Данное изобретение относится к химическому процессу и, в частности, к способу получения 2-оксиарилальдоксимов.
Известно использование 2-оксиарилальдоксимов в качестве экстрагентов при гидрометаллургическом извлечении металлов из металлических руд. Это процесс описан, например, в заявке Великобритании GB-A-1421766 и несколько лет применяется в промышленности.
2-оксиарилальдоксимы могут быть получены обычным способом при реакции соответствующего 2-оксиарилальдегида с гидроксиламином. На практике гидроксиламин обычно применяют в виде соли, например, сульфата или хлорида гидроксиламмония. Эта реакция проводится в присутствии вещества, связывающего кислоту, например, карбоната натрия, которое реагирует с выделяющейся кислотой с образованием сульфата или хлорида натрия, подвергающегося разложению, и двуокиси углерода. Поскольку реакция обычно проводится в двухфазной среде воды и органического растворителя, то, если не принять соответствующие и часто дорогостоящие меры предосторожности, выделение двуокиси углерода может вызвать потерю органического растворителя с экономическими и экологическими последствиями.
Как теперь установлено, проблемы, связанные с современными процессами получения 2-оксиарилальдоксимов, могут быть устранены или сведены к минимуму, если 2-оксиарилальдегид использовать по крайней мере частично в виде соли и/или комплексного соединения определенных металлов, описанных далее, или в присутствии соединения указанных металлов. В некоторых случаях реакция протекает немного быстрее, чем в случае обычного процесса, описанного выше, и, кроме того, объединение реакции оксимирования с реакцией формирования при получении альдегида позволяет достигнуть исполнительных производственных выгод.
Согласно данному изобретению предлагается способ получения 2-оксиарилальдоксимов, который включает в себя реакцию гидроксиламина с 2-оксиарилальдегидом, причем указанная реакция проводится в присутствии соединения металла группы II, группы III, группы IVA или группы VIA Периодической таблицы элементов и/или при таких условиях, что 2-оксиарилальдегид присутствует по крайней мере частично в виде соли и/или комплекса металла группы II, группы III, группы IVA или группы VIA Периодической таблицы элементов.
В качестве примеров 2-оксиарилальдегидов, которые могут использоваться при способе по данному изобретению, можно упомянуть соединения с формулой: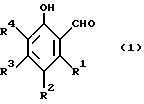
где каждый из R1, R2, R3 и R4 в отдельности обозначает атом водорода или галогена или алкил-, циклоалкил, аралкил-, арил-, алкарил-, алкокси-, арилокси-, ацил- или оксигруппу. Каждая из различных гидрокарбил-, гидрокарбилокси- и ацилгрупп, которые могут быть обозначены как R1, R2, R3 и R4, может содержать до 36 атомов углерода, например 5-22 атомов углерода.
Особенно можно упомянуть о 2-оксиарилальдегидах с формулой: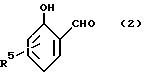
где
R5 обозначает водород или C1-12-алкил-радикал.
Указанные соединения используются при получении 2-оксиарилальдоксимов с формулой: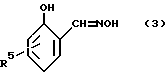
Предпочтительно, R5 является C7-12-алкил-радикалом, особенно в положении 4-относительно гидроксильной группы.
Из-за присутствия соединения металла группы II, группы III, группы IVA или группы VIA Периодической таблицы элементов, 2-оксиарилальдегид, по-видимому, будет находиться в реакционной смеси по меньшей мере частично в виде соли, то есть арилоксида и/или комплекса указанного металла. В качестве примеров особенно подходящих металлов можно упомянуть магний (группа IIA), алюминий (группа IIIB), титан и цирконий (группа IVA) и хром (группа VIA). Соль, содержащая металл, и/или комплекс оксиарилальдегида могут быть приготовлены заранее или могут образовываться в реакционной смеси, может быть, только временно и, возможно, с одним или большим числом производных металла.
Условия реакции, подходящие для получения 2-оксиарилальдегидов в виде солей магния, описаны в нашей заявке EP-A-0529870. Условия, при которых 2-оксиарилальдегиды могут быть получены в присутствии соединений алюминия, титана, циркония и хрома, описаны в заявках EP-A-0077279, EP-A-0106653 и US-A-4231967. Предполагается, что эти условия могут привести к образованию 2-оксиарилальдегида по крайней мере частично в виде солей и/или комплексов указанных металлов.
При осуществлении способа по этому изобретению гидроксиламин можно успешно использовать в виде соли, например, водного раствора соли. В число подходящих солей входят бромид, фосфат, нитрат и ацетат, но особенно сульфат гидроксиламмония.
Если гидроксиламин будет применяться в виде соли, а оксиарилальдегид будет использоваться частично в виде соли, при этом соединения металла будут присутствовать меньше, чем величина химического эквивалента относительно оксиарилальдегида, например каталитического количества соединения титана, то обычно потребуется проводить реакцию в присутствии основания. В число подходящих оснований входят гидроокиси, карбонаты, ацетаты щелочных металлов и т. п. и азотистые основания. Если металл, например магний, используется по крайней мере в количестве, соответствующем химическому эквиваленту относительно оксиарилальдегида, то обычно не требуется добавление дополнительного основания в качестве вещества, связывающего кислоту.
Реакцию, на которой основан способ согласно данному изобретению, можно успешно проводить в среде подходящего растворителя при температурах от 30 до 150oC, хотя при желании могут применяться несколько меньшие или большие температуры. К числу подходящих растворителей относятся органические растворители, например спирты, в которых в значительной степени растворимы как оксиарилальдегид, так и гидроксиламин. Однако гидроксиламин и его соль предпочтительно применять в виде водного раствора. Оксиальдегид, который по крайней мере частично присутствует в виде соли и/или комплекса металла, может в зависимости от его структуры и также от степени ионизации использоваться как таковой или в виде раствора или дисперсии в воде или в органическом растворителе, смешивающемся или несмешивающемся с водой. К числу предпочтительных систем растворителей относятся смеси воды и ароматических углеводородов, как например, толуола или ксилола.
Продукт реакции получения 2-оксиарилальдоксима может быть любым обычным способом извлечен из реакционной смеси, в которой он получен.
Способ по данному изобретению представляет особую ценность для получения 2-оксиарилальдоксимов при реакции гидроксиламина или его соли с бис-арилоксидом магния - производным 2-оксиарилальдегида, особенно 2-оксиарилальдегида вышеприведенных формулы 1 или формулы 2, и особенно для получения экстрагента металла - 5-нонилсалицилальдоксима из соответствующего бис-(2-формил-4-нонилфенолята)магния.
Способ по данному изобретению является также ценным для получения 2-оксиарилальдоксимов при реакции гидроксиламина или его соли с 2-оксиарилальдегидом, особенно с 2-оксиарилальдегидом вышеприведенных формулы 1 или формулы 2 в присутствии производного титана (IV). В число подходящих производных титана (IV) входят соединения с формулой: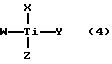
где каждое из W, X, Y и Z в отдельности обозначает атом галогена или алкокси-, арилокси-, алкарилокси-, аралкокси- или циклопентадиенилгруппу или остаток β- дикетона, оксихинолина или произвольно замещенного 2-оксибензальдегида или где два обозначения из W, X, Y и Z вместе обозначают атом кислорода, а каждое из оставшихся двух обозначений в отдельности - атом галогена или алкокси-, арилокси-, аралкокси-, алкарилокси- или ацилоксигруппу или остаток β- дикетона, оксихинолина или произвольно замещенного 2-оксибензальдегида. В общем, алкильная или ацильная часть группы W, X, Y или Z будет содержать до 22 атомов углерода, а арильной частью будет фенил. Конкретными примерами титансодержащих (IV) производных являются тетраизопропилат титана, тетрабутилат титана и тетрафенолят титана.
Способы получения 2-оксиарилальдегидов орто-формилированием произвольно замещенных фенолов в присутствии различных металлсодержащих производных описаны в вышеупомянутых патентных заявках. В соответствии с этими способами 2-оксиарилальдегиды, кажется, получают по крайней мере отчасти в виде солей или комплексов металлов, из которых сам альдегид может быть извлечен обычными методами, например подкислением или экстракцией. Исключительно полезной особенностью данного изобретения является то, что 2-оксиарилальдегиды, полученные при указанных процессах формилирования, могут быть использованы непосредственно как исходные материалы без необходимости выделения их из реакционных смесей, содержащих металлосодержащие производные.
Следовательно, еще одной стороной данного изобретения является способ получения 2-оксиарилальдоксимов при реакции гидроксиламина с 2-оксиарилальдегидом, который является прямым продуктом реакции фенола, имеющего по крайней мере одно свободное орто-положение, с формальдегидом или соединением, выделяющим формальдегид в свободном состоянии при по существу безводных условиях в присутствии соединения металла группы II, группы III, группы IVA или группы VIA Периодической таблицы элементов.
В предпочтительном варианте осуществления этой стороны изобретения гидроксиламин или соль гидроксиламина реагирует с 2-формилфенолятом магния, который получен при реакции бис-гидрокарбилоксида магния, производного по крайней мере частично от оксиароматического соединения, по крайней мере одним свободным орто-положением к гидроксильной группе, с формальдегидом или соединением, выделяющим формальдегид в свободном состоянии, при, по существу, безводных условиях.
