Изобретение относится к молекулярной биологии, диагностике и созданию лекарственных средств.
Противовирусная химиотерапия остается одним из важных средств контроля вирусных инфекций. Несмотря на значительные успехи в этой области необходимы новые препараты, особенно для лечения инфекций, которые до настоящего времени не контролируются в должной мере. К ним относятся грипп, гепатиты, гастроэнтериты, респираторные инфекции, иммунодефицит человека и некоторые другие.
Имеется ряд стадий вирусной репродукции, позволяющих осуществлять химиотерапию, используя различия в вирусспецифических и клеточных процессах. К ним относится адсорбция вируса, проникновение вируса в клетку, "раздевание" вируса, транскрипция, трансляция и выделение вирусных частиц. Разработанные к настоящему времени противовирусные препараты относятся к пуринпиримидиновым антиметаболитам, влияющим на процесс вирусной репликации.
Наиболее актуальной проблемой при применении химиотерапии является специфичность препарата. Многие противоопухолевые и противовирусные препараты, влияющие на процесс репликации и транскрипции ДНК, практически не имеют специфичности. Использование вирусом биохимических процессов "хозяйской" клетки чрезвычайно затрудняет дискриминацию вирусспецифического и клеточного процессов. Применение "антисмысловых" олигонуклеотидов позволяет селективно воздействовать на вирусную ДНК [1, 2]. Недостатки метода, использующего "антисмысловые" олигонуклеотиды, связаны с недостаточной стабильностью образующихся олигонуклеотидных дуплексов, содержащих "антисмысловой" олигодезоксирибонуклеотид. Существует способ повышения стабилизации олигонуклеотидных дуплексов, образованных "антисмысловым" олигонуклеотидом и ДНК мишени, основанный на использовании интеркалирующих соединений, ковалентно связанных с олигонуклеотидом [3] (прототип). Метод базируется на способности плоских ароматических лигандов встраиваться между соседними парами оснований олигонуклеотидного дуплекса.
Способ имеет следующие недостатки - недостаточную стабильность олигонуклеотидных дуплексов из-за относительно низкой энергии связывания интеркалирующего агента с дуплексом. Константа диссоциации интеркалирующего агента и олигонуклеотидного дуплекса составляет 104-106 M-1 [4-6]. Кроме того, интеркалятор не обладает селективностью и стабилизирует в одинаковой мере любой, в том числе "неправильный" дуплекс, если есть возможность лиганду встроиться между парами комплементарных оснований.
Технической задачей изобретения является увеличение стабильности олигонуклеотидных дуплексов правильной структуры.
Поставленная задача решается путем использования коньюгатов олигодезоксирибонуклеотидов с лигандами, связывающимися в малой бороздке олигонуклеотидного дуплекса. При связывании в малой бороздке дуплекса остов этих лигандов образует спираль, изогеометричную B-форме ДНК. При этом специфичность связывания достигается посредством образования водородных связей между атомами азота амидных групп лигандов и атомами кислорода O2 тиминов, а также атомами N3 аденинов, выходящими в узкую бороздку олигонуклеотидного дуплекса. Поэтому различные малобороздочные лиганды связываются с олигонуклеотидным дуплексом только в определенных сайтах, характерных для этого лиганда, но не связываются с любыми, как интеркалятор, олигонуклеотидным дуплексом.
Для решения поставленной задачи получают коньюгаты малобороздочных лигандов с олигонуклеотидами следующей структуры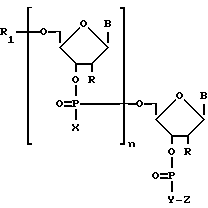
где
радикалы B - природные или модифицированные основания нуклеиновых кислот;
радикалы X - оксаанион O-, тиоанион S-, алкильная группа, алкоксильная группа, арилоксидная группа, аминоалкильная группа, аминоалкоксильная группа, тиоалкильная группа или группировка -Y-Z;
радикалы R и R1- атом водорода и/или группировка Y-Z;
радикал Y - полиметиленовое звено или аминоалкильный, аминоалкоксильный, тиоалкальный радикал;
радикал Z - соответствует малобороздочному лиганду. В качестве малобороздочных лигандов могут быть: производные нетропсина, дистамицина, дауномицина, хромомицина, митрамицина, беренила, стильбамидина, CC-1065, Хехст 33258, SN6999, лекситропсины, содержащие имидазольные пиррольные и тиазольные звенья.
n - число нуклеотидных звеньев в олигонуклеотиде,
и определяют стабильность олигонуклеотидных дуплексов, образованных этими коньюгатами.
