Изобретение относится к классу соединений пиримидина, которые пригодны для лечения заболеваний и расстройств центральной нервной системы (ЦНС), например для предотвращения церебральных ишемических поражений, к содержащим их фармацевтическим композициям и к способам их получения.
Глутамат является возбуждающей аминокислотой, которая действует как нейтротрансмиттер. Однако, если его внеклеточная концентрация достаточно высока, глутамат действует как сильный нейтротоксин, способный убивать нейроны в центральной нервной системе (Rothman & Olney (1986) Prog. Brain. Res., 63, 69).
Нейротическое действие глутамата включено в ряд заболеваний и расстройств центральной нервной системы, включая церебральное ишемическое поражение, эпилепсию и такие хронические нейродегенеративные расстройства, как болезнь Алцмейера, расстройства двигательной системы и хорея Хантингтона (Meldrum Clinical Science (1985) 68, 113-122). Кроме того, глутамат был включен в другие нейрологические расстройства, такие как маниакальная депрессия, депрессия, шизофрения, неврологический синдром высокого давления, хроническая боль, тригеминальная невралгия и мигрень.
В Европейской патентной заявке N 21121 раскрыта группа 3,5-диамино-6-(замещенный фенил)-1,2,4-триазинов, которые активны при лечении ЦНС заболеваний, например при лечении эпилепсии. Одно из соединений, описанных в этой заявке, 3,5-диамино-6-(2,3-дихлорфенил)-1,2,4-триазин (ламотригин), как было показано, ингибирует высвобождение возбуждающих аминокислот, глутамата и аспартата (Leach et al. Epilepsia 27, 490-497, 1986, A.A. Miller et al. New anticonvulsant drugs. Ed. Meldrum and Porter, 165-177, 1987).
Некоторые фенилпиримидины известны специалистам как обладающие противомалярийной активностью, см., например, Brit. J. Pharmacol. 6, 185-200 (1951; JACS, 73, 3763-70 (1951). Другие фенилпиримидины известны из Chem. Biol. Pteridines, 463-468 (1982) and Pharmacotherap. Budesinsky, p. 129-141 (1963), ed. Oldrich Hanc.
Авторы настоящего изобретения обнаружили, что ряд замещенных соединений пиримидина, как определено в формуле I, являются потенциальными ингибиторами высвобождения глутамата, эти соединения полезны при лечении вышеуказанных ранее заболеваний и расстройств центральной нервной системы, которые вызывает действие глутамата. Соединения пиримидина формулы I также являются ингибиторами высвобождения аспартата.
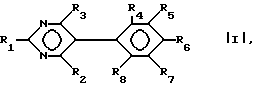
Таким образом, в первом аспекте настоящего изобретения предложен пиримидин формулы I: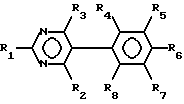
где
R1 - NH2, N-(алкил)амино или N,N-ди(алкил)амино;
R2 - NH2;
R3 является трифторметилом или группой CH2X, где
X - гидрокси, C1-C6-алкокси, фенокси, бензилокси или галоид;
R4 и R5 - каждый галоид;
R6 - R8 являются водородом,
и его фармацевтически пригодные соли присоединения кислоты.
Некоторые соединения формулы I являются хиральными, и следует учитывать, что в этих случаях формула I включает как рацемическую смесь, так и индивидуальные энантиомеры таких соединений.
В настоящем изобретении
R1 предпочтительно является амино;
R3 предпочтительно является метоксиметилом, трифторметилом, бензилоксиметилом или феноксиметилом.
Предпочтительно, чтобы в формуле I R3 являлся трифторметилом или метоксиметилом или же R3 может быть фторметилом, R4 предпочтительно хлор, R5 предпочтительно хлор.
Предпочтительно, чтобы алкильный фрагмент содержал от
1 до 4 атомов углерода.
Наиболее предпочтительным классом соединений формулы I являются те, в которых
R1 выбирают из амино, N,N-диметиламино и N-этиламино;
R2 является аминогруппой,
R3 выбирают из трифтометила, бензилоксиметила и метоксиметила,
R4 и R5 - оба хлор.
Соединения формулы I, являются потенциальными ингибиторами высвобождения глутамата, демонстрируют лишь слабые (то есть ИК50 более 20 мкм) или незначительные ингибирующие эффекты на фермент дигидрофолатредуктазу.
Соединения формулы I можно использовать при лечении или профилактике острых и хронических нарушений центральной нервной системы млекопитающих, вызванных действием глутамата.
Острые состояния включают церебральную ишемию, которая может возникать по разным причинам из различных случаев, включая удары, остановку сердца, послеоперационные состояния, неонатальную гипоксию и гипогликемию, а также физические поражения или травмы позвоночника или мозга. Хронические нейродегенеративные расстройства, которые можно лечить, включают болезнь Алцхеймера, хорею Хантингтона, оливопонтоцеребральную атропию, расстройства двигательной системы. Другие неврологические состояния, которые можно лечить соединениями формулы I, включают депрессии, маниакальные депрессии, шизофрению, хронические боли, эпилепсию, тригеминальную невралгию и мигрени.
