Изобретение относится к новым фторсодержащим органическим соединениям, более конкретно к производным амина, обладающим биологической активностью.
Известны азотсодержащие органические соединения, в частности N-гетероциклические соединения, которые обладают биологической активностью и могут применяться в качестве активного вещества фармацевтической композиции, проявляющей, например, средство в отношении рецепторов 5-HT1A и D2 (см. заявку EP N 0054304, опубликованную 23-го июня 1982 г.).
Задачей изобретения является расширение ассортимента высокоэффективных производных амина со сродством в отношении рецепторов 5-HT1 и D2.
Поставленная задача решается предлагаемыми производными амина формулы (I)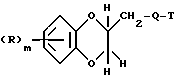
где R - галоид, гидроксил, алкил с 1 - 3 атомами углерода, алкоксил с 1 - 3 атома ми углерода,
Q - двухвалентная группа формулы (IIa), (IIб),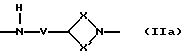
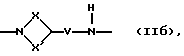
где V - метилен или этилен,
X - алкиленовая цепь с 0 - 2 атомами углерода,
X' - алкиленовая цепь с 1 - 4 атомами углерода,
при этом общее число атомов углерода в остатках Х и X' составляет 3 или 4,
T - фенил, пиридил, пиразинил, бензо[b]фуранил, 1,4-бензодиоксанил, и хиназолинил, незамещенные или замещенные галоидом, метоксилом или трифторметилом,
m - 0,1,
и их фармацевтически приемлемые соли.
В первую группу предпочтительных соединений входят соединения, у которых R означает гидроксил, метокси, фтор или хлор.
Во вторую группу предпочтительных соединений входят соединения, у которых T означает 2-пиридил, 2-пиразинил, фенил, 2,3-дигидро-бензо[b]фуран-7-ил, 1,4-бензодиоксан-5-ил или 4-хиназолинил, незамещенные или замещенные метоксилом, трифторметилом или галоидом.
В третью группу предпочтительных соединений входят соединения, выбранные из группы, включающей: N-(1,4-бензодиоксан-2-илметил)-1-[1-(пиразин-2-ил)пиперид-4-ил] метиламин; N-(1,4-бензодиоксан-2-илметил)-1-[1-(2-метоксифенил)пиперид-4-ил] метиламин; N-(1,4-бензодиоксан-2-илметил)-1-[1-(3-хлорпирид-2-ил)пиперид-4-ил] метиламин; N-(1,4-бензодиоксан-2-илметил)-1-[1-(хиназолин-4-ил)пиперид-4-ил] метиламин; N-(1,4-бензодиоксан-2-илметил)-1-[1-(пирид-2-ил)пиперид-4-ил] метиламин; N-(8-метокси-1,4-бензодиоксан-2-илметил)-1-[1-(2-метоксифенил)пиперид-4-ил] метиламин; N-(1,4-бензодиоксан-2-илметил)-1-(1-фенилпиперид-4-ил)метиламин; N-(1,4-бензодиоксан-2-илметил)-1-[1-(1,4-бензодиоксан-5-ил)пиперид-4-ил] метиламин; 1-[1-(1,4-бензодиоксан-2-илметил)пиперид-4-ил] -N-(2-метоксифенил)метиламин; N-(1,4-бензодиоксан-2-илметил)-1-[1-(4-метоксифенил)пиперид-4-ил] метиламин; N-(8-метокси-1,4-бензодиоксан-2-илметил)-N'-(2-метоксифенил)-1,3-пропандиамин; N-(1,4-бензодиоксан-2-илметил)-1-[1-(3-метоксифенил)пиперид-4-ил] метиламин; N-(1,4-бензодиоксан-2-илметил)-1-[1-(2-хлорфенил)пиперид-4-ил] метиламин; N-(5-фтор-1,4-бензодиоксан-2-илметил)-1-[1-(2-метоксифенил)пиперид-4-ил] метиламин; N-(8-фтор-1,4-бензодиоксан-2-илметил)-1-[1-(2-метоксифенил)пиперид-4-ил] метиламин; 1-[1-(2,3-дигидро-бензо[b]фуран-7-ил)пиперид-4-ил] -N-(8-метокси-1,4-бензодиоксан-2-илметил)метиламин; N-(6-хлор-1,4-бензодиоксан-2-илметил)-1-[1-(2-метоксифенил)пиперид-4-ил] метиламин; N-(7-хлор-1,4-бензодиоксан-2-илметил)-1-[1-(2-метоксифенил)пиперид-4-ил] метиламин; N-(8-окси-1,4-бензодиоксан-2-илметил)-1-[1-(2-метоксифенил)пиперид-4-ил] метиламин; и их фармацевтически приемлемые соли, которые могут иметься в виде отдельных энантиомеров, рацематов или других смесей энантиомеров.
B четвертую группу предпочтительных соединений входят соединения, выбранные из группы, включающей:
(S)-(-)-N-(1,4-бензодиоксан-2-илметил)-1-[1-(2-метоксифенил)-пиперид-4-ил]метиламин;
(R)-(+)-N-(1,4-бензодиоксан-2-илметил)-1-[1-(2-метоксифенил)-пиперид-4-ил]метиламин;
(-)-N-(1,4-бензодиоксан-2-илметил)-1-[1-(пирид-2-ил)пиперид-4-ил] метиламин в виде дигидрохлорида;
(+)-N-( 1,4-бензодиоксан-2-илметил)-1-[1-(пирид-2-ил)пиперид-4-ил] метиламин в виде дигидрохлорида.
Соединения общей формулы (I) могут иметься в виде соли с фармацевтически приемлемыми кислотами. Примером таких кислот являются гидрохлориды, гидробромиды, сульфаты, метансульфонаты, нитраты, малеаты, ацетаты, цитраты, фумараты, тартраты [например (+)-тартраты, (-)-тартраты и их смеси, а также рацематы], сукцинаты, бензоаты и соли с аминокислотой, такой, как, например, глютаминовая кислота. Соединения общей формулы (I) и их соли могут иметься в виде сольватов, например, гидратов.
Соединения общей формулы (I) могут иметь один или несколько хиральных центров и иметься в виде различных оптически активных форм. При наличии одного хирального центра соединения формулы (I) имеются в виде двух энантиомерных форм и поэтому настоящее изобретение охватывает оба энантиомера и их смеси. Отдельные энантиомеры могут получатся известными специалисту методами. Примерами таких методов являются образование соли диастереомеров, которые можно разделять, например, путем кристаллизации, образование производных или комплексов диастереомеров, которые можно разделять, например, путем кристаллизации, газо-жидкостной хроматографии или жидкостной хроматографии, избирательное взаимодействие одного энантиомера с подходящим реагентом, например, энзиматическая этерификация, или же газо-жидкостная или жидкостная хроматография в хиральной среде например, на хиральном носителе, например, двуокиси кремния, со связанным хиральным лигандом, или же в присутствии хирального растворителя. Если желаемый энантиомер переводят в другую химическую форму одним из вышеуказанных методов, то необходимо последующее выделение желаемой энантиомерной формы. Также возможно синтезировать энантиомеры путем асимметричного синтеза с использованием оптически активных реагентов, субстратов, катализаторов или растворителей, или же путем перевода одного энантиомера в другой энантиомер за счет асимметричной перегруппировки.
Если соединения общей формулы (I) имеют по меньшей мере два хиральных центра, то они имеются в виде диастереомеров, которые разделяются известными специалисту методами, например, хроматографией или кристаллизацией.
При этом отдельные изомеры любой диастереомерной пары можно выделять вышеуказанными методами. Данное изобретение охватывает любой диастереоизомер соединений формулы (I), а также любые смеси диастереомеров.
Некоторые соединения общей формулы (I) и их соли могут иметься в виде разных кристаллических форм и поэтому данное изобретение охватывает любую кристаллическую форму и смеси этих форм. Некоторые соединения общей формулы (I) и их соли также могут иметься в виде сольватов, например, гидратов, и поэтому данное изобретение охватывает любой сольват и смеси сольватов.
Дальнейшим объектом настоящего изобретения является фармацевтическая композиция со сродством в отношении рецепторов 5-HT1A и D2, содержащая терапевтически эффективное количество соединения общей формулы (I) или его соли и фармацевтически приемлемый разбавитель или носитель.
Применяемый здесь термин "активное начало" означает соединение формулы (I) или его соль. В рамках терапии активное начало дают орально, ректально, парентерально или местно, предпочтительно орально. Таким образом предлагаемая фармацевтическая композиция может представлять собой любую стандартную форму терапевтического препарата для оральной, ректальной, парентеральной или местной аппликации. В качестве фармацевтически приемлемого разбавителя или носителя предлагаемая композиция содержит любое известное в данной области вещество. Как правило, предлагаемая композиция содержит 0,1 - 99 вес.% активного начала. Как правило, она имеется в виде дозировочной единицы. Дозировочная единица активного начала предпочтительно составляет 1 - 500 мг. Целевыми добавками в содержащей предлагаемые соединения композиции являются общеизвестные вещества.
Примерами предпочтительной композиции для оральной дачи являются таблетки, капсулы, сиропы, водные и масляные суспензии. Целевыми добавками в такой композиции являются общеизвестные вещества. Таблетки готовят путем смешивания активного начала с инертными наполнителями, такими, как, например, фосфат кальция, дезинтегрирующими агентами, такими, как, например, кукурузный крахмал, смазочными веществами, такими, как, например, стеарат магния, обеспечивающими таблетирование предлагаемой композиции. Таблетки могут иметь продленное действие, т.е. могут быть выполнены с таким расчетом, что активное начало высвобождается в течение определенного времени. Для этой цели на таблетки можно наносить известными приемами покрытие, например, из фталата ацетата целлюлозы. Кроме того, возможно переводить предлагаемую композицию в капсулы, например, из твердой или мягкой желатины, которую приготовляют известными приемами и при необходимости на них наносят покрытия желаемой характеристики. Как правило, таблетки и капсулы содержат 1 - 500 мг активного начала.
Другой композицией для оральной дачи является, например, водная суспензия, содержащая активное начало в водной среде в присутствии нетоксичного суспендирующего агента такого, как например карбоксиметилцеллюлоза натрия, и масляная суспензия, содержащая соединение вышеприведенной общей формулы (I) в среде подходящего растительного масла, такого, как, например, арахисовое масло.
Активное начало можно также переводить в гранулы, при необходимости с применением дополнительных вспомогательных веществ. Гранулы могут непосредственно применяться пациентом, или же они могут добавляться к подходящему жидкому носителю, такому, как, например, вода, перед применением. Гранулы могут также содержать фармацевтически приемлемые дезинтегрирующие агенты, такие, как, например, сыпучая смесь, состоящая из кислоты и соли углекислоты или бикарбоната, которая облегчает диспергирование в жидкой среде.
Подходящей для ректальной дачи формой предлагаемой композиции являются, например, суппозитории, содержащие масло какао или полиэтиленгликолевое основание.
Подходящей для парентеральной дачи формой предлагаемой композиции являются, например, стерильные суспензии или стерильные растворы в подходящем растворителе.
Композиции для местной дачи могут содержать матрицу, в которой активное начало диспергировано с таким расчетом, что оно находится в контакте с кожей с тем, чтобы активное начало может проникать в организм трансдермально. Подходящая композиция для трансдермальной аппликации можно готовить путем смешивания фармацевтически активного начала с пригодным носителем, таким, как, например, минеральное масло, петролатум и/или воск, например, парафин или пчелиный воск, и пригодным для трансдермальной аппликации ускорителем, таким, как, например, диметилсульфоксид или пропиленгликоль. Но активное начало также может быть диспергировано в фармацевтически приемлемом креме или в основе для мази. В случае местной аппликации активное начало должно содержаться в подходящем препарате с таким расчетом, что терапевтически эффективное количество активного начала высвобождается в течение желаемого периода времени.
Соединения вышеприведенной общей формулы (I) могут также даваться путем непрерывной инфузии, например, путем внутривенного вливания, или же с применением источника, размещенного внутри тела пациента. Такими внутренними источниками являются, например, имплантированные емкости, которые содержат подлежащее инфузии активное начало. В таком случае активное начало непрерывно высвобождается, например, путем осмоза. Кроме того, имплантированные препараты могут представлять собой жидкость, такую, как, например, суспензия или раствор в фармацевтически приемлемом масле подлежащего инфузии соединения, например, в виде труднорастворимого в воде производного, такого, как, например, соль или эфир с додекановой кислотой, или же служащее в качестве основы для подлежащего инфузии соединения твердое вещество, например, синтетическая смола или воск. При этом основание может представлять собой единичное тело, содержащее активное начало, или же ряд тел, каждое из которых содержит часть апплицируемого активного начала. Активное начало должно иметься во внутреннем источнике с таким расчетом, что терапевтически эффективное количество активного начала отдается в течение желаемого периода времени.
Для некоторых целей может быть целесообразным то, что композиция содержит соединения вышеприведенной общей формулы (I) в виде мельчайших частиц, получаемых, например, путем распыления.
Предлагаемая фармацевтическая композиция может также содержать другие фармакологически активные начала, которые совместимы с соединениями вышеприведенной общей формулы (I).
Как уже указывалось выше, предлагаемая фармацевтическая композиция проявляет сродство в отношении рецепторов 5-HT1A и D2 и поэтому может применяться для лечения психозов, в частности шизофрении. Необходимое для успешного лечения количество активного начала зависит от ряда факторов таких, как, например, возраст пациента, серьезность заболевания, история болезни, поэтому в каждом конкретном случае врач должен подбирать подходящую дозировку. Как правило, новые соединения дают в количестве 1 - 1000 мг/сутки, предпочтительно 5 - 500 мг/сутки, в качестве единичной дозы или же нескольких доз один раз или несколько раз в сутки.
Ниже описываются способы получения соединений вышеприведенной общей формулы (I).
Соединения формулы (I), где Q - группа формулы (IIа), могут получаться путем взаимодействия соединения формулы (III)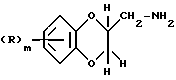
где R и m имеют вышеуказанные значения,
с соединением формулы (IV)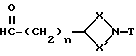
где n - 1 или 2, а X, X', и T имеют вышеуказанные значения,
с последующим взаимодействием получаемого при этом имина с восстановителем, таким, как, например, боргидрид натрия.
Соединения формулы (I), где Q - группа формулы (IIа), могут также получаться путем взаимодействия соединения вышеприведенной формулы (III) с соединением формулы (V)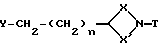
где X, X' и n имеют вышеуказанные значения, a Y означает удаляемую группу, такую, как, например, толуол-4-сульфонилокси,
при необходимости в среде подходящего растворителя и в присутствии основания, например, карбоната калия.
Кроме того, соединения общей формулы (I), где Q - группа формулы (IIa), могут получаться путем взаимодействия соединения формулы (VI)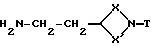
где X, X' и T имеют вышеуказанные значения,
с соединением формулы (VII)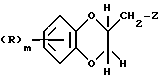
где R и m имеют вышеуказанные значения, a Z означает удаляемую группу, такую, как, например, толуол-4-сульфонилокси.
при необходимости в среде подходящего растворителя и в присутствии основания, например, карбоната калия.
Соединения общей формулы (I) могут также получаться путем взаимодействия соединения формулы (VIII)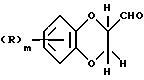
где R и m имеют вышеуказанные значения,
с соединением формулы (VI), с последующим восстановлением получаемого при этом имина подходящим восстановителем, таким, как, например, боргидрид натрия.
Соединения общей формулы (III) могут получаться путем восстановления со единения формулы (IX)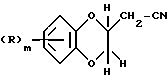
где R и m имеют вышеуказанные значения,
восстановителем, таким, как, например, алюмогидрид лития.
Соединения формулы (IX) могут получаться путем взаимодействия соединения формулы (X)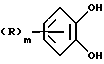
где R и m имеют вышеуказанные значения,
с нитрилом формулы (XI)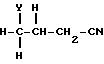
где Y - удаляемая группа, такая, как, например, галоид, бром,
в присутствии основания, такого, как, например, карбонат калия.
Соединения формулы (III) могут также получаться из соединений формулы (XII)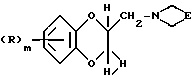
где R и m имеют вышеуказанные значения, а заместитель E вместе с атомом азота, с которым он связан, образует циклический имид, такой, как, например, фталимид, путем катализированного кислотой или основанием гидролиза, или путем расщепления реагентом, например, гидратом гидразина.
Соединения формулы (XII), где заместитель E вместе с атомом азота, с которым он связан, представляет собой фталимид, могут получаться путем взаимодействия соединения формулы (VII), где Z - удаляемая группа, такая, как, например, толуол-4-сульфонилокси, с фталимидом калия.
Соединения формулы (III) могут также получаться путем восстановления соединения формулы (XIII)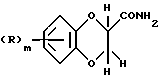
где R и m имеют вышеуказанные значения, подходящим восстановителем, таким, как, например, алюмогидрид лития.
Соединения формулы (XIII) могут получаться путем взаимодействия соединения формулы (XIV)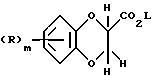
где R и m имеют вышеуказанные значения, a L означает алкильную группу с 1 - 6 атомами углерода, с аммонием.
Соединения формулы (IV) могут получаться путем взаимодействия соединения формулы (XV)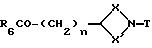
где X, X', T и n имеют вышеуказанные значения, а R6 - алкоксильная группа с 1 - 4 атомами углерода, с восстановителем, таким, как, например, бис(2-метоксиэтокси)алюмогидрид натрия, в среде растворителя, например, толуола.
Соединения формулы (XV) могут получатся путем взаимодействия соединения формулы (XVI)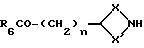
где R6, X, X' и n имеют вышеуказанные значения, с галоидзамещенным ароматическим соединением, таким, как, например, 2-галопиридин, например, 2-хлорпиридин, при необходимости в присутствии основания, такого, как, например, триэтиламин.
Соединения формулы (IV) могут также получатся путем окисления соединения формулы (XVII)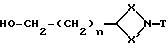
где X, X', T и n имеют вышеуказанные значения, подходящим окислителем, таким, как, например, оксалилхлорид в среде диметилсульфоксида.
Соединения формулы (V), где Y - толуол-4-сульфонилокси, могут получаться путем взаимодействия соединения формулы (XVII) с агентом тозилирования, таким, как, например, толуол-4-сульфонилхлорид.
Соединения формулы (XVII) могут получаться путем восстановления соединения формулы (XV) восстановителем, таким, как, например, алюмогидрид лития, или путем взаимодействия с соединением формулы (XVIII)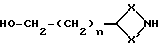
где X, X' и n имеют вышеуказанные значения, с галоидзамещенным ароматическим соединением, таким, как, например, 2-галопиридин, например, 2-хлорпиридин, при необходимости в присутствии основания, такого, как, например, триэтиламин.
