Изобретение относится к области биоорганической химии, а именно к получению ковалентно-связанных комплексов олигонуклеотид-нуклеиновая кислота, содержащих ковалентную связь в определенном месте нуклеотидной последовательности нуклеиновой кислоты (НК), общей формулы: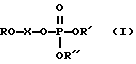
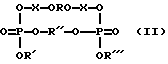
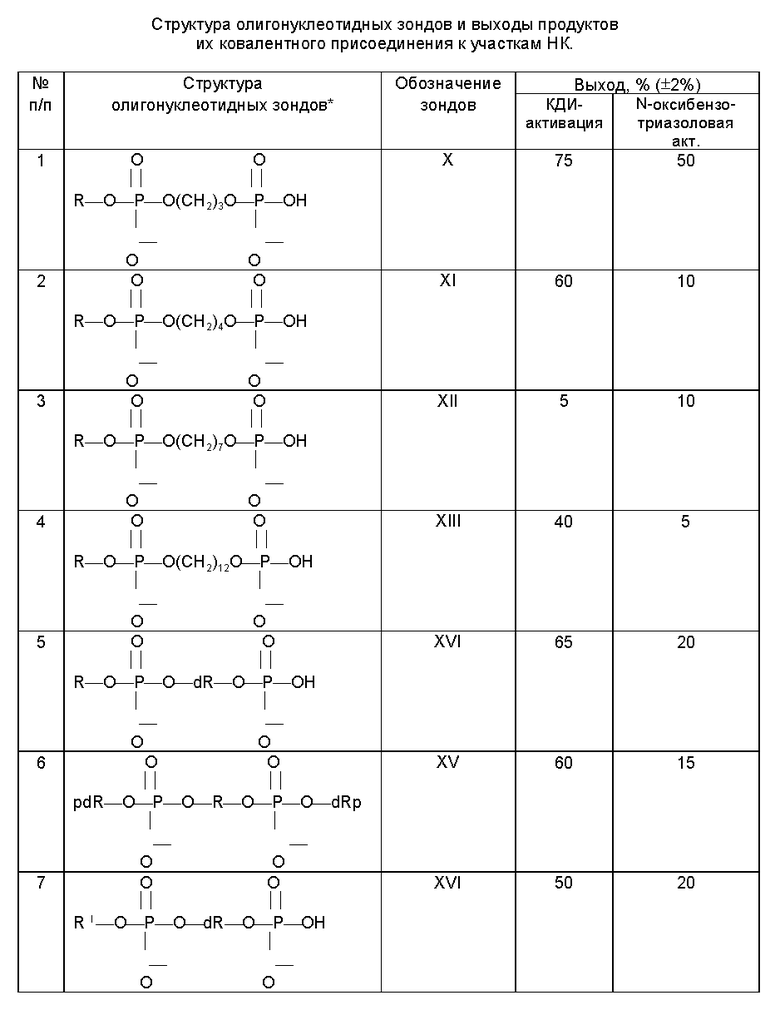
где
R - остатки олигодезоксирибонуклеотидов
R', R'', R''' - остатки нуклеиновой кислоты;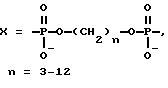
которые могут использоваться в качестве новых аналогов субстратов для изучения механизмов действия ферментов нуклеинового обмена в энзимологии; в структурно-функциональных исследованиях белков и нуклеиновых кислот; в гибридизационном анализе нуклеиновых кислот; а также в других различных молекулярно-биологических исследованиях биополимеров.
Аналогами сходной структуры являются ковалентно-связанные комплексы олигонуклеотид-нуклеиновая кислота типа (III):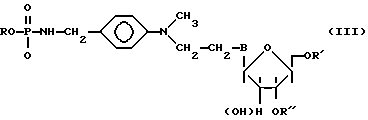
где
R - остаток олигодезоксирибонуклеотида;
R', R'' - остатки нуклеиновой кислоты;
B - остатки гетероциклических оснований G, C, A.