Лечение или предотвращение расстройств или заболеваний ЦНС у млекопитающих, включая человека, вызванных действием глутамата, может быть осуществлено путем введения млекопитающему эффективного количества соединения формулы I или его фармацевтически приемлемой соли присоединения кислоты.
В частности, млекопитающим, предрасположенным к имеющим нейротоксичные уровни внеклеточного глутамата в центральной нервной системе, может быть введено нетоксичное эффективное количество соединения формулы I или его фармацевтически приемлемой соли присоединения кислоты.
Предпочтительные новые соединения настоящего изобретения включают нижеследующие, причем номера соответствуют номерам примеров, в которых они появляются.
Пример N
1. 2,4-диамино-5-(2,3-дихлорфенил)-6-трифторметилпиримидин
2. 2,4-диамино-5-(2,3-дихлорфенил)-6-метоксиметилпиримидин
3-3. 2,4-диамино-5-(2,3-дихлорфенил)-6-гидроксиметилпиримидин
3-4. 2,4-диамино-5-(2,3-дихлорфенил)-6-фторметилпиримидин
4. 2,4-диамино-5-(2,3-дихлорфенил)-6-феноксиметилпиримидин
или их фармацевтически приемлемые соли присоединения кислоты.
Подходящие фармацевтически приемлемые соли присоединения кислоты соединений формулы I включают те, которые образуются как с органическими, так и с неорганическими кислотами.
Так, предпочтительные соли включают те, которые получены из соляной, бромистоводородной, серной, лимонной, винной, фосфорной, молочной, пировиноградной, уксусной, янтарной, фумаровой, малеиновой, оксалоуксусной, метансульфокислоты, этансульфокислоты, пара-толуолсульфокислоты, бензолсульфокислоты и изетионовой кислоты. Эти соли можно получить при взаимодействии соединений в виде свободных оснований с соответствующими кислотами.
Хотя соединения формулы I можно вводить в виде чистых химических соединений, предпочтительно включать их в фармацевтические композиции. Композиции настоящего изобретения включают новые соединения формулы I, определенной ранее, или их фармацевтически приемлемые соли вместе с одним или более носителями и необязательно другими терапевтическими ингредиентами. Носитель (носители) должен быть "приемлем" в том смысле, что он должен быть совместим с другими ингредиентами композиции и не должен причинять вред тому, кто их принимает.
Эти композиции включают такие, которые пригодны для орального, парэнтерального (включая подкожные, внутримышечные и внутривенные инъекции), ректального и поверхностного (включая нанесение на кожу, через нос или под язык) введения, хотя наиболее подходящий способ будет зависеть от вида, состояния и степени заболеваний пациента. Композиции обычно можно изготовить в единичной дозированной форме и можно получить любыми способами, известными фармацевтам. Все способы включают стадию соединения формулы I или его фармацевтически приемлемой соли присоединения кислоты ("активного ингредиента") с носителем, который состоит из оного или более вспомогательных ингредиентов. Обычно композиции приготавливают, тщательно перемешивая активный ингредиент с жидкими носителями или тонко измельченными твердыми носителями, или обоими, а затем, при необходимости, придавая композиции нужную форму.
Композиции настоящего изобретения, пригодные для орального введения, могут быть представлены в дискретном виде, например в виде капсул, лепешек или таблеток, причем каждая содержит определенное количество активного ингредиента в виде порошка или гранул, в виде раствора или суспензии в водном разбавителе или в неводной жидкости или в виде жидкой эмульсии типа масло в воде или вода в масле, активный ингредиент может быть также представлен в виде шариков, электуария или пасты.
Таблетки можно изготавливать прессованием или из расплава необязательно с одним или более из вспомогательных ингредиентов. Прессованные таблетки можно получить прессованием в подходящем устройстве активного ингредиента в свободнопересыпающейся форме, например в виде порошка или гранул, необязательно в смеси со связующим, смазывающим агентом, инертным разбавителем, поверхностно-активным или диспергирующим агентом. Таблетки из расплава можно получить, расплавляя в соответствующем устройстве смесь порошкообразного соединения, смоченного инертным жидким разбавителем. Таблетки могут быть необязательно с нанесенным покрытием, а композиции могут быть такими, чтобы обеспечить медленное и контролируемое высвобождение из него активного ингредиента.
Композиции для парэнтерального введения включают водные и неводные стерильные растворы для инъекций, которые могут содержать антиоксиданты, буферирующие агенты, бактериостаты и растворенные вещества, которые придают композициям изотоничность с кровью пациента, и водные и неводные стерильные суспензии, которые могут включать суспендирующие и загущающие агенты. Такие композиции могут существовать в виде единичных доз или в упаковках для многих доз, например в запаянных ампулах и пробирках, и могут храниться в лиофилизированных состояниях, которые требуют только добавления стерильного жидкого носителя, например воды для инъекций, непосредственно перед использованием.
