Изобретение относится к биохимии, в частности, к применению известных производных 1,2,5-оксадиазоло[3,4-d]пиридазин-5,6-диоксида общей формулы I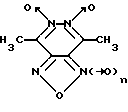
где n = 0 или 1, в качестве специфических регуляторов (активаторов или ингибиторов) активности нуклеотид-зависимых ферментов.
Согласно данному изобретению предпочтительно применение соединений общей формулы I для регуляции активности следующих нуклеотид-зависимых ферментов: растворимой формы гуанилатциклазы (гуанозин-5'-трифосфат-зависимый фермент), глицеральдегид-3-фосфат дегидрогеназы и алкогольдегидрогеназы /никотинамидадениндинуклеотид-зависимые ферменты/, а также H,K-аденозин-5'-трифосфатазы (аденозин-5'-трифосфат-зависимый фермент).
Гуанилатциклаза /КФ 4.6.1.2; гуанозин-5'-трифосфат-пирофосфатлиаза (циклизующая)/ является ферментом, катализирующим биосинтез гуанозин-3', 5'-циклофосфата (цГМФ) - универсального регулятора внутриклеточного метаболизма [1]. Растворимая форма фермента (рГЦ) активируется оксидом азота в результате его взаимодействия с атомом железа гема, входящего в состав фермента, и образования комплекса нитрозил-гем.
В настоящее время показано влияние доноров оксида азота и его биологически активных форм (восстановленной формы NO-/HNO, пероксинитрита, нитрозотиолов) на активность ряда других ферментов. В число таких ферментов входят глицеральдегид-3-фосфат дегидрогеназа и алкогольдегидрогеназа.
Глицеральдегид-3-фосфат дегидрогеназа /никотинамидадениндинуклеотид (НАД)-зависимая /(КФ 1.2.1.12) (ГАФД) является ферментом, катализирующим окислительное фосфорилирование 3-фосфоглицеринового альдегида и играющим важную роль в различных метаболических и регуляторных процессах (например, гликолиз, глюконеогенез, репарация ДНК, гибель клеток, секреция нейромедиаторов, фосфорилирование белков и др.). Показано ингибирование активности ГАФД под действием NO и его доноров (например, 3-(4-морфолино)сиднонимина) [2]. Кроме того, NO, нитропруссид натрия, пероксинитрит и другие доноры активных форм оксида азота также активируют НАД-зависимую ковалентную модификацию фермента [3].
Алкогольдегидрогеназа (НАД-зависимая) (КФ 1.1.1.1) - фермент, катализирующий окисление этанола до ацетальдегида. Показано ингибирование активности алкогольдегидрогеназы под действием NO и его доноров (например, комплекса NO с диэтиламином, S-нитрозо-N-ацетилпеницилламина) [4].
Тест на ингибирование активности алкогольдегидрогеназы предложен для использования при создании фармакологических средств для лечения алкоголизма [5] . Запатентовано применение фуроксанов в ряду других доноров оксида азота для профилактики и лечения заболеваний печени, вызванных действием алкоголя или других токсических веществ [6].
H,K-аденозин-5'-трифосфатаза (H,K-АТФаза) (КФ 3.6.1.36) - фермент, катализирующий гидролиз АТФ, сопряженный с транспортом ионов H+ и K+ через мембрану. Влияние NO и/или его доноров на активность H,K-АТФазы не изучено.
Известны различные N-оксиды и близкие к ним по строению соединения, являющиеся донорами оксида азота и/или его вышеуказанных биологически активных форм и активаторами рГЦ [7].
Так, известны 3,4-дизамещенные фуроксаны, в частности, 1,2,5-оксадиазол-3,4-динитрил-2-оксид, 3-фенил-1,2,5-оксадиазол-4-нитрил-2-оксид и его изомер общей формулы II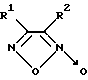
где R1 = CN и R2 = CN или C6H5 или R1 = C6H5 и R2 = CN, являющиеся донорами оксида азота и активаторами рГЦ из легких крысы в присутствии цистеина [8]. Действие данных соединений на другие ферменты не изучено.
Известны соли диазенийдиолатов (NONOаты) общей формулы III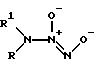
где R и R1 - алкил, замещенный алкил, представляющие собой димерные аддукты NO с азотсодержащими нуклеофилами [9]. Данные соединения являются спонтанными донорами NO или NO- и повышают уровень цГМФ в клетке. Кроме этого, как указывалось выше, одно из соединений общей формулы III (R = R1 = C2H5) вызывало ингибирование активности алкогольдегидрогеназы [4]. Влияние данных соединений на активность ГАФД и H,K-АТФазы не изучено.
Недостатками диазенийдиолатов общей формулы III являются относительная сложность синтеза (автоклавирование под давлением в атмосфере NO в течение 1-3 дней), сравнительная неустойчивость в водной среде при физиологических значениях pH, а также возможность образования канцерогенных N-нитрозодиалкиламинов при их разложении в аэробных условиях [9].
Известны замещенные 1,2-диазетин-1,2-диоксиды общей формулы IV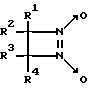
где R1 - H или Br, R2 - алкил или фенил, R3 - алкил и R4 - H или алкил, генерирующие оксид азота при нагревании (80oC), активирующие рГЦ из тромбоцитов человека [10, 11]. Действие данных соединений на другие ферменты не исследовано.
В целом 1,2-диазетин-1,2-диоксиды обладают пониженной устойчивостью при хранении при комнатной температуре, что связано с их способностью спонтанно генерировать оксид азота.
Известны 3,6-дизамещенные пиридазин-1,2-диоксиды общей формулы V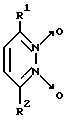
где R1 и R2 - CH3 или C6H5 [12].
Однако биохимические свойства данных соединений не изучены.