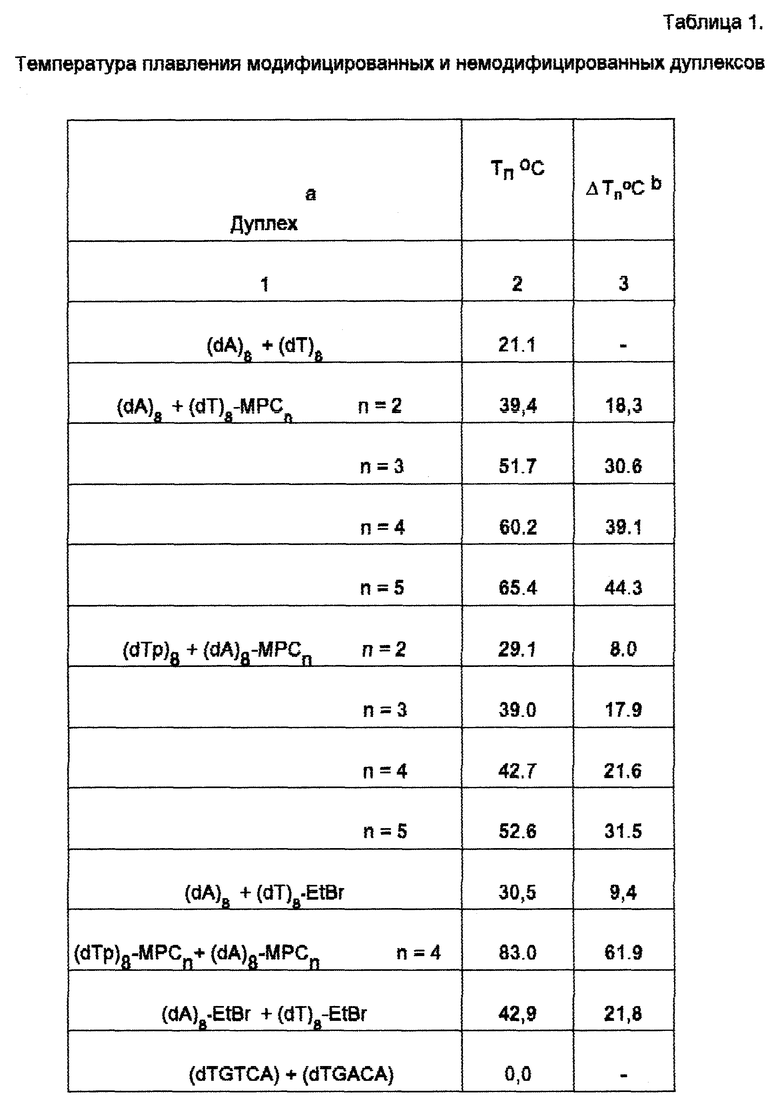
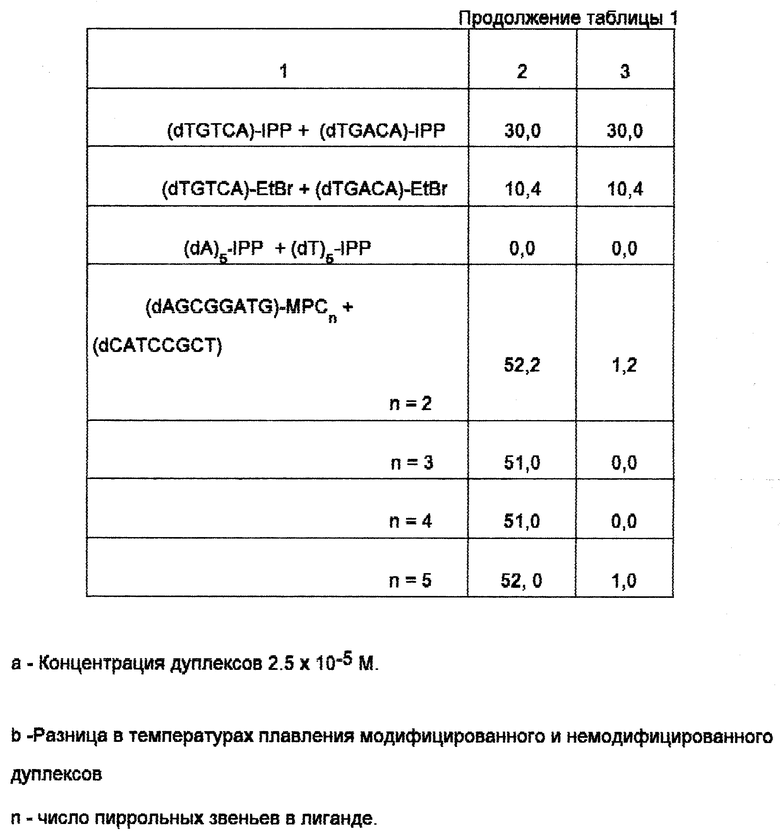
Данные о стабильности модифицированных и немодифицированных дуплексов приведены в таблице.
Константа диссоциации лигандов, связывающихся в малой бороздке олигонуклеотидных дуплексов, составляет 107-109 М-1 [7-9]. Поэтому дуплексы, образованные с использованием коньюгатов олигодезоксирибонуклеотидов и малобороздочных лигандов более стабильны по сравнению с дуплексами, образованными с использованием коньюгатов олигодезоксирибонуклеотидов и интеркалирующих соединений. Например, температура плавления дуплекса (dTp)8 + (dAp)8-MPCn, где MPCn полипиррольный малобороздочный лиганд, связывающийся с A-T-трактом, составляет 39,0, 42,7, и 52,6oC в зависимости от числа полипиррольных звеньев в лиганде. Температура плавления этого же дуплекса, содержащего коньюгат октатимидилата с этидий бромидом (наиболее эффективным из интеркалирующих соединений), составляют 30,5oC, т.е. значительно меньше. Температура плавления немодифицированного дуплекса составляет 21,1oC.
При введении в состав дуплекса двух молекул лиганда стабильность дуплекса увеличивается. Например, (Tp)8-MPC4 + (dAp)8-MPC4 плавится уже на 61,9oC выше, чем немодифицированный дуплекс. Два остатка этидий бромида в этом случае увеличивают температуру плавления дуплекса только на 21,8oC. Аналогично 1-метилимидазол-2-карбоксамидонетропсин стабилизирует дуплекс (dTGTCA) + (dTGACA). Температура плавления модифицированного дуплекса равняется 30,0oC, немодифицированного - около 0oC. Этидий бромидные группы стабилизируют этот дуплекс значительно слабее. Температура плавления дуплекса, стабилизированного интеркалирующими группами составляла 10,4oC.
