Изобретения относится к новым серосодержащим органическим соединениям, обладающим биологической активностью, в частности к сульфидам, сульфоксидам или сульфонам и их фармацевтически приемлемым солям, а также к фармацевтической композиции, их содержащей.
Известны сульфиды, сульфоксиды и сульфоны и их фармацевтически приемлемые соли, которые могут представлять активное начало многоцелевой фармацевтической композиции (см. заявку ЕР N 0 111 994, опуб. 27.06.84).
Задачей изобретения является расширение ассортимента сульфидов, сульфоксидов и сульфонов, обладающих биологической активностью.
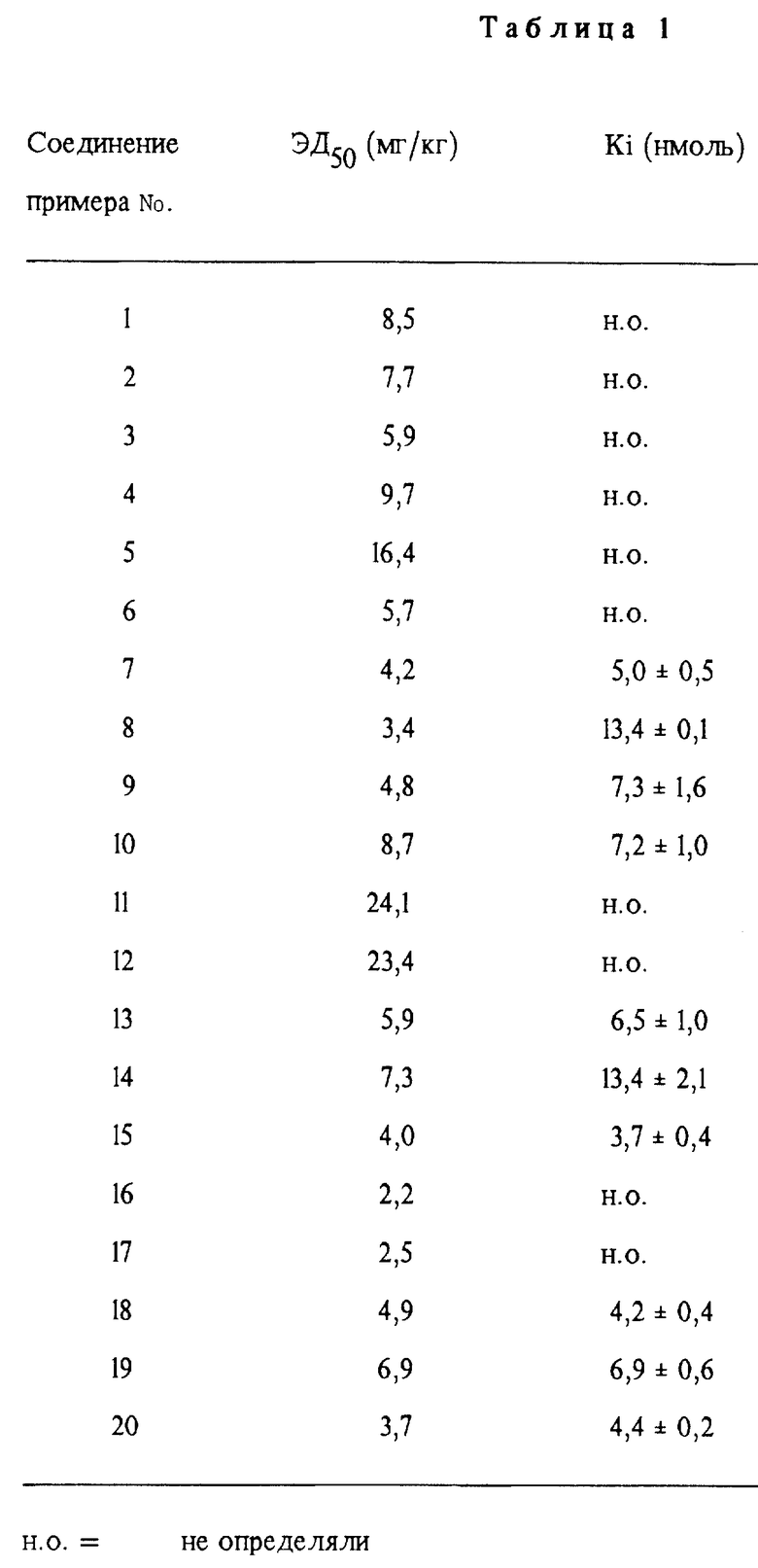
Поставленная задача решается сульфидами, сульфоксидами или сульфонами общей формулы (I)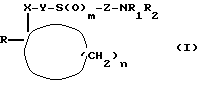
где X - карбонил или группа формулы (II)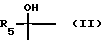
где R5 означает водород или алкил с 1 - 4 атомами углерода,
Y - алкиленовая цепь с 1 или 2 атомами углерода,
Z - алкиленовая цепь с 2-5 атомами углерода, незамещенная или замещенная по меньшей мере одной алкильной группой с 1 - 3 атомами углерода,
R означает группу формулы (III)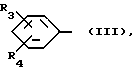
где R3 означает галоид, а R4 - водород или галоид, или же R3 или R4 вместе с атомами углерода, с которыми они связаны, образуют приконденсированное бензольное кольцо,
R1 и R2 одинаковы или различны и означают водород, разветвленную или неразветвленную алкильную группу с 1 - 4 атомами углерода, фенилалкил с 1 - 3 атомами углерода в алкильной части, при этом если R1 означает бензил, то R2 означает водород или метил,
m - 0, 1 или 2
n - 3 или 4,
и их фармацевтически приемлемыми солями.
В первую группу предпочтительных соединений входят сульфиды, сульфоксиды и сульфоны вышеприведенной общей формулы (I), где R3 означает хлор, a R4 - водород, или каждый из радикалов R3 и R4 означают хлор, или R3 и R4 вместе с атомами углерода, с которыми они связаны, образуют приконденсированное бензольное кольцо.
Во вторую группу предпочтительных соединений входят сульфиды, сульфоксиды и сульфоны вышеприведенной общей формулы (I), где R3 означает хлор, находящийся в положении 3 фенила, a R4 - водород, или каждый из радикалов R3 и R4 означает хлор, при этом атомы хлора находятся в положениях 3 и 4 фенильного кольца, или R3 и R4 вместе с фенильным кольцом, с которым они связаны, образуют 2-нафтил.
В третью группу предпочтительных соединений входят сульфиды, сульфоксиды и сульфоны вышеприведенной общей формулы (I), где X означает карбонил или группу формулы (II), где R5 означает водород.
В четвертую группу предпочтительных соединений входят сульфиды, сульфоксиды и сульфоны вышеприведенной общей формулы (I), где Y означает метилен.
В пятую группу предпочтительных соединений входят сульфиды, сульфоксиды и сульфоны вышеприведенной общей формулы (I), где Z означает алкиленовую цепь с 2-4 атомами углерода, незамещенную или замещенную по меньшей мере одной алкильной группой с 1 - 3 атомами углерода.
В шестую группу предпочтительных соединений входят сульфиды, сульфоксиды и сульфоны вышеприведенной общей формулы (I), где Z означает алкиленовую цепь с 2 - 4 атомами углерода, незамещенную или замещенную по меньшей мере одной метальной группой.
В седьмую группу предпочтительных соединений входят сульфиды, сульфоксиды и сульфоны вышеприведенной общей формулы (I), где R означает фенил, замещенный одним или двумя атомами хлора, или R означает нафтил.
В восьмую группу предпочтительных соединений входят сульфиды, сульфоксиды и сульфоны вышеприведенной общей формулы (I), где R1 означает алкил с 1 -3 атомами углерода или бензил, R2 - алкил с 1-3 атомами углерода, причем, если R1 означает бензил, то R2 означает метил.
В девятую группу предпочтительных соединений входят сульфиды, сульфоксиды и сульфоны вышеприведенной общей формулы (I), где каждый из радикалов R1 и R2 означают метил или этил или R1 означает бензил, a R2 - метил.
В частности предпочитаются сульфиды, сульфоксиды или сульфоны вышеприведенной общей формулы (I), выбранные из группы, включающей
1-[1-(3,4-дихлорфенил)циклобутил]-2-[2- (диметиламино)этилтио]этанон;
1-[1-(3,4-дихлорфенил)циклобутил] -2- [2-(диметиламино)этилсульфинил]-этанон;
1-[1-(3,4-дихлорфенил)циклобутил] -2-[2-(диметиламино) этилсульфонил]-этанон;
1-[1-(3,4-дихлорфенил)циклобутил]-2-[2-(диэтиламино)этилтио]этанон;
2-[2-(N-бензил-N-метиламино)этилтио] -1-[1-(3,4-дихлорфенил) циклобутил] -этанон;
1-[1-(3,4-дихлорфенил)циклобутил]-2-[2-(диметиламино)этилтио]этанол;
1-[1-(3,4-дихлорфенил)циклобутил]-2-[3- (диметиламино)пропилтио]этанон;
1-[1-(3,4-дихлорфенил)циклобутил] -2-[3- (диметиламино)пропилсульфонил] -этанон;
1-[1-(3,4-дихлорфенил)циклобутил]-2-[3- (диметиламино)пропилтио]этанол;
1-[1-(3,4-дихлорфенил)циклобутил]- 2-[3-(диметиламино)-2-метилпропилтио] -этанон;
2-[2-(диметиламино)этилтио]-1-[1-(2-нафтил)циклобутил]этанон;
1-[1-(3-хлорфенил)циклобутил]-2-[3-(диметиламино)пропилтио]этанон;
1-[1-(3,4-дихлорфенил)циклобутил]-2-[4- (диметиламино)бутилтио]этанон;
1-[1-(3,4-дихлорфенил)циклобутил]-2- [3-(дипропиламино)пропилтио]этанон;
1-[1-(3,4-дихлорфенил)циклобутил]-2-[3-(диметиламино)- 2-метилпропилтио] -этанол;
1-[1-(3,4-дихлорфенил)циклопентил]-2-[3- (диметиламино)пропилтио]этанон;
или их фармацевтически приемлемые соли в виде отдельных энантиомеров, рацематов или других смесей энантиомеров.
Как уже указывалось выше, соединения вышеприведенной общей формулы (I) могут иметься в виде соли с фармацевтически приемлемыми кислотами. Примером таких кислот являются гидрохлориды, гидробромиды, сульфаты, метансульфонаты, нитраты, малеаты, ацетаты, цитраты, фумараты, тартраты [например, (+)-тартраты, (-)-тартраты или их смеси, а также рацематы], сукцинаты, бензоаты и соли с аминокислотой, такой, как, например, глутаминовая кислота. Соединения общей формулы (I) и их соли могут иметься в виде сольватов, например, гидратов, которые также охватываются данным изобретением.
Некоторые соединения общей формулы (I) могут иметься в виде кристаллических форм и поэтому данное изобретение охватывает любую кристаллическую форму и смеси этих форм.
По формуле (I) и вышеприведенному значению указанных в ней радикалов можно заключить, что новые соединения могут иметь один или несколько хиральных центров. При наличии одного хирального центра соединения формулы (I) имеются в виде двух энантиомерных форм и поэтому настоящее изобретение охватывает оба энантиомера и их смеси. Отдельные энантиомеры могут получаться известными специалисту методами. Примерами таких методов являются образование соли диастереомеров, которые можно разделять, например, путем кристаллизации, образование производных или комплексов диастереомеров, которые можно разделять, например, путем кристаллизации, газожидкостной хроматографии или жидкостной хроматографии, избирательное взаимодействие одного энантиомера с подходящим реагентом, например, энзиматическая этерификация, энзиматическое окисление или восстановление или же газожидкостная или жидкостная хроматография в хиральной среде, например, на хиральном носителе, например, двуокиси кремния, со связанным хиральным лигандом, или же в присутствии хирального растворителя. Если желаемый энантиомер переводят в другую химическую форму одним из вышеуказанных методов, то необходимо последующее выделение желаемой энантиомерной формы. Также возможно синтезировать энантиомеры путем асимметричного синтеза с использованием оптически активных реагентов, субстратов, катализаторов или растворителей или же путем перевода одного энантиомера в другой энантиомер за счет асимметричной трансформации.
