Настоящая заявка является частичным продолжением заявки N 08/034455 на патент США, поданной 19 марта 1993 года.
Сущность изобретения
Настоящее изобретение касается непептидных антагонистов рецептора эндотелина, представленных соединением формулы I, фармацевтических композиций, содержащих эти соединения, а также методов лечения с использованием соединений настоящего изобретения. Соединения настоящего изобретения представляют терапевтические, в частности, пригодные для лечения астмы, повышенного давления, легочной гипертонии, артериосклероза, застойной сердечной недостаточности, почечной недостаточности, в особенности постишемической почечной недостаточности, циклоспориновой нефротоксичности, вазоспазма, сосудистого рестеноза, церебральной и сердечной ишемии и других ишемических состояний, инфаркта миокарда, болезни Рейно, доброкачественной простатической гиперплазии, воспалительных кишечных заболеваний, включая болезнь Крона и неспецифические язвенные колиты, а также других воспалительных заболеваний, или эндотоксинового бактериально-токсического шока, вызванных эндотелином или связанных с ним.
В это изобретение, кроме того, входит способ антагонизации с эндотелиновых рецепторов у млекопитающих, включая людей, который включает введение млекопитающему нуждающемуся в таком лечении, эффективного количества соединения формулы I.
Предпосылки изобретения
Эндотелин представляет собой пептид, состоящий из 21-аминокислоты, производимый эндотелиальными клетками. Пептид является не только эндотелиальными клетками, но также трахеальными эпителиальными клетками или почечными клетками. Эндотелин (ET-I) обладает сильным сосудосуживающим действием. Сосудосуживающее действие является следствием связывания эндотелина с его рецептором на клетках сосудистых гладких мышц.1-3
Эндотелин (ET-I) является одним из трех недавно идентифицированных сильнодействующих сосудосуживающих пептидов, который также включает эндотелин-2(ET-2) и эндотелин-3(ET-3), последовательности которых отличаются от ET-1 соответственно двумя или шестью аминокислотами4.
Повышенные уровни эндотелина по сравнению с обычными уровнями были обнаружены в крови больных гипертонической болезнью, острым инфарктом миокарда, легочной гипертензией, болезнью Рейно или атеросклерозом или в промывных жидкостях дыхательных путей больных астмой.5-8
Экспериментальная модель церебрального вазоспазма и вторая модель острой почечной недостаточности позволили сделать заключение, что эндотелина является одним из медиаторов, вызывающих церебральный вазоспазм, приводящий к субарахноидальному кровоизлиянию, и почечную недостаточность.9-10
Было также найдено, что эндотелин контролирует выделение многих физиологических веществ, например ренина, предсердного мочевого пептида, производимого эндотелием фактора релаксации (EDRF), тромбоксана A2, 14, простациклина, норпинефрина, ангиотензина II и вещества P11-16.
Кроме того, эндотелин вызывает сокращение гладких мышц желудочно-кишечного тракта и гладких мышц матки.17-19
Было также показано, что эндотелин способствует росту клеток сосудистых гладких мышц у крыс, что, как предполагают, возможно вызывает артериальную гипертрофию.20
Эндотелиновые рецепторы присутствуют в высоких концентрациях в периферических тканях, а также в центральной нервной системе, и было показано, что церебральное введение эндотелина вызывает изменение поведения у животных, что приводит к предположению о том, что эндотелин может играть важную роль при регуляции нервных функций.21
Было показано, что эндотоксин способствует выделению эндотелина. Исходя из этого, можно предположить, что эндотелин является важным медиатором для болезней, вызванных эндотоксином.22-23
Изучение показало, что циклоспорин, добавленный к почечной культуре клеток, вызывает секрецию эндотелина.24 Другое значение показало, что введение циклоспорина крысам приводит к уменьшению гломерулярной скорости фильтрации и увеличению давления крови, связанным с заметным увеличением уровня циркуляции эндотелина. Эта почечная недостаточность, связанная циклоспорином, может быть подавлена введением антитела анти-эндотелина.25
Эти изучения показали, что эндотелин в значительной степени включен в патогенез почечной болезни, вызванной циклоспорином.
Недавнее изучение больных с застойной сердечной недостаточностью проиллюстрировало прямую взаимосвязь между повышенными уровнями эндотелина в плазме и тяжестью заболевания.26
Эндотелин представляет эндогенное вещество, которое прямо или косвенно (через регулируемое выделение других различных эндогенных веществ) вызывает непрерывное сокращение сосудистых или несосудистых гладких мышц. Полагают, что избыточное воспроизведение или избыточная секреция является одним из факторов, ответственных за гипертензию, легочную гипертензию, болезнь Рейно, бронхиальную астму, острую почечную недостаточность, инфаркт миокарда, стенокардию, атеросклероз, церебральный вазоспазм и церебральный инфаркт. A. M. Loherty, Endothelyn. A new Chalenge, J. Med. Chem., 1493 - 1508 (1992).
Полагают, что вещества, которые специфически подавляют связывание эндотелина с его рецептором, блокируют физиологическое действие эндотелина и являются полезными для лечения больных с нарушениями, связанными с эндотелином.
Новые соединения настоящего изобретения являются пригодными в качестве непептидных антагонистов эндотелина и не были раскрыты в опубликованных патентах или опубликованных заявках на патент. Среди опубликованных заявок на патент, раскрывающих линейные и циклические пептидные соединения в качестве антагонистов эндотелина, можно представить следующие: Fujisawa в заявке на Европейский патент EP 457195 и международной заявке (PCT) N WO 93/10144, Banyu в EP-436189 и 460679, Immunopharmaceutics Inc. в WO 93/225580, Warner Lambert Co. , WO 92/20706 и Takeda Chemical Ind. в EP 528312, EP-543425, EP-547317 и WO 91/13089.
Fujisawa также раскрыл два непептидных антагонистических соединения эндотелина: антрахиноновые производные, полученные ферментацией с использованием Streptomyces sp. N 89009 в EP 405421 и патенте США N 5187195; и производные 4-феноксифенола, полученные ферментацией с использованием Penicillium citreonigrum F-12880 в заявке Великобритании 2259450 на патент UK. Shionogi и Co. также раскрыли непептидные антагонисты эндотелина, являющиеся тритерпеновыми соединениями, которые получают ферментацией с использованием Мyrica cerifera в WO 92/12991.
Непептидными антагонистами эндотелина, которые известны в патентной литературе, являются: 1) ряд замещенных (1,4-хинолинокси)метилбифенилкарбоновых кислот, представленных Roussel-Uclaf в EP 498723; 2) ряд N-(4-пиримидинил)бензолсульфонамидов с различным видом замещения из EP 510526 и EP 526708, опубликованных Hoffmann-La Roche; 3) ряд нафталинсульфонамидов и бензолсульфонамидов, раскрытые E.R. Squibb & Sons соответственно EP 558258 и EP 569193; 4) ряд соединений, представленных 3-(3-индолилметил)-1,4-диаза-2,5-диоксобицикло[4.3.0] нонан-9-карбоновой кислотой от Immunopharmaceutics Inc. в WO 93/23404; 5) ряд сконденсированных [1,2,4]тиадиазолов, замещенных иминосульфонильным заместителем, от Такеда Кемикал Индастриз, раскрытые в EP 582599; и 6) ряд производных индана и индена от Smith-kline Beеcham. Corp., раскрытые в WO 93/08779.
Youssefyeh, et al. в патенте США N 4748272 раскрыл феноксифенилацетаты в качестве пригодных ингибиторов липоксигеназы для лечения воспалительных заболеваний и аллергических реакций.
Talet, et al. в патенте США N 3499008 раскрыл производные бифенилацетата, пригодные в качестве гербицидов, фунгицидов и бактерицидов, а также в качестве ингредиента в термоотверждающихся акриловых красках или в качестве исходного материала для получения эпоксидных смол.
Ссылки
1. Nature, 332, 411 - 415 (1988).
2. FEBS Letters, 231, 440 - 444 (1988).
3. Biochem. Biophys. Res. Commun., 154, 868 - 875 (1988).
4. TiPS, 13, 103 - 108, March 1992.
5. Japan J. Hypertension, 12, 79 (1989).
6. J. Vascular Medicine Biology, 2, 207 (1990).
7. J. Am. Ved. Association, 264 2868 (1990).
8. The Lancet, ii, 207 (1990) и The Lancet, ii, 747 - 748 (1989).
9. Japan Soc. Cereb. Blood Flow & Metabol, 1, 78 (1989).
10. J. Clin. Invest., 83, 1762 - 1767 (1989).
11. Biochem. Biophys. Res. Comm., 157, 1164 - 1168 (1988).
12. Biochem. Biophys. Res. Comm., 155, 167 - 172 (1989).
13. Proc. Natl. Acad. Sci. USA, 85, 9797 - 9800 (1989).
14. J. Cardiovasc. Pharmacol., 13, 589 - 592 (1989).
15. Japan J. Hypertension 12, 76 (1989).
16. Neuroscience Letters, 102, 179 - 184 (1989).
17. FEBS Letters, 246, 337 - 340 (1989).
18. Eur. J. Pharmacol., 154, 227 - 228 3(1989).
19. Biochem. Biophys. Res. Comm., 159, 317 - 323 (1988).
20. Atherosclerosis, 78, 225 - 228 (1989).
21. Neuroscience Letters, 97, 276 - 279 (1989).
22. Biochem. Biophys. Res. Commun., 161, 1220 - 1227 (1989).
23. Acta. Physiol. Scand., 137, 317 - 318 (1989).
24. Eur. J. Pharmacol., 180, 191 - 192.
25. Kidney Int., 37, 1487 - 1491 (1990).
26. Mayo Clinic. Proc., 67, 719 - 724 (1992).
Изобретение касается новых соединений, имеющих структурную формулу I:
или их фармацевтически приемлемой соли,
в которой R1, R2, R3a и R3b независимо являются:
(a) H,
(b) F, Cl, Br или I,
(c) -NO2,
(d) -NH2,
(e) -NH(C1-C4)-алкилом,
(f) -N[(C1-C4)-алкилом]2,
(g) -SO2NHR7,
(h) -CF3,
(i) -(C1-C6)-алкилом,
(j) -OR7,
(k) -S(O)n-(C1-C4)-алкилом,
(l) -NHCO-(C1-C4)-алкилом,
(m) -NHCO-O(C1-C4)-алкилом,
(n) -CH2O-(C1-C4)-алкилом,
(o) -O-(CH2)m-OR7,
(p) -CONR7R11,
(q) -COOR7, или
(r) - фенилом,
R1 и R2 у соседних атомов углерода могут соединяться с образованием циклической структуры:
A представляет:
a) -Y-C(R4) = C(R5)-, -
b) -Y-C(R4)=N-,
c) -Y-N=C(R4)-,
d) -Y-[C(R6)(R6)]s-Y-,
e) -Y-C(R6)(R6)-C(R6)(R6)-,
f) -C(R4) = C(R5)-Y-,
g) -N=C(R4)-Y-,
h) -C(R6)(R6)-C(R6)(R6)-Y-, или
i) -C(R4) = C(R5)-C(R4) = C(R5)-,
n равно 0, 1 или 2;
m равно 2, 3 или 4;
s равно 1 или 2;
Y представляет -O-, -S(O)n- и NR7;
R4 и R5 независимо являются:
(a) H,
(b) (C1-C6)-алкилом или (C2-C6)-алкенилом, каждый из которых не замещен или замещен одним или двумя заместителями, выбранными из группы состоящей из:
(i) -OH,
(ii) -O-(C1-C4)-алкила,
(iii) -S(O)n-(C1-C4)-алкила,
(iv) -NR7-(C1-C4)-алкила,
(v) -NHR7,
(vi) -COOR7,
(vii) -CONHR7,
(viii) -OCOR11, или
(ix) -CONR7R11,
(c) (C3-C7)циклоалкилом,
(d) F, Cl, Br, I,
(e) CF3,
(f) -COOR7,
(g) -CONR7R11,
(h) -NR7R11,
(i) -NR7CONR7R11,
(j) -NR7COOR11,
(k) -SO2NR7R11,
(l) -O-(C1-C4)-алкилом,
(m) -S(O)n-(C1-C4)-алкилом, или
(n) -NHSO2R11;
R6 является:
(a) H,
(b) (C1-C4)-алкилом, незамещенным или замещенным одним из следующих заместителей:
(i) -OH,
(ii) -NR7R11,
(iii) -COOR7,
(iv) -CONHR7, или
(v) -CONR7R11;
R7 является:
(a) водородом,
(b) (C1-C6)-алкилом,
(c) фенилом,
(d) (C1-C6)-алкилфенилом, или
(e) (C3-C7)-циклоалкилом;
R8 является:
(a) H,
(b) (C1-C6)-алкилом, незамещенным или замещенным заместителем, выбранным из группы, состоящей из:
(i) -фенила,
(ii) -(C3-C7)-циклоалкила,
(iii) -NR7R11,
(iv) -морфолин-4-ила,
(v) -OH,
(vi) -CO2R7, или
(vii) -CON(R7)2,
(c) фенилом, незамещенным или замещенным заместителем, выбранным из группы, состоящей из:
i) C1-C4-алкила
ii) -O-(C1-C4)-алкила,
iii) -CONR7R11,
iv) F, Cl, Br или I, или
v) -COOR7;
R9 и R10 независимо являются:
(a) H,
(b) (C1-C6)-алкилом, незамещенным или замещенным (C3-C7)-циклоалкилом или -CO2R7,
(c) (C2-C6)-алкенилом,
(d) (C2-C6)-алкинилом,
(e) Cl, Br, F, I,
(f) (C1-C6)-алкокси,
(g) перфтор-(C1-C6)-алкилом,
(h) (C3-C7)-циклоалкилом, незамещенным или замещенным (C1-C6)-алкилом,
(i) фенилом,
(j) (C1-C6)-алкил-S(O)n- (CH2)n-,
(k) гидрокси-(C1-C6)алкилом,
(l) -CF3,
(m) -CO2R7,
(n) -OH,
(o) -NR7R11,
(p) -[(C1-C6)-алкил]NR7R11,
(q) -NO2,
(r) -(CH2)n-SO2-N(R7)2,
(s) -NR7CO(C1-C4)-алкилом, или
(t) -CON(R7)2
R9 и R10, расположенные у соседних атомов углерода, могут соединяться с образованием сконденсированного фенильного кольца, незамещенного или замещенного заместителем, выбранным из группы, состоящей из:
(C1-C6)-алкила, (C1-C6)алкокси, (C3-C7)-циклоалкила и (C1-C6)-алкил-(C3-C7)-циклоалкила,
R11 является:
(a) (C1-C6)-алкилом, незамещенным или замещенным заместителем, выбранным из группы, состоящей из:
(i) -OR7,
(ii) -N[R7]2,
(iii) -NH2,
(iv) -COOR7, или
(v) -N[CH2CH2]2Q;
(b) арилом, где арил определяют как фенил или нафтил, который является незамещенным или замещенным одним или двумя заместителями, выбранными из группы, состоящей из:
i) (C1-C4)-алкила,
ii) -O-(C1-C4)-алкила,
iii) -CO[NR7]2,
iv) F, Cl, Br или I,
v) -COOR7,
vi) -NH2,
vii) -NH[(C1-C4)-алкила],
viii) -N[(C1-C4)-алкила]2, или
ix) -C0N[CH2CH2]2Q;
c) (C1-C4)-алкиларилом, где арил является таким, как он определен выше,
(d) (C3-C7)-циклоалкилом,
(e)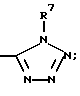
R7 и R11 у одного и того же атома азота, могут соединяться с образованием кольца, выбранного из группы, состоящей из: морфолина, пиперазинила, или пирролила, или
Q является O, S или -NR7;
R12 является:
a) H,
b) (C1-C6)-алкилом, незамещенным или замещенным одним или двумя заместителями, выбранными из группы, состоящей из:
i) -OH,
ii) -O-(C1-C4)-алкила,
iii) -O-(C1-C4)-циклоалкила,
iv) -S(O)n-(C1-C4)-алкила,
v) -NR7R11,
vi) -COOR7,
vii) -CONHR7,
viii) -OCOR11,
ix) -CONR7R11,
x) -NR7CONR7R11,
xi) -NR7COOR11,
xii) -C(R6)(OH)-C(R6)(R7)(OH), или
xiii) -SO2NR7R11,
(c) (C3-C7)-циклоалкилом,
(d) -OR7,
(e) -COOR7,
(f) -CONH2,
(g) -CONR7R11,
(h) -CONR7CO2R7,
(i) -NH2,
(j) -NR7R11,
(k) -NR7CONR7R11,
(l) -NR7COOR11,
(m) -C(R6)(OH)-C(R6)(R7)(OH),
(n) -SO2NR7R11,
(o) -S(O)2NR7COR11,
(p) -S(O)2NR7CO2R11,
(q) -S(O)2NR7CONR7R11,
(r) -NHSO2R11,
(s) -NR7SO2NR7R11,
(t) -CONHSO2R11,
(u) - CO-аминокислотой, где аминокислоту определяют как L- и D-аминокислоту, выбранную из группы, состоящей из Ala, Ile, Phe, Asp, Pro и Val и которая может быть, кроме того, замещена (C1-C6)-алкиловым эфиром, или амидом, или
(v)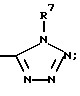
X является:
(a) -O-,
(b) -S(O)n-,
(c) -NR7,
(d) -CH2O-,
(e) -CH2S(O)n-,
(f) -CH2NR7-,
(g) -OCH2-,
(h) -N(R7)CH2-,
(i) -S(O)nCH2-, или
(j) - простой связью.
Z является:
(a) -CO2H,
(b) -CO2R13,
(c) -CONH-(тетразол-5-ил),
(d) -CONHSO2OR11,
(e) -CONHSO2-арилом, где арил определяют как фенил или нафтил, который не замещен или замещен одним или двумя заместителями, выбранными из группы, состоящей из:
i) (C1-C4)-алкила,
ii) -O-(C1-C4)-алкила,
(iii) -CONR7R11,
iv) F, Cl, Br или I,
v) -COOR7,
vi) -NH2,
vii) -NH[(C1-C4)-алкила],
viii) -N[(C1-C4)-алкила]2,
ix) -фенила,
x) -OH,
xi) -OCH2CH2OH,
xii) -CF3;
(f) -CONHSO2-(C1-C8)-алкилом, где алкильная группа является незамещенной или замещенной, как это определено в R4(b).
(g) -CONHSO2-(C1-C4)-перфторалкилом,
(h) -тетразол-5-илом,
(i) -CONHSO2-гетероарилом, где гетероарил определяют как карбазолил, фурил, тиенил, пирролил, изотиазолил, имидазолил, изоксазолил, тиазолил, оксазолил, пиразолил, пиразинил, пиридил, пиримидил, пуринил или хинолинил, который является незамещенным или замещенным одним или двумя заместителями, выбранными из группы, состоящей из:
i) (C1-C4)-алкила,
ii) -O-(C1-C4)-алкила,
iii) -CONR7R11,
iv) F, Cl, Br или I,
v) -COOR7,
vi) -NR7CONR7R11 и
vii) -NR7COOR11,
(j) -SO2NHCO-арилом, где арил является таким, как он определен выше в Z(d),
(k) -SO2NHCO-(C1-C8)-алкилом, где алкильная группа является незамещенной или замещенной, как это определено в R4(b),
(l) -SO2NHCO-(C1-C4)-перфторалкилом,
(m) -SO2NHCO-гетероарилом, где гетероарил является таким, как он определен выше в Z(g),
(n) -SO2NHCON(R11)2, где группы R11 являются одинаковыми или разными,
(o) -PO(OR7)2, где группы R7 являются одинаковыми или разными, или
(p) -PO(R11)OR7;
R13 является:
(a) (C1-C4)-алкилом;
(b) CHR14-O-COR15,
(c) CH2CH2-N[(C1-C4)- алкилом]2,
(d) CH2CH2-N[CH2CH2]2O,
(e) (CH2CH2O)y-O- [(C1-C4)-алкилом], в котором y равно 1 или 2,
(f) фенилом, нафтилом, CH2-фенилом или CH2-нафтилом, где фенил или нафтил замещен или не замещен CO2-(C1-C4)-алкилом,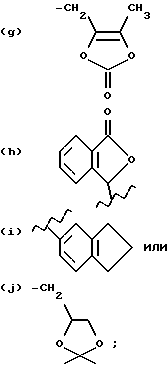
R14 и R15 независимо являются (C1-C6)-алкилом или фенилом.
Одним из объектов настоящего изобретения является соединение со структурной формулой II
или его фармацевтически приемлемая соль,
в которой R1, R2, R3a и R3b независимо являются:
(a) H,
(b) F, Cl, Br или I,
(c) -NO2,
(d) -NH2,
(e) -NH(C1-C4)-алкилом,
(f) -N[(C1-C4)-алкилом]2,
(g) -SO2NHR7,
(h) -CF3,
(i) -(C1-C6)-алкилом,
(j) -OR7,
(k) -S(O)n-(C1-C4)-алкилом,
(l) -NHCO-(C1-C4)-алкилом,
(m) -NHCO-O(C1-C4)-алкилом,
(n) -CH2O-(C1-C4)-алкилом,
(o) -O-(CH2)m-OR7,
(p) -CONR7R11, или
(q) -COOR7,
R1 и R2 у соседних атомов углерода могут соединиться с образованием циклической структуры
A представляет:
a) -Y-C(R4)=C(R5)-,
b) -Y-C(R4)=N-,
c) -Y-N=C(R4)-,
d) -Y-[C(R6)(R6)]s-Y-,
e) -Y-C(R6)(R6)-C(R6)(R6)-,
f) -C(R4)=C(R5)-Y-,
g) -N=C(R4)-Y-,
h) -C(R6)(R6)-C(R6)(R6)-Y-, или
i) -C(R4)=C(R5)-C(R4)=C(R5)-,
m равно 2, 3 или 4,
n равно 0, 1 или 2,
s равно 1 или 2,
Y равно -O-, -S(O)n и NR7;
R4 и R5 независимо являются:
(a) H,
(b) C1-C6-алкилом или (C1-C6)-алкенилом, каждый из которых не замещен или замещен одним или двумя заместителями, выбранными из группы, состоящей из:
i) -OH,
ii) -O-(C1-C4)-алкила,
iii) -S(O)n-(C1-C4)-алкила,
iv) -NR7-(C1-C4)-алкила,
v) -NHR7,
vi) -COOR7,
vii) -CONHR7,
viii) -OCOR11, или
ix) -CONR7R11,
c) (C3-C7)-циклоалкилом,
d) F, Cl, Br, I,
(e) CF3,
(f) -COOR7,
(g) -CONR7R11,
(h) -NR7R11,
(i) NR7CONR7R11,
(j) -NR7COOR11,
(k) -SO2NR7R11,
(l) -O(C1-C4)алкилом,
(m) -S(O)n-(C1-С4)-алкилом, или
(n) -NHSO2R11;
R6 является:
(a) H,
(b) (C1-C4)-алкилом, незамещенным или замещенным одним или двумя заместителями, выбранными из группы, состоящей из:
i) -OH,
ii) -NR7R11,
iii) -COOR7,
iv) -CONHR7 или
v) -CONR7R11,
R7 является:
(a) H,
(b) (C1-C6)-алкилом,
(c) фенилом,
(d) (C1-C6)-алкилфенилом,
(e) (C3-C7)-циклоалкилом,
R8 является:
(a) H,
(b) (C1-C6)-алкилом, незамещенным или замещенным одним или двумя заместителями, выбранными из группы, состоящей из:
(i) -фенила,
(ii) -(C3-C7)-циклоалкила,
(iii) -NR7R11,
(iv) - морфолин-4-ила,
(v) -OH,
(vi) -CO2R7, или
(vii) -CON(R7)2 или
(c) фенилом;
R9 и R10 независимо являются:
(a) H,
(b) (C1-C6)-алкилом, незамещенным или замещенным (C3-C7)-циклоалкилом или -CO2R7,
(c) (C2-C6)-алкенилом,
(d) (C2-C6)-алкинилом,
(e) Cl, Br, F, I,
(f) (C1-C6)-алкокси,
(g) перфтор-(C1-C6)-алкилом,
(h) (C3-C7)-циклоалкилом, незамещенным или замещенным (C1-C6)-алкилом,
(i) фенилом,
(j) (C1-C6)-алкил-S(O)n- (CH2)n-,
(k) гидрокси-(C1-C6)-алкилом,
(l) -CF3,
(m) -CO2R7,
(n) -OH,
(o) -NR7R11,
(p) -[(C1-C6-алкил]NR7R11,
(q) -NO2,
(r) -(CH2)n-SO2-N(R7)2,
(s) -NR7CO-(C1-C4)-алкилом, или
(t) -CON(R7)2;
R9 и R10 у соседних атомов углерода могут соединяться с образованием фенильного кольца, незамещенного или замещенного заместителем, выбранным из группы, состоящей из:
(C1-C6)-алкила, (C1-C6)-алкокси, (C3-C7)-циклоалкила и (C1-C6)-алкил-(C3-C7)-циклоалкила;
R11 является:
(a) (C1-C6)-алкилом, незамещенным или замещенным заместителем, выбранным из группы, состоящей из:
i) -OR7,
ii) -N[R7]2,
iii) -NH2,
iv) -COOR7 или
v) -N[CH2CH2]Q;
(b) арилом, где арил определяют как фенил или нафтил, который незамещен или замещен одним или двумя заместителями, выбранными из группы, состоящей из:
i) (C1-C4)-алкила,
ii) -O-(C1-C4)-алкила,
iii) -CO[NR7]2,
iv) F, Cl, Br или I,
v) -COOR7,
vi) -NH2,
vii) -NH[(C1-C4)-алкила],
viii) -N[(C1-C4)-алкила]2, или
ix) -CОN[CH2CH2]2Q;
c) -(C1-C4)-алкиларилом, где арил является таким, как он определен выше,
(d) (C3-C7)-циклоалкилом,
(e)
R7 и R11 у одного и того же атома азота могут соединяться с образованием кольца, выбранного из группы, состоящей из: морфолина, пиперазинила или пирролила, или
Q является O, или -NR7;
R12 является:
a) H,
b) (C1-C4)-алкилом, незамещенным или замещенным одним или двумя заместителями, выбранными из группы, состоящей из:
i) -OH,
ii) -O-(C1-C4)-алкила,
iii) -O-(C1-C4)-циклоалкила,
iv) -S(O)n-(C1-C4)-алкила,
v) -NR7R11,
vi) -COOR7,
vii) -CONHR7,
viii) -OCOR11,
ix) -CONR7R11,
x) -NR7CONR7R11,
xi) -NR7COOR11,
xii) -C(R6)(OH)-C(R6)(R7)(OH), или
xiii) -SO2NR7R11,
(c) (C3-C7)-циклоалкилом,
(d) -OR7,
(e) -COOR7,
(f) -CONH2,
(g) -CONR7R11,
(h) -CONR7CO2R7,
(i) -NH2,
(j) -NR7R11,
(k) -NR7CONR7R11,
(l) -NR7COOR11,
(m) -C(R6)(OH)-C(R6)(R7)(OH),
(n) -SO2NR7R11,
(o) -S(O)2NR7COR11,
(p) -S(O)2NR7CO2R11,
(q) -S(O)2NR7CONR7R11,
(r) -NHSO2R11,
(s) -NR7SO2NR7R11,
(t) -CONHSO2R11,
(u) - CO-аминокислотой, где аминокислоту определяют как L- и D-аминокислоту, выбранную из группы, состоящей из Ala, Ile, Phe, Asp, Pro и Val и которая может быть замещена (C1-C6)-алкиловым эфиром или
(v)
X является:
(a) -O-,
(b) -S(O)n-,
(c) -NR7,
(d) -CH2O-,
(e) -CH2S(O)n-,
(f) -CH2NR7-,
(g) -OCH2-,
(h) -N(R7)CH2-,
(i) -S(O)nCH2-, или
(j) - простой связью.
Z является:
(a) -CO2H,
(b) -CO2R13,
(c) -CONH-(тетразол-5-илом),
(d) -CONHSO2OR11,
(e) -CONHSO2-арилом, где арил определяют как фенил или нафтил, который незамещен или замещен одним или двумя заместителями, выбранными из группы, состоящей из:
i) (C1-C4)-алкила,
ii) -O-(C1-C4)-алкила,
(iii) -CONR7R11,
iv) F, Cl, Br или I,
v) -COOR7,
vi) -NH2,
vii) -NH[(C1-C4)-алкила],
viii) -N[(C1-C4)-алкила]2,
ix) -фенила,
x) -OH,
xi) -OCH2CH2OH,
xii) -CF3;
(f) -CONHSO2-(C1-C8)-алкилом, где алкильная группа является незамещенной или замещенной, как это определено в R4 (b).
(g) -CONHSO2-(C1-C4)-перфторалкилом,
(h) -тетразол-5-илом,
(i) -CONHSO2-гетероарилом, где гетероарил определяют как карбазолил, фурил, тиенил, пирролил, изотиазолил, имидазолил, изоксазолил, тиазолил, оксазолил, пиразолил, пиразинил, пиридил, пиримидил, пуринил или хинолинил, который является незамещенным или замещенным одним или двумя заместителями, выбранными из группы, состоящей из:
i) (C1-C4)-алкила,
ii) -O-(C1-C4)-алкила,
iii) -CONR7R11,
iv) F, Cl, Br или I,
v) -COOR7,
vi) -NR7CONR7R11 и
vii) -NR7COOR11,
(j) -SO2NHCO-арилом, где арил является таким, как он определен выше в Z(d),
(k) -SO2NHCO-(C1-C8)-алкилом, где алкильная группа является незамещенной или замещенной, как это определено в R4(b).
(l) -SO2NHCO-(C1-C4)-перфторалкилом,
(m) -SO2NHCO-гетероарилом, где гетероарил является таким, как он определен выше в Z(g),
(n) -SO2NHCON(R11)2, где группы R11 являются одинаковыми или разными,
(o) -PO(OR7)2, где группы R7 являются одинаковыми или разными, или
(p) -PO(R11)OR7;
R13 является:
(a) (C1-C4)-алкилом;
(b) CHR14-O-COR15,
(c) CH2CH2-N[(C1-C2)- алкилом]2,
(d) CH2CH2-N[CH2CH2]2O,
(e) (CH2CH2O)y-O- [(C1-C4)-алкилом], в y котором равно 1 или 2,
(f) фенилом, нафтилом, CH2-фенилом или с CH2-нафтилом, где фенил или нафтил замещен или не замещен CO2-(C1-C4)-алкилом,
R14 и R15 независимо являются (C1-C6)-алкилом или фенилом.
Вариантом соединений формулы (II) являются соединения формулы (III):
или их фармацевтически приемлемая соль,
где R1, R2, R3a и R3b независимо являются:
(a) H,
(b) F, Cl, Br или I,
(c) -NO2,
(d) -(C1-C6)-алкилом,
(e) -OR7,
(f) -NHCO-(C1-C4)-алкилом,
(g) -NHCO-O(C1-C4)-алкилом,
(h) -O-(CH2)m-OR7
(i) -CONR7R11, или
(j) -COOR7;
R1 и R2 у смежных атомов углерода могут объединиться с образованием циклической структуры:
A представляет:
a) -Y-C(R4)=C(R5)-,
b) -Y-C(R4)=N-,
c) -Y-N=C(R4)-,
d) -Y-[C(R6)(R6)]s-Y-,
e) -Y-C(R6)(R6)-C(R6)(R6)-,
f) -C(R4)=C(R5)-Y-,
g) -N=C(R4)-Y-,
h) -C(R6)(R6)-C(R6)(R6)-Y-, или
i) -C(R4)=C(R5)-C(R4)=C(R5)-,
m равно 2, 3 или 4;
n равно 0, 1 или 2,
s равно 1 или 2,
Y равен -O-, -S- и NR7;
R4 и R5 независимо являются:
(a) H,
(b) (C1-C6)-алкилом,
(c) (C3-C7)-циклоалкилом,
(d) F, Cl, Br, I,
(e) -NR7COOR11,
(f) -SO2NR7R11,
(g) -O-(C1-C6)-алкилом,
(h) -S(O)n-(C1-C6)-алкилом, или
(i) -NHSO2R11;
R6 является:
(a) H, или
(b) (C1-C4)-алкилом;
R7 является:
(a) H,
(b) (C1-C6)-алкилом,
(c) фенилом, или
(d) бензилом;
R8 является:
(a) H,
(b) (C1-C6)-алкилом, или
(c) фенилом;
R9 и R10 независимо являются:
(a) H,
(b) (C1-C6)-алкилом, незамещенным или замещенным (C3-C7)-циклоалкилом,
(c) Cl, Br, F, I,
(d) (C1-C6)-алкокси, или
(e) гидрокси-(C1-C6)-алкилом.
R11 является
(a) (C1-C6)-алкилом, незамещенным или замещенным заместителем, выбранным из группы, состоящей из:
i) -OR7,
ii) -N[R7]2,
iii) -NH2,
iv) -COOR7, или
v) -N[CH2CH2]2Q;
(b) арилом, где арил определяют как фенил или нафтил, который не замещен или замещен одним или двумя заместителями, выбранными из группы, состоящей из:
i) (C1-C4)-алкила,
ii) -O-(C1-C4)-алкила,
iii) -CO[NR7]2,
iv) F, Cl, Br или I,
v) -COOR7,
vi) -NH2,
vii) -NH[(C1-C4)-алкила],
viii) -N[(C1-C4)-алкила]2, или
ix) -CОN[CH2CH2]2Q;
c) (C1-C4)-алкиларилом, где арил является таким, как он определен выше,
(d) (C3-C7)-циклоалкилом,
(e) 
R7 и R11 у одного и того же атома азота могут соединяться с образованием кольца, выбранного из группы, состоящей из:
морфолинила, пиперазинила или пирролила, или
Q является O, S или -NR7,
R12 является:
(a) H,
(b) (C1-C4)-алкилом, незамещенным или замещенным одним или двумя заместителями, выбранными из группы, состоящей из:
i) OH,
ii) -O-(C1-C4)-алкила,
iii) -O-(C1-C4)-циклоалкила,
iv) -S(O)n-(C1-C4)-алкила,
iv) -NR7-(C1-C4)-алкила,
v) -NR7R11,
vi) -COOR7,
vii) -CONHR7,
viii) -OCOR11,
ix) -CONR7R11,
x) -NR7CONR7R11,
xi) -NR7COOR11,
xii) -C(R6)(OH)-C(R6)(R7(OH); или
xiii) -SO2NR7R11,
(c) -COOR7,
(d) -CONH2,
(e) -CONR7R11,
(f) -CONR7CO2R7,
(g) -C(R6)(OH)-C(R6)(R7)(OH),
(h) -CONHSO2R11,
(i) -SO2NR7R11,
(j) -NR7SO2NR7R11,
(k) - CO-аминокислотой, где аминокислоту определяют как L- и D-аминокислоту, выбранную из группы, состоящей из Ala, Ile, Phe, Asp, Pro и Val и которая может быть замещена (C1-C6)-алкиловым эфиром, или амидом
или
(l)
X является:
(a) -O-,
(b) -NR7-, или
(c) - простой связью,
Z является:
(a) -CO2H,
(b) -CO2R13,
(c) -CONH-(тетразол-5-илом);
(d) -CONHSO2-арилом, где арил определяют как фенил или нафтил, который является незамещенным или замещенным одним или двумя заместителями, выбранными из группы, состоящей из:
i) (C1-C4)-алкила,
ii) -O-(C1-C4)-алкила,
(iii) -CONR7R11,
iv) F, Cl, Br или I,
v) -COOR7,
vi) -NH2,
vii) -NH[(C1-C4)-алкила],
viii) -N[(C1-C4)-алкила]2,
ix) -фенила,
(e) -CONHSO2-(C1-C8)-алкилом, где алкил является незамещенным или замещенным, как это определено в R4(b),
(f) -CONHSO2-гетероарилом, где гетероарил определяют как карбазолил, фурил, тиенил, пирролил, изотиазолил, имидазолил, оксазолил, изоксазолил, тиазолил, пиразолил, пиразинил, пиридил, пиримидил, пуринил, или хинолинил, который не замещен или замещен одним или двумя заместителями, выбранными из группы, состоящей из:
i) (C1-C4)-алкила,
ii) -O-(C1-C4)-алкила,
iii) -CONR7R11,
iv) F, Cl, Br или I,
v) -COOR7,
vi) -NR7CONR7R11, или
vii) -NR7COOR11,
(g) -тетразол-5-илом; и
R13 является: (C1-C4)-алкилом.