Указанные соединения (III) образуются при взаимодействии алкилирующих производных олигонуклеотидов (IV) с комплементарными участками НК по следующей схеме [I]: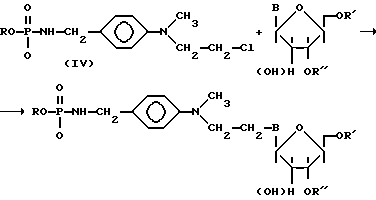
Недостатком ковалентно-связанных комплексов олигонуклеотид-НК типа (III) является то, что при образовании этих соединений происходит алкилирование гетероциклических оснований, входящих в состав НК, что приводит к повышению лабильности N-гликозидных связей. В результате этого процесса возможно выщепление алкилированных гетероциклических оснований из полинуклеотидной цепи с последующим ее разрушением [2], что ограничивает сферу применения указанных соединений.
Для получения соединений типа (III) проводят реакцию между НК и комплементарным ее участку алкилирующим производным олигонуклеотида в 10 мМ трис HCl (pH 7,6), содержащем 0,1 М NaCl и 1 мМ EDTA, при 10 - 25oC в течение 0,5 - 3 суток. Выходы целевого продукта достигают 70 - 90%. При этом алкилированию подвергаются остатки гетероциклических оснований, отстоящих на 1 - 3 нуклеотида от конца комплементарного дуплекса.
Недостатком описанного способа получения является то, что используемые в реакции алкилирующие производные олигонуклеотидов (IV) обладают высокой реакционной способностью, приводящей к снижению ее селективности [3]. Кроме того, алкилированию подвергаются как остатки гуанина, так и остатки цитидина и аденина полинуклеотидной цепи, что приводит к образованию смеси продуктов ковалентного присоединения алкилирующих производных олигонуклеотидов к НК [4] . Это может затруднять интерпретацию результатов, полученных при использовании такого рода соединений для решения различных молекулярно-биологических задач. Еще одним недостатком является то, что синтез алкилирующих производных олигонуклеотидов представляет собой достаточно трудоемкий многостадийный процесс, требующий использования дорогостоящих и труднодоступных реагентов [5].
Задача изобретения - получение ковалентно-связанных комплексов олигонуклеотид-НК общей формулы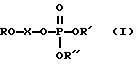
где
R - остатки олигодезоксирибонуклеотидов
R', R'', R''' - остатки нуклеиновой кислоты;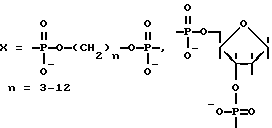
отличающихся от аналога (III) легкостью получения, более высокой селективностью образования ковалентной связи и возможностью ее избирательного расщепления с образованием исходных соединений, что делает эти соединения более доступными и удобными для практического использования и расширяет сферу их применения, и разработка способа их получения.
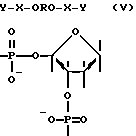
Поставленная задача достигается тем, что эквимолярную смесь нуклеиновой кислоты и активного олигонуклеотидного зонда выдерживают в водном буферном растворе в условиях устойчивости комплементарного комплекса при pH 6,0 - 9,0 с последующим выделением образующихся продуктов. Вместо алкилирующих производных олигонуклеотидов используются специально сконструированные олигонуклеотидные зонды, несущие активированную фосфатную группу, связанную с 3'-, 5'- или одновременно с 3', 5'-концами олигонуклеотида через гибкое спейсерное звено (V - VII).
RO-X-Y (V)
Y-X-OR (VI)
Y-X-ORO-X-Y- (VII)
где
R и X те же, что и для соединений (I) и (II)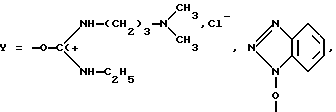
Соединения (V - VII) получают в процессе автоматического синтеза олигонуклеотидов на полимере, содержащем β-этилсульфоновые группы, путем постадийных конденсаций стандартных мономерных компонентов амидофосфитного синтеза олигонуклеотидов, используя наряду с ними диметокситритильные производные O-β-цианэтил-N, N-диизопропиламидофосфитов алифатических гликолей и 1,2-дидезокси-D-рибофуранозы, причем эти производные добавляют либо (и) вначале постадийной конденсации, либо (и) в конце ее, а в качестве активирующих зонд соединений используют 1-этил-3(3'-диметиламинопропил)карбодиимид или N-оксибензотриазол.
