Изобретение относится к химико-фармацевтической промышленности, конкретно к получению гидробромида лаппаконитина (аллапинина), который применяется в медицинской практике в качестве лекарственного противоаритмического средства.
Известны способы получения лаппаконитина - полупродукта производства лаппаконитина гидробромида (аллапинина):
из корней Aconitum septentrionale [1] четырехкратной экстракцией измельченного сырья метанолом при комнатной температуре и перемешивании, упаривании в вакууме объединенного экстракта, растворения остатка в 2 н. серной кислоте, фильтрования кислого раствора, промывки фильтрата эфиром, подщелачивания карбонатом натрия, экстракции хлороформом, упаривания экстракта, растворения остатка в бензоле, хроматографировании на Al2O3, элюируя бензолом, упаривания элюата, кристаллизации остатка из эфира;
из корней Aconitum septentrionale путем обработки измельченного сырья гексаном, экстракцией 80%-ным этанолом, отгонкой растворителя, распределением остатка между хлороформом и 2%-ной серной кислотой, подщелачиванием кислого слоя едким натром до pH 12, экстракцией хлороформом, упариванием, растворением остатка в ацетоне, охлаждением, отделением кристаллического осадка, получением лаппаконитина двойной перекристаллизацией из смеси хлороформ-ацетон, а также хроматографированием упаренных объединенных маточников на Al2O3 в системах гексан - диэтиловый эфир - этанол с увеличивающейся полярностью и выделением лаппаконитина из фракций 60-80% эфир-гексан [2];
из корней Aconitum excelsum (синоним Aconitum leucostomum) путем обработки измельченного сырья 5%-ным раствором соды, исчерпывающей экстракцией дихлорэтаном, экстракцией дихлорэтанового раствора 5%-ной серной кислотой, прибавлением к сернокислому экстракту эфира и подщелачиванием 5%-ным раствором соды, фильтрованием выпавшего осадка (часть А), экстракцией маточника эфиром (часть Б), растворением части А в 5%-ной соляной кислоте и прибавлением избытка насыщенного раствора перхлората натрия, кристаллизацией перхлората лаппаконитина из метанола; растворением части Б в 5%-ной серной кислоте, дробным подщелачиванием с последующим извлечением эфиром и упариванием получают дополнительное количество лаппаконитина [3].
Однако рассмотренные работы носят научно-исследовательский характер, в них отсутствует детальное описание экстракции, сведения о чистоте продукта, процесс выделения лаппаконитина является многостадийным.
Известны способы получения лаппаконитина гидробромида (аллапинина) из надземной части Aconitum septentrionale 8-кратной экстракцией хлороформом при комнатной температуре с последующим выделением гидробромида лаппаконитина, однако детальное описание экстракции и сведения, в частности, выхода и чистоты лаппаконитина гидробромида отсутствуют [4].
Наиболее близким к изобретению является способ получения лаппаконитина гидробромида (аллапинина) путем пятикратной обработки травы аконита белоустого (Aconitum leucostomum) 80%-ным раствором спирта, сгущения объединенного экстракта до 10% от первоначального объема, подщелачивания до pH 9 и четырехкратного извлечения алкалоидов хлороформом, пятикратной экстракцией 5%-ным раствором серной кислоты, двухкратного промывания объединенного кислого экстракта хлороформом, подщелачивания содой до pH 9, четырехкратного извлечения суммы алкалоидов хлороформом, упаривания до сухого остатка, обработки остатка в спиртовом растворе 5%-ной HBr до pH 2, отделение осадка технического аллапинина [5]. К недостаткам этого способа следует отнести многостадийность процесса, возможность протекания побочных реакций гидролиза, необходимость не менее трехкратной перекристаллизации технического продукта до аллапинина фармакопейной чистоты, отсутствие данных по выходу фармакопейного аллапинина.