Если соединения общей формулы (I) имеют по меньшей мере два хиральных центра, то они имеются в виде диастереомеров, которые разделяются известными специалисту методами, например, хроматографией или кристаллизацией. При этом отдельные изомеры любой диастереомерной пары можно выделять вышеуказанными методами. Данное изобретение охватывает любой диастереоизомер соединений формулы (I), а также любые смеси диастереомеров.
Новые соединения вышеприведенной общей формулы (I) обладают антидепрессивной активностью и способностью к торможению поглощения допамина. Поэтому дополнительным объектом изобретения является фармацевтическая композиция, обладающая антидепрессивной активностью и способностью к торможению поглощения допамина, содержащая активное начало и по меньшей мере один фармацевтически приемлемый разбавитель или носитель, при этом в качестве активного начала она содержит сульфид, сульфоксид или сульфон вышеприведенной общей формулы (I).
В качестве фармацевтически приемлемого разбавителя или носителя предлагаемая композиция содержит любое известное в данной области вещество. Как правило, предлагаемая композиция содержит 0,1 - 99 вес.% активного начала. Как правило, она имеется в виде дозировочной единицы.
Примерами предпочтительной композиции для оральной дачи являются таблетки, капсулы, гранулы, сиропы, растворы и водные или масляные суспензии. Целевыми добавками в такой композиции являются общеизвестные вещества. Таблетки готовят путем смешивания активного начала с наполнителями, такими, как, например, фосфат кальция, дезинтегрирующими агентами, такими, как, например, кукурузный крахмал, смазочными веществами, такими, как, например, стеарат магния, связующими, такими, как, например, микрокристаллическая целлюлоза или поливинилпирролидон, и другими известными целевыми добавками, обеспечивающими таблетирование предлагаемой композиции. Таблетки могут быть снабжены покрытием известными приемами. Для выполнения покрытия можно применять широко известные вещества, например, фталат гидроксипропилметилцеллюлозы. Таблетки могут также иметь продленное действие, т. е. могут быть выполнены с таким расчетом, что активное начало высвобождается в течение определенного времени. Для этой цели на таблетки можно наносить известными приемами покрытие, например, из ацетатфталата целлюлозы. Кроме того, возможно переводить предлагаемую композицию в капсулы, например, из твердой или мягкой желатины, которую приготовляют известными приемами и при необходимости на них наносят покрытия желаемой характеристики. Ингредиенты капсулы можно составлять с таким расчетом, что она обеспечивает высвобождение активного начала в течение определенного времени. Как правило, таблетки и капсулы содержат 1 - 500 мг активного начала.
Другой композицией для оральной дачи является, например, водная суспензия, содержащая активное начало в водной среде в присутствии нетоксичного суспендирующего агента, такого, как, например, карбоксиметилцеллюлоза натрия, и масляная суспензия, содержащая соединение вышеприведенной общей формулы (I) в среде подходящего растительного масла, такого, как, например, арахисовое масло. Активное начало можно также переводить в гранулы, при необходимости с применением дополнительных вспомогательных веществ. Гранулы могут непосредственно применяться пациентом, или же они могут добавляться к подходящему жидкому носителю, такому, как, например, вода, перед применением. Гранулы могут также содержать дезинтегрируюшие агенты, такие, как, например, сыпучая смесь, состоящая из кислоты и соли углекислоты или бикарбоната, которая облегчает диспергирование в жидкой среде.
Подходящей для ректальной дачи формой предлагаемой композиции являются, например, суппозитории, содержащие масло какао или полиэтиленгликолевое основание.
Подходящей для парентеральной дачи формой предлагаемой композиции являются, например, стерильные суспензии или стерильные растворы в подходящем растворителе.
Композиции для местной дачи могут содержать матрицу, в которой активное начало диспергировано с таким расчетом, что оно находится в контакте с кожей с тем, чтобы активное начало могло проникать в организм трансдермально. Но активное начало может быть также диспергировано в фармацевтически приемлемом креме или в основе для мази. В случае местной аппликации активное начало должно содержаться в подходящем препарате с таким расчетом, что терапевтически эффективное количество активного начала высвобождается в течение желаемого периода времени.
Соединения вышеприведенной общей формулы (I) могут также даваться путем непрерывной инфузии, например путем внутривенного вливания, или же с применением источника, размещенного внутри тела пациента. Такими внутренними источниками являются, например, имплантированные емкости, которые содержат подлежащее инфузии активное начало. В таком случае активное начало непрерывно высвобождается, например, путем осмоса. Кроме того, имплантированные препараты могут представлять собой жидкость, такую, как, например, суспензия или раствор в фармацевтически приемлемом масле подлежащего инфузии соединения, например, в виде труднорастворимого в воде производного, такого, как, например, соль соединения вышеприведенной формулы (I) или (III) с додекановой кислотой, или же служащее в качестве основы для подлежащего инфузии соединения твердое вещество, например, синтетическая смола или воск. При этом основание может представлять собой единичное тело, содержащее активное начало, или же ряд тел, каждое из которых содержит часть апплицируемого активного начала. Активное начало должно иметься во внутреннем источнике с таким расчетом, что терапевтически эффективное количество активного начала отдается в течение требуемого периода времени.
Для некоторых целей может быть целесообразным то, что композиция содержит соединения вышеприведенной общей формулы (I) в виде мельчайших частиц, получаемых, например, путем распыления.
Предлагаемая фармацевтическая композиция может также содержать другие фармакологически активные начала, которые совместимы с соединениями вышеприведенной общей формулы (I).
Предлагаемая фармацевтическая композиция может применяться для лечения депрессий, страха, болезни Паркинсона, ожирения, расстройств познавательной способности, приступов, припадков и неврологических нарушений, таких, как, например, эпилепсия, а также в качестве нейрозащитных средств для защиты человека от вызывающих, например, удары состояний. Как правило, новые соединения дают в количестве 1 - 1000 мг/сутки, предпочтительно 5 - 500 мг/сутки в качестве единичной дозы или же нескольких доз один раз или несколько раз в сутки. Само собой разумеется, что необходимое для успешного лечения количество активного начала зависит от ряда факторов, таких, как, например, возраст пациента, серьезность заболевания и т.п. факторов. Поэтому в каждом конкретном случае врач должен подбирать подходящую дозировку.
В случае лечения болезни Паркинсона соединения вышеприведенной общей формулы (I) могут даваться в сочетании с предшественником допамина, таким, как, например, леводопамин, и/или ингибитором декарбоксилазы допамина, таким, как, например, карбидоп или бензеразид.
Соединения вышеприведенной общей формулы (I) могут получаться широко известными методами.
Так, например, соединения формулы (I), где X означает карбонил, Y - метилен, a m - 0, можно получать путем взаимодействия соединения формулы (IV)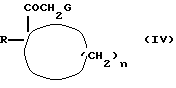
где R и n имеют вышеуказанные значения, a G означает удаляемую группу, например, галоид, такой, как, например, хлор, бром или йод,
с соединением формулы (V)
HS-Z-NR1R2 (V)
или с его солью в присутствии основания, такого, как, например, этилат натрия.
Соединение формулы (IV), где G означает галоид, можно получать путем взаимодействия соединения формулы (VI)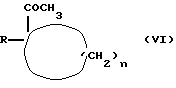
где R и n имеют вышеуказанные значения,
с агентом галогенирования, таким, как, например, бром.
Соединения формулы (VI) можно получать путем взаимодействия соединения формулы (VII)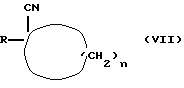
где R и n имеют вышеуказанные значения,
с металлоорганическим агентом, таким, как, например, метиллитий или соединение Гриньяра, такое, как, например, йодид метилмагния, с последующим гидролизом.
Соединения формулы (VII), где R имеет вышеуказанное значение, можно получать путем взаимодействия соединения формулы (VIII)
R-CH2CN, (VIII)
где R имеет вышеуказанное значение,
с соединением формулы (IX)
Y-(CH2)n-Y, (IX)
где Y означает удаляемую группу, например, бром, а n - 2, 3, 4 или 5, в присутствии основания, такого, как, например, гидрид натрия, гидроокись натрия или гидроокись калия, при необходимости в присутствии катализатора переноса фаз, такого, как, например, хлорид бензилтриэтиламмония.
Соединения формулы (V) можно получать путем гидролиза, например, основного гидролиза, соединения формулы (X)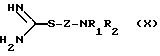
где R1, R2 и Z имеют вышеуказанные значения, или его соли.
Соединения формулы (X) можно получать путем взаимодействия соединения формулы (XI)
A-Z-NR1R2, (XI)
где Z, R1 и R2 имеют вышеуказанные значения, а А означает удаляемую группу, например, хлор, бром или йод, с
тиомочевиной.
Соединение формулы (XI) можно получать путем взаимодействия соединения формулы (XII)
HO-Z-NR1R2, (XII)
где Z, R1 и R2 имеют вышеуказанные значения,
с агентом галогенирования, таким, как, например, тионилхлорид.
Соединения формулы (I), где X означает группу формулы (II), можно получать путем восстановления соединения формулы (I), где X означает карбонил, например, бораном натрия. При этом получают соединение формулы (I), у которого R5 означает водород. Соединения формулы (I), где X означает группу формулы (II) можно также получать путем взаимодействия соединения формулы (I), где X означает карбонил, с металлоорганическим соединением, таким, как, например, литийорганическое соединение формулы R5Li, где R5 означает алкил. При этом получают соединение формулы (I), где R5 означает низший алкил.
Соединения формулы (I), где m означает 1, можно получать путем окисления соединения формулы (I), у которого m означает 0, подходящим агентом окисления, таким, как, например, монопероксифталат магния.
Соединения формулы (I), где m означает 2, можно получать путем окисления соединения формулы (I), у которого m означает 0 или 1, подходящим агентом окисления, таким, как, например, перманганат калия.
Соединения формулы (I), где m означает 0, X - карбонил, а Y - этилен, можно получать путем взаимодействия соединения вышеприведенной формулы (V) с соединением формулы (XIII)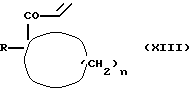
где R и n имеют вышеуказанные значения.
Соединения формулы (XIII) можно получать путем взаимодействия соединения формулы (XIV)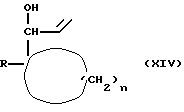
где R и n имеют вышеуказанные значения,
с агентом окисления, таким, как, например, двуокись марганца.
Соединение формулы (XIV) можно получать путем взаимодействия соединения формулы (XV)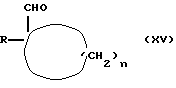
где R и n имеют вышеуказанные значения,
с металлоорганическим соединением, таким, как, например, соединение Гриньяра формулы CH2=CHMgCl.
Соединения формулы (XV) можно получать путем восстановления соединения формулы (VII) подходящим агентом, таким, как, например, гидрид диизобутилалюминия, с последующим гидролизом.
Соединение формулы (III) можно получать тем же образом, что и соединение формулы (I).
