Настоящее изобретение относится к способу получения пикриновой кислоты.
Пикриновая кислота, или 2,4,6-тринитрофенол, широко используется в промышленности, в частности при изготовлении взрывчатых веществ, лекарственных средств, красителей и в других областях [Tadeusz Urbanski, Chemistry and Technology of Explosives, Pergamon Press, с.499 (1964)].
Существуют два пути получения пикриновой кислоты, описанные в литературе, а именно с помощью сульфонитрования фенола или с помощью нитрования динитрофенола, полученного путем гидролиза хлординитробензола.
Первый путь заключается в осуществлении двух стадий - стадии сульфирования фенола, а затем стадии нитрования полученного сульфированного фенола.
Однако этому способу свойственны несколько недостатков: сульфирование является продолжительной и низко производительной стадией, а нитрование проводится уже в разбавленной среде, что ведет к снижению производительности способа.
Другой путь получения пикриновой кислоты включает также несколько стадий: нитрование монохлорбензола в хлординитробензол, последующий гидролиз полученного продукта, а затем нитрование динитрофенола с помощью смеси азотной кислоты и олеума.
Этот способ также не удовлетворяет требованиям, так как является длинным, сложным и приводит к образованию хлорида натрия на стадии гидролиза. В результате возникает проблема по обработке водных солевых отходов.
В основу заявленного изобретения положена задача создать способ, позволяющий избежать вышеперечисленные недостатки.
Для решения этой задачи предлагается способ получения пикриновой кислоты, заключающийся в том, что осуществляют нитрование о-нитрофенола и/или п-нитрофенола до динитрофенола таким образом, что этот образовавшийся промежуточный продукт остается растворимым в реакционной среде, а затем продолжают нитрование динитрофенола до получения пикриновой кислоты, которая выпадает в осадок.
В способе согласно изобретению образуют динитрофенол как промежуточный продукт. Более точно, речь идет о 2,4-динитрофенолe, когда исходным субстратом является паранитрофенол и смесь изомеров, а именно 2,4-динитрофенол и 2,6-динитрофенол, полученные из ортонитрофенола.
Совершенно неожиданно было найдено, что возможно получить пикриновую кислоту из ортонитрофенола и/или паранитрофенола при условии, что образующееся промежуточное вещество не выпадает в осадок в реакционной среде, так как оказывается, что выпавший в осадок продукт невозможно далее подвергать нитрованию.
Таким образом, важно осуществлять контроль за отсутствием осаждения 2,4-динитрофенола или осаждения смеси 2,4- и 2,6-динитрофенолов.
В соответствии с изобретением один или более образующихся промежуточных продуктов должны оставаться растворенными в реакционной среде, что вызывает необходимость в контроле за концентрацией исходного субстрата, которая зависит от метода осуществления способа - непрерывного или периодического. Хотя и допускается небольшое содержание промежуточных продуктов в виде осадка, предпочтительно менее 3, более предпочтительно менее 1, однако это снижает отчасти выход реакции.
Первый вариант осуществления изобретения состоит в том, что вводят азотную кислоту или нитрующую смесь в о-нитрофенол или/и в п-нитрофенол.
Под термином "нитрующая смесь" понимается азотная кислота в смеси с сильной кислотой.
О-нитрофенол может применяться в твердой форме, в виде расплава или раствора в сокислоте. Что касается п-нитрофенола, то это соединение может применяться в твердой форме, в растворе в сокислоте или в виде смеси с водой, в виде эвтектической смеси.
В описанном варианте осуществления изобретения необходимо следить, чтобы концентрация дифенола в реакционной среде была такой, чтобы дифенол оставался растворимым в реакционных условиях.
Таким образом, при заданной температуре концентрация дифенола должна поддерживаться на уровне более низком, чем концентрация, при которой начинается осаждение дифенола.
Количество солюбилизированного фенола зависит от значения реакционной температуры и кислотности среды.
Для сведения следует уточнить, не ограничивая при этом изобретение, что растворимость 2,4-динитрофенола при температуре 65oC в растворе 95%-ной серной кислоты составляет 12%.
Необходимо отметить, что контролирование концентрации промежуточного продукта распространяется также на другие варианты введения реагентов. Однако поскольку концентрация является фактором, определяющим производительность, могут быть использованы другие методы контроля, более предпочтительные.