Соединения формулы (XVIII) могут получаться путем восстановления соединения формулы (XVI), где R6 - алкоксильная группа с 1 - 4 атомами углерода, с восстановителем, таким, как, например, алюмогидрид лития.
Соединения формулы (VI) могут получаться путем взаимодействия соединения формулы (XIX),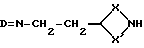
где X и X' имеют вышеуказанные значения, a D - защитная группа, например, a-метилбензилидин или 4-нитробензилидин, с галоидзамещенным ароматическим соединением, таким, как, например, 2-галопиридин, например 2-хлорпиридин, при необходимости в присутствии основания, такого, как, например, триэтиламин, с последующим удалением защитной группы путем, например, катализированного кислотой гидролиза.
Соединения формулы (XIX) могут получаться путем взаимодействия соединения формулы (XX)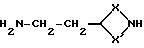
где X и X' имеют вышеуказанные значения, с защитным реагентом, таким, как, например, ацетофенон или 4-нитробензальдегид.
Соединения формулы (VI) можно получать непосредственно путем взаимодействия соединения формулы (XX) с галоидзамещенным ароматическим соединением, таким, как, например, галопиридин, 2-хлорпиридин, при необходимости в присутствии основания, такого, как, например, карбонат калия.
Соединения формулы (XX) могут получаться путем восстановления соединения формулы (XVI), где R6 - амино-группа, восстановителем, таким, как, например, алюмогидрид лития.
Соединения формулы (VI) могут также получаться путем взаимодействия соединения формулы (XXI)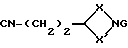
где X и X' имеют вышеуказанные значения, a G означает водород, с галоидзамещенным ароматическим соединением, таким, как, например, 2-хлорпиридин, при необходимости в присутствии основания, например, триэтиламина, с последующим восстановлением с применением восстановителя, такого, как, например, алюмогидрид лития.
Соединения формулы (XXI), где G означает водород, могут получаться путем взаимодействия соединения формулы (XXI), где G означает алкильную или арилалкильную группу, например, бензил, с агентом деалкилирования, таким, как, например, 1-хлорэтиловый эфир хлормуравьиной кислоты, с последующим расщеплением получаемого в качестве промежуточного продукта сложного эфира карбаминовой кислоты.
Соединения формулы (XX) могут также получаться путем взаимодействия соединения формулы (XXI), где G - защищающая амин группа, например, бензил, с восстановителем, таким, как, например, алюмогидрид лития, с последующим удалением защитной группы, например путем взаимодействия с муравьиной кислотой в присутствии палладия на угле в качестве катализатора.
Соединения формулы (VI) могут также получаться путем восстановления соединения формулы (XXII)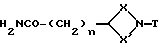
где X, X', T и n имеют вышеуказанные значения, восстановителем, таким, как, например, комплекс борана и диметилсульфида или алюмогидрид лития.
Соединения формулы (XXII), где общее число атомов углерода в радикалах X и X' составляет 4, и T - ароматическая группа, не содержащая атом азота, могут получаться путем восстановления соединения формулы (XXIII)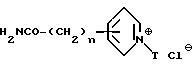
где n имеет вышеуказанное значение, а T - ароматическая группа, не содержащая атом азота, восстановителем, таким, как, например, формиат аммония или водород, в присутствии палладия на угле в качестве катализатора.
Соединения формулы (XXIII) могут получаться в результате взаимодействия соединения формулы (XXIV)
где n имеет вышеуказанное значение, с соединением формулы (XXV)
T-NH2
где T - ароматическая группа, не содержащая атом азота, при этом происходит перегруппировка 2,4-динитроанилина.
Соединения формулы (XXIV) могут получаться путем взаимодействия 2,4-динитрохлорбензола с соединением формулы (XXVI)
где n имеет вышеуказанное значение.
Соединения формулы (VII), где Z означает толуол-4-сульфонилокси, могут получаться путем взаимодействия соединения формулы (XXVII)
где R и m имеют вышеуказанное значение, с толуол-4-сульфонилхлоридом, при необходимости в присутствии основания, например, пиридина.
Соединения формулы (XXVII) могут получатся путем восстановления соединения формулы (XIV), где L означает алкильную группу с 1 - 4 атомами углерода, восстановителем, таким, как, например, алюмогидрид лития.
Соединения формулы (XXVII) могут также получаться путем взаимодействия соединения формулы (XXVIII)
где Z - удаляемая группа, например, хлор или толуол-4-сульфонилокси, с соединением формулы (X) в среде пригодного растворителя, такого, как, например, вода или диметилформамид, в присутствии основания, например, гидроокиси натрия. В случае использования энантиомерно чистой формы соединения формулы (XXVIII), такой, как, например, (R)-глицидол-O-толуол-4-сульфонат, можно получать отдельный энантиомер соединения формулы (XXVII).
Соединения формулы (XXVII), где R означает алкоксильную группу с 1 - 3 атомами углерода, могут получаться в результате алкилирования соответствующего соединения формулы (XXVII), где R означает гидроксил, путем взаимодействия с агентом алкилирования, например, метилйодидом, в присутствии основания, такого, как, например, гидроокись натрия.
Соединения формулы (VIII) могут получаться путем окисления соединения формулы (XXVII), где U - метилен, подходящим окислителем, таким, как, например, хлорхромат пиридиния, или путем восстановления соединения формулы (XIV), где m - 0, подходящим восстановителем, таким, как, например, бис(2-метоксиэтокси)алюмогидрид натрия, в среде растворителя, например, толуола.
Соединения формулы (XIV) могут получаться путем взаимодействия соединения формулы (XXIX)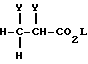
где Y - удаляемая группа, например, бром, a L - алкильная группа с 1 - 6 атомами углерода, с соединением формулы (X) в присутствии основания, например, карбоната калия.
Соединения формулы (I), где Q - группа формулы (IIб), могут получаться путем взаимодействия соединения формулы (XXX)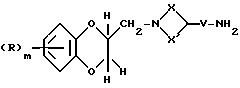
где R, X, X', H и V имеют вышеуказанные значения, с галоидзамещенным ароматическим соединением, таким, как, например, 2-галопиридин, например, 2-хлорпиридин, при необходимости в присутствии основания, такого, как, например, триэтиламин.
Соединения формулы (XXX) могут получаться из соединений формулы (XXXI)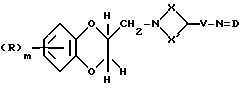
где R, X, X' V и m имеют вышеуказанные значения, a D - защитная группа, например, 5-хлор-2-оксибензилиден, путем катализированного кислотой или основанием гидролиза.
Соединения формулы (XXXI) могут получаться путем взаимодействия соединения формулы (XXXII)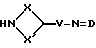
где X, X', V и D имеют вышеуказанные значения, с соединением формулы (VII), при необходимости в присутствии основания, такого, как, например, триэтиламин.
Соединения формулы (XXXII) могут получаться путем взаимодействия соединения формулы (XXXIII)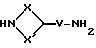
где X, X' и V имеют вышеуказанные значения, с защитным реагентом, таким, как, например, 5-хлорсалицилальдегид.
Соединения формулы (I), где Q - группа формулы (IIб), могут также получаться путем взаимодействия соединения формулы (XXXIV)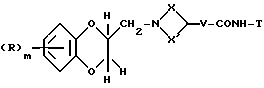
где R, X, X', V и m имеют вышеуказанные значения, с восстановителем, например, комплексом борана и диметилсульфида.
Соединения формулы (XXXIV) могут получаться путем взаимодействия соединения формулы (XXXV)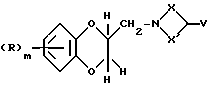
где R, X, X' V и m имеют вышеуказанные значения, с агентом образования смешанного ангидрида, таким, как, например, сложный этиловый эфир хлормуравьиной кислоты, при необходимости в присутствии основания, такого, как, например, триэтиламин, с последующим взаимодействием с соединением формулы (XXV).
Соединения формулы (XXXV) могут получаться путем гидролиза соединения формулы (XXXVI)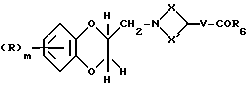
где R6 - алкоксильная группа с 1 - 4 атомами углерода, с основанием, таким, как, например, гидроокись калия.
Соединения формулы (XXXVI) могут получаться путем взаимодействия соединения формулы (VII) с соединением формулы (XVI), где R6 - алкоксильная группа с 1 - 4 атомами углерода.
Способность соединений общей формулы (I) к взаимодействию с рецепторами 5-окситриптамина (далее: 5-НТ) выявляли с помощью следующего опыта, направленного на определение способности соединений к торможению in vitro связывания меченого тритием лиганда с рецепторами 5-НТ, в частности с рецепторами 5-HT1A.
Гиппокамповую ткань головного мозга крыс-самцев штамма Charles River CD весом 150 - 250 г гомогенизировали в холодном как лед 50-мм. буфере Трис-HCl значением pH, равным 7,7 (измерение при температуре 25oC), взятом в весовом соотношении к ткани, равном 1:40 в пересчете на объем и центрифугировали со скоростью 30.000 g при температуре 4oC в течение 10 минут. Осадок вновь гомогенизировали в том же буфере, инкубировали при температуре 37oC в течение 10 минут и центрифугировали со скоростью 30.000 g при температуре 4oC в течение 10 минут. Получаемый при этом осадок суспендировали в 50-мм. буфере Трис-HCl значением pH, равным 7,7, содержащем 4 ммоль дихлорида кальция, 0,1% L-аскорбиновой кислоты и 10 мкмоль паргилина в виде гидрохлорида (что является эквивалентным 6,25 мг влажной ткани/мл) и сразу использовали в опыте для определения связывания. Аликвотные количества (400 мкл, что является эквивалентным 2,5 мг влажной ткани на трубку) получаемой суспензии подавали в трубки, содержащие 50 мкл (2 нмоль) лиганда и 50 мкл дистиллированной воды (полное связывание), или 50 мкл (10 мкмоль) 5-НТ (неспецифичное связывание) или 50 мкл исследуемого соединения (взятого в единой концентрации 10-6 моль или в 10 различных концентрациях, равных 10-11 - 10-3 моль). В качестве лиганда использовали [3H]8-окси-2-(дипропиламино)тетралин. Смесь инкубировали при температуре 25oC в течение 30 минут, после чего инкубацию прекращали путем быстрой фильтрации.
Фильтры промывали холодным как лед буфером Трис-HCl и высушивали. Фильтры вводили в пробирки, куда добавляли сцинтиляционную жидкость, после чего радиоактивность определяли путем жидкостного сцинтиляционного подсчета, %-ное замещение специфичного связывания меченого тритием лиганда рассчитывали для единой концентрации (10-6 моль) исследуемого соединения. Составляли кривые замещения для тех соединений, которые замещали ≥ 50% специфичного связывания меченого тритием лиганда при концентрации 10-6 моль с использованием соединения в разных концентрациях. Концентрацию соединения, обеспечивающую 50%-ное торможение специфичного связывания (IC50) получали из кривой. Коэффициент торможения (Ki) определяли с применением следующего уравнения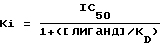
где [лиганд] означает концентрацию меченого тритием лиганда, a KD означает константу равновесия диссоциации лиганда.
Способность соединений формулы (I) к взаимодействию с сайтами связывания с адреноцепторами выявляли с помощью следующего опыта, направленного на определение способности соединений к торможению in vitro связывания меченого тритием лиганда с адреноцепторами, в частности с α1-адреноцептором.
Целую кортикальную ткань головного мозга крыс-самцев штамма Charles River CD весом 150 - 250 г гомогенизировали в холодном как лед 50-мм. буфере Трис-HCl значением pH, равным 7,6 (измерение при температуре 25oC), взятом в весовом соотношении к ткани, равном 1:40 в пересчете на объем и центрифугировали со скоростью 1.000 g при температуре 4oC в течение 10 минут. Надосадочную жидкость центрифугировали со скоростью 30.000 g при температуре 4oC в течение 10 минут. Остаток гомогенизировали в холодном как лед 50-мм. буфере Трис-HCl значением pH, равным 7,6, взятом в весовом соотношении к ткани, равном 1:40 в пересчете на объем, и центрифугировали со скоростью 30.000 g при температуре 4oC в течение 10 минут. Получаемый окончательный остаток гомогенизировали в 50-мм. буфере Трис-HCl значением pH, равным 7,6, (эквивалентно 12,5 мг влажной ткани/мл) и сразу использовали в опыте для опеределения связывания. Аликвотные количества (400 мкл, что является эквивалентным 5 мг влажной ткани на трубку) получаемой суспензии подавали в трубки, содержащие 50 мкл (0,1 нмоль) лиганда и 50 мкл дистиллированной воды (полное связывание), или 50 мкл (5 мкмоль) фентоламина (неспецифичное связывание) или 50 мкл исследуемого соединения (взятого в единой концентрации 10-6 моль или в 10 различных концентрациях, равных 10-11-10-3 моль). В качестве лиганда использовали [7-метокси-3H]празосин. Смесь инкубировали при температуре 30oC в течение 30 минут, после чего инкубацию прекращали путем быстрой фильтрации.
Фильтры промывали холодным как лед буфером Трис-HCl и высушивали. Фильтры вводили в пробирки, куда добавляли сцинтиляционную жидкость, после чего радиоактивность определяли путем жидкостного сцинтиляционного подсчета, %-ное замещение специфичного связывания меченого тритием лиганда рассчитывали для единой концентрации (10-6 моль) исследуемого соединения. Составляли кривые замещения для тех соединений, которые замещали ≥ 50% специфичного связывания меченого тритием лиганда при концентрации 10-6 моль с использованием соединения в разных концентрациях. Концентрацию соединения, обеспечивающую 50%-ное торможение специфичного связывания (IC50) получали из кривой. Коэффициент торможения (Ki) определяли с применением следующего уравения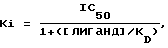
где [лиганд] означает концентрацию меченого тритием лиганда, а KD означает константу равновесия диссоциации лиганда.
Способность соединений формулы (I) к взаимодействию с сайтами связывания с адреноцепторами выявляли с помощью следующего опыта, направленного на определение способности соединений к торможению in vitro связывания лиганда с адреноцепторам, в частности α2- адреноцептором.
Ткань передней коры головного мозга крыс-самцев штамма Charles River CD весом 150 - 250 г гомогенизировали в холодной как лед 0,25-м. сахарозе, взятой в весовом соотношении к ткани, равном 1:30 в пересчете на объем и центрифугировали со скоростью 1.000 g при температуре 4oС в течение 12 минут. Надосадочную жидкость хранили на льду, а остаток регомогенизировали в 0,25-м. сахарозе, взятой в весовом соотношении к ткани, равном 1:15 в пересчете на объем, и центрифугировали со скоростью 850 g при температуре 4oC в течение 12 минут. Объединенные надосадочные жидкости разбавляли 5-мм. буфером трис-HCl значением pH, равным 7,5, содержащем 0,5 моль этилендиаминотетрауксусной кислоты, pH вновь доводили до 7,5 (при температуре 25oC) добавкой 1-м. гидроокиси натрия до весового соотношения, равном 1:80 в пересчете на объем, и центрифугировали со скоростью 30.000 g при температуре 4oC в течение 10 минут. Получаемый остаток ресуспендировали в 50-мм. буфере Трис-HCl значением pH, равным 7,5, содержащем 5,68 ммоль L-аскорбиновой кислоты и 5 ммоль этилендиаминотетрауксусной кислоты, pH вновь доводили до 7,5 (при температуре 25oC) путем добавки 1-молярной гидроокиси натрия, и центрифугировали со скоростью 30.000 g в течение 10 минут. Получаемый окончательный остаток ресуспендировали в 50-мм. буфере Трис-HCl значением pH, равным 7,5, содержащем 5,68 ммоль L-аскорбиновой кислоты и 5 ммоль этилендиаминотетрауксусной кислоты, (что является эквивалентным 12,5 мг влажной ткани/мл) и сразу использовали в опыте для определения связывания. Аликвотные количества (400 мкл, что является эквивалентным 5 мг влажной ткани на трубку) получаемой суспензии подавали в трубки, содержащие 50 мкл (1 нмоль) лиганда и 50 мкл дистиллированной воды (полное связывание), или 50 мкл (5 мкмоль) фентоламина (неспецифичное связывание) или 50 мкл исследуемого соединения (взятого в единой концентрации 10-6 моль или в 10 различных концентрациях, равных 10-11-10-3 моль). В качестве лиганда использовали меченый тритием идазоксан ((1,4-[6,7(н)-3H] бензодиоксан-2-ил)-2-имидазолин в виде гидрохлорида). Смесь инкубировали при температуре 0oC в течение 75 минут, после чего инкубацию прекращали путем быстрой фильтрации.
Фильтры промывали холодным как лед буфером Трис-HCl и высушивали. Фильтры вводили в пробирки, куда добавляли сцинтиляционную жидкость, после чего радиоактивность определяли путем жидкостного сцинтиляционного подсчета, %-ное замещение специфичного связывания меченого тритием лиганда рассчитывали для единой концентрации (10-6 моль) исследуемого соединения. Составляли кривые замещения для тех соединений, которые замещали ≥ 50% специфичного связывания меченого тритием лиганда при концентрации 10-6 моль с использованием соединения в разных концентрациях. Концентрацию соединения, обеспечивающую 50%-ное торможение специфичного связывания (IC50) получали из кривой. Коэффициент торможения (Ki) определяли с применением следующего уравнения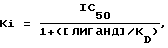
где [лиганд] означает концентрацию меченого тритием лиганда, а KD означает константу равновесия диссоциации лиганда.
Способность соединений формулы (I) к взаимодействию с рецепторами допамина выявляли с помощью следующего опыта, направленного на определение способности соединений к торможению in vitro связывания меченого тритием лиганда с рецепторами допамина, в частности с рецепторами допамина D2.
Разрезанную на полосы ткань мозга крыс-самцев штамма Charles River CD весом 140 - 250 г гомогенизировали в холодном как лед 50-ммолярном буфере Трис-HCl значением pH, равным 7,7 при температуре 25oC, и центрифугировали со скоростью 40.000 g в течение 10 минут. Остаток ресуспендировали в Трис-солевом буфере (50 ммоль буфера Трис-HCl, содержащего 120 ммоль хлорида натрия, 5 ммоль хлорида калия, 2 ммоль дихлорида кальция, 1 ммоль дихлорида магния, а также 6 ммоль аскорбиновой кислоты; pH буфера при температуре 25oC равен 7,7) и снова центрифурировали со скоростью 40.000 g в течение 10 минут. Получаемый окончательный остаток хранили при температуре -80oC. Перед каждым испытанием твердую массу ресуспендировали в Трис-солевом буфере (эквивалентном 2 мг веса влажной ткани/мл). Аликвотные количества (720 мкл, что является эквивалентным 1,44 мг влажной ткани на трубку) получаемой суспензии подавали в трубки, содержащие 40 мкл (1 нмоль) лиганда и 40 мкл Трис-солевого буфера (полное связывание), или 40 мкл (10 нмоль) спироперидола (неспецифичное связывание) или 40 мкл исследуемого соединения (взятого в единой концентрации 10-6 моль или в 6 различных концентрациях, равных 10-11 - 10-4 моль). В качестве лиганда использовали меченый тритием (S)-сульпирид. Смесь инкубировали при температуре 4oC в течение 40 минут, после чего инкубацию прекращали путем быстрой фильтрации.
