Это изобретение относится к области химии нуклеиновых кислот и биохимических анализов. Точнее, оно касается крупных гребневидно-разветвленных полинуклеотидов, которые используются в качестве накопительных мультимеров в гибридизационных исследованиях нуклеиновых кислот.
Состояние вопроса
Гибридизационный анализ нуклеиновых кислот обычно используется в исследованиях в области генетики и медицинской биохимии и в клинической диагностике. В гибридизационном анализе основной нуклеиновой кислоты из одноцепочечной исследуемой нуклеиновой кислоты образуют гибрид с меченым одноцепочечным зондом нуклеиновой кислоты и исследуют образовавшийся меченый дуплекс. Чтобы облегчить разделение дуплекса для распознавания постороннего материала и/или усилить сигнал, подлежащий исследованию, были разработаны варианты этой основной схемы. Один способ усиления сигнала для исследования описан в заявке в Европейское патентное бюро (EPA) 883096976 (соответствующей заявке США, серийный N 340031, поданной 18 апреля 1989 г.). В ней сигнал усиливается при помощи накопительных мультимеров. Эти мультимеры являются полинуклеотидами, которые сконструированы так, чтобы у них были первый сегмент, который специфически образует гибрид с анализируемой нуклеиновой кислотой или с нитью нуклеиновой кислоты, связанной с анализируемой, и повторения второго сегмента, который специфически образует гибрид с меченым зондом. Амплификация теоретически пропорциональна числу повторений второго сегмента. Мультимеры могут быть либо линейными, либо разветвленными. Описаны два главных типа разветвленных мультимеров: вилкообразные и гребневидные.
При исследовании двух типов разветвленных мультимеров было обнаружено, что вилкообразные структуры с большим числом ветвей, чем около 8, создают пространственную преграду, которая мешает фиксации меченого зонда на мультимерах. С другой стороны, у гребневидных структур не было пространственных препятствий, и поэтому их сочли предпочтительным типом разветвленного мультимера. Однако, к сожалению, повторные попытки создать гребневидные структуры с более чем 10 ветвями оказались безуспешными. Заявители разработали в настоящее время способы получения крупных гребневидно-разветвленных мультимеров. Эти крупные гребневидные структуры позволяют достичь большей степени амплификации, чем было возможно раньше.
Раскрытие изобретения
Одним аспектом изобретения является крупный, гребневидно-разветвленный полинуклеотид, включающий:
а) скелет полинуклеотида, имеющий:
(I) не менее 15 мультифункциональных нуклеотидов, каждый из которых определяет место боковой цепи, и
(II) первую одноцепочечную олигонуклеотидную единицу, которая может специфически связываться с интересующей первой одноцепочечной полинуклеотидной последовательностью; и
б) подвесные полинуклеотидные боковые цепи, отходящие от указанных мультифункциональных нуклеотидов, каждая из которых включает повторения второй одноцепочечной олигонуклеотидной единицы, которая способна специфически связываться со второй интересующей одноцепочечной нуклеотидной последовательностью.
Другим аспектом этого изобретения является способ создания крупного гребневидно-разветвленного полинуклеотида, пригодного в качестве амплификационного мультимера при анализе гибридизации нуклеиновой кислоты, включающей:
а) синтезирование скелета одноцепочечного полинуклеотида, включающего:
(I) не менее 15 мультифункциональных нуклеотидов, каждый из которых имеет защищенную функциональную группу, которая служит местом отхождения нуклеотидов боковой цепи, и
(II) первый сегмент места сшивания;
б) снятие защиты с указанных функциональных групп;
в) удлинение каждого из указанных мест, по крайней мере, до 5 нуклеотидов, чтобы создать вторые сегменты места сшивания;
г) сшивание первой единицы одноцепочечного олигонуклеотида с первым местом сшивания так, чтобы указанная первая единица одноцепочечного олигонуклеотида оказалась способной образовывать специфическую связь с первой последовательностью одноцепочечной нуклеиновой кислоты, представляющей интерес; и
д) сшивание вторых единиц одноцепочечного олигонуклеотида со вторыми сегментами места сшивания так, чтобы указанные вторые единицы одноцепочечного олигонуклеотида включали повторения последовательности, способной образовывать специфическую связь со вторым одноцепочечным олигонуклеотидом, представляющим интерес.
Еще одним аспектом этого изобретения является альтернативный способ создания крупного гребневидно-разветвленного полинуклеотида, пригодного в качестве амплификационного мультимера для анализа нуклеиновой кислоты методом гибридизации; этот способ включает:
а) синтезирование скелета одноцепочечного полинуклеотида, включающего:
(I) не менее 15 мультифункциональных нуклеотидов, каждый из которых имеет защищенную функциональную группу, служащую местом наращивания боковой цепи нуклеотидов, и
(II) первую единицу одноцепочечного олигонуклеотида, способную образовывать специфическую связь с первой последовательностью одноцепочечного полинуклеотида, представляющего интерес;
б) снятие защиты с указанных функциональных групп;
в) удлинение каждого из указанных мест, по крайней мере, до 5 нуклеотидов, чтобы создать сегменты мест сшивания; и
г) сшивание вторых единиц одноцепочечного олигонуклеотида с сегментами места сшивания указанных вторых единиц одноцепочечного олигонуклеотида, включающих повторения последовательности, способной связываться со вторым одноцепочечным олигонуклеотидом, представляющим интерес.
Еще одним аспектом этого изобретения является использование этих крупных гребневидно-разветвленных полинуклеотидов в анализах нуклеиновой кислоты методом гибридизации. В этих анализах:
а) разветвленный полинуклеотид образует гибрид, через первую олигонуклеотидную единицу, с одноцепочечной анализируемой нуклеиновой кислотой, связанной с твердой фазой, или с одноцепочечным олигонуклеотидом, связанным с предметом анализа;
б) несвязанный разветвленный полинуклеотид удаляется;
в) одноцепочечный меченый олигонуклеотид образует гибрид с разветвленным полинуклеотидом через вторые олигонуклеотидные единицы;
г) несвязанный меченый олигонуклеотид удаляется и
д) определяется наличие метки, связанной с разветвленным полинуклеотидом.
Термины, употребляемые в описании изобретения
"Крупный" применяется здесь для описания гребневидно-разветвленных полинуклеотидов изобретения и означает молекулу, имеющую не менее 15 мест ветвления и не менее 20 повторений последовательности, связывающей меченый зонд.
"Гребневидный" применяется здесь для описания структуры разветвленных полинуклеотидов изобретения и означает полинуклеотид, имеющий линейный скелет с множеством отходящих от него боковых ветвей.
"Мультифункциональный" или "модифицированный" нуклеотид означает мономер нуклеотида, который может стабильно включаться в полинуклеотид, имеющий добавочную функциональную группу (преимущественно - цитозин, у которого 4 положение изменено на функциональную гидроксигруппу), с которой нуклеотид может образовывать ковалентную связь для формирования боковой цепи.
"Расщепляемая молекула линкера" означает молекулу, которая может стабильно встраиваться в цепь полинуклеотида и образующая ковалентную связь, которая может разрываться или расщепляться химической обработкой или физическим воздействием, таким как облучение.
"Амплификационный мультимер" означает полинуклеотид, который способен, прямо или косвенно, образовывать гибрид с анализируемой нуклеиновой кислотой и создавать множество копий с меченых зондов.
Характеристика крупных гребневидно-разветвленных полинуклеотидов
Полинуклеотидные мультимеры изобретения состоят из линейного скелета и подвесных боковых цепей. Скелет включает сегмент, который предоставляет специфическое место гибридизации для исследуемой нуклеиновой кислоты или нуклеиновой кислоты, связанной с анализируемой, тогда как подвешенные боковые цепи включают повторения сегмента, который обеспечивает специфические места гибридизации для меченого зонда.
Предпочтительные воплощения этих гребневидных полинуклеотидов могут быть представлены следующей схематической формулой: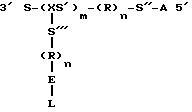
где S - первый спейсерный сегмент из, по крайней мере, 15 нуклеотидов, предпочтительно из 15-50 нуклеотидов;
X - мультифункциональный нуклеотид, который обеспечивает место ветвления;
S' - спейсерный сегмент места ветвления из 0-15 нуклеотидов, предпочтительно 0-10 нуклеотидов;
m - целое число, равное или более 15, предпочтительно 15-100;
R - молекула отщепляемого линкера;
n - 0 или 1;
S'' - второй спейсерный сегмент из 0-10 нуклеотидов, предпочтительно 5-10 нуклеотидов;
A - сегмент, способный образовывать гибрид специфически с анализируемой нуклеиновой кислотой или с нуклеиновой кислотой, связанной с анализируемой;
S''' - третий спейсерный сегмент из 0-10 нуклеотидов;
L - сегмент, содержащий 2-10 повторений, предпочтительно 3-6 повторений, нуклеотидной последовательности, способной образовывать гибрид специфически с меченым олигонуклеотидным зондом;
E - олигонуклеотидный удлиняющий сегмент из 5-10 нуклеотидов.
Весь скелет мультимера или его часть от S до S'', включительно, и часть боковой цепи, исключая L, обычно синтезируются химическими методами и при помощи оборудования для общепринятых способов автоматизированного твердофазного синтеза олигонуклеотидов, как единое целое. В этом отношении спейсерный сегмент S служит разъединению части молекулы, которая содержит места ветвления, с твердой фазой (S' конец S связан с поверхностью твердой фазы).
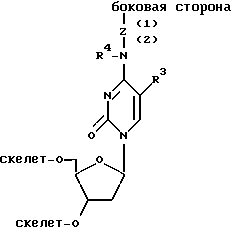
Модифицированные нуклеотиды или ветвящиеся мономеры, обозначенные буквой X в приведенной формуле, являются мультифункциональными нуклеотидами, у которых одна функциональная группа используется для наращивания боковой цепи, а другие используются для связей скелета. Примеры мультифункциональных нуклеотидов описаны в EPA 883096976 (США, серийный N 340031), раскрытие которых включено сюда в порядке ссылки. Эти модифицированные нуклеотиды по преимуществу имеют формулу
где R3 - водород, метил, I, Br или F;
R4 - водород или метил;
Z - выбирается из группы, состоящей из: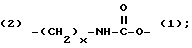
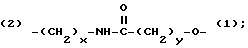

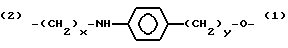
(2)-(CH2-CH2-O)x-(1) и (2)-(CH2)x-O-(1),
где x и y могут быть одними и теми же или разными целыми числами в диапазоне от 1 до 8, включительно. (Обозначения (1) и (2) на Z связи указывают на ориентацию Z половины линкера).