Подклассом соединений формулы III являются соединения формулы IV:
или их фармацевтически приемлемая соль,
в которой R1 и R2, взятые вместе, образуют циклическую структуру:
A представляет:
a) -Y-[C(R6)(R6)]s-Y-, или
b) -C(R4)=C(R5)-C(R4)=C(R5)-;
s равно 1 или 2;
Y равен -O-;
R3a является:
(a) H,
(b) F, Cl, Br или I,
(c) (C1-C6)-алкилом,
(d) -OR7,
(e) -O-(CH2)m-OR7,
(f) -CONR7R11, или
(g) -COOR;
m равно 2, 3 или 4;
R4 и R5 независимо являются:
(a) H,
(b) (C1-C6)-алкилом,
(c) (C3-C7)-циклоалкилом,
(d) F, Cl, Br, I,
(e) -NR7COOR11,
(f) -SO2NR7R11,
(g) -O-(C1-C6)-алкилом,
(h) -S(O)n-(C1-C6)-алкилом, или
(i) -NHSO2R11;
n равно 0, 1 или 2,
R6 является:
(a) H, или
(b) (C1-C4)-алкилом;
R7 является:
(a) H,
(b) (C1-C6)-алкилом,
(c) фенилом, или
(d) бензилом;
R8 является:
(a) H,
(b) (C1-C6)-алкилом, или
(c) фенилом;
R9 является:
(a) H,
(b) (C1-C6)-алкилом, незамещенным или замещенным (C3-C7)-циклоалкилом,
(c) Cl, Br, F, I,
(d) (C1-C6)-алкокси, или
(e) гидрокси-(C1-C6)-алкилом,
R11 является:
(a) (C1-C6)-алкилом, незамещенным или замещенным заместителем, выбранным из группы, состоящей из:
i) -OR7,
ii) -N[R7]2,
iii) -NH2,
iv) -COOR7, или
v) -N[CH2CH2]2Q;
(b) арилом, где арил определяют как фенил или нафтил, который не замещен или замещен одним или двумя заместителями, выбранными из группы, состоящей из:
i) (C1-C4)-алкила,
ii) -O-(C1-C4)-алкила,
iii) -CO[NR7]2,
iv) F, Cl, Br или I,
v) -COOR7,
vi) -NH2,
vii) -NH[(C1-C4)-алкила],
viii) -N[(C1-C4)-алкила]2, или
ix) -CОN[CH2CH2]2Q,
c) -(C1-C4)-алкиларилом, где арил является таким, как он определен выше,
(d) -(C3-C7)-циклоалкилом,
(e)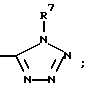
R7 и R11 у одного и того же атома азота могут соединяться с образованием кольца, выбранного из группы, состоящей из: морфолинила, пиперазинила или пирролила, или
Q является O, S или -NR7,
R12 является:
(a) H,
(b) (C1-C4)-алкилом, незамещенным или замещенным одним или двумя заместителями, выбранными из группы, состоящей из:
i) OH,
ii) -O-(C1-C4)-алкила,
iii) -O-(C1-C4)-циклоалкила,
iv) -S(O)n-(C1-C4)-алкила,
iv) -NR7-(C1-C4)-алкила,
v) -NR7R11,
vi) -COOR7,
vii) -CONHR7,
viii) -OCOR11,
ix) -CONR7R11,
x) -NR7CONR7R11,
xi) -NR7COOR11,
xii) -C(R6)(OH)-C(R6)(R7(OH), или
xiii) -SO2NR7R11,
(c) -COOR7,
(d) -CONH2,
(e) -CONR7R11,
(f) -CONR7CO2R7,
(g) -C(R6)(OH)-C(R6)(R7)(OH), или
(h) -CONHSO2R11,
(i) -SO2NR7R11,
(j) -NR7SO2NR7R11,
(k) -CO-аминокислотой, где аминокислоту определяют как L- и D-аминокислоту, выбранную из группы, состоящей из Ala, Ile, Phe, Asp, Pro и Val и которая может быть замещена (C1-C6)-алкиловым эфиром или амидом
(l)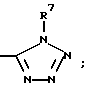
X является:
(a) -O-,
(b) -NR7-, или
(c) -простой связью,
Z является:
(a) -CO2H,
(b) -CO2R
(c) -CONH-(тетразол-5-ил);
(d) -CONHSO2-арилом, где арил определяют как фенил или нафтил, который является незамещенным или замещенным двумя заместителями, выбранными из группы, состоящей из:
i) (C1-C4)-алкила,
ii) -O-(C1-C4)-алкила,
(iii) -CONR7R11,
iv) F, Cl, Br или I,
v) -COOR7,
vi) -NH2,
vii) -NH[(C1-C4)-алкила],
viii) -N[(C1-C4)-алкила]2,
ix) -фенила,
(e) -CONHSO2-(C1-C8)-алкилом, где алкил является незамещенным или замещенным, как это определено в R4 (b).
(f) -CONHSO2-гетероарилом, где гетероарил определяют как карбазолил, фурил, тиенил, пирролил, изотиазолил, имидазолил, оксазолил, изоксазолил, тиазолил, пиразолил, пиразинил, пиридил, пиримидил, пуринил, или хинолинил, который не замещен или замещен одним или двумя заместителями, выбранными из группы, состоящей из:
i) (C1-C4)-алкила,
ii) -O-(C1-C4)-алкила,
iii) -CONR7R11,
iv) F, Cl, Br или I,
v) -COOR7,
vi) -NR7CONR7R11, или
vii) -NR7COOR11,
(g) -тетразол-5-илом; и
R13 является: (C1-C4)-алкилом.
Вторым вариантом соединений формулы II являются соединения формулы V
или их фармацевтически приемлемая соль,
где R1, R2, R3a и R3b независимо являются:
(a) H,
(b) F, Cl, Br или I,
(C) -NO2,
(d) (C1-C6)-алкилом,
(e) -OR7,
(f) -NHCO-(C1-C4)-алкилом,
(g) -NHCO-O(C1-C4)-алкилом,
(h) -O-(CH2)m-OR7,
(i) -CONR7R11, или
(j) -COOR7;
m равно 2, 3 или 4;
R4 и R5 независимо являются:
(a) H,
(b) (C1-C6)-алкилом,
(c) (C3-C7)-циклоалкилом,
(d) F, Cl, Br, I,
(e) -NR7COOR11,
(f) -SO2NR7R11,
(g) -O-(C1-C6)-алкилом,
(h) -S(O)n-(C1-C6)-алкилом, или
(i) -NHSO2R11;
n равно 0, 1 или 2,
R6 является:
(a) H, или
(b) (C1-C4)-алкилом;
R7 является:
(a) H,
(b) (C1-C6)-алкилом,
(c) фенилом, или
(d) бензилом;
R8 является:
(a) H,
(b) (C1-C6)-алкилом, или
(c) фенилом;
R9 и R10 независимо являются:
(a) H,
(b) (C1-C6)-алкилом, незамещенным или замещенным (C3-C7)-циклоалкилом,
(c) Cl, Br, F, I,
(d) (C1-C6)-алкокси, или
(e) гидрокси-(C1-C6)-алкилом,
R11 является:
(a) (C1-C6)-алкилом, незамещенным или замещенным заместителем, выбранным из группы, состоящей из:
i) -OR7,
ii) -N[R7]2,
iii) -NH2,
iv) -COOR7, или
v) -N[CH2CH2]2Q;
(b) арилом, где арил определяют как фенил или нафтил, который не замещен или замещен одним или двумя заместителями, выбранными из группы, состоящей из:
i) (C1-C4)-алкила,
ii) -O-(C1-C4)-алкила,
iii) -CO[NR7]2,
iv) F, Cl, Br или I,
v) -COOR7,
vi) -NH2,
vii) -NH[(C1-C4)-алкила],
viii) -N[(C1-C4)-алкила]2, или
ix) -CОN[CH2CH2]2Q, или
c) (C1-C4)-алкиларилом, где арил является таким, как он определен выше,
(d) (C3-C7)-циклоалкилом,
(e)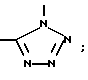
R7 и R11 у одного и того же атома азота могут соединяться с образованием кольца, выбранного из группы, состоящей из: морфолинила, пиперазинила или пирролила, или
Q является O, S или -NR7,
R12 является:
(a) H,
(b) (C1-C4)-алкилом, незамещенным или замещенным одним или двумя заместителями, выбранными из группы, состоящей из:
i) OH,
ii) -O-(C1-C4)-алкила,
iii) -O-(C1-C4)-циклоалкила,
iv) -S(O)n-(C1-C4)-алкила,
iv) -NR7-(C1-C4)-алкила,
v) -NR7R11,
vi) -COOR7,
vii) -CONHR7,
viii) -OCOR11,
ix) -CONR7R11,
x) -NR7CONR7R11,
xi) -NR7COOR11,
xii) -C(R6)(OH)-C(R6)(R7(OH), или
xiii) -SO2NR7R11,
(c) -COOR7,
(d) -CONH2,
(e) -CONR7R11,
(f) -CONR7CO2R7,
(g) -C(R6)(OH)-C(R6)(R7)(OH) или
(h) -CONHSO2R11,
(i) -SO2NR7R11,
(j) -NR7SO2NR7R11,
(k) - CO-аминокислотой, где аминокислоту определяют как L- и D-аминокислоту, выбранную из группы, состоящей из Ala, Ile, Phe, Asp, Pro и Val и которая может быть замещена (C1-C6)-алкиловым эфиром или амидом или
(l)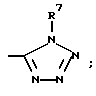
X является:
(a) -O-,
(b) -NR7-, или
(c) - простой связью;
Z является:
(a) -CO2H,
(b) -CO2R13,
(c) -CONH-(тетразол-5-илом),
(d) -CONHSO2-арилом, где арил определяют как фенил или нафтил, который является незамещенным или замещенным двумя заместителями, выбранными из группы, состоящей из:
i) (C1-C4)-алкила,
ii) -O-(C1-C4)-алкила,
(iii) -CONR7R11,
iv) F, Cl, Br или I,
v) -COOR7,
vi) -NH2,
vii) -NH[(C1-C4)-алкила],
viii) -N[(C1-C4)-алкила]2, или
ix) -фенила,
(e) -CONHSO2-(C1-C8)-алкилом, где алкил является незамещенными или замещенными, как это определено в R4 (b),
(f) -CONHSO2-гетероарилом, где гетероарил определяют как карбазолил, фурил, тиенил, пирролил, изотиазолил, имидазолил, оксазолил, изоксазолил, тиазолил, пиразолил, пиразинил, пиридил, пиримидил, пуринил или хинолинил, который не замещен или замещен одним или двумя заместителями, выбранными из группы, состоящей из:
i) (C1-C4)-алкила,
ii) -O-(C1-C4)-алкила,
iii) -CONR7R11,
iv) F, Cl, Br или I,
v) -COOR7,
vi) -NR7CONR7R11, или
vii) -NR7COOR11,
(g) -тетразол-5-илом, и
R13 является: (C1-C4)-алкилом.
Третьим вариантом соединений формулы II являются соединения формулы VI: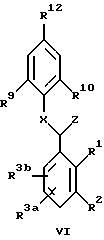
или их фармацевтически приемлемая соль,
в которой R1 и R2 представлены следующей циклической структурой:
A представляет:
(a) -Y-[C(R6)(R6)]s-Y, или
(b) -C(R4)=C(R5)-C(R4)=C(R5)-;
s равно 1 или 2;
Y равен -O-, -S-; и NR7
R3a и R3b независимо являются:
(a) H,
(b) F, Cl, Br или I,
(c) -NO2,
(d) (C1-C6)-алкилом,
(e) -OR7,
(f) -NHCO-(C1-C4)-алкилом,
(g) -NHCO-O(C1-C4)-алкилом,
(h) -O-(CH2)m-OR7,
(i) -CONR7R11, или
(j) -COOR7;
m равно 2, 3 или 4,
R4 и R5 независимо являются:
(a) H,
(b) (C1-C6)-алкилом,
(c) (C3-C7)-циклоалкилом,
(d) F, Cl, Br, I,
(e) -NR7COOR11,
(f) -SO2NR7R11,
(g) -O-(C1-C6)-алкилом,
(h) -S(O)n-(C1-C6)-алкилом, или
(i) -NHSO2R11;
n равно 0, 1 или 2,
R6 является:
(a) H, или
(b) (C1-C4)-алкилом;
R7 является:
(a) H,
(b) (C1-C6)-алкилом,
(c) фенилом, или
(d) бензилом;
R8 является:
(a) H,
(b) (C1-C6)-алкилом, или
(c) фенилом,
R9 и R10 независимо являются:
(a) H,
(b) (C1-C6)-алкилом, незамещенным или замещенным (C3-C7)-циклоалкилом,
(c) Cl, Br, F, I,
(d) (C1-C6)-алкокси, или
(e) гидрокси-(C1-C6)-алкилом,
R11 является:
(a) (C1-C6)-алкилом, незамещенным или замещенным заместителем, выбранным из группы, состоящей из:
i) -OR7,
ii) -N[R7]2,
iii) -NH2,
iv) -COOR7, или
v) -N[CH2CH2]2Q,
(b) арилом, где арил определяют как фенил или нафтил, который не замещен или замещен одним или двумя заместителями, выбранными из группы, состоящей из:
i) (C1-C4)-алкила,
ii) -O-(C1-C4)-алкила,
iii) -CO[NR7]2,
iv) F, Cl, Br или I,
v) -COOR7,
vi) -NH2,
vii) -NH[(C1-C4)-алкила],
viii) -N[(C1-C4)-алкила]2, или
ix) -CОN[CH2CH2]2Q,
c) -(C1-C4)-алкиларилом, где арил является таким, как он определен выше,
(d) (C3-C7)-циклоалкилом,
(e)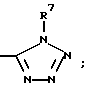
R7 и R11 у одного и того же атома азота могут соединяться с образованием кольца, выбранного из группы, состоящей из: морфолинила, пиперазинила или пирролила, или
Q является O, S или -NR7,
R12 является:
(a) H,
(b) (C1-C4)-алкилом, незамещенным или замещенным одним или двумя заместителями, выбранными из группы, состоящей из:
i) OH,
ii) -O-(C1-C4)-алкила,
iii) -O-(C1-C4)-циклоалкила,
iv) -S(O)n-(C1-C4)-алкила,
v) -NR7R11,
vi) -COOR7,
vii) -CONHR7,
viii) -OCOR11,
ix) -CONR7R11,
x) -NR7CONR7R11,
xi) -NR7COOR11,
xii) -C(R6)(OH)-C(R6)(R7(OH), или
xiii) -SO2NR7R11,
(c) -COOR7,
(d) -CONH2,
(e) -CONR7R11,
(f) -CONR7CO2R7,
(g) -C(R6)(OH)-C(R6)(R7)(OH), или
(h) -CONHSO2R11,
(i) -SO2NR7R11,
(j) -NR7SO2NR7R11,
(k) -CO-аминокислотой, где аминокислоту определяют как L- и D-аминокислоту, выбранную из группы, состоящей из Ala, Ile, Phe, Asp, Pro и Val и которая может быть замещена (C1-C6)-алкиловым эфиром или амидом или
(l)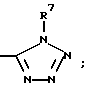
X является:
(a) -O-,
(b) -NR7, или
(c) - простой связью;
Z является:
(a) -CO2H,
(b) -CO2R13,
(c) -CONH-(тетразол-5-ил),
(d) -CONHSO2-арилом, где арил определяют как фенил или нафтил, который является незамещенным или замещенным одним или двумя заместителями, выбранными из группы, состоящей из:
i) (C1-C4)-алкила,
ii) -O-(C1-C4)-алкила,
(iii) -CONR7R11,
iv) F, Cl, Br или I,
v) -COOR7,
vi) -NH2,
vii) -NH[(C1-C4)-алкила],
viii) -N[(C1-C4)-алкила]2,
ix) -фенила,
(e) -CONHSO2-(C1-C8)-алкилом, где алкил является незамещенным или замещенным, как это определено в R4 (b).
(f) -CONHSO2-гетероарилом, где гетероарил определяют как карбазолил, фурил, тиенил, пирролил, изотиазолил, имидазолил, оксазолил, изоксазолил, тиазолил, пиразолил, пиразинил, пиридил, пиримидил, пуринил или хинолинил, который не замещен или замещен одним или двумя заместителями, выбранными из группы, состоящей из:
i) (C1-C4)-алкила,
ii) -O-(C1-C4)-алкила,
(iii) -CONR7R11,
iv) F, Cl, Br или I,
v) -COOR7,
vi) -NR7CONR7R11, или
vii) -NR7COOR11,
(g) -тетразол-5-илом, и
R13 является: (C1-C4)-алкилом.
Вариантом соединений формулы (I) являются следующие соединения:
2-[(2,6-дипропил-4-гидроксиметил)фенокси] -2-(3-метилфенил)- уксусная кислота;
2-[(2,6-дипропил-4-гидроксиметил)фенокси] -2-(4-феноксифенил)- уксусная кислота;
2-[(2,6-дипропил-4-гидроксиметил)фенокси] -2-(4-фенилфенил)- уксусная кислота;
2-[(2,6-дипропил-4-гидроксиметил)фенил] -2-(3-карбоксифенил)- уксусная кислота;
2-[(2,6-дипропил-4-гидроксиметил)фенокси] -2- (3,4-этилендиоксифенил)уксусная кислота;
2-[(2,6-дипропил-4-гидроксиметил)фенокси] -2- (3,4,5-триметоксифенил)уксусная кислота;
2-[(2,6-дипропил-4-гидроксиметил)фенокси] -2- (3,4-метилендиоксифенил)уксусная кислота;
2-[(2,6-дипропил-4-гидроксиметил)фенокси]-2- (3,4-диметоксифенил)уксусная кислота;
2-[(2,6-дипропил-4-гидроксиметил)фенокси]-2- (3,5-диметоксифенил)уксусная кислота;
2-((2,6-дипропил-4-тетразол-5-ил))фенокси-2- (3-бромфенил)уксусная кислота;
2-[(2,6-дипропил-4-гидроксиметил)фенокси] -2- (3-бромфенил)уксусная кислота;
2-[(2,6-дипропил-4-гидроксиметил)фенокси]-2- (2-нафтил)уксусная кислота;
2-[(2,6-дипропил-4-(2-гидроксиэтил)фенокси]-2- (2-нафтил)уксусная кислота,
2-[(2,6-дипропил-4-(2-гидроксиэтил)фенокси] -2- (3,4-метилендиоксифенил)уксусная кислота;
2-[(2,6-дипропил-4-(2-гидроксиэтил)фенокси] -2- (3-метоксифенил)уксусная кислота;
2-[(2,6-дипропил-4-(1,2-дигидроксиэтил)фенокси] -2- (2-нафтил)уксусная кислота;
2-[(2,6-дипропил-4-(1-гидроксипентил)фенокси] -2- (2-нафтил)уксусная кислота;
2-[(4-карбокси-2,6-дипропил)фенокси]-2-фенилуксусная кислота;
2-[(4-карбокси-2,6-дипропил)фенокси] -2-(3,4-дихлорфенил)- уксусная кислота;
2-[(4-карбокси-2,6-дипропил)фенокси]-2- (3-бромфенил)уксусная кислота;
2-[(4-карбокси-2,6-дипропил)фенокси]-2- [3,4-метилендиоксифенил]уксусная кислота;
2-[(4-карбокси-2,6-дипропил)фенокси] -2- (3-метоксифенил]уксусная кислота,
(N-бензолсульфонил)-2-[4-(N-бензолсульфонил)карбоксамидо- 2,6-дипропилфенокси]-2-(3-бромфенил)ацетамид,
(N-трет-бутилбензолсульфонил)-2-(4-метоксикарбонил-2- пропилфенокси)-2-(3,4-метилендиоксифенил)ацетамид,
(N-бензолсульфонил)-2-[4-метоксикарбонил-2- пропилфенокси)-2-(3,4-метилендиоксифенил)ацетамид,
N-(4-фенилбензолсульфонил)-2-(4-метоксикарбонил-2- пропилфенокси)-2-(3,4-метилендиоксифенил)ацетамид;
N-(4-хлорбензолсульфонил)-2-(4-метоксикарбонил-2- пропилфенокси)-2-(3,4-метилендиоксифенил)ацетамид,
N-(4-метилбензолсульфонил)-2-(4-метоксикарбонил-2- пропилфенокси)-2-(3,4-метилендиоксифенил)ацетамид;
N-(5-изо-бутилтиен-2-илсульфонил)-2-(4-метоксикарбонил- 2-пропилфенокси)-2-(3,4-метилендиоксифенил)ацетамид;
N-(4-метоксибензолсульфонил)-2-(4-метоксикарбонил-2- пропилфенокси)-2-(3,4-метилендиоксифенил)ацетамид;
N-(4-диметиламинобензолсульфонил)-2-(4-метоксикарбонил-2- пропилфенокси)-2-(3,4-метилендиоксифенил)ацетамид;
N-(2-метилбензолсульфонил)-2-(4-метоксикарбонил-2- пропилфенокси)-2-(3,4-метилендиоксифенил)ацетамид;
N-(2-метоксикарбонилбензолсульфонил)-2-(4-метоксикарбонил)-2- пропилфенокси)-2-(3,4-метилендиоксифенил)ацетамид,
N-(2-хлорбензолсульфонил)-2-(4-метоксикарбонил-2- пропилфенокси)-2-(3,4-метилендиоксифенил)ацетамид,
N-(3-хлорбензолсульфонил)-2-(4-метоксикарбонил-2- пропилфенокси)-2-(3,4-метилендиоксифенил)ацетамид,
N-(фенилметансульфонил)-2-(4-метоксикарбонил-2- пропилфенокси)-2-(3,4-метилендиоксифенил)ацетамид,
N-(дансилсульфонил)-2-(4-метоксикарбонил-2- пропилфенокси)-2-(3,4-метилендиоксифенил)ацетамид,
N-(8-хинолинсульфонил)-2-(4-метоксикарбонил-2- пропилфенокси)-2-(3,4-метилендиоксифенил)ацетамид;
N-(4-третбутилбензолсульфонил)-2-(4-карбокси-2- пропилфенокси)-2-(3,4-метилендиоксифенил)ацетамид,
N-(бензолсульфонил)-2-(4-карбокси-2-пропилфенокси)-2- (3,4-метилендиоксифенил)ацетамид,
N-(4-фенилбензолсульфонил)-2-(4-карбокси-2-пропилфенокси)- 2-(3,4-метилендиоксифенил)ацетамид;
N-(4-хлорбензолсульфонил)-2-(4-карбокси-2-пропилфенокси)-2- (3,4-метилендиоксифенил)ацетамид,
N-(4-метилбензолсульфонил)-2-(4-карбокси-2-пропилфенокси)-2- (3,4-метилендиоксифенил)ацетамид;
N-(5-изобутилтиен-2-илсульфонил)-2-(4-карбокси-2-пропилфенокси)- 2-(3,4-метилендиоксифенил)ацетамид,
N-(4-метоксибензолсульфонил)-2-(4-карбокси-2-пропилфенокси)- 2-(3,4-метилендиоксифенил)ацетамид;
N-(4-диметиламинобензолсульфонил)-2-(4-карбокси-2-пропилфенокси)- 3,4-метилендиоксифенил)ацетамид;
N-(2-метилбензолсульфонил)-2-(4-карбокси-2-пропилфенокси)- 2-(3,4-метилендиоксифенил)ацетамид;
N-(2-метоксикарбонилбензолсульфонил)-2-(4-карбокси-2- пропилфенокси)-2-(3,4-метилендиоксифенил)ацетамид,
N-(2-хлорбензолсульфонил)-2-(4-карбокси-2- пропилфенокси)-2-(3,4-метилендиоксифенил)ацетамид,
N-(3-хлорбензолсульфонил)-2-(4-карбокси-2- пропилфенокси)-2-(3,4-метилендиоксифенил)ацетамид;
N-(фенилметансульфонил)-2-(4-карбокси-2- пропилфенокси)-2-(3,4-метилендиоксифенил)ацетамид,
N-(дансульфонил)-2-(4-карбокси-2-пропилфенокси)-3,4- метилендиоксифенил)ацетамид;
N-(8-хинолинсульфонил)-2-(4-карбокси-2-пропилфенокси)-2- (3,4-метилендиоксифенил)ацетамид;
N-(8-хинолинсульфонил)-2-(4-карбоксамидо-2-пропилфенокси)-2- (3,4-метилендиоксифенил)ацетамид,
α- (4-карбометокси-2-н-пропилфенокси)-3,4- метилендиоксифенил)уксусная кислота;
N-(4-изо-пропилбензолсульфонил) -α- (4-карбометокси-2-н- пропилфенокси)-3,4-метилендиоксифенил)ацетамид;
Двукалиевая соль N-(4-изо-пропилбензосульфонил) -α- (4-карбокси-2-н-пропилфенокси)-3,4-метилендиоксифенилацетамида;
α- (2-изо-изобутил-4-карбометоксифенокси)- 3,4-метилендиоксифенил)уксусная кислота;
N-(4-изо-пропилбензосульфонил) -α- (2-изо-бутил-4- карбометоксифенокси)-3,4-метилендиоксифенилацетамид;
N-(4-изо-пропилбензосульфонил) -α- (2-изо-бутил-4- карбоксифенокси)-3,4-метилендиоксифенилацетамид;
N-(4-изо-пропилбензолсульфонил) -α- (2-н-пропил-4- метоксикарбонилфенокси) α- метил-3,4-метилендиоксифенилацетамид;
Двукалиевая соль N-(4-изо-пропилбензолсульфонил) -α- (2-н-пропил-4-карбоксифенокси) -α- метил 3,4-метилендиоксифенилацетамида;
N-(4-изо-пропилбензосульфонил) -α- (2-н-пропил-4- карбоксамидофенокси)-3,4-метилендиоксифенилацетамид;
N-(4-изо-пропилбензолсульфонил) -α- (2-н-пропил-4- гидроксиметилфенокси)-3,4-метилендиоксифенилацетамид;
N-(4-изо-пропилбензолсульфонил) -α- (4-формил-2-н- пропилфенокси)-3,4-метилендиоксифенилацетамид,
α- (4-ацетил-2-н-пропилфенокси)- 3,4-метилендиоксифенил)-уксусная кислота;
N-(4-изо-пропилбензолсульфонил) -α- (4-ацетил-2-н- пропилфенокси)-3,4-метилендиоксифенилацетамид;
α- (2-н-пропилфенокси)-3,4-метилендиоксифенил)уксусная кислота;
N-(4-изо-пропилбензолсульфонил) -α- (2-н-пропилфенокси)-3,4-метилендиоксифенилацетамид,
α- (3-метоксифенокси)-3,4-метилендиоксифенилуксусная кислота;
α- (2-(2-гидроксиэтил)фенокси)-3,4- метилендиоксифенилуксусная кислота;
α- (2-(2-карбометоксиэтил)фенокси)- 3,4-метилендиоксифенилуксусная кислота;
α- (4-гидроксиметил-2-н-пропилфенокси)-3,4- метилендиоксифенилуксусная кислота;
α- (4-(2-гидроксиэтил)-2н-пропилфенокси)- 3,4-метилендиоксифенилуксусная кислота;
N-(4-изо-пропилбензолсульфонил) -α- (2-(2- карбометоксиэтил)-фенокси)-3,4-метилендиоксифенилацетамид;
N-(4-изо-пропилбензолсульфонил) -α- (2-(2- карбоксиэтил)-фенокси)-3,4-метилендиоксифенилацетамид;
α- (2-(2-карбоксиэтил)фенокси)-3,4- метилендиоксифенилуксусная кислота;
N-(4-изо-пропилбензолсульфонил)-2-(4-карбометокси-2н- пропилфенокси)-2-(5-метокси-3,4-метилендиоксифенил)ацетамид;
N-(4-изо-пропилбензолсульфонил)-2-(4-карбокси-2н- пропилфенокси)-2-(5-метокси-3,4-метилендиоксифенил)ацетамид;
N-(4-изо-пропилбензолсульфонил)-2-(4-(N-изо- пропилбензолсульфонил)карбоксамидо)-2-пропилфенокси)-2- (5-метокси-3,4-метилендиоксифенил)ацетамид;
N-(4-изо-пропилбензолсульфонил)-2-(4-карбоксамидо-2- пропилфенокси)-2-(5-метокси-3,4-метилендиоксифенил)ацетамид;
N-(4-изо-пропилбензолсульфонил)-2-(4-(N- метилкарбоксамидо)-2-пропилфенокси)-2-(5-метокси-3,4- метилендиоксифенил)ацетамид;
N-(4-изо-пропилбензолсульфонил)-2-(4-(N-2-гидроксиэтил- карбоксамидо)-2-пропилфенокси)-2-(5-метокси-3,4- метилендиоксифенил)ацетамид;
N-(4-изо-пропилбензолсульфонил)-2-(4-(N-морфолинил- карбоксамидо)-2-пропилфенокси)-2-(5-метокси-3,4- метилендиоксифенил)ацетамид;
N-(4-изо-пропилбензолсульфонил)-2-(4-N-3- метилбутилкарбоксамидо)-2-пропилфенокси)-2-(5-метокси-3,4- метилендиоксифенил)ацетамид;
N-(4-изо-пропилбензолсульфонил)-2-(4-(N- карбоксиметилкарбоксамидо)-2-пропилфенокси)-2-(5-метокси-3,4- метилендиоксифенил)ацетамид;
N-(4-изо-пропилбензолсульфонил)-2-(4-(N-(L-Ala-OEt) карбоксамидо)-2-пропилфенокси)-2-(5-метокси-3,4- метилендиоксифенил)ацетамид;
N-(4-изо-пропилбензолсульфонил)-2-(4-(N-2- этоксикарбонилэтилкарбоксамид-2-пропилфенокси)-2-(5-метокси-3,4- метилендиоксифенил)ацетамид;
N-(4-изо-пропилбензолсульфонил)-2-(4-(N-(L-Ala) карбоксамидо)-2-пропилфенокси)-2-(5-метокси-3,4- метилендиоксифенил)ацетамид;
N-(4-изо-пропилбензолсульфонил)-2-(4-(N-2- карбоксиэтилкарбоксамидо)-2-пропилфенокси)-2-(5-метокси-3,4- метилендиоксифенил)ацетамид;
N-(4-изо-пропилбензолсульфонил)-2-(4-(N-3- гидроксипропилкарбоксамидо)-2-пропилфенокси)-2-(5-метокси-3,4- метилендиоксифенил)ацетамид;
N-(4-изо-пропилбензолсульфонил)-2-(4-(N-тетразол-5- илкарбоксамидо)-2-пропилфенокси)-2-(5-метокси-3,4- метилендиоксифенил)ацетамид;
N-(4-изо-пропилбензолсульфонил)-2-(4-(N-3-(морфолин-4-ил)- пропилкарбоксамидо)-2-пропилфенокси)-2-(5-метокси-3,4- метилендиоксифенил)ацетамид;
N-(4-изо-пропилбензолсульфонил)-2-(4-(N-(D-Ala-OMe) карбоксамидо)-2-пропилфенокси)-2-(5-метокси-3,4- метилендиоксифенил)ацетамид;
N-(4-изо-пропилбензолсульфонил)-2-(4-(N-(D-Ala) карбоксамидо)-2-пропилфенокси)-2-(5-метокси-3,4- метилендиоксифенил)ацетамид;
N-(4-изо-пропилбензолсульфонил)-2-(4-(N-3- карбоксиметилпропил)карбоксамидо)-2-пропилфенокси)-2-(5-метокси-3,4- метилендиоксифенил)ацетамид;
N-(4-изо-пропилбензолсульфонил)-2-(4-(N-(3- карбоксипропил)карбоксамидо)-2-н-пропилфенокси)-2-(5-метокси-3,4- метилендиоксифенил)ацетамид;
N-(4-изо-пропилбензолсульфонил)-2-(4-(N-изопропилкарбамоил)- амино-2-пропилфенокси)-2-(3,4-метилендиоксифенил)ацетамид;
α- (2-н-пропил-4-метиламиносульфонил)фенокси)- (3,4-метилендиоксифенил)уксусная кислота;
Калиевая соль N-(4-изо-пропилбензолсульфонил) -α- 2-н-пропил-4-метиламиносульфонилфенокси)- 3,4-метилендиоксифенилацетамида;
Предпочтительными соединениями этого изобретения являются:
Двукалиевая соль N-(4-изо-пропилбензолсульфонил) -α- (4-карбокси-2н-пропилфенокси)-3,4-метилендиоксифенилацетамида;
N-(8-хинолинсульфонил)-2-(4-карбокси-2-пропилфенокси)-2- (3,4-метилендиоксифенил)ацетамид;
N-(4-диметиламинобензолсульфонил)-2-(4-карбокси-2-н- пропилфенокси)-2-(3,4-метилендиоксифенил)ацетамид;
Алкильные заместители, перечисленные выше, представляют собой углеводороды с прямой и разветвленной цепью определенной длины, например метил, этил, изопропил, изобутил, неопентил, изопентил и т.д.
Алкенильные заместители представляют собой описанные выше алкильные группы, которые модифицируют таким образом, чтобы каждая содержала углерод-углеродную двойную связь, например винил, аллил и 2-бутенил.
Циклоалкил означает кольца, состоящие из метиленовых групп в количестве от 3-х до 8-ми, каждая из которых может быть замещена или не замещена другими углеводородными заместителями, и включающие например, циклопропил, циклопентил, циклогексил и 4-метилциклогексил.
Алкоксигруппа представляет собой алкильную группу, которая описана выше, присоединенную через кислородный мостик.
Гетероарил определяют как карбазолил, фурил, тиенил, пирролил, изотиазолил, имидазолил, изоксазолил, тиазолил, оксазолил, пиразолил, пиразинил, пиридил, пиримидил, пуринил или хинолинил.
Хотя реакционные схемы, описанные ниже, являются общими, специалистам в области органического синтеза понятно, что присутствие одной или нескольких функциональных групп, содержащихся в данном соединении формулы I, могут представлять молекулу, получение которой не описано данной последовательностью синтеза. В таком случае, может быть использован альтернативный путь синтеза, альтернативный порядок стадий или методика защиты или освобождения от защиты.
Во всех случаях особенности реакционных условий, включая реагенты, растворитель, температуру и время, следует выбирать таким образом, чтобы они были совместимы с природой функциональных групп, присутствующих в молекуле.
Соединения формулы I, и в особенности соединения формулы III, можно синтезировать с использованием реакций и методик, описанных для синтеза негетероциклических компонентов в заявке на патент WO 91/11999 (Мерк и Ко., опубликованной 22 августа 1991), а также в патенте США N 5177095 (Мерк и Ко. , 5 января 1993).
Реакционные схемы, описанные ниже, для простоты были обобщены. Кроме того, следует понимать, что в обобщенных схемах, приведенных ниже, алкильные и арильные группы представляют нефункциональные или функциональные производные, которые описаны выше, если в тексте не указано более узко. Уходящей группой Q, присутствующей в алкилирующих агентах, является либо хлор, бром, иод, метансульфонат, р-толуолсульфонат или трифлат.
Схема 1.
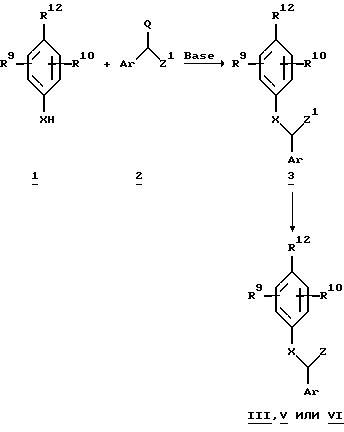

Q = Cl, Br, I, OMs, OTs или OTf
Z1 = предшественник Z
Более конкретно, соединения формулы III, V или VI (где X является кислородом, серой или соответствующим образом замещенным азотом) можно синтезировать, как показано на схеме 1. Замещенное соединение 1 может взаимодействовать с алкилирующим агентом 2 в соответствующем растворителе, например спиртах (метаноле, этаноле, изопропаноле и т.п.), диметилформамиде (DMF), диметилсульфоксиде (DMSO), тетрагидрофуране (THF) и ацетоне, в присутствии соли щелочного металла, например алкоксидов, карбонатов, гидроксидов и гидридов, или органических оснований, например триалкиламинов или алкиллития, для получения соединения 3. Группу, присутствующую в соединении 3, можно затем превратить для получения соответствующих соединений формулы III, V или VI.
В общем, алкилирующий агент 2 можно получить с использование способов и методик, приведенных в патенте США N 5177095. Более конкретно, соединение 2 (где Z1 представляет COOR, а Q является Br) можно синтезировать из замещенных арилуксусных кислот 4, которые приведены в схеме 2. Замещенную арилуксусную кислоту 4 превращают в соответствующий эфир или путем кипячения с обратным холодильником кислоты в соответствующем спирте в присутствии каталитического количества концентрированной серной кислоты, или при использовании других общепринятых способов этерификации. Полученный эфир затем кипятят с обратным холодильником в тетрахлориде углерода с N-бромсукцинимидом и каталитическим количеством инициатора радикалов (например, AIBN-азобисизобутиронилтрила или бензоилпероксида) для получения эфира 2-бром-арилусусной кислоты 5.
Схема 2.

Альтернативно эфир 5 можно также получить из соответствующих арилальдегидов (Схема 3.). Альдегид 6 можно реагировать с триметилсилилцианидом и каталитическими количествами KCN и 18-краун-6-эфира для получения соответствующего триметилсилилцианогидрина 7, который при дальнейшей обработке газообразным HCl и спиртом дает 2-гидроксиэфир 8.
Эфир 8 обрабатывают трифенилфосфеном и тетрабромидом углерода в метиленхлориде для получения производных 2-бромарилацетата.
Схема 3.
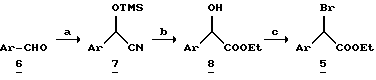
a. TMSCN, Кат. KCN, CH2Cl2, C8-краун-6;
b. HCl (г), этиловый спирт;
Схема 4 иллюстрирует обычный синтез алкилирующего агента 12 (где Ar представляет гетероцикл, например индол). Соответственным образом замещенный цианиндол 9 (общий синтез замещенных индолов представлен в R.K.Brown, Indoles, Part One, Ed. W.J. Houlihan, Vol. 25, Chapter 11, Wiley Interscience, New Jork, 1972) восстанавливают DIBAL-6 для получения соответствующего альдегида, который затем превращают в производную N-Boc 10.