Произвольные растворы для инъекций и суспензий можно приготовить из стерильных порошков, гранул и таблеток описанных ранее типов.
Композиции для ректального введения можно приготовить в виде суппозиториев с такими обычными носителями, как масло какао или полиэтиленгликоль.
Композиции для поверхностного нанесения, например в рот или под язык, включают лепешки, содержащие активный ингредиент с такими вкусовыми добавками, как сахароза и акация или трагакант, и пастилки, содержащие активный ингредиент на базе желатина и глицерина или сахарозы и акации.
Предпочтительные композиции единичных доз содержат эффективную дозу, как будет указано далее, или соответствующую ее часть активного ингредиента.
Следует учитывать, что в дополнение к указанным конкретно ранее ингредиентам композиции настоящего изобретения могут включать и другие агенты, обычные для практики и касающиеся рассматриваемого типа композиции, например композиции для орального введения могут включать вкусовые агенты или отдушки.
Таблетки или другие формы, предложенные в дискретном виде, могут содержать такие количества соединения формулы I, которые эффективны в таких дозах или в кратных им дозах, как, например, единичные дозы могут составлять от 5 до 500 мг и обычно от 10 до 250 мг.
Соединения формулы I предпочтительно используют для лечения расстройств и заболеваний ЦНС путем орального приема или путем инъекций (интрапарэнтеральных или подкожных). Точные количества вводимого соединения зависят от лечащего врача. Однако прописываемая доза должна зависеть от ряда факторов, включая возраст и пол пациента, точный диагноз и степень заболевания подлежащего лечению. Так, например, при лечении больного эпилепсией интервал доз должен быть, по-видимому, существенно ниже, чем при лечении пациента после удара для снятия церебральных ишемических поражений. Точно также и способ введения, по-видимому, должен зависеть от состояния и его тяжести.
Соединения формулы I можно вводить орально или путем инъекций в дозах от 0,1 до 30 мг/кг в день. Интервал доз для взрослых обычно от 8 до 2400 мг/день и предпочтительно от 35 до 1050 мг/день. Некоторые соединения формулы I являются соединениями длительного действия, и может оказаться важным принять в первый день начальную дозу от 70 до 2400 мг, а затем снизить дозу в последующие дни до 20-1200 мг.
Соединения с длительным действием особенно важны клинически, так как с ними легче работать. При хронических ситуациях их можно вводить без вливаний, и таким образом свести к минимуму медицинское вмешательство, также и в острых состояниях успокоить больного, снижая дневную дозу. Напротив, такие быстродействующие соединения позволяют обеспечивать клинический контроль за фармакологическим действием соединения с высокой точностью, так как такие соединения удаляются из центральной нервной системы быстро.
Соединения настоящего изобретения можно получить любым способом, известным для аналогичных соединений (см. JACS vol 73 (1951) 3763-70).
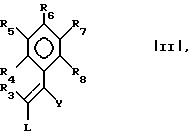
В настоящем изобретении предложен также способ получения пиримидина формулы I или его фармацевтически приемлемой соли присоединения кислоты взаимодействием соединения формулы II: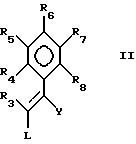
где
R3 до R8 имеют указанные ранее значения, L является отщепляемой группой, а Y является цианогруппой,
с соединением формулы III: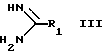
где
R1 имеет указанные ранее значения,
выделяя соединение формулы I в виде свободного основания или его фармацевтически приемлемой соли присоединения кислоты и, необязательно, превращая основание в его фармацевтически приемлемую соль присоединения кислоты или в другой пиримидин формулы I или его фармацевтически приемлемую соль присоединения кислоты.
Если в продукте вышеуказанного процесса R3 является группой CH2OR, где R является алкилом, этот продукт можно превратить в CH2X взаимодействием с HX (X - галоид) в, например, уксусной кислоте. Далее его можно превратить во фторметил обработкой, например, фторидом цезия (CsF).
В другом варианте группу CH2OR можно деалкилировать до получения соответствующего спирта, например Me3SiI, а его далее превратить во фторметил с помощью трифторида диэтиламиносеры (DAST).
В воплощении указанного: A) соединение формулы II, где R3 группа CH2OR, в которой R представляет C1-C6-алкил, подвергают взаимодействию с соединением формулы III, получая пиримидин формулы I, где R3 - указанная группа CH2OR;
B) полученный таким образом пиримидин отделяют и деалкилируют для получения другого пиримидина формулы I, где R3 группа CH2OH; и
C) полученный таким образом пиримидин подвергают взаимодействию с трифторидом диэтиламиносеры, получая пиримидин формулы I, где R3 - фторметил.
Следует учитывать, что можно провести и другие взаимопревращения, как ясно специалистам, в соответствии со стандартной методологией.
Примеры подходящих отщепляемых групп (L) включают C1-4-алкокси, галоид, анилино, морфолино, C1-4-алкиламино, бензиламино или алкилтио.