Терапевтическая активность соединений вышеприведенной общей формулы (I) исследовалась путем определения их способности к предотвращению птоза (закрытия глаз), вызываемого резерпином следующим образом. Крысы-самцы штамма Charles River CD весом 140 - 180 г подразделяли на группы по 5 животных, которые посадили в клетки, в которых им давали корм и воду в желаемом количестве. За 18 часов до начала опыта 4 крысы каждой группы отмечали карандашом с таким расчетом, что каждую крысу можно было идентифицировать. Затем корма больше не давали. На следующее утро за два часа до начала опыта крысы взвешивали, после чего произвольно определяли, какому из следующих опытов подвергали крысы.
а) Оральная дача раствора исследуемого соединения в деионизированной воде в дозе 10 мл/кг веса тела с последующим незамедлительным внутривенным впрыскиванием 1 мл/кг веса тела резерпина (0.75 мг/кг) в виде раствора в деионизированной воде, содержащего 238 ммоль лимонной кислоты, 1,02 об.% Твина 80 и 0,2 об.% бензилового спирта (крысы обработанной группы).
б) Оральная дача деионизированной воды в дозе 10 мл/кг веса тела с последующим незамедлительным внутривенным впрыскиванием 1 мл/кг веса тела резерпина (0,75 мг/кг) в виде раствора в деионизированной воде, содержащем 238 ммоль лимнонной кислоты, 1,02 об. % Твина 80 и 0,2 об.% бензилового спирта (крысы положительной контрольной группы).
в) Оральная дача деионизированной воды в дозе 10 мл/кг веса тела с последующим незамедлительным внутривенным впрыскиванием 1 мл/кг веса тела деионизированной воды, содержащей 238 ммоль лимонной кислоты, 1,02 объем.%) Твина 80 и/или 0,2 объем.% бензилового спирта (крысы отрицательной контрольной группы).
Через три часа каждую крысу помещали в прозрачную клетку размерами 42 х 42 х 22 см и за ними наблюдало лицо, которое не знало, какому опыту крысы подвергали. Степень птоза определяли по истечении 45 и 75 сек, соответственно, по следующей шкале баллов: 0 = глаз полностью открыт, 1 = глаз закрыт на 25%, 2 = глаз закрыт на 50%, 3 = глаз закрыт на 75%, 4 = глаз полностью закрыт. Затем для каждой крысы определяли среднюю степень птоза исходя из группы, обычно включающей 8 крыс. При этом среднюю степень птоза у крыс отрицательной контрольной группы высчитывали из средней степени птоза у крыс положительной контрольной группы с тем, чтобы получить степень птоза, вызываемого резерпином в отсутствии исследуемого соединения. В основу определения средней степени птоза для каждой группы обработанных крыс положено несколько доз исследуемых соединений с тем, чтобы определить дозу ЭД50, т.е. дозу, обеспечивающую 50%-ное предотвращение вызываемого резерпином птоза. Соединения с дозой ЭД50, равной ≤ 30 мг/кг, сведены в нижепредставленной таблице 1. Данный опыт является стандартным для выявления антидепрессивной активности соединений.
Способность соединений формулы (I) к взаимодействию с сайтами повторного поглощения допамина выявляли с помощью следующего опыта, направленного на определение способности соединений к торможению ин витро поглощения допамина.
Разрезанную на полосы ткань мозга крыс-самок Charles River весом 150 - 250 г гомогенизировали в холодной как лед 0,32-молярной сахарозе, при этом ткань и сахарозу берут в соотношении, равном 1 вес. часть ткани на 10 объемов сахарозы. Остатки ядер и клеток удаляли путем центрифугирования со скоростью 1500 g при температуре 4oC в течение 10 минут. Осадок удаляли, а надосадочную жидкость центрифугировали со скоростью 18 000 g при температуре 4oC в течение 10 минут. Получаемый при этом сырой синаптосомальный остаток повторно суспендировали в буфере Кребса-Гензелайта (количество эквивалентно 4,2 мг влажного веса ткани/мл).
Сырые синаптосомы инкубировали при встряхивании на водяной бане при температуре 37oC в течение 15 минут. Аликвотные количества (150 мкл, что является эквивалентным 0,625 мг влажного веса ткани/на трубку) подавали в трубки, содержащие 275 мкл буфера Кребса-Гензелайта и 50 мкл буфера Кребса-Гензелайта (общее поглощение) или 50 мкл исследуемого соединения (применяемого в 10 различных концентрациях от 4-11 моль) или же 50 мкл GBR 12909 (в концентрациях от 5-10 моль; неспецифичное поглощение). Поглощение вызывали путем добавления 25 мкл (2,5 ммоль) свежеполученного [3H]допамина с последующим перемешиванием и подачей трубок на 5 минут в качающуюся водяную баню при температуре 37oC.
Поглощение заканчивали путем вакуумной фильтрации с помощью фильтров марки Скатрон 11735 с применением аппарата марки Скатрон для сбора клеток. Затем фильтры промывали холодным как лед рассолом, взятым в количестве 8 мл. Фильтры в виде диска вводили в пробирки, куда добавляли сцинтилляционную жидкость, после чего радиоактивность определяли путем жидкостного сцинтилляционного подсчета.
%-ное торможение специфичного поглощения меченного тритием лиганда рассчитывали для каждой концентрации исследуемого соединения. Полученные при этом данные используют для составления кривых торможения, по которым можно было определять концентрацию соединений, которые обеспечивают 50%-ное торможение специфичного поглощения (KT50). Затем константу торможения (Ki) определяли с применением следующего уравнения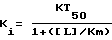
где [L] означает концентрацию применяемого меченного тритием лиганда, а Km - сродство сайта поглощения к лиганду. Значение Ki соединений формулы (I), которые сведены в таблице 1 (см. в конце описания), представляют собой среднее значение (± стандартное отклонение) трех независимых опытов.
Настоящее изобретение далее иллюстрируется следующим и примерами, в которых целевые продукты характеризуют данными следующих анализов: газово-жидкостной хроматографии, высокопроизводительной жидкостной хроматографии, элементарного анализа, спектроскопии ЯМР и ИК спектроскопии.
Пример 1
Йодид метилмагния получают в атмосфере азота путем прикапывания раствора 93,8 г метилйодида в 100 мл диэтилового эфира к перемешиваемой суспензии 15,1 г магниевых стружек в 100 мл диэтилового эфира при комнатной температуре. Когда начинается экзотермическая реакция смесь нагревают с обратным холодильником. После окончания добавления раствора метилйодида реакционную смесь перемешивают в течение 30 минут, после чего каплями добавляют при комнатной температуре раствор 100 г 1-(3,4-дихлорфенил)- циклобутанкарбонитрила в 80 мл диэтилового эфира. Получаемую при этом суспензию перемешивают, нагревают с обратным холодильником в течение трех часов и затем перемешивают еще при комнатной температуре в атмосфере азота в течение 16 часов. Получаемое при этом твердое вещество собирают путем фильтрации, тщательно промывают диэтиловым эфиром, порциями добавляют к холодной как лед смеси 400 мл воды и 25 мл концентрированной соляной кислоты. Получаемую смесь нагревают при температуре 95oC в течение 1 часа при случайном перемешивании и затем охлаждают до комнатной температуры. Получаемый продукт экстрагируют 6 раз диэтиловым эфиром, взятым в количестве по 150 мл, экстракты сушат над сульфатом магния и растворитель удаляют в вакууме. Получают 105 г масла, которое в результате перегонки дает 89,6 г 1-[1-(3,4- дихлорфенил)-циклобутил]этанона с точкой кипения 116 - 118oC/0,13 мбар.
Раствор 18 мл брома в 80 мл хлороформа каплями добавляют при температуре 10 - 15oC в течение 90 минут к перемешиваемому раствору 89,6 г 1-[1-(3,4-дихлорфенил)-циклобутил] этанона в смеси 120 мл метанола и 20 мл хлороформа. После окончания процесса добавления реакционную смесь перемешивают при комнатной температуре в течение 1 часа и затем подают в избыток ледяной воды. Получаемый водный слой отделяют и продукт экстрагируют два раза дихлорметаном, взятым в количестве по 150 мл. Объединенные органические растворы промывают два раза насыщенным раствором бикарбоната натрия, взятым в количестве по 200 мл, и затем водой, сушат над сульфатом кальция и растворитель удаляют в вакууме. Получаемое при этом масло подвергают перегонке с получением 88,31 г 2-бром-1-[1-(3,4- дихлорфенил)- циклобутил]этанона с точкой кипения 148 - 154oC/0,66 мбар.
Раствор этилата натрия, получаемый из 0,69 г натрия и 60 мл этанола, добавляют к перемешиваемой суспензии 2,12 г 2-(диметиламино)этантиола в виде гидрохлорида в 30 мл этанола и получаемую смесь перемешивают при комнатной температуре в течение 1 часа. Добавляют раствор 4,8 г 2-бром-1-[1-(3,4-дихлорфенил)циклобутил] этанона, полученного описанным выше образом, в 30 мл этанола и получаемую смесь перемешивают при комнатной температуре в течение дальнейших 2 часов. Затем реакционную смесь перемешивают при температуре 50oC в течение 1 часа и растворитель удаляют в вакууме. Получаемый остаток разбавляют 30 мл воды и получаемый продукт два раза экстрагируют диэтиловым эфиром, взятым в количестве по 50 мл. Экстракты сушат над сульфатом магния и растворитель удаляют в вакууме. Получают 5,1 г масла, которое растеряют в диэтиловом эфире, и получаемый раствор насыщают хлористым водородом. Растворитель удаляют в вакууме с получением 5,1 г масла, которое очищают путем флеш-хроматографии на силикагеле с применением в качестве элюента сначала дихлорметана и затем смеси этилацетата и метанола в соотношении 1 : 1. Продуктсодержащие фракции объединяют и растворитель удаляют в вакууме. Получают 2,9 г гидрохлорида 1-[1-3,4-дихлорфенил)циклобутил]-2-[2- (диметиламино)этилтио]этанона в качестве масла.
Пример 2
1,7 г 1-[1-(3,4-дихлорфенил)циклобутил]-2-[2- (диметиламино)этилтио]этанона (получаемого из соответствующего гидрохлорида, получаемого аналогично примеру 1) растворяют в 10 мл этанола и добавляют раствор 1,6 г 87 %-ного монопероксифталата магния в виде гексагидрата в 75 мл воды. Добавляют еще 20 мл этанола и получаемую реакционную смесь перемешивают при комнатной температуре в течение 1 часа. Растворитель удаляют в вакууме, остаток разбавляют водой и продукт экстрагируют этилацетатом. Получаемый экстракт сушат над сульфатом магния и растворитель удаляют в вакууме. При этом получают 1,8 г масла, которое растворяют в этаноле и получаемый раствор насыщают хлористым водородом. Получаемое при этом твердое вещество собирают путем фильтрации и кристаллизуют из этанола. Получают 0,5 г гидрохлорида 1-[1-(3,4-дихлорфенил)циклобутил)-2-[2-(диметиламино) этилсульфинил]этанона в качестве белого твердого вещества с точкой плавления 184 - 185oC.