В отличие от аналога (III), образование ковалентной связи происходит селективно при взаимодействии активированной фосфатной группы зонда с пространственно сближенной межнуклеотидной фосфатной группой НК в месте, непосредственно примыкающем к двуспиральному участку олигонуклеотидный зонд - НК, а введение в качестве ковалентной связи замещенной пирофосфатной дает возможность направленного контролируемого расщепления указанных комплексов по этой связи с образованием исходных соединений и тем самым использовать предлагаемые соединения для зондирования активных центров ферментов нуклеинового обмена и их аффинной модификации.
Соединения (I) и (II) получаются при взаимодействии активированной концевой фосфатной группы олигонуклеотидных зондов с пространственно сближенной межнуклеотидной фосфатной группой комплементарного участка НК по схеме: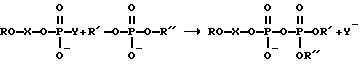
или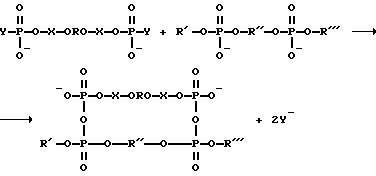
где
R - остатки олигодезоксирибонуклеотидов;
R', R'', R''' -остатки нуклеиновой кислоты;
X - спейсерная группы;
Y - активирующая фосфат группа.
Реакция протекает при выдерживании смеси НК и комплементарного ее участку активного олигонуклеотидного зонда, растворенных в эквимолярных соотношениях в водных буферных растворах при pH 6,0 - 9,0. При этом образуется замещенная пирофосфатная межнуклеотидная связь. Выход целевого продукта достигает 75%.
Отличие предлагаемого способа получения ковалентно-связанных комплексов (I) и (II) от способа получения аналога (III) состоит в использовании более простых по конструкции и способу получения компонентов реакции - активных олигонуклеотидных зондов, а также тем, что данную реакцию проводят в более широком интервале pH (6,0 - 9,0).
Использование этих зондов позволяет получать ковалентно-связанные комплексы с новым типом ковалентной связи. При этом исключается дополнительная стадия введения реакционноспособной группы, представляющей собой аналог азотистого иприта, который является достаточно дорогостоящим и труднодоступным препаратом. Это делает предлагаемые ковалентно-связанные комплексы более доступными и расширяет сферу их применения. Особенность конструкции олигонуклеотидных зондов дает возможность получать ковалентно-связанные комплексы с двумя ковалентными связями - одновременно по 3'- и 5'-концам олигонуклеотидных зондов и повысить селективность их образования, что также расширяет сферу применения этих соединений. Образование замещенной пирофосфатной связи в процессе получения ковалентно-связанных комплексов дает возможность их контролируемого избирательного расщепления под действием нуклеофильных агентов, в том числе и нуклеофильных групп ряда аминокислот, входящих в активные центры белков нуклеинового обмена, и тем самым открывает перспективы аффинной модификации и более детального изучения последних.
Предлагаемый способ получения ковалентно-связанных комплексов позволяет существенно расширить интервал значений pH, в котором возможно протекание данной реакции, вплоть до 9,0. Важно отметить, использование таких значений pH невозможно для аналога, поскольку в этом случае наблюдается гидролиз гликозидной связи с последующим разрушением углеводофосфатного остова нуклеиновых кислот [2].
Совокупность указанных отличительных признаков позволяет упростить процесс, сократить длительность приготовления ковалентно-связанных комплексов олигонуклеотид-НК, снизить их стоимость за счет использования недорогостоящих компонентов.
Изобретение иллюстрируется следующими примерами.