Как указывалось, спейсейрный сегмент S' является факультативным и может использоваться по желанию для разделения каждого места ветвления от предшествующего-последующего фланговых мест ветвления или ряда прилежащих мест ветвления от фланговых рядов мест ветвления. Второй спейсерный сегмент S'' также является факультативным и может использоваться для разделения разветвленной части молекулы с сегментом A, с которым, в конце концов, связывается анализируемый материал. Было обнаружено, что такое разделение улучшает фиксацию анализируемого материала на мультимере. Таким же образом является факультативным третий спейсерный сегмент S'''. Он является преимущественно поли-T.
Сегмент A имеет последовательность и длину, позволяющие ему образовывать специфическую и устойчивую связь с анализируемой нуклеиновой кислотой или нуклеиновой кислотой, соединенной с анализируемым материалом. Для достижения такой специфичности и стабильности сегмент A нормально должен состоять из 15-50, предпочтительно из 15-30, нуклеотидов в длину и иметь содержание GC в диапазоне от 40 до 60%. Специфическая длина и последовательность этого сегмента будут, конечно, варьировать в зависимости от нуклеиновой кислоты, с которой он должен образовать гибрид.
Сегмент E является удлиняющим сегментом боковой цепи, который синтезируется при помощи оборудования и технологии автоматизированного твердофазного химического синтеза олигонуклеотидов. Обычно он длиной около 5-10 нуклеотидов и служит местом, с которым может быть ферментативно сшит сегмент L.
Сегмент L включает повторения олигомерной единицы, которая может образовывать специфический и устойчивый гибрид с меченым олигонуклеотидным зондом. Эти единицы также обычно длиной в 15-150 нуклеотидов, предпочтительно в 20-120 нуклеотидов, и имеют содержание GC в диапазоне от 40 до 60%. Каждый сегмент L содержит в норме от 2 до 10 повторений единицы, предпочтительно от 3 до 6 повторений. Некоторые боковые цепи могут не включать L сегмент. Нормально не менее 50% боковых цепей, предпочтительно не менее 70% боковых цепей, должны включать L сегмент.
Расщепляемые молекулы линкера (R) в скелете и/или боковых цепях являются факультативными, но предпочтительными. Они содержат места расщепления, которые можно выбрать так, чтобы образцы крупного гребневидного полинуклеотида можно было отщепить для анализа и характеристики. В этом отношении предпочтительно, чтобы места расщепления имелись в каждой боковой цепи и место расщепления приходилось на положение 5', когда оно приходится на место ветвления. Примеры расщепляемых молекул линкера, которые могут встраиваться в полинуклеотиды, раскрыты в EPA 883096976 и в примерах, данных ниже.
Синтез крупных гребневидно-разветвленных мультимеров
Сборка полинуклеотидов изобретения осуществляется сочетанием твердофазного прямого синтеза олигонуклеотидов с ферментативным сшиванием.
Тело гребня, включающее спейсер на стороне 3' (S), места ветвления (X), факультативно - сегменты S', S'' и S''', сегмент A, факультативно - заданные молекулы линкера (R) и удлиняющий сегмент боковой цепи E, синтезируется по технологии автоматизированного твердофазного синтеза олигонуклеотидов. Предпочтительной твердой фазой является стекло с заданным размером пор, не менее 2000 ангстрем. В этом синтезе спейсерный сегмент S наращивается от твердой фазы. Для удобства этот сегмент является поли-T. Затем к цепи добавляются мультифункциональные нуклеотиды, содержащие места ветвления, с или без вставочных нуклеотидов в виде спейсеров между местами ветвления. На модифицированных нуклеотидах используются ортогональные защитные или блокирующие группы так, что защитная группа, которая позволяет наращивать скелет, может удаляться, не повреждая защитную группу, которая позволяет наращивать боковую цепь.
Примеры подходящих защитных групп также описаны в EPA 883096976. В качестве блокирующей группы на углеводной части нуклеотида предпочтительно используется диметокситритил (ДМТ). В качестве блокирующей группы на гидроксильной части модифицированного нуклеотида предпочтительно используется левулинил или антрахинонильная группа следующей формулы: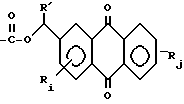
где R' - водород, арил или аралкил;
Ri может быть одним и тем же или различным и избирается из группы, состоящей из амино-, нитро-, гало-, гидроксил-, низший алкил- и низший алкокси-;
Rj может быть одним и тем же или различным и избирается из группы, состоящей из амино-, нитро-, гало-, гидроксил-, низший алкил- и низший алкокси-;
i - 0, 1, 2 или 3;
j - 0, 1, 2, 3 или 4.
После того, как было встроено заданное количество мест ветвления, 5' конец молекулы наращивается сегментами S'' (факультативный) и A или просто коротким сегментом S'' (5-10 нуклеотидов), содержащим место для энзиматического сшивания с сегментом A. Как указывалось выше, избираемое место расщепления предпочтительно встраивать в удлиняющий сегмент. Если сегмент A синтезируется прямо, а не добавляется сшиванием, для защиты мест боковых цепей модифицированных нуклеотидов нужно использовать защитную группу, такую как 2-метилантрахинонил, которую можно удалить избирательно, без нежелательного повреждения остатка молекулы.
После заданного удлинения конца 5' тела гребня удаляются группы, защищающие гидроксильную часть модифицированных нуклеотидов, и одновременно удлиняются места ветвления предпочтительно с включением избираемого места расщепления так, чтобы каждое место ветвления имело удлиняющий сегмент (E) из не мене 5-10 нуклеотидов, служащий местом сшивания.
Сегмент L (и также сегмент A, если он не синтезируется непосредственно) затем сшивается с удлиняющими сегментами боковых цепей при помощи добавления T4-лигазы и подходящих матриц линкера. Сегменты A и L так же можно синтезировать, используя доступные методы и оборудование для автоматизированного твердофазного синтеза олигонуклеотидов.
Анализы методом гибридизации
В анализах нуклеиновой кислоты методом гибридизации крупный гребневидный мультимер изобретения связывается с анализируемой нуклеиновой кислотой или с одноцепочечным олигонуклеотидом, связанным с исследуемым материалом. Так как мультимер включает большое число (20 или более) повторений последовательности, предназначенной для специфической гибридизации с меченым олигонуклеотидом, с анализируемым материалом может связаться намного больше меченых групп, чем при аналогичных методиках. Большое количество меченых групп снижает порог обнаружения исследуемого материала.
Мультимеры могут использоваться, в сущности, в любых известных типах гибридизации нуклеиновых кислот, как при тех, где исследуемый материал связывается непосредственно с твердой фазой, так и при гибридизации методом сэндвича, при котором анализируемый материал связывается с олигонуклеотидом, который, в свою очередь, связывается с твердой фазой. Это особенно полезно при сэндвич-гибридизации в жидкой фазе, методе анализа, описанном в EPA 883096976.
В таких анализах сэндвич-гибридизацией в жидкой фазе мультимер используется следующим образом. Одноцепочечная анализируемая нуклеиновая кислота инкубируется в условиях для гибридизации с избытком двух наборов зондов одноцепочечной нуклеиновой кислоты: (1) набора захватывающих зондов, имеющих каждый по первой фиксирующей последовательности, комплементарной к анализируемому материалу, и второй фиксирующей последовательности, комплементарной к одноцепочечному олигонуклеотиду, связанному с твердой фазой, и (2) набора усилительных зондов, имеющих каждый по первой фиксирующей последовательности, которая способна специфически связываться с анализируемым материалом, по второй фиксирующей последовательности, которая способна специфически связываться с A сегментом мультимера. При помощи усилительного зонда мультимер можно превратить в "универсальный" реагент, так что не нужно делать разные мультимеры для каждого анализируемого материала. Образовавшийся продукт является трехкомпонентным комплексом нуклеиновой кислоты из двух зондов, скрещенных с исследуемым материалом своими первыми фиксирующими последовательностями. Вторые фиксирующие последовательности зондов сохраняются в виде одноцепочечных хвостов, так как они не комплементарны анализируемому материалу.
Затем этот комплекс добавляется к твердой фазе, с которой связан одноцепочечный олигонуклеотид, комплементарный второй фиксирующей последовательности захватывающего зонда, и проводится гибридизация. Образовавшийся продукт включает комплекс, связанный с твердой фазой дуплексом, образованным олигонуклеотидом, связанным с твердой фазой, и второй фиксирующей последовательностью захватывающего зонда. Затем, твердая фаза с фиксированным комплексом отделяется от несвязанных материалов.
Затем, в условиях гибридизации к комплексу твердой фазы с анализируемым материалом и зондом добавляют крупный гребневидный амплификационный мультимер, чтобы дать мультимеру образовать гибрид с доступной второй фиксирующей последовательностью усилительного зонда комплекса. Образовавшийся твердофазный комплекс затем отделяется промыванием от несвязанного мультимера. Затем добавляют меченый олигонуклеотид в условиях, позволяющих ему образовать гибрид с олигонуклеотидными единицами на боковых цепях мультимера. Образовавшийся комплекс твердой фазы с меченой нуклеиновой кислотой отделяется затем от избытка меченого олигонуклеотида промыванием, удаляющим несвязанный меченый олигонуклеотид, и считывается.
Анализируемые нуклеотидные кислоты могут быть из разных источников, например биологические жидкости или твердые вещества, пищевые продукты, материалы из окружающей среды и т.п., и их можно приготовить для гибридизационного анализа различными способами, например протеиназа K/додецилсульфат натрия, хаотропные соли и т.п. Также может быть выгодно уменьшить среднюю величину анализируемых нуклеиновых кислот ферментными, физическими или химическим способами, например ферментами рестрикции, ультразвуковой дезинтеграцией, химической деградацией (например, ионами металлов) и т.п. Фрагменты могут быть такими маленькими, как на 0,1 тыс. оснований, обычно бывают размером не менее 0,5 тысяч оснований и могут быть на 1 тыс. оснований или больше. Для анализа исследуемая последовательность должна быть в одноцепочечной форме. Если последовательность имеет естественную одноцепочечную форму, денатурация не требуется. Однако, когда последовательность представлена двухцепочечной формой, ее нужно денатурировать. Денатурацию можно проводить различными способами, такими как воздействие щелочью, обычно от приблизительно 0,05 до 0,2 М гидроксидом, формамидом, солями, нагреванием или их сочетаниями.