Запатентованы замещенные пирроло[2,3-d] пиридазин-5,6-диоксиды в ряду других аналогов общей формулы Vl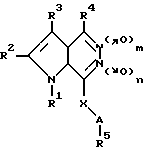
где A, R1 - R5, X имеют указанные в патенте значения, m = 0 или 1 и n = 0 или 1, в качестве средств для лечения язвы желудка, вызванной Helicobacter pylori [13].
Биохимические свойства вышеуказанных диоксидов не изучены.
Известен 4,4,7,7-тетраметил-4,7-дигидро- 1,2,5-оксадиазоло-[3,4-d] пиридазин-1,5,6-триоксид формулы VII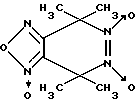
в качестве фотоактивируемого донора оксида азота [14].
Однако биохимические свойства данного соединения не изучены. Наиболее близкими к производным 1,2,5-оксадиазоло[3,4-d]пиридазин-5,6-диоксида вышеуказанной общей формулы I являются 3,5-дизамещенные пиразол-3-он-1,2-диоксиды общей формулы VIII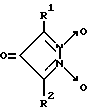
где R1 - алкил (CH3), арил (C6H5), замещенный арил и др., R2 - арил (C6H5), замещенный арил и др., являющиеся тиол-зависимыми донорами NO, активаторами рГЦ [15].
Недостатками данных соединений, в частности, 3,5-дифенилпиразол-3-он-1,2-диоксида (соединение 3) являются невысокая степень активации рГЦ (в 2,5 раза в концентрации 10 мкМ, см. пример 6) и крайне низкая растворимость в воде. Кроме того отсутствуют данные о влиянии соединения 3 на активность других ферментов.
Известны производные 1,2,5-оксадиазоло[3,4-d] пиридазин-5,6-диоксида общей формулы I в качестве продуктов химического синтеза [16-20]. Биохимические свойства данных соединений до настоящего времени не изучены.
Целью изобретения является поиск новых регуляторов активности нуклеотид-зависимых ферментов, обладающих более выраженными биохимическими свойствами.
Указанная цель достигается применением известных производных 1,2,5-оксадиазоло[3,4-d] пиридазин-5,6-диоксида вышеуказанной общей формулы I в качестве регуляторов нуклеотид-зависимых ферментов.
Конкретными соединениями согласно общей формуле I являются:
4,7-диметил-1,2,5-оксадиазоло[3,4-d] пиридазин-1,5,6-триоксид (4,7-диметилфуразано[3,4-d]пиридазин-1,5,6-триоксид; соединение 1),
4,7-диметил-1,2,5-оксадиазоло[3,4-d]пиридазин-5,6-диоксид (4,7-диметилфуразано[3,4-d]пиридазин-5,6-диоксид; соединение 2).
Предпочтительно применение производных 1,2,5-оксадиазоло[3,4-d]пиридазин-5,6-диоксида общей формулы I в качестве активаторов рГЦ и ингибиторов ГАФД.
Предпочтительно, применение 4,7-диметил-1,2,5- оксадиазоло[3,4-d]пиридазин-1,5,6-триоксида в качестве активатора H,K-АТФазы и ингибитора алкогольдегидрогеназы.
Соединение 1 было получено окислением диоксима 3,4-диацетилфуроксана четырехокисью азота, азотной кислотой или разбавленным раствором перманганата калия [16,17,18] , а также при взаимодействии 1-галоген-2-метилглиоксимов с нитритом серебра [19, 20].
Соединение 2 было получено обработкой диоксима 3,4-диацетилфуроксана водным раствором гидроокиси натрия [21, 18].
Описание данного изобретения содержит 11 примеров, иллюстрирующих экспериментальный материал.
Примеры 1-5 отражают химические основы молекулярного механизма действия соединений 1 и 2, преимущественно заключающиеся в генерации оксида азота и его активных форм (восстановленной формы и S-нитрозотиолов), модификации сульфгидрильных групп и взаимодействии с атомом железа гемовой группы белков.
Примеры 6-10 иллюстрируют биохимические эффекты соединений 1 и 2, заключающиеся в специфической регуляции активности нуклеотид-зависимых ферментов, а также отсутствии влияния на активность нуклеотид-независимого фермента.
Пример 1. Образование оксида азота из производных 1,2,5-оксадиазоло[3,4-d]пиридазин-5,6-диоксида.
Для определения оксида азота использовали известный способ, основанный на реакции оксида азота с кислородом воздуха в водной среде с образованием нитрита, количество которого измеряли по интенсивности окрашивания пробы продуктом реакции азосочетания с помощью спектрофотометра. В качестве доказательства того, что данный способ дает возможность измерить именно оксид азота, образующийся из соединений в результате химической реакции, а не нитрит, реакцию проводят в присутствии оксигемоглобина, который количественно реагирует с оксидом азота (но не нитритом), образуя нитрат, который не вступает в реакцию азосочетания.
Проба конечным объемом 1 мл содержала 50 мМ калий-фосфатный буфер (pH 7,4) или 20 мМ калий-цитратный буфер (pH 5,0), 0,5 мМ цистеин или глутатион, изучаемое соединение в концентрации 0,1 мМ и 0,2% диметилсульфоксид (ДМСО), а при инкубации с оксигемоглобином его концентрация составляла 0,1 мМ. В качестве отрицательного контроля использовали водный раствор ДМСО в концентрации 0,2%, а в качестве положительного контроля 0,1 мМ нитрит натрия, содержащий 0,2% ДМСО. Пробы инкубировали 30 мин при 37oC и добавляли последовательно 100 мкл 3 М ацетата натрия, 400 мкл 0,92% раствора сульфаниловой кислоты в 30% уксусной кислоте и 400 мкл 0,05% N-нафтилэтилендиамина. Затем пробы инкубировали 10 мин и измеряли оптическую плотность при длине волны 554 нм на спектрофотометре.