Пример 1
Синтез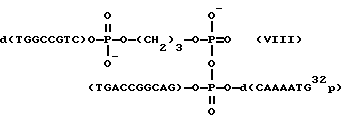
0,04 о.е. смеси эквимолярных количеств олигонуклеотидного зонда (X) (см. таблицу) и гептадекануклеотида
d(GTAAAACGACGGCCAGT) (IX)
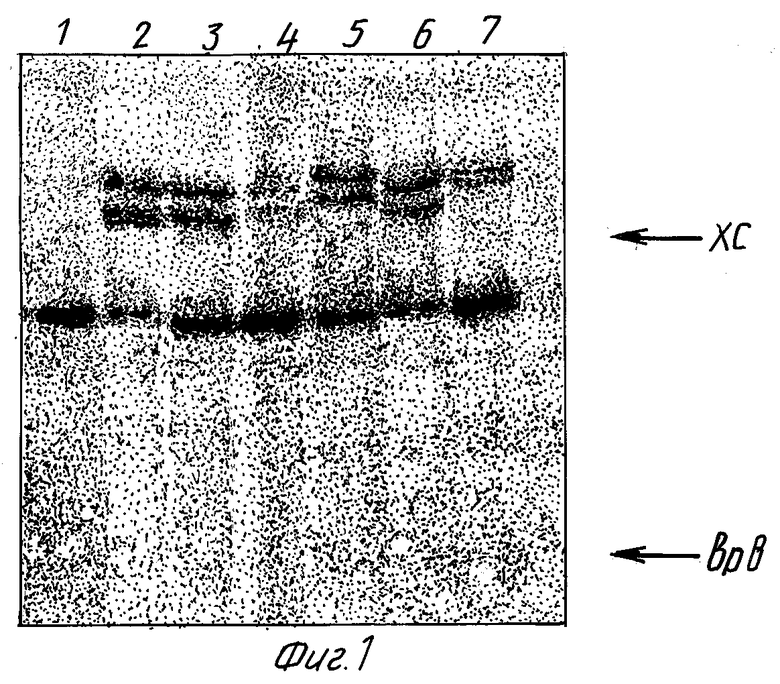
(суммарная нуклеотидная концентрация 10-3 М) обрабатывали 0,2 М раствором КДИ в морфолиноэтансульфонатном буфере (pH 6,0), содержащем ионы двухвалентного металла, при 10oC в течение 2 суток. Контроль за ходом реакции осуществляли методом гель-электрофореза в 20% полиакриламидном геле (ПААГ), содержащем 7 М мочевину. Радиоавтограмма гель-электрофореза реакционных смесей, образующихся при получении ковалентно-связанных комплексов олигонуклеотид-НК приведена на фиг. 1. Выделение продуктов реакции из реакционной смеси проводили элюцией с ПААГ. Выход целевого продукта составил 75% (см. таблицу).
Образование ковалентно-связанных комплексов олигонуклеотидных зондов (XI - XV) с гептадекануклеотидом (IX) под действием КДИ проводили аналогично. Радиоавтограмма гель-электрофореза реакционных смесей приведена на фиг. 1. Выходы целевых продуктов приведены в таблице. Снижение температуры до 0oC не оказало существенного влияния на выходы образующихся продуктов.
Пример 2
0,1 о. е. олигонуклеотидного зонда (X) растворяли в 10 мкл 50% водного раствора диметилформамида (ДМФА). К раствору добавляли 2 М раствор N-оксибензотриазола в 50% ДМФА и 7 мг КДИ. Реакцию проводили в течение 2,5 ч при 8oC. Синтезированный с количественным выходом N-оксибензотриазоловый эфир выделяли высаживанием 2% раствором перхлората лития в ацетоне и затем к нему добавляли эквимольное количество гептадекануклеотида (IX), растворенного в N-(2-гидроксиэтил)пиперазин-N'-2-этаносульфонатном буфере (pH 8,75), содержащем ионы двухвалентного металла. Смесь инкубировали при 10oC в течение 2 суток. Контроль за ходом реакции и выделение продуктов осуществляли как в примере 1. Радиоавтограмма гель-электрофореза реакционных смесей, образующихся при получении ковалентно-связанных комплексов олигонуклеотид-НК приведена на фиг. 2. Выход целевого продукта составил 50% (см. таблицу).
