Изобретение относится к содержащим серебро катализаторам, подходящим для экспонирования олефинов, не содержащих аллильный водород, в частности этилена, и к применению катализаторов для получения этиленоксида. Катализаторы получают при использовании уникального носителя на основе α- оксида алюминия.
Катализаторы для получения этиленоксида из этилена и молекулярного кислорода в большинстве случае представляют серебряные катализаторы на носителе. Такие катализаторы обычно промотируют щелочными металлами. Использование незначительных количеств щелочных металлов калия, рубидия и цезия в качестве пригодных промоторов в серебряных катализаторах на носителе упоминалось в патенте США N 3962136, опубликованном 8 июня 1976, патенте США N 4010115, опубликованном 1 марта 1977. Использование других со-промоторов, таких как рений, или совместно с серой, молибденом, вольфрамом и хромом описано в патенте США N 4766105, опубл. 23 августа 1988 г, и патенте США N 4808738, опубл. 28 февраля 1989 г. В патенте США N 4908343, опубл. 13 марта 1990 г. , представлен серебряный катализатор на носителе, содержащий смесь цезиевой соли и одну или несколько солей щелочного и щелочноземельного металла.
В патенте США N 4897498, опубл. 30 января 1990 г, описано применение катализаторов на основе серебра, промотированных щелочным металлом, нанесенных на носитель, при эпоксидировании олефинов, не имеющих аллильного водорода.
Использование носителей катализаторов на основе оксида алюминия было ранее описано в ряде патентов, например, таких как: патент США N 5100859, опубл. 31 марта 1992 г., патент США N 5055442, опубл. 8 октября 1991, патент США N 5037794, опубл. 6 августа 1991 и патент США N 4874739, опубл. 17 октября 1989 г. Эти носители из оксида алюминия широко применяются в области катализа, и они являются в особенности пригодными, когда основным оксидом алюминия является α- -оксид алюминия, и при применении желательно сопротивление истиранию.
В патенте США N 5063195, опубл. в ноябре 1991 г., описаны катализаторы для получения этиленоксида на носителе, который приготовлен из тригидрата оксида алюминия, бемита (который также представляет гидратированный оксид алюминия), фторида и наполнителей.
Данное изобретение относится к катализатору, пригодному для эпоксидирования этилена с кислородом в паровой фазе, который содержит каталитически эффективное количество серебра и промотирующее количество щелочного металла, нанесенные на носитель на основе α- оксида алюминия, имеющий прочность на раздавливание по крайней мере 2,3 кг и определенную объемную плотность по крайней мере 0,48 кг/л, который включает первый и второй компоненты α- оксида алюминия, при этом первый компонент α- оксида алюминия в форме частиц, имеющих средний размер кристаллитов от 0,4 до 4 мкм, обеспечивает от 95% до 40% от общего веса α- оксида алюминия в носителе, и второй компонент α- оксида алюминия, полученный in situ золь-гелевым процессом, дополняет остальное количество α- оксида алюминия в носителе.
Было найдено, что катализаторы, имеющие такой уникальный носитель из оксида алюминия, обладают улучшенной селективностью и/или активностью по сравнению с катализаторами, содержащими обычные носители из оксида алюминия. Эти катализаторы также обладают улучшенной стабильностью селективности и/или стабильностью активности.
Далее будут подробно описаны носитель, катализатор, содержащий этот носитель, и применение катализатора.
Носитель катализатора в настоящем изобретении представляет новый носитель катализатора на основе α- оксида алюминия, имеющий прочность на раздавливание (которая измерена на динамометре Комптона, модель 50-ОР) по крайней мере 2,3 кг и определенную объемную плотность (которая измерена ASTM D-4699-87 (Американским стандартным методом испытаний), модифицированным путем использования цилиндра с внутренним диаметром 9,52 см и длиной 45,7 см) по крайней мере 0,48 кг/л, предпочтительно по крайней мере 0,56 кг/л и более предпочтительно по крайней мере 0,6 кг/л, который содержит первый и второй компоненты α- оксида алюминия, при этом первый компонент α- оксида алюминия в форме частиц, имеющих средний размер кристаллита от 0,4 до 4 мкм, обеспечивает от 95% до 40%, предпочтительно от 95% до 65% от общего веса α- оксида алюминия в носителе, и второй компонент α- оксида алюминия, полученный in situ зольгелевым процессом, дополняет остальное количество α- оксида алюминия в носителе.
Использован здесь термин "зольгелевый процесс" относится к процессу, который включает нагревание золя и/или геля оксида алюминия (т.е. гидратированного оксида алюминия) до температуры, при которой происходит превращение по крайней мере части золя и/или геля оксида алюминия в оксид алюминия, имеющий корундовую кристаллическую структуру (т.е. гексагональную структуру с плотной упаковкой).
Превращение обычно осуществляют при температуре по крайней мере 400oC, предпочтительно выше 1100 oC и более предпочтительно от 1100 до 1500oC.
Носитель катализатора получают способом, который включает:
а) приготовление смеси, содержащей:
(I) по крайней мере, дин компонент α- оксида алюминия со средним размером частицы 3 до 8 мкм и средним размером кристаллита от 0.4 до 4 мкм;
(II) гидратированный предшественник α- оксида алюминия (золь и/или гель) в количестве, достаточном для обеспечения от 5% по весу до 60% по весу от общего веса α- оксида алюминия в носителе катализатора,
(III) выгорающий материал в количестве от 5% до 40% относительно веса α- оксида алюминия, и
(IV) воду в количестве, достаточном для экструдирования вышеприведенной
(b) экструдирование смеси в формованные изделия требуемого вида и
(c) обжиг для превращения предшественника α- оксида алюминия в α- оксид алюминия с получением носителя для катализатора, в котором частицы α- оксида алюминия со средним размером частицы от 3 до 8 мкм и средним размером кристаллита от 0.4 до 4 мкм диспергированы в матрице α- оксида алюминия, полученной из материала предшественника.