В вышеуказанных условиях при pH 7,4 не наблюдалось образование заметного количества нитрита (< 0,01 моль нитрита/моль исходного соединения (исх.с.)) в отсутствии тиолов. В присутствии тиолов соединение 1 генерировало 0,32 моль нитрита на моль исх.с. в присутствии цистеина и 0,43 моль нитрита на моль исх. с. в присутствии глутатиона; соединение 2 генерировало 0,02 моль нитрита на моль исх.с. в присутствии цистеина и 0,01 моль нитрита на моль исх. с. в присутствии глутатиона. В тех же условиях, но при pH 5,0 происходило образование из соединения 1 0,72 моль нитрита на моль исх.с. в присутствии цистеина и 0,75 моль нитрита на моль исх.с. в присутствии глутатиона; в случае соединения 2 соответствующие значения составляли 0,13 и 0,14 моль нитрита на моль исх.с. Дальнейшая инкубация не приводила к дополнительному образованию нитрита. При инкубации в вышеуказанных условиях при pH 7.4 в присутствии оксигемоглобина соединение 1 генерировало 0,04 и 0,05 моль нитрита на моль исх.с. в присутствии цистеина и глутатиона, соответственно, а оптическая плотность положительной контрольной пробы не изменялась. Это свидетельствует о том, что до 90% нитрита, определяемого по этому способу, является продуктом окисления оксида азота при pH 7,4. При pH 5,0 нитрит реагирует с оксигемоглобином, что затрудняет однозначное определение образования оксида азота при этом значении pH. Известный аналог (соединение 3) в указанных условиях в присутствии тиолов генерировало 0,08 моль нитрита на моль исх.с.
Пример 2. Образование S-нитрозотиолов (S-нитрозоглутатиона) производными производных 1,2,5-оксадиазоло[3,4-d]пиридазин-5,6-диоксида.
Для определения S-нитрозотиолов использовали известный способ, основанный на их реакции с хлоридом ртути (II), в ходе которой происходит образование нитрита. Для учета образования нитрита в ходе реакции соединения 1 с глутатионом реакцию проводили как описано в примере 1, а затем определяли нитрит по способу, описанному в примере 1, в отсутствии и в присутствии хлорида ртути (II) в концентрации 0,1%. Разность полученных значений соответствует количеству S-нитрозоглутатиона.
В вышеуказанных условиях происходит образование 0,17 моль S-нитрозоглутатиона на моль соединения 1. Образование S-нитрозотиолов известным аналогом (соединение 3) не исследовано.
Пример 3. Образование восстановленной формы оксида азота производными 1,2,5-оксадиазоло[3,4-d]пиридазин-5,6-диоксида.
Для определения восстановленной формы оксида азота (NO-/HNO) использовали известный способ, основанный на том, что образовавшийся в ходе реакции нитроксил реагирует с тиолами с образованием гидроксиламина (или происходит конкурентное образование N2O), который после окисления ионами I
В вышеуказанных условиях происходит образование 0,18 моль гидроксиламина на моль соединения 1 в присутствии цистеина и 0,34 моль гидроксиламина на моль соединения 1 в присутствии глутатиона. В данных условиях из соединения 2 в присутствии или в отсутствии тиолов не происходит образования гидроксиламина. Возможность генерации известным аналогом (соединением 3) восстановленной формы оксида азота не изучена.
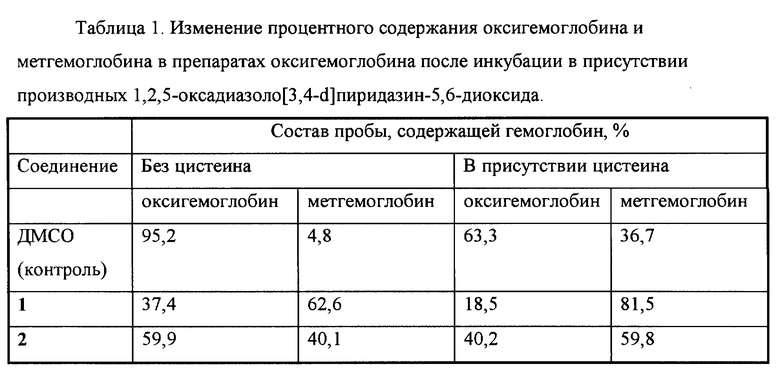
Пример 4. Образование метгемоглобина из оксигемоглобина под действием производных 1,2,5-оксадиазоло[3,4-d]пиридазин-5,6-диоксида.
Образование метгемоглобина из оксигемоглобина изучали известным способом с помощью спектрофотометра.
Пробы конечным объемом 1 мл содержали 50 мМ калий-фосфатный буфер (pH 7,4), 40 мкМ оксигемоглобин, 50 мкМ соединения 1 или 2 (в контрольные пробы добавляли ДМСО до концентрации 0,2%) и 0,25 мМ цистеин или воду. Пробы инкубировали 60 мин при 37oC и измеряли оптическую плотность при длинах волн 577, 630 и 700 нм, рассчитывая концентрации оксигемоглобина, метгемоглобина и холеглобина.
Изменение процентного содержания оксигемоглобина и метгемоглобина в препаратах оксигемоглобина после инкубации в присутствии производных 1,2,5-оксадиазоло[3,4-d]пиридазин-5,6-диоксида показано в табл.1.
В вышеуказанных условиях соединения 1 и 2 вызывали образование метгемоглобина из оксигемоглобина, причем данный эффект усиливался в присутствии цистеина. Это свидетельствует о способности указанных соединений взаимодействовать с атомом железа гемовой группы белков. Аналогичный эффект известного аналога (соединения 3) не исследовался.
Пример 5. Модификация сульфгидрильных групп низкомолекулярных тиолов под действием производных 1,2,5-оксадиазоло[3,4-d]пиридазин-5,6-диоксида.