Образование ковалентно-связанных комплексов олигонуклеотидных зондов (XI - XV) с гептадекануклеотидом (IX) методом N-оксибензотриазоловых эфиров проводилось аналогично. Радиоавтограмма гель-электрофореза реакционных смесей приведена на фиг. 2. Выходы целевых продуктов приведены в таблице.
Пример 3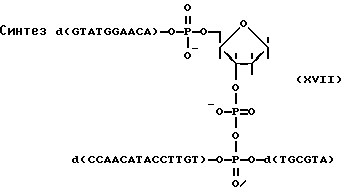
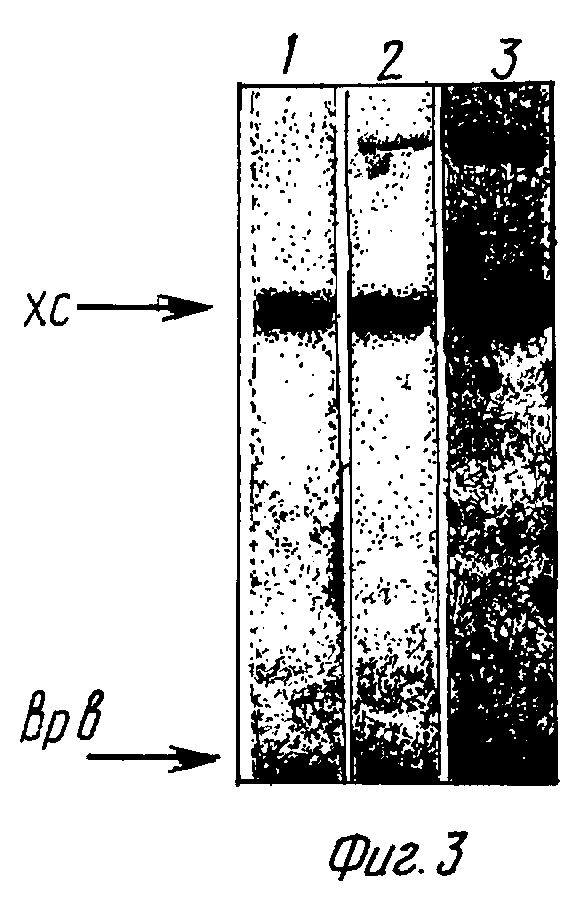
Синтез соединения (XVII) под действием КДИ и методом N-оксибензотриазоловых эфиров проводили как описано в примерах 1 и 2 соответственно, используя в реакциях олигонуклеотидный зонд (XIII) (см. таблицу) и комплементарный ему 20-звенный олигонуклеотид d(ATGCGTTGTTCCATACAACC) (XVIII). Радиоавтограмма гель-электрофореза реакционных смесей приведена на фиг. 3. Выход целевого продукта достигал 40%.
Пример 4
Синтез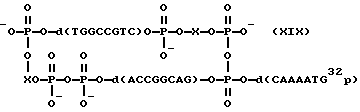
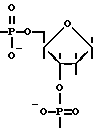
где X= 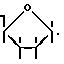
Синтез соединения (XIX) под действием КДИ и методом N-оксибензотриазоловых эфиров проводили как описано в примерах 1 и 2 соответственно, используя в реакциях олигонуклеотидный зонд (XV) (см. таблицу) и гептадекануклеотид (IX). Радиоавтограмма гель-электрофореза реакционных смесей приведена на фиг. 2. Выход целевого продукта достигал 65%.
В таблице приведены выходы продуктов реакций образования ковалентно-связанных комплексов олигонуклеотид-НК, полученных при взаимодействии олигонуклеотидных зондов (X - XVI) с комплементарными участками НК.
