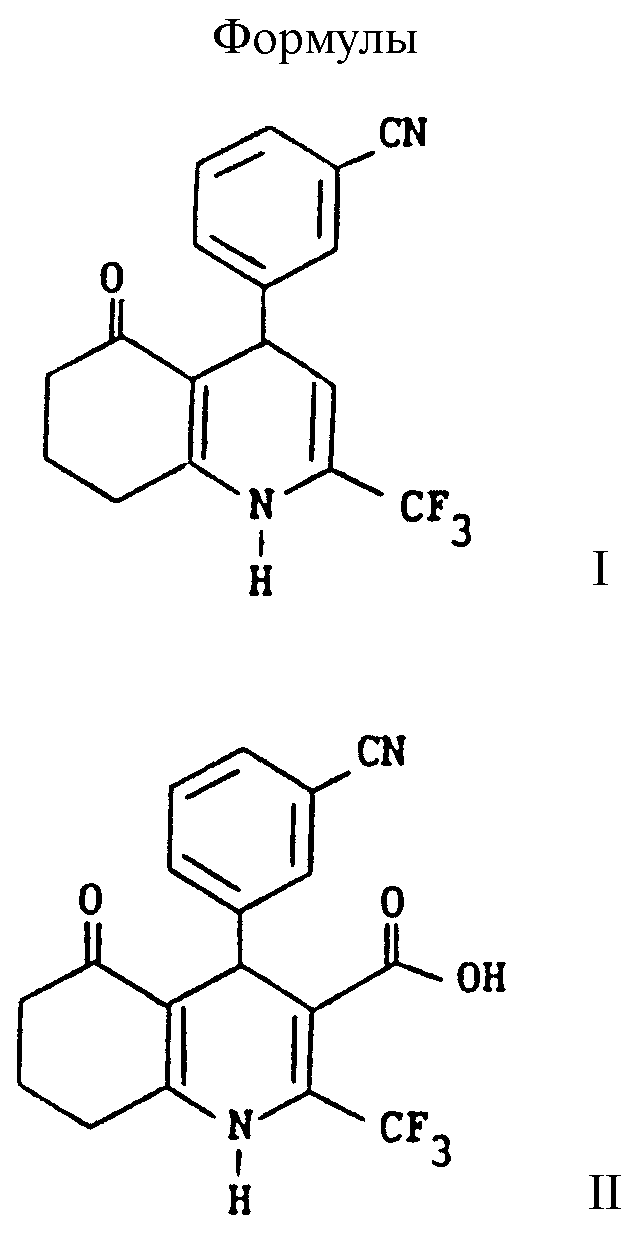
Изобретение относится к новому соединению, полезному при лечении нестабильности мочевого пузыря у млекопитающих, таких как человек. Более конкретно, данное изобретение относится к (S)-(-)-4-(3-цианофенил)-2-трифторметил-4,6,7,8-тетрагидро-5(1H)-хинолону (соединение), применению соединения в лечении недержания мочи у млекопитающих (включая человека), способу получения соединения и фармацевтическим композициям, содержащим соединение.
Существующие способы лечения недержания мочи в основном плохие и основаны на лекарственных средствах, первоначально разработанных для других показаний. Одна группа таких лекарственных средств включает блокаторы кальциевых каналов, такие как нифедипин, которые были первоначально разработаны и использовались главным образом как сердечно-сосудистые средства.
Нифедипин принадлежит к классу соединений, известных как дигидропиридины. Этот структурный класс широко исследован и структурные требования для блокирующего кальций воздействия в настоящее время вполне хорошо установлены. Итак, как описано в главе 14.1 медицинского химического руководства, Comprehensive Medicinal Chemistry, том 3, под редакцией John C.Emmett и изданного Pergamon Press в 1990, соединения обладают 1,4- дигидропиридиновым циклом, имеющим, оптимально, арильную группу в 4-положении и сложноэфирные группы в 3- и 5-положениях. Удаление сложноэфирных групп или замена их на ацетильные или цианогруппы связаны с ослаблением активности. В большинстве случаев соединения имеют метильные группы в 2- и 6-положениях.
Grinshteins и др. , Khim. Geterotsicl. Soedin. (6), 1118-20, 1967 описывают соединения; 3-циано-4-фенил-2,7,7-триметил-4,6,7,8- тетрагидро-5(1H)-хинолон и 3-этаноил-4-фенил-2,7,7-триметил-1,4,6,7,8- тетрагидро-5(1H)-хинолон. Vitolinya et. al., Khim.-Farm. Zh., 15(1), 39-42, 1981 описывают исследование воздействия некоторых 4,6,7,8-тетрагидро-5(1H)-хинолонов, имеющих сложноэфирную или цианогруппу в 3-положении, на сердечно-сосудистую систему и на гладкую мышцу кишечника. Сообщается, что 3-циано-4-фенил-2,7,7-триметил-4,6,7,8- тетрагидро-5(1H)-хинолон обладает гипотензивными свойствами и способен блокировать спазмогенное действие как ацетилхолина, так и хлорида бария на гладкую мышцу кишечника.
DE 2003148 раскрывает группу 1,4-дигидропиридиновых производных, включающую некоторые 4,6,7,8-тетрагидро-5(1H)-хинолоны, которые имеют сложноэфирную или кето-группу в 3-положении и, как указано, проявляют широкий и многогранный фармакологический спектр воздействия. Указано, что основные воздействия, проявляемые соединениями, включают сильные мышечные спазмолитические действия, которые становятся очевидными в гладкой мускулатуре желудочно-кишечного тракта, мочеполового тракта и дыхательной системы. Установлено, что другие основные воздействия состоят в действии на сердце (эффект "успокаивания сердца") и в снижении кровяного давления у животных с нормальным артериальным давлением и животных с гипертонией, так что они могут быть использованы в качестве гипотензивных средств.
Известно, что ткань мочевого пузыря является возбудимой и что недержание мочи может быть вызвано контролируемыми или нестабильными сокращениями мочевого пузыря. Неожиданно было найдено, что соединение способно расслаблять гладкую мышцу мочевого пузыря, таким образом предупреждая или уменьшая интенсивность неконтролируемых или нестабильных сокращений мочевого пузыря. Следовательно, соединение может быть полезным при лечении недержания с позывами, которое включает, например, детрусорную нестабильность, что может быть следствием цистита, уретрита, опухолей, камней, затрудненности дивертикула или оттока; и детрусорную гиперрефлексию, которая может быть следствием приступа, деменции, болезни паркинсона, супрасакрального спинно-мозгового повреждения или супрасакрального спинно-мозгового заболевания.
Также неожиданно было найдено, что соединение служит веществом, открывающим калиевые каналы. Известно, что, вызывая открытие калиевых каналов, открывающие калиевые каналы соединения могут тем самым вызывать расслабление гладкой мышцы. Не претендуя на теоретическое обоснование, соответственно полагают, что соединение действует, открывая калиевые каналы в клетках мочевого пузыря, тем самым вызывая расслабление гладкой мышцы и таким образом предупреждая или уменьшая интенсивность неконтролируемых сокращений мочевого пузыря, которые могут вызывать недержание мочи. Nurse D.A., Restorick J.M., and Mundy A.R., British Journal of Urology, (1991), 68, 27-31 описывают, что кромакалим, хорошо известный как вещество, открывающее калиевые каналы, как было найдено, является эффективным в предварительном клиническом испытании при лечении недержания мочи.