Первые фиксирующие последовательности захватывающего и усилительного зондов, комплементарные исследуемой последовательности, должны иметь каждая не менее 15 нуклеотидов, обычно не менее 25 нуклеотидов, и не более 5 тыс. оснований, обычно не более 1 тыс. оснований, предпочтительно не более 100 нуклеотидов. Типично, чтобы они имели приблизительно 30 нуклеотидов. В номер они должны выбираться для связи с различными последовательностями анализируемого материала. Первые фиксирующие последовательности могут выбираться по разным соображениям. В зависимости от природы анализируемого материала можно интересоваться обобщающей типичной последовательностью, последовательностью, ассоциированной с полиморфизном, особенным фенотипом или генотипом, особенным штаммом или т.п.
Соответствующим подбором первых фиксирующих последовательностей усилительных и захватывающих зондов можно пользоваться для идентификации молекулы конкретной нуклеиновой кислоты, включающей отдельный ген, или другой последовательности, являющейся частью различных молекул нуклеиновой кислоты. Для того, чтобы отличить интересующую молекулу нуклеиновой кислоты от других молекул, также содержащих данную последовательность, один из зондов делается комплементарным к данной последовательности, а другой - к другой последовательности, которая является уникальной для этой молекулы (т.е. не присутствует в других молекулах, содержащих данную последовательность).
Вторые фиксирующие последовательности захватывающих и усилительных зондов выбираются по признаку комплементарности соответственно к олигонуклеотиду, прикрепленному к твердой фазе, и к сегменту A мультимера таким образом, чтобы они не пришли в соприкосновение с эндогенными последовательностями в этом образце/анализируемом материале. Вторая фиксирующая последовательность может контактировать с первой фиксирующей последовательностью или отделяться от нее промежуточной некомплементарной последовательностью. По желанию зонды могут включать другие некомплементарные последовательности. Эти некомплементарные последовательности не должны мешать связи фиксирующих последовательностей или вызывать неспецифическое связывание.
Захватывающие и усилительные зонды можно получить методами синтеза олигонуклеотидов или клонированием, предпочтительнее первое.
Нужно отдавать себе отчет в том, что фиксирующая последовательность не нуждается в совершенной комплементарности для образования гомодуплексов. Во многих ситуациях достаточно образования гетеродуплексов, где ошибочно спарены менее 10% оснований, игнорируя петли из 5 или более нуклеотидов. Соответственно, использующийся здесь термин "комплементарный" означает степень комплементарности, достаточную для того, чтобы обеспечить образование стабильной структуры дуплекса.
Твердая фаза, применяющаяся для анализа, может быть в виде частиц или быть поверхностью стенки любого из множества контейнеров, например центрифужной пробирки, колонки, лунки титрационного микропланшета, фильтра, трубки т. д. Когда пользуются частицами, предпочтительно, чтобы они имели размеры в диапазоне 0,4-200 мкм, чаще 0,8-4,0 мкм. Частицы могут быть из любого общепринятого материала, такого как латекс или стекло. Титрационные микропланшеты представляют предпочтительную твердую поверхность. Олигонуклеотиды, комплементарные ко второй фиксирующей последовательности захватывающего зонда, могут стабильно прикрепляться к твердой поверхности функциональными группами по известным методикам.
Необходимо отдавать себе отчет в том, что возможно замещение второй фиксирующей последовательности захватывающего зонда и олигонуклеотида, прикрепленного к твердой фазе, соответствующей парой лиганд-рецептор, которая потом образует стабильную связь, соединяющую твердую фазу с первой фиксирующей последовательностью захватывающего зонда. Примерами таких пар являются биотин/авидин, тироксин/тироксин-связывающий глобулин, антиген/антитело, углевод/лектин и т.п.
Меченый олигонуклеотид должен включать последовательность, комплементарную олигонуклеотидным единицам на боковых цепях мультимера. Меченый олигонуклеотид должен включать одну или более молекул ("меток"), которые прямо или косвенно дают поддающийся обнаружению сигнал. Метки могут быть связаны с отдельными членами комплементарной последовательности или могут быть представлены концевым членом или хвостом, имеющим множество меток. В литературе описаны различные способы создания связи метки с последовательностью. См., например, Leary et al., Proc. Nat. Acad. Sci. USA (1983) 80:4045; Renz and Kurz, Nucl. Acids Res. (1984) 12:3435; Richardson and Gumport, Nucl. Acids Res. (1983) 11:6167; Smitf et al., Nucl. Acids Res. (1985) 13:2399; Meinkoth and Wahl, Anal. Biochem. (1984) 138:267. Метки могут связываться с комплементарной последовательностью ковалентно или нековалентно. В качестве меток могут использоваться радионуклиды, флуоресценты, хемилюминесценты, красители, ферменты, субстраты ферментов, кофакторы ферментов, ингибиторы ферментов, субъединицы ферментов, ионы металлов и т.п. Иллюстративные специфические метки включают флуоресцеин, родамин, техасский красный, фикоэритрин, умбеллиферон, люминол, НАД.Н, альфа-бета-галактозидазу, перексидазу хрена и т.п.
Отношение захватывающего зонда и усилительного зонда к предполагаемому количеству молей анализируемого вещества должно быть у каждого, по крайней мере, стехиометрическим, а предпочтительно - избыточным. Предпочтительно соотношение не менее 1,5:1 и еще предпочтительнее - не менее 2:1. Нормальными были бы пределы от 2:1 до 10.000:1. Концентрация каждого из зондов, в общем, должна быть в пределах от около 10-10 до 10-6 М при концентрации образца нуклеиновой кислоты, варьирующей от 10-21 до 10-12 М. Стадии гибридизации в анализе занимают, в общем, от 10 минут до 2 часов, чаще завершаясь в пределах одного часа. Гибридизация может проводиться при слегка повышенной температуре, в общем, в пределах от около 20oC до 80oC, чаще - от около 35oC до 70oC, особенно - 65oC.
Реакцию гибридизации обычно проводят в водной среде, особенно в забуференной водной среде, которая может включать различные добавки. Добавки, которыми можно пользоваться, включают низкие концентрации детергентов (0,1-1%), соли, например лимоннокислый натрий (0,017-0,17 М), фиколл, поливинилпирролидин, носители нуклеиновых кислот, носители белков и т.д. К водной среде можно добавить неводные растворители, такие как диметилформамид, диметилсульфоксид, спирты и формамид. Эти и другие растворители могут присутствовать в количестве от 2 до 50%.
Строгость среды гибридизации может регулироваться температурой, концентрациями солей, системой растворителя и т.п. Поэтому, в зависимости от длины и природы интересующей последовательности строгость среды должна изменяться.
Методика стадий разделения в анализе должна меняться в зависимости от природы твердой фазы. Когда используют частицы, для разделения используют центрифугирование или фильтрацию, сливая или удаляя супернатант. Когда анализируют частицы, они должны быть тщательно отмыты, обычно 1-5 раз, подходящей забуференной средой, например ЗФР, содержащей такой детергент, как додецилсульфат натрия. Когда разделительным средством служит стенка или опорная подложка, супернатант можно слить или удалить, и стенку отмывают так же, как частицы.
В зависимости от природы метки можно пользоваться различными методиками для выявления присутствия метки. Для флуоресцентов имеется множество флуорометров. Для хемилюминесцентов имеются люминометры или фотопленки. С ферментами можно получить флуоресцирующие, хемилюминесцентные или окрашенные продукты, которые выявляются флуорометрически, люминометрически, спектрофотометрически или визуально. Различные метки, использующиеся для иммуноанализа, и методики иммуноанализа можно использовать для данных анализов.
В гибридизационном анализе, при котором анализируемая нуклеиновая кислота непосредственно связана с твердой фазой так, как в анализе дот-блоттингом, мультимер образует гибрид непосредственно со связанным анализируемым материалом. В этих случаях сегмент A мультимера комплементарен последовательности исследуемого материала, а олигонуклеотидные единицы на боковых цепях комплементарны меченому олигонуклеотиду. Несвязанный мультимер удаляют с твердой фазы, после чего меченый олигонуклеотид образует гибрид со связанным комплексом: анализируемый материал-мультимер. Удаляют избыток меченого олигомера и считывают меченый, связанный комплекс.
Мультимеры можно также применять в других анализах, таких как прямой, непрямой и сэндвич-иммуноанализы. В этих случаях реагент, играющий роль меченого антитела или другого лиганда, связанного с анализируемым материалом прямо или косвенно, имеет олигонуклеотид, комплементарный A сегменту мультимера, больше связанный с ним, чем с меткой. Например, в сэндвич-иммуноанализе на антигенный анализируемый материал анализируемый образец инкубируют с твердой фазой, с которой связано первое антитело к этому антигену. Несвязаннный образец удаляют с твердой фазы, и второе антитело к этому антигену, которое связано с олигонуклеотидом, комплементарным единице мультимера, вступает в реакцию со связанным комплексом, образуя трехчленный комплекс. После удаления избытка второго антитела мультимер образует гибрид с комплексом через олигонуклеотид, связанный со вторым антителом. Избыток мультимера удаляют, и меченый олигонуклеотид образует гибрид с другими олигонуклеотидными единицами мультимера. После удаления избытка меченого олигонуклеотида считывают комплекс.
Наборы для проведения анализов гибридизации амплифицированной нуклеиновой кислоты согласно изобретению должны включать сочетание следующих реагентов в одной упаковке: мультимер; соответствующий меченый олигонуклеотид; твердая фаза, способная связывать анализируемый материал; факультативно-захватывающий зонд, если тип анализа относится к одному из тех, в которых анализируемый материал фиксируется к твердой фазе через промежуточный олигонуклеотид или другой лиганд; и факультативно-усилительный зонд, если тип анализа относится к одному из тех, в которых мультимер не образует гибрид прямо с анализируемым материалом. Каждый из этих реагентов должен находиться в отдельном контейнере. В набор также можно включить реактив для денатурации анализируемого материала, буферы для гибридизации, растворы для промывания, субстраты для ферментов, материалы для положительного и отрицательного контроля и письменные инструкции по проведению анализа.