Предпочтительно реакцию соединения формул II и III вести в неводных растворителях, например в алканоле, например в этаноле, при повышенных температурах (например, между 50 и 110oC) в основании, предпочтительно алканоксиде, предпочтительно при температуре кипения с обратным холодильником, используя в качестве основания этилат натрия.
Соединения формулы II можно получить способами, известными специалистам (JACS, 1951, 73, 3763-3770), например взаимодействием соединения формулы IV: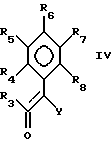
где
Y - циано, R4-R8 определены выше,
с диазометаном или с алкилортоэфиром (JACS 1952, 74, 1310-1313), или конденсацией с амином.
Соединение формулы IV может быть получено способом, известным в данной области
(JACS, 1951, 73, 3763-70).
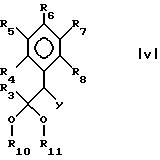
В настоящем изобретении предложен также способ получения пиримидина формулы I взаимодействием соединения формулы V: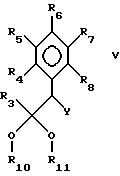
где
R3-R8 имеют указанные ранее значения, а R10 и R11 являются алкилом или вместе образуют -(C(R)2)n-, где n имеет значения от 2 до 4, а R является H или алкилом,
с соединением формулы III. Более предпочтительно, чтобы R был амино.
Предпочтительно реакцию вести в неводном растворителе, например этаноле, при кипячении с обратным холодильником, используя в качестве основания этилат натрия.
В приводимых далее примерах настоящего изобретения сокращения и обозначения химических соединений соответствуют общепринятым у специалистов и имеют следующие значения:
DMF - диметилформамид
Et2O - диэтиловый эфир
NaOEt -тилат натрия
EtOH - этанол
AcOH - уксусная кислота
MeOH - метанол
DMSO - диметилсульфоксид
DME - диметоксиэтан
Et2N - триэтиламин
Пример 1. Получение 2,4-диамино-5-(2,3-дихлорфенил)-6- трифторметилпиримидина.
1. Получение 2,3-дихлорбензилового спирта.
К раствору 2,3-дихлорбензальдегида (Aldrich, 50 мг) в 800 мл спирта при комнатной температуре добавляют NaBH4 (8,54 г) и полученную смесь перемешивают в течение 1,5 часа. Реакцию гасят водой, а растворитель выпаривают в вакууме до разделения остатка между CHCl3 и насыщенным раствором NaHCO3. Органическую фазу промывают рассолом, сушат над сульфатом магния, фильтруют и растворитель выпаривают в вакууме до получения белого твердого продукта, 48,38 г, Т.плавления 87-87,5oC.
2. Получение 2,3-дихлорбензилбромида.
К раствору спирта в бензоле (500 мл) в атмосфере азота добавляют PBr3 (167,8 г) и полученную смесь перемешивают при 55-60oC в течение 3,5 часа. После охлаждения полученную смесь выливают на измельченный лед (2 л) и выделяют бензольный слой. Водную фазу промывают бензолом (x3) и объединенные бензольные экстракты промывают насыщенным NaHCO3 раствором и водой, сушат над MgSO4, фильтруют и растворитель выпаривают, оставляя коричневатую жидкость, которая отверждается при стоянии. Получают 37,53 г, Т.пл. 31-32oC.
3. Получение 2,3-дихлорфенилацетонитрила.
Бромид суспендируют в DMCO (155 мл)/вода (105 мл) при 0oC и KCN (20,24 г) добавляют порциями. После перемешивания при 30-35oC в течение 2 часов суспензию разбавляют водой и экстрагируют Et2O. Объединенные эфирные экстракты промывают водой, сушат над сульфатом магния, фильтруют и растворитель выпаривают в вакууме до получения твердого продукта белого цвета, 27,52 г, т.пл. 64-67oC.
4. Получение 2-(2,3-дихлорфенил)-4,4,4-трифтор-3-оксо- бутиронитрила.
К раствору NaOEt (из 1,48 г Na) в EtOH (25 мл) при комнатной температуре в атмосфере азота добавляют 10,0 г нитрила, а затем 9,3 г этилтрифторацетата и полученную смесь перемешивают при кипячении с обратным холодильником в течение 5 часов. После охлаждения растворитель удаляют в вакууме, а остаток растворяют в воде. Водную фазу промывают Et2O (сливают), подкисляют серной кислотой и экстрагируют Et2O. Объединенные экстракты Et2O промывают водой, сушат над сульфатом магния, фильтруют и растворитель выпаривают в вакууме до получения масла. Его тщательно растирают c петролейным эфиром, отфильтровывают твердую часть и сушат. Получают 9,56 г. Т.пл. 74-75oC.
5. Получение 2-(2,3-дихлорфенил)-4,4,4-трифтор-3- метоксибут-2-енонитрила.
