Изобретение относится к новому способу контрацепции на основе конкурентных антагонистов прогестерона с использованием новых антагонистов прогестерона.
При ингибировании формирования желез эндометрия и разрастания эпителиальной ткани имплантация оплодотворенной яйцеклетки в матке становится невозможной (вследствие ингибирования рецептивной способности матки). Поэтому конкурентные антагонисты прогестерона, в соответствии с данным изобретением, могут использоваться для контрацепции у особей женского пола.
RU 486 ( 11β -[4-N,N-(диметиламино)фенил]- 17β- гидрокси- 17α -пропинил-эстра-4,9(10)- диен-3-он; EP-A-0057115) и другие 11β -арил или 11β , 19-арилен-замещенные стероиды являются соединениями, способными вытеснять прогестерон и глюкокортиксиды из соответствующих рецепторов. Фармакологически эти вещества отличаются сильно выраженными антагонистическими свойствами по отношению к прогестерону и глюкортикоидам. Эти свойства легли в основу их использования в медицинской практике. Например, RU 486 применяется в качестве антагониста прогестерона для медицинского прерывания беременности, а так же как антагонист глюкокортикоидов при лечении синдрома Кушинга на фоне патологически усиленной активности коры надпочечников. RU 486 в дозировке 200 - 600 мг вызывает аборт у женщин.
Давно известно, что конкурентные антагонисты прогестерона способны ингибировать овуляцию у различных видов животных у человеческих особей женского пола (Collins et al., "Blockade of the spontaneous mid-cycle gonadotropin surge in moukeys by RU 486; A. progesterone autagonist or agonist", J. Chin. Metab., 63:1270-1276 (1986); Croxatto, H.B., "Salvatierra 1990 Cyclic use of antigestagens for fertility control", IIIrd Internasional Symposium on Coutraception, Heidelberg, June 19 - 23, 1990; Danford et al., "Coutraceptive potential of RU 486 by ovulation inhibition III Preliminary observations on once weekly administration", Coutraception 40: 195 - 200 (1989);
Kekkonen et al. , "Lahteonenmaki P 1990 Interference with ovulation by sequential treatment with the ahtiprogesterone RU 486 abd synthetic progestin", Fertil Sterile [Fertile Sterile] 53, 4747;
Puri et al., "Gonadal and pituitary respon ses to progesterone antagonist LK 98 299 during follicular phase of the menstrual cycle in bonnet monkeys", Coutraception 39,2:22 - 243 (1989);
Puri et al., "Coutraceptive potential of a progesterone antagonist LK 98 734:Effect on folliculogenesis, ovulation abd corpus luteum functoin in bonnet monkeys". In Moudgal et al., (eds) (1990).
Согласно патенту США 4 764 513, для повышения вероятности успешной имплантации оплодотворенной в лабораторных условиях (in vitro) яйцеклетки период восприимчивости эндометрия к имплантации (имплантационное окно может быть сдвинуто (растянуто) путем введения женщине конкурентных антагонистов прогестерона.
Стероиды с 11β 19-o-фенилен-мостиковыми связями, демонстрирующие особенно сильно выраженную антагонистическую активность по отношению к прогестерону при антиглюкокортикоидной активности, значительно меньшей по сравнению со взятым для сопоставления соединением 11β -(4-диметиламинофенил) 17β гидрокси- 17α (пропил-1-ил)-4,9(10)-эстрадиен-3-оном (RU 486; EP-A-O 057115), впервые описаны в патенте США 5095129. Новые химические соединения (соединения I и II), описанные в данной заявке на изобретение, подпадают под объем притязаний, заявленных в формуле США 595129, но не описаны ни по названию, ни в примере.
Дозировка конкурентных антагонистов прогестерона, оказывающего ингибирующий эффект на овуляцию, в большой степени зависит от вида живых организмов, подвергаемых его воздействию. В случае использования RU 486 эта дозировка составляет 50-100 мг для человеческой особи женского пола (Croxatto et al.; Loc. cit., Ledge et al. (1992). Inhibition of ovulation using very low dose mifepristone;
Abstract: Second Cougress of the Europen Society of (Coutraception).