В особо предпочтительном варианте осуществления этой стороны изобретения гидроксиламин или соль гидроксиламина с бис(2-формилфенолятом) магния, который получен при реакции бис-фенолята магния, производного от фенола по крайней мере с одним свободным орто-положением, с формальдегидом или соединением, выделяющим формальдегид в свободном состоянии, при, по существу, безводных условиях.
По существу безводные условия, необходимые для получения бис(2-формилфенолята) магния путем реакции формилирования, обычно могут быть обеспечены использованием безводных реагентов наряду с применением общепринятых методов удаления случайной влаги, например перегонкой. Обычно полезно проводить реакцию в присутствии по существу безводного растворителя. В число подходящих растворителей обычно входят инертный неполярный или низкополярный органический растворитель и/или полярный органический растворитель, способный действовать как лиганд в отношении атомов магния.
Подходящие инертные неполярные или низкополярные органические растворители будут жидкостями при температуре реакции и будут действовать как растворители для бис-гидрокарбоксида магния. Предпочтительно, чтобы они давали возможность удалять перегонкой один или большее число летучих побочных продуктов. Примерами подходящих инертных растворителей являются ароматические углеводороды, например толуол, ксилол, мезитидол, кумол, цимол, тетралин, и хлорированные ароматические углеводороды, например хлорбензол и ортодихлорбензол. Можно использовать смеси инертных растворителей.
Подходящие полярные растворители будут жидкостями при температуре реакции и могут рассматриваться как совместные растворители при использовании вместе с неполярными или низкополярными растворителями. В качестве примеров подходящих полярных совместных растворителей могут быть упомянуты полярные апротонные растворители, такие, как диметилсульфоксид, сульфолан, диметил-ацетамид, N-формилпиперидин, N-метилпирролидинон, тетраметилмочевина и особенно диметилформамид, третичные основания, такие как триэтиламин, триоктиламин, тетраметилэтилендиамин и пиридин, эфиры, такие, как диэтиловый эфир, дифениловый эфир, тетрагидрофуран, глим, диглим, триглим, три[2-(2-метоксиэтокси)этил] амин и кронэфиры, и другие полярные растворители, такие как "Полимег" 1000 и "Целлосолв" и т.п. К числу особенно полезных совместных растворителей относятся низшие спирты, такие как этанол и особенно метанол. Можно использовать смеси совместных растворителей. Совместный растворитель может быть введен в реакционную смесь как таковой или в виде лиганда, уже образовавшего комплекс с атомами магния в бис-диарилоксиде.
При осуществлении способа по данному изобретению некоторые растворители могут обладать способностью действовать в качестве и "растворителя" и "совместного растворителя". Например, вещество, такое как тетрагидрофуран, может быть использовано в качестве растворителя вместе с более полярным совместным растворителем или в качестве совместного растворителя вместе с менее полярным растворителем либо оно может быть использовано в качестве единственного растворителя - совместного растворителя.
Реакцию формирования, используемую для получения бис-/2-формилфенолята/магния лучше проводить при температуре обратного стока в интервале от около 60oC до около 130oC. Побочные продукты реакции, например метанол, метиловый эфир муравьиной кислоты и метилаль, лучше удалять из реакционной смеси по мере их образования. В любом конкретном случае температура нагревания с обратным холодильником будет зависеть от состава системы растворителей и от давления, созданного в реакционной зоне. Формилирование можно успешно проводить при атмосферном или повышенном давлениях, но в некоторых случаях его предпочитается осуществлять при пониженных давлениях, т.е. при давлениях ниже нормального атмосферного давления, например при давлениях 50-700 мм рт. ст. (абсолютное). В частности, установлено, что кроме способствования удалению летучих побочных продуктов реакции, значительное улучшение выхода и/или чистоты альдегида и заметное уменьшение образования побочных продуктов наблюдались при проведении реакции при пониженном давлении (и следовательно, при меньшей температуре) по сравнению с проведением такой же реакции в аналогичном растворителе при атмосферном давлении.
В некоторых случаях может оказаться предпочтительным проведение реакции при температуре обратного стока в интервале от около 70o до около 80oC, например при около 75oC. Давление реакции выбирается с учетом необходимости обеспечения перегонки побочных продуктов реакции. Давления в интервале от около 50 до около 500 мм рт.ст. (абсолютное) обычно будут обеспечивать предпочитаемые температуры обратного стока.
Бис-гидрокарбилоксиды магния, которые можно использовать при реакции формилирования, являются соединениями, содержащими два гидрокарбилоксильных остатка на атом магния, при этом по крайней мере один из указанных гидрокарбилоксильных остатков является арилокси, например фенокси или нафтилокси, имеющими по крайней мере одно свободное орто-положение по отношению к атому кислорода. Особенно подходящими являются бис-феноляты магния, в которых фенолятные остатки могут быть незамещенными или могут быть замещены в любом или во всех положениях, других, чем оба 2- и 6-положения, заместителями, которые не препятствуют ходу реакции и которые предпочтительно являются отдающими электрон или слабоотнимающими электрон.
Особенно полезными бис-фенолятами магния являются производные фенолов формулы:
где каждый из R1, R2, R3 и R4 имеет вышеуказанные значения.
Можно особенно упомянуть о бис-фенолятах магния, являющихся производными фенолов формулы: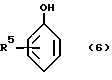
где
R5 имеет вышеуказанное значение.
Бис-феноляты магния, являющиеся производными фенолов формулы 5 или формулы 6, могут рассматриваться как составы, содержащие структуры соответственно формулы 7 или формулы 8, а так же как родственные, но более сложные структуры, содержащие более одного атома магния на одну молекулу.
В структурах формулы 7: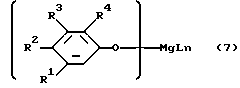
каждый из R1, R2, R3 и R4 имеет вышеуказанное значение;
L - обозначает молекулу лиганда, полученную из другого компонента реакционной смеси:
n - целое число от 1 до 6.
В структуре формулы 8: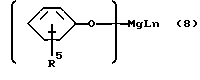
где
R5, L и n имеют вышеуказанные значения.
При реакции формилирования компонентами реакционной смеси, которая предоставляет молекулы лиганда L, могут быть совместные растворители, формальдегид и метанольный побочный продукт и их смеси.
Однако особенно удобно использовать бис-арилоксид магния, который благодаря методу его получения уже содержит соответствующие молекулы лиганда.
Таким образом, лучше использовать бис-гидрокарбилоксид магния, который получен по методу, описанному Рамицем и другими в "Synthesis", 1979 г., 71, т.е. при реакции алкоголята магния формулы:
Mg(OR6)2, (9)
где
R6-алкил, например C1-4-алкил-радикал, особенно метил, с вплоть до двух молей фенола, который имеет по крайней мере одно незамещенное положение, примыкающее к фенольной гидроксигруппе, например, фенола формулы 5 или формулы 6. Предпочтительные отношения молей фенола на моль алкоголята магния составляет от 0,9 до 2, особенно от 1,5 до 2, типично около 1,66.
Бис-арилоксиды магния, которые используются при реакции формилирования, содержат два арилоксильных остатка на атом магния и, по-видимому, также содержат один или большее число молекул или групп лиганда, например молекулы метанола, так что они соответствуют или структурно аналогичны формуле 7. Однако следует понять, что данное изобретение не основывается на какой-либо теории относительно точной структуры бис-фенолятов магния и должно рассматриваться как относящееся к использованию указанных бис-фенолятов так или иначе в виде комплексов формулы 7.
В число других бис-гидрокарбилоксидов магния, которые могут быть использованы при способе согласно данному изобретению, входят соединения, содержащие один арилоксильный и один другой гидрокарбилоксильный, например алкоксильный, остаток на атом магния. Такие бис-гидрокабилоксиды могут быть получены, например, при реакции одного моля алкоголята магния формулы 9 с приблизительно одним молем фенола, который имеет по крайней мере одно незамещенное положение, примыкающее к фенольной гидроксильной группе, и, при желании, могут быть использованы в отдельности или в смеси с вышеупомянутыми бис-диарилоксидами.
Формальдегид, используемый при способе по данному изобретению, может быть в виде свободного газообразного формальдегида или его раствора в безводном растворителе либо в виде выделяющего формальдегид соединения, т.е. соединения, способного выделять формальдегид в свободном состоянии при условиях, применяемых при способе по данному изобретению. В число подходящих соединений, способных выделять формальдегид в свободном состоянии, входят полимерные формы формальдегида, например параформальдегид, формальдегид или соединение, выделяющее формальдегид в свободном состоянии, рекомендуется постепенно (непрерывно или прерывисто) добавлять к бис-диарилоксиду в системе растворителей.
При способе по данному изобретению формальдегид или соединение, выделяющее формальдегид в свободном состоянии, обычно применяется в количестве по крайней мере двух молей, отнесенных к формальдегиду (HCHO), на моль фенола, присутствующего в бис-гидрокарбилоксиде. Предпочтительные отношения составляют от 2 до 3, типично около 2,75 молей формальдегида на моль фенола в бис-гидрокарбилоксиде. Совместный растворитель лучше использовать в количестве, не превышающем 5 молей на моль бис-гидрокабилоксида, причем предпочтительные количества находятся в интервале от 1 до 2 молей на моль бис-гидрокарбоксилоксида. В эти количества входит количество любого совместного растворителя, уже присутствующего в качестве лиганда в бис-гидрокарбиоксиде. Поскольку метанол является побочным продуктом реакции, то степень конверсии и выход продукта можно увеличить до максимума путем удаления перегонкой этого метанола и любых других летучих побочных продуктов в ходе реакции, поддерживая тем самым на оптимальном уровне отношение совместного растворителя к бис-феноляту.