Следующие примеры изобретения предлагаются в порядке иллюстрации, а не ограничения.
Пример 1
Этот пример иллюстрирует синтез гребневидно-разветвленного полинуклеотида, имеющего 15 мест ветвления и области удлинения боковых цепей, имеющие три места фиксации меченого зонда. Этот полинуклеотид был сконструирован для использования при гибридизации в жидкой фазе, как описано в EPA 883096976.
Все олигонуклеотиды синтезированы химически на автоматическом синтезаторе ДНК (Applied Biosystems Inc. (ABI) модель 380 A/B). Использовался фосфорамидитный метод типа метокси, за исключением S'-фосфорилирования, в котором пользовались реактивом PhostelTM (ABN). Применялись стандартные методики ABI с указанными исключениями. По показаниям пользовались мультипликацией цикла (например, 1,5 х цикл, 4,5 х цикл), в конкретном цикле применялось кратное увеличение стандартного количества амидита, рекомендованного ABI. Здесь приводятся программы проведения циклов 0,4, 1,5, 4,5 и CAP-PRIM для управления синтезатором Applied Biosystems модель 380 A/B.
Сначала получали тело гребня следующей структуры:
3'T20-X15(GTCAGTp5')15-(GTTTGTp-5')1
где X является модифицированным нуклеотидом, описанным ранее.
Первой синтезировали часть тела гребня с 15 повторениями при помощи стекла с регулируемыми порами (СРП) на 40 мг тимидина (3000 ангстрем, 3 мкМ тимидина на грамм подложки) в режиме 1,5 х циклов. Место ветвления нуклеотида имело формулу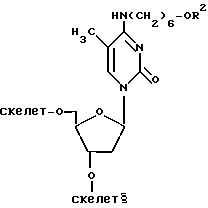
где R2 представлен  или
или
Мономер, где R2 представляет MAC, сделан следующим образом. К раствору N-4-(6-гидроксигексил)-5'-DMT-5-метил-2' дезоксицитидина (17 ммоль), приготовленному как описывалось ранее (Horn and Urdea, NAR vol. 17:17, p. 6959-6967 (1989)), в 200 мл метиленхлорида добавляли пиридин (40 ммоль) и смесь охлаждали до 0oC. По каплям добавляли раствор 2-антрахинонметоксихлорформата (MAC-Cl) (20 ммоль) в 200 мл CH2Cl2 и перемешивали 10 минут. Анализ ТСХ (силиконовые пластинки, обработанные 10% метанол/CH2Cl2) показал, что исходный материал полностью израсходован. Реакционную смесь разводили 400 мл этилацетата и органическую фазу экстрагировали 2 раза по 300 мл 5% NaHCO3 и 80% насыщенным водным раствором NaCl. После осушения органической фазы над Na2SO4 в течение 30 минут с последующей фильтрацией растворитель удаляли в вакууме. Продукт очищали хроматографией на силикагеле с градиентом метанола (0-6%) в CH2Cl2, чтобы получить 13 г чистого продукта (85% выхода).
Получали 0,1 М раствор 2-(гидроксиметил)-антрахинона (MAG-OH) растворяя 25 ммоль (5,95 г) в 250 мл диоксана. Желтый раствор фильтровали и растворитель выпаривали до удаления влаги. Остаток повторно растворяли в 200 мл диоксана и добавляли пиридин (2 мл, 25 ммоль). Этот раствор добавляли каплями в перемешиваемый раствор трифосгена (2,5 г, 25 Мэкв) в 50 мл CH2Cl2. Кончив добавлять, смесь перемешивали при 20oC в течение 18 часов. Смесь разводили 800 мл этилацетата и органическую фазу промывали 3 раза 60 мл 80% насыщенного водного раствора NaCl. После осушения органической фазы над Na2SO4 растворитель выпаривали в вакууме до получения желтого сухого остатка, который растворяли в CH2Cl2 (250 мл, 0,1 М). Этим раствором пользовались без дальнейшей очистки.
Нуклеозид N-4-(O-антрахинонметоксикарбонил-6-оксигексил)-5'-DMT-5- метил-2'дезоксицитидин (14,4 ммоль) растворяли в CH2Cl2 (50 мл), содержащем 70 ммоль DiPEA. После охлаждения до 4oC добавляли N,N-диизопропиламинометоксихлорфосфин (2,72 мл, 14 ммоль). Фосфитилирующий реактив добавляли маленькими порциями до тех пор, пока не поглощались 95% исходного материала. Затем реакционную смесь разводили этилацетатом (300 мл), экстрагировали 5% раствором NaHCO3 (2 х 300 мл), затем - 2 х 300 мл 80% насыщенного водного раствора NaCl и, наконец, осушали над твердым Na2SO4. Растворитель удаляли в вакууме.
Неочищенный фосфорамидит очищали хроматографией на силикагеле. Очищенный фосфорамидит растворяли в толуоле и добавляли, быстро помешивая, до 800 мл холодных гексанов (-50oC). Образовавшийся преципитат быстро собирали фильтрацией и высушивали в высоком вакууме в течение 18 часов, чтобы получить 12,4 г (4,5 ммоль, 80% выхода) слегка желтоватого сухого продукта. Снятие защиты с защищенного MAC нуклеотида осуществляется обработкой дитионитом натрия в нейтральных условиях.
Для синтеза тела гребня (не включающего боковые цепи) концентрация мономеров метилфосфорамидита составляет 0,1 М для A, C, G и T, 0,15 М для мономера X места ветвления и 0,2 М для реагента PhostelTM. Детритилирование осуществлялось 3% трихлоруксусной кислоты в метиленхлориде в непрерывном потоке в течение всего периода снятия защиты. В заключение 5' DMT замещался ацетильной группой.
Области удлинения боковой цепи из шести оснований формулы 3'-GTCAGTp синтезировали в каждом месте ветвления мономера следующим образом. Удаление группы, защищающей основание (R2 в вышеуказанной формуле), осуществляли вручную, в то время как сохранялась подложка СРП, на той же самой колонке, где синтезировали тело гребня. Если R2 - ливулинил, вводили раствор 0,5 М гидразингидрата в смеси пиридин/ледяная уксусная кислота (объемное соотношение 1: 1) и оставляли в контакте с подложкой СРП на 90 минут, каждые 15 минут меняя жидкость. После обильного промывания смесью пиридин/ледяная уксусная кислота (объемное соотношение 4:1) и потом ацетонитрилом фильтры в колонке заменяли. Если R2 - 2-метилантрахинонил, вводили раствор дитионита натрия (1 г дитионита натрия, растворенный в 20 мл 1 М бикарбоната триметиламмония) с последующим добавлением 20 мл диоксана и оставляли в контакте с подложкой СРП на 90 минут. После снятия защиты добавляли области удлинения боковых цепей из 6 оснований, пользуясь режимом 4,5 х циклов и концентрациями мономера 0,2 М.
В этих реакциях синтеза концентрации мономеров составляли 0,2 М (включая R и реагент PhostelTM). Детритилирование осуществляли 2,5% раствором дихлоруксусной кислоты в толуоле/30% трихлоруксусной кислоты в метиленхлориде (объемное соотношение 1:1) в непрерывном потоке. Защитные группы удалялись следующим образом. Фосфатные защитные группы удалялись с фрагментов продукта на твердой подложке обработкой СРП раствором: тиофенол/триэтиламин/ацетонитрил (объемные соотношения 1:1:2) в течение 1 часа при 20oC с последующим промыванием ацетонитрилом (10 х 1 мл) и метанолом. Фрагмент продукта удаления с СРП подложки обрабатывали 0,5 мл концентрированного гидроксида аммония в течение 20 мин и удалением супернатанта. Обработку повторяли дважды с общей продолжительностью экспозиции в пределах одного часа. Объединенные супернатанты переносили во флакон с завинчивающейся крышкой и нагревали при 60oC в течение 18 часов. После охлаждения до комнатной температуры растворитель удаляли в испарителе Speed-Vac и остаток растворяли в 100 мкл воды.
При помощи автоматического синтезатора были собраны 5'-области удлинения скелета (сегмента A), области удлинения боковых цепей и матрица/линкеры сшивания следующей структуры:
Область удлинения скелета 5'
3'-AGGTGCTCCGTATCCTGGGCACAG-5'
Область удлинения боковой цепи
3'-GATGCGR(TTCATGCTGTTGGTGTAG)3-5'
Матрица лигирования для сшивания областей удлинения скелета 5'
3'-GCACCTACAAAC-5'
Матрица лигирования для сшивания областей удлинения боковой цепи
3'-CGCATCACTGAC-5'
R в области удлинения боковой цепи представлен следующим линкером с избирательным расщеплением: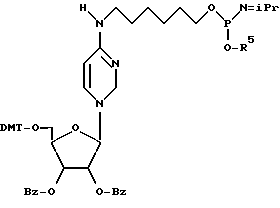
где DMT - диметокситритил,
Bz - бензоил,
R5 - метил или бета-цианоэтил и
iPr - изопропил.
Расщепление в области R достигается двустепенной химической реакцией: (1) окислением водным NaIO4 в течение 1 часа с последующей (2) обработкой водным н-пропиламином.
Неочищенное тело гребня очищали обычным способом в полиакриламидном геле (10% с 7 М мочевиной).
5'-область удлинения скелета и области удлинения боковых цепей сшивались с телом гребня T4-лигазой по стандартной прописи (Urdea (1987) Methods in Enzymol. 146: 22-41), за исключением более продолжительного времени реакции (>8 часов), пользовались 14% полиэтиленгликолем при нейтральной температуре.
После сшивания и очистки часть продукта метили 32P и проводили ступенчатое расщепление, описанное выше. Затем проводили анализ пробы электрофорезом в полиакриламидном геле, чтобы определить количество встроенных областей удлинения боковых цепей, подсчитывая число полос на геле. Обнаруживалось, что продукт имеет 24 меченых места связи в целом.