Селективность связывания коньюгата олигодезоксирибонуклеотида и малобороздочного лиганда с целевым олигонуклеотидным дуплексом обусловлена, в отличие от олигодезоксирибонуклеотидов, содержащих интеркалятор, как нуклеотидной, так и лигандной частью молекулы. Так, лиганды, стабилизирующие A-T-пары, не стабилизируют G-C-содержащие сайты. Например, температура плавления олигонуклеотидного дуплекса, образованного из коньюгатата (dCATCCGCCT)-MPCn и немодифицированного олигодезоксинуклеотида (dAGCGGATG) составляет 51oC. Аналогичную температуру плавления имеет и дуплекс, полученный из немодифицированных олигонуклеотидов. 1-метилимидазол-2-карбоксамидонетросин, стабилизирующий гетерогенный дуплекс (dTGTCA) + (dTGACA), не стабилизирует дуплекс, содержащий только A-T-пары. Температуры плавления дуплексов (dA)5 + (dT)5 и (dA)5•IPP + (dT)5•IPP практически одинаковы.
Существенным отличием в предложенном способе является использование ковалентно связанного лиганда, связывающегося в малой бороздке олигонуклеотидного дуплекса.
Коньюгаты олигодезоксирибонуклеотидов с малобороздочными лигандами в литературе не описаны и впервые получены в настоящей работе.
Полученные коньюгаты могут найти применение в гибридизационной диагностике для повышения порога чувствительности метода, в медицине - в качестве антигенных и "антисмысловых" препаратов.
Сущность изобретния иллюстрируется следующими схемами:
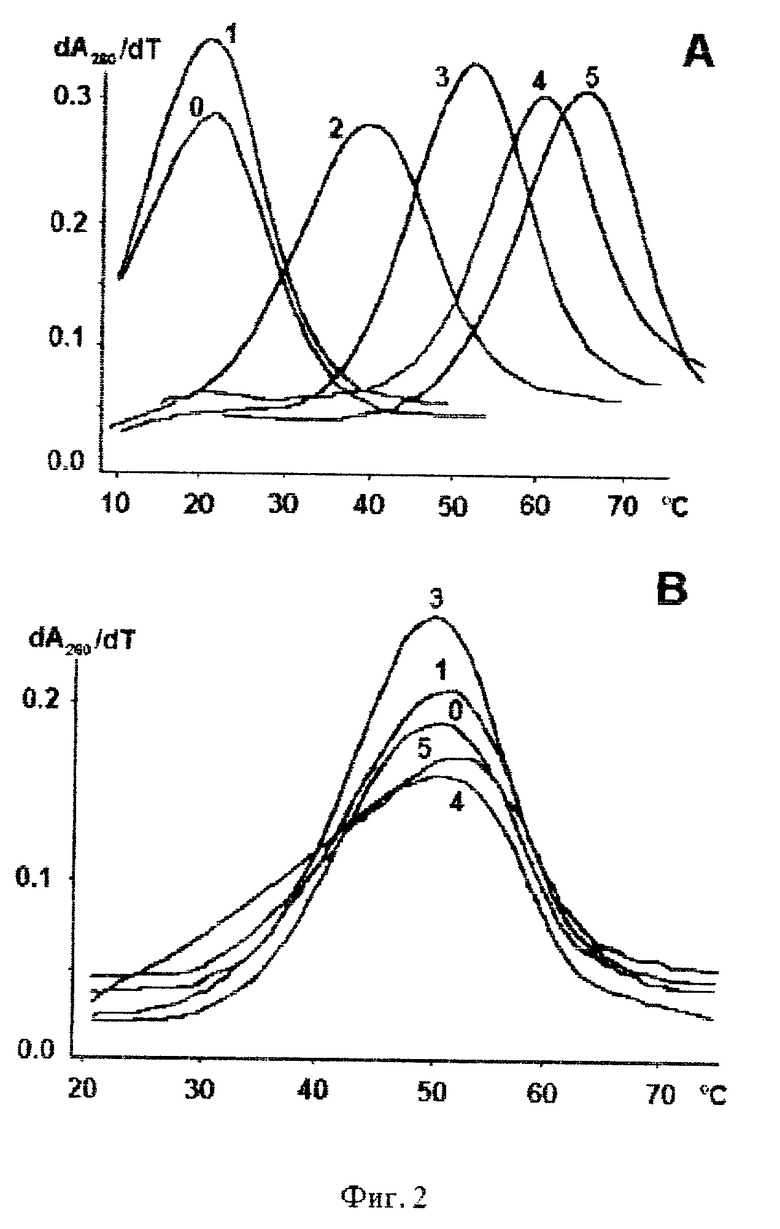
фиг. 1 Структура полипиррольных (MPC4) и имидазолпиррольного (IPP) малобороздочных лигандов.
фиг.2A Дифференциальные кривые плавления при 260 нм дуплекса (dTp)8-MPCn + (dAp)8.
фиг. 2B Профиль первой производной кривой плавления при 260 нм дуплекса (dAGCGGATG) + (dCATCCGCT)-MPCn
Для лучшего понимания сущности предлагаемого технического решения ниже следуют примеры его осуществления.
Пример 1. Синтез 2-[4-(Фенилазо)бензилтио]этил 5-[(третбутилокси)карбоксамидо]пентилкарбоксилата
К 4,16 г (18 ммоль) 6-[(трет-бутилокси)карбоксамидо]гексановой кислоты добавляют 4,08 г (15 ммоль) Pse-OH в 25 мл ДМФА, реакционную смесь охлаждают льдом и добавляют 3,71 г (18 ммоль) ДЦК и 1,83 г (15 ммоль) ДМАР, перемешивают 2 ч при охлаждении, затем 4 ч при комнатной температуре, оставляют на ночь при 6oC. Затем упариванием с бутилацетатом удаляют ДМФА, добавляют этилацетат (150 мл) и экстрагируют 30 мл 0,7 н. HCI и 100 мл 5% NaHCO3, затем водой, сушат над Na2SO4. Экстракт упаривают, осадок промывают эфиром. Выход 6,91 г (89%) г.
ПМР-спектр (CDCI3, δ, м.д.): 7,91 (m, 4H), 7,52 (m, 5H), 4,48 (t, 2H), 4,34 (s, 2H), 3,20 (t, 2H), 3,08 (m, 2H), 2,35 (t, 2H), 1,64-1,2 (m, 7H), 1,41 (s, 9H).
Пример 2. Синтез 2-[4-(Фенилазо)бензилтио]этил 5-[1-метил- 4-[1-метил-4-(трет-бутилокси)пиррол-2-карбоксамидо]пиррол-2-карбоксамидо] пентилкарбоксилата
К 728 мг (1,5 ммоля) 2-[4-(Фенилазо)бензилтио]этил 5-[(трет-бутилокси)карбоксамидо)пентилкарбоксилата при 0oC добавляют 5 мл охлажденной CF3COOH, выдерживают 20 минут (контроль TCX SiO2, бутилацетат), затем несколько раз упаривают с CHCl3, растворяют в 15 мл CH2Cl2, добавляют 590 мг (1,65 ммоль) 1,2,3-бензотриазолил-1 1-метил-4-(трет-бутилокси)карбоксамидопиррол-2-карбоксилата. Реакционную смесь титруют NEt3 до полного исчезновения аминопиррола. По окончании реакционную смесь разбавляют CHCl3, промывают 2х20 мл 5% NaHCO3 и 2х20 мл H2O, сушат над Na2SO4, упаривают до объема 3 мл и хроматографируют на 50 мл SiO2 (элюент CHCl3). Выход 0,88 г (91,8 %). ПМР-спектр (CDCl3,δ, м.д.): 7,88 (m, 4H), 7,46 (m, 5H), 6,74 (s, 1H), 6,38 (s, 1H), 6,26 (s, 1H), 5,87 (t, 1H), 4,18 (t, 2H, J = 6 Hz), 3,82 (s, 3H), 3,79 (s, 2H) 3,3 (m,2H), 2,63 (t, 2H J=6 Hz), 2,30 (t, 2H J=6 Hz), 1,64-1,2 (m, 6H), 1,46 (s, 9H).
Пример 3. Синтез 2-[4-(Фенилазо)бензилтио]этил 5-[1-метил-4-[1-метил-4-[1-метил-4-(трет-бутилокси)пиррол-2-карбоксамидо] пиррол-2-карбоксамидо] пиррол-2-карбоксамидо]пентилкарбоксилата
Аминопиррол, полученный из 2432 мг (4 ммоля) 2-[4(Фенилазо)бензилтио]- этил 5-[1-метил-4-[1-метил-4-(трет-бутилокси)пиррол-2-карбоксамидо] пиррол- 2-карбоксамидо]пентилкарбоксилата (8 мл CH2Cl2, 4 мл CF3COOH, 60 мин, 20oC), растворяют в 10 мл CH2Cl2, добавляют 1430 мг (4 ммоль) 1,2,3-бензотриазо-лил-1 1-метил-4-(трет-бутилокси)карбоксамидопиррол-2-карбоксилата и титруют NEt3 до исчезновения аминопиррола. По окончании реакционную смесь разбавляют CHCl3, промывают 2х20 мл 5% NaHCO3 и 2х20 мл H2O, сушат над Na2SO4, упаривают до объема 3 мл и хроматографируют на 50 мл SiO2(элюент CHCl3). Выход 1,95 г (66,8 %). ПМР-спектр (CDCl3, δ, м.д.): 7,87 (m, 4H), 7,46 (m, 5H), 7,04 (d, 1H, J=1,5 Hz), 6,77 (br s, 1H), 6,52 (br s, 1H), 6,50 (d, 1H, J=1,5 Hz), 6,31 (br s, 1H), 5,95 (t, 1H), 4,19 (t, 2H, J = 6 Hz), 3,85 (s, 6H), 3,78 (s, 2H), 3,32 (m, 2H), 2,64 (t, 2H, J = 6 Hz), 2,31 (t, 2H, J = 6 Hz), 1,64-1,2 (m, 6H), 1,48 (s, 9H).
Пример 4. Синтез 2-[4-(Фенилазо)бензилтио]этил 5-[1-метил-4-[1-метил- 4-[1-метил-4-[1-метил-4-(трет-бутилокси)пиррол-2-карбоксамидо]пиррол- 2-карбоксамидо]пиррол-2-карбоксамидо]пиррол-2-карбоксамидо]пентилкарбоксилата
Аминопиррол, полученный из 1900 мг (2,6 ммоля) 2-[4-(Фенилазо)бензилтио] этил 5-[1-метил-4-[1-метил-4-[1-метил- 4-(трет-бутилокси)пиррол-2-карбоксамидо]пиррол-2-карбоксамидо]пиррол- 2-карбоксамидо]пентилкарбоксилата (6 мл CH2Cl2, 3 мл CF3COOH, 60 мин, 20oC), растворяют в 5 мл CH2Cl2, добавляют 2,0 мл NEt3 и 1395 мг (3,9 ммоля) 1,2,3-бензотриазолил-1 1-метил-4-(трет-бутилокси)карбоксамидопиррол- 2-карбоксилата и выдерживают при 20oC 1 ч. По окончании реакционную смесь разбавляют CHCl3, промывают 2х20 мл 5% NaHCO3 и 2х20 мл H2O, сушат над Na2SO4, упаривают до объема 3 мл и хроматографируют на 50 мл SiO2, используя градиент метанола от 0 до 1,5% в CHCl3. Выход 1,56 г (70,5%) TCX SiO2 CHCl3 - CH3OH 20:1. Rf 0,3. ПМР-спектр (CDCl3, δ, м.д.): 7,87 (m, 4H), 7,68 (br s, 1H), 7,60 (br s, 1H), 7,46 (m, 5H), 7,08 (br s, 2H), 6,78 (br s, 1H), 6,56 (d, 1H, J=1,5 Hz), 6,60 (br s, 1H), 6,55 (d, 1H, J= 1,5 Hz), 6,03 (t, 1H), 4,18 (t, 2H, J=6 Hz), 3,86 (m, 9H), 3,78 (s, 2H), 3,32 (m, 2H), 2,63 (t, 2H, J=6 Hz), 2,30 (t, 2H, J=6 Hz), 1,64-1,2 (m, 6H), 1,48 (s, 9H).
Пример 5. Синтез 2-[4-(Фенилазо)бензилтио]этил 5-[1-метил-4-[1-метил- 4-[1-метил-4-[1-метил-4-(трет-бутилокси)карбоксамидо] пиррол-2-карбоксамидо] пиррол-2-карбоксамидо]пиррол-2-карбоксамидо]пентилкарбоксилата
Аминопиррол, полученный из 320 мг (0,32 ммоля) 2-[4-(Фенилазо)бензилтио] этил 5-[[1-метил-4-[1-метил-4-[1-метил- 4-(трет-бутилокси)карбоксамидо]пиррол-2-карбоксамидо] пиррол- 2-карбоксамидо] пентилкарбоксилата (5 мл CH2Cl2, 2,5 мл CF3COOH, 60 мин, 20oC), растворяют в 1 мл CH2Cl2, добавляют 0,1 мл NEt3 и 110 мг (0,32 ммоля) 1,2,3-бензотриазолил-1 1-метил-4-(трет-бутилокси)карбоксамидопиррол- 2-карбоксилата и перемешивают при 20oC 1,5 ч. По окончании реакционную смесь разбавляют CHCl3, промывают 2х20 мл 5% NaHCO3 и 2х20 мл H2O, сушат над Na2SO4, упаривают досуха. Выход 250 мг (80%). ПМР-спектр (CDCl3, δ, м.д.): 8,17 (br s, 1H), 7,98 (br s), 7,96 (br s, 1H), 7,85 (m, 4H), 7,44 (m, 5H), 7,09 (br s, 2H), 7,02 (s, 1H), 6,78 (br s, 1H), 6,74 (br s, 1H), 6,66 (s, 1H), 6,58 (s, 3H), 6,29 (t, 1H), 4,18 (d, 2H, J=6 Hz), 3,78 (m, 14H), 3,28 (m, 2H), 2,60 (d, 2H, J=6 Hz), 2,26 (t, 2H, J=6 Hz), 1,64-1,2 (m, 6H), 1,48 (s, 9H).
Пример 6. Синтез 2-[4-(Фенилазо)бензилтио]этил 5-[1-метил-4-[1-метил- 4-[1-метил-4-[1-метил-4-(трет-бутилокси)карбоксамидо]пиррол- 2-карбоксамидо] пиррол-2-карбоксамидо] пиррол-2-карбоксамидо] пиррол- 2-карбоксамидо]пентилкарбоксилата
Аминопиррол, полученный из 650 мг (0,67 ммоля) 2-[4-(Фенилазо)бензилтио] этил 5-[1-метил-4-[1-метил-4-[1-метил- 4-[1-метил-4-(трет-бутилокси)карбоксамидо] пиррол-2-карбоксамидо] пиррол- 2-карбоксамидо]пиррол-2-карбоксамидо] пентилкарбоксилата (10 мл CH2Cl2, 5 мл CF3COOH, 90 мин, 20oC), растворяют в 1 мл ДМФА, добавляют 0,18 мл NEt3 и 240 мг (0,67 ммоля) 1,2,3-бензотриазолил-1 1-метил-4-(трет-бутилокси)карбоксамидопиррол- 2-карбоксилата и перемешивают при 20oC 3 ч. По окончании реакционную смесь упаривают досуха, растворяют в 2,5% ДМФА в CHCl3 и хроматографируют на 50 мл SiO2 используя градиент метанола от 0 до 2,5% в CHCl3-ДМФА 40:1. Выход 670 мг (45%). ПМР-спектр (CDCl3, δ, м.д.): 8,35 (s, 1H), 8,20 (br s, 1H), 8,12 (br s, 2H), 8,02 (br s, 1H), 7,98 (br s, 1H), 7,86 (m, 4H), 7,45 (m, 5H), 7,07 (br s, 2H), 7,00 (s, 1H), 6,80 (br s, 1H), 6,75 (br s, 1H), 6,62 (s, 1H), 6,58 (s, 3H), 6,30 (t, 1H), 4,20 (t, 2H, J=6 Hz), 3,80 (s, 15H), 3,78 (s, 2H), 3,30 (m, 2H), 2,58 (t, 2H, J=6 Hz), 2,26 (t, 2H, J=6 Hz), 1,64-1,2 (m, 6H), 1,48 (s, 9H).
Пример 7. Синтез этил 5-[1-метил-4-нитропиррол-2- карбоксамидо]пентилкарбоксилата
Суспензию 1-метил-4-нитропиррол-2-карбоновой кислоты 0,5 г (0,29 ммоль) в 3 мл SO2Cl2 кипятят 3 ч, затем упаривают в вакууме досуха и дважды с хлористым метиленом. Полученный хлорангидрид растворяют в 5 мл хлористого метилена, добавляют хлоргидрат 6-аминогексановой кислоты 0,65 г (3,3 ммоль). К реакционной смеси, охлажденной до 0oC, прибавляют 1,2 мл этилдиизопропиламина в 3 мл сухого ацетонитрила. Реакционную массу перемешивают при комнатной температуре 1 ч, затем упаривают досуха, экстрагируют 1% HCl (3х60 мл), водой (1х60 мл), сушат Na2SO4. Остаток после концентрирования хроматографируют на 140 мл SiO2 (элюент CHCl3). Получено 0,783 г (85,5%). ПМР-спектр (CD3COCD3, δ, м.д.): 7,87 (d, 1H, J=1,5 Hz), 7,69 (br s, 1H), 7,28 (d, 1H, J= 1,5 Hz), 4,05 (q, 2H, J=6 Hz), 4,00 (s, 3H), 3,33 (q, 2H, J=6 Hz), 2,28 (t, 2H, J=6 Hz), 1,59-1,41 (m, 6H), 1,18 (t, 3H, J=6 Hz).
Пример 8. Синтез этил 5-[[1-метил-4-[1-метил-4-нитропиррол- 2-карбоксамидо]пиррол-2-карбоксамидо]пентилкарбоксилата
Суспензию 350 мг (0,28 ммоль) 1-метил-4-нитропиррол-2-карбоновой кислоты в 2 мл SO2Cl2 кипятят 3 ч, затем упаривают в вакууме досуха и дважды с хлористым метиленом. Полученный хлорангидрид растворяют в 5 мл сухого ацетонитрила.
716 мг (2,3 ммоля) этил 5-[1-метил-4-нитропиррол-2-карбоксамидо] пентилкарбоксилата в смеси 9 мл 95% этанола и 5 мл циклогексена кипятят в присутствии 720 мг 10% Pd/C в течение 9 часов. Катализатор отфильтровывают, промывают этанолом, фильтрат упаривают досуха, затем дважды с абс. ацетонитрилом, растворяют в 4 мл сухого ацетонитрила с добавлением 0,8 мл диизопропилэтиламина. К полученному раствору на холоду прибавляют раствор хлорангидрида, реакционную смесь выдерживают 30 мин при комнатной температуре, упаривают досуха, растворяют в CHCl3 (150 мл), экстрагируют водой (2х30 мл) и хроматографируют на 140 мл SiO2 (CHCl3). Получено 0,76 г (85,2%). ПМР-спектр (CD3COCD3, δ, м. д.): 7,89 (d, 1H, J=2 Hz), 7,44 (d, 1H, J=2 Hz), 7,23 (d, 1H, J=2 Hz), 6,82 (d, 1H, J=2 Hz), 4,14 (q, 2H, J=6 Hz), 4,02 (s, 3H), 3,89 (s, 3H), 3,33 (q, 2H, J=6 Hz), 2,36 (t, 2H, J=6 Hz), 1,59-1,41 (m, 6H), 1,26 (t, 3H, J=6 Hz).
Пример 9. Синтез этил-5-[1-метил-4-[1-метил-4-(1-метилимидазол- 2-карбоксамидо)пиррол-2-карбоксамидо]пиррол-2-карбоксамидо]пентилкарбоксилата
433 мг (1 ммоль) этил 5-[[1-метил-4-[1-метил-4-нитропиррол- 2-карбоксамидо] пиррол-2-карбоксамидо]пентилкарбоксилата в смеси 10 мл 95% этанола и 5 мл циклогексена кипятят в присутствии 700 мг 10% Pd/C в течение 9 часов. Катализатор отфильтровывают, промывают этанолом, фильтрат упаривают досуха, затем дважды с абс. ацетонитрилом. Одновременно смесь 188 мг (2 ммоль) 1-метилимидазол-2-карбоновой кислоты, 300 мг (2,22 ммоль) N-гидроксибензотриазола и 430 мг (2,22 ммоль) дициклокарбодиимида в 12 мл сухого ДМФА перемешивают при комнатной температуре в течение 6 ч. Затем осадок дициклогексилмочевины отфильтровывают и фильтрат добавляют к аминопроизводному. Реакционную смесь перемешивают 20 ч при комнатной температуре и упаривают досуха. Остаток растворяют в 100 мл хлороформа и экстрагируют водой (4х30 мл), концентрируют и хроматографируют на 140 мл SiO2 используя градиент метанола от 0 до 1,5% в CHCl3. Выход 300 мг (58,7%). ПМР-спектр (CD3COCD3, δ, м.д.): 9,56 (s, 1H), 9,31 (s, 1H), 7,34 (d, 1H, J=2 Hz), 7,29 (s, 1H), 7,24 (d, 1H, J=1 Hz), 7,20 (d, 1H, J=2 Hz), 7,06 (d, 1H, J=2 Hz), 6,97 (d, 1H, J=1 Hz), 6,82 (s, 1H, J=2 Hz), 4,07 (s, 3H), 4,05 (q, 2H, J=6 Hz), 3,95 (s, 3H), 3,88 (s, 3H), 3,29 (q, 2H, J=6 Hz), 2,28 (t, 2H, J=6 Hz), 1,59-1,41 (m, 6H), 1,19 (t, 3H, J=6 Hz).
Пример 10. Синтез 6-[1-метил-4-[1-метил-4-(1-метилимидазол- 2-карбоксамидо)пиррол-2-карбоксамидо]пиррол-2-карбоксамидо]гексанкарбоновой кислоты
К суспензии 223 мг (0,44 ммоль) этил 5-[1-метил-4-[1-метил-4-(1- метилимидазол-2-карбоксамидо)пиррол-2-карбоксамидо] пиррол- 2-карбоксамидо] пентилкарбоксилата в 1 мл этилового спирта добавляют 0,5 мл 10% NaOH и перемешивают до полного этилового эфира. Реакционную смесь нейтрализуют разб. HCl, полученный осадок карбоновой кислоты отфильтровывают и сушат в вакууме. Выход 180 мг (85,4%). ПМР-спектр (DMFA-d7): 10,27 (s, 1H), 10,08 (s, 1H), 7,40 (m, 2H), 7,32 (m, 2H), 7,04 (s, 2H), 7,00 (dr s, 1H), 4,09 (s, 3H), 3,97 (s, 3H), 3,91 (s, 3H), 3,42 (m, 2H), 2,59 (t, 2H), 1,99-1,84 (m, 6H).
Пример 11. Синтез коньюгатов олигонуклеотид-пептид
10 о. е. олигодезоксирибонуклеотида, содержащего на 3'-конце фосфатную группу, растворяют в 50 мкл дистиллированной воды. К раствору прибавляют 8 мкл 8% водного раствора гексадецилтриметиламмоний бромида, полученную суспензию центрифугируют, осадок сушат в течение ночи над SiO2 в вакууме. К высушенной цетавлоновой соли олигодезоксирибонуклеотида прибавляют 10 мг 2,2'-дипиридиндисульфида, 10 мг трифенилфосфина, 10 мг 4,4'-диметиламинопиридина, 2 мг соответствующего пептида и 100 мкл ДМФА. Реакционную смесь перемешивают в течение 20 мин при 20oC, затем прибавляют 1,4 мл 2% раствора LiClO4 в ацетоне, перемешивают и центрифугируют. Супернатант отделяют, осадок суспендируют в 1,5 мл 2% LiClO4 и снова центрифугируют. Осадок растворяют в 1 мл воды и хроматографируют на хроматографе Altex (США) на колонке с сорбентом LiChrosorb RP-18 (Мерк, Германия, 4,6х250 мм), используя градиент ацетонитрила от 5 до 60% в 0,05 М LiClO4. Фракцию, содержащую целевой коньюгат упаривают до объема 50 мкл и разбавляют 1,5 мл ацетона, суспензию центрифугируют. Осадок промывают ацетоном и сушат в вакууме. Выход целевого коньюгата 30-60%.
Пример 12. Определение термодинамической стабильности олигонуклеотидных дуплексов.
Оптические кривые плавления регистрируют на специальной установке, созданной на базе спектрофотометрического УФ-детектора жидкостного хроматографа "Миллихром". Установка имеет термостатированную кювету объемом 2 мкл. Температуру кюветы поддерживают за счет прокачивания теплоносителя жидкостным термостатом через кюветодержатель. Температуру измеряют медьконстантановой термопарой, откалиброванной с абсолютной погрешностью ±0,1oC, и подключенной к цифровому вольтметру Щ-1516. Данные накапливают и обрабатывают с помощью ПВЭМ. Каждую экспериментальную точку получают интегрированием сигнала за 10 с. Каждая кривая плавления содержит 500-600 точек. Данные сглаживают центральной линейной интерполяцией в интервале 2oC, делают поправку на тепловое расширение воды. Скорость нагрева растворов во всех случаях составляла 0,6-0,7oC/мин. Дифференциальные кривые плавления при 260 нм дуплексов (dTp)8-MPCn+(dAp)8, (dAGCGGATG)+(dCATCCGCT)-MPCn, приведены на фиг. 2.
Температура плавления модифицированных и немодифицированных дуплексов приведена в таблице.
Литература
1. Zamecnik P.C., Stephenson M.L. Proc. Natl. Acad. Sci. USA, 1978, v. 75, p. 280-294.
2. Izant J.G., Weitraub H. Cell, 1984, v. 36, p. 1007-1015.
3. Патент Франции N 2586704, C 12 Q, 1/68, 1987.
4. Isaacs S.T., Shen C.K., Hearst J.E., Rapoport H. Biochemistry, 1977, v. 16, p. 1058.
5. Reinhard C.G., Krugh T.R. Biochemistry, 198, v. 17, p. 4845.
6. Hansen J.B., Koch T., Buchardt O., Nielsen P.E., Wirth M., Norden B. Biochemistry, 1983, v. 22, p. 4878.
7. Luck G., Triebel H., Waring M., Zimmer C. Nucleic Acids Res., 1974, v. 1, p. 503.
8. Wartell R.M., Larson J.E., Wells R.D., J.Biol. Chem., 1974, v. 249, p. 6719.
9. Marky L. A., Breslauer K.J. Proc. Natl. Acad. Sci. U.S.A., 1987, v. 84, p. 4359.
| название | год | авторы | номер документа |
|---|---|---|---|
| 2`-АМИНО-2`-ДЕЗОКСИНУКЛЕОЗИДЫ - ИНГИБИТОРЫ РЕПРОДУКЦИИ ВИРУСОВ КОРИ И МАРБУРГ | 2003 |
|
RU2264409C2 |
| АНТАГОНИСТЫ CXCR7 | 2013 |
|
RU2649004C2 |
| НОВОЕ ПРОИЗВОДНОЕ ПИРРОЛА, ИМЕЮЩЕЕ В КАЧЕСТВЕ ЗАМЕСТИТЕЛЕЙ УРЕИДОГРУППУ, АМИНОКАРБОНИЛЬНУЮ ГРУППУ И БИЦИКЛИЧЕСКУЮ ГРУППУ, У КОТОРЫХ МОГУТ БЫТЬ ЗАМЕСТИТЕЛИ | 2009 |
|
RU2500669C2 |
| 6-СУЛЬФАМОИЛХИНОЛИН-4-КАРБОНОВЫЕ КИСЛОТЫ, ИХ ПРОИЗВОДНЫЕ И КОМБИНАТОРНАЯ БИБЛИОТЕКА | 2003 |
|
RU2229475C1 |
| БИЦИКЛИЧЕСКИЕ АМИНЫ В КАЧЕСТВЕ НОВЫХ ИНГИБИТОРОВ JAK-КИНАЗЫ | 2018 |
|
RU2764980C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ БЕНЗОДИАЗЕПИНА | 2019 |
|
RU2799340C2 |
| ПРОИЗВОДНЫЕ ЦИКЛИЧЕСКИХ АМИНОВ В КАЧЕСТВЕ АНТАГОНИСТОВ РЕЦЕПТОРА ЕР4 | 2011 |
|
RU2565596C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ БЕНЗОДИАЗЕПИНА | 2010 |
|
RU2545080C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ БЕНЗОДИАЗЕПИНА | 2010 |
|
RU2683325C2 |
| ИНГИБИТОРЫ GCN2 И ИХ ПРИМЕНЕНИЕ | 2019 |
|
RU2811408C2 |
Изобретение относится к молекулярной биологии, диагностике и созданию лекарственных средств. Для стабилизации олигонуклеотидных дуплексов предложено использовать взаимодействие коньюгатов олигодезоксирибонуклеотидов с малобороздочными лигандами. Стабилизированные дуплексы могут быть использованы в гибридизационной диагностике для повышения порога чувствительности метода, а также в медицине - в качестве антигенных и "антисмысловых" препаратов. 1 з.п.ф-лы, 2 ил., 1 табл.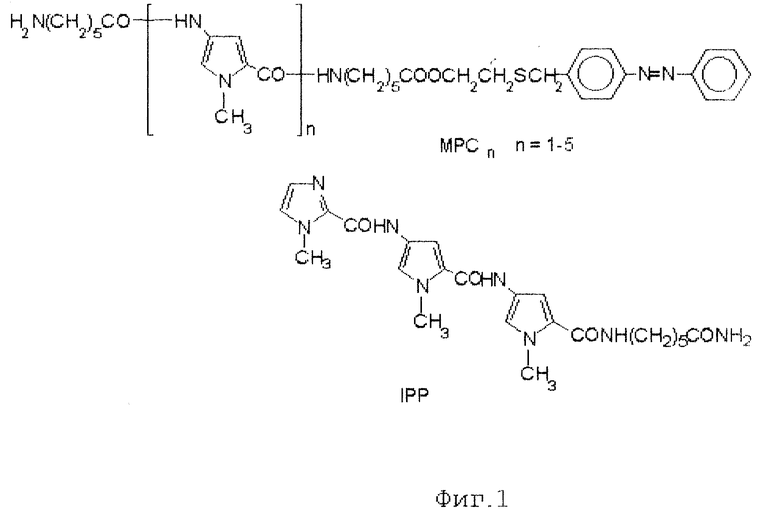
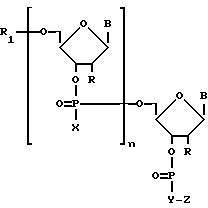
где радикалы В - природные или модифицированные основания нуклеиновых кислот;
радикалы Х - оксаанион О-, тиоанион S-, алкильная группа, алкоксильная группа, арилоксидная группа, аминоалкильная группа, аминоалкоксильная группа, тиоалкильная группа или группировка -Y-Z;
радикалы R и R1 - атом водорода и/или группировка Y - Z;
радикал Y - полиметиленовое звено или аминоалкильный, аминоалкоксильный, тиоалкильный радикал;
радикал Z - соответствует малобороздочному лиганду.
| СПОСОБ ПОЛУЧЕНИЯ ХЛЕБНОГО КВАСА | 2015 |
|
RU2586704C1 |
| Способ получения динуклеотидов | 1977 |
|
SU730691A1 |
| Способ получения динуклеотидов | 1983 |
|
SU1404512A1 |