Пример 3
Раствор 1,2 г перманганата калия в 40 мл воды добавляют к раствору 1,4 г 1-[(3,4-дихлорфенил)циклобутил] -2-[2- (диметиламино)-этилтио] этанона (получаемого из соответствующего гидрохлорида, получаемого аналогично примеру 1), 0,1 г бромида тетра-н-бутиламмония и 10 мл уксусной кислоты в 30 мл толуола и получаемую при этом смесь перемешивают при комнатной температуре в течение 22 часов. Насыщенный водный раствор бисульфита натрия добавляют к реакционной смеси до исчезновения пурпурной окраски, после чего получаемый прозрачный раствор нейтрализуют путем добавления твердого карбоната калия. Получаемый продукт экстрагируют толуолом, экстракты сушат над сульфатом магния и растворитель удаляют в вакууме. При этом получают 1,7 г масла, которое растворяют в этаноле и добавляют избыток эфирного раствора соляной кислоты. Получаемый раствор упаривают с получением масла, которое с помощью дихлорметана растирают в порошок. Получают 0,1 г гидрохлорида 1-[1-(3,4- дихлорфенил)циклобутил]-2-[2-(диметиламино)этилсульфонил]этанона в качестве белого твердого вещества с точкой плавления 208 - 210oC.
Пример 4
Раствор этилата натрия, получаемый из 0,5 г натрия и 40 мл этанола, добавляют к раствору гидрохлорида 1,7 г 2-(диэтиламино)этантиола в 30 мл этанола и получаемую реакционную смесь перемешивают при комнатной температуре в течение 1 часа. Раствор 3,2 г 2-бром-1-[1-(3,4-дихлорфенил)циклобутил]этанона (получаемого аналогично примеру 1) в 30 мл этанола добавляют к реакционной смеси, после чего перемешивают при комнатной температуре в течение 90 минут. Растворитель удаляют в вакууме и остаток разбавляют 25 мл воды. Продукт экстрагируют два раза диэтиловым эфиром, взятым в количестве 50 мл, экстракты сушат над сульфатом магния и раствор упаривают в вакууме. Получают 3,5 г масла, которое очищают флеш-хроматографии на силикагеле. При этом соблюдают следующую последовательность элюентов: смесь дихлорметана и этилацетата в соотношении 1 : 1, этилацетат и смесь этилацетата и метанола в соотношении 9 : 1. Продуктсодержащие фракции объединяют и растворители удаляют в вакууме. Получаемое при этом масло растворяют в 15 мл диэтилового эфира и раствор насыщают хлористым водородом. Получаемый твердый осадок собирают путем фильтрации, промывают небольшим количеством диэтилового эфира и сушат в вакууме. Получают 1,6 г гидрохлорида 1-[1-(3,4-дихлорфенил)циклобутил] -2-[2- (диметиламино)этилтио] этанона с точкой плавления 109-110oC.
Пример 5
Раствор этилата натрия, получаемого из 0,2 г натрия и 25 мл этанола, добавляют к раствору 1,8 г 2-(N-бензил-N- метиламино)этантиола в 20 мл этанола и получаемую при этом смесь перемешивают при комнатной температуре в течение 1 часа. Добавляют раствор 3.2 г 2-бром-[1-(3,4-дихлорфенил)-циклобутил] этанона, получаемого аналогично примеру 1, в 20 мл этанола и получаемую смесь перемешивают при комнатной температуре в течение 3 часов. Растворитель удаляют в вакууме и остаток разбавляют 15 мл воды. Получаемый при этом продукт экстрагируют дихлорметаном, экстракты сушат над хлоридом кальция и растворитель удаляют в вакууме. Получаемое при этом масло растворяют в этаноле и получаемый раствор насыщают хлористым водородом. Получаемое при этом твердое вещество собирают путем фильтрации, промывают небольшим количеством метанола и сушат в вакууме. Получают 1,2 г гидрохлорида 2-[2-(N-бензил-N- метиламино)этилтио]-1-(3,4-дихлорфенил)циклобутил)этанона с точкой плавления 159 - 163oC.
Пример 6
Смесь 2,0 г 1-[1-(3,4-дихлорфенил)циклобутил]-2-[2- (диметиламино)этилтио]этанона, получаемого из соответствующего гидрохлорида, получаемого аналогично примеру 1, и 2,2 г борана натрия в 80 мл пропан-2-ола перемешивают при комнатной температуре в течение 16 часов.
Получаемую при этом суспензию осторожно разбавляют 15 мл ацетона с последующим добавлением избытка насыщенного водного раствора хлористого аммония. Получаемую смесь сгущают в вакууме, остаток разбавляют водой и получаемый продукт экстрагируют диэтиловым эфиром. Экстракты сушат над сульфатом магния и растворитель удаляют в вакууме. При этом получают масло, которое растворяют в этилацетате и раствор насыщают хлористым водородом. Получаемый при этом раствор разбавляют диэталовым эфиром. Образующуюся при этом смолу и надосадочную жидкость удаляют путем декантирования, после чего смоле дают концентрироваться при комнатной температуре. Образующееся при этом масло отделяют путем декантирования и растворяют в метаноле. В результате упаривания растворителя получают 1,55 г гидрохлорида 1-[1-(3,4-дихлорфенил)циклобутил] -2- [2-(диметиламино)этилтио]этанола в качестве бесцветного масла.
Пример 7
Смесь 200 г гидрохлорида 1-хлор-3-(диметиламино)пропана, 98,1 тиомочевины и 1 л этанола перемешивают и нагревают с обратным холодильником в течение 25 часов. Получаемый раствор охлаждают до комнатной температуры и добавляют этилацетат до постоянной опалесценции. Реакционную смесь оставляют стоять в течение ночи при температуре 4oC, после чего фильтруют. Получают 283 г дигидрохлорида S-[3-(диметиламино)пропил]изотиомочевины в качестве бесцветного твердого вещества с точкой плавления 155 - 159oC. Данный гидрохлорид растворяют в 340 мл воды и на получаемый при этом раствор подают слой диэтилового эфира. Реакционную смесь охлаждают на льду и каплями добавляют 97 мл 25-молярного раствора гидроокиси натрия. После окончания добавления гидроокиси натрия реакционную смесь перемешивают и нагревают с обратным холодильником в течение 2 часов. Раствор охлаждают до комнатной температуры и продукт экстрагируют диэтиловым эфиром. Экстракт сушат над сульфатом магния и растворитель удаляют в вакууме. При этом получают 70,6 г прозрачного масла, 35 г которого растворяют в диэтиловом эфире. Получаемый при этом раствор насыщают хлористым водородом, в результате чего получают бесцветное твердое вещество, которое собирают путем фильтрации, промывают диэтиловым эфиром и сушат в вакууме. Получают 21,2 г гидрохлорида 3-(диметиламино) пропантиола с точкой плавления 103 - 107oC.
Раствор этилата натрия, получаемый из 1,0 г натрия и 60 мл этанола, добавляют к суспензии 3,5 г гидрохлорида 3-(диметаламино)пропантиола в 50 мл этанола и получаемую при этом реакционную смесь перемешивают при комнатной температуре в течение 1 часа, после чего добавляют 9,35 г 2-бром-1-[1-(3,4- дихлорфенил)циклобутил] этанона, получаемого аналогично примеру 1, в 30 мл этанола и смесь перемешивают при комнатной температуре в течение 25 часов. Растворитель удаляют в вакууме и к получаемому твердому остатку добавляют 30 мл воды. Экстрагируют этилацетатом, экстракт сушат над сульфатом магния и растворитель удаляют в вакууме. Получают 10,5 г масла, которое растворяют в этилацетате, и получаемый раствор насыщают хлористым водородом. В результате вакуумного удаления растворителя получают 9,1 г масла, которое очищают путем флеш-хроматографии на силикагеле с применением в качестве элюента смеси этилацетата и метанола в соотношении 1 : 1. Содержащие продукт фракции объединяют и растворитель удаляют в вакууме. Получают 5 г масла, к которому добавляют избыток 5-молярного водного раствора гидроокиси натрия, и получаемое при этом свободное основание экстрагируют два раза диэтиловым эфиром, взятым в количестве по 25 мл. Экстракты промывают водой, сушат над сульфатом магния и растворитель упаривают в вакууме. Получаемое при этом масло растворяют в диэтиловом эфире и насыщают хлористым водородом. Получаемое при этом белое твердое вещество собирают путем фильтрации, промывают небольшим количеством диэтилового эфира и сушат в вакууме. Получают 1,6 г гидрохлорида 1-[1-(3,4-ди- хлорфенил)-циклобутил]-2-[3-(диметиламино)пропилтио]этанона с точкой плавления 115 - 118oC.
Пример 8
Раствор 3,1 г перманганата калия в 95 мл воды добавляют к смеси 3,4 г 1-[1-(3,4-дихлорфенил)циклобутил] -2-[3- (диметиламино)-пропилтио] этанона, получаемого из соответствующего гидрохлорида, получаемого аналогично примеру 7, 0,3 г бромида тетра-н-бутиламмония, 25 мл уксусной кислоты и 80 мл толуола и получаемую при этом смесь перемешивают при комнатной температуре в течение 72 часов. К получаемому при этом коричневому раствору добавляют насыщенный раствор метабисульфита натрия до изменения на оранжевую окраску (после добавления примерно 100 мл указанного раствора), после чего реакционную смесь нейтрализуют путем добавления твердого карбоната калия. Продукт три раза экстрагируют этилацетатом, взятым в количестве по 300 мл (фильтрация необходима для удаления твердых веществ на границе раздела фаз), экстракты объединяют, сушат над сульфатом магния и растворитель удаляют в вакууме. Получают 3,7 г коричневого масла, которое очищают путем флеш-хроматографии на силикагеле с применением в качестве элюента сначала смеси толуола и триэтиламина в соотношении 4 : 1 и затем смеси толуола и триэтиламина в соотношении 1:1. Содержащие продукт фракции объединяют и растворитель удаляют в вакууме. Получают 0,7 г коричневого масла, которое кристаллизует при стоянии.
Получаемое твердое вещество растворяют в смеси 50 мл горячего диэтилового эфира и 8 мл этилацетата и получаемый при этом раствор фильтруют, охлаждают и насыщают хлористым водородом. Получаемое при этом твердое вещество собирают путем фильтрации, сушат в вакууме при температуре 40oC в течение 24 часов, измельчают и снова сушат в вакууме при температуре 40oC в течение дальнейших 24 часов. Получают 0,4 г гидрохлорида 1-[1-(3,4-дихлорфенил)циклобутил] -2-[3- (диметиламино)пропилсульфонил]этанона в качестве белого твердого вещества с точкой плавления 168 - 176oC.
Пример 9
Раствор 4,6 г 1-[1-(3,4-дихлорфенил)циклобутил]-2-[3- (диметиламино)пропилтио]этанона, получаемого из соответствующего гидрохлорида, получаемого аналогично примеру 7, в 75 мл пропан-2-ола каплями добавляют к перемешиваемой суспензии 4,8 г борана натрия в 100 мл пропан-2-ола при комнатной температуре в атмосфере азота и получаемую при этом смесь перемешивают при комнатной температуре в течение 93 часов. Получаемую при этом суспензию осторожно разбавляют 33 мл ацетона с последующим добавлением 100 мл насыщенного водного раствора хлористого аммония. Получаемую при этом нейтральную смесь сгущают в вакууме, остаток разбавляют 100 мл воды и продукт экстрагируют три раза диэтиловым эфиром, взятым в количестве по 200 мл. Экстракты объединяют, сушат над сульфатом магния и растворитель удаляют в вакууме. Получают 4 г желтого масла, которое очищают путем флеш-хроматографии на силикагеле с последовательным применением в качестве элюента следующих смесей: смеси дихлорметана и промышленных метилированных спиртов в соотношении 9 : 1, смеси дихлорметана и промышленных метилированных спиртов в соотношении 4 : 1, смеси дихлорметана и промышленных метилированных спиртов в соотношении 1: 1. Продуктсодержащие фракции объединяют и растворитель удаляют в вакууме. Получают 1,44 г 1-[1-(3,4- дихлорфенил)циклобутил]-2-[3-(диметиламино)пропилтио]этанола в качестве светло-зеленого масла.