Таким образом, изобретение дает соединение (S)-(-)-4-(3-цианофенил)-2-трифторметил-4,6,7,8-тетрагидро-5(1H)-хинолон и его фармацевтически приемлемые соли.
Следует понимать, что соединение может проявлять полиморфизм и может образовывать сольваты. Должно быть понятно, что данное изобретение включает любую полиморфную форму, или сольват, или их смеси, которые полезны для расслабления гладкой мышцы мочевого пузыря, в соответствующей области хорошо известно, как определить, является ли соединение способным к расслаблению гладкой мышцы мочевого пузыря на основании стандартных испытаний, описанных ниже. Стереохимия соединения была первоначально обозначена как (-), после этого было установлено, что абсолютная стереохимия представляет собой (S). Таким образом, может быть предпочтительно применять соединение в форме, которая характеризуется как содержащая, например, по крайней мере, 95%, 98% или 99% энантиомерный избыток (ее) (S)-(-)-формы.
Соединение может быть получено способами, которые включают способы, известные в химической области для получения структурно аналогичных соединений. Такие способы промышленного получения (S)-(-)-4-(3-цианофенил)-2-трифторметил-4,6,7,8-тетрагидро-5(1H)хинолона составляют дальнейшие аспекты изобретения и иллюстрируются приведенными ниже методиками. Такой способ может обычно выражаться в декарбоксилировании соответствующей (S)-(-)-формы карбоновой кислоты формулы II.
Реакцию декарбоксилирования удобно выполнять при повышенной температуре, например, в интервале от 50oC до 250oC, предпочтительно, в интервале от 190oC до 220oC в отсутствие кислотного катализатора, особенно от 130oC до 185oC (например, используя нагревающую "рубашку") и предпочтительно, в интервале от 90oC до 120oC с кислотным катализатором. Реакция декарбоксилирования с применением бескислотного катализатора может быть выполнена как в чистом расплаве, так и в инертном растворителе с подходящей температурой кипения, таком как простой дифениловый эфир или N-метил-пирролидин-2-он. Предпочтительным растворителем является N-метилпирролидин-2-он. Подходящие растворители для катализируемого кислотой декарбоксилирования включают спирты, например, метанол или этанол; диметилсульфоксид; ароматические углеводороды, такие как толуол; простые эфиры, такие как, например, 1,2-диметоксиэтан или диглим; и N-метилпирролидин-2-он. В качестве кислотного катализатора обычно могут быть использованы концентрированная серная кислота, фосфорная кислота, соляная кислота, бромистоводородная кислота, йодистоводородная кислота, сильные органические кислоты, такие как трифторуксусная кислота, органическая сульфокислота, такая как метансульфокислота или п-толуолсульфокислота. Реакция может обычно быть проведена точно при температуре плавления или при температуре выше температуры плавления вещества.
(S)-(-)-форму карбоновой кислоты формулы II обычно получают разделением соответствующей рацемической карбоновой кислоты формулы II. Удобный способ разделения соединения формулы II состоит в получении разделяемой соли кислоты, например, использованием S-(-)- α- метилбензиламина. Соль восстанавливают и затем подкисляют для выделения разделяемой карбоновой кислоты формулы II. Перекристаллизацию выполняют предпочтительно при температуре 75oC или ниже и желательно без перемешивания.
Или же иначе, неочищенное рацемическое соединение формулы I может быть разделено использованием хиральной колонки, что дает соединение формулы I.
При отсутствии коммерческой доступности, материалы, необходимые для способов, таких как описаны ниже, могут быть получены способами, которые выбирают из стандартных органических химических методик, методик, которые являются аналогичными методикам, используемым для синтезов известных, структурно подобных соединений, или методик, которые являются аналогичными для вышеописанного способа или способов, описанных в примерах.
Промежуточный продукт формулы II может быть получен, как показано в схеме 1, путем взаимодействия эфира ацетоуксусной кислоты формулы III или его полуацеталей, в которых OPa обозначает спиртовой остаток, образующий сложный эфир, который может быть расщеплен в слабощелочной среде, такого как, например, сложный 2-цианэтоксиэфир, с 3-цианбензальдегидом и 1,3-циклогександионом с образованием спирта формулы IVa. Дегидратация спирта дает сложный эфир формулы IV, который может быть подвергнут омылению с образованием кислоты формулы II.
Иначе, промежуточный продукт формулы II может быть получен, как показано в схеме II, путем взаимодействия эфира ацетоуксусной кислоты формулы III или его полуацеталей, в которых ORa обозначает спиртовой остаток, образующий сложный эфир, который может быть легко расщеплен кислотой, такого, как, например, изоборниловый эфир, с 3-цианбензальдегидом и 1,3-циклогександионом с образованием спирта формулы IVa.
Кислотно-катализируемая дегидратация и гидролиз соединения формулы IVa, например в условиях, аналогичных описанным в примерах, дают соединение формулы II. Кислотным катализатором для получения соединения формулы IV из IVa может служить любой из упомянутых ранее кислотных катализаторов и, предпочтительно, п-толуолсульфокислота. Кислотным катализатором для получения соединения формулы II из IVa служит, предпочтительно, п-толуолсульфокислота. Реакцию желательно проводить в присутствии инертного растворителя, такого как толуол или уксусная кислота. В целях предотвращения декарбоксилирования полезно использовать безводный растворитель, например, ледяную уксусную кислоту.
Для дальнейшего сведения к минимуму декарбоксилирования следует поддерживать температуру реакционной смеси максимум при 104oC, предпочтительно, в интервале от 100oC до 104oC, особенно желательно 102-104oC.
Изоборниловый эфир может быть получен путем обмена сложноэфирных групп изоборнила и этил-4,4,4-трифторацетоацетата, и полуацеталя 2-цианоэтила и 2-цианоэтил-4,4,4-трифторацетоацетата путем сложноэфирного взаимообмена 3-гидроксипропионитрила и этил-4,4,4-трифторацетоацетата.
Дальнейшие аспекты данного изобретения составляют способы получения соединения формулы I и его промежуточных продуктов.
Фармацевтически приемлемые соли могут быть получены использованием стандартных способов, хорошо известных в соответствующей области, например взаимодействием соединения с подходящей кислотой или основанием, дающими физиологически приемлемый противоион.
При использовании для лечения недержания мочи соединение обычно вводят в виде фармацевтической композиции, которая включает соединение вместе с фармацевтически приемлемым разбавителем или носителем, композиция адаптирована для конкретного выбранного пути введения. Такие композиции также составляют предмет изобретения.
Следовательно, согласно другому аспекту, изобретение дает фармацевтическую композицию, которая включает соединение или его фармацевтически приемлемую соль, как определено выше, и фармацевтически приемлемый разбавитель или носитель.
Композиции могут быть получены использованием обычных способов и эксципиентов и связывающих веществ и могут быть представлены различными дозированными формами. Например, они могут быть в форме таблеток, капсул, растворов или суспензий для перорального применения: в форме суппозиториев для ректального введения; в форме стерильных растворов или суспензий для внутривенной, внутрипузырной, подкожной или внутримышечной инъекции или инфузии; или в форме пластыря для чрескожного введения.
Далее изобретение дает способ лечения недержания мочи, включающий введение нуждающемуся в таком лечении млекопитающему эффективного количества соединения или его фармацевтически приемлемой соли.
Обработка с применением соединения может быть лечебной или терапевтической в зависимости от введения соединения сразу в начале или после развития недержания мочи у пациента. Обработка также может быть профилактической или перспективной при введении соединения в ожидании возможного развития недержания мочи, например пациенту, в прошлом страдавшему недержанием.
