Изобретение относится к новым производным изохинолина, обладающим фармакологической активностью, более конкретно к новым производным тетрагидроизохинолина.
Известны производные изохинолина, обладающие фармакологической активностью (см. Европейскую заявку N 0419247, опубл. в 1991 г.).
Задачей изобретения является разработка новых производных изохинолина, обладающих фармакологической активностью.
Данная задача решается предлагаемыми производными тетрагидроизохинолина общей формулы (I)
Данная задача решается предлагаемыми производными тетрагидроизохинолина общей формулы (I)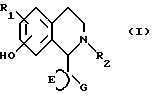
в которой R1 - водород или алкил с 1 - 3 атомами углерода;
R2 - алкил с 1 - 3 атомами углерода,
E - алкиленовая цепь, содержащая 2 или 3 атома углерода,
G - насыщенная или ненасыщенная алициклическая группа, содержащая 3 - 8 атомов углерода, алкил с 1 - 6 атомами углерода, незамещенный или замешенный алкилом с 1 - 3 атомами углерода или циклоалкилом с 3 - 7 атомами углерода, пиридил, фурил, тиенил или нафтил,
и их фармацевтически приемлемыми солями в форме индивидуальных энантиомеров, рацематов или других смесей энантиомеров.
В первую группу предпочтительных производных тетрагидроизохинолина вышеприведенной формулы (I) входят соединения, у которых
R1 - водород или метил.
Во вторую группу предпочтительных производных тетрагидроизохинолина вышеприведенной формулы (I) входят соединения, у которых
R2 - метил.
В третью группу предпочтительных производных тетрагидроизохинолина вышеприведенной формулы (I) входят соединения, у которых
G - метилалкил, циклоалкил, циклоалкенил, 1,2,3,4-тетрагидронафтил, тиенил, фурил или пиридил.
В четвертую группу предпочтительных производных тетрагидроизохинолина вышеприведенной формулы (I) входят соединения, у которых гидроксильная группа находится в положении 7 тетрагидроизохинолинового кольца.
Эти соединения имеют общую формулу (II)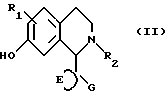
в которой R1, R2, E и G имеют значения, указанные выше.
В пятую группу предпочтительных производных тетрагидроизохинолина вышеприведенной формулы (I) входят соединения, выбранные из группы, включающей: 7-гидрокси-2-метил-1-[1-(2-метилпропил)-циклобутил] - 1,2,3,4-тетрагидроизохинолин; 1-[1-(циклопентилметил)-циклопропил]-7-гидрокси-2,6-диметил- 1,2,3,4-тетрагидроизохинолин; 1-[1-(циклогекс-1-ен-3-ил)-циклобутил] -7- гидрокси-2,6-диметил- 1,2,3,4-тетрагидроизохинолин; 7-гидрокси-2,6-диметил-1-[1-( 1,2,3,4-тетрагидронафт-1-ил)- циклопропил]-1,2,3,4-тетрагидроизохинолин; 7-гидрокси-2,6-диметил-1-[1-(2-тиенил)-циклопропил] -1,2,3,4- тетрагидроизохинолин; 7-гидрокси-2,6-диметил-1-[1-(3-тиенил)-циклопропил] - 1,2,3.4-тетрагидроизохинолин; 1-[1-(2-фурил)-циклопропил] -7-гидрокси-2,6- диметил-1,2,3,4-тетрагидроизохинолин; 7-гидрокси-2-метил-1-[1 -(2-пиридил)-циклобутил] -1,2,3,4-тетрагидроизохинолин; 7-гидрокси-2,6-диметил-1-[1-(2-пиридил)-циклопропил]-1,2,3,4- тетрагидроизохинолин
и их фармацевтически приемлемые соли в форме индивидуальных энантиомеров, рацематов или других смесей энантиомеров.
Характерными формами энантиомеров соединений формулы (I) являются:
(-)-7-гидрокси-2,6-диметил-1-[1 -(3-тиенил)-циклопропил] - 1,2,3,4-тетрагидроизохинолин и его фармацевтически приемлемые соли.
Производные тетрагидроизохинолина вышеприведенной общей формулы (I) и их фармацевтически приемлемые соли могут применяться как таковые или в виде стандартных фармацевтических композиций для аналгезии или лечения психозов, болезни Паркинсона, синдрома Леша-Найхана, расстройства познавательной способности, лекарственной зависимости или поздней дискинезии.
Соединения общих формул (I), (II) могут находиться в виде солей фармацевтически приемлемых кислот. Примеры таких солей включают гидрохлориды, гидробромиды, гидройодиды, сульфаты, нитраты, малеаты, ацетаты, цитраты, фумараты, тартраты, сукцинаты, бензоаты, пальмоаты, метилсульфаты, додеканоаты и соли аминокарбоновых кислот, таких как глутаминовая кислота.
Соединения формул (I), (II) и их соли могут также находиться в форме сольватов (например, гидратов).
Для специалиста в данной области понятно, что соединения формул (I), (II) содержат хиральный центр. Когда соединение формулы (I), (II) содержит один хиральный центр, оно существует в форме двух энантиомеров. Объем настоящего изобретения охватывает индивидуальные энантиомеры и смеси таких энантиомеров. Энантиомеры можно получать по методикам, известным в данной области. Такие известные типичные способы включают разделение путем образования диастереоизомерных солей, которые можно разделить, например, путем кристаллизации; разделение путем образования диастереоизомерных производных или комплексов, которые можно разделить, например, путем кристаллизации, газожидкостной или жидкостной хроматографии; разделение путем селективного взаимодействия одного энантиомера со специфичным для этого энантиомера реагентом, например, энзиматической этерификацией, окислением или восстановлением; или разделение путем газо-жидкостной или жидкостной хроматографии в хиральной среде, например, на хиральном носителе, таком как силикагель со связью хирального лиганда или в присутствии хирального растворителя. Разумеется, когда определенный энантиомер превращается в другую химическую структуру по одному из способов разделения, описанных выше, необходим следующий этап для выделения целевой энантиомерной формы. С другой стороны, некоторые энантиомеры можно синтезировать путем асимметричного синтеза с использованием оптически активных реагентов, подложек, катализаторов или растворителей или превращением одного энантиомера в другой путем асимметричной трансформации.
Когда соединение формулы (I), (II) содержит больше, чем один хиральный центр, оно может находиться в диастереоизомерных формах. Диастереоизомерные формы можно разделять по методикам, известным в данной области, например, хроматографией или кристаллизацией, и индивидуальные энантиомеры в каждой паре можно разделить, как описано выше. Настоящее изобретение включает каждый диастереоизомер соединений формулы (I), (II) и их смеси.
Определенные соединения формулы (I), (II) могут находиться в более, чем одной кристаллической форме и настоящее изобретение охватывает каждую кристаллическую форму и их смеси.
Стандартные фармацевтические композиции включают терапевтически эффективное количество соединения формулы (I), (II) наряду с фармацевтически приемлемым разбавителем или носителем.
Использованный в данном тексте термин "активное соединение" означает соединение формулы (I), (II). При терапевтическом применении активное соединение можно принимать орально, ректально, парентерально или местным употреблением, предпочтительно орально. Таким образом, терапевтическая композиция может находиться в виде любой известной фармацевтической формы для орального, ректального, парентерального или местного употребления. Фармацевтически приемлемые носители, пригодные для применения в таких композициях, хорошо известны в области фармацевтики.
Фармацевтическая композиция может содержать 0,1-90 вес.% активного соединения. Обычно композиции готовят в виде разовых дозировочных форм.
Композиции для орального употребления являются предпочтительными и такие фармацевтические формы хорошо известны для такого употребления, например, таблетки, капсулы, гранулы, сиропы, растворы и водные или масляные суспензии. Наполнители, используемые при приготовлении этих композиций, являются хорошо известными для фармацевтов. Таблетки можно приготовить из смеси активного соединения с твердыми наполнителями, такими как фосфат кальция: дезинтегрирующими агентами, например, кукурузный крахмал; агентами-смазками, например, стеарат магния; связующими веществами, например, микрокристаллическая целлюлоза или поливинилпирролидон, и другими необязательными компонентами, известными из уровня техники для содействия таблетированию смеси известными методами. Если необходимо, на таблетки можно наносить покрытия, используя известные методики и вещества, которые могут включать покрытие энтеросолюбильной оболочкой с использованием, например, фталата гидроксипропилметилцеллюлозы. Таблетки могут быть приготовлены известным образом, так чтобы было обеспечено устойчивое выделение соединения по данному изобретению. Если необходимо, такие таблетки можно снабжать внешним покрытием известным способом, например, с применением фталата-ацетата целлюлозы. Подобным образом, капсулы, например, твердые или мягкие желатиновые капсулы, содержащие активное соединение согласно данному изобретению с добавлением наполнителей или без них, можно приготавливать известными способами и, при необходимости, известным образом снабжать внешним покрытием. Содержание капсулы можно готовить известным образом при достижении устойчивого выделения активного соединения. Таблетки или капсулы могут приемлемо содержать от 1 до 500 мг активного соединения.
Другие композиции для орального применения могут включать, например, водные суспензии, содержащие активное соединение в водной среде в присутствии нетоксичного суспендирующего агента, такого как натрий-карбоксиметилцеллюлоза, и водные суспензии, содержащие соединения согласно данному изобретению в подходящем растительном масле, например, арахисовом масле. Активное соединение может находиться в виде гранул с добавлением наполнителей или без них. Гранулы можно вводить пациенту непосредственно или их можно добавлять в подходящий жидкий носитель (например, воду) перед введением пациенту. Гранулы могут содержать дезинтеграторы, например, выделяющую пузырьки пару, состоящую из кислоты и карбонатной или бикарбонатной соли, для содействия распределению в жидкой среде.
Композиции, пригодные для ректального употребления, являются известными фармацевтическими формами для такого употребления, например, свечки с твердым жиром или на основе полиэтиленгликоля.
Композиции, пригодные для парентерального употребления, являются известными фармацевтическими формами для такого употребления, например, стерильные суспензии или стерильные растворы в подходящем растворителе.
Композиции для местного употребления могут включать матрицу, в которой фармакологически активные соединения согласно изобретению могут быть диспергированы таким образом, что соединения поддерживаются в контакте с кожей для введения в организм через кожу. С другой стороны, активные соединения могут быть диспергированы в фармацевтически приемлемом креме, геле или мази. Количество активного соединения, находящееся в составе для местного употребления, должно быть таким, чтобы терапевтически эффективное количество соединения выделялось в течение периода времени, за которое черезкожный состав находится на коже.
Соединения согласно данному изобретению можно также употреблять непрерывным введением или из внешнего источника, например, внутривенным вливанием, или из источника соединения, находящегося внутри организма. Внутренние источники включают имплантированные резервуары, содержащие требуемое соединение, которое постоянно выделяется, например, путем осмоса, и имплантанты, которые могут быть (а) жидкими, такими как суспензия или раствор в фармацевтически приемлемом масле соединения, которое должно быть введено, например, в форме очень умеренно растворимого в воде производного, такого как соль - додеканат, или соединения формулы (III), как описано выше, или (б) твердыми в форме имплантированного носителя, например, синтетической смолы или воскоподобного вещества, для соединения, которое должно быть введено. Все количество соединения может одновременно содержаться в одном носителе или несколько носителей могут содержать частичные количества соединения. Количество активного соединения должно быть таким, чтобы терапевтически эффективное количество соединения выделялось в течение долгого периода времени.
В некоторых составах может быть полезным использовать соединения согласно данному изобретению в форме частиц очень маленького размера, например, получаемые при жидкостном размалывании.
В фармацевтических композициях активное соединение может быть ассоциировано с другими совместимыми фармакологически активными компонентами.