Пример 10
Смесь 200 г гидрохлорида 1-хлор-3-(диметиламино)-2- метилпропана, 97,3 г тиомочевины и 950 мл этанола перемешивают и нагревают с обратным холодильником в течение 72 часов. Получаемому раствору дают охлаждаться и растворитель удаляют в вакууме. Получаемый при этом остаток растворяют в небольшом количестве этанола и диэтиловый эфир добавляют до первой постоянной опалесценции. Реакционную смесь оставляют стоять при температуре 4oC в течение 16 часов, после чего растворитель удаляют в вакууме. Получаемое при этом восково-маслянистое вещество сушат в вакууме над хлоридом кальция в течение 48 часов и затем растирают в порошок с помощью пропан-2-ола. Получаемое при этом твердое вещество собирают путем фильтрации, промывают пропан-2-олом и сушат в вакууме. Получают 90 г дигидрохлорида S-[3- (диметиламино)2-метилпропил]изотиомочевины в качестве светло-коричневого твердого вещества.
Раствор 19,3 г гидроокиси натрия в 20 мл воды каплями добавляют при температуре к перемешиваемому раствору 60 г дигидрохлорида S-[3-(диметиламино)-2-метил-пропил)изотиомочевины в 100 мл воды. Перемешиваемую смесь нагревают при температуре 95oC в течение 2 часов, после чего ей дают охлаждаться. Продукт экстрагируют 4 раза диэтиловым эфиром, взятым в количестве по 70 мл, объединенные экстракты сушат над сульфатом натрия и растворитель удаляют в вакууме. Маслянистый остаток растворяют в диэтиловом эфире и раствор насыщают хлористым водородом. Получаемое при этом твердое вещество собирают путем фильтрации, промывают диэтиловым эфиром и сушат в вакууме над пятиокисью фосфора в течение 24 часов. При этом получают 30 г гидрохлорида 3-(диметиламино)-2-метил-пропантиола в качестве белого твердого вещества.
Раствор этилата натрия, получаемый из 3 г натрия и 300 мл этанола, добавляют к суспензии 10,3 г 3-(диметиламино)-2- метилпропантиола в виде гидрохлорида в 150 мл этанола при комнатной температуре в атмосфере азота, после чего реакционную смесь перемешивают при комнатной температуре в течение 1 часа. Добавляют 20,4 г раствора 2-бром-1-[1-(3,4-дихлорфенил)циклобутил]этанона, получаемого аналогично примеру 1, в 130 мл этанола и получаемую при этом смесь перемешивают при комнатной температуре в течение 24 часов. Растворитель удаляют в вакууме и остаток разбавляют 150 мл воды. Продукт экстрагируют 4 раза дихлорметаном, взятым в количестве по 100 мл, объединенные экстракты сушат над сульфатом натрия и растворитель удаляют в вакууме. Получаемый при этом остаток растворяют в диэтиловом эфире и раствор насыщают хлористым водородом. Получаемое при этом твердое вещество собирают путем фильтрации, промывают диэтиловым эфиром и сушат в вакууме в течение 24 часов. В результате кристаллизации из смеси этилацетата и пропан-2-ола в соотношении 2 : 1 получают 0,55 г гидрохлорида 1-[1-(3,4- дихлорфенил)-2-[3-(диметиламино)-2-метилпропилтио)этанона в качестве белого твердого вещества с точкой плавления 136 - 137oC.
Пример 11
Йодид метилмагния получают путем прикапывания в атмосфере азота раствора 48,3 г метилйодида в 72 мл диэтилового эфира к перемешиваемой суспензии 8,2 г магниевых стружек в 60 мл диэтилового эфира, причем процесс сначала осуществляют при комнатной температуре и, когда начинается экзотермическая реакция, смесь нагревают с обратным холодильником. После окончания добавления раствора метилйодида реакционную смесь перемешивают при комнатной температуре в течение 30 минут, после чего каплями добавляют при комнатной температуре раствор 48,2 г 1-(2- нафтил)циклобутанкарбонитрила в 100 мл толуола. Получаемую при этом смесь перемешивают при комнатной температуре в течение 16 часов. Получаемое твердое вещество собирают путем фильтрации, промывают диэтиловым эфиром и порциями добавляют к смеси 125 мл концентрированной соляной кислоты и 200 мл воды. Получаемую при этом смесь нагревают при температуре примерно 95oC в течение 10 минут, охлаждают и продукт экстрагируют три раза толуолом, взятым в количестве по 200 мл. Экстракты промывают 200 мл воды, сушат над сульфатом магния и растворитель удаляют в вакууме. Получают 45 г масла, которое растирают в порошок с помощью петролейного эфира, имеющего пределы кипения 45 - 60oC. Получаемое при этом твердое вещество собирают путем фильтрации и сушат в вакууме. Получают 35 г 1-[1-(2-нафтил)циклобутил]этанона.
Раствор 4,3 мл брома в 20 мл хлороформа каплями добавляют при температуре 10 - 15oC в течение 30 минут к перемешиваемому раствору 20 г 1-[1-(2-нафтил)циклобутил] -этанона в смеси 20 мл метанола и 30 мл хлороформа. Получаемую при этом смесь перемешивают при комнатной температуре в течение 90 минут, подают в 300 мл ледяной воды и продукт экстрагируют три раза дихлорметаном, взятым в количестве по 150 мл. Экстракты последовательно промывают насыщенным водным раствором бикарбоната натрия и водой, сушат над хлоридом кальция и растворитель удаляют в вакууме. При этом получают 24,0 г 2-бром-1- [1-(2-нафтил)циклобутил]-этанона в качестве масла.
Раствор этилата натрия, получаемого из 6,5 г натрия и 100 мл этанола, добавляют к суспензии 6,4 г 2-(диметиламино)этантиола в виде гидрохлорида в 10 мл этанола и получаемую смесь перемешивают при комнатной температуре в течение 1 часа. Добавляют раствор 19,5 г 2-бром-1-[1-(2-нафтил)циклобутил] этанона, получаемого описанным выше образом, в 50 мл этанола, после чего реакционную смесь перемешивают при комнатной температуре в течение 18 часов. Растворитель удаляют в вакууме и остаток разбавляют 200 мл воды. Продукт экстрагируют два раза этилацетатом, взятым в количестве по 200 мл, экстракты сушат над сульфатом магния и растворитель удаляют в вакууме. Получают 11,0 г масла, которое растворяют в этилацетате и получаемый раствор насыщают хлористым водородом. В результате вакуумного удаления растворителя получают 1,5 г масла, которое растирают в порошок с помощью смеси пропан-2-ола, диэтилового эфира и этилацетата. Получаемое при этом твердое вещество собирают путем фильтрации и сушат в вакууме. Получают 1,7 г гидрохлорида 2-[2- (диметиламино)этилтио]-1-[1-(2-нафтил)циклобутил]этанона в качестве кремнеобразного твердого вещества с точкой плавления 95 - 102oC.
Пример 12
138 мл 3-молярного раствора йодида метилмагния в диэтиловом эфире каплями добавляют к перемешиваемому раствору 53 г 1-(3- хлорфенил)циклобутанкарбонитрила в 100 мл диэтилового эфира при температуре ОoC в атмосфере азота. Реакционную смесь перемешивают при комнатной температуре в течение 24 часов. Получаемое при этом твердое вещество собирают путем фильтрации, тщательно промывают диэтиловым эфиром и затем порциями добавляют к холодной как лед смеси 200 мл воды и 125 мл концентрированной соляной кислоты. Получаемую желтую суспензию нагревают при температуре 95oC в течение 1 часа при случайном перемешивании и затем охлаждают до комнатной температуры. Продукт экстрагируют 5 раз диэтиловым эфиром, взятым в количестве по 100 мл, объединенные экстракты промывают 2 раза водой, взятой в количестве по 100 мл, сушат над сульфатом магния и растворитель удаляют в вакууме. В результате перегонки получаемого при этом масла получают 47,5 г 1-[1-(3-хлорфенил)циклобутил]этанона с точкой кипения 108 - 109oC/2 мбар.
Раствор 9,9 мл брома в 50 дихлорметана прикапывают при температуре 10 - 15oC в течение трех часов к перемешиваемому раствору 38 г 1-[1-(3-хлорфенил)циклобутил] -этанона в смеси 75 мл метанола и 15 мл дихлорметана. После окончания процесса добавления реакционную смесь перемешивают при комнатной температуре в течение 150 минут, после чего ее подают в избыток ледяной воды. Водный слой отделяют и продукт экстрагируют три раза дихлорметаном, взятым в количестве по 90 мл. Объединенные органические растворы промывают два раза насыщенным водным раствором бикарбоната натрия, взятым в количестве по 100 мл, и затем 100 мл воды, сушат над хлоридом кальция и растворитель удаляют в вакууме. Получают 47 г 2-бром-1- [1-(3-хлорфенил)циклобутил]этанона в качестве масла.
Раствор этилата натрия, получаемого из 5,3 г натрия и 500 мл этанола, добавляют к перемешиваемой суспензии 16,2 г 3-(диметиламино)пропантиола в виде гидрохлорида, получаемого аналогично примеру 7, в 250 мл этанола в атмосфере азота и получаемую при этом смесь перемешивают при комнатной температуре в течение 1 часа. Добавляют раствор 30 г 2-бром-1-[1-(3- хлорфенил)циклобутил] этанона в 130 мл этанола и получаемую смесь перемешивают при комнатной температуре в течение дальнейших 24 часов. Растворитель удаляют в вакууме и остаток разбавляют 200 мл воды. Продукт экстрагируют четыре раза дихлорметаном, взятым в количестве по 100 мл. Объединенные экстракты сушат над сульфатом магния и растворитель удаляют в вакууме. Получают 31 г красно- коричневого масла, 8 г которого растворяют в диэтиловом эфире, и получаемый раствор насыщают хлористым водородом. Растворитель удаляют в вакууме и остаток растирают в порошок с помощью диэтилового эфира. Получаемое при этом твердое вещество собирают путем фильтрации и кристаллизуют из смеси пропан-2-ола и диэтилового эфира. Получают 1,7 г гидрохлорида 1-[1-(3- хлорфенил)циклобутил]-2-[3-(диметиламино)пропилтио]этанона в качестве белого твердого вещества с точкой плавления 66 - 68oC.
Пример 13
88,5 г 4-(диметиламино)бутанола прикапывают при температуре 0oC в течение 2 часов к перемешиваемому тионилхлориду, взятому в количестве 93,4 г, после чего реакционную смесь перемешивают при комнатной температуре в течение 1 часа и подают в 500 мл этанола. Перемешиваемый раствор нагревают с обратным холодильником в течение 10 минут и затем растворитель удаляют в вакууме. Получаемый при этом твердый остаток кристаллизуют из этанола с получением белого твердого вещества, которое собирают путем фильтрации, промывают этанолом и сушат в вакууме при комнатной температуре в течение 24 часов. Получают 115 г гидрохлорида 1-хлор-4-(диметиламино)бутана в качестве белого твердого вещества с точкой плавления 100 - 105oC.
Перемешиваемую смесь 115 г гидрохлорида 1-хлор-4- (диметиламино)-бутана, 51,9 г тиомочевины и 500 мл этанола нагревают с обратным холодильником в течение 24 часов и затем ее оставляют стоять при температуре 4oC в течение 24 часов. Получаемое при этом твердое вещество собирают путем фильтрации, промывают диэтиловым эфиром и сушат в вакууме при комнатной температуре в течение 24 часов. Получают 140 г дигидрохлорида S-[4-(диметиламино)бутил]изотиомочевины в качестве беловатого твердого вещества с точкой плавления 179 - 182oC.