В другом ценном варианте осуществления изобретения гидроксиламин или его соль могут непосредственно реагировать с алюминий-, титан-, цирконий- или хромсодержащими производными 2-оксиарилальдегидов, которые получены при реакциях формилирования, описанных в заявках EP-A-0077279, EP-A-0106653 и US-A-4231967, без необходимости выделения самих оксиальдегидов из реакционных смесей, в которых они образовались.
Изобретение иллюстрируется, но не ограничивается следующими примерами.
Пример 1.
В 2-х литровый стеклянный реакционный сосуд загружали метанол (224 г) и толуол (98 г), а затем помещали магниевые опилки (2,92 г). Добавляли раствор активатора (10 г) для активизирования магния и нагревали смесь до температуры обратного стока (65oC), для растворения магния с выделением газообразного водорода. Смесь в течение 0,5 часа выдерживали при температуре обратного стока и затем еще добавляли магний четырьмя порциями (4х2,92 г) за общий период времени в 1,5 часа. Каждая порция добавлялась после прекращения выделения водорода от прежней порции. Затем смесь нагревали с обратным холодильником еще час, чтобы обеспечить полное растворение магния, добавляли 4-нонилфенол (224 г) и смесь нагревали с обратным холодильником в течение 1 часа для образования нонилфенольной соли магния. Отбирали раствор активатора из состава (1116 г), содержащего нонилфенольную соль магния (461 г), метилат магния (17,3 г), толуол (194 г) и метанол (443,7 г).
Добавляли толуол (175 г) и удаляли метанол-толуольную азеотропную смесь (292 г) перегонкой до тех пор, пока температура реакционной смеси не достигала 90-95oC. Перемешанную суспензию параформальдегида (85 г) в толуоле (120 г) добавляли к получаемому толуольному раствору нонилфенольной соли магния в течение 3 часов при температуре 90-95oC с удалением дистиллатов толуола и летучих побочных продуктов (100 г). По окончании добавления параформальдегида реакционную смесь в течение 1 часа нагревали до 95-100oC, чтобы обеспечить завершение реакции, и затем смесь охлаждали до 45-50oC.
Раствор сульфата гидроксиламина (98,5 г) в воде (300 г) в течение 1 часа и при 45-50oC добавляли к реакционной смеси, имеющейся при реакции формилирования. Перемешивание продолжали при этой температуре еще в течение 1,5 часов, после чего давали смеси отстояться и разделяли фазы.
К органической фазе добавляли кислотную промывочную жидкость, состоящую из воды (250 г) и серной кислоты (16 г), и перемешивали смесь при 45oC в течение 0,5 часа. Смеси давали отстояться и промывали органическую фазу водой (2х125 г) при 50oC. Затем удаляли толуол из органической фазы путем испарения при пониженном давлении, после чего оставался неочищенный 5-нонилсалицальдоксим в виде желтого масла (271 г). Этот оксим очищали перегонкой при 180oC и давлении 0,555 мм рт. ст.
Пример 2.
В 500-мл трехгорлышковую колбу с круглым дном помещали 65,9 г 5-нонилсалициальдегида при 90%-ной концентрации в 48 мл толуола. К этому раствору добавляли 2 мл тетраизопропилата титана. Сразу же после добавления этого комплекса титана раствор приобретал красновато-коричневый цвет. Нагревали содержимое до 45oC и в течение 30 секунд добавляли раствор 21,8 г сульфата гидроксиламина в 35 мл воды (предварительно подогретый по 40-45oC). Перемешивали реакционную смесь при частоте вращения мешалки 300 об/мин и в течение 5 минут добавляли раствор 14,5 г карбоната натрия в 30 мл воды. После окончания его добавления температуру реакции поддерживали на уровне 45oC. Периодически брали пробы органической фазы и с помощью газовой хроматографии делали их анализ на присутствие альдегида (колонка 6х1/8 дюйма с 2%-ным полибутандиолсукцинатом на хромосорбе-WHP). После двух часов анализ показал наличие 0,7 альдегида, оставшегося в растворе. После трех часов содержание альдегида не изменилось значительно. Прекращали перемешивание и удаляли водную фазу. Органическую фазу промывали 25 мл 5,6%-ной серной кислоты при 45oC. Удаляли водную фазу и дважды промывали органическую фазу 25 мл воды до конечной pH, равной 2,6. С помощью роторного испарителя удаляли толуол (2 мм рт. ст., температура ванны 60oC) и получали 69,3 г красно-коричневого масла. Анализ загрузкой Cu и титрованием показал концентрацию оксима в 86,1%.
Пример 3.
Повторяли методику, описанную в примере 1, за исключением того, что сульфат гидроксиламина заменяли эквивалентным количеством самого гидроксиламина (в виде 50%-ного раствора в воде). При добавлении раствора гидроксиламина к реакционной смеси реакции формилирования получали желто-молочную суспензию, которая медленно становилась белой. Через 1 час при 45oC газохроматографический анализ показал завершение реакции оксимации.
После опускания в водном растворе серной кислоты слой толуола был сильномолочного цвета, но становился прозрачным после перемешивания в течение 3 часов. Слой толуола отделяли от водного слоя, промывали кислотой и затем водой и фильтровали. С помощью роторного испарителя удаляли толуол и получали 5-нонилсалицилальдоксим (концентрация 87,9%) при выходе в 87%.
Пример 4.
В 250-мл трехгорлышковую колбу с круглым дном, снабженную механической мешалкой, термометром и обратным холодильником, загружали магниевые опилки (2,95 г, 0,12 моля), безводный метанол (75 мл, 1,85 моля) и безводный толуол (25 мл). К этому добавляли 8%-ный раствор метилата магния в метаноле (3 мл, 0,002 моля) и реакционную смесь затем нагревали до обратного тока. Через несколько минут наблюдалось выделение водорода. Смесь нагревали с обратным холодильником в течение 1 часа до растворения всего магния, в результате чего получали мутный белый раствор/суспензию при никаком дальнейшем выделении водорода.
Добавляли пара-додецилфенол (46,8 г, 0,179 моля) и полученный желтый раствор нагревали с обратным холодильником в течение 1 часа, после чего оставляли на ночь под осушительной трубкой для охлаждения до комнатной температуры. Загружали толуол (120 мл) и переоборудовали аппаратуру на дробную перегонку. Смесь нагревали для удаления метанола, образующего азеотропную смесь с толуолом, пока не достигали внутренней температуры в 102oC. При перегонке (при приблизительно 97oC) заметно возрастала вязкость раствора. Затем удаляли колонку для дробной перегонки и при 100-105oC в течение 1 часа добавляли порциями суспензию параформальдегида (18 г, 0,6 моля) в толуоле (40 мл) при одновременной перегонке растворителя и низкокипящих побочных продуктов (59 мл). Реакцию поддерживали при 100-105oC в течение 1 часа, после чего охлаждали до 55oC для реакции оксимации.
При 40-50oC приготавливали раствор сульфата гидроксиламмония (19,7 г, 0,12 моля) в воде (70 мл) и затем в течение 30 минут добавляли его в реакционный сосуд при быстром перемешивании. Реакцию продолжали в течение 3 часов при 55oC, а затем содержимое реакционного сосуда охлаждали до 30-40oC.
Останавливали мешалку и содержимое реакционного сосуда перемещали в делительную воронку. Удаляли водный слой и зелено-черный слой органических веществ затем перемещали обратно в реакционный сосуд. В сосуд загружали разбавленный раствор серной кислоты (13 г, 0,13 моля) в воде (100 мл) и перемешивали его в течение 20 минут при 50oC. В первую же минуту происходило быстрое изменение цвета на желтый. После этой кислотной обработки содержимое сосуда вновь перемещали в делительную воронку и удаляли кислый водный слой. После этого проводили двухкратную промывку горячей (приблизительно 50oC) водой (2х100 мл). С помощью роторного испарителя удаляли растворитель до получения 53,25 г бледно-желтого масла. Затем с использованием аппаратуры Лейбольда часть этого масла перегоняли при следующих условиях: температура стенки - 230oC, вакуум - 2,0 мм рт.ст., скорость добавления - 8,0 мл/мин.
В результате получали сильно бледно-желтое масло, которое, как оказалось, имело концентрацию 95,3% по 1H ЯМР с использованием бензилацетата в качестве стандартного вещества при ошибке ±2%. Это затем использовали в качестве стандартного вещества для анализа неочищенного продукта с помощью газовой хроматографии. Как оказалось, неочищенный продукт имел концентрацию 89,3% при выходе 5-дидецилсалицилальдоксима в 87,3%±2%.
Пример 5.
В 1,0-л трехгорлышковую колбу с круглым дном, снабженную механической мешалкой, термометром и обратным холодильником, загружали магниевые опилки (555 г, 0,228 моля), безводный метанол (150 мл, 3,7 моля) и безводный толуол (50 мл). К этому добавляли 8%-ный раствор метилата магния в метаноле (5 мл, 0,004 моля) и затем нагревали реакционную смесь до обратного стока. Через несколько минут замечалось выделение водорода. Смесь нагревали с обратным холодильником в течение 1 часа до растворения всего магния, в результате чего получали мутный белый раствор/суспензию при никаком дальнейшем выделении водорода.