Как уже указывалось выше, фармацевтическая композиция, содержащая терапевтически эффективное количество соединения формулы (I), (II), может быть использована в аналгезии или для лечения психозов (например, шизофрении), болезни Паркинсона, синдрома Леша-Найхана, расстройства познавательной способности, лекарственной зависимости или поздней дискинезии.
При таком лечении количество соединения формулы (I), (II), которое может употребляться орально, ректально или парентерально, составляет ежедневно от 0,1 до 5000 мг, предпочтительно от 5 до 500 мг. взятое в виде одной дозы или разделенное на несколько доз, принимаемых один или несколько раз в течение дня.
Соединения формулы (I), (II) можно употреблять для лечения болезни Паркинсона или сами по себе, или в сочетании с предшественником допамина, таким как ингибитор леводопа и/или допа декарбоксилазы, такой как карбидопа или бензеразид.
Ниже представлено описание способов получения соединений формулы (1).
Соединения формулы (I) могут быть получены путем превращения соединений формулы (III).
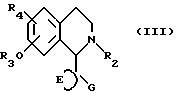
в которой R3 означает алкильную группу (например, метил или бензил), которая может быть замещена и R4 имеет значения группы R1 (см. выше) или это группа, которая может быть превращена в группу R1.
Деметилирование может быть осуществлено путем реакции с бромистоводородной кислотой, при необходимости в присутствии ледяной уксусной кислоты, с трибромидом бора, с гидрохлоридом пиридина, с этанэтиолатом натрия, с цианидом натрия или с триметил-йод-силаном. Дебензилирование можно осуществлять путем гидролиза, например, кислотным гидролизом или гидрогенолизом, например, с использованием катализатора, например, палладия на активном угле.
Соединения формулы (I) можно также получать алкилированием соединений формулы (IV)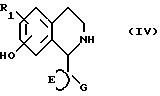
осуществляемым при условиях, при которых не происходит алкилирование гидроксильной группы. Например, соединения формулы (I), в которой R2 означает метил, можно получить путем метилирования соединений формулы (IV), например, используя формальдегид и муравьиную кислоту или формальдегид и циан-боргидрид натрия.
Соединения формулы (II) можно получать по методикам, аналогичным описанным выше для получения соединений формулы (I).
Соединения формулы (III) можно получать алкилированием соединений формулы (V)
например, путем реакции с алкил-галоидом (например, метил-йодидом) или алкенил-галоидом (например, аллил-йодидом или аллил-бромидом).
Соединения формулы (III) можно также получать путем восстановительного алкилирования соединений формулы (V), например, путем реакции с альдегидом или кетоном и восстанавливающим агентом. Например, соединения формулы (III), в которой R2 означает метил, можно получить метилированием соединений формулы (V), например, с использованием формальдегида и муравьиной кислоты, формальдегида и первичного кислого фосфита натрия или формальдегида и цианборгидрида натрия.
Соединения формулы (III), в которой R2 означает метил, можно получать реакцией соединений формулы (VI)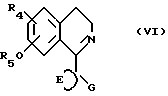
в которой R5 имеет те же значения, что и группа R3, при условиях, при которых происходит восстановление и метилирование соединения формулы (VI), например, путем реакции соединения формулы (VI) с формальдегидом и восстанавливающим агентом, таким как циан-боргидрид натрия, или с муравьиной кислотой и восстанавливающим агентом, таким как боргидрид натрия.
Соединения формулы (III) можно также получать реакцией соединения формулы (VII)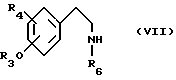
в которой R6 имеет значения группы R2, с соединением формулы (VIII)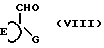
в присутствии кислоты, например, хлористоводородной кислоты.
Соединения формулы (III) можно получать и восстановлением соединений формулы (IX)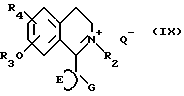
в которой Q- является подходящим анионом, таким как йодид или метилсульфат, например, с боргидридом натрия, цианборгидридом натрия, бором, комплексом бор-диметилсульфид, литий-алюминий-гидридом или путем каталитического гидрирования. Для получения одного из энантиомеров соединения формулы (III) можно использовать хиральные восстанавливающие агенты, такие как хиральные триацилоксиборгидриды натрия (например, подходящие энантиомеры трис-(N-бензилоксикарбонил-пропилокси)-боргидрида натрия или трис-[N-(2-метилпропилоксикарбонил)-пропилокси] -боргидрида натрия), хиральные диалкилокси-бораны или хиральные оксазаборолидины. Один из энантиомеров соединений формулы (III) можно получать каталитическим гидрированием с использованием хирильного катализатора. Подходящим катализатором является комплекс, образованный реакцией хирального фосфина (например, 2,3-0-изопропилиден-2,3-дигидрокси-1,4-бис-(дифенилфосфин)-бутан) с комплексом переходного металла (например, димер хлор-(1,5-циклооктадиен)-родия(1)).
Соединения формулы (IV) можно получать превращением соединений формулы (V), в которой R4 имеет те же значения, что и R1, или означает группу, которую можно превратить в группу R1 подобно тому, как описано выше в отношении соединений формулы (I).
Соединения формулы (IV) можно получать восстановлением соединений формулы (VI), в которой R5 означает водород, например, с использованием реакций восстановления, подобных описанным выше для восстановления соединений формулы (IX). Можно использовать восстанавливающие агенты для получения одного из энантиомеров соединения формулы (IV) таким же образом, как это описано выше для восстановления соединений формулы (IX).
Соединения формулы (V) можно получать восстановлением соединений формулы (VI), в которой R5 имеет значения группы R3, таким же образом, как описано выше для получения соединений формул (III) и (IV).
Соединения формулы (V) можно получать восстановлением соединений формулы (X)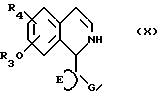
например, при использовании каталитического гидрирования.
Соединения формулы (V) можно получать реакцией соединения формулы (VII), в которой R6 означает водород, с соединением формулы (VIII) присутствии кислоты, например, хлористоводородной кислоты.
Соединения формулы (VI) можно получать циклизацией соединений формулы (XI)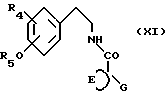
в которой R5 означает водород или R3. Циклизацию можно осуществлять в присутствии конденсирующего агента, такого как оксихлорид фосфора, пятиокись фосфора, пентахлорид фосфора, сложный эфир полифосфорной кислоты, полифосфорная кислота, хлорид цинка, хлористоводородная кислота, тионилхлорид или серная кислота.
Соединения формулы (VI) можно получать реакцией соединения формулы (XII)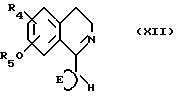
с основанием, таким как диизопропиламид лития, и соединением формулы X-G, в которой X является отщепляемой группой, такой как тозилокси-группа или галоген.
Соединения формулы (VIII) можно получать восстановлением циклоалканкарбонитрилов формулы (XIII)
с использованием ди-трет. -бутилалюминий-гидрида или диизобутил-алюминий-гидрида или восстановлением циклоалканкарбонил-хлоридов формулы (XIV)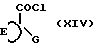
с использованием три-трет.-бутокси-алюминий-гидрида.
Соединения формулы (IX) можно получать реакцией соединения формулы (VI), в которой R5 имеет те же значения, что и R3, с алкилирующим агентом формулы R2Q, например, метилйодидом или диметил-сульфатом.
Соединения формулы (X) можно получать циклизацией соединений формулы (XV)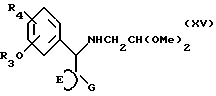
Циклизацию можно осуществлять в присутствии кислоты, такой как серная кислота.
Соединения формулы (XI) можно получать реакцией фенэтиламина формулы (XVI)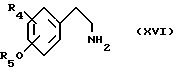
в которой R5 означает водород или R3, с циклоалканкарбонил-хлоридом формулы XIX, например, в присутствии органического основания, такого как триэтиламин. Соединения формулы (XI) можно также получать конденсацией фенэтиламина формулы (XVI) с циклоалканкарбоновой кислотой формулы (XVII)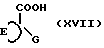
или ее сложным эфиром, например, путем приконденсирования или действием агента конденсации, такого как 1,1-карбонилдиимидазол или 1,3-дициклогексилкарбодиимид.
Соединения формулы (XII) можно получать циклизацией соединений формулы (XVIII)
при условиях, указанных выше для циклизации соединений формулы (XI).
Циклоалканкарбонитрилы формулы (XIII) можно получать реакцией карбонитрилов формулы (XIX)
G - CH2 - CN (XIX)
с дизамещенным соединением формулы (XX)
Z - E - Z' (XX),
в которой Z и Z', которые могут быть одинаковыми или различными, означают отщепляемые группы, такие как галоген, например, хлор или бром, в присутствии основания, такого как гидрид натрия или гидроокись калия.
Циклоалканкарбонитрилы формулы (XIII) можно также получать реакцией карбонитрила формулы (XXI)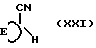
с основанием, таким как диизопропиламид лития и соединения формулы X-G, в которой X означает отщепляемую группу (например, галоген).
Циклоалканкарбонил-хлориды формулы (XIV) можно получать, исходя из циклоалканкарбоновых кислот формулы (XVII) по методикам, которые известны в данной области, например, путем реакции с тионилхлоридом.
Соединения формулы (XV) можно получать реакцией соединения формулы (XXII)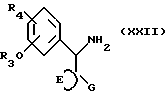
с галоидацетальдегид-диметилацеталем, например, хлорацетальдегид-диметилацеталем.
Циклоалканкарбоновые кислоты формулы (XVII) можно получать гидролизом например, основным гидролизом циклоалканкарбонитрилов формулы (XIII) или реакцией перекиси водорода с циклоалканкарбонитрилами формулы (XIII) в присутствии основания с последующей реакцией с азотной кислотой с образованием целевой карбоновой кислоты.
Соединения формулы (XVIII) можно получать реакцией фенилэтиламина формулы (XVI) с циклоалкан-карбонил-хлоридом формулы (XXIII)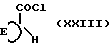
Соединения формулы (XXII) можно получать реакцией соединения формулы (XXIV)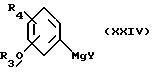
в которой Y означает галоген (например, хлор или бром), с цикло-алканкарбонитрилом формулы (XIII) с последующим восстановлением, например, боргидридом натрия.
Соединения формулы (XXIV) можно получать реакцией магния с соединением формулы (XXV)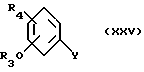
в которой Y означает галоген (например, хлор или бром).
Способность соединений формулы I или формулы II взаимодействовать с рецепторами допамина была продемонстрирована в испытаниях, описанных ниже, в которых определяли способность соединений ингибировать меченый тритием лиганд, связанный с рецепторами допамина "в пробирке" и, в частности, с рецепторами допамина D1 и D2.
Стриарные образцы из мозга самца крысы семейства Charles River CD весом 140-250 r гомогенизировали в охлаждаемом льдом буфере, содержащем 50 мм трис-HCl (pH 7,4 при измерении при 25oC для образца, связанного с D1, и pH 7,7 при измерении при 25oC для образца, связанного с D2) и центрифугировали в течение 10 минут (при 21000g в случае образца, связанного с D1, 40000 g в случае образца, связанного с D2), г). Осадок от центрифугирования ресуспендировали в таком же буфере, снова центрифугировали и перед каждым испытанием полученный осадок центрифугирования выдерживали при -80oC. Перед каждым испытанием твердую массу ресуспендировали в буфере, содержащем 50 мМ трис-HCl 120 мМ NaCl, 5 мМ KCl, 2 мМ CaCl2 и 1мМ MgCl2 при pH 7,4 для образцов, связанных с D1, и при pH 7,7 для образцов, связанных с D2. Аликвоты этой суспензии затем добавляли в колонки, содержащие лиганд и испытуемое соединение или лиганд и буфер. В случае образцов, связанных с D1, лигандом был меченый тритием SCH 23390 и смесь инкубировали при 37oC в течение 30 минут, после чего инкубирование заканчивали быстрой фильтрацией. В случае образца, связанного с D2, лигандом был меченый тритием (S)-сульпирид и смесь инкубировали в течение 40 минут, затем инкубацию заканчивали быстрой фильтрацией. Heспецифичное связывание определяли экспериментально путем добавления хлорпромазина или спироперидола для рецепторов D1 и D2 соответственно в концентрации насыщения.