В заявленном техническом решении предложено получать лаппаконитин гидробромид (аллапинин) по упрощенной технологии с хорошим выходом и фармакопейной чистотой из корней или травы аконита белоустого (Aconitum leucostomum) или корней или травы аконита северного (Aconitum septentrionale) экстракцией 50-80%-ным (по объему) водным ацетоном с удалением примесей неалкалоидного характера из подкисленного водного остатка (после отгонки ацетона из объединенного экстракта) однократной обработкой дихлорэтаном, выделением алкалоидов из раствора 2-3-кратной экстракцией дихлорэтаном или этилацетатом. Дальнейшее понижение концентрации ацетона ниже 50% приводит к образованию устойчивых эмульсий, а ее повышение выше 80% нецелесообразно по экономическим соображениям, так как увеличивает расход ацетона. Использование при экстракции из водно-щелочного слоя дихлорэтана или этилацетата обеспечивает полноту выделения лаппаконитина за 2-3 экстракции. Предложенная совокупность отличительных признаков позволяет сократить две стадии процесса (четырехкратная экстракция хлороформом щелочного упаренного экстракта, пятикратная экстракция 5%-ной серной кислотой), уменьшить протекание побочных реакций, связанных с легкостью гидролиза сложноэфирной группы лаппаконитина в кислой (до дезацетилаппаконитина) и щелочной (до лаппаконина) средах за счет исключения одной стадии подщелачивания и 5-кратной экстракции алкалоидов раствором серной кислоты. Кроме того, спиртовая экстракция в известном, по сравнению с ацетоновой в заявленном, обладает меньшей избирательностью и извлекает из растительного сырья большее количество балластных веществ, сильно окрашена. Отгонка спирта требует более высоких температур, что приводит к частичному осмолению. В заявленном применении на начальном этапе экстракции растительного сырья водно-ацетоновых растворов, а для извлечения алкалоидов из щелочного раствора этилацетата или дихлорэтана, позволяет получать экстракты с меньшим содержанием балластных веществ. Все вышеперечисленные факторы приводят к тому, что в предлагаемом методе получаемый аллапининин после однократной перекристаллизации дает продукт, удовлетворяющий требованиям фармакопейной статьи [6], отпадает необходимость в многократной перекристаллизации сырца (потери на каждой стадии очистки составляют 15 - 20%), что повышает выход фармакопейного аллапинина.
Сущность заявленного технического решения состоит в следующем: измельченное растительное сырье экстрагируют 50-80%-ным водным ацетоном. Из объединенных экстрактов удаляют ацетон, водный остаток подкисляют раствором серной кислоты до pH 2-3, однократно промывают дихлорэтаном, подщелачивают до pH 9 и извлекают алкалоиды органическим растворителем (дихлорэтаном, этилацетатом).
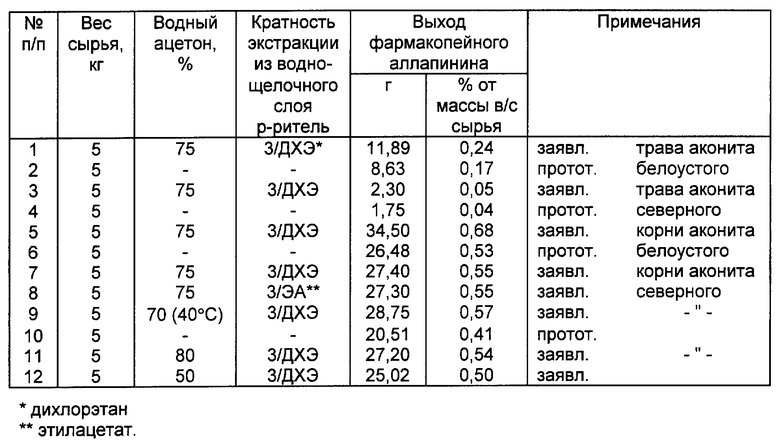
Пример 1. Экстрагируют четырехкратно 5 кг измельченного сырья (травы аконита белоустого) 75%-ным водным ацетоном (ацетон - вода 75 : 25 по объему) методом настаивания. На первую экстракцию используют 20 литров растворителя, после 7 часового настаивания сливают 10 л экстракта. Повторно заливают 10 л экстрагента, через 7 часов сливают 10 л экстракта. Соответственно проводят 3 и 4-е извлечение алкалоидов. Из объединенного водно-ацетонового экстракта отгоняют ацетон, водный остаток (8 л) подкисляют 20% серной кислотой до pH 3 и однократно экстрагируют 3 л дихлорэтана, затем подщелачивают содой до pH 9 и трижды экстрагируют щелочной раствор дихлорэтаном порциями по 4 л. Объединенный дихлорэтановый экстракт из щелочного раствора упаривают, к остатку прибавляют 100 мл ацетона (в соотношении остаток:ацетон приблизительно 1:3 по объему), через сутки отфильтровывают 13,0 г кристаллического лаппаконитина. К суспензии 13 г лаппаконитина в 50 мл спирта прибавляют при перемешивании 10%-ный спиртовый раствор HBr до pH 3, выдерживают в течение 10 часов при комнатной температуре и отфильтровывают 14,75 г аллапинина. Однократная перекристаллизация 14,75 г технического аллапинина из водного спирта по методике, описанной в технологическом регламенте по получению аллапинина (спирт-вода, 80:20 по объему; соотношение продукт-растворитель 1: 15) дает 11,89 г аллапинина, отвечающего требованиям фармакопейной статьи [6].