Концентрацию SH-групп определяли известным способом Эллмана. Пробы конечным объемом 0,2 мл содержали 50 мМ калий-фосфатный буфер (pH 7,4), тиолы в концентрации 0,5 мМ и соединения 1 или 2 в концентрации 0,05 мМ. Контрольные пробы содержали соответствующее количество ДМСО (0,2%). Инкубацию проводили в течение 20 мин при 37oC и добавляли 1,8 мл 0,5 мМ раствора 5,5'-дитиобис(2-нитробензойной) кислоты в 50 мМ калий-фосфатном буфере (pH 7,4). Через 5 мин измеряли оптическую плотность при длине волны 412 нм и рассчитывали концентрацию SH-групп.
Уменьшение количества свободных SH-групп при взаимодействии производных 1,2,5-оксадиазоло[3,4-d] пиридазин-5,6-диоксида с цистеином, глутатионом и 2-меркаптоэтанолом дано в табл.2.
Взаимодействие известного аналога (соединения 3) с низкомолекулярными тиолами не исследовано.
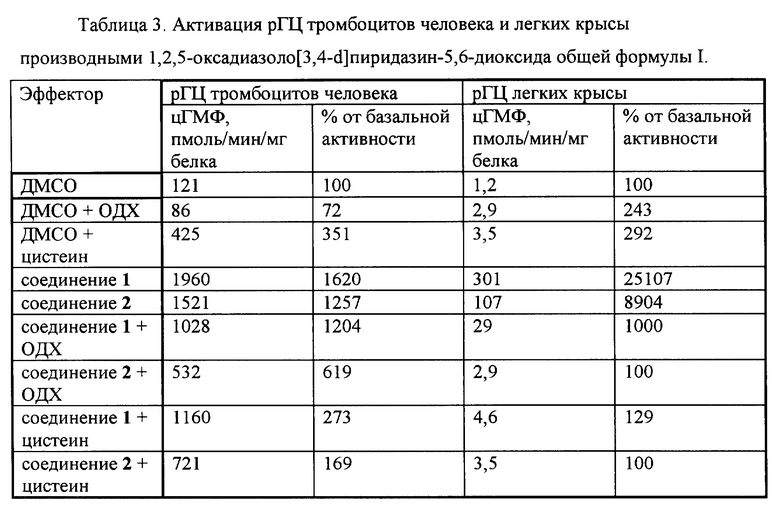
Пример 6. Активация растворимой формы гуанилатциклазы производными 1,2,5-оксадиазоло[3,4-d]пиридазин-5,6-диоксида общей формулы I.
Активность рГЦ измеряли на препаратах цитозоля тромбоцитов человека и легких крысы известными способами.
В первом случае для получения препарата рГЦ выделяли тромбоциты из венозной крови здоровых доноров известным способом с применением разделения форменных элементов крови в градиенте плотности фиколла. Тромбоциты промывали 3 раза 20 мМ трис-HCl-буфером (pH 7,5), содержащим 150 мМ NaCl и 5 мМ ЭДТА, центрифугируя при 1000 g в течение 15 мин, а затем суспендировали в 50 мМ трис-HCI-буфере (pH 7,6), содержащем 0,2 мМ дитиотреитол, и озвучивали ультразвуком с помощью ультразвукового генератора MSE 7-78 (Великобритания) в течение 20 сек при 4oC. Суспензию разрушенных клеток центрифугировали при 100000 g в течение 1 ч, супернатант разводили до концентрации белка 1 мг/мл, добавляли дитиотреитол до концентрации 0,2 мМ и определяли активность фермента по количеству цГМФ, образовавшегося из гуанозин-5'-трифосфата (ГТФ), иммуноферментным способом с использованием наборов реактивов для количественного определения цГМФ (АО "Биоген", Россия). Инкубационная смесь для определения активности конечным объемом 0,15 мл готовилась при 4oС и содержала 50 мМ трис-HCl (pH 7,6), 1 мМ ГТФ, 4 мМ MgCl2, 4 мМ креатинфосфат, 100 мкг (50 ед/мг) креатинфосфокиназы, 10 мМ теофиллин и ферментный препарат (10-20 мкг белка супернатанта). При определении активирующего действия в среду инкубации вносили изучаемое соединение в концентрации 10 мкМ в виде раствора в водном ДМСО и, при необходимости, другие соединения, например, 0,3 мкМ 1H-[1,2,4] оксадиазол[4,3-α]хиноксалин-1-он (ОДХ, специфический ингибитор гем-зависимой активации рГЦ) или 1 мМ цистеин, а в контрольные пробы добавляли ДМСО до концентрации 0,02%. Контрольная проба показала отсутствие влияния ДМСО в указанной концентрации на базальную активность рГЦ. Пробы инкубировали при 37oС в течение 15 мин. Реакцию останавливали кипячением проб в течение 2 мин с последующим охлаждением в ледяной бане. После отделения денатурировавшего белка при центрифугировании (10 мин при 1 500 g) в супернатанте определяли количество образовавшегося цГМФ вышеуказанным способом.
Для получения препарата рГЦ из легких крысы ткань гомогенизировали в 5 объемах 50 мМ трис-HCl-буфера (pH 7,6), содержащего 10 мМ MgCI2, с помощью гомогенизатора "стекло-стекло". Гомогенат центрифугировали в течение 30 мин при 30 000 g, супернатант отбирали и определяли активность фермента по количеству [32P]цГМФ, образовавшегося из [ α-32P] ГТФ, известным способом. Инкубационная смесь конечным объемом 100 мкл содержала 50 мМ трис-HCl-буфер (pH 7,6), 5 мМ MgCl2, 5 мМ креатинфосфат, 0,4 мг/мг креатинфосфокиназы, 1 мМ 3-изобутил-1-метилксантин, 2 мМ цГМФ, 0,2 мМ ГТФ (около 200 000 имп/мин на пробу) и ферментный препарат (40-50 мкг белка). При определении активирующего действия в среду инкубации вносили изучаемое соединение в концентрации 10 мкМ в виде раствора в водном ДМСО и, при необходимости, другие соединения, например, 3 мкМ ОДХ или 5 мМ цистеин, а в контрольные пробы добавляли ДМСО до концентрации 0,02%. Контрольная проба показала отсутствие влияния ДМСО в указанной концентрации на базальную активность рГЦ. Пробы инкубировали при 37oC в течение 15 мин. Реакцию останавливали кипячением проб в течение 2 мин. После охлаждения до комнатной температуры в пробы добавляли 0,5 мл 30 мМ Na2CO3 и 0,6 мл 36 мМ Zn(CH3COO)2, перемешивали, инкубировали при 4oC в течение 10 мин и центрифугировали при 15 000 g в течение 5 мин. Супернатант наносили на колонки с подкисленной известным способом окисью алюминия, которые промывали водой. Элюцию [32P] цГМФ проводили 0,2 М формиатом аммония во флаконы для сцинтилляционного счета, и радиоактивность измеряли с помощью жидкостного сцинтилляционного счетчика известным способом Черенкова.