Носитель катализатора может состоять из нескольких компонентов α- оксида алюминия, выбранных для обеспечения необходимых физических свойств, включая пористость, объем пор, прочность на раздавливание и т.п. Часто предпочтительным является сочетание двух различных α- оксидов алюминия, при этом первый компонент с частицами большого размера смешивают со вторым компонентом с частицами малого размера при весовом соотношении от 10:90 до 90:10. Первый обычно включает от 10% до 90%, предпочтительно от 40% до 20% по весу первого исходного α- оксида алюминия, и второй компонент обычно включает от 10 до 90%, предпочтительно от 20% до 60% по весу исходного α- оксида алюминия. Целью вышеуказанного является получение удельной поверхности в конечном обожженном носителе от 0.4 до 5 м2/г. Используемый здесь термин "удельная поверхность" означает удельную поверхность, измеренную по методу БЭТа (Брунауэроа, Эммета, Теллера) с использованием в качестве поглощенного газа азота или криптона. Удельная поверхность в конечном носителе отчасти меньше, чем у частиц свободного оксида алюминия. Таким образом, подходящая смесь может включать, например, два вида части α- оксида алюминия, при этом первый имеет удельную поверхность от 0,9 до 1,4 и предпочтительно 1 м2/г, средний размер частиц от 2 до 4 и предпочтительно от 3 до 3,4 мкм и средний размер кристаллитов 1,6 до 2,2 мкм и второй имеет удельную поверхность от 3 до 5 м2/г, средний размер частиц от 4 до 8 мкм и средний размер кристаллитов 0,4 - 0,7 мкм.
Гидратированный предшественник α- оксида алюминия основан предпочтительно на моногидрате, например бемите, но хорошие результаты получают также, если предшественник содержит смесь бемита с тригидратом алюминия, например гиббситом или байеритом. Когда используют такую смесь, предпочтительно использовать весовое отношение моногидрата (бемита) к тригидрату от 1: 10 до 1:3 и более предпочтительно от 1:8 до 1:4. Когда предшественник α- оксида алюминия содержит тригидрат оксида алюминия, он обычно содержит от 10% до 35% по весу тригидрата оксида алюминия от общего веса α- оксида алюминия в носителе. Хотя могут быть использованы другие тригидраты оксида алюминия, наиболее часто используемым тригидратом оксида алюминия является гиббсит, имеющий средний размер частицы от 4 до 20 мкм.
По предпочтительному варианту в гидратированном предшественнике α- оксида алюминия используют затравку, которой может быть любой материал, эффективный для получения центров зародышеобразования в предшественнике для того, чтобы понизить температуру перехода, при которой оксид алюминия превращается в α- оксид алюминия. Затравки, с которыми можно достигнуть этой цели, обычно имеют тот же самый тип кристаллической решетки, что и сам α- оксид алюминия, при этом размеры решетки не слишком сильно отличаются от размеров решетки α- оксида алюминия. Понятно, что наиболее подходящей затравкой является сам α- оксид алюминия и предпочтительной затравкой являются частицы альфа-оксида алюминия с размером менее 1 мкм. В предпочтительном варианте затравка α-оксида алюминия имеет средний размер частиц менее чем 0,2 микрометр и составляет от 0.2% до 5% по весу относительно общего веса оксида алюминия, определенного в виде α- оксида алюминия, в носителе катализатора. Тем не менее можно использовать другие затравки, например, α- оксид железа (II), оксид хрома и определенные комплексные оксиды титана.
α- Оксид алюминия, образованный из предшественника, содержащего затравку, когда экструдированную смесь обычно обжигают, имеет значительно меньший размер кристаллов, чем частицы α- оксида алюминия, с которыми смешивают предшественник, содержащий затравку, если во время обжига не поддерживают высокую температуру в течение длительного периода времени. После завершения содержащий затравку зольгелевый материал имеет кристаллическую структуру с размером кристаллита меньше мкм, но его выдерживают при температурах выше 1400oC в течение длительного периода времени, при этом начинается рост кристаллов, и дифференцирование может стать менее явным.
Конечный обожженный носитель имеет предпочтительно пористость по крайней мере 50% и более, желательно от 60 до 75%, прочность на раздавливание по крайней мере 2,3 кг и определенную объемную плотность по крайней мере 0,5 мГ/л, предпочтительно по крайней мере 0,6 кг/л. Удельная поверхность конечного обожженного продукта составляет предпочтительно от 0,4 до 5 м2/г и более предпочтительно от 0,6 до 1,2 м2/г.
Было найдено, что часто является выгодным добавить к экструдируемой смеси оксид титана в количестве от 0,05 до 1%, предпочтительно от 0,05 до 0,5%, более предпочтительно 0,08 до 0,4% и наиболее предпочтительно от 0,08 до 0,25 от веса обожженного носителя.
Определенные формы оксида алюминия и связующего могут также содержать оксид титана в виде примесей или компонентов. В этом случае оксид титана не учитывается в количествах, указанных выше. Оксид титана может быть добавлен в виде диоксида, в виде титаната или в виде предшественника оксида титана. В дальнейшем в описании все указанные виды следует понимать под термином "оксид титана". Полагают, что оксид титана может действовать как ингибитор роста кристаллов в α- оксиде алюминия, образованном в результате превращения предшественника, содержащего затравку. Следовательно, можно ожидать, что другие такие материалы, например такие, как оксид циркония или магния, которые могут действовать в этой емкости, могут быть полезны в качестве заменителей оксида титана. Полагают, что происходят сложные многостадийные реакции в твердой фазе между оксидом алюминия/связующим, примесями и оксидом титана, добавленным к носителю, что приводит к возрастанию прочности и плотности носителя.
Оксид титана находится предпочтительно в форме порошка с относительно высокой удельной поверхностью, т.е. по крайней мере 8 и предпочтительно от 8 до 300 м2/г.
На практике предпочтительные оксиды титана имеют аморфную структуру или структуру анатаза. Без отсылки к какой-либо теории, можно предположить, что рутильная структура оксида титана обычно не дает преимуществ, которые можно получить от аморфной и анатазной структур оксида титана, потому что он обычно имеет при этом гораздо меньшую удельную поверхность. Промышленные пигментные сорта оксида титана могут также давать хорошие результаты.
Компоненты оксида алюминия для носителя затем обычно смешивают с выгорающим материалом и/или связующим и водой, формуют в различные формы и обжигают.
Выгорающий материал представляет такой материал, который добавляют к смеси при обжиге для того, чтобы его полностью удалять из носителя, оставив при этом регулируемую пористость в носителе. Эти материалы представляют углеродистые материалы, например уголь, угольные порошки, графит, порошковые пластики, например полиэтилен, полистирол и поликарбонат, канифоль, целлюлозу, и материалы на основе целлюлозы, опилки и другие материалы растительного происхождения, такие как скорлупа земляного ореха, например, американского ореха, кешью, грецкого ореха и фундука.
Выгорающие материалы на основе углерода могут также служить в качестве связующего вещества. Выгорающие материалы применяют в таком количестве и при таком распределении по размерам, которые обеспечивают в конечном носителе объем пор по воде в диапазоне от 0.2 до 0.6 мг/л. Выгорающие материалы обычно берут в количестве от 5 до 40% по весу от веса α- оксида алюминия в носителе. Предпочтительными выгорающими материалами являются материалы, полученные из целлюлозы, например скорлупа земляного ореха.