Фильтры промывали холодным как лед буфером Трис-HCl и высушивали. Фильтры вводили в пробирки, куда добавляли сцинтиляционную жидкость, и оставляли в течение 20 часов, после чего радиоактивность определяли путем сцинтиляционной спектрофотометрии, %-ное замещение специфичного связывания меченого тритием лиганда рассчитывали для единой концентрации (10-6 моль) исследуемого соединения. Составляли кривые замещения для тех соединений, которые замещали ≥ 50% специфичного связывания меченого тритием лиганда при концентрации 10-6 моль с использованием соединения в разных концентрациях. Концентрацию соединения, обеспечивающую 50%-ное торможение специфичного связывания (IC50) получали из кривой. Коэффициент торможения (Ki) определяли с применением следующего уравения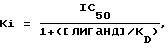
где [лиганд] означает концентрацию меченого тритием лиганда, а KD означает константу равновесия диссоциации лиганда.
Величины Ki, определенные в описанных выше испытаниях по связыванию к 5-HT1A, α1,α2 и D2 для всех целевых веществ, полученных по приведенным ниже примерам 1 - 32, даны в таблице 1. В нескольких случаях определение Ki не было возможно, поэтому значение Ki дано как "больше чем" (>), полученное из применения вышеприведенной формулы к самой высокой концентрации, обеспечивающей замещение лиганда ≤ 50%.
%-ные величины в таблице 1 относятся к %-ному замещению при концентрации соединения, равной 10-6 моль.
Изобретение иллюстрируется следующими примерами, которые не ограничивают его объем. Целевые продукты в каждом из этих примеров были характеризованы одним или несколькими из следующих методов исследования: газо-жидкостная хроматография, высокопроизводительная жидкостная хроматография, элементный анализ, спектроскопия ядерного магнитного резонанса, инфракрасная спектроскопия.
Пример 1.
Смесь 14,3 г хлорпиразина, 14,25 г 1-(пиперид-4-ил)метиламина, 11,4 г карбоната натрия и 100 мл 3-метил-1-бутанола перемешивают и нагревают с обратным холодильником в течение 64 часов, после чего охлаждают до комнатной температуры и фильтруют. Растворитель удаляют в вакууме, а оставшееся масло перегоняют с получением 16,2 г 1-[1-(пиразин-2-ил)пиперид-4-ил]метиламина в качестве бледно-желтого масла с пределами кипения 136 -160oC/0,6 мбар.
Раствор 60,25 г толуол-4-сульфонилхлорида в 75 мл пиридина по каплям при температуре 20oC подают в перемешиваемый раствор 50 г 1-(1,4-бензодиоксан-2-ил)метанола в 100 мл пиридина, и смесь перемешивают при комнатной температуре в течение 18 часов, затем наливают на избыток льда и хлористоводородной кислоты (5 моль). Получаемое твердое вещество собирают путем фильтрации, тщательно промывают водой и сушат в вакууме с получением 82,3 г 1,4-бензодиоксан-2-илметил-толуол-4-сульфоната в качестве беловатого твердого вещества с точкой плавления: 75 - 78oC.
Смесь 6,4 г получаемого твердого вещества, 3,8 г 1-[1-(пиразин-2-ил)пиперид-4-ил] метиламина, 50 г карбоната калия, 0,05 йодида калия и 50 мл ацетонитрила перемешивают и нагревают с обратным холодильником в течение 70 часов, затем охлаждают до комнатной температуры и фильтруют. Твердое вещество промывают небольшим количеством метанола, а фильтрат и промывочную жидкость объединяют. Растворители удаляют в вакууме, при этом получают масло, которое растирают в порошок с помощью диэтилового эфира, и раствор фильтруют. Растворитель удаляют в вакууме с получением 6,2 г масла, которое очищают путем перегонки по Кюгельреру, при этом получают 4 г масла с температурой кипения ~ 220oC/0,13 мбар. Масло растворяют в метаноле, а раствор насыщают хлористым водородом. Растворитель удаляют в вакууме с получением смолы, которую растворяют в 25 мл горячего этанола и охлаждают на льду с получением 4 г желтого твердого вещества, которое собирают путем фильтрации и сушат в вакууме. Твердое вещество растворяют в воде, а раствор промывают этилацетатом и подщелачивают 5-молярным водным раствором гидроокиси натрия. Свободное основание экстрагируют этилацетатом, и получаемые экстракты сушат над сульфатом магния. Растворитель удаляют в вакууме с получением 2,55 г масла, которое очищают путем флеш-хроматографии при пониженном давлении на силикагеле с применением в качестве элюента смеси диэтилового эфира и метанола в соотношении 9:1. Соответствующие фракции объединяют, и растворитель удаляют в вакууме с получением 2 г бесцветного масла. Масло растворяют в 50 мл этанола, и раствор насыщают хлористым водородом с получением твердого вещества, которое собирают путем фильтрации и сушат в вакууме при температуре 80oC. Получают 1,4 г 1, . 4-гидрохлорид N-(1,4-бензодиоксан-2-илметил)-1- [1-(пиразин-2-ил)пиперид-4-ил] метиламина в виде желтого твердого вещества с точкой плавления 242 - 245oC.
Пример 2.
Смесь 8 г пиридин-4-карбоксамида и 40 г 2,4-динитрохлорбензола нагревают при температуре 95oC в течение 1 часа, охлаждают и суспендируют в смеси 60 мл метанола и 600 мл диэтилового эфира. Надосадочную жидкость декантируют, а твердый остаток еще дважды суспендируют в той же смеси растворителей.
Твердый остаток кипятят в 100 мл метанола, после чего реакционной смеси дают охлаждаться. Продукт собирают путем фильтрации и сушат в вакууме с получением 15,35 г хлорида 4-карбамоил-1-(2,4-динитрофенил)пиридиния в виде желтоватого порошка с точкой плавления 236 - 238oC (разлож.)
Смесь 13,6 г хлорида 4-карбамоил-1-(2,4-динитрофенил)пиридиния, 10 мл 2-метоксианилина и 500 мл метанола перемешивают при комнатной температуре в течение 16 часов, после чего растворитель удаляют в вакууме. Добавляют диэтиловый эфир, содержащий небольшое количество ацетона, смесь нагревают с обратным холодильником до завершения отверждения продукта, которое собирают путем фильтрации. Получают 11,5 г хлорида 4-карбамоил-1-(2-метоксифенил)-пиридиния в качестве желтого твердого вещества с точкой плавления 229 - 230oС (разлож.).
Смесь 10,3 г хлорида 4-карбамоил-1-(2-метоксифенил)- пиридиния, 10 г катализатора в виде 10%-го палладия на угле, 20 г формиата аммония и 200 мл метанола перемешивают при комнатной температуре в течение 20 минут, затем нагревают с обратным холодильником в течение 3,5 часов. Охлажденную смесь фильтруют, а растворитель удаляют в вакууме с получением 8,8 г синевато-серого твердого вещества.
Пробу 0,75 г синевато-серого твердого вещества перемешивают с горячей водой, а получаемое при этом твердое вещество собирают путем фильтрации, сушат в вакууме и кристаллизуют из 2-пропанола с получением 0,13 г серого твердого вещества [A] с точкой плавления 176 - 180oC.
Остальные 8,05 г синевато-серого твердого вещества суспендируют в воде, а продукт 5 раз экстрагируют этилацетатом, взятым в количестве по 100 мл. Экстракты промывают водой, сушат над сульфатом магния, и растворитель удаляют в вакууме с получением 5,0 г светло-коричневого твердого вещества с точкой плавления 167 - 172oC, которое кристаллизуют из 60 мл 2-пропанола. При этом получают 2,5 г светло-коричневого твердого вещества [Б] с точкой плавления 176 - 180oC.
Вязкие жидкости, оставшиеся после кристаллизации, упаривают, и остаток кристаллизуют из 10 мл 2-пропанола с получением 0,5 г светло-коричневого твердого вещества [В] с точкой плавления 176 - 180oC.
Продукты [А] , [Б] и [В] объединяют с получением всего 3,1 г 1-(2-метоксифенил)пиперидин-4-карбоксамида.
4 мл 10-м. раствора комплекса борана и диметилсульфида в диметилсульфиде по каплям при температуре 15 - 20oC в атмосфере азота подают в перемешиваемый раствор 2,6 г 1-(2-метоксифенил)пиперидин-4-карбоксамида в 25 мл тетрагидрофурана, и перемешиваемую смесь нагревают с обратным холодильником в течение 6 часов. Смесь оставляют стоять при комнатной температуре в течение 16 часов, после чего реакцию прекращают путем медленной подачи реакционной смеси по каплям в избыток льда и воды. Водную смесь подкисляют 5-м. хлористоводородной кислотой, затем подщелачивают 5-м. водным раствором гидроокиси натрия, и продукт экстрагируют диэтиловым эфиром. Экстракты промывают водой, после чего продукт три раза экстрагируют 5-м. хлористоводородной кислотой, два раза взятой в количестве по 100 мл, а один раз - 50 мл. Кислотные экстракты подщелачивают 5-м. водным раствором гидроокиси натрия, и продукт экстрагируют диэтиловым эфиром. Экстракты сушат над сульфатом магния и растворитель удаляют в вакууме с получением 1,05 г масла, которое очищают от побочных продуктов путем хроматографии на силикагеле с применением в качестве элюента смеси диэтилового эфира и метанола в соотношении 9:1. Силикагель суспендируют в метаноле, смесь нагревают с обратным холодильником в течение 10 минут, дают охлаждаться и фильтруют. Растворитель удаляют в вакууме с получением 0,65 г 1-[1-(2-метоксифенил)пиперид-4-ил]метиламина в виде масла.
Смесь 0,65 г 1-[1-(2-метоксифенил)пиперид-4-ил]метиламина, 0,95 г 1,4-бензодиоксан-2-илметил-толуол-4-сульфоната (полученного аналогичным примеру 1 приемом), 0,8 г карбоната калия, 0,01 г йодида калия и 20 мл ацетонитрила перемешивают и нагревают с обратным холодильником в течение 20 часов, затем фильтруют.
Растворитель удаляют в вакууме с получением 1 г масла, которое очищают путем флеш-хроматографии на силикагеле при пониженном давлении с применением в качестве элюента смеси диэтилового эфира и метанола в соотношении 9:1. Соответствующие фракции объединяют, растворители удаляют в вакууме с получением 0,6 г масла, которое растворяют в диэтиловом эфире. Раствор насыщают хлористым водородом, при этом получают твердое вещество, которое собирают путем фильтрации и сушат в вакууме с получением 0,5 г дигидрохлорида N-(1,4-бензодиоксан-2-илметил)-1-[1-(2-метоксифенил)-пиперид-4-ил]метиламина в качестве белого твердого вещества с точкой плавления 219 - 223oC.
Пример 3.
Перемешиваемую смесь 20 г 2,3-дихлорпиридина, 30 г 1-(пиперид-4-ил)метиламина, 30 г карбоната натрия и 100 мл 3-метил-1-бутанола нагревают при температуре 95oC в течение 16 часов, затем охлаждают до комнатной температуры, разбавляют 200 мл этилацетатом и фильтруют. Растворители удаляют в вакууме, а остаточное масло перегоняют с получением 22,2 г 1-[1-(3-хлорпирид-2-ил)пиперид-4-ил] метиламина в качестве бесцветного масла с пределами кипения 115 - 140oC/0,13 мбар.
Перемешиваемую смесь 3,5 г получаемого на предыдущей стадии масла, 5 г 1,4-бензодиоксан-2-илметил-толуол-4-сульфоната, полученного аналогичным примеру 1 приемом, 10 г карбоната калия и 50 мл ацетонитрила нагревают с обратным холодильником в течение 24 часов, растворитель удаляют в вакууме, и остаток разбавляют 100 мл этилацетата. Получаемую суспензию фильтруют, растворитель удаляют в вакууме с получением 6,6 г коричневого масла, которое очищают путем флеш-хроматографии на силикагеле с применением в качестве элюента смеси диэтилового эфира и метанола в соотношении 9:1. Соответствующие фракции объединяют, растворители удаляют в вакууме с получением двух продуктов:
(1) 2,2 г бледно-желтого масла, и
(2) 2,2 г бледно-желтого масла.
Продукт (1) растворяют в 50 мл диэтилового эфира, и раствор насыщают хлористым водородом. Получаемое твердое вещество собирают путем фильтрации, промывают диэтиловым эфиром, сушат в вакууме при комнатной температуре с получением 2,0 г белого твердого вещества с точкой плавления 241 - 243oC.
Продукт (2) обрабатывают аналогичным приемом с получением 1,9 г белого твердого вещества с точкой плавления 250 - 252oC.
Два продукта в качестве твердого вещества объединяют и суспендируют в смеси 20 мл этанола и 20 мл этилацетата. Смесь нагревают с обратным холодильником в течение 5 минут, после чего ей дают охлаждаться до комнатной температуры. Твердое вещество собирают путем фильтрации, промывают этилацетатом и сушат в вакууме при температуре 60oC с получением 3 г 1,4-гидрохлорида N-(1,4-бензодиоксан-2-илметил)-1-[1-(3-хлорпирид-2-ил)пиперид-4-ил] метиламина в качестве белого твердого вещества с точкой плавления: 251 - 253oC.
Пример 4. Раствор 2,15 г 1-(пиперид-4-ил)метиламина в 20 мл этанола подают в раствор 3,1 г 4-хлорхиназолина и 6 мл триэтиламина в 50 мл этанола. Смесь перемешивают при комнатной температуре в течение 100 минут, после чего продукт собирают путем фильтрации, промывают небольшим количеством этанола и сушат в вакууме при температуре 60oC с получением 3 г твердого вещества с точкой плавления 226 - 227oC. Твердое вещество растирают в порошок с помощью 100 мл горячего этилацетата, получаемое твердое вещество собирают путем фильтрации и сушат в вакууме при температуре 60oC с получением 2,8 г гидрохлорида 1-[1-(хиназолин-4-ил)пиперид-4-ил] метиламина в качестве белого твердого вещества с точкой плавления 231 - 232oC.
Раствор 1,2 г получаемого на предыдущей стадии твердого вещества в 250 мл воды подщелачивают 5-м. водным раствором гидроокиси натрия, и свободное основание экстрагируют диэтиловым эфиром. Экстракт сушат над сульфатом магния, а растворитель удаляют в вакууме с получением 1 г масла.
Смесь 1 г получаемого на предыдущей стадии масла, 1,3 г 1,4-бензодиоксан-2-илметил-толуол-4-сульфоната, получаемого аналогичным примеру 1 приемом, 1,1 г карбоната калия и 15 мл ацетонитрила перемешивают и нагревают с обратным холодильником в течение 64 часов, затем охлаждают и фильтруют. Твердый остаток промывают диэтиловым эфиром, а фильтрат и промывочную жидкость объединяют. Растворители удаляют в вакууме с получением оранжевого масла, которое очищают путем флеш-хроматографии на силикагеле под пониженным давлением с применением в качестве элюента смеси диэтилового эфира и метанола в соотношении 9:1.
Соответствующие фракции объединяют, и растворители удаляют в вакууме с получением 1,2 г масла, которое растворяют в диэтиловом эфире. Раствор насыщают хлористым водородом с получением твердого вещества, которое собирают путем фильтрации и сушат в вакууме с получением 1 г белого твердого вещества с точкой плавления 90 - 150oC.
Твердое вещество растворяют в этаноле, раствор насыщают хлористым водородом, и растворитель удаляют в вакууме. Остаток растворяют в как можно меньшем количестве горячего 2-пропанола, а раствору дают охлаждаться до комнатной температуры, причем осаждается смесь белого твердого вещества и смолы. Смесь осторожно нагревают и растирают в порошок с помощью до того, как вся коричневая смола переведена в белое твердое вещество. Смесь охлаждают, твердое вещество собирают путем фильтрации, промывают диэтиловым эфиром и сушат в вакууме с получением 0,6 г дигидрохлорида N-(1,4-бензодиоксан-2-илметил)-1-[1-(хиназолин-4-ил)пиперид-4-ил] метиламина в виде белого твердого вещества с точкой плавления 210 - 216oC.
Пример 5. Смесь 19,4 г 2-хлорпиридина, 14,25 г 1-(пиперид-4-ил)метиламина, 11,4 г карбоната натрия и 100 мл 3-метил-1-бутанола перемешивают и нагревают с обратным холодильником в течение 16 часов, затем фильтруют. Растворитель удаляют в вакууме с получением масла, которое перегоняют с получением 5,65 г 1-[1-(пирид-2-ил)пиперид-4-ил]метиламина в виде бледно-желтого масла с пределами кипения 126 - 130oC/0,6 мбар.
Смесь 3,4 г масла, 5,7 r 1,4-бензодиоксан-2-илметил-толуол-4-сульфоната, полученного аналогичным примеру 1 приемом, 4,5 г карбоната калия и 45 мл ацетонитрила перемешивают и нагревают с обратным холодильником в течение 48 часов, затем фильтруют. Растворитель удаляют в вакууме с получением масла, которое очищают путем флеш-хроматографии на силикагеле под пониженным давлением с применением в качестве элюента смеси диэтилового эфира и метанола в соотношении 9:1. Соответствующие фракции объединяют, и растворители удаляют в вакууме с получением масла, которое растворяют в диэтиловом эфире. Раствор насыщают хлористым водородом, при этом получают твердое вещество, которое собирают путем фильтрации, промывают диэтиловым эфиром и сушат в вакууме с получением 3,65 г дигидрохлорида N-(1,4-бензодиоксан-2-илметил)-1-[1-(пирид-2-ил)пиперид-4-ил] метиламина в качестве белого твердого вещества с точкой плавления 217 - 228oC.