Определение белка проводили по известному способу Лоури с использованием бычьего сывороточного альбумина в качестве стандарта.
Активация рГЦ тромбоцитов человека и легких крысы производными 1,2,5-оксадиазоло[3,4-d]пиридазин-5,6-диоксида общей формулы I показано в табл.3.
Соединения 1 и 2 активировали рГЦ из тромбоцитов человека и легких крысы. Активация снижалась в присутствии ОДХ, что указывает на гем-зависимую природу эффекта. Активация практически полностью ингибировалась в присутствии цистеина, что указывает на предпочтительное действие указанных производных 1,2,5-оксадиазоло[3,4-d] пиридазин-5,6-диоксида общей формулы I на рГЦ в клетках с пониженным содержанием свободных тиолов. В вышеописанных условиях известный аналог (соединение 3) активировал рГЦ тромбоцитов человека в 2,5 раза (250%).
Пример 7. Ингибирование глицеральдегид-3-фосфат дегидрогеназы производными 1,2,5-оксадиазоло[3,4-d]пиридазин-5,6-диоксида общей формулы I.
Активность ГАФД определяли известным способом с помощью спектрофотометра. В качестве источника фермента использовали эритроциты человека и, кроме этого, изучали действие соединения 1 на очищенный препарат фермента из скелетных мышц кролика.
Эритроциты выделяли из венозной крови здоровых доноров при центрифугировании (1000 g в течение 10 мин), удаляя верхний слой лейкоцитов. Затем эритроциты промывали 0,9% раствором NaCl три раза, осаждали центрифугированием и добавляли к 0,5 мл осадка клеток соединения 1 и 2 до концентрации 10 мкМ. Пробы инкубировали 3 мин при 37oC, и клетки промывали три раза 0,9% NaCl. Эритроциты лизировали в 5 мл холодной воды на льду в течение 1 ч, лизат разделяли центрифугированием при 20 000 g (15 мин) на цитозольную и мембранную фракции. Мембраны промывали 0,9 % NaCl и измеряли активность ГАФД в пробе объемом 3 мл, содержащей 0,1 М глициновый буфер (pH 8,9), 0,5 мМ НАД, 0,5 мМ 3-фосфоглицериновый альдегид и 20 мМ арсенат натрия. Активность фермента определяли по изменению оптической плотности при длине волны 340 нм с помощью спектрофотометра в течение 45 сек, регистрируя начальную скорость реакции. Активность фермента выражали в мкмоль НАДН/мин/0,5 мл осадка эритроцитов.
Фермент из мышц кролика (1,23 мкМ) инкубировали в течение 10-40 мин в присутствии соединения 1 в концентрации 12,3 мкМ или ДМСО в концентрации 0,025% в 50 мМ HEPES-NaOH-буфере (pH 7,5) при 25oC. Активность измеряли так же, как описано выше для фермента из эритроцитов человека.
Влияние производных 1,2,5-оксадиазоло[3,4-d] пиридазин-5,6- диоксида общей формулы I на активность ГАФД в эритроцитах человека показано в табл.4.
Из данных табл. 4 видно, что соединение 1 преимущественно ингибирует мембранную форму фермента, а соединение 2 - цитоплазматическую. Однако сравнение общей активности указывает на то, что она одинаково снижается в случае соединений 1 и 2.
Соединение 1 также ингибировало активность очищенной ГАФД из мышц кролика. При инкубации в течение 20 мин активность снижалась со 103 мкмоль/мин/мг (ед.) до 74 ед. Инкубация в течение 55 мин снижала активность до 35 ед. Цистеин в концентрации 1 мМ ускорял инактивацию, так как уже через 20 мин активность ГАФД составляла 39 ед. НАД в концентрации 0,5 мМ не защищал фермент от инактивации как в присутствии только соединения 1, так и смеси соединения 1 и цистеина. Однако практически полная защита (на 97%) наблюдалась при добавлении в среду инкубации НАД и 3-фосфоглицеринового альдегида (0,5 мМ). Следовательно, соединения 1 и 2 являются специфическими ингибиторами внутриклеточной ГАФД, а соединение 1 способно специфически инактивировать и очищенный фермент. Влияние известного аналога (соединения 3) на активность ГАФД не исследовано.
Пример 8. Ингибирование алкогольдегидрогеназы производными 1,2,5-оксадиазоло[3,4-d]пиридазин-5,6-диоксида общей формулы 1.