Другой вариант выполнения изобретения состоит в постепенном введении п-нитрофенола и/или о-нитрофенола в реакционную среду, содержащую азотную кислоту или нитрующую смесь, или состоит во введении одновременно п-нитрофенола и/или о-нитрофенола и азотной кислоты или нитрующей смеси в донную часть начальной реакционной емкости, содержащей воду или сокислоту.
Под постепенным введением понимают непрерывную подачу реагентов или подачу их несколькими порциями.
О-нитрофенол может использоваться как в твердом состоянии, так и в состоянии расплава или раствора в сокислоте. Что касается п-нитрофенола, то его можно использовать в твердом виде, в растворе в сокислоте или в смеси с водой, в виде эвтектической смеси.
В подобном случае скорость введения о-нитрофенола и/или п-нитрофенола должна быть выбрана более низкой, чем скорость нитрования динитрофенола.
Скорость нитрования зависит от многих параметров, а именно от температуры, концентрации азотной кислоты, сокислоты и динитрофенола.
В этом случае можно иметь концентрацию исходного субстрата более высокую, сохраняя при этом растворимость динитрофенола в реакционной среде.
Можно выбрать значительную скорость нитрования, осуществляя при этом обычное дозирование и анализируя полученный осадок, который должен представлять собой пикриновую кислоту.
Исходные продукты, используемые в способе согласно заявляемому изобретению представляют собой о-нитрофенол или п-нитрофенол или смесь названных соединений.
Когда исходный субстрат вводят в растворе сокислоты, его концентрация обычно составляет от 20 до 80 мас.%, предпочтительно 25 - 35 мас.%.
В соответствии с заявляемым изобретением стадия нитрования может быть проведена согласно двум вариантам осуществления.
Первый вариант состоит в использовании только азотной кислоты.
Используют водный раствор азотной кислоты, имеющий различную концентрацию, которая может изменяться в пределах от 30 до 100%. Однако предпочтительной является концентрация, выбранная в пределах от 68 до 100%.
В подобном случае азотная кислота вводится с большим избытком. Количество азотной кислоты в 4 - 10 раз превышает массу о-нитрофенола и/или п-нитрофенола.
Другой вариант выполнения изобретения состоит в том, что азотную кислоту соединяют с другой сильной кислотой, которую далее будут называть просто "сокислота".
Под сильной кислотой в настоящем изобретении понимают кислоту, имеющую pKa в воде менее -0,1, а предпочтительно менее -1,0.
Величина pKa определяется как константа ионной диссоциации пары кислота/основание, когда вода используется в качестве растворителя.
Среды кислот, отвечающих этому определению, предпочтительно использовать те, которые стабильны в отношении азотного окисления.
Более конкретно, можно назвать галогенсодержащие и не содержащие галоген оксикислоты, например серную кислоту, пиросерную кислоту, фосфорную кислоту, полифосфорные кислоты, хлорную кислоту, галогенсульфокислоты, например, фторсульфокислоту, хлорсульфокислоту или трифторметансульфокислоту, метансульфокислоту, этансульфокислоту, этандисульфокислоту, бензолсульфосиклоту, бензолдисульфокислоты, толуолсульфокислоты, нафталинсульфокислоты и нафталиндисульфокислоты.
Среды этих кислот выбирают преимущественно серную кислоту или фосфорную кислоту.
Предпочтительной сокислотой является серная кислота, причем предпочтение отдают концентрированному раствору серной кислоты: концентрация серной кислоты преимущественно выбирается выше 90 мас.%, а еще более предпочтительно выше 98 мас.%.
Возможен вариант замены серной кислоты на олеумы: при этом олеумы содержат SO3 в различных количествах, но предпочтительно от 20 до 50%.
Количество используемой азотной кислоты, выраженное соотношением количества азотной кислоты и количества молей о-нитрофенола и/или п-нитрофенола, может изменяться между примерно 2 и примерно 3, а более точно между 2 и 2,2.
Количество используемой сокислоты может изменяться в широких пределах. В основном оно составляет 1 - 10-кратную величину от массы о-нитрофенола и/или п-нитрофенола.