Пример 6. Смесь 10 г 8-окси-1,4-бензодиоксан-2-илметанола, полученного приемом, аналогичным тому, описанному в патенте США N 3101354, 2,2 г гидроокиси натрия и 150 мл воды перемешивают в течение 30 минут, затем охлаждают до температуры 0oC. По каплям добавляют 3,42 мл метилйодида, после чего смесь нагревают до температуры 80oC и перемешивают в течение 4 часов. Смесь охлаждают до комнатной температуры и перемешивают в течение 16 часов. Продукт дважды экстрагируют этилацетатом, взятым в количестве по 200 мл, объединенные экстракты дважды промывают насыщенным раствором тиосульфата натрия, взятым в количестве по 200 мл, сушат над сульфатом магния, и растворитель удаляют в вакууме с получением 6,89 г 1-(8-метокси-1,4-бензодиоксан-2-ил)метанола в качестве твердого вещества.
Раствор 5,84 г толуол-4-сульфонилхлорида в 10 мл пиридина каплями при температуре 10 - 15oC подают в раствор 5,0 г 1-(8-метокси-1,4-бензодиоксан-2-ил)метанола в 30 мл пиридина, и смесь перемешивают при комнатной температуре в течение 20 часов. Смесь подают на избыток льда и разбавленной хлористоводородной кислоты. Получаемое твердое вещество собирают путем фильтрации, промывают водой и сушат в вакууме с получением 6,61 г 8-метокси-1,4-бензодиоксан-2-илметил-толуол-4-сульфоната в качестве белого твердого вещества с точкой плавления 66 - 68oC.
12,53 г 1-(2-метоксифенил)пиперидин-4-карбоксамида, полученного приемом, аналогичным методу, описанному в примере 2, порциями подают в перемешиваемую суспензию 4,36 г алюмогидрида лития в 900 мл тетрагидрофурана в атмосфере азота. Получаемую смесь перемешивают при комнатной температуре в течение 72 часов. Добавляют 5 мл 5-м. водного раствора гидроокиси натрия и 5 мл воды, а получаемое твердое вещество удаляют путем фильтрации. Растворитель удаляют из фильтрата в вакууме, и остаток растворяют в 500 мл этилацетата. Органическую фазу дважды промывают водой, взятой в количестве по 300 мл, сушат над сульфатом магния, и растворитель удаляют в вакууме с получением 13 г коричневого масла. Масло очищают путем флеш-хроматографии на силикагеле с применением в качестве элюента смеси этилацетата и метанола в соотношении 1:1. Силикагель из колонки суспендируют в метаноле, а смесь нагревают с обратным холодильником в течение 3 часов, фильтруют, и растворитель удаляют в вакууме с получением 4,8 г 1-[1-(2-метоксифенил)пиперид-4-ил]метиламина в качестве светло-коричневого масла.
Перемешиваемую смесь 1,12 г 8-метокси-1,4-бензодиоксан-2-илметил-толуол-4-сульфоната, 0,7 г 1-[1-(2-метоксифенил)пиперид-4-ил]метиламина, 1 г карбоната калия и каталитического количества йодида калия в 50 мл ацетонитрила нагревают с обратным холодильником в течение 120 часов. Растворитель удаляют в вакууме, и остаток растворяют в 100 мл этилацетата. Раствор промывают 50 мл воды, и продукт три раза экстрагируют 5-м. хлористоводородной кислотой, взятой в количестве по 70 мл. Кислотные экстракты объединяют, промывают 50 мл диэтилового эфира, подщелачивают 5-м. водным раствором гидроокиси натрия и три раза экстрагируют этилацетатом, взятом в количестве по 200 мл. Органические экстракты объединяют, сушат над сульфатом магния, а растворитель удаляют в вакууме с получением 0,85 г светло-коричневого масла, которое растворяют в 30 мл диэтилового эфира. В раствор подают поток газообразного хлористого водорода. Получаемое твердое вещество собирают путем фильтрации и сразу сушат в вакууме с получением 0,24 г дигидрохлорида N-(8-метокси-1,4-бензодиоксан-2-илметил)-1-[1-(2-метоксифенил)пиперид-4-ил] метиламина в качестве белого твердого вещества с точкой плавления 160oC (разлож.)
Пример 7. Смесь 7,25 г карбоната калия и 200 мл диметилформамида обрабатывают ультразвуком до того, как получают мутную, содержащую мельчайшие частицы суспензию. Добавляют 5,8 г катехина и 10 г (R)-глицидил-толуол-4-сульфоната, и получаемую смесь перемешивают и нагревают при температуре 60oC в течение 36 часов. Смеси дают охлаждаться, после чего ее подают в 200 мл ледяной воды, и продукт три раза экстрагируют диэтиловым эфиром, взятым в количестве по 300 мл. Объединенные вытяжки три раза промывают рассолом, взятым в количестве по 200 мл, сушат над сульфатом магния, а растворитель удаляют в вакууме с получением 7 г белого твердого вещества. Твердое вещество кристаллизуют из смеси этилацетата и петролейного эфира пределами кипения 40 - 60oC, при этом получают 3,47 г (S)-1,4-бензодиоксан-2-илметанола в качестве белого кристаллического вещества.
4,2 г толуол-4-сульфонилхлорида при температуре -10oC в атмосфере азота подают в перемешиваемый раствор 3,3 г (S)-1,4-бензодиоксан-2-илметанола в 50 мл пиридина. Смеси дают нагреваться до комнатной температуры, перемешивают в течение еще 60 часов, затем подают в 200 мл ледяной воды. Продукт три раза экстрагируют этилацетатом, взятым в количестве по 200 мл, объединенные экстракты промывают 100 мл разбавленной хлористоводородной кислоты и 100 мл воды, сушат над сульфатом магния, и растворитель удаляют в вакууме. Получают 5,84 г оранжевого масла, которое медленно отвердевается. Масло очищают путем кристаллизации из смеси диэтилового эфира и петролейного эфира с пределами кипения 40 - 60oC, при этом получают две порции белого твердого вещества весом 2,4 г и 1,2 г. 2,4 г получаемого твердого вещества 3 раза рекристаллизуют из этанола, при этом содержание желаемого (R)-изомера продукта в твердом веществе каждый раз становится меньше, поэтому твердое вещество не обрабатывают дальше. Маточные растворы трех рекристаллизаций, которые теперь в основном содержат желаемый (R)-изомер продукта, объединяют, растворитель удаляют в вакууме и остаток рекристаллизуют из этанола с получением 1,2 г (R)-1,4-бензодиоксан-2-илметил-толуол-4-сульфоната в виде белых кристаллов с точкой плавления 105 - 107oC.
Перемешиваемую смесь 0,84 г 1-[1-(2-метоксифенил)пиперид-4-ил]метиламина, полученного аналогичным примеру 2 приемом, 1,2 г (R)-1,4-бензодиоксан-2-илметил-толуол-4-сульфоната, 1 г карбоната калия и каталитического количества йодида калия в 50 мл ацетонитрила в течение 90 часов нагревают с обратным холодильником. Растворитель удаляют в вакууме, а остаток разбавляют 100 мл этилацетата. Продукт три раза экстрагируют 5-м. хлористоводородной кислотой, взятой в количестве по 70 мл, экстракты объединяют и подщелачивают 5-м. водным раствором гидроокиси натрия. Продукт три раза экстрагируют этилацетатом, взятым в количестве по 150 мл, объединенные экстракты дважды промывают водой, взятой в количестве по 100 мл, сушат над
сульфатом магния, а растворитель удаляют в вакууме. Остаток очищают путем флеш-хроматографии на силикагеле с применением этилацетата в качестве элюента. Соответствующие фракции объединяют, а растворитель удаляют в вакууме с получением желтого масла, которое кристаллизуется, когда оставляют стоять. Продукт затем рекристаллизуют из этилацетата, при этом получают 0,41 г (S)-(-)-N-(1,4-бензодиоксан-2-илметил)-1-[1-(2-метоксифенил)пиперид-4-ил] метиламина в качестве светло-коричневых кристаллов с точкой плавления 78 - 79oC, [α]
Пример 8. 8 г 1,4-бензодиоксан-2-илметил-толуол-4-сульфоната, полученного аналогичным примеру 1 приемом, порциями в течение 30 минут при температуре 100 - 140oC подают в раствор 4,9 г N-[3-(трифторметил)пирид-2-ил]этандиамина в 36 мл ксилола, смесь перемешивают при температуре 140oC в течение 6 часов и оставляют стоять при комнатной температуре в течение 48 часов.
Смесь подают в 200 мл воды, и продукт экстрагируют дихлорметаном. Экстракты промывают водой, сушат над сульфатом магния, и растворители удаляют в вакууме с получением 7,4 г коричневого масла, которое очищают путем флеш-хроматографии при пониженном давлении на силикагеле с применением этилацетата в качестве элюента. Соответствующие фракции объединяют, и растворитель удаляют в вакууме с получением двух фракций коричневого масла (фракция 1 - 1,35 г; фракция 2 - 0,6 г).
1,35 г фракции 1 растворяют в диэтиловом эфире, раствор насыщают хлористым водородом с получением твердого вещества, которое собирают путем фильтрации, суспендируют в 2-пропаноле, в течение 5 минут нагревают с обратным холодильником, охлаждают, собирают путем фильтрации, промывают этилацетатом и сушат в вакууме при температуре 40oC, при этом получают 1,4 г бежевого твердого вещества с точкой плавления 220 - 224oC.
0,6 г фракции 2 растворяют в диэтиловом эфире, а раствор насыщают хлористым водородом с получением твердого вещества, которое собирают путем фильтрации и сушат в вакууме при температуре 45oC, при этом получают 0,7 г коричневого твердого вещества. Твердое вещество суспендируют в этилацетате, в течение 5 минут нагревают с обратным холодильником, охлаждают, собирают путем фильтрации, суспендируют в пропан-2-оле, в течение 5 минут нагревают с обратным холодильником, охлаждают, собирают путем фильтрации, промывают этилацетатом и сушат в вакууме при температуре 45oC, при этом получают 0,55 г бежевого твердого вещества с точкой плавления 220 - 224oC.
Две порции получаемого твердого вещества объединяют, растирают в порошок и сушат в вакууме с получением 1,75 г дигидрохлорида N-(1,4-бензодиоксан-2-илметил)-N'-[3-(трифторметил)-2-(пиридил)] этандиамина в качестве бежевого твердого вещества с точкой плавления 222 - 224oC.
Пример 9. 120 мл диметилсульфата при температуре 70oC в атмосфере азота в течение 1 часа по каплям подают в перемешиваемую смесь 100 г 1,6-диоксинафталина, 75 г гидроокиси натрия и 600 мл воды. Смесь перемешивают при температуре 70oC еще в течение 3 часов. Дополнительно подают 30 мл диметилсульфата, и смесь перемешивают при комнатной температуре в течение 16 часов, после чего смесь разбавляют 1000 мл воды. Продукт три раза экстрагируют диэтиловым эфиром, взятым в количестве по 500 мл, экстракты три раза промывают водой, взятой в количестве по 500 мл, сушат над сульфатом магния, и растворитель удаляют в вакууме. Остаток перегоняют с получением 95 г 1,6-диметоксинафталина в качестве масла с пределами кипения 100 - 130oC/0,6 мбар, которое, когда его оставляют стоять, отверждается при комнатной температуре.
220 мл 2,5-м. раствора н-бутиллия в гексанах каплями при температуре 70oС в атмосфере азота подают в перемешиваемый раствор 95 г 1,6-диметоксинафталина в 1000 мл тетрагидрофурана. Добавляют 150 мл N,N,N',N'-тетраметилэтилендиамина, смесь перемешивают при комнатной температуре в течение 20 часов, затем охлаждают до температуры -20oC и выливают на 500 г твердой двуокиси углерода. По окончании выделения газа смесь разбавляют 1000 мл воды, подщелачивают 100 г карбоната натрия, дважды промывают диэтиловым эфиром, взятым в количестве по 100 мл, и подкисляют концентрированной хлористоводородной кислотой. Продукт экстрагируют этилацетатом, экстракты промывают рассолом, сушат над сульфатом магния, и растворители удаляют в вакууме. Остаток кристаллизуют из этилацетата с получением твердого вещества, которое собирают путем фильтрации, сушат в вакууме и растирают с получением 45,6 г 3,8-диметокси-2-нафтойной кислоты в виде бежевого порошка с точкой плавления 151 - 153oC. При концентрации жидкости получают вторую порцию 5,4 г 3,8-диметокси-2-нафтойной кислоты.
1,2 г лития в виде проволоки длиной 27 см и диаметром 3,2 мм небольшими порциями подают в перемешиваемую смесь 5 г 3,8-диметокси-2-нафтойной кислоты, 45 мл тетрагидрофурана, 10 мл т-бутанола и 130 мл жидкого аммиака до того, как синий цвет остается в течение 5 минут. Добавляют 10 г хлорида аммония, а аммиаку дают испарятся. Остаток подкисляют 5-м. хлористоводородной кислотой, и продукт экстрагируют этилацетатом. Экстракты промывают водой, сушат над сульфатом магния, а растворители удаляют в вакууме с получением желтого твердого вещества, которое кристаллизуют из водного метанола в качестве 3,2 г 8-метокси-1,2,3,4-тетрагидронафталин-2-карбоновой кислоты в виде блестящих, желтоватых плиток с точкой плавления 136 - 138oC.
Вышеописанный прием повторяют с применением 46 г 3,8-диметокси-2-нафтойной кислоты, 400 мл тетрагидрофурана, 100 мл т-бутанола, 1200 мл жидкого аммиака и 11 г лития в виде проволоки длиной 250 см и диаметром 3,2 мм. Получают 29,35 г продукта в виде желтоватых плиток.
Раствор 5 г 8-метокси-1,2,3,4-тетрагидронафталин-2-карбоновой кислоты в 50 мл тетрагидрофурана в атмосфере азота нагревают до температуры кипячения, и по каплям добавляют 5 мл 10-м. раствора комплекса борана и диметилсульфида в диметилсульфиде. Смесь в течение 5 часов осторожно нагревают с обратным холодильником, после чего ее оставляют стоять при комнатной температуре в течение 16 часов, охлаждают на льду и прекращают реакцию путем осторожного добавления воды. Смесь подкисляют 5-м. хлористоводородной кислотой, нагревают с удалением диметилсульфида, охлаждают, подщелачивают 5-молярным водным раствором гидроокиси натрия, и продукт экстрагируют этилацетатом. Экстракты промывают водой, сушат над сульфатом магния, а растворители удаляют с получением 3,8 г 1-(8-метокси-1,2,3,4-тетрагидронафт-2-ил)метанола в виде бледно-желтого сиропа, который при комнатной температуре медленно отверждается.
Раствор 1,3 г толуол-4-сульфонилхлорида в 1,4 мл пиридина по каплям подают в перемешиваемый раствор 1,0 г 1-(8-метокси-1,2,3,4-тетрагидронафт-2-ил)-метанола в 2,4 мл пиридина, и смесь перемешивают в течение 64 часов при комнатной температуре, и еще в течение 4 часов при температуре 35 - 52oC. Смесь подают в ледяную воду, и продукт экстрагируют дихлорметаном. Экстракты промывают холодной как лед 5-м. хлористоводородной кислотой и холодной как лед водой, затем сушат над сульфатом магния. Растворители удаляют в вакууме с получением 1,15 г оранжевого масла.
Вышеописанный прием повторяют с добавлением раствора 2,75 г толуол-4-сульфонилхлорида в 3,0 мл сухого пиридина в раствор 2,2 г спирта в 5,15 мл пиридина. Смесь перемешивают в течение 6 часов при температуре 35 - 50oC, оставляют стоять в течение ночи при комнатной температуре, и обрабатывают вышеописанным приемом с получением 2,8 г оранжевого масла.
Получаемые оранжевые масла весом всего 3,95 г объединяют и растворяют в 75 мл пиридина. Добавляют раствор 4,1 г толуол-4-сульфонилхлорида в 4,5 мл пиридина, смесь перемешивают при комнатной температуре в течение 24 часов и подают в холодную как лед 5-м. хлористоводородную кислоту. Добавляют петролейный эфир с пределами кипения 40 - 60oC, и смесь растирают в порошок до завершения отверждения продукта. Твердое вещество собирают путем фильтрации, промывают водой и петролейным эфиром с пределами кипения 40 - 60oC, и сушат в вакууме при температуре 45 - 50oC, с получением 2,9 г 8-метокси-1,2,3,4-тетрагидронафт-2-илметил-толуол-4-сульфоната в виде твердого вещества с точкой плавления 53 - 58oC.
15,4 г мелкого порошкообразного 2-хлорпиримидина в течение 40 минут порциями подают в горячую, имеющую температуру 95oC, перемешиваемую смесь 30 г 1-(пиперид-4-ил)метиламина, 30 г карбоната натрия и 100 мл 3-метил-1-бутанола. Смесь перемешивают при температуре 95oC в течение 18 часов, охлаждают, разбавляют этилацетатом и фильтруют. Растворители удаляют в вакууме с получением темно-коричневого масла, которое подвергают перегонке с получением 12,2 г бледно-желтого масла с пределами кипения 126-130oC/1 мбар. Масло растворяют в диэтиловом эфире, и раствор насыщают хлористым водородом. Добавляют этанол, и смесь растирают в порошок с получением твердого вещества, которое собирают путем фильтрации, промывают диэтиловым эфиром и сушат в вакууме при температуре 40oC с получением 9,75 г дигидрохлорида 1-[1-(пиримидин-2-ил)пиперид-4-ил] метиламина в виде бледно-желтого твердого вещества с точкой плавления 240-245oC.
2,2 г дигидрохлорида 1-[1-(пиримидин-2-ил)пиперид-4-ил] метиламина подщелачивают 5-м. водным раствором гидроокиси натрия, а свободное основание дважды экстрагируют этилацетатом, взятым в количестве по 25 мл. Экстракты промывают водой, сушат над сульфатом магния, а растворители удаляют в вакууме с получением 1,6 г оранжевого масла. Масло одной порцией подают в смесь 2,9 г 8-метокси-1,2,3,4-тетрагидронафт-2-илметил-толуол-4-сульфоната, 2,5 г карбоната калия и 25 мл ацетонитрила, смесь перемешивают и нагревают с обратным холодильником в атмосфере азота в течение 30 часов. Смеси дают охлаждаться, фильтруют, и отфильтрованный осадок промывают ацетонитрилом. Фильтрат и промывочную жидкость объединяют, и растворитель удаляют в вакууме с получением 3,2 г оранжевого масла.
Масло очищают путем флеш-хроматографии под пониженным давлением на силикагеле с применением в качестве элюента смеси диэтилового эфира и метанола в соотношении 9:1. Соответствующие фракции объединяют, и растворитель удаляют в вакууме с получением 2,1 г желтого масла.