RU 486 проявляет слабую диссоциацию (или не проявляет ее вообще) центрального и эндометриального эффектов у человека (Ledge WL et al., Terra Symposium on Progesteron Antagonists, May 25-29, 1992 Mohouk, N.Y.)
Для ингибирования имплантации было предложено лечение "ЛГ+2" (Swahn et al. , "The effect of RU 486 administration during the early buteal phase on bleeding pattern, hormonal parameters and endometrium," Human Reproduction,5 4: 402-408 (1990): через два дня после наступления пиковой активности ЛГ (ЛГ-лютенизирующий гормон) в менструальном цикле женщины (пиковая активность ЛГ соответствует времени овуляции), т.е. на 14 -, 15 - или 16-й день однократно вводят ингибирующую овуляцию дозу RU 486. Таким образом, активное соединение вводят только после овуляции в лютеальной фазе менструального цикла (лютеальная контрацепция).
Недавно в печати появились сообщения о том, что десинхронизация эндометрия у женщин без гормональных изменений (концентраций прогестерона и эстрадиола) может быть достигнута путем введения конкурентного антагониста прогестерона RU 486 в том случае, если последний вводят на 5 - и 8 -й день после наступления пика ЛГ в менструальном цикле (дозировка в каждом случае - 10 мг, перорально) (Kettel et al, 1992). Надежная контрацепция без ингибирования овуляции не может быть достигнута, если конкурентный антагонист прогестерона вводят только после наступления пика ЛГ в менструальном цикле.
В соответствии с данным изобретением, конкурентные антагонисты прогестерона в дозировке, не ингибирующей овуляцию и не вызывающей аборт, способны ингибировать формирование желез эндотермия в фазе пролиферации, а также функции желез в лютеальной фазе менструального цикла, и тем самым способствовать контрацепции, если введение соответствующей дозировки происходит, по меньшей мере, один раз до, а желательно и после наступления пика ЛГ.
В том его аспекте, который касается способа, данное изобретение относится к способу контрацепции у женщин, включающему введение в фолликулярной фазе ее менструального цикла, и желательно и в лютеальной фазе последнего, конкурентного антагониста прогестерона, ингибирующего образование желез эндометрия, являющихся необходимым условием для имплантации оплодотворенной яйцеклетки в матке, причем конкурентный антагонист прогестерона вводят в количестве меньшем, чем доза, ингибирующая овуляцию, и меньшем, чем доза, вызывающая аборт.
В том аспекте, который касается композиции, данное изобретение относится к 11β , 19-о-фенилен- 17β -гидрокси - 17α (3-гидроксипроп-1(Z)-енил1-4-андростен-3-он, формулы:
где 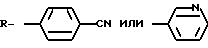
В другом касающемся композиции аспекте данное изобретение относится к фармацевтическому составу, включающему в смеси с фармацевтически приемлемой основой конкурентный антагонист прогестерона, ингибирующий формирование желез эндометрия и разрастание эпителиальной ткани, причем конкурентный антагонист прогестерона вводят в такой дозировке, которая меньше дозы, ингибирующей овуляцию, и меньше дозы, вызывающий аборт.
В фазе пролиферации при нормальном менструальном цикла в эндометрии происходит индуцированное экстрогеном формирование секреторных желез, тогда как в лютеальной фазе (называемой также секреторной фазой) наблюдается секреторная активность желез, индуцированная прогестероном. Следовательно, в основу описываемого здесь действия конкурентных антагонистов прогестерона в фазе пролиферации, т. е. до овуляции, положен не эффект ингибирования прогестерона, так как пролиферация желез эндометрия является экстроген-зависимой. Кроме того, в фазе пролиферации прогестерона в крови являются очень низкими. При применении согласно данному изобретению конкурентных антагонистов прогестерона достигается избирательное ингибирование маточной восприимчивости без оказания какого-либо вредного воздействия на менструальный цикл, причем вводимые дозы являются слишком низкими, чтобы вызвать аборт, если имплантация яйцеклетки уже произошла.