Способ согласно настоящему изобретению целесообразно проводить при температуре, равной от 40 до 100oC, но предпочтительно при температуре, выбранной в интервале от 40 до 70oC.
Способ в основном осуществляют при атмосферном давлении, но возможно также вести процесс при несколько пониженном давлении, составляющем, например, 500 - 760 мм рт.ст.
При осуществлении заявляемого способа нет необходимости в создании атмосферы благородного газа.
С практической точки зрения заявляемый способ легко осуществим, так как не связан с необходимостью использования специального оборудования.
Практический способ может быть осуществлен нижеследующим образом.
В аппарат загружают различные составляющие реакционной смеси.
Используя несколько вариантов, описанных выше, осуществляют следующие операции введения реагентов:
- введение азотной кислоты или нитрующей смеси в о-нитрофенол и/или п-нитрофенол;
- введение о-нитрофенола и/или п-нитрофенола в азотную кислоту или нитрующую смесь;
- введение параллельно о-нитрофенола и/или п-нитрофенола и азотной кислоты или нитрующей смеси в донную часть реакционной емкости, содержащей воду или сокислоту.
Последний вариант является предпочтительным, поскольку в донной части реакционной емкости находится раствор сокислоты, предпочтительно раствор концентрированной серной кислоты, имеющей предпочтительно концентрацию, превышающую 90%.
Продолжительность подачи о-нитрофенола и/или п-нитрофенола может изменяться, например, в пределах от 30 мин до 4 ч, а предпочтительно от 1 до 2 ч.
После введения реагентов выдерживают реакционную смесь в температурном режиме, указанном выше, в течение некоторого времени (15 - 30 мин).
В конце реакции пикриновая кислота оседает в виде осадка в реакционной среде.
Осадок может быть отделен в соответствии с известными методами разделения твердых и жидких веществ, но предпочтительно путем фильтрования.
Полученный продукт промывают один или несколько раз, предпочтительно 2 раза, используя воду.
Таким образом, выделяют пикриновую кислоту.
Преимущество способа согласно изобретению состоит в том, что получение пикриновой кислоты осуществляется, минуя образование расплавленной фазы, что позволяет избежать опасность взрыва на стадиях производства пикриновой кислоты.
Примеры, которые следуют ниже, иллюстрируют настоящее изобретение, но никак не ограничивают его.
В примерах упомянутыe выходы соответствуют следующему определению:
Сокращения, использованные в примерах, имеют следующие значения:
- ONP = о-нитрофенол.
- ONP = п-нитрофенол.
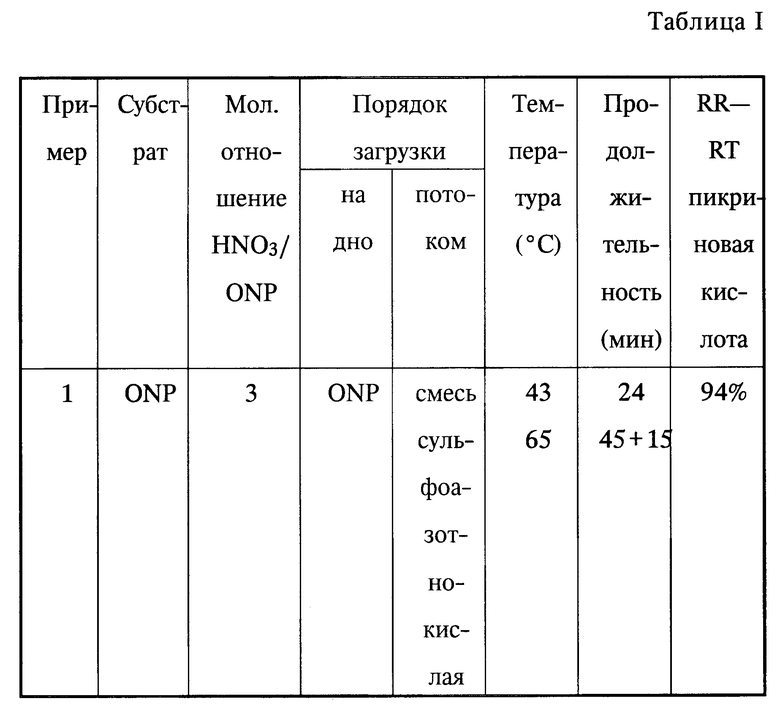
Пример 1.