Термин "связующее вещество", как используется здесь, относится к веществу, которое удерживает вместе различные компоненты носителя после того, как они были сформованы в конечную форму посредством экструзии или гранулирования Эти связующие вещества обеспечивают сушку и обжиг формованных материалов без разрушения. Такие связующие вещества обычно представляют "клейкие" органические материалы, например поливиниловые спирты или целлюлозные материалы. Связующие вещества могут также служить в качестве вспомогательного средства для экструзии. В определенных случаях вместо связующих веществ могут быть использованы пептизирующие кислоты.
Несмотря на то, что можно предположить, что α- оксид алюминия, образованный из содержащего затравку предшественника, действует в некотором смысле как матричное связующее, удерживающее вместе остальные частицы α- оксида алюминия, обычно предпочтительно добавить к смеси для придания дополнительной прочности обожженному носителю керамический связующий материал.
Керамический связующий материал обычно присутствует в количестве от 1 до 3% по весу от общего веса компонентов оксида алюминия, выраженного в виде α- оксида алюминия. Общепринятые связующие керамические материалы можно использовать и после обжига, они обычно включают компоненты (в виде оксидов), например диоксид кремния, оксид алюминия, оксиды щелочного металла, оксиды щелочноземельного металла, оксид железа и оксид титана, при этом первые два компонента являются преобладающими.
В предпочтительном варианте керамический связующий материал включает следующие компоненты в виде оксидов при следующих приблизительных содержаниях компонентов: 60 вес. % диоксида кремния, 29 вес.% оксида алюминия, 3 вес. % оксида кальция, 2 вес.% оксида магния, 4 вес.% оксидов щелочного металла и менее чем 1 вес.% каждого из оксидов железа (III) и оксида титана.
После того, как компоненты носителя смешаны посредством измельчения, смешанный материал экструдируют в формованные гранулы, например цилиндры, кольца, трехлепестковые и четырехлепестковые частицы и т.р. Для способствования экструзии могут быть использованы "вспомогательные средства для экструзии", например Vaseline Petroleum Jelly (вазелин) и другие органические смазочные материалы. Для удаления воды, которая может превратиться в пар во время обжига и разрушать экструдатные формы, экструдированный материал сушат. После сушки до низкого содержания воды, т.е. менее 2%, экструдированный материал обжигают при условиях, достаточных для удаления выгорающих материалов, вспомогательных средств для экструзии и связующих веществ и для расплавления части α- оксида алюминия в пористую, твердую массу. Обжиг обычно осуществляют в окислительной атмосфере, например кислородном газе или предпочтительно воздухе, и при максимальной температуре более чем 1300oC, предпочтительно в диапазоне от 1350oC до 1500oC. Время при таких максимальных температурах обычно составляет от 1 до 10, предпочтительно от 0,5 до 5 часов.
Обожженные носители и катализаторы, полученные из них, обычно имеют объем пор по воде в диапазоне от 0.2 до 0.6, предпочтительно от 0.3 до 0.5 мл/г и удельную поверхность в диапазоне от 0.15 до 3, предпочтительно от 0.3 до 2 м2/г.
Состав носителя предпочтительно имеет низкое содержание соды, которое составляет менее чем 0.06% по весу. На практике очень трудно получить состав, не содержащий натрия; было найдено, что обычно является приемлемым содержание соды от 0.02 до 0.06% по весу.
Носители, описанные выше, являются в особенности подходящими для получения катализаторов, применимых для получения этиленоксида, которые имеют высокую начальную селективность.
Катализатор настоящего изобретения включает каталитически эффективное количество серебра и промотирующее количество щелочного металла(ов), осажденных на носителе, который описан выше. Необязательно могут присутствовать другие промоторы в промотирующих количествах, например редкие земли, магний, рений и со-промоторы рения, выбранные из серы, хрома, молибдена, вольфрама и их смесей.
В общем случае катализатор настоящего изобретения получают путем пропитки пористого огнеупорного носителя, включающего α- оксид алюминия, ионами серебра или соединением(ями), комплексным соединением(ями) и/или солью(ями), растворенными в соответствующем растворителе в количестве, достаточном для нанесения на носитель серебра от 1 до 40, предпочтительно от 1 до 30% по весу от общего веса катализатора. Затем пропитанный носитель отделяют от раствора и нанесенное соединение серебра восстанавливают до металлического серебра.
Также подходящие ионы или соединение (я), и/или соль(и) щелочного металла, растворенные в соответствующем растворителе, могут быть нанесены на носитель перед нанесением серебра, или одновременно, или после нанесения серебра. Также подходящие необязательно применяемые промоторное соединение(я), комплексное(ые) соединение(я) и/или соль(и), растворенная(ые) в соответствующем растворителе, могут быть нанесены на носитель вместе с серебром и/или щелочным металлом.
Катализаторы настоящего изобретения получают посредством методики, по которой промотор из щелочного металла, а также любые дополнительные промоторы в форме растворимых солей и/или соединений наносят на катализатор и/или носитель перед нанесением, одновременно с нанесением или после нанесения серебра и каждого другого из вышеуказанных веществ. Предпочтительным способом является одновременное нанесение серебра и щелочного металла на носитель, т.е. на одной стадии пропитки, хотя полагают, что отдельное или совместное нанесение щелочного металла перед нанесением серебра и/или после нанесения серебра будет также приводить к получению соответствующих катализаторов.
Промотирующие количества щелочного металла или смесей щелочного металла наносят на пористый носитель при использовании соответствующего раствора. Хотя щелочные металлы существуют в чисто металлическом состоянии, они не являются подходящими для использования в такой форме. Их используют в виде ионов или соединений щелочного металла, растворенного в подходящем растворителе с целью пропитки. Носитель пропитывают раствором, содержащим ионы промотора из щелочного металла, солью(ями) и/или соединением(ями), перед пропиткой, во время пропитки или после пропитки ионами серебра или солью(ями), комплексным(и) соединением(ями), и/или соединением(ями). Промотор из щелочного металла может быть нанесен на носителе даже после восстановления до металлического серебра. Промотирующее количество использованного щелочного металла будет зависеть от нескольких факторов, например удельной поверхности, структуры пор и поверхностных химических свойств используемого носителя, содержания серебра в катализаторе и особенностей ионов, используемых в сочетании с катионом щелочного металла, необязательных со-промоторов.
