Настоящее изобретение относится к способу синтеза моногидрата и кристаллическим модификациям флуконазола формулы I

Здесь и далее термины «кристаллическая модификация» и «полиморфная модификация» имеют одно и тоже значение и используются как синонимы.
Патент Великобритании №2078719 А описывает очень эффективные фунгицидные соединения, которые обладают также существенным эффектом, регулирующим рост растений. Вышеуказанные соединения проиллюстрированы формулой (А)

где значением R являются алкильная, циклоалкильная, арильная или аралкильная группа или их производные, содержащие один или два атома галогена, либо замещенные алкокси, фенилом, фенокси или трифторметилом арильную и бензильную группы, a Y1 и Y2 независимо представляют собой группу -N= или -СН=.
Согласно патенту Великобритании №2099818 А соединение 2-(2,4-дифторфенил)-1,3-бис(1,2,4-триазол-1-ил)-пропан-2-ол, принадлежащее к вышеуказанной группе (далее - флуконазол), может быть также использовано в качестве фунгицида у человека. Флуконазол является среди других активным ингредиентом дифлукана, который является очень эффективным фунгицидным лекарством для людей на рынке.
Согласно патенту Великобритании №2078719 А, производные пропан-2-ола формулы (А) синтезируют с помощью взаимодействия реактива Гриньяра формулы R-Mg-галоген, где значение R такое, как определено выше, с дихлорацетоном. Образованное таким образом производное 1,3-дихлорпропан-2-ола формулы (VI)
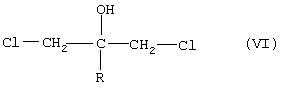
подвергают взаимодействию с избытком соли имидазола или триазола, например натриевой соли, в протонной или апротонной среде (например, в диметилформамиде). Данную реакцию можно осуществлять также с эпокси-производными, которые образуются in situ из упомянутого дигалоген-соединения в присутствии основания при удалении хлористого водорода. Желаемые соединения также могут быть синтезированы с помощью взаимодействия подходящего 1,3-бисимидазолила или 1,3-бис(1,2,4-триазол-1-ил)-ацетона с реактивом Гриньяра формулы R-Mg-галоген. В соответствии с другим путем синтеза соединения формулы (VII), где значения R и Y1 такие, как определено выше,
превращают в соединения формулы (IV), содержащие заместитель R вместо R1, с помощью диметил-оксосульфония метилида,