К раствору трифторметилкетона в Et2O (90 мл) при комнатной температуре добавляют диазометан (из 19,35 г Diazald) в Et2O (180 мл) и полученную смесь оставляют выстаиваться при комнатной температуре в течение ночи. Затем избыток диазометана удаляют в вакууме в AcOH и остаток растворяют в Et2O, сушат над сульфатом магния, фильтруют и растворитель выпаривают в вакууме до получения коричневатого твердого вещества, 6,44 г.
6. Получение 2,4-динамино-5-(2,3-дихлорфенил)-6- трифторметилпиримидина.
К раствору вышеуказанного енольного эфира в этаноле (37 мл) добавляют гуанидингидрохлорид (1,92 г), а затем раствор NaOEt (из 540 мг Na) в EtOH (90 мл) и полученную смесь перемешивают при кипячении с обратным холодильником в течение 3 часов. После охлаждения суспензию фильтруют и полученный фильтрат выпаривают досуха в вакууме. После хроматографирования на силикагеле при элюировании CHCl3 до 2% MeOH-CHCl3 получают целевой продукт, который тщательно растирают с Et2O и сушат в вакууме до получения 673 мг, Т.пл. 218-219oC.
7. 2,4-диамино-5-(2,3-дихлорфенил)-6-трифторметилпиримидин-метансульфона.
К суспензии свободного основания (100 мг) в этаноле добавляют метансульфокислоту (30 мг) и полученный прозрачный раствор перемешивают при комнатной температуре в течение 2 часов. Этот раствор выпаривают досуха, а оставшуюся твердую часть тщательно растирают с эфиром, фильтруют и сушат в вакууме, 107 мг, Т.пл. 253-256oC.
8. 2,4-диамино-5-(2,3-дихлорфенил)-6- трифторметилпиримидингидрохлорид.
К раствору свободного основания (150 мг) в метаноле добавляют эфирный раствор хлористого водорода. После перемешивания растворитель выпаривают досуха и полученный твердый продукт тщательно растирают с эфиром, фильтруют и сушат в вакууме, 160 мг. Т.пл. 233-236oC.
Пример 2. Получение 2,4-диамино-5-(2,3-дихлорфенил)-6-метоксиметилпиримидина.
1. Получение 2-(2,3-дихлорфенил)-4-метокси-3-оксо- бутиронитрила.
К перемешиваемому кипящему с обратным холодильником раствору NaOEt (из 1,38 г Na) в EtOH (25 мл) добавляют смесь этилметоксиацетата (8,85 г) и 2,3-дихлорфенилацетонитрил (пример 1.3) (9,3 г), растворенный в 20 мл ДМЕ за 5 минут. Спустя 5 часов появляется осадок (натриевая соль продукта). Полученную смесь охлаждают и фильтруют, полученный фильтрат выпаривают досуха в вакууме и остаток разделяют между эфиром и водой (эфирную фазу сливают). Водный остаток подкисляют 2 н. H2SO4 и экстрагируют эфиром (х2). Объединенные экстракты Et2O промывают водой, сушат над сульфатом магния, фильтруют и выпаривают в вакууме до получения желтого твердого вещества (a). Вышеуказанную натриевую соль растворяют в воде и раствор экстрагируют эфиром и сливают. Водный раствор подкисляют 2 н. H2SO4 и экстрагируют эфиром. Эфирные растворы промывают водой, сушат над сульфатом магния, фильтруют и выпаривают в вакууме до получения твердого продукта белого цвета (b).
Вышеуказанные продукты (a и b) объединяют до получения 10,4 г, которые используют без дальнейшей очистки. На ТСХ одно пятно (19:1 CH2Cl2:MeOH), Rf 0,35.
2. Получение 2-(2,3-дихлорфенил(-3,4-диметоксибут-2-енонитрила.
К перемешиваемому раствору вышеуказанного нитрила (9,4 г) в эфире добавляют порциями диазометан (0,4 - 0,45 M) в эфире. Вначале наблюдается интенсивное вспенивание, и после дополнительного добавления немедленной реакции не происходит. Полученную смесь оставляют выстаиваться при перемешивании при комнатной температуре в течение 3 часов и выпаривают в вакууме в AcOH до получения енольного эфира.
3. Получение 2,4-диамино-5-(2,3-дихлорфенил)-6- метоксиметилпиримидина.
К раствору NaOEt (из 0,92 г Na) в EtOH (40 мл) добавляют гуанидингидрохлорид (3,44 г). Добавляют вышеуказанный раствор енольного эфира в EtOH (30 мл) и полученную смесь кипятят с обратным холодильником приблизительно 3 часа. После охлаждения растворитель выпаривают в вакууме и остаток обрабатывают 5 н. NaOH (примерно 50 мл). Красный раствор фильтруют, растворяют в AcOH (примерно 20 мл), разбавляют 40 мл воды, обрабатывают древесным углем и фильтруют. Полученный фильтрат (желтый раствор) подщелачивают 2 н. NaOH и белы осадок отфильтровывают, сушат и перекристаллизовывают из EtOH. Получают 4,39 г. Т.плавления 237 - 240oC.
Пример 3. Получение 2,4-диамино-5-(2,3-дихлорфенил)-6-фторметилпиримидина.
