом в зависимости от концентрации анализируемых компонентов, а также в зависимости от оснащенности аналитическим оборудованием. Известно, что распределение компонентов в фазе сорбента симбатно их распределению в равновесном растворе, соответствующем данному сечению сорбционной колонны. Использование специальной колонны с пробоотборниками, показанной на фиг. 3 и фиг. 4, позволяет отбирать пробы растворов, соответствующих разным сечениям по высоте колонны. Анализ этих проб на содержание макрокомпонента B позволяет построить кривую его распределения вдоль сорбционного слоя, далее по средней точке этой кривой можно определять значение lВ, а затем <ν(t)> и Ci, как показано выше.
Целесообразно, таким образом, проводить анализ распределения в слое сорбционного материала концентрации слабее сорбируемого иона из числа макрокомпонентов контролируемой воды посредством анализа состава раствора, равновесного с каждой из частей слоя сорбционного материала после пропускания через него контролируемой воды.
В ряде случаев, при высоких уровнях загрязнений или при наличии высокочувствительного аналитического оборудования, возможно проведение анализа накопления загрязняющего микрокомпонентов в слое сорбента так же, как и макрокомпонентов, по анализу проб равновесного раствора. В иных условиях и при наличии стандартного оборудования (типа обычных атомно-абсорбционных, эмиссионных, фотоколориметрических или электрохимических и других средств для анализа жидких сред) такой способ оказывается неэффективным, загрязняющие микрокомпоненты из сорбционного слоя необходимо количественно переносить в концентрированные растворы.
Целесообразно в этом случае проводить определение содержания загрязняющих ионов посредством регенерации сорбционного материала и последующего анализа регенерата. При таком способе анализа пробы сорбентов не разрушаются, но анализ является более трудоемким.
При наличии атомно-абсорбционных и атомно-эмиссионных спектрометров, оснащенных графитовой печкой, а также при использовании специальных химических агентов, растворяющих или разрушающих сорбенты, нет необходимости проводить длительную по времени трудоемкую регенерацию сорбента.
Целесообразно в этом случае определение содержания загрязняющих ионов проводить посредством химического анализа известного количества усредненной пробы сорбента.
Установка для осуществления способа сорбционного контроля загрязнений водных объектов (фиг. 3) состоит из сорбционной колонны 1, наполненной сорбентом 2. С двух сторон сорбента 2 установлены дренажные устройства 3 и 4. С внешней стороны колонны размещены пробоотборники 5. На входе колонны 1 установлен раструб 6, через который проходят основной поток контролируемой воды 7.
При принудительной прокачке контролируемой воды (фиг. 4) используют насос 8.
Примеры осуществления процесса.
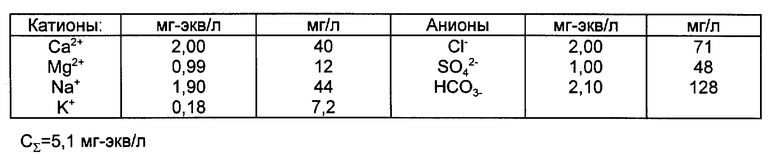
Пример 1. Контролируют сточные воды предприятия на содержание меди. Постоянный солевой фон воды соответствует составу Москвы - реки и содержит макрокомпоненты в концентрациях, приведенных в таблице.
Ионообменную колонку сечением S=0,79 см2, снабженную пробоотборниками по всей длине на расстоянии 2 см друг от друга, устройством для выгрузки сорбента, а также раструбом для выхода воды, как показано на фиг. 3, загружают промышленным катионитом КУ-2х8 в Li-форме. Высота слоя сорбента в колонке L= 25 см. Параметры взятого сорбента, определенные независимыми опытами, а также известные из справочной литературы следующие: полная обменная емкость aΣ= 3,5 мг-экв/г=1,6•103 мг-экв/л в расчете на единицу объема сорбционного слоя; порозность слоя сорбента (доля свободного объема между зернами ионита): ε = 0,35; значения констант равновесия ионного обмена K и коэффициентов селективности K: KNa Ca =(aCa/cCa)1/2(aNa/cNa)=1,5; KNa Mg =0,9; KNa K = KNa K = (aK/cK)/(aNa/cNa)=1,25; KNa Li = KNa Li =0,8.