Реакция 10 с анионом трихлорметила (генерированного из KOH и CHCl3; J.M. Wyvratt et al. , J. Org. Chem., 52, 944-945 (1987), после которой следует обработка водным раствором NaOH в DMF (диметилформамиде) дает спирт 11. Обработка спирта 11 диазометаном, после которой следует реакция с CBr4/Ph3P, приводит к получению алкилирующего агента 12.
Схема 4
a. (i) DIBALH, Толуол; (ii) BOC2O, DMAP, CH2Cl2
b. CHCl3, KOH, DMF, 0oC; (ii) NaOH, DMF/H2O
(c) (i) CH2N2; (ii) CBr4/Ph3P, CH2Cl2.
Обычный синтез алкилирующих агентов, содержащих замещенный бензоксазол или бензотиазольное кольцо, представлен на схеме 5. Замещенный бензоксазол 14 получают из соответствующего о-аминофенола 13 путем реакции соответствующего ортоэфира в условиях дефлегмации (другие способы синтеза бензоксазолов смотри: S.A.Lang и Y. Lin, Comprehensive Heterocyclic Chemistry, Vol. 6, 1-130, Ed. C.W.Rees, и ссылки, приведенные в этом источнике информации). Восстановление замещенного бензоксазола 14 NaBH4 дает спирт 15, который затем окисляют пиридинийдихроматом (PDC) для получения соответствующего альдегида 16. Дальнейшая обработка альдегида 16 дает ключевое промежуточное соединение 17. Подобным образом может быть получен бензотиазол 19 из соответствующим образом замещенного о-аминотиофенола 18.
Схема 5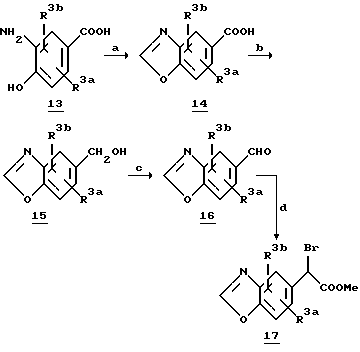
a. CH(OEt)3, EtOH, кипячение
b. (i) ClCOOEt, Et3N, THF; (ii) NaBH4, THF-H2O
c. сихромат пиридиния CH2Cl2
d. (i) CHCl3, KOH, DMF, 0oC; (ii) NaOH, DME/H2O;
(iii) HCl/MeOH; (iv) CBr4/Ph3P, CH2Cl2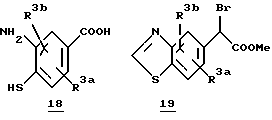
Схема 6 показывает синтез алкилирующих агентов бензофурана и дигидробензофурана 23 и 25. Бензофуран 21 можно получить из α -феноксикарбонильного соединения 20 реакцией замыкания цикла [Stoemer и Wehin, Chem. Ber., 35, 3549 (1902)] (общие способы синтеза бензофурана и дигидробензофуранов смотри в: R.C. Elderfield и V.B. Meyer, Heterocyclic Compounds, Vol. 2, Chapter 1, Ed. R.C. Elderfield, Wiley; и в ссылках, приведенных в этом источнике информации). Для получения альдегида 22, который затем превращают в соответствующий алкилирующий агент 23, эфир 21 восстанавливают.
Дигидробензофурановый эфир 24, полученный путем каталитического восстановления эфира 21, можно затем превратить в соответствующий алкилирующий агент 25 использованием последовательности реакций, приведенных в схеме 6.
Бензотиофен 26 можно синтезировать из соответствующего альдегида 26b способом, подобным тому, который представлен на схеме 6 для бензофурана 23. Бензотиофен 26b моно получить окислительной циклизацией (с использованием щелочного раствора феррицианида калия) соответствующим образом замещенной о-меркаптокоричной кислоты 26a [C. Chmelewsky и P. Friedlander, Chem. Ber., 46, 1903 (1913)]. Общие способы синтеза бензотиофена смотри в Comprehensive Heterocyclic Chemistry, vol. 4, Chapter 3-15; Eds. A. Katritzky и C.W. Rees).
На схеме 7 представлен обычный синтез α -бромарилацетатов 30 и 32, содержащих соответствующим образом замещенные метилендиокси- или 1,4-диоксановые кольца. Для получения соединения 28 замещенную производную катехина 27 обрабатывают соответствующим дибромидом (где m равно 1 или 2) в присутствии карбоната цезия в диметилформамиде. Обработка 28 DIBALH приводит к получению альдегида 29, который затем превращают в желаемый алкилбромид.
Схема 6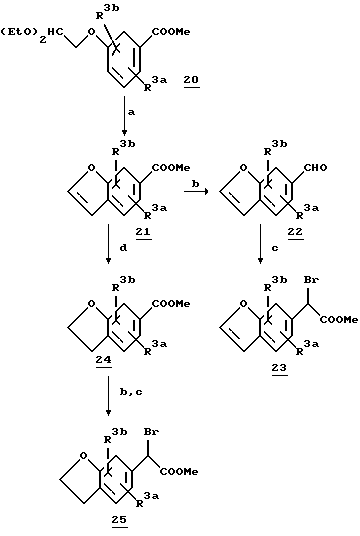
a. ZnCl2 толуол
b. DIBALH,
c. (i) CHCl3, KOH, DMF, 0oC;
(ii) NaOH, DME/H2O;
(iii) HCl/MeOH;
(iv) CBr4/Ph3P, CH2Cl2;
d. Ra-Ni/H2
Схема 7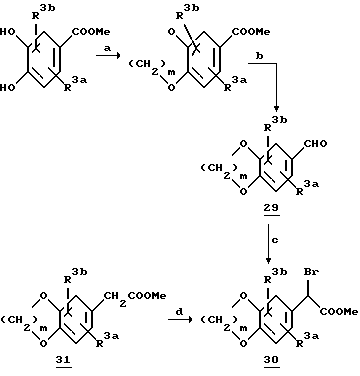
a. Br-(CH2)m-Br, Cs2CO3, DMF
b. DIBALH, толуол
c. (i) CHCl3, KOH, DMF, 0oC; (ii) NaOH, DME/H2O; (iii) HCl/MeOH; (iv) CBr4/Ph3P, CH2Cl2;
d. NBS, AIBN, CCl4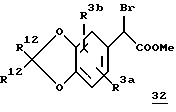
Реакции осуществляли в растворителе, подходящем для используемых реагентов и материалов, а также походящем для осуществления превращения. Специалистам в области органического синтеза понятно, что функциональные группы, имеющиеся у гетероцикла и используемых реагентов, должны быть совместимы с проводимыми химическими превращениями. В зависимости от применяемых реакций и методик оптимальные выходы могут потребовать изменений последовательности стадий синтеза или использования защитных групп с последующим освобождением от защиты.
Соединения, используемые в новом способе лечения этого изобретения, образуют соли с различными неорганическими и органическими кислотами и основаниями, которые также включены в объем изобретения. Такие соли включают аммониевые соли, соли щелочного металла, например соли натрия и калия, соли щелочноземельного металла, например кальция и магния, соли органических оснований, например соли дициклогексиламина, N-метил-D-глюкамина, соли аминокислот, например аргинина, лизина и т.п. Могут быть также получены соли органических и неорганических кислот, например HCl, HBr, H2SO4, H3PO4, метансульфоновой, толуолсульфоновой, малеиновой, фумаровой, камфорсульфоновой.
Соли могут быть образованы обычным способом, например, путем взаимодействия свободнокислотных или свободно-основных форм продукта с одним или несколькими эквивалентами соответствующего основания или кислоты в растворителе и среде, в котором(ой) соль нерастворима, или в растворителе, таком как вода, которую затем удаляют в вакууме или посредством сушки вымораживанием, или путем обмена катионов существующей соли на другой катион на соответствующей ионообменной смоле.
Следует принимать во внимание, что в этом изобретении могут быть получены соединения производных функциональных групп общей формулы I с получением производных пролекарства, которое способно к обратному превращению в исходные соединения in vivo. Концепция введения пролекарства широко освещена (например, A.A. Sinkula и Annual Reports в Medical Chemistry, vol. 10, R.V. Heinzelman, Ed. , Academic Press, New York, London, 1975, Ch. 31, pp. 306-326, H. Ferres, Drugs of Today, vol. 19, 499-538 (1983) и J. Med. Chem., 18, 172 (1975). Примеры таких пролекарств включают производные физиологически приемлемых и метаболически нестойких сложных эфиров, например низшего алкилового (например, метилового или этилового эфиров), арильных (например, 5-инданильных эфиров), алкенильных (например, виниловых эфиров), алкоксиалкиловых (например, метоксиметиловых эфиров), алкилтиоалкиловых (например, метилтиометиловых эфиров), алканоил оксиалкиловых (например, пивалоилоксиметиловых эфиров), и замещенные или незамещенные аминоэтиловые эфиры (например, 2-диметиламиноэтиловые эфиры). Кроме того, в объем настоящего изобретения входят любые физиологически приемлемые эквиваленты соединений общей формулы I, подобные метаболически нестойким эфирам, которые способны к продуцированию исходных соединений общей формулы I in vivo.
Кроме того, следует учитывать, что большинство соединений общей формулы I, представленных в формуле изобретения, являются асимметричными и получаются в виде рацемических смесей энантиомеров, и что как рацемические соединения, так и выделенные отдельные энантиомеры следует рассматривать как входящие в объем этого изобретения. Рацемические соединения этого изобретения можно разделить для получения отдельных энантиомеров с использованием методов, известных специалистам в области органического синтеза. Диастереоизомерные соли, сложные эфиры или имиды можно получить, например, из рацемического соединения общей формулы I и подходящего оптически активного амина, аминокислоты, спирта или т.п.
Диастереоизомерные соли, сложные эфиры или имиды разделяют и очищают, оптически активные энантиомеры регенерируют, при этом предпочтительным энантиомером является более сильнодействующий изомер. Выделенные энантиомеры соединений общей формулы I, их фармацевтически приемлемые соли и их пролекарственные формы также включены в объем этого изобретения.
Эндотелин (ET-1) и два близкородственных биоактивных пептида ET-2 и ET-3 широко распространены в тканях млекопитающих, и они могут вызвать многочисленные биологические реакции в несосудистых, а также в сосудистых тканях при связывании с по крайней мере двумя различными подтипами рецепторов эндотелина. Рецепторы эндотелина, помимо сердечно-сосудистых гладких мышц, нервных и предсердных участков, могут быть также обнаружены в желудочно-кишечных, почечных, легочных, мочеполовых, маточных и плацентарных тканях.
Эндотелин является сильнодействующим сосудосуживающим пептидом и поэтому играет роль in vivo в гомеостазе артериального давления-объема. Посредством эндотелина увеличивается не только периферическое сопротивление сосудов, но и коронарное сопротивление сосудов; минутный объем сердца уменьшается, в то время как активность ренина плазмы возрастает. Происходит снижение почечного кровотока и гломерулярной скорости фильтрации, в то время как уровни предсердного мочевого фактора, вазопрессина и альдостерона возрастают.
В соответствии с настоящим изобретением полагают также, что антагонисты рецепторы эндотелина могут быть пригодны для предотвращения или уменьшения рестеноза после денудации и последующей пластической операции на сосудах. Такая денудация вследствие повышенного выделения эндотелина приводит к миоинтимному уплотнению и последующей пластической операции на сосудах. Эндотелин действует как фактор роста по отношению к клеткам гладких мышц и фибропластов, и также возможно по отношению к другим типам клеток.
Эндотелин является также нейропептидом, действуя на нейрогипофиз, где он модулирует выделение нейросекреторных гормонов вазопрессина и окситоцина. Эндотелин, выделенный из нейрогипофиза, также действует как циркулирующий гормон, обладающий широким спектром действия, который обсуждался выше. Он включает воздействие на эндокринную систему, главным образом на надпочечники. Эндотелин увеличивает уровни плазмы эпинефрина.
Следовательно, новые соединения настоящего изобретения, которые являются антагонистами рецептора эндотелина, обладают терапевтической полезностью при предотвращении, уменьшении или модуляции различных физиологических воздействий эндотелина, обсужденных выше, путем полного или частичного блокирования доступа эндотелина к его рецептору.
Анализ связывания эндотелинового рецептора
Связывание новых соединений этого изобретения с рецептором эндотелина определяли в соответствии с анализом, описанным подробно ниже. Он подобен анализу, описанному в Ambar et al. (1989) Biochem. Biophys. Res. Commun., 158, 195-201; и Khoog et al. (1989) FEBS Letters, 253, 199-202.
Эндотелины (ET-ы) обладают рядом сильных воздействий на множество клеток и осуществляют свое действие путем взаимодействия со специфическими рецепторами, присутствующими на клеточных мембранах.
Соединения, описанные в настоящем изобретении, действуют как антагонисты ET на рецепторы. Для того, чтобы идентифицировать антагонисты ET и определить их эффективность in vivo, проводили следующие анализы рецептора.
Анализ связывания рецептора с использованием препарирования мембраны аорты коровы
Грудную аорту получили от свежеубойных телят и поместили в лаборатории на влажный лед. Удалили адвентициальную оболочку и аорту раскрыли вдоль. Люминальную поверхность ткани очистили марлей, чтобы удалить эндотелиальный слой. Ткань измельчили в мясорубке и суспендировали в охлажденной льдом 0,25 М сахарозе, 5 мМ трис-HCl, pH 7,4, содержащей 0,5 мг/мл лейпептина и 7 мг/мл пепстатина A. Ткань гомогенизировали дважды и затем центрифугировали в течение 10 минут при 750 xg и 4oC. Надосадочную жидкость отфильтровали через марлю и центрифугировали опять в течение 30 минут при 48000 xg и 4oC. Полученную таким образом гранулу повторно суспендировали в буферном растворе, описанном выше (включая ингибиторы протеазы) и аликвоты быстро заморозили и хранили до использования при -70oC. Мембраны разбавили в 50 мМ KPi, 5 мМ EDTA (этилендиаминтетрауксусной кислоты) pH 7,5, содержащих 0,01% сывороточный альбумин человека. Анализы проводили трижды. Испытуемые соединения и 100 pM [125I]-эндотелина-1 (2000-2200 Ci/милимоль, полученного от Нью Инглэнд Ньюклиа или Амершема) поместили в трубку, содержащую этот буфер и добавили после этого мембраны, полученные выше. Пробы инкубировали в течение 60 минут при 37oC. В конце этой инкубации пробы отфильтровывали на предварительно увлажненных (2% BSA в воде) фильтрах из стекловолокна и промыли 150 мМ NaCl, 0,1% BSA.
Фильтр анализировали на радиоактивность 125I в счетчике гамма-лучей. В присутствии 100 нМ немеченного эндотелина-1 измерили не способное к замещению связывание [125I-эндотелина-1. [Эндотелин-1 (ET-1) приобрели от Амершем (Арлингтон Хейтс, IL)].
Специфическое связывание представляет разность между общим связыванием и связыванием, не способным к замешению. Ингибирующая концентрация (IC50), которая дает 50% замещение от общего специфического связывания [125I]-эндотелина-1, является показателем эффективности таких соединений в качестве антагонистов ET.
Анализ связывания рецептора с использованием препарирования мембраны гиппокампа крысы
Гиппокамп крысы получили от свежеумерщвленных крыс мужского пола Sprague-Lawley и поместили в охлажденную льдом 0,25 М сахарозу, 5 мМ трис-HCl, pH 7,4, содержащую 0,5 мг/мл лейпептина, 7 мг/мл пепстатина A. Гиппокамп взвесили и поместили в гомогенизатор Dounce (Дунца) (стекло-стекло) с пестиком типа A, при этом гомогенизатор находился во льду. Гомогенат ткани центрифугировали при 750xg в течение 10 мин при 4oC. Надосадочную жидкость отфильтровали через увлажненную марлю и центрифугировали опять при 48000xg в течение 30 минут при 4oC. Гранулы повторно суспендировали в буфере сахарозы с ингибиторами протеазы.
Аликвоты этого препарирования быстро заморозили и хранили до использования при -70oC. Мембраны разбавили в 50 мМ KPi, 5 мМ EDTA, pH 7,5, содержащих 0,01% сывороточный альбумин человека. Анализы проводили трижды. Испытуемые соединения и 25 pM [125I]-эндотелина-1 (2000-2200 Ci/ммоль, полученного от Нью Инглэнд Ньюклиа или Амершема) поместили в трубку, содержащую этот буфер, и добавили после этого мембраны, полученные выше. Пробы инкубировали в течение 60 минут при 37oC. В конце этой инкубации пробы отфильтровали через предварительно увлажненные фильтры из стекловолокна и промыли 150 мМ NaCl, 0,1% BSA.
Фильтры анализировали на радиоактивность 125I в счетчике гамма-лучей.
В присутствии 100 нМ немеченного эндотелина-1 измерили не способное к замещению связывания [125I] -эндотелина-1. [Эндотелин-1(ET-1) приобрели от Пептидз Интернэшнл (Lowisville, KY). 125I-ET-1 (2000 CI/Моль приобрели от Амершем (Арлингтон Хейтс, IL)]. Специфическое связывание представляет разность между общим связыванием и связыванием, не способным к замещению. Ингибирующая концентрация (IC50), которая дает 50% замещение от общего специфического связывания [125I]-эндотелина-1, является показателем эффективности таких соединений в качестве антагонистов эндотелина.
Анализ связывания рецептора с использованием клонированных рецепторов эндотелина человека, представленных в клетках яичника китайского хомяка.
Оба подтипа рецепторов эндотелина клонировали из человеческой библиотеки cDNA и индивидуально выразили в клетках яичника китайского хомяка. Клетки собрали путем добавления 126 мМ NaCl, 5 мМ KCl, 2 мМ EDTA, 1 мМ NaH2PO4, 15 мМ глюкозы, 10 мМ трис/HEPES pH 7,4. Клетки центрифугировали при 250xg в течение 5 минут. Надосадочную жидкость отсосали и клетки повторно суспендировали в 50 мМ KPi, 5 мМ EDTA pH 7,5, содержащих 0,01% сывороточный альбумин человека. Анализ проводили трижды. Испытаемые соединения и 25-100 pM[125I] -эндотелина-1 (2000-2200 Ci/ммоль, полученного от Нью Инглэнд Ньюкалиа или Амершема) поместили в трубку, содержащую 50 мМ KPi, 50 мМ EDTA pH 7,5, содержащие 0,01% сывороточный альбумин человека и добавили после этого клетки, полученные выше. Пробы инкубировали в течение 60 минут при 37oC.
В конце этой инкубации пробы отфильтровали через предварительно увлажненные (2% BSA в воде) фильтры из стекловолокна и промыли 150 мМ NaCl, 0,1% BSA. Фильтры анализировали на радиоактивность 125I в счетчике гамма-лучей. В присутствии 100 нМ немеченного эндотелина-1 измерили не способное к замещению связывание [125I]-эндотелина-1 [Эндотелин-1 (ET-1) приобрели от Пептидз Интернэшнл (Lowisville, KY)]. [125I]-ET-1 (2000 Ci/милимоль), приобрели от Амершем (Арлингтон Хейтс, IL).
Специфическое связывание представляет разность между общим связыванием и связыванием, не способным к замещению. Ингибирующая концентрация [IC50], которая дает 50% замещение от общего специфического связывания [125I-эндотелина-1, является показателем эффективности таких соединений в качестве антагонистов индотелина.
Анализа связывания, описанные выше, использовали для оценки эффективности взаимодействия характерных соединений изобретения с рецепторами эндотелина. Чтобы определить, являются ли эти соединения антагонистами эндотелина, проводили анализы, которые измеряли способность соединений ингибировать гидролиз фосфатидилинозитола, стимулированный эндотелином. Матка крыс содержит один из известных подтипов рецептора эндотелина (ETA), в преобладающем количестве.
Анализ гидролиза фосфатидилинозитола с использованием срезов матки крыс
Крыс-самок Sprague-Dawley, усыпленных диэтилстильбэстролом, умерщвили и собрали их матки, отделили жир и соединительную ткань и измельчили. Измельченную ткань добавили к насыщенному кислородом (95% O2, 5% CO2) 127 мМ NaCl, 25 мМ KCl, 1,2 мМ KH2PO4, 1,2 мМ MgSO4, 1,8 мМ CaCl2. К измельченной ткани добавили 1,2 мМ мио-[3H]-инозитола (Амершем). Измельченную ткань инкубировали 90 мин при 37oC при постоянном насыщении кислородом. После инкубации насыщенную измельченную ткань промыли пять раз тем же самым буфером, насыщенным кислородом, для удаления избытка инозитола, меченного радиоактивным изотопом. Измельченную ткань повторно суспендировали в вышеуказанном буфере, содержащем 10 мМ LiCl, помещенном в трубки в виде аликвот, и 3 нМ эндотелина-1 и начали проводить анализ с добавлением и без добавления испытуемых соединений. Анализ проводили 4 раза. Пробы инкубировали при 37oC при продувке кислорода в водяной бане, закрытой крышкой, в течение 30 минут.
Реакцию завершили путем добавления трихлоруксусной кислоты 6% концентрации. Пробы подвергали звуковой обработке в течение 10 минут, центрифугировали в течение 20 минут, затем этиловым эфиром, насыщенным водой, экстрагировали трихлоруксусную кислоту. Аликвоту каждой пробы нейтрализовали и разбавили путем добавления 50 мМ трис-HCl pH 7,4. 100 мл аликвоту этого раствора анализировали на радиоактивность в счетчике бета-частиц. Разбавленную нейтрализованную пробу поместили в колонки Дауэкса (Dowex) размером 1х8, промыли водой, затем промыли 60 мМ формиата аммония, 5 мМ тетрабората натрия. Пробы элюировали 200 мМ формиата аммония, 5 мМ тетрабората натрия. В счетчике бета-частиц измерили радиоактивность каждой элюированной пробы. Радиоактивность стандартизировали путем деления радиоактивности в послеколоночной пробе на радиоактивность в предколоночной пробе. Контрольные величины (выраженные на 100%) представляли величины в присутствии эндотелина за вычетом величин в отсутствии эндотелина (основных). Величины испытуемой пробы представляют величины в присутствии эндотелина и испытуемой пробы за вычетом основных. Ингибирующая концентрация (IC50) представляет концентрацию испытуемого соединения, необходимую для получения активности пробы, равной 50% от контрольной величины.
Сарафотоксин S6c явлется членом семейства эндотелинов, который предпочтительно связывается с одним из известных подтипов эндотелинового рецептора (ETB).
Анализ гидролиза фосфатидилинозитола с использованием срезов легкого крыс
Умерщвили крыс-самцов Sprague-Dawley, собрали их легкие, отделили жир и соединительную ткань и измельчили. Измельченную ткань добавили к насыщенному кислородом (95% O2, 5% CO2) 127 мМ NaCl, 1,2 мМ NaHCO3, 10 мМ глюкозы, 2,5 мМ KCl, 1,2 мМ KH2PO4, 1,2 мМ MgSO4, 1,8 мМ CaCl2. К измельченной ткани добавили 1,2 мкм мио-[3H]-инозитола. Измельченную ткань инкубировали 60 мин при 37oC при постоянном насыщении кислородом. После инкубации насыщенную измельченную ткань промыли пять раз тем же самым буфером, насыщенным кислородом, для удаления избытка инозитола, меченного радиоактивным изотопом. Измельченную ткань повторно суспендировали в вышеприведенном буфере, содержащем 10 мМ LiCl, помещенном в трубки в виде аликвот, добавили 3 нМ сарафотоксина S6c и начали проводить анализ с добавлением и без добавления испытуемых соединений. Анализ проводили 4 раза. Пробы инкубировали при 37oC при продувке кислорода в водяную баню, закрытую крышкой, в течение 30 минут. Реакцию завершили путем добавления 0,5 мл 18% трихлоруксусной кислоты до 6%-ной концентрации. Пробы подвергали звуковой обработке в течение 10 минут, центрифугировали 20 минут, затем этиловым эфиром, насыщенным водой, экстрагировали трихлоруксусную кислоту. Аликвоты каждой пробы нейтрализовали и разбавили путем добавления 50 мМ трис-HCl pH 7,4. 100 мл аликвоту этого раствора анализировали на радиоактивность в счетчике бета-частиц. Разбавленную нейтрализованную пробу поместили в колонки Дауэкса (Dowex) размером 1х8, промыли водой, затем промыли 60 мМ формиата аммония, 5 мМ тетрабората натрия. Пробы элюировали 200 мМ формиата аммония, 5 мМ тетрабората натрия. В счетчике бета-частиц измерили радиоактивность каждой элюированной пробы. Радиоактивность стандартизировали путем деления радиоактивности в послеколоночной пробе на радиоактивность в предколоночной пробе. Контрольные значения (стимулирование на 100%) представляли значения в присутствии сарафотоксина за вычетом значений в отсутствии сарафотоксина (основных). Значения испытуемой пробы представляли значения в присутствии сарафотоксина и испытуемой пробы за вычетом основных. Ингибирующая концентрация (IC50) представляла концентрацию испытуемого соединения, необходимую для получения активности пробы, равной 50% от контрольного значения.
Анализ гидролиза фосфатидилинозитола с использованием клонированных рецепторов эндотелина человека, выраженных в клетках яичника китайского хомяка
Рецепторы эндотелина обоих подтипов клонировали из человеческой библиотеки cDNA и индивидуально выразили в клетках яичника китайского хомяка. Клетки насыщали всю ночь добавлением к питательной среде 1.2 мкм мио-[3H]-инозитола. Клетки собирали путем добавления 126 мМ NaCl, 5 мМ KCl, 2 мМ EDTA, 1 мМ NaH2PO4, 15 мМ глюкозы, 10 мМ трис/HEPES pH 7.4. Клетки промыли пять раз путем центрифугирования при 250xg в течение 5 минут для удаления избытка инозитола, меченного радиоактивным изотопом. Надосадочную жидкость отсосали и клетки повторно суспендировали в том же самом насыщенном кислородом (95% O2, 5% CO2) буфере, содержащем 10 мМ LiCl, аликвоты поместили в трубки, добавили 0,3 нМ эндотелина-1 и начали проводить анализ с добавлением испытуемых соединений.
Анализ проводили четыре раза. Пробы инкубировали при 37oC при продувании кислорода в закрытой крышкой водяной бане в течение 30 минут. Реакцию завершили путем добавления 0,5 мл 18% трихлоруксусной кислоты до 6%-ной концентрации. Пробы подвергали звуковой обработке в течение 10 минут, центрифугировали 20 минут, затем этиловым эфиром, насыщенным водой, экстрагировали трихлоруксусную кислоту. Аликвоту каждой пробы нейтрализовали и разбавили путем добавления 50 мМ трис-HCl pH 7.4. 100 мл аликвоту этого раствора анализировали на радиоактивность в счетчике бета-частиц. Разбавленную нейтрализованную пробу поместили в колонки Дауэкса (Dowex) размером 1х8, промыли водой, затем промыли 60 мМ формиата аммония, 5 мМ тетрабората натрия. Пробы элюировали 200 мМ формиата аммония, 5 мМ тетрабората натрия. В счетчике бета-частиц измерили радиоактивность каждой элюированной пробы. Радиоактивность стандартизировали путем деления радиоактивности в послеколоночной пробе на радиоактивность в предколоночной пробе. Контрольные значения (стимулирование на 100%) представляли значения в присутствии эндотелина за вычетом значений в отсутствии эндотелина (основных). Значения испытуемой пробы представляли значения в присутствии эндотелина и испытуемой пробы за вычетом основных. Ингибирующая концентрация (IC50) представляла концентрацию испытуемого соединения, необходимую для получения активности пробы, равной 50% от контрольного значения.
При использовании методики, описанной выше, оценили соединения, представленные в настоящем изобретении, и нашли, что они показывают значения IC50, равные по крайней мере < 50 мкМ, что демонстрирует и подтверждает полезность соединений изобретения в качестве эффективных антагонистов эндотелина.
Внутривенное воздействие антагониста эндотелина-1, двукалиевой соли N-(4-изо-пропилбензолсульфонил) -α- (4-карбокси-2-н- пропилфенокси)-3,4-метилендиоксифенилацетамида (Пример 58) на изменения диастолического кровяного давления и уретрального давления у анестезированных самцов собак, вызванные эндотелином-1
Методика определения способности селективного антагониста ET-1 ингибировать простатические уретральные сокращения, связанные с ET-1, у модели собаки-дворняжки.
В разные дни двух закрепленных дворняжек-кобелей HRP Inc.), весом 11,0 и 12,4 кг, анестезировали внутривенно пентобербиталом натрия (Steris Laboratories, Inc. ) в количестве 35 мг/кг, после чего осуществили внутривенное вливание 4 мг/кг/час. Вставили инкубационную трубку с надувной манжетой и каждого животного вентилировали комнатным воздухом, используя нагнетательный перемещающийся вентилятор для больших животных (аппаратура Харварда), имеющий скорость 18 вдохов/минуту и средний дыхательный объем 18 мл/кг веса тела.
Температуру тела поддерживали грелкой-подушкой и нагревательной лампой, используя регулятор температуры (VSI) и эзофагеальный зонд. В аорту через бедренные артерии (по одному в каждую артерию) поместили два катетера для введения эндотелина или фенилэфрина и для непрерывного непосредственного контроля кровяного давления и частоты сердечных сокращений использовали датчик кровяного давления Statham (Стахэма) и компьютерную систему Modular Instruments, Inc. (Модула Инструментс, Инк.). Для введения пентобарбитала и двукалиевой соли N-(4-изо-пропил-бензолсульфонил) -α- (4-карбокси-2-н- пропилфенокси)-3,4-метилендиоксифенилацетамида (пример 58) два других катетера поместили в полую вену через бедренные артерии (один катетер в каждую вену). С боковой стороны полового члена сделали надлобковый надрез длиной в 1,27 см для обнажения мочеточников, мочевого пузыря и уретры. Для облегчения вскрытия мочеточников уменьшили купол мочевого пузыря. Мочеточники канюлировали PE90 и отсоединили от мочевого пузыря. Пуповину обмотали под уретрой в шейке мочевого пузыря, а другую часть пуповины пропустили к простате приблизительно на 1-2 см от края. Надрезали купол мочевого пузыря и в уретру переместили датчик катетера Micro-tip®. (Миллар Инструментс, Инк.). Чтобы удержать датчик, шейку мочевого пузыря перевязали пуповиной. На рассечение мочевого пузыря наложили шов из 3-0-шелка (кисетный шов). До помещения в предстательную часть мужского испускательного канала датчик убрали. Положение катетера Micro-tip® проверили путем осторожного сжатия простаты, при этом перед лигированием периферической уретры отмечали большие изменения уретрального давления.
Протокол эксперимента
Ввели фенилэфрин (PE) (10 мкг/кг внутриартериально) и отметили вазопрессорное воздействие на диастолическое кровяное давление (DBP) и внутриуретральное давление (IUP). Когда кровяное давление возвратилось к норме, ввели эндотелин-1 (ET-I) (1 нмоль/кг внутриартериально).
Изменения DBP и IUP контролировали в течение одного часа и ввели селективный антагонист эндотелина ET-I, например соединение примера 58, двукалиевую соль N-(4-изопропилбензолсульфонил) -α- (4-карбокси-2-н-пропилфенокси)-3,4-метилендиоксифенилацетамида (30 мг/кг внутривенно). Через 10-15 минут, когда кровяное давление стабилизировалось, ET-I ввели опять и отметили ингибирование эффектов, вызванных ET-I. В конце эксперимента для проверки специфичности блокады ET-I ввели PE. Собак умерщвили передозировкой пентобарбитала, затем насыщенным KCl.
Лекарствами, используемыми в вышеописанном эксперименте, были следующие:
1) Фенилэфрин, HCl (pE) (Сигма Кэмикал, Ко) вводили в количестве 0,05 мл/кг;
2) Эндотелин-1 (ET-I) (человека, свиньи, собаки, крысы, мыши, быка) вводили в количестве 0,05 мл/кг;
3) Селективный антагонист ET-I, например соединение примера 58, двукалиевую соль N-(4-изо-пропилбензолсульфонил) -α- (4-карбокси-2- н-пропилфенокси)-3,4-метилендиоксифенилацетамида вводили в количестве 0,3 мл/кг.
Все лекарства растворили в изотоническом солевом растворе.
Результаты
ET-I вызвал начальное депрессорное воздействие и затем продолжительное вазопрессорное воздействие. У одной собаки вазопрессорное воздействие было двухфазным. Уменьшение DBP у обоих собак в среднем составило 13 мм Hg, в то время как пиковое вазопрессорное воздействие составило 26 мм Hg. Среднее увеличение IUP, вызванное эндотелином ET-I, составило 15 мм Hg. Через 10-14 минут после введения двукалиевой соли N-(4-изо-пропилбензолсульфонил) -α- (4-карбокси-2-н-пропилфенокси)-3,4-метилендиоксифенилацетамида (пример 58) собакам опять ввели ET-I и ингибировали депрессорное и вазопрессорное воздействие на DBP соответственно на 69 и 76%. Вазопрессорное воздействие на IUP ингибировали на 93% (таблица 1. Увеличения DBP и IUP, вызванные внутриартериальным введением PE, значительно не изменились после введения одной из подопытных собак двукалиевой соли N-(4-изопропилбензолсульфонил)- -α- -(4-карбокси-2-н-пропилфенокси)-3,4-метилендиоксифенилацетамида (пример 58). Учитывая DBP и IUP ингибировали соответственно на 35 и 13% (таблица 2).
Заключения
ET-I вызывает сокращение предстательной части мужского мочеиспускательного канала, а также комплексную реакцию кровяного давления, включающую начальную депрессорную реакцию и последующую вазопрессорную реакцию у анестезированных собак. Реакцию кровяного давления и реакцию предстательной части мужского мочеиспускательного канала на ET-1 специфически посредством двукалиевой соли N-(4-изо- пропилбензолсульфонил)- -α- (4-карбокси-2-н-пропилфенокси)-3,4- метилендиоксифенилацетамида. Эффективность двукалиевой соли N-(4-изо- пропилбензолсульфонил) -α- (4-карбокси-2-н-пропилфенокси)-3,4- метилендиоксифенилацетамида при ингибировании вазопрессорного воздействия ET-1 на предстательную часть мужского мочеиспускательного канала означает, что селективные антагонисты ET-1 будут пригодны при лечении необходимости мочевых путей при доброкачественной гиперплазии простаты.
Простата крысы in situ
Крыс-самцов Sprague-Dawley (Taconic Farms), весящих 300-400 г, анестезировали уретаном (1,75 г/кг, внутрибрюшинно), вставили трахеотомическую трубку (канюлю) и канюлировали бедренную артерию. Внутреннюю температуру тела поддерживали при 37 +0,5oC.
Для обнажения мочевого пузыря и простаты осуществили срединную лапаратомию на 4-5 см. Простату отделили от оболочки посредством отслаивания с помощью щипцов. Отрезок хирургического шелка осторожно закрепили вокруг верхних передних концов долей простаты. Второй отрезок хирургического щелка, прикрепленный к атравматической игле, пропустили и привязали к основанию простаты позади первого отрезка приблизительно на расстоянии 10-12 мм. Заднюю лигатуру соединили с датчиком Grass FTO3 (Грас Инструментс, Квинси, МА) и сохраняли при давлении 1 г. Сигналы от датчика усилили и зарегистрировали на полиграфе (усилители Хьюлетта-Паккарда 8805В и регистрирующий прибор 7758А, Пало Альта, Канада). После уравновешивания в течение ~ 15 минут крысам предварительно вводили лекарства (1 мг/кг атропина (+) 1 мг/кг пропанолола) через внутриартериальную (1A) канюлю в течение 10 минут. Через 30 минут, каждые 30 минут, 3 раза внутриартериально впрыскивали ET -1 (0,3 моля/кг). За 5 минут до третьего впрыскивания ET-1 впрыскивали внутриартериально растворитель с антагонистом эндотелина или растворитель без эндотелина. Реакцию простаты ET-1 определяли количественно путем измерения изменения (Δ) от базисного давления до пика реакции во время 5-минутного периода после третьего впрыскивания ET-1.
Протокол простаты крысы in situ использовали для определения антагонистической активности и действенности соединений этого изобретения в отношении блокирования влияния ET-1 на сокращаемость простаты крыс in vivo. В этом протоколе было проиллюстрировано, что двукалиевая соль N-(4-изо-пропилбензолсульфонил) -α- (4-карбокси-2-н-пропилфенокси)-3,4-метилендиоксифенилацетамида вызывает специфическое ингибирование ET-1 и будет пригодна при лечении непроходимости мочевых путей при доброкачественной гиперплазии простаты.
Соответственно новые соединения настоящего изобретения являются пригодными для терапии человека при лечении астмы, гипертензии, почечной недостаточности, в особенности постишемической почечной недостаточности, циклоспориновой нефротоксичности, вазоспазма, церебральной и сердечной ишемии, инфаркта миокарда, или эндоксинового бактериально-токсического шока, вызванного эндотелином или связанного с ним, путем введения больному, в случае необходимости такого лечения, терапевтически эффективного их количества.