Добавляли пара-хлорфенол (48,7 г, 0,38 моля) и полученный желтый раствор нагревали с обратным холодильником в течение 1,5 часа, после чего оставляли на ночь под осушительной трубкой для охлаждения до комнатной температуры. Загружали толуол (240 мл) и переоборудовали аппаратуру на дробную перегонку. Смесь нагревали для удаления метанола, образующего азеотропную смесь с толуолом, пока не достигали внутренней температуры в 100oC. При перегонке (приблизительно при 87oC) выпадал осадок с образованием бледной суспензии. Реакционную смесь затем охлаждали до 90-95oC и снимали колонку для дробной перегонки перед тем, как начать порциями добавлять суспензию параформальдегида (34,1 г, 1,14 моля) в толуоле (80 мл) в течение 1 часа при 90-95oC с одновременной перегонкой растворителя и низкокипящих побочных продуктов (100 мл). Реакцию поддерживали при 90-95oC в течение 1 часа, после чего реакционную смесь охлаждали до 55oC и аппаратуру переделывали на работу с обратным холодильником для реакции оксимации. Образовывалась желтая суспензия.
При 40-50oC приготавливали раствор сульфата гидроксиламмония (37,3 г, 0,227 моля) в воде (120 мл) и затем в течение 30 минут добавляли его в реакционный сосуд при быстром перемешивании. Реакцию продолжали 4,5 часа при 55oC, а затем содержимое реакционного сосуда охлаждали до 30-40oC. В водном слое образовывался белый осадок, который растворяли при добавлении 0,5%-ного раствора серной кислоты (200 мл).
Останавливали мешалку и содержимое реакционного сосуда перемещали в делительную воронку. Удаляли водный слой и пурпурно-черный слой органических веществ перемещали обратно в реакционный сосуд. В сосуд загружали разбавленный раствор серной кислоты (16,6 г, 0,166 моля) в воде (250 мл) и перемешивали в течение 20 минут при 50oC. В первую минуту происходило быстрое изменение цвета на желтый. После этой кислотной обработки содержимое вновь перемещали в делительную воронку и удаляли кислый водный слой. После этого проводили двухкратную промывку горячей (приблизительно 50oC) водой (2х100 мл). С помощью роторного испарителя удаляли растворитель и получали 49,6 г желтого воскообразного твердого вещества, которое, как оказалось, имело концентрацию 42,8% по 1H ЯМР с использованием бензилацетата в качестве стандартного вещества при выходе 5-хлорсалицилальдоксима в 32,6%.
Пример 6.
В 1,0-л трехгорлышковую колбу с круглым дном, снабженную механической мешалкой, термометром и обратным холодильником, загружали магниевые опилки (5,85 г, 0,24 моля), безводный метанол (150 мл, 3,7 моля) и безводный толуол (50 мл). К этому добавляли 8%-ный раствор метилата магния в метаноле (5 мл, 0,004 моля) и затем нагревали реакционную смесь до обратного стока. Через несколько минут замечали выделение водорода. Смесь нагревали с обратным холодильником в течение 1 часа до растворения всего магния, в результате чего получали мутный белый раствор/суспензию при никаком дальнейшем выделении водорода.
Добавляли пара-метоксифенол (49,6 г, 0,4 моля) и полученный желтый раствор нагревали с обратным холодильником в течение 2 часов до загрузки толуола (240 мл), а затем переоборудовали аппаратуру на дробную перегонку. Удаляли метанол, образующий азеотропную смесь с толуолом, пока не достигали внутренней температуры в 100oC. При перегонке (приблизительно при 78oC) выпадал осадок с образованием бледной суспензии. Реакционную смесь затем охлаждали до 90-95oC и снимали колонку для дробной перегонки перед тем, как начать порциями добавлять суспензию параформальдегида (36/0 г, 1,2 моля) в толуоле (80 мл) в течение 1 часа при 90-95oC и с одновременной перегонкой растворителя и низкокипящих побочных продуктов (68 мл). Реакцию поддерживали при 90-95oC в течение 1 часа, после чего реакционную смесь охлаждали до 55oC, а аппаратуру переделывали на работу с обратным холодильником для реакции оксимации. Образовывался оранжевый раствор.
При 40-50oC приготавливали раствор сульфата гидроксиламмония (39,4 г, 0,24 моля) в воде (120 мл) и затем в течение 30 минут добавляли его в реакционный сосуд при быстром перемешивании. Реакцию продолжали 4,5 часа при 55oC, а затем содержимое реакционного сосуда оставляли на ночь для охлаждения по комнатной температуре под азотом. В водном слое образовывался белый осадок, который растворяли при добавлении 0,5% об. раствора серной кислоты (200 мл). В слое органических веществ также образовывался коричневый осадок. Пытались растворить его добавлением толуола (100 мл) и нагреванием до 50oC, но твердое вещество оставалось.
Останавливали мешалку и содержимое реакционного сосуда перемещали в делительную воронку. Удаляли водный слой и затем слой органических веществ в виде суспензии коричневых кристаллов и раствора перемещали обратно в реакционный сосуд, а твердые частицы, оставшиеся в делительной воронке, смывали в колбу толуолом (80 мл). В сосуд загружали разбавленный раствор серной кислоты (16,6 г, 0,166 моля) в воде (250 мл) и перемешивали его в течение 20 минут при 50oC. В первую минуту происходило быстрое изменение цвета на желтый. После этой кислотной обработки содержимое вновь перемещали в разделительную воронку и осаждали. Отделяли мутный кислый водный слой и удаляли раствор органических веществ. Для растворения твердых частиц добавляли дихлорметан и затем раствор объединяли с вышеуказанным раствором толуола. С использованием дихлорметана (200 мл) раздельно экстрагировали мутный кислый водный слой и водный слой из реакционной смеси. Объединяли все слои органических веществ и с помощью роторного испарителя удаляли растворитель с получением 60,5 г желтого воскообразного твердого вещества. Затем это твердое вещество перекристаллизовывали из толуола (250 мл) нагреванием до ниже 70oC, после чего охлаждали в ледяной ванне до 0oC и фильтровали осадок. Это давало 37,1 г от бледного до палевого твердого вещества, которое, как оказалось, имело концентрацию 90,4% по 1H ЯМР с использованием бензилацетата в качестве стандартного вещества. С помощью роторного испарителя удаляли толуоловые фильтраты и получали 24,45 г коричневого масла, которое позже затвердевало. Оно имело концентрацию 26,7% по 1H ЯМР с использованием бензилацетата в качестве стандартного вещества при общем выходе 5-метоксисалицилальдоксиме в 60%.
Пример 7.
В 1,0-л трехгорлышковую колбу с круглым дном, снабженную механической мешалкой, термометром и обратным холодильником, загружали магниевые опилки (5,85 г, 0,24 моля), безводный метанол (150 мл, 3,7 моля) и безводный толуол (50 мл). К этому добавляли 8%-ный раствор метилата магния в метаноле (5 мл, 0,004 моля) и реакционную смесь затем нагревали до обратного стока. Через несколько минут замечали выделение водорода. Смесь нагревали с обратным холодильником в течение 1 часа до растворения всего магния, в результате чего получали мутный белый раствор /суспензию при никаком дальнейшем выделении водорода.
Добавляли орто-крезол (43,2 г, 0,4 моля) и полученный желтый раствор нагревали с обратным холодильником в течение 2 часов, после чего оставляли на ночь под осушительной трубкой для охлаждения до комнатной температуры. Добавляли толуол (240 мл) и аппаратуру переоборудовали на дробную перегонку. Удаляли метанол, образующий азеотропную смесь с толуолом, пока не достигали внутренней температуры в 97oC. При перегонке (приблизительно при 80oC) происходило осаждение с образованием коричневой суспензии. Реакционную смесь затем охлаждали до 90-95oC и удаляли колонку для дробной перегонки перед тем, как начать порциями добавлять суспензию параформальдегида (36,0 г, 1,2 моля) в толуоле (80 мл) в течение 1 часа при 90-95oC с одновременной перегонкой растворителя и низкокипящих побочных продуктов (89 мл). Реакцию поддерживали при 90-95oC в течение 1 часа, после чего реакционную смесь охлаждали до 55oC, а аппаратуру переоборудовали на работу с обратным холодильником для реакции оксимации. Образовывалась желтая суспензия.
При 40-50oC приготавливали раствор сульфата гидроксиламмония (39,4 г, 0,24 моля) в воде (120 мл) и затем в течение 30 минут добавляли его в реакционный сосуд при быстром перемешивании. Реакцию продолжали 1,5 часа при 55oC. В водном слое образовывался белый осадок, который растворяли при добавлении 0,5% об. раствора серной кислоты (250 мл). Останавливали мешалку и раствор перемещали в делительную воронку.
Удаляли водный слой и зелено-черный слой органических веществ затем перемещали обратно в реакционный сосуд. В сосуд загружали разбавленный раствор серной кислоты (33,12 г, 0,331 моля) в воде (250 мл) и перемешивали в течение 20 минут при 50oC. В первую минуту происходило быстрое изменение цвета на желтый. После этой кислотной обработки содержимое вновь перемещали в делительную воронку и удаляли кислый водный слой. После этого проводили двухкратную промывку горячей (приблизительно 50oC) водой (2х100 мл). С помощью роторного испарителя удаляли растворитель и получали 61,1 г желтого твердого вещества, которое, как оказалось, имело концентрацию 55,1% по 1H ЯМР с использованием бензилацетата в качестве стандартного вещества при выходе 3-метилсалицилальдоксима в 55,7%.