Пример 2. В условиях прототипа экстрагируют 5 кг измельченной травы аконита белоустого путем 5-ти кратного настаивания 80%-ным раствором этилового спирта. Для первой экстракции используют 15,7 л экстрагента, настаивают 8 часов, сливают 7,5 литров экстракта. Повторно заливают 7,5 л экстрагента, через 6 часов сливают 7,5 л экстракта, соответственно проводят 3, 4 и 5 извлечения алкалоидов с настаиванием в течение 6, 4 и 4 часов. Объединенный экстракт сгущают до 3,8 л, подщелачивают до pH 9 и извлекают алкалоиды хлороформом 4 раза по 1,3 л. Из хлороформного извлечения алкалоиды экстрагируют 5 раз по 0,3 л 5%-ным раствором серной кислоты. Кислые извлечения объединяют, промывают хлороформом 2 раза по 0,5 л, подщелачивают содой до pH 9 и извлекают алкалоиды хлороформом 4 раза по 0,5 л, упаривают и получают сухой остаток суммы алкалоидов в количестве 36,75 г, который растворяют в 125 мл спирта и прибавляют 5%-ную HBr до pH 2. После выдерживания реакционной массы в течение 10 часов отфильтровывают осадок аллапинина в количестве 14,38 г.
Для получения фармакопейного аллапинина технический продукт перекристаллизовывают трехкратно по описанной выше методике примера 1 и получают 8,63 г фармакопейного аллапинина. Проверка соответствия препарата аллапинина фармакопейной статье после каждой перекристаллизации проводилась по методике, описанной в [6].
Пример 3. Экстрагируют траву аконита северного по методике примера 1, причем для экстракции используют 5 кг травы и 40 литров экстрагента - 75%-ного ацетона. После соответствующей обработки выделяют 2,5 г лаппаконитина, из которого получают 2,8 г аллапинина, однократная перекристаллизация которого по методике примера 1 дает 2,3 г аллапинина фармакопейной чистоты.
Пример 4. Экстрагируют траву аконита северного в условиях прототипа по методике примера 2, причем для экстракции используют 5 кг травы и 45,7 литров 80%-ного спирта, выделяют 14,75 г суммы алкалоидов, из которой в условиях прототипа получают 3,0 г аллапинина. Трехкратной перекристаллизацией последнего по методике примера 1 получают 1,75 г продукта фармакопейной чистоты.
Пример 5. Экстрагируют корни аконита белоустого по методике примера 1, причем для экстракции используют 5 кг измельченных корней и 40 литров экстрагента - 75%-ного ацетона. После соответствующей обработки по методике примера 1 выделяют 36 г лаппаконитина, из которого получают 41 г аллапинина. Однократной перекристаллизацией по методике примера 1 получают 34,5 г аллапинина фармакопейной чистоты.
Пример 6. Экстрагируют корни аконита белоустого в условиях прототипа по методике примера 2, причем для экстракции используют 5 кг измельченных корней и 45,7 литров 80%-ного спирта, выделяют 110 г суммы алкалоидов, из которой в условиях прототипа получают 41,5 г аллапинина. Трехкратной перекристаллизацией по методике примера 1 получают 26,48 г аллапинина фармакопейной чистоты.
Пример 7. Экстрагируют корни аконита северного по методике примера 1, причем в экстракции используют 5 кг измельченных корней и 40 литров экстрагента - 75%-ного ацетона. Обработку проводят по методике примера 1 за исключением того, что экстракцию 8 л водно-щелочного раствора проводят трехкратно дихлорэтаном порциями по 4 л и каждую порцию обрабатывают отдельно. Суммарно выделяют 29 г лаппаконитина, причем экстракционное извлечение последнего по 1-3 ступеням экстракции составляет соответственно 79,17 и 4% (22,9; 4,9 и 1,2 г). По методике примера 1 из 29 г лаппаконитина получают 32,9 г аллапинина, однократной перекристаллизацией которого получают 27,4 г фармакопейного аллапинина.