Активность алкогольдегидрогеназы из пекарских дрожжей определяли известным способом с помощью спектрофотометра. Фермент инкубировали при концентрации 0,1 мг/мл в 50 мМ калий-фосфатном буфере (pH 7,4) с 15-50 мкМ соединения 1, вносимого в пробу в виде 50 мМ раствора в диметилформамиде (ДМФА), в течение 5-15 мин при 25oC в присутствии различных добавок. Контрольные пробы содержали ДМФА в соответствующей концентрации (0,03-0,1%). По окончании инкубации 25 мкг белка переносили в кювету спектрофотометра конечным объемом 3 мл, содержащую 50 мМ калий-фосфатный буфер (pH 7,4), 0,1 М этанол и 0,5 мМ НАД. Регистрировали изменение оптической плотности при длине волны 340 нм с помощью спектрофотометра. Количество свободных сульфгидрильных групп белка определяли известным способом Эллмана. Для этого белок в концентрации 0,1 мг/мл в 50 мМ трис-HCl-буфере (pH 7,6) инкубировали в присутствии 25 мкМ соединения 1 20 мин при 25oC, добавляли додецилсульфат натрия до концентрации 2% и 5,5'-дитиобис(2-нитробензойную) кислоту до концентрации 0,5 мМ. Через 20 мин измеряли оптическую плотность при длине волны 412 нм с помощью спектрофотометра.
Инкубация алкогольдегидрогеназы в присутствии соединения 1 приводила к ингибированию активности фермента, которое зависело от времени инкубации и концентрации соединения 1, а инкубация в присутствии ДМФА не влияла на активность фермента. Временная зависимость позволила рассчитать константу скорости инактивации, которая, например, при концентрации соединения 1, равной 25 мкМ, составляла 0,031 мин-1, а зависимость константы скорости инактивации от концентрации соединения 1 позволила рассчитать константу ингибирования, которая составляла 91 мкМ. Константа скорости инактивации на 30% снижалась в присутствии НАД при концентрации соединения 1 25 мкМ, а цистеин и глутатион в концентрации 0,15 мМ защищали фермент от инактивации в присутствии тех же концентраций соединения 1 на 75%. Цистеин и глутатион в концентрации 2 мМ защищали фермент от инактивации в тех же условиях на 100%. Соединение 1 снижает количество SH-групп фермента, реагирующих с реактивом Эллмана, на 94%. Таким образом соединение 1 вызывает специфическое ингибирование алкогольдегидрогеназы путем модификации сульфгидрильных групп аминокислотных остатков цистеина фермента. Влияние известного аналога (соединение 3) на активность алкогольдегидрогеназы не исследовано.
Пример 9. Активация H,K-АТФазы производными 1,2,5-оксадиазоло [3,4-d]пиридазин-5,6-диоксида общей формулы I.
Активность H, K-АТФазы, полученной известным способом из слизистой желудка кролика, измеряли известным способом по скорости гидролиза АТФ с использованием спектрофотометрии. Пробы конечным объемом 0,3 мл содержали 0,3 М сахарозу, 4 мМ PIPES, 8 мМ трис (pH 7,4), 0,26 мг белка, 0,05% ДМФА и 25 мкМ соединение 1 (контрольные пробы имели тот же состав за исключением соединения 1). Инкубацию проводили при 37oC в течение 30 мин. Каждые 10 мин из реакционной смеси отбирали по 30 мкл и вносили в среду для определения активности конечным объемом 1 мл, содержащую 0,3 М сахарозу, 20 мМ HCl, 5 мМ PIPES, 10 мМ трис (pH 7,4), 3 мМ АТФ, 3 мМ MgCl2, 1,5 мМ фосфоенолпируват, 0,2 мМ НАДН, 20 ед. лактатдегидрогеназы (5-10 мкг белка), 20 ед. пируваткиназы (5-10 мкг белка). Регистрировали снижение оптической плотности при 20oC при длине волны 340 нм, которое соответствует гидролизу АТФ до АДФ.
В вышеуказанных условиях активность фермента в присутствии ДМФА составляла 56,5 мкмоль НАДН/ч/мг белка, и эта величина не менялась в ходе инкубации. Активность H,K-АТФазы, инкубированной в присутствии соединения 1, изменялась с течением времени и составляла 78,8, 88,2, 106,4, 65,3 и 59,0 мкмоль НАДН/ч/мг белка при инкубации в течение 0,33, 3, 10, 30 и 40 мин соответственно. Активация имеет двухфазный характер, повышаясь в течение 10 мин до максимального уровня 188%, а затем снижаясь до уровня контрольной пробы, содержащей ДМФА. Эффект активации H,K-АТФазы соединениями, способными генерировать оксид азота и/или его биологически активные формы, до настоящего времени не описан.
Пример 10 (сравнительный). Влияние производных 1,2,5-оксадиазоло [3,4-d] пиридазин-5,6-диоксида общей формулы I на активность каталазы.
Активность фермента определяли известным способом, измеряя разложение пероксида водорода с использованием спектрофотометра.
Каталазу (КФ 1.11.1.6) из печени лошади в концентрации 10 мкг/мл инкубировали в 20 мМ калий-фосфатном буфере (pH 7,4) в присутствии соединения 1 или ДМСО (0,05%) и 10 мМ пероксида водорода или воды при комнатной температуре в течение 60 мин. По окончании инкубации 0,1 мкг белка отбирали и вносили в пробу для измерения активности, содержащую 20 мМ калий-фосфатный буфер (pH 7,4) и 10 мМ пероксид водорода. Измеряли снижение оптической плотности при длине волны 230 нм и для расчета скорости реакции использовали данные начального линейного участка.
В вышеуказанных условиях активность фермента, инкубированного в присутствии ДМСО или соединения 1, составляла 38,1 и 37,9 ммоль/мин/мг белка, соответственно. При инкубации фермента в присутствии 10 мМ пероксида водорода и ДМСО или соединения 1 активность составляла 26,9 и 27,2 ммоль/мин/мг белка. Таким образом, соединение 1 не влияет на активность каталазы, не являющейся нуклеотид-зависимым ферментом.
Пример 11. Растворимость производных 1,2,5-оксадиазоло[3,4-d]пиридазин-5,6-диоксида общей формулы I и стабильность водных растворов и растворов, приготовленных с использованием различных органических растворителей.