1. 2,4-диамино-5-(2,3-дихлорфенил)-6-(диэтоксиметил)-пиримидин.
К перемешиваемому при кипячении с обратным холодильником раствору NaOEt (1,38 г натрия) в 25 мл этанола добавляют за 5 минут смесь этилдиэтоксиацетата (13,21 г, 75 ммолей) и (пример 1.3) 2,3-дихлорфенилацетонитрил (9,3 г, 50 ммолей) в сухом диметоксиэтане (20 мл). Спустя 4 часа охлаждают и выпаривают в вакууме. Остаток разделяют между водой (100 мл) и эфиром (100 мл) и эфирную фазу сливают, а водный остаток подкисляют 1 н. серной кислотой. После экстрагирования CH2Cl2 получают ацилацетонитрил (13,47 г), который используют без дальнейшей очистки.
К перемешиваемому раствору вышеуказанного ацетонитрила в эфире (100 мл), охлажденному на льду, добавляют порциями раствор диазометана (примерно 3 г) в эфире. Спустя 2 часа этот раствор выпаривают в вакууме до получения целевого енольного эфира в виде масла, которое используют без дальнейшей очистки.
К раствору NaOEt (из 1,4 г натрия) в этаноле (50 мл) добавляют гуанидингидрохлорид (4,8 г, 50 ммолей). Добавляют раствор вышеуказанного енольного эфира в этаноле (20 мл) и полученную смесь кипятят с обратным холодильником в течение 4 часов, охлаждают и концентрируют в вакууме до приблизительно 30 мл, и разбавляют водой до получения темно-пурпурного твердого вещества, которое фильтруют, растворяют в CH2Cl2, промывают водой, сушат над сульфатом магния и выпаривают в вакууме. Остаток тщательно растирают с 50 мл этанола и отфильтровывают, получая целевой продукт (8,4 г), который используют без дальнейшей очистки (т.пл. 214 - 217oC).
2. 2,4-диамино-5-(2,3-дихлорфенил)пиримидин-6-карбоксальдегид.
Смесь вышеуказанного ацеталя (7 г) и 0,4 M HCl (150 мл) кипятят с обратным холодильником при перемешивании в течение 1 часа, охлаждают на льду и нейтрализуют 2 M NaOH. Полученную смесь фильтруют, промывают водой и сушат на воздухе до получения целевого продукта (6,2 г), который используют без дальнейшей очистки.
3. 2,4-диамино-5-(2,3-дихлорфенил)-6-гидроксиметилпиримидин.
К перемешиваемому раствору вышеуказанного альдегида (2,8 г, 10 ммолей) в смеси диметоксиэтана (15 мл) и этанола (15 мл) добавляют порциями боргидрид натрия (110 мг, 3 ммоля). Спустя 30 минут раствор обрабатывают водой (50 мл) и добавляют несколько капель уксусной кислоты для разрушения остатков боргидрида. После экстрагирования дихлорметаном (2 х 50 мл) промывают водой и экстракт сушат над сульфатом магния. После выпаривания растворителя получают твердое вещество розового цвета, которое тщательно растирают с эфиром, фильтруют и сушат (1,6 г). После перекристаллизации из метанола (50 мл) получают целевой продукт в виде тонких бесцветных кристаллов, 0,65 г. Т.пл. 173 - 176oC.
4. 2,4-диамино-5-(2,3-дихлорфенил)-6-фторметилпиримидин.
К перемешиваемой суспензии 2,4-диамино-5-(2,3-дихлорфенил)-6-гидроксиметилпиримидина (185 мг, 1 ммоль) в сухом дихлорметане (25 мл) в атмосфере азота при -70oC, добавляют по каплям трифторид диэтиламиносеры (263 мкл, 2 ммоля). Полученной смеси дают нагреваться до 0oC, и поддерживают эту температуре в течение 4 часов. После охлаждения до -70oC смесь гасят водным бикарбонатом натрия, экстрагируют дихлорметаном (2 х 50 мл), промывают насыщенным рассолом и сушат над сульфатом магния. После концентрирования получают бесцветную смолу (0,2 г). После хроматографирования на силикагеле при элюировании 0,01:1:19 Et3N:MeOH:CH2Cl2 получают целевой продукт, который тщательно растирают с CCl4 и сушат в вакууме, получают 111 мг, т.плавления 224 - 226oC.
Пример 4. 2,4-диамино-5-(2,3-дихлорфенил)-6-феноксиметилпиримидин.
К перемешиваемому раствору NaOEt (из 1,38 г натрия) в этаноле (70 мл) при кипении с обратным холодильником добавляют за 10 минут смесь 2,3-дихлорфенилацетонитрила (9,3 г) и этилфеноксиацетат (13,5 г) в сухом диметоксиэтане (50 мл). После перемешивания при кипячении с обратным холодильником в течение 3 часов полученную смесь охлаждают, фильтруют и растворитель выпаривают в вакууме. Остаток растворяют в воде, промывают эфиром (сливают), подкисляют 2 н. соляной кислотой и экстрагируют дихлорметаном. Объединенные экстракты промывают рассолом, сушат над сульфатом магния и выпаривают в вакууме, получая желто-коричневый твердый продукт (8 г), который используют без дальнейшей очистки.