Масло растворяют в 20 мл 2-пропанола и добавляют 5 мл концентрированной хлористоводородной кислоты. Растворители удаляют с получением очень гигроскопического твердого вещества, которое суспендируют в 5-м. водном растворе гидроокиси натрия. Продукт экстрагируют этилацетатом. Экстракты сушат над сульфатом магния и подают в раствор 0,7 г фумаровой кислоты в 10 мл метанола. Получаемый раствор разбавляют этилацетатом и сгущают при пониженном давлении до осаждения белого твердого вещества. Твердое вещество собирают путем фильтрации и кристаллизуют из 50 мл смеси этилацетата и денатурированного спирта в соотношении 4:1, при этом получают 1,1 г белого твердого вещества с точкой плавления 184 - 186oC, представляющего собой N-(8-метокси-1,2,3,4-тетрагидронафт-2-илметил)-1-[1-(пиримидин-2-ил)пиперид-4-ил]метиламин в виде 1,1-гидрохлорида вместо ожидаемой соли фумаровой кислоты.
Пример 10. Рекристаллизационные жидкости, оставшиеся после выделения продукта, описанного в примере 9, вновь подщелачивают 5-м. водным раствором гидроокиси натрия, а свободное основание экстрагируют этилацетатом. Экстракты промывают водой, сушат над сульфатом магния, фильтруют, и растворители удаляют в вакууме с получением 0,35 г N-(8-метокси-1,2,3,4-тетрагидронафт-2-ил-метил)-1-[1-(пиримидин-2-ил)пиперид-4-ил]метиламина в качестве коричневого масла. Масло растворяют в 14 мл дихлорметана, а в атмосфере азота добавляют 3,5 мл 1-м. раствора трехбромистого бора в дихлорметане. Смесь перемешивают в атмосфере азота при комнатной температуре в течение 24 часов, после чего добавляют 5 мл метанола. Смесь перемешивают при комнатной температуре в течение еще 10 минут, после чего растворитель удаляют путем перегонки при атмосферном давлении с получением зеленого масла, которое растирают в порошок с помощью горячего 2-пропанола, при этом получают 0,5 г бледно-коричневого твердого вещества с точкой плавления 85 - 90oC.
При попытке кристаллизации из смеси денатурированного спирта и этилацетата в соотношении 1:2 получают смолу. В результате декантирования надосадочной жидкости и титрования остатка с ацетоном получают твердое вещество, которое собирают путем фильтрации и сушат в вакууме с получением 0,14 г дигидробромида 7-{ N-[1-(пиримидин-2-ил)пиперид-4-илметил]аминометил}-5,6,7,8-тетрагидронафт-1-ола в виде бледно-коричневого твердого вещества с точкой плавления > 110oC (разлож.)
Пример 11. Смесь 50 г 3-метоксифенола, 60 мл этилвинилового эфира, 250 мл дихлорметана и 1,3 г трихлоруксусной кислоты перемешивают при комнатной температуре в течение 20 часов, разбавляют 1000 мл диэтилового эфира, дважды промывают 0,5-м. водным раствором гидроокиси натрия, взятым в количестве по 100 мл, и один раз - 100 мл рассола, сушат над карбонатом калия, а растворитель удаляют в вакууме. Получают 3-(1-этоксиэтокси)анизол, который применяют без дальнейшей очистки.
3-(1-этоксиэтокси)анизол растворяют в 750 мл диэтилового эфира, и каплями при комнатной температуре в атмосфере азота добавляют 500 мл 1,6-м. раствора н-бутиллития в гексанах. Смесь перемешивают при комнатной температуре в течение 2 часов, охлаждают до температуры 10oC и каплями добавляют раствор 94 г диметилформамида в 150 мл диэтилового эфира. Смесь перемешивают при комнатной температуре в течение 2 часов, затем наливают на лед. Продукт экстрагируют диэтиловым эфиром, объединенные экстракты сушат над сульфатом магния, и растворители удаляют в вакууме. Остаточное масло растворяют в 500 мл метанола, и перемешиваемый раствор охлаждают во льду и сильно подкисляют 2-м. хлористоводородной кислотой. Получаемое твердое вещество собирают путем фильтрации, промывают водой, сушат в вакууме над пятиокисью фосфора и кристаллизуют из петролейного эфира с пределами кипения 60 - 80oC, при этом получают 16 г 2-окси-6-метокси-бензальдегида в виде бледно-желтого игольчатого вещества с точкой плавления 69 - 71oC.
Весь процесс повторяют с использованием в качестве исходного вещества 111,2 г 3-метоксифенола. На второй стадии процесса перед добавлением льда реакционную смесь перемешивают в течение 16 часов при комнатной температуре. В результате кристаллизации из петролейного эфира с пределами кипения 40 - 60oC получают продукт в качестве двух партий бледно-желтого игольчатого вещества. Две партии объединяют и рекристаллизуют из петролейного эфира с пределами кипения 40 - 60oC, при этом получают 40 г 2-окси-6-метоксибензальдегида в виде бледно-желтого игольчатого вещества.
Смесь 29,2 г 2-окси-6-метоксибензальдегида, 7,5 г 1,4-диазабицикло[2.2.2] -октана и 100 мл этилакрилата в течение 16 часов нагревают при температуре 95oC. Смесь разбавляют 500 мл этилацетата, охлаждают до комнатной температуры, дважды промывают насыщенным водным раствором бикарбоната натрия, взятым в количестве по 100 мл, дважды - 5-м. хлористоводородной кислотой, взятой в количестве по 100 мл, и дважды водой, взятой в количестве по 100 мл, сушат над сульфатом магния, а растворитель удаляют в вакууме с получением коричневого масла.
Масло растворяют в 500 мл этанола, и добавляют 30 г гидроокиси калия и 500 мл воды. Смесь в течение 3 часов нагревают с обратным холодильником и оставляют стоять при комнатной температуре в течение 16 часов. Смесь разбавляют водой, дважды промывают диэтиловым эфиром, взятым в количестве по 200 мл, и концентрированной хлористоводородной кислотой подкисляют до pH 1. Осаждается твердое вещество, которое собирают путем фильтрации, промывают водой и растирают в порошок с помощью диэтилового эфира, при этом получают 4,3 г бледно-коричневого твердого вещества. Вязкому остатку от титрования дают сгущаться в течение 16 часов при комнатной температуре, при этом получают еще 0,9 г бледно-коричневого твердого вещества. Две партии получаемого твердого вещества объединяют. Выход: 5,2 г 5-метокси-2H-1-бензопиран-3-карбоновой кислоты с точкой плавления 209 - 211oC.
Смесь 12,25 г 5-метокси-2H-1-бензопиран-3-карбоновой кислоты, полученной приемом, аналогичным тому, описанному выше, 3 г 10%-ного палладия на угле в качестве катализатора, и 250 мл этанола гидрируют при давлении 1 атм в течение 1 часа. Поглощение водорода осуществляется только медленно, по этому добавляют еще 3 г катализатора, и гидрирование продолжается в течение 5 часов. Смесь фильтруют, и растворитель удаляют в вакууме с получением полутвердого вещества, которое кристаллизуют из 2-пропанола, при этом получают массу бледно-желтых кристаллов, которую измельчивают, собирают путем фильтрации, промывают холодным как лед 2-пропанолом, сушат в вакууме, растирают в порошок, и вновь сушат в вакууме с получением 7,4 г 5-метокси-3,4-дигидро-2H-1-бензопиран-3-карбоновой кислоты в качестве желтоватого порошка с точкой плавления 147 - 150oC.
1 мл 1-м. раствора комплекса борана и диметилсульфида в диметилсульфиде при комнатной температуре в атмосфере азота подают в перемешиваемый раствор 1,0 г 5-метокси-3,4-дигидро-2H-1-бензопиран-3-карбоновой кислоты в 10 мл тетрагидрофурана, и смесь в течение 5 часов нагревают с обратным холодильником, после чего в течение 16 часов ее оставляют стоять при комнатной температуре в атмосфере азота.
Смесь охлаждают на льду, и реакцию прекращают тем, что по каплям добавляют воду до прекращения выделения газа, после чего смесь подкисляют 5-м. хлористоводородной кислотой и нагревают до температуры примерно 95oC с удалением диметилсульфида. Смесь охлаждают на льду и подщелачивают 5-м. водным раствором гидроокиси натрия. Продукт экстрагируют этилацетатом, экстракты промывают насыщенным солевым раствором и сушат над сульфатом магния. Растворители удаляют в вакууме с получением 1 г 1-(5-метокси-3,4-дигидро-2H-1-бензопиран-3-ил)метанола в качестве бледно-желтого масла.
Вышеописанную реакцию повторяют с применением остальных 6,0 г 5-метокси-3,4-дигидро-2H-1-бензопиран-3-карбоновой кислоты в 60 мл тетрагидрофурана и 6 мл комплекса борана и диметилсульфида. При этом получают еще 5,6 г 1-(5-метокси-3,4-дигидро-2H-1-бензопиран-3-ил)метанола.
Раствор 8,1 г толуол-4-сульфонилхлорида в 9 мл пиридина по каплям при комнатной температуре добавляют в раствор 6,5 г 1-(5-метокси-3,4-дигидро-2H-1-бензопиран-3-ил)метанола в 15,2 мл пиридина, после чего смесь в течение 3 часов перемешивают при комнатной температуре и оставляют стоять в течение 16 часов при комнатной температуре. Добавляют еще 2,0 г толуол-4-сульфонилхлорида, смесь перемешивают при комнатной температуре в течение 6 часов и оставляют стоять в течение 72 часов при комнатной температуре. Смесь подают в избыток 5-м. хлористоводородной кислоты и льда, и продукт экстрагируют этилацетатом. Экстракты промывают холодной как лед водой, сушат над сульфатом магния, а растворитель удаляют в вакууме с получением 6,6 г желтого масла, которое растворяют в 50 мл дихлорметана. Добавляют 3,35 г 4-(диметиламин)пиридина. Раствор охлаждают до температуры 0oC, и одной порцией добавляют 4,8 г толуол-4-сульфонилхлорида. Смесь перемешивают при температуре 0oC, в то время как толуол-4-сульфонилхлорид растворяется, затем прекращают охлаждение, и раствор перемешивают при комнатной температуре в течение 16 часов. Смесь в вакууме сгущают до объема 20 мл, затем разбавляют этилацетатом, при этом осаждается белое твердое вещество. Смесь фильтруют, фильтрат разбавляют дополнительным этилацетатом с получением второй партии твердого вещества, и снова фильтруют. Фильтрат второй партии оставляют стоять при комнатной температуре в течение 72 часов, при этом получают третий осадок твердого вещества и снова фильтруют. Растворители удаляют в вакууме с получением 7,0 г 5-метокси-3,4-дигидро-2H-1-бензопиран-3-илметил-толуол-4-сульфоната в качестве желтого масла, которое используют без дальнейшей очистки.
Смесь 7,0 г 5-метокси-3,4-дигидро-2H-1-бензопиран-3-илметил-толуол-4-сульфоната, 5,6 г карбоната калия и 57 мл ацетонитрила перемешивают при температуре 60oC, и добавляют 3,1 г 1-[1-(пиримидин-2-ил)-пиперид-4-ил]-метиламина, полученного аналогичным примеру 9 приемом. Смесь перемешивают и нагревают с обратным холодильником в атмосфере азота в течение 24 часов, после чего смеси дают охлаждаться и фильтруют. Фильтровальную прокладку промывают ацетонитрилом, а фильтрат и промывочную жидкость объединяют. Растворитель удаляют в вакууме с получением 7,5 г оранжевого масла, которое очищают путем флеш-хроматографии при пониженном давлении на силикагеле с применением в качестве элюента смеси диэтилового эфира и метанола в соотношении 9:1. Соответствующие фракции объединяют, и растворители удаляют в вакууме с получением двух фракций масла в виде 3,6 г продукта с небольшим количеством примесей, и 1,5 г чистого продукта.
1,5 г получаемого чистого масла растворяют в 40 мл этилацетата и подают в раствор 0,45 г фумаровой кислоты и 20 мл денатурированного спирта. Получаемый раствор разбавляют этилацетатом до общего объема 100 мл и охлаждают до температуры 0oC. Получаемый осадок собирают путем фильтрации, промывают этилацетатом и сушат в вакууме с получением 1,5 г N-(5-метокси-3,4-дигидро-2H-1-бензопиран-3-илметил)-1-[1-(пиримидин-2-ил)пиперид-4-ил] -метиламина в виде монофумарата в качестве белого твердого вещества с точкой плавления 202 - 204oC.
Пример 12. Смесь 50 г хлорида 4-карбамоил-1-(2,4-динитрофенил)пиридиния, полученного аналогичным примеру 2 приемом, 35 мл анилина и 1 л метанола перемешивают при комнатной температуре в течение 48 часов. Получаемую суспензию в течение 1 часа нагревают при температуре 50oC, охлаждают, и растворитель удаляют в вакууме. Твердый остаток дважды растирают в порошок с помощью ацетона, взятого в количестве по 1 л, после чего собирают путем фильтрации с получением 33,24 г хлорида 4-карбамоил-1-фенилпиридиния с точкой плавления 290 - 292oC.
Смесь 13 г хлорида 4-карбамоил-1-фенилпиридиния, 260 мг 10%-ного палладия на угле в качестве катализатора и 250 мл этанола гидрируют при комнатной температуре и атмосферном давлении в течение 2 дней. Раствор фильтруют на силикате марки Celite и фильтрат сгущают до объема примерно 50 мл. Раствор охлаждают и твердый осадок собирают путем фильтрации с получением 5,99 г 1-фенилпиперидин-4-карбоксамида.
Состоящую из Celite фильтровальную прокладку тщательно промывают горячим этанолом, и растворитель удаляют в вакууме. Остаток объединяют с полученным на вышеописанной стадии фильтратом, и смесь в течение 0,5 часов нагревают с обратным холодильником. Осадившееся при последующем охлаждении твердое вещество собирают путем фильтрации с получением еще 3,42 г 1-фенилпиперидин-4-карбоксамида. Общий выход: 9,41 г.
1,5 г 1-фенилпиперидин-4-карбоксамида порциями подают в перемешиваемую суспензию 0,5 г алюмогидрида лития в 100 мл сухого тетрагидрофурана в атмосфере азота. Получаемую суспензию перемешивают при комнатной температуре в течение 2 часов, после чего нагревают с обратным холодильником в течение 2 часов. Смесь охлаждают и последовательно добавляют 0,5 мл воды и 0,5 мл концентрированного раствора гидроокиси натрия. Получаемый осадок удаляют путем фильтрации на силикате марки Celite. Фильтрат сушат над сульфатом магния, и растворитель удаляют в вакууме с получением 1,15 г 1-(1-фенилпиперид-4-ил)метиламина.
Вышеописанный процесс повторяют с использованием исходных веществ в указанном выше количестве, умноженном на фактор 4,33, при этом получают 5,1 г 1-(1-фенилпиперид-4-ил)метиламина.
Перемешиваемую смесь 6 г 1-(1-фенилпиперид-4-ил)метиламина, 10,1 г 1,4-бензодиоксан-2-илметил-толуол-4-сульфоната, полученного аналогичным примеру 1 приемом, 15 г карбоната калия и ацетонитрила в течение 72 часов нагревают с обратным холодильником. Получаемую смесь охлаждают и фильтруют. Растворитель удаляют в вакууме, и остаток растворяют в этилацетате. Раствор фильтруют на силикагеле с применением в качестве элюента 500 мл этилацетата. Растворитель удаляют в вакууме. Остаток растворяют в диэтиловом эфире, и в раствор подают газообразный хлористый водород. Получаемое твердое вещество собирают путем фильтрации, растирают в порошок с помощью 250 мл смеси этилацетата и метанола в соотношении 25:1 и вновь фильтруют с получением 5,64 г дигидрохлорида N-(1,4-бензодиоксан-2-ил-метил)-1-(1-фенилпиперид-4-ил)метиламина в качестве белого твердого вещества с точкой плавления 278 - 280oC.
Пример 13. Смесь 3,5 г 1,4-бензодиоксан-5-иламина (полученного приемом, аналогичным тому, описанному в Ж. Орг. Хим., 1967 г., 3, стр. 1121), 6,8 г хлорида 4-карбамоил-1-(2,4-динитрофенил)пиридиния (полученного аналогичным примеру 2 приемом) и 150 мл метанола перемешивают в течение 16 часов при комнатной температуре. Добавляют 100 мл метанола, и получаемую смесь нагревают с обратным холодильником в течение 3 часов, затем оставляют стоять в течение 2,5 дней при комнатной температуре. Добавляют еще 0,35 г 1,4-бензодиоксан-5-иламина, и смесь нагревают с обратным холодильником в течение 24 часов. Растворитель удаляют в вакууме, и остаток растирают в порошок с помощью диэтилового эфира. Получаемое твердое вещество кристаллизуют из смеси 2-пропанола и этилацетата в соотношении 1:1, при этом получают бледно-желтое твердое вещество. Растворители в вакууме удаляют из фильтрата, и остаток растирают в порошок с помощью ацетона с получением дополнительного желтого твердого вещества, которое объединяют с полученным при кристаллизации твердым веществом, при этом получают 3,57 г партии (1).
Смесь 3,0 г 1,4-бензодиоксан-5-иламина, 5,8 г хлорида 4-карбамоил-1-(2,4-динитрофенил)пиридиния и 150 мл метанола перемешивают и нагревают с обратным холодильником в течение 16 часов. Растворитель удаляют в вакууме, и остаток растирают в порошок с помощью диэтилового эфира. Диэтиловый эфир декантируют, и твердый остаток растирают в порошок с помощью горячего ацетона. Получаемую смесь охлаждают и фильтруют, при этом получают партию (2) в виде 5,30 г желтого твердого вещества.
Партии (1) и (2) объединяют и растирают в порошок с помощью горячего ацетона. Смесь охлаждают, и получаемое твердое вещество собирают путем фильтрации и сушат в вакууме с получением 8,7 г хлорида 1-(1,4-бензодиоксан-5-ил)-4-карбамоилпиридиния в качестве желтого твердого вещества с точкой плавления 256 - 258oC.
В 1,8 г 10%-ного палладия на угле в качестве катализатора в атмосфере азота подают 2,0 г хлорида 1-(1,4-бензодиоксан-5-ил)-4-карбамоилпиридиния. Добавляют 3,5 г формиата аммония, и при перемешивании получаемой смеси по каплям подают 35 мл метанола. Получаемую смесь в течение 3,5 часов нагревают с обратным холодильником. Формиат аммония, который кристаллизуется в конденсаторе, добавлением 35 мл метанола вновь подают в смесь. Смесь охлаждают и фильтруют на силикате марки Celite в атмосфере азота. Фильтрат подщелачивают твердым бикарбонатом натрия, и растворитель удаляют в вакууме. Остаток разбавляют 100 мл воды, и продукт три раза экстрагируют дихлорметаном, взятым в количестве по 100 мл. Объединенные экстракты промывают 100 мл воды, сушат над сульфатом магния, фильтруют, а растворитель удаляют в вакууме с получением 1,27 г 1-(1,4-бензодиоксан-5-ил)пиперидин-4-карбоксамида в качестве белого твердого вещества с точкой плавления ~194oC.