Раствор 16,4 г гидроокиси натрия в 16,5 мл воды прикапывают при температуре 0oC в атмосфере азота к перемешиваемому раствору 51 г дигидрохлорида S-[4-(диметиламино)бутил]-изотиомочевины в 60 мл воды, получаемую смесь нагревают при температуре 95oC в течение 2 часов и затем ей дают охлаждаться до комнатной температуры. Добавляют 100 мл воды и продукт последовательно экстрагируют 50 мл диэтилового эфира, 3 раза дихлорметаном, взятым в количестве по 50 мл, и 2 раза диэтиловым эфиром, взятым в количестве по 50 мл. Объединенные органические растворы сушат над сульфатом магния и растворители удаляют в вакууме. Остаток растворяют в диэтиловом эфире и раствор насыщают хлористым водородом. Получаемое при этом твердое вещество собирают путем фильтрации и сушат в вакууме при комнатной температуре. Получают 21 г гидрохлорида 4-(диметиламино) бутантиола в качестве белого твердого вещества, которое без дальнейшей очистки применяют на следующей стадии.
Раствор этилата натрия, получаемый из 1,6 г натрия и 175 мл этанола, добавляют при комнатной температуре в атмосфере азота к перемешиваемой суспензии 5,5 г гидрохлорида 4-(диметиламино)бутантиола в 75 мл этанола и получаемую смесь перемешивают при комнатной температуре в течение 1 часа. Добавляют раствор 11 г 2-бром-1-[1-(3,4-дихлорфенил)циклобутил]этанона, получаемого аналогично примеру 1, в 40 мл этанола и получаемую при этом смесь перемешивают при комнатной температуре в течение 48 часов. Растворитель удаляют в вакууме и остаток разбавляют 200 мл воды. Продукт экстрагируют 4 раза дихлорметаном, взятым в количестве по 90 мл, объединенные экстракты сушат над сульфатом натрия и растворитель удаляют в вакууме. Получаемое при этом масло очищают путем колоночной хроматографии на силикагеле с применением в качестве элюента смеси толуола и триэтиламина в соотношении 9 : 1. Содержащие продукт фракции объединяют и растворитель удаляют в вакууме. Остаток растворяют в диэтиловом эфире и получаемый раствор насыщают хлористым водородом. Получаемое при этом твердое вещество собирают путем фильтрации, промывают диэтиловым эфиром, сушат в вакууме над пятиокисью фосфора в течение 24 часов и перекристаллизовывают из смеси этилацетата и этанола в соотношении 4 : 1. Получаемое твердое вещество собирают путем фильтрации, промывают этилацетатом и сушат в вакууме над пятиокисью фосфора при комнатной температуре в течение 48 часов. При этом получают 566 г гидрохлорида 1-[1-(3,4-дихлорфенил)циклобутил] -2- [4-диметиламино)-бутилтио] этанона в качестве белого твердого вещества с точкой плавления 129 - 130oC.
Пример 14
32,9 г 3-(дипропиламино)пропанола прикапывают при температуре 0oC в течение 90 минут к перемешиваемому тионилхлориду, взятому в количестве 15,7 мл. После окончания процесса добавления получаемую смесь перемешивают при комнатной температуре в течение 2 часов и затем подают в 250 мл этанола. Перемешиваемую смесь нагревают с обратным холодильником в течение 10 минут и затем растворитель удаляют в вакууме. Получают 42 г гидрохлорида 1-хлор-3- (дипропиламино)-пропана в качестве беловатого твердого вещества, которое без дальнейшей очистки используют на следующей стадии.
Перемешиваемую смесь 240 г 1-хлор-3-(дипропиламино)-пропана в виде гидрохлорида, 15,5 г тиомочевины и 250 мл этанола нагревают с обратным холодильником в течение 24 часов, после чего ей дают охлаждаться до комнатной температуры. Добавляют этилацетат до слабой опалесценции, после чего реакционную смесь оставляют стоять при температуре 4oC в течение 24 часов. Образующееся при этом масло отделяют путем декантирования с последующим удалением остаточного растворителя в вакууме. Масло растирают в порошок с помощью этанола, растворитель удаляют путем декантирования и остаток сушат в вакууме при комнатной температуре в течение 24 часов с получением светло-коричневого твердого вещества. Раствор этанола, который декантируют от масла, сгущают в вакууме и остаток растирают в порошок с помощью этанола с тем, чтобы получить вторую фракцию светло-коричневого твердого вещества. Оставшийся после выделения второй фракции светло-коричневого твердого вещества этанольный раствор сгущают в вакууме и остаток растирают в порошок с помощью пропанол-2-ола. При этом получают третью фракцию твердого вещества. Все три фракции твердого вещества объединяют и получают 51 г дигидрохлорида S-[3- (дипропиламино)пропил] изотиомочевины в качестве светло-коричневого вещества с точкой плавления 143 - 145oC.
11 мл 25-молярного водного раствора гидроокиси натрия прикапывают при температуре 0oC в атмосфере азота к перемешиваемому раствору 40 г дигидрохлорида S-[3- (дипропиламино)пропил]изотиомочевины в 100 мл воды и получаемую при этом смесь перемешивают при температуре 95oC в течение 2 часов, после чего ей дают охлаждаться до комнатной температуры. Продукт экстрагируют 4 раза диэтиловым эфиром, взятым в количестве по 70 мл, экстракты сушат над сульфатом магния и растворитель удаляют в вакууме. Остаток растворяют в диэтиловом эфире и получаемый раствор насыщают хлористым водородом. Получаемое при этом небольшое количество белого твердого вещества собирают путем фильтрации, фильтрат сгущают в вакууме и остаток объединяют с белым твердым веществом и растворяют в этаноле. Получаемый раствор насыщают хлористым водородом и растворитель удаляют в вакууме. При этом получают 18,3 г сырого гидрохлорида 3-(дипропиламино)пропантиола в качестве бесцветного полутвердого вещества, которое без дальнейшей очистки используют на следующей стадии. Раствор этилата натрия, получаемого из 1,6 г натрия и 175 этанола, при комнатной температуре в атмосфере азота добавляют к перемешиваемой суспензии 7 г сырого дигидрохлорида 3-(дипропиламино)пропантиола в 75 мл этанола и получаемую смесь перемешивают при комнатной температуре в течение 1 часа. Добавляют раствор 10,6 г 2-бром-1-[1-(3,4-дихлорфенил)циклобутил]этанона, получаемого аналогично примеру 1, в 40 мл этанола и получаемую при этом смесь перемешивают при комнатной температуре в течение 24 часов. Растворитель удаляют в вакууме, остаток разбавляют 100 мл воды и продукт экстрагируют 4 раза дихлорметаном, взятым в количестве по 75 мл. Экстракты сушат над сульфатом натрия, растворитель удаляют в вакууме и остаток очищают путем колоночной хроматографии на силикагеле с применением в качестве элюента смеси толуола и триэтиламина в соотношении 19 : 1. Продуктсодержащие фракции объединяют и растворители удаляют в вакууме. При этом получают 5,5 г 1-[1-(3,4-дихлорфенил)циклобутил] -2-[3- (дипропиламино)пропилтио] этанона в качестве желтоватого масла.
Пример 15
Раствор 0,65 г фумаровой кислоты в 20 мл горячего этанола добавляют к раствору 2,1 г 1-[1-(3,4-дихлорфенил)циклобутил]-2-[3- (диметиламино)пропилтио]этанона, получаемого из соответствующего гидрохлорида, получаемого аналогично примеру 7, в 10 мл диэтилового эфира и получаемую смесь оставляют стоять при температуре 4oC в течение 96 часов. Твердое вещество не образуется и поэтому растворитель удаляют в вакууме. При этом получают коричневое масло, которое растирают в порошок с помощью петролейного эфира с пределами кипения 40 - 60oC. Получаемое при этом твердое вещество собирают путем фильтрации, промывают диэтиловым эфиром, сушат в вакууме при комнатной температуре в течение 18 часов и перекристаллизовывают из смеси этилацетата и петролейного эфира с пределами кипения 60 - 80oC в соотношении 3 : 2. Получаемое при этом твердое вещество собирают путем фильтрации, промывают петролейным эфиром с пределами кипения 60-80oC и сушат в вакууме при комнатной температуре в течение 18 часов. При этом получают 1,1 г фумарата 1-[1-(3,4-дихлорфенил)циклобутил] -2-[3- (диметиламино)пропилтио] этанона в качестве белого твердого вещества с точкой плавления 100 - 10ЗoC.
Пример 16
3,2 г борана натрия порциями добавляют при температуре 0oC в атмосфере азота к перемешиваемому раствору 16 г 1-[1-(3,4- дихлорфенил)циклобутил]-2-[3-(диметиламино)-пропилтио)этанона, получаемого из соответствующего гидрохлорида аналогично примеру 7, в 200 мл метанола и получаемую при этом смесь перемешивают при комнатной температуре в течение 7 дней, после чего разбавляют 350 мл воды. Продукт экстрагируют 4 раза дихлорметаном, взятым в количестве по 100 мл, экстракты последовательно промывают 100 мл воды и 100 мл насыщенного водного раствора хлористого натрия, сушат над сульфатом натрия и растворитель удаляют в вакууме. Получаемое при этом зеленое масло очищают путем хиральной высокопроизводительной жидкостной хроматографии. При этом получают 3,5 г (-)-1-[1-(3,4-дихлорфенил)циклобутил]-2-[3- (диметиламино)пропилтио)этанола в качестве масла, имеющего показатель вращения 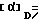 = -8,615o (с = 1; этанол; комнатная температура) и 3,4 г (+)-1- [1-(3,4-дихлорфенил)циклобутил]-2-[3- (диметиламино)пропилтио]этанола в качестве масла, имеющего показатель вращения [α]D= = +9,740o (с = 1; этанол; комнатная температура).
= -8,615o (с = 1; этанол; комнатная температура) и 3,4 г (+)-1- [1-(3,4-дихлорфенил)циклобутил]-2-[3- (диметиламино)пропилтио]этанола в качестве масла, имеющего показатель вращения [α]D= = +9,740o (с = 1; этанол; комнатная температура).
Раствор 1,77 г лимонной кислоты в 10 мл горячего этанола добавляют к раствору 3,42 г (-)-1-[1-(3,4- дихлорфенил)циклобутил]-2-[3-(диметиламино)-пропилтио)-этанола в 30 мл диэтилового эфира и получаемую смесь оставляют стоять при температуре 4oC в течение 18 часов. Получаемое твердое вещество собирают путем фильтрации, промывают диэтиловым эфиром и сушат в вакууме при комнатной температуре в течение 24 часов. При этом получают 3,9 г цитрата (-)-1-[1-(3,4-дихлорфенил)циклобутил]- 2-[3-(диметиламино)пропилтио]этанола в качестве белого твердого вещества с точкой плавления 109 - 115oC.
[α]D= -10,17o (с = 1; метанол; комнатная температура).
Пример 17
Раствор 1,73 г лимонной кислоты в 10 мл горячего этанола добавляют к раствору 3.29 г (+)-1-[1-(3,4-дихлорфенил)циклобутил] - 2-[3-диметиламино)пропилтио] -этанола, получаемого описанным в примере 16 образом, в 30 мл диэтилового эфира и получаемую смесь оставляют стоять при температуре 4oС в течение 18 часов. Получаемое твердое вещество собирают путем фильтрации, промывают диэтиловым эфиром и сушат в вакууме при комнатной температуре в течение 24 часов. При этом получают 3,8 г цитрата (+)-1-[1-(3,4- дихлорфенил)циклобутил]-2-[3-(диметиламино)-пропилтио]этанола в качестве белого твердого вещества с точкой плавления 109 - 115oC
[α]D= +10,88o (с = 1; метанол; комнатная температура).