Растворимость соединений 1 и 2 и известного аналога (соединения 3) определяли путем внесения аликвоты концентрированного 50 мМ раствора в смешивающемся с водой органическом растворителе (ДМСО, ДМФА, диоксане, ацетоне, метаноле) в воду до той конечной концентрации, при которой происходило образование нерастворившихся частиц и хлопьев.
Установлено, что в указанных условиях растворимость в воде соединения 1 составляет 250 мкмоль/л, соединения 2 - 1000 мкмоль/л, известного аналога (соединения 3) - 50 мкмоль/л.
Стабильность в растворах органических растворителей изучалась на примере 50 мМ раствора соединения 1 в ДМСО и 0,25 мМ раствора в воде, содержащей 0,5% ДМСО. Снижение концентрации соединения оценивали с помощью спектроскопии, используя величины молярного коэффициента поглощения при 353 нм ( ε = 5320 М-1•см-1) в воде.
Раствор соединения 1 в ДМСО был стабилен в течение 1 месяца при комнатной температуре при хранении в защищенном от света месте и в замороженном виде при -20oC до 6 месяцев. Водный раствор соединения 1 достаточно стабилен при комнатной температуре при хранении в защищенном от света месте при нейтральных или слабокислых значениях pH ( τ 1/2 > 4,2 мес). Водный раствор соединения 1 нестабилен при слабощелочных и щелочных значениях pH (pH > 9), на ярком свету и в присутствии тиолов. В сухом виде соединение 1 устойчиво при комнатной температуре при хранении в защищенном от света месте в течение как минимум двух лет.
Из вышеприведенных примеров 1-5 следует вывод о том, что соединения 1 и 2 являются значительно более эффективным донором оксида азота, чем известный аналог (соединение 3). Кроме того, соединение 1 генерирует биологически активные формы оксида азота, включая восстановленную форму NO и нитрозотиолы, а также, как и соединение 2, является модификатором SH-групп и атома железа гемовой группы белков. Из примера 6 следует, что производные 1,2,5-оксадиазоло[3,4-d] пиридазин-5,6-диоксида общей формулы I гем-зависимо активируют рГЦ значительно эффективнее, чем их известный аналог (соединение 3). Кроме того, соединения 1 и 2 в отличие от их аналога (соединения 3) оказывают выраженное специфическое действие на активность других нуклеотид-зависимых ферментов: ГАФД (пример 7), алкогольдегидрогеназы (пример 8), H,K-АТФазы (пример 9).
Таким образом, применение производных 1,2,5-оксадиазоло[3,4-d]пиридазин-5,6-диоксида общей формулы I в биохимических исследованиях позволяет расширить ассортимент эффективных регуляторов активности ферментов.
Источники информации
1. Murad, F. "Regulation of cytosolic guanylyl cyclase by nitric oxide: The NO-cGMP signal transduction system" Adv. Pharmacol. 1994, v.26, p. 19-33.
2. Dimmeler, S., Lottspeich, F., Brune, B. "Nitric oxide causes ADP-ribosylation and inhibition of glyceraldehyde-3-phosphate dehydrogenase" J. Biol. Chem. 1992, v.267, p. 16771-16774.
3. Kots, A. Ya. , Skurat, A.V., et al. "Nitroprusside stimulates the cysteine-specific mono(ADP-ribosylation) of glyceraldehyde-3-phosphate dehydrogenase from human erythrocytes" FEBS Lett., 1992, v.300, p.9-12.
4. Gergel, D., Cederbaum, A.I. "Inhibition of the catalytic activity of alcohol dehydrogenase by nitric oxide is associated with S nitrosylation and the release of zinc" Biochemistry, 1996, v.35, p. 16186-16194.
5. Г. А. Пхакадзе, К.Л. Коноплицкая и др. "Активность ферментов обмена этанола как тест для определения эффективности антиалкогольных препаратов сенсибилизирующего действия" Укр. Биох. Журнал, 1987, т. 59, N 4, С.25-29.
6. Выложенная заявка PCT N 94/16740 (A 16 K 49/00) оп. 1994 г.
7. "Methods in nitric oxide research" Ed. Feelisch, M., Stamler, J., J. Wiley & Sons, 1996, P.71-118.
8. Ferioli R., Foico G.C. et al. "A new class of furoxan derivatives as NO donors: mechanism of action and biological activity" Brit. J. Pharmacol. 1995, v.ll4, p.816-820.
9. "Methods in nitric oxide research" Ed. Feelisch, M., Stamler, J., J. Wiley & Sons, 1996, P.89-90.
10. Severina, I.S., Ryaposova, I.K., et al. "Derivatives of l,2-diazetidine-l,2-di-N-oxides - a new class of soluble guanylate cyclase activators with vasodilatory properties" Biochem. Mol. Biol. Internat., 1993, v.30, p. 357-366.
11. Г. Я. Шварц, Н.Б. Григорьев и др. "Производные 1,2-диазетин-1,2-диоксида - новый класс генераторов оксида азота, обладающих сосудорасширяющей активностью". - Хим.- фарм. Журнал, 1994, т.28, N 4, С.38-42.
12. Severina, I.S., Belushkina, N.N., et al. "Inhibition of ADP-induced human platelet aggregation by a new class of soluble guanylate cyclase activators capable of nitric oxide generation" Biochem. Mol. Biol. Internal., 1994, v.33, p.957-967.
13. Европейский патент N 742218 (C 07 D 487/04) оп. 1996 г.
14. Paschenko, S.V., Khramtsov, V.V., et al. "EPR and laser flash photolysis studies of the reaction of nitric oxide with water soluble NO trap Fe(ll)-proline-dithiocarbamate complex" Biochem. Biophys. Res. Commun., 1996, v.225, p.577-584.
15. Выложенная заявка ФРГ N 4322545 (C 07 D 231/18) оп. 1995 г. - прототип.