Колонку устанавливают в створе потока сточной воды, как показано на фиг. 3, и оставляют там на время tВ = 36 часов. Все пробоотборники в ходе этого процесса держат закрытыми. После окончания сорбционного процесса все устройство и небольшое дополнительное количество контролируемой воды (200 мл) забирают в аналитическую лабораторию. Устройство устанавливают в вертикальном положении, заливают сверху взятый дополнительный объем воды, закрывают выход из колонны и поочередно открывают пробоотборники, начиная с низа колонки, через них обирают по 10 мл пробы раствора. Пробы анализируют на содержание макрокомпонентов и, воспользовавшись приведенными выше параметрами, получают картину их распределения, как показано на фиг. 2. В ходе выполнения этой операции дополнительно удостоверяются в том, что слабее сорбируемым компонентом из числа макрокомпонентов контролируемой воды является ион натрия, что в процессе сорбции формируется "чистая зона" этого компонента в колонке, что натрий, в свою очередь, сильнее сорбируется, чем ион исходной формы - ион лития, так как последний вытесняется из колонны в виде неразмывающегося сорбционного фронта и, наконец, что середина фронта Na+ и Li+ обмена не успела "покинуть" пределы колонки, а находится внутри анализируемого сорбционного слоя. Вся указанная процедура выполняется один раз для данного сорбента и данного типа контролируемой воды и для дальнейших анализов достаточно знать распределения по слою ("фронт") только иона Na+. С помощью графика на фиг. 2 определяют точное положение середины фронта натрия, которое равно lВ=20,8 см. По формуле: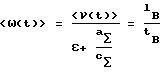 определяют значение средней линейной скорости фильтрации контролируемой воды за время проведения сорбционного процесса: <ν(t)> = 240 см/час. По формуле: V = <ν(t)>•S•tB определяют объем контролируемой воды, прошедшей через колонку: V=6,7 л. Проводят десорбцию меди из катионита пропусканием через него 5 M раствора HCl до момента, когда следы меди перестают обнаруживаться на выходе по данным атомно-абсорбционного анализа. Всего пропускают 250 мл 5 M раствора HCl. Полученный концентрат упаривают в 10 раз, доводят дистиллированной водой до объема 100 мл в аналитической колбе и определяют содержание меди на атомно-абсорбционном спектрометре в пламени ацетилен-воздух. Общее количество меди в указанном объеме 100 мл конечного раствора: MCu=0,06 мг-экв. По формуле Ci=Mi/V определяют содержание меди в сточной воде предприятия: CCu=9•10-3 мг-экв/л=0,3 мг/л.
определяют значение средней линейной скорости фильтрации контролируемой воды за время проведения сорбционного процесса: <ν(t)> = 240 см/час. По формуле: V = <ν(t)>•S•tB определяют объем контролируемой воды, прошедшей через колонку: V=6,7 л. Проводят десорбцию меди из катионита пропусканием через него 5 M раствора HCl до момента, когда следы меди перестают обнаруживаться на выходе по данным атомно-абсорбционного анализа. Всего пропускают 250 мл 5 M раствора HCl. Полученный концентрат упаривают в 10 раз, доводят дистиллированной водой до объема 100 мл в аналитической колбе и определяют содержание меди на атомно-абсорбционном спектрометре в пламени ацетилен-воздух. Общее количество меди в указанном объеме 100 мл конечного раствора: MCu=0,06 мг-экв. По формуле Ci=Mi/V определяют содержание меди в сточной воде предприятия: CCu=9•10-3 мг-экв/л=0,3 мг/л.
Проводят регенерацию катионита пропусканием через колонну 100 мл 0,5 M раствора NaOH, затем 250 мл 1 M раствора NaCl и затем 100 мл дистиллированной воды. Колонку устанавливают для следующего цикла проведения сорбционного контроля водного объекта.
Пример 2. Проводят процесс как описано в примере 1, за исключением того, что при определении содержания макрокомпонентов в пробах, отобранных через пробоотборники, наряду с определением содержания макрокомпонентов проводят определение содержания меди с использованием атомно-абсорбционного спектрометра с индуктивно связанной плазмой. Получают кривую распределения концентрации меди в катионите вдоль сорбционной колонки, показанную на фиг. 2. Определяют общую массу меди и концентрацию меди в сточной воде предприятия: CCu= 0,35 мг/л.
Пример 3. Проводят процесс как описано в примере 1, за исключением того, что после анализа проб растворов, отобранных через пробоотборники, на содержание макрокомпонентов выгружают сорбент из ионообменной колонки и сушат до воздушно-сухого состояния. Вес всего катионита 9,03 г. Проводят аналитический отбор усредненной пробы сорбента в количестве 0,1 г. Проводят прямое определение содержания в нем меди с использованием атомно-абсорбционного спектрометра с графитовой печкой. Содержание меди: aCu=0,21 мг/г. Масса меди MCu=1,82 мг, откуда CCu=0,32 мг/л.