Пример 2
Этот пример иллюстрирует получение того же самого мультимера, как в примере 1, используя подложку СРП со средним размером пор/более нагруженную подложку СРП, которую сначала доводят до подходящего уровня нагрузки. Первичный синтез производился начиная с подложки СРП на 30 мг тимидина (1000 ангстрем; 20 ммоль тимидина на грамм подложки). Первые 20 циклов соединения с T производились в режиме 0,4 х цикл, чтобы снизить нагрузку ниже 10 ммоль на грамм подложки. За этим следовали 20 циклов соединения с T, 15 циклов с X (модифицированным нуклеотидом) и, наконец, встраивание последовательности 3'-GTTTGTGGp при помощи 1,5 х цикла. Удаляли терминальную 5-ДMT-группу и завершали последовательность по программе CAP-PRIM цикла на аппарате ABI. Колонку вынимали из аппарата и дальнейшие манипуляции производили вручную. Удаление левулинатных защитных групп с точки ветвления производилось так, как описано выше, после чего подложка СРП переносилась в новую колонку ABI. Наращивание боковых цепей осуществлялось так, как описано выше, чтобы встроить последовательность 3'-GTCAGTp, используя 4,5 х цикл. Защитные группы удалялись так, как описано в примере 1 (см. выше), и неочищенный продукт растворяли в 100 мкл воды.
Сшивание групп A и L производили так, как в примере 1.
Пример 3
Полинуклеотид из примера 1 с 24 местами гребневидного разветвления использовался для сэндвич-анализа в жидкой фазе на N.gonorrhoeae с помощью полинового ген-специфического захватывающего и усилительного зондов и зондов, меченых и щелочной фосфатазой так, как описано в примере 5 из EPA 883096976. Эти два типа меток использовались для того, чтобы оценить, создает ли какие-либо пространственные проблемы гребневидная структура с 24 ветвями для применения зондов, меченых щелочной фосфатазой. Результаты сопоставлялись с полученными при использовании структуры гребня с 5 ветвями, которая не обнаруживала каких-либо пространственных препятствий.
Когда применялся зонд, меченый 32P, молекула с 24 разветвлениями давала увеличение относительного выпуска в 4,76 по сравнению со стандартным гребнем с 5 ветвями (теоретически - 4,8; 195.000 ± 10.000 импульсов в минуту против 41.000 ± 1.200 импульсов в минуту на 10 атом-молей соответственно). Когда применялся зонд, меченый щелочной фосфатазой, молекула с 24 разветвлениями давала увеличение относительной продукции в 3,94 раза по сравнению со стандартным гребнем с 5 ветвями (50,1 ± 1,7 вспышек против 12,7 ± 0,2 на 10 атом-молей соответственно). Различие в эффективности меток у этих двух типов зондов указывает, что ферментная метка хорошо приспособлена к структуре гребня.
Анализы на другие нуклеиновые кислоты, описанные в примерах EPA 883096976, могут проводиться таким же образом.
Модификации вышеописанных способов осуществления изобретения, которые очевидны для специалистов в области химии нуклеиновых кислот и гибридизации защиты нижеследующей формулы изобретения: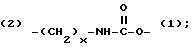
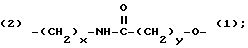
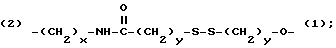
(2)-(CH2-CH2-O)x-(1) and
(2)-(CH2)x-O-(1).
Пример 4
Синтез гребнеобразного разветвленного полинуклеотида
В этом примере иллюстрируется синтез гребнеобразного разветвленного полинуклеотида, имеющего 15 сайтов ветвления и удлиняющие участки боковой цепи, содержащие три сайта связывания с меченым зондом. Указанный полинуклеотид был сконструирован для использования в жидкофазной гибридизации, как описано в EPA 883096976.
Все процедуры химического синтеза олигонуклеотидов осуществляли на автоматическом ДНК-синтезаторе (Applied Biosystems Inc. (ABI) model 380 B). При этом был использован фосфорамидитный метод типа бета-цианоэтилирования, предусматривающий 5'-фосфорилирование с применением реагента  (ABN). За исключением конкретно указанных изменений были использованы стандартные протоколы (ABI). В случае использования кратного цикла (например, цикла 1,2), в данном конкретном цикле использовали соответствующее кратное стандартное количество амидита, рекомендованное ABI. Предлагаемые в настоящем описании программы для проведения циклов 1,2 и 6,4 осуществляли на ДНК-синтезаторе (Applied Biosystems Model 380 B).
(ABN). За исключением конкретно указанных изменений были использованы стандартные протоколы (ABI). В случае использования кратного цикла (например, цикла 1,2), в данном конкретном цикле использовали соответствующее кратное стандартное количество амидита, рекомендованное ABI. Предлагаемые в настоящем описании программы для проведения циклов 1,2 и 6,4 осуществляли на ДНК-синтезаторе (Applied Biosystems Model 380 B).
Сначала получали следующую гребнеобразную структуру: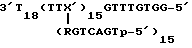
где X' представляет собой мономер ветвления, а R представляет собой расщепляемый периодатом линкер.
В первую очередь синтезировали ту часть гребнеобразной структуры, которая соответствует 15 повторам, используя для этого стекло с контролируемым размером пор, на которое было нанесено 33,8 мг аминопропил-дериватизированного тимидина (2000  , 7,4 мкМ тимидина на 1 г носителя) в соответствии со схемой осуществления цикла 1,2. Нуклеотид сайта ветвления имел следующую формулу:
, 7,4 мкМ тимидина на 1 г носителя) в соответствии со схемой осуществления цикла 1,2. Нуклеотид сайта ветвления имел следующую формулу: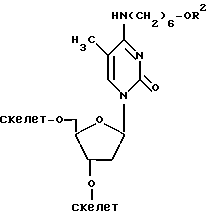
где R2 представляет 
Для синтеза гребнеобразной структуры (не включая боковых цепей) концентрация бета-цианоэтилфосфорамидитных мономеров составляет 0,1 М для A, C, G и T; 0,15 М для мономера E, имеющего сайт ветвления; и 0,2 М для реагента PhostelTM. Детритилирование проводили с использованием 3%-ной трихлоруксусной кислоты в метиленхлориде и с применением ступенчатого поточного метода в процессе деблокирования. В заключение 5'-ДМТ замещали ацетильной группой.
Расщепляемый линкер R и удлиняющие участки боковой цепи, состоящие из шести оснований и имеющие формулу 3'-RGTCAGTp (SEQ ID N 1), синтезировали в каждом мономерном сайте ветвления следующими образом. Защитную группу удаляли вручную (R2 в вышеуказанной формуле), а CPG-носитель (CPG - стекло с контролируемым размером пор) оставляли в той же самой колонке, которую использовали для синтеза гребнеобразной структуры. В случае, если R2 представлял собой левулинил, то вводили раствор 0,5 М гидрата гидразина в пиридине/ледяной уксусной кислоте (1: 1, об. /об.) и поддерживали в контакте с CPG-носителем в течение 90 минут с обновлением жидкости каждые 15 минут, после чего экстенсивно промывали пиридином/ледяной уксусной кислотой (1:1, об. /об.), а затем ацетонитрилом. После деблокирования расщепляемого линкера R присоединяли удлиняющие участки боковой цепи, состоящие из 6 оснований, с использованием цикла 6,4.
В этом синтезе концентрация фосфорамидитов составляла 0,1 М (за исключением 0,2 М R и реагента PhostelTM, где R представлял собой 2-(4-(4-(2-диметокситритилокси)этил)-фенокси-2,3-ди(бензоилокси)- бутилокси)фенил)этил-2-цианоэтил-N,N-диизопропилфосфорамидит).
Детритилирование осуществляли с использованием раствора 3%-ной трихлоруксусной кислоты в метиленхлориде в непрерывном сквозном потоке с последующим промыванием раствором толуола/хлорометана (1:1, об./об.). Разветвленные полинуклеотидные цепи отделяли от твердых носителей автоматически в 380 B с использованием цикла "CENH3". Раствор гидроксида аммония собирали в 4-миллитровые склянки Wheaton с завинчивающейся крышкой и нагревали в течение 12 часов при 60oC для удаления всех нуклеотид-защищающих групп. После охлаждения до комнатной температуры, растворитель удаляли в вакуумном скоростном испарителе (Speed-Vac), а остаток растворяли в 100 мкл воды.
Удлиняющие сегменты 3'-конца каркаса (сегмент A), удлиняющие сегменты боковой цепи и матрицы/линкеры для лигирования, имеющие нижеприведенные структуры, синтезировали также с использованием автоматического синтезатора:
Удлиняющий сегмент 3'-конца каркаса:
3'-TCCGTATCCTGGGCACAGAGGTGCp-5' (SEQ ID N 2)
Удлиняющий сегмент боковой цепи: 3'-GATGCG(TTCATGCTGTTGGTGTAG)3-5' (SEQ ID N 3)
Лигирующая матрица для присоединения удлиняющего сегмента к 3'-концу каркаса: 3'-AAAAAAAAAACCACCTp-5' (SEQ ID N 4)
Лигирующая матрица для присоединения удлиняющего сегмента к боковой цепи: 3'-CGCATCACTCAC-5' (SEQ ID N 5)
Неочищенный гребнеобразный полинуклеотид очищали стандартным методом с использованием полиакриламидного геля (7% с 7 М мочевины и IX буфера TBE для электрофореза).