Возможность достижения контрацепции при дозах, более низких, чем дозы, вызывающие аборт и ингибирующие овуляцию, представляется очень важным соображением для некоторых женщин, отрицательно относящихся к аборту или озабоченных долгосрочными медицинскими последствиями ингибирования овуляции.
Серьезное преимущество предлагаемого способа заключается в очень высокой контрацептивной надежности применения конкурентного антагониста прогестерона, поскольку эндометрий не может принять оплодотворенную яйцеклетку после введения относительно очень низкой дозировки конкурентного антагониста прогестерона до овуляции, а желательно и после. Имплантация не может быть исключена и в пролиферационной фазе нормального менструального цикла. В связи с тем, что секреция желез эндометрия является необходимым условием для восприимчивости эндометрия, в случае атрофии желез и эпителиальной ткани эндометрия имплантация окажется невозможной. Следовательно, надежность контрацепции достигается и у женщин с нерегулярным менструальным циклом.
Новые конкурентные антагонисты прогестерона, а именно 11β , 19-[4-(4-(4-цианофенил)-о-фенилен] - 17β -гидрокси- 17α -(3-гидроксипроп-1 (Z)-енил-4-андростен-3-он (1) и 11β , 19-[4-(3-пиридинил)-о-фенилен]- 17β -гидрокси- 17α -(3-гидроксипроп-1 (Z)-енил)- 4-андростен-3-он (II) отличается очень сильно выраженной периферической избирательной эффективностью, т.е. действие на эндометрий соединений I и II является очень сильно выраженным, тогда как центральное действие, а именно действие на систему гипофиз-яичники при той же дозировке выражено очень слабо.
Такие конкурентные антагонисты прогестерона могут также быть названы диссоциированными, так как при определенной пороговой дозе овуляция (центральный эффект) не ингибируется, хотя в эндометрии и наблюдаются изменения. Отношение дозы, ингибирующей овуляцию, к дозе, ингибирующей имплантацию (как было установлено в опытах на крысах при пероральном введении) может быть использовано для измерения диссоциации (коэффициент диссоциации). Это отношение может быть различным у разных видов живых организмов, но выражается числом 30 или больше при введении диссоциированного или конкурентного антагониста прогестерона.
Преимущество диссоциированных или конкурентных антагонистов прогестерона заключается в том, что их можно вводить в дозах, достаточно высоких для того, чтобы вызвать изменения в эндометрии без ингибирования овуляции. В результате сохраняется нормальный менструальный цикл.
Конкурентные антагонисты прогестерона вводят предпочтительно отдельными лечебными дозами, распределенными во времени, например предпочтительно в течение 4 - 10 дней с правильными интервалами, например, в каждой неделе менструального цикла, и каждый раз в такой дозировке, которая недостаточна для ингибирования овуляции или для индуцирования аборта, если имплантация уже произошла. Аналогичное воздействие на восприимчивость эндометрия может быть достигнуто ежедневным введением более низких доз перорально. Возможно также применение систем медленного поступления препарата (микрокристаллические суспензии, трансдермальные пластыри и подкожные имплантаты), при условии, что выделяемое из них количество антагониста прогестерона будет достаточным для ингибирования имплантации яйцеклетки на протяжении всего периода действия системы, но меньшим, чем доза, препятствующая овуляции, которая должна произойти в течение этого периода времени.
Описанное выше применение конкурентных антагонистов прогестерона иллюстрируется испытаниями на взрослой самке обезьяны Боннета (Macaca radiata), которой вводился онапристон [ 11β -(4-диметиламинофенил 1)- 17α -гидрокси 17β -(3-гидроксипропил 1)- 13α метил-4,9-гонадиен-3-он; EP-A-0129499], типичный конкурентный антагонист прогестерона.
Методика проведения испытания приведена ниже (табл. 1).
