Изобретение относится к области химии биологически активных соединений, в частности к получению соединений из группы тетрааминов, обладающих селективным сродством к M2-холинорецепторам.
Известно, что соли полиметилентетраминов общей формулы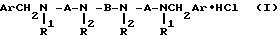
где A = (CH2)n
n = 4 - 8;
B = (CH2)m;
m = 4 - 12;
Ar = C6H5, o-CH3OC6H4, гетарил и т.п.;
R1, R2 = H или CH3 в разных сочетаниях,
способны блокировать M-холинореактивные системы, причем в большинстве случаев сродство их к M2 - подтипу рецепторов выше, чем к M1 и M3 [Minarini A. , Quaglia W., Tumiatti V., Melchiorre C.// Chem. and Ind. - 1989. -N 19. P. 652-653; Melchiorre C. , Quaglia W., Picchio M.T., Giardino P.// J.Med. Chem. -1989.V. 32, N 1.-P. 79-84.]
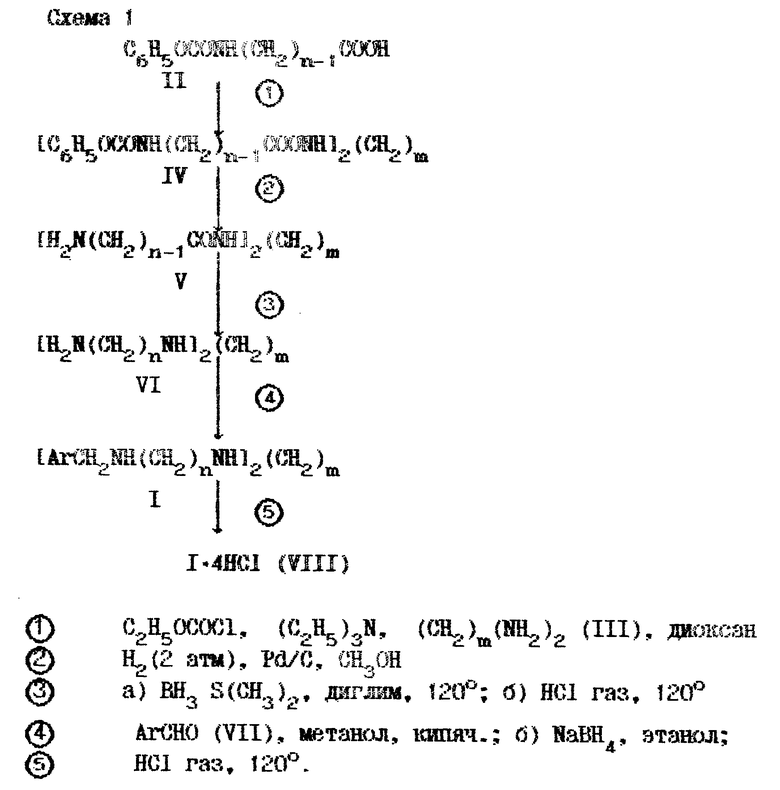
Синтез таких препаратов был осуществлен в соответствии со схемой 1, приведенной в конце описания. В ней на первой стадии карбобензилоксипроизводное ω-аминоалкановой кислоты (II) с помощью этилхлоругольного эфира превращается в смешанный ангидрид, которым затем без выделения ацилируют соответствующий полиметилендиамин (III). В образующемся тетраамиде (IV) защитные концевые группы удаляют гидролизом на палладиевом катализаторе и в диаминодиамиде (V) обе амидные группы восстанавливают комплексом боран-диметилсульфид. Образующийся тетраамин (VI) подвергают арилметилированию путем реакции с замещенным бензальдегидом (VII) и восстанавливают получающийся бис-азометил борогидридом натрия в целевой тетраамин I. В последней стадии основание превращают в тетрагидрохлорид (VIII).
Один из препаратов этого ряда (VIII с m = 8, Ar = 2-CH3OC6H4, R1 = R2 = H) под названием "метоктрамин" был отобран для клинических испытаний [Minarini A. , Quaglia W., Tumiatti V., Melchiorre C.// -Chem. and Ind. - 1989. -N 19. P.652-653; Melchiorre C., Quaglia W., Picchio M.T., Giardino P. // J. Med. Chem.-1989.-V. 32, N 1. - P. 79-84.]; он выбирается нами в качестве прототипа.