3'-удлиняющий сегмент каркаса и удлиняющие сегменты боковой цепи присоединяли к гребнеобразной структуре следующим образом. Гребнеобразную структуру (4 пМ/мкл), 3'-удлиняющий сегмент каркаса (6,25 пМ/мкл), удлиняющий сегмент боковой цепи (93,75 пМ/мкл), матрицу для присоединения к боковой цепи (75 пМ/мкл) и матрицу для присоединения к каркасу (5 пМ/мкл) объединяли в 1 мМ АТР/5 мМ ДТТ/50 мМ Трис-HCl, pH 8,0/10 мМ MgCl2/2 мМ спермидина с 0,5 ед./мкл полинуклеотид-киназой T4. Полученную смесь инкубировали в течение 2 часов при 37oC, после чего нагревали в водяной бане до 95oC и медленно в течение 1 часа охлаждали до температуры ниже 35oC. Затем добавляли 2 мМ АТР, 10 мМ ДТТ, 14% полиэтиленгликоля и 0,21 ед./мкл лигазы T4, и полученную смесь инкубировали в течение 16-24 часов при 23oC. ДНК осаждали в NaCl/этаноле, ресуспендировали в воде и подвергали второй процедуре лигирования следующим образом. Смесь доводили до следующих уровней концентраций: 1 мМ АТР, 5 мМ ДТТ, 14% полиэтиленгликоля, 50 мМ Трис-HCl, pH 7,5, 10 мМ MgCl2, 2 мМ спермидина, 0,5 ед./мкл полинуклеотид-киназы T4 и добавляли 0,21 ед. /мкл лигазы T4, после чего смесь инкубировали в течение 16-24 часов при 23oC. Затем продукты лигирования очищали с помощью электрофореза на полиакриламидном геле.
После лигирования и очистки часть продукта подвергали 32P-мечению и расщеплению в сайте R посредством окисления водным NaIO4 в течение 1 часа. Затем, для определения числа включенных удлиняющих сегментов боковой цепи, образец анализировали с помощью электрофореза в ПААГ путем количественной оценки радиоактивной метки в полосах на геле. Было установлено, что полученный продукт имел, в целом, 45 сайтов связывания с меченым зондом.
Пример 5
"Сэндвич"-гибридизационный анализ на ДНК HBV с использованием мультимера
В этом примере проиллюстрировано использование большого гребнеобразного мультимера, полученного как описано в примере 1, в гибридизационном анализе на присутствие ДНК HBV. Зонды и условия гибридизации описаны в заявке на патент России N 93004859.13.
В этом примере был использован жидкофазный метод "сэндвич"-гибридизационного анализа с использованием "15 х 3"-амплифицированной нуклеиновой кислоты. "15 х 3" означает, что в данном методе использовали два мультимера: (1) зонд-амплификатор, имеющий первый сегмент (A), который связывается с нуклеиновой кислотой HBV, и второй сегмент (B), который гибридизируется с (2) мультиметром-амплификатором, имеющим первый сегмент (B*), который гибридизируется с сегментом (B), и 15 итераций сегмента (C), где указанный сегмент C гибридизируется с тремя мечеными олигонуклеотидами.
В данном анализе были использованы сегменты зонда-амплификатора и зонда захвата, приведенные в конце описания.
Каждый зонд-амплификатор, помимо последовательностей, в основном, комплементарных HB-последовательностям, содержал следующий 5'-удлиняющий сегмент, комплементарный сегменту мультимера-амплификатора:
AGGCATAGGACCCGTGTCTT (SEQ ID N 54).
Каждый зонд захвата, помимо последовательностей, в основном, комплементарных HBV-ДНК, содержал расположенную ниже последовательность, комплементарную ДНК, связанной с твердой фазой (т. е. комплементарную XT1*): CTTTCTTTGGAGAAAGTGGTG (SEQ ID N 55).
Планшеты для микротитрования получали следующим образом. Полистироловые 96-луночные планшеты для микротитрования (White Microlitell Removawell) были закуплены у фирмы Dynatech Inc. Каждую лунку наполняли 200 микролитрами 1 н. HCl и инкубировали в течение 15-20 мин при комнатной температуре. Затем планшеты 4 раза промывали 1X PBS, и из лунок удаляли жидкость путем отсасывания. После этого лунки наполняли 200 микролитрами 1 н. NaOH и инкубировали в течение 15-20 минут при комнатной температуре. Затем планшеты снова 4 раза промывали 1X PBS, и удаляли жидкость из лунок путем отсасывания.
Poly(phe-lys) закупали у фирмы Sigma Chemicals, Inc. Этот полипептид имел молярное отношение phe:lys 1:1; средневесовую молекулярную массу 47900 г/М; среднюю длину в 309 аминокислот и содержал 155 аминов/М. 1 мг/ил-раствор указанного полипептида смешивали с 2 М NaCl/1X PBS до конечной концентрации 0,1 мг/мл (pH 6,0). В каждую лунку добавляли 10 мкл этого раствора. Во избежание высыхания планшеты обертывали полиэтиленовой пленкой и инкубировали в течение ночи при 30oC. Затем планшеты 4 раза промывали 1X PBS, и из лунок удаляли жидкость путем отсасывания.
Для связывания олигонуклеотида XT1* с планшетами осуществляли следующую процедуру. XT1* синтезировали как описано в EPA 883096976. 20 мг дисукцинимидилсуберата растворяли в 300 мкл диметилформамида (ДМФ). К 100 мкл буфера для связывания (50 мМ фосфата натрия, pH 7,8) добавляли 26 (ОП260) единиц XT1*. Затем к DSS-ДMФ-раствору добавляли смесь для связывания, и эту смесь размешивали в магнитной мешалке в течение 30 минут. Колонку NAP-25 уравновешивали 10 мМ фосфата натрия, pH 6,5. К 2 мл 10 мМ фосфата натрия (pH 6,5, 4oC) добавляли DSS-ДМФ-раствор смеси для связывания. Эту смесь интенсивно перемешивали и загружали в уравновешенную колонку NAP-25. DSS-активированную XT1*-ДНК элюировали с колонки 3,5 миллилитрами 10 мМ натрийфосфата (pH 6,5). Затем 5,6 (ОП260) единиц элюированной DSS-активированной XT1*-ДНК добавляли к 1500 мл 50 мМ натрийфосфата (pH 7,8). После этого 50 мкл этого раствора добавляли в каждую лунку, и планшеты инкубировали в течение ночи. Затем планшеты 4 раза промывали 1X PBS, и из лунок удаляли жидкость путем отсасывания.
Конечную очистку планшетов осуществляли следующим образом. В каждую лунку добавляли 200 мкл 0,2 н. NaOH, содержащего 0,5% (мас./об.) ДСН. Затем планшеты обертывали полиэтиленовой пленкой и инкубировали в течение 60 минут при 65oC. После этого планшеты 4 раза промывали 1X PBS, и жидкость из лунок удаляли путем отсасывания. Очищенные таким образом планшеты хранили при 2-8oC в присутствии осушающих шариков (сиккатив).
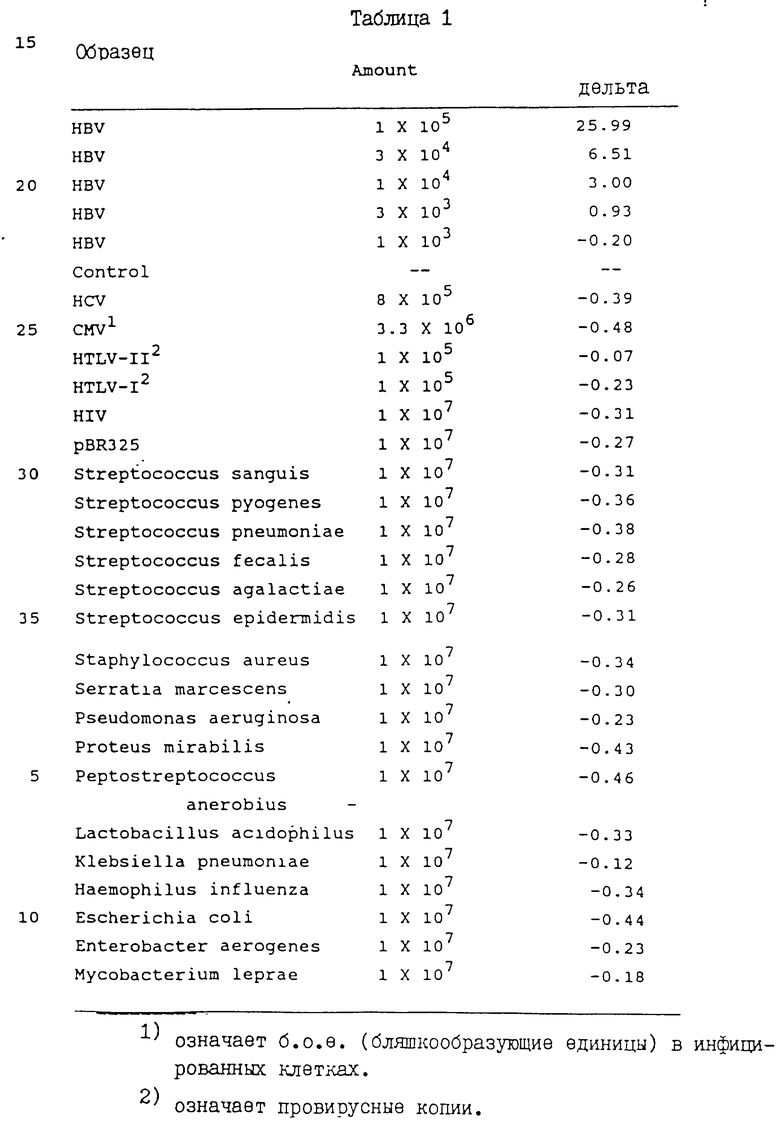
Образец получали путем добавления в каждую лунку 12,5 мкл буфера P-K (2 мг/мл протеиназы K в 10 мМ Трис-HCl, pH 8,0/0,15 М aCl/10 мМ E TA, pH 8,0/1% ДСН/40 мг/мл обработанной ультразвуком ДНК спермы лосося). Стандартную ДНК HBV получали путем разведения клонированной ДНК HBV, подтипа adw, в HBV-негативной сыворотке человека и введения в каждую лунку аликвот разведений, соответствующих 1000, 3000, 10000, 30000 или 100000 молекулам. Тесты на перекрестную гибридизацию с гетерологичными ДНК осуществляли путем добавления в каждую лунку либо очищенную ДНК, либо инфицированные клетки. Количество каждого из микроорганизмов указано в таблице.
Планшеты покрывали и размешивали для смешивания образцов, после чего планшеты инкубировали при 65oC для высвобождения нуклеиновых кислот.
В каждую лунку добавляли смесь, состоящую из HBV-специфического зонда-амплификатора и зонда захвата (5 фМ каждого зонда на лунку, разведенного в 1 н. NaOH). После этого планшеты покрывали и слегка помешивали для размешивания реагентов, а затем инкубировали в течение 30 минут при 65oC.