К суспензии неочищенного ацилацетонитрила (8 г) и 150 мл эфира добавляют порциями избыток раствора диазометана в эфире. После перемешивания в течение 1 часа при комнатной температуре раствор концентрируют в вакууме до получения енольного эфира, который используют без дальнейшей очистки.
К раствору этинолята натрия (из 0,63 г натрия) в этаноле (29 мл) при комнатной температуре добавляют гуанидингидрохлорид (2,39 г). Спустя 15 минут добавляют раствор вышеуказанного енольного эфира в этаноле (25 мл) и полученную смесь перемешивают при кипячении с обратным холодильником в течение 4 часов. После охлаждения растворитель выпаривают в вакууме. Остаток суспендируют в 2 н. NaOH (75 мл), фильтруют, промывают водой, сушат на воздухе и перекристаллизовывают из этанола до получения целевого продукта в виде бесцветного твердого продукта, 3,82 г. Т.плавления 211 - 213oC.
Предпочтительными среди соединений формулы (I) являются пиримидины вышеприведенных примеров с их фармацевтически приемлемыми солями присоединения кислоты.
Эти соединения имеют следующую двумерную структуру:
Пример 1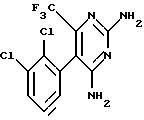
Пример 2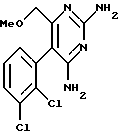
Приведена таблица 1Н ЯМР (см. табл. 1).
Фармакологические активности
Ингибирование высвобождения глутамата и ингибирование DHFR печени крыс
Соединения формулы (I) тестировали на предмет их эффективности на вызванное вератрином высвобождение глутамата из срезов мозга крыс по протоколу, описанному в Epilepsia 27(5):490 - 497, 1986. Протокол для тестирования ингибирования активности DHFR является модификацией способа, описанного в Pharmacology Vol. 20, pp. 561 - 574, 1971.
Полученные результаты приведены в таблице 2, причем IC50 является концентрацией соединения, вызывающей 50% ингибирования (a), вызванного вератрином высвобождения глутамата, и (b) активности фермента DHFR (ДИГИДРОФОЛАТ РЕДУКТАЗЫ).
Пример фармацевтической композиции
A: Инъекции
Соль соединения формулы I растворяют в стерильной воде для инъекций.
В нижеследующих примерах в качестве активного соединения можно использовать любое соединение формулы (I) или его фармацевтической соли.
B: Композиции для капсул
Композиция для капсул A
Композицию A можно получить, смешивая ингредиенты и заполняя состоящие из двух частей твердые желатиновые капсулы полученной смесью.
Композиция для капсул А - мг/капсулу
(a) Активный ингредиент - 250
(b) Лактоза В.Р. - 143
(c) Натрийкрахмалгликоллят - 25
(d) Стеарат магния - 2
Итого - 420
Композиция для капсул В - мг/капсулу
(а) Активный ингредиент - 250
(b) Макрогель 4000ВР - 350
Итого - 600
Капсулы можно изготовить, расплавляя макрогель 4000BP, диспергируя активный ингредиент в расплав и заполняя им желатиновые капсулы, состоящие из двух частей.
Композиция для капсул B (капсулы с регулируемым высвобождением) - мг/капсулу
(a) Активный ингредиент - 250
(b) Микрокристаллическая целлюлоза - 125
(c) Лактоза В.Р. - 125
(d) Этилцеллюлоза - 13
Итого - 513
Композиции для капсул с контролируемым высвобождением можно получить, экстрагируя смешанные ингредиенты (a) до (c), используя экструдер, придавая им сферическую форму и высушивая экструдат. Высушенные таблетки покрывают этилцеллозой (d) в качестве контролирующей высвобождение мембраны и заполняют в твердые желатиновые капсулы, состоящие из двух частей.
Композиция сиропа - г/мл
Активный ингредиент - 0,2500 г
Раствор сорбитола - 1,5000 г
Глицерин - 1,0000 г
Бензоат натрия - 0,0050 г
Вкусовой агент - 0,0125 г
Очищенная вода - до 5,0 мл
Бензоат натрия растворяют в порции очищенной воды и добавляют раствор сорбитола. Добавляют активный ингредиент и растворяют. Полученный раствор смешивают с глицерином, а затем доводят до нужного объема очищенной водой.
Композиции суппозиториев - мг/суппозиторий
Активный ингредиент (63 мкм)х - 250
Твердый жир, BP (Witepsol H15 - Dynamit Nobel) - 1770
Итого - 2020
х Активный ингредиент используют в виде порошка, в котором по крайней мере 90% частиц имеют диаметр 63 мкм или менее.