При лечении гипертензии и клинических состояний, указанных выше, соединения этого изобретения могут быть использованы, например, в виде таблеток, капсул или эликсиров для перорального приема, суппозиториев для ректального введения, стерильных растворов или суспензий для парентерального введения или внутримышечного введения и т.п. Соединения этого изобретения можно вводить больным (животным и человеку) в случае необходимости такого лечения, в дозах, обеспечивающих оптимальную фармацевтическую эффективность. Хотя доза будет изменяться от больного к больному в зависимости от природы и тяжести заболевания, веса пациента, режима питания больного, одновременно принимаемых лекарственных средств и других факторов, специалисты в данной области признают, что диапазон дозы составляет для больного обычно от ~0,5 мг до 1,0 г в день, и ее можно вводить в виде однократной дозы или многократных доз. Предпочтительно диапазон дозировки больному составляет от ~ 0,5 мг до 500 мг в день; более предпочтительно от около 0,5 мг до 200 мг в день.
Соединения этого изобретения можно вводить в сочетании с агонистами рецептора A2 - адренозина, α- адренергическими антагонистами, антагонистами ангиотензина II, ингибиторами ангиотензинпревышающего фермента, β- адренергическими антагонистами, ингибиторами атриопептидазы (отдельно или с ANP), блокаторами кальциевого канала, диуретиками, агонистами калиевого канала, ингибиторами ренина, антагонистами серотонина, симпатолитическими средствами, а также другими антигипертензивными средствами.
Соединения этого изобретения можно вводить в сочетании, например, с такими соединениями, как A-69729, FК 906, FК 744, UK-73900, CSG 22492C, амилоридом, атенололом, атриопептином, бендрофлуметиазидом, хлорталидоном, хлортиазидом, клонидином, кромакалином, ацетатами, криптенамина и таннатами криптенамина, дезерпидином, диазоксидом, доксазосином, гуанабензом, гуанетидином, сульфатом гуанетидина, гидрохлоридом гидралазина, гидрохлортиазидом, израдипином, кетансерином, лозартаном, метолазоном, метопрололом, тартатом метопролола, метиклотиазидом, метилдопой, гидрохлоридом метилдопата, миноксидилом, надололом, паргилингидрохлоридом, пинакидилом, пиндололом, политиазидом, празосином, пропранололом, rauwolfia serpentina, ресциннамином, резерпином, нитропруссидом натрия, спиронолактоном, теразосином, тимололмалеатом, трихлорметиазидом, триметофанкамсилатом, верапамилом, бензтиазидом, хинетазоном, тикринафаном, триамтереном, ацетазоламидом, аминофиллином, циклотиазидом, этакриновой кислотой, фуроземидом, меретоксилинпрокаином, этакринатом натрия, каптоприлом, делаприлгидрохлоридом, энанлаприлом, эналаприлатом, фозиноприлом натрия, лизиноприлом, пентоприлом, хинаприлом, хинаприлгидрохлоридом, рамаприлом, тепротидом, зофеноприлом, кальцийзофеноприлом, дифлузиналом, дильтиаземом, фелодипином, никардипином, нифедипином, нилудипином, нимодипином, нисолдипином, нитрендипином и т.п., а также с их смесями и сочетаниями. Сочетания, пригодные при лечении застойной сердечной недостаточности, включают, кроме этого, соединения этого изобретения с кардиотоническими стимуляторами, например добутамином и ксамотеролом, и ингибиторами фосфодиэстеразы, включающими амринон и милринон.
Обычно индивидуальные суточные дозы для этих сочетаний могут колебаться в пределах от около одной пятой от минимальных рекомендованных клинических доз до максимальных рекомендованных доз для таких форм, взятых в отдельности. Для иллюстрации этих сочетаний один из клинически эффективных антагонистов эндотелина этого изобретения при данном диапазоне суточной дозы можно эффективно сочетать при уровнях, которые являются меньшими, чем диапазон суточной дозы, со следующими соединениями при указанном суточном диапазоне дозы: гидрохлортиазидом (6-100 мг), хлортиазидом (125-500 мг), фуросемидом (5-8 мг), этакриновой кислотой (5-200 мг), амилоридом (5-20 мг), дильтиаземом (30-540 мг), фелодипином (1 - 20 мг), нифедипином (5 - 120 мг), нитрендипином (5 - 60 мг), тимололмалеатом (1 - 20 мг), пропанололом (10 - 480 мг) и метилдопой (125 - 2000 мг).
Кроме этого, эффективными сочетаниями для регулирования кровяного давления у больных гипертензией являются тройные лекарственные комбинации из гидрохлортиазида (6 - 100 мг), амилорида (5 - 20 мг) и антагонистов эндотелина этого изобретения, или из гидрохлортиазида (6 - 100 мг), тимололмалеата (1 - 20 мг) и антагонистов эндотелина этого изобретения, или из гидрохлортиазида (6 - 100 мг), нифедипина (5 - 60 мг) и антагонистов этого изобретения. Конечно, эти диапазоны дозы можно регулировать, когда необходимо, посредством единиц, чтобы обеспечить разделенную суточную дозу, при этом доза будет изменяться в зависимости от природы и тяжести заболевания, веса пациента, режима питания и других факторов.
Настоящее изобретение также относится к лекарственным препаратам для лечения астмы, гипертензии, почечной недостаточности, в особенности постишемической почечной недостаточности, циклоспориновой нефротоксичности, вазоспазма, цебральной и сердечной ишемии, доброкачественной гиперплазии простаты, инфаркта миокарда или эндотоксинового бактериально-токсического шока, вызванных эндотелином или связанных с ним, включающим терапевтически эффективное количество нового соединения этого изобретения вместе с фармацевтически приемлемым его носителем.
В унифицированной лекарственной форме, предназначенной для приемлемой фармацевтической практики, смешивают от около 0,5 мг до 1,0 г соединения или смеси соединений формулы I, или физиологически приемлемой соли с физиологически приемлемым растворителем, носителем, наполнителем, связующим, консервантом, стабилизатором, корригентом и т.д. Количество активного вещества в этих составах или препаратах является таковым, что получают соответствующую дозировку в указанном диапазоне.
Примерами синергистов, которые можно включить в таблетки, капсулы и т.п. , являются следующие вещества: связующее, например трагакантовая камедь, аравийская камедь, кукурузный крахмал или желатин; наполнитель, например микрокристаллическая целлюлоза; разрыхляющий агент, например кукурузный крахмал, предварительно желатинированный крахмал, альгиновая кислота и т.п.; смазывающие вещества, например стеарат магния; подсластители, например сахароза, лактоза или сахарин; вкусовые добавки, например мята перечная, масло грушанки или вишня. Когда единичной формой дозировки является капсула, она может содержать, кроме материалов вышеперечисленного типа, жидкий носитель, например жирное масло. В качестве покрытий или для изменения физической формы дозированных единиц могут присутствовать различные другие материалы. Таблетки, например, могут быть покрыты шеллаком, сахаром или тем и другим. Сироп или эликсир могут содержать активное соединение, в качестве подсластителя сахарозу, в качестве консервантов метил- и пропилпарабены, краситель и вкусовые добавки, например вишню или апельсиновую вкусовую добавку.
В соответствии с традиционной фармацевтической практикой стерильные композиции для инъекции могут быть получены путем растворения или суспендирования активного вещества в растворителе, например воде для инъекции, растительном масле естественного происхождения, подобном кунжутному маслу, кокосовому маслу, арахисовому маслу, хлопковому маслу и т.д. или в синтетическом жирном растворителе, подобном этилолеату или т.п. Когда необходимо, могут быть включены буферы, консерванты, антиоксиданты и т.п.
Последующие примеры иллюстрируют получение соединений формулы I и их включение в лекарственные композиции, при этом их не следует рассматривать как ограничивающие изобретение.
Пример 1.
2-(2,6-дипропил-4-гидроксиметил)феноксифенилуксусные кислоты
Стадия А:
Получение алкил-2-бром-2-фенилацетатов
Способ А:
Замещенную фенилуксусную кислоту превратили в соответствующий метиловый эфир нагреванием с обратным холодильником кислоты в метаноле в присутствии каталитического количества концентрированной серной кислоты. Полученный таким образом эфир затем кипятили с обратным холодильником в тетрахлориде углерода с N-бромсукцинимидом (1,1 экв.) и AIBN (азобисизобутиронитрилом (0,05 - 0,1 экв. ). По завершении реакции для обеспечения желаемого алкилбромида полученный продукт очистили тонкослойной колоночной хроматографией с использованием силикагеля и в качестве элюента этилацетата в гексане.
Способ B
Смесь арилальдегида с триметилсилилцианидом в присутствии каталитических количеств KCN и 18-краун-6 в метиленхлориде выдерживали в течение ночи. Реакционную смесь резко охладили водой и экстрагировали смесью CH2Cl2 (этилацетата) сложного эфира (1/2/2). Органическую фазу промывали насыщенным водным раствором NaHCO3. После сушки и концентрирования органической фазы полученный триметилсилилцианогидрин гидролизовали до получения соответствующей оксикислоты.
Обработка газообразным HCl в метаноле или этаноле при 0oC в течение 0,5 часа и затем в течение ночи при комнатной температуре привела к получению сырого 2-гидроксиэфира. Затем эфир обрабатывали трифенилфосфином и тетрабромидом углерода в метиленхлориде при 0oC в течение ночи.
Метиленхлорид удалили и посредством тонкослойной колоночной хроматографии сырого продукта с использованием силикагеля и в качестве элюента этилацетата/гексана получили желаемые 2-бромфенилацетаты.
Стадия B: Алкирование фенола
2,3-(Дипропил-4-гидроксиметил)фенол (полученный, как описано в заявке WO 91/11999 на патент), алкилировали 2-бром-2-арилэфирами в DMF (N,N-диметилформамиде) с использованием или карбоната цезия (Cs2CO3), или карбоната калия (K2CO3), или гидрида натрия NaH. Алкилированный продукт очистили тонкослойной колоночной хроматографией с использованием силикагеля и, в качестве элюента, смеси этилацетата/гексана, обеспечив при этом желаемые замещенные эфиры 2-фенокси-2-фенил-уксусной кислоты.
Стадия C: Общая методика гидролиза сложного эфира.
Продукт стадии B растворили в метаноле или этаноле, и он взаимодействовал с водным раствором NaOH или LiOH, или KOH при комнатной температуре в течение 1 - 6 часов, затем его нейтрализовали до pH 7 IN HCl и концентрировали в вакууме. Остаток очистили тонкослойной колоночной хроматографией на силикагеле, получив при этом соответствующую карбоновую кислоту.
Перечисленные ранее производные феноксифенилуксусной кислоты получили с использованием общих методик, представленных в примере 1.
Пример 2.
2-[(2,6-Дипропил-4-гидроксиметил)фенокси] -2-(3-метилфенил)- уксусная кислота.
1H ЯМР (400 мГц, CD3OD, част. на миллион): δ 7,15 - 6,95 (m, 4H), 6,86 (s, 2H), 4,92 (широк. s, 1H), 4,5 (s, 2H), 2,3 - 2,1 (m, 4H), 2,2 (s, 3H), 1,5 - 1,35 (m, 2H), 1,32 - 1,18 (m, 2H), 0,7 (t, 6H).
Пример 3.
2-[(2,6-Дипропил-4-гидроксиметил)фенокси] -2-(4-феноксифенил)уксусная кислота.
1H ЯМР (400 мГц, CD3OD, част. на миллион): δ 7,42 (d, 2H, J = 8,4 Гц), 7,33 (dd, 2H, J = 7,4 Гц), 8,5, 7,09 (t, 1H, J = 7,4 Гц), 6,97 - 6,95 (m, 4H), 6,91 (d, 2H, J = 8,4 Гц), 4,85 (s, 1H), 4,47 (s, 2H), 2,38 (t, 4H, J = 8,0 Гц), 1,56 (sx, 2H, J = 7,1 Гц), 1,42 (sx, 2H, J = 7,1 Гц), 0,85 (t, 6H, J = 7,3 Гц).
FAB-MSm/e = 435 (M + 1)
Пример 4.
2-[(2,6-Дипропил-4-гидроксиметил)фенокси] -2-(4-фенилфенил)- уксусная кислота.
1H ЯМР (400 мГц, CD3OD, част. на миллион): δ 7,62 - 7,60 (m, 4H), 7,51 (широк. , 2H), 7,44 (t, 2H, J = 7,5 Гц), 7,34 (t, 1H, J = 7,4 Гц), 6,99 (s, 2H), 4,83 (s, 1H), 2,40 (широк., 4H), 1,53 (широк., 2H), 1,42 (широк., 2H), 0,82 (t, 6H, J = 6,2 Гц).
FAB-MSm/e = 419 (M + 1)
Пример 5.
2-[(2,6-Дипропил-4-гидроксиметил)фенокси] -2- (3-карбоксифенил)уксусная кислота.
1H ЯМР (400 мГц, CD3OD, част. на миллион): δ 8,18 (s, 1H), 8,04 (d, 1H, J = 7,5 Гц), 7,70 (d, 1H, J = 7,4 Гц), 7,51 (d, 1H, J = 7,6 Гц), 6,99 (s, 2H), 5,15 (s, 1H), 4,48 (s, 2H), 2,37 (m, 4H), 1,52 (m, 2H), 1,44 (m, 2H), 0,80 (t, 6H, J = 7,3 Гц).
FAB-MSm/e = 387 (M + 1)
Пример 6.
2-[(2,6-Дипропил-4-гидроксиметил)фенокси] -2-(3,4-этилендиоксифенил)уксусная кислота
1H ЯМР (200 мГц, CD3OD, част. на миллион): δ 6,95 (m, 3H), 6,85 (dd, 1H, J = 8,3, 2,0 Гц), 6,72 (d, 1H, J = 8,3, Гц), 4,76 (s, 1H), 4,46 (s, 2H), 4,20 (s, 4H), 2,37 (t, 4H, J = 7,9 Гц), 1,44 (m, 4H), 0,83 (t, 6H, J = 7,3 Гц).
FAB-MSm/e = 401 (M + 1).
Пример 7.
2-[(2,6-Дипропил-4-гидрокси)фенокси] -2-(3,4,5-триметоксифенил)уксусная кислота.
1H ЯМР (400 мГц, CD3OD, част. на миллион): δ 6,97 (s, 2H), 6,80 (s, 2H), 4,88 (s, 1H), 4,48 (s, 1H), 3,81 (s, 6H), 3,75 (s, 3H), 2,39 (t, 3H, J = 8,1 Гц), 1,55 (m, 2H), 0,82 (t, 6H, J = 7,3 Гц).
FAB-MSm/e = 433 (M + 1).
Пример 8.
2-[(2,6-Дипропил-4-гидроксиметил)фенокси] -2- (3,4-метилендиоксифенил)уксусная кислота.
1H-ЯМР (200 мГц, CD3OD, част. на миллион): δ 7,03 (s, 1H), 6,97 (s, 2H), 6,83 (d, 1H, J = 7,7 Гц), 6,73 (d, 2H, J = 7,7 Гц), 5,94 (s, 2H), 4,84 (s, 1H), 4,48 (s, 2H), 2,38 (t, 4H, J = 8,0 Гц), 1,46 (m, 4H), 0,85 (t, 6H, J = 7,5 Гц).
FAB-MS m/e = 387 (M+1).
Пример 9.
2-[(2,6-Дипропил-4-гидроксиметил)фенокси]-2- (3,4-диметоксифенил)уксусная кислота.
1H-ЯМР (400 мГц, CD3OD, част. на миллион): δ 7,15 (d, 1H), 6,95 (d, 2H), 6,85 (dd, 2H), 4,8 (широк. s, 1H), 4,46 (широк. s, 2H), 3,84 (s, 3H), 3,8 (s, 3H), 2,35 (t, 4H), 1,62-1,47 (m, 2H), 1,45 - 1,3 (m, 2H), 0,85 (t, 6H).
Пример 10.
2-[(2,6-Дипропил-4-гидроксиметил)фенокси]-2- (3,5-диметоксифенил)уксусная кислота.
1H-ЯМР (400 мГц, CD3OD, част. на миллион): δ 6,954 (s, 2H), 6,66 (d, 2H), 6,39 (t, 1H), 4,747 (s, 1H), 4,47 (s, 2H), 3,74 (s, 6H), 2,39 (t, 4H), 1,60 - 1,51 (m, 2H), 1,437-1,35 (m, 2H), 0,82 (t, 6H).
Пример 11.
2-[(2,6-Дипропил-4-тетразол-5-ил)фенокси] -2-(3-бромофенил)- уксусная кислота.
1H-ЯМР (400 мГц, CD3OD, част. на миллион): δ 7,73 (s, 2H), 7,67 (t, 1H, J = 1,8 Гц), 7,57 (m, 1H), 7,46 (m, 1H), 7,33 (t, 1H, J = 7,9 Гц), 5,89 (ddd, 1H, J = 1,6, 10,1, 17,1 Гц), 5,41 (s, 1H), 5,41 (s, 1H), 5,08 (dd, 1H, J = 10,1, 1,6 Гц), 5,01 (dd, 1H, J = 1,7, 17,1 Гц), 4,93 (s, 2H), 3,72 (s, 3H), 3,36-3,30 (m, 2H)
FAB-MS m/e = 470 (M+1)
Пример 12.
2-[(2,6-Дипропил-4-гидроксиметил)фенокси] -2- (3-бромофенил)уксусная кислота.
1H-ЯМР (400 мГц, CD3OD/CDCl3, 2/1 част. на миллион): δ 7,667 (s, 1H), 7,496 (d, 1H), 7,496 (d, 1H), 7,3925 (d, 1H), 7,252 (t, 1H), 6,964 (s, 2H), 4,995 (s, 1H), 4,485 (s, 2H), 2,342 (t, 4H), 1,65 - 1,35 (m, 2H), 0,803 (t, 6H).
Пример 13.
2-[(2,6-Дипропил-4-гидроксиметил)фенокси]-2-(2-нафтил)-уксусная кислота.
1H-ЯМР (400 мГц, CD3OD, част. на миллион): δ 8,56-8,52 (m, 1H), 7,86 - 7,79 (m, 2H), 7,47 - 7,43 (m, 2H), 7,35 - 7,32 (m, 1H), 6,91 (s, 2H), 5,36 (s, 1H), 4,44 (s, 1H), 1,46 - 1,41 (m, 2H), 1,2 - 1,16 (m, 2H), 0,58 (t, J = 7,37, 6H).
Пример 14.
2-[(2,6-Дипропил-4-(2-гидроксиэтил)фенокси]-2-(нафтил)-уксусная кислота.
Стадия A.
Трет-бутилдиметилсилилокси-2,6-дипропил-4-формилбензол
К раствору 5,03 г (15,4 ммоль) трет-5-бутилдиметилсилилокси- 2,6-дипропил-4-гидроксиметилбензола в 30 мл метиленхлорида добавили 8,7 г пиридинийдихромата (PDC).
Реакционную смесь перемешивали в течение 3-х часов и затем разбавили 300 мл этилового эфира. Затем раствор отфильтровали через подушку, состоящую из смеси флоризила и целита 1:1. Путем концентрирования фильтрата получили 4,85 г названного соединения.
1H-ЯМР (400 мГц, CDCl3, част. на миллион): δ 9,8 (s, 1H), 7,51 (s, 2H), 2,59 - 2,55 (m, 2H), 1,59 - 1,55 (m, 2H), 0,99 (s, 9H), 0,91 (t, J = 7,28 Гц, 3H), 0,20 (s, 6H).
Стадия B:
Трет-бутилдиметилсилилокси-2,6-дипропил-4-винилбензол.
К раствору 1,0 г (2,80 ммоль) метилтрифенилфосфонийбромида в 5,0 мл эфира при 0oC добавили 1,12 мл (2,5 М, 2,80 ммоль) бутиллития. Реакционную смесь перемешали в течение 30 минут при 0oC и затем добавили 756 мг (2,33 ммоль) соединения из примера 14 (стадия A). После перемешивания в течение одного часа при комнатной температуре реакционную смесь влили в этилацетат и промыли водой и затем насыщенным водным раствором хлорида натрия. Органический слой сушили над безводным сульфатом натрия, фильтровали и концентрировали. Путем очистки тонкослойной хроматографией (силикагель, гексан/этилацетат, (7:3) получили 411 мг названного соединения.
1H-ЯМР (400 мГц, CDCl3, част. на миллион): δ 7,02 (s, 2H), 6,6 (dd, J = 17,6, J = 10,7 Гц, 1H), 5,55 (d, J = 17,6 Гц, 1H), 5,08 (d, J = 10,7 Гц, 1H), 2,53 - 2,50 (m, 4H), 1,59 - 1,53 (m, 4H), 0,996 (s, 9H), 0,90 (t, J = 7,33 Гц, 6H), 0,16 (s, 6H).
Стадия C:
Трет-бутилдиметилсилилокси-2,6-дипропил-4-(2-гидроксиэтил)бензол
К раствору 475 мг (1,48 ммоль) продукта стадии B в 3 мл THF (тетрагидрофурана) при 0oC добавили 1,6 мл (1,62 ммоль) IN раствора борана/THF. Через 1 час TLC (тонкослойная хроматография) показала, что исходный материал израсходован.
Реакционную смесь резко охладили 3 каплями метанола и затем добавили 0,70 мл (6,22 ммоль) 30% пероксида натрия и 6,2 мл (6,2 ммоль) IN гидроксида натрия. Через 2 часа реакционную смесь разбавили этилацетатом и промыли водой и рассолом. Затем органический слой сушили над сульфатом натрия, фильтровали и концентрировали. Тонкослойной хроматографией (силикагель, гексан/этилацетат (4: 1) получили 265 мг названного соединения в виде светлого масла.
1H-ЯМР (400 мГц, CDCl3, част. на миллион): δ 6,79 (s, 2H), 3,79 (m, 2H), 2,75 - 2,72 (m, 2H), 2,51 - 2,48 (m, 4H), 1,58 - 1,51 (m, 6H), 0,99 (s, 9H), 0,90 (t, J = 7,33, 6H), 0,16 (s, 6H).
Стадия D.
2,6-Дипропил-4-(2-гидроксиэтил)фенол
К раствору 1,0 г (2,98 ммоль) продукта стадии C в 30 мл THF добавили 3,57 мл (3,57 ммоль) 1,0N раствора тетрабутиламмонийфторида в THF. Через 15 минут TLC (тонкослойная хроматография) показала, что реакция завершилась. Реакционную смесь концентрировали и затем очистили тонкослойной хроматографией (силикагель, гексан/этилацетат 3: 1), получив при этом 1,13 г названного соединения.
1H-ЯМР (400 мГц, CDCl3, част. на миллион): δ 6,8 (s, 2H), 3,78 (t, J = 6,5 Гц, 2H), 2,73 (t, J = 6,5 Гц, 2H), 2,54-2,50 (m, 4H), 1,66 - 1,56 (m, 4H), 0,96 (t, J = 7,3 Гц, 6H).
Стадия E.
Метил-2-[(2,6-дипропил-4-2-гидроксиэтил)фенокси]-2- (2-нафтил)ацетат
Названное соединение получили из 2,6-дипропил-4-(2-гидроксиэтил)фенола (стадия D) путем алкилирования метил-2-бром-2-(2-нафтил)ацетатом при использовании карбоната цезия или карбоната калия в DMF. Реакционную смесь отфильтровали через целит и осадок промыли метиленхлоридом. Фильтрат концентрировали и полученный материал очистили тонкослойной колоночной хроматографией до получения названного сложного эфира.
1H-ЯМР (400 мГц, CDCl3, част. на миллион): δ 7,90 - 7,82 (m, 4H), 7,69-7,67 (m, 1H), 7,49-7,47 (m, 2H), 6,8 (s, 2H), 2,74 (t, J = 6,2 Гц, 2H), 2,36 - 2,32 (m, 4H), 1,49 - 1,41 (m, 4H), 0,72 (t, J = 7,3, 6H).
Стадия F.
2-[(2,6-Дипропил-4-(2-гидроксиэтил)фенокси] -2-(2-нафтил)- уксусная кислота
Названное соединение получили из продукта стадии E путем омыления IN водным KOH в метаноле, как описано в стадии C примера 1.
1H-ЯМР (400 мГц, CD3OD, част. на миллион): δ 7,84 - 7,7 (m, 4H), 7,73 (d, J = 6,8 Гц, 1H), 7,45 - 7,43 (m, 2H), 6,79 (s, 2H), 5,01 (s, 2H), 3,66 (t, J = 7,2 Гц, 2H), 2,68 (t, J = 7,2 Гц, 2H), 2,33 - 2,29 (m, 4H), 1,55 -1,45 (m, 2H), 1,40 - 1,28 (m, 2H), 0,69 (t, J = 7,3 Гц, 6H).
FAB-MS: m/e = 445 (M+K), 429 (M+Na), 407 (M+I).
Перечисленные далее производные феноксифенилуксусной кислоты получили с использованием общих методик, представленных в примере 14.
Пример 15.
2-[(2,6-Дипропил-4-(2-гидроксиэтил)фенокси] -2- (3,4-метилендиоксифенил)уксусная кислота
1H ЯМР (200 мГц, CD3OD, част. на миллион): δ 7,03 (s, 1H), 6,83 (m, 3H), 6,72 (d, 1H, J = 7,8 Гц), 5,93 (s, 2H), 4,80 (s, 1H), 3,68 (t, 2H, J = 7,1 Гц), 2,69 (t, 2H, J = 7,1 Гц), 2,35 (t, 4H, J = 7,0 Гц), 1,42 (m, 4H), 0,83 (t, 6H, J = 7,3 Гц).
FAB-MS: m/e = 401 (M+I).
Пример 16.
2-[(2,6-Дипропил-4-(2-гидроксиэтил)фенокси] -2- (3-метоксифенил)уксусная кислота
1H ЯМР (400 мГц, CD3OD, част. на миллион): δ 7,28 (t, 1H, J = 7,9 Гц), 7,07 (m, 1H), 7,15 (m, 1H), 6,94 (m, 1H), 6,84 (s, 2H), 4,99 (s, 1H), 3,79 (s, 3H), 3,68 (t, 2H, J = 7,1 Гц), 2,34 (t, 4H, J = 8,0 Гц), 1,54 - 1,40 (m, 4H), 0,80 (t, 3H, J = 7,3 Гц).
FAB-MS: m/e = 387 (M+I).
Пример 17.
2-[(2,6-Дипропил-4-(1,2-дигидроксиэтил)фенокси] -2- (2-нафтил)уксусная кислота.
Стадия A:
2,6-дипропил-4-винилфенол
Названное соединение получили из трет-бутилдиметилсилилокси-2,6- дипропил-4-винилбензола (стадия B, пример 14) обработкой тетрабутиламмонийфторидом в THF в течение нескольких часов. Его влили в смесь эфира/этилацетата и промыли рассолом. После удаления растворителя сырой продукт очистили тонкослойной колоночной хроматографией с использованием в качестве элюента этилацетат/гексана.
1H ЯМР (400 мГц, CDCl3, част. на миллион): δ 7,01 (s, 2H), 6,55 (dd, J = 17,6, J = 11,0 Гц, 1H), 5,55 (d, J = 17,6 Гц, 1H), 5,06 (d, J = 11,0 Гц, 1H), 2,64 (t, J = 7,7 Гц, 4H), 1,65 - 1,60 (m, 4H), 0,96 (t, J = 7,2 Гц, 6H).
Стадия B:
Метил-2-[(2,6-дипропил-4-винил)фенокси]-2-(2-нафтил)ацетат
Названное соединение получили путем алкилирования 2,6-дипропил-4-винилфенола (стадия A) метил-2-бром-2-(2-нафтил)-ацетатом с использованием методики алкилирования, описанной в стадии B примера 1.
1H ЯМР (400 мГц, CDCl3, част. на миллион): δ 7,9 - 7,8 (m, 4H), 7,68 (d, J = 6,7 Гц, 1H), 7,50 - 7,48 (m, 2H), 7,02 (s, 2H), 6,57 (dd, J = 18,4, 10,8 Гц, 1H), 5,61 (d, J = 18,4, 1H), 5,26 (s, 1H), 5,14 (d, J = 10,8, 1H), 3,73 (s, 1H), 2,38 - 2,34 (m, 4H), 1,54-1,43 (m, 4H), 0,74 (t, J = 7,33 Гц, 6H).
Стадия C:
Метил-2-[(2,6-дипропил-4-(1,2-дигидроксиэтил)фенокси] -2- (2-нафтил)ацетат
К раствору 6 мл (0,024 ммоль) OsO4 и 31 мг (0,263 ммоль) N-метилморфолин-N-оксида (NMO) в 3 мл ацетона и 2 каплях воды добавили 96 мг (0,239 ммоль) продукта стадии B. Через 90 минут реакционную смесь влили в смесь эфира и воды. Слои разделили и водный слой экстрагировали дважды простым эфиром. Объединенные водные слои промыли насыщенным хлоридом натрия, сушили над безводным сульфатом магния, фильтровали и концентрировали. Путем очистки тонкослойной хроматографии (силикагель, гексан/этилацетат, 1:1) получили 63 мг названного соединения.
1H ЯМР (400 мГц, CDCl3, част. на миллион): δ 7,89 - 7,80 (m, 4H), 7,67 (d, J = 8,4 Гц, 1H), 7,50 - 7,48 (m, 2H), 6,9 (s, 2H), 5,25 (s, 1H), 4,75 - 4,68 (m, 1H), 3,72 (s, 3H), 3,75 - 3,61 (m, 2H), 2,38 - 2,34 (m, 4H), 1,53 - 1,46 (m, 4H), 0,75 - 0,71 (m, 6H).
Стадия D:
2-[(2,6-Дипропил-4-(1,2-дигидроксиэтил)фенокси] -2- (2-нафтил)уксусная кислота.
Названное соединение получили из продукта стадии C путем омыления IN водным раствором KOH, как описано выше.
1H ЯМР (400 мГц, CD3OD, част. на миллион): δ 7,84 - 7,71 (m, 5H), 7,46 - 7,42 (m, 2H), 6,96 (s, 2H), 5,03 (s, 1H), 4,55 (t, J = 7,2 Гц, 1H), 3,54 (t, J = 7,2 Гц, 2H), 2,34 (t, J = 7,9 Гц, 4H), 1,51 - 1,30 (m, 4H), 0,70 (t, J = 7,3 Гц, 6H).
Пример 18.
2-[(2,6-Дипропил-4-(1-гидроксипентил)фенокси] -2- (2-нафтил)уксусная кислота
Стадия A:
Метил-2-[(2,6-дипропил-4-формил)фенокси]-2-(2-нафтил)ацетат
К раствору 262 мг (0,645 ммоль) метил-2-[(2,6-дипропил-4- формил)фенокси] -2-(2-нафтил)ацетата в 2 мл метиленхлорида добавили 404 мг (0,968 ммоль) PDC. Через 4 часа реакционную смесь разбавили 20 мл эфира и фильтровали через подушку флоризила/целита и концентрировали до получения 235 мг названного соединения.
1H ЯМР (400 мГц, CDCl3, част. на миллион): δ 9,86 (s, H), 7,89 - 7,80 (m, 4H), 7,65 (m, 1H), 7,52 - 7,49 (m, 4H), 5,35 (s, 1H), 3,73 (s, 3H), 2,47 - 2,43 (m, 4H), 1,54 - 1,43 (m, 4H), 0,77 (t, J = 7,3 Гц, 6H).
Стадия B:
Метил-2-[(2,6-дипропил-4-(1-гидроксипентил)фенокси]-2- (2-нафтил)ацетат
К раствору 56 мг (0,143 ммоль) продукта стадии A в 1 мл THF при -78oC добавили 0,075 мл (2,0 М в THF, 0,150 ммоль) н-бутилмагнийхлорида. Анализ TLC показал, что исходный материал остался неизрасходованным, поэтому добавили 0,020 мл н-бутилмагнийхлорида. Через 1 час реакционную смесь разбавили насыщенным водным раствором хлорида аммония и затем экстрагировали дважды этилацетатом. Органические слои сушили над безводным сульфатом натрия, фильтровали и концентрировали.
Путем очистки тонкослойной колоночной хроматографией (силикагель, гексан/этилацетат 6:1) получили 32 мг названного соединения.
1H ЯМР (400 мГц, CDCl3, част. на миллион): δ 7,89 - 7,82 (m, 4H), 4,67 (m, 1H), 7,49 - 7,47 (m, 2H), 6,9 (s, 2H), 5,26 (s, 1H), 2,36 (t, J = 8,0 Гц, 4H), 1,75 - 1,2 (m, 8H), 0,86 (t, J = 7,2 Гц, 3H), 0,72 (t, J = 7,3 Гц, 6H).
Стадия C:
2-[(2,6-Дипропил-4-(1-гидроксипентил)фенокси]-2- (2-нафтил)уксусная кислота
Названное соединение получили из продукта стадии B путем омыления водным IN KOH в метаноле, как описано выше.
1H ЯМР (400 мГц, CD3OD, част. на миллион): δ 7,84 - 7,72 (m, 5H), 7,45 - 7,43 (m, 2H), 6,91 (s, 2H), 5,03 (s, 1H), 4,45 (t, 1H), 2,36 - 2,32 (m, 4H), 1,75 - 1,45 (m, 4H), 1,35 - 1,29 (m, 4H), 0,87 (t, J = 7,2 Гц, 3H), 0,70 (t, J = 7,2 Гц, 6H).
FAB-MS: m/e = 487 (M+K), 469 (M+Na).
Пример 19.
2-[(4-карбокси-2,6-дипропил)фенокси]-2-фенилуксусная кислота
Стадия A:
Трет-бутил-2-[(4-карбометокси-2,6-дипропил)фенокси]-2- фенилацетат
Метил-2,6-дипропил-4-гидроксибензоат (1,5 г, 6,383 ммоль) кипятят с обратным холодильником с K2CO3 (1,5 экв) и треб-бутил-α- бромфенилацетатом (2,4 г, 8,856 ммоль) в ацетоне в течение 16 часов.
Реакционную смесь фильтровали через целит, осадок промыли ацетоном и объединенный фильтрат и промывки концентрировали. Полученное сырое масло подвергли хроматографии (тонкослойной колоночной) с использованием силикагеля и 10% этилацетата в гексане для получения названного соединения (2,7 г).
1H ЯМР (400 мГц, CD3OD, част. на миллион): δ 7,665 (s, 2H), 7,443 (dd, 2H), 7,345 (dd, 3H), 5,019 (s, 1H), 3,851 (s, 3H), 2,49 - 2,335 (m, 4H), 1,63 - 1,4 (m, 4H), 1,364 (s, 9H), 0,803 (t, 6H).
Стадия B:
Трет-бутил-2-[(4-карбокси-2,6-дипропил)фенокси]-2-фенилацетат
Названное соединение (125 мг) получили омылением вышеописанного трет-бутил-2-[(4-карбометокси-2,6-дипропил)-фенокси]-2-фенилацетата (200 мг, 0,47 ммоль) IN водным раствором LiOH в метаноле.
1H ЯМР (400 мГц, CD3OD, част. на миллион): δ 7,66 (s, 2H), 7,5 - 7,4 (dd, 2H), 7,43 - 7,36 (dd, 3H), 4,88 (s, 1H), 2,5-2,35 (m, 4H), 1,63 - 1,33 (m, 4H), 1,38 (t, 9H), 0,83 (t, 6H).
Стадия C:
2-[(4-карбометокси-2,6-дипропил)фенокси]-2-фенилуксусная кислота
Трет-бутил-2-[(4-карбокси-2,6-дипропил)фенокси]-2-фенилацетат (стадия A) (125 мг, 0,293 ммоль) обработали 3 мл трифторуксусной кислоты (TFA) в метиленхлориде в течение 2-х часов. Для получения названного соединения (90 мг) удалили летучие компоненты.
1H ЯМР (400 мГц, CD3OD, част. на миллион): δ 7,67 (s, 2H), 7,463-7,44 (m, 2H), 7,387-7,362 (m, 3H), 5,177 (s, 1H), 3,856 (s, 3H), 2,377 (t, 4H), 1,6-1,366 (m, 4H), 0,773 (t, 6H).
Стадия D:
2-[(4-карбокси-2,6-дипропил)фенокси]-2-фенилуксусная кислота
Продукт стадии C (70 мг, 0,19 ммоль) обработали 1N водным раствором LiOH в метаноле. Реакцию контролировали посредством TCX. Когда исходный материал был полностью израсходован, смесь подкислили при 0oC до pH 5 путем добавления IN HCl. Водную фазу экстрагировали этилацетатом, сушили и фильтровали. Фильтрат концентрировали до получения названного соединения (25 мг).
1H ЯМР (400 мГц, CD3OD, част. на миллион): δ 7,68 (s, 2H), 7,52-7,45 (m, 2H), 7,43-7,365 (m, 3H), 5,175 (s, 1H), 2,43 (t, 4H), 1,64-1,4 (m, 4H), 0,83 (t, 6H).
Пример 20.
2-[(4-карбокси-2,6-дипропил)фенокси]-2-(3,4-дихлорфенил) уксусная кислота.
Названное соединение получили с использованием методик, подобных той, которая описана в примере 19.
1H ЯМР (400 мГц, CD3OD, част. на миллион): δ 7,72 (d, 1H, J=2,0 Гц), 7,52-7,45 (m, 2H), 7,69 (s, 2H), 7,56 (d, 1H, J=8,3 Гц), 7,43 (dd, 1H, J= 8,3, 1,9 Гц), 5,18 (s, 1H), 2,45 (m, 4H), 1,58-1,43 (m, 4H), 0,84 (t, 6H, J= 7,3 Гц).