Согласно дальнейшему аспекту изобретение представляет применение соединения при промышленном способе изготовления лекарственного средства для лечения недержания мочи.
Поскольку соединение действует, открывая клеточные калиевые каналы, оно может также быть полезным в качестве терапевтического средства при лечении других состояний или заболеваний, при которых действие терапевтического средства, открывающего калиевые каналы, желательно, или известно, что оно дает улучшение. Такие состояния или заболевания включают гипертензию, астму, заболевание периферических сосудов, недостаточность правых отделов сердца, застойную сердечную недостаточность, ангину, ишемическую болезнь сердца, нарушения сосудов головного мозга, глаукому, почечные колики, нарушения, связанные с камнями в почках, слизистый колит, гнездовую мужскую плешивость, преждевременные роды и пептические язвы.
Вводимая доза соединения обязательно должна варьироваться в соответствии с принципами, хорошо известными в соответствующей области, принимающими во внимание путь введения, тяжесть состояния недержания и размер и возраст пациента. В основном соединение вводится теплокровным животным (таким, как человек), так что эффективная доза является общепринятой, обычно суточная доза выше 0,005, например, приблизительно в интервале от 0,01 до 10 мг/кг веса тела. Предпочтительно, соединение в этом интервале доз вводится перорально.
Для специалиста в соответствующей области очевидно, что соединение может быть введено совместно с другими терапевтическими или профилактическими средствами и/или лекарственными препаратами, для которых отсутствует медицинская несовместимость.
Действие соединения как вещества, расслабляющего гладкую мышцу, полезного в качестве терапевтического средства для лечения недержания мочи, состоящее в его воздействии на открытие калиевых каналов и гиперполяризацию мембранного потенциала в детрусорной гладкой мышце мочевого пузыря, может быть показано применением соответственно предназначенных тестов in vitro, таких как тест, описанный ниже. Соединение показывает в испытании IC50=4,2 микромол. "IC50" является хорошо известным термином и означает концентрацию испытуемого соединения, которая вызывает 50% снижение в сокращении in vitro ткани мочевого пузыря, как описано в приведенном ниже испытании.
Самцов морских свинок (альбинос Hartley) (450-500 г) забивают, используя двуокись углерода, чтобы вызвать асфиксию, и быстро выпускают кровь. Нижнюю абдоминальную полость раскрывают и отделяют мочевой пузырь. Пузырь очищают от окружающей соединительной и жировой ткани, и часть, расположенную выше отверстия мочеточника, удаляют и промывают в буферном растворе Krebs-Henseleit'a следующего состава (в мМ): NaCl 118.0, KCl 4.7, MgSO4 1.2, KH2PO4 1.2, CaCl2 2.5, NaHCO3 25.0 и d-глюкоза 11.1. Раствор нагревают до 37oC и насыщают газообразной смесью: 95% O2 и 5% CO2. При сильном барботировании, раствор должен достигать значения pH, близкого к 7.4.
Купол промытого мочевого пузыря отсекают и отбрасывают: оставшийся мочевой пузырь помещают на марлю в чашку Петри, содержащую буферный раствор. Делают с помощью ножниц средневентральный продольный разрез, чтобы открыть мочевой пузырь. Из купола нарезают полоски, и кромку дна мочевого пузыря отбрасывают. Оставшуюся детрусорную среднюю секцию разрезают на две горизонтальные полоски приблизительно 2,0 мм шириной. Эти две полоски затем делят пополам по среднедорсальному отделу, получая четыре полоски одинакового размера.
Два конца каждой отдельной полоски привязывают к стеклянному поддерживающему стержню и преобразователю силы-смещения (Grass model FR 03), соответственно, с помощью 4-0 черной плетеной шелковой нити.
Датчики соединяют с полиграфом (Grass model 7E), который калибруют при 5 мВ/см и калибровку проверяют на линейность с грузами 5 и 0,5 граммов. Аналоговые электрические сигналы, выходящие из полиграфа, преобразуют в цифры с помощью Modular Instrument Micro 5000 сигнальной воспроизводящей системы, используя системную биологическую программу Biowindow Data Acquisition Software, которую проводят под контролем Microsoft OS/2 оперативной системы на машинах IBM-PC-совместимых.
Детрусорные полоски закрепляют на стеклянных стержнях в 20 мл ваннах для ткани и дают уравновеситься при предварительной нагрузке в 2 г. Во время периода уравновешивания, которое достигается через 45 - 60 мин, ткань промывают свежим буферным раствором с 15-минутным интервалом, используя при необходимости перед промыванием доведение нагрузки до 2 г.
Когда ткань расслабляется до устойчивого состояния, после окончательного промывания, снова применяют 15 мМ KCl. Как только миогенная активность ткани достигает устойчивого состояния, нулевые данные получают с помощью Biowindows Data Acquisition системы путем усреднения 5-минутных миогенных данных, полученных при 32 Гц. После установления нулевой линии экспериментальное соединение дозированно вводят кумулятивным способом, используя инкременты полу-логарифмов. Время контакта каждой дозы составляет 10 мин, причем последние 5 мин являются периодом времени, за который получают данные о зависимости доза-отклик. Если 30 мкМ испытуемого соединения не устраняет детрусорную механическую активность, тогда дозированно вводят 30 мкМ кромакалима, вещества, предполагаемо открывающего калиевые каналы, для установления максимального отклика. Эффект соединения при каждой дозе выражают в % максимально ингибируемого отклика, который затем соотносят с соответствующим эффектом контроля-носителя соединения. Нормализованный отклик используют затем для получения IC50 расслабляющей активности соединения, применяя способ Marquardt'a подгонки нелинейной повторяющейся кривой к стандартной функции доза-эффект лекарственного вещества.
Кроме того, способность соединения открывать калиевые каналы в детрусорной гладкой мышце может быть продемонстрирована вторым испытанием in vitro. Этот второй тест in vitro аналогичен описанному выше испытанию в отношении подгонки ткани и обработки данных. Однако следует отметить следующие исключения. В этом втором испытании сокращение детрусорных полосок в начальный период и после периода уравновешивания достигается использованием 80 мМ вместо 15 мМ KCl (общая концентрация в ванне). После этой высокой KCl стимуляции длительное (остаточное) напряжение в ткани делается очевидным, поскольку чувствительные к напряжению кальциевые каналы оказываются открытыми для обеспечения притока кальция к клеткам и развития тонизирующего напряжения. Это напряжение обычно снимается 300 мкМ папаверина, который тем самым используют для установления максимальной реакции в данном испытании.
Характерные блокаторы кальциевых каналов, такие как нифедипин, нимодипин, испадипин и верапамил способны уменьшать напряжение и восстанавливать миогенную активность детрусорных полосок морской свинки в обоих испытаниях благодаря их блокирующему действию на кальциевые каналы. Однако все вышеупомянутые блокаторы кальциевых каналов более сильно действуют во втором испытании, когда используют 80 мкМ KCl, чем в первом испытании, где используют 15 мкМ KCl. В противоположность этому, несмотря на то, что кромакалим, вещество предполагаемо открывающее калиевые каналы, обладает мощной расслабляющей активностью в первом испытании с IC50 в интервале от 0,6 до 0,9 мкМ, он проявляет незначительную расслабляющую активность во втором испытании при таких высоких концентрациях как 30 мкМ. Таким образом, показатели более высокой расслабляющей активности в первом испытании по сравнению со вторым испытанием для соединений по изобретению указывают на то, что соединения функционируют как вещества, открывающие калиевые каналы. Соединение показывает во втором испытании IC50 41,1 мкМ. Способность соединения действовать на ткань мочевого пузыря как вещества, открывающего калиевые каналы, может быть дополнительно продемонстрирована стандартным испытанием, в котором измеряют воздействие испытуемых соединений на скорость истечения рубидия (86Rb) или калия (42K) из ткани.