Пример 8.
В 1,0-л трехгорлышковую колбу с круглым дном, снабженную механической мешалкой, термометром и обратным холодильником, загружали магниевые опилки (5,85 г, 0,24 моля), безводный метанол (150 мл, 3,7 моля) и безводный толуол (50 мл). К этому добавляли 8%-ный раствор метилата магния в метаноле (5 мл, 0,004 моля) и затем нагревали реакционную смесь до обратного стока. Через несколько минут замечали выделение водорода. Смесь нагревали с обратным холодильником в течение 1 часа до растворения всего магния, в результате чего получали мутный белый раствор/суспензию при никаком дальнейшем выделении водорода.
Добавляли мета-крезол (43,2 г, 0,4 моля) и полученный желтый раствор нагревали с обратным холодильником в течение 2 часов, после чего оставляли на ночь под осушительной трубкой для охлаждения до комнатной температуры. Добавляли толуол (240 мл) и аппаратуру переоборудовали на дробную перегонку. Удаляли метанол, образующий азеотропную смесь с толуолом, пока не достигали внутренней температуры в 97oC. При перегонке (приблизительно при 87oC) происходило осаждение с образованием суспензии кремового цвета. Реакционную смесь затем охлаждали до 95oC и удаляли колонку для дробной перегонки перед тем, как начать порциями добавлять суспензию параформальдегида (36,0 г, 1,2 моля) в толуоле (30 мл) в течение 1 часа при 95oC с одновременной перегонкой растворителя и низкокипящих побочных продуктов (79 мл). Реакцию поддерживали при 95oC в течение 1 часа, после чего реакционную смесь охлаждали до 55oC, аппаратуру переоборудовали на работу с обратным холодильником для реакции оксимации. Образовывалась желтая суспензия.
При 40-50oC приготавливали раствор сульфата гидроксиламмония (39,4 г, 0,24 моля) в воде (120 мл) и затем в течение 30 минут добавляли его в реакционный сосуд при быстром перемешивании. Реакцию продолжали в течение 4 часов при 55oC. Останавливали мешалку и содержимое перемещали в делительную воронку.
Удаляли водный слой и зелено-черный слой органических веществ затем перемещали обратно в реакционный сосуд. В сосуд загружали разбавленный раствор серной кислоты (33,12 г, 0,331 моля) в воде (250 мл) и перемешивали его в течение 20 минут при 50oC. В первую минуту цвет быстро изменялся на желтый. После этой кислотной обработки содержимое вновь перемещали в делительную воронку и удаляли кислый водный слой. После этого проводили двухкратную промывку горячей (приблизительно 50oC) водой (2х250 мл). С помощью роторного испарителя удаляли растворитель и получали 60,1 г желтого твердого вещества, которое, как установлено, имело концентрацию 61,3% по 1H ЯМР с использованием бензилацетата в качестве стандартного вещества, что означало выход в 61,0%.
Пример 9.
В 1,0-л трехгорлышковую колбу с круглым дном, снабженную механической мешалкой, термометром и обратным холодильником, загружали магниевые опилки (5,85 г, 0,24 моля), безводный метанол (150 мл) и безводный толуол (50 мл). К этому добавляли 8%-ный раствор метилата магния в метаноле (5 мл, 0,004 моля) и затем нагревали реакционную смесь до обратного стока. Через несколько минут замечали выделение водорода. Смесь нагревали с обратным холодильником в течение 1 часа до растворения всего магния, в результате чего получали мутный белый раствор/суспензию при никаком дальнейшем выделении водорода.
Добавляли пара-крезол (43,2 г, 0,4 моля) и полученный желтый раствор нагревали с обратным холодильником в течение 2 часов, после чего оставляли на ночь под осушительной трубкой для охлаждения до комнатной температуры. Добавляли толуол (240 мл) и аппаратуру переоборудовали на дробную перегонку. Удаляли метанол, образующий азеотропную смесь с толуолом, пока не достигали внутренней температуры в 98oC. При перегонке (приблизительно при 87oC) происходило осаждение с образованием суспензии кремового цвета. Реакционную смесь затем охлаждали до 95oC и удаляли колонку для дробной перегонки перед тем, как начать порциями добавлять суспензию параформальдегида (36,0 г, 1,2 моля) в толуоле (80 мл) в течение 1 часа при 95oC с одновременной перегонкой растворителя и низкокипящих побочных продуктов (79 мл). Реакцию поддерживали при 95oC в течение 1 часа, после чего реакционную смесь охлаждали до 55oC, а аппаратуру переоборудовали на работу с обратным холодильником для реакции оксимации. Образовывалась желтая суспензия. (При реакции физическая потеря из-за пенообразования составляла около 5% при соответствующем уменьшении выхода).
При 40-50oC приготавливали раствор сульфата гидроксиламмония (39,4 г, 0,24 моля) в воде (120 мл) и затем в течение 30 минут добавляли его в реакционный сосуд при быстром перемешивании. Реакцию продолжали в течение 1,25 часа при 55oC. Останавливали мешалку и содержимое перемещали в делительную воронку.
Удаляли водный слой и зелено-черный слой органических веществ затем перемещали обратно в реакционный сосуд. В сосуд загружали разбавленный раствор серной кислоты (33,12 г, 0,331 моля) в воде (250 мл) и перемешивали его в течение 20 минут при 50oC. В первую минуту происходило быстрое изменение цвета на желтый. После этой кислотной обработки содержимое вновь перемещали в делительную воронку и удаляли кислый водный слой. После этого проводили двухкратную промывку горячей (приблизительно 50oC) водой (2х125 мл). С помощью роторного испарителя удаляли испаритель и получали 57,65 г желтого твердого вещества, которое, как установлено, имело концентрацию 77,3% по 1H ЯМР с использованием бензилацетата в качестве стандартного вещества, что означало выход 5-метилсалицилальдоксима в 73,8%.
Пример 10.
В 1,0-л трехгорлышковую колбу с круглым дном, снабженную механической мешалкой, термометром и обратным холодильником, загружали магниевые опилки (5,85 г, 0,24 моля), безводный метанол (150 мл, 3,7 моля) и безводный толуол (50 мл). К этому добавляли 8%-ный раствор метилата магния в метаноле (5 мл, 0,004 моля) и затем нагревали реакционную смесь до обратного стока. Через несколько минут замечали выделение водорода. Смесь нагревали с обратным холодильником в течение 1 часа до растворения всего магния, в результате чего получали мутный белый раствор/суспензию при никаком дальнейшем выделении водорода.
Добавляли 2,5-диметилфенол (48,8 г, 0,4 моля) и полученный желтый раствор нагревали с обратным холодильником в течение 2 часов, после чего оставляли на ночь под осушительной трубкой для охлаждения до комнатной температуры. Добавляли толуол (240 мл) и аппаратуру переоборудовали на дробную перегонку. Удаляли метанол, образующий азеотропную смесь с толуолом, пока не достигали внутренней температуры в 95oC. При перегонке (приблизительно при 92oC) повышалась вязкость. Реакционную смесь затем охлаждали до 93oC и удаляли колонку для дробной перегонки перед тем, как начать порциями добавлять суспензию параформальдегида (36,0 г, 1,2 моля) в толуоле (80 мл) в течение 1 часа при 95oC с одновременной перегонкой растворителя и низкокипящих побочных продуктов (67 мл). Реакцию поддерживали при 95oC в течение 1,5 часа, после чего реакционную смесь охлаждали до 55oC, а аппаратуру переоборудовали на работу с обратным холодильником для реакции оксимации. Образовывался желтый раствор.
При 40-50oC приготавливали раствор сульфата гидроксиламмония (39,4 г, 0,24 моля) в воде (120 мл) и затем в течение 30 минут добавляли его в реакционный сосуд при быстром перемешивании. Реакцию продолжали в течение 1,5 часа при температуре, после чего реакционную смесь оставляли на ночь для охлаждения до комнатной температуры. В водном слое образовывался белый осадок, который растворяли при добавлении 0,5% об. раствора серной кислоты (200 мл). В слое органических веществ также образовывался бледно-коричневый осадок. Твердое вещество в органической фазе повторно растворяли при нагревании до 50oC.
Останавливали мешалку и содержимое перемещали в делительную воронку. Быстро удаляли водный слой ввиду образования осадка в слое органических веществ. Слой органических веществ с коричневой кристаллической суспензией/раствором затем перемещали обратно в реакционный сосуд, а твердые частицы, оставшиеся в делительной воронке, смывали толуолом (50 мл) в колбу. В сосуд загружали разбавленный раствор серной кислоты (33,12 г, 0,331 моля) в воде (250 мл) и перемешивали его в течение 20 минут при 50oC. В первую минуту происходило быстрое изменение цвета на желтый. После этой кислотной обработки содержимое вновь перемещали в делительную воронку и удаляли кислый водный слой. Затем проводили двухкратную промывку горячей (приблизительно 65oC) водой (2х125 мл). Слой органических веществ, который уже начал осаждаться, затем перемещали в коническую колбу, которую устанавливали в ледяной ванне для достижения полного осаждения. После этого отфильтровывали твердое вещество, при этом выход составлял около 29 г. С помощью роторного испарителя фильтраты затем уменьшали по объему почти наполовину. Их вновь фильтровали и в результате получали второй выход, равный около 13 г. Эту операцию повторили с получением третьего выхода, равного около 5 г. Эти твердые вещества объединяли с получением белого твердого вещества весом 47,8 г, которое, как установлено, имело концентрацию по 1H ЯМР с использованием бензилацетата в качестве стандартного вещества. Это давало общий выход 3,5-диметилсалицилальдоксима в 32,8%.