и полученное подвергают взаимодействию с натриевой солью имидазола или триазола по аналогии с вышеприведенным способом. Исходные материалы получают в соответствии с известными методиками.
В способе синтеза активного вещества флуконазола, описанном в патенте Великобритании №2099818 А, используются соединения формул (VI) и (IV), которые содержат заместитель R вместо R1, в качестве исходных материалов, но в качестве реагентов вместо триазолата натрия используют основание и триазол.
Общим признаком способов обоих патентов является то, что выделение продуктов реакции осуществляют путем экстракции после разбавления реакционной смеси водой, затем следует очистка с помощью колоночной хроматографии, или перегонки под вакуумом, или другими методами. Выход полученного продукта составляет 30-50%.
Согласно патенту Испании № ES 549020 А1, 1 моль 1,3-дихлорацетона подвергают взаимодействию с 2 молями 1,2,4-триазола, затем полученный с низким выходом 1,3-бис(1,2,4-триазол-1-ил)-пропан-2-он подвергают взаимодействию с 2,4-дифторфенилмагнийбромидом с получением флуконазола. Выход составляет примерно 45% в расчете на реактив Гриньяра.
Общим признаком способов, описанных в патентах Испании №№ ES 549021 А1, ES 549022 А1 и ES 549684 А1, является то, что одну или обе триазольных группы флуконазола вводят в молекулу с использованием (1,2,4-триазол-1-ил)-метилмагнийгалогенида. В соответствии с описаниями выходы составляют примерно 45-55%. Известно, что реактивы Гриньяра, содержащие триазольные группы, являются нестабильными или иногда неактивными, поэтому они дают реакции с низкими выходами. При воспроизведении способов, описанных в этих патентах, выход всегда составлял ниже 10%.
В патенте Испании № ES 2026416 описан лучший способ, чем в вышеуказанных патентах. В соответствии с ним 1-(1,2,4-триазол-1-ил)-2-(2,4-дифторфенил)-3-галоген-пропан-2-ол подвергают взаимодействию с 4-амино-1,2,4-триазолом, и полученный 1-(1,2,4-триазол-1-ил)-2-(2,4-дифторфенил)-3-(4-амино-1,2,4-триазол-1-ил)-пропан-2-ол диазотируют и образованную таким образом соль диазония гидролизуют для удаления аминогруппы. Приведенные выходы составляют 78% для первой стадии и 85% для второй стадии. Этот способ имеет несколько недостатков с промышленной точки зрения. Первым является то, что производное З-галоген-пропан-2-ола, используемое в качестве исходного материала, синтезируют из эпокси-производного формулы (IV) путем кипячения с обратным холодильником в агрессивной среде галогеноводорода. Другим недостатком является то, что используемый в качестве реагента 4-амино-1,2,4-триазол может быть куплен только как продукт тонкого органического синтеза. Реакция диазотирования и гидролиз соли диазония в промышленном масштабе являются очень опасными процессами. Наконец, суммарный выход многостадийного способа составляет лишь 42-43%.
В выпуске Journal of Ph. Sciences (Vol. 84, №12) за декабрь 1995 г. кристаллические формы I и II флуконазола, а также данные рентгеновской порошковой дифракции и спектры комбинационного рассеяния света для различных кристаллических модификаций описаны без способа их синтеза.
В патенте GB 2270521 описан синтез моногидрата флуконазола из безводного флуконазола. В соответствии с данными порошковой дифракции рентгеновских лучей безводный флуконазол, используемый в качестве исходного материала, идентичен кристаллической модификации II. В описании этого патента дается ссылка на патент US 4404219 в отношении синтеза этой кристаллической модификации, но в нем отсутствует указание кристаллической модификации продукта.
В задачу данного изобретения входит экономичный синтез чистого или легкоочищаемого конечного продукта флуконазола без использования реагентов, трудных при применении в промышленном масштабе, и выделение образованного флуконазола в его желаемой кристаллической модификации I или II, а также в задачу входит сделать возможным превращение этих различных кристаллических модификаций друг в друга.
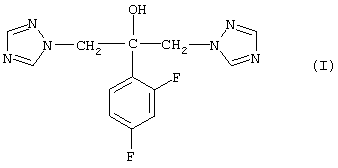
Таким образом, в настоящем изобретении предложен способ синтеза моногидрата флуконазола формулы (I)

отличающийся тем, что
осуществляют гидролиз производного силилового эфира формулы (II)

где значение R2 представляет собой водород либо С1-С10алкильную или фенильную группу, R3 и R4 независимо друг от друга представляют собой C1-С10алкильную или фенильную группу, при рН либо ниже 3, либо выше 8, в водном растворе,
затем охлаждают полученную реакционную смесь, содержащую флуконазол формулы (I), и выделяют осажденный моногидрат флуконазола.
Предпочтительно гидролиз производного силилового эфира формулы (II), где значения R2, R3 и R4 такие, как определено выше, осуществляют в водном метанольном растворе в присутствии гидроксида натрия.
Предпочтительно гидролиз производного силилового эфира формулы (II), где значения R2, R3 и R4 такие, как определено выше, осуществляют в водном растворе гидроксида натрия.
Предпочтительно в качестве исходного материала используют производное силилового эфира формулы (II), где R2, R3 и R4 представляют собой метильные группы.
Кроме того, в настоящем изобретении предложен способ синтеза кристаллической модификации II флуконазола формулы I