Количество промотора из щелочного металла, осажденного на носителе или присутствующего на катализаторе, обычно составляет от 10 до 3000, предпочтительна от 15 до 2000, и более предпочтительно от 20 до 1500 частями на миллион по весу от общего веса катализатора. Наиболее предпочтительно количество промотора находится в диапазоне от 50 до 1000 частей на миллион по весу от общего веса катализатора.
С целью удобства количество щелочного металла, нанесенного на носитель или присутствующего в катализаторе, выражают в пересчете на металл. Без ограничения объема изобретения полагают, что соединениями щелочного металла являются оксидные соединения. В частности полагают, что соединения щелочного металла находятся, вероятно, в форме смешанных поверхностных оксидов или двойных поверхностных оксидов, или комплексных поверхностных оксидов с алюминием носителя и/или серебром катализатора, возможно в сочетании с разновидностями, содержащимися в реакционной смеси или образованными из реакционной смеси, например такими, как хлориды или карбонаты или остаточные разновидности из пропиточного(ых) раствора(ов).
В предпочтительном варианте по крайней мере основную часть (более чем 50% вес. %) щелочных металлов выбирают из группы, состоящей из калия, рубидия, цезия и их смесей.
Предпочтительный промотор, которым является щелочной металл, представляет цезий. В особенности предпочтительный промотор из щелочного металла представляет цезий плюс, по крайней мере, один дополнительный щелочной металл. Дополнительный щелочной металл выбирают предпочтительно из натрия, лития и их смесей, при этом литий является предпочтительным.
Следует понимать, что количества промоторов из щелочного металла в катализаторах не являются неизбежно общими количествами этих металлов, присутствующих в катализаторе. Скорее всего они являются количествами промоторов из щелочного металла, которые добавляют к катализатору посредством пропитки соответствующим раствором ионов, солей и/или соединений и/или комплексных соединений щелочных металлов. Эти количества не включают количества щелочных металлов, которые закреплены на носителе, например, путем обжига, или которые не экстрагируются соответствующим растворителем, например вода или низший спирт или амин или их смеси, и не обеспечивают промотирующее действие. Полагают также, что источником ионов промотора из щелочного металла, солей и/или соединений, использованных для промотирования катализатора, может быть носитель. Носитель может содержать экстрагируемые количества щелочного металла, которые можно экстрагировать соответствующим растворителем, например водой или низшим спиртом, получая таким образом пропиточный раствор, из которого осаждаются или переосаждаются на носитель ионы щелочного металла, соли и/или соединения.
Катализатор может также содержать умеренные количества хлорида с целью ускорения начала процесса для катализаторов. Когда в катализатор добавляют хлорид, носитель может быть пропитан раствором, содержащим ионы хлоридного соединения, соль(и), и/или соединения, и перед пропиткой, во время пропитки или после пропитки ионами серебра или солью(ями), комплексным соединением(ями) и/или соединением(ями), перед пропиткой, во время пропитки или после пропитки ионами промотора или солью(ями), комплексным(и) соединением(ями), и/или соединением(ями). Хлоридное соединение может быть нанесено на носитель даже после восстановления до металлического серебра. Подходящие хлоридсодержащие соли, использованные для получения пропиточного раствора, включают хлориды промотора, например, хлорид лития, хлорид натрия, хлорид калия, хлорид рубидия и хлорид цезия, а также хлорид аммония. Хлорид аммония является предпочтительной солью для использования при получении хлоридсодержащих пропиточных растворов. Подходящими являются также другие соединения, которые разлагаются при обработке катализатора до хлорид-иона. Хлоридсодержащие пропиточные растворы обычно содержат по крайней мере незначительные количества воды для повышения растворимости хлоридсодержащей соли или соединения. В сочетании с серебром и промоторами из щелочного металла могут быть использованы другие промоторы и со-промоторы.
Неограничивающие примеры других промоторов включают рений, сульфат, молибдат, вольфрамат и хромат (см. патент США N 4766105); сульфатный анион, фторидный анион, оксианионы групп 3b-6b (см. патент N 5102848); (i) оксианионы элемента, выбранного из групп 3-7 (ii) и соли щелочного металла с галогенидными анионами, и оксианионы, выбранные из групп 3а-7a и 3b-7b (см. патент США N 4908343).
Полученный таким образом пропитанный носитель нагревают для восстановления серебра до металлического серебра. Его обычно нагревают до температуры в диапазоне от 50oC до 600oC в течение периода времени, достаточного для восстановления соли серебра, соединения или комплекса до металлического серебра и для образования слоя тонкодиспергированного серебра, который связан с поверхностью носителя как с наружной геометрической, так и с поверхностью пор.
Во время стадии нагрева над носителем может быть пропущен воздух или другой окислительный газ, восстановительный газ, инертный газ или их смеси.
Один способ получения содержащего серебро катализатора описан в патенте CIF N 3702259. Другие способы получения содержащих серебро катализаторов, которые, коме того, содержат более высокие количества промоторов из щелочного металла, описаны в патенте США N 4010115, патенте США N 4356312, патенте США N 3962136 и патенте США N 4012425. Способы получения содержащих серебро катализаторов, содержащих более высокие количества щелочного металла и промоторов рения, раскрыты в патенте США N 4761394, а способы получения содержащих серебро катализаторов, содержащих более высокие количества щелочного металла, промоторов рения и сопромоторов рения раскрыты в патенте США N 4766105. Способы получения серебросодержащих катализаторов с множеством различных промоторов представлены в патентах США N 4908343 и 5057481.
Концентрация серебра (в пересчете на металл) в содержащем серебро растворе будет составлять от 1 г/л до предела растворимости в том случае, когда используют одну пропитку. Когда используют одну стадию пропитки, концентрация щелочного металла (в пересчете на металл) составляет от 1•10-3 г/л до 12 г/л и предпочтительно от 10•10-3 г/л до 12 г/л.
Концентрации, выбранные в пределах вышеуказанных диапазонов, зависят от объема пор катализатора, требуемого содержания в конечном катализаторе и от того, осуществляют ли пропитку одно- или многократно.
Независимо от формы, в которой присутствует серебро в растворе перед осаждением на носитель, используют обычно термин "восстановление до металлического серебра", хотя, между тем, часто происходит разложение посредством нагрева. В данном тексте предпочитают использовать термин "восстановление", так как ион серебра (Ag+) превращается в атом металлического серебра. Время восстановления обычно может изменяться в зависимости от обстоятельств от 0,5 минуты до около 8 часов.