Кроме того, специалисту в соответствующей области понятно, что эффективность соединения может быть продемонстрирована стандартным испытанием in vivo. Далее приводится описание такого стандартного испытания, которое может быть использовано для того, чтобы убедиться, является ли испытуемое соединение активным и, кроме того, обладает ли испытуемое соединение селективностью в отношении мочевого пузыря без существенных сердечно-сосудистых воздействий при пероральном введении.
Самцов крыс Wistar (400-500 г) подвергают анестезии, используя 50 мг/кг нембутала, внутрибрюшинно. Каждой крысе выбривают область живота и переднюю и заднюю часть шеи и на кожу наносят йод-повидон. Для каротидной катетеризации, левую сонную артерию раскрывают с помощью небольшого вентрального цервикального разреза. Открытую поверхность промывают 2% лидокаин-HCl-раствором, чтобы расслабить сосуд. Катетер, заполненный 0,9% физиологическим раствором, вводят приблизительно на 2,4 см в артерию, так, чтобы его конец находился в дуге аорты. Дистальный конец катетера выводят на заднюю часть шеи, заполняют гепарином (1000 ед/мл) и запаивают.
Для катетеризации мочевого пузыря мочевой пузырь раскрывают абдоминальным разрезом по средней линии. Троакар продвигают через абдоминальную мышцу приблизительно на 1 см от верхнего конца разреза и затем осуществляют подкожную туннелизацию, чтобы он выступал наружу через кожу задней части шеи. Заполненный физиологическим раствором катетер пропускают через троакар. Небольшое отверстие в своде мочевого пузыря выполняют с помощью Accu-Temp каутера. Катетер вводят в мочевой пузырь и закрепляют 4-0-шелковой лигатурой. Катетер промывают физиологическим раствором и раскрытое состояние отмечают. Наружный конец катетера запечатывают при нагреве для предупреждения утечки мочи. Абдоминальные мышцы и кожу сшивают. Оба катетера продевают через узелок фиксатора из нержавеющей стали (Instech), который затем пришивают к подкожной мышце в точке выворачивания наружу. Кожу плотно зашивают над узлом. Животным дают восстановиться после анестезии.
Через 24-48 часов после операции каждую крысу помещают в камеру для исследования метаболизма и соединяют с помощью узла фиксатора с Instech пружинным креплением и шарнирной системой для защиты катетеров от повреждения, чтобы позволить животным свободно перемещаться в камере. Каротидный катетер присоединяют к Gould P23XL датчику для измерения кровяного давления. Катетер мочевого пузыря присоединяют к насосу для вливания физиологического раствора и к датчику давления с помощью PE50 трубок и 4-ходового запорного крана. Весы с верхней загрузкой и с собирающей чашкой помещают под камерой для измерения выделяемой мочи.
Крыс взвешивают, перорально имитируют дозирование (вводят дозирующие иглы, но не удаляют жидкость) и начинают и продолжают во время эксперимента трансвезикальное вливание физиологического раствора. Изменения кровяного давления, частоты сердечных сокращений, давления внутри мочевого пузыря и диурез регистрируют либо на Grass полиграфе, либо на Gould TA4000 регистрирующей системе. Животным дают восстановиться, пока режим мочеиспускания не станет нормальным (приблизительно 45-90 мин). В этой точке регистрируют нулевую линию каждого экспериментального параметра и крысам вводят через оральный зонд соответствующую дозу соединения (в 75% PE 400-физиологический раствор растворителей) в таких концентрациях, что объем составляет 1 мл/кг веса тела. Воздействия соединений на экспериментальные параметры наблюдаются в течение пяти часов после введения.
Экспериментальные результаты как для интервала концентрации, так и для частоты сердечных сокращений выражают как среднее значение ±SEM (стандартная ошибка измерений) % изменения от нулевого уровня, для каждого животного используется его собственный контроль. MAP (ср. артериальное давление) выражают как среднее значение ±SEM мм Hg изменения относительно нулевого уровня.
Соединение по данному изобретению активно в вышеуказанных испытаниях и селективно для мочевого пузыря при отсутствии значительных сердечно-сосудистых воздействий в случае перорального введения, например, при дозе 3 мг/кг в приведенном выше испытании in vivo.
Далее изобретение иллюстрируется следующими нелимитирующими примерами, в которых, если не оговорено особо:
(I) Температуры даны в градусах Цельсия (oC); работы проводятся при комнатной температуре или температуре окружающей среды, т.е. при температуре в интервале 18-25oC;
(II) органические растворители сушат над безводным сульфатом магния; упаривание растворителя осуществляют, используя роторный испаритель при пониженном давлении (600-4000) паскалей; 4,5-30 мм Hg) при температуре бани 60oC;
(III) под хроматографией имеется в виду "флэш" хроматография; обращенная фазовая хроматография обозначает флэш хроматографию на покрытом октадесилсиланом (ODS) носителе, имеющем диаметр частиц 32-74 мк, известном как "PREP-40-ODS" (Art. 731740-100 from Bodman Chemicals, Aston, PA, USA); тонкослойную хроматографию (TCX) выполняют на пластинах из силикагеля;
(IV) в основном за ходом реакции следят TCX и время реакции дается только для иллюстрации;
(V) температуры плавления приведены без поправок и (разл.) означает разложение; температуры плавления даны такие, как получены для продуктов, синтезированных согласно описанию; полиморфизм может выражаться в получении в некоторых способах веществ с различными температурами плавления;
(VI) конечные продукты подтверждаются спектрами протонного ядерного магнитного резонанса (ПМР);
(VII) выходы даны только для иллюстрации и не являются такими, которые неизбежно могут быть получены при тщательном выполнении процесса; синтезы повторяют, если требуется большее количество вещества;
(VIII) когда они приведены, ЯМР-данные представлены в форме дельта-величин для основных определяемых протонов, приведенных в миллионных долях (м.д. ) относительно тетраметилсилана (TMS) в качестве внутреннего стандарта, определенных при 300 МГц использованием пердейтерированного диметил сульфоксида (ДМСО-d6) в качестве растворителя; для формы сигналов использованы общепринятые обозначения; постоянные связи (J) даны в Гц; Ar обозначает ароматический протон, там, где дается такое отнесение;
(IX) химические символы имеют их обычные значения; использованы единицы и символы системы СИ:
(X) пониженное давление приведено как абсолютное давление в паскалях (Pa); повышенное давление приведено как давление, измеренное в барах;
(XI) соотношения растворителей даны в объем/объем (V/V) терминах и
(XII) масс спектр (МС) снят при энергии электронов 70 электроновольт химических ионизирующих колебаниях (CI) с использованием проб прямой экспозиции; где указанная ионизация вызывается соударением электронов (EI) или быстрой атомной бомбардировкой (FAB); приведены значения для m/z, обычно приведены только ионы, которые указывают материнскую массу.
Пример 1
(S)-(-)-4-(3-Цианофенил)-2-трифторметил-4,6,7,8-тетрагидро- 5(1H)-хинолон.