Одну пятую часть Witepsol H15 расплавляют в сосуде с паровой рубашкой при 45oC максимум. Активный ингредиент пропускают через сито 200 мкм и добавляют к расплавленному основанию, используя Silverson с режущей головкой, до получения однородной дисперсии. Поддерживая температуру смеси 45oC, добавляют остальной Witepsol H15 к суспензии, которую перемешивают для обеспечения гомогенной смеси. Затем всю суспензию пропускают через сито из нержавеющей стали 250 мкм и при непрерывном перемешивании дают остыть до 40oC. При температуре 38 - 40oC по 2,02 г аликвот смеси заполняют в подходящие пластиковые формы и суппозиториям дают остыть до комнатной температуры.
Раскрыт класс замещенных фенилпиримидиновых веществ формулы I, где R1 представляет собой NH2, N-алкиламино, N, N-диалкиламино; R2 представляет собой NH2; R3 представляет собой трифторметил или CH2X, в котором X представляет собой C1-C6-алкил, фелокси, бензилокси, гидрокси или галоген; R4 и R5 каждый означает галоген; R6 - R8 каждый представляет водород, и его фармацевтические приемлемые кислотно-аддитивные соли, которые являются сильными ингибиторами возбуждающей аминокислоты, глютамата. Такие вещества являются полезными при лечении или предупреждении ряда нарушений центральной нервной системы, включая церебральное ишемическое повреждение и эпилепсию. Способ получения соединений формулы I основан на взаимодействии соединений формулы III: R1C(=NH) NH2 с соединениями формул II, V. Предложена фармкомпозиция, обладающая активностью, в качестве ингибитора выделения глутамата, включающая в качестве активного ингредиента эффективное количество соединений формулы I или его фармацевтически приемлемой соли. 3 с. и 6 з.п. ф-лы, 2 табл.
 0
0
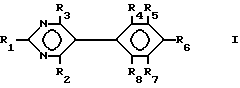
где R1 представляет собой NH2, N-алкиламино; N,N-диалкиламино;
R2 представляет собой NH2;
R3 представляет собой трифторметил или CH2X, в котором X представляет собой C1-C6-алкокси, фенокси, бензилокси, гидрокси или галоген;
R4 и R5 каждый означает галоген;
R6-R8 каждый представляет водород,
и его фармацевтические приемлемые кислотно-аддитивные соли.
2,4-диамино-5-(2,3-дихлорфенил)-6-трифторметилпиримидин;
2,4-диамино-5-(2,3-дихлорфенил)-6-метоксиметилпиримидин,
2,4-диамино-5-(2,3-дихлорфенил)-6-фторметилпиримидин,
2,4-диамино-5-(2,3-дихлорфенил)-6-феноксиметилпиримидин,
2,4-диамино-5-(2,3-дихлорфенил)-6-гидроксиметилпиримидин и их фармацевтически приемлемые кислотно-аддитивные соли.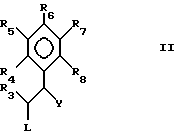
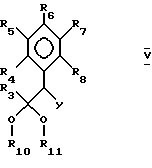
где R3-R8 принимают значения, определенные в п.1;
R10 и R11 оба представляют собой алкил или вместе образуют группу -(C(R)2)n-, в которой R представляет собой водород или алкил и n принимает значение целых чисел от 2 до 4;
L представляет собой удаляемую группу;
Y представляет собой цианогруппу,
с соединением формулы III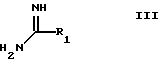
где R1 принимает значения, определенные в п.1,
выделяют образующийся пиримидин формулы I в виде свободного основания или в виде его кислотно-аддитивной соли и необязательно превращают основание в его кислотно-аддитивную соль или в другой пиримидин формулы I или его фармацевтически приемлемую кислотно-аддитивную соль.
Приоритет по признакам и пунктам:
07.12.88 - по п.1, где пиримидин формулы I имеет значение R3, представляющее собой трифторметил, и по п.3;
18.08.89 - по п.2;
07.12.88 - по п.4, где пиримидин формулы I имеет значение R1 = NH2 и R3 - трифторметил;
14.04.89 - если значение R1 = NH2 и R3 - метоксиметил;
07.12.88 - по п.5 - 2,4-диамино-5-(2,3-дихлорфенил)-6-трифторметилпиридин;
14.04.89 - по п.5 - 2,4-диамино-5-(2,3-дихлорфенил)-6-метоксиметилпиримидин;
18.08.89 - по п.5 - 2,4-диамино-5-(2,3-дихлорфенил)-6-фторметилпиридин и 2,4-диамино-5-(2,3-дихлорфенил)-6-гидроксиметилпиримидин;
07.12.88 - по п.6 - при использовании соединений формулы II;
14.04.89, 18.08.89 - в зависимости от приоритета получаемых соединений формулы I по пп.1 - 5.
| Leach et al | |||
| Epilepsia, 27, 1986, p.490 - 497 | |||
| A.A | |||
| Miller et al | |||
| New anticonvulsant drugs | |||
| Ed | |||
| Meldrum and Porter, 1987, p | |||
| Устройство для отыскания металлических предметов | 1920 |
|
SU165A1 |