Пример 11.
В 500-мл трехгорлышковую колбу с круглым дном, снабженную механической мешалкой, термометром и обратным холодильником, загружали магниевые опилки (1,92 г, 0,12 моля), безводный метанол (80 мл, 2,0 моля) и безводный толуол (20 мл). К этому добавляли 8%-ный раствор метилата магния в метаноле (2 мл, 0,0015 моля) и затем нагревали реакционную смесь до обратного стока. Через несколько минут замечали выделение водорода. Смесь нагревали с обратным холодильником в течение 1 часа до растворения всего магния, в результате чего получали мутный белый раствор/суспензию при никаком дальнейшем выделении водорода.
Добавляли орто-вторичный-бутилфенол (30,0 г, 0,2 моля) и полученный желтый раствор нагревали с обратным холодильником в течение 3 часов, после его оставляли на ночь под осушительной трубкой для охлаждения до комнатной температуры. Загружали толуол (120 мл) и аппаратуру переоборудовали на дробную перегонку. Смесь нагревали для удаления метанола, образующего азеотропную смесь с толуолом, пока не достигали внутренней температуры в 102oC. При перегонке смесь оставалась в виде слабого зеленого раствора, способного к перемешиванию. Затем охлаждали реакционную смесь до 90-95oC и удаляли перегонную колонку до того, как начать порциями добавлять суспензию параформальдегида (18,0 г, 0,6 моля) в толуоле (40 мл) в течение 1 часа при 95-98oC с одновременной перегонкой растворителя и низкокипящих побочных продуктов (62 мл). Реакцию поддерживали при 99oC в течение 40 минут, после чего реакционную смесь охлаждали до 55oC, а аппаратуру переоборудовали на работу с обратным холодильником для реакции оксимации. Образовывался желтый раствор.
При 40-50oC приготавливали раствор сульфата гидроксиаммония (19,7 г, 0,12 моля) в воде (60 мл) и затем в течение 30 минут добавляли его в реакционной сосуд при быстром перемешивании. Реакцию продолжали 2,5 часа при 55oC, после чего реакционную смесь оставляли на ночь для охлаждения до комнатной температуры. В одном слое образовывался белый осадок, который растворяли при добавлении 0,5% об. раствора серной кислоты (200 мл) и нагревали до 45oC.
Останавливали мешалку и содержимое перемещали в делительную воронку. Удаляли водный слой и пурпурно-черный слой органических веществ затем перемещали обратно в реакционный сосуд. В сосуд загружали разбавленный раствор серной кислоты (16,6 г, 1,166 моля) в воде (100 мл) и перемешивали его в течение 30 минут при 50oC. В первую минуту происходило быстрое изменение цвета на желтый. После этой кислотной обработки содержимое вновь перемещали в делительную воронку и удаляли кислый водный слой, после этого проводили двухкратную промывку горячей (приблизительно 50oC) водой (2х100 мл). С помощью роторного испарителя удаляли растворитель и получали 38,6 г оранжевого масла, которое, как установлено, имело концентрацию 14,4% по 1H ЯМР с использованием бензилацетата в качестве стандартного вещества, что означало выход вторичного 3-бутилсалицилальдоксима в 14,4%.
Пример 12.
В 500-мл трехгорлышковую колбу с круглым дном, снабженную механической мешалкой, термометром и обратным холодильником, загружали магниевые опилки (1,46 г, 0,06 моля), безводный метанол (50 мл, 1,23 моля) и безводный толуол (10 мл). К этому добавляли 8%-ный раствор метилата магния в метаноле (1 мл, 0,00074 моля) и затем реакционную смесь нагревали до обратного стока. Через несколько минут замечали выделение водорода. Смесь нагревали с обратным холодильником в течение 1 часа до растворения всего магния, в результате чего получали мутный белый раствор/суспензию при никаком дальнейшем выделении водорода.
Добавляли мета-третичный бутилфенол (15,0 г, 0,1 моль) и полученный желтый раствор нагревали с обратным холодильником в течение 1,5 часа, после чего оставляли на ночь под осушительной трубкой для охлаждения до комнатной температуры. Загружали толуол (70 мл) и аппаратуру переоборудовали на дробную перегонку. Смесь нагревали для удаления метанола, образующего азеотропную смесь с толуолом, пока не достигали внутренней температуры в 97oC. При перегонке (приблизительно при 93oC) увеличивалась вязкость раствора. Затем охлаждали реакционную смесь до 95oC и удаляли перегонную колонку перед тем, как начать порциями добавлять суспензию параформальдегида (9,0 г, 0,3 моля) в толуоле (20 мл) в течение 50 минут при 93-95oC с одновременной перегонкой растворителя и низкокипящих побочных продуктов (34 мл). Реакцию поддерживали при 95oC в течение 40 минут, после чего реакционную смесь охлаждали до 55oC, а аппаратуру переоборудовали на работу с обратным холодильником для реакции оксимации. Образовывалась желтая суспензия.
При 40-50oC приготавливали раствор сульфата гидроксиламмония (9,85 г, 0,06 моля) в воде (30 мл) и затем в течение 30 минут добавляли его в реакционный сосуд при быстром перемешивании. Реакция продолжалась 2 часа при 55oC, после чего реакционную смесь оставляли на ночь для охлаждения до комнатной температуры. В водном слое образовывалось небольшое количество осадка, который растворяли при добавлении 0,5% об. раствора серной кислоты (100 мл) и нагревании до 50oC.
Останавливали мешалку и содержимое перемещали в делительную воронку. Удаляли водный слой и пурпурно-черный слой органических веществ затем перемещали обратно в реакционный сосуд. В сосуд загружали разбавленный раствор серной кислоты (11,05 г, 0,11 моля) в воде (50 мл) и перемешивали в течение 20 минут при 50oC. В первую минуту происходило быстрое изменение цвета на желтый. После этой кислотной обработки содержимое вновь перемещали в делительную воронку и удаляли кислый водный слой. После этого проводили двухкратную промывку горячей (приблизительно 50oC) водой (2х75 мл). С помощью роторного испарителя удаляли растворитель и получали 16,7 г бледно-желтого масла, которое позже затвердевало. Как установлено, оно имело концентрацию 73,0% по 1H ЯМР с использованием бензилацетата в качестве стандартного вещества, что означало выход в 63,2%. С помощью газохроматографического анализа был обнаружен только один изомер (4-третичный-бутилсалицилальдоксим).
Пример 13.
В 1,0-л трехгорлышковую колбу с круглым дном, снабженную механической мешалкой, термометром и обратным холодильником, загружали магниевые опилки (5,85 г, 0,24 моля), безводный метанол (150 мл, 3,7 моля) и безводный толуол (50 мл). К этому добавляли 8%-ный раствор метилата магния в метаноле (5 мл, 0,004 моля) и затем реакционную смесь нагревали до обратного стока. Через несколько минут замечали выделение водорода. Смесь нагревали с обратным холодильником в течение 1 часа до растворения всего магния, в результате чего получали мутный белый раствор/суспензию при никаком дальнейшем выделении водорода.
Добавляли фенол (37,6 г, 0,40 моля) и полученный желтый раствор нагревали с обратным холодильником в течение 45 минут, после чего оставляли на ночь под осушительной трубкой для охлаждения до комнатной температуры. Загружали толуол (240 мл), а аппаратуру переоборудовали на дробную перегонку. Смесь нагревали для удаления метанола, образующего азеотропную смесь с толуолом, пока не достигали внутренней температуры в 95oC. При перегонке (приблизительно при 90oC) происходило осаждение с образованием суспензии бледного цвета. Затем охлаждали реакционную смесь до 90-95oC и удаляли колонку для дробной перегонки перед тем, как начать порциями добавлять суспензию параформальдегида (36,0 г, 1,2 моля) в толуоле (80 мл) в течение 1 часа при 95oC с одновременной перегонкой растворителя и низкокипящих побочных продуктов (100 мл). Реакцию поддерживали при 90-95oC в течение 1 часа, после чего реакционную смесь охлаждали до 55oC, а аппаратуру переоборудовали на работу с обратным холодильником для реакции оксимации. Образовывалась желтая суспензия.
При 40-45oC приготавливали раствор сульфата гидроксиламмония (37,3 г, 0,227 моля) в воде (120 мл) и затем в течение 30 минут добавляли его в реакционный сосуд при быстром перемешивании. Реакцию продолжали 2 часа при 55oC, после чего охлаждали реакционную смесь до 30-40oC и останавливали мешалку. Наблюдалось плохое разделение, которое было улучшено при добавлении 1,0% об. раствора серной кислоты (100 мл).