FAB-MS m/e=426 (M+1)
Пример 21.
2-[(4-карбокси-2,6-дипропил)фенокси]-2-(3-бромфенил)уксусная кислота
Названное соединение получили с использованием методик, подобной той, которая описана в примере 19.
1H ЯМР (200 мГц, CD3OD, част. на миллион): δ 7,73 (d, 1H, J=1,8 Гц), 7,69 (s, 2H), 7,56 (dd, 1H, J=7,8, 1,9 Гц), 7,46 (d, 1H, J=7,9 Гц), 7,32 (t, 1H, J=7,8 Гц), 5,19 (s, 1H), 2,44 (t, 4H, J=7,6 Гц), 1,70-1,34 (m, 4H), 0,84 (t, 6H, J=7,3 Гц).
FAB-MS m/e=436 (M+1)
Пример 22.
2-[(4-карбокси-2,6-дипропил)фенокси] -2-(3,4-метилендиоксифенил)уксусная кислота
Названное соединение получили с использованием методик, подобных той, которая описана в примере 19.
1H ЯМР (200 мГц, CD3OD, част. на миллион): δ 7,68 (s, 2H), 7,02 (d, 1H, J=1,6 Гц), 6,84 (m, 2H), 5,98 (s, 2H), 5,08 (s, 1H), 2,44 (t, 4H, J=7,9 Гц), 1,52 (m, 4H), 0,86 (t, 6H, J=7,3 Гц).
FAB-MS m/e = 401 (M+1).
Пример 23.
2-[(4-карбокси-2,6-дипропил)фенокси]-2-(3-метоксифенил)уксусная кислота.
Названное соединение с использованием методик, подобных той, которая описана в примере 19.
1H ЯМР (200 мГц, CD3OD, част. на миллион): δ 7,68 (s, 2H), 7,29 (t, 1H, J= 7,9 Гц), 7,08 (d, 1H, J=2,3 Гц), 7,03 (d, 1H, J=7,7 Гц), 6,95 (dd, 1H, J= 0,9, 8,3 Гц), 5,14 (s, 1H), 3,79 (s, 3H), 2,43 (t, 4H, J=7,9 Гц), 1,58-1,42 (m, 4H), 0,82 (t, 6H, J=7,3 Гц)
FAB-MS m/e = 387 (M+1)
Пример 24.
(N-Бензолсульфонил)-2-[(4-(N-бензолсульфонил)карбоксамидо-2,6- дипропилфенокси]-2-(3-бромфенил)ацетамид
Названное соединение получили с использованием методики, описанной для синтеза N-сульфонилкарбоксамидов в патенте США N 5177095. Двухосновную 2-[(4-карбокси-2,6-дипропил)-фенокси] -2- (3-бромфенил)уксусная кислоту (200 мг, 0,46 ммоль, из примера 21) кипятили с обратным холодильником с карбонилдиимидазолом (1,5 экв) в THF в течение 3-4 часов. При комнатной температуре смесь 1,5 экв. бензолсульфонамида и 1,5 экв. DBU в THF добавили в вышеприведенной реакционной смеси и смеси перемешивали всю ночь.
Реакционную смесь разбавили этилацетатом и промыли 5% водным раствором лимонной кислоты. Растворитель удалили и сырой продукт очистили тонкослойной колоночной хроматографией, получив при этом 238 мг названного соединения.
1H ЯМР (400 мГц, CD3OD, част. на миллион): δ 8,07 (dd, 2H, J=1,4, 7,2 Гц), 7,91-7,88 (m, 2H), 7,67-7,49 (m, 8H), 7,46 (s, 2H), 7,28-7,25 (m, 2H), 5,01 (s, 1H), 2,28-2,23 (m, 4H), 1,49-1,29 (m, 4H), 0,71 (t, 6H, J=7,4 Гц).
FAB-NS m/e = 713 (M+1)
Пример 25.
N-(4-трет-бутилбензолсульфонил)-2-(4-метоксикарбонил-2- пропилфенокси)-2-(3,4-метилендиоксифенил)ацетамид
Стадия A:
Получение 2-/4-карбометокси-2-пропилфенокси/-3,4- метилендиоксифенилуксусной кислоты
К этил-2-(4-карбометокси-2-пропилфенокси)-3,4- метилендиоксифенилацетату (стадия A примера 56) (2,04 г, 5,10 ммоль) в MeOH (40 мл) добавили 5N NaOH (8 мл).
Тотчас же последовала быстрая реакция монодеэтерификации, которую контролировали посредством TCX. После удаления этилового эфира и перед омылением метилового эфира реакционную смесь резко охладили 9N HCl (4,5 мл). К реакционной смеси, пока она была основной, добавили насыщенный раствор NaHCO3 и в вакууме удалили MeOH. Остаток разделили между Et2O и H2O, собрали продукт в водной фазе и удалили примеси у органической фазы. Затем водную фазу подкислили 9N HCl (pH 1) и продукт экстрагировали в EtOAc. Раствор сушили над MgSO4, фильтровали и растворитель удалили. Выход составил 1,78 г (4,78 ммоль, 94%), rfg=0,16 (CHCl3:MeOH:NH4OH/80:10:1)
Стадия B:
Получение предшественника сульфонамида
К дихлорметановому раствору сульфонилхлорида, охлажденному до 0oC, добавили трет-бутиламин (3 экв). Через 3-5 часов CH2Cl2 удалили и заменили EtOAc. Реакционный раствор промыли 1N HCl, водой, 1N NaOH и рассолом. Полученный раствор сушили над MdSO4 и фильтровали. Удалили растворитель. К полученному веществу для удаления трет-бутильной группы добавили пару капель анизола и затем TFA. После всего освободили от защиты сульфонамид, удалили в вакууме TFA и остаток поместили в EtOAc/Et2O. Раствор промыли насыщенным раствором NaHCO3 для удаления остаточного TFA, затем рассолом, сушили над MgSO4, фильтровали и растворитель удалили.
Предшественники сульфонамида, используемые при получении соединений примеров 28, 29, 31, 32, 37, 38 и 39, получили из соответствующих сульфонилхлоридов с использованием методики, описанной выше. Предшественники сульфонамида, используемые при получении соединений примеров 26 и 33-36, являются коммерчески доступными.
Предшественники сульфонамида, используемые в примерах 27, 30 и 32, сульфонилхлориды которых являются коммерчески недоступными, получили с использованием стандартных химических методик:
Получение предшественника сульфонамида для примера 27
Трет-бутилсульфонамид из 4-бромбензолсульфонилхлорида получили с использованием методики, описанной выше. Затем трет-бутилсульфонамид соединили с фенилбороновой кислотой в реакции поперечного сочетания с NaOH, EtOH, толуолом и Pd(PPh3)4, катализируемой палладием, при 100oC для получения бифенилсульфонамида. Освобождение от защиты третбутилсульфонамида TFA и анизолом привело к получению освобожденного сульфонамида.
Получение предшественника сульфонамида для примера 30
Трет-бутилсульфонамид из 2-тиофенсульфонилхлорида получили с использованием методики, описанной выше. Обработкой трет-бутилсульфонамида BuLi и затем изобутилиодидом получили 5-изобутил-2-тиофен-трет-бутилсульфонамид, который затем освободили от защиты TFA и анизолом для получения свободного сульфонамида.
Получение предшественника сульфонамида для примера 32
Трет-бутилсульфонамид из р-нитробензолсульфонилхлорида получили с использованием методики, описанной выше. Восстановление нитрогруппы до амина осуществляли водородом в MeOH над Pd-C.
Обработкой свободного амина LiBr и (MEO)3PO и затем NaOH получили диметиламин и для получения свободного сульфонамида трет-бутилсульфонамид освободили от защиты TFA и анизолом.
Стадия C:
Получение N-((4-трет-бутилбензолсульфонил)-2- (4-карбометокси-2-пропилфенокси)-3,4-метилендиоксифенилацетамида
К продукту стадии A (57,2 мг, 0,154 ммоль) в сухом THF (0,75 мл) добавили CDl (76,0 мг, 0,469 ммоль) и реакционную смесь нагревали до 50oC в течение 2,5 часов. К этому раствору добавили раствор п-трет-бутилфенилсульфонамида (131,7 мг, 0,618 ммоль) и DBU (92,1 мкл, 0,594 ммоль) в сухом THF (0,75 мл). Реакционную смесь продолжали перемешивать при 50oC, осуществляя контроль посредством тонкослойной хроматографии до тех пор, пока израсходовали всю монокислоту (приблизительно 3 часа). Реакционную смесь концентрировали в вакууме и остаток поместили в Et2O : EtOAc /50:50. Органическую фазу промыли 10% лимонной кислотой, 2 раза водой и рассолом, затем сушили над MgSO4, фильтровали и удалили растворитель. Очистку осуществляли посредством радиальной хроматографии при элюировании гексаном: EtOAc /3:2.
Выход составил 74,3 мг (0,131 ммоль, 85%) rf = 0,32 масс-спектр FAB (CHCl3: MEOH: NH4OH/80:10:1) m/e 590,0 (M+ Na вычислено для C30H33NSO8 590).
Смотри Drummond, J.T.; Johnson, G. Tetrahedron Lett., 1988, 29, 1653.
Примеры 26 - 39.
Примеры 26 - 39 выполняли с использованием методик, описанных выше в примере 25 (см. табл. 3).
Данные протонного ядерно-магнитного резонанса для примера 29 представлены ниже:
Пример 29.
N-(4-метилбензолсульфонил)-2-[(4-метоксикарбонил-2- пропилфенокси] -2-(3,4-метилендиоксифенил)ацетамид
1H ЯМР (400 мГц, CD3OD, час. на миллион): δ 0,89 (t, 3H), 1,59 (m, 2H), 2,34 (s, 3H), 2,63 (m, 2H), 3,85 (s, 3H), 5,49 (s, 1H), 5,97 (s, 2H), 6,55 (d, 1H), 6,79 (d, 1H), 6,96 (dd, 1H), 7,26 (d, 2H), 7,58 (dd, 1H), 7,70 (d, 2H), 7,74 (d, 1H).
Пример 40.
N-(4-трет-бутилбензолсульфонил)-2-[(4-карбокси-2- пропилфенокси)] -2-(3,4-метилендиоксифенил)ацетамид
К продукту примера 25 (51,1 мг, 0,090 ммоль) в MeOH (2 мл) добавили 5 N NaOH (0,5 мл). Реакцию контролировали посредством ТСХ. Когда реакция завершалась, MeOH удалили и остаток разделили между водой и Et2O : EtOAc. Водный слой подкислили раствором HCl и продукт экстрагировали в органическую фазу. Органическую фазу промыли рассолом, затем сушили над MgSO4, фильтровали и растворитель удалили. Путем растирания в порошок с Et2O/Гекс. получили белое твердое вещество. Выход составил 25,8 мг (0,047 ммоль, 52%) масс-спектр FAB, m/e 554,2 (M+1 вычислено для C29H31NSO8 554).
Примеры 41 - 54.
Примеры 41 - 54 выполняли с использованием методик, описанных выше в примере 40 (см. табл. 4).

Данные протонного ядерного-магнитного резонанса для нескольких примеры даны ниже.
Пример 47.
N-(4-диметиламинобензолсульфонил)-2-(4-карбокси-2- пропилфенокси)-2-(3,4-метилендиоксифенил)ацетамид
1H ЯМР (300 мГц, CD3OD, част. на миллион): δ 0,89 (t, 3H), 1,59 (m, 2H), 2,34 (s, 3H), 2,62 (m, 2H), 3,03 (s, 6H), 5,48 (s, 1H), 5,96 (s, 2H), 6,43 (d, 1H), 6,64 (dd, 2H), 6,79 (d, 1H), 6,91 (m, 2H), 7,55 (dd, 1H), 7,64 (dd, 2H), 7,74 (d, 1H).
Пример 49.
N-(2-карбоксибензолсульфонил)-2-(4-карбокси-2- пропилфенокси)-2-(3,4-метилендиоксифенил)ацетамид
1H ЯМР (400 мГц, CD3OD, част. на миллион): δ 0,92 (t, 3H), 1,62 (m, 2H), 2,67 (m, 2H), 5,66 (s, 1H), 5,95 (s, 2H), 6,74 (d, 1H), 6,77 (d, 1H), 6,93 (d, 1H), 6,98 (dd, 1H), 7,69 (m, 1H), 7,75 (m, 2H), 8,09 (d, 1H).
Пример 53.
N-(дансульфонил)-2-(4-карбокси-2-пропилфенокси)-2-(3,4- метилендиоксифенил)ацетамид
1H ЯМР (400 мГц, CD3OD, част. на миллион): δ 0,83 (t, 3H), 1,51 (m, 2H), 2,57 (m, 2H), 2,88 (s, 6H), 5,40 (s, 1H), 5,95 (dd, 2H), 6,26 (d, 1H), 6,65 (d, 1H), 6,79 (d, 1H), 6,85 (dd, 1H), 7,19 (dd, 1H), 7,25 (d, 1H), 7,48 (t, 1H), 7,54 (t, 1H), 7,66 (d, 1H), 8,13 (d, 1H), 8,30 (dd, 1H), 8,53 (d, 1H).
Пример 54.
N-(8-хинолинсульфонил)-2-(4-карбокси-2-пропилфенокси)-2- (3,4-метилендиоксифенил)ацетамид
1H ЯМР (300 мГц, CD3OD, часть на миллион): δ 0,85 (t, 3H), 1,54 (m, 2H), 2,59 (m, 2H), 5,57 (s, 1H), 5,94 (s, 2H), 6,47 (d, 1H), 6,66 (d, 1H), 6,71 (d, 1H), 3,87 (dd, 1H), 7,34 (dd, 1H), 7,58 (dd, 1H), 7,68 (m, 2H), 8,20 (dd, 1H), 8,39 (dd, 1H), 8,47 (dd, 1H), 8,83 (dd, 1H).
Пример 55.
N-(8-хинолинсульфонил)-2-(4-карбоксамидо-2-пропилфенокси)-2- (3,4-метилендиоксифенил)ацетамид
К N-(8-хинолинсульфонил)-2-(4-карбокси-2-пропилфенокси)-2- (3,4-метилендиоксифенил)ацетамиду, пример 54 (25,8 мг, 0,047 ммоль) в сухом DMF (0,5 мл) добавили гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида, EDC (18,4 мг, 0,096 ммоль), NH4Cl (6,7 мг, 0,125 ммоль) и TEA (триэтаноламин) (17,5 мл, 0,125 ммоль). После реакции осуществили тонкослойную хроматографию (CH2Cl2 : MeOH : HOAc/100:15:1,5).
После завершения реакции DMF удалили в вакууме и остаток поместили в Et2O/EtOAc. Раствор промыли 10% лимонной кислотой, водой и рассолом, затем сушили над MgSO4, фильтровали и растворитель удалили. Продукт очистили хроматографией при элюировании CH2Cl2 : MeOH : HOAc/200:5:1,5, rf = 0,35. Масс-спектр FAB (CH2Cl2 : MeOH : HOAc/100:5:1,5), m/e 548,0 (M+1 = вычисленный для C28H25N3SO7 548).
Пример 56.
α- (4-карбометокси-2-н-пропилфенокси)-3,4-метилендиоксифенил) уксусная кислота.
Стадия A.
Получение этил -α- (4-карбометокси-2-н-пропилфенокси)- 3,4-метилендиоксифенолацетата.
В 2 л трехгорлую 24/40 круглодонную колбу, снабженную механической мешалкой, отверстием для впуска азота и капельной воронкой, добавили сначала раствор 36,0 г (0,185 моль) метил-4-гидрокси-3-н-пропилбензоата, растворенного в 700 мл безводного DMF, после чего добавили 66,4 г (0,204 моль) карбоната цезия. Колбу продули азотом и реакционную смесь перемешивали при комнатной температуре в течение 2-х часов. Затем через капельную воронку в течение 15 минут добавили раствор 58,5 г (0,204 моль) этил- α -бром-3,4-метилендиоксифенилацетата, растворенного в 100 мл DMF. Реакционную смесь перемешивали при комнатной температуре еще в течение одного часа, затем резко охладили путем добавления к 5 л 5% водного раствора лимонной кислоты. Органический продукт экстрагировали в диэтиловый эфир (2х4 л), органические слои разделили, промыли насыщенным водным NaCl, сушили (MgSO4), фильтровали и выпаривали. Остаток поместили в колонку с силикагелем (2 кг, 70 - 230 меш), уравновешенную в 10% CH2Cl2-гексане. Затем колонку последовательно элюировали 12 л 10% CH2Cl2-гексана. 12 л 5% EtOAc-гексана, 4 л 7,5% EtOAc-гексана, 12 л 10% EtOAc-гексана и в конце 8 л 20% EtOAc-гексана. Объединение очищенных фракций и выпаривание в вакууме привело к получению 76,3 г (74,2 теоретически) названного соединения в виде бледно-желтого масла, которое использовали на следующей стадии без дальнейшей очистки.
Стадия B:
Получение α- (4-карбометокси-2-н-пропилфенокси)-3,4- метилендиоксифенилуксусной кислоты
1 л трехгорлую 24/40 круглодонную колбу, снабженную механической мешалкой, капельной воронкой и отверстием для впуска азота, загрузили раствором 76,3 г (0,185 моль) полуочищенного продукта стадии A, растворенного в 500 мл метанола. Колбу продули азотом, включили мешалку и через капельную воронку в течение 30 минут добавили 37 мл 5,0 N водного раствора гидроксида натрия. Реакционную смесь перемешивали при комнатной температуре в течение еще 30 минут, после чего анализ ТСХ (CH2Cl2-MeOH-NH4OH 90:10:1) показал, что исходный материал израсходован. С помощью 6 N HCl установили pH реакционной смеси, равный 4, и фазу органического растворителя удалили в вакууме. Затем осажденный органический продукт и водный слой разделили между CH2Cl2 (1 л) и водой (1 л), что привело к получению обильной эмульсии. Затем реакционную смесь подвергли старению в течение ночи в холодильнике, при этом произошла кристаллизация органического продукта. Кристаллический осадок отделили от двухфазной смеси посредством фильтрации и промыли CH2Cl2. Осадок опять суспендировали в диэтиловом эфире, фильтровали, промыли гексаном и затем сушили в вакууме до получения 65 г (94%) названного соединения в виде белого твердого кристаллического вещества.
1H ЯМР (400 мГц, CD3OD, част. на миллион): δ 0,93 (t, J = 7,2 Гц, 3H), 1,62 - 1,75 (m, 2H), 2,63 - 2,70 (m, 1H), 2,77 - 2,81 (m, 1H), 3,84 (s, 3H), 5,54 (s, 1H), 5,94 (s, 2H), 6,81 (d, J = 7,60 Гц, 1H), 6,89 (d, J = 9,20 Гц, 1H), 7,08 (d, J = 1,6 Гц, 1H), 7,11 (шир. s, 1H) 7,78 - 7,81 (m, 2H).
Пример 57.
N-(4-изопропилбензолсульфонил) -α- (4-карбометокси-2-н-пропилфенокси)-3,4-метилендиоксифенил ацетамид
Стадия A.
Получение N-(4-изопропилбензолсульфонил) -α- (4-карбометокси-2-н-пропилфенокси)-3,4-метилендиоксифенилацетамида
Высушенную в печи трехгорлую 24/40 1 л круглодонную колбу снабдили механической мешалкой, отверстием для впуска азота и мембраной. Колбу продули азотом, затем загрузили 20,06 г (53,9 ммоль) продукта примера 56,400 мл безводного THF и 9,76 мл (70,0 ммоль) триэтиламина. Колбу продули азотом, затем загрузили 20,06 г (53,9 ммоль) продукта примера 56, 400 мл безводного THF и 9,76 мл (70,0 ммоль) триэтиламина. Колбу и ее содержимое перемешали и охладили до -78oC с помощью бани со смесью сухого льда с ацетоном и затем медленно через шприц добавили 7,30 мл (59,3 ммоль) триметилацетилхлорида.
После завершения добавления баню со смесью сухого льда с ацетоном заменили баней со смесью льда с водой и реакционную смесь перемешали при 0oC в течение 1 часа. Отдельную высушенную в печи трехгорлую 24/40 2 л круглодонную колбу снабдили механической мешалкой, мембраной и отверстием для впуска азота. Колбу промыли азотом, затем загрузили 16,102 г (80,8 ммоль) 4-изопропилбензолсульфонамида и 300 мл безводного метилсульфоксида. Включили мешалку и через шприц через мембрану медленно добавили 162 мл 1,0 М раствора литий-бис(триметилсилиламида) в THF.
После завершения добавления реакционную смесь перемешивали при комнатной температуре в течение еще 30 минут. Содержимое первой реакционной смеси, включая мелкокристаллический белый осадок, который суспендировали в реакционной смеси, затем медленно перенесли через канюлю с широким диаметром в перемешанный раствор депротонированного сульфонамида во второй колбе. Объединенную реакционную смесь затем перемешивали в атмосфере азота в течение еще 14 часов. Реакционную смесь резко охладили 1,0 N HCl и большинство летучих растворителей удалили в вакууме. Остаток разделили между EtOAc и 1,0 NHCl, затем органический слой отделили, промыли насыщенным водным NaCl, сушили (MgSO4), фильтровали и выпаривали в вакууме. Остаток очистили колоночной хроматографией (15 см х 150 см) на силикагеле (3 кг, 70-230 меш) при элюировании (CH2Cl2-MeOH-NH4OH 90:10:1). Объединение очищенных фракций и выпаривание в вакууме привело к получению 18,367 г (62%) названного соединения.
1H ЯМР (400 мГц, CD3OD, част. на миллион): δ 0,88 (t, J = 7,60 Гц, 3H), 1,24 (d, J = 7,00 Гц, 3H), 1,25 (t, J = 7,00 Гц, 3H), 1,55 - 1,60 (m, 2H), 2,59 - 2,66 (m, 2H), 2,97 (sept, J = 7,00 Гц, 1H), 3,83 (s, 3H), 5,52 (s, 1H), 5,97 (s, 2H), 6,50 (d, J = 8,80 Гц, 1H), 6,80 (d, J = 8,00 Гц, 1H), 6,89 (d, J = 1,60 Гц, 1H), 6,94 (dd, J = 2,00, 8,00 Гц, 1H), 7,14 (d, J = 8,80 Гц, 2H), 7,59 (dd, J = 2,20, 8,80 Гц, 1H), 7,75 (d, J = 2,20, 1H), 7,79 (d, J = 8,80 Гц, 2H).
Пример 58.
Двукалиевая соль N-(4-изо-пропилбензолсульфонил) -α- (4-карбокси-2-н-пропилфенокси)-3,4-метилендиоксифенилацетамида
Стадия A:
Получение двукалиевой соли N-(4-изо-пропилбензолсульфонил) -α- (4-карбокси-2-н-пропилфенокси)-3,4-метилендиоксифенилацетамида
К раствору 18,367 г (33,2 ммоль) продукта примера 57, растворенного в 100 мл метанола, добавили раствор 6,56 г (116,9 ммоль) гидроксида калия в 25 мл воды, и реакционную смесь перемешали при 60oC в атмосфере азота. Через 6 часов анализ ТСХ (CHCl3-MeOH-NH4OH 80:15:1) показал, что гидролиз сложного эфира завершился. Реакционную смесь охладили до комнатной температуры, разбавили 100 мл воды, фильтровали через 0,45 микронный фильтр и затем разделили на две равные по объему порции. Фракции отдельно обессолили и очистили на жидкостном хроматографе Waters Millipore Delta Prep (Вотез Милипор Дельта Преп), снабженном модулем М 1000 Prep-Pak, содержащем катушку с фотографическими пленками 47 х 30 мм Delta-Pak C18 15 мкм 100А. Использовали две емкости для растворителя: систему растворителя A (вода-ацетонитрил 95-5) и систему растворителя B (вода-ацетонитрил 5-95), и поток, вытекающий из колонны, одновременно контролировали при 210 и 280 нм детектором видимых ультрафиолетовых лучей модели Waters 490.
Каждую фракцию инжектировали насосом в колонку и обессоливали путем элюирования (50 мл/мин) несколькими колоночными объемами системы растворителя A.
Затем начали градиентное элюирование, начальные условия которого были: 100% системы растворителя A-0% системы растворителя B и через 30 минут достигли: 50% системы растворителя A-50% системы растворителя B, и фракции собрали с помощью сборника фракций ISCO Foxy 200.Очищенные фракции соединили в круглодонных колбах, заморозили при -78oC в бане со смесью сухого льда с ацетоном и подвергли лиофилизации. Объединение очищенного продукта дало 18,719 г (82%) названного соединения в виде белого лиофилизованного порошка.
1H ЯМР (400 мГц, CD3OD, част. на миллион): δ 0,88 (t, J = 7,20 Гц, 3H), 1,21 (d, J = 7,00 Гц, 3H), 1,22 (d, J = 7,00 Гц, 3H), 1,56 - 1,63 (m, 2H), 2,52 - 2,59 (m, 1H), 2,67 - 2,74 (m, 1H), 2,91 (sept, J = 7,00 Гц, 1H), 5,33 (s, 1H), 5,92 (d, J = 1,20 Гц, 1H), 6,72 (d, J = 8,50 Гц, 1H), 6,76 (d, J = 8,50 Гц, 1H), 7,04 (dd, J = 2,00, 8,50 Гц, 1H), 7,67 (d, J = 8,50 Гц, 2H), 7,73 (d, J = 2,0 Гц, 1H).
Микроанализ C28H27NSO8K2 • H2O:
Вычислено: C = 53,06, H = 4,61, N = 2,21, K = 12,34
Найдено: C = 52,81, H = 4,56, N = 2,17, K = 12,02.
Пример 59.
α- (2-изо-бутил-4-карбометоксифенокси)-3,4- метилендиоксифенилуксусная кислота
Стадия A:
Получение этил -α- (2-изо-бутил-4-карбометоксифенокси)- 3,4-метилендиоксифенилацетата
К раствору 1,008 г (4,84 ммоль) метил-3-изо-бутил-4- гидроксибензоата и 1,737 г (6,05 ммоль) этил -α- бром-3,4- метилендиоксифенилацетата в 10 мл ацетона добавили 1,338 г (10 ммоль) тонкоизмельченного порошка карбоната калия. Реакционную смесь перемешивали магнитной мешалкой, нагревали в колбе с обратным холодильником в течение 4-х часов, затем охладили до комнатной температуры, фильтровали и выпаривали. Остаток очистили тонкослойной колоночной хроматографией на силикагеле при элюировании 10% EtOAc-гексаном; объединение очищенных фракций и сушка в вакууме привели к получению 1,518 г (78%) названного соединения в виде аморфного порошка.
1H ЯМР (400 мГц, CDCl3, част. на миллион): δ 0,90 (d, J = 6,60 Гц, 3H), 0,94 (d, J = 6,60 Гц, 3H), 1,17 (t, J = 7,20 Гц, 3H), 2,02 - 2,08 (m, 2H), 2,55 (dd, J = 7,20, 13,20 Гц, 1H), 2,64 (dd, J = 7,20; 13, 20 Гц, 1H), 3,85 (s, 3H), 4,11 - 4,19 (m, 2H), 5,56 (s, 1H), 5,96 (s, 2H), 6,70 (d, J = 9,20 Гц, 1H), 6,68 (d, J = 7,60 Гц, 1H), 7,02 (dd, J = 1,60, 8,00 Гц, 1H), 7,05 (d, J = 2,00 Гц, 1H), 7,78-7,81 (m, 2H).
Стадия B:
Получение α- (2-изо-бутил-4-карбометоксифенокси)-3,4- метилендиоксифенилуксусной кислоты
К раствору 1,518 г (3,66 ммоль) продукта стадии A, растворенного в 8,0 мл метанола, добавили 1,0 мл 5,0 М водного раствора гидроксида натрия. Реакционную смесь перемешивали при комнатной температуре и контролировали посредством ТСХ (CHCl3-MeOH-NH4OH 80: 15: 1). Через 1,5 часа реакцию посчитали завершенной и pH реакционной смеси установили с помощью 1,0 N HCl равным 5. Затем реакционную смесь разделили между EtOAc и водой, разделили, сушили (MgSO4), фильтровали и выпаривали. Остаток очистили тонкослойной колоночной хроматографией на силикагеле при элюировании CHCl3-MeOH-NH4OH (80:15:1), выпаривание очищенных фракций и сушка в вакууме привели к получению названного соединения в виде аморфной пены.
1H ЯМР (400 мГц, CD3OD, част. на миллион): δ 0,86 (d, J = 6,80 Гц, 3H), 0,89 (d, J = 6,80 Гц, 3H), 1,96-2,04 (m, 1H), 2,49 (dd, J = 7,20, 12,80 Гц, 1H), 2,69 (dd, J = 7,20, 12,80 Гц, 1H), 3,84 (s, 3H), 5,49 (s, 1H), 5,92 (d, J = 1,20 Гц, 1H), 5,93 (d, J = 1,20 Гц, 1H), 6,79 (d, J = 8,00 Гц, 1H), 6,89 (d, J = 8,80 Гц, 1H), 7,08 (dd, J = 1,60, 8,00 Гц, 1H), 7,11 (d, J = 1,60 Гц, 1H), 7,74 (d, J = 2,40 Гц, 1H), 7,78 (dd, J = 2,40, 8,80 Гц, 1H).
C1-MS m/e = 386,2 (M+).
Пример 60.
N-(4-изо-пропилбензолсульфонил) -α- (2-изобутил-4-карбометоксифенокси)-3,4-метилендиоксифенилацетамид
Стадия A.
Получение N-(4-изопропилбензолсульфонил) -α- (2-изобутил-4- карбометоксифенокси)-3,4-метилендиоксифенилацетамида
К раствору 0,727 г (1,88 ммоль) продукта стадии B в примере 58, растворенного в 4 мл безводного THF, добавили 0,458 г (2,82 ммоль), 1,1'-карбонил-диимидазола, смесь перемешали магнитной мешалкой и нагревали в колбе с обратным холодильником в течение 2 часов. Затем реакционную смесь охладили до комнатной температуры и добавили 0,562 г (2,82 ммоль) 4-изо-пропилбензолсульфонамид и 0,42 мл (2,82 ммоль) 1,8-диазабицикло[5,4,0]ундец-7-ена. Реакционную смесь перемешали при комнатной температуре в течение еще 3-х часов, затем выпарили в вакууме. Остаток разделили между EtOAc и 1,0 N HCl и экстрагировали. Отделили органический слой, сушили (MgSO4), фильтровали и выпаривали, и остаток очистили тонкослойной колоночной хроматографией на силикагеле при элюировании CHCl3-MeOH-NH4OH (80:15:1). Выпаривание очищенных фракций и сушка в вакууме привели к получению 0,666 г (63%) названного соединения.
1H ЯМР (400 мГц, CD3OD, част. на миллион): δ 0,81 (d, J = 6,80 Гц, 3H), 0,84 (d, J = 6,80 Гц, 3H), 1,23 (d, J = 6,80 Гц, 3H), 1,24 (d, J = 6,80 Гц, 3H), 1,88-1,94 (m, 1H), 2,45 (dd, J = 7,00, 13,00 Гц 1H), 2,58 (dd, J = 7,00, 13,00 Гц, 1H), 2,95 (sept, J = 6,80 Гц, 1H), 3,84 (s, 3H), 5,46 (s, 1H), 5,95 (d, J = 1,20 Гц, 1H), 5,96 (d, J = 1,20 Гц, 1H), 6,59 (d, J = 8,60 Гц, 1H), 6,79 (d, J = 8,00 Гц, 1H), 6,98 (широк. s, 1H), 6,99 (dd, J = 1,60, 8,00 Гц, 1H), 7,30 (d, J = 8,40 Гц, 2H), 7,60 (dd, J = 2,00, 8,60 Гц, 1H), 7,70 (d, J = 2,00 Гц, 1H), 7,72 (d, J = 8,40 Гц, 2H).
Пример 61.
N-(4-изо-пропилбензолсульфонил) -α- (2-изобутил-4-карбоксифенокси)-3,4-метилендиоксифенилацетамид
Стадия A.
Получение N-(4-изо-пропилбензолсульфонил) -α- (2-изобутил-4- карбоксифенокси)-3,4-метилендиоксифенилацетамида
К раствору 0,294 г (0,52 ммоль) продукта примера 60, растворенного в 3,0 мл метанола, добавили 1,0 мл 5,0 N водного раствора гидроксида натрия. Реакционную смесь перемешали магнитной мешалкой при 60oC. Через 3 часа анализ TLC (CHCl3-MeOH-NH4OH 80:15:1) показал завершение гидролиза сложного эфира. Реакционную смесь охладили до комнатной температуры, путем добавления по каплям 1,0 N HCl установили pH, равный 5, затем разделили между EtOAc и водой. Органический слой отделили, промыли насыщенным водным NaCl, сушили (MgSO4), фильтровали и выпаривали. Остаток сушили в вакууме до получения 0,238 г (83%) названного соединения в виде аморфного порошка.
1H ЯМР (400 мГц, CD3OD, част. на миллион): δ 0,82 (d, J = 6,80 Гц, 3H), 0,85 (d, J = 6,80 Гц, 3H), 1,24 (d, J = 7,20 Гц, 3H), 1,25 (d, J = 7,20 Гц, 3H), 1,91 (sept, J = 6,80 Гц, 1H), 2,48 (dd, J = 7,20, 13,20 Гц, 1H), 2,56 (dd, J = 7,20, 13,20 Гц, 1H), 2,97 (sept, J = 7,20 Гц, 1H), 5,51 (s, 1H), 5,97 (s, 1H), 6,50 (d, J = 8,40 Гц, 1H), 6,81 (d, J = 8,00 Гц, 1H), 6,91 (d, J = 1,60 Гц, 1H), 6,95 (dd, J = 1,60, 8,00 Гц, 1H), 7,36 (d, J = 8,40, 2H), 7,62 (dd, J = 2,20, 8,40 Гц, 1H), 7,72 (d, J = 2,20 Гц, 1H), 7,79 (d, J = 8,40, 2H).
FAB-MS m/e = 554 (M+1).
Пример 62.
N-(4-изо-пропилбензолсульфонил) -α- (2-н-пропил-4- метоксикарбонилфенокси) -α- метил-3,4-метилендиоксифенилацетамид
Стадия A.
Получение N-(4-изо-пропилбензолсульфонил) -α- (2-н-пропил- 4-метоксикарбонилфенокси) -α- метил-3,4-метилендиоксифенилацетамид
К раствору 0,516 г (0,93 ммоль) продукта примера 57,
растворенного в 1,0 мл безводного THF, добавили 2,80 мл (2,80 ммоль) 1,0 М раствора литий-бис(триметилсилиламида) в THF при -78oC в атмосфере азота. Реакционную смесь перемешивали магнитной мешалкой при -78oC в течение одного часа, затем через шприц добавили 174 мкл (2,80 ммоль) иодометана. Реакционная смесь нагревалась до комнатной температуры и ее перемешивали еще 14 часов. Затем реакционную смесь резко охладили избытком 10% водного NaHSO4 и разделили между EtOAc и водой. Органический слой промыли насыщенным водным NaCl, сушили (MgSO4), фильтровали и выпаривали. Остаток очистили тонкослойной колоночной хроматографией на силикагеле при элюировании CHCl3-MeOH-NH4OH (90 : 10 : 1).
Выпаривание очищенных фракций и сушка в вакууме дали 0,293 г (55%) названного соединения в виде аморфного твердого вещества.
1H ЯМР (400 мГц, CD3OD, част. на миллион): δ 0,99 (d, J = 7,20 Гц, 3H), 1,31 (d, J = 6,80 Гц, 6H), 1,64 - 1,72 (m, 2H), 1,66 (s, 3H), 2,64 - 2,73 (m, 1H), 2,81 - 2,88 (m, 1H), 3,02 (sept, J = 6,80 Гц, 1H), 3,85 (s, 3H), 5,96 (s, 2H), 6,36 (d, J = 8,40 Гц, 1H), 6,77 (d, J = 8,40 Гц, 2H), 6,99 (m, 1H), 7,05 (широк. s, 1H), 7,37 (d, J = 7,60 Гц, 1H), 7,43 (dd, J = 2,40, 8,60 Гц, 1H), 7,76 (d, J = 8,40 Гц, 2H), 7,81 (широк. s, 1H).
FAB-MS m/e = 568 (M + 1)
Пример 63.
Двукалиевая соль N-(4-изо-пропилбензолсульфонил) -α- (2-н-пропил-4-карбоксифенокси) -α- метил-3,4-метилендиоксифенилацетамида
Стадия A:
Получение двукалиевой соли N-(4-изо-пропилбензолсульфонил) -α- (2-н-пропил-4-карбоксифенокси) -α- метил-3,4-метилендиоксифенилацетамида
К раствору 0,293 г (0,52 ммоль) продукта примера 62, растворенного в 2,0 мл метанола, добавили раствор 0,143 г (2,54 ммоль) гидроксида калия, растворенного в 1,0 мл воды. Реакционную смесь перемешивали магнитной мешалкой при 60oC в течение 4 часов до тех пор, пока анализ ТСХ (CDCl3-MeOH-NH4OH 80 : 15 : 1) показал, что исходный материал полностью гидролизовался. Затем реакционную смесь охладили до комнатной температуры, разбавили 5,0 мл воды и отфильтровали через 0,45 микронный фильтр. Затем фильтрат очистили на жидкостном хроматографе Waters Millipore Delta Prep (Вотез Милипор Дельта Преп), снабженном двумя последовательно соединенными колонками 21,2 мм x 25 см ODS DU Pont Zorbax® (Ду Понт Зорбакс), для проведения высокоэффективной жидкостной хроматографии с обращенной фазой. Использовали две емкости для растворителя: систему растворителя A (вода - ацетонитрил 95 - 5) и систему растворителя B (вода - ацетонитрил 5 - 95) и поток, вытекающий из колонны, одновременно контролировали при 210 и 280 нм детектором видимых ультрафиолетовых лучей модели Waters 490.