В промышленном масштабе этилен и кислород превращают в этиленоксидном реакторе, который содержит большой неподвижный трубчатый теплообменник, содержащий несколько тысяч трубок, заполненных катализаторами. В межтрубной зоне реактора для удаления тепла реакции используют охладитель. Температуры охладителя часто используют в качестве указателя каталитической активности, при этом высокие температуры охладителя соответствуют пониженной каталитической активности.
В реакции этилена с кислородом с получением этиленоксида этилен обычно присутствует в по крайней мере двойном избытке (в молях) по отношению к кислороду, но обычно количество используемого этилена гораздо выше. Поэтому степень превращения обычно вычисляют в соответствии с молярной концентрацией кислорода, который расходуется а реакции образования этиленоксида и окисленных побочных продуктов.
Степень превращения кислорода зависит от температуры реакции, а температура реакции является мерой активности применяемого катализатора. Значение T40 указывает температуру при 40%-ном превращении кислорода в реакторе, и значение T выражено в oC. Эта температура для любого данного катализатора будет выше, когда будет выше степень превращения кислорода. Более того эта температура сильно зависит от используемого катализатора и условий реакции. Селективность (по этиленоксиду) указывает молярное количество этиленоксида в продукте реакции по сравнению с общим молярным количеством превращенного этилена.
В данном тексте селективность указана в виде S40, что означает селективность при 40%-ной степени превращения кислорода.
Условия проведения такой окислительной реакции в присутствии серебряных катализаторов в соответствии с настоящим изобретением в значительной степени включают условия, которые уже были описаны в литературе по известному уровню техники. Это относится, например, к соответствующим температурам, давлениям, времени реакции разбавителям, таким как азот, диоксид углерода, водяной пар, аргон, метан или другим насыщенным углеводородом, к присутствию замедлителей для регулирования каталитического действия, например, 1-2-дихлорэтана, винилхлорида, этилхлорида или хлорированных полифенильных соединений, к желательности использования рециркуляции или к использованию последовательных превращений в различных реакторах для увеличения выходов этиленоксида и к любым другим специальным условиям, которые могут быть выбраны в способе получения этиленоксида. Обычно используют давление в диапазоне от атмосферного до 3500 кРа. Более высокие давления, однако, не исключаются. Молекулярный кислород, используемый в качестве реагента, может быть получен из обычных источников. Подходящее кислородсодержащее сырье может, по существу, состоять или из относительно чистого кислорода, концентрированного кислородного потока, включающего кислород в преобладающем количестве, с меньшими количествами одного или нескольких разбавителей, таких как азот или аргон, или из другого кислородсодержащего потока, например воздуха. Поэтому использование серебряных катализаторов в этиленоксидных реакциях никоим образом не ограничивается применением конкретных условий из числа тех, которые известны как эффективные.
Таблицы, приведенные только с целью иллюстрации, показывают диапазон условий, которые часто используют в общепринятых промышленных этиленоксидных реакторах и которые являются также подходящими для настоящего способа.
При предпочтительном применении содержащих серебро катализаторов в соответствии с изобретением этиленоксид получают, когда кислородсодержащий газ контактирует с этиленом в присутствии этих катализаторов при температуре в диапазоне от 180oC до 330oC и предпочтительно от 200oC до 325oC.
Хотя катализаторы настоящего изобретения используют предпочтительно для превращения этилена в этиленоксид, их можно использовать также для эпоксидирования других олефинов, не имеющих аллильного водорода, например таких, которые широко представлены в патенте США N 4897498. Примерами таких олефинов могут служить бутадиен, третичный бутилэтилен, винилфуран, метилвинилкетон, N-винлипирролидон и т. п. В настоящее время предпочтительным олефином для использования в этом способе является бутадиен, вследствие его доступности, относительно низкой стоимости и широкого диапазона возможных применений. В патенте США N 5081096, опубл. 14 января 1992, описан промотированный щелочным металлом катализатор на основе серебра, на носителе, который применяется для эпоксидирования бутадиена, путем обработки предшественника катализатора, после его пропитки серебросодержащим соединением, водородсодержащим газом при температуре не выше 350oC. То же самое можно осуществить с катализаторами в соответствии с настоящим изобретением.
Изобретение проиллюстрировано примерами выполнения изобретения.
Получение носителя.
Носитель A:
Керамические компоненты смешивали в течение одной минуты с выгорающим материалом (порошком из скорлупы грецкого ореха) и борной кислотой.
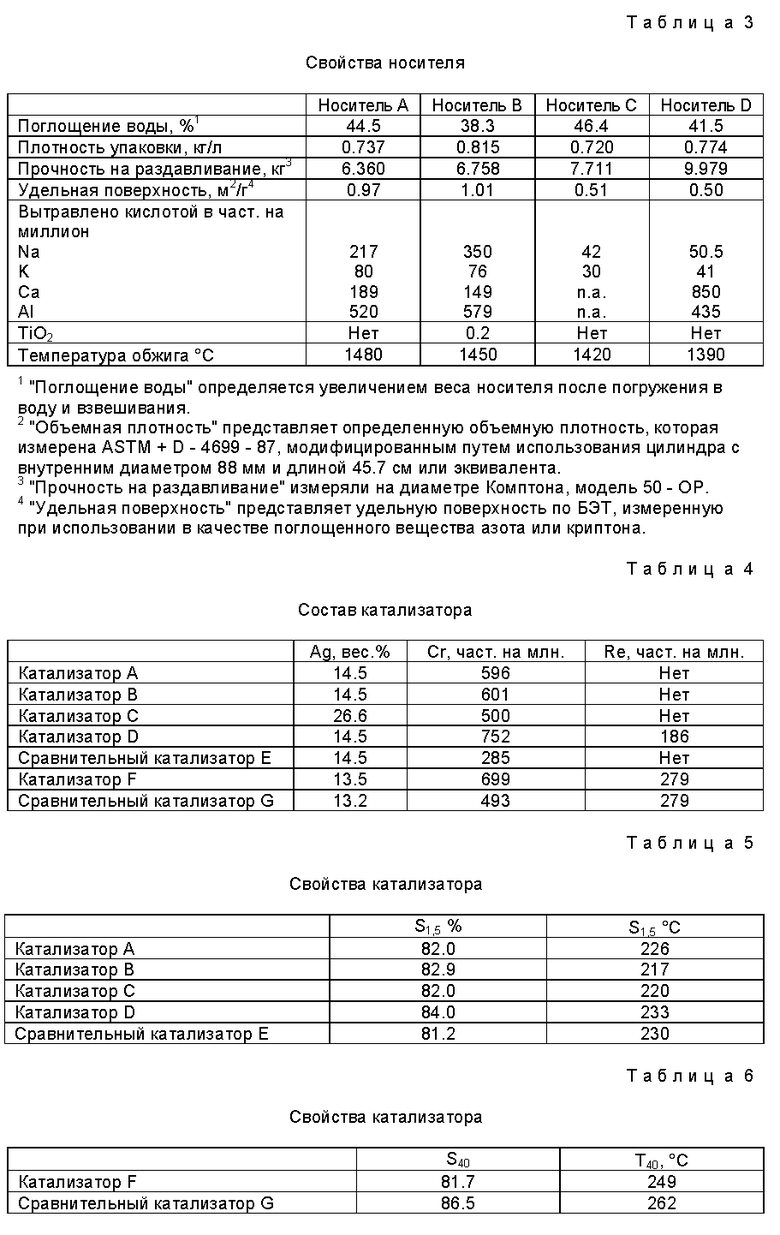
Затем добавили воду и затравочный компонент, при этом воду добавили в количестве, которое необходимо для того, чтобы сделать смесь экструдируемой. Обычно количество воды составляет около 30% по весу относительно общего количества присутствующих твердых веществ. Смесь перемешивали в течение времени от 2-х до 4-х минут и затем в качестве вспомогательного средства для экструзии добавили 5% по весу петролатума (вазелина) (Vaseline - фабричная марка) относительно веса керамических компонентов. Затем смесь перемешивали в течение от 2-х до 4-х минут, после чего экструдировали в форме полых цилиндров и сушили до содержания несвязанной воды менее 2%. Затем смесь обжигали в тунельной печи при максимальной температуре около 1500oC в течение около 4-х часов. Состав носителя представлен в табл. 2, а его физические свойства - в табл. 3.