Описанным способом было синтезировано много представителей тетрааминов I, но во всех случаях остатки A и B, соединяющие атомы азота аминогрупп, представляли собой только полиметиленовые цепочки различной длины. Можно было ожидать, что включение в радикалы A и B арильных групп или гетероатомов должно существенно изменить свойства получаемых препаратов. Благодаря увеличению жесткости и полярности молекул в первом случае, а также по причине роста полярности и возрастания гидрофильности во втором, конформационные свойства соединений должны были измениться, что в свою очередь не могло бы сказаться на их биологической активности, в том числе и специфичности к подтипам рецептора.
Учитывая эти соображения, задачей настоящего изобретения является получение неизвестных ранее соединений из группы тетрааминов общей формулы I, где в радикале B часть группы CH2 заменена арильным остатком либо одним или двумя атомами кислорода. Его результатом явилось получение 8 неизвестных ранее солей тетрааминов общей формулы I, где Ar = C6H5, 2-CH3OC6H4 и B = 1,4-C6H4, 1,3-C6H4, (CH2)2O(CH2)2, CH2O(CH2)2OCH2, (CH3)3O(CH3)3,
(CH2)2O(CH2)2O(CH2)2; R1 = R2 = H. Их свойства приведены в табл.1, биологическая активность - в табл.2. Полученные данные позволяют предположить возможность применения синтезированных препаратов в медицине.
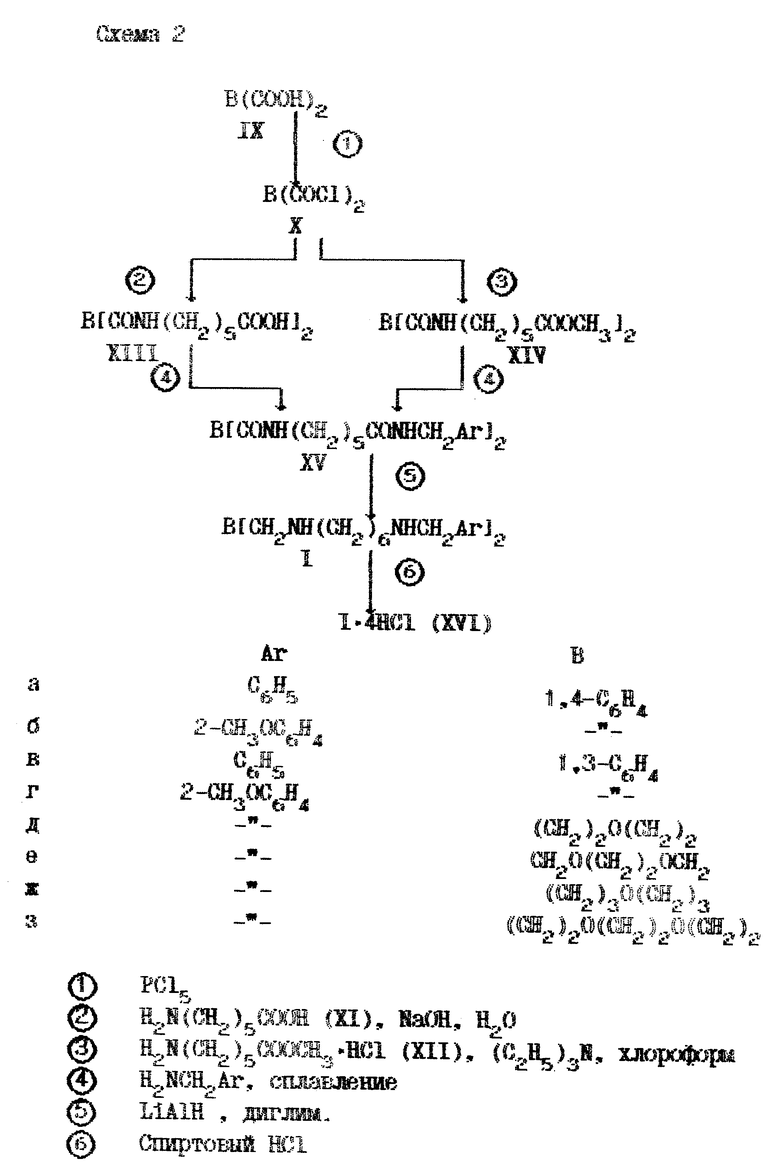
Для получения целевых соединений была разработана последовательность реакций, приведенная на схеме 2, представленной в конце описания. Как показано на ней, арилдикарбоновая кислота (IX) переводится в дихлорангидрид (X), которым ацилируется либо аминокапроновая кислота (XI) по Шоттен-Бауману, либо гидрохлорид ее метилового эфира (XII) в присутствии триэтиламина в органическом растворителе. Полученная при этом дикислота (XIII) или ее диметиловый эфир (XIV) в идентичных условиях сплавляют с подходящим бензиламином, что приводит к тетраамиду (XV). Восстановление последнего алюмогидридом лития в диглиме позволяет синтезировать целевой тетраамин (I), из которого обычным образом получают соль (XVI).