Затем содержимое реакционного сосуда перемещали в делительную воронку. Удаляли водный слой и затем пурпурно-черный слой органических веществ перемещали обратно в реакционный сосуд. В сосуд загружали разбавленный раствор серной кислоты (36,8 г, 0,368 моля) в воде (250 мл) и перемешивали в течение 20 минут при 50oC. В первую минуту происходило быстрое изменение цвета на желтый. После этой кислотной обработки содержимое вновь перемещали в делительную воронку и удаляли кислый водный слой. После этого проводили двухкратную промывку горячей (приблизительно 50oC) водой (2х100 мл). С помощью роторного испарителя удаляли растворитель и получали 51,3 г желтого масла, которое позже частично затвердевало. Как установлено, оно имело концентрацию 63,0% по 1H ЯМР с использованием бензилацетата в качестве стандартного вещества, что давало выход салицилальдоксима в 59,0%,
Пример 14.
В 1,0-л трехгорлышковую колбу с круглым дном, снабженную механической мешалкой, термометром и обратным холодильником, загружали магниевые опилки (5,85 г, 0,24 моля), безводный метанол (150 мл, 3,7 моля) и безводный толуол (50 мл). К этому добавляли 8%-ный раствор метилата магния в метаноле (5 мл, 0,004 моля) и реакционную смесь затем нагревали до обратного стока. Через несколько минут замечали выделение водорода. Смесь нагревали с обратным холодильником в течение 1 часа до растворения всего магния, в результате чего получали мутный белый раствор/суспензию при никаком дальнейшем выделении водорода.
Добавляли α-нафтол (57,6 г, 0,4 моля) и полученный желтый раствор нагревали с обратным холодильником в течение 1 часа, после чего оставляли на ночь под осушительной трубкой для охлаждения до комнатной температуры. Загружали толуол (240 мл) и аппаратуру переоборудовали на дробную перегонку. Удаляли метанол, образующий азеотропную смесь с толуолом, пока не достигали внутренней температуры в 99oC. При перегонке смесь оставалась в виде слабого темно-коричневого раствора, способного к перемешиванию. Затем охлаждали реакционную смесь до 90oC и удаляли колонку для дробной перегонки перед тем, как начать добавлять частями суспензию параформальдегида (36,0 г, 1,2 моля) в толуоле (80 мл) в течение 1,5 часов при 95-99oC с одновременной перегонкой растворителя и низкокипящих побочных продуктов (95 мл). Реакцию поддерживали при 97oC в течение 1,5 часов, после чего реакционную смесь охлаждали до 55oC. а аппаратуру переоборудовали на работу с обратным холодильником для реакции оксимации. Образовывалась темно-зеленая суспензия.
При 40-50oC приготавливали раствор сульфата гидроксиламмония (39,4 г, 0,24 моля) в воде (120 мл) и затем в течение 30 минут добавляли его в реакционный сосуд при быстром перемешивании. Реакцию продолжали в течение 5,5 часа при 55oC, после через реакционную смесь оставляли на ночь для охлаждения до комнатной температуры. Темно-зеленый осадок оставался без изменений, но теперь был преимущественно в водной фазе. От двух фаз отфильтровывали твердое вещество и высушивали его, в результате чего получали неочищенный 1-оксинафталин-2-карбоксальдегидоксим (соль магния) весом 82,9 г.
Пример 15.
В 1,0-л трехгорлышковую колбу с круглым дном, снабженную механической мешалкой, термометром и обратным холодильником, загружали магниевые опилки (5,85 г, 0,24 моля), безводный метанол (150 мл, 3,7 моля) и безводный толуол (50 мл). К этому добавляли 8%-ный раствор метилата магния в метаноле (5 мл, 0,004 моля) и затем реакционную смесь нагревали до обратного стока. Через несколько минут замечали выделение водорода. Смесь нагревали с обратным холодильником в течение 1 часа до растворения всего магния, в результате чего получали мутный белый раствор/суспензию при никаком дальнейшем выделении водорода.
Добавляли β-нафтол (57,6 г, 0,4 моля). Полученная смесь затвердевала. Добавляли толуол (200 мл) и получали густую суспензию, к которой добавляли метанол (50 мл). Суспензию нагревали с обратным холодильником в течение 1 час, после чего оставляли на ночь под осушительной трубкой для охлаждения до комнатной температуры. Загружали толуол (240 мл) и аппаратуру переоборудовали на дробную перегонку. Удаляли метанол, образующий азеотропную смесь с толуолом, пока не достигали внутренней температуры в 99oC. При перегонке увеличивалась вязкость суспензии до тех пор, пока поверхность смеси не становилась неперемешиваемой. Затем охлаждали реакционную смесь до 93-95oC и удаляли колонку для дробной перегонки перед тем, как начать порциями добавлять суспензию параформальдегида (36,0 г, 1,2 моля) в толуоле (80 мл) в течение 1,5 часа при 95-99oC с одновременной перегонкой растворителя и низкокипящих побочных продуктов (72 мл). Реакцию поддерживали при 99oC в течение 2 часов, после чего реакция, как показала взятая проба, была неоконченной. Поэтому в течение 25 минут еще добавляли параформальдегид (20 г, 0,66 моля) в виде суспензии в толуоле (40 мл). Реакционную смесь перемешивали при 99oC еще 2 часа перед удалением избытка толуола (120 мл) перегонкой, после чего охлаждали реакционную смесь до 55oC и переделывали аппаратуру на работу с обратным холодильником для реакции оксимации. Образовывалась суспензия горчично-желтого цвета.
При 40-50oC приготавливали раствор сульфата гидроксиламмония (39,4 г, 0,24 моля) в воде (120 мл) и затем добавляли его в реакционный сосуд в течение 30 минут при быстром перемешивании. Реакцию продолжали 3 часа при 55oC, после чего реакционную смесь оставляли на ночь для охлаждения до комнатной температуры. Смесь становилась зеленой. В водном слое образовался белый осадок, который растворяли при добавлении 0,5% об. раствора серной кислоты (200 мл) и нагревании до 50oC. Останавливали мешалку, при этом происходило плохое разделение, которое улучшали добавлением поваренной соли (25 г).
Содержимое реакционного сосуда затем перемещали в делительную воронку. Удаляли водный слой, а коричневый слой органических веществ затем перемещали обратно в реакционный сосуд. В сосуд загружали разбавленный раствор серной кислоты (36,8 г, 0,368 моля) в воде (250 мл) и перемешивали в течение 20 минут при 50oC. Не происходило никакого изменения цвета. Затем перемещали коричневый раствор обратно в делительную воронку и удаляли кислый слой. Затем проводили двухкратную промывку горячей (приблизительно 50oC) водой (2х100 мл). С помощью роторного испарителя удаляли растворитель и получали 70,4 г неочищенного 2-оксинафталин-1-карбоксальдегидоксима.
Пример 16.
В 1-л трехгорлышковую колбу с круглым дном, снабженную механической мешалкой, термометром и обратным холодильником, загружали магниевые опилки (14,6 г, 0,6 ммоля), безводный метанол (284 мл, 7,0 моля) и безводный толуол (112 мл). К этому добавляли 8%-ный раствор магния в метаноле (5 мл, 0,004 моля) и реакционную смесь затем нагревали до обратного стока. Через несколько минут замечали выделение водорода. Реакционную смесь нагревали с обратным холодильником в течение 1,5 часа до растворения всего магния, в результате чего получали мутный белый раствор/суспензию при никаком дальнейшем выделении водорода.
Добавляли пара-нонилфенол (220,0 г, 1,0 моля) и полученный желтый раствор нагревали с обратным холодильником в течение 1 часа, после чего оставляли на ночь под осушительной трубкой для охлаждения до комнатной температуры. Загружали толуол (240 мл) и переоборудовали аппаратуру на дробную перегонку под вакуумом. Смесь нагревали при давлении 380 мм рт.ст. для удаления метанола, образующего азеотропную смесь с толуолом, пока не достигали внутренней температуры в 75oC. При перегонке (приблизительно при 71oC) заметно увеличивалась вязкость раствора. Затем при 75-77oC в течение 2 часов порциями добавляли суспензию параформальдегида (90 г, 3,0 моля) в толуоле (150 мл) с одновременной перегонкой растворителя и низкокипящих побочных продуктов (210 мл). Внутреннюю температуру реакции поддерживали на 75-77oC путем постепенного снижения давления до 270 мм рт.ст. на всем протяжении добавления этой суспензии. Реакцию поддерживали при 75oC и 270 мм рт. ст. , после чего снимали вакуум, переоборудовали аппаратуру для работы с обратным холодильником и охлаждали реакционную смесь до 55oC для реакции оксимации.
При 40-50oC приготавливали раствор сульфата гидроксиламмония (98,5 г, 0,6 моля) в воде (300 мл) и затем в течение 30 минут добавляли его при быстром перемешивании. Реакция продолжалась 3 часа при 55oC, после чего реакционную смесь охлаждали до 30-40oC.
Останавливали мешалку и содержимое перемещали в делительную воронку. Удаляли водный слой и затем пурпурно-черный слой органических веществ перемещали обратно в реакционный сосуд. В сосуд загружали разбавленный раствор серной кислоты (18,4 г, 0,184 моля) в воде (250 мл) и перемешивали в течение 20 минут при 50oC. В первую минуту происходило быстрое изменение цвета на желтый. После этой кислотной обработки содержимое вновь перемещали в делительную воронку и удаляли водный слой. Затем проводили двухкратную промывку горячей (приблизительно 50oC) водой (2х250 мл). С помощью роторного испарителя удаляли растворитель и получали 261,8 г бледно-желтого масла, которое, как установлено, газохроматографическим анализом, имело концентрацию в 87,0%, что означало выход 5-нонилсалицилальдоксима в 86,6%.