Пример 4. Проводят процесс как описано в примере 1, за исключением того, что в схеме, показанной на фиг. 3, дополнительно устанавливают подавительную колонку с катионитом в H+-форме, расположенную после основной колонны последовательно по ходу фильтруемого потока воды. Параметры слоя сорбента в подавительной колонне: L=20 см, S=5 см2. В ходе сорбционного процесса литий, выходящий из первой колонны, не попадает в водную среду, а накапливается в подавительной колонне. Подавительная колонна используется в течение 5 циклов сорбционного контроля. При этом сорбент в подавительной колонне переходит в литиевую форму, и его далее используют в качестве катионита для проведения сорбционного анализа в основной колонне.
Пример 5. Проводят процесс как описано в примере 1, за исключением того, что вместо схемы, показанной на фиг. 3, используют схему с применением насоса, показанную на фиг. 4. Начальную скорость фильтрации устанавливают на уровне 200 мл/час. Сорбционный процесс проводят в течение 30 часов. Конечная скорость фильтрации, как показывают испытания перед снятием сорбционной колонны, составляет 165 мл/час. Падение скорости происходит в результате кальматации слоя сорбента из-за механических примесей, содержащихся в сточной воде. Не интересуясь функцией падения скорости в ходе проведения сорбционного процесса, проводят все операции по построению кривой распределения концентрации ионов натрия по слою сорбента и определению суммарного содержания меди в слое сорбента, как описано в примере 1 находят конечный результат: CCu=0,32 мг/л.
Пример 6. Проводят процесс как описано в примере 1, за исключением того, что анализируют не медь, а кадмий. Получают результат: CCd=0,12 мг/л.
Пример 7. Проводят процесс как описано в примере 6, за исключением того, что после снятия очередной колонны для проведения лабораторных анализов сразу же устанавливают вторую свежую колонну и продолжают сорбционный процесс. Таким образом осуществляют непрерывный контроль качества водного объекта.
Пример 8. Проводят процесс как описано в примере 6, за исключением того, что осуществляют сорбционные процессы с использованием трех колонн с "перекрытием" по времени анализа. Колонны "запускают" с интервалом в 12 часов. При этом получены следующие результаты в трех последовательных анализах, общая продолжительность сорбционного процесса в каждом из которых составила 36 часов: 1) CCd=0,96 мг/л; 2) CCd=0,96 мг/л; 3) CCd=0,12 мг/л.
Делают вывод о том, что в течение 12-часового промежутка времени между запусками первой и третьей колонн предприятие осуществило залповый (скрытый) сброс сточной воды с минимальным содержанием кадмия 3 мг/л.
Предложенный способ сорбционного контроля загрязнения водных объектов за счет создания специальных условий, обеспечивающих возможность использования в качестве внутренних стандартов объективно-правильной оценки скорости фильтрации через колонну ионообменных характеристик сопутствующих макрокомпонентов, образующих постоянный солевой фон сбросных вод, позволяет многократно упрощать и удешевлять экологический мониторинг, а также повышать эффективность сорбционного способа контроля водных объектов.
Источники информации, использованные при составлении заявки
1. Г.В. Мясоедова, Н.И. Щербинина, З.С. Сванидзе, Г.М. Варшал, Б.Ф. Мясоедов. Сорбционное концентрирование и атомно-абсорбционное определение кадмия в минеральных водах. Ж. аналит.химии, 1986, т. 14, вып. 3, стр. 477 - 480.