Носитель B:
Носитель B получили способом, подобным тому, которым получили носитель A, за исключением того, что к составу носителя добавили оксид титана. Состав носителя представлен в табл. 2, а его физические свойства - в табл. 3.
Носитель C:
Носитель C получили способом, подобным тому, которым получили носитель A, за исключением того, что носитель не содержал компонент α-оксида алюминия, полученного зольгелевым процессом, и к составу носителя не добавили затравочный компонент, т.е. α-оксид алюминия N 5. Состав носителя представлен в табл. 2, а его физические свойства - в табл. 3.
Носитель D:
Носитель D получили способом, подобным тому, которым получили носитель A, за исключением того, что носитель не содержал компонент α- оксида алюминия, полученного зольгелевым процессом, и к составу носителя не добавили затравочный компонент, т. е. α- оксид алюминия N 5. Состав носителя представлен в таблице II, а его физические свойства - в таблице III.
Получение катализатора.
Следующий пояснительный вариант выполнения изобретения описывает подготовительные методики для получения катализаторов настоящего изобретения (катализаторов A, B, C, D и F) и сравнительных катализаторов (Сравнительных катализаторов E и G) и методику измерения свойств этих катализаторов.
Часть A: Получение исходного раствора оксалата серебра/этилендиамина для использования при получении катализатора:
1) Растворили 415 г гидроксида натрия химической чистоты в 2340 мл деионизированной воды. Установили температуру 50oC.
2) Растворили 1699 г (высокочистого) нитрата серебра в 2100 мл деионизированной воды. Установили температуру 50oC.
3) К раствору нитрита серебра при перемешивании и поддержании температуры 50oC медленно добавили раствор гидроксида натрия. После завершения добавления осуществляли перемешивание в течение 15 минут и затем температуру понизили до 410oC.
4) Вставили чистые фильтровальные палочки и удалили из осадка, полученного на стадии (3), столько воды, сколько было возможно для того, чтобы затем удалить ионы натрия и нитрат-ионы. Измерили электропроводность удаленной воды и добавили столько же свежей деионизированной воды, сколько удалили посредством фильтровальных палочек. Осуществили перемешивание в течение 15 минут при 40oC. Этот процесс повторяли до тех пор, пока электропроводность удаленной воды стала меньше, чем 90 мкм ho/см. Затем добавили 1500 мл деионизированной воды.
5) Добавили 630 г дигидрата высокочистой щавелевой кислоты порциями приблизительно 100 г. Поддерживали температуру при 40oC и смесь тщательно перемешали. Медленно добавили последнюю порцию дигидрата щавелевой кислоты и отрегулировали pH таким образом, чтобы он не упал ниже значения, равного 7,8.
6) Из смеси удалили максимально возможное количество воды при использовании чистых фильтровальных папочек для того, чтобы образовалась высококонцентрированная серебросодержащая суспензия. Суспензию оксалата серебра охладили до 30oC.
7) Добавили 699 г 92 вес.% этилендиамина (8% деионизированной воды). Обеспечили, чтобы температура во время добавления не превосходила 30oC.
Вышеприведенная процедура привела к получению раствора, содержащего приблизительно 27-33 вес.% серебра, что обеспечило получение "исходного раствора", использованного при получении катализаторов A, B, C, D и F и сравнительных катализаторов E и G.
Часть B: Получение растворов для пропитки.
Для катализатора AA:
161.3 г содержащего серебро исходного раствора с относительной плотностью 1.543 разбавили 4.2 г воды и 13.5 г моноэтаноламина. 0.0350 г NH4F растворили в 2 мл воды и добавили к раствору серебра. К 60 г вышеприведенного разбавленного раствора серебра добавили CsOH (50%-раствор в воде) в количестве 0.1367 г, и полученную смесь использовали для пропитки носителя.
Для катализатора B:
175.4 г содержащего серебро раствора с относительной плотностью 1.53 разбавили 3,6 г воды. 0.0387 г NH4F растворили в 2 см3 воды и добавили к раствору серебра. К 60 г вышеупомянутого разбавленного раствора серебра добавили CsOH (50% раствор в воде) в количестве 0.1536 г, и полученную смесь использовали для пропитки носителя.
Для катализатора C:
165.3 г содержащего серебро исходного раствора с относительной плотностью 1.55 разбавили 13.9 г моноэтаноламина. 0.0426 г NH4F растворили в 2.5 г воды и добавили к раствору серебра. К 60 вышеприведенного разбавленного раствора серебра добавили CsOH (50% раствор в воде) в количестве 0.1406 г, и полученную смесь использовали для пропитки носителя.
Для катализатора D:
161.2 г содержащего серебро исходного раствора с относительной плотностью 1.555 разбавили 17.8 г воды. 0.0868 г (NH4)ReO4 растворили в 2 см3 смеси воды/ЭDA (50/50 по весу) и добавили к раствору серебра. К 60 г вышеприведенного разбавленного раствора серебра добавили CsOH (50% раствор в воде) в количестве 0.1743 г, и полученную смесь использовали для пропитки носителя.