Пример 17.
В 1,0-л трехгорлышковую колбу с круглым дном, снабженную механической мешалкой, термометром и обратным холодильником, загружали магниевые опилки (5,85 г, 0,24 моля), безводный метанол (150 мл, 3,7 моля) и безводный толуол (50 мл). К этому добавляли 8%-ный раствор метилата магния в метаноле (5 мл, 0,004 моля) и реакционную смесь затем нагревали до обратного стока. Через несколько минут замечали выделение водорода. Смесь нагревали с обратным холодильником в течение 1 часа до растворения всего магния, в результате чего получали мутный белый раствор/суспензию при никаком дальнейшем выделении водорода.
Добавляли пара-метоксифенол (48,6 г, 0,39 моля) и полученный желтый раствор нагревали с обратным холодильником в течение 1 часа, после чего оставляли на ночь под осушительной трубкой для охлаждения до комнатной температуры. Загружали толуол (240 мл), а аппаратуру переоборудовали на дробную перегонку под вакуумом. При давлении 380 мм рт.ст. нагревали смесь для удаления метанола, образующего азеотропную смесь с толуолом, пока не достигали внутренней температуры в 75oC. При перегонке (приблизительно при 64oC) происходило осаждение с образованием суспензии бледного цвета. Затем при 75-77oC в течение 1,5 часа добавляли порциями суспензию параформальдегида (36 г, 1,2 моля) в толуоле (80 мл) при одновременной перегонке растворителя и низкокипящих побочных продуктов (63 мл). Внутреннюю температуру реакции поддерживали при 75-77oC в течение 1,5 часа добавляли порциями суспензию параформальдегида (36 г, 1,2 моля) в толуоле (80 мл) при одновременной перегонке растворителя и низкокипящих побочных продуктов (63 мл). Внутреннюю температуру реакции поддерживали при 75-77oC путем постепенного снижения давления до 270 мм рт.ст. на всем протяжении добавления этой суспензии. Реакцию продерживали при 75oC и 270 мм рт.ст. в течение 1 часа, после чего снимали вакуум, переоборудовали аппаратуру на работу с обратным холодильником и охлаждали реакционную смесь до 55oC для реакции оксимации. При 40-50oC приготавливали раствор сульфата гидроксиламмония (39,4 г, 0,24 моля) в воде (120 мл) и затем добавляли его в реакционный сосуд в течение 30 минут при быстром перемешивании. Реакция продолжалась 3 часа при 55oC, после чего реакционную смесь оставляли на ночь для охлаждения до комнатной температуры под азотом. В водном слое образовывался коричневый осадок.
Из двух фаз отфильтровывали твердое вещество, а фильтраты затем перемещали в делительную воронку. Удаляли водный слой. Затем твердый фильтровальный осадок и слой органических веществ перемещали обратно в реакционный сосуд, загружали разбавленный раствор серной кислоты (36,8 г, 0,368 моля) в воде (250 мл) и затем перемешивали его в течение 20 минут при 50oC. В первую минуту происходило быстрое изменение цвета на желтый. После этой кислотной обработки содержимое перемещали в делительную воронку. Мутный кислый водный слой быстро отделяли ввиду образования осадка, а органический раствор/суспензию дважды промывали горячей (приблизительно 65oC) водой (2х100 мл). Раствор толуола/суспензию затем перемещали в коническую колбу и устанавливали ее в ледяную ванну для завершения осаждения. Затем отфильтровывали и высушивали бледно-желтое твердое вещество (27,9 г). С использованием дихлорметана (200 мл) из реакционной смеси раздельно экстрагировали мутный кислый водный слой и водный слой. Объединяли слои органических веществ (дихлорметан из водных экстрактов и толуольные фильтраты) и с помощью роторного испарителя удаляли растворитель, получая 33,6 г желтого твердого вещества. Оба твердых вещества анализировали хроматографическим методом с использованием стандартного вещества известной концентрации и получали совокупный выход 5-метоксисалицилальдоксима в 41%.
Пример 18.
В 1,0-л трехгорлышковую колбу с круглым дном, снабженную механической мешалкой, термометром и обратным холодильником, загружали магниевые опилки (5,85 г, 0,24 моля), безводный метанол (150 мл, 3,7 моля) и безводный толуол (50 мл). К этому добавляли 8%-ный раствор метилата магния в метаноле (5 мл, 0,004 моля) и затем реакционную смесь нагревали до обратного стока. Через несколько минут замечали выделение водорода. Смесь нагревали с обратным холодильником в течение 1 часа до растворения всего магния, в результате чего получали мутный белый раствор/суспензию при никаком дальнейшем выделении водорода.
Добавляли 2,4-диметилфенол (48,8 г, 0,4 моля) и полученный желтый раствор нагревали с обратным холодильником в течение 1,5 часа, после чего оставляли на ночь под осушительной трубкой для охлаждения до комнатной температуры. Добавляли толуол (240 мл) и переоборудовали аппаратуру на дробную перегонку под вакуумом. Под пониженным давлением 380 мм рт.ст. нагревали смесь для удаления метанола, образующего азеотропную смесь с толуолом, пока не достигали внутренней температуры в 75oC. При перегонке (приблизительно при 71oC) заметно увеличивалась вязкость раствора. При 75-77oC в течение 2 часов порциями добавляли суспензию параформальдегида (36,0 г, 1,2 моля) в толуоле (80 мл) с одновременной перегонкой растворителя и низкокипящих побочных продуктов (100 мл). Внутреннюю температуру реакции поддерживали при 75-77oC путем постепенного снижения давления до 245 мм рт.ст. на всем протяжении добавления этой суспензии. Реакцию поддерживали при 75oC и 245 мм рт. ст. в течение 1 часа, после чего снимали вакуум, переоборудовали аппаратуру на работу с обратным холодильником и охлаждали реакционную смесь до 55oC для реакции оксимации. Образовывался желтый раствор.
При 40-50oC приготавливали раствор сульфата гидроксиламмония (39,4 г, 0,24 моля) в воде (120 мл) и затем в течение 30 минут добавляли его в реакционный сосуд при быстром перемешивании. Реакцию продолжали 3 часа при 55oC, после чего реакционную смесь оставляли на ночь для охлаждения до комнатной температуры. В водном слое образовывался белый осадок, который растворяли при добавлении 0,5% об. раствора серной кислоты (200 мл). В слое органических веществ также образовывался бледно-коричневый осадок. Твердое вещество в органической фазе повторно растворяли при нагревании до 50oC.
Останавливали мешалку и содержимое перемещали в делительную воронку. Водный слой быстро удаляли ввиду образования осадка в слое органических веществ. Затем слой органических веществ с коричневой кристаллической суспензией/раствором перемещали обратно в реакционный сосуд, а некоторое количество твердых частиц, оставшихся в делительной воронке, смывали толуолом в колбу. В сосуд загружали разбавленный раствор серной кислоты (33,12 г, 0,331 моля) в воде (250 мл) и перемешивали в течение 20 минут при 50oC. В первую минуту происходило быстрое изменение цвета на желтый. После этой кислотной обработки содержимое вновь перемещали в делительную воронку и удаляли кислый водный слой. Затем проводили двухкратную промывку горячей (приблизительно 65oC) водой (2х200 мл). Слой органических веществ, который уже начал осаждаться, затем перемещали в коническую колбу и ее устанавливали в ледяной ванне для завершения осаждения. Отфильтровывали твердое вещество с выходом в 41,5 г. С помощью роторного испарителя выпаривали фильтраты и получали 26,9 г желтого твердого вещества. Оба твердых вещества анализировали хроматографическим методом с использованием стандартного вещества известной концентрации и получали совокупный выход 3,5-диметилсалицилальдоксима в 57,5%.
2-Оксиарилальдоксимы могут, например, использоваться в качестве экстрагентов при гидрометаллургическом извлечении металлов из металлических руд. Способ получения 2-гидроксиарилальдоксимов включает взаимодействие гидроксиламина с 2-гидроксиарильдегидом в присутствии соединения металла групп II, III, IVA Периодической системы и в таких условиях, когда 2-гидроксиарилальдегид находится по крайней мере частично в виде соли и/или комплексе металла группы II, III, IVA или VIA Периодической системы. Использование 2-оксиарилальдегида частично в виде соли и/или комплексного соединения ускоряет процесс получения 2-гидроксиарилальдоксима. 29 з.п.ф-лы.
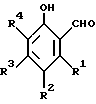
и каждый из R1, R2, R3, R4 независимо представляют собой водород, или атом галогена, или алкил-, циклоалкил-, аралкил-, арил-, алкарил-, алкокси-, арилокси-, ацил- или гидроксигруппу.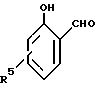
R5 представляет собой водород или C1 - C22 алкил.
где каждый из R1, R2, R3, R4 независимо представляет собой водород, или атом галогена, или алкил, циклоалкил-, аралкил-, арил-, алкарил-, алкокси-, арилокси-, ацил- или гидроксигруппу.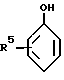
где R5 - водород или C1 - C22-алкил.
Mg(OR6)2,
где R6 представляет алкил, содержащий до двух молей фенола, имеющего по крайней мере одно незамещенное ортоположение по отношению к гидроксильной группе.
| US 4020106 A1, 1977 | |||
| Способ получения оксимов | 1969 |
|
SU315434A1 |