Пример 18
Перемешиваемую смесь 305,7 г гидрохлорида 1-хлор-3- (диметиламино)пропана, 150 г тиомочевины и 1530 мл этанола нагревают с обратным холодильником в течение 24 часов, после чего смесь охлаждают до комнатной температуры. Добавляют этилацетат до слабой опалесценции, после чего смесь оставляют стоять при температуре 4oC в течение 72 часов. Получаемое при этом твердое вещество собирают путем фильтрации, промывают этилацетатом и сушат в вакууме при температуре 40oC. Получают 403 г дигидрохлорида S-[3-(диметиламино)пропил]изотиомочевины в качестве белого твердого вещества с точкой плавления 155 - 157oC.
Раствор 250 г гидроокиси натрия в 250 мл воды добавляют при температуре до 25oC в течение 10 минут к перемешиваемому раствору 731 г дигидрохлорида S-[3-(диметиламино)пропил] изотиомочевины, получаемого аналогично указанным выше образом, в 880 мл воды и получаемую при этом смесь перемешивают при температуре 95oC в течение 3 часов с последующим охлаждением до температуры 10oC. Продукт экстрагируют 4 раза дихлорметаном, взятым в количестве по 500 мл, экстракты объединяют и растворитель удаляют в вакууме. Остаток растворяют в 1 л диэтилового эфира, получаемый раствор отделяют от белого твердого остатка путем декантирования и растворитель удаляют в вакууме. Получают 362,7 г бесцветного масла, которое прикапывают при температуре до 15oC в течение 20 минут к 650 мл перемешиваемой 5-молярной соляной кислоты и получаемую смесь сгущают в вакууме при температуре 70oC. Получаемое при этом белое твердое вещество разбавляют 1 л пропан-2-ола и растворитель удаляют в вакууме. Получаемый остаток разбавляют 1 л толуола и растворитель удаляют в вакууме. Получаемый при этом остаток растирают в порошок с помощью взятого в количестве 1 л диэтилового эфира и получаемое твердое вещество собирают путем фильтрации, промывают диэтиловым эфиром и последовательно сушат в вакууме над пятиокисью фосфора, сначала при температуре 40oC в течение 3 дней и затем при комнатной температуре в течение 5 дней. При этом получают 405,9 г гидрохлорида 3-(диметиламино)пропантиола в качестве белого твердого вещества с точкой плавления 81 - 84oC.
Раствор 163,6 г 1-(3,4-дихлорфенил)циклобутанкарбонитрила в 130 мл диэтилового эфира прикапывают при комнатной температуре в течение 30 минут в атмосфере азота к 300 мл перемешиваемого 3-молярного раствора йодида метилмагния в диэтиловом эфире, получаемую смесь нагревают с обратным холодильником в течение 1 часа, разбавляют 100 мл диэтилового эфира, нагревают с обратным холодильником в течение дальнейших 150 минут и затем перемешивают при комнатной температуре в течение 18 часов. Получаемое твердое вещество собирают путем фильтрации, тщательно промывают диэтиловым эфиром и порциями добавляют при температуре до 20oC к перемешиваемой смеси 410 мл концентрированной соляной кислоты и 650 мл воды. Получаемую смесь нагревают при температуре 95oC в течение 30 минут при случайном перемешивании и затем охлаждают до комнатной температуры. Продукт экстрагируют 3 раза дихлорметаном, взятым в количестве по 200 мл, объединенные экстракты сушат над сульфатом магния и растворитель удаляют в вакууме. Получают 772,7 г 1-[1-(3,4-дихлорфенил)-циклобутил] этанона в качестве темно-красного масла, которое без дальнейшей очистки используют на следующей стадии.
Раствор 96 мл брома в 427 мл хлороформа прикапывают при температуре 10 - 15oC в течение 90 минут к перемешиваемому раствору 477,2 г 1-[1-(3,4-дихлорфенил)циклобутил] этанона, получаемого аналогично описанному выше методу, в смеси 643 мл метанола и 107 мл хлороформа. После окончания процесса добавления реакционную смесь перемешивают при комнатной температуре в течение 3 часов и затем подают в 2 л ледяной воды. Водный слой отделяют и три раза промывают дихлорметаном, взятым в количестве по 500 мл, объединенные органические растворы последовательно промывают два раза насыщенным водным раствором бикарбоната натрия, взятым в количестве по 400 мл, и 500 мл воды, сушат над хлоридом кальция и растворитель удаляют в вакууме. Получают 593,5 г 2-бром-1-[1-(3,4- дихлорфенил)циклобутил] этанона в качестве оранжевого масла, которое без дальнейшей очистки применяют на следующей стадии.
Раствор 235,6 г 3-(диметиламино)пропантиола, получаемого из соответствующего гидрохлорида, в 1 л этанола прикапывают при комнатной температуре к перемешиваемому раствору этилата натрия, получаемого из 50 г натрия и 2 л этанола, получаемую при этом смесь перемешивают при комнатной температуре в течение 1 часа, после чего добавляют раствор 817,4 г 2-бром-1-[1-(3,4-дихлорфенил)циклобутил]этанона (полученного вышеописанным образом) в 1,5 л этанола и получаемую реакционную смесь перемешивают при комнатной температуре в течение 18 часов. Растворитель удаляют в вакууме и получаемый твердый остаток разбавляют 2 л воды. Продукт экстрагируют три раза дихлорметаном, взятым в количестве по 500 мл, объединенные экстракты сушат над сульфатом магния и растворитель удаляют в вакууме. Получаемый при этом остаток растворяют в диэтиловом эфире, получаемый раствор отделяют от нерастворимой смолы путем декантирования и растворитель удаляют в вакууме с получением 713 г оранжевого масла, которое растворяют в 3,5 л петролейного эфира с пределами кипения 60 - 80oC, добавляют активный уголь и сульфат магния, смесь фильтруют на силикагеле марки Целите, после чего растворитель удаляют в вакууме. При этом получают 713 г оранжевого масла, которое при температуре до 20oC добавляют к перемешиваемой смеси 255 мл концентрированной соляной кислоты и 1750 мл воды и получаемую при этом смесь сгущают в вакууме при температуре 50oC. Получаемый при этом остаток неоднократно разбавляют толуолом и сгущают в вакууме до полного удаления воды, после чего получаемый остаток растирают в порошок с помощью диэтилового эфира, взятого в количестве 2,5 л. Диэтиловый эфир удаляют путем декантирования, остаток растворяют в 2,5 л этилацетата, добавляют 5 л диэтилового эфира, получаемое при этом твердое вещество собирают путем фильтрации, промывают диэтиловым эфиром, суспендируют в 2,5 л этилацетата, собирают путем фильтрации, промывают этилацетатом и сушат в вакууме при температуре 5oC. При этом получают 425 г гидрохлорида 1-[1-(3,4- дихлорфенил)циклобутил] -2-[3-(диметиламино)-пропилтио]этанона в качестве кремообразного твердого вещества.
Если остаток от данного эксперимента подвергать повторной переработке описанным выше образом, то получают еще 313 г дальнейшей фракции сырого продукта в качестве кремообразного твердого вещества. Обе фракции сырого твердого вещества объединяют, растворяют в 2,5 л воды и подщелачивают до величины pH 9 путем добавления твердого карбоната натрия. Получаемое при этом свободное основание экстрагируют три раза дихлорметаном, взятым в количестве по 500 мл, после чего растворитель удаляют в вакууме. Получаемую при этом смолу распределяют между водой и этилацетатом, получаемую при этом эмульсию фильтруют на силикагеле марки Целите, органический слой отделяют, сушат над сульфатом натрия и растворитель удаляют в вакууме. При этом получают 638 г коричневого масла, которое порциями по 50 г очищают путем фильтрации на силикагеле с применением в качестве элюента смеси дихлорметана и метанола в соотношении 9 : 1. Продуктсодержащие фракции объединяют и растворители удаляют в вакууме с получением 484,3 г оранжеватого масла, которое при температуре до 20oC добавляют к перемешиваемой смеси 173 мл концентрированной соляной кислоты и 415 мл воды, после чего получаемую смесь сгущают в вакууме. Остаток сушат путем неоднократного разбавления толуолом и вакуумного удаления растворителя, получаемую при этом смолу растворяют в 500 мл этилацетата и разбавляют 2,5 л диэтилового эфира. Получаемое при этом твердое вещество собирают путем фильтрации, промывают диэтиловым эфиром и сушат в вакууме при температуре 45oС. Получают 507,1 г гидрохлорида 1-[1-(3,4-дихлорфенил)циклобутил] -2-[3- (диметиламино)пропилтио] этанона в качестве кремообразного твердого вещества.
480 г гидрохлорида 1-[1-(3,4-дихлорфенил)циклобутил]-2-[3- (диметиламино)пропилтио)этанона суспендируют в избыточном насыщенном водном растворе бикарбоната натрия, получаемую при этом смесь перемешивают при комнатной температуре в течение 30 минут и получаемое свободное основание экстрагируют дихлорметаном. Экстракты сушат над сульфатом натрия и растворитель удаляют в вакууме с получением 415 г коричневого масла, которое растворяют в 1700 мл диэтилового эфира и получаемый при этом раствор добавляют к раствору 210 г лимонной кислоты в 320 мл горячего этанола. Получаемой при этом смеси дают охлаждаться до комнатной температуры и оставляют стоять при температуре 4oC в течение 48 часов. При этом образуется коричневатое твердое вещество. Надосадочную жидкость удаляют путем декантирования, остаток разбавляют 300 мл этанола и получаемую смесь умеренно нагревают до исчезновения кристаллической массы. Получаемый продукт собирают путем фильтрации, промывают диэтиловым эфиром, сушат в вакууме и перекристаллизовывают из этанола. Получаемое при этом твердое вещество собирают путем фильтрации, промывают этанолом и сушат в вакууме при температуре 50oC в течение 4 часов. Получают 410 г цитрата 1-[1-(3,4-дихлорфенил)- циклобутил] -2-[3-(диметиламино)пропилтио] этанона в качестве кремообразного твердого вещества с точкой плавления 103 - 105oC.
Пример 19
Маточные растворы, получаемые в результате выделения целевого продукта примера 10, сгущают в вакууме и получаемый при этом остаток разбавляют водой и подщелачивают путем добавления биполярного водного раствора гидроокиси натрия. Получаемый продукт экстрагируют диэтиловым эфиром, экстракты промывают водой, сушат над сульфатом магния и растворитель удаляют в вакууме. При этом получают 1-[1-(3,4-дихлорфенил)циклобутил]-2-[3-(диметиламино)-2- метилпропилтио] этанона в качестве желтоватого масла. 4 г этого масла растворяют в 40 мл этанола, после чего в атмосфере азота при температуре 0oC порциями добавляют 0,8 г борана натрия. Получаемую при этом смесь перемешивают при комнатной температуре в течение 7 дней и затем разбавляют 120 мл воды. Получаемый продукт экстрагируют 4 раза дихлорметаном, взятым в количестве по 50 мл, объединенные экстракты промывают 50 л воды и 50 мл насыщенного водного раствора хлористого натрия, сушат над сульфатом натрия и растворитель удаляют в вакууме. При этом получают 4,2 г бесцветного масла, которое растворяют в диэтиловом эфире, и получаемый раствор насыщают хлористым водородом. При этом получают твердое вещество, которое собирают путем фильтрации, промывают диэтиловым эфиром и сушат в вакууме над пятиокисью фосфора в течение 72 часов. Получаемое при этом белое твердое вещество является гигроскопическим и поэтому его растворяют в воде и подщелачивают путем добавления насыщенного водного раствора бикарбоната натрия. Получаемый при этом продукт экстрагируют три раза дихлорметаном, взятым в количестве по 50 мл, объединенные экстракты промывают водой, сушат над сульфатом натрия и растворитель удаляют в вакууме. При этом получают 3 г 1-[1-(3,4- дихлорфенил)циклобутил]-2-[3-(диметиламино)-2-метилпропилтио]этанола в качестве зеленоватого масла.