Фильтры промывали охлаждаемым льдом трис-HCl буфером и высушивали. Фильтры измельчали в пробирки, содержащие жидкость сцинтилляции и выдерживали в течение 20 часов, затем подсчитывали сцинтилляцию спектрофотометрически. Сравнивали кривые связывания в области концентраций испытуемого соединения и рассчитывали коэффициент ингибирования Ki из данных с использованием нелинейного графика соответствующей программы компьютера EBDA ("Биософт").
Величины Ki, определенные в описанных выше испытаниях по связыванию с D1 и D2 для всех целевых веществ, полученных по приведенным ниже примерам, даны в таблице, в которой дано также отношение между этими двумя величинами.
Изобретение иллюстрируется следующими примерами, которые не ограничивают его объем. В этих примерах все температуры даны в градусах Цельсия. Конечные продукты в каждом из этих примеров были охарактеризованы одним или несколькими из следующих методов исследования: элементный анализ, спектроскопия ядерного магнитного резонанса и инфракрасная спектроскопия.
Пример 1. Циклобутанкарбонил-хлорид (5,5 г) добавляли к раствору 2-(4-метоксифенил)-этиламина (7 г) и триэтиламина (6,5 мл) в эфире (300 мл) при комнатной температуре. Смесь перемешивали в течение ночи, выливали в воду и подкисляли 2 М хлористоводородной кислотой, затем смесь экстрагировали этилацетатом (3 х 100 мл). Объединенные экстракты промывали рассолом, высушивали и растворители удаляли в вакууме с образованием осадка. Осадок промывали петролейным эфиром и высушивали в вакууме с получением N-[2-(4-метоксифенил)-этил] -циклобутанкарбоксиамида в виде твердой массы (9,54 г), темп.плавл. 118-120oC.
Порцию твердой массы (9 г) в осушенном ацетонитриле (170 мл), содержащем хлорокись фосфора (23,7 мл), нагревали с обратным холодильником в течение 43 часов. Охлажденную смесь затем выливали в разбавленный раствор аммиака и смесь экстрагировали этилацетатом (3 х 150 мл). Раствор этилацетата затем экстрагировали разбавленной хлористоводородной кислотой (3 х 100 мл). Водные кислотные экстракты нейтрализовали добавлением водного раствора аммиака и экстрагировали этилацететом. Органические экстракты промывали рассолом, высушивали и концентрировали с образованием масла, которое дистиллировали при 190oC/0,2 мбар с получением 1-цикло-бутил-7-метокси-3,4-дигидроизохинолина в виде бесцветной твердой массы (4 г), темп.плавл. 44-46oC.
Н. бутиллитий (2,83 мл, 1,8 М в гексане) добавляли по каплям к раствору диизопропиламина (0,71 мл) в сухом тетрагидрофуране (5 мл) при комнатной температуре. Через 15 минут раствор охлаждали до -23oC и медленно обрабатывали раствором 1-циклобутил-7-метокси-3,4-дигидроизохинолина (1 г) в тетрагидрофуране (11 мл). Раствор темно-зеленого цвета перемешивали в течение 30 минут, затем охлаждали до -78oC и обрабатывали при этой температуре 1-бром-2-метилпропаном (5,1 мл) в течение 1 часа. Затем смесь самопроизвольно нагревалась до комнатной температуры, затем ее нагревали с обратным холодильником в течение 1 часа. Смесь выливали в воду, подкисляли и промывали эфиром. Водную фазу затем подщелачивали добавлением водного раствора гидроокиси натрия и экстрагировали этилацетатом. Растворитель удаляли в вакууме с получением оранжевой смолы, которую очищали (флэш-хроматографией на силикагеле с использованием смеси этилацетата и легкого петролейного эфира (1:4) в качестве элюэнта с получением 7-метокси-1-1-(2-метилпропил)-циклобутил-3,4-дигидро-изохинолина в виде масла (0,85 г).
Смесь масла (0,98 г, полученное аналогично описанному выше), тетрагидрофурана (15 мл) и боргидрида натрия (1 г) охлаждали до 0oC и обрабатывали очень медленным прикапыванием муравьиной кислоты (10,3 мл) Смесь самопроизвольно нагревалась до комнатной температуры, затем ее перемешивали в течение 2 дней. Смесь выливали в воду и подщелачивали добавлением водного раствора гидроокиси натрия, экстрагировали эфиром. Экстракты осушали и растворитель удаляли в вакууме с образованием смолы, которая очень медленно отверждалась. Этот твердый продукт растворяли в эфире и обрабатывали эфирным раствором рацематной дибензоилвинной кислоты, осадок отфильтровывали и высушивали в вакууме. Соль перекристаллизовывали из изопропилового спирта с получением дибензоилтартрата 7-метокси-2-метил-1-[1-(2-метилпропил)-циклобутил]-1,2,3,4-дигидроизохинолина (0,8 г), темп. плавл. 135-136oC.
Указанную выше соль нейтрализовали и свободное основание (0,3 г) нагревали с обратным холодильником в мягких условиях в течение 3 часов в смеси 48%-ной бромистоводородной кислоты (12 мл) и уксусной кислоты (12 мл). Растворитель удаляли в вакууме и остаток осушали азеотропной дистилляцией с пропанолом-2. Остаток суспендировали в изопропропиловом спирте и собирали путем фильтрации. Осадок на фильтре промывали следующей порцией изопропилового спирта и затем высушивали в вакууме с получением чистого гидробромида 7-гидрокси-2-метил-1-[1-(2-метилпропил)-циклобутил-1,2,3,4 тетрагидроизохинолина (0,3 г), темп. плавл. 241-244oC (разл.).
Пример 2. Раствор бутиллития в гексане (2,5 М; 80 мл) добавляли к раствору диизопропиламина (27,8 мл) и 1,3-диметил-3,4,5,6-тетрагидро-2(1Н)-пиримидинона (28,6 мл) в тетрагидрофуране (188 мл) с перемешиванием при 0oC. Перемешивание продолжали в течение 0,5 часа, затем смесь охлаждали до -78oC и добавляли циклопропанкарбонитрил (13,4 г). После перемешивания при -78oC в течение следующего 1 часа медленно добавляли циклопентилметилбромид (37,6 г), затем в смеси самопроизвольно температура повышалась до комнатной и смесь перемешивали в течение следующих 18 часов. Смесь выливали в воду (1 л), затем экстрагировали этилацетатом (4 х 150 мл). Объединенные экстракты промывали рассолом, затем осушали над сульфатом магния и затем растворитель удаляли в вакууме. Твердое вещество удаляли фильтрацией и оставшееся масло тритурировали с эфиром, причем твердое вещество снова удаляли фильтрацией. Оставшееся масло затем очищали дистилляцией под пониженным давлением с получением 1-(циклопентилметил)-циклопропанкарбонитрила (8,9 г) в виде масла, темп. кип. 50oC/1 мбар.
Продукт предыдущей реакции (8,9 г), порошок гидроокиси калия (85%; 6,6 г) и 1,2-этандиола (65 мл) нагревали вместе с обратным холодильником в течение 50 часов. Смесь выливали в воду (200 мл), затем промывали эфиром. Водную фазу подкисляли концентрированной хлористоводородной кислотой, затем экстрагировали эфиром (5 х 50 мл). Объединенные экстракты осушали над сульфатом магния и растворитель удаляли в вакууме с получением 1-(циклопентилметил)-циклопропанкарбоновой кислоты (8,2 г) в виде рыжевато-коричневой твердой массы, темп. плавл. 42-45oC.
Раствор 1,1-карбонилдиимидазола (8,05 г) в тетрагидрофуране (125 мл) добавляли по каплям к раствору продукта предыдущей реакции (8,2 г) в тетрагидрофуране (125 мл) с перемешиванием при 0oC. Смесь перемешивали при комнатной температуре в течение 18 часов, затем добавляли раствор гидрохлорида 2-(4-метокси-З-метилфенил)-этиламина (8,9 г) и триэтиламина (6,27 мл) в тетрагидрофуране (200 мл). Смесь перемешивали при комнатной температуре в течение 18 часов, затем выливали на воду (500 мл) и подщелачивали водным раствором аммиака. Затем смесь экстрагировали этилацетатом (5 х 100 мл), объединенные экстракты промывали разбавленной хлористоводородной кислотой, затем рассолом, затем осушали над сульфатом магния. Растворитель удаляли в вакууме с образованием N-[2-(4-метокси-3-метилфенил)-этил] -1-(циклопентилметил)-циклопропанкарбоксамида (10,6 г) в виде смолы.
Смесь продукта предыдущей реакции (10,55 г), хлорокиси фосфора (21,3 мл) и ацетонитрила (170 мл) нагревали с обратным холодильником в течение 4,5 часов, затем охлаждали и выливали в ледяную воду (200 мл). Смесь подщелачивали водным раствором аммиака, затем экстрагировали этилацетатом (4 х 60 мл). Объединенные экстракты промывали рассолом, высушивали над сульфатом магния и растворитель удаляли в вакууме с образованием смолы, которую частично очищали флэш-хроматографией на силикагеле с использованием смеси петролейного эфира (темп. кип. 60-80oC) и этилацетата (2:1) в качестве растворителя для элюирования с получением сырого 1-[1-(циклопентилметил)-циклопропил] -7-метокси-6-метил-3,4-дигидроизохинолина (5,0 г), который использовали без дальнейшей очистки.
Цианоборгидрид натрия (2,13 г) добавляли одной порцией в охлаждаемый льдом раствор сырого продукта предыдущей реакции (5,0 г) в смеси метанола (25 мл) и уксусной кислоты (50 мл). После перемешивания при комнатной температуре в течение 18 часов смесь выливали на разбавленный водный раствор гидроокиси натрия (150 мл) и экстрагировали этилацетатом (4 х 50 мл). Объединенные экстракты промывали рассолом, затем высушивали над сульфатом магния и растворитель удаляли в вакууме с образованием смолы, которую очищали флэш-хроматографией на силикагеле с использованием смеси петролейного эфира (темп. кип. 60-80oC) и триэтиламина (5:1) в качестве растворителя для элюирования с получением 1-[1-(циклопентилметил)-циклопропил]-7-метокси-6-метил-1,2,3,4-тетрагидроизохинолин (2,65 г) в виде смолы.
Цианоборгидрид натрия (2,37 г) добавляли в раствор продукта предыдущей реакции (2,65 г), а также водный раствор формальдегида (37%; 4,65 мл) в метаноле (80 мл) и смесь перемешивали в течение 5 часов. Смесь выливали в разбавленный водный раствор аммиака (150 мл) и экстрагировали этилацетатом (4 х 60 мл). Объединенные экстракты высушивали над сульфатом магния и растворитель удаляли в вакууме. Остаток растворяли в эфире (150 мл) и через раствор пропускали газообразный хлористый водород; полученный осадок отфильтровывали и высушивали с получением гидрохлорида 1-[1-(циклопентилметил)-циклопропил] -7-метокси-2,6-диметил-1,2,3,4-тетрагидроизохинолина (2,1 г), темп. плавл. 166-169oC.