Испытания ковалентно-связанных комплексов олигонуклеотид-НК
Проведенные исследования по изучению гидролитической устойчивости и свойств синтезированных ковалентно-связанных комплексов олигонуклеотид-НК показали, что эти соединения устойчивы в водных буферных растворах (pH 6,0 - 8,5, 20oC), не содержащих сильных нуклеофилов, по крайней мере в течение 48 часов, а при -20oC могут храниться в течение нескольких месяцев. В то же время в присутствии таких нуклеофилов, как первичные, вторичные и третичные амины происходит расщепление замещенной пирофосфатной связи с образованием исходных соединений по схеме: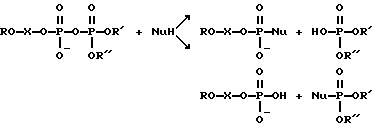
При обработке этих соединений 15% водным раствором уксусной кислоты или 0,1 н водным раствором NaOH также наблюдается образование исходных соединений.
Свойства полученных ковалентно-связанных комплексов олигонуклеотид-НК иллюстрируются следующими примерами.
Пример 5
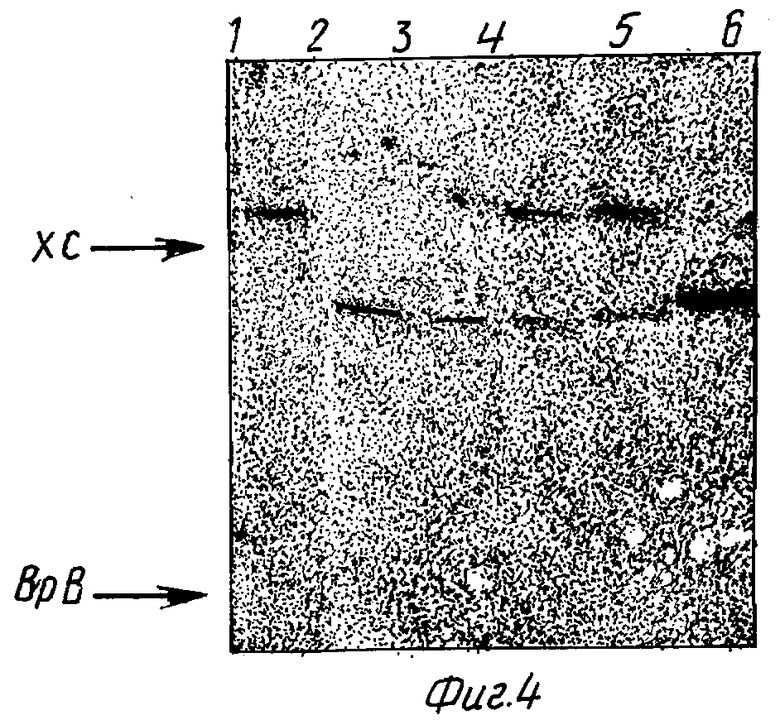
0,01 о. е. соединения (VIII) обрабатывали 15% водным раствором CH3COOH при 50oC в речение 2 ч. На фиг. 4. приведен радиоавтограф гель-электрофореза продуктов этой реакции. Гидролиз изучаемого соединения проходил на 10%.
Пример 6
0,1 о. е. соединения (VIII) выдерживали в 0,1 н NaOH при 20oC в течение 12 ч. Радиоавтограмма гель-электрофореза продуктов этой реакции приведена на фиг. 4. В результате реакции наблюдается количественный гидролиз (VIII) с образованием исходных соединений.
Пример 7
0,01 о. е. соединения (VIII) выдерживали в 0,5 М водном растворе этилендиамина (ЭДА) (pH 8,0) при 37oC в течение 1 сут. На фиг. 4 приведен радиоавтограф гель-электрофореза продуктов реакции аминолиза этого соединения. Как видно из этой фиг., при обработке соединения (VIII) ЭДА происходит количественное расщепление модифицированной связи и наблюдается образование ЭДА-производного гептадекануклеотида (IX).