отличающийся тем, что
безводный флуконазол или его моногидрат растворяют в С1-С4-спирте с прямой или разветвленной цепью при температуре кипения и охлаждают этот раствор со скоростью 5-15°С/час с получением кристаллической модификации II флуконазола.
Предпочтительно моногидрат флуконазола, используемый для синтеза кристаллической модификации II флуконазола, получают путем гидролиза силил-флуконазола, как определено выше.
Предпочтительно раствор безводного флуконазола или его моногидрата в изопропаноле, полученный при температуре кипения, охлаждают со скоростью 10°С/час.
Предпочтительно раствор безводного флуконазола или его моногидрата в этаноле, полученный при температуре кипения, охлаждают со скоростью 10°С/час.
Предпочтительно раствор безводного флуконазола или его моногидрата во втор-бутаноле, полученный при температуре кипения, охлаждают со скоростью 10°С/час.
Более предпочтительно указанные растворы охлаждают до 0°С.
Кроме того, в настоящем изобретении предложен способ синтеза кристаллической модификации II флуконазола формулы (I)

отличающийся тем, что
моногидрат флуконазола сушат при 30-70°С с получением кристаллической модификации II флуконазола.
Предпочтительно моногидрат флуконазола сушат в присутствии затравочных кристаллов кристаллической модификации II.
Более предпочтительно указанную сушку осуществляют при перемешивании в вакууме при 40°С в течение 2 часов, а затем при 70°С в течение 4 часов.
Кроме того, в настоящем изобретении предложен способ синтеза кристаллической модификации I флуконазола формулы (I)

отличающийся тем, что
моногидрат флуконазола сушат при 80°С с получением кристаллической модификации I флуконазола.
Предпочтительно моногидрат флуконазола сушат в присутствии затравочных кристаллов кристаллической модификации I.
Более предпочтительно указанную сушку осуществляют при перемешивании в вакууме при 80°С в течение 4 часов, до тех пор, пока масса не станет постоянной.
Основой данного изобретения является открытие того, что силиловые эфиры формулы (II), которые представляют собой желаемые соединения патента США №5707976,

где значение R2 представляет собой водород либо C1-С10алкильную или фенильную группу, R3 и R4 независимо друг от друга представляют собой С1-С10алкильные или фенильные группы, в водных кислых или основных условиях могут быть количественно гидролизованы до флуконазола формулы (I). Соединения формулы (II) могут быть получены в соответствии с патентом США №5707976, например, из подходящим образом замещенных эпокси-производных формулы (IV) с использованием подходящим образом замещенного силилтриазола формулы (V), где значения R2, R3 и R4 такие, как описано выше, в присутствии сильного основания в качестве катализатора.