Результаты наблюдений за изменениями в эндометрии и уровнями гормонов
В табл. 2 показано, что протяженность менструального цикла и менструального периода при лечении препаратами в соответствии с данным изобретением не изменились по сравнению с контрольной группой.
Уровни эстрадиола и прогестерона свидетельствуют о том, что у всех животных фолликулогенез и овуляция прошли нормально: нормальный предовуляторный пик эстрадиола, нормальный уровень прогестерона в лютеальной фазе (см. чертеж).
Гистологическое исследование показывает, что, в отличие от контрольных животных у животных из обеих подвергнутых лечению групп эндометрий оказался атрофированным. В наибольшей степени изменения наблюдались в железах эндометрия: они были атрофированными и бездействующими, с разросшейся стромальной тканью.
Описанные выше результаты наблюдений ясно показывают, что конкурентные антагонисты прогестерона могут применяться для контрацепции (ингибирования имплантации) в дозировках, не оказывающих ингибирующего овуляцию и индуцирующего аборт действия, при условии, что эти дозировки вводят до, а желательно и после овуляции во время каждого менструального цикла.
В качестве конкурентных антагонистов прогестерона могут использоваться все соединения, имеющие сродство с рецептором гестагена (рецептором прогестерона), но не проявляющие собственной гестагенной активности. Например, могут считаться подходящими следующие стероиды:
11β [(4-N, N-диметиламино)-фенил] 17β -гидрокси 17α -пропинил-4,9(10)-эстрадиен-3-он (RU-38486);
11β [(4-N,N-диметиламино)-фенил] 17β- гидрокси-18-метил 17α -пропинил-4,9(10)-эстрадиен-3-он и
11β -[(4-N, N-диметиламино)-фенил] 17aβ- гидрокси- 17aα -пропинил-D-гомо-4,9(10),16-эстратриен-3-он (все эти соединения описаны в EP-A-0057115);
11β -n-метоксифенил 17β- гидрокси 17α -этинил-4,9(10)-эстрадиен-3-он (Steroids 37 (1981)3, 361 - 382), и
11β -(4-ацетилфенил) 17β- гидрокси- 17α- (проп-1-инил)-4,9(10)- эстрадиен-3-он (ЕП-A 0190759); и
11β -арил 14β -эстрадиены и эстратриены, описанные в ЕП-A 0277676; 19, 11β -стероиды с мостиковыми связями (патент США 5095129); 11β- арил-6-алкил (или 6-алкенил или 6-алкинил)-эстрадиены и прегнадиены, известные из EP-A 0289073; 11β -арил-7-метил (или 7-этил)-эстрадиены, известные из ЕП-A 0321010, а также стероиды 10β -H (EP-A 0404283). Этот перечень является далеко не полным, так как в вышеназванных целях могут использоваться и другие конкурентные антагонисты прогестерона, описанные в упомянутых публикациях, а также в других публикациях, здесь не приведенных.
В соответствии с данным изобретением, предпочтительно использовать те конкурентные антагонисты прогестерона, которые отличаются избирательными периферическим действием, т.е. те, которые дают сильно выраженный эндометриальный эффект в дозировке, при которой наблюдается очень незначительный центральный эффект (воздействие на системы гипофиз-яичники).
Такие конкурентные антагонисты прогестерона могут быть также названы диссоциированными, так как при определенной пороговой дозе происходят изменения в эндометрии, но овуляция (центральный эффект) не ингибируется. Отношение дозы, ингибирующей овуляцию, к дозе, ингибирующей имплантацию (коэффициент диссоциации), может использоваться для определения величины диссоциации. Это отношение бывает различным в зависимости от вида живых организмов, которым вводят антагонисты прогестерона, и составляет величину порядка 30 или больше для диссоциированного конкурентного антагониста прогестерона (у крысы после перорального введения), предложенного в данном изобретении.
Преимущество конкурентных антагонистов прогестерона при их использовании в соответствии с данным изобретением состоит в том, что для большей надежности получения желаемого эффекта они могут вводиться в более высоких дозах без ингибирования овуляции, т.е. при сохранении "нормального" менструального цикла.