Реакционную смесь инжектировали на колонку и обессоливали путем элюирования (50 мл/мин) приблизительно 1 л системы растворителя A. Затем начали градиентное элюирование, начальные условия которого были: 100% системы растворителя A - 0% системы растворителя B и через 30 минут достигли: 50% системы растворителя A - 50% системы растворителя B, и фракции собирали с помощью сборника фракций ISCO Foxy 200. Очищенные фракции соединили в круглодонных колбах, заморозили при -78oC в бане со смесью сухого льда с ацетоном и подвергли лиофилизации. Объединение очищенного продукта дало 0,273 г (84%) названного соединения в виде белого лиофилизованного порошка.
1H ЯМР (400 мГц, CD3OD, част. на миллион): δ 0,96 (t, J = 7,20 Гц, 3H), 1,25 (d, J = 7,20 Гц, 3H), 1,26 (d, J = 7,20 Гц, 3H), 1,64 - 1,71 (m, 2H), 1,67 (s, 3H), 2,58 - 2,65 (m, 1H), 2,74 - 2,82 (m, 1H), 2,96 (sept, J = 7,20 Гц, 1H), 5,91 (d, J = 1,20 Гц, 1H), 5,92 (d, J = 1,20 Гц, 1H), 6,52 (d, J = 8,40 Гц, 1H), 6,72 (d, J = 8,00 Гц, 1H), 7,12 (dd, J = 1,80, 8,00 Гц, 1H), 7,17 (d, J = 2,00 Гц, 1H), 7,28 (d, J = 8,00 Гц, 2H), 7,50 (dd, J = 2,20, 8,40 Гц, 1H), 7,72 (d, J = 8,80 Гц, 2H), 7,74 (d, J = 2,80 Гц, 1H).
FAB-MS m/e = 591,6 (M + K+)
Пример 64.
N-(4-изо-пропилбензолсульфонил) -α- (2-н-пропил-4- карбоксамидофенокси)-3,4-метилендиоксифенилацетамид
Стадия A.
Получение N-(4-изо-пропилбензолсульфонил) -α- (2-н-пропил-4- карбоксамидофенокси)-3,4-метилендиоксифенилацетамида
К раствору 0,162 г (0,30 ммоль) N-(4-изо-пропилбензолсульфонил) -α- (4-карбокси-2-н-пропилфенокси)-3,4-метилендиоксифенилацетамида (свободная кислотная форма продукта примера 58), растворенного в 4 мл безводного THF, добавили 0,073 мл (0,45 ммоль) 1,1'-карбонилдиимидазола, полученную смесь перемешивали магнитной мешалкой и нагревали в колбе с обратным холодильником в течение 50 минут. Реакционную смесь охладили до комнатной температуры и затем при 0oC добавили к избытку THF, который предварительно насытили безводным газообразным аммиаком. Реакционную смесь герметизировали и затем перемешивали при комнатной температуре в течение 14 часов. Затем реакционную смесь влили в воду (70 мл) и экстрагировали EtOAc (150 мл). Органический слой отделили, промыли насыщенным водным NaCl, сушили (MgSO4), фильтровали и выпаривали в вакууме до получения названного соединения в виде аморфного твердого вещества
1H ЯМР (400 мГц, CD3OD, част. на миллион): δ 0,88 (t, J = 7,60 Гц, 3H), 1,21 (d, J = 6,80 Гц, 1H), 1,55 - 1,66 (m, 2H), 2,54 - 2,62 (m, 1H), 2,70 - 2,72 (m, 1H), 2,89 (sept, J = 6,80 Гц, 1H), 5,36 (s, 1H), 5,93 (d, J = 1,20 Гц, 1H), 5,94 (d, J = 1,20 Гц, 1H), 6,75 (d, J = 8,40 Гц, 1H), 6,78 (d, J = 8,80 Гц, 1H), 7,02 - 7,04 (m, 2H), 7,06 (широк. s, 2H), 7,20 (d, J = 8,40 Гц, 2H), 7,55 (dd, J = 2,20, 8,60 Гц, 1H), 7,62 - 7,66 (m, 2H), 7,71 (s, 1H):
FAB-MS m/e = 539 (M + 1).
Пример 65.
N-(4-изо-пропилбензолсульфонил) -α- (2-н-пропил-4- гидроксиметилфенокси)-3,4-метилендиоксифенилацетамид
Стадия A:
Получение метил -α- (4-гидроксиметил-(2-н-пропилфенокси)- 3,4-метилендиоксифенилацетата
К раствору 3,84 г (23,13 ммоль) 4-гидрокси-3-н-пропилбензолового спирта, растворенного в 70 мл безводного DMF, добавили 9,04 г (27,7 ммоль) карбоната цезия и реакционную смесь перемешивали магнитной мешалкой при комнатной температуре в течение 15 минут. Добавили метил -α- бром-3,4-метилендиоксифенилацетат (7,58 г, 27,7 ммоль) и затем реакционную смесь перемешивали при комнатной температуре под атмосферой азота в течение еще 14 часов. Затем реакционную смесь разделили между 5% водной лимонной кислотой (700 мл) и EtOAc (100 мл) и экстрагировали. Органический слой отделили, промыли насыщенным водным NaCl, сушили (MgSO4), фильтровали и выпаривали. Остаток очистили флэш колоночной хроматографией на силикагеле при элюировании 40% EtO-гексаном. Очищенные фракции соединили, выпаривали и сушили в вакууме до получения 6,74 г (81%) названного соединения в виде желтого масла.
1H ЯМР (200 мГц, CDCl3, част. на миллион): δ 0,97 (t, J = 7,60 Гц, 3H), 2,55 - 2,75 (m, 2H), 2,71 (t, J = 7,20 Гц, 2H), 3,71 (s, 3H), 4,59 (s, 2H), 5,55 (s, 1H), 5,97 (s, 2H), 6,69 (d, J = 8,20 Гц, 1H), 6,82 (d, J=7,80 Гц, 1H), 7,02-7,28 (m, 4H).
FAB-MS m/e =359 (M+1).
Стадия B:
Получение метил -α- (4-трет-бутилдиметилсилилоксиметил- 2-н-пропилфенокси)-3,4-метилендиоксифенилацетата
К раствору 2,50 г (6,98 ммоль) продукта стадии А, растворенного в 20 мл дихлорметана, добавили 1,95 мл (14,0 ммоль) триэтиламина, 1,26 г (8,38 ммоль) трет-бутилдиметилхлорсилана, 85 мг (0,1 экв) 4-диметиламинопиридина и реакционную смесь перемешивали при комнатной температуре в течение 30 минут в атмосфере азота. Реакционную смесь разбавили 100 мл EtOAc, промыли водой, 1,0 N HCl, насыщенным водным NaHCO3, насыщенным NaCl, сушили (MgSO4), фильтровали и выпаривали в вакууме до получения 3,20 г (97%) названного соединения EI-MS m/e = 472 (M+).
Стадия C:
Получение α- (4-трет-бутилметилсилилоксиметил-2-н- пропилфенокси)-3,4-метилдиоксифенилуксусной кислоты
К раствору 3,20 г (6,78 ммоль) продукта стадии B, растворенного в 10 мл метанола и 3 мл дихлорметана, добавили 1,42 (7,12 ммоль) 5,0 N водного раствора гидроксида натрия и реакционную смесь перемешивали магнитной мешалкой при комнатной температуре. Через 4 часа анализ ТСХ (CHCl3-MeOH-NH4OH 80:15: 1) показал, что гидролиз прошел полностью и pH реакционной смеси с помощью 1,0 N HCl установили равным 4. Затем реакционную смесь полностью выпарили и сушили в вакууме до получения сырого продукта, который непосредственно использовали на следующей стадии.
FAB-MS m/e = 481 (M + Na+).
Стадия D.
Получение N-(4-изо-пропилбензолсульфонил) -α- (4-трет- бутилдиметилсилилоксиметил-2-н-пропилфенокси)-3,4-метилендиоксифенилацетамида
К раствору 3,30 г (7,21 ммоль) сырого продукта со стадии C, растворенного в 40 мл безводного THF, добавили 1,75 г (10,8 ммоль) 1,1'-карбонилимидазола и реакционную смесь перемешивали магнитной мешалкой в течение 10 минут. Затем реакционную смесь охладили до комнатной температуры, добавили 2,15 г (10,8 ммоль) 4-изо-пропилбензолсульфонамида и 1,61 мл (10,8 ммоль) 1,8-диазабицикло [5.4.0] -ундец-7-ена и реакционную смесь перемешивали в течение еще 30 минут при комнатной температуре. Затем смесь разбавили EtOAc (80 мл), промыли 10% водным раствором лимонной кислоты, насыщенным водным NaCl, сушили (MgSO4), фильтровали и выпаривали. Остаток частично очистили тонкослойной хроматографией на силикагеле при элюировании CHCl-MeOH-NH4OH (92:8:0,5). Полуочищенный материал соединили и еще раз очистили флэш колоночной хроматографией на силикагеле во второй колонке при элюировании первоначально 35% EtOAc-гексаном, затем 50% EtOAc-гексаном и в конце 70% EtOAc-гексаном. Объединение очищенных фракций и выпаривание дало 3,20 г (69%) названного соединения в виде желтого масла.
FAB-MS m/e = 678 (M+K+)
Стадия E.
Получение N-(4-изо-пропилбензолсульфонил) -α- (4-гидроксиметил- 2-н-пропилфенокси)-3,4-метилендиоксифенилацетамида
К раствору 3,2 г (5,01 ммоль) продукта стадии D, растворенного в 50 мл безводного THF, добавили 5,06 мл (5,06 ммоль) 1,0 M раствора тетрабутиламмонийфторида в THF и реакционную смесь перемешивали при комнатной температуре в атмосфере азота. Через 2,5 часа добавили еще 1,0 мл тетрабутиламмонийфторида в THF и реакционную смесь перемешивали в течение еще 14 часов. Затем реакционную смесь концентрировали в вакууме, подвергали флэш колоночной хроматографии на силикагеле при элюировании 60% EtOAc-гексаном. Объединение очищенных фракций и сушка в вакууме дали 0,691 г (26%) названного соединения в виде аморфного порошка.
1H ЯМР (400 мГц, CD3OD, част.на миллион): δ 0,87 (t, J = 7,60 Гц, 3H), 1,26 (d, J = 6,80 Гц, 3H), 1,27 (d, J = 6,80 Гц, 3H), 1,51 - 1,63 (m, 2H), 2,54 - 2,68 (m, 2H), 2,98 (sept, J = 6,80 Гц, 1H), 4,46 (s, 2H), 5,37 (s, 1H), 5,95 (s, 1H), 6,51 (d, J = 8,40 Гц, 1H), 6,77 (d, J = 8,00 Гц, 1H), 6,88 - 6,95 (m, 3H), 7,10 (d, J = 2,00 Гц, 1H), 7,36 (d, J = 8,40 Гц, 2H), 7,77 (d, J = 8,40 Гц, 2H).
FAB-MS m/e = 548 (M + Na+)
Пример 66.
N-(4-изо-пропилбензолсульфонил)- -α- (4-формил-2-н-пропилфенокси)-3,4-метилендиоксифенилацетамид
Стадия A:
Получение N-(4-изо-пропилбензолсульфонил) -α- (4-формил-2-н-пропилфенокси)-3,4-метилендиоксифенилацетамида
К раствору 0,573 г (1,09 ммоль) продукта примера 65, растворенного в 5,0 мл дихлорметана, добавили 2,86 г (32,9 ммоль) диоксида марганца и 1,15 г тонкоизмельченных в порошок 3A молекулярных сит и реакционную смесь перемешивали магнитной мешалкой при комнатной температуре в течение 14 часов. Затем реакционную смесь фильтровали через слой целита и MgSO4 и фильтрат выпаривали в вакууме. Остаток растворили в дихлорметане, подвергали флэш колоночной хроматографии на силикагеле и затем элюировали 3% MeOH-CHCl2. Выпаривание очищенных фракцией и сушка в вакууме дали 0,149 г (26%) названного соединения.
1H ЯМР (400 мГц, CD3OD, част.на миллион): δ 0,89 (t, J = 7,60 Гц, 3H), 1,24 (d, J = 7,20 Гц, 3H), 1,25 (d, J = 7,20 Гц, 3H), 1,57-1,68 (m, 2H), 2,63 - 2,74 (m, 2H), 2,96 (sept, J = 7,20 Гц, 1H), 5,56 (s, 1H), 5,97 (s, 2H), 6,70 (d, J = 8,40 Гц, 1H), 6,80 (d, J = 8,00 Гц, 1H), 6,91 (d, J = 1,60 Гц, 1H), 6,96 (dd, J = 1,60, 8,00 Гц, 1H), 7,34 (d, J = 8,40 Гц, 2H), 7,53 (dd, J = 2,0, 8,40 Гц, 1H), 7,66 (d, J = 2,00 Гц, 1H), 7,76 (d, J = 8,40 Гц, 2H), 9,77 (s, 1H).
FAB-MS m/e = 546 (M+Na+).
Пример 67.
α- (4-ацетил-2-н-пропилфенокси)-3,4-метилендиоксифенилуксусная кислота
Стадия A:
Получение 4-гидрокси-2-н-пропилацетофенона
Аппарат Парра для гидрирования загрузили раствором 2,00 г (11,36 ммоль) 3-аллил-4-гидроксиацетофенона, растворенного в 10 мл этанола, и 200 мг 10% палладия на углеродном катализаторе.
Колбу установили в аппарат Парра и встряхивали в атмосфере водорода 46 фунтов/дюйм2 (3,2343 кг/см2) в течение 15 минут. В конце этого периода анализ ТСХ (15% EtOAc-гексан) показал, что исходный материал полностью израсходован, реакционную смесь фильтровали и выпаривали. Остаток очистили флэш колоночной хроматографией при элюировании 25% EtOAc-гексаном. Выпаривание очищенных фракций и сушка в вакууме дали 1,83 г (91%) названного соединения.
1H ЯМР (200 мГц, CDCl3, част.на миллион): δ 0,98 (t, J = 7,40 Гц, 3H), 1,56 - 1,78 (m, 2H), 2,57 (s, 3H), 2,63 (t, J = 7,20 Гц, 2H), 6,08 (шир., s, 1H), 6,84 (d, J = 8,20 Гц, 1H), 7,74 (dd, J = 2,20, 8,20 Гц, 1H), 7,79 (d, J = 2,20 Гц, 1H).
FAB-MS m/e = 178 (M+).
Стадия B:
Получение метил -α- (4-ацетил-2-н-пропилфенокси)-3,4-метилендиоксифенилацетата
К раствору 0,250 г (1,40 ммоль) продукта стадии A, растворенного в 3 мл DMF, добавили 0,504 г (1,54 ммоль) карбоната цезия и реакционную смесь перемешивали магнитной мешалкой при комнатной температуре в атмосфере азота в течение 15 минут. Затем добавили метил- α -бром-3,4-метилендиоксифенилацетат (0,422 г, 1,54 ммоль) и полученную смесь перемешивали при комнатной температуре в течение еще 24-х часов. Затем реакционную смесь разделили между 10% водным раствором лимонной кислоты и ETOAc. Органический слой промыли насыщенным водным NaCl, сушили (MgSO4), фильтровали и выпаривали в вакууме до получения названного соединения.
1H ЯМР (300 мГц, CDCl3, част. на миллион): δ 0,96 (t, J = 7,50 Гц, 3H), 1,62 - 1,74 (m, 2H), 2,52 (s, 3H), 2,68-2,75 (m, 2H), 3,71 (s, 3H), 5,61 (s, 1H), 5,98 (s, 2H), 6,81 (d, J = 8,20 Гц, 1H), 7,02 (dd, J = 1,80, 8,20 Гц, 1H), 7,04 (d, J = 1,80 Гц, 1H), 7,73 (dd, J = 2,20, 8,60 Гц, 1H), 7,79 (d, J=2,20 Гц, 1H).
FAB-MS m/e = 371 (M+1).
Стадия C:
Получение α- (4-ацетил-2-н-пропилфенокси)-3,4- метилендиоксифенилуксусной кислоты
К раствору 0,556 г (1,50 ммоль) продукта стадии B, растворенного в 4,0 мл метанола, добавили 0,45 мл (2,25 ммоль) 5,0 N водного раствора гидроксида натрия. Реакционную смесь перемешивали при комнатной температуре и контролировали ТСХ (CHCl3-MeOH-NH4OH 80: 15: 1). Через 4 часа реакцию посчитали завершенной и 6,0 N HCl установили pH реакционной смеси, равным 7. Затем смесь выпаривали в вакууме и остаток очистили флэш колоночной хроматографией на силикагеле при элюировании CHCl3-MeOH-NH4OH (80:15:1). Выпаривание очищенных фракций и сушка в вакууме дали 0,416 г (78%) названного соединение.
1H ЯМР (400 мГц, CD3OD, част. на миллион): δ 0,94 (t, J = 7,60 Гц, 3H), 1,62-1,70 (m, 2H), 2,53 (s, 3H), 2,61-2,69 (m, 1H), 2,80-2,88 (m, 1H), 5,39 (s, 1H), 5,93 (d, J = 1,20 Гц, 1H), 5,94 (d, J = 1,20 Гц, 1H), 6,79 (d, J = 8,00 Гц, 1H), 6,91 (d, J = 8,80 Гц, 1H), 7,10 (dd, J = 1,60, 8,00 Гц, 1H), 7,15 (d, J = 1,60 Гц, 1H), 7,78 (d, J = 2,40 Гц, 1H), 7,81 (dd, J = 2,40, 8,80 Гц, 1H).
Пример 68.
N-(4-изо-пропилбензолсульфонил) -α- (4-ацетил-2-н-пропилфенокси)-3,4-метилендиоксифенилацетамид.
Стадия A:
Получение N-(4-изо-пропилбензолсульфонил) -α- (4-ацетил-2-н-пропилфенокси)-3,4-метилендиоксифенилацетамида
К раствору 0,181 г (0,51 ммоль) продукта примера 67, растворенного в 25 мл безводного DMF, добавили 0,248 г (1,53 ммоль) 1,1'-карбонилдиимидазола и реакционную смесь перемешали магнитной мешалкой и нагрели при 80oC под атмосферой азота в масляной бане. Через 20 минут реакционную смесь охладили до комнатной температуры и добавили 0,152 г (0,77 ммоль) 4-изо-пропилбензолсульфонамида и 381 мкл (2,55 ммоль). Реакционную смесь нагрели при 80oC в течение еще 10 минут, затем опять охладили до комнатной температуры и разделили между EtOAc и 10% водным раствором лимонной кислоты. Органический слой отделили, промыли насыщенным водным NaHCO3, насыщенным водным NaCl, сушили (MgSO4), фильтровали и выпаривали. Остаток очистили тонкослойной колоночной хроматографией на силикагеле при элюировании CHCl3-MeOH-NH4OH (80: 15: 1), выпаривание очищенных фракций и сушка в вакууме привели к получению 0,128 г (47%) названного соединения в виде аморфного твердого вещества.
1H ЯМР (400 мГц, CD3OD, част. на миллион): δ 0,88 (t, J = 7,60 Гц, 3H), 1,21 (d, J = 6,80 Гц, 3H), 1,22 (d, J = 6,80 Гц, 3H), 1,55-1,65 (m, 2H), 2,51 (s, 3H), 2,54-2,64 (m, 1H), 2,67-2,75 (m, 1H), 2,92 (sept, J = 6,80 Гц, 1H), 5,43 (s, 1H), 5,94 (s, 2H), 6,75 (d, J = 8,80 Гц, 1H), 6,77 (d, J = 8,40 Гц, 1H), 7,01-7,03 (m, 2H), 7,23 (d, J = 8,40 Гц, 2H), 7,66 (dd, J = 2,40, 8,80 Гц, 1H), 7,67 (d, J = 8,40 Гц, 2H), 7,73 (d, J = 2,40 Гц, 1H).
FAB-MS m/e = 538 (M+I).
Пример 69.
α- (2-н-пропилфенокси)-3,4-метилендиоксифенилуксусная кислота
FAB-MS для C18H18O5: m/e = 337 (M + Na+)
Пример 70.
N-(4-изо-пропилбензолсульфонил) -α- (2-н-пропилфенокси)-3,4-метилендиоксифенилацетамид
FAB-MS для C27H29NSO6: m/e = 534 (M+K+)
Пример 71.
α- (3-метоксифенокси)-3,4-метилендиоксифенилуксусная кислота.
EI-MS для C16H14O6: m/e = 302 (M+).
Пример 72.
α- (2-(2-гидроксиэтил)фенокси)-3,4-метилендиоксифенилуксусная кислота.
FAB-MS для C17H16O6: m/e = 317 (M+I).
Пример 73.
α- (2-(2-карбометоксиэтил)фенокси)-3,4-метилендиоксифенилуксусная кислота.
CI-MS для C19H18O7: m/e = 359 (M+I).
Пример 74.
α- (4-гидроксиметил-2-н-пропилфенокси)-3,4- метилендиоксифенилуксусная кислота.
CI-MS для C19H20O6: m/e = 326 (M+-H2O).
Пример 75.
α- (4-(2-гидроксиэтил)-2-н-пропилфенокси)-3,4- метилендиоксифенилуксусная кислота.
CI-MS для C20H22O6: m/e = 359 (M+I).
Пример 76.
N-(4-изо-пропилбензолсульфонил) -α- (2-(2-карбометоксиэтил)фенокси)-3,4-метилендиоксифенилацетамид.
ESI-MS для C28H29NSO8: m/e = 540 (M+I)
Пример 77.
N-(4-изо-пропилбензолсульфонил) -α- (2-(2-карбоксиэтил)фенокси)-3,4-метилендиоксифенилацетамид.
CI-MS для C27H27NSO8: m/e = 526 (M+I)
Пример 78.
α- (2-(2-карбоксиэтил)фенокси)-3,4-метилендиоксифенилуксусная кислота.
CI-MS для C18H16O7: m/e = 345 (M+I)
Пример 79.
N-(4-изо-пропилбензолсульфонил)-2-(4-карбометокси-2-н- пропилфенокси)-2-(5-метокси-3,4-метилендиоксифенил)ацетамид.
Стадия A:
Этил-2-(4-карбометокси-2-н-пропилфенокси)-2-(5-метокси-3,4- метилендиоксифенил)ацетат
К смеси метил-4-гидрокси-3-н-пропилбензоата (3,0 г, 15,46 ммоль) и Cs2CO3 (5,1 г, 16 ммоль) в сухом диметилформамиде (50 мл) добавили этил-2-бром-2-(5-метокси-3,4-метилендиокси)фенилацетата (4,3 г, 15,56 ммолей) и полученную смесь перемешивали при комнатной температуре в течение 6-ти часов. В конце этого периода реакционную смесь разбавили смесью воды со льдом (300 мл) и экстрагировали этилацетатом (3х60 мл). Объединенную органическую фазу промыли водой и рассолом и затем сушили над безводным MgSO4, фильтровали и для получения сырого продукта удалили растворитель. Очистка сырого продукта флэш хроматографией на силикагеле при использовании этилацетата-гексана (1: 9) дала названный продукт в виде масла (5,1 г).
1H ЯМР (200 мГц, CDCl3, част. на миллион): δ 7,82 (m, 2H), 6,61 (d, 1H, J = 1,5 Гц) 5,93 (s, 1H), 5,54 (s, 1H), 4,15 (m, 2H), 3,84 (s, 3H), 3,83 (s, 3H), 2,68 (m, 2H), 1,69 (m, 2H), 1,20 (t, 3H, J = 7,4 Гц), 0,90 (t, 3H, J = 7,4 Гц).
Стадия B:
2-(4-карбометокси-2-н-пропилфенокси)-2-(5-метокси-3,4- метилендиоксифенил)уксусная кислота
К раствору продукта стадии A (4,3 г, 12 ммоль) в метаноле (25 мл) добавили водный 2N NaOH (10 мл) и реакционную смесь перемешивали при комнатной температуре. Быстрое течение моно-деэтерификации контролировали путем анализа ТСХ с использованием CHCl3-MeOH-NH4OH (80:15:1). Через 15 минут реакционную смесь охладили до 0oC и нейтрализовали водной 2N HCl. Осажденный маслянистый продукт экстрагировали в метиленхлорид (3х40 мл) и объединенную органическую фазу промыли водой, рассолом и затем сушили над MgSO4. Удаление растворителя в вакууме дало сырой продукт, который затем очистили тонкослойной хроматографией на силикагеле при использовании CHCl3-MeOH-NH4OH (80: 10:1) для получения желательного продукта в виде аммониевой соли. Соль обработали водной 1N HCl (20 мл) для обеспечения названного соединения в виде белого твердого вещества (3,4 г).
1H ЯМР (200 мГц, CD3OD, част. на миллион): δ 7,78 (m, 2H), 6,77 (m, 3H), 6,61 (d, 1H, J = 1,5 Гц), 5,93 (s, 2H), 5,54 (s, 1H), 3,84 (s, 3H), 3,83 (s, 3H), 2,68 (m, 2H), 1,69 (m, 2H), 0,90 (t, 3H, J = 7,4 Гц).
Стадия C:
N-(4-изопропилбензолсульфонил)-2-(4-карбометокси-2-н- пропилфенокси)-2-(5-метокси-3,4-метилендиоксифенил)ацетамид
К продукту стадии B (0,12 г, 0,30 ммоль) в сухом THF (1,5 мл) добавили 1,1'-карбонилдиимидазол (0,1 г, 0,61 ммоль) и реакционную смесь перемешивали при 50oC в течение 3-х часов. К этому раствору добавили раствор 4-изо-пропилбензолсульфонамида (0,17 г, 0,9 ммоль) и DBU (0,14 мл, 0,94 ммоль) в сухом THF (1,5 мл) и реакцию продолжили при 50oC в течение 4 часов.
Реакционную смесь разбавили смесью воды со льдом и подкислили водной 1N HCl. Осажденный материал поместили в EtOAc и органическую фазу промыли водой, рассолом и затем сушили над MgSO4, фильтровали и удалили растворитель. Продукт очистили тонкослойной хроматографией на силикагеле при использовании в качестве элюента CHCl3-MeOH-NH4OH (80:10:1) для получения названного продукта в виде аммониевой соли. Подкисление аммониевой соли привело к получению названного продукта в виде белого твердого вещества (0,14 г).
1H ЯМР (400 мГц, CD3OD, част. на миллион): δ 7,78 (d, 2H, J = 8,4 Гц), 7,76 (d, 1H, J = 2,3 Гц), 7,62 (dd, 1H, J = 8,6, 2,2 Гц), 7,37 (d, 2H, J = 8,4 Гц), 6,70 (d, 1H, J = 1,4 Гц), 6,61 (d, 1H, J = 1,5 Гц), 5,97 (s, 2H), 5,49 (s, 1H), 3,84 (s, 3H), 3,83 (s, 3H), 2,98 (sept, 1H, J = 6,9 Гц), 2,65 (m, 2H), 1,59 (m, 2H), 1,25 (dd, 6H, J = 7,0, 2,5 Гц), 0,90 (t, 3H, J = 7,4 Гц).
C30H32NO9N:
Вычислено: C 59,50 H 5,33 N 2,31
Найдено: C 59,60 H 5,34 N 2,59
Пример 80.
N-(4-изо-пропилбензолсульфонил)-2-(4-карбокси-2-пропилфенокси)- 2-(5-метокси-3,4-метилендиоксифенил)ацетамид
К продукту примера 79 (0,6 г, 1,02 ммоль) в MeOH (15 мл) добавили водный 2N NaOH (5 мл) и реакционную смесь перемешали при 60oC в течение 3 часов. Когда реакция завершилась, MeOH удалили в вакууме и водную фазу подкислили 2N HCl. Осажденный продукт экстрагировали в метиленхлорид (3х50 мл) и объединенную органическую фазу промыли рассолом, затем сушили над MgSO4, фильтровали и растворитель удалили. Осадок растерли в порошок с простым эфиром, что обеспечило получение названного продукта в виде белого твердого вещества (0,45 г).
1H ЯМР (400 мГц, DMSO-d6, част. на миллион): δ 12,67 (широк., 1H), 12,63 (широк. , 1H), 7,70 (d, 2H J = 8,4 Гц), 7,66 (d, 1H, J = 2,1 Гц), 7,58 (dd, 1H, J = 8,5, 2,2 Гц), 7,40 (d, 2H J = 8,4 Гц), 6,78 (d, 1H, J = 1,2 Гц), 6,66 (d, 1H, J = 1,2 Гц), 6,51 (d, 1H, J = 8,5 Гц), 6,02 (d, 2H J = 3,1 Гц), 5,69 (s, 1H), 3,79 (s, 3H), 2,93 (sept, 1H, J = 6,9 Гц), 2,56 (m, 2H), 1,53 (m, 2H), 1,17 (d, 6H, J = 6,9 Гц), 0,83 (t, 3H, J = 7,4 Гц)
масс-спектр FAB: m/e = 570 (M + 1).
C29H31NO9S:
Вычислено: C 61,15 H 5,49 N 2,46
Найдено: C 60,86 H 5,64 N 2,71
Пример 81.
N-(4-изо-пропилбензолсульфонил)-2-(4-(N-(4-изо- пропилбензолсульфонил)карбоксамидо)-2-пропилфенокси)-2- (5-метокси-3,4-метилендиоксифенил)ацетамид.
Названное соединение получили из N-(4-изо-пропилбензолсульфонил)- 2-(4-карбокси-2-пропилфенокси)-2-5-метокси-3,4- метилендиоксифенилацетамида (пример 80) с использованием методики, подобной той, которая описана в стадии C примера 79.
Масс-спектр FAB: m/e 751 (M+I)
C38H42N2O10S2 • 0,5H2O:
Вычислено: C 60,06, H 5,70 N 3,69
Найдено: C 60,15, H 5,73 N 3,58.
Пример 82.
N-(4-изо-пропилбензолсульфонил)-2-(4-карбоксамидо)-2- пропилфенокси)-2-(5-метокси-3,4-метилендиоксифенил)ацетамид
К N-(4-изо-пропилбензолсульфонил))-2-(4-карбокси-2-пропилфенокси)- 2-(5-метокси-3,4-метилендиоксифенил)ацетамиду (пример 80) (0,12 г, 0,21 ммоль) в сухом THF (1,5 мл) добавили 1,1'-карбонилдиимидазол (0,1 г, 0,61 ммоль) и смесь перемешали при 50oC в течение 2 часов. Реакционную смесь перемешали при комнатной температуре в течение 1 часа и затем подкислили. Выделенный сырой продукт очистили флэш колоночной хроматографией на силикагеле при использовании CHCl3-MeOH-NH4OH (40:10:1) для получения продукта в виде аммониевой соли. Подкисление дало желательный названный продукт в виде белого твердого вещества (0,06 г).
1H ЯМР (300 мГц, DMSO-d6, част. на миллион): δ 7,76 (широк., 1H), 7,68 (d, 2H, J = 8,4 Гц), 7,64 (d, 1H, J = 2,1 Гц), 7,55 (dd, 1H, J = 8,6, 2,3 Гц), 7,40 (d, 2H J = 8,4 Гц), 7,16 (широк., 1H), 6,76 (s, 1H), 6,65 (d, 1H, J = 1,2 Гц), 6,53 (d, 1H, J = 8,7 Гц), 6,01 (d, 2H, J = 2,9 Гц), 5,67 (s, 1H), 3,789 (s, 3H), 2,93 (sept, 1H, J = 6,8 Гц), 2,55 (m, 2H), 1,54 (m, 2H), 1,17 (d, 6H, J = 6,9 Гц), 0,84 (t, 3H, J = 7,3 Гц).
Масс-спектр FAB: m/e 569 (M+I).
Пример 83.
N-(4-изо-пропилбензолсульфонил)-2-(4-(N-метил)карбоксамидо- 2-пропилфенокси)-2-(5-метокси-3,4-метилендиоксифенил)ацетамид
Названное соединение получили с использованием методик, подобных тем, которые описаны в пример 82.
1H ЯМР (300 мГц, DMSO-d6, част. на миллион): δ 8,21 (q, 1H, 4,7 Гц), 7,68 (d, 2H, J = 8,4 Гц), 7,59 (d, 1H, J = 1,9 Гц), 7,49 (dd, 1H, J = 8,6, 2,1 Гц), 7,40 (d, 2H, J = 8,4 Гц), 6,77 (s, 1H), 6,65 (s, 1H), 6,53 (d, 1H, J = 8,7 Гц), 6,01 (d, 2H, J = 2,9 Гц), 5,67 (s, 1H), 3,78 (s, 3H), 2,93 (sept, 1H, J = 6,8 Гц), 2,73 (d, 3H, J = 4,4 Гц), 2,56 (m, 2H), 1,54 (m, 2H), 1,16 (d, 6H, J = 6,9 Гц), 0,84 (t, 3H, J = 7,3 Гц).
C30H34N2O8S:
Вычислено: C 61,84, H 5,88 N 4,81
Найдено: C 61,84 H 6,03 N 4,59
Пример 84.
N-(4-изо-пропилбензолсульфонил)-2-(4-(N-2-гидроксиэтилкарбоксамидо)- 2-пропилфенокси)-2-(5-метокси-3,4-метилендиоксифенил)ацетамид
Названное соединение получили с использованием методик, подобных тем, которые описаны в примере 82.
1H ЯМР (400 мГц, CD3OD, част. на миллион): δ 7,77 (d, 2H, J = 8,4 Гц), 7,64 (d, 1H, J = 2,3 Гц), 7,51 (dd, 1H, J = 8,5, 2,4 Гц), 7,36 (d, 2H, J = 8,5 Гц), 6,68 (d, 1H, J = 1,4 Гц), 6,60 (m, 2H), 5,96 (s, 2H), 5,48 (s, 1H), 3,82 (s, 3H), 3,68 (t, 2H, J = 5,9 Гц), 3,46 (t, 2H, J = 5,9 Гц), 2,97 (sept, 1H, J = 6,9 Гц), 2,66 (m, 2H), 1,62 (m, 2H), 1,25 (d, 6H, J = 6,9, 1,2 Гц), 0,90 (t, 3H, J = 7,4 Гц).
C31H36N2O9S:
Вычислено: C 60,77, H 5,92, N 4,57
Найдено: C 60,49, H 6,04, N 4,45.
Пример 85.
N-(4-изо-пропилбензолсульфонил)-2-(4-(N-морфолинилкарбоксамидо)- 2-пропилфенокси)-2-(5-метокси-3,4-метилендиоксифенил)ацетамид
Названное соединение получили с использованием методик, подобных тем, которые описаны в пример 82.
1H ЯМР (400 мГц, CD3OD, част. на миллион): δ 7,77 (d, 2H, J = 8,5 Гц), 7,37 (d, 2H, J = 8,4 Гц), 7,22 (d, 1H, J = 2,1 Гц), 7,09 (dd, 1H, J = 8,4, 2,2 Гц), 6,66 (d, 1H, J = 1,5 Гц), 6,62 (d, 1H, J = 8,5 Гц), 6,57 (d, 1H, J = 1,5 Гц), 5,95 (s, 2H), 5,46 (s, 1H), 3,81 (s, 3H), 3,65 (m, 8H), 2,98 (m, 1H), 2,66 (m, 2H), 1,60 (m, 2H), 1,26 (d, 6H, J = 7,1 Гц), 0,90 (t, 3H, J = 7,4 Гц).
C33H38N2O9S:
Вычислено: C 62,05, H 6,00, N 4,39
Найдено: C 61,96, H 5,98, N 4,55.
Пример 86.
N-(4-изо-пропилбензолсульфонил)-2-(4-(N-3-метилбутилкарбоксамидо)- 2-пропилфенокси)-2-(5-метокси-3,4-метилендиоксифенил)ацетамид
Названное соединение получили с использованием методик, подобной тем, которые описаны в пример 82.
1H ЯМР (400 мГц, CD3OD, част. на миллион): δ 7,77 (d, 2H, J = 8,5 Гц), 7,60 (d, 1H, J = 2,3 Гц), 7,47 (dd, 1H, J = 2,3, 8,5 Гц), 7,36 (dd, 2H, J = 8,5 Гц), 6,68 (d, 1H, J = 1,5 Гц), 6,59 (d, 1H, J = 1,4 Гц), 6,58 (d, 1H, J = 8,6 Гц), 5,96 (s, 2H), 5,48 (s, 1H), 3,82 (s, 3H), 3,36 (t, 2H, J = 7,5 Гц), 2,97 (m, 1H), 2,66 (m, 2H), 1,62 (m, 3H), 1,49 (q, 2H, J = 7,2 Гц), 1,25 (dd, 2H, J = 1,2, 6,9 Гц), 0,95 (d, 6H, J = 6,6 Гц), 0,90 (t, 3H, J = 7,4 Гц).