Продукт предыдущей реакции (2,0 г) и тетрабутилфосфорбромид (0,19 г) нагревали вместе с бромистоводородной кислотой (48%; 15 мл) при 95oC в течение 24 часов, затем выливали на воду (100 мл) и смесь подщелачивали водным раствором аммиака. Смесь экстрагировали этилацетатом (4 х 40 мл) и объединенные экстракты промывали рассолом, затем осушали над сульфатом магния. Растворитель удаляли в вакууме с получением остаточного красного масла, которое, по-видимому, еще содержало исходное вещество. Остаток затем растворяли в смеси уксусной кислоты (20 мл) и бромистоводородной кислоты (48%; 20 мл) и смесь нагревали при 95oC, затем выливали на воду (150 мл) и промывали эфиром. Водную фазу подщелачивали водным раствором аммиака, затем экстрагировали этилацетатом (4 х 50 мл), объединенные экстракты промывали рассолом, затем осушали над сульфатом магния. Растворитель удаляли в вакууме с образованием коричневого масла, которое растворяли в эфире (150 мл), затем обрабатывали избытком насыщенного раствора щавелевой кислоты в эфире. Образовавшийся осадок рекристаллизовали из ацетонитрила, но продукт оставался неочищенным. Свободное основание регенерировали распределением между разбавленным водным раствором аммиака (50 мл) и этилацетатом (50 мл). Органическую фазу отделяли и водную фазу экстрагировали этилацетатом (3 х 20 мл). Объединенные органические экстракты промывали рассолом, высушивали над сульфатом магния. Растворитель удаляли в вакууме и остаток тритурировали с эфиром с образованием твердой массы, которую рекристаллизовали из ацетонитрила с получением 1-[1-(циклопентилметил)-циклопропил] -7-гидрокси-2,6-диметил-1,2,3,4-тетрагидроизохинолина (0,15 г), темп. плавл. 136-138oC.
Пример 3. Циклобутанкарбонил-хлорид (7 г) добавляли по каплям к раствору 2-(4-метокси-3-метилфенил)-этиламина (11,36 г) и триэтиламина (13 мл) в тетрагидрофуране (150 мл) и полученную суспензию перемешивали в течение 16 часов. Смесь выливали в воду (150 мл), подкисляли разбавленной хлористоводородной кислотой и экстрагировали этилацетатом (4 х 50 мл). Объединенные экстракты промывали рассолом, высушивали и растворители удаляли в вакууме. Осадок промывали петролейным эфиром (темп.кип. 40-60oC) с получением сырого N-[2-(4-метокси-3-метилфенил)-этил]-циклобутанкарбоксиамида (12 г). Образец, рекристаллизованный из ацетонитрила, имел темп. плавл. 103-104oC.
Раствор сырого продукта предыдущей реакции (12 г) и хлорокиси фосфора (28 мл) в ацетонитриле (240 мл) нагревали с обратным холодильником в мягких условиях в течение 2,75 часа, охлаждали, затем выливали в разбавленный раствор аммиака (700 мл). Смесь экстрагировали этилацетатом (4 х 80 мл). Объединенные экстракты промывали рассолом, высушивали над сульфатом магния и растворитель удаляли в вакууме. Оставшуюся смолу распределяли между эфиром (150 мл) и хлористоводородной кислотой (3 М; 150 мл). Органическую фазу затем экстрагировали хлористоводородной кислотой (3 М; 3 х 60 мл). Объединенные водные кислые растворы промывали эфиром, подщелачивали водным раствором аммиака, затем экстрагировали этилацетатом (6 х 80 мл). Объединенные экстракты промывали рассолом, затем высушивали над сульфатом магния и растворитель удаляли в вакууме с получением 1-циклобутил-7-метокси-6-метил-3,4-дигидроизохинолина (8,4 г) в виде смолы, которую использовали без дальнейшей очистки.
Раствор трет.бутиллития в пентане (1,7 М, 25,8 мл) добавляли по каплям к раствору сырого продукта предыдущей реакции (8,4 г) в тетрагидрофуране (130 мл) при -78oC под азотом, смесь перемешивают при этой температуре в течение 1 часа 3-бромциклогексен (11,76 г) добавляли по каплям и раствор перемешивали в течение 1 часа при -78oC, затем смесь самопроизвольно нагревалась до комнатной температуры.
Раствор выливали в разбавленную хлористоводородную кислоту (300 мл) и промывали эфиром. Водную фазу затем подщелачивали добавлением водного раствора аммиака и экстрагировали этилацетатом (4 х 60 мл). Объединенные экстракты промывали рассолом, затем высушивали над сульфатом магния и растворитель удаляли в вакууме с получением оранжевого масла, которое растворяли в эфире (250 мл) и добавляли раствор (±)-дибензоилвинной кислоты (0,3 М; 25 мл). После перемешивания в течение 20 минут осадок отфильтровывали и выбрасывали. Фильтрат нейтрализовали концентрированным водным раствором аммиака и экстрагировали эфиром (4 х 50 мл). Растворитель удаляли в вакууме с получением масла, которое очищали флэш-хроматографией на силикагеле с использованием смеси петролейного эфира (темп. кип. 60-80oC) и триэтиламина (5:1) в качестве растворителя для элюирования с получением 1-[1-(циклогексен-1-ил-3)-циклобутил] -7-метокси-6-метил-3,4-дигидроизохинолина в виде смолы (5,9 г), которая медленно отверждалась, темп. плавл. 70-73oC.
Цианоборгидрид натрия (0,6 г) добавляли в раствор продукта предыдущей реакции (1,5 г) в смеси метанола (8 мл) и уксусной кислоты (16 мл) и раствор перемешивали в течение 16 часов. Добавили водный раствор гидроокиси натрия (20%; 150 мл), затем смесь экстрагировали этилацетатом (4 х 50 мл). Объединенные органические экстракты промывали водным раствором гидроокиси натрия (0,1 М), водой, затем рассолом, затем высушивали над сульфатом магния. Растворитель удаляли в вакууме с образованием 1-[1-(циклогексен-1-ил-3)-циклобутил] -7-метокси-6-метил-1,2,3,4-тетрагидроизохинолина (1,47 г) в виде бесцветной смолы, которую использовали без дальнейшей очистки.
Добавляли в раствор продукта предыдущей реакции (1,47 г) в ацетонитриле (60 мл) водный раствор формальдегида (37 вес.%; 1,8 мл) с образованием бесцветного осадка. Добавили цианоборгидрид натрия (0,47 г) и смесь перемешивали в течение 20 минут, затем нейтрализовали уксусной кислотой. Через 50 минут добавили водный раствор гидроокиси натрия (10%; 200 мл), затем смесь экстрагировали этилацетатом (4 х 50 мл). Объединенные органические экстракты промывали рассолом, затем высушивали над сульфатом магния и растворитель удаляли в вакууме. Оставшуюся твердую массу перекристаллизовали из ацетонитрила (50 мл) с образованием 1-[1-(циклогексен-1-ил-3)-циклобутил]-7-метокси-2,6-диметил-1,2,3,4-тетрагидроизохинолина (1,1 г) в виде бесцветной твердой массы, темп. плавл. 124-125oC.
Гидрид натрия (60%-ная дисперсия в масле; 0,51 г) добавляли порциями в охлаждаемый льдом раствор этантиола (0,94 мл) в сухом диметилформамиде (7,5 мл) и смесь перемешивали в течение 20 минут, за которые температура массы повышается до комнатной. Медленно добавляли суспензию продукта предыдущей реакции (0,9 мл) в сухом диметилформамиде (25 мл), затем смесь нагревали при 140oC в течение 6 часов. После выдерживания при комнатной температуре в течение 2,5 дней смесь выливали на ледяную воду (150 мл), промывали петролейным эфиром (темп. кип. 60-80oC), затем устанавливали pH 6 хлористоводородной кислотой (2 М) и смесь снова промывали петролейным эфиром. Смесь подщелачивали водным раствором аммиака и экстрагировали дихлорметаном (3 х 40 мл), затем экстрагировали этилацетатом (4 х 60 мл). Этилацетатные экстракты объединяли, высушивали над сульфатом магния и растворитель удаляли в вакууме. Остаток растворяли в эфире и раствор обрабатывали избытком раствора щавелевой кислоты в эфире. Полученный осадок перекристаллизовывали из промышленно полученного метилированного спирта с получением оксалата 1-[1-(циклогексен-1-ил-3)-циклобутил] -7-гидрокси-2,6-диметил-1,2,3,4-тетрагидроизохинолина (0,28 г) в виде смеси диастереоизомеров, темп. плавл. 194-196oC.
Пример 4.
Циклопропанкарбонил-нитрил (17,14 г) добавляли по каплям к смеси раствора диизопропиламида лития (2 М в смеси гептана, тетрагидрофурана и этилбензола; 128 мл ), 1,3-диметил-3,4,5,6-тетрагидро-2(1Н)-пиримидинона (30 мл) и тетрагидрофурана (240 мл) с перемешиванием при -78oC под азотом. Перемешивание продолжали в течение 1 часа при этой температуре, затем медленно добавляли раствор 1-хлортетралина (48,4 г) в тетрагидрофуране (50 мл), выдерживали еще 1 час и температуру в смеси доводили до комнатной и ее нагревали с обратным холодильником в мягких условиях в течение 2 часов. Растворитель удаляли в вакууме и остаток подкисляли концентрированной хлористоводородной кислотой и экстрагировали этилацетатом (4 х 100 мл). Объединенные экстракты высушивали над сульфатом магния и растворитель удаляли в вакууме с образованием полутвердой массы, которую отфильтровывали и промывали ацетонитрилом, затем перекристаллизовывали из ацетона с получением 1-(1,2,3,4-тетрагидронафтил-1)-циклопропанкарбонитрила (8,3 г), темп. плавл. 91-93oC.
Смесь продукта предыдущей реакции (8,3 г), порошок гидроокиси калия (85%; 4,66 г) и 1,2-этандиола (50 мл) нагревали с обратным холодильником в мягких условиях в течение 7 часов. Смесь разбавляли водой (200 мл) и хорошо промывали эфиром. Водную фазу подкисляли концентрированной хлористоводородной кислотой, затем экстрагировали этилацетатом (5 х 150 мл). Экстракты осушали и растворитель удаляли в вакууме. Полученную твердую массу перекристаллизовывали из ацетона с получением 1-(1,2,3,4-тетрагидронафтил-1)-циклопропанкарбоновой кислоты (2,25 г), темп. плавл. 140-142oC. В эфирной промывной жидкости определили содержание непрореагировавшего исходного нитрила, который нагревали с порошком гидроокиси калия (85%; 1,8 г) в 1,2-этандиоле (20 мл) в течение 2,5 дней. Поступая аналогично вышесказанному с последующей перекристаллизацией из ацетонитрила получили дополнительное количество целевого продукта (2,45 г).
Раствор 1,1-карбонилдиимидазола (3,2 г) в тетрагидрофуране (50 мл) добавляли медленно к раствору продукта предыдущей реакции (4,3 г) в тетрагидрофуране (50 мл) и смесь перемешивали в течение 18 часов. Добавляли гидрохлорид 2-(4-метокси-3-метилфенил)-этиламина (4,0 г) и триэтиламина (2,8 мл) и смесь перемешивали в течение 18 часов. Поскольку реакция оказалась незавершенной, в раствор добавили следующую порцию исходного амина (3,09 г) и триэтиламина (2,1 мл) в тетрагидрофуране (20 мл), перемешивание продолжали в течение 3 дней. Т. к. реакция оставалась незавершенной, добавляли разбавленный водный раствор гидроокиси натрия (200 мл) и продолжали перемешивание в течение 18 часов. Органический слой отделяли и водный слой экстрагировали этилацетатом (5 х 150 мл). Объединенные органические экстракты промывали рассолом, затем осушали над сульфатом магния и растворитель удаляли в вакууме. Остаток хорошо промывали петролейным эфиром (темп. кип. 60-80oC) с получением сырого N-[2-(4-метокси-3-метилфенил)-этил] -1-(1,2,3,4-тетрагидронафтил-1)-циклопропанкарбоксамид в виде коричневой смолы, которую используют без дальнейшей очистки.