Поскольку полученные силил-флуконазольные производные являются очень аполярными вследствие присутствия триалкилсилильной группы, их легко отделить от примесей и они могут быть экономично синтезированы в очень чистой форме.
В соответствии с настоящим изобретением моногидрат флуконазола формулы (I) синтезируют путем осуществления гидролиза производного силилового эфира формулы (II) в водном растворе при рН предпочтительно либо ниже 3 либо выше 8.
Гидролиз представляет собой быстрый процесс. Например, триметилсилиловый эфир флуконазола полностью гидролизуется при рН выше 10 в 10% водном растворе диметилформамида при комнатной температуре в течение 10 минут. В сходных условиях, но при рН ниже 2 гидролиз полностью проходит за 0,5-1 ч.
Гидролиз можно осуществить в нейтральных условиях в гомогенной фазе в присутствии воды при повышенной температуре, предпочтительно при температуре дефлегмации. Быстрый и промышленно эффективный гидролиз предпочтительно осуществляют либо при рН меньше 3 либо при рН больше 8. Этот гидролиз является очень мягким, нежелательные побочные продукты не образуются даже в следовых количествах, следовательно, при гидролизе надлежащим образом очищенных производных силиловых эфиров формулы (II) может быть синтезирован очень чистый флуконазол и он может быть выделен из реакционной смеси в виде моногидрата.
Гидролиз предпочтительно осуществляют в гомогенной фазе, в смеси протонного или апротонного диполярного растворителя, смешивающегося с водой, и воды при рН, указанном выше. Флуконазол, образовавшийся в этой реакции, предпочтительно выделяют путем разбавления реакционной смеси водой и охлаждения. Вследствие охлаждения образовавшийся флуконазол кристаллизуется из реакционной смеси в виде очень чистого моногидрата и может быть выделен, например, путем фильтрования.
Моногидрат является стабильным при комнатной температуре; он превращается в безводный флуконазол, так называемый ангидрат, между 40 и 90°С со скоростью, зависящей от условий дегидратации.
Полиморфные модификации имеют различную кристаллическую структуру, кристаллографические константы (расстояния и энергии кристаллической решетки) и, следовательно, имеют разную скорость растворения. Различные полиморфные модификации могут быть дифференцированы друг от друга по их спектрам комбинационного рассеяния. На фиг.1 и 2 показаны спектры комбинационного рассеяния кристаллической модификации I и II флуконазола между 3500,0 и 200,0 см-1, в то время как на фиг.3 и 4 показана область между 3300,0 и 2800,0 см-1, где могут быть обнаружены характеристические отличия для кристаллических модификаций I и II флуконазола.
В терапии непременным условием воспроизводимого перманентного эффекта твердых фармацевтических лекарственных форм (например, пероральных лекарственных форм) является то, что растворение активного ингредиента должно быть постоянным в случае различных партий. По этой причине желательно всегда использовать одну и ту же кристаллическую модификацию тех активных ингредиентов, которые имеют несколько кристаллических модификаций, например флуконазола.
При приготовлении препарата условия образования кристаллической модификации I и II были подробно изучены для удовлетворения морфологическим требованиям флуконазола.
Неожиданно было обнаружено, что если раствор безводного флуконазола или моногидрата флуконазола, полученный путем растворения их в С1-С4-спирте с прямой или разветвленной цепью при температуре кипения, медленно охлаждать, предпочтительно со скоростью 5-15°С/час, тогда осажденные и высушенные кристаллы являются идентичными кристаллической модификации II флуконазола.