Описанным методом было синтезировано 8 соединений различного строения, их структура и некоторые свойства приведены в табл.1.
Для получения соединений было определено сродство к подтипам M-холинорецептора.
Оно определялось in vitro с помощью метода радиолигандного анализа [Yamamura H.J., Snyder H.S.// Proc. Natl. Acad. Sci USA.- 1974.-V. 71, N 5. - P. 1725-1729]. Для этого навески тканей больших полушарий мозга (M1-подтип), миокарда (M2-подтип) и слюнных желез (M3-подтип) крысы гомогенизировали в 10 объемах ледяной дистиллированной воды, фильтровали через 2 слоя марли, быстро замораживали и использовали в течение 4 дней. Для идентификации мускариновых рецепторов применяли меченный тритием высокоспецифичный мускариновый антагонист - хинуклидинилбензилат ([3H]-ХНБ) фирмы "Amerscham" с удельной активностью 37,0 Ки/ммоль. Гомогенат ткани из расчета 200 мкг белка мозга, 2 мг белка миокарда или 5 мг белка слюнной железы инкубировали в течение часа при 30oC в 50 мМ Na, K-фосфатного буфера с pH=7.4, содержащего [3H]-ХНБ или [3H]-ХНБ совместно с немеченным холинергическим лигандом. Неспецифическое связывание определяли в присутствии 6 мкМ атропина. Оно представляло собой разность между общим и специфическим связыванием и не превышало 15% от общего. После инкубации пробы фильтровали под вакуумом через задерживающие белок фильтры Watman, GF-c, дважды промывали ледяным фосфатным буфером для удаления несвязывающейся метки и помещали в виалы для счета радиоактивности. Пробы выдерживали в сцинтилляторе 24 часа при комнатной температуре и помещали на счет.
Счет радиоактивности проводили на жидкостно-сцинтилляционном счетчике "Марк-111" с использованием стандартного диоксанового сцинтиллятора с добавлением 10% Тритона Х-100. Эффективность счета составляла 40%.
Концентрацию М-ХР рассчитывали по величине максимального связывания (Bmax) меченого хинуклидинилбензилата на 1 мг белка. О сродстве меченого лиганда к рецептору судили по величине кажущейся константы диссоциации (KD) в условиях равновесного связывания. Величина KD определялась по графику Скетчарда [Scatchard G.// Ann. N.Y. Acad. Sci. - 1946. - N 51. - P. 660.] и была численно равна концентрации [3H]-ХНБ, при которой оккупировано 50% М-ХР. Сродство немеченных холинотропных лигандов к М-ХР характеризовалось их способностью конкурировать с [3H]-ХНБ за специфические участки связывания. Конкурентное вытеснение [3H] -ХНБ немеченными лигандами проводили при его насыщенной концентрации, равной 0,25 нМ для препаратов больших полушарий и миокарда и 0,5 нМ для препарата слюнной железы. Константы сродства холинотропных соединений к М-ХР рассчитывали по методу Ченга [Cheng J.C., Prusoff W. H. // Biochem. Pharmacol. - 1973. N 22. - P. 3099]. Сродство характеризовалось величиной I50, численно равной концентрации немеченного лиганда, необходимой для 50% вытеснения с рецептора [3H]-ХНБ, и значением константы ингибирования Ki: Ki = I50/(1 + D/KD), где D - насыщающая концентрация [3H]-ХНБ, KD - равновесная константа диссоциации [3H]-ХНБ.