Смесь сырого продукта предыдущей реакции (7,0 г), хлорокиси фосфора (11 мл) и ацетонитрила (90 мл) нагревали с обратным холодильником в течение 3 часов. Охлажденный раствор осторожно выливали на разбавленный водный раствор аммиака (200 мл) и экстрагировали этилацетатом (4 х 150 мл). Объединенные экстракты промывали рассолом, затем осушали над сульфатом магния, растворитель удаляли в вакууме и остаток очищали флэш-хроматографией на силикагеле с использованием смеси петролейного эфира (темп. кип. 60-80oC) и этилацетата (2: 1) в качестве растворителя для элюирования с получением 7-метокси-6-метил-1-[1-(1,2,3,4-тетрагидронафтил-1)-циклопропил] -3,4-дигидроизохинолина в виде темной смолы (1,5 г).
Цианоборгидрид натрия (0,53 г) добавляли одной порцией в раствор продукта предыдущей реакции (1,46 г) в смеси метанола (7 мл) и уксусной кислоты (14 мл) и всю массу перемешивали в течение 18 часов, затем выливали в воду (150 мл); полученную смесь подщелачивали концентрированным водным раствором аммиака и экстрагировали этилацетатом (4 х 60 мл). Объединенные экстракты промывали рассолом, затем осушали над сульфатом магния и растворитель удаляли в вакууме с получением 7-метокси-6-метил-1-[1-(1,2,3,4-тетрагидронафтил-1)-циклопропил] -1,2,3,4-тетрагидроизохинолин в виде желтого масла (1,58 г), которое используют без дальнейшей очистки.
Цианоборгидрид натрия (0,47 г) добавляли в перемешиваемый раствор продукта предыдущей реакции (1,5 г) и водный раствор формальдегида (37%; 1,8 мл) в ацетонитриле (60 мл). Через 20 минут добавляли уксусную кислоту (2 мл), перемешивание продолжали в течение 1 часа, смесь выливали на разбавленный водный раствор аммиака (150 мл) и экстрагировали этилацетатом (4 х 100 мл). Объединенные экстракты высушивали над сульфатом магния и растворитель удаляли в вакууме. Остаток очищали флэш-хроматографией на силикагеле с использованием смеси петролейного эфира (темп. кип. 60-80oC) и этилацетата (2:1) в качестве растворителя для элюирования с получением 7-метокси-2,6-диметил-1-[1-(1,2,3,4-тетрагидронафтил-1)-циклопропил] -3,4-дигидроизохинолина (1,2 г) в виде бесцветного масла.
Продукт предыдущей реакции (1,2 г) в смеси уксусной кислоты (40 мл) и бромистоводородной кислоты (48%; 40 мл) нагревали при 95oC в течение 3 дней. Охлажденную смесь выливали на воду (150 мл), затем промывали эфиром. Водную фазу подщелачивали концентрированным водным раствором аммиака и экстрагировали этилацетатом (5 х 100 мл). Объединенные экстракты промывали рассолом, затем осушали над сульфатом магния и растворитель удаляли в вакууме с образованием темно-коричневой смолы. Очисткой путем флэш-хроматографии на силикагеле с использованием смеси петролейного эфира (темп. кип. 60-80oC) и этилацетата (4: 1) в качестве растворителя для элюирования с получением смолы, которую растворяли в эфире (150 мл) и обрабатывали хлористым водородом и образовавшийся осадок отфильтровывали и высушивали с получением (1,05) . гидрохлорида-(0,7) . гидрата 7-гидрокси-2,6-диметил-1-[1-(1,2,3,4-тетрагидронафтил-1)-циклопропил] -1,2,3,4-тетрагидроизохинолина (0,74 г), темп. плавл. 160-163oC.
Пример 5. К интенсивно перемешиваемой смеси 50 вес.%-ного раствора гидроокиси натрия (190 мл), 1,2-дибромэтана (150 г) и бензилтриэтиламмоний-хлорида (10 г) добавляли по каплям 2-тиофенацетонитрил (50 г). Перемешиваемую смесь нагревали при 75oC в течение 3 часов, затем охлаждали и подкисляли 5 М хлористоводородной кислотой, смесь профильтровывали и фильтрат экстрагировали этилацетатом (4 х 100 мл). Объединенные экстракты промывали рассолом, высушивали и растворители удаляли в вакууме. Полученную черную смолу обесцвечивали активированным углем в кипящем метаноле с получением коричневой смолы (53,6 г). Смолу промывали петролейным эфиром с получением 1-(2-тиенил)-циклопропанкарбонитрила в виде твердой массы, темп. плавл. 118-122oC.
Твердую массу (42 г) добавили в суспензию гидроокиси калия (31,2 г) в этиленгликоле (300 мл), перемешивали и нагревали с обратным холодильником в мягких условиях в течение 16 часов. Охлажденную смесь выливали на воду (1000 мл). Этот раствор хорошо промывали эфиром, затем подкисляли и экстрагировали этилацетатом (4 х 150 мл). Объединенные органические экстракты промывали водой, затем рассолом и осушали. Растворитель удаляли в вакууме с получением 1-(2-тиенил)-циклопропанкарбоновой кислоты (37,6 г), темп. плавл. 116-118oC.
Смесь 2-(4-метокси-3-метилфенил)-этиламина (16 г), триэтиламина (1,74 мл), 1,3-дициклогексил-карбодиимида (8,75 г), 1-(2-тиенил)-циклопропанкарбоновой кислоты (7 г) и 1-гидроксибензотриазола (5,6 г) в тетрагидрофуране (95 мл) перемешивали в течение 16 часов. Смесь выливали на воду, подщелачивали водным раствором аммиака и фильтровали. Фильтрат экстрагировали этилацетатом (3 х 50 мл) и объединенные экстракты промывали 1 М хлористоводородной кислотой и рассолом, затем осушали и растворитель удаляли в вакууме. Полученную смолу обрабатывали ацетонитрилом и нерастворимое вещество отфильтровывали. При выпаривании фильтрата получили сырого N-[2-(4-метокси-3-метилфенил)-этил] -1-(2-тиенил)-циклопропанкарбоксамид в виде твердой массы (10,7 г), которую используют без дальнейшей очистки.
Порцию этого карбоксамида (3,5 г) в растворе ацетонитрила (52 мл) и хлорокись фосфора (6,5 мл) нагревали с обратным холодильником в течение 2,5 часов. Охлажденный реакционный раствор затем выливали на разбавленный водный раствор аммиака и продукт экстрагировали этилацетатом. Объединенные экстракты промывали рассолом, затем осушали над сульфатом магния, растворители удаляли в вакууме с получением темной смолы. Его очищали флэш-хроматографией на силикагеле с использованием смеси легкого петролейного эфира и триэтиламина (5:1) в качестве растворителя для элюирования с получением сырого 7-метокси-6-метил-1-[1-(2-тиенил)-циклопропил]-3,4-дигидроизохинолина (4,4 г).
Раствор этого дигидроизохинолина (1,85 г) в ледяной уксусной кислоте (20 мл) и метаноле (10 мл) охлаждали льдом и обрабатывали порциями цианоборгидрида натрия (0,77 г). Реакционную смесь перемешивали в течение 16 часов и выливали на воду, подщелачивали концентрированным раствором гидроокиси натрия и экстрагировали этилацетатом (3 х 50 мл). Объединенные экстракты промывали 1 М раствором гидроокиси натрия, водой и рассолом, затем осушали и растворитель удаляли в вакууме с получением 7-метокси-6-метил-1-[1-(2-тиенил)-циклопропил]-1,2,3,4-тетрагидроизохинолин в виде бесцветной смолы.
Смолу (2 г, получена, как описано выше) в ацетонитриле (80 мл) и водный раствор формальдегида (37 вес.%; 2,6 мл) обрабатывали цианборгидридом натрия (0,67 г). Через 15 минут смесь нейтрализовали уксусной кислотой и перемешивание продолжали в течение следующих 45 минут. Смесь подщелачивали добавлением 2М раствора гидроокиси натрия и продукт экстрагировали этилацетатом (4 х 50 мл). Объединенные органические промывали 0,1 М водным раствором гидроокиси натрия, водой и рассолом, высушивали и концентрировали с получением смолы. Ее очищали флэш-хроматографией на силикагеле с использованием смеси метанола и дихлорметана (1: 49) в качестве растворителя для элюирования с получением сырого 7-метокси-2,6-диметил-1-[1-(2-тиенил)-циклопропил] -1,2,3,4-тетрагидроизохинолина в виде твердой массы (1,1 г).
Раствор этого 2,6-диметилтетрагидроизохинолина (2,07 г, получен, как описано выше) в дихлорметане (10 мл) охлаждали до -78oC и обрабатывали по каплям 1 М раствором трибромида бора в дихлорметане (6,6 мл). После перемешивания при комнатной температуре в течение 2 дней смесь выливали на метанол и концентрировали почти досуха. Добавляли метанол и выпаривали снова трижды, затем добавляли пронанол-2 и весь растворитель удаляли в вакууме. Остаток перекристаллизовывали из промышленно метилированного спирта с получением гидробромида 7-метокси-6-метил-1-[1-(2-тиенил)-циклопропил] -1,2,3,4-тетрагидроизохинолина (1,1 г), темп. плавл. 245-246oC.
Пример 6. Раствор тиофен-3-ацетонитрила (30 г) и 1-бром-2-хлорэтана (40,8 мл) в диметилсульфоксиде (120 мл) добавляли медленно по каплям к интенсивно перемешиваемой суспензии гидрида натрия (60%-ная в минеральном масле; 38,8 г) в диметилсульфоксиде (800 мл) при 30-35oC. Раствор перемешивали при комнатной температуре в течение 21 часа, затем осторожно добавляли воду (100 мл) и хлористоводородную кислоту (2М; 100 мл). Смесь экстрагировали эфиром (6 х 80 мл) и объединенные экстракты промывали водой (2 х 100 мл), затем рассолом и затем высушивали над сульфатом магния. Растворитель удаляли в вакууме и остаток промывали петролейным эфиром (темп. кип. 60-80oC) с последующей дистилляцией под пониженным давлением с получением 1-(3-тиенил)-циклопропанкарбонитрила в виде масла (26,2 г), темп. кип. 82-86oC/2 мбар.
Продукт предыдущей реакции (26,0 г) и порошок гидроокиси калия (85%; 19,0 г) нагревали в 1,2-этандиоле (190 мл) с обратным холодильником в мягких условиях в течение 3 дней. Охлажденную реакционную смесь выливали на воду (600 мл) и промывали эфиром. Водную фазу подкисляли концентрированной хлористоводородной кислотой и экстрагировали этилацетатом (6 х 200 мл). Объединенные экстракты промывали рассолом, затем осушали над сульфатом магния и растворитель удаляли в вакууме. Остаток тритурировали с эфиром и получали 1-(3-тиенил)-циклопропанкарбоновой кислоты (20 г) в виде расслаивающейся твердой массы, темп. плавл. 132-133oC.
Раствор N,N'-дициклогексилкарбодиимида (24,76 г) в тетрагидрофуране (30 мл) добавляли медленно к раствору продукта предыдущей реакции (20 г), гидрохлорида 2-(4-метокси-3-метилфенил)-этиламина (22,2 г) и 4-(диметиламино)-пиридина (13,4 мл) в тетрагидрофуране (150 мл) и смесь перемешивали в течение 18 часов. Смесь разбавляли водой, подкисляли концентрированной хлористоводородной кислотой и экстрагировали этилацетатом (4 х 100 мл). Объединенные экстракты промывали разбавленным водным раствором гидроокиси натрия и рассолом, затем осушали над сульфатом магния и растворитель удаляли в вакууме с получением сырого N-[2-(4-метокси-3-метилфенил)-этил]-1-(3-тиенил)-циклопропанкарбоксамида в виде светло-желтой смолы, которую используют без дальнейшей очистки.