Пример 8
Аналогичным образом, как описано в примерах 5 и 7, был проведен кислотный гидролиз и аминолиз ковалентно-связанного комплекса олигонуклеотид-НК следующей структуры: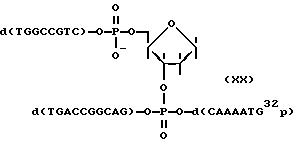
Радиоавтограмма гель-электрофореза продуктов этих реакций
Пример 9
Синтез
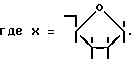
0,04 о.е. смеси эквимольных количеств олигонуклеотидного зонда d(AGTGTGGTTAATGATCTACAGTT)pdRp и олигонуклеотидного зонда pdRpd(CACCAATTACTAGATGTCAATAATTT)p32, где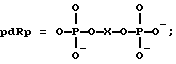
(суммарная нуклеотидная концентрация 10-3 М) обрабатывали 0,2 М раствором КДИ в морфолиноэтансульфонатном буфере (pH 6,0), содержащем ионы двухвалентного металла, при 10oC в течение 2 суток. Контроль за ходом реакции осуществляли методом гель-электрофореза в 20% полиакриламидном геле (ПААГ), содержащем 7 М мочевину. Выделение продуктов реакции из реакционной смеси проводили элюцией с ПААГ.
Пример 10
0,01 о.е. соединения (XXI) обрабатывали 15% водным раствором CH3COOH при 50oC в течение 2 ч. В ходе реакции проходил гидролиз изучаемого соединения с образованием исходных соединений.
Пример 11
0,1 о.е. соединения (XXI) выдерживали в 0,1 н NaOH при 20oC в течение 12 ч. В результате реакции наблюдается количественный гидролиз (XXI) с образованием исходных соединений.
Пример 12
0,01 о. е. соединения (XXI) выдерживали в 0,5 М водном растворе этилендиамина (ЭДА) (pH 8,0) при 37oC в течение 1 сут. В результате реакции происходит количественное расщепление модифицированной связи и наблюдается образование ЭДА-производного исходного олигонуклеотида.
Возможность избирательного расщепления полученных ковалентно-связанных комплексов олигонуклеотид-НК является важным преимуществом этих соединений и открывает новые перспективы их использования в молекулярной биологии и генной инженерии. Таким образом, предложенное техническое решение неизвестно из уровня техники, явным образом не следует из него и может быть без изменения использовано в отраслях народного хозяйства.
Источники информации
1. Власов В.В., Кнорре Д.Г., Кутявин И.В., Мамаев С.В., Подуст Л.М., Федорова О.С. Биоорган. химия. 1987, т. 13, N 9, с. 1221 - 1229.
2. Шабарова З. А. , Богданов А.А. Химия нуклеиновых кислот и их компонентов. М., 1978, с. 360 - 361.
3. Кнорре Д. Г., Зарытова В.Ф., Бадашкеева А.Г., Федорова О.С. Реакционноспособные производные олигонуклеотидов как геннаправленные биологически активные вещества. М.: "Итоги науки и техники". Сер. Биотехнология. 1991. т. 37, с. 1 - 182.
4. Vlasov V.V., Zarytova V.F., Kutiavin I.V, Mamaev S.V., Podyminogin M. A. Nucl. Acids Res., 1986, v. 14, N 10, p. 4065 - 4076.
5. Годовикова Т. С. , Зарытова В.Ф., Халимская Л.М. Биоорганич. химия, 1986, т. 12, N 4, с. 475 - 481.