График Скетчарда теряет свою линейность, если лиганд способен различать в препарате подтипы М-ХР и взаимодействовать с ними с различным сродством. В этом случае рассчитывали константы высокого (Khigh) и низкого (Klow) сродства к рецепторам и процентное содержание участков соответствующего сродства в общей популяции ХР методом нелинейного регрессивного анализа по специально разработанной программе.
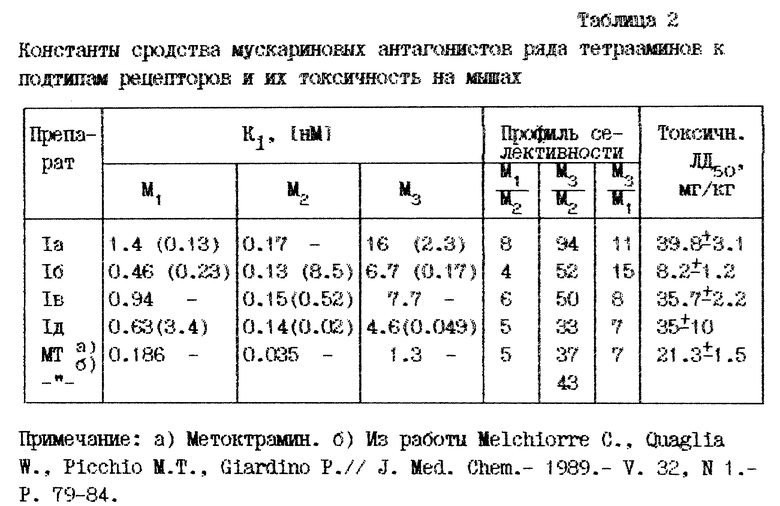
В табл. 2 помещены результаты определения специфичности к подтипам рецептора для некоторых из синтезированных препаратов: в каждом столбце приведена величина Khigh и в скобах величина Klow. В ней же помещены наши данные по метоктрамину и данные по этому препарату, приведенные в литературе [Melchiorre C. , Quaglia W. , Picchio M.T., Glardino P.// J. Med. Chem. - 1989. - V. 32, N 1. - P. 79 - 84.1. Как видно из их сравнения два из полученных тетрааминов не уступают по селективности метоктрамину, а соединение XVIIa существенно селективнее.
Токсичность полученных препаратов определена классическим способом на мышах при внутрибрюшинном введении: она приведена в последнем столбце табл. 2. Эти данные демонстрируют, что большинство из полученных тетрааминов существенно менее токсичны, чем метоктрамин, причем наиболее селективный препарат XVIIa превосходит последний по этому показателю вдвое.
Возможность осуществления изобретения подтверждается следующими примерами.
Примеры
Соли тетрааминов получали смешением спиртовых HCl или щавелевой кислоты и спиртового основания. Раствор соли упаривали в вакууме досуха и остаток кристаллизовали.
1. 1,4-Бис-(2',9'-диаза-10'-фенил-1'-децил)бензол (1a).
1.1 1,4-Бис-(2',9'-аза-8'-карбокси-1'-октанон-1'-ил)бензол (XIIIa).
2.03 г (10 ммоль) дихлорангидрида терефталевой кислоты растворяли в 20 мл сухого хлороформа, добавляли 2.62 г (20 ммоль) 6-аминокапроновой кислоты и к размешиваемой суспензии приливали по каплям 15 мл 20% водного КОН. Смесь оставляли при комнатной температуре на ночь, слои разделяли, водный слой подкисляли и выделившийся продукт отфильтровывали. После сушки и кристаллизации из диметилформамида получали 2.74 г (71%) вещества с т.пл. 218.5 - 220oC.
Спектр ПМР (CDCl3, δ, м.д.): 7.90 с. (4H, C6H4), 3.25 кв.(д.тр.) (4H, NCH2), 2.20 тр. (4H, COCH2), 1.70 - 1.30 м. (12 H, (CH2 )3).