Для сравнительного катализатора E:
129.7 г содержащего серебро исходного раствора, содержащего 29.7% Ag, разбавили 14 г воды и 6.3 г моноэтаноламина. 0.0285 г NH4F растворили в 2 мл воды и добавили к раствору серебра. К 50 г вышеприведенного разбавленного раствора серебра добавили CsOH (50% раствор в воде) в количестве 0.0582 г, и полученную смесь использовали для пропитки носителя.
Для катализатора F:
168 г содержащего серебро исходного раствора с относительной плотностью 1.546 разбавили 10.9 г воды 0.1442 г (NH4)ReO4, 0,0704 г Li2SO4 • H2O, 0.303 г LiNO3 растворили в смеси приблизительно 2 мл этилендиамина/воды (50/50 по весу) и добавили к раствору серебра. К 50 г вышеприведенного разбавленного серебра добавили CsOH (50% раствор в воде) в количестве 0.1985 г, и полученную смесь использовали для пропитки носителя.
Для сравнительного катализатора G:
101 г содержащего серебро исходного раствора с относительной плотностью 1.558 разбавили 12.9 г воды. 0.0766 г (NH4)ReO4, 0.0374 г Li2SO4 • H2O, 0.1616 г LiNO3 растворили в приблизительно 2.0 г этилендиамина/воды (50/50 по весу) и добавили к раствору серебра. К 50 г вышеприведенного разбавленного раствора серебра добавили CsOH (50% раствор в воде) в количестве 0.111 г, и полученную смесь использовали для пропитки носителя.
Часть C. Пропитка катализатора и отверждение.
Катализатор A:
Приблизительно 30 г носителя A, охарактеризованного в табл. 2 и 3), выдерживали под вакуумом 25 мм в течение 3 минут при комнатной температуре. Затем для пропитки носителя ввели приблизительно от 50 до 60 г пропиточного раствора (который описан выше в части B под названием "Для катализатора A"), и в течение 3-х минут сохраняли вакуум 25 мм. В конце этого периода вакуум отключили и из носителя центрифугированием в течение 2-х минут при 500 оборотах в минуту удалили избыток пропиточного раствора. Если пропиточный раствор получали без моноэтанолиамина, тогда пропитанный носитель затем отверждали путем непрерывного встряхивания в 850 л/ч воздушном потоке, протекающем через поперечное сечение приблизительно 19.4 - 32.3 см3 при 240-270oC в течение 3-6 минут. Если в пропиточном растворе присутствует значительное количество моноэтаноламина, тогда пропитанный носитель отверждали при непрерывном встряхивании в 850 л/час воздушном потоке при температуре от 250 до 270oC в течение 4-8 минут. После этого отвержденный катализатор был готов для испытаний. Свойства катализатора A показаны в табл. 4.
Катализатор B:
Катализатор B получили тем же самым образом, что и катализатор A, за исключением того, что вместо носителя катализатора A использовали носитель катализатора B и пропиточным применяемым раствором был раствор, описанный в части B под названием "Для катализатора B". Свойства катализатора B показаны в табл. 4.
Катализатор C:
Катализатор C получили с использованием двойной методики пропитки. В этой методике 120 г носителя катализатора B пропитали 240 г содержащего серебро исходного раствора с относительной плотностью 1.555. Пропитанный носитель сушили и прокаливали для разложения солей серебра до металлического серебра. Водный объем пор определяли после первой пропитки и использовали для вычисления концентраций поглощенного вещества.
Вторую пропитку осуществляли пропиточным раствором, описанным в части B под названием "Для катализатора C". Катализатор отверждали способом, подобным тому, который описан выше. Свойства катализатора C показаны в табл. 4.
Катализатор D:
Приблизительно 30 г носителя A, (описанного в табл. 2 и 3 выдерживали под вакуумом 25 мм в течение 3 минут при комнатной температуре. Затем для пропитки носителя ввели приблизительно от 50 до 60 г пропиточного раствора (который описан в части B под названием "Для катализатора D") и в течение еще 3-х минут сохраняли вакуум 25 мм. В конце этого периода вакуум отключили, и из носителя центрифугированием в течение 2-х минут при 500 оборотах в минуту удалили избыток пропиточного раствора. Если пропиточный раствор получали без моноэтаноламина, тогда пропитанный носитель затем отверждали при непрерывном встряхивании в воздушном потоке, протекающем через поперечное сечение приблизительно 19.4-32.3 см2 со скоростью 850 л/час при 240-270oC в течение 3-6 минут. Если в пропиточном растворе присутствует значительное количество моноэтаноламина, тогда пропитанный носитель отверждали при непрерывном встряхивании в воздушном потоке со скоростью 850 л/час при температуре от 250oC до 270oC в течение 4-8 минут. После этого отвержденный катализатор был готов для испытаний. Свойства катализатора D показаны в табл. 4.
Сравнительный катализатор E:
Сравнительный катализатор E получили тем же самым образом, что и катализатор A, за исключением того, что вместо носителя катализатора A использовали носитель катализатора C и используемым пропиточным раствором был раствор, описанный в части B под названием "Для сравнительного катализатора E".
Свойства сравнительного катализатора E показаны в табл. 4.
Катализатор F:
Катализатор F получили тем же самым образом, что и катализатор D, за исключением того, что вместо носителя катализатора A использовали носитель катализатора B, и используемым пропиточным раствором был раствор, который описан выше в части B под названием "Для катализатора F". Свойства катализатора F показаны ниже в таблице IV.
Сравнительный катализатор G:
Сравнительный катализатор G получили тем же самым образом, что и катализатор D, за исключением того, что вместо носителя катализатора A использовали носитель катализатора D, и используемым пропиточным раствором был раствор, который описан выше в части B под названием "Для катализатора G". Свойства катализатора G показаны ниже в табл. 4.
Действительное содержание серебра в катализаторе может быть определено посредством любой из ряда стандартных опубликованных методик. Действительный уровень цезия в катализаторе может быть определен путем использования исходного раствора гидроксида цезия, который метили радиоактивным изотопом цезия, при получении катализатора. Содержание цезия в катализаторе может быть затем определено путем измерения радиоактивности катализатора. Кроме того, содержание цезия в катализаторе может быть определено путем выщелачивания катализатора кипящей деионизированной водой. В этом способе содержание цезия, а также других щелочных металлов измеряли посредством извлечения из навески 10 г катализатора при кипении 20 мл воды в течение 5 минут, повторения вышеописанного более 2-х раз, смешения экстрактов и определения присутствующего количества щелочного металла путем сравнения со стандартными растворами сравнительных щелочных металлов при использовании атомной абсорбционной спектроскопии (при применении Varian Techtron Model 1200 или эквивалента).