Кристаллические модификации I и II можно получать путем высушивания моногидрата флуконазола при различных температурах. В этом случае применение соответствующих затравочных кристаллов способствует образованию желаемой модификации.
Если моногидрат флуконазола сушат после затравливания кристаллами кристаллической модификации II медленно, предпочтительно в вакууме, при температуре между 30 и 70°С, тогда образуется кристаллическая модификация II. Если сушку осуществляют быстро при 80°С, тогда из моногидрата флуконазола образуется кристаллическая модификация I.
Спирты, используемые при кристаллизации, могут представлять собой спирты с разветвленной цепью, предпочтительно изопропанол или втор-бутанол, либо спирты с прямой цепью, предпочтительно этанол. Содержание воды в С1-С4-спиртах с прямой или разветвленной цепью, используемых при кристаллизации, может даже достигать 5%. Таким образом, качество чистоты является достаточным в случае 96%-ного этанола. Наилучшие результаты получают при использовании изопропанола.
В таблице 1 показаны данные рентгеновской порошковой дифракции (РПД) кристаллических модификаций I и II флуконазола, как измерено на образцах Примеров 2 и 5. (Рентгеновский порошковый дифрактометр Philips PW 1840; CuKα излучение 30 кВ и 30 мА; скорость гониометра 0,05 °2θ/с; чувствительность: 2·103 имп/с; Т.С.: 5 с.; ширина щели 0,05 мм).
На Фиг.5 и 6 показаны картины рентгеновской порошковой дифракции (РПД) образцов Примеров 2 и 5. Картина рентгеновской порошковой дифракции образца Примера 3 является такой же, как у образца Примера 2; картины рентгеновской порошковой дифракции образцов Примеров 6, 7 и 8 являются такими же, как у образца Примера 5.
Способ по изобретению проиллюстрирован следующими неограничивающими Примерами.
Пример 1
Моногидрат 2-(2,4-дифторфенил)-1,3-бис(1,2,4-триазол-1-ил)-пропан-2-ола
Смесь 7,50 г (0,02 моль) 2-(2,4-дифторфенил)-1,3-бис(1,2,4-триазол-1-ил)-2-(триметилсилилокси)пропана, 25 мл метанола, 2 мл воды и 1,0 мл конц. соляной кислоты перемешивали при 30°С в течение 1 часа. Реакционную смесь концентрировали до объема 10 мл и после добавления 50 мл воды рН горячего раствора подводили до 8 с помощью 10% водного раствора гидроксида натрия. После охлаждения осажденные кристаллы отфильтровывали и сушили при 40°С до тех пор, пока масса не стала постоянной, с выходом 6,06 г (93,5%) соединения, указанного в заголовке. Т. пл. 139-140°С.
Пример 2
Синтез кристаллической модификации I флуконазола
Смесь 7,5 г (0,02 моль) 2-(2,4-дифторфенил)-1,3-бис(1,2,4-триазол-1-ил)-2-(триметилсилилокси)пропана, 40 мл метанола, 3 мл воды и 0,1 г гидроксида натрия перемешивали при комнатной температуре в течение 1 часа. После добавления 300 мл воды раствор концентрировали до объема 50 мл с помощью вакуумной перегонки. Полученную суспензию охлаждали до 0°С и фильтровали. Масса полученного продукта составляла 6,12 г, содержание воды - 11,5%. После сушки при 80°С получали 5,35 г соединения, указанного в заголовке. Выход: 87,4%. Т. пл. 139-141°С.
Пример 3
Синтез кристаллической модификации I флуконазола