1.2 1,4-Бис-(2'-аза-9'-окса-1',8'-декандион-1'-ил)бензол (XIVa).
2.03 г (10 ммоль) дихлорангидрида терефталевой кислоты и 3.64 г (20 ммоль) гидрохлорида метилового эфира 6-аминокапроновой кислоты смешивали с 20 мл сухого ацетонитрила и к суспензии прибавляли по каплям 6 мл триэтиламина. Осадок быстро почти полностью растворялся, но через несколько минут выпадал вновь. Массу перемешивали при комнатной температуре 2 часа, разбавляли водой, отфильтровывали осадок, промывали его 0,4 н. HCl и водой, сушили и кристаллизовали из хлороформа. Получали 2.02 г (48%) продукта с т.пл. 161 - 162oC.
Спектр ПМР (CDCl3, δ, м.д.): 7.75 с. (4H, C6H4), 6.91 уш.с. (2H, NH), 3.62 (6H, CH3), 3.43 кв. (д.тр.)(4H, NCH2), 2.30 тр. (4H, COCH2), 1.70 - 1.30 м. (12H, (CH2)3)
1.3 1,4-Бис-(2',9'-диаза-10'-фенил-1',8'-декандион-1'-ил)- бензол (XVa)
а) 3.0 г (72 ммоль) диэфира (XIVa) и 2.46 г (2.5 мл, 23 ммоль) бензиламина нагревали в колбе с обратным холодильником 2 часа до 200 - 210oС. Масса вначале плавилась, но через несколько минут закристаллизовывалась вновь. По окончании выдержки плав растворяли в спирте, разбавляли водой, осадок отфильтровывали, промывали водой и сушили. Получали 3.73 г (90%) продукта с т. пл. 223 - 225oC. Перекристаллизованный из диметилформамида тетраамид плавился при 233 - 235oC.
Спектр ПМР (ДМСО-d6) δ, м.д.): 7.87 с. (4H, C6H4), 7.27 м. (10H, C6H5, 4.25 д. (4H, ArCH2), 2.18 тр. (4H, COCH2), 1.70 - 1.30 м. (12H, (CH2)3). Сигналы групп NCH2 скрыты сигналом остаточной воды в растворителе.
б) Аналогичное сплавление кислоты (XIIIa) с небольшим избытком бензиламина дало 89% тетраамида (XIII) с теми же характеристиками, которые приведены выше.
1.4 1,4-Бис-(2',9'-диаза-10'-фенил-1'-децил)бензол (Ia)
5.5 г (9,6 ммоль) тетрааамида (XVa) прибавляли за один прием к суспензии 6,5 г (170 ммоль) алюмогидрида лития в сухом диглиме. Массу перемешивали, осторожно нагревали до 70oC и выдерживали при этой температуре 5 часов, после чего охлаждали, добавляли по каплям 2 - 3 мл этанола и 20 мл 20% NaOH. Диглим сливали с образовавшегося шлама, который еще дважды тщательно промывали эфиром. После сушки объединенного экстракта и удаления растворителя получили 5.57 г слегка увлажненного диглимом осадка. Кристаллизация его из ацетона дала 2.86 г (56%) вещества с т.пл. 61 - 62oC. Дополнительная кристаллизация повысила ее до 65 - 66oC. Основание, выделенное из очищенного гидрохлорида, плавилось при 71 - 72oC.
Спектр ПМР (CDCl3, δ, м.д.): 7.3 м. (C6H4, C6H5), 3.80 с. (4H, ArCH2), 3.62 тр. (8H,NCH2), 1.60 - 1.25 м. (16H, (CH2)4).
2. 13-Окса-2,9,17,24-тетрааза-1,25-бис(2-метоксифенил)пентакозан (Iд).
2.1 2,13,24-триокса-9,17-диазапентакозан-3,10,16,23-тетрон (XIVд)
К суспензии 8.4 г (46 ммоль) гидрохлорида метилового эфира 6-аминокапроновой кислоты в 75 мл сухого хлороформа, охлажденного до -10oC, прибавляли при перемешивании 12.8 мл (92 ммоль) триэтиламина, а затем медленно при температуре не выше 0oC добавляли дихлорангидрид 4-окса-1,7-гександионовой кислоты (получен обычным образом из 3.25 г (20 ммоль) соответствующей кислоты и SOCl2) и смесь оставили на ночь. На следующий день массу промыли 10% HCl, 10% K2CO3, водой и высушивали. После удаления хлороформа получено 6.0 г (72%) сырого кристаллического продукты. Перекристаллизованный из смеси спирта и эфира диэфир плавился при 83 - 84oC.