Перемешиваемый раствор (S)-(-)-(3-цианофенил)-2-трифторметил-5- оксо-1,4,5,6,7,8-гексагидрохинолин-3-карбоновой кислоты (3,93 г, 10,85 ммоль) в N-метилпирролидин-2-оне (24 мл) помещают в предварительно нагретую до 210oC масляную баню на 20 минут. Затем охлажденную реакционную смесь выливают в воду (200 мл) и дважды экстрагируют диэтиловым эфиром. Объединенные эфирные экстракты дважды промывают водой, сушат (MgSO4), фильтруют и удаляют растворитель, получая не совсем белый твердый продукт. Хроматография (элюент: метиленхлорид/диэтиловый эфир 9:1) и растирание с диэтиловый эфир/гексаном дают указанное в заглавии соединение (3,42 г, 79%) в виде белого твердого вещества. Т. пл. 187-189oC. ЯМР 1,88-1,91 (м, 2H, CH2), 2,21-2,25 (м, 2H, CH2), 2,53-2,64 (м, 2H, CH2), 4,68 (д, 1H, J = 5,3 Гц, CH), 5,61 (д, 1H, J = 5,3 Гц, CH), 7,50-7,54 (м, 2H, Ar), 7,61-7,66 (м, 2H, Ar), 9,64 (с, 1H, NH); МС: m/z = 319 (M + 1); [α]
Рассчитано: C, 64.14; H, 4.12; N, 8.80.
Найдено: C, 64.07; H, 4.24; N, 8.78.
19F-ЯМР-анализ этого вещества в присутствии реагента хирального сдвига (R)-(-)-(9-антрил)-2,2,2-трифторэтанол-d11 (CDCl3) при -30oC) показывает присутствие (S)-(-)-энантиомера приблизительно в 99% ее.
Промежуточную (S)-(-)-4-(3-цианофенил)-2-трифторметил-5-оксо- 1,4,5,6,7,8-гексагидрохинолин-3-карбоновую кислоту получают, как описано ниже.
A. (+)-Изоборнил 4,4,4-трифторацетоацетат.
Смесь этил 4,4,4-трифторацетоацетата (160,28 г, 870 ммоль) и (±)-изоборнеола (86,81 г, 563 ммоль) перемешивают при 130oC (температура бани) 18 часов под 4-дюймовой (1 дюйм = 25,4 мм) колонкой Вигре, позволяющей отогнать этанол. Температуру бани увеличивают до 150oC, чтобы отогнать оставшийся этанол; всего собирают 29 мл (88% теоретич.). Оставшуюся смесь фракционируют затем при пониженном давлении, получая (±)-изоборнил 4,4,4-трифторацетоацетат в виде бесцветного масла (124,6 г, 76%); т.кип. 84-92o/0,4 торр; ЯМР (CDCl3): 0,82-1,17 (м, 11H, CH3, CH2), 1,55-1,84 (м, 5H, CH2, CH), 3,72 (с, CH2-дикето форма), 4,74-4,81 (м, 1H, -OCH), 5,59 (с, энольная форма CH), 11,97 (с, энольная форма OH).
B. (±)-Изоборнил 4-(3-цианофенил)-2-трифторметил-2- гидрокси-5-оксо-1,2,3,4,5,6,7,8-октагидрохинолин-3-карбоксилат.
Перемешиваемую смесь (±)-изоборнил 4,4,4-трифторацетоацетата (82,6 г, 282,8 ммоль), 1,3-циклогександиона (31,7 г, 282,8 ммоль), 3-цианобензальдегида (37,1 г, 282,8 ммоль) и ацетата аммония (54,4 г, 706,3 ммоль) в этаноле (2070 мл) нагревают при температуре кипения с обратным холодильником 4 часа. После удаления осадившегося 9-(3-цианофенил)-3,4,6,7,9,10-гексагидро-1,8-(2H, 5H)-акридиндиона фильтрацией, фильтрат концентрируют в вакууме. Хроматография (элюент: этилацетат/гексан 7:3) дает (±)-изоборнил 4-(3-цианофенил)-2-трифторметил-2-гидрокси-5-оксо-1,2,3,4,5,6,7,8- октагидрохинолин-3-карбоксилат 92 г (63%) в виде белого твердого продукта; т.пл. 209-213oC разл. ; ЯМР: 0,63-0,73 (м, 9H, CH3), 0,86-1,64 (м, 7H, CH2, CH), 1,84-1,88 (м, 2H, CH2), 2,03-2,07 (м, 2H, CH2), 2,34-2,63 (м, 2H, CH2), 2,64-2,72 (м, 1H, CH), 3,92-3,96 (д, 1H, CH), 4,10-4,16 (м, 1H, CH), 7,22-7,25 (д, 1H, OH), 7,39-7,45 (м, 2H, Ar), 7,49 (с, 1H, Ar), 7,57-7,61 (м, 1H, Ar), 8,11-8,13 (д, 1H, Ar); МС: m/z = 517 (M + 1), Анализ для C28H31F3N2O4:
Рассчитано: C, 65.10; H, 6.05; N, 5,42.
Найдено: C, 64.93; H, 6.05; N, 5.22.
C. 4-(3-Цианофенил)-2-трифторметил-5-оксо-1,4,5,6,7,8- гексагидрохинолин-3-карбоновая кислота.
Перемешиваемую смесь (±)-изоборнил 4-(3-цианофенил)-2- трифторметил-2-гидрокси-5-оксо-1,2,3,4,5,6,7,8-октагидрохинолин-3- карбоксилата (8,5 г, 16,5 ммоль), п-толуолсульфокислоты (1,05 г, 5,5 ммоль) и толуола (170 мл) нагревают при температуре кипения с обратным холодильником 3 часа. Реакционная смесь состоит из нерастворимой смолы, которая содержит заданную кислоту и толуольный раствор. В целях предотвращения декарбоксилирования допускается только нагрев, обеспечивающий слабое кипение реакционной смеси. После удаления растворителя, остаток распределяют между этилацетатом и водой. Этилацетат промывают водой, отделяют и дважды экстрагируют насыщенным водным бикарбонатом натрия. Перемешиваемые объединенные экстракты бикарбоната натрия охлаждают на ледяной бане и добавляют по каплям концентрированную соляную кислоту до тех пор, пока раствор не станет сильнокислым. Смесь экстрагируют диэтиловым эфиром и сушат, фильтруют и концентрируют в вакууме, получая желтую масляную смолу. Растирание с метиленхлоридом/гексаном дает карбоновую кислоту (1,8 г, 31%) в виде не совсем белого твердого вещества. Вещество идентично по ЯМР и ТСХ (силикагель - 10% метанол в хлороформе, содержащий несколько капель уксусной кислоты) веществу, описанному и охарактеризованному в подразделе 1 примера 1.
Этилацетатный слой сушат и упаривают, получая чистый (±)-изоборнил 4-(3-цианофенил)-2-трифторметил-5-оксо-1,4,5,6,7,8- гексагидрохинолин-3-карбоксилат, который идентичен ранее полученному и охарактеризованному образцу; белый твердый продукт: т.пл. 170-180oC; ЯМР: 0,61-0,72 (м, 9H, CH3), 1,00-2,6 (м, 13H, CH2, CH), 4,45-4,57 (м, 1H, CH), 4,88-4,92 (д, 1H, CH), 7,46-7,55 (м, 3H, Ar), 7,65-7,69 (м, 1H, Ar), 9,70-9,71 (д, 1H, NH), МС (CI, CH4): 517 (M + 1). Анализ для C28H29F3N2O4:
Рассчитано: C, 67.46; H, 5,86; N, 5.62.