Смесь 4,1 г 1-(1,4-бензодиоксан-5-ил)пиперидин-4-карбоксамида, полученного вышеописанным приемом, и 400 мл тетрагидрофурана по каплям подают в перемешиваемую смесь 1,2 г алюмогидрида лития в 100 мл тетрагидрофурана в атмосфере азота. Получаемую смесь перемешивают в течение 2 часов при комнатной температуре. Добавляют еще 0,4 г алюмогидрида лития, и получаемую смесь перемешивают в течение еще 1,5 часов.
Добавляют 3 мл воды и 3 мл концентрированного раствора гидроокиси натрия, смесь перемешивают в течение 15 минут и фильтруют на силикате марки Celite.
Прокладку Celite промывают этилацетатом. Фильтраты объединяют, и растворители удаляют в вакууме. Остаток растворяют в дихлорметане, раствор сушат над сульфатом магния, фильтруют, и растворитель удаляют в вакууме с получением 4,19 г 1-[1-(1,4-бензодиоксан-5-ил)пиперид-4-ил]метиламина в качестве оранжевого масла, которое используют без дальнейшей очистки.
Смесь 4,19 г оранжевого масла, 5,0 г 1,4-бензодиоксан-2-илметил-толуол-4-сульфоната, полученного аналогичным примеру 1 приемом, 4,32 г карбоната калия и 100 мл ацетонитрила перемешивают при нагревании с обратным холодильником в течение 30 часов, после чего смесь оставляют стоять при комнатной температуре в течение 2,5 дней. Получаемую смесь фильтруют, и растворитель удаляют в вакууме с получением 8,0 г оранжевого масла.
Получаемое масло очищают путем флеш-хроматографии на силикагеле, при этом в качестве элюента последовательно применяют смесь петролейного эфира с пределами кипения 40 - 60oC и этилацетата в соотношении 1:1, смесь петролейного эфира с пределами кипения 40 - 60oC и этилацетата в соотношении 1:2, этилацетат, а затем метанол. Растворители удаляют из соответствующих фракций в вакууме, а 2,7 г получаемого при этом остатка растворяют в смеси этилацетата и метанола. Раствор насыщают хлористым водородом, растворители удаляют в вакууме, и остаток кристаллизуют из смеси метанола и этилацетата с получением 2,6 г дигидрохлорида N-(1,4-бензодиоксан-2-илметил)-1-[1-(1,4-бензодиоксан-5-ил)пиперид-4-ил] метиламина в качестве светло-розового твердого вещества с точкой плавления 226 - 228oC.
Пример 14. Перемешиваемую смесь 0,89 г 1-[1-(2-метоксифенил)пиперид-4-ил] метиламина, полученного аналогичным примеру 2 приемом, 1,3 г (S)-1,4-бензоди-оксан-2-илметил-толуол-4-сульфоната, 1 г карбоната калия и несколько кристаллов йодида калия в 50 мл ацетонитрила в течение 72 часов нагревают с обратным холодильником. Растворитель удаляют в вакууме, добавляют 50 мл воды, и продукт три раза экстрагируют этилацетатом, взятым в количестве по 50 мл. Объединенные органические экстракты сушат над сульфатом магния, и растворитель удаляют в вакууме. Продукт очищают путем флеш-хроматографии на силикагеле с применением этилацетата в качестве элюента. Соответствующие фракции объединяют, и растворитель удаляют в вакууме, при этом получают 0,69 г белого кристаллического твердого вещества. Твердое вещество 6 раз рекристаллизуют из этанола с получением 0,4 г (R)-(+)-N-(1,4-бензодиоксан-2-илметил)-1-[1-(2-метоксифенил)пиперид-4-ил] метиламина с точкой плавления 79 - 82oC. [α]
Пример 15. Смесь 20,0 г 2-(хлорметил)-1,4-бензодиоксана и 34,04 г этил-пиперидин-4-карбоксилата перемешивают и нагревают при температуре 130oC в течение 3,5 часов. При охлаждении смесь частично отверждается. Добавляют 100 мл воды, и продукт три раза экстрагируют этилацетатом, взятым в количестве по 100 мл. Объединенные экстракты сушат над сульфатом магния, фильтруют, и растворитель удаляют в вакууме с получением 35 г коричневого масла, которое очищают путем перегонки по Кюгельреру. Фракция, получаемая после нагревания до температуры 250oC/5,3 мбар представляет собой 31,5 г сложного этилового эфира 1-(1,4-бензодиоксан-2-илметил)пиперидин-4-карбоновой кислоты.
Раствор 5 г сложного этилового эфира 1-(1,4-бензодиоксан-2-илметил)пиперидин-4-карбоновой кислоты в 100 мл денатурированного спирта подают в раствор 4 г гидроокиси калия в 50 мл воды. Смесь перемешивают при комнатной температуре в течение 1 часа, после чего в течение 6 часов нагревают с обратным холодильником. Раствор охлаждают, и растворитель удаляют в вакууме. Остаток разбавляют водой, затем нейтрализуют разбавленной хлористоводородной кислотой. Продукт экстрагируют этилацетатом, и экстракт сушат над сульфатом магния. Растворитель удаляют в вакууме с получением 0,28 г сырой 1-(1,4-бензодиоксан-2-илметил)пиперидин-4-карбоновой кислоты. Водную фазу упаривают досуха, и остаток дважды растирают в порошок с помощью смеси дихлорметана и метанола в соотношении 25:1, взятой в количестве по 100 мл. Неорганические остатки удаляют путем фильтрации, и растворитель удаляют из фильтрата в вакууме с получением еще 5,03 г сырого целевого продукта, который используют без дальнейшей очистки.
0,1 г сложного этилового эфира хлормуравьиной кислоты по каплям подают в перемешиваемый раствор 0,28 г 1-(1,4-бензодиоксан-2-илметил)пиперидин-4-карбоновой кислоты и 0,12 г триэтиламина в 10 мл хлороформа при температуре 0oC. Спустя 30 минут добавляют раствор 0,12 г 2-метоксианилина в 5 мл хлороформа, и смесь перемешивают при комнатной температуре в течение 16 часов. Реакционную смесь последовательно промывают водой, 1-м. водным раствором гидроокиси натрия, 1-м. хлористоводородной кислотой и водой. Раствор сушат над сульфатом магния, растворитель удаляют в вакууме, а остаток подвергают элюации на силикагельной колонке с применением в качестве элюента смеси петролейного эфира с пределами кипения 40 - 60oC и этилацетата в соотношении 1: 1. Растворитель удаляют в вакууме с получением 50 мг 1-(1,4-бензодиоксан-2-илметил)-N-(2-метоксифенил)пиперидин-4-карбоксамида в качестве твердого вещества. Описанный здесь процесс повторяют с применением исходных веществ, взятых в указанном выше количестве, умноженном на фактор 9, со следующими изменениями: после перемешивания при комнатной температуре в течение 16 часов, реакционную смесь в течение 1 часа нагревают до температуры 50oC. Растворитель удаляют в вакууме, и остаток элюируют на силикагеле как описано выше. При этом получают еще 1,05 г целевого продукта.
0,5 мл 10-м. раствора комплекса борана и диметилсульфида в диметилсульфиде по каплям в атмосфере азота подают в раствор 0,35 г 1-(1,4-бензодиоксан-2-илметил)-N-(2-метоксифенил)пиперидин-4-карбоксамида в 25 мл сухого тетрагидрофурана, после чего смесь в течение 2 часов нагревают с обратным холодильником. Реакционную смесь охлаждают, и растворители удаляют в вакууме. Остаток осторожно разбавляют 70 мл 1-м. хлористоводородной кислоты и в течение 1,5 часов нагревают при температуре 95oC. Водную смесь охлаждают и подщелачивают 2,5-м. водным раствором гидроокиси натрия, после чего продукт экстрагируют дихлорметаном. Органический экстракт промывают водой, сушат над сульфатом магния, фильтруют, и растворитель удаляют в вакууме с получением 0,32 г 1-[1-(1,4-бензодиоксан-2-илметил)пиперид-4-ил] -N-(2-метоксифенил)метиламина. Процесс повторяют с применением 1,37 г 1-(1,4-бензодиоксан-2-илметил)-N-(2-метоксифенил)пиперидин-4-карбоксамида, полученного аналогично вышеописанному приему. Продукты двух реакций объединяют и растворяют в диэтиловом эфире. В раствор подают газообразный хлористый водород, и 1,2 г получаемого твердого осадка собирают путем фильтрации. Твердое вещество растворяют в 100 мл насыщенного водного раствора бикарбоната натрия, и получаемое масло экстрагируют этилацетатом, взятым в количестве 100 мл. Добавляют избыток насыщенного раствора щавелевой кислоты в этилацетате, твердый осадок собирают путем фильтрации и сушат в вакууме при температуре 50oC с получением 0,78 г 1-[1-(1,4-бензодиоксан-2-илметил)пиперид-4-ил]-N-(2-метоксифенил)метиламина в виде моно-оксалата с точкой плавления 215 - 218oC.
Пример 16. Смесь 50 г хлорида 4-карбамоил-1-(2,4-динитрофенил)-пиридиния, полученного аналогичным примеру 2 приемом, 40 г 4-метоксианилина и 1800 мл метанола перемешивают при комнатной температуре в течение 16 часов.
Растворитель удаляют в вакууме, а остаток растирают в порошок с помощью 1200 мл горячего диэтилового эфира. Суспензию охлаждают и фильтруют с получением 67,2 г твердого вещества золотого цвета. Твердое вещество рекристаллизуют из 1500 мл денатурированного спирта, при этом получают 45,6 г темно-желтых кристаллов. В денатурированный спирт подают диэтиловый эфир, и получаемое твердое вещество собирают путем фильтрации с получением 3 г хлорида 4-карбамоил-1-(4-метоксифенил)пиридиния. 45,6 г кристаллов суспендируют в 500 мл ацетона, и смесь нагревают с обратным холодильником в течение несколько минут, после чего смесь фильтруют. Получаемое при этом твердое вещество в течение 16 часов экстрагируют ацетоном в экстракторе Сокслета, при этом удаляют непрореагировавшиеся исходные продукты. Получаемое твердое вещество сушат в вакууме с получением еще 31,8 г хлорида 4-карбамоил-1-(4-метоксифенил)пиридиния. Общий выход: 34,8 г.
Суспензию 22,0 г хлорида 4-карбамоил-1-(4-метоксифенил)пиридиния в 250 мл метанола в присутствии 1,0 г 10%-ного палладия на угле в качестве катализатора в течение 2 дней при комнатной температуре и атмосферном давлении подвергают гидрированию. Добавляют еще 0,75 г катализатора, а после окончания гидрирования катализатор удаляют путем фильтрации. Фильтрат подщелачивают добавлением избытка триэтиламина, и растворитель удаляют в вакууме. Остаток распределяют между водой и смесью хлороформа и метанола в соотношении 19:1. Растворитель из органической фазы удаляют в вакууме, и остаток сушат в вакууме над пятиокисью фосфора. При этом получают 10 г 1-(4-метоксифенил)пиперидин-4-карбоксамида в качестве белого твердого вещества с точкой плавления 178 - 181oC. 15 мл 10-м. раствора комплекса борана и диметилсульфида в диметилсульфиде по каплям при температуре 15 - 20oC в атмосфере азота подают в перемешиваемую суспензию 10 г 1-(4-метоксифенил)пиперидин-4-карбоксамида в 95 мл тетрагидрофурана. Смесь в течение 4 часов нагревают с обратным холодильником, после чего ее оставляют стоять при комнатной температуре в течение 16 часов. Добавляют ледяную воду для разрушения избыточного восстановителя, после чего смесь подкисляют 5-м. хлористоводородной кислотой и промывают 200 мл диэтилового эфира. Водную фазу подщелачивают концентрированным водным раствором гидроокиси натрия, после чего продукт три раза экстрагируют диэтиловым эфиром, взятым в количестве по 250 мл. Объединенные экстракты сушат над сульфатом магния и растворитель удаляют в вакууме. Остаток очищают путем флеш-хроматографии на силикагеле с применением в качестве элюента смеси дихлорметана и денатурированного спирта в соотношении 9:1. Растворители удаляют из соответствующих фракций в вакууме, при этом получают 0,5 г 1-[1-(4-метоксифенил)-пиперид-4-ил]метиламина в качестве масла.
Перемешиваемую смесь 0,42 г 2-хлорметил-1,4-бензодиоксана, 0,5 г 1-[1-(4-метоксифенил)пиперид-4-ил] метиламина, 1,0 г карбоната калия в 50 мл ацетонитрила нагревают с обратным холодильником в течение 7 часов. Добавляют 0,32 мл триэтиламина, и перемешивание продолжают при нагревании с обратным холодильником в течение 6 часов. Добавляют 0,73 г 1,4-бензодиоксан-2-илметил-толуол-4-сульфоната, полученного аналогичным примеру 1 приемом, и смесь перемешивают в течение 7 часов при нагревании с обратным холодильником. Растворитель удаляют в вакууме с получением желтого масла, которое очищают путем флеш-хроматографии на силикагеле с применением в качестве элюента сперва дихлорметана, а затем смеси дихлорметана и денатурированного спирта в соотношении 19:1. Продуктсодержащие фракции собирают, и растворители удаляют в вакууме с получением 0,20 г масла золотого цвета. Масло растворяют в 50 мл диэтилового эфира, и в раствор подают газообразный хлористый водород. Получаемое белое твердое вещество собирают путем фильтрации, промывают диэтиловым эфиром и сразу сушат в вакууме, при этом получают 0,13 г дигидрохлорида N-(1,4-бензодиоксан-2-илметил)-1-[1-(4-метоксифенил)пиперид-4-ил] метиламина с точкой плавления 268 - 270oC.
Пример 17. Перемешиваемую смесь 26,8 г N-(3-бромпропил)фталимида, 24,6 г 2- метокси-анилина и 60 мл ксилола в течение 4 часов нагревают при температуре 140oC. После охлаждения растворитель удаляют в вакууме. Смесь получаемого масла, 100 мл денатурированного спирта и 6,9 г гидрата гидразина в течение 1 часа при перемешивании нагревают с обратным холодильником. Смесь охлаждают и подкисляют 5-м. хлористоводородной кислотой. Белый осадок удаляют путем фильтрации, и растворитель удаляют из фильтрата в вакууме. Остаток растворяют в 15 мл воды и подщелачивают 5-м. водным раствором гидроокиси натрия. Получаемое масло дважды экстрагируют дихлорметаном, взятым в количестве по 50 мл. Экстракты сушат, и растворитель удаляют в вакууме с получением 16,5 г маловязкого красного масла. Получаемое масло очищают путем флеш-хроматографии на силикагеле, при этом в качестве элюента последовательно применяют дихлорметан, смесь дихлорметана и денатурированного спирта в соотношении 9: 1, а затем метанол. Соответствующие фракции собирают и растворители удаляют в вакууме с получением 8,5 г N-(2-метоксифенил)-1,3-пропандиамина в качестве маловязкого масла золотого цвета.
Перемешиваемую смесь 2,4 г 8-метокси-1,4-бензодиоксан-2-илметил-толуол-4-сульфоната, полученного аналогичным примеру 6 приемом, 1,30 г N-(2-метоксифенил)-1,3-пропандиамина, каталитического количества йодида калия и 1,8 г карбоната калия в 120 мл ацетонитрила в течение 24 часов нагревают с обратным холодильником. После охлаждения смесь фильтруют и растворитель удаляют из фильтрата в вакууме, при этом получают 3,57 г красноватого масла. Получаемое масло очищают путем флеш-хроматографии на силикагеле с применением в качестве элюента смеси дихлорметана и денатурированного спирта в соотношении 19: 1. Продуктсодержащие фракции собирают и растворитель удаляют в вакууме с получением 1,4 г желтого масла. Масло растворяют в 50 мл диэтилового эфира и в раствор подают газообразный хлористый водород, при этом получают гигроскопическое твердое вещество. Масляное твердое вещество собирают путем фильтрации и распределяют между разбавленным водным раствором гидроокиси натрия и дихлорметаном. Органическую фазу отделяют, сушат над сульфатом магния, и растворитель удаляют в вакууме с получением желтого масла. Получаемое масло растворяют в 50 мл диэтилового эфира, и в раствор по каплям подают избыток насыщенного раствора малеиновой кислоты в диэтиловом эфире. Получаемое при этом твердое вещество собирают путем фильтрации и сушат в вакууме с получением 0,35 г N-(8-метокси-1,4-бензодиоксан-2-илметил)-N'-(2-метоксифенил)-1,3-пропандиамина в виде малеата в качестве твердого вещества с точкой плавления 59 - 62oC.
Пример 18. Смесь 43,2 г хлорида 4-карбамоил-1-(2,4-динитрофенил)пиридиния, полученного аналогичным примеру 2 приемом, 34 г 3-метоксианилина и 1500 мл метанола перемешивают при комнатной температуре в течение 16 часов. После этого смесь в течение 30 минут нагревают при температуре 40oC, и перемешивание продолжают в течение 2 часов при комнатной температуре. Растворитель удаляют в вакууме. В получаемое масляное твердое вещество добавляют диэтиловый эфир, затем твердое вещество собирают путем фильтрации, растирают в порошок с помощью 1 л горячего 2-пропанола, собирают путем фильтрации и сушат. Твердое вещество растирают в порошок с помощью горячего ацетона, собирают путем фильтрации и сушат с получением 32,4 г хлорида 4-карбамоил-1-(3-метоксифенил)пиридиния в качестве бежевого твердого вещества с точкой плавления 272oC (разлож.).
Суспензию 26 г хлорида 4-карбамоил-1-(3-метоксифенил)пиридиния в 300 мл метанола гидрируют до окончания поглощения водорода (т.е. в течение примерно 2 дней) при комнатной температуре и атмосферном давлении в присутствии в качестве катализатора 2,0 г 10%-ного палладия на угле. Смесь фильтруют, добавляют избыток триэтиламина, и растворитель удаляют в вакууме с получением розового твердого вещества, которое распределяют между водой и хлороформом. Органическую фазу сушат, и растворитель удаляют в вакууме с получением 19 г 1-(3-метоксифенил)пиперидин-4-карбоксамида в качестве розового твердого вещества с точкой плавления 153 - 156oC.
12,2 мл 10-м. раствора комплекса борана и диметилсульфида в диметилсульфиде по каплям при температуре 15 - 20oC в атмосфере азота подают в перемешиваемую суспензию 8,0 г 1-(3-метоксифенил)пиперидин-4-карбоксамида в 75 мл тетрагидрофурана. Суспензия постепенно растворяется. Когда все 12,2 мл вышеуказанного раствора подано, смесь нагревают с обратным холодильником в течение 4 часов, после чего ее оставляют стоять при комнатной температуре в течение 16 часов. Реакцию прекращают путем добавки реакционной смеси по каплям в перемешиваемую смесь льда и воды. Водную смесь подкисляют разбавленной хлористоводородной кислотой и промывают 200 мл диэтилового эфира. Нерастворимое вещество удаляют путем фильтрации и не обрабатывают дальше. Водную фазу подщелачивают концентрированным водным раствором гидроокиси натрия, охлаждают, и продукт три раза экстрагируют диэтиловым эфиром, взятым в количестве по 250 мл. Объединенные экстракты сушат над сульфатом магния, и растворитель удаляют в вакууме с
получением 4,7 г непрозрачного масла. Получаемое масло очищают путем флеш-хроматографии на силикагеле, при этом в качестве элюента последовательно применяют смесь дихлорметана и денатурированного спирта в соотношении 9: 1, а затем метанол. Продуктсодержащие фракции собирают, и растворитель удаляют в вакууме с получением 2,9 г 1-[1-(3-метоксифенил)-пиперид-4-ил]метиламина в качестве масла золотого цвета.
Перемешиваемую смесь 4,2 г 1,4-бензодиоксан-2-илметил-толуол-4-сульфоната, полученного аналогичным примеру 1 приемом, 2,9 г 1-[1-(3-метоксифенил)пиперид-4-ил] метиламина и 4 г карбоната калия в 250 мл ацетонитрила нагревают с обратным холодильником в течение 3 дней. Карбонат калия удаляют путем фильтрации и промывают ацетонитрилом. Растворитель удаляют из фильтрата в вакууме, при этом получают 6,0 г мутного масла, которое очищают путем флеш-хроматографии на силикагеле с применением в качестве элюента смеси дихлорметана и денатурированного спирта в соотношении 19:1. Продуктсодержащие фракции собирают и растворители удаляют в вакууме с получением 2,3 г желтого масла. Получаемое масло растворяют в диэтиловом эфире, и в раствор подают газообразный хлористый водород. Получаемое белое твердое вещество собирают путем фильтрации, промывают диэтиловым эфиром и сушат в вакууме с получением 0,93 г гидрохлорида N-(1,4-бензодиоксан-2-илметил)-1-[1-(3-метоксифенил)пиперид-4-ил] метиламина в качестве твердого вещества с точкой плавления 186 - 188oC.
Пример 19. 1,5 г рацемического N-(1,4-бензодиоксан-2-илметил)-1-[1-(пирид-2-ил)пиперид-4-ил] метиламина, полученного путем подщелачивания дигидрохлорида, полученного аналогичным примеру 5 приемом, разделяют на отдельные энантиомеры с помощью препаративной высокопроизводительной жидкостной хроматографии на колонке марки Chiralcel ОС внутренними габаритами 25 см x 2 см, с применением в качестве элюента смеси изогексана и этанола в соотношении 1:1. Соответствующие фракции объединяют, и растворители удаляют в вакууме.
Первую элюированную фракцию растворяют в диэтиловом эфире, раствор насыщают хлористым водородом, и получаемое твердое вещество собирают путем фильтрации и сушат в вакууме с получением 0,5 г дигидрохлорида (-)-N-(1,4-бензодиоксан-2-илметил)-1-[1-(пирид-2-ил)пиперид-4-ил] метиламина в качестве белого твердого вещества с точкой плавления 228 - 232oC. [α]
Пример 20. Вторую элюированную фракцию, полученную в результате описанного в примере 19 разделения путем препаративной высокопроизводительной жидкостной хроматографии, растворяют в диэтиловом эфире, раствор насыщают хлористым водородом, а получаемое твердое вещество собирают путем фильтрации и сушат в вакууме с получением 0,55 г дигидрохлорида (+)-N-(1,4-бензодиоксан-2-илметил)-1-[1-(пирид-2-ил)пиперид-4-ил] метиламина в качестве белого твердого вещества с точкой плавления 227 - 230oC. [α]
Пример 21. Второй раствор 12,88 г 1-(6,7-дихлор-1,4-бензодиоксан-2-ил)метанола, полученного приемом, аналогичным тому, описанному в Gazz.Chim. Ital, 1965 г, 95, стр. 1447, в 9 мл пиридина по каплям подают в охлажденный льдом перемешиваемый раствор 12 г толуол-4-сульфонилхлорида в 21 мл пиридина. Смеси дают охлаждаться до комнатной температуры и перемешивают в течение 60 часов, после чего ее подают в 150 мл воды, подкисляют концентрированной хлористоводородной кислотой, и продукт 4 раза экстрагируют этилацетатом, взятым в количестве по 80 мл. Объединенные экстракты дважды промывают водой, взятой в количестве по 100 мл, и рассолом, сушат над сульфатом магния, и растворитель удаляют в вакууме. При этом получают 17.5 г оранжевого масла, которое медленно отверждается. Твердое вещество дважды растирают в порошок с помощью диэтилового эфира, взятого в количестве по 100 мл, собирают путем фильтрации и рекристаллизуют из ацетонитрила с получением 6,6 г 6,7-дихлор-1,4-бензодиоксан-2-илметил-толуол-4-сульфоната в качестве белых кристаллов с точкой плавления 124 - 125oC.
Смесь 3 г 6,7-дихлор-1,4-бензодиоксан-2-илметил-толуол-4-сульфоната, 1,65 г 1-[1-(2-метоксифенил)пиперид-4-ил]метиламина, полученного аналогичным примеру 2 приемом, 2,1 г карбоната калия и 25 мл диметилформамида перемешивают в течение 16 часов при температуре 85oC в атмосфере азота, затем подают в 150 мл воды. Продукт 4 раза экстрагируют этилацетатом, взятым в количестве по 80 мл. Объединенные экстракты дважды промывают водой, взятой в количестве по 100 мл, и рассолом, сушат над сульфатом магния, и растворитель удаляют в вакууме. При этом получают беловатую смолу, которую очищают путем флеш-хроматографии на силикагеле с применением в качестве элюента смеси метанола и дихлорметана в соотношении 1:20. Соответствующие фракции объединяют, и растворители удаляют в вакууме. При этом получают 0,54 г твердого вещества, которое растворяют в диэтиловом эфире. В раствор подают избыток эфирного раствора хлористого водорода, после чего растворитель удаляют в вакууме. Остаток сушат путем азеотропной перегонки с использованием 2-пропанола, растирают в порошок с помощью горячего этанола, а получаемое твердое вещество собирают путем фильтрации и сушат в вакууме с получением 280 мг гидрохлорида N-(6,7-дихлор-1,4-бензодиоксан-2-илметил)-1-[1-(2-метоксифенил)пиперид-4-ил] метиламина с точкой плавления 248 - 252oC (разлож.).
Пример 22. Смесь 41 г хлорида 4-карбамоил-1-(2,4-динитрофенил)пиридиния и 20 г 2-хлоранилина в 500 мл метанола перемешивают в течение 24 часов при комнатной температуре. Смесь нагревают с обратным холодильником в течение еще 24 часов, после чего подают еще 10 г 2-хлоранилина. Продолжают нагревать с обратным холодильником в течение еще 48 часов, после чего подают еще 10 г 2-хлоранилина. Смесь нагревают в течение еще 48 часов, охлаждают, и растворитель удаляют в вакууме. Остаток дважды растирают в порошок с помощью горячего ацетона, взятого в количестве по 1 л, охлаждают, и твердое вещество собирают путем фильтрации. Твердое вещество растворяют в 100 мл горячего метанола, обрабатывают активным углем и фильтруют в горячем состоянии. В фильтрат подают 500 мл горячего этилацетата, и раствору дают охлаждаться. Получаемое твердое вещество собирают путем фильтрации, промывают этилацетатом и сушат с получением 23,69 г сырого хлорида 4-карбамоил-1-(2-хлорфенил)пиридиния, который используют без дальнейшей очистки.
2,5 г сырого продукта предыдущей реакции в этаноле в присутствии 0,25 г 10%-ного родия на угле в качестве катализатора гидрируют при комнатной температуре и атмосферном давлении в течение 7 часов, после чего подают еще 0,5 г катализатора, и гидрирование продолжают в течение 8 часов. Добавляют еще 0,5 г катализатора, и гидрирование продолжают в течение 6 часов. Катализатор удаляют путем фильтрации на силикате марки Celite, а фильтрат подщелачивают насыщенным водным раствором бикарбоната натрия. Растворитель удаляют в вакууме, и твердое вещество кристаллизуют из этилацетата, а нерастворимые неорганические вещества удаляют путем фильтрации. Получают 0,5 г 1-(2-хлорфенил)пиперидин-4-карбоксамида.
0,25 г 1-(2-хлорфенил)пиперидин-4-карбоксамида, полученного аналогично вышеописанному приему, растворяют в 25 мл тетрагидрофурана, и в раствор подают 0,4 мл 10-м. раствора комплекса борана и диметилсульфида в диметилсульфиде. Получаемый раствор нагревают с обратным холодильником в течение 2 часов, после чего растворитель удаляют в вакууме, добавляют 50 мл 1-м. хлористоводородной кислоты, и смесь перемешивают при комнатной температуре в течение 1 часа. Раствор подщелачивают 5-м. водным раствором гидроокиси натрия, и продукт экстрагируют диэтиловым эфиром. Экстракты промывают водой, сушат над сульфатом магния, и растворитель удаляют в вакууме с получением 0,19 г 1-[1-(2-хлорфенил)пиперид-4-ил]метиламина в качестве масла. Реакцию повторяют с использованием исходных веществ, взятых в указанном выше количестве, умноженном на фактор 4,4, при этом получают еще 1,0 г 1-[1-(2-хлорфенил)пиперид-4-ил]метиламина.
Смесь 1,68 г 1,4-бензодиоксан-2-илметил-толуол-4-сульфоната, полученного аналогичным примеру 1 приемом, 1,8 г 1-[1-(2-хлорфенил)пиперид-4-ил]метиламина и 10 г карбоната калия в 150 мл ацетонитрила при перемешивании в течение 24 часов нагревают с обратным холодильником. В реакционную смесь подают 0,2 г йодида калия, при перемешивание при нагревании с обратным холодильником продолжают в течение еще 24 часов. Охлажденную смесь фильтруют, а растворитель в вакууме удаляют из фильтрата. Остаток подвергают хроматографии на силикагеле с применением в качестве элюента смеси петролейного эфира с пределами кипения 40 - 60oC и этилацетата в соотношении 1:1. Растворитель в вакууме удаляют из элюата, а прозрачный масляный остаток растворяют в диэтиловом эфире. Добавляют избыток эфирного раствора хлористого водорода, а растворитель удаляют в вакууме с получением 0,37 г гидрохлорида N-(1,4-бензодиоксан-2-илметил)-1-[1-(2-хлорфенил)пиперид-4-ил] метиламина с точкой плавления 275 - 278oC (разлож.).
Пример 23. 18,3 мл эпихлоргидрина по каплям в атмосфере азота подают в раствор 10 г 3-фторкатехина и 4,56 г гидроокиси калия в 45 мл воды, и смесь нагревают с обратным холодильником в течение 4,5 часов. Смесь охлаждают и продукт три раза экстрагируют этилацетатом, взятым в количестве по 50 мл. Органический экстракт последовательно промывают 25%-ным водным раствором гидроокиси натрия и рассолом, и сушат над сульфатом магния. Растворитель удаляют в вакууме с получением 15,5 г сырой смеси 1-(5-фтор-1,4-бензодиоксан-2-ил)-метанола и 1-(8-фтор-1,4- бензодиоксан-2-ил)метанола.
Раствор 15,5 г сырой смеси изомеров, полученной в результате предыдущей реакции, и 14,88 г толуол-4-сульфонилхлорида в 20 мл пиридина перемешивают при комнатной температуре в течение 18 часов. Добавляют 100 мл воды, и продукты три раза экстрагируют этилацетатом, взятым в количестве по 50 мл. Органический экстракт последовательно промывают разбавленной хлористоводородной кислотой и рассолом, и сушат над сульфатом магния. Растворитель удаляют в вакууме, а остаток очищают путем флеш-хроматографии на силикагеле с применением в качестве элюента смеси циклогексана и диэтилового эфира в соотношении 1:1. Соответствующие фракции объединяют, и остаток, получаемый в результате удаления растворителя в вакууме, далее очищают путем флеш-хроматографии на силикагеле с применением в качестве элюента смеси циклогексана и диэтилового эфира в соотношении 7:3. Растворитель удаляют из соответствующих фракций в вакууме, при этом получают 4,5 г смеси 5-фтор-1,4-бензодиоксан-2-илметил-толуол-4-сульфоната и 8-фтор-1,4-бензодиоксан-2-илметил-толуол-4-сульфоната.
Перемешиваемую смесь 4,5 г изомеров, полученных в результате предыдущей реакции, 2,93 г 1-[1-(2-метоксифенил)пиперид-4-ил]метиламина, 3,68 г карбоната калия и 100 мл ацетонитрила в течение 18 часов нагревают с обратным холодильником. Охлажденный раствор очищают с использованием прокладки силикагеля, применяя в качестве элюента сперва смесь этилацетата и петролейного эфира с пределами кипения 40 - 60oC в соотношении 2:1 для удаления оставшегося исходного толуол-4-сульфоната, а затем чистый этилацетат для элюирования целевого продукта. Растворитель удаляют в вакууме, остаток растворяют в диэтиловом эфире, после чего добавляют избыток эфирного раствора хлористого водорода. Получаемый осадок собирают путем фильтрации и сушат в вакууме с получением 260 мг смеси гидрохлорида N-(5-фтор-1,4-бензодиоксан-2-илметил)-1-[1-(2-метоксифенил)пиперид-4-ил] метиламина и гидрохлорида N-(8-фтор-1,4-бензодиоксан-2-илметил)-1-[1-(2-метоксифенил)-пиперид-4-ил] метиламина с точкой плавления 228 - 230oC.
Пример 24. 2,32 г эпихлоргидрина по каплям подают в перемешиваемый раствор 4,0 г 1,2-диоксинафталина и 4,0 г гидроокиси калия в 32 мл воды, и получаемую при этом смесь перемешивают при температуре 60 - 80oC в течение 2,5 часов, после чего смесь подают в 100 мл воды. Продукт 6 раз экстрагируют диэтиловым эфиром, взятым в количестве по 100 мл, причем для повышения растворимости добавляют небольшое количество метанола. Объединенные экстракты последовательно промывают 1-м. водным раствором гидроокиси натрия и водой, и сушат над сульфатом магния. Растворитель удаляют в вакууме с получением 4,0 г сырого 1-(нафто[1,2-b] диоксан-2-ил)метанола в качестве темного масла, которое используют без дальнейшей очистки.
Раствор 4,0 г сырого продукта предыдущей реакции и 3,53 г толуол-4-сульфонил-хлорида в 10 мл пиридина перемешивают при комнатной температуре в течение 3 часов, после чего добавляют еще 200 мг толуол-4-сульфонилхлорида и смесь оставляют стоять в течение 18 часов. Смесь подают в 100 мл воды, и продукт три раза экстрагируют этилацетатом, взятым в количестве по 100 мл. Объединенные экстракты дважды промывают 5-м. хлористоводородной кислотой, взятой в количестве по 100 мл, дважды - насыщенным водным раствором бикарбоната натрия, взятым в количестве по 100 мл, и рассолом, после чего сушат над сульфатом магния. Растворитель удаляют в вакууме с получением красного масла, которое очищают путем флеш-хроматографии на силикагеле с применением в качестве элюента смеси циклогексана и диэтилового эфира в соотношении 85: 15. Растворитель удаляют из продуктсодержащих фракций в вакууме, и остаток кристаллизуют из смеси диэтилового эфира и этилацетата с получением 0,48 г почти чистого нафто[1,2-b]диоксан-2-илметил-толуол-4-сульфоната в качестве розового твердого вещества.
Реакцию повторяют с использованием исходных веществ, взятых в указанном выше количестве, умноженном на фактор 4,15, при этом получают еще 1,75 г продукта. Целевые продукты двух реакций объединяют и рекристаллизуют из этилацетата с получением 1,38 г чистого нафто[1,2-b]диоксан-2-илметил-толуол-4-сульфоната в качестве белого твердого вещества с точкой плавления 122 - 124oC.
Смесь 1,32 г продукта предыдущей реакции, 0,79 г 1-[1-(2- метоксифенил)-пиперид-4-ил]метиламина, 1,0 г карбоната калия и 50 мл ацетонитрила перемешивают и нагревают с обратным холодильником в течение 40 часов. Охлажденную смесь фильтруют, растворитель удаляют в вакууме, и остаток очищают путем флеш-хроматографии на силикагеле с применением этилацетата в качестве элюента. Растворитель удаляют из соответствующей фракции в вакууме, и получаемое при этом белое твердое вещество растворяют в этилацетате. Раствор насыщают хлористым водородом, и получаемое твердое вещество собирают путем фильтрации и сушат в вакууме с получением 0,86 г 1,3-гидрохлорида 1-[1-(2-метоксифенил)пиперид-4-ил] -N-(нафто[1,2-b] диоксан-2-илметил)метиламина в виде гидрата с точкой плавления 182 - 185oC.
Пример 25. Смесь 6,8 г 2,3-дигидробензо[b]фуран-7-иламина, полученного приемом, аналогичным тому, описанному в Tetrahedron Letters 1982 г., 23, стр. 147, 14,7 г хлорида 4-карбамоил-1-(2,4-динитрофенил)пиридиния, полученного аналогичным примеру 2 приемом, и 400 мл метанола в течение 5 часов перемешивают при нагревании с обратным холодильником, после чего оставляют стоять при комнатной температуре и нагревают в течение 16 часов. Растворитель удаляют в вакууме, и остаток растирают в порошок с помощью горячего ацетона, охлаждают и фильтруют с получением 11,5 г хлорида 1-(2,3-дигидробензо[b] фуран-7-ил)-4-карбамоилпиридиния в качестве желтого твердого вещества с точкой плавления 289 - 290oC.
12,7 г хлорида 1-(2,3-дигидробензо[b]фуран-7-ил)-4-карбамоилпиридиния, полученного аналогично вышеописанному приему, в атмосфере азота подают в 12,3 г 10%-ного палладия на угле в качестве катализатора. Добавляют 23,5 г формиата аммония, и при перемешивании получаемой смеси по каплям добавляют 250 мл метанола. Смесь в течение 4 часов нагревают с обратным холодильником (в кристаллизованный в конденсаторе формиат аммония добавляют ~150 мл метанола, и получаемую смесь опять подают в реакцию). После охлаждения смесь фильтруют на силикате марки Celite в атмосфере азота, при этом уделяют внимание на то, что катализатору нельзя высыхать, подщелачивают насыщенным раствором бикарбоната натрия, растворитель удаляют в вакууме, и остаток разбавляют водой. Продукт три раза экстрагируют дихлорметаном, взятым в количестве по 100 мл. Объединенные органические экстракты промывают водой, сушат над сульфатом магния, фильтруют, и растворитель удаляют в вакууме с получением 7,75 г розового твердого вещества. Получаемое твердое вещество растирают в порошок с помощью горячего этилацетата, охлаждают и фильтруют с получением 6,64 г сырого 1-[2,3-дигидробензо[b]фуран-7-ил]пиперидин-4-карбоксамида в качестве светло-розового твердого вещества, которое используют без дальнейшей очистки.
Раствор 6,6 г светло-розового твердого вещества в 500 мл тетрагидрофурана по каплям в атмосфере азота подают в перемешиваемую смесь 2,05 г алюмогидрида лития в 250 мл тетрагидрофурана. Получаемую смесь перемешивают при комнатной температуре в течение 16 часов, после чего добавляют 4 мл воды и 4 мл концентрированного водного раствора гидроокиси натрия. Полу чаемую смесь перемешивают в течение 30 минут и фильтруют на силикате марки Celite. Растворитель удаляют в вакууме, и остаток растворяют в дихлорметане. Раствор сушат над сульфатом магния, фильтруют, и растворитель удаляют в вакууме с получением 6,24 г 1-[1-(2,3-дигидробензо[b]фуран-7-ил)-пиперид-4-ил]метиламина в качестве оранжевого масла, которое используют без дальнейшей очистки.
Смесь 2,12 г 8-метокси-1,4-бензодиоксан-2-илметил-толуол-4-сульфоната, полученного аналогичным примеру 6 приемом, 1,4 г 1-[1-(2,3-дигидробензо[b] -фуран-7-ил)пиперид-4-ил] метиламина, 1,67 г карбоната калия и 50 мл ацетонитрила перемешивают при нагревании с обратным холодильником в течение 4 дней. Смесь подают в 50 мл воды, и продукт три раза экстрагируют этилацетатом, взятым в количестве по 50 мл. Экстракты промывают 50 мл рассола, сушат над сульфатом магния, фильтруют, и растворитель удаляют в вакууме с получением смолы, которую очищают путем флеш-хроматографии на силикагеле, при этом в качестве элюента последовательно применяют смеси этилацетата и петролейного эфира с пределами кипения 60 - 80oC в соотношении 2:1, а затем в соотношении 1: 1. Растворитель удаляют из соответствующей фракции в вакууме, и остаток растворяют в диэтиловом эфире. Добавляют избыток эфирного раствора хлористого водорода, после чего растворитель удаляют в вакууме с получением 180 мг гидрохлорида 1-[1-(2,3-дигидробензо-[b]фуран-7-ил)пиперид-4-ил]-N-(8-метокси-1,4-бензодиоксан-2-илметил)метиламина в качестве твердого вещества с точкой плавления 194 - 196oC.
Пример 26. Смесь 5,0 г 4-хлоркатехина в 20 мл 10%-ного водного раствора гидроокиси калия перемешивают в атмосфере азота, добавляют 9,6 г эпихлоргидрина, и смесь нагревают при температуре 95oC в течение 4 часов. Cмесь охлаждают до комнатной температуры, и продукт дважды экстрагируют диэтиловым эфиром, взятым в количестве по 50 мл. Экстракты промывают 50 мл 5-м. водного раствора гидроокиси натрия и 50 мл воды, сушат над сульфатом магния, фильтруют, и растворители удаляют в вакууме с получением 8,24 г желтого масла.
Получаемое масло очищают путем флеш-хроматографии на силикагеле, при этом в качестве элюента последовательно применяют смеси петролейного эфира с пределами кипения 40 - 60oC и этилацетата в соотношении 10:1, 4:1, затем 1: 1, и в конце - этилацетат. Соответствующие фракции объединяют, и растворители удаляют в вакууме с получением 6,0 г бежевого твердого вещества.
Смесь 6,0 г получаемого на предыдущей стадии твердого вещества, 40 мл пиридина и 17,1 г толуол-4-сульфонилхлорида перемешивают при комнатной температуре в течение 16 часов, после чего смесь подают в 200 мл воды. Продукт три раза экстрагируют этилацетатом, взятым в количестве по 100 мл, экстракты последовательно дважды промывают 5-м. хлористоводородной кислотой, взятой в количестве по 100 мл, дважды - насыщенным водным раствором бикарбоната натрия, взятым в количестве по 100 мл и один раз - 100 мл рассола. Экстракты сушат над сульфатом магния, фильтруют, и растворитель удаляют в вакууме с получением 11,39 г желтого масла. Масло очищают путем флеш-хроматографии на силикагеле с применением в качестве элюента смеси циклогексана и диэтилового эфира в соотношении 85:15. Соответствующие фракции объединяют, и растворители удаляют в вакууме с получением 8 фракций.
Фракцию 4 рекристаллизуют из диэтилового эфира с получением 0,44 г белого твердого вещества, представляющего собой смесь 7-хлор-1,4-бензодиоксан-2-илметил-толуол-4-сульфоната и 6-хлор-1,4-бензодиоксан-2-илметил-толуол-4-сульфоната в соотношении 9:1.
Фракции 5 и 6 объединяют и очищают путем флеш-хроматографии на силикагеле с применением в качестве элюента смеси циклогексана и диэтилового эфира в соотношении 85:15, при этом получают 3 фракции А, Б и В.
Фракцию А рекристаллизуют из диэтилового эфира с получением 1,2 г белого твердого вещества, которое объединяют с рекристаллизированным продуктом фракции 4. При этом получают общий выход 1,64 г смеси изомеров в соотношении 9:1.
Перемешиваемую смесь 1,5 г вышеуказанного объединенного твердого вещества, 0,93 г 1-[1-(2-метоксифенил)пиперид-4-ил] метиламина, 1,2 г карбоната калия и 50 мл ацетонитрила в течение 40 часов нагревают с обратным холодильником. Смесь охлаждают, фильтруют, и растворители удаляют в вакууме. Остаточное масло очищают путем флеш-хроматографии на силикагеле с применением этилацетата в качестве элюента. Получаемое при этом масло растворяют в диэтиловом эфире, и в раствор подают газообразный хлористый водород, при этом получают бесцветное твердое вещество, которое собирают путем фильтрации, промывают этилацетатом и сушат с получением 0,87 г смеси гидрохлорида N-(7-хлор-1,4-бензодиоксан-2-илметил)-1-[1-(2-метоксифенил)пиперид-4-ил] метиламина в виде гидрата и гидрохлорида N-(6-хлор-1,4-бензодиоксан-2-илметил)-1-[1-(2-метоксифенил)пиперид-4-ил] метиламина в виде гидрата в соотношении примерно 9:1. Точка плавления: 237 - 240oC.
Пример 27. Фракцию 7, получаемую в результате первой очистки путем хроматографии, описанной на второй стадии примера 26, рекристаллизуют из диэтилового эфира с получением 0,21 г белого твердого вещества, которое объединяют с фракцией В, полученной в результате очистки путем хроматографии, описанной в примере 26, с получением общего выхода 1,40 г смеси 7-хлор-1,4-бензодиоксан-2-илметил-толуол-4-сульфоната и 6-хлор-1,4-бензодиоксан-2-илметил-толуол-4-сульфоната в соотношении 4:6.
Перемешиваемую смесь 1,3 г смеси полученных в результате предыдущей реакции п-толуолсульфонатов в соотношении примерно 4:6, 1,5 г 1-[1-(2-метоксифенил)пиперид-4-ил] метиламина, 1,01 г карбоната калия и 80 мл ацетонитрила нагревают с обратным холодильником в течение 30 часов. Охлажденную смесь фильтруют, после чего растворитель удаляют в вакууме с получением масла, которое очищают путем флеш-хроматографии на силикагеле с применением этилацетата в качестве элюента. Растворитель удаляют из соответствующих фракций в вакууме, получаемое при этом масло растворяют в диэтиловом эфире. Раствор насыщают хлористым водородом, и получаемое твердое вещество собирают путем фильтрации и сушат в вакууме с получением 0,69 г смеси 1,7-гидрохлорида N-(7-хлор-1,4-бензодиоксан-2-илметил)-1-[1-(2-метоксифенил)пиперид-4-ил] метиламина в виде гидрата и 1,7-гидрохлорида N-(6-хлор-1,4-бензодиоксан-2-илметил)-1-[1-(2- метоксифенил)пиперид-4-ил] метиламина в виде гидрата в соотношении примерно 4:6, с точкой плавления 237-240oC.
Пример 28. 10 г толуол-4-сульфонилхлорида порциями при температуре -10oC подают в перемешиваемый раствор 4,9 г 1-(8-окси-1,4-бензодиоксан-2-ил)метанола в 50 мл сухого пиридина в атмосфере азота. Раствор перемешивают в течение 4 часов, оставляют стоять при комнатной температуре в течение 48 часов, после чего раствор подают в 100 мл ледяной воды. Воду декантируют с получаемой смолы, которую растворяют в 200 мл дихлорметана. Раствор дважды промывают водой, взятой в количестве по 200 мл, сушат над сульфатом магния, и растворитель удаляют в вакууме с получением коричневого масла, которое кристаллизуется, когда его оставляют стоять. В результате титрования с применением смеси этанола и метанола в соотношении 1:1 получают 1,6 г сырого [8-(толуол-4-сульфонат)-1,4-бензодиоксан-2-ил] метил-толуол-4-сульфоната в качестве беловатого твердого вещества, которое используют без дальнейшей очистки.
Смесь 1,6 г сырого продукта предыдущей реакции, 0,72г 1-[1-(2-метокси-фенил)пиперид-4-ил] метиламина, 1,0 г карбоната калия и каталитического количества йодида калия в 50 мл ацетонитрила нагревают с обратным холодильником в течение 72 часов. Растворитель удаляют в вакууме, и остаток растворяют в 100 мл этилацетата. Раствор промывают водой, после чего продукт три раза экстрагируют 5-м. хлористоводородной кислотой, взятой в количестве по 70 мл. Объединенные экстракты дважды промывают этилацетатом, взятом в количестве по 30 мл, подщелачивают 5-м. водным раствором гидроокиси натрия, и продукт три раза экстрагируют этилацетатом, взятым в количестве по 150 мл. Экстракты объединяют, сушат над сульфатом магния, и растворитель удаляют в вакууме с получением желтого масла, которое очищают путем флеш-хроматографии на силикагеле с применением этилацетата в качестве элюента. Соответствующие фракции объединяют и растворитель удаляют в вакууме. Остаточное масло растворяют в 20 мл диэтилового эфира, и в раствор подают газообразный хлористый водород. При этом получают беловатое твердое вещество, которое собирают путем фильтрации и сразу сушат в вакууме с получением 0,15 г дигидрохлорида [2-({ [1-(2-метоксифенил)пиперид-4-ил] метиламино} метил)-1,4-бензодиоксан-8-ил] толуол-4-сульфоната с точкой плавления 225oC.
Раствор 1,0 г гидроокиси калия в 19,7 мл воды и 19,7 мл этанола порциями по примерно 10 мл в интервалах 15 минут подают в 0,15 г дигидрохлорида [2-({ [1-(2-метоксифенил)пиперид-4-ил] метиламино} метил)-1,4-бензодиоксан-8-ил]-толуол-4-сульфоната. Смесь нагревают с обратным холодильником в течение 2,5 часов, охлаждают и нейтрализуют ледяной уксусной кислотой. Продукт три раза экстрагируют диэтиловым эфиром, взятым в количестве по 100 мл, и объединенные экстракты оставляют стоять в течение 16 часов. Получаемый при этом осадок собирают путем фильтрации и сушат с получением 0,2 г N-(8-окси-1,4-бензодиоксан-2-илметил)-1-[1-(2-метоксифенил)пиперид-4-ил] метиламина в качестве бесцветного твердого вещества с точкой плавления 116 - 119oC.
Пример 29. Данный пример иллюстрирует применение предлагаемых соединений для изготовления фармацевтических композиций. При этом под термином "активное начало" понимается любое соединение настоящего изобретения, а в частности любое соединение, представляющее собой целевой продукт одного из предыдущих примеров.
а) Капсулы
10 вес. частей активного начала интенсивно смешивают в 240 вес. частями лактозы и получаемую смесь измельчают, загружают в капсулы из твердой желатины с таким расчетом, что каждая капсула содержит либо единичную дозу, либо часть единичной дозы активного начала.
б) Таблетки.
Таблетки состава (вес. части): 10 активного начала, 190 лактозы, 22 кукурузного крахмала, 10 поливинилпирролидона и 3 стеарата магния приготовляют следующим образом.
Активное начало, лактозу и часть кукурузного крахмала измельчают, перемешивают и получаемую при этом смесь гранулируют с помощью раствора поливинилпирролидона в этаноле. После сушки гранулы смешивают с стеаратом магния и остаточным количеством кукурузного крахмала. Получаемую при этом смесь подают в таблетировочную машину с тем, чтобы получить таблетки, каждая из которых содержит либо единичную дозу, либо часть единичной дозы активного начала.
в) Таблетки с покрытием.
Таблетки приготовляют тем же образом, что и описано в п.б). Затем на таблетки известными приемами наносят раствор 20%-ного фталата ацетата целлюлозы и 3%-ного диэтилфталата в смеси этанола и дихлорметана в соотношении 1:1.
г) Cуппозитории.
100 вес. частей активного начала вырабатывают в 1300 вес.частей смеси триглицеридов и получаемую при этом смесь переводят в суппозитории, каждый из которых содержит терапевтически эффективное количество активного начала.
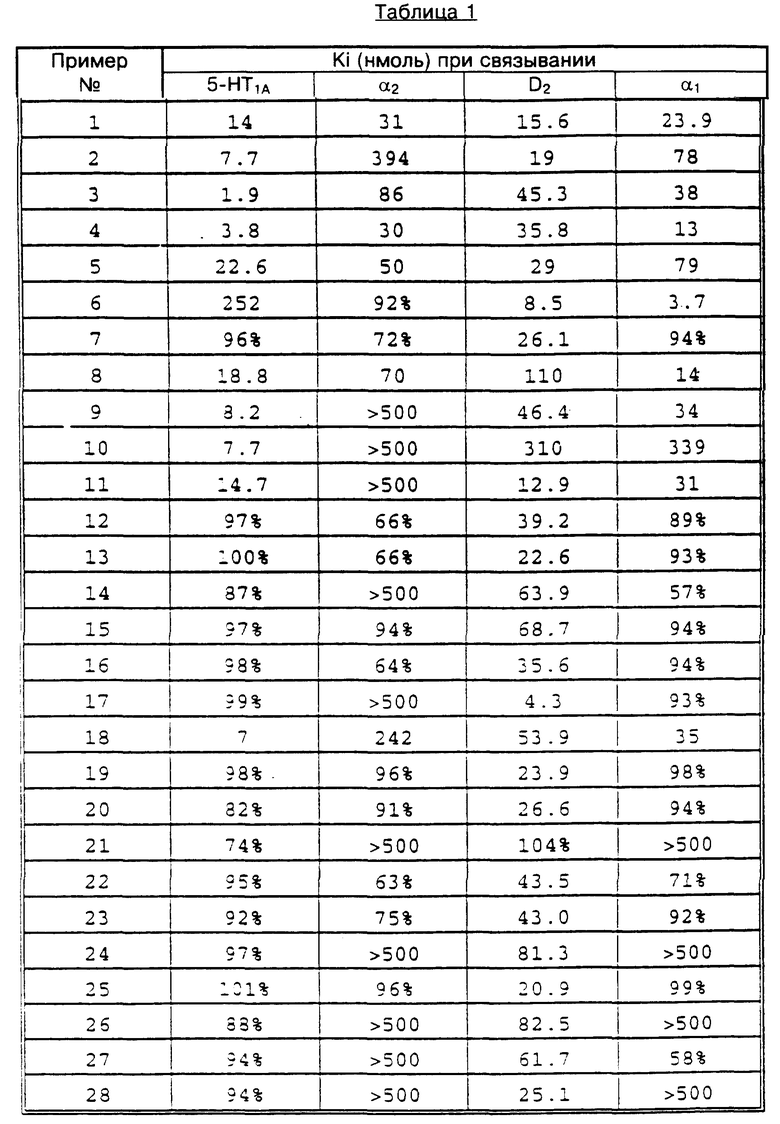
Производные амина формулы I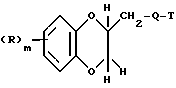
где R - галоид, гидроксил, C1-3-алкил, C1-3-алкоксил; Q - остаток формулы (IIа), (IIб)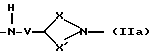
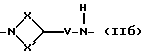
V - метилен или этилен, Х - C0-2-алкиленовая цепь, X' - C1-4-алкиленовая цепь, Т-фенил, пиридил, пиразинил, бензо[b]фуранил, 1,4-бензодиоксанил, хиназолинил, возможно замещенные галоидом, метоксилом или трифторметилом, m = 0 или 1, и их фармацевтически приемлемые соли, обладают высоким сродством в отношении рецепторов 5-HT1 и D2. 2 с. и 4 з.п. ф-лы, 1 табл.
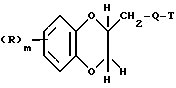
где R - галоид, гидроксил, алкил с 1 - 3 атомами углерода, алкоксил с 1 - 3 атомами углерода;
Q - двухвалентная группа формулы (IIa), (IIб)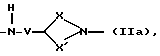
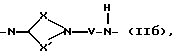
где V - метилен или этилен;
X - алкиленовая цепь с 0 - 2 атомами углерода;
X' - алкиленовая цепь с 1 - 4 атомами углерода, при этом общее число атомов углерода в остатках X и X' составляет 3 или 4;
T - фенил, пиридил, пиразинил, бензо[b]фуранил, 1,4-бензодиоксанил и хиназолинил, незамещенные или замещенные галоидом, метоксилом или трифторметилом;
m - 0, 1,
и их фармацевтически приемлемые соли.
| Способ получения производных хиназолина или их солей | 1979 |
|
SU1098521A3 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ КУМЕРМИЦИНОВ | 0 |
|
SU342339A1 |
| Машина для чистки и смазки внутренней поверхности изложниц | 1938 |
|
SU54304A1 |
| КОМБИНИРОВАННАЯ МАШИНА ДЛЯ ОДНОВРЕМЕННОГО ЛЕНТОЧНОГО ВНЕСЕНИЯ ГЕРБИЦИДОВ, ВЫСЕВА МИНЕРАЛЬНЫХ УДОБРЕНИЙ И ПОСЕВА | 0 |
|
SU352613A1 |
| US 4569933 A, 1986 | |||
| WO 9317017 A, 02.09.93. | |||