Пример 20
106 мл 1,4-дибромбутана прикапывают при температуре 70 - 80oC в течение 1 часа в атмосфере азота к перемешиваемой смеси 150 г 3,4-дихлорфенилацетонитрила, 2 г хлорида бензилтриэтиламмония и 300 мл 50%-ного водного раствора гидроокиси натрия. После окончания процесса добавления реакционную смесь перемешивают при температуре 70 - 80oC в течение 2 часов и затем охлаждают до комнатной температуры. Добавляют 400 мл диэтилового эфира и 200 мл воды и образующиеся слои отделяют. Водный слой промывают два раза диэтиловым эфиром, взятым в количестве по 200 мл, объединенные органические растворы сушат над сульфатом магния и растворитель удаляют в вакууме. В результате перегонки получаемого при этом остатка получают 135 г 1-(3.4-дихлорфенил)циклопентанкарбонитрила в качестве желтоватого масла с точкой кипения 132 - 140oC/0,4 мбар.
100 мл 3-молярного раствора йодида метилмагния в диэтиловом эфире прикапывают при температуре ОoC в атмосфере азота к перемешиваемому раствору 48 г 1-(3,4- дихлорфенил)циклопентанкарбонитрила в 100 мл диэтилового эфира и получаемую при этом смесь перемешивают при комнатной температуре в течение 24 часов. Получаемое твердое вещество собирают путем фильтрации, промывают диэтиловым эфиром и порциями добавляют к холодной как лед смеси 200 мл воды и 124 мл концентрированной соляной кислоты. Получаемую смесь нагревают при температуре 95oC в течение 1 часа, после чего ей дают охлаждаться до комнатной температуры. Получаемый продукт экстрагируют 5 раз диэтиловым эфиром, взятым в количестве по 100 мл, объединенные экстракты промывают два раза водой, взятой в количестве по 100 мл, сушат над сульфатом магния и растворитель удаляют в вакууме. В результате перегонки остатка получают 31,9 г 1-[1-(3,4- дихлорфенил)-циклопентил] этанона в качестве желтоватого масла с точкой кипения 125 - 128oC/0,5 мбар.
Раствор 6,1 мл брома в 50 мл дихлорметана прикапывают при температуре 10 - 15oC в течение 3 часов в атмосфере азота к перемешиваемому раствору 31,9 г 1-[1-(3,4- дихлорфенил)циклопентил] этанона в смеси 60 мл этанола и 10 мл дихлорметана и получаемую при этом смесь перемешивают при комнатной температуре в течение 150 минут и затем подают в избыток ледяной воды. Водный слой отделяют и промывают три раза дихлорметаном, взятым в количестве по 100 мл, объединенные органические растворы последовательно промывают два раза насыщенным водным раствором бикарбоната натрия, взятым в количестве по 100 мл, и 100 мл воды, сушат над хлоридом кальция и растворитель удаляют в вакууме. Получаемый при этом остаток подвергают вакуумной перегонке, получаемую при этом фракцию с точкой кипения свыше 174oC/1,3 мбар собирают и подвергают повторной перегонке. Получаемую в результате повторной перегонки фракции с точкой кипения свыше 182oC/2,6 мбар собирают и подвергают повторной перегонке. При этом получают 11,8 г 2-бром-1-[1-(3,4-дихлорфенил)циклопентил] этанона в качестве желтоватого масла с точкой кипения 156 - 162oC/0,4 мбар.
Раствор этилата натрия, получаемый из 1,4 г натрия и 175 мл этанола, добавляют при комнатной температуре в атмосфере азота к перемешиваемой суспензии 4,5 г гидрохлорида 3-(диметиламино)пропантиола, получаемого аналогично примеру 7, в 75 мл этанола и получаемую при этом смесь перемешивают при комнатной температуре в течение 1 часа. Добавляют раствор 10,5 г 2-бром-1- [1-(3,4-дихлорфенил)-циклопентил] этанона в 40 мл этанола и получаемую при этом смесь перемешивают при комнатной температуре в течение 24 часов. Растворитель удаляют в вакууме, остаток разбавляют 100 мл воды и получаемый при этом продукт 4 раза экстрагируют дихлорметаном, взятым в количестве по 75 мл. Экстракты сушат над сульфатом натрия, растворитель удаляют в вакууме и остаток очищают путем колоночной хроматографии на силикагеле с применением в качестве элюента смеси толуола и триэтиламина в соотношении 19 : 1. Продуктсодежащие фракции объединяют и растворители удаляют в вакууме с получением 7 г коричневатого масла, которое растворяют в диэтиловом эфире, получаемый раствор насыщают хлористым водородом и растворитель удаляют в вакууме. Остаток растирают в порошок с помощью диэтилового эфира и получаемое при этом твердое вещество собирают путем фильтрации, промывают диэтиловым эфиром и сушат в вакууме при комнатной температуре над пятиокисью фосфора в течение 48 часов. При этом получают 5,6 г гидрохлорида 1- [1-(3,4-дихлорфенил)циклопентил] - 2- [3- (диметиламино)-пропилтио]этанона в качестве белого твердого вещества с точкой плавления 77 - 80oC.
Пример 21
Данный пример иллюстрируют составы возможных фармацевтических композиций. При этом под используемым термином "активное начало" понимается любое соединение вышеприведенной общей формулы (I).
а) Капсулы
10 вес. частей активного начала интенсивно смешивают с 240 вес. частями лактозы и получаемую смесь измельчают, загружают в капсулы из твердой желатины с таким расчетом, что каждая капсула содержит либо единичную дозу, либо часть единичной дозы активного начала.
б) Таблетки
Таблетки состава (вес. части): 10 активного начала, 190 лактозы, 22 кукурузного крахмала, 10 поливинилпирролидона и 3 стеарата магния приготовляют следующим образом.
Активное начало, лактозу и часть кукурузного крахмала измельчают, перемешивают и получаемую при этом смесь гранулируют с помощью раствора поливинилпирролидона в этаноле. После сушки гранулы смешивают с стеаратом магния и остаточным количеством кукурузного крахмала. Получаемую при этом смесь подают в таблетировочную машину с тем, чтобы получить таблетки, каждая из которых содержит либо единичную дозу, либо часть единичной дозы активного начала.
в) Таблетки с покрытием
Таблетки приготовляют тем же образом, что и описано в п. б). Затем на таблетки известными приемами наносят раствор 20%-ного фталата ацетата целлюлозы и 3%-ного диэтилфталата в смеси этанола и дихлорметана в соотношении 1 : 1.
г) Суппозитории
100 вес. частей активного начала вырабатывают в 1300 вес. частей смеси триглицеридов и получаемую при этом смесь переводят в суппозитории, каждый из которых содержит терапевтически эффективное количество активного начала.
Сульфиды, сульфоксиды или сульфоны формулы I, где Х - карбонил или остаток формулы II; R5 - водород или алкил Y - С1-2алкиленовая цепь; Z-С2-5алкиленовая цепь, возможно замещенная алкилом; R - остаток формулы III; R3 - галоид; R4 - Н или галоид; или R3 или R4 вместе с атомами углерода, с которыми они связаны, образуют приконденсированное бензольное кольцо; R1 и R2 - Н, алкил, фенилалкил; m = 0, 1, 2; n = 3 или 4, и их фармацевтически приемлемые соли обладают антидепрессивной активностью и способностью к торможению поглощения допамина. 2 с. и 10 з.п. ф-лы, 1 табл.
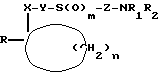
где X - карбонил или группа формулы II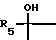
где R5 означает водород или алкил с 1-4 атомами углерода;
Y - алкиленовая цепь с 1 или 2 атомами углерода,
Z - алкиленовая цепь с 2-5 атомами углерода, незамещенная или замещенная по меньшей мере одной алкильной группой с 1-3 атомами углерода;
R - означает группу формулы III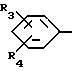
где
R3 означает галоид, а R4 - водород или галоид или же R3 или R4 вместе с атомами углерода, с которыми они связаны, образуют приконденсированное бензольное кольцо;
R1 и R2 одинаковы или различны и означают водород, разветвленную или неразветвленную алкильную группу с 1-4 атомами углерода, фенилалкил с 1-3 атомами углерода в алкильной части, при этом если R1 означает бензил, то R2 означает водород или метил;
m = 0, 1 или 2;
n = 3 или 4,
и их фармацевтически приемлемые соли.
1-[1-(3,4-дихлорфенил)циклобутил]-2-(2-(диметиламино)этилтио] этанон;
1-[1-(3,4-дихлорфенил)циклобутил]-2-[2-(диметиламино)этил-сульфинил]-этанон;
1-[1-(3,4-дихлорфенил)циклобутил]-2-[2-(диметиламино)этил-сульфонил]-этанон;
1-[1-(3,4-дихлорфенил)циклобутил]-2-[2-(диэтиламино)этилтио]этанон;
2-[2-(N-бензил-N-метиламино)этилтио] -1-[1-(3,4-дихлорфенил)циклобутил] этанол;
1-[1-(3,4-дихлорфенил)циклобутил]-2-[2-(диметиламино)этилтио]этанол;
1-[1-(3,4-дихлорфенил)циклобутил] -2-[3-(диметиламино)пропилтилтио] этанон;
1-[1-(3,4-дихлорфенил)циклобутил] -2-[3-(диметиламино)пропилсульфонил] этанон;
1-[1-(3,4-дихлорфенил)циклобутил]-2-[3-(диметиламино)пропилтио]этанол;
1-[1-(3,4-дихлорфенил)циклобутил]-2-[3-(диметиламино)-2-метил-пропилтио] этанон;
2-[2-(диметиламино)этилтио]-1-[1-(2-нафтил)циклобутил]этанон;
1-[1-(3-хлорфенил)циклобутил]-2-[3-(диметиламино)пропилтио]этанон;
1-[1-(3,4-дихлорфенил)циклобутил]-2-[4-(диметиламино)бутилтио]этанон;
1-[1-(3,4-дихлорфенил)циклобутил]-2-[3-(дипропиламино)пропилтио]этанон;
1-[1-(3,4-дихлорфенил)циклобутил]-2-[3-(диметиламино)-2-метил-пропилтио] -этанол;
1-[1-(3,4-дихлорфенил)циклопентил]-2-[3-(диметиламино)пропилтио]этанон,
или их фармацевтически приемлемые соли в виде отдельных энантиомеров, рацематов или других смесей энантиомеров.
| Способ получения гетероциклических производных циклобутиламинометана | 1983 |
|
SU1274622A3 |
| Способ получения 4-окси-2-оксопирролидин-1-ил-ацетамида | 1986 |
|
SU1482525A3 |
| Способ получения (1-арилциклобутил)-алкиламинов или их фармакологически приемлемых солей | 1985 |
|
SU1461372A3 |
| US 5047432 A, 10.09.91 | |||
| УСТРОЙСТВО для АВТОМАТИЧЕСКОГО УПРАВЛЕНИЯ ПРОЦЕССОМ ПОКУСКОВОЙ СОРТИРОВКИ МИНЕРАЛЬНОГОСЫРЬЯ | 0 |
|
SU282206A1 |