В короткую трубку, снабженную перемешивателем, холодильником, счетчиком пузырьков и двумя шприцами и нагреваемую с помощью термостатируемой ванны, загружают 15 г водного раствора 95%-ной серной кислоты, 2,74 г (20 ммоль) о-нитрофенола.
При перемешивании вводят 9,58 г (или 60,8 ммоль азотной кислоты) сульфоазотнокислой смеси, содержащей 57% серной кислоты, 40% азотной кислоты и 3% воды.
Сначала прибавляют треть названной смеси при температуре 43oC в течение 24 мин, затем прибавляют две трети названной смеси при температуре 65oC в течение 45 мин. Затем поддерживают нагрев на уровне 65oC в течение 15 мин.
Реакционную смесь охлаждают, после чего ее выливают на смесь 300 мл вода-лед/сульфамиловая кислота (1 г) и достаточное количество метанола для получения общего объема в 1000 мл.
Реагенты, которые не прореагировали, и полученные продукты анализировали с помощью жидкостной высокоэффективной хроматографии.
Полученные результаты представлены в таблице I.
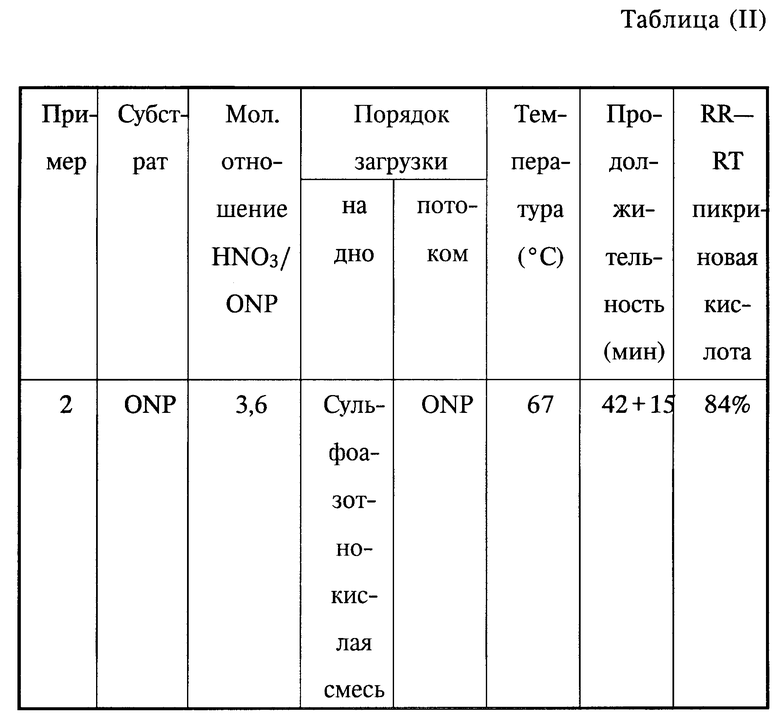
Пример 2.
В трехгорлую колбу емкостью 50 мл, снабженную перемешивателем, холодильником, счетчиком пузырьков и подающим шприцем и нагреваемую с помощью термостатируемой ванны, загружают 20 г сульфоазотнокислой смеси, содержащей 57% серной кислоты, 39% азотной кислоты и 4% воды (или 124 ммоль азотной кислоты).
При перемешивании нагревают до 67oC, затем вливают в течение 42 мин 23,76 г (34,2 ммоль) о-нитрофенола в виде 20%-ного раствора в 95%-ной серной кислоте.
После этого поддерживают нагревание на уровне 67oC в течение 15 мин.
Реакционную смесь охлаждают, а затем выливают на 300 мл смеси вода-лед/сульфаминовая кислота (1 г) и метанол, взятый в количестве, достаточном для получения общего объема в 1000 мл.
Реагенты, которые не прореагировали, и полученные продукты анализировали с помощью высокоэффективной жидкостной хроматографии.
Полученные результаты приведены в таблице II.
Пример 3.
В трехгорлую колбу емкостью 100 мл, снабженную перемешивателем, холодильником, счетчиком пузырьков и двумя шприцами и нагреваемую с помощью термостатируемой ванны, загружают 18,3 г водного раствора 95%-ной серной кислоты.