Спектр ПМР (CDCl3) δ, м.д.): 6.51 уш.с. (2H, NH), 3.65 т. (4H, OCH2), 3.60 с. (6H, OCH3), 3.17 д.т. (4H, NCH2), 2.38 т. (4H, NCOCH2), 2.25 т. (4H, OCOCH2), 1.65 - 1.20 м. (12H, (CH2)3).
2.2 13-Окса-2,9,17,24-тетрааза-1,25-бис-(2-метоксифенил)-пентакозан- (57) 3,10,16,23-тетрон (XVд)
2.1 г (5 ммоль) диэфира (XIVд) и 1.5 г (11 ммоль) о-метоксибензиламина нагревали с обратным холодильником 3 ч при 160 - 170oC. Вязкое масло растворили в хлороформе, промыли 10% HCl, водой и после сушки раствора хлороформ удаляли. Выход сырого продукта 38%, т.пл. 115 - 117oC.
Спектр ПМР (CDCl3 δ, м.д.): 7.31 - 7.18 м., 6.35 - 6.82 м. (8H, C6H4), 6.78 уш. с. (2H, ArCNH), 6.38 уш.с. (2H, NH), 4.40 д. (4H, ArCH2), 3.83 с. (6H, OCH3), 3.67 тр. (4H, OCH2), 3.20 д.тр. (4H, NCH2), 2.43 тр. (4H, COCH2), 2.19 тр. (4H, COCH2), 1.69 - 1.25 м. (12H, (CH2)3).
2.3 13-Окса-2,9,17,24-тетрааза-1,25-бис-(2-метоксифенил)пентакозан (Iд).
Получен так же, как продукт (Ia), из тетраамида XVд и LiA1H4 в диглиме. Выход сырого продукта (не закристаллизовался) 87%.
Спектр ПМР (CDCl3, δ, м.д.): 7.31 - 7.15 м., 6.95 - 6.79 (8H, C6H4), 3.80 с. (6H, OCH3), 3.73 с. (4H, ArCН2), 3.51 тр. (4H, OCH2), 2.56 м. (12H, NCH2), 1.58 - 1.20 м. (20H, (CH2)4, CH2).
Остальные продукты, приведенные в табл. 1, получены по той же схеме.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ S-ДИАЛКИЛ, АЛКИЛФЕНИЛ- ИЛИ ДИФЕНИЛАРСИНИСТЫХ ЭФИРОВ 4-МЕТОКСИФЕНИЛДИТИОФОСФОНОВЫХ КИСЛОТ | 1997 |
|
RU2124520C1 |
| МАКРОЦИКЛИЧЕСКИЕ АЛКИЛАММОНИЕВЫЕ ПРОИЗВОДНЫЕ 6-МЕТИЛУРАЦИЛА, ОБЛАДАЮЩИЕ АНТИХОЛИНЭСТЕРАЗНОЙ АКТИВНОСТЬЮ | 2013 |
|
RU2534903C1 |
| ПРОИЗВОДНЫЕ N-АЦИЛПРОЛИЛДИПЕПТИДОВ | 1993 |
|
RU2119496C1 |
| СПОСОБ ПОЛУЧЕНИЯ СУЛЬФАНИЛПРОИЗВОДНЫХ АНТИПИРИНА | 2020 |
|
RU2740911C1 |
| СРЕДСТВО, ОБЛАДАЮЩЕЕ КАРДИОПРОТЕКТОРНЫМ ДЕЙСТВИЕМ, И ГАЛОГЕНИДЫ 1,3-ДИЗАМЕЩЕННЫХ 2-АМИНОБЕНЗИМИДАЗОЛИЯ | 2013 |
|
RU2526902C1 |
| ПРИМЕНЕНИЕ ГИДРОХЛОРИДА 1-ФЕНИЛ-1-(1`-МЕТИЛЦИКЛОПЕНТИЛ)-4-ПИПЕРИДИНО-2-БУТИН-1-ОЛА В КАЧЕСТВЕ СРЕДСТВА, ОБЛАДАЮЩЕГО М1 ХОЛИНОБЛОКИРУЮЩЕЙ АКТИВНОСТЬЮ | 2004 |
|
RU2273478C2 |
| Низкотоксичные аминофосфониевые соли, обладающие противоопухолевой активностью, и способ их получения | 2024 |
|
RU2836726C1 |
| ПРОИЗВОДНЫЕ ЯНТАРНОЙ КИСЛОТЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ ДЛЯ ИХ СИНТЕЗА И СПОСОБ ПОЛУЧЕНИЯ ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ | 1997 |