После этого в каждую лунку добавляли буфер для нейтрализации (0,77 М 3-(N-морфолино)пропансульфоновая кислота/1,845 М NaCl/0,185 цитрат натрия). Затем планшеты покрывали и инкубировали в течение 12-18 часов при 65oC.
После выдерживания планшетов в течение еще 10 минут при комнатной температуре содержимое каждой лунки отсасывали для удаления всей жидкости, а лунки промывали 2X-буфером для промывания (0,1% ДСН/0,15 М NaCl/0,0015 цитрата натрия).
Затем в каждую лунку добавляли амплификатор-мультимер (30 фМ на лунку). Планшеты покрывали и содержимое лунок перемешивали, после чего планшеты инкубировали в течение 30 минут при 55oC.
После выдерживания планшетов в течение еще 5-10 минут при комнатной температуре лунки промывали как описано выше.
Затем в каждую лунку добавляли меченый щелочной фосфатазой зонд, описанный в EP 883096976 (40 мкл/лунка 2,5 фМ/мкл). После инкубирования в течение 15 минут при 55oC и выдерживания в течение 5 минут при комнатной температуре лунки дважды промывали как описано выше, а затем промывали 3X-смесью 0,015 М NaCl/0,0015 М цитрата натрия.
Был использован стимулированный ферментом диоксетан (Schaap et al., Tet. Lett. (1987) 28:1159-1162; и публ. EPA N 0254051), полученный от фирмы Lumigen Inc. В каждую лунку добавляли 30 мкл Lumiphos 530 (Lumigen). По лункам слегка постукивали так, чтобы реагент осаждался на дно, а для того, чтобы реагент равномерно распределялся по дну лунки, эти лунки слегка вращали круговыми движениями. Затем лунки покрывали и инкубировали в течение 40 минут при 37oC.
Считывание планшетов проводили на люминометре Dynatech ML 1000. Выходные данные получали в виде полной интегральной величины количеств света, продуцируемого в процессе реакции.
Результаты эксклюзионного исследования HBV-зондов представлены в таблице. Результаты для каждого стандартного образца выражали как разность между средним значением для негативного контроля плюс два стандартных отклонения и средним значением для образца минус два стандартных отклонения (дельта). Если дельта больше нуля, то образец считался положительным. Эти результаты показали, что описанная серия зондов способна отличать ДНК HBV от гетерологических микроорганизмов, а также что чувствительность описанного анализа составляет около 1000-3000 молекул HBV.
Пример 6
Разветвленный нуклеиновокислотный полимер с двумя 5'-концами
В этом примере гребнеобразный мультимер, описанный выше, продуцировали путем лигирования гребнеобразного каркаса с L при помощи разветвленного нуклеиновокислотного полимера, имеющего 3'-конец и два 5'-конца, которые вводятся ветвью, имеющей следующую структуру: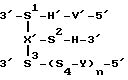
где S1 представляет собой T6, H' представляет собой 5'-CGCATC-3'; V' представляет собой 5'-ACTGAC-3' (комплементарную сайту лигирования E в гребнеобразной структуре, описанной в примере 1); X' представляет собой модифицированный нуклеотид, описанный выше в примере 1; H представляет собой 5'-GCGTAG-3', S3 представляет собой T2; S4 отсутствует (т.е. S4 = 0), Y эквивалентен L в примере 1 и имеет последовательность 5'-GACGTGGTTGTCGTACTT-3' (SEQ ID N 56) (B A3c), а n = 3.
Для лигирования разветвленного нуклеиновокислотного полимера с "15х3"-гребнеобразной структурой получали следующую смесь из 15х3-гребнеобразной структуры (1500 пМ), линкера последовательности 5-TCCACGAAAAAAAAAA-3 (SEQ ID N 57) (1875 пМ), 3'-удлиняющего сегмента каркаса (пример 1) (2344 пМ) и разветвленного нуклеиновокислотного полимера (35,137 аМ) в 50 мМ Трис-HCl, pH 7,5/10 мМ NgCl2/2 мМ спермидина/1 мМ АТР/5 мМ дитриотреитола (ДТТ). После легкого размешивания добавляли полинуклеотид-киназу T4 (0,74 ед./мкл), и реакционную смесь (полный объем 255 мкл) инкубировали в течение 2 часов при 37oC. Реакционную смесь нагревали в водяной бане до 95oC, а затем медленно в течение 60 минут охлаждали до 37oC. Смесь доводили до 2 мМ АТР, 5 мМ ДТТ, 14% полиэтиленгликоля, после чего добавляли лигазу T4 (0,21 ед./мкл), и инкубировали (полный объем смеси составлял 375 мкл) в течение 16-24 часов при 23oC. Затем добавляли NaCl до конечной концентрации 0,25 М, и для осаждения нуклеиновой кислоты добавляли 2,5 объемов этанола (95-100%).
После этого ДНК ресуспендировали в воде и подвергали второй процедуре лигирования. Смесь доводили до концентрации 1 мМ АТР, 5 мМ ДТТ, 14% полиэтиленгликоля, 50 мМ Трис-HCl, pH 7,5, 10 мМ MgCl2, 2 мМ спермидина, а затем добавляли полинуклеотид-киназу T4 (0,5 ед./мкл) и лигазу T4 (0,21 ед./мкл). Реакционную смесь (полный объем 375 мкл) инкубировали в течение 16-24 часов при 23oC и осаждали как описано выше с использованием NaCl/этанола. Осадок растворяли в буфере для загрузки геля (90% об./об формамида/1% мас./об. фиколла/0,05% мас. /об. бромофенолового синего) и подвергали электрофорезу на 5%-ном денатурирующем акриламидном геле. ДНК визуализировали путем УФ-оттенения, и полосу, содержащую лигированный продукт, вырезали из геля. ДНК подвергали электроэлюированию из геля и осаждали NaCl/этанолом, как описано выше.
Амплификатор-мультимер, полученный таким образом, тестировали в анализе на вирус гепатита C (HCV) следующим образом. Условия анализа и последовательности зондов для HCV раскрываются в одновременно рассматриваемой заявке тех же авторов рег. N 07/697326, которая вводится в настоящее описание посредством ссылки.
Стандартную кривую РНК HCV получали путем серийного разведения РНК HCV в буфере РК (2 мг/мл протеиназы K/40 мМ Трис-HCl, pH 8/8 мМ EDTA/1% ДСН/12 мкг/мл обработанной ультразвуком ДНК спермы лосося/4X SSC/5% формамида) до 500; 250; 50; 25; 12,5; или 0 тМ/150 мкл (1 тМ = 602 молекулы или 10-21 М). Зонды захвата и амплификации для HCV добавляли до конечной концентрации 0,83 фМ/мкл. HCV-специфические участки зондов-амплификаторов и зондов захвата показаны в конце описания.
Каждый зонд-амплификатор, помимо последовательностей, в основном, комплементарных HCV-последовательностям, содержал нижеуказанный 5'-удлиняющий сегмент, комплементарный сегменту мультимера-амплификатора:
AGGCATAGGACCCGTGTCTT (SEQ ID N 54).
Каждый зонд захвата, помимо последовательностей, в основном комплементарных ДНК HCV, содержал расположенную ниже последовательность, комплементарную ДНК, связанной с твердой фазой (XT1*):
CTTCTTTGGAGAAAGTGGTG (SEQ ID N 55).
В планшеты для микротитрования, подготовленные как описано выше в примере 1, добавляли реакционную смесь и 50 мкл негативной сыворотки человека. Эти планшеты инкубировали в течение ночи при 65oC. Затем планшеты убирали из инкубатора и оставляли на 10 минут при комнатной температуре для охлаждения. Лунки промывали 2X-буфером для промывки (1% ДСН/0,015 М NaCl/0,0015 М цитрата натрия). В каждую лунку добавляли 100 фМ (в 50 мкл) амплификатора мультимера, и планшеты инкубировали в течение 30 минут при 55oC. Затем в каждую лунку добавляли 100 фМ меченого щелочной фосфатазой (AP) зонда, описанного выше, и планшеты инкубировали в течение 30 минут при 55oC. После этого планшеты охлаждали в течение 10 минут при комнатной температуре, а затем промывали 2X-раствором 0,015 М NaCl/0,0015 цитрата натрия.
Был использован стимулированный ферментом диоксетан (Schaap et al., Tet. Lett. (1987) 28:1159-1162 и публ. EPA 0254051), полученный от фирмы Lumigen Inc. В каждую лунку добавляли 50 мкл Лумифоса 530 (Lumigen). При этом по лункам слегка постукивали, чтобы реагент осаждался на дно, а для того, чтобы реагент равномерно распределялся по дну лунки, эти лунки слегка вращали круговыми движениями. Затем лунки покрывали и инкубировали в течение 40 минут при 37oC.
Считывание планшетов проводили на люминометре Dynatech ML 1000. Выходные данные получали в виде полной интегральной величины количества света, продуцируемого в процессе реакции.
Полученные результаты представлены ниже. Результаты для каждого стандартного образца выражали как разность между средним значением для негативного контроля плюс два среднеквадратичных отклонения и средним значением для образца минус два среднеквадратичных отклонения (дельта). Если "дельта" больше нуля, то образец считался положительным. Эти результаты, полученные при проведении описанного анализа, показали, что чувствительность этого анализа составляет менее 12,5 тМ ДНК HCV.
тМ HC - дельта
500 - 27,24
250 - 8,07
50 - 0,81
25 - 1,13
12,5 - 0,50
0 - 0
Пример 7
Присоединение "flex extender" к зонду-амплификатору
Присоединение нуклеиновой кислоты "flex extender" осуществляли как описано ниже. Последовательность "flex extender" имеет 5'-сегмент, включающий в себя последовательность 5'-GCGTAG-3'; четыре итерации второго сегмента, который является, в основном, комплементарным последовательности, присутствующей в мультимере-амплификаторе; и третий сегмент у 3'-конца, включающий в себя последовательность, в основном комплементарную последовательности нуклеиновой кислоты, присутствующей в линкерной молекуле. Итерации второго сегмента отделены друг от друга спейсерным сегментом из шести тимидиновых остатков (T6). 3'-сегмент указанного "flex extender" служит в качестве матрицы для лигирования с зондом-амплификатором. Лигирование осуществляли с помощью "линкерной" молекулы, включающей в себя сегмент, в основном, комплементарный 3'-последовательности конца молекулы "extender"; и сегмент, в основном, комплементарный уникальной последовательности у 5'-конца зонда-амплификатора. В этом примере 3'-сегмент "flex extender" представляет собой 5'-TGAXTG-3', а сегмент с итерациями представляет собой 5'-AGGCATAGGACCCGTGTC-3' (SEQ ID N 86). Линкерная молекула имеет последовательность 5'-ATGCCTCAGTCA-3' (SEQ ID N 87).