| название | год | авторы | номер документа |
|---|---|---|---|
| ОЛИГОДЕЗОКСИРИБОНУКЛЕОТИДЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1992 |
|
RU2117011C1 |
| СПОСОБ АМПЛИФИКАЦИИ НУКЛЕИНОВЫХ КИСЛОТ С ИСПОЛЬЗОВАНИЕМ ФОСФОРИЛГУАНИДИНОВЫХ ОЛИГОНУКЛЕОТИДОВ | 2017 |
|
RU2698134C2 |
| СПОСОБ ВЫЯВЛЕНИЯ АНАЛИЗИРУЕМОЙ ПОСЛЕДОВАТЕЛЬНОСТИ НУКЛЕИНОВЫХ КИСЛОТ | 1996 |
|
RU2146707C1 |
| СПОСОБ ДЕТЕКЦИИ СПЕЦИФИЧЕСКИХ ПОСЛЕДОВАТЕЛЬНОСТЕЙ НУКЛЕИНОВЫХ КИСЛОТ (ВАРИАНТЫ) И УСТРОЙСТВО ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ | 2011 |
|
RU2509157C2 |
| ПОЛИНУКЛЕОТИДЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ГИБРИДИЗАЦИОННЫЙ АНАЛИЗ НУКЛЕИНОВОЙ КИСЛОТЫ | 1991 |
|
RU2142467C1 |
| СПОСОБ ВЫЯВЛЕНИЯ АНАЛИЗИРУЕМОЙ ПОСЛЕДОВАТЕЛЬНОСТИ ДНК | 2003 |
|
RU2259402C2 |
| Способ получения конъюгатов олигонуклеотидов с кластерами бора | 2022 |
|
RU2786533C1 |
| Способ направленного истощения олигонуклеотидных библиотек для снижения неспецифической адсорбции при твердофазной селекции аптамеров на основе нуклеиновых кислот | 2015 |
|
RU2618872C1 |
| Способ получения 5-или-3-фосфодиэфиров моно-или олигонуклеотидов | 1983 |
|
SU1121266A1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ПОСЛЕДОВАТЕЛЬНОСТИ НУКЛЕИНОВОЙ КИСЛОТЫ (ВАРИАНТЫ) И НАБОР ДЛЯ ИСПОЛЬЗОВАНИЯ ПРИ ОПРЕДЕЛЕНИИ ПОСЛЕДОВАТЕЛЬНОСТИ НУКЛЕИНОВОЙ КИСЛОТЫ | 1994 |
|
RU2143004C1 |

Изобретение относится к биоорганической химии. Ковалентно-связанные комплексы олигонуклеотид-нуклеиновая кислота общей формулы I и II, где R - остатки олигодезоксирибонуклеотидов, R', R", R"' -остатки нуклеиновой кислоты, n = 3-12, могут использоваться для изучения механизмов действия ферментов нуклеинового обмена в энзимологии. Соединения формулы I и II получают выдержкой эквимолекулярной смеси нуклеиновой кислоты и активного олигонуклеотидного зонда в водном буферном растворе в условиях устойчивости комплементарного комплекса при рН 6,0-9,0. Использование в качестве активных олигонуклеотидных зондов олигодезоксирибонуклеотидов (III-V) позволяет исключить дополнительную стадию введения реакционноспособной группы и повысить селективность образования комплексов I и II. 3 c. и 1 з.п. ф-лы, 4 ил., 1 табл.

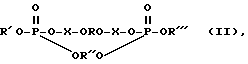
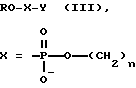
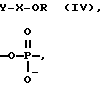
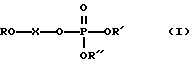
где R - остатки олигодезоксирибонуклеотидов;
R', R'' - остатки нуклеиновой кислоты;
n = 3 - 12.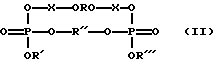
где R - остатки олигодезоксирибонуклеотидов;
R', R'', R''' - остатки нуклеиновой кислоты;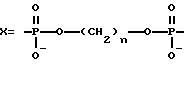
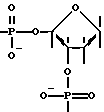
n = 3 - 12.
RO - X - Y (III)
Y - X - OR (IV)
Y - X - OPO - X - Y - (V)
где R, X имеют вышеуказанные значения, а
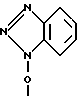
выдерживают в водном буферном растворе, содержащем ионы двухвалентного металла, при рН 6,0 - 9,0 и температуре 0 - 10oC с последующим выделением целевых продуктов.
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| S.A | |||
| Kuznetsova, M.G | |||
| Jvanovskay, L.A | |||
| Shabarova, Bioorgan khimija, 16, 219 (1990) 2 | |||
| L.A | |||
| Shabarova, M.G | |||
| Jvanovskay, M.G | |||
| Isaguliants, FEBS, Lett., 154, 288 (1983). | |||