Смесь сырого продукта предыдущей реакции (38,4 г), хлорокиси фосфора (60 мл) и ацетонитрила (500 мл) нагревали с обратным холодильником в течение 3,5 часов. Охлажденный раствор выливали в воду (1 л) и осторожно подщелачивали концентрированным водным раствором аммиака. Смесь экстрагировали эфиром (5 х 100 мл). В первом из экстрактов при выдерживании образовывались кристаллы целевого продукта (3,8 г), которые отфильтровывали и высушивали. Остальные экстракты объединяли и высушивали над сульфатом магния и растворитель удаляли в вакууме. Остаток перекристаллизовывали из ацетонитрила с получением дополнительного количества продукта (8,8 г). Маточную жидкость выпаривали и остаток очищали флэш-хроматографией на силикагеле с использованием смеси (2: 1) этилацетата и петролейного эфира (темп. кип. 60-80oC) в качестве растворителя для элюирования с последующей перекристаллизацией из ацетонитрила с получением третьего дополнительного количества продукта (2,0 г). Все три порции объединяют с получением 7-метокси-6-метил-1-[1-(3-тиенил)-циклопропил]-3,4-дигидроизохинолина (14,6 г), темп. плавл. 116-118oC.
Цианоборгидрид натрия (0,84 г) добавляли одной порцией в раствор продукта предыдущей реакции (2,0 г) в смеси метанола (10 мл) и уксусной кислоты (20 мл) и смесь перемешивали в течение 3 часов. Смесь выливали в воду (200 мл), подщелачивали концентрированным водным раствором аммиака и экстрагировали этилацетатом (4 х 60 мл). Объединенные экстракты промывали рассолом, затем осушали над сульфатом магния и растворитель удаляли в вакууме с получением сырого 7-метокси-6-метил-1-[1-(3-тиенил-циклопропил]-1,2,3,4-тетрагидроизохинолин (2,1 г) в виде бесцветной смолы.
Цианоборгидрид натрия (0,67 г) добавляли в раствор сырого продукта предыдущей реакции (2 г) в смеси водного раствора формальдегида (37%; 2,6 мл) и ацетонитрила (80 мл). После перемешивания в течение 15 минут подкисляли уксусной кислотой и перемешивание продолжали в течение 1 часа, затем смесь выливали в воду, подщелачивали концентрированным водным раствором аммиака и экстрагировали этилацетатом (4 х 60 мл). Объединенные экстракты промывали рассолом, высушивали над сульфатом магния и растворитель удаляли в вакууме. Остаток перекристаллизовывали из ацетонитрила с получением 7-метокси-2,6-диметил-1-[1-(3-тиенил)-циклопропил] -1,2,3,4-тетрагидроизохинолина (1,7 г) в виде бесцветной твердой массы, темп. плавл. 103-105oC.
Смесь продукта предыдущей реакции (1,7 г), бромистоводородной кислоты (48%; 60 мл) и уксусной кислоты (60 мл) перемешивали и нагревали при 150oC в течение 3 часов. Смесь выливали в воду (300 мл) и подщелачивали концентрированным водным раствором аммиака, затем экстрагировали этилацетатом (4 х 150 мл). Объединенные экстракты промывали рассолом, затем осушали над сульфатом магния и растворитель удаляли в вакууме с образованием 7-гидрокси-2,6-диметил-1-[1-(3-тиенил)-циклопропил]-1,2,3,4-тетрагидроизохинолина (1,3 г) в виде смолы. Следующую порцию этого соединения получили аналогичным образом с использованием тех же реактивов, взятых в количестве, в 1,47 раз большем, чем указано выше. Объединенное количество вещества затем разделяли на энантиомеры путем жидкостной высокоэффективной хирально-проградуированной хроматографии с использованием (95:5) - смеси гексана и 2-пропанола в качестве растворителя для элюирования при 30 мл/мин и УФ-детектировании при 254 нм. Индивидуальный (-)-энантиомер, [α]D = -19,4o (C = 0,95, CH2Cl2), обрабатывали избытком насыщенного раствора щавелевой кислоты в эфире и полученную соль перекристаллизовывали из промышленно метилированного спирта с получением оксалата (-)-7-гидрокси-2,6-диметил-1-[1-(3-тиенил)-циклопропил] -1,2,3,4-тетрагидроизохинолина, (140 г), темп. плавл. 222oC [α]D = -118,6o (c = 1, MeOH).
Пример 7. Раствор 7-метокси-6-метил-1-[1-(3-тиенил)-циклопропил]-3,4-дигидроизохинолина (2,0 г, получен аналогично описанному в примере 6) в дихлорметане (87 мл) медленно добавляли в охлаждаемую льдом смесь трис-[(S)-N-(трет. бутоксикарбонил)-пропилокси] -боргидрид натрия (11,4 г) в дихлорметане (70 мл). После выдерживания при ок. 4oC в течение 2 дней добавляли следующую порцию восстанавливающего агента (2,0 г) и смесь перемешивали при комнатной температуре в течение 3 часов, затем добавляли насыщенный водный раствор щавелевой кислоты (150 мл). Эту смесь перемешивали в течение 1 часа и подщелачивали концентрированным водным раствором аммиака, экстрагировали дихлорметаном (4 х 100 мл). Объединенные экстракты промывали водой (100 мл), затем рассолом (100 мл), высушивали над сульфатом магния и растворитель удаляли в вакууме с получением светло-оранжевой смолы. Смолу растворяли в эфире (150 мл) и обрабатывали раствором щавелевой кислоты в эфире (0,43 М; 20 мл) и полученный осадок отфильтровывали, затем перекристаллизовывали из этанола с получением энантиомера оксалата 7-метокси-6-метил-1-[1-(3-тиенил)-циклопропил] -1,2,3,4-тетрагидроизохинолина (1,2 г), который используют без дальнейших исследований.
Цианоборгидрид натрия (0,28 г) добавляли в перемешиваемый раствор продукта предыдущей реакции (1,1 г) в смеси ацетонитрила (35 мл) и водного раствора формальдегида (37%; 1,1 мл). Через 3 часа смесь выливали на воду (150 мл), подщелачивали концентрированным раствором аммиака и экстрагировали этилацетатом (5 х 50 мл). Объединенные экстракты промывали водой (100 мл), затем рассолом (100 мл) и высушивали над сульфатом магния. Растворитель удаляли в вакууме и твердый остаток перекристаллизовывали из ацетонитрила с получением (-)-7-метокси-2,6-диметил-1-[1-(3-тиенил)-циклопропил]-1,2,3,4-тетрагидроизохинолина (0,75 г) в виде бесцветных кристаллов, [α]D= - 44,8o (c = 0,5, CH2Cl2).
Смесь продукта предыдущей реакции (0,65 г), бромистоводородной кислоты (48%; 9 мл) и уксусной кислоты (9 мл) нагревали при 150oC в течение 3 часов под азотом. Смесь распределяли между эфиром (50 мл) и водой (100 мл), в водном слое устанавливали pH 8 медленным прибавлением концентрированного водного раствора аммиака, затем отделяли эфирный слой и водный слой экстрагировали эфиром (2 х 100 мл), затем этилацетатом (3 х 100 мл). Объединенные экстракты промывали рассолом, затем осушали над сульфатом магния и растворитель удаляли в вакууме с образованием светло-желтой смолы, которую растворяли в эфире (150 мл), обрабатывали раствором щавелевой кислоты в эфире (0,43 М; 7 мл) и полученный гелеподобный осадок собирали фильтрованием. Это вещество перекристаллизовывали из промышленно метилированного спирта с получением двух порций оксалата (-)-7-гидрокси-2,6-диметил-1-[1-(3-тиенил)-циклопропил] -1,2,3,4-тетрагидроизохинолина (0,53 г), идентичного полученному в примере 6.
Оксалат распределяли между эфиром (50 мл) и водой (100 мл) и в водном слое устанавливали pH 8 медленным добавлением концентрированного водного раствора аммиака. Эфирный слой отделяли и водный слой экстрагировали эфиром (2 х 100 мл), затем этилацетатом (3 х 100 мл). Объединенные экстракты промывали рассолом, высушивали над сульфатом магния и растворитель удаляли в вакууме с получением светло-желтой, которую тритурировали с дихлорметаном (2 мл) и получили соответствующее свободное основание в виде светло-желтой твердой массы (0,42 г), темп. плавл. 155-157oC, [α]D = -19,4o (c = 0,95, CH2Cl2). Свободное основание растворяли в эфире (120 мл) и обрабатывали газообразным хлористым водородом в течение 1 минуты. Осадок собирали фильтрацией, высушивали в вакууме с получением гидрохлорида (0,8 г) гидрата (-)-7-гидрокси-2,6-диметил-1-[1-(3-тиенил)-циклопропил]-1,2,3,4-тетрагидроизохинолина (0,41 г), темп. плавл. 42-45oC (разл.), [α]D = -124,3o (c = 0.6, MeOH).
Пример 8. Повторили процедуру деметилирования, описанную в примере 6, причем использовали вещества в количестве, увеличенном в 1,2 раза, растворитель из охлажденной реакционной смеси удаляли в вакууме. Остаток перекристаллизовывали из ацетона с получением гидробромида (-)-7-гидрокси-2,6-диметил-1-[1-(3-тиенил)-циклопропил] -1,2,3,4-тетрагидроизохинолина (1,1 г), в виде белой твердой массы, темп. плавл. 230oC (разл.).
Пример 9. Раствор фуран-2-ацетонитрила (28,4 г) и 1-бром-2-хлорэтана (33,1 мл) в диметилсульфоксиде (50 мл) медленно добавляли к перемешиваемой суспензии гидрида натрия (60%-ная дисперсия в минеральном масле; 31,35 г) в сухом диметилсульфоксиде (300 мл) и поддерживали внешнюю температуру ниже 35oC периодическим охлаждением. После перемешивания в течение 18 часов при комнатной температуре смесь разбавляли водой (500 мл) и экстрагировали эфиром (1 х 200 мл, затем 2 х 100 мл). Объединенные экстракты высушивали над карбонатом калия и выпаривали до образования масла, которое дистиллировали под пониженным давлением с получением 1-(2-фурил)-циклопропанкарбонитрила (28,0 г) в виде бесцветного масла, темп. кип. 54oC/0,55 мбар.
Смесь продукта предыдущей реакции (28,0 г), гидроокиси калия (25,0 г), метанола (20 мл) и воды (250 мл) нагревали с обратным холодильником в течение 90 минут. Метанол отгоняли под пониженным давлением и водный раствор промывали эфиром, затем подкисляли охлаждаемой льдом хлористоводородной кислотой (2М). Осадок отфильтровывали, хорошо промывали водой и сушили на воздухе с получением 1-(2-фурил)-циклопропанкарбоновой кислоты (27,5 г) в виде бесцветной твердой массы, темп. плавл. 113-114oC.
Раствор продукта предыдущей реакции (7,6 г) в сухом тетрагидрофуране (125 мл) добавляли к раствору 1,1-карбонилдиимидазола (8,1 г) в сухом тетрагидрофуране (125 мл) и смесь выдерживали в течение 18 часов при исключении увлажнения. Затем добавляли раствор гидрохлорида 2-(4-метокси-3-метилфенил)-этиламина (9,8 г) и триэтиламина (14,4 мл) и смесь перемешивали при исключении влаги в течение 18 часов. Затем добавляли воду (200 мл) и подщелачивали водным раствором аммиака (2М). Затем смесь экстрагировали эфиром (3 х 80 мл), объединенные экстракты сушили над карбонатом калия, затем растворитель удаляли в вакууме с образованием сырого 1-(2-фурил)-N-[2-(4-метокси-3-метилфенил)-этил]-циклопропанкарбоксамида (13,4 г) в виде масла.