"Flex extender" присоединяли к зонду-амплификатору. Для этого получали смесь зондов амплификатров для HBV (как описано в примере 2, всего 15625 пМ), линкера (12500 пМ) и "flex extender" (10000 пМ). К этой смеси добавляли 50 мМ Трис-HCl, pH 7,5/10 мМ MgCl2/ 2 мМ тригидрохлорида спермидина/1 мМ АТР/50 мМ ДТТ и 250 единиц киназы T4 до конечного объема 250 мкл. Смесь инкубировали в течение 1-2 часов при 37oC, а затем охлаждали до комнатной температуры. После этого добавляли 90 единиц лигазы T4, и смесь инкубировали в течение ночи при комнатной температуре.
Продукты лигирования очищали следующим образом. ДНК осаждали в NaCl/этаноле и подвергали электрофорезу на препаративном денатурирующем геле. ДНК-полосу визуализировали путем УФ-оттенения и вырезали из геля. Затем ДНК подвергали электроэлюции, ресуспендировали в 10 мМ Трис-HCl, pH 8,0/ 1 мМ EDTA и разводили до 100 фМ/мкл.
Этот удлиненный зонд был предназначен для использования в анализе на ДНК HBV, описанном в примере 2 (см. выше). Зонды-амплификаторы примера 2 были использованы вместе с "flex extender", описанным выше, а зонды захвата использовали как описано в примере 2.
К каждой лунке добавляли смесь HBV-специфических зонда "амплификатор-flex extender" и зонда захвата (25 фМ в 5 мкл/лунка). Планшеты покрывали и слегка помешивали для смешивания реагентов, после чего инкубировали в течение 30 минут при 65oC.
Затем в каждую лунку добавляли буфер для нейтрализации (0,77 М 3-(N-морфолино)пропансульфоновая кислота/1,845 М NaCl/0,185 цитрата натрия, 3 мкл/лунка). После этого планшеты покрывали и инкубировали в течение 12-18 часов при 65oC.
После выдерживания в течение еще 10 минут при комнатной температуре содержимое каждой лунки отсасывали для удаления всей жидкости, и лунки промывали 2X-буфером для промывания (200 мкл) (0,1% ДСН/0,015 М NaCl/0,0015 цитрата натрия).
Затем в каждую лунку добавляли мультимер- амплификатор (25 фМ в 40 мкл буфера для гибридизации/лунка). После этого планшеты покрывали и размешивали содержимое, а затем инкубировали в течение 30 минут при 55oC.
После выдерживания в течение еще 5-10 минут при комнатной температуре лунки промывали как описано выше.
Затем в каждую лунку добавляли зонд, меченый щелочной фосфатазой, описанный в EP 883096976 (40 мкл/лунка, 2,5 фМ/мкл). После инкубирования в течение 15 минут при 55oC и 5 минут при комнатной температуре лунки дважды промывали, как описано выше, а затем промывали 3X-буфером (200 мкл 0,015 М NaCl/0,0015 М цитрата натрия).
Был использован стимулирующий ферментом диоксетан (Schaap et al., Tet. Lett. (1987) 28:1159-1162 и публ. EPA N 0254051), полученный от фирмы Lumigen Inc. В каждую лунку добавляли 30 мкл Lumiphos 530 (Lumigen). По лункам слегка постукивали, чтобы реагент осаждался на дно, а для того, чтобы реагент равномерно распределялся по дну лунки, эти лунки слегка вращали круговыми движениями. Затем лунки покрывали и инкубировали в течение 40 минут при 37oC.
Считывание планшетов проводили на люминометре Dynatech ML 1000. Выходные данные получали в виде полной интегральной величины количества света, продуцируемого в процессе реакции.
Результаты (см. ниже) для каждого стандартного образца выражали как разность между средним значением для негативного контроля плюс два стандартных отклонения и средним значением для образца минус два стандартных отклонения (дельта). Если дельта больше нуля, то образец считался положительным. Эти результаты показали, что чувствительность анализа составляет 5-10 тМ ДНК.
тМ HB - Дельта
500 - 155,59
250 - 87,87
50 - 19,61
10 - 5,54
5 - -0,73
0 - 0о






Изобретение относится к химии нуклеиновых кислот и биохимическим анализам, точнее гребневидно-разветвленным полинуклеотидам, которые используются в качестве накопительных мультимеров в гибридизационных исследованиях нуклеиновых кислот. Описывается новый крупный гребневидно-разветвленный полинуклеотид, включающий: а) скелет полинуклеотида, имеющий 1) не менее 15-мультифункциональных нуклеотидов, каждый из которых имеет место прикрепления боковой цепи, и 2) первую единицу одноцепочечного олигонуклеотида, способную специфически связываться с первой одноцепочечной полинуклеотидной последовательностью, представляющей интерес, и б) подвесные полинуклеотидные боковые цепи, простирающиеся от указанных мультифункциональных нуклеотидов, каждая из которых включает повторения второй одноцепочечной олигонуклеотидной единицы, которая способна специфически связываться со второй одноцепочечной полинуклеотидной последовательностью, представляющей интерес, при общем количестве повторений во всех боковых цепях не менее 20. Описываются способ его получения, а также гибридизационный анализ нуклеиновых кислот. 7 с. и 18 з.п.ф-лы, 1 табл.
где R3 - водород, метил, I, Br или F;
R4 - водород или метил;
Z выбирается из группы, состоящей из: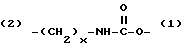
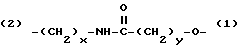
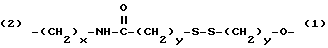
(2) - (CH2 - CH2 - O)x - (1)
(2) - (CH2)x - O - (1)
где Х и У могут быть одинаковыми или разными и являются целыми числами в диапазоне от 1 до 8 включительно.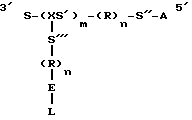
где S - первый спейсерный сегмент из по крайней мере 15 нуклеотидов;
Х - мультифункциональный нуклеотид, предоставляющий место для ветвления;
S' - спейсерный сегмент места ветвления, содержащий от 0 до примерно 15 нуклеотидов;
m - целое число, равное или более 15;
R - молекула расщепляемого линкера;
n - 0 или 1;
S'' - второй спейсерный сегмент, содержащий от 0 до примерно 10 нуклеотидов;
А - нуклеотидный сегмент, способный специфически образовывать гибрид с анализируемой нуклеиновой кислотой или с нуклеиновой кислотой, связанной с анализируемым материалом;
S''' - третий спейсерный сегмент, содержащий от 0 до 10 нуклеотидов,
Е - удлиняющий олигонуклеотидный сегмент, состоящий из 5 - 10 нуклеотидов;
L - сегмент, содержащий от 2 до 10 повторений нуклеотидной последовательности, способный к специфической гибридизации с меченым олигонуклеотидным зондом.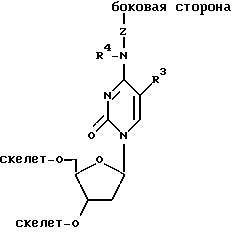
где R3 - водород, метил, I, Br или F;
R4 - водород или метил;
Z выбирается из группы, состоящей из: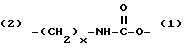
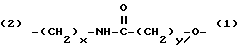
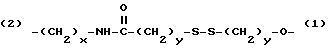
(2) - (CH2 - CH2 - O)x - (1)
(2) - (CH2)x - O - (1)
где Х и У могут быть одинаковыми или различными и являются целыми числами от 1 до 8;
S' - содержит от 5 до 10 нуклеотидов;
L - содержит от 3 до 6 повторений.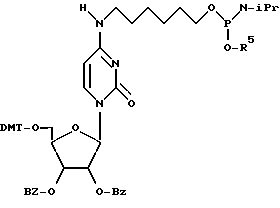
где ДМТ - диметокситритил;
Bz - бензоил;
R5 - метил, или бета-цианоэтил;
iPr - изопропил.
15. Способ по п.14, отличающийся тем, что указанная область нуклеотидного удаления удлиняется на 5 - 10 нуклеотидов.
где R2 представляет 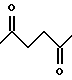
или 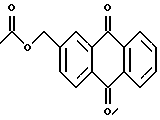
17. Способ получения крупного гребневидно разветвленного полинуклеотида, пригодного в качестве амплификационного мультимера в гибридизационном анализе нуклеиновой кислоты, отличающийся тем, что осуществляют а) синтез одноцепочечного полинуклеотидного скелета, включающего I) не менее 15 мультифункциональных нуклеотидов, каждый из которых имеет защищенную функциональную группу, служащую местом наращивания нуклеотидов боковой цепи, и II) первую одноцепочечную олигонуклеотидную единицу, способную специфически связываться с первой одноцепочечной полинуклеотидной последовательностью, представляющей интерес; б) снятие защиты с указанных функциональных групп; в) удлинение каждого из указанных мест, по крайней мере, на 5 нуклеотидов, чтобы создать сегменты места сшивания; и г) сшивание вторых одноцепочечных олигонуклеотидных единиц с сегментами области лигирования указанных вторых одноцепочечных олигонуклеотидных единиц, включающих повторения последовательности, способной связываться со вторым одноцепочечным олигонуклеотидом, представляющим интерес.
где R2 представляет 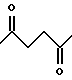
или 
19. Способ по п.18, отличающийся тем, что синтез и наращивание проводятся твердофазным способом, в котором 3'-конец скелета фиксируется к твердой фазе, и скелет включает 3'-спейсерную последовательность, состоящую из по крайней мере 15 нуклеотидов.
21. Способ по п.18, отличающийся тем, что указанные места наращивания нуклеотидов удлиняются на 5 - 10 нуклеотидов.
| US 4843122, 1989 | |||
| US 4910300. |