|
RU2125040C1 |
| Способ получения гидроксамовых кислот, производных 2-арил-2,3-дигидрохиназолин-4(1Н)-онов | 2020 |
|
RU2744750C1 |
| АНАЛОГИ ВИТАМИНА D, СПОСОБ ИХ ПОЛУЧЕНИЯ, ДИАСТЕРЕОИЗОМЕР, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1993 |
|
RU2163234C2 |


Новые гидрохлориды или оксалаты N, N'-бис-(6-арилметил-аминогексил)-1,4- и -1,3-диаминометилбензолов или α,w- полиметилен- и α,w- полиоксаполиметилендиаминов общей формулы
ArCH2NH(CH2)6 NHCH2BCH2NH(CH2)6 NHCH2 Ar•4X
где Ar = C6H5, 2-CH3OC6H4; B = 1,4-C6H4, 1,3-C6H4, (CH2)2O(CH2)2, CH2O(CH2)2, OCH2, (CH2)3O(CH2)3, (CH2)2O(CH2)2O(CH2)2, (CH2)6, X = HCl, H2C2O4/2, обладающих избирательным сродством к М-холинорецепторам. 2 табл.
Новые гидрохлориды или оксалаты N,N'-бис-(6-арилметиламиногексил)-1,4- и -1,3-диаминометилбензолов или -α,w-полиметилен- и -α,w-полиоксаполиметилендиаминов общей формулы
ArCH2NH(CH2)6NHCH2 BCH2NH(CH2)6NHCH2Ar • 4X
где Ar = C6H5, 2-CH3OC6H4;
B = 1,4-C6H4, 1,3-C6H4, (CH2)2O(CH2)2, CH2O(CH2)2OCH2, (CH2)3O(CH2)3,
(CH2)2O(CH2)2O(CH2)2, (CH2)6;
X = HCl, H2C2O4/2,
обладающих избирательным сродством к M2-холинорецепторам.
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Minarini A., Quaglia W., Tumiatti V., Melchiorre C | |||
| Chem | |||
| and Ind, 1989, N 19, P | |||
| Оконное сигнальное приспособление | 1923 |
|
SU652A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Yamamura H.J., Snyder H., S | |||
| Proc | |||
| Nati | |||
| Acad | |||
| Sci USA, 1974, V | |||
| Контрольный стрелочный замок | 1920 |
|
SU71A1 |
| Телефонная трансляция с катодным реле | 1920 |
|
SU1725A1 |
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Scatchard G | |||
| Ann.N.Y | |||
| Acad | |||
| Sci, 1946, N 51, P | |||
| Льновыдергивающая машина | 1923 |
|
SU660A1 |
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |
| Cheng J.C., Prusoff W.H | |||
| Приспособление для склейки фанер в стыках | 1924 |
|
SU1973A1 |
| КАССОВЫЙ КОНТРОЛЬНО-СЧЕТНЫЙ ПРИБОР | 1924 |
|
SU3099A1 |
| Кипятильник для воды | 1921 |
|
SU5A1 |
| Melchiorre C., Quaglia W., Picchio M.T., Giardino P | |||
| J | |||
| Med | |||
| Chem., 1989, V | |||
| Способ образования коричневых окрасок на волокне из кашу кубической и подобных производных кашевого ряда | 1922 |
|
SU32A1 |
| Цилиндрический сушильный шкаф с двойными стенками | 0 |
|
SU79A1 |