Найдено: C, 67.29; H, 5.99; N, 5.71.
Выделенный (±)-изоборнил 4-(3-цианофенил)-2-трифторметил-5- оксо-1,4,5,6,7,8-гексагидрохинолин-3-карбоксилат обрабатывают в условиях, аналогичных описанным в примере 1С, получая дополнительное количество 4-(3-цианофенил)-2-трифторметил-5-оксо- 1,4,5,6,7,8-гексагидрохинолин-3-карбоновой кислоты, общий выход 2,9 г (49%).
D. Соль S-(-)- α- Метилбензиламин (S)-(-)-4-(3-цианофенил)-2- трифторметил-5-оксо-1,4,5,6,7,8-гексагидрохинолин-3-карбоновой кислоты.
К перемешиваемому раствору рацемической 2-трифторметил-4-(3-цианофенил)-5-оксо-1,4,5,6,7,8-гексагидрохинолин- 3-карбоновой кислоты (28,2 г, 77,84 ммоль) в н-бутаноле (211 мл) добавляют толуол (1000 мл) и впоследствии раствор S-(-)- α- метилбензиламина (9,4 г, 77,84 ммоль) в толуоле (198 мл). После выдерживания при температуре окружающей среды в течение ночи, полученный осадок отфильтровывают и промывают толуолом и диэтиловым эфиром, получая 32 г белой соли. Пятикратная перекристаллизация из толуол/н-бутанола (3: 1), при нагревании смеси до растворения соли, дает соль в виде белого твердого вещества (7,77 г); т.пл. размягчение и превращение в стеклообразную массу 112-115oC, расплавление до жидкого состояния 148-150oC; ЯМР: 1,36-1,38 (д, 3H, CH3), 1,75-1,88 (м, 2H, CH2), 2,19-2,23 (м, 2H, CH2), 2,49-2,61 (м, 2H, CH2), 3,35-3,42 (м, способный к обмену, вода), 4,24-4,26 (кв, 1H, J = 6,8 Гц, CH), 4,91 (с, 1H, CH), 7,16-7,60 (м, 9H, Ar); МС: m/z = 363 (M + 1); [α]D = -180.5o (c = 1.075, метанол, 23oC); 99% ее по 19F-ЯМР в CDCl3. Анализ для C26H24F3N3O3• 1.0C4H9OH•0.5H2O:
Рассчитано: C, 63.59; H, 6.23; N, 7.42.
Найдено: C, 63.66; H, 6.15; N, 7,06.
Дальнейшие исследования показывают, что максимальная температура во время перекристаллизации должна составлять 75oC для предотвращения декарбоксилирования и что во время перекристаллизации не должно применяться перемешивание, иначе энантиомерное обогащение (enhancement) слабое.
E. (S)-(-)-4-(3-Цианофенил)-2-трифторметил-5-оксо-1,4,5,6,7,8- гексагидрохинолин-3-карбоновая кислота.
К охлаждаемой (ледяная баня) перемешиваемой суспензии соли S-(-)- α- метилбензиламин (S)-(-)-4-(3-цианофенил)-2-трифторметил-5- оксо-1,4,5,6,7,8-гексагидрохинолин-3-карбоновой кислоты (7,5 г, 15,5 ммоль) в воде (150 мл) добавляют по каплям концентрированную соляную кислоту до тех пор, пока смесь не станет сильнокислой. Смесь экстрагируют дважды диэтиловым эфиром и объединенный эфирный слой сушат, фильтруют и растворитель удаляют, получая желтую пену. Растирание с дихлорметаном дает 4,9 г (88%) карбоновой кислоты в виде бледно-желтого твердого вещества; т.пл. 206-208oC; ЯМР: 1,74-1,95 (м, 2H, CH2), 2,18-2,32 (м, 2H, CH2), 2,54-2,73 (м, 2H, CH2), 4,92 (с, 1H, CH), 7,47-7,52 (м, 3H, Ar), 7,64-7,68 (м, 1H, Ar), 9,60 (с, 1H, NH), 13,07 (с, 1H, COOH); МС: m/z = 363 (M + 1).
Промежуточная 4-(3-цианофенил)-2-трифторметил-5-оксо-1,4,5,6,7,8- гексагидрохинолин-3-карбоновая кислота, описанная в примере 1C, может быть получена иначе, как описано в примере 6.
F. 2-Цианоэтил полуацеталь 2-цианоэтил 4,4,4-трифторацетоацетата.
Смесь этил 4,4,4-трифторацетоацетата (30 мл, 205 ммоль) и 3-гидроксипропионитрила (7,11 г, 100 ммоль) перемешивают при 150oC (температура бани) в течение 18 часов под 4-дюймовой колонкой Вигре, позволяющей отогнать этанол. Затем оставшуюся смесь фракционируют при атмосферном давлении. Определено, что фракция, перегоняющаяся при 222-228oC (11,71 г), содержит приблизительно 50 мольн. % 2-цианоэтил полуацеталя 2-цианоэтил 4,4,4-трифторацетоацетата согласно ЯМР и масс-спектральным анализам: ЯМР (CDCl3): 2,43-2,65 (м, 2H), 2,76-2,99 (м, 4H), 3,78-3,82 (м, 1H), 4,13-4,15 (м, 1H), 4,33-4,57 (м, 2H), 5,96 (с, 1H, OH).
G. 2-Цианоэтил 4-(3-цианофенил)-2-трифторметил-2-гидрокси-5- оксо-1,2,3,4,5,6,7,8-октагидрохинолин-3-карбоксилат.
Перемешиваемую смесь вещества из примера 1 наряду с 3-цианобензальдегидом (7,34 г, 56 ммоль), 1,3-циклогександионом (6,83 г, 56 ммоль) и ацетатом аммония (13,10 г, 170 ммоль) в этаноле (400 мл) перемешивают при нагревании до температуры кипения с обратным холодильником в течение 10 часов. Охлажденный раствор фильтруют для удаления осадившегося 9-(3-цианофенил)-3,4,6,7,9,10-гексагидро- 1,8-(2H,5H)-акридиндиона. Фильтрат упаривают досуха и остаток хроматографируют (элюент - метиленхлорид, 1:1 этилацетат/метиленхлорид и этилацетат), получая 4,14 г 2-цианоэтил 4-(3-цианофенил)-2-трифторметил-2-гидрокси-5-оксо-1,2,3,4,5,6,7,8- октагидрохинолин-3-карбоксилата; ЯМР: 1,86-1,90 (м, 2H, CH2), 1,94-2,18 (м, 2H, CH2), 2,31-2,38 (м, 1H, алифатический), 2,50-2,72 (м, 3H, алифатический), 2,82 (д, 1H, алифатический, J = 11,9), 3,35 (ушир. с, 1H, алифатический), 3,94-4,02 (м, 2H, алифатический), 7,37-7,59 (м, 5H, Ar, OH), 8,16 (с, 1H, NH); МС: m/z = 434 (M + 1).