C34H42N2O8S:
Вычислено: C 63,93, H 6,63, N 4,39
Найдено: C 63,81, H 6,73, N 4,44.
Пример 87.
N-(4-изо-пропилбензолсульфонил)-2-(4-(N-карбоксиметилкарбоксамидо)- 2-пропилфенокси)-2-(5-метокси-3,4-метилендиоксифенил)ацетамид.
Стадия A:
N-(4-изо-пропилбензолсульфонил)-2-(4-(N-трет- бутоксикарбонилметилкарбоксамидо)-2-пропилфенокси)-2- (5-метокси-3,4-метилендиоксифенил)ацетамид.
Названное соединение получили с использованием методик, подобных тем, которые описаны в примере 82, при этом глицин-трет-бутиловый эфир был исходным аминовым материалом.
1H ЯМР (300 мГц, CDCl3, част. на миллион): δ 7,70 (d, 2H, J = 8,2 Гц), 7,66 (d, 1H, J = 1,3 Гц), 7,56 (m, 1H), 7,41 (d, 2H, J = 8,2 Гц), 6,79 (s, 1H), 6,67 (s, 1H), 6,59 (d, 1H, J = 8,5 Гц), 6,03 (s, 2H), 5,71 (s, 1H), 3,88 (d, 2H, J = 5,5 Гц), 3,80 (s, 3H), 2,95 (sept, 1H, J = 6,9 Гц), 2,78 (m, 2H), 1,32 (s, 9H), 1,17 (d, 6H, J = 6,8 Гц), 0,86 (t, 3H, J = 7,3 Гц).
Стадия B:
N-(4-изо-пропилбензолсульфонил)-2-(4-(N- карбоксиметилкарбоксамидо)-2-пропилфенокси)-2- (5-метокси-3,4-метилендиоксифенил)ацетамид
Раствор продукта стадии A (0,069, 0,1 ммоль) в безводной трифторуксусной кислоте (1,5 мл) перемешивали при комнатной температуре в течение 4-х часов. Избыток реагента выпаривали в вакууме и полученный остаток растирали в порошок с сухим эфиром до получения названного продукта в виде белого твердого вещества (0,6 г).
1H ЯМР (300 мГц, DMSO-d6, част. на миллион): δ 7,70 (d, 2H, J = 8,2 Гц), 7,66 (d, 1H, J = 1,3 Гц), 7,56 (m, 1H), 7,41 (d, 2H, J = 8,2 Гц), 6,79 (s, 1H), 6,67 (s, 1H), 6,59 (d, 1H, J = 8,5 Гц), 6,03 (s, 2H), 5,71 (s, 1H), 3,88 (d, 2H, J = 5,5 Гц), 3,80 (s, 3H), 2,95 (sept, 1H, J = 6,9 Гц), 2,58 (m, 2H), 1,56 (m, 2H), 1,17 (d, 6H, J = 6,8 Гц), 0,86 (t, 3H, J = 7,3 Гц).
C31H34N2O10S:
Вычислено: C 58,74, H 5,53, N 4,42
Найдено: C 58,79, H 5,83, N 4,37.
Пример 88.
N-(4-изо-пропилбензолсульфонил)-2-(4-(N-(L-Ala-OEt) карбоксамидо)-2-пропилфенокси)-2-(5-метокси-3,4- метилендиоксифенил)ацетамид
Названное соединение получили с использованием методик, подобных тем, которые описаны в примере 82, при этом исходным аминовым материалом был L-аланинэтиловый эфир.
1H ЯМР (400 мГц, DMSO-d6, част. на миллион): δ 8,55 (d, 1H, J = 6,1 Гц), 7,69 (m, 3H), 7,57 (q, 1H, J = 9,2 Гц), 7,40 (m, 2H), 6,78 (d, 1H, J = 3,8 Гц), 6,63 (s, 1H), 6,55 (m, 1H), 6,02 (s, 2H), 5,70 (s, 1H), 4,39 (m, 1H), 4,08 (q, 2H, J = 6,8 Гц), 3,79 (d, 3H, J = 2,9 Гц), 2,93 (sept, 1H, J = 6,9 Гц), 2,57 (m, 2H), 1,55 (m, 2H), 1,37 (d, 3H, J = 5,5 Гц), 1,16 (m, 9H), 0,85 (t, 3H, J = 7,5 Гц).
Пример 89.
N-(4-изо-пропилбензолсульфонил)-2-(4-(N-2- этоксикарбонилэтилкарбоксамидо)-2-пропилфенокси)-2-(5-метокси- 3,4-метилендиоксифенил)ацетамид
Названное соединение получили с использованием методик, подобных тем, которые описаны в примере 82, при этом исходным аминовым материалом был β-аланинэтиловый эфир.
1H ЯМР (400 мГц, DMSO-d6, част. на миллион): δ 8,34 (t, 1H, J = 5,4 Гц), 7,68 (d, 2H, J = 8,3 Гц), 7,58 (d, 1H, J = 2,2 Гц), 7,49 (dd, 1H, J = 8,6, 2,3 Гц), 7,39 (d, 2H, J = 8,4 Гц), 6,77 (d, 1H, J = 1,4 Гц), 6,65 (d, 1H, J = 1,3 Гц), 6,53 (m, 1H, J = 8,8 Гц), 6,01 (s, 2H), 5,67 (s, 1H), 4,05 (q, 2H, J = 7,1 Гц), 3,78 (s, 3H), 3,44 (m, 2H), 2,92 (sept, 1H, J = 6,9 Гц), 2,53 (m, 2H), 1,54 (m, 2H), 1,16 (m, 9H), 0,85 (t, 3H, J = 7,4 Гц).
Пример 90.
N-(4-изо-пропилбензолсульфонил)-2-(4-(N-(L-Ala)-карбоксамидо)- 2-пропилфенокси)-2-(5-метокси-3,4-метилендиоксифенил)ацетамид
Продукт из примера 88 подвергли омылению для получения названного продукта.
1H ЯМР (400 мГц, DMSO-d6, част. на миллион): δ 12,64 (широк., 1H), 12,51 (широк. , 1H), 8,44 (dd, 1H, J = 7,1, 2,7 Гц), 7,69 (m, 3H), 7,56 (m, 1H), 7,40 (m, 2H), 6,77 (d, 1H, J = 1,6 Гц), 6,66 (d, 1H, J = 1,7 Гц), 6,55 (m, 1H), 6,01 (d, 2H, J = 2,6 Гц), 5,69 (s, 1H), 4,37 (pn, 1H, J = 7,4 Гц), 3,79 (d, 3H, J = 1,9 Гц), 2,93 (sept, 1H, J = 6,9 Гц), 2,57 (m, 2H), 1,54 (m, 2H), 1,36 (dd, 3H, J = 7,3, 2,7 Гц), 1,16 (d, 6H, J = 6,8 Гц), 0,85 (t, 3H, J = 7,2 Гц).
Пример 91.
N-(4-изо-пропилбензолсульфонил)-2-(4-(N-(2- карбоксиэтилкарбоксамидо)-2-пропилфенокси)-2-(5-метокси-3,4- метилендиоксифенил)ацетамид
Продукт из примера 89 подвергали омылению для получения названного продукта.
1H ЯМР (400 мГц, DMSO-d6, част. на миллион): δ 12,64 (широк., 1H), 12,21 (широк. , 1H), 8,32 (t, 1H, J = 5,5 Гц), 7,68 (d, 2H, J = 8,4 Гц), 7,59 (d, 1H, J = 1,9 Гц), 7,49 (dd, 1H, J = 8,5, 2,1 Гц), 7,40 (d, 2H, J = 8,4 Гц), 6,77 (s, 1H), 6,65 (d, 1H, J = 1,2 Гц), 6,01 (d, 2H, J = 2,9 Гц), 5,68 (s, 1H), 3,79 (s, 3H), 3,39 (m, 2H), 2,93 (sept, 1H, J = 6,8 Гц), 2,55 (m, 2H), 1,54 (m, 2H), 1,16 (d, 6H, J = 6,9 Гц), 0,84 (t, 3H, J = 7,3 Гц).
C32H36N2O10S:
Вычислено: C 59,99, H 5,66, N 4,37
Найдено: C 59,72, H 5,77, N 4,49.
Пример 92.
N-(4-изо-пропилбензолсульфонил)-2-(4-(N-3- гидроксипропилкарбоксамидо)-2-пропилфенокси)-2- (5-метокси-3,4-метилендиоксифенил)ацетамид
Названное соединение получили с использованием методик, подобных тем, которые описаны в примере 82, при этом исходным аминовым материалом был 3-аминопропанол.
1H ЯМР (400 мГц, CD3OD, част. на миллион): δ 8,33 (m, 1H), 7,77 (d, 2H, J = 8,5 Гц), 7,60 (d, 1H, J = 2,3 Гц), 7,48 (dd, 2H, J = 8,5, 2,3 Гц), 7,36 (d, 2H, J = 8,4 Гц), 6,68 (d, 1H, J = 1,5 Гц), 6,60 (d, 1H, J = 1,4 Гц), 6,59 (d, 1H, J = 8,6 Гц), 5,96 (s, 2H), 5,48 (s, 1H), 3,82 (s, 3H), 3,63 (t, 2H, J = 6,3 Гц), 3,43 (t, 2H, J = 5,8 Гц), 2,97 (sept, 1H, J = 7,0 Гц), 2,66 (m, 2H), 1,80 (pn, 2H, J = 6,7 Гц), 1,61 (m, 2H), 1,25 (dd, 6H, J = 6,9, 1,3 Гц), 0,90 (t, 3H, J = 7,4 Гц).
C32H38N2O9S:
Вычислено: C 61,33, H 6,11, N 4,47
Найдено: C 61,07, H 6,09, N 4,48.
Пример 93.
N-(4-изо-пропилбензолсульфонил)-2-(4-тетразол-5-илкарбоксамидо)- 2-пропилфенокси)-2-(5-метокси-3,4-метилендиоксифенил)ацетамид
Названное соединение получили с использованием методик, подобных тем, которые описаны в примере 82, при этом исходным аминовым материалом был 5-аминотетразол.
FAB-MS m/e = 640 (M+1).
Пример 94.
N-(4-изо-пропилбензолсульфонил)-2-(4-(N-3-(морфолин-4-ил)- пропилкарбоксамидо)-2-пропилфенокси)-2-(5-метокси-3,4- метилендиоксифенил)ацетамид
Названное соединение получили с использованием методик, подобных тем, которые описаны в примере 82, при этом исходным аминовым материалом был 3-(N-морфолинил)аминопропан.
1H ЯМР (400 мГц, CD3OD, част. на миллион): δ 7,65 (d, 2H, J = 8,3 Гц), 7,65 (s, 1H), 7,58 (dd, 1H, J = 2,4, 8,6 Гц), 7,24 (d, 2H, J = 8,4 Гц), 6,81 (d, 1H, J = 8,6 Гц), 6,78 (d, 1H, J = 1,4 Гц), 6,69 (d, 1H, J = 1,4 Гц), 5,94 (s, 2H), 5,40 (s, 1H), 3,82 (s, 7H), 3,54 (m, 2H), 3,12 (m, 6H), 2,92 (sept, 1H, J = 6,9 Гц), 2,66 (m, 2H), 1,62 (m, 2H), 1,22 (d, 6H, J = 6,9 Гц), 0,90 (t, 3H, J = 7,4 Гц).
C35H43N3O9S:
Вычислено: C 60,46, H 6,45, N 6,04
Найдено: C 60,39, H 6,43, N 5,93.
Пример 95.
N-(4-изо-пропилбензолсульфонил)-2-(4-(N-(D-Ala-OMe)- карбоксамидо)-2-пропилфенокси)-2-(5-метокси-3,4- метилендиоксифенил)ацетамид.
Названное соединение получили с использованием методик, подобных тем, которые описаны в примере 82, при этом исходным аминовым материалом был D-аланинметиловый эфир.
1H ЯМР (400 мГц, CD3OD, част. на миллион): δ 8,54 (d, 1H, J = 6,8 Гц), 7,77 (d, 2H, J = 8,3 Гц), 7,66 (s, 1H), 7,53 (m, 1H), 7,36 (d, 2H, J = 8,3 Гц), 6,68 (s, 1H), 6,60 (n, 2H), 5,96 (s, 2H), 5,49 (s, 1H), 4,57 (m, 1H), 3,82 (s, 3H), 3,72 (s, 3H), 2,97 (sept. 1H, J = 6,8 Гц), 2,67 (m, 2H), 1,62 (m, 2H), 1,47 (d, 3H, J = 7,4 Гц), 1,24 (d, 6H, J = 7,0 Гц), 0,91 (t, 3H, J = 7,4 Гц).
Пример 96.
N-(4-изо-пропилбензолсульфонил)-2-(4-(N-(D-Ala)- карбоксамидо)-2-пропилфенокси)-2-(5-метокси-3,4- метилендиоксифенил)ацетамид
Продукт из примера 95 подвергали омылению для получения названного продукта.
1H ЯМР (400 мГц, DMSO-d6, част.на миллион): δ 12,64 (широк., 1H), 12,48 (широк. , 1H), 8,44 (dd,, 1H, J = 7,3 и 2,6 Гц), 7,68 (m, 3H), 7,56 (m, 1H), 7,40 (dd, 2H, J = 4,0, 8,4 Гц), 6,77 (d, 1H, J = 2,3 Гц), 6,66 (m, 1H), 6,55 (dd, 1H, J = 21,0, 8,8 Гц), 6,01 (d, 2H, J = 3,6 Гц), 5,70 (s, 1H, J = 3,8 Гц), 4,37 (pn, 1H, J = 7,3 Гц), 3,78 (d, 3H, J = 1,8 Гц), 2,93 (sept, 1H, J = 7,0 Гц), 2,57 (m, 2H), 1,55 (m, 2H), 1,36 (dd, 3H, J = 7,3, 2,7 Гц), 1,16 (d, 6H, J = 6,9 Гц), 0,85 (t, 3H, J = 7,3 Гц).
Пример 97.
N-(4-изо-пропилбензолсульфонил)-2-(4-(N-(3-карбоксиметилропил) карбоксамидо)-2-пропилфенокси)-2-(5-метокси-3,4- метилендиоксифенил)ацетамид
Названное соединение получили с использованием методик, подобных тем, которые описаны в примере 82, при этом исходным аминовым материалом был метил -γ- аминобутират.
1H ЯМР (400 мГц, CD3OD, част. на миллион): δ 7,77 (d, 2H, J = 8,5 Гц), 7,61 (d, 1H, J = 2,2 Гц), 7,48 (dd, 1H, J = 8,5, 2,3 Гц), 7,36 (d, 2H, J = 8,5 Гц), 6,68 (s, 1H), 6,59 (d, 1H, J = 8,3 Гц), 5,95 (s, 2H), 5,48 (s, 1H), 4,09 (q, 2H, J = 7,1 Гц), 3,82 (s, 3H), 3,37 (m, 2H), 2,97 (sept, 1H, J = 6,9 Гц), 2,66 (m, 2H), 2,38 (t, 2H, J = 7,4 Гц), 1,89 (pn, 2H, J = 7,1 Гц), 1,61 (m, 2H), 1,23 (d, 6H, J = 6,9 Гц), 0,90 (t, 3H, J = 7,3 Гц).
Пример 98.
N-(4-изо-пропилбензолсульфонил)-2-(4-(N-(3-карбоксипропил)- карбоксамидо)-2-н-пропилфенокси)-2-(5-метокси-3,4-метилен- диоксифенил)ацетамид.
Для получения названного продукта продукт из примера 97 подвергали омылению.
1H ЯМР (400 мГц, DMSO-d6, част. на миллион): δ 12,63 (широк., 1H), 12,06 (широк. , 1H), 8,27 (t, 1H, J = 5,5 Гц), 7,68 (d, 2H, J = 8,4 Гц), 7,60 (d, 1H, J = 2,2 Гц), 7,50 (dd, 1H, J = 8,5, 2,2 Гц), 7,40 (d, 2H, J = 8,4 Гц), 6,77 (d, 1H, J = 1,2 Гц), 6,66 (d, 1H, J = 1,3 Гц), 6,52 (d, 1H, J = 8,8 Гц), 6,01 (d, 2H, J = 2,9 Гц), 5,68 (s, 1H), 3,22 (q, 2H, J = 6,5 Гц), 2,92 (sept, 6,8 Гц), 2,54 (m, 2H), 2,25 (t, 2H, J = 7,4 Гц), 1,71 (pn, 7,1 Гц), 1,54 (m, 2H), 1,16 (d, 6H, J = 6,8 Гц), 0,84 (t, 3H, J = 7,3 Гц).
C33H38N2O10S:
Вычислено: C 60,54, H 5,85, N 4,28
Найдено: C 60,26, H 6,17, N 4,02.
Пример 99.
N-(4-изо-пропилбензолсульфонил)-2-(4-(N-изо- пропилкарбамоил)амино-2-н-пропилфенокси)-2-(3,4- метилендиоксифенил)ацетамид
Стадия A:
4-нитро-2-(пропен-3-ил)фенол
Смесь 4-нитрофеноксиаллилового эфира (4,0 г, 22,35 ммоль) и 1,2-дихлорбензола (15 мл) нагревали в колбе с обратным холодильником в течение 6 часов. Реакционную смесь охладили и очистили тонкослойной колоночной хроматографией на силикагеле при использовании в качестве элюентов соответственно гексана и EtOAc-гексана (1:6). Чистый продукт получили в виде желтого масла (2,6 г).
1H ЯМР (200 мГц, CDCl3, част. на миллион): δ 8,05 (d, 2H), 6,92 (d, 1H), 6,01 (m, 1H), 5,18 (m, 2H), 3,42 (d, 2H, J = 7,3 Гц).
Стадия B:
Метил-2-(4-нитро-2-(пропен-3-ил)фенокси)-2- (3,4-метилендиоксифенил)ацетат
Названное соединение получили с использованием методик, подобных той, которая была описана в стадии A примера 79.
В качестве алкилирующего агента использовали метил-2-бром-2-(3,4-метилендиоксифенил)ацетат. Очистку сырого продукта осуществляли тонкослойной колоночной хроматографией на силикагеле с использованием этилацетата-гексана (1:5).
1H ЯМР (200 мГц, CDCl3, част.на миллион): δ 8,05 (m, 2H), 7,02 (m, 2H), 6,78 (m, 2H), 5,96 (s, 2H), 5,6 (s, 1H), 5,15 (m, 2H), 4,15 (m, 2H), 3,75 (s, 3H), 3,47 (m, 2H).
Стадия C:
2-(4-(N-изо-пропилкарбамоил)амино-2-н-пропилфенокси)- 2-(3,4-метилендиоксифенил)уксусная кислота
К раствору продукта стадии B (0,5 г) в метаноле (6 мл) добавили Pd-C (10%) (0,05 г) и реакционную смесь перемешивали при комнатной температуре в течение 6 часов в атмосфере водорода.
Катализатор отфильтровали и фильтрат концентрировали в вакууме до получения названного метил -α- (4-амино-2-н-пропилфенокси-2-(3,4- метилендиоксифенил)ацетата (0,5 г) в виде твердого вещества. Этот материал без дальнейшей очистки растворили в сухом THF (5 мл), и он взаимодействовал с N-изо-пропилизоцианатом (0,1 мл) при комнатной температуре в течение 12-ти часов. Очистка сырого продукта посредством тонкослойной хроматографии с использованием EtOAc-гексана (1:2) привела к получению названного соединения в виде белого твердого вещества (0,36 г).
1H ЯМР (300 мГц, CDCl3, част.на миллион): δ 7,1 - 6,77 (m, 6H), 6,61 (d, 1H, J = 1,5 Гц), 5,93 (s, 2H), 5,54 (s, 1H), 3,98 (m, 1H), 3,78 (s, 3H), 3,63 (m, 1H), 2,68 (m, 2H), 1,69 (m, 2H), 1,15 (dd, 6H, J = 7,0, 0,2,5 Гц), 0,90 (t, 3H, J = 7,4 Гц).
Стадия D:
N-(4-изо-пропилбензолсульфонил)-2-(4-(N-изо-пропилкарбамоил)амино-2-н-пропилфенокси)-2-(3,4-метилендиоксифенил)ацетамид
Названный продукт получили из продукта, полученного на стадии C, с использованием методик, подобных той, которые описаны в стадиях B и C примера 79.
1H ЯМР (300 мГц, CD3OD, част. на миллион): δ 7,78 (d, 2H, J = 8,4 Гц), 7,76 (m, 1H), 7,52 (m, 1H), 7,32 (d, 2H, J = 8,4 Гц), 7,07 (d, 1H, J = 1,4 Гц), 6,75 (d, 1H, J = 8,2 Гц), 6,42 (d, 1H, J = 8,2 Гц), 5,97 (s, 2H), 5,21 (s, 1H), 3,88 (m, 1H), 2,82 (m, 1H), 2,54 (m, 2H), 1,69 (m, 2H), 1,26 (dd, 6H, J = 7,0, 2,5 Гц), 1,15 (dd, 6H, J = 7,0, 2,5 Гц), 0,90 (t, 3H, J = 7,4 Гц),
FAB-MS: m/e 596 (M+1).
Пример 100.
α- (2-н-пропил-4-метиламиносульфонилфенокси)-3,4- метилендиоксифенилуксусная кислота
Стадия A: Получение 3-аллил-4-гидроксибензолсульфонамида
К раствору 5,00 г (28,9 ммоль) 4-гидроксибензолсульфонамида, растворенного в 30 мл безводного DMF, добавили 10,36 г (31,8 ммоль) карбоната цезия и реакционную смесь перемешивали магнитной мешалкой при комнатной температуре в атмосфере азота в течение 10 минут. Добавили аллилбромид (2,75 мл, 31,8 ммоль) и затем реакционную смесь разделили между EtOAc (60 мл) и 10% водным раствором лимонной кислоты (20 мл) и экстрагировали. Органический слой отделили, промыли насыщенным водным NaCl, сушили (MgSO4), фильтровали, выпаривали и сушили в вакууме до получения 5,40 г (88%) желтого твердого вещества. Затем сырой O-аллиловый эфир (5,36 г, 25,2 ммоль) растворили в 10 мл 1,2-дихлорбензола в 50 мл круглодонной колбе и осуществляли перемешивание магнитной мешалкой при нагревании с обратным холодильником под атмосферой азота в течение 15 часов. Затем реакционную смесь охладили до комнатной температуры и разбавили метанолом.
1,2-дихлорбензол удалили посредством экстракции слоя метанола гексаном, слой метанола отделили, затем выпарили. Затем остаток очистили флэш колоночной хроматографией на силикагеле при элюировании 6% MeOH-CH2Cl2. Объединение очищенных фракций, выпаривание и сушка в вакууме привели к получению 3,04 г (57%) названного соединения.
1H ЯМР (400 мГц, CD3OD, часть на миллион): δ 3,38 (d, J=6,4 Гц, 2H), 5,02 - 5,10 (m, 2H), 5,94 - 6,04 (m, 1H), 6,84 (d, J=8,40, 1H), 7,58 (dd, J= 2,40, 8,40 Гц, 1H), 7,61 (d, J=2,40 Гц, 1H).
CI-MS m/e = 213 (M+).
Стадия B:
Получение 4-гидрокси-3-н-пропилбензолсульфонамида
Колбу Парра для гидрирования загрузили раствором 3,04 г (14,30 ммоль) продукта стадии A, растворенного в 25 мл этанола, и добавили 0,300 г 10% палладия на углеродном катализаторе.
Колбу поместили в аппарат для гидрирования, не содержащий воздуха, создали повышенное давление посредством водорода (40 фунтов/дюйм2) (2,8124 кг/см2) и встряхивали в течение 15 минут. В конце этого периода TCX (3% MeOH-CH2Cl2, 2 элюирования) показал, что реакция завершилась. Реакционную смесь отфильтровывали и выпаривали. Продукт сушили в вакууме до получения 3,06 г (99%) названного соединения.
1H ЯМР (400 мГц, CD3OD, част. на миллион): δ 0,94 (t, J=7,20 Гц, 3H), 1,58 - 1,68 (m, 2H), 2,01 - 2,62 (m, 2H), 6,82 (d, J=8,40 Гц, 1H), 7,55 (dd, J=2,40, 8,40 Гц, 1H), 7,60 (d, J=2,40 Гц, 1H).
FAB-MS m/e = 216 (M+1).
Стадия C:
Получение метил -α- (2-н-пропил-4-аминосульфонилфенокси-3,4- метилендиоксифенилацетата
К раствору 3,06 г (14,23 ммоль) продукта стадии B, растворенного в 25 мл безводного DMF, добавили 4,87 г (15,0 ммоль) карбоната цезия и реакционную смесь перемешивали магнитной мешалкой в атмосфере азота при комнатной температуре в течение 15 минут. Затем добавили метил -α- бром-3,4-метилендиоксифенилацетат (4,08 г, 15,0 ммоль) и реакционную смесь перемешивали в течение еще 3 часов. Затем реакционную смесь разделили между EtOAc (80 мл) и 10% водным раствором лимонной кислоты (300 мл). Органический слой отделили, промыли насыщенным водным NaHCO3 насыщенным водным NaCl, сушили (MgSO4), фильтровали и выпаривали. Остаток сушили в вакууме до получения 5,90 г (5,79 теоретически) названного соединения, которое использовали на следующей стадии без дальнейшей очистки.
1H ЯМР (400 мГц, CD3OD, част. на миллион): δ 0,97 (t, J=7,20, 3H), 1,64 - 1,76 (m, 2H), 2,74 (t, J=7,20 Гц, 2H), 3,70 (s, 3H), 5,87 (s, 1H), 5,97 (S, 2H), 6,85 (d, J=8,0 Гц, 1H), 6,93 (d, J=8,40 Гц, 1H), 7,03 (d, J=1,60 Гц, 1H), 7,06 (dd, J=1,60, 8,00 Гц, 1H), 7,65 (dd, J=2,40, 8,40 Гц, 1H), 7,69 (d, J=2,40 Гц, 1H).
FAB-MS m/e = 408 (M+1).
Стадия D:
Получение метил -α- (2-н-пропил-4-метиламиносульфонилфенокси- 3,4-метилендиоксифенилацетата
К раствору 2,19 (5,38 ммоль) продукта стадии C, растворенного в 20 мл безводного THF, добавили 2,41 мл (16,1 ммоль) 1,8-диазабицикло [5,4,0]-ундец-7-ена и реакционную смесь перемешивали магнитной мешалкой в атмосфере азота при комнатной температуре в течение 25 минут. Добавили иодометан (1,0 мл, 16,1 ммоль) и реакционную смесь перемешивали еще 15 часов при комнатной температуре. Реакционную смесь разбавили EtOAc и образованный осадок вновь растворили добавлением метанола. Затем смесь разбавили теплым соединением EtOAc (150 мл в целом), охлаждали всю ночь и выделенное твердое вещество удалили фильтрацией. Фильтрат выпарили в вакууме и остаток очистили флэш колоночной хроматографией на силикагеле при элюировании 5% EtOAc-CHCl3. Объединение очищенных фракций и выпаривание в вакууме привели к получению 0,164 г названного соединения и ряда примесей, которые были предназначены для очистки.
1H ЯМР (400 мГц, CD3OD, част. на миллион): δ 0,97 (t, J=7,20 Гц, 3H), 1,65 - 1,77 (m, 2H), 2,48 (s, 3H), 2,74 (t, J=7,20 Гц, 2H), 3,71 (s, 3H), 5,87 (s, 1H), 5,98 (s, 2H), 6,85 (d, J=8,00 Гц, 1H), 6,96 (d, J=8,80 Гц, 1H), 7,04 (d, J=1,60 Гц, 1H), 7,07 (dd, J=1,60, 8,0 Гц, 1H), 7,58 - 7,61 (m, 2H).
FAB-MS m/e = 421 (M+).
Стадия E:
Получение α- (2-н-пропил-4-метиламиносульфонилфенокси-3,4- метилендиоксифенилуксусной кислоты.
К раствору 0,372 г (0,884 ммоль) продукта стадии D, растворенного в 3,0 мл метанола, добавили 212 мкл (1,06 ммоль) 5,0 N водного раствора гидроксида натрия, что привело к получению мутной суспензии. Для получения раствора реакционную смесь нагрели, добавили метанол (1 мл), затем дихлорметан (0,5 мл), однако прозрачный раствор не получили. Добавили дополнительно 5N раствор гидроксида натрия (212 мкл) и в конечном счете 0,5 мл THF, что привело к получению прозрачного раствора. После перемешивания в течение 15 часов при комнатной температуре анализ TCX (CHCl3-MeOH-NH4OH 80:15:1) показал завершение гидролиза исходного материала, и затем с помощью 6N HCl установили pH реакционной смеси, равным 7. Затем реакционную смесь концентрировали в вакууме и остаток очистили тонкослойной колоночной хроматографией на силикагеле при элюировании CHCl3-MeOH-NH4OH (92:7:1). Объединение очищенных фракций и сушка в вакууме привели к получению 0,335 г (93%) названного соединения в виде аморфного твердого вещества.
1H ЯМР (400 мГц, CD3OD, част. на миллион): δ 0,96 (t, J=7,20 Гц, 3H), 1,66 - 1,78 (m, 2H), 2,48 (s, 3H), 2,73 - 2,77 (m, 2H), 5,74 (s, 1H), 5,97 (s, 2H), 6,85 (d, J=7,60 Гц, 1H), 6,98 (d, J=7,60 Гц, 1H), 6,98 (d, J=9,20 Гц, 1H), 7,07 - 7,10 (m, 2H), 7,59 - 7,62 (m, 2H).
FAB-MS m/e = 407 (M+).
Пример 101.
Двукалиевая соль N-(4-изо-пропилбензолсульфонил) -α- (2-н- пропил-4-метиламиносульфонилфенокси)-3,4-метилендиоксифенил-ацетамида
К раствору 0,298 г (0,73 ммоль) продукта примера 100, растворенного в 4,0 мл безводного THF, добавили 0,237 г (1,46 ммоль) 1,1'-карбонилдиимидазола и реакционную смесь перемешивали магнитной мешалкой и нагревали в колбе с обратным холодильником в атмосфере азота в течение 2-х часов.
Затем реакционную смесь охладили до комнатной температуры, добавили 0,219 г (1,10 ммоль) 4-изо-пропилбензолсульфонамида и 164 мкл (1,10 ммоль) 1,8-диазабицикло[5.4.0] -ундец-7-ена и реакционную смесь перемешивали и нагревали в колбе с обратным холодильником еще в течение 10-ти минут. Затем реакционную смесь охладили до комнатной температуры, разделили между 10% водным раствором лимонной кислоты и EtOAc и экстрагировали. Отделенный органический слой промывали насыщенным водным NaCl, сушили (MgSO4), фильтровали и выпаривали. Остаток повторно растворили в 1,0 мл метанола и обработали 2,20 мл (3 эквивалента) 1,1 М водным раствором гидроксида калия. Затем смесь разбавили 5 мл воды и фильтровали через 0,45-микронный фильтр. Фильтрат обессолили и очистили на жидкостном хроматографе Waters Millipolre Delta Prep 300 (Вотез Милипор Дельта Преп), снабженным модулем M1000 Prep-Pak, содержащим катушку с фотографическими пленками 47х300 мм Delta-Pak C 18 15 мкм, 100А. Использовали две емкости для растворителя: систему растворителя A (вода - ацетонитрил 95:5) и систему растворителя B (вода - ацетонитрил 5: 95), и поток, вытекающий из колонны, одновременно контролировали при 210 и 280 нм детектором видимых ультрафиолетовых лучей модели Waters 490. Колонку предварительно уравновесили системой растворителя A и инжектировали фильтрат. Продукт обессолили посредством элюировали 0,5 л системы растворителя A (50 мл/мин), затем начали градиентное элюирование, начальные условия которого были: 100% системы растворителя A - 0% системы растворителя B и через 15 минут достигли: 60% системы растворителя A - 40% системы растворителя B, и фракции собрали с помощью сборника фракций ISCO Foxy 200. Очищенные фракции соединили в круглодонных колбах, заморозили при -78oC в бане со смесью сухого льда с ацетоном и подвергли лиофилизации. Объединение очищенного продукта дало 0,284 г (62%) названного соединения в виде белого лиофилизованного порошка.
1H ЯМР (400 мГц, CD3OD, част. на миллион): δ 0,89 (t, J=7,60 Гц, 3H), 1,21 (d, J=6,80 Гц, 3H), 1,22 (d, О=6,80 Гц, 3H), 1,57 - 1,64 (m, 2H), 2,45 (s, 3H), 2,56 - 2,63 (m, 1H), 2,70 - 2,76 (m, 1H), 5,37 (s, 1H), 5,93 (d, J= 1,20 Гц, 1H), 5,94 (d, J=1,20 Гц, 1H), 6,76 (d, J=8,40 Гц, 1H), 6,85 (d, J= 8,80 Гц, 1H), 7,02 - 7,04 (m, 2H), 7,21 (d, J=8,40 Гц, 2H), 7,47 (dd, J= 2,40, 8,80 Гц, 1H), 7,52 (d, J=2,40 Гц, 1H), 7,65 (d, J=8,40 Гц, 2H).
ESI-MS m/e = 627 (M+1).
1. Примеры препаративных форм фармацевтической композиции
Соединение настоящего изобретения может вводиться перорально в твердых дозированных формах, таких как капсулы, таблетки, пастилки, драже, гранулы и порошки, или жидких дозированных формах, таких как эликсиры, сиропы, эмульсии, дисперсии и суспензии. Соединение может также вводиться парентерально, в виде стерильных жидких дозированных форм, таких как дисперсии, суспензии или растворы. Могут использоваться другие дозированные формы для введения активного ингредиента, такие как мази, кремы, чрезкожные пластыри или порошки для местного введения, а также офтальмологические растворы или суспензии, например глазные капли для глазного введения, а также аэрозоли или порошки для ингаляций или интраназального введения, а также кремы, мази, спрей или суппозитории для ректального или вагинального введения.
Желатиновые капсулы содержат активный ингредиент и порошкообразные носители, такие как лактоза, крахмал, производные целлюлозы, стеарата магния, стеариновую кислоту и подобные. Такие разбавители могут использоваться для производства таблеток. Как таблетки, так и капсулы могут быть приготовлены в виде препаратов с пролонгированным выделением для обеспечения непрерывного выделения активного ингредиента во времени. Таблетки могут иметь сахарную оболочку или пленочное покрытие для устранения неприятного вкуса и защиты таблеток от атмосферного влияния, или энтеросолюбильную оболочку для селективного растворения в желудочно-кишечном тракте.
Жидкие дозированные формы для перорального введения могут содержать красители и вкусовые добавки для стимулирования приема пациентами.
Обычно подходящими носителями для парентеральных растворов являются вода, подходящее масло, физраствор, водная декстроза (глюкоза) и растворы родственных сахаров и гликолей, таких как пропиленгликоль или полиэтиленгликоль. Растворы для парентерального введения предпочтительно содержат водорастворимую соль активного ингредиента, подходящий стабилизирующий агент и, если необходимо, буферные вещества. Подходящими стабилизирующими агентами являются противоокислительные агенты, такие как бисульфит натрия, сульфит натрия или аскорбиновая кислота, как таковые или в смеси. Также используют лимонную кислоту и ее соли и натриевую соль этилендиаминтетрауксусной кислоты. Кроме того, парентеральные растворы могут дополнительно содержать консерванты, такие как хлорид бензалкония, метил- или пропилпарабен и хлорбутанол.
Подходящие фармацевтические носители описаны в Remington's Pharmaceutical Sciences, A. Osol, обычно используемом в данной области ссылочном источнике.
Для введения в виде ингаляций соединение данного изобретения обычно приготавливают в виде аэрозоля в баллончике или распылителя. Соединение также может вводиться в виде порошков и композиций порошков, которые могут быть введены с помощью инсуффлятора. Предпочтительной системой для ингаляций является аэрозоль для ингаляций в дозирующем устройстве (МДИ), который может быть приготовлен в виде суспензии или раствора соединения формулы I в подходящих пропеллентах, таких как фторуглероды или углеводороды.
Для глазного введения может быть приготовлена глазная препаративная форма в виде раствора или суспензии соединения формулы I в подходящем весовом отношении в подходящем офтальмологическом растворителе, таким образом, чтобы соединение находилось в контакте с глазной поверхностью достаточное время для того, чтобы соединение проникло внутрь роговичной и внутренней области глаза.
Предпочтительными фармацевтическими формами для введения соединений данного изобретения являются следующие формы.
Пример 1. Капсулы
Единичные капсулы готовят заполнением каждой стандартной твердой желатиновой капсулы, состоящей из двух частей, 500 мг порошкообразного активного ингредиента, 150 мг лактозы, 50 мг целлюлозы и 6 мг стеарата магния.
Пример 2. Мягкие желатиновые капсулы
Готовят смесь активного ингредиента и стимулирующего пищеварение масла, такого как соевое масло, хлопковое масло или оливковое масло, и вводят с помощью насоса с вытесняющим механизмом в желатин для образования мягких желатиновых капсул, содержащих около 500 мг активного ингредиента. Капсулы промывают и сушат.