Пример 8. Экстрагируют 5 кг корней аконита северного по методике примера 1, за исключением того, что экстракцию водно-щелочного слоя проводят трехкратно этилацетатом по 4 л. Выделяют 28,9 г лаппаконитина, экстракционное извлечение которого по 1-3 ступеням экстракции составляет 73,21 и 6% (21,1; 6,1 и 1,7 г соответственно). По методике примера 1 из 28,9 г лаппаконитина получают 32,8 г аллапинина, однократной перекристаллизацией которого получают 27,3 г фармакопейного аллапинина.
Пример 9. Экстрагируют 5 кг измельченных корней аконита северного 70%-ным водным ацетоном при 40o и перемешивании. На первую экстракцию подают 20 литров экстрагента, перемешивают при 40o в течение 7 часов, после чего сливают 20 л экстракта. Повторно заливают 10 литров экстрагентов и после 7 часовой экстракции сливают 10 литров экстракта. Соответственно проводят 3 и 4-е извлечение алкалоидов. Водно-ацетоновые экстракты объединяют, ацетон отгоняют, водный остаток (11,5 л) подкисляют 20%-ной серной кислотой до pH 3 и однократно экстрагируют 3 л дихлорэтана, затем подщелачивают до pH 9, пропуская через раствор газообразный аммиак. Щелочной раствор трижды экстрагируют дихлорэтаном порциями по 3 литра. Объединенный экстракт упаривают, к остатку прибавляют 250 мл ацетона, через сутки отфильтровывают 30 г лаппаконитина. К суспензии 30 г лаппаконитина в 100 мл спирта прибавляют при перемешивании 10%-ный спиртовый раствор HBr до pH 3, выдерживают в течение 10 часов при комнатной температуре и отфильтровывают 34 г аллапинина, после однократной перекристаллизации которого по методике примера 1 получают 28,75 г фармакопейного аллапинина.
Пример 10. Экстрагируют корни аконита северного в условиях прототипа, причем, для экстракции используют 5 кг корней и 45,7 л 80%-ного спирта, после соответствующей обработки выделяют 102,5 г суммы алкалоидов, из которых в условиях прототипа получают 32,5 г аллапинина. Для получения фармакопейного аллапинина технический продукт трижды перекристаллизовывают по методике примера 1, получают 20,51 г продукта, удовлетворяющего требованиям фармакопеи.
Пример 11. Экстрагируют корни аконита северного по методике примера 1, причем для экстракции используют 5 кг корней и 40 л 80%-ного ацетона. После соответствующей обработки выделяют 28,5 г лаппаконитина, из которых получают 32,4 г технического аллапинина, однократной перекристаллизацией последнего по методике примера 1 получают 27,2 г аллапинина фармакопейной чистоты.
Пример 12. Экстрагируют корни аконита северного по методике примера 1, причем в экстракции используют 5 кг корней и 40 л 50%-ного ацетона. После соответствующей обработки по методике примера 1 выделяют 27,4 г лаппаконитина, из которого получают 30,2 г технического аллапинина. После однократной перекристаллизации по методике примера 1 получают 25,02 г фармакопейного аллапинина.
В таблице представлены данные по выходу фармакопейного аллапинина при экстракции растительного сырья 50-80%-ным водным ацетоном в сравнении с экстракцией в условиях прототипа. Во всех случаях для сравнительного изучения экстракции использовался один и тот же образец растительного сырья.
Результаты опытов позволяют сделать следующие выводы: предлагаемый способ обеспечивает получение продукта фармакопейной частоты с выходом, существенно превышающим выход в условиях прототипа. Дальнейшее понижение концентрации водного ацетона ниже 50% приводит к образованию устойчивых эмульсий, а ее повышение выше 80% нецелесообразно по экономическим соображениям, так как увеличивает расход ацетона. Использование при экстракции из водно-щелочного раствора дихлорэтана или этилацетата обеспечивает полноту выделения лаппаконитина за 2-3 экстракции. Проведение одной экстракции как дихлорэтаном, так и этилацетатом приводит к существенной потере (21-27%) целевого продукта, а проведение четвертой экстракции нецелесообразно по экономическим соображениям.
Технико-экономические преимущества.
Предложенный способ получения лаппаконитина гидробромида сокращает число стадий процесса, обеспечивает высокий выход и степень чистоты.
Список литературы
1. Marion L. , Fonzer L., Wilkins C.K., Boca Jr and J.P., Sandberg F., Thorsen R., Linden E. //Can J. Chim. 1967. Vol. 45. P. 969-973.
2. Ross A., Pelletier S.W. //Tetrahedron. 1992. Vol. 48. N 7. P. 1183-1192.
3. Платонова Т. Ф. , Кузовков А.Д. Массагетов П.С. //ЖОХ. - 1958, с. 258-261.