Смесь 7,5 г (0,02 моль) 2-(2,4-дифторфенил)-1,3-бис(1,2,4-триазол-1-ил)-2-(триметилсилилокси)пропана, 40 мл метанола, 3 мл воды и 0,1 г гидроксида натрия перемешивали при комнатной температуре в течение 1 часа. После добавления 300 мл воды раствор концентрировали до объема 50 мл с помощью вакуумной перегонки. Полученную суспензию охлаждали до 0°С и фильтровали. Получили 6,12 г продукта, содержание воды составляло 11,5%. Его помещали в колбу на 100 мл и добавляли 0,1 г затравочных кристаллов кристаллической модификации I флуконазола. Соединение сушили на роторном испарителе при 80°С в течение 3-4 часов, до тех пор, пока масса не стала постоянной. Получали 5,45 г соединения, указанного в заголовке. Выход: 87,4%. Т. пл. 139-141°С.
Пример 4
Синтез моногидрата флуконазола
Смесь 7,58 г (0,02 моль) 2-(2,4-дифторфенил)-1,3-бис(1,2,4-триазол-1-ил)-2-(триметилсилилокси)пропана, 0,04 г гидроксида натрия и 70 мл воды перемешивали при 80°С в течение 10 минут. Затем добавляли 0,5 г угля и горячий раствор фильтровали. Фильтрат охлаждали до 0°С. Осажденные кристаллы отфильтровывали и сушили при 40°С до тех пор, пока масса не стала постоянной, с выходом 5,98 г (92,1%) соединения, указанного в заголовке. Содержание воды 5,6%, Т. пл. 139-140°С.
Пример 5
Синтез кристаллической модификации II флуконазола
6,12 г (0,02 моль) безводного флуконазола растворяли в 60 мл изопропанола с перемешиванием при 70°С, а затем раствор охлаждали. После того как температура достигла 50°С, скорость охлаждения составляла 10°С/час. Осаждение кристаллов началось примерно при 40°С. Через 5 часов, когда температура достигла 0°С, кристаллическую модификацию II флуконазола фильтровали и сушили при 50°С до тех пор, пока масса не стала постоянной, с получением 5,85 г (91,2%) соединения, указанного в заголовке. Т. пл. 139-141°С.
Пример 6
Синтез кристаллической модификации II флуконазола
6,12 г (0,02 моль) флуконазола растворяли в 25 мл этанола с перемешиванием при 50°С, затем раствор медленно охлаждали с постоянной скоростью (10°С/час) до °С. Осаждение кристаллов началось примерно при 40°С. Осажденную кристаллическую модификацию II флуконазола отфильтровывали и сушили при 50°С до тех пор, пока масса не стала постоянной, с получением 5,23 г (85,5%) соединения, указанного в заголовке. Т. пл. 139-140°С.
Пример 7
Синтез кристаллической модификации II флуконазола
6,12 г (0,02 моль) флуконазола растворяли в 60 мл втор-бутанола при 60°С, затем раствор охлаждали до 0°С со скоростью 10°С/час. Осаждение началось примерно при 42°С. Кристаллы фильтровали и сушили при 50°С до тех пор, пока масса не стала постоянной, с получением 5,70 г (93,1%) соединения, указанного в заголовке. Т. пл. 139-140°С.
Пример 8
Синтез кристаллической модификации II флуконазола
Смесь 7,5 г (0,02 моль) 2-(2,4-дифторфенил)-1,3-бис(1,2,4-триазол-1-ил)-2-(триметилсилилокси)пропана, 40 мл метанола, 3 мл воды и 0,1 г гидроксида натрия перемешивали при комнатной температуре в течение 1 часа. После добавления 300 мл воды раствор концентрировали до объема 50 мл с помощью вакуумной перегонки. Полученную суспензию охлаждали до 0°С и фильтровали. Получили 6,00 г моногидрата флуконазола, содержание воды составляло 11,0%. Его помещали в колбу на 100 мл и добавляли 0,1 г затравочных кристаллов кристаллической модификации II флуконазола. Соединение сушили на роторном испарителе при давлении 13,3 кПа, так что кристаллы моногидрата нагревались в течение 3 часов от 30 до 70°С, и удерживали при этой температуре до тех пор, пока масса не стала постоянной (приблизительно 2 часа). Получили 5,4 г соединения, указанного в заголовке. Т. пл. 139-141°С. На фиг.7 приведены данные рентгеновской порошковой дифракции для моногидрата.