16. Ponzio, G., Bernardi, V. "Ricerche sulle dioccime.-XXII" Gazz. Chim. Ital., 1925, v.55, p.67-72.
17. Ю. А. Стреленко, О.А. Ракитин и др. "Спектры ЯМР и строение N,N'-диоксида 4,7-диметилпиридазино[4,5-с]фуроксана "Изв. АН СССР, сер. Химическая, 1988, с.2848-2850.
18. Fruttero, R., Ferrarotti, B., et al. "A structural study of Tryller's and Schmitz's compounds and related substances" Liebigs Ann. Chem., 1988, p. 1017-1023.
19. Ponzio, G. "Ricerche sulle dioccime.-LXXXVIII" Gazz. Chim. Ital., 1932, v.62, p.424-427.
20. Carbone, G. "Ricerche sulle dioccime.- LXXXIX" Gazz. Chim. Ital., 1932, v.62, p.428-431.
21. Steffens, С., Behrend, R. "Zur Kenntniss der Isomeren Verbindungen C6H8N404 aus Acetylmethyinitrolsaure" Liebigs Ann. Chem., 1899, В.309, p. 241-253.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ 1,2,5-ОКСАДИАЗОЛО-[3,4-D]-ПИРИДАЗИН-5,6-ДИОКСИДА В КАЧЕСТВЕ АКТИВАТОРОВ РАСТВОРИМОЙ ФОРМЫ ГУАНИЛАТЦИКЛАЗЫ И СРЕДСТВ ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ СЕРДЕЧНО-СОСУДИСТОЙ СИСТЕМЫ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ НА ИХ ОСНОВЕ | 1997 |
|
RU2165256C2 |
| ДОНОР ОКСИДА АЗОТА И АКТИВАТОР РАСТВОРИМОЙ ФОРМЫ ГУАНИЛАТЦИКЛАЗЫ | 1998 |
|
RU2139932C1 |
| ПРОИЗВОДНЫЕ 1,4,2,5-ДИОКСАДИАЗИНА | 2001 |
|
RU2212409C1 |
| ДОНОР ОКСИДА АЗОТА И АКТИВАТОР РАСТВОРИМОЙ ФОРМЫ ГУАНИЛАТЦИКЛАЗЫ | 1997 |
|
RU2123046C1 |
| КОМПЛЕКСЫ ВКЛЮЧЕНИЯ ПРОИЗВОДНЫХ 1,2,5-ОКСАДИАЗОЛ-2-ОКСИДА С ПОЛИЦИКЛИЧЕСКИМИ ПРОИЗВОДНЫМИ ГЛЮКОПИРАНОЗЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2001 |
|
RU2186782C1 |
| ДОНОР ОКСИДА АЗОТА, АКТИВИРУЮЩИЙ РАСТВОРИМУЮ ФОРМУ ГУАНИЛАТЦИКЛАЗЫ, ИНГИБИРУЮЩИЙ АГРЕГАЦИЮ ТРОМБОЦИТОВ И ОБЛАДАЮЩИЙ СПАЗМОЛИТИЧЕСКИМ И СОСУДОРАСШИРЯЮЩИМ ДЕЙСТВИЕМ | 2001 |
|
RU2208438C1 |
| АКТИВАТОР РАСТВОРИМОЙ ФОРМЫ ГУАНИЛАТЦИКЛАЗЫ | 1997 |
|
RU2122582C1 |
| ИНГИБИТОР NO-ЗАВИСИМОЙ АКТИВАЦИИ РАСТВОРИМОЙ ФОРМЫ ГУАНИЛАТЦИКЛАЗЫ | 2001 |
|
RU2188865C1 |
| ИНГИБИТОР РАСТВОРИМОЙ ФОРМЫ ГУАНИЛАТЦИКЛАЗЫ | 1999 |
|
RU2151799C1 |
| ИНГИБИТОР NO-ЗАВИСИМОЙ АКТИВАЦИИ РАСТВОРИМОЙ ФОРМЫ ГУАНИЛАТЦИКЛАЗЫ | 2001 |
|
RU2189392C1 |


Изобретение относится к биохимии, в частности, может быть использовано для изучения механизма регуляции активности ферментов - растворимой формы гуанилатциклазы (рГЦ) и глицеральдегид-3-фосфат дегидрогеназы (ГАФД) и Н, К-аденозин-5-трифосватазы (Н,К-АТФаза). Изобретение заключается в применении производных 1,2,5-оксадиазоло[3,4-d]пиридазин-5,6-диоксида общей формулы I в качестве активаторов рГЦ и ингибиторов ГАФД и применении 4,7-диметил-1,2,5-оксадиазоло[3,4-d] пиридазин-1,5,6-триоксида в качестве активатора Н, К-АТФазы и ингибитора алкогольдегидрогеназы. Производные 1,2,5-оксадиазоло[3,4-d] пири-дазин-5,6-диоксида общей формулы I, приведенной в описании, оказывают регулирующее действие на нуклеотид-зависимые ферменты более выраженное, чем эффект прототипа. 2 з.п.ф-лы, 4 табл.
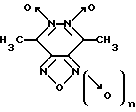
где n = 0 или 1, для специфической регуляции активности нуклеотид-зависимых ферментов.
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| DE 4322545 A1, 12.01.95 | |||
| Gergel, D., Cederbaum, A.I | |||
| ''Inhibition of the catalytic activity of alcohol dehydrogenase by nitric oxide is associated with S nitrosytaion and the release of zinc" Biochemistry, 1996, v.35, p.16186-16194 | |||
| Severina, I.S., Belushkina, N.N., et al | |||
| "Inhibition of ADP-induced human platelet aggregation by a new class of soluble guanylate cyclase activators capable of nitric oxide generation" | |||
| - Biochem.Mol.Internat., 1994,v.33, p.957-967 | |||
| Ferioli R., Folco G.C.et al | |||
| "A new class of furoxan derivatives as NO donors: mechanism of action and biological activity." - Brit.J.Pharmacol | |||
| Топка с качающимися колосниковыми элементами | 1921 |
|
SU1995A1 |