2. И.Ю. Андреева, И.Ф. Германова, Н.Г. Поливанова, Е.Я. Данилова, Групповое концентрирование ионов тяжелых металлов с помощью волокон-ионитов на основе полиакрилонитрила. Вестник ЛГУ, сер. 4, 1990, вып. 3, N 18, стр. 57 - 61 - прототип.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ СОРБЦИОННОГО КОНТРОЛЯ ЗАГРЯЗНЕНИЯ ВОДНЫХ ОБЪЕКТОВ | 2000 |
|
RU2176788C1 |
| СПОСОБ КОМПЛЕКСНОЙ ПЕРЕРАБОТКИ МОРСКОЙ ВОДЫ И УСТАНОВКА ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ | 1996 |
|
RU2104969C1 |
| СПОСОБ ПОЛУЧЕНИЯ МИНЕРАЛЬНЫХ ВЕЩЕСТВ ИЗ МОРСКОЙ ВОДЫ | 1992 |
|
RU2006476C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ РАДИОНУКЛИДОВ СТРОНЦИЯ В ПРИРОДНЫХ ОБЪЕКТАХ | 1993 |
|
RU2069868C1 |
| СПОСОБ КОНЦЕНТРИРОВАНИЯ ВЕЩЕСТВ ИЗ РАСТВОРОВ И УСТАНОВКА ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ (ВАРИАНТЫ) | 1992 |
|
RU2034651C1 |
| СПОСОБ ТВЕРДОФАЗНОГО РАЗДЕЛЕНИЯ И ОПРЕДЕЛЕНИЯ ИОНОВ И ЭЛЕКТРОХИМИЧЕСКАЯ ЯЧЕЙКА ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ | 1999 |
|
RU2150107C1 |
| Способ очистки никелевого электролита | 1990 |
|
SU1794115A3 |
| СПОСОБ ОЧИСТКИ ПИТЬЕВОЙ ВОДЫ ОТ СТРОНЦИЯ | 1991 |
|
RU2032626C1 |
| Способ ионообменной очистки сточных вод от никеля | 1990 |
|
SU1738758A1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ХРОМА (VI) | 1991 |
|
RU2024848C1 |
Изобретение используется для экологического контроля природных и техногенных вод, включая речные водоемы, шахтные и карьерные воды, а также промышленные стоки различной природы. В способе сорбционного контроля загрязнения водных объектов пропускают контролируемую воду через слой сорбционного материала. В качестве сорбционного материала используют ионообменный материал в исходной форме иона, слабее сорбируемого этим материалом по сравнению с ионами макрокомпонентов контролируемой воды. Пропускание контролируемой воды производят до обнаружения на выходе из слоя сорбционного материала наиболее слабо сорбируемого данным материалом иона из числа макрокомпонентов контролируемой воды. По результатам измерений определяют содержание загрязняющих ионов. Технический результат данного изобретения выражается в упрощении и повышении эффективности сорбционного способа контроля загрязняющих микрокомпонентов. 3 з.п.ф-лы, 4 ил., 1 табл.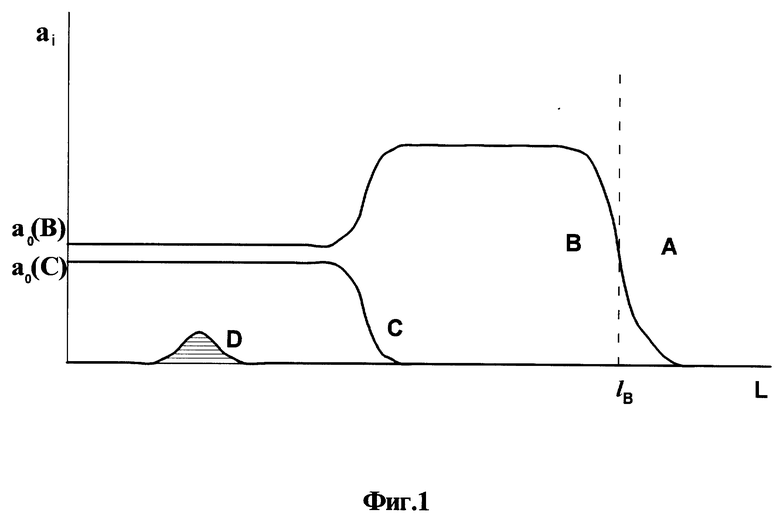
Cv = Mi/V,
где Mi - общее количество иона, накопленного в сорбционном материале за время пропускания контролируемой воды;
V - объем контролируемой воды, пропущенный через материал, определяемый как функция скорости перемещения в слое материала концентрационного профиля слабее сорбируемого иона из числа макрокомпонентов контролируемой воды.
| Андреева И.Ю | |||
| и др | |||
| Групповое концентрирование ионов тяжелых металлов с помощью волоконионитов на основе полиакрилонитрила | |||
| Вестник ЛГУ, сер | |||
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |
| Аширов А | |||
| Ионообменная очистка сточных вод, растворов, газов | |||
| - Л.: Химия, 1983, с.240-242, 113-117 | |||
| RU 2001883 C1, 30.10.93 | |||
| Способ извлечения стронция из высокоминерализованных растворов, содержащих натрий и кальций | 1988 |
|
SU1590441A1 |
| Пожарный двухцилиндровый насос | 0 |
|
SU90A1 |
| Мясоедова Г.В | |||
| и др | |||
| Сорбционное концентрирование и атомно-абсорбционное определение кадмия в минеральных водах | |||
| - Ж | |||
| аналитической химии, 1986, т | |||
| Паровоз для отопления неспекающейся каменноугольной мелочью | 1916 |
|
SU14A1 |
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |

