Изобретение относится к новым азотсодержащим гетероциклическим соединениям, обладающим биологической активностью, более конкретно к производным 1,2,4-триазоло[1,5-а]пиримидинов, их фармацевтически приемлемым солям и стереоизомерам, фармацевтической композиции, их содержащей, и способу ингибирования припадков.
Известны производные арил- и аралкилтиоимидазо[1,2-b]пиридазинов, которые обладают биологической активностью, в частности, антисудорожной активностью (см. заявку WO-A-89/01478, опуб. 23.02.1989 г.)
Задачей изобретения является расширение ассортимента азотсодержащих гетероциклических соединений, обладающих антисудорожной активностью.
Поставленная задача решается предлагаемыми производными 1,2,4- триазоло[1,5-а]пиримидинов общей формулы (I)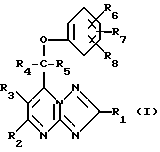
где
R1, R4 и R5 являются атомом водорода или алкилом с 1 - 6 атомами углерода:
R2 и R3 являются атомом водорода;
R6, R7 и R8 независимо друг от друга являются атомом водорода, галоидом, циано-группой, алканоилом с 1 - 6 атомами углерода, алкоксилом с 1 - 6 атомами углерода, незамещенным или замещенным галоидом, алкилтио с 1 - 6 атомами углерода, алкилсульфонилом с 1 - 6 атомами углерода, алкилсульфинилом с 1 - 6 атомами углерода, алкилом с 1 - 6 атомами углерода, незамещенным или замещенным галоидом,
при этом, если R1, R2, R3, R4 и R8 - атомы водорода, R5 - метил, R6 и R7 - атомы водорода, или R6 - 4-хлор и R7 - атом водорода или 2-хлор, то соединение формулы (I) не представляет собой рацемат, и их фармацевтически приемлемыми солями и стереоизомерами.
Вторым объектом изобретения является фармацевтическая композиция, обладающая противосудорожной активностью, которая включает активное начало на основе азотсодержащего гетероциклического соединения и фармацевтически приемлемый разбавитель или носитель, отличительная особенность которой заключается в том, что в качестве активного начала на основе азотсодержащего гетероциклического соединения она содержит соединение общей формулы (II)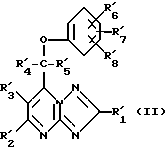
где R'1, R'4 и R'5 являются атомом водорода или алкилом с 1 - 6 атомами углерода;
R'2 и R'3 являются атомом водорода;
R'6, R'7 и R'8 независимо друг от друга являются атомом водорода, галоидом, циано-группой, алканоилом с 1 - 6 атомами углерода, алкоксилом с 1 - 6 атомами углерода, незамещенным или замещенным галоидом, алкилтио с 1 - 6 атомами углерода, алкилсульфинилом с 1 - 6 атомами углерода, алкилсульфонилом с 1 - 6 атомами углерода, алкилом с 1-6 атомами углерода, незамещенным или замещенным галоидом,
или его фармацевтически приемлемой солью или стереоизомером в терапевтически эффективном количестве.
В первую группу предпочтительных производных 1,2,4-триазоло [1,5-а]пиримидинов общей формулы (I) и (II) входят соединения, у которых R1, R'1, R4, R'4, R5 и R'5 являются атомом водорода или алкилом с 1 - 4 атомами углерода;
R2, R'2, R3, и R'3 являются атомом водорода;
R6, R'6, R7, R'7, R8, R'8 - независимо друг от друга являются атомом водорода, галоидом, циано-группой, алкилом с 1 - 4 атомами углерода, незамещенным или замещенным галоидом, алкоксилом с 1 - 4 атомами углерода, незамещенным или замещенным галоидом, алканоилом с 1 - 4 атомами углерода, алкилтио с 1 - 4 атомами углерода, алкилсульфинилом с 1 - 4 атомами углерода и алкилсульфонилом с 1 - 4 атомами углерода.
Во вторую группу предпочтительных производных 1,2,4-триазоло [1,5-а]пиримидинов общей формулы (I) и (II) входят соединения, у которых
R1 и R'1 является атомом водорода или метилом,
R4, R'4, R5 и R'5 независимо друг от друга являются атомом водорода, метилом или этилом,
R6, R'6, R7, R'7, R8 и R'8 независимо друг от друга являются атомом водорода, фтором, хлором, бромом, цианогруппой, трифторметилом, метоксилом, трифторметоксилом, ацетилом, метилтиогруппой, этилтиогруппой, метилсульфинилом или метилсульфонилом.
В частности предпочитаются соединения общих формул (I) и (II), которые представляют собой:
7-[1-(4-фторфенокси)этил]-1,2,4-триазол[1,5-а]-пиримидин;
7-[1-(4-хлорфенокси)этил]-1,2,4-триазол[1,5-а]-пиримидин;
7-[1-(4-бромфенокси)этил]-1,2,4-триазол[1,5-а]-пиримидин;
7-[1-(4-цианофенокси)этил]-1,2,4-триазол[1,5-а]-пиримидин;
7-[1-(4-трифторметилфенокси)этил]-1,2,4-триазол[1,5-а]-пиримидин;
7-[1-(4-метоксифенокси)этил]-1,2,4-триазол[1,5-а]-пиримидин;
7-[1-(4-трифторметоксифенокси)этил]-1,2,4-триазол[1,5-а]-пиримидин;
7-[1-(4-ацетилфенокси)этил]-1,2,4-триазол[1,5-а]-пиримидин;
7-{1-[4-(метилтио)фенокси]этил}-1,2,4-триазол[1,5-а]-пиримидин;
7-[1-(4-метилсульфинилфенокси)этил]-1,2,4-триазол[1,5-а]-пиримидин;
7-[1-(4-метилсульфонилфенокси)этил]-1,2,4-триазол[1,5-а]-пиримидин;
7-{1-[4-(этилтио)фенокси]этил}-1,2,4-триазол[1,5-а]-пиримидин;
7-[1-(3-хлорфенокси)этил]-1,2,4-триазол[1,5-а]-пиримидин;
7-[1-(2,4-дифторфенокси)этил]-1,2,4-триазол[1,5-а]-пиримидин;
7-[1-(2,4-дихлорфенокси)этил]-1,2,4-триазол[1,5-а]-пиримидин;
7-[1-(3,4-дихлорфенокси)этил]-1,2,4-триазол[1,5-а]-пиримидин;
7-[1-(2-хлор-4-фторфенокси)этил]-1,2,4-триазол[1,5-а]-пиримидин;
7-[1-(4-хлорфенокси)этил]-2-метил-1,2,4-триазол[1,5-а]-пиримидин;
7-(4-хлорфеноксиметил)-1,2,4-триазол[1,5-а]-пиримидин;
7-[1-(4-хлорфенокси)-1-метилэтил]-1,2,4-триазол[1,5-а]-пиримидин;
7-[1-(4-хлорфенокси)пропил]-1,2,4-триазол[1,5-а]-пиримидин.
(+)-7-[1-(4-фторфенокси)этил]-1,2,4-триазол[1,5-а]-пиримидин,
(-)-7-[1-(4-фторфенокси)этил]-1,2,4-триазол[1,5-а]-пиримидин,
(+)-7-[1-(4-хлорфенокси)этил]-1,2,4-триазол[1,5-а]-пиримидин,
(-)-7-[1-(4-хлорфенокси)этил]-1,2,4-триазол[1,5-а]-пиримидин.
Пригодными фармацевтически приемлемыми солями соединений общей формулы (I) или (II) в частности являются соли с кислотами, включающие соли с неорганической кислотой, такой, как, например, хлористоводородная, бромистоводородная, йодистоводородная, азотная, сульфурная и/или фосфорная кислоты; соли с органической кислотой, такой, как, например, малеиновая, уксусная, лимонная, фумаровая. винная, янтарная, бензойная, памовая, пальмитиновая, метилсерная и/или додекановая кислоты; или соли с аминокислотой, такой, как, например, глютаминовая кислота. Такие соли включают все фармацевтически приемлемые соли с многовалентными кислотами, например, бикарбонаты и/или ортофосфаты.
Фармацевтически приемлемые соли соединений общей формулы (I) или (II) в терапии можно применять вместо соответствующего соединения общей формулы (I) или (II). Такие соли могут получаться путем взаимодействия соответствующего соединения общей формулы (I) или (II) с подходящей кислотой или подходящим основанием стандартным приемом.
Некоторые соединения общей формулы (I) или (II) могут иметься в разных физических формах, например, в виде разных кристаллических форм, и поэтому данное изобретение охватывает любую физическую, например, кристаллическую, форму соединений общей формулы (I) или (II) и их смеси.
Соединения общей формулы (I) или (II) могут иметься в виде сольватов, например, гидратов, и поэтому данное изобретение охватывает каждый сольват соединений формулы (I) или (II) и их смеси. Степень сольватации может быть не-стихиометрическая. Если растворителем является вода, то гидрат может представлять собой, например, полугидрат, моногидрат или дигидрат.
Некоторые соединения общей формулы (I) или (II) могут иметь один или несколько хиральных центров и иметься в виде различных оптически активных форм. Так, например, соединения общей формулы (I) или (II), где радикалы R4 и R5 различны, имеют хиральный центр у асимметрично замещенного атома углерода. При наличии одного хирального центра соединения формулы (I) или (II) имеются в виде двух энантиомерных форм, и поэтому настоящее изобретение охватывает оба энантиомера соединений формулы (I) или (II) и их смеси. Отдельные энантиомеры могут получатся известными специалисту методами. Примерами таких методов являются образование соли диастереоизомеров или комплексов диастереоизомеров, которые можно разделять, например, путем кристаллизации, образование производных или комплексов диастереоизомеров, которые можно разделять, например, путем кристаллизации, газожидкостной хроматографии или жидкостной хроматографии с последующим выделением желаемого энантиромера из произодного, избирательное взаимодействие одного энантиомера с подходящим реагентом, например, энзиматическая этерификация, окисление или восстановление с последующим разделением модифицированного и немодифицированного энантиромеров, газо-жидкостная или жидкостная хроматография в хиральной среде, например, на хиральном носителе, например, силикагель, со связанным хиральным лигандом и/или в присутствии хирального растворителя, асимметричный синтез определенного энантиомера с использованием оптически активных реагентов, субстратов, катализаторов или растворителей, и/или энзиматических процессов, или же перевод одного энантиомера в другой энантиомер за счет асимметричной перегруппировки.
Если соединения общей формулы (I) или (II) имеют по меньшей мере два хиральных центра, то они могут иметься в виде диастереоизомеров, которые можно разделять известными специалисту методами, например, хроматографией или кристаллизацией. При этом отдельные изомеры любой диастереоизомерной пары можно выделять вышеуказанными методами. Данное изобретение охватывает любой диастереоизомер соединений формулы (I) или (II), а также их смеси.
Если активная часть соединения преобразуется в ходе вышеуказанных процессов разделения, то на дополнительной стадии продукт преобразования опять переводят в активную форму.
Некоторые соединения формулы (I) или (II) могут иметься в разных таутомерных формах или в виде разных геометрических изомеров, поэтому данное изобретение охватывает любой таутомер и/или геометрический изомер соединений формулы (I) или (II), а также их смеси.
Предлагаемая фармацевтическая композиция может представлять собой любую стандартную форму терапевтического препарата для оральной, ректальной, парентеральной или местной аппликации. Композицию известными приемами можно изготовлять так, что обеспечивается контролированное, т.е. либо быстрое, либо продленное выделение активного вещества. В качестве фармацевтически приемлемого разбавителя или носителя предлагаемая композиция содержит любое известное в данной области вещество. Как правило, предлагаемая композиция содержит примерно 0,1 - 99 вес.% активного начала. Как правило, она имеется в виде дозировочной единицы. Дозировочная единица активного начала предпочтительно составляет примерно 1 - 1000 мг.
Предлагаемую композицию предпочтительно дают орально в известных для такой аппликации фармацевтических препаратах. Препаратами, пригодными для оральной дачи, являются таблетки, драже, капсулы, гранулы, порошки, жгуты, эликсиры, сиропы, растворы, водные и масляные суспензии.
Твердые препараты для оральной дачи, например таблетки, готовят путем смешивания активного начала с по меньшей мере одним компонентом и/или смесью компонентов, включающих:
инертные наполнители, такие, как, например, лактоза, порошковый сахар, крахмал, каолин, маннитол, фосфат кальция, сульфат кальция;
дезинтегрирующие агенты, такие, как, например, кукурузный крахмал, метилированная целлюлоза, агар, бентонит, целлюлоза, древесные продукты, альгиновая кислота, гуаран, цитрусовая мякоть, карбоксиметицеллюлоза и/или лаурилсульфат натрия;
смазочные вещества, такие, как, например, стеарат магния, борная кислота, натриевая соль бензойной кислоты, натриевая соль уксусной кислоты, хлорид натрия, лейцингликоль, полиэтиленгликоль,
связующие, такие, как, например, крахмал, желатина, сахары, например сахароза, меласса и/или лактоза, и/или натуральные и/или синтетические смолы, например, акация, альгинат натрия, экстракт кагарена, карбоксиметилцеллюлоза, метилцеллюлоза, этилцеллюлоза, полиэтиленгликоль, воски, микрокристаллическая целлюлоза и/или поливинилпирролидон,
красящие вещества, такие, как, например, стандартные, фармацевтически приемлемые красители,
подслащивающие и/или вкусовые вещества,
консервирующие вещества,
по меньшей мере одна фармацевтически приемлемая пара, включающая, например, кислоту и карбонат и/или бикарбонат, которая путем вскипания способствуют растворению в случае подачи твердого препарата в воду,
и другие известные компоненты, обеспечивающие изготовление препаратов для оральной дачи известными способами, например, таблетированием.
Твердые препараты для оральной дачи могут быть выполнены с таким расчетом, что активное начало высвобождается в течение определенного времени. Содержащие новые соединения твердые препараты для оральной дачи, выполненные с покрытием, в зависимости от активного начала могут оказаться преимущественными. Разные вещества, такие, как, например, шеллак и/или сахар, могут применяться в качестве покрытия или для другого вида модификации физической формы препарата, предназначенного для оральной дачи. Например, на таблетки или драже при необходимости можно наносить известными приемами покрытие, например, из фталата ацетата целлюлозы и/или фталата оксипропилметилцеллюлозы.
Капсулы, например, из твердой или мягкой желатины, содержащие активное начало и при необходимости наполнители, например, масло, приготовляют известными приемами и при необходимости на них наносят покрытие желаемой известными методами. Содержимое капсулы составляют известными приемами с таким расчетом, что получают продленное выделение активного начала.
Жидкими препаратами для оральной дачи, содержащими предлагаемые соединения, являются эликсир, суспензия и/или сироп, например, водная суспензия, содержащая активное начало в водной среде в присутствии нетоксичного суспендирующего агента, такого, как, например, карбоксиметилцеллюлоза натрия, и/или масляная суспензия, содержащая активное начало в пригодном растительном масле, таком, как, например, арахисовое масло и/или подсолнечное масло. Жидкие препараты для оральной дачи могут также содержать подслащивающие вещества, вкусовые вещества, консервирующие вещества и/или их смеси.
Активное начало можно также переводить в гранулы и порошки, при необходимости с применением дополнительных вспомогательных веществ. Гранулы и порошки могут непосредственно применяться пациентом, или же они могут добавляться к подходящему жидкому носителю, такому, как, например, вода, перед применением. Гранулы и/или порошки могут также содержать фармацевтически приемлемые дезинтегрирующие агенты, такие, как, например, сыпучая смесь, состоящая из кислоты и соли углекислоты или бикарбоната, которая облегчает диспергирование в жидкой среде.
Каждый из вышеуказанных препаратов предпочтительно содержит примерно 1 - 1000 мг, более предпочтительно примерно 5 - 500 мг активного вещества.
Подходящей для ректальной дачи формой предлагаемой композиции являются известные фармацевтические формы, например, суппозитории, содержащие отвержденный жир, полусинтетические глицериды, масло какао и/или полиэтиленгликолевое основание.
Подходящей для парентеральной дачи путем, например, внутривенной инъекции, формой предлагаемой композиции являются известные фармацевтические формы, например, стерильные суспензии в водном и/или масляном среде или стерильные растворы в подходящем растворителе.
Композиции для местной дачи могут содержать матрицу, в которой активное начало диспергировано с таким расчетом, что оно находится в контакте с кожей с тем, чтобы активное начало могло проникать в организм трансдермально. В случае местной аппликации активное начало должно содержаться в подходящем препарате с таким расчетом, что терапевтически эффективное количество активного начала высвобождается в течение желаемого периода времени.
Подходящую композицию для трансдермальной аппликации можно готовить путем смешивания или диспергирования фармацевтически активного начала в пригодном носителе и с пригодным для трансдермальной аппликации ускорителем, таким, как, например, диметилсульфоксид и/или пропиленгликоль. В качестве носителя могут применяться фармацевтически приемлемые пена, паста, мазь, лосьон, крем, эмульсия и/или гель, а также композиция, пригодная для аппликации распылителем. Средства для трансдермальной аппликации могут также представлять собой, например, припарки, пластыри и/или пропитанные повязки.
Подходящий крем можно изготовлять путем включения активного начала в петролатум и/или жидкий парафин, который с использованием поверхностно-активных веществ диспергируют в водной среде. Мазь можно изготовлять путем смешивания активного начала с минеральным маслом, петролатумом и/или воском, таким, как, например, парафин или пчелиный воск. Гель можно изготовлять путем смешивания активного начала с загустителем, таким, как, например, подщелачиванный торговый продукт Carbomer BP, в присутствии воды. Прозрачный гель может содержать осветляющее вещество, такое, как например, денатурированный спирт, например, денатурированный этанол.
Предлагаемая фармацевтическая композиция для местной аппликации предпочтительно содержит сгуститель и/или вещество для регулирования величины pH, совместимые с активным началом. Вещество для регулирования величины pH предпочтительно применяется в количестве, достаточном для активации сгустителя, если такой имеется, и сохраняет pH композиции внутри фармацевтически и косметически приемлемых пределах с таким расчетом, что кожа не повреждается. В частности предпочитается величина pH композиции, равная примерно 5,0 - 9,0.
Если фармацевтической композицией для местной аппликации по данному изобретению является эмульсия, то она может представлять собой либо эмульсию типа "масло в воде" либо эмульсию типа "вода в масле". В масляной фазе такой эмульсии содержится по меньшей мере один из следующих компонентов: углеводородные масла, воски, натуральные масла, силиконовые масла, сложные эфиры жирных кислот, спирты жирного ряда и/или их смеси. Фармацевтические композиции данного изобретения в качестве эмульсий можно изготовлять с использованием эмульгатора или смеси эмульгаторов, пригодных для применения в эмульсиях типа "масло в воде" или типа "вода в масле" и приемлемых для использования в фармацевтических композициях, предназначенных для местной аппликации. Пригодные эмульгаторы представляют собой любые известные специалисту эмульгаторы и/или их смеси.
Если фармацевтическая композиция данного изобретения, предназначенная для местной аппликации, не представляет собой эмульсию, то все-таки можно применять эмульгатор в качестве поверхностно-активного вещества для повышения терапевтической активности местно апплицируемой фармацевтической композиции.
Фармацевтическая композиция данного изобретения, предназначенная для местной аппликации, дополнительно может содержать дальнейший компонент или же дальнейшие компоненты, известные специалисту, такие, как, например: стабилизаторы эмульсии, в том числе в виде соли, средства против раздражения кожи, увлажнители, вещества для образования пленки, отдушки, консерванты, красящие вещества и/или их смеси.
Предлагаемые соединения могут также даваться путем непрерывной инфузии либо снаружи, например, путем внутривенного вливания, либо с применением источника активного начала, размещенного внутри тела пациента. Такими внутренними источниками являются, например, имплантированные емкости, которые содержат подлежащее инфузии активное начало. В таком случае активное начало непрерывно высвобождается, например, путем осмоза. Кроме того, имплантированные препараты могут представлять собой жидкость, такую, как, например, суспензия или раствор в фармацевтически приемлемом растворителе подлежащего инфузии соединения, например, в виде труднорастворимого в воде производного, такого, как, например, соль или эфир с додекановой кислотой, или же служащее в качестве основы для подлежащего инфузии соединения твердое вещество, например, синтетическая смола или воск. При этом основание может представлять собой единичное тело, содержащее все активное начало, или же ряд тел, каждое из которых содержит часть апплицируемого активного начала. Активное начало должно иметься во внутреннем источнике с таким расчетом, что терапевтически эффективное количество активного начала отдается в течение желаемого периода времени.
Для некоторых целей может быть целесообразным то, что композиция содержит активное начало в виде мельчайших частиц, получаемых, например, путем распыления.
Предлагаемая фармацевтическая композиция может также содержать другие фармакологически активные начала, которые совместимы с предлагаемыми соединениями.
Третьим объектом изобретения является способ ингибирования припадков путем дачи людям или животным азотсодержащего гетероциклического соединения, который заключается в том, что в качестве азотсодержащего гетероциклического соединения используют соединение общей формулы (I)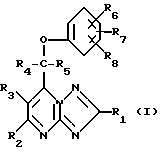
где R1, R4 и R5 являются атомом водорода или алкилом с 1 - 6 атомами углерода;
R2 и R3 являются атомом водорода;
R6, R7 и R8 независимо друг от друга являются атомом водорода, галоидом, циано-группой, алканоилом с 1 - 6 атомами углерода, алкоксилом с 1 - 6 атомами углерода, незамещенным или замещенным галоидом, алкилтио с 1 - 6 атомами углерода, алкилсульфинилом с 1 - 6 атомами углерода, алкилсульфонилом с 1 - 6 атомами углерода, алкилом с 1-6 атомами углерода, незамещенным или замещенным галоидом,
или его фармацевтически приемлемую соль или стереоизомер в дневной дозе, равной примерно 1 - 1000 мг.
Хотя точный механизм действия активного начала еще неизвестен, предполагается, что фармакологическая активность активного начала в указанных здесь условиях базируется на способности потенцирования передачи нейротрансмиттера γ- аминомасляной кислоты и/или способности активации калиевых (K+) канальцев в нейронах. Поэтому дальнейшим объктом изобретения является описанный здесь способ лечения, где активное начало представляет собой вещество, потенцирующее передачу γ- аминомасляной кислоты и/или активатор калиевых канальцев в нейронах. Однако изобретение не ограничивается активным началом, обладающим указанной здесь фармакологической активностью.
Необходимое для успешной терапии количество активного начала зависит от ряда факторов, таких, как, например, серьезность заболевания, возраст и/или история болезни пациента, поэтому в каждом конкретном случае аптекарь, врач и/или ветеринарный врач должен подбирать подходящую дозировку. Как правило, новые соединения человеку или животным дают в количестве 1 - 1000 мг/сутки, предпочтительно 5 - 500 мг/сутки, в качестве единичной дозы или же нескольких доз один раз или несколько раз в сутки, при этом предпочитается оральная дача.
Активное вещество может применяться вместе с одним или несколькими известными соединениями той же биологической активности для достижения синергистического эффекта.
Ниже описываются возможные способы получения соединений вышеприведенных общих формул (I) и (II).
Соединения формулы (I) могут получаться путем взаимодействия соединения формулы (III)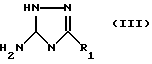
с соединением формулы (IV)
где Y - подходящая удаляемая группа, такая, как, например, атом хлора, диметиламин или алкокси-группа.
Соединения формулы (I) могут получаться путем взаимодействия соединения формулы (V)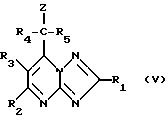
где Z означает удаляемую группу, такую, как, например, атом брома или хлора, с анионом формулы (VI)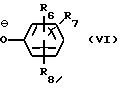
Соединения формулы (IV), где Y - диметиламин, могут получаться путем взаимодействия соединения формулы (VII)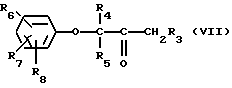
с реагентом Гольда, представляющим собой соединение формулы Me2NCH=NCH= NMe2Cl (Me - метил).
Соединения формулы (V) могут получаться путем взаимодействия соединения формулы (III) с соединением формулы (VIII)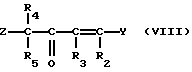
Соединения формулы (IX), где Y - диметиламин, могут получаться путем взаимодействия соединения формулы ZCR4R5COCH2R3 с реагентом Гольда.
Соединения формулы (V), где Z - галоид, могут получаться путем взаимодействия соединения формулы (IX)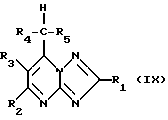
с агентом галогенирования, таким, как, например, N-бромсукцинимид.
Соединения формулы (IX) могут получаться путем взаимодействия соединения формулы (III) с соединением формулы (X)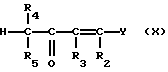
Соединения формулы (X), где Y - диметиламин, могут получаться путем взаимодействия соединения формулы CHR4R5COCH2R3 с вышеуказанным реагентом Гольда.
Соединения формулы (IX), где R1 не означает атом водорода, могут получаться путем взаимодействия соединения формулы (XI)
с соединением формулы R1CN--->O, при этом получают промежуточный продукт, который подвергают циклизации с применением подходящего кислотного катализатора.
Соединения формулы (IX) могут получаться путем взаимодействия соединения формулы (XII)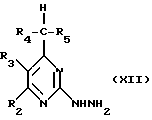
с карбоновой кислотой формулы R1CO2H или соединением формулы R1C(OR11)3, где R11 означает метил или этил.
Соединения формулы (IX) могут также получаться путем декарбоксилирования кислоты формулы (XIII)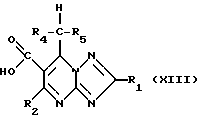
с помощью, например, тепла и/или подходящего кислотного катализатора.
Соединения формулы (XIII) могут получаться путем гидролиза сложного эфира формулы (XIV)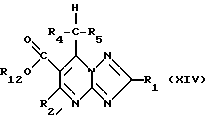
где заместитель R12 означает незамещенный или замещенный алкил или незамещенный или замещенный арил.
Соединения формулы (XIV) могут получаться путем взаимодействия соединения формулы (III) с соединением формулы (XV)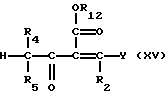
Соединения формулы (XV), где Y - диметиламин, могут получаться путем взаимодействия соединения формулы CHR4R5COCHR3CO2R12 с вышеуказанным реагентом Гольда.
Соединения формулы (I) или (II) могут также получаться путем сочетания спирта формулы (XVI)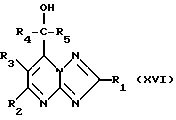
с фенолом формулы (XVII)
в присутствии способствующего сочетанию вещества, представляющего собой, например, в реакции по Митсунобу диэтилазодикарбоксилат с трифенилфосфином.
Если R4 и R5 различны, то стереоспецифичная реакция по Митсунобу представляет собой возможность получения отдельных энантиомеров соединений формулы (I) или (II).
Соединения формулы (V), где Z - галоид, могут получаться путем взаимодействия спирта формулы (XVI) с агентом галогенирования, таким, как, например, тионилхлорид, или трифенилфосфин с бромом.
Спирты формулы (XVI), где R5 - атом водорода, могут получаться путем восстановления соединения формулы (XVIII)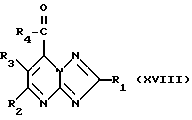
восстановителем, таким, как, например, боргидрид натрия, или же с хиральным восстановителем, с получением отдельных энантиомеров спирта формулы (XVI).
Соединения формулы (XVIII) могут получаться путем снятия защитной группы с соединения формулы (XIX)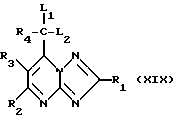
где L1 и L2 означают алкокси-группу или алкилтио-группу
или вместе с атомом углерода, с которым они связаны, представляют собой диоксолановое, диоксановое, дитиолановое или дитиановое кольцо. Например, если соединение формулы (XIX) представляет собой дитиолан или дитиан, то в качестве агента, способствующего удалению защитной группы, можно использовать нитрат серебра с N-хлорсукцинимидом или цериевый нитрат аммония. Если L1 и L2 оба означают метокси, то в качестве отщепляющего агента можно использовать подходящий ионообменник марки "Amberlyst" (торговый продукт фирмы Aldrich Chemicals).
Соединения формулы (IX), где R5 - атом водорода, могут получаться путем восстановления соединения формулы (XVIII).
Соединения формулы (XIX) могут получатся путем взаимодействия соединения формулы (III) с соединением формулы (XX)
Соединения формулы (XX), где Y - диметиламин, могут получатся путем взаимодействия соединения формулы (XXI)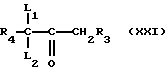
с вышеуказанным реагентом Гольда.
Спирты формулы (XVI) могут также получаться путем взаимодействия соединения формулы (V) гидроксильным ионом, например, с использованием подходящей щелочи.
Кроме того, спирты формулы (XVI) могут получаться путем гидролиза соединения формулы (XXII)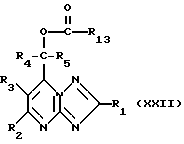
где остаток R13 - незамещенный или замещенный алкил, или незамещенный или замещенный арил, например, с карбонатом калия. Гидролиз можно осуществлять в таких условиях, что получают отдельные энантиомеры спирта формулы (XVI), путем, например, использования соответствующего гидролитического энзима.
Соединения формулы (XXII) могут получаться путем взаимодействия соединения формулы (V) с карбоксилатными анионами формулы R13CO2 -, которые могут представлять собой любую ацилатную группу, такую, как, например, ацетат или бензоат, и которые также могут представлять собой хиральную группу, такую, как, например, манделат [PhCH(OH)CO2]. Если отдельные энантиомеры формулы R13CO2 - используют для получения соединений формулы (XXII), где R4 и R5 различны, то можно получать смесь диастереомерных сложных эфиров, которые можно разделять, например, путем избирательной рекристаллизации, и гидрировать желаемые диастереоизомеры с получением отдельных энантиомеров спирта формулы (XVI).
Соединения формулы (XXII) могут получаться путем взаимодействия соединения формулы (XVI) с карбоновой кислотой формулы R13CO2H в присутствии способствующего сочетание вещества, такого, как, например, дициклогексилкарбодиимид, или трифенилфосфин с диэтилазодикарбоксилатом.
Соединения формулы (XXII) могут также получаться путем взаимодействия соединения формулы (III) с соединением формулы (XXIII)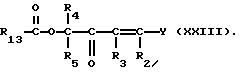
Соединения формулы (XXIII), где Y - диметиламин, могут получаться путем взаимодействия соединения формулы (XXIV)
с вышеуказанным реагентом Гольда.
Соединения формулы (XXIV) могут получаться путем взаимодействия соединения формулы (XXV)
с анионом формулы R13CO2 -.
Соединения формулы (I) или (II), где R1 не означает атом водорода, могут также получаться путем взаимодействия соединения формулы (XXVI)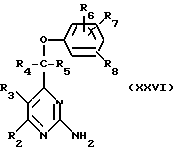
с соединением формулы R1CN--->O, при этом получают промежуточный продукт, который подвергают циклизации с применением подходящего кислотного катализатора.
Соединения формулы (I), где по меньшей мере один из остатков R6, R7 и/или R8 выбран из группы, включающей алкилсульфинил и алкилсульфонил, могут получаться путем окисления соединения формулы (I), где R6, R7 и/или R8 означают алкилтио-группу, с использованием, например, надуксусной кислотой или 3-хлорнадбензойной кислотой.
Соединения формулы (I) могут также получаться путем декарбоксилирования кислоты формулы (XXVII)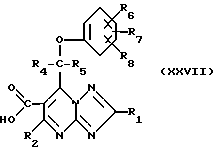
с использованием, например, тепла и/или подходящего кислотного катализатора.
Если R4 и R5 различны, то это представляет собой возможность получения отдельных энантиомеров соединения (I).
Соединения формулы (XXVII) могут получаться путем гидрирования сложного эфира формулы (XXVIII)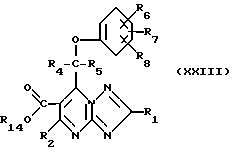
где R14 означает незамещенный или замещенный алкил или незамещенный или замещенный арил.
Соединения формулы (XXVIII) могут получаться путем взаимодействия соединения формулы (III) с соединением формулы (XXIX)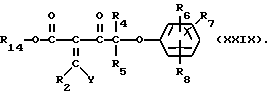
Соединения формулы (XXIX) могут получаться путем взаимодействия соединения формулы (VIII) с соединением формулы (XXX)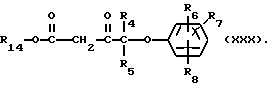
Кроме того, соединения формулы (I) или (II) могут получаться путем восстановления соединения формулы (XXXI)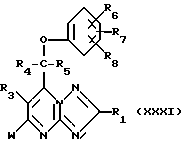
где W - подходящая удаляемая группа, например, галоид, восстановителем. Если W - галоид, то в качестве восстановителя можно применять, например, водород, при необходимости в присутствии катализатора, такого, как, например, палладий.
Если R4 и R5 различны, то это представляет собой возможность получения отдельных энантиомеров соединения (I).
Соединения формулы (XXXI), где W - галоид, могут получаться путем взаимодействия соединения формулы (XXXI), где W - гидрокси-группа, с агентом галогенирования, таким, как, например, фосфорилхлорид.
Соединения формулы (XXXI), где R3 - атом водорода, а W - гидрокси-группа, могут получаться путем взаимодействия соединения формулы (III) с соединением формулы (XXXI).
Соединения формулы (II) могут получаться тем же путем, что и соединения формулы (I).
Соединения формулы (I) или (II) проявляют противосудорожную активность, что выявилось с помощью следующих фармакологических опытов.
Первый опыт был направлен на определение способности соединений формулы (I) или (II) к проявлению действия, антагонистического миоклоническим припадкам, вызываемым в мышах дачей торгового продукта (+)-бикукуллина (см. Buckett W.R.; J. Pharmacol.Meth.; 1981 г., 5; стр. 35 -41). Бикукуллин, представляющий собой избирательный антагонист рецепторов γ-аминомасляной кислоты A, при интравенной дачи вызывает характерный судорожный синдром.
Этот синдром можно предотвращать дачей антиэпилептических лекарств, потенцирующих передачу нейротрансмиттера γ- аминомасляной кислоты. Опыт в следующем обозначается сокращением "BICM".
В опыте "BICM" использовались мыши-самки весом 25-30 г. За 2 часа до начала опыта мыши больше не получали корма, а лишь воду в желаемом количестве. Мыши подразделяли на две группы, т.е. контрольную группу и подопытную группу. Контрольной группе орально давали дозу 10 мл/кг 1%-ного водного раствора метилцеллюлозы. Подопытной группе орально давали суспендированное в той же дозе раствора метилцеллюлозы соединение формулы (I) или (II) либо в дозе 100 мг/кг для начального опыта, либо - при наличии соединения в достаточном количестве - в разных дозах с целью определения ЭД50 (см. ниже). Спустя 1 час после дачи всех препаратов всем мышам из обеих групп внутривенно в вену хвоста дали (+)-бикукуллин в дозе, равной 0,55 мг/кг. (+)-бикукуллин в такой дозе, как правило, у мышей вызывает припадок.
В течение следующих двух минут наблюдали каждую группу мышей, записывали число мышей, у которых проявлялись судороги, и таким образом определяли процентное число мышей из подопытной группы, у которых предотвратили судороги. Чем сильнее антисудорожная активность соединения формулы (I) или (II), тем выше процентное число, полученное в результате опыта "BICM". При наличии результатов, полученных с разными дозами соединения, дозу, ингибирующую припадки в 50% мышей (ЭД50), рассчитывали путем регрессионного анализа. При этом для каждой дозы формулы (I) или (II) определяли процентное число животных, у которых ингибировали припадки.
Второй опыт по определению антисудорожной активности был направлен на определение способности соединения формулы (I) или (II) к проявлению действия, антагонистического припадкам, вызываемым в мышах путем максимального электрошока. Опыт в следующим обозначается сокращением "MESM".
В опыте "MESM" использовали мыши-самцы весом 25-30 г, которым до начала опыта давали корм и воду в желаемом количестве. Мыши подразделяли на две группы, т. е. контрольную группу и подопытную группу. Контрольной группе орально давали дозу 10 мл/кг 1%-ного водного раствора метилцеллюлозы. Подопытной группе орально давали суспендированное в той же дозе раствора метилцеллюлозы соединение формулы (I) или (II) либо в дозе 100 мг/кг для начального опыта, либо - при наличии соединения в достаточном количестве - в разных дозах с целью определения ЭД50 (см. ниже). Спустя 1 час после дачи всех препаратов всем мышам из обеих групп с помощью электродов, закрепленных на ушах и увлажненных солевым раствором, в течение 1 секунды давали электрошок интенсивностью 99 мА, частотой 50 Гц и длительностью импульсов 0,4 мсек. Такой электрошок, как правило, у мышей вызывает припадок.
В течение следующих двух минут наблюдали каждую группу мышей, записывали число мышей, у которых проявлялось тонизирующее выпрямление задних конечностей, и таким образом определяли процентное число мышей из подопытной группы, у которых предотвратили судороги.
Чем сильнее антисудорожная активность соединения формулы (I) или (II), тем выше процентное число, полученное в результате опыта "MESM". При наличии результатов, полученных с разными дозами соединения, дозу, ингибирующую припадки в 50% мышей (ЭД50), рассчитывали путем регрессионного анализа. При этом для каждой дозы формулы (I) или (II) определяли процентное число животных, у которых ингибировали припадки.
Описанные в нижеследующих примерах 1 - 25а соединения формулы (I) или (II) в по меньшей мере одном из опытов "BICM" и "MESM" проявляли противосудорожную активность.
Изобретение иллюстрируется следующими примерами, которые не ограничивают его объем. Целевые продукты в каждом из этих примеров были характеризованы одним или несколькими из следующих методов исследования: элементный анализ, инфракрасная спектроскопия, спектроскопия ядерного магнитного резонанса, газожидкостная хроматография, жидкостная хроматография. Температуры указаны в "oC".
Пример 1.
1,12 г 4-фторфенола подают в перемешиваемую суспензию 0,48г гидрида натрия в 35 мл сухого 1,2-диметоксиэтана. Смесь перемешивают при комнатной температуре в течение 30 минут, после чего по каплям добавляют раствор 2,27 г 7-(1-бромэтил)-1,2,4-триазол [1,5-а]пиримидина, полученного аналогичным примеру 6 приемом, в 85 мл сухого 1,2-диметоксиэтана. Смесь перемешивают при комнатной температуре в течение 24 часов. Бромид натрия удаляют из смеси путем фильтрации. Растворитель упаривают из фильтрата и остаток растворяют в дихлорметане и последовательно промывают 200 мл 5%-ного водного раствора гидроокиси натрия и водой. Органическую фазу сушат над сульфатом магния. Растворитель упаривают, при этом получают сырой продукт, который очищают путем колоночной хроматографии на силикагеле с применением в качестве элюента смеси петролейного эфира и этилацетата в соотношении 6:4, с последующей рекристаллизацией из смеси этилацетата и гексана. Получают 1,03 г 7-[1-(4-фторфенокси)этил] - 1,2,4-триазол[1,5-а]-пиримидина с точкой плавления 106 - 108oC.
ЭД50 данного соединения в вышеописанном опыте "BICM": 13,9 мг/кг.
ЭД50 данного соединения в вышеописанном опыте "MESM": 21,4 мг/кг.
Пример 2.
Смесь 34,50 г 3-(4-хлорфенокси)-2-бутанона и 20,70 г N,N-диметилформамид-диметилацеталя в атмосфере аргона на бане масла нагревают при температуре 120oC в течение 13 часов. Получаемый метанол удаляют при пониженном давлении и остаточное масло с помощью н-гексана растирают в порошок. Твердое вещество собирают путем фильтрации и промывают холодным диэтиловым эфиром. Получают 32,50 г 4-(4-хлорфенокси)-1-(диметиламино)-1-пентен-3-она.
Раствор 9,80г 4-(4-хлорфенокси)-1-(диметиламино)-1-пентен-3-она в 50 мл ледяной уксусной кислоты добавляют в перемешиваемый раствор 3,25 г 3-амино-1,2,4-триазола в 50 мл ледяной уксусной кислоты. Смесь в течение 5 часов нагревают с обратным холодильником, после чего охлаждают до комнатной температуры. Смесь подают в 300 мл ледяной воды и экстрагируют толуолом. Экстракты последовательно промывают 10%-ным водным раствором бикарбоната натрия и водой, сушат над неводным сульфатом магния, растворитель упаривают при пониженном давлении. Остаток с помощью холодного диэтилового эфира растирают в порошок, получаемое твердое вещество собирают путем фильтрации и рекристаллизуют из смеси этилацетата и петролейного эфира с пределами кипения 40 - 60oC, при этом получают 6,91 г 7-[1-(4-хлорфенокси)этил]-1,2,4-триазол[1,5-а]пиримидина с точкой плавления 111 - 112oC.
ЭД50 данного соединения в вышеописанном опыте "BICM": 12,7 мг/кг.
ЭД50 данного соединения в вышеописанном опыте "MESM": 64,1 мг/кг.
Пример 3.
Раствор 1,73 г 4-бромфенола в сухом 1,2-диметоксиэтане медленно подают в перемешиваемую суспензию 0,48 г гидрида натрия в 35 мл сухого 1,2-диметоксиэтана. Смесь перемешивают в течение 30 минут, после чего по каплям добавляют раствор 2,27 г 7-(1-бромэтил)-1,2,4-триазол [1,5-а]пиримидина, полученного аналогичным примеру 6 приемом, в 85 мл сухого 1,2-диметоксиэтана. Реакционную смесь перемешивают при комнатной температуре в течение 1,5 часов. Бромид натрия удаляют из смеси путем фильтрации. Растворитель упаривают из смеси, и остаток растворяют в дихлорметане и последовательно промывают 200 мл 5%-ного водного раствора гидроокиси натрия и водой. Органическую фазу сушат над сульфатом магния. Растворитель упаривают, при этом получают сырой продукт, который очищают путем колоночной хроматографии на силикагеле с применением в качестве элюента смеси этилацетата и петролейного эфира в соотношении 4:6, с последующей рекристаллизацией из смеси этилацетата и гексана. Получают 2,28 г 7-[1-(4-бромфенокси)этил]-1,2,4-триазол[1,5-а]-пиримидина с точкой плавления 121 - 124oC.
ЭД50 данного соединения в вышеописанном опыте "BICM": 18,9 мг/кг.
ЭД50 данного соединения в вышеописанном опыте "MESM": 73,7 мг/кг.
Пример 4.
Раствор 1,19 г 4-цианофенола в сухом 1,2-диметоксиэтане медленно подают в перемешиваемую суспензию 0,48 г гидрида натрия в 35 мл сухого 1,2-диметоксиэтана. Смесь перемешивают в течение 30 минут, после чего по каплям добавляют раствор 2,27 г 7-(1-бромэтил)-1,2,4-триазол [1,5-а]пиримидина, полученного аналогичным примеру 6 приемом, в 85 мл сухого 1,2-диметоксиэтана. Реакционную смесь перемешивают при комнатной температуре в течение ночи. Бромид натрия удаляют из смеси путем фильтрации. Растворитель упаривают из смеси, и остаток растворяют в дихлорметане и последовательно промывают 200 мл 5%-ного водного раствора гидроокиси натрия и водой. Органическую фазу сушат над сульфатом магния. Растворитель упаривают, при этом получают сырой продукт, который очищают путем колоночной хроматографии на силикагеле с применением в качестве элюента смеси этилацетата и петролейного эфира в соотношении 6:4, с последующей рекристаллизацией из этилацетата. Получают 1,07 г 7-[1-(4-цианофенокси)этил] -1,2,4-триазол[1,5-а] -пиримидина с точкой плавления 163 - 164oC.
Процентное число подопытных мышей, у которых в опыте "MESM" не проявлялось припадков, при дозировке данного соединения, равной 100 мг/кг, составляло 60%.
Пример 5.
1,62 г 4-трифторметилфенола подают в перемешиваемую суспензию 0,48г гидрида натрия в 35 мл сухого 1,2-диметоксиэтана. Смесь перемешивают при комнатной температуре в течение 30 минут, после чего по каплям добавляют раствор 2,27 г 7-(1-бромэтил)-1,2,4-триазол[1,5-а]пиримидина, полученного аналогичным примеру 6 приемом, в 85 мл сухого 1,2-диметоксиэтана. Смесь перемешивают при комнатной температуре в течение 24 часов. Бромид натрия удаляют из смеси путем фильтрации. Растворитель упаривают из фильтрата и остаток растворяют в дихлорметане и последовательно промывают 200 мл 5%-ного водного раствора гидроокиси натрия и водой. Органическую фазу сушат над сульфатом магния. Растворитель упаривают, при этом получают сырой продукт, который очищают путем колоночной хроматографии на силикагеле с применением в качестве элюента смеси петролейного эфира и этилацетата в соотношении 6:4, с последующей рекристаллизацией из гексана. Получают 1,1 г 7-[1-(4-трифторметилфенокси)этил]-1,2,4- триазол[1,5-а]-пиримидина с точкой плавления 100 - 102oC.
ЭД50 данного соединения в вышеописанном опыте "BICM": 29,8 мг/кг.
ЭД50 данного соединения в вышеописанном опыте "MESM": 52,1 мг/кг.
Пример 6.
Смесь 11,74 r 3-амино-1,2,4-тоиазола и 16,5 r 1-хлор-1-пентен-3-она в 225 мл уксусной кислоты нагревают с обратным холодильником в течение 45 минут. Реакционную смесь охлаждают, наливают на лед и экстрагируют дихлорметаном. Органическую фазу сушат и растворитель упаривают при пониженном давлении с получением 11,72 г 7-этил-1,2,4-триазол[1,5-а]пиримидина.
Раствор 10,05 г 7-этил-1,2,4-триазол[1,5-а]пиримидина, 12.63 г N-бромсукцинимида, 0,3 г дибензоилпероксида и 270 мл тетрахлорметана в течение 5 часов при перемешивании нагревают с обратным холодильником. Смесь фильтруют и растворитель упаривают из фильтрата, при этом получают сырой продукт, который очищают путем рекристаллизации из тетрахлорметана. Получают 10,8 г 7-(1-бромэтил)-1,2,4-триазол [1,5-а]пиримидина.
Раствор 1,24 г 4-метоксифенола в сухом 1,2-диметоксиэтане медленно подают в перемешиваемую суспензию 0,48 г гидрида натрия в 35 мл сухого 1,2-диметоксиэтана. Смесь перемешивают в течение 30 минут, после чего по каплям добавляют раствор 2,27 r 7-(1-бромэтил)-1,2,4-триазол [1,5-а]пиримидина в 85 мл сухого 1,2-диметоксиэтана. Реакционную смесь перемешивают при комнатной температуре в течение ночи, фильтруют, и растворитель упаривают из фильтрата при пониженном давлении. Остаток очищают путем флеш-хроматографии с применением в качестве элюента смеси этилацетата и петролейного эфира, с последующей рекристаллизацией из смеси этилацетата и гексана. Получают 1,67 г 7-[1-(4-метоксифенокси)этил] -1,2,4-триазол[1,5-а]- пиримидина с точкой плавления 112 - 114oC.
ЭД50 данного соединения в вышеописанном опыте "BICM": 93,3 мг/кг.
Пример 7.
Раствор 1,78 г 4-трифторметоксифенола в сухом 1,2-диметоксиэтане медленно подают в перемешиваемую суспензию 0,48 г гидрида натрия в 35 мл сухого 1,2-диметоксиэтана. Смесь перемешивают в течение 30 минут, после чего по каплям добавляют раствор 2,27г 7-(1-бромэтил)-1,2,4- триазол[1,5-а]пиримидина, полученного аналогичным примеру 6 приемом, в 85 мл сухого 1,2-диметоксиэтана. Реакционную смесь перемешивают при комнатной температуре в течение 4 часов. Бромид натрия удаляют из смеси путем фильтрации. Растворитель упаривают из смеси и остаток растворяют в дихлорметане и последовательно промывают 200 мл 5%-ного водного раствора гидроокиси натрия и водой. Органическую фазу сушат над сульфатом магния. Растворитель упаривают, при этом получают сырой продукт, который очищают путем колоночной хроматографии на силикагеле с применением в качестве элюента смеси петролейного эфира и этилацетата в соотношении 6:4, с последующей рекристаллизацией из смеси этилацетата и гексана. Получают 2,69 г 7-[1-(4-трифторметоксифенокси)этил]-1,2,4-триазол[1,5-а]-пиримидина с точкой плавления 91 - 93oC.
ЭД50 данного соединения в вышеописанном опыте "BICM": 11,4 мг/кг.
ЭД50 данного соединения в вышеописанном опыте "MESM": 52,8 мг/кг.
Пример 8.
Раствор 1,36 г 4-оксиацетофенона в сухом 1,2-диметоксиэтане медленно подают в перемешиваемую суспензию 0,48 г гидрида натрия в 35 мл сухого 1,2-диметоксиэтана. Смесь перемешивают в течение 30 минут, после чего по каплям добавляют раствор 2,27 г 7-(1-бромэтил)-1,2,4- триазол[1,5-а]пиримидина, полученного аналогичным примеру 6 приемом, в 85 мл сухого 1,2-диметоксиэтана. Смесь перемешивают при комнатной температуре в течение 24 часов. Бромид натрия удаляют из смеси путем фильтрации. Растворитель упаривают из смеси и остаток растворяют в дихлорметане и последовательно промывают 200 мл 5%-ного водного раствора гидроокиси натрия и водой. Растворитель упаривают, при этом получают сырой продукт, который рекристаллизуют из этилацетата. Получают 0,87 г 7-[1-(4-ацетилфенокси)этил]-1,2,4-триазол[1,5-а]- пиримидина с точкой плавления 136 - 138oC.
ЭД50 данного соединения в вышеописанном опыте "BICM": 105,8 мг/кг.
Пример 9.
Раствор 2,80 г 4-(метилтио)фенола в сухом 1,2-диметоксиэтане медленно подают в перемешиваемую суспензию 0,87 г гидрида натрия в 50 мл сухого 1,2-диметоксиэтана. Реакционную смесь перемешивают в течение 30 минут, после чего по каплям добавляют раствор 4,54 г 7-(1-бромэтил)-1,2,4-триазол[1,5-а]- пиримидина, полученного аналогичным примеру 6 приемом, в 150 мл сухого 1,2-диметоксиэтана. Смесь перемешивают при комнатной температуре в течение ночи. Бромид натрия удаляют из смеси путем фильтрации. Растворитель упаривают из смеси и остаток растворяют в дихлорметане и последовательно промывают 200 мл 5%-ного водного раствора гидроокиси натрия и водой. Органическую фазу сушат над сульфатом магния. Растворитель упаривают, при этом получают сырой продукт, который очищают путем колоночной хроматографии на силикагеле с применением в качестве элюента смеси этилацетата и петролейного эфира в соотношении 4:6, с последующей рекристаллизацией из смеси этилацетата и гексана. Получают 3,66 г 7-{1-[4-(метилтио)фенокси]этил}- 1,2,4-триазол[1,5-а]-пиримидина с точкой плавления 84 - 86oC.
Процентное число подопытных мышей, у которых в опыте "MESM" не проявлялось припадков, при дозировке данного соединения, равной 50 мг/кг, составляло 50%.
Пример 10.
Раствор 0,63 г 3-хлорнадбензойной кислоты в 30 мл дихлорметана по каплям при температуре -78oC подают в перемешиваемый раствор 0,89 г 7-{1-[4-(метилтио)фенокси]этил}-1,2,4-триазол[1,5-а]-пиримидина, полученного аналогичным примеру 9 приемом, в 30 мл дихлорметана. Реакционную смесь перемешивают при температуре -78oC в течение 2 часов, промывают 10%-ным водным раствором бикарбоната натрия и водой. Органическую фазу сушат и растворитель упаривают при пониженном давлении. Остаток очищают путем флеш-хроматографии с применением в качестве элюента смеси дихлорметана и этанола в соотношении 95: 5, с последующей рекристаллизацией из смеси этилацетата и гексана. Получают 0,76 г 7-[1-(4-метилсульфинилфенокси)этил]-1,2,4-триазол[1,5-а]-пиримидина с точкой плавления 89 - 102oC.
Процентное число подопытных мышей, у которых в опыте "BICM" не проявлялось припадков, при дозировке данного соединения, равной 100 мг/кг, составляло 60%.
Пример 11.
Раствор 2,13 г 3-хлорнадбензойной кислоты в 50 мл дихлорметана по каплям при комнатной температуре подают в перемешиваемый раствор 1,2 г 7-{1-[4-(метилтио)фенокси]этил}-1,2,4-триазол[1,5-а]-пиримидина, полученного аналогичным примеру 9 приемом, в 70 мл дихлорметана. Реакционную смесь перемешивают в течение 3 часов, промывают 10%-ным водным раствором бикарбоната натрия и водой. Органическую фазу сушат и растворитель упаривают при пониженном давлении. Остаток очищают путем флеш-хроматографии с применением этилацетата в качестве элюента, с последующей рекристаллизацией из этанола. Получают 0,72 г 7-[1-(4-метилсульфонилфенокси)этил]-1,2,4-триазол[1,5-а]-пиримидина с точкой плавления 163 - 164oC.
Процентное число подопытных мышей, у которых в опыте "BICM" не проявлялось припадков, при дозировке данного соединения, равной 100 мг/кг, составляло 60%.
Пример 12.
Раствор 1,54 г 4-(этилтио)фенола в сухом 1,2-диметоксиэтане медленно подают в перемешиваемую суспензию 0,48 г гидрида натрия в 35 мл сухого 1,2-диметоксиэтана. Смесь перемешивают в течение 30 минут, после чего по каплям добавляют раствор 2,27г 7-(1-бромэтил)-1,2,4- триазол[1,5-а]пиримидина, полученного аналогичным примеру 6 приемом, в 85 мл сухого 1,2-диметоксиэтана. Смесь перемешивают при комнатной температуре в течение ночи. Бромид натрия удаляют из смеси путем фильтрации. Растворитель упаривают из смеси и остаток растворяют в дихлорметане и последовательно промывают 200 мл 5%-ного водного раствора гидроокиси натрия и водой. Органическую фазу сушат над сульфатом магния. Растворитель упаривают, при этом получают сырой продукт, который очищают путем колоночной хроматографии на силикагеле с применением в качестве элюента смеси диэтилового эфира и этил- ацетата в соотношении 6:4, с последующей рекристаллизацией из смеси этилацетата с гексаном. Получают 2,28 г 7-{1-[4-(этилтио)фенокси]этил}- 1,2,4-триазол[1,5-а]-пиримидина с точкой плавления 65 - 67oC.
ЭД50 данного соединения в вышеописанном опыте "BICM": 48,9 мг/кг.
Пример 13.
Раствор 1,28 г 3-хлорфенола в сухом 1,2-диметоксиэтане медленно подают в перемешиваемую суспензию 0,48 г гидрида натрия в 35 мл сухого 1,2-диметоксиэтана. Смесь перемешивают в течение 30 минут, после чего по каплям добавляют раствор 2,27 г 7-(1-бромэтил)-1,2,4-триазол [1,5-а]пиримидина, полученного аналогичным примеру 6 приемом, в 85 мл сухого 1,2-диметоксиэтана. Реакционную смесь перемешивают при комнатной температуре в течение ночи, фильтруют и растворитель под пониженным давлением упаривают из смеси. Остаток очищают путем флеш-хроматографии с применением в качестве элюента смеси этилацетата и петролейного эфира. Получают 2,11 г 7-[1-(3-хлор-фенокси)этил]-1,2,4- триазол[1,5-а]-пиримидина с точкой плавления 124 -126oC.
Процентное число подопытных мышей, у которых в опыте "BICM" не проявлялось припадков, при дозировке данного соединения, равной 100 мг/кг, составляло 78%.
Процентное число подопытных мышей, у которых в опыте "MESM" не проявлялось припадков, при дозировке данного соединения, равной 100 мг/кг, составляло 60%.
Пример 14.
Раствор 1,30 г 2,4-дифторфенола в сухом 1,2-диметоксиэтане медленно подают в перемешиваемую суспензию 0,48 г гидрида натрия в 35 мл сухого 1,2-диметоксиэтана. Смесь перемешивают в течение 30 минут, после чего по каплям добавляют раствор 2,27 г 7-(1-бромэтил)-1,2,4- триазол[1,5-а]пиримидина, полученного аналогичным примеру 6 приемом, в 85 мл сухого 1,2-диметоксиэтана. Реакционную смесь перемешивают при комнатной температуре в течение ночи. Бромид натрия удаляют из смеси путем фильтрации. Растворитель упаривают из смеси и остаток растворяют в дихлорметане и последовательно промывают 200 мл 5%-ного водного раствора гидроокиси натрия и водой. Растворитель упаривают, при этом получают сырой продукт, который очищают путем колоночной хроматографии на силикагеле с применением в качестве элюента смеси дихлорметана и этанола в соотношении 97:3, с последующей рекристаллизацией из смеси этилацетата и гексана. Получают 1,8 г 7-[1- (2,4-дифторфенокси)этил]-1,2,4-триазол[1,5-а]-пиримидина с точкой плавления 96 - 97oC.
ЭД50 данного соединения в вышеописанном опыте "BICM": 37,5 мг/кг.
Пример 15.
Смесь 4,66 г 3-(2,4-дихлорфенокси)-2-бутанона и 2,38 г N,N-диметилформамид-диметилацеталя в атмосфере аргона на бане масла нагревают при температуре 120oC в течение 11 часов. Получаемый при этом метанол удаляют при пониженном давлении и остаток с помощью н-гексана растирают в порошок. Твердое вещество собирают путем фильтрации и промывают холодным диэтиловым эфиром. Получают 4,07 г 4-(2,4-дихлорфенокси)-1-(диметиламино)-1-пентен-З-она.
Раствор 2,9 г 4-(2,4-дихлорфенокси)-1-(диметиламино)-1-пентен-3-она в 25 мл ледяной уксусной кислоты добавляют в перемешиваемый раствор 0,93 г 3-амино-1,2,4-триазола в 25 мл ледяной уксусной кислоты. Смесь в течение 5 часов нагревают с обратным холодильником, после чего охлаждают до комнатной температуры. Смесь подают в 200 мл ледяной воды и экстрагируют толуолом. Экстракты последовательно промывают 10%-ным водным раствором бикарбоната натрия и водой, сушат над неводным сульфатом магния, растворитель упаривают при пониженном давлении. Остаток с помощью холодного диэтилового эфира растирают в порошок, получаемое твердое вещество собирают путем фильтрации и рекристаллизуют из смеси этилацетата и петролейного эфира с пределами кипения 40 - 60oC. при этом получают 2,02 г 7-[1-(2,4-дихлорфенокси)этил]-1,2,4-триазол[1,5-а]пиримидина с точкой плавления 137 - 138oC.
ЭД50 данного соединения в вышеописанном опыте "BICM": 39,7 мг/кг.
ЭД50 данного соединения в вышеописанном опыте "MESM": 109,7 мг/кг.
Пример 16
Раствор 1,22 г 3,4-дихлорфенола в сухом 1,2-диметоксиэтане медленно подают в перемешиваемую суспензию 0,33 г гидрида натрия в 30 мл сухого 1,2-диметоксиэтана. Смесь перемешивают в течение 30 минут, после чего по каплям добавляют раствор 1,68 г 7-(1-бромэтил)-1,2,4- триазол[1,5-а]пиримидина, полученного аналогичным примеру 6 приемом, в 60 мл сухого 1,2-диметоксиэтана. Реакционную смесь перемешивают при комнатной температуре в течение ночи. Бромид натрия удаляют из смеси путем фильтрации. Растворитель упаривают из смеси и остаток растворяют в дихлорметане и последовательно промывают 200 мл 5%-ного водного раствора гидроокиси натрия и водой. Органическую фазу сушат над сульфатом магния. Растворитель упаривают, при этом получают сырой продукт, который очищают путем колоночной хроматографии на силикагеле с применением в качестве элюента смеси этилацетата и петролейного эфира в соотношении 4:6, с последующей рекристаллизацией из смеси этилацетата и гексана. Получают 1,45 г 7-[1-(3,4-дихлорфенокси)этил]- 1,2,4-триазол[1,5-а]-пиримидина с точкой плавления 146 - 149oC.
Процентное число подопытных мышей, у которых в опыте "BICM" не проявлялось припадков, при дозировке данного соединения, равной 100 мг/кг, составляло 50%.
Пример 17.
Смесь 0,65 г 2-хлор-4-фторфенола и 210 мг гидрида натрия в 15 мл сухого 1,2-диметоксиэтана перемешивают при комнатной температуре в течение 30 минут. Раствор 1 г 7-(1-бромэтил)-1,2,4-триазол[1,5-а]- пиримидина, полученного аналогичным примеру 6 приемом, в 35 мл 1,2- диметоксиэтана по каплям подают в вышеуказанную перемешиваемую смесь. Смесь перемешивают при комнатной температуре в течение 21 часов, фильтруют и растворитель под пониженным давлением упаривают из фильтрата. Остаток растворяют в дихлорметане, последовательно промывают 5%-ным водным раствором гидроокиси натрия и водой, сушат над неводном сульфате магния и растворитель удаляют при пониженном давлении. Твердый остаток очищают путем флеш-хроматографии на силикагеле с применением в качестве элюента смеси этилацетата и петролейного эфира в соотношении 1:1, с последующей рекристаллизацией из смеси этилацетата и гексана. Получают 0,99 г 7-[1-(2-хлор-4-фторфенокси)-этил]-1,2,4-триазол[1,5-а]-пиримидина с точкой плавления 89 - 91oC.
Процентное число подопытных мышей, у которых в опыте "BICM" не проявлялось припадков, при дозировке данного соединения, равной 100 мг/кг, составляло 50%.
ЭД50 данного соединения в вышеописанном опыте "MESM": 66,0 мг/кг.
Пример 18.
Раствор 1,58 г 4-(4-хлорфенокси)-1-(диметиламино)-1-пентен-3-она, полученного аналогичным примеру 2 приемом, в 5 мл ледяной уксусной кислоты добавляют в перемешиваемый раствор 0,62г 3-амино-5-метил- 1,2,4-триазола в 10 мл ледяной уксусной кислоты. Смесь в течение 2,5 часов нагревают с обратным холодильником, после чего охлаждают до комнатной температуры. Смесь подают в 50 мл ледяной воды и экстрагируют толуолом. Экстракты последовательно промывают 10%-ным водным раствором бикарбоната натрия и водой, сушат над неводным сульфатом магния, растворитель упаривают при пониженном давлении. Остаток с помощью холодного диэтилового эфира растирают в порошок, получаемое твердое вещество собирают путем фильтрации и рекристаллизуют из смеси этилацетата и петролейного эфира с пределами кипения 40 - 60oC, при этом получают 1,13 г 7-[1-(4-хлорфенокси)этил]- 2-метил-1,2,4-триазол[1,5-а]пиримидина с точкой плавления 138oC.
ЭД50 данного соединения в вышеописанном опыте "BICM": 78,2 мг/кг.
ЭД50 данного соединения в вышеописанном опыте "MESM": 107,7 мг/кг.
Пример 19.
10,9 г 1,4-дихлор-1-бутен-3-она, полученного аналогичным примеру 20 приемом получения 1-хлор-4-метил-1-пентен-3-она, 6,5 г 3-амино-1,2,4-триазола и ледяной уксусной кислоты в течение 1,5 часов нагревают с обратным холодильником. Реакционную смесь наливают на лед и экстрагируют дихлорметаном. Органическую фазу сушат над сульфатом магния, и растворитель упаривают с получением твердого вещества. Сырой продукт очищают путем флеш-хроматографии с применением в качестве элюента смеси дихлорметана и этанола в соотношении 97:3, с последующей рекристаллизацией из тетрахлорметана. Получают 10,15 г 7-хлорметил- 1,2,4-триазол[1,5-а]пиримидина.
2,18 г 4-хлорфенола, 0,92г метилата натрия и 150 мл сухого метанола при перемешивании в течение 1 часа нагревают с обратным холодильником. Растворитель удаляют при пониженном давлении. 2,8 г 7-хлорметил-1,2,4-триазол-[1,5-а]пиримидина и 170 мл сухого 1,2- диметоксиэтана подают в сырую реакционную смесь. Смесь при перемешивании нагревают с обратным холодильником в течение 10 часов. Растворитель упаривают при пониженном давлении и сырой продукт очищают путем флеш-хроматографии с применением этилацетата в качестве элюента с последующей рекристаллизацией из этилацетата. Получают 0,26 г 7-(4-хлорфеноксиметил)-1,2,4-триазол[1,5-а]пиримидина с точкой плавления 193 - 194oC.
Процентное число подопытных мышей, у которых в опыте "BICM" не проявлялось припадков, при дозировке данного соединения, равной 100 мг/кг, составляло 70%.
Пример 20.
27,2 г неводного хлорида алюминия подают в перемешиваемый раствор 23,4 г 2-метилпропионилхлорида в 100 мл сухого трихлорметана, при этом реакционную смесь снаружи охлаждают до температуры 0 - 15oC. В течение 1 часа через смесь, которую сохраняют при температуре 24 -26oC, подают 20 г хлорэтена, после чего перемешивание продолжают в течение еще 40 минут. Реакционную смесь наливают на лед, органическую фазу отделяют, сушат над сульфатом магния и перегоняют при пониженном давлении с получением 23 г 1,1-дихлор-4-метил-3-пентанона в качестве бесцветной жидкости.
12,53 г 1,1-дихлор-4-метил-3-пентанона смешивают с 6,23 г бикарбоната натрия и 30 мл воды. Смесь нагревают с обратным холодильником в течение 4 часов, охлаждают и экстрагируют трихлорметаном. Органическую фазу сушат над сульфатом магния и перегоняют под пониженным давлением. При этом получают 5,93 г 1-хлор- 4-метил-1-пентен-3-она в качестве бесцветной жидкости.
Смесь 5,83 г 1-хлор-4-метил-1-пентен-3-она, 3,69 г 3-амино-1,2,4- триазола и ледяной уксусной кислоты в течение 1,5 часов нагревают с обратным холодильником. Реакционную смесь наливают на лед и экстрагируют дихлорметаном. Органическую фазу сушат над сульфатом магния и растворитель упаривают с получением промежуточного продукта, который рекристаллизуют из петролейного эфира с пределами кипения 100 - 140oC. При этом получают 4,18 г 7-(1-метилэтил)-1,2,4-триазол[1,5-а]пиримидина.
Смесь 4,18 г 7-(1-метилэтил)-1,2,4-триазол[1,5-а]пиримидина, 4,59 г N-бром-сукцинимида и 70 мг дибензоилпероксида в 105 мл тетрахлорметана при перемешивании в течение 11 часов нагревают с обратным холодильником. Смесь фильтруют и растворитель удаляют из фильтрата с получением 2,94 г 7-(1-бром-1-метилэтил)-1,2,4-триазол[1,5-а]пиримидина.
Смесь 2,95 г 7-(1-бром-1 -метилэтил)-1,2,4-триазол[1,5-а]пиримидина, полученного аналогично вышеописанному приему, 1,56 г 4-хлорфенола, 1 г бикарбоната натрия, 5 мг ацетилацетона никеля d 80 мл сухого толуола при перемешивании в течение 7 дней нагревают с обратным холодильником. Растворитель упаривают и сырой продукт очищают путем флеш-хроматографии с применением в качестве элюента смеси толуола и этилацетата в соотношении 5:1, с последующей рекристаллизацией из н-гексана. Получают 0,67 г 7-[1-(4-хлорфенокси)-1-метилэтил]-1,2,4- триазол[1,5-а]пиримидина с точкой плавления 132 - 135oC.
ЭД50 данного соединения в вышеописанном опыте "MESM": 45,0 мг/кг.
Пример 21.
Раствор 10 г 3-хлор-2-пентанона в 50 л ацетона по каплям подают в перемешиваемую смесь 11 г 4-хлорфенола, 20 г бикарбоната калия и 1 г йодида калия в 100 мл ацетона. После окончания добавления смесь в течение 6 часов нагревают с обратным холодильником. Смесь фильтруют промывают ацетоном, а ацетон упаривают при пониженном давлении. Остаток растворяют в 150 мл диэтилового эфира и эфирную смесь последовательно промывают 300 мл 10%-ного водного раствора гидроокиси натрия и 300 мл воды. Смесь сушат над сульфатом магния. Растворитель упаривают и остаток перегоняют при пониженном давлении. При этом получают 10,96 г 3-(4-хлорфенокси)-2-пентанона.
Раствор 10,96 3-(4-хлорфенокси)-2-пентанона и 6 г N,N- диметилформамид-диметиацеталя в атмосфере аргона на бане масла нагревают при температуре 120oC в течение 24 часов. Получаемый в ходе реакции метанол удаляют при пониженном давлении, и остаточное масло в качестве 12,71 г 4-(4-хлорфенокси)-1-(диметиламино)-1-гексен-3-она непосредственно используют на следующей стадии.
Раствор 12,67 г 4-(4-хлорфенокси)-1-(диметиламино)-1-гексен-3-она в 75 мл ледяной уксусной кислоты добавляют в перемешиваемый раствор 3,78 г 3-амино-1,2,4-триазола в 75 мл ледяной уксусной кислоты. Раствор в течение 2 часов нагревают с обратным холодильником, после чего смесь подают в 200 мл воды и экстрагируют толуолом. Органическую фазу последовательно промывают 10%-ным водным раствором бикарбоната натрия и водой, сушат над неводным сульфатом магния, растворитель упаривают при пониженном давлении. Остаток с помощью холодного диэтилового эфира растирают в порошок, получаемое твердое вещество собирают путем фильтрации и рекристаллизуют из смеси этилацетата и гексана, при этом получают 4,54 г 7-[1-(4-хлорфен-оксипропил)]-1,2,4- триазол[1,5-а]пиримидина с точкой плавления 108 - 109oC.
ЭД50 данного соединения в вышеописанном опыте "BICM": 38,6 мг/кг.
ЭД50 данного соединения в вышеописанном опыте "MESM": 79,4 мг/кг.
Пример 22.
30 г рацемического 7-[1-(4-фторфенокси)этил]-1,2,4-триазол [1,5-а]-пиримидина, полученного аналогичным примеру 1 приемом, разделяют на отдельные энантиомеры путем высокопроизводительной жидкостной хроматографии на колонке типа Chiralcel OD внутренными габаритами, равными 50 см х 10 см, с применением в качестве элюента смеси изогексана и изопропанола в соотношении 1:1. При этом в качестве первой элюированной фракции получают (+)-7-[1-(4-фторфенокси)этил] -1,2,4-триазол[1,5-а]-пиримидин с оптической чистотой выше 99% и удельным вращением [α]
ЭД50 данного соединения в вышеописанном опыте "BICM": 48,3 мг/кг.
ЭД50 данного соединения в вышеописанном опыте "MESM": 56,9 мг/кг.
Пример 23.
(-)-7-[1-(4-фторфенокси)этил] -1,2,4-триазол[1,5-а]-пиримидин выделяют в качестве второй элюированной фракции из рацемического 7-[1-(4-фторфен-окси)этил] -1,2,4-триазол[1,5-а] -пиримидина путем высокопроизводительной жидкостной хроматографии аналогичным примеру 22 приемом. Оптическая чистота продукта - выше 99%, удельное вращение [α]
ЭД50 данного соединения в вышеописанном опыте "BICM": 12,7 мг/кг.
ЭД50 данного соединения в вышеописанном опыте "MESM": 79,8 мг/кг.
Пример 24.
5 г рацемического 7-[1-(4-хлорфенокси)этил]-1,2,4-тоиазол[1,5-а]- пиримидина, полученного аналогичным примеру 2 приемом, разделяют на отдельные энантиомеры путем высокопроизводительной жидкостной хроматографии на колонке типа Chiralcel OD внутренними габаритами, равными 50 см х 10 см, с применением в качестве элюента смеси изогексана и изопропанола в соотношении 1:1. При этом в качестве первой элюированной фракции получают (+)-7-[1-(4-хлорфенокси)этил] -1,2,4-триазол[1,5-а]-пиримидин с оптической чистотой выше 99% и удельным вращением [α]
ЭД50 данного соединения в вышеописанном опыте "BICM": 28,1 мг/кг.
ЭД50 данного соединения в вышеописанном опыте "MESM": 80,8 мг/кг.
В следующих примерах 24а и 246 иллюстрируются дальнейшие методы получения (+)-7-[1 -(4-хлорфенокси)этил]-1,2,4-триазол[1,5-а]-пиримидина.
Пример 24a.
9,8 г 7-(1-бромэтил)-1,2,4-триазол[1,5-а]-пиримидина, полученного аналогичным примеру 6 приемом, 32,8 г (R)-миндальной кислоты, 30 мл триэтиламина и 500 мл диоксана смешивают и нагревают на паровой бане в течение 2,5 часов. Смесь охлаждают и растворитель упаривают при пониженном давлении. Твердый остаток растворяют в 400 мл этилацетата и промывают 300 мл воды. Водные промывочные жидкости экстрагируют 200 мл этилацетата, а органический слой последовательно промывают 200 мл воды, 10 мл триэтиламина, 100 мл воды и 100 мл рассола. Органический слой сушат над сульфатом магния, нагревают в присутствии активного угля, фильтруют и растворитель упаривают из фильтрата. При этом получают 10,75 г смеси двух диастереоизмеров: (R) сложный этиловый эфир (+)-1-(1,2,4-триазол[1,5-а] -пиримидин-7-ил)-миндальной кислоты и (R) сложный этиловый эфир (-)-1-(1,2,4-триазол[1,5-а] -пиримидин-7- ил)-миндальной кислоты.
Смесь двух диастереоизмеров, полученных вышеописанным приемом, подвергают фракционированной кристаллизации с применением этилацетата в качестве растворителя. Получают две фракции: первую фракцию в качестве 2,8 г менее полярного диастереоизомера, а вторую фракцию в качестве 3,3 г более полярного диастереоизомера.
0,7 г менее полярного диастереоизомера, полученного вышеописанным приемом, 1,4 г карбоната калия, 10 мл воды и 25 мл метанола перемешивают при комнатной температуре в течение 2 часов. Раствор разбавляют 100 мл воды и в течение ночи непрерывно экстрагируют дихлорметаном. Экстракты сушат над сульфатом магния и растворитель упаривают с получением 0,27 г (+)-1-(1,2,4-триазол[1,5-а]-пиримидин-7-ил)этанола.
Смесь 0,27г (+)-1-(1,2,4-триазол[1,5-а]-пиримидин-7-ил)этанола, 0,29 г диэтилазодикарбоксилата, 0,44 г трифенилфосфина и 0,22 г 4-хлорфенола в 40 мл сухого тетрагидрофурана перемешивают при комнатной температуре в течение 2 дней. Добавляют еще 0,15 г диэтилазодикарбоксилата и 0,22 г трифенилфосфина и раствор перемешивают в течение ночи до окончания реакции. Растворитель удаляют при пониженном давлении, а остаток растворяют в 100 мл этилацетата. Получаемый раствор последовательно промывают 40 мл 1-м. раствора гидроокиси натрия и 20 мл рассола. Растворитель упаривают. Остаток очищают путем флеш-хроматографии на силикагеле с применением в качестве элюента смеси триэтиламина и этилацетата в соотношении 1:100. Получают 0,33 г (+)-7-[1-(4-хлорфенокси)-этил]-1,2,4-триазол- [1,5-а]пиримидина.
Пример 24б.
Смесь 10,6 г 3-хлорбутанона, 15,0 г бензойной кислоты, 30 мл триэтиламина и 100 мл ацетонитрила нагревают с обратным холодильником в течение 1,5 часов. Смесь охлаждают и фильтруют для удаления осадившейся гидроокиси триэтиламина. Ацетонитрил удаляют из фильтрата при пониженном давлении, а остаток растворяют в этилацетате и промывают водой. Органические экстракты сушат над сульфатом магния, фильтруют на активном угле, а растворитель удаляют из фильтрата при пониженном давлении. Получают 16,7 г 3-бензоилоксибутан-2-она в качестве желтого масла, которое используют на следующей стадии без дальнейшей очистки.
Смесь 15,5 г 3-бензоилоксибутан-2-она и 14,4 г диметилформамид-диметилацеталя в течение 2,5 часов нагревают на паровой бане. Растворитель удаляют из смеси при пониженном давлении. В остаток при стряхивании подают петролейный эфир с пределами кипения 60 - 80oC. Эфирный слой отделяют от нерастворимого красного масла. При стряхивании в масло подают больше петролейного эфира и избыточный эфир удаляют при пониженном давлении. При этом получают красновато-коричневое масло, которое, когда оставляют стоять в течение ночи, частично кристаллизуется. Добавляют петролейный эфир, и твердое вещество собирают путем фильтрации и промывают петролейным эфиром. При этом получают 5,6 г 4-бензоилокси-1-диметиламино-1-пентен-3-она. Получаемое твердое вещество используют на следующей стадии без дальнейшей очистки.
Смесь 1,78 г аминотриазола и 5,0 г 4-бензоилокси-1-диметиламино- 1-пентен-З-она в 25 мл уксусной кислоты нагревают с обратным холодильником в течение 1,5 часов. Смесь наливают на лед, нейтрализуют добавлением твердого бикарбоната натрия и экстрагируют 200 мл этилацетата. Органические экстракты сушат над сульфатом магния, фильтруют и растворитель удаляют из фильтрата при пониженном давлении. К твердому остатку добавляют диэтиловый эфир. Твердый остаток собирают путем фильтрации с получением 3,8 г 1-(1,2,4-триазол-[1,5-а] пиримидин-7-ил)этил-бензоата в виде бледно-коричневого твердого вещества.
Смесь 1 г 1-(1,2,4-триазол-[1,5-а] пиримидин-7-ил)этил-бензоата, 2 г карбоната калия, 25 мл метанола и 20 мл воды перемешивают при комнатной температуре в течение 1,5 часов. Метанол удаляют из смеси при пониженном давлении, после чего смесь разбавляют 20 мл рассола и в течение ночи непрерывно экстрагируют дихлорметаном. Экстракты сушат над сульфатом магния и растворитель упаривают при пониженном давлении с получением 0,39 г 1-(1,2,4-триазол-[1,5-а]пиримидин-7-ил)этанола.
Раствор 1 г (1,2,4-триазол-[1,5-а]пиримидин-7-ил)этанола, полученного аналогично вышеописанному приему, в 30 мл дихлорметана в течение 30 минут по каплям подают в раствор 0,5 мл тионилхлорида в 50 мл дихлорметана, который нагревают с обратным холодильником и нагревание продолжают в течение еще 2 часов. Добавляют еще 0,5 мл тионилхлорида, и нагревание продолжают в течение ночи. Дихлорметан удаляют путем перегонки и остаток растворяют в 50 мл дихлорметана. Раствор промывают смесью 20 мл воды и 3 мл 1-м. раствора бикарбоната натрия, водную промывочную жидкость экстрагируют 20 мл дихлорметана. Дихлорметановые растворы объединяют, промывают 40 мл рассола, сушат над сульфатом магния, а растворитель упаривают. При этом получают 1,0 г 7-(1-хлорэтил)-1,2,4-триазол-[1,5-а] пиримидина в качестве светло-коричневого твердого вещества. 3,81 мл триэтиламина подают в смесь 4,16 г (R)-миндальной кислоты в 50 мл сухого ацетонитрила, сушенного над молекулярной сите размером отверстий, равном 4А. По истечении 5 минут в смесь подают 0,90 г 7-(1-хлорэтил)-1,2,4-триазол-[1,5-а]пиримидина. Смесь в течение ночи нагревают с обратным холодильником и растворитель упаривают под пониженным давлением при температуре 60oC. Остаток экстрагируют 100 мл этилацетата и 50 мл воды. Этилацетатный слой последовательно промывают смесью 20 мл воды и 3 мл 1-м. раствора бикарбоната натрия, и 20 мл рассола. Раствор сушат над сульфатом магния и растворитель удаляют под пониженным давлением при температуре 60oC. При этом получают 1,33 г смеси двух диастереоизмеров: (R) сложный этиловый эфир (+)-1-(1,2,4-триазол[1,5-а] -пиримидин-7-ил)-миндальной кислоты и (R) сложный этиловый эфир (-)-1-(1,2,4-триазол[1,5-а] -пиримидин-7-ил)- миндальной кислоты. Менее полярный диастереоизомер можно выделять путем фракционированной кристаллизации и приемом, аналогичным, описанному в примере 24а, переводить в (+)-7-[1-(4-хлорфенокси)этил]- 1,2,4-триазол[1,5-а]-пиримидин (через (+)-1-(1,2,4-триазол[1,5-а]- пиримидин-7-ил)этанол в качестве промежуточного продукта).
Пример 25.
Из рацемического 7-[1-(4-хлорфенокси)этил]-1,2,4-триазол [1,5-а]-пиримидина путем высокопроизводительной жидкостной хроматографии приемом, аналогичным, описанному в примере 24 в качестве второй элюированной фракции выделяют 1,9 г (-)-7-[1-(4-хлорфенокси)этил]- 1,2,4-триазол[1,5-а]-пиримидина. Оптическая чистота выше 98%; удельное вращение [α]
ЭД50 данного соединения в вышеописанном опыте "BICM": 9,4 мг/кг.
ЭД50 данного соединения в вышеописанном опыте "MESM": 77,2 мг/кг.
В следующем примере 25а иллюстрируется дальнейший метод получения (-)-7-[1-(4-хлорфенокси)этил]-1,2,4-триазол[1,5-а]-пиримидина.
Пример 25а.
Раствор 10,75 г смеси двух диастереоизомеров (R) сложных эфиров (+) и (-) 1-(1,2,4-триазол[1,5-а] -пиримидин-7-ил)-миндальной кислоты, полученной аналогичным примеру 24а или 24б приемом, в этилацетате подвергают фракционированной кристаллизации с получением двух фракций: 2,8 г менее полярного диастереоизомера в качестве первой фракции и 3,3 г более полярного диастереоизомера в качестве второй фракции. 0,7 г более полярного диастереоизомера из второй фракции, 1,61 г карбоната калия, 10 мл воды и 25 мл метанола перемешивают в течение 1,5 часов, после чего упаривают метанол. Смесь разбавляют 50 мл воды и в течение 2 часов непрерывно экстрагируют дихлорметаном. Экстракты сушат над сульфатом магния и растворитель упаривают с получением 0,28 г (-)-1-(1,2,4- триазол[1,5-а]-пиримидин-7-ил)этанола.
Смесь 0,27 г (-)-1-(1,2,4-триазол[1,5-а]-пиримидин-7-ил)этанола, 0,44 г диэтилазодикарбоксилата, 0,68 г трифенилфосфина и 0,22 г 4-хлорфенола в 20 мл сухого тетрагидрофурана оставляют стоять в течение ночи при комнатной температуре до окончания реакции. Растворитель удаляют при пониженном давлении и остаток растворяют в 100 мл этилацетата. Раствор последовательно промывают 40 мл 1-м. раствора гидроокиси натрия и 20 мл рассола и растворитель упаривают. Остаток очищают путем флеш-хроматографии на силикагеле с применением в качестве элюента смеси триэтиламина и этилацетата в соотношении 1:100. Получают 0,26 г (-)-7-[1-(4-хлорфенокси)этил]-1,2,4- триазол-[1,5-а]-пиримидина.
Пример 26
A.
20 г рацемического 7-[1-(4-трифторметоксифенокси)этил]-1,2,4-триазоло [1,5-а] пиримидина разделяют на энантиомеры высокоэффективной жидкостной хроматографии на силикагеле типа хирацель в колонке с внутренними размерами 50 х 10 см с использованием в качестве элюента смеси изогексана и этанола в соотношении 9 : 1. В качестве первой фракции получают (+)-7-[1-(4- трифторметоксифенокси)этил] -1,2,4-триазоло[1,5-а] пиримидина с 100%- ной оптической чистотой. Свободное основание представляет собой оранжевое масло, которое растворяют в диэтиловом эфире, и получаемый раствор насыщают газообразным хлористым водородом. При этом получают 9,46 г гидрохлорида (+)-7-[1-(4-трифторметоксифенокси)этил] -1,2,4- триазоло[1,5-а]пиримидина с т.пл. 156-158oC. [α]
Б.
В результате вышеуказанной жидкостной хроматографии получают в качестве второй фракции (-)-7-[1-(4-трифторметоксифенокси)этил] - 1,2,4-триазоло[1,5-а] пиримидина с 98%-ной оптической чистотой. Свободное основание представляет собой оранжевое масло, которое растворяют в диэтиловом эфире, и получаемый раствор насыщают газообразным хлористым водородом. При этом получают 7,2 г гидрохлорида (-)-7-[1-(4-трифторметоксифенокси)этил]-1,2,4-триазоло [1,5-а]пиримидина с т.пл. 156 - 158oC [α]
Примеры фармацевтических композиций.
Пример А.
Таблетки состава (вес. части): 10 активного начала, 190 лактозы, 22 кукурузного крахмала, 10 поливинилпирролидона и 3 стеарата магния приготовляют следующим образом.
Активное начало, лактозу и часть кукурузного крахмала измельчают, перемешивают и получаемую при этом смесь гранулируют с помощью раствора поливинилпирролидона в этаноле. После сушки гранулы смешивают с стеаратом магния и остаточным количеством кукурузного крахмала. Получаемую при этом смесь подают в таблетировочную машину с тем, чтобы получить таблетки, каждая из которых содержит 10 мг активного начала.
Пример Б.
Таблетки приготовляют тем же образом, что и описано в примере А. Затем на таблетки известными приемами наносят покрытие, состоящее из раствора 20%-ного фталата ацетата целлюлозы и 3%-ного диэтилфталата в смеси этанола и дихлорметана в соотношении 1:1 по объему в качестве растворителя.
Пример В.
При приготовлении капсул 10 вес. частей активного начала и 240 вес. частей лактозы измельчают и смешивают. Получаемую смесь загружают в капсулы из твердой желатины с таким расчетом, что каждая капсула содержит 10 мг активного начала.
Пример Г.
При приготовлении капсул 50 вес. частей активного начала, 300 вес. частей лактозы и 3 вес. части стеарата магния измельчают и смешивают. Получаемую смесь загружают в капсулы из твердой желатины с таким расчетом, что каждая капсула содержит 50 мг активного начала.
Пример Д.
При приготовлении суппозиторий 100 вес.частей активного начала срабатывают в 1300 вес.частей смеси полусинтетических триглицеридов и получаемую при этом смесь переводят в суппозитории, каждый из которых содержит 100 мг активного начала.
Пример E.
При приготовлении основы для мази 0,1 г активного начала врабатывают в основу, состоящую из 9,9 г белого мягкого парафина путем гомогенизации до того, как активное начало равномерно распределено. 10 г мази загружают в янтарные банки с навертными крышками.
Изобретение относится к новым азотсодержащим гетероциклическим соединениям, обладающим биологической активностью, более конкретно к производным 1,2,4-триазоло[1,5-a]пиримидинов, их фармацевтически приемлемым солям и стереоизомерам, фармацевтической композиции, их содержащей, и способу ингибирования припадков. Описываются производные 1,2,4-триазоло-[1,5-a]пиримидинов общей формулы I, где R1, R4 и R5 являются атомом водорода или алкилом с 1 - 6 атомами углерода; R2 и R3 являются атомом водорода; R6, R7 и R8 независимо друг от друга являются атомом водорода, галоидом, цианогруппой, алканоилом с 1 - 6 атомами углерода, алкоксилом с 1 - 6 атомами углерода, незамещенным или замещенным галоидом, алкилтио с 1 - 6 атомами углерода, алкилсульфонилом с 1 - 6 атомами углерода, алкилсульфонилом с 1 - 6 атомами углерода, алкилом с 1 - 6 атомами углерода, незамещенным или замещенным галоидом, при этом, если R1, R2, R3, R4 и R8 - атомы водорода, R5 - метил, R6 и R7 - атомы водорода или R6 - 4-хлор- и R7 - атом водорода или 2-хлор, то соединение формулы I не представляет собой рацемат, и их фармацевтически приемлемыми солями и стереоизомерами. Описывается фармацевтическая композиция, обладающая противосудорожной активностью на основе соединений формулы I и способ ингибирования припадков с использованием соединений формулы I. 3 с. и 8 з.п. ф-лы.
 где R1, R4 и R5 являются атомом водорода или алкилом с 1 - 6 атомами углерода;
где R1, R4 и R5 являются атомом водорода или алкилом с 1 - 6 атомами углерода;
R2 и R3 являются атомом водорода;
R6, R7 и R8 независимо друг от друга являются атомом водорода, галоидом, цианогруппой, алканоилом с 1 - 6 атомами углерода, алкоксилом с 1 - 6 атомами углерода, незамещенным или замещенным галоидом, алкилтио с 1 - 6 атомами углерода, алкилсульфинилом с 1 - 6 атомами углерода, алкилсульфонилом с 1 - 6 атомами углерода, алкилом с 1 - 6 атомами углерода, незамещенным или замещенным галоидом,
при этом, если R1, R2, R3, R4 и R8 - атомы водорода, R5 - метил, R6 и R7 атомы водорода или R6 - 4-хлор и R7 - атом водорода или 2-хлор, то соединение формулы I не представляет собой рацемат,
и их фармацевтически приемлемые соли и стереоизомеры.
7-[1-(4-фторфенокси)этил]-1,2,4-триазоло[1,5-a]пиримидин;
7-[1-(4-хлорфенокси)этил]-1,2,4-триазоло[1,5-a]пиримидин;
7-[1-(4-бромфенокси)этил]-1,2,4-триазоло[1,5-a]пиримидин;
7-[1-(4-цианофенокси)этил]-1,2,4-триазоло[1,5-a]пиримидин;
7-[1-(4-трифторметилфенокси)этил]-1,2,4-триазоло[1,5-a]пиримидин;
7-[1-(4-метоксифенокси)этил]-1,2,4-триазоло[1,5-a]пиримидин;
7-[1-(4-трифторметоксифенокси)этил]-1,2,4-триазоло[1,5-a]пиримидин;
7-[1-(4-ацетилфенокси)этил]-1,2,4-триазоло[1,5-a]пиримидин;
7-{1-[4-(метилтио)фенокси]этил}-1,2,4-триазоло[1,5-a]пиримидин;
7-[1-(4-метилсульфинилфенокси)этил]-1,2,4-триазоло[1,5-a]пиримидин;
7-[1-(4-метисульфонилфенокси)этил]-1,2,4-триазоло[1,5-a]пиримидин;
7-{1-[4-(этилтио)фенокси]этил}-1,2,4-триазоло[1,5-a]пиримидин;
7-[1-(3-хлорфенокси)этил]-1,2,4-триазоло[1,5-a]пиримидин;
7-[1-(2,4-дифторфенокси)этил]-1,2,4-триазоло[1,5-a]пиримидин;
7-[1-(2,4-дихлорфенокси)этил]-1,2,4-триазоло[1,5-a]пиримидин;
7-[1-(3,4-дихлорфенокси)этил]-1,2,4-триазоло[1,5-a]пиримидин;
7-[1-(2-хлор-4-фторфенокси)этил]-1,2,4-триазоло[1,5-a]пиримидин;
7-[1-(4-хлорфенокси)этил]-2-метил-1,2,4-триазоло[1,5-a]пиримидин;
7-(4-хлорфеноксиметил)-1,2,4-триазоло[1,5-a]пиримидин;
7-[1-(4-хлорфенокси)-1-метилэтил]-1,2,4-триазоло[1,5-a]-пиримидин;
7-[1-(4-хлорфенокси)пропил]-1,2,4-триазоло[1,5-a]-пиримидин.
(+)-7-[1-(4-фторфенокси)этил]-1,2,4-триазоло[1,5-a]пиримидин;
(-)-7-[1-(4-фторфенокси)этил]-1,2,4-триазоло[1,5-a]пиримидин;
(+)-7-[1-(4-хлорфенокси)этил]-1,2,4-триазоло[1,5-a]пиримидин;
(-)-7-[1-(4-фторфенокси)этил]-1,2,4-триазоло[1,5-a]пиримидин.
где R'1, R'4 и R'5 являются атомом водорода или алкилом с 1 - 6 атомами углерода;
R'2 и R'3 являются атомом водорода;
R'6, R'7 и R'8 независимо друг от друга являются атомом водорода, галоидом, цианогруппой, алканоилом с 1 - 6 атомами углерода, алкоксилом с 1 - 6 атомами углерода, незамещенным или замещенным галоидом, алкилтио с 1 - 6 атомами углерода, алкилсульфинилом с 1 - 6 атомами углерода, алкилсульфонилом с 1 - 6 атомами углерода, алкилом с 1 - 6 атомами углерода, незамещенным или замещенным галоидом,
или его фармацевтически приемлемую соль или стереоизомер в терапевтически эффективном количестве.
R'2 и R'3 являются атомом водорода;
R'6, R'7 и R'8 независимо друг от друга являются атомом водорода, галоидом, цианогруппой, алкилом с 1 - 4 атомами углерода, незамещенным или замещенным галоидом, алкоксилом с 1 - 4 атомами углерода, незамещенным или замещенным галоидом, алкилтио с 1 - 4 атомами углерода, незамещенным или замещенным галоидом, алканоилом с 1 - 4 атомами углерода, алкилтио с 1 - 4 атомами углерода, алкилсульфинилом с 1 - 4 атомами углерода и алкилсульфонилом с 1 - 4 атомами углерода.
7-[1-(4-фторфенокси)этил]-1,2,4-триазоло[1,5-a]пиримидин;
7-[1-(4-хлорфенокси)этил]-1,2,4-триазоло[1,5-a]пиримидин;
7-[1-(4-бромфенокси)этил]-1,2,4-триазоло[1,5-a]пиримидин;
7-[1-(4-цианофенокси)этил]-1,2,4-триазоло[1,5-a]пиримидин;
7-[1-(4-трифторметилфенокси)этил]-1,2,4-триазоло[1,5-a]пиримидин;
7-[1-(4-метоксифенокси)этил]-1,2,4-триазоло[1,5-a]пиримидин;
7-[1-(4-трифторметоксифенокси)этил]-1,2,4-триазоло[1,5-a]пиримидин;
7-[1-(4-ацетилфенокси)этил]-1,2,4-триазоло[1,5-a]пиримидин;
7-{1-[4-(метилтио)фенокси]этил}-1,2,4-триазоло[1,5-a]пиримидин;
7-[1-(4-метилсульфинилфенокси)этил]-1,2,4-триазоло[1,5-a]пиримидин;
7-[1-(4-метилсульфонилфенокси)этил]-1,2,4-триазоло[1,5-a]пиримидин;
7-{1-[4-(этилтио)фенокси]этил}-1,2,4-триазоло[1,5-a]пиримидин;
7-[1-(3-хлорфенокси)этил]-1,2,4-триазоло[1,5-a]пиримидин;
7-[1-(2,4-дифторфенокси)этил]-1,2,4-триазоло[1,5-a]пиримидин;
7-[1-(2,4-дихлорфенокси)этил]-1,2,4-триазоло[1,5-a]пиримидин;
7-[1-(3,4-дихлорфенокси)этил]-1,2,4-триазоло[1,5-a]пиримидин;
7-[1-(2-хлор-4-фторфенокси)этил]-1,2,4-триазоло[1,5-a]пиримидин;
7-[1-(4-хлорфенокси)этил]2-метил-1,2,4-триазоло[1,5-a]пиримидин;
7-(4-хлорфеноксиметил)-1,2,4-триазоло[1,5-a]пиримидин;
7-[1-(4-хлорфенокси)-1-метилэтил]-1,2,4-триазоло[1,5-a]пиримидин;
7-[1-(4-хлорфенокси)пропил]-1,2,4-триазоло[1,5-a]пиримидин.
(+)-7-[1-(4-фторфенокси)этил]-1,2,4-триазоло[1,5-a]-пиримидин;
(-)-7-[1-(4-фторфенокси)этил]-1,2,4-триазоло[1,5-a]-пиримидин;
(+)-7-[1-(4-хлорфенокси)этил]-1,2,4-триазоло[1,5-a]-пиримидин;
(-)-7-[1-(4-фторфенокси)этил]-1,2,4-триазоло[1,5-a]-пиримидин.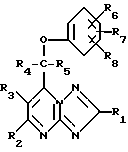
где R1, R4 и R5 являются атомом водорода или алкилом с 1 - 6 атомами углерода;
R2 и R3 являются атомом водорода;
R6, R7 и R8 независимо друг от друга являются атомом водорода, галоидом, цианогруппой, алканоилом с 1 - 6 атомами углерода, алкоксилом с 1 - 6 атомами углерода, незамещенным или замещенным галоидом, алкилтио с 1 - 6 атомами углерода, алкилсульфинилом с 1 - 6 атомами углерода, алкилсульфонилом с 1 - 6 атомами углерода, алкилом с 1 - 6 атомами углерода, незамещенным или замещенным галоидом, или его фармацевтически приемлемую соль или стереоизомер в дневной дозе, равной примерно 1 - 1000 мг.
| Способ получения производных триазолопиримидина или их солей с метансульфокислотой | 1984 |
|
SU1347865A3 |
| Способ размножения копий рисунков, текста и т.п. | 1921 |
|
SU89A1 |
| Машковский М.Д | |||
| Лекарственные средства | |||
| - М.: Медицина, ч.1, с.112-120. | |||