H. 2-Цианоэтил 4-(3-цианофенил)-2-трифторметил-5-оксо- 1,4,5,6,7,8-гексагидрохинолин-3-карбоксилат.
Смесь 2-цианоэтил 4-(3-цианофенил)-2-трифторметил-2-гидрокси-5- оксо-1,2,3,4,5,6,7,8-октагидрохинолин-3-карбоксилата (4,14 г, 9,6 ммоль), п-толуолсульфоновой кислоты (0,61 г, 3,2 ммоль) и толуола (100 мл) перемешивают под насадкой Дина-Старка в течение 2 часов. Реакционную смесь, состоящую из темного масла и толуольной фазы охлаждают и выливают на хроматографическую колонку диаметром 1,5 дюйма, содержащую 90 г силикагеля. Смесь промывают на колонке небольшим количеством этилацетата. Элюирование этилацетатом и растирание полученного твердого вещества с диэтиловым эфиром дает сложный эфир в виде желто-оранжевого твердого вещества (2,84 г, 72%); т.пл. 148-151oC ЯМР: 1,79-1,99 (м, 2H, CH2), 2,19-2,33 (м, 2H, CH2), 2,50-2,74 (м, 2H, CH2), 2,85 (т, 2H, CH2), 4,15-4,27 (м, 2H, CH2), 4,94 (с, 1H, CH), 7,47-7,63 (м, 3H, Ar), 7,65-7,67 (м, 1H, Ar), 9,84 (с, 1H, NH); МС: m/z = 416 (M + 1). Анализ для C21H16F3N3O3:
Рассчитано: C, 60.72; H, 3.88; N, 10.12.
Найдено: C, 60.63; H, 3.80; N, 9.89.
I. 4-(3-Цианофенил)-2-трифторметил-5-оксо-1,4,5,6,7,8- гексагидрохинолин-3-карбоновая кислота.
К охлажденной (ледяная баня) перемешиваемой суспензии 2-цианоэтил-2-трифторметил-4-(3-цианофенил)-5-оксо-1,4,5,6,7,8- гексагидрохинолин-3-карбоксилата (2,80 г, 6,74 ммоль) в 1,2-диметоксиэтане (8,5 мл) добавляют по каплям за 10 минут предварительно охлажденный раствор гидроокиси натрия (0,80 г, 20,0 ммоль) в воде (6,5 мл). Получают исходный растворенный сложный эфир в виде темно-коричневого раствора. После перемешивания при комнатной температуре в течение 2 часов желтовато-коричневый раствор разбавляют водой (16 мл), возвращают в ледяную баню и перемешивают во время обработки смеси концентрированной соляной кислотой (2 мл). Осаждается коричневое масло, которое при перемешивании затвердевает, образуя твердый продукт кремового цвета. Твердое вещество фильтруют и промывают охлажденной водой. Продукт сушат при 50o/0,1 торр в течение ночи, получая карбоновую кислоту (2,33 г, 95%); т. пл. 209-211oC разл. с выделением газа; ЯМР: 1,75-1,88 (м, 1H, CH2), 1,89-1,95 (м, 1H, CH2), 2,18-2,32 (м, 2H, CH2), 2,54-2,73 (м, 2H, CH2), 4,92 (с, 1H, CH), 7,47-7,54 (м, 3H, Ar), 7,65-7,67 (м, 1H, Ar), 9,62 (с, 1H, NH), 13,10 (с, 1H, CO2H); МС: m/z = 363 (M + 1). Анализ для C18H13F3N2O3:
Рассчитано: C, 59.67; H, 3.62; N, 7,73.
Найдено: C, 59.53; H, 3,84; N, 7.69.
Пример 2
(S)-(-)-4-(3-Цианофенил)-2-трифторметил-4,6,7,8-тетрагидро- 5(1H)-хинолон.
Продукт примера 1 получают в больших масштабах следующим образом.
Перемешиваемый раствор (S)-(-)-4-(3-цианофенил)-2-трифторметил-5- оксо-1,4,5,6,7,8-гексагидрохинолин-3-карбоновой кислоты (50,5 г, 139,4 ммоль) в N-метилпирролидин-2-оне (325 мл) нагревают быстро (нагревающая рубашка) за 20 минут до 180oC (внутренний термометр), затем выдерживают при этой температуре дополнительно 20 минут. Охлажденную реакционную смесь выливают в воду (1200 мл) и экстрагируют, дважды промывают водой, сушат (MgSO4), фильтруют и растворитель удаляют, получая не совсем белое твердое вещество. Хроматография (элюент: метиленхлорид/этилацетат 85:15) и перекристаллизация из ацетонитрила дают указанное в заглавии соединение (32,85 г, 74%) в виде белого твердого вещества, которое идентично продукту примера 1.
Пример 3
Промежуточный (±)-изоборнил 4,4,4-трифторацетоацетат (из примера 1А) получают в больших масштабах следующим образом:
Перемешанную смесь этил 4,4,4-трифторацетоацетата (2,75 кг, 14,91 моль) и (±)-изоборнеола (1,53 кг, 9,93 моль) перемешивают при 105oC (внутренний термометр) 20 часов с установленной сверху насадкой для перегонки, позволяющей отогнать этанол. Температуру затем постепенно увеличивают за 10 часов до 155oC, чтобы отогнать оставшийся этанол: всего собирают 650 мл (114% теоретич.). Оставшуюся смесь фракционируют, затем при пониженном давлении, получая (1)-изоборнил 4,4,4-трифторацетоацетат в виде бесцветного масла (2,21 кг, 76%), который идентичен продукту примера 1A.
Пример 4
Промежуточный (±)-изоборнил 4-(3-цианофенил)-2-трифторметил- 2-гидрокси-5-оксо-1,2,3,4,5,6,7,8-октагидрохинолин-3-карбоксилат (из примера 1B) получают в больших масштабах следующим образом.
Перемешанную смесь (±)-изоборнил 4,4,4-трифторацетоацетата (830,6 г, 2,86 моль), 1,3-циклогександиона (320,7 г, 2,86 моль), 3-цианобензальдегида (375,0 r, 2,86 моль) и ацетата аммония (551,0 г, 7,15 моль) в этаноле (15,0 л) нагревают при температуре кипения с обратным холодильником в течение 8 часов. Охлажденную смесь фильтруют для удаления 9-(3-цианофенил)-3,4,6,7,9,10-гексагидро-1,8- (2H,5H)-акридиндиона. Фильтрат концентрируют до половины объема в вакууме. Первую порцию заданного продукта собирают фильтрацией и промывают диэтиловым эфиром (657,0 г, 44,5 %). Фильтраты и промывные воды концентрируют в вакууме и остаток растирают с диэтиловым эфиром и фильтруют, получая вторую порцию продукта (225,0 г, 15,2%). Этот объединенный продукт используют без дальнейшей очистки. Образец чистят хроматографически (элюент: этилацетат/гексан 7:3), что дает (±)-изоборнил 4-(3-цианофенил)-2-трифторметил-2-гидрокси-5-оксо- 1,2,3,4,5,6,7,8-октагидрохинолин-3-карбоксилат в виде белого твердого вещества, которое идентично веществу, полученному в примере 1.
Пример 5
Получение 4-(3-Цианофенил)-2-трифторметил-5-оксо-1,4,5,6,7,8- гексагидрохинолин-3-карбоновой кислоты (из примера 1C) в больших масштабах выполняют следующим образом.