Пример 3. Таблетки
Таблетки получают обычными способами, так, чтобы они содержали около 500 мг активного ингредиента, 0,2 мг коллоидного диоксида кремния, 5 мг стеарата магния, 275 мг микрокристаллической целлюлозы, 11 мг крахмала и 98,8 мг лактозы. Для придания приятного вкуса или замедления абсорбции таблетки могут быть покрыты подходящей оболочкой.
Пример 4. Инъекционные формы
Парентеральную композицию, подходящую для введения в виде инъекций, получают, смешивая 1,5 мас.% активного ингредиента в 10 об.% пропиленгликоля. Раствор доводят до соответствующего объема водой для инъекций и стерилизуют.
Пример 5. Суспензии
Водные суспензии готовят для перорального введения таким образом, чтобы каждые 5 мл содержали около 500 мг тонко измельченного активного ингредиента, 100 мг натрий карбоксиметилцеллюлозы, 5 мг бензоата натрия, 1,0 г раствора сорбитола, U.S.P., и 0,025 мл ванилина.
Подобные лекарственные формы обычно используют, если соединение данного изобретения вводят ступенчато или в сочетании с другими терапевтическими агентами. Если препараты вводят в виде физической смеси, лекарственные формы и способ введения должен выбираться в зависимости от совместимости объединенных препаратов. Таким образом, термин совведение подразумевает введение одновременно, или последовательно, или альтернативно, в виде фиксированной дозированной комбинации двух активных компонентов.
2. Данные по биологической активности
В Таблице представлены данные тестов, проведенных по следующим методикам:
1) Human ETA и Human ETB (нМ) - анализ связывания рецептора с использованием клонированных рецепторов эндотелина человека, представленных в клетках яичника китайского хомячка;
2) Rat Hip ETB (нМ) - анализ связывания рецепторов с использованием препарата мембраны гиппокампа крысы;
3) Cow Aorta (нМ) - анализ связывания рецептора с использованием препарата мембраны аорты коровы.



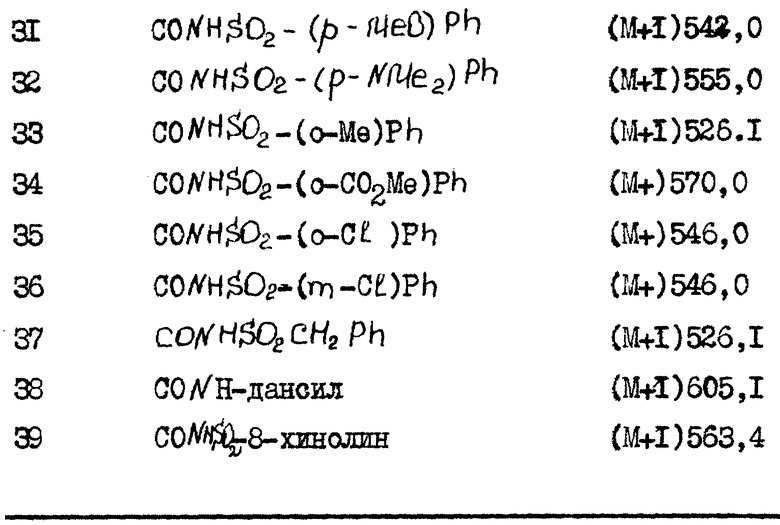

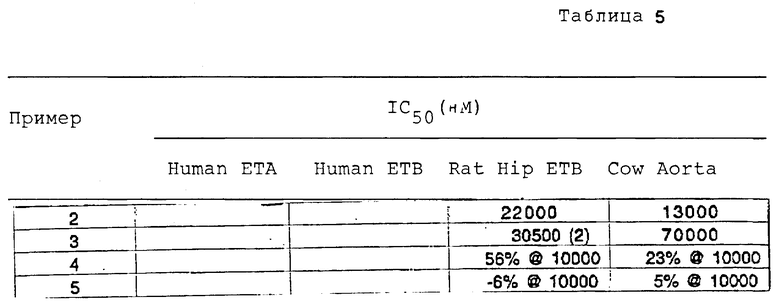



Производные феноксифенилуксусной кислоты формулы I, где R1, R2, R3a, R3b - водород, F, С1, Вr, J, ОR7 или R1 и R2 у соседних атомов углерода соединяются с образованием циклической структуры  где А представляет собой -Y-[С(R6)(R6)]s-Y-, -С(R4)=С(R5)-С(R4)=С(R5)-, s = 1, 2; Y представляет -О-; R4, R5, R6 - водород ( другие обозначения см. в п. 1 ф-лы изобретения) или их фармацевтически приемлемые соли пригодны в качестве непептидных антагонистов эндотелина. 3 c. и 10 з.п. ф-лы, 5 табл.
где А представляет собой -Y-[С(R6)(R6)]s-Y-, -С(R4)=С(R5)-С(R4)=С(R5)-, s = 1, 2; Y представляет -О-; R4, R5, R6 - водород ( другие обозначения см. в п. 1 ф-лы изобретения) или их фармацевтически приемлемые соли пригодны в качестве непептидных антагонистов эндотелина. 3 c. и 10 з.п. ф-лы, 5 табл.

где R1, R2, R3a, R3b независимо являются а) Н, b) F, Cl, Br, J, c) -OR7,
или R1 и R2 у соседних атомов углерода могут соединяться с образованием циклической структуры:
где А представляет
а) -Y-[C(R6)(R6)]s-Y-,
b) -С(R4) = C(R5) - С(R4) = С(R5)-,
s равно 1 или 2;
Y представляет -О-;
R4 и R5 независимо являются водородом;
R6 является водородом;
R7 является: а) водородом; b) (C1-C6) алкилом; c) фенилом,
R8 является водородом;
R9 и R10 независимо являются: a) H, b) C1-C6-алкилом; с) C1-C6-алкокси;
R11 является: a) C1-C6-алкилом, b) арилом, где арил определяют как фенил, который является незамещенным или замещенным одним или двумя заместителями, выбранными из C1-C4-алкила;
R12 является: а) H, b) C1-C6-алкилом, незамещенным или замещенным одним или двумя заместителями, выбранными из группы, состоящей из i) -ОН, ii) -COOR7, iii) -CONHR7, iv) -NR7CONR7R11, v) -NR7COOR11,
или представляет собой с) - COOR7; d) -CONH2; e) -CONR7R11; f) -NR7CONR7R11; g) -SO2NR7R11, h) -NR7SO2NR7R11; i) -CONHSO2R11; j) CO-аминокислотой, где аминокислоту определяют как L или D-Аlа, которая может быть замещена С1-C6-алкиловым эфиром; к)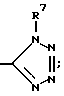
Х является -О-;
Z является а) -СО2Н; b) -CONHSO2-арилом, где арил определяют как фенил, нафтил, которые могут быть замещены или не замещены одним или двумя заместителями, выбранными из группы, состоящей из i) С1-C4-алкила; ii) -O-(С1-C4)-алкила; iii) F, Cl, Br или J; iv) -COOR7; v) -NH2; vi) -NH(C1-C4)-алкила; vii) N[(C1-C4)-алкила]2; viii) фенила,
или представляет собой с) -CONHSO2-(C1-C8)алкил, где алкильная группа является незамещенной или замещенной фенилом; d) -CONHSO2-гетероарил, где гетероарил определяют как тиенил, хинолинил, при условии, что Z в соединении структурной формулы I является СО2Н,
Х является -О-, R9 является Н, С1-С6-алкилом, R10 является водородом, R3a является Н, F, Cl, J и R3b является F, Cl, Br, J, OR7, где R7 является С1-С6-алкилом, тогда R1 и R2 не являются оба водородом или вместе у соседних атомов углерода не образуют циклическую группу  где А представляет собой -CR4=CR5-CR4=CR5-, где R4 и R5 являются водородом;
где А представляет собой -CR4=CR5-CR4=CR5-, где R4 и R5 являются водородом;
или их фармацевтически приемлемые соли.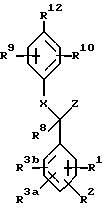
где R1, R2, R3a, R3b, R8, R9, R10 и R12 принимают значения, определенные в п.1,
или его фармацевтически приемлемая соль.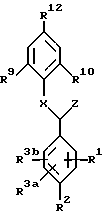
в которой R1, R2, R3a и R3b независимо являются: (а) Н; (b) F, Cl, Br или J, с) OR7,
R1 и R2 у соседних атомов углерода могут соединяться с образованием циклической структуры
А представляет a) -Y-[C(R6)(R6)]s-Y-; b) -С(R4)=C(R5)-C(R4)=C(R5)-;
s равно 1 или 2,
Y равен -О-,
R4 и R5 независимо являются водородом;
R6 является водородом;
R7 является: (а) Н; (b) (С1-С6)-алкилом; (с) фенилом,
R8 является водородом;
R9 и R10 независимо являются: (a) H; (b) (С1-С6)-алкилом; (с) (С1-С6)-алкокси;
R11 является (а) (С1-С6)-алкилом; (b) арилом, где арил определяют как фенил, который не замещен или замещен одним или двумя заместителями, выбранными из (С1-С4)-алкила;
R12 является: (а) Н; (b) (С1-С4)-алкилом, незамещенным или замещенным одним или двумя заместителями, выбранными из группы, состоящей из: i) -ОН; ii) -COOR7; iii) -CONHR7; iv) -NR7CONR7R11; v) -NR7COOR11; (c) -COOR7; (d) -CONH2; (e) -CONR7R11; (f) -CONHSO2R11; (g) -SO2NR7R11; (h) -NR7SO2NR7R11; (j) -CO- аминокислотой, где аминокислоту определяют как L- или D-Ala и которая может быть замещена (С1-С6)-алкиловым эфиром; (k)
X является -O-;
Z является: (a) -CO2H; (b) -CONHSO2 - арилом, где арил определяют как фенил или нафтил, который является незамещенным или замещенным одним или двумя заместителями, выбранными из группы, состоящей из: i) (С1-С4)-алкила; ii) -О-(С1-С4)-алкила; iii) F, Cl, Br или J, iv) -COOR7; v) NH2; vi) -NH[(С1-С4)-алкила]; vii) -N[(С1-С4)-алкила]2; viii) фенила
или представляет собой (c) -СОNНSO2-(С1-С8)-алкил, где алкил является незамещенным или замещенным фенилом, (d) -СОNНSO2-гетероарил, где гетероарил определяют как тиенил, хинолинил;
или его фармацевтически приемлемые соли.
в которой R1 и R2, взятые вместе, образуют циклическую структуру:
А представляет (a) -Y -[C(R6)(R6)]s-Y-; b) -С(R4)=С(R5)-С(R4)=С(5)-;
s равно 1 или 2;
Y равен -O-;
R3a является: (а) Н; (b) F, Cl, Br или I, (с) -OR7;
R4 и R5 независимо являются вoдopoдoм;
R6 является водородом;
R7 является: (а) Н; (b) (С1-С6)-алкилом; (с) фенилом;
R9 является: (a) H; (b) (С1-С6)-алкилом; (c) (С1-С6)-алкокси;
R11 является: (а) (С1-С6)-алкилом; (b) арилом, где арил определяют как фенил, который не замещен или замещен одним или двумя заместителями, выбранными из (С1-С4)-алкила,
R12 является: (а) Н; (b) (С1-С6)-алкилом, незамещенным или замещенным одним или двумя заместителями, выбранными из группы, состоящей из: i) -ОН; ii) -COOR7, iii) -CONHR7; iv) -NR7CONR7R11; (v) -NR7COOR11; (с) -СOOR7; (d) -CONH2; (e) -CONR7R11; (i) -CONHSO2R11; (g) -SO2NR7R11; (h) -NR7SO2NR7R11; (j) -СО-аминокислотой, где аминокислоту определяют как L- или D-Ala, которая может быть замещена (С1-С6) алкиловым эфиром; (k)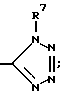
X является -О-,
Z является: (а) -СО2Н; (b) -CONHSO2-арилом, где арил определяют как фенил, который является незамещенным или замещенным двумя заместителями, выбранными из группы, состоящей из: i) (С1-С4)-алкила; ii) -О-(С1-С4)-алкила; iii) F, Cl, Br или J; iv) -COOR7; v) -NH2; vi) -NH[(С1-С4)-алкила] ; vii) -N[(С1-С4)-алкила] 2; viii) фенила; (с) -СОNНSO2-(С1-С8)-алкилом, где алкил является незамещенным или замещенным фенилом; (d) -СОNНSO2-гетероарилом, где гетероарил определяют как тиенил, хинолинил;
или его фармацевтически приемлемые соли.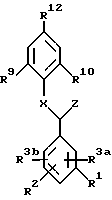
в которой R1, R2, R3a и R3b независимо являются: (а) Н; (b) F, Cl, Br или J, (с) -OR7,
R4 и R5 независимо являются водородом;
R6 является водородом;
R7 является: (а) Н; (b) (С1-С6)-алкилом; (с) фенилом;
R9 и R10 независимо являются: (а) Н; (b) (С1-С6)-алкилом; (с) (С1-С6)-алкокси;
R11 является: (а) (С1-С6)-алкилом; (b) арилом, где арил определяют как фенил, который не замещен или замещен одним или двумя заместителями, выбранными из (С1-С4)-алкила,
R12 является: (a) H; (b) (С1-С6)-алкилом, незамещенным или замещенным одним или двумя заместителями, выбранными из группы, состоящей из: i) -ОН; ii) -COOR7; iii) -CONHR7, iv) -NR7CONR7R11; v) -NR7COOR11; (c) -COOR7; (d) -CONH2; (e) -CONR7R11; (i) -CONHSO2R11; g) -SO2NR7R11; (h) -NR7SO2NR7R11; (j) -CO-аминокислотой, где аминокислоту определяют как L- или D-Ala, которая может быть замещена (С1-С6)-алкиловым эфиром; (k)
Х является -О-,
Z является: (а) -СО2Н; (b) -СОNНSO2-арилом, где арил определяют как фенил или нафтил, которые являются незамещенными или замещенными двумя заместителями, выбранными из группы, состоящей из: i) (С1-С4)-алкила; ii) -О-(С1-С4)-алкила; iii) F, Cl, Br или J; iv) -COOR7; v) -NH2; vi) -NН[(С1-С4)-алкила] ; vii) -N[(С1-С4)-алкила] 2; viii) фенила; (с) -CONHSO2-(C1-C8)-алкилом, где алкил является незамещенным или замещенным фенилом, (d) -CONHSO2-гетероарилом, где гетероарил определяют как тиенил, хинолинил
или его фармацевтически приемлемые соли.
где R1 и R2 представлены следующей кольцевой структурой
А представляет: (а) -Y-[C(R6)(R6)] s-Y-, или (b) - С(R4)=С(R5)-С(R4)= С(R5)-;
s равно 1 или 2;
Y равен -O-;
R3a и R3b независимо являются: (а) H; (b) F, Cl, Br или J; (с) -OR7;
R4 и R5 независимо являются водородом;
R6 является водородом;
R7 является: (а) Н; (b) (С1-С6)-алкилом; (с) фенилом;
R9 и R10 независимо являются: (а) Н; (b) (С1-С6)-алкилом; (с) (С1-С6)-алкокси;
R11 является: (а) (С1-С6)-алкилом; (b) арилом, где арил определяют как фенил, который не замещен или замещен одним или двумя заместителями, выбранными из (С1-С4)-алкила,
R12 является (а) Н; (b) (С1-С4)-алкилом, незамещенным или замещенным одним или двумя заместителями, выбранными из группы, состоящей из i) -OH; ii) - СOOR7; iii) -CONHR7; iv) -NR7CONR7R11; v) -NR7COOR11; (c) -СOOR7; (d) -CONH2; (e) -CONR7R11; (i) -CONHSO2R11; (g) -SO2NR7R11; (h) -NR7SO2NR7R11; (j) -CO-аминокислотой, где аминокислоту определяют как L- или D-Ala, которая может быть замещена (С1-С6)-алкиловым эфиром; (k)
Х является -О-,
Z является: (а) -СО2Н; (b) -CONHSO2- арилом, где арил определяют как фенил или нафтил, которые являются незамещенными или замещенными двумя заместителями, выбранными из группы, состоящей из: i) (С1-С4)-алкила; ii) -О-(С1-С4)-алкила; iii) F, Cl, Br или J; iv) -COOR7; v) -NH2; vi) -NH[(С1-С4)-алкила] ; vii) -N[(С1-С4)-алкила] 2; viii) фенила; (с) -СОNНSO2-(С1-С6)-алкилом, где алкил является незамещенным или замещенным фенилом; (d) - СONНSO2-гетероарилом, где гетероарил определяют как тиенил, хинолинил;
или его фармацевтически приемлемые соли.
(1) 2-[(2,6-дипропил-4-гидроксиметил)фенокси]-2-(3-метилфенил) уксусной кислоты;
(2) 2-[(2,6-дипропил-4-гидроксиметил)фенокси]-2-(4-феноксифенил) уксусной кислоты;
(3) 2-[(2,6-дипропил-4-гидроксиметил)фенокси]-2-(4-фенилфенил) уксусной кислоты;
(4) 2-[(2,6-дипропил-4-гидроксиметил)фенокси]-2-(3-карбоксифенил) уксусной кислоты;
(5) 2-[(2,6-дипропил-4-гидроксиметил)фенокси]-2-(3,4-этилендиоксифенил) уксусной кислоты;
(6) 2-[(2,6-дипропил-4-гидроксиметил)фенокси]-2-(3,4,5-триметоксифенил) уксусной кислоты;
(7) 2-[(2,6-дипропил-4-гидроксиметил)фенокси]-2-(3,4-метилендиоксифенил) уксусной кислоты;
(8) 2-[(2,6-дипропил-4-гидроксиметил)фенокси]-2-(3,4-диметоксифенил) уксусной кислоты;
(9)2-[(2,6-дипропил-4-гидроксиметил)фенокси] -2-(3,5-диметоксифенил) уксусной кислоты;
(10) 2-((2,6-дипропил-4-тетразол-5-ил)фенокси)-2-(3-бромфенил) уксусной кислоты;
(11) 2-[(2,6-дипропил-4-гидроксиметил)фенокси]-2-(3-бромфенил) уксусной кислоты;
(12) 2-[(2,6-дипропил-4-гидроксиметил)фенокси]-2-(2-нафтил)уксусной кислоты;
(13) 2-[(2,6-дипропил-4(2-гидроксиэтил)фенокси]-2-(2-нафтил)уксусной кислоты;
(14) 2-[(2,6-дипропил-4(2-гидроксиэтил)фенокси] -2-(3,4-метилендиоксифенил)уксусной кислоты;
(15) 2-[(2,6-дипропил-4(2-гидроксиэтил)фенокси] -2-(3-метоксифенил)уксусной кислоты;
(16) 2-[(2,6-дипропил-4(1,2-дигидроксиэтил)фенокси]-2-(2-нафтил)уксусной кислоты;
(17) 2-[(2,6-дипропил-4(1-гидроксипентил)фенокси] -2-(2-нафтил)уксусной кислоты;
(18) 2-[(4-кар6окси-2,6-дипропил)фенокси]-2-фенилуксусной кислоты;
(19) 2-[(4-карбокси-2,6-дипропил)фенокси] -2-(3,4-дихлорфенил)уксусной кислоты;
(20) 2-[(4-карбокси-2,6-дипропил)фенокси] -2-(3-бромфенил)уксусной кислоты;
(21) 2-[(4-кар6окси-2,6-дипропил)фенокси]-2-[3,4-метилендиоксифенил]уксусной кислоты;
(22) 2-[(4-карбокси-2,6-дипропил)фенокси] -2-(3-метоксифенил)уксусной кислоты;
(23) (N-бензолсульфонил)-2-[(4-(N-6ензолсульфонил)карбоксамидо-2,6-дипропилфенокси]-2-(3-бромфенил)ацетамида;
(24) (N-4-трет-бутилбензолсульфонил)-2-(4-метоксикарбонил-2-пропилфенокси)-2-(3,4-метилендиоксифенил)ацетамида;
(25) N-(бензолсульфонил)-2-(4-метоксикарбонил-2-пропилфенокси)-2-(3,4-метилендиоксифенил)ацетамида;
(26) N-(4-фенилбензолсульфонил)-2-(4-метоксикарбонил-2-пропилфенокси)-2-(3,4-метилендиоксифенил)ацетамида;
(27) N-(4-хлорбензолсульфонил)-2-(4-метоксикарбонил-2-пропилфенокси)-2-(3,4-метилендиоксифенил)ацетамида;
(28) N-(4-метилбензолсульфонил)-2-(4-метоксикарбонил-2-пропилфенокси)-2-(3,4-метилендиоксифенил)ацетамида;
(29) N-(5-изо-бутилтиен-2-илсульфонил)-2-(4-метоксикарбонил-2-пpoпилфeнoкcи)-2-(3,4-мeтилeндиoкcифeнил)aцeтaмидa;
(30) N-(4-метоксибензолсульфонил)-2-(4-метоксикарбонил-2-пропилфенокси)-2-(3,4-метилендиоксифенил)ацетамида;
(31) N-(4-диметиламинобензолсульфонил)-2-(4-метоксикарбонил-2-пропилфенокси)-2-(3,4-метилендиоксифенил)ацетамида;
(32) N-(2-метилбензолсульфонил)-2-(4-метоксикарбонил-2-пропилфенокси)-2-(3,4-метилендиоксифенил)ацетамида;
(33) N-(2-метоксикарбонилбензолсульфонил)-2-(4-метоксикарбонил-2-пропилфенокси)-2-(3,4-метилендиоксифенил)ацетамида;
(34) N-(2-хлорбензолсульфонил)-2-(4-метоксикарбонил-2-пропилфенокси)-2-(3,4-метилендиоксифенил)ацетамида;
(35) N-(3-xлopбeнзoлcульфoнил)-2-(4-мeтoкcикapбoнил-2-пропилфенокси)-2-(3,4-метилендиоксифенил)ацетамида;
(36) N-(фенилметансульфонил)-2-(4-метоксикарбонил-2-пропилфенокси)-2-(3,4-метилендиоксифенил)ацетамида;
(37) N-(данзилсульфонил)-2-(4-метоксикарбонил-2-пропилфенокси)-2-(3,4-метилендиоксифенил)ацетамида;
(38) N-(8-хинолинсульфонил)-2-(4-метоксикарбонил-2-пропилфенокси)-2-(3,4-метилендиоксифенил)ацетамида;
(39) N-(4-трет-бутилбензолсульфонил)-2-(4-карбокси-2-пропилфенокси)-2-(3,4-метилендиоксифенил)ацетамида;
(40) N-(бензолсульфонил)-2-(4-карбокси-2-пропилфенокси)-2-(3,4-метилендиоксифенил)ацетамида;
(41) N-(4-фенилбензолсульфонил)-2-(4-карбокси-2-пропилфенокси)-2-(3,4-метилендиоксифенил)ацетамида;
(42) N-(4-хлорбензолсульфонил)-2-(4-карбокси-2-пропилфенокси)-2-(3,4-метилендиоксифенил)ацетамида;
(43) N-(4-мeтилбeнзoлcульфoнил)-2-(4-кapбoкcи-2-пpoпилфeнoкcи)-2-(3,4-метилендиоксифенил)ацетамида;
(44) N-(5-изобутилтиен-2-илсульфонил)-2-(4-карбокси-2-пропилфенокси)-2-(3,4-метилендиоксифенил)ацетамида;
(45) N-(4-метоксибензолсульфонил)-2-(4-карбокси-2-пропилфенокси)-2-(3,4-метилендиоксифенил)ацетамида;
(46) N-(4-диметиламинобензолсульфонил)-2-(4-карбокси-2-пропилфенокси)-2-(3,4-метилендиоксифенил)ацетамида;
(47) N-(2-метилбензолсульфонил)-2-(4-карбокси-2-пропилфенокси)-2-(3,4-метилендиоксифенил)ацетамида;
(48) N-(2-метоксикарбонилбензолсульфонил)-2-(4-карбокси-2-пропилфенокси)-2-(3,4-метилендиоксифенил)ацетамида;
(49) N-(2-хлорбензолсульфонил)-2-(4-карбокси-2-пропилфенокси)-2-(3,4-метилендиоксифенил)ацетамида;
(50) N-(3-хлорбензолсульфонил)-2-(4-карбокси-2-пропилфенокси)-2-(3,4-метилендиоксифенил)ацетамида;
(51) N-(фeнилмeтaнcульфoнил)-2-(4-кapбoкcи-2-пpoпилфeнoкcи)-2-(3,4-метилендиоксифенил)ацетамида;
(52) N-(данзилсульфонил)-2-(4-карбокси-2-пропилфенокси)-2-(3,4-метилендиоксифенил)ацетамида;
(53) N-(8-хинолинсульфонил)-2-(4-карбокси-2-пропилфенокси)-2-(3,4-метилендиоксифенил)ацетамида;
(54) N-(8-хинолинсульфонил)-2-(4-карбоксамидо-2-пропилфенокси)-2-(3,4-метилендиоксифенил)ацетамида;
(55) α-(4-карбметокси-2-н-пропилфенокси)-3,4-метилендиоксифенилуксусной кислоты;
(56) N-(4-изo-пpoпилбeнзoлcульфoнил)-α-(4-кapбмeтoкcи-2-н-пропилфенокси)-3,4-метилендиоксифенилацетамида;
(57) двукалиевая соль N-(4-изо-пропилбензолсульфонил)-α-(4-карбокси-2-н-пропилфенокси)-3,4-метилендиоксифенилацетамида;
(58) α-(2-изо-бутил-4-карбометоксифенокси)-3,4-метилендиоксифенилуксусной кислоты;
(59) N-(4-изо-пропилбензолсульфонил)-α-(2-изо-бутил-4-карбометоксифенокси)3,4-метилендиоксифенилацетамида;
(60) N-(4-изо-пропилбензолсульфонил)-α-(2-изо-бутил-4-карбоксифенокси)3,4-метилендиоксифенилацетамида;
(61) N-(4-изo-пpoпилбeнзoлcульфoнил)-α-(2-н-пpoпил-4-метоксикарбонилфенокси)-α-метил-3,4-метилендиоксифенилацетамида;
(62) двукалиевой соли N-(4-изо-пропилбензолсульфонил)-α-(2-н-пропил-4-карбоксифенокси)-α-метил-3,4-метилендиоксифенилацетамида;
(63) N-(4-изо-пропилбензолсульфонил)-α-(2-н-пропил-4-карбоксамидофенокси)3,4-метилендиоксифенилацетамида;
(64) N-(4-изо-пропилбензолсульфонил)-α-(2-н-пропил-4-гидроксиметилфенокси)3,4-метилендиоксифенилацетамида;
(65) N-(4-изо-пропилбензолсульфонил)-α-(4-формил-2-н-пропилфенокси)3,4-метилендиоксифенилацетамида;
(66) α-(4-ацетил-2-н-пропилфенокси)-3,4-метилендиоксифенилуксусной кислоты;
(67) N-(4-изо-пропилбензолсульфонил)-α-(4-ацетил-2-н-пропилфенокси)3,4-метилендиоксифенилацетамида;
(68) α-(2-н-пропилфенокси)-3,4-метилендиоксифенилуксусной кислоты;
(69) N-(4-изо-пропилбензолсульфонил)-α-(2-н-пропилфенокси)3,4-метилендиоксифенилацетамида;
(70) α-(3-метоксифенокси)-3,4-метилендиоксифенилуксусной кислоты;
(71) α-(2-(2-гидроксиэтил)фенокси)-3,4-метилендиоксифенилуксусной кислоты;
(72) α-(2-(2-карбметоксиэтил)фенокси)-3,4-метилендиоксифенилуксусной кислоты;
(73) α-(4-гидроксиметил-2-н-пропилфенокси)-3,4-метилендиоксифенилуксусной кислоты;
(74) α-(4-(2-гидроксиэтил)-2-н-пропилфенокси)-3,4-метилендиоксифенилуксусной кислоты;
(75) N-(4-изо-пропилбензолсульфонил)-α-(2-(2-карбметоксиэтил)фенокси)3,4-метилендиоксифенилацетамида;
(76) N-(4-изо-пропилбензолсульфонил)-α-(2-(2-карбоксиэтил)фенокси)3,4-метилендиоксифенилацетамида;
(77) α-(2-(2-карбоксиэтил)фенокси)-3,4-метилендиоксифенилуксусной кислоты;
(78) N-(4-изо-пропилбензолсульфонил)-2-(4-карбометокси-2-н-пропилфенокси)-2-(5-метокси-3,4-метилендиоксифенил)ацетамида;
(79) N-(4-изo-пpoпилбeнзoлcульфoнил)-2-(4-кapбoкcи-2-пpoпилфeнoкcи)-2-(5-метокси-3,4-метилендиоксифенил)ацетамида;
(80) N-(4-изo-пpoпилбeнзoлcульфoнил)-2-(4-(N-(4-изo-пропилбензолсульфонил)карбоксамидо)-2-пропилфенокси)-2-(5-метокси-3,4-метилендиоксифенил)ацетамида;
(81) N-(4-изо-пропилбензолсульфонил)-2-(4-карбоксамидо-2-пропилфенокси)-2-(5-метокси-3,4-метилендиоксифенил)ацетамида;
(82) N-(4-изo-пpoпилбeнзoлcульфoнил)-2-(4-(N-мeтилкapбoкcaмидo)-2-пропилфенокси)-2-(5-метокси-3,4-метилендиоксифенил)ацетамида;
(83) N-(4-изо-пропилбензолсульфонил)-2-(4-(N-2-гидроксиэтилкарбоксамидо)-2-пропилфенокси)-2-(5-метокси-3,4-метилендиоксифенил)ацетамида;
(85) N-(4-изо-пропилбензолсульфонил)-2-(4-(N-3-метилбутилкарбоксамидо)-2-пропилфенокси)-2-(5-метокси-3,4-метилендиоксифенил)ацетамида;
(86) N-(4-изо-пропилбензолсульфонил)-2-(4-(N-карбоксиметилкарбоксамидо)-2-пропилфенокси)-2-(5-метокси-3,4-метилендиоксифенил)ацетамида;
(87) N-(4-изo-пpoпилбeнзoлcульфoнил)-2-(4-(N-(L-Ala-OEt)кapбoкcaмидo)-2-пропилфенокси)-2-(5-метокси-3,4-метилендиоксифенил)ацетамида;
(88) N-(4-изо-пропилбензолсульфонил)-2-(4-(N-2-этоксикарбонилэтилкарбоксамидо)-2-пропилфенокси)-2-(5-метокси-3,4-метилендиоксифенил)ацетамида;
(89) N-(4-изo-пpoпилбeнзoлcульфoнил)-2-(4-(N-(L-Ala)кapбoкcaмидo)-2-пропилфенокси)-2-(5-метокси-3,4-метилендиоксифенил)ацетамида;
(90) N-(4-изо-пропилбензолсульфонил)-2-(4-(N-2-карбоксиэтилкарбоксамидо)-2-пропилфенокси)-2-(5-метокси-3,4-метилендиоксифенил)ацетамида;
(91) N-(4-изо-пропилбензолсульфонил)-2-(4-(N-3-гидроксипропилкарбоксамидо)-2-пропилфенокси)-2-(5-метокси-3,4-метилендиоксифенил)ацетамида;
(92) N-(4-изо-пропилбензолсульфонил)-2-(4-(N-тетразол-5-илкарбоксамидо)-2-пропилфенокси)-2-(5-метокси-3,4-метилендиоксифенил)ацетамида;
(94) N-(4-изо-пропилбензолсульфонил)-2-(4-(N-(D-Аlа-ОМе)карбоксамидо)-2-пропилфенокси)-2-(5-метокси-3,4-метилендиоксифенил)ацетамида;
(95) N-(4-изo-пpoпилбeнзoлcульфoнил)-2-(4-(N-(D-Ala)кapбoкcaмидo)-2-пропилфенокси)-2-(5-метокси-3,4-метилендиоксифенил)ацетамида;
(96) N-(4-изо-пропилбензолсульфонил)-2-(4-(N-(3-карбоксиметилпропил)карбоксамидо)-2-пропилфенокси)-2-(5-метокси-3,4-метилендиоксифенил)ацетамида;
(97) N-(4-изо-пропилбензолсульфонил)-2-(4-(N-(3-карбоксипропил)карбоксамидо)-2-н-пропилфенокси)-2-(5-метокси-3,4-метилендиоксифенил)ацетамида;
(98) N-(4-изо-пропилбензолсульфонил)-2-(4-(N-изо-пропилкарбамоил)амино-2-н-пропилфенокси)-2-(3,4-метилендиоксифенил)ацетамида;
(99) α-(2-н-пропил-4-метиламиносульфонилфенокси)-3,4-метилендиоксифенилуксусной кислоты;
(100) калиевой соли N-(4-изо-пропилбензолсульфонил)-α-(2-н-пропил-4-метиламиносульфонилфенокси)-3,4-метилендиоксифенил)ацетамида;
(106) N-(2-мeтил-3-xинoлинcульфoнил)-2-(4-кapбoкcи-2-пpoпилфeнoкcи)-2-(3,4-метилендиоксифенил)ацетамида;
(107) N-(3-хинолинсульфонил)-2-(4-карбокси-2-пропилфенокси)-2-(3,4-метилендиоксифенил)ацетамида;
(115) N-[(2,5-диметокси)бензолсульфонил] -2-(4-карбокси-2-пропилфенокси)-2-(3,4-метилендиоксифенил)ацетамида;
(116) N-[(3,4-диметокси)бензолсульфонил] -2-(4-карбокси-2-пропилфенокси)-2-(3,4-метилендиоксифенил)ацетамида;
(122) N-[(2,4,6-триметокси)бензолсульфонил] -2-(4-карбокси-2-пропилфенокси)-2-(3,4-метилендиоксифенил)ацетамида;
(123) N-(8-xинoлинcульфoнил)-2-(4-кapбoкcи-2-пpoпилфeнoкcи)-2-(3,4-метилендиоксифенил)ацетамида;
(124) N-(3-xинoлинcульфoнил)-2-(4-кapбoкcи-2-пpoпилфeнoкcи)-2-(3,4-метилендиоксифенил)ацетамида;
(125) N-(8-хинолинсульфонил)-2-(4-карбоксамидо-2-пропилфенокси)-2-(5-метокси-3,4-метилендиоксифенил)ацетамида;
(128) N-[4-изо-пропилбензолсульфонил] -2-(4-карбокси-2-пропилфенокси)-2-(3,4-метилендиоксифенил)ацетамида;
(133) двукалиевой соли (-)-N-(4-изо-пропилбензолсульфонил)-α-(4-карбокси-2-н-пропилфенокси)-3,4-метилендиоксифенилацетамида;
(134) двукалиевой соли N-(4-изo-пpoпилбeнзoлcульфoнил)-α-(4-кapбoкcи-2-н-пропилфенокси)-3,4-метилендиоксифенилацетамида;
(135) N-(8-хинолинсульфонил)-2-(4-карбокси-2-пропилфенокси)-2-(3,4-метилендиоксифенил)ацетамида, и
(136) N-(4-диметиламинобензолсульфонил)-2-(4-карбокси-2-н-пропилфенокси)-2-(3,4-метилендиоксифенил)ацетамида.
(1) двукалиевой соли (-)-N-(4-изо-пропилбензолсульфонил)-α-(4-карбокси-2-н-пропилфенокси)-3,4-метилендиоксифенилацетамида;
(2) двукалиевой соли N-(4-изо-пропилбензолсульфонил)-α-(4-карбокси-2-н-пропилфенокси)-3,4-метилендиоксифенилацетамида;
(3) N-(8-хинолинсульфонил)-2-(4-карбокси-2-пропилфенокси)-2-(3,4-метилендиоксифенил)ацетамида; и
(4) N-(4-диметиламинобензолсульфонил)-2-(4-карбокси-2-н-пропилфенокси)-2-(3,4-метилендиоксифенил)ацетамида.
Приоритет по пунктам:
19.03.93 - по пп. 1-6, кроме R11 в) фенил, который замещен одним или двумя С1-С4 алкилами; R12- d) CONH2, h) -NR7SO2NR7R11, j) СО-аминокислотой, где аминокислоту определяют как L- или D-Ala, которая может быть замещена (C1-С6) алкиловым эфиром; k)
Z- b) -CONНSO2 - фенил, который является замещенным v) -NH2, vi) -NH (С1-С4)-алкилом, vii) -N(С1-С4)2-алкилом, viii) фенилом; -CONHSO2 - нафтил, который является незамещенным или замещен двумя заместителями, выбранными из группы, состоящей i) С1-С4-алкила, ii) -O-(С1-С4)-алкила, iii) F, Cl, Br, J, iv) -COOR7, v) -NH2, vi) -NH С1-С4-алкила, vii) -N(С1-С4)2-алкила, viii) фенила; с) -CONHSO2-(С1-С8)-алкил, где алкил замещен фенилом; d) -CONHSO2 - хинолинил;
24.02.94 - по пп. 1-17 и для перечисленных выше значений, кроме Z означает с) CONHSO2-(С1-С8)-алкил, который замещен фенилом.
| EP 0460679, 1991 | |||
| US 47448272, 1988 | |||
| Домовый номерной фонарь, служащий одновременно для указания названия улицы и номера дома и для освещения прилежащего участка улицы | 1917 |
|
SU93A1 |
| Машковский М.Д | |||
| Лекарственные средства | |||
| -М.: Медицина, 1993, ч | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