Конкурентные антагонисты прогестерона могут вводиться, например, местно, локально, энтерально, трансдермально или парентерально. Предпочтительным является пероральное введение.
Для перорального введения препарат может быть в виде таблеток, таблеток с оболочками, капсюлей, пилюль, суспензий или растворов, которые изготавливают обычным способом - с добавками и носителями, обычно используемыми в медицинской практике. Например, при местном и топикальном применении возможны вагинальные суппозитории, вагинальные гели, имплантаты, вагинальные кольца или трансдермальные системы, такие, как кожные пластыри. Можно также использовать вагинальные кольца, снимаемые через определенный промежуток времени, например через 14 дней после введения, а затем вводимые вновь в соответствии со схемой лечения.
Единичная доза обычно содержит 0,25 - 50 мг 11β -[(4-N,N-диметиламино)-фенил] 17α-гидрокси 17β - (3-гидроксипропил)- 13α -метил-4,9(10)-гонадиен-3-она или биологически эквивалентное количество другого конкурентного антагониста прогестерона.
Если введение фармацевтического агента, изготовленного в соответствии с данным изобретением, осуществляют посредством имплантата, вагинального кольца или трансдермальной системы, то все они должны быть рассчитаны таким образом, чтобы ежедневно поступающая в организм доза конкурентного антагониста прогестерона при данной схеме лечения находилась в пределах 0,25 - 50 мг.
Вводимая по данному изобретению доза конкурентного антагониста прогестерона ниже дозы упомянутого антагониста, ингибирующей овуляцию, и дозы, вызывающей аборт.
В общем случае однократная доза составляет 0,25 - 20 мг; в особых случаях применения конкурентных антагонистов прогестерона с периферически избирательным действием однократная доза может достигать 0,5 - 50 мг, так как вещества с периферически избирательным действием могут вводиться и в более высоких дозировках без риска ингибирования овуляции. Термин "однократная" доза или "однократное" введение включает в себя системы непрерывного действия, выделяющие конкурентный антагонист прогестерона со скоростью, соответствующей ежедневной дозе в 0,25 - 20 мг или единичной дозе 0,5 - 50 мг.
Предполагается, что специалист не нуждается в дальнейшем изложении всех деталей и что приведенного выше описания будет достаточно для применения настоящего изобретения во всем его объеме. Поэтому представленные ниже конкретные примеры следует рассматривать лишь как иллюстрирующие данное изобретение и ни в коей мере не ограничивающие объем заявленных притязаний.
В следующем и в дальнейших примерах температура приведена в градусах Цельсия и, если не указано иначе, все части и проценты взяты по весу.
Все упомянутые в описании заявки, патенты и публикации, а также приоритетные заявки ФРГ P 42 16 003.0 и P 42 16 004.9 приведены в качестве ссылок.
Пример приготовления 1
11β , 19-[4-(4-цианофенил)-о-фенилен] - 17β -гидрокси 17α- (3-гидроксипроп-1(Z)-енил)-4-андростен-3-он
a) 3,3-диметилтриметилендиокси 11β , 19-(4-нонафторбутилсульфонилокси-о-фенилен)-андростан- 5α,17β- диол
50 г 3,3-диметилтриметилендиокси 11β , 19-(4-гилрокси-о-фенилен)-андростан - 5α,17β - диоля (пример 18а заявки PCT (PCT/DE88/00150), растворяют под защитным газом в 1,75 л тетрагидрофурана (слегка замутненный раствор) и смешивают при 0oC с 71,3 мл раствора литий-бутила (1,6 М в гексане). После перемешивания в течение 30 минут вливают по каплям 22,8 мл. 1,1,2,2,3,3,4,4,4-нонафторо-1-бутансульфонилфторида (~ 90%). После перемешивания в течение 1 часа с охлаждением на ледяной бане реакционную смесь помещают, не переставая перемешивать, в насыщенный раствор бикарбоната натрия и интенсивно перемешивают в течение еще 1 часа. Затем после добавления этилацетата, водную фазу отделяют и несколько раз экстрагируют с этилацетатом. Комбинированные органические фазы промывают до нейтрального состояния в насыщенном растворе хлорида натрия, высушивают на сульфате натрия и концентрируют путем испарения в вакууме. 90,9 г искомого соединения получают в виде сырого продукта.