Перемешиваемую смесь (±)-изоборнил 4-(3-цианофенил)-2- трифторметил-2-гидрокси-5-оксо-1,2,3,4,5,6,7,8-октагидрохинолин-3- карбоксилата (800,0 г, 1,55 моль), п-толуолсульфокислоты (148,0 г, 0,78 моль) и ледяной уксусной кислоты (7,5 л) нагревают (нагревающая рубашка) за 1 час 15 минут до 100oC. Смесь выдерживают при температуре 102-104oC 6 часов, к этому времени ТСХ (силикагель - этилацетат/гексан 6:4) показывает, что реакция завершилась. Чтобы свести к минимуму декарбоксилирование, применяют только достаточный нагрев, выдерживая реакционную смесь максимум при 104oC. После удаления растворителя остаток распределяют между этилацетатом и водой. Этилацетат промывают водой, отделяют и трижды экстрагируют насыщенным водным бикарбонатом натрия. Перемешанные объединенные экстракты бикарбоната натрия охлаждают на ледяной бане и добавляют по каплям концентрированную соляную кислоту, пока раствор не станет сильнокислым. Смесь экстрагируют диэтиловым эфиром и объединенные экстракты сушат (MgSO4), фильтруют и концентрируют в вакууме, получая желтую пену. Растирание с метиленхлорид/гексаном дает карбоновую кислоту (289,0 г, 51,5%) в виде светло-желтого твердого вещества. Вещество идентично по ЯМР и ТСХ (силикагель - 10% метанол в хлороформе, содержащий несколько капель уксусной кислоты) продукту, описанному и охарактеризованному в подразделе I примера 1.
Этилацетатный слой сушат (MgSO4), фильтруют и упаривают в вакууме, получая чистый (±)-4-(3-цианофенил)-2-трифторметил- 4,6,7,8-тетрагидро-5(1H)-хинолон (490 г) в виде воскообразного светло-желтого твердого вещества. Этот продукт далее отнесен к рацемату.
Пример 6
Промежуточную (S)-(-)-4-(3-цианофенил)-2-трифторметил-5-оксо- 1,4,5,6,7,8-гексагидрохинолин-3-карбоновую кислоту (из примера 1E) получают в больших масштабах, как описано далее.
К охлаждаемой (ледяная баня) перемешиваемой суспензии соли S-(-)- α- метилбензиламин (S)-(-)-4-(3-цианофенил)-2-трифторметил-5- оксо-1,4,5,6,7,8-гексагидрохинолин-3-карбоновой кислоты (68,0 г, 140,6 ммоль) в воде (700 мл) добавляют по каплям концентрированную соляную кислоту до тех пор, пока смесь не станет сильнокислой. Смесь экстрагируют четыре раза диэтиловым эфиром и объединенные экстракты сушат (MgSO4), фильтруют и растворитель удаляют, получая желтую пену (50,6 г, 99%). Растирание с метиленхлорид/гексаном дает чистый образец карбоновой кислоты в виде бледно-желтого твердого вещества, которое идентично продукту примера 1E.
Пример 7
Указанное в заглавии соединение (S)-(-)-4-(3-цианофенил)-2- трифторметил-4,6,7,8-тетрагидро-5(1H)-хинолон выделяют из очищенного рацемата препаративной хиральной хроматографией.
Неочищенный рацемат (2,95 кг) - вещество, объединенное из 6 равных по величине партий, выделенное, как описано в примере 5, чистят хроматографически (элюент: метиленхлорид/этилацетат 85: 15), чтобы получить очищенный рацемат (580,8 г), который разделяют, как описано ниже. Процесс выделения этого продукта во время начальной очистки не оптимизирован.
Препаративную хиральную хроматографию очищенного рацемата (580,8 г) выполняют на CHIRALPACKR ADTM (торговая марка) колонке (носитель - амилоза, 10 х 50 см, элюент: гексан/этанол 85:15). Исходный раствор образца получают первоначальным растворением соединения в этаноле, затем разбавлением раствора гексаном до тех пор, пока конечная композиция раствора не станет эквивалентной составу элюента.
Конечная концентрация образца основного раствора составляет 6 г/литр. Каждое хроматографическое разделение проводится впрыскиванием 500 мл исходного раствора и элюированием при скорости потока 200 мл/мин, (S)-(-)-энантиомер (указанное в заглавии соединение) является первым пиком, вымываемым из колонки, (+)-энантиомер вымывается вторым. Упаривание элюата в вакууме дает (S)-(-)-энантиомер (281,0 г, 99,7% ее, 98,6% химическая чистота) в виде не совсем белого твердого вещества. Перекристаллизация из ацетонитрила (1000 мл) дает 261,7 г указанного в заглавии соединения в виде белого кристаллического твердого вещества, идентичного во всех отношениях веществу, полученному, как описано в примере 1.
Пример 8
Следующий далее иллюстративный пример представляет фармацевтические дозированные формы, содержащие соединение (далее обозначено как "соединение X"), предназначенные для терапевтического и профилактического применения человеком:
(а) Таблетка - мг/таблетка
Соединение X - 50,0
Маннитол, фармакопея США - 223,75
Натрий кроскармелоза - 6,0
Маисовый крахмал - 15,0
Гидроксипропилметилцеллюлоза (HPMC), фармакопея США - 2,25
Стеарат магния - 3,0
(б) Капсула
Соединение X - 10,0
Маннитол, фармакопея США - 488,5
Натрий кроскармелоза - 15,0
Стеарат магния - 1,5
Приведенные выше составы могут быть получены обычными способами, хорошо известными в фармацевтической технике. Таблетки могут быть покрыты энтеросолюбильной оболочкой обычными способами, например покрытием из фталата ацетата целлюлозы.


Изобретение относится к (S)-(-)-2-трифторметил-4-(3-цианофенил)-4,6,7,8-тетрагидро-5(1Н)-хинолону и его фармацевтически приемлемым солям, которые могут использоваться для лечения недержания мочи. Описаны 6 способов его получения. 9 с. и 14 з.п. ф-лы, 3 ил.
1) взаимодействие сложного ацетоуксусного эфира формулы III или его полуацеталя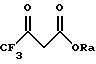
где ORа обозначает спиртовой остаток,
с ацетатом аммония, 3-цианобензальдегидом и 1,3-циклогександионом с получением соединения формулы IVа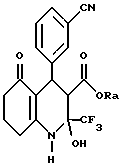
2) дегидратируют соединение формулы IVа до соединения формулы IV
3) омыляют полученное соединение IV с образованием кислоты формулы II
4) выделяют соединение формулы II в виде соли (S)-(-)-α-метилбензиламина путем перекристаллизации из растворителя, и 5) декарбоксилируют выделенное соединение формулы II.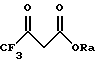
где ORа обозначает спиртовой остаток,
с ацетатом аммония, 3-цианобензальдегидом и 1,3-циклогександионом с получением соединения формулы IVа
2) полученное соединение формулы IVа подвергают катализируемой кислотой дегидратации и гидролизу с образованием кислоты формулы II
3) выделяют соединение формулы II в виде соли (S)-(-)-α-метилбензиламина путем перекристаллизации из растворителя, и 4) декарбоксилируют выделенное соединение формулы II.
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ 5-КЕТО-5,6,7,8-ТЕТРАГИДРОХИНОЛИНА | 1972 |
|
SU435237A1 |
| US 3991060, 09.11.1976 | |||
| Устройство для крепления цилиндрических образцов с головками при испытаниях на прочность | 1986 |
|
SU1432379A1 |
| Устройство для автоматического регулирования энергоблока | 1974 |
|
SU539153A1 |
| Машковский М.Д | |||
| Лекарственные средства | |||
| - М.: Медицина, 1985, ч.1, с | |||
| Кладка стен из фасонного кирпича | 1922 |
|
SU542A1 |