При перемешивании нагревают до 67oC, а затем в названный раствор вливают одновременно в течение 60 мин 28,2 (40,6 ммоль) о-нитрофенола в виде 20%-ного раствора в 95%-ной серной кислоте и 19,52 г сульфоазотнокислой смеси, содержащей 57% серной кислоты, 39% азотной кислоты и 4% воды (или 121 ммоль азотной кислоты).
После этого нагрев до 67oC поддерживают еще 15 мин.
Охлаждают реакционную смесь и выливают ее на 300 мл смеси вода-лед/сульфаминовая кислота (1 г) и на метанол, взятый в количестве, достаточном для получения общего объема, равного 1000 мл.
Реагенты, которые не прореагировали, и полученные продукты анализируют с помощью высокоэффективной жидкостной хроматографии.
Полученные результаты приведены в таблице III.
Пример 4.
В трехгорлую колбу емкостью 100 мл, снабженную перемешивателем, холодильником, счетчиком пузырьков и двумя шприцами и нагреваемую с помощью термостатируемой ванны, загружают 18,3 г водного раствора 95%-ной серной кислоты.
При перемешивании нагревают до 67oC, после чего в названный раствор вливают одновременно в течение 56 мин 28,34 г (40,8 ммоль) о-нитрофенола в виде 20%-ного раствора в 95%-ной серной кислоте и 13,58 г сульфоазотнокислой смеси, содержащей 57% серной кислоты, 39% азотной кислоты и 4% воды (или 84 ммоль азотной кислоты).
После этого нагрев до 67oC поддерживают в течение 19 мин.
Реакционную смесь охлаждают, а затем выливают на 300 мл смеси вода-лед/сульфаминовая кислота (1 г) и на метанол, взятый в количестве, достаточном для получения общего объема в 1000 мл.
Реагенты, которые не прореагировали, и полученные продукты анализируют путем высокоэффективной жидкостной хроматографии.
Полученные результаты приведены в таблице IV.
Пример 5.
В трехгорлую колбу емкостью 100 мл, снабженную перемешивателем, холодильником, счетчиком пузырьков и двумя шприцами и нагреваемую с помощью термостатируемой ванны, вводят 18,3 г водного раствора 95%-ной серной кислоты.
При перемешивании нагревают до 67oC, а затем в указанный раствор одновременно вливают в течение 60 мин 28,2 г (40,6 ммоль) п-нитрофенола в виде 20%-ного раствора в 95%-ной серной кислоте и 19,52 г сульфоазотнокислой смеси, содержащей 57% серной кислоты, 39% азотной кислоты и 4% воды (или 121 ммоль азотной кислоты).
После этого нагрев до 67oC поддерживают в течение 15 мин.
Реакционную среду охлаждают, а затем выливают на 300 мл смеси вода-лед/сульфаминовая кислота (1 г) и на метанол, взятый в количестве, достаточном для получения общего объема в 1000 мл.
Реагенты, которые не прореагировали, и полученные продукты анализируют с помощью высокоэффективной жидкостной хроматографии.
Полученные результаты приведены в таблице V.



Пикриновая кислота, или 2,4,6-тринитрофенол, широко используется при изготовлении взрывчатых веществ, лекарственных средств, красителей и в других областях. Способ получения пикриновой кислоты заключается в нитровании о-нитрофенола и/или п-нитрофенола таким образом, чтобы образующийся продукт был растворим в реакционной среде, и продолжают нитрование до получения пикриновой кислоты, которая выпадает в осадок. При осуществлении способа нет необходимости в создании атмосферы благородного газа, следовательно, нет необходимости использовать специальноe оборудованиe. 15 з.п. ф-лы, 5 табл.
| Способ получения пикриновой кислоты нитрованием динитрофенола | 1949 |
|
SU82150A1 |
| ГАЗОВАЯ ГОРЕЛКА ДЛЯ РЕЗКИ С ЗАЩИТНОЙ ГАЗОВОЙ | 0 |
|
SU368770A1 |
| Экономайзер | 0 |
|
SU94A1 |
| Усилительное устройство низкой частоты | 1926 |
|
SU11005A1 |