4. Усманова С.К., Тельнов В.А., Юнусов М.С., Абдуллаев Н.Д., Шретер А.И. , Филипова Г.Б. //ХПС. - 1987. с. 879-883.
5. Авторское свидетельство СССР N 1196004, кл. A 61 K 35/78, 1985.
6. Временная фармакопейная статья 42-1667-86 от 11.12.86 и изменение N 1 от 26.02.88.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ЛАППАКОНИТИН ГИДРОБРОМИДА И ЛАППАКОНИТИНА | 2016 |
|
RU2641967C1 |
| Способ получения лаппаконитин гидробромида | 2016 |
|
RU2646794C1 |
| СПОСОБ ПОЛУЧЕНИЯ ЛАППАКОНИТИНА ГИДРОБРОМИДА (ВАРИАНТЫ) | 2012 |
|
RU2545799C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРЕПАРАТОВ ИЗ РАСТИТЕЛЬНОГО ЛЕКАРСТВЕННОГО СЫРЬЯ | 1999 |
|
RU2176919C2 |
| АНТИАРИТМИЧЕСКОЕ СРЕДСТВО, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ОСНОВЕ АНТИАРИТМИЧЕСКОГО СРЕДСТВА | 2022 |
|
RU2825632C2 |
| СПОСОБ ПОЛУЧЕНИЯ ЛАППАКОНИТИН ГИДРОБРОМИДА | 2017 |
|
RU2676304C1 |
| СПОСОБ ПОЛУЧЕНИЯ "ЛАППАКОНИТИНА ГИДРОБРОМИДА" | 2004 |
|
RU2295355C2 |
| СПОСОБ ПОЛУЧЕНИЯ АЛЛАПИНИНА | 2003 |
|
RU2238103C1 |
| СПОСОБ ПРОМЫШЛЕННОГО ПОЛУЧЕНИЯ АЛЛАПИНИНА | 2012 |
|
RU2518742C2 |
| СПОСОБ ПОВЫШЕНИЯ СОДЕРЖАНИЯ АЛКАЛОИДА ЛАППАКОНИТИНА В ЗАГОТОВЛЕННЫХ КОРНЕВИЩАХ ACONITUM SEPTENTRIONALE KOELLE | 2014 |
|
RU2580042C2 |
Изобретение относится к химико-фармацевтической промышленности, конкретно к получению лаппаконитина гидробромида ( аллапинина), который применяется в качестве лекарственного противоаритмического средства. Экстракцию корней или травы аконита северного ( Aconitum Septenrionale), корней или травы аконита белоустого (Aconitum leucostomum) проводят 50-80%- ным по объему водным ацетоном с удалением примесей из упаренного и подкисленного экстракта однократной обработкой дихлорэтаном, выделением алкалоидов из щелочного раствора 2-3-кратной экстракцией дихлорэтаном или этилацетатом. Способ обеспечивает получение лаппаконитина гидробромида по упрощенной технологии с высоким выходом и фармакопейной степенью чистоты. 1 табл.
Способ получения лаппаконитина гидробромида путем экстракции, упаривания экстракта, обработки его водным раствором серной кислоты, подщелачивания, извлечения алкалоидов органическим растворителем и обработки их спиртовым раствором бромистоводородной кислоты, отличающийся тем, что проводят экстракцию корней или травы аконита северного (Aconitum septentrionale) или корней или травы аконита белоустого (Aconitum leucostomum) 50 - 80%-ным по объему водным раствором ацетона с удалением примесей из упаренного и подкисленного экстракта однократной обработкой дихлорэтаном, выделение алкалоидов из щелочного раствора осуществляют 2 - 3-кратной экстракцией дихлорэтаном или этилацетатом.
| Способ получения лаппаконитина гидробромила | 1984 |
|
SU1196004A1 |
| СПОСОБ ПОЛУЧЕНИЯ СРЕДСТВА, ОБЛАДАЮЩЕГО ПРОТИВОАТЕРОСКЛЕРОТИЧЕСКИМ, АНТИЛИТОГЕННЫМ, КАРДИОТОНИЧЕСКИМ, АНТИАЛЛЕРГИЧЕСКИМ И ИММУНОМОДУЛИРУЮЩИМ ДЕЙСТВИЕМ | 1992 |
|
RU2036657C1 |
| Устройство для усиления микрофонного тока с применением самоиндукции | 1920 |
|
SU42A1 |