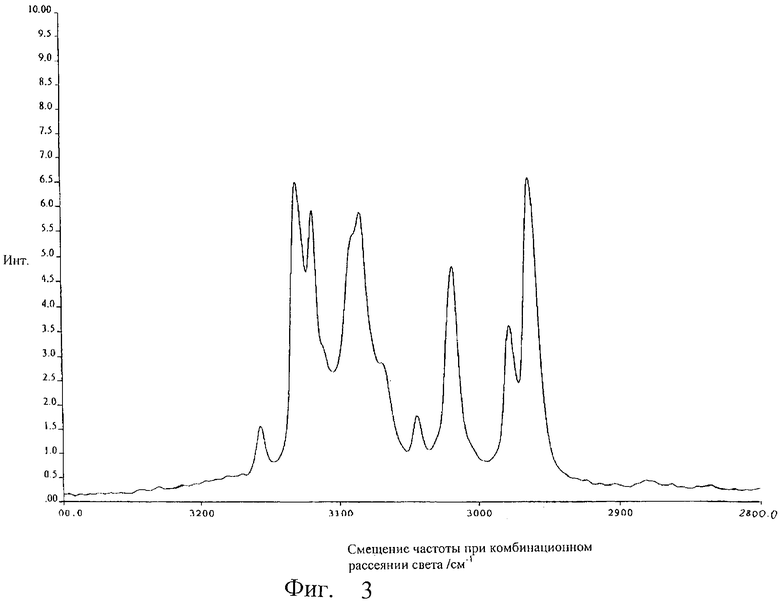




Описывается способ синтеза моногидрата флуконазола формулы (I),  путем гидролиза производного силилового эфира формулы (II),
путем гидролиза производного силилового эфира формулы (II), 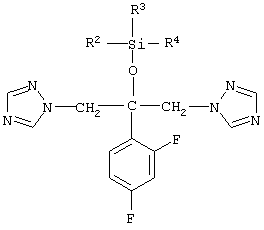 где R2 означает водород либо C1-С10алкильную или фенильную группу, R3 и R4 независимо друг от друга означают C1-С10алкильную или фенильную группу, при рН либо ниже 3, либо выше 8, в водном растворе, с последующим охлаждением полученной реакционной смеси и выделением осажденного моногидрата флуконазола. Описываются также способы синтеза кристаллической модификации II флуконазола формулы I, в котором безводный флуконазол или его моногидрат растворяют в С1-С4-спирте с прямой или разветвленной цепью при температуре кипения и охлаждают этот раствор со скоростью 5-15°С/час или моногидрат флуконазола сушат при 30-70°С. Кроме того описывается способ синтеза кристаллической модификации I флуконазола формулы I, в котором моногидрат флуконазола сушат при 80°С. Описываемое изобретение позволяет получить чистый или легкоочищаемый флуконазол в кристаллических модификациях, позволяющих легко получать из них удобные лекарственные формы. 4 н. и 12 з.п. ф-лы, 1 табл., 7 ил.
где R2 означает водород либо C1-С10алкильную или фенильную группу, R3 и R4 независимо друг от друга означают C1-С10алкильную или фенильную группу, при рН либо ниже 3, либо выше 8, в водном растворе, с последующим охлаждением полученной реакционной смеси и выделением осажденного моногидрата флуконазола. Описываются также способы синтеза кристаллической модификации II флуконазола формулы I, в котором безводный флуконазол или его моногидрат растворяют в С1-С4-спирте с прямой или разветвленной цепью при температуре кипения и охлаждают этот раствор со скоростью 5-15°С/час или моногидрат флуконазола сушат при 30-70°С. Кроме того описывается способ синтеза кристаллической модификации I флуконазола формулы I, в котором моногидрат флуконазола сушат при 80°С. Описываемое изобретение позволяет получить чистый или легкоочищаемый флуконазол в кристаллических модификациях, позволяющих легко получать из них удобные лекарственные формы. 4 н. и 12 з.п. ф-лы, 1 табл., 7 ил.

отличающийся тем, что осуществляют гидролиз производного силилового эфира формулы (II)

где значение R2 представляет собой водород либо C1-С10алкильную или фенильную группу, R3 и R4 независимо друг от друга представляют собой C1-С10алкильную или фенильную группу, при рН либо ниже 3, либо выше 8, в водном растворе,
затем охлаждают полученную реакционную смесь, содержащую флуконазол формулы (I), и выделяют осажденный моногидрат флуконазола.
отличающийся тем, что
безводный флуконазол или его моногидрат растворяют в С1-С4-спирте с прямой или разветвленной цепью при температуре кипения и охлаждают этот раствор со скоростью 5-15°С/ч.

отличающийся тем, что
моногидрат флуконазола сушат при 30-70°С.
отличающийся тем, что моногидрат флуконазола сушат при 80°С.
| ЭКРАННАЯ ПРОТИВОМЕТЕОРИТНАЯ ЗАЩИТА КОЛЛЕКТОРОВ ТЕПЛОНОСИТЕЛЯ ХОЛОДИЛЬНИКА-ИЗЛУЧАТЕЛЯ | 1995 |
|
RU2078719C1 |
| ПРЕОБРАЗОВАТЕЛЬ СВЕТОВОЙ ЭНЕРГИИ В ЭЛЕКТРИЧЕСКУЮ НА ОСНОВЕ P-N-ПЕРЕХОДА С ПОВЕРХНОСТНЫМ ИЗОТИПНЫМ ГЕТЕРОПЕРЕХОДОМ | 1996 |
|
RU2099818C1 |
| US 5707976 А, 13.01.1998 | |||
| WO 9832744 A, 30.07.1998 | |||
| СПОСОБ ПОЛУЧЕНИЯ ФЛУКОНАЗОЛА | 2000 |
|
RU2163804C1 |
| ЛЕКАРСТВЕННОЕ СРЕДСТВО, СОДЕРЖАЩЕЕ ФЛУКОНАЗОЛ И СПОСОБ ПОЛУЧЕНИЯ ФЛУКОНАЗОЛА | 2000 |
|
RU2173994C1 |