Часть D: Условия испытаний катализатора в стандартном микрореакторе/методика.
A. Для катализаторов A, B, C и сравнительного катализатора Е.
От 1 до 3 г измельченного катализатора с размером частиц 0.841 - 0.595 мм (20 - 30 меш) загрузили в U-образную трубку из нержавеющей стали диаметром 6.4 мм. U-образную трубку погрузили в баню с металлическим расплавом (нагревающая среда) и концы присоединили к системе газового потока. Для достижения среднечасовой скорости подачи газа 6800 отрегулировали массу используемого катализатора и скорость потока газа на входе. Давление газа на выходе составило 1550 кРа.
Газовая смесь, пропущенная через слой катализатора (один раз) во время всего испытания (включая начальную), состояла из 25% этилена, 7% кислорода, 5% диоксида углерода, от 1.25 до 5 част, на миллион по весу этилхлорида, при этом остальным был азот/аргон.
Начало процесса включало подъем температуры от 180oC до 230oC следующим образом: 1 час до 180oC, 1 час до 190oC, 1 час до 200oC, 1 час до 210oC, 2 часа до 220oC, 2 часа до 225oC, 2 часа до 230oC, и затем температуру регулировали таким образом, чтобы обеспечить 1.5% этиленоксида на выходе из реактора. При этих условиях измерили селективность катализатора (S1.5) и активность катализатора (T1,5).
Для возможности сравнения свойств катализаторов, испытанных в различное время, катализаторы A, B, C и сравнительный катализатор E испытывали одновременно со стандартным катализатором сравнения, который имеет S1,5=81.7% и T1,5=235oC.
Катализаторы A, B, C и сравнительный катализатор E, полученные выше, испытывали при использовании вышеприведенной методики, результаты приведены в табл. 5.
B. Для катализатора D:
Катализатор D испытывали способом, подобным тому, который описан выше для катализаторов A, B, C и сравнительного катализатора E, за исключением того, что начало процесса было следующим.
Первоначальная температур реактора (нагревающая среда) составила 225oC. Через 3 часа нахождения под потоком азота при этой первоначальной температуре температуру увеличили до 235oC в течение 1 часа, затем до 245oC в течение 1 часа. Затем температуру отрегулировали таким образом, чтобы обеспечить на выходе из реактора 1,5% этиленоксида. Результаты приведены в табл. 5.
Как можно видеть из таблицы, начальные селективности катализаторов A, B, C и D по сравнению с начальной селективностью сравнительного катализатора E улучшились. Можно также видеть, что начальные активности катализаторов A, B и C также улучшены по сравнению с начальной активностью катализатора E.
C. Для катализатора F и сравнительного катализатора G:
От 3 до 5 г измельченного катализатора (14-20 меш) загрузили в U-образную трубку, выполненную из нержавеющей стали диаметром 1/4 дюйма (6.350 мм). U-лобразную трубку погрузили в баню с металлическим расплавом (нагревающая среда), и концы присоединили к системе газового потока. Для достижения среднечасовой скорости подачи газа 3300 отрегулировали массу используемого катализатора и скорость потока газа на входе. Давление газа на выходе составило 210 psig (7.031 • 2.10 = 14.765 кг/см2).
Газовая смесь, пропущенная через слой катализатора (один раз) во время испытания (включая начальную) состояла из 30% этилена, 8,5% кислорода, 5% диоксида углерода, от 1.5 до 5 част. на миллион по объему этилхлорида, при этом остальным был азот/аргон.
Перед контактированием с реагирующими газами катализаторы обычно предварительно обрабатывали газообразным азотом в течение 3-х часов при температуре 225oC для свежих катализаторов или в течение более длительного периода времени для старевших, но не подвергаемых испытанию катализаторов (контрольных).
Первоначальная температура реактора (нагревающая среда) составила 225oC. Через 1 час выдерживания при этой начальной температуре температуру увеличили до 235oC, затем в течение часа до 245oC. Затем температуру отрегулировали таким образом, чтобы, достигнуть степени превращения кислорода, равной 40% (T40).
Для определения оптимального количества замедлителя количество замедлителя изменяли в течение 4-24 часов, и когда катализатор находился в потоке времени около 24 часов, обычно получали T40 и S40, которые приведены в табл. 5.
Благодаря незначительной разнице в составе исходного газа, скоростях потока газа и градуировке аналитических инструментов, используемых для определения составов исходного и полученного газа, измеренная селективность и активность данного катализатора могут изменяться в незначительной степени при переходе от одного испытания к другому.
Для возможности значительного сравнения свойств катализаторов, испытанных в различное время, все катализаторы, описанные в этом иллюстративном варианте реализации способа, испытывали одновременно со стандартным катализатором сравнения, который имеет T40=81% и T40=230oC.
Катализатор F и сравнительный катализатор G, полученные выше, испытывали с использованием вышеприведенной методики, результаты приведены в табл. 6.
Как можно видеть из табл. 6, начальная активность и начальная селективность катализатора F улучшена по сравнению с начальной активностью и начальной селективностью сравнительного катализатора G.


Изобретение относится к катализатору для получения этиленоксида, который содержит серебро и один или несколько промоторов из щелочного металла, нанесенных на носитель, имеющий прочность на раздавливание по крайней мере 2,3 кг и определенную объемную плотность, по крайней мере около 0,48 кг/л, который включает первый и второй компоненты α-оксида алюминия, при этом первый компонент α-оксида алюминия в форме частиц, имеющих средний размер кристаллитов 0,4 - 4 мкм, составляет от 95% до 40% от общего веса α-оксида алюминия в носителе, и второй компонент α-оксида алюминия, полученный in situ зольгелевым процессом, составляет остальное количество α-оксида алюминия в носителе. К носителю необязательно добавляют оксид титана. Описывается также способ получения носителя для вышеуказанного катализатора. Катализаторы, имеющие уникальный носитель из оксида алюминия, обладают улучшенной селективностью и/или активностью по сравнению с катализаторами, содержащими обычные носители из оксида алюминия. 2 с. и 15 з.п. ф-лы, 6 табл.
| US 5063195 А, 05.11.91 | |||
| SU 18737959 А3, 30.08.93 | |||
| СПОСОБ ПОЛУЧЕНИЯ КРЕМНИЙОРГАНИЧЕСКИХ ОЛИГОМЕРОВ | 0 |
|
SU199396A1 |