Раствор сырого продукта предыдущей реакции (8,4 г) в этилполифосфате (40 г) взвихряли при 95oC под азотом в течение 40 минут и смесь выливали на смесь льда (200 г) и водного раствора аммиака (60 мл), затем ее экстрагировали эфиром (3 х 70 мл). Из объединенных экстрактов растворитель удаляли в вакууме и остаток очищали флэш-хроматографией на силикагеле с использованием смеси петролейного эфира (темп. кип. 40-60oC), эфира и триэтиламина (17:2:1) в качестве растворителя для элюирования с получением 1-[1-(2-фурил)-циклопропил]-7-метокси-6-метил-3,4-дигидроизохинолина (1,36 г) в виде бесцветной кристаллической массы, темп. плавл. 98-100oC.
Боргидрид натрия (2,0 г) добавляли малыми порциями в перемешиваемый раствор продукта предыдущей реакции (2,65 г) в промышленно метилированном спирте (100 мл). Смесь выдерживали в течение 18 часов и затем ее нагревали с обратным холодильником в течение 1 часа. Растворитель удаляли в вакууме, к остатку добавляли воду (100 мл) и затем экстрагировали эфиром (2 х 50 мл). Объединенные экстракты высушивали над карбонатом калия и растворитель удаляли в вакууме с получением 1-[1-(2-фурил)-циклопропил]-7-метокси-6-метил-1,2,3,4-тетрагидроизохинолина (1,2 г) в виде масла.
Цианоборгидрид натрия (0,7 г) добавляли в перемешиваемый раствор продукта предыдущей реакции (1,2 г) в смеси метанола (60 мл) и водного раствора формальдегида (37%; 3,0 мл). После перемешивания в течение 18 часов метанол удаляли в вакууме при температуре ниже 40oC и водный остаток подщелачивали смесью льда (50 г) и водного раствора аммиака (20 мл), затем экстрагировали эфиром (3 х 40 мл). Объединенные экстракты высушивали над карбонатом калия и растворитель удаляли в вакууме с получением 1-[1-(2-фурил)-циклопропил]-7-метокси-2,6-диметил-1,2,3,4-тетрагидроизохинолина (1,3 г) в виде смолы.
Этантиолат натрия (2,0 г) добавляли к перемешиваемому раствору продукта предыдущей реакции (1,3 г) в диметилформамиде (25 мл), затем смесь нагревали при 180oC в течение 1,5 часа. Охлажденную смесь разбавляли водой (200 мл), подкисляли охлаждаемой льдом хлористоводородной кислотой (5 М), затем промывали эфиром (3 х 80 мл). Водный слой подщелачивали водным раствором аммиака, затем экстрагировали эфиром (3 х 80 мл). Объединенные экстракты осушали над сульфатом натрия и растворитель удаляли в вакууме с получением коричневого масла, которое очищали флэш-хроматографией на силикагеле с использованием смеси петролейного эфира (темп. кип. 40-60oC), эфира и триэтиламина (50:45: 5) в качестве растворителя для элюирования. Продукт обрабатывали раствором малеиновой кислоты в эфире. Растворитель декантировали из образовавшейся смолы, которую затем тритурировали с кипящим этилацетатом. Полученную бесцветную твердую массу сушили с получением малеата 1-[1-(2-фурил)-циклопропил] -7-гидрокси-2,6-диметил-1,2,3,4-тетрагидроизохинолина (0,77 г), темп. плавл. 155oC.
Пример 10. Циклобутанкарбонил-хлорид (5,5 г) добавляли к раствору 2-(4-метоксифенил)-этиламина (7 г) и триэтиламина (6,5 мл) в эфире (300 мл) при комнатной температуре. Смесь перемешивали в течение ночи, выливали в воду и подкисляли (2 М) хлористоводородной кислотой, затем продукт экстрагировали этилацетатом (3 х 100 мл). Объединенные экстракты промывали рассолом, высушивали и растворители удаляли в вакууме с получением остатка. Его промывали легким петролейным эфиром и высушивали в вакууме с получением N-[2-(4-метоксифенил)-этил] -циклобутанкарбоксиамида (9,54 г), темп. плавл. 118-120oC.
Раствор этого амида (9 г) в сухом ацетонитриле (170 мл), содержащий хлорокись фосфора (23,7 мл) нагревали с обратным холодильником в течение 43 часов. Охлажденную смесь выливали в разбавленный раствор аммиака и эту смесь экстрагировали этилацетатом (3 х 150 мл). Этилацетатный раствор затем экстрагировали разбавленной хлористоводородной кислотой (3 х 100 мл). Водные кислые экстракты подщелачивали добавлением водного раствора аммиака, затем экстрагировали этилацетатом. Органические экстракты промывали рассолом и концентрировали масло, которое дистиллировали при 190oС/0,2 мбар с получением 1-циклобутил-7-метокси-3,4-дигидроизохинолина (4 г) в виде бесцветной твердой массы, темп. плавл. 44-46oC.
Н. бутиллитий (7,3 мл, 2,09 М в гексане) добавляли по каплям при комнатной температуре к раствору диизопропиламина (2,12 мл) в сухом тетрагидрофуране (14 мл). Через 15 минут раствор охлаждали до -23oC и добавляли раствор 1-циклобутил-7-метокси-3,4-дигидроизохинолина (3 г) в тетрагидрофуране (34 мл). Через 30 минут смесь охлаждали до -78oC и обрабатывали 2-хлорпиридином (1,56 мл), перемешивали в течение 1 часа, затем нагревали до комнатной температуры. Смесь нагревали с обратным холодильником в течение 5 минут, перемешивали в течение 16 часов при комнатной температуре, затем нагревали с обратным холодильником в течение 30 минут. Реакционную смесь выливали в воду и экстрагировали этилацетатом. Экстракты сушили и концентрировали и оставшееся оранжевое масло нагревали при 90oC/13,3 мбар до удаления избытка 2-хлорпиридина. Остаток очищали флэш-хроматографией на силикагеле с использованием смеси легкого петролейного эфира и триэтиламина ( 5:1 ) в качестве элюэнта с получением 7-метокси-1-[1-(2-пиридил)-циклобутил]-3,4-дигидроизохинолина в виде твердой массы (0,58 г).
Смесь дигидроизохинолина (1,06 г, полученный аналогично описанному выше), ледяной уксусной кислоты (12 мл) и метанола (6 мл) обрабатывали цианборгидридом натрия (0,47 г) при 0-10oC и перемешивали в течение 16 часов, затем выливали в водный раствор гидроокиси натрия. Продукт экстрагировали этилацетатом (3 х 100 мл) и объединенные экстракты промывали разбавленным водным раствором аммиака и рассолом. Органический слой осушали, растворитель удаляли в вакууме и образовавшееся масло растворяли в эфире и обрабатывали эфирным раствором щавелевой кислоты. Собирали образующуюся твердую массу путем фильтрования с получением оксалата 7-метокси-1-[1-(2-пиридил)-циклобутил]-1,2,3,4-тетрагидроизохинолина (1,06 г), темп. плавл. 135-138oC.
Порцию продукта предыдущей реакции (0,95 г) в метаноле (32 мл), содержащую водный раствор формальдегида (37 вес.%; 1,9 мл), обрабатывали цианоборгидридом натрия (1 г) и реакционную смесь перемешивали в течение 16 часов. Смесь затем концентрировали и остаток подщелачивали водным раствором гидроокиси натрия. Смесь экстрагировали этилацетатом (3 х 100 мл) и объединенные экстракты промывали разбавленным водным раствором аммиака и рассолом. Экстракты высушивали и растворитель удаляли в вакууме с получением масла, которое очищали флэш-хроматографией на силикагеле с использованием смеси легкого петролейного эфира и триэтиламина (5:1) в качестве элюэнта с получением 7-метокси-2-метил-1-[1-(2-пиридил)-циклобутил] -1,2,3,4-тетрагидроизохинолина (0,52 г) в виде масла.
Раствор 2-метилтетрагидроизохинолина (0,52 г) в водном растворе бромистоводородной кислоты (48%; 15 мл) и ледяной уксусной кислоте (15 мл) нагревали с обратным холодильником в течение 4 часов. Растворители удаляли в вакууме и остаток осушали азеотропной дистилляцией с пропанолом-2. Остаток затем перекристаллизовывали из изопропилового спирта с образованием бромистоводородной соли в виде твердой массы. Ее собирали фильтрованием, нейтрализовали и превращали (обработкой эфирным раствором щавелевой кислоты) с получением оксалата 7-гидрокси-2-метил-1-[1-(2-пиридил)-циклобутил] -1,2,3,4-тетрагидроизохинолина (0,15 г), темп. плавл. 95-97oC (разл.).
Пример 11. 50%-ный водный раствор гидроокиси натрия (100 мл) добавили к перемешиваемой смеси 2-(2-пиридил)-ацетонитрила (25 г), 1-бром-2-хлорэтана (26,5 мл), бензил-триэтиламмоний-хлорида (1 г) и толуола (100 мл) при 25oC, затем смесь нагревали при 70-75oC в течение 2 часов. Раствор охлаждали до комнатной температуры, добавляли активированный уголь и раствор фильтровали (Целит). Этот продукт экстрагировали эфиром (2 х 100 мл). Объединенные органические экстракты осушали над карбонатом калия и растворитель удаляли в вакууме с получением красно-оранжевой твердой смолы (28 г), которую обрабатывали при 150oС/10 мбар с получением 1-(2-пиридил)-циклопропанкарбонитрила (26,1 г) в виде твердой массы.
1-(2-пиридил)-циклопропанкарбонитрил (26 г) нагревали с обратным холодильником в течение 2 часов с 10%-ным водным раствором гидроокиси калия (140 мл). После охлаждения раствор промывали толуолом (2 х 100 мл) и подкисляли добавлением смеси концентрированной серной кислоты (5,7 мл) и воды (50 мл). Растворитель удаляли в вакууме и остаток осушали азеотропной дистилляцией с метанолом. Остаток суспендировали в метаноле (100 мл), раствор фильтровали и растворитель удаляли в вакууме с получением 1-(2-пиридил)-циклопропанкарбоновой кислоты (28 г) в виде масла.
Триметил-ортоацетат (38,0 г) добавили в раствор 1-(2-пиридил)-циклопропанкарбоновой кислоты (17,2 г) в толуоле (200 мл) под азотом. Смесь перемешивали с обратным холодильником в течение 24 часов, затем охлаждали до комнатной температуры и промывали водным раствором гидроокиси натрия (2 М; 2 х 100 мл), затем водой (2 х 100 мл). Смесь высушивали над сульфатом натрия и растворитель удаляли в вакууме с образованием коричневого масла (12,8 г). Продукт очищали флэш-хроматографией на силикагеле с использованием смеси (5: 1) триэтиламина и петролейного эфира (темп. кип. 40-60oC) в качестве элюэнта с получением метил-1-(2-пиридил)-циклопропанкарбоксилата (11,5 г) в виде желто-зеленого масла.
Метил-1-(2-пиридил)-циклопропанкарбоксилат (9,4 г) и 2-(4-метокси-3-метилфенил)-этиламина (8,8 г) перемешивали при 95oC в течение 16 часов. Смесь перемешивали при 110oC под азотом в течение 24 часов, затем растворяли в дихлорметане (100 мл) и промывали хлористоводородной кислотой (2 М; 2 х 100 мл). Смесь осушали над сульфатом магния и растворитель удаляли в вакууме с получением сырого N-[2-(4-метокси-3-метилфенил)-этил]-1-(2-пиридил)-циклопропанкарбоксамида (2,1 г). Водный слой подщелачивали водным раствором гидроокиси натрия, затем экстрагировали дихлорметаном (2 х 10 мл). Органический слой осушали над сульфатом магния, объединяли с исходным карбоксамидом и концентрировали в вакууме. Продукт очищали флэш-хроматографией на силикагеле с использованием смеси (1:1) петролейного эфира (темп. кип. 40-60oC) и этилацетата в качестве растворителя для элюирования с получением N-[2-(4-метокси-3-метил-фенил)-этил] -1-(2-пиридил)-циклопропанкарбоксамида (4,4 г), темп. плавл. 65-66oC.
Раствор амида (1,0 г, получен аналогично описанному выше) в сложном полифосфатном эфире (50 вес.% в CHCl3; 10 г) нагревали с перемешиванием при 95oC под азотом в течение 16 часов. Смесь резко охлаждали смешением с ледяной водой (100 мл), подщелачивали водным раствором аммиака (25 об.%) и экстрагировали этилацетатом (3 х 100 мл). Объединенные органические слои осушали над сульфатом натрия и растворитель удаляли в вакууме с получением 7-метокси-6-метил-1-[-1-(2-пиридил)-циклопропил]-3,4-дигидроизохинолина (0,9 г).
Раствор дигидроизохинолина (3,1 г, получен аналогично описанному выше) в метаноле (18 мл) и ледяной уксусной кислоты (36 мл) перемешивали при 0-10oC и обрабатывали цианоборгидридом натрия (1,38 г) и образовавшийся раствор перемешивали при комнатной температуре в течение 14 часов. Раствор подщелачивали добавлением водного раствора гидроокиси натрия (2N), затем экстрагировали этилацетатом (2 х 100 мл). Объединенные органические слои промывали водным раствором аммиака (25 об.%; 100 мл) и рассолом, затем высушивали над сульфатом натрия и растворитель удаляли в вакууме с получением коричневого масла (2,5 г).
Коричневое масло (2,5 г) растворяли в эфире и добавляли эфирную щавелевую кислоту. Осаждается белая смола, которую отделяют декантированием, затем очищают повторяющимся тритурированием с эфиром и смесью 3:1 эфира и этилацетата. Образующийся белый порошок подщелачивали путем обработки водным раствором карбоната натрия и экстрагировали этилацетатом. Органический слой высушивали над сульфатом натрия и растворитель удаляли в вакууме с получением 7-метокси-6-метил-1-[-1-(2-пиридил)-циклопропил] -1,2,3,4-тетрагидроизохинолина в виде коричневого масла (0,7 г ).
Раствор тетрагидроизохинолина (0,7 г, получен аналогично описанному выше) в метаноле и формальдегиде (37-40 вес.%; 1,9 мл) перемешивали и обрабатывали цианборгидридом натрия (1 г). Смесь перемешивали, затем добавляли ледяную уксусную кислоту до установления в смеси pH 6. Смесь перемешивали следующие 40 минут, затем концентрировали в вакууме и подщелачивали добавлением водного раствора гидроокиси натрия (2N). Продукт экстрагировали этилацетатом (2 x 100 мл) и объединенные органические экстракты промывали водным раствором аммиака (25 об.%; 100 мл) и рассолом (100 мл), высушивали над сульфатом натрия и растворитель удаляли в вакууме с получением коричневого масла. Масло растворяли в эфире и добавляли эфирную щавелевую кислоту. Образовавшийся осадок тритурировали с эфиром с получением 7-метокси-2,6-диметил-1-[1-(2-пиридил)-циклопропил]-1,2,3,4-тетрагидроизохинолина (0,7 г).
Раствор 7-метокси-2,6-диметил-1-[1-(2-пиридил)-циклопропил] -1,2,3,4-тетрагидроизохинолина (0,7 г) в 48%-ной бромистоводородной кислоте (10 мл) и ледяной уксусной кислоте (10 мл) нагревали с обратным холодильником под азотом в течение 5 часов. Смесь охлаждали до комнатной температуры и концентрировали в вакууме. Добавляли воду (50 мл) и раствор подщелачивали добавлением водного раствора гидроокиси натрия (2N) до установления в растворе pH 8. Продукт экстрагировали эфиром (100 мл) и этилацетатом (200 мл) и объединенные органические фракции промывали водой (100 мл). Органический слой высушивали над сульфатом магния и растворитель удаляли в вакууме с образованием коричневой твердой массы, которую тритурировали с эфиром с получением 7-гидрокси-2,6-диметил-1-[1-(2-пиридил)-циклопропил] -1,2,3,4-тетрагидроизохинолина (0,3 г) в виде твердой массы, темп. плавл. 213oC (разл.).
Пример 12. Использование соединений согласно настоящему изобретению для приготовления фармацевтических композиций иллюстрируется ниже. В данном тексте термин "активное соединение" означает любое соединение согласно изобретению, но в особенности, любое соединение, которое является конечным продуктом приведенных выше примеров.
а) Капсулы.
Для приготовления капсул 10 весовых частей активного соединения и 240 весовых частей лактозы измельчали и перемешивали. Смесью заполняли твердые желатиновые капсулы, причем каждая капсула содержала единичную дозу активного соединения.
б) Таблетки.
Таблетки готовили из следующих ингредиентов, вес.ч.:
Активное соединение - 10
Лактоза - 190
Кукурузный крахмал - 22
Поливинилпирролидон - 10
Стеарат магния - 3
Активное соединение, лактозу и некоторое количество крахмала измельчали, перемешивали и полученную смесь гранулировали с раствором поливинилпирролидона в этаноле. Сухой гранулят перемешивали со стеаратом магния и остаточным количеством крахмала. Затем смесь прессовали на таблеточной машине с получением таблеток, причем каждая таблетка содержала единичную дозу активного соединения.
в) Покрытые энтеросолюбильной оболочкой таблетки.
Таблетки приготавливали по методике, описанной выше в пункте (б). На таблетки наносили энтеросолюбильную оболочку известным образом с использованием 20%-ного раствора ацетата-фталата целлюлозы и 3%-ного раствора диэтилфталата в смеси этанола/дихлорметана (1:1).
г) Суппозитории.
Для приготовления суппозиториев 100 весовых частей активного соединения вводили в 1300 весовых частей триглицеридной основы суппозиториев и смесь формовали в суппозитории, причем каждый суппозиторий содержал терапевтически эффективное количество активного вещества.
д) Составы для инъекции.
Составы для инъекции приготавливали из следующих ингредиентов, вес.%/об. %:
Активное соединение - 0,4
Кислый фосфат натрия ВР - 0,8
Фосфат натрия ВР - 0,02
Динатрий-эдетат ВР - 0,05
Хлорид натрия ВР - 0,1
Вода для инъекций ВР - До 100
Активное соединение растворяли в воде для инъекций, при необходимости для установления pH добавляли буферирующие агенты. Затем добавляли другие агенты и затем - воду до требуемого объема. Раствор для инъекций фильтровали для удаления частиц и стерилизовали подходящими средствами (например, нагревом в автоклаве или асептической фильтрацией). Раствор загружали в виде единичных доз в ампулы или шприцы.
e) Составы для депо-инъекций.
Составы для инъекций готовили из следующих ингредиентов, вес.%/об.%:
Активное соединение - 2,5
Кунжутное масло - До 100
Активное соединение растворяли в кунжутном масле, затем стерилизовали фильтрацией и загружали асептически единичными дозами в ампулы или шприцы.
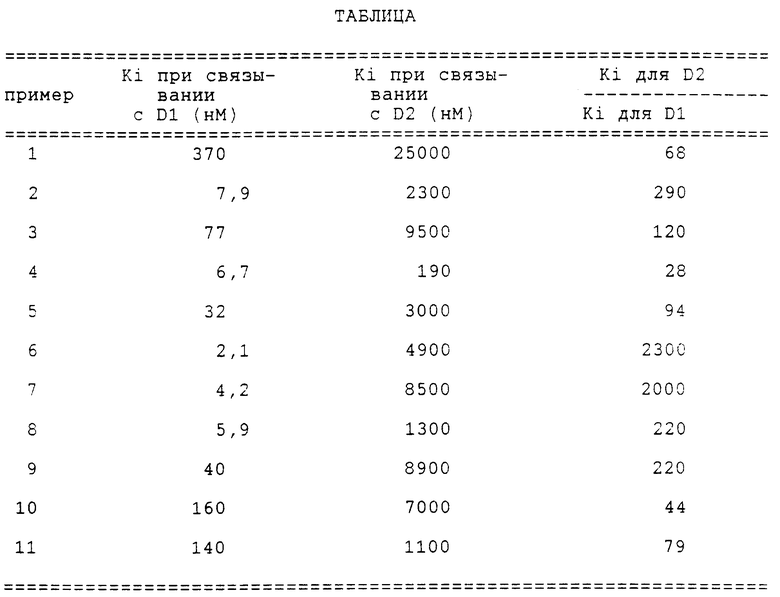
Производные тетрагидроизохинолина общей формулы I, где R1 - водород или C1-3-алкил, R2 - C1-3-алкил; Е - алкиленовая цепь, содержащая 2 или 3 атома углерода; G - насыщенная или ненасыщенная С3-8-алициклическая группа, C1-6-алкил, пиридил, фурил, тиенил или нафтил, или их фармацевтические соли могут применяться для аналгезии или лечения психозов, болезни Паркенсона. 5 з.п. ф-лы, 1 табл.
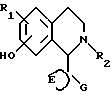 (I)
(I)
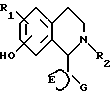
в которой R1 - водород или алкил c 1-3 атомами углерода;
R2 - алкил с 1-3 атомами углерода;
Е - алкиленoвая цепь, содержащая 2 или 3 атома углерода;
G-насыщенная или ненасыщенная алициклическая группа, содержащая 3-8 атомов углерода, алкил с 1-6 атомами углерода, незамещенный или замещенный алкилом с 1-3 атомами углерода или циклоалкилом с 3-7 атомами углерода, пиридил, фурил, тиенил или нафтил,
и их фармацевтически приемлемые соли в форме индивидуальных энантиомеров, рацематов или других смесей энантиомеров.
7-гидрокси-2-метил-1-[1-(2-метилпропил)-циклобутил] -1,2,3,4-тетрагидроизохинолин;
1-[1-(циклопентилметил)циклопропил] -7-гидрокси-2,6-диметил-1,2,3,4-тетрагидроизохинолин;
1-[1-(циклогекс-1-ен-3-ил)циклобутил] -7-гидрокси-2,6-диметил-1,2,3,4-тетрагидроизохинолин;
7-гидрокси-2,6-диметил-1-[1-(1,2,3,4-тетрагидронаф-1-ил)циклопропил] -1,2,3,4-тетрагидроизохинолин;
7 -гидрокси-2,6-диметил-1-[1-(2-тиенил)циклопропил] -1,2,3,4-тетрагидроизохинолин;
7-гидрокси-2,6-диметил-1-[1-(3-тиенил)циклопропил] -1,2,3,4-тетрагидрсизохинолин; 1-[1-(2-фурил)циклопропил]-7-гидрокси-2,6-диметил 1,2,3,4-тетрагидроизохинолин;
[7-гидрокси-2-метил-1-[1-(2-пиридил)циклобутил] -1,2,3,4-тетрагидроизохинолин;
7-гидрскси-2,6-диметил-1-[1-(2-пиридил)циклопропил] -1,2,3,4-тетрагидроизохинолин и их фармацевтически приемлемые соли в форме индивидуальных энантиомеров, рацематов или других смесей энантиомеров.
| СОЛИ 2-[2-(ДИЭТИЛАМИНО)ЭТИЛТИО] -5,6-ДИМЕТИЛБЕНЗИМИДАЗОЛА, ОБЛАДАЮЩИЕ ПРОТИВОИШЕМИЧЕСКОЙ, АНТИГИПОКСИЧЕСКОЙ И АНТИАРИТМИЧЕСКОЙ АКТИВНОСТЬЮ И 2-[2-(ДИЭТИЛАМИНО)ЭТИЛТИО] -5,6-ДИМЕТИЛБЕНЗИМИДАЗОЛ В КАЧЕСТВЕ ПРОМЕЖУТОЧНОГО ПРОДУКТА ДЛЯ СИНТЕЗА СОЛЕЙ 2-[2-(ДИЭТИЛАМИНО)ЭТИЛТИО] -5,6-ДИМЕТИЛБЕНЗИМИДАЗОЛА | 1991 |
|
RU2027709C1 |
| 1972 |
|
SU419247A1 |