1,9 г полученного при этом нонафлата (C4F9SO3) хроматографируют на силикагеле со смесью этилацетат/гексан.
1,27 г чистого искомого продукта получают в виде белой пены.
Точка плавления: 132 - 133oC; [α]
б) 3,3-диметилтриметилендиокси 5α - гидрокси 11β- , 19-(4-нонафторобутилсульфонилокси-о-фенилен)-андростан-17-он
63 г трехокиси хрома добавляют дробными порциями при 0oC к смеси, состоящей из 210 мл пиридина и 600 мл хлористого метилена. Затем 89 г нонафлата, полученного на стадии а) растворенного в 250 мл хлористого метилена, инстиллируют при той же температуре. После этого реакционную смесь медленно нагревают до комнатной температуры и перемешивают в течение 2 часов. После перемешивания всплывающую фазу переливают в другой сосуд, а осадок несколько раз тщательно промывают хлористым метиленом. Комбинированные органические фазы освобождают от остаточных неорганических компонентов промывкой в 0,5 М растворе гидроокиси натрия, промывают в воде до нейтрального состояния, высушивают на сульфате натрия и концентрируют путем испарения в вакууме (пиридин удаляют путем азеотропной дистилляции с толуолом). Посредством хроматографии осадка на окиси алюминия (нейтральном, этап III) со смесью этилацетат/гексана получают 61,9 г искомого продукта в виде желтоватой пены. После кристаллизации из этилацетата получают 55,7 г .
Точка плавления: 176 - 177oC; [α]
в) 3,3-диметилтриметилендиокси - 11β , 19-(4-нонафтор-бутилсульфонилокси-о-фенилен) 17α -[3-(тетрагидропиран-2-илокси)-проп-1-инил]-андростан - 5α,17β -диол
1 л абсолютного тетрагидрофурана смешивают при 0oC под защитным газом с 73,5 мл 2-(2-пропинилокси)тетрагидро-2H-пирана. Затем медленно вливают по каплям 328 мл 1,6 М раствора n-литий-бутила(гексан) без ощутимого подъема температуры. После 30-минутного перемешивания 50 г кетона, полученного на стадии б) и растворенного в 500 мл абсолютного тетрагидрофурана, медленно вливают в эту реакционную смесь и перемешивают еще в течение 30 минут. Затем реакционную смесь смешивают с насыщенным раствором хлорида аммония и экстрагируют водную фазу этилацетатом. Комбинированные органические фазы промывают раствором хлористого натрия, высушивают на сульфате натрия и концентрируют испарением в вакууме. Осадок хроматографируют на окиси алюминия (нейтральном, этап III). 50,3 г искомого соединения получают в виде белой пены.
г) 11β,19-[4-(4-цианофенил)-о-фенилен]-3,3-диметилтриметилендиокси- 17α -[3-(тетрагидропиран-2-илокси)-проп-1-инил]-андростан- 5α,17β -диол
50 г нонафлата, полученного на стадии в), растворяют в смеси из 400 мл толуола и 155 мл этанола и последовательно смешивают под защитным газом с 1,44 г тетракиса (трифенилфосфин) палладия (O), 5,33 г хлорида лития, 78 мл 2 М раствора карбоната натрия и 13,1 г.
4-(1,3,2-диоксаборинан-2-ил)бензонитрила. (S. Takahashi et al., Bil. Chem. Soc. Jpn., 62, 3896, 1989).
После этого реакционную смесь перемешивают в течение 3 часов при температуре масляной бани 95oC, охлаждают до комнатной температуры и смешивают с водой и этилацетатом. Водную фазу отделяют и экстрагируют с этилацетатом. Комбинированные органические фазы высушивают на сульфате натрия и концентрируют испарением в вакууме. Осадок хроматографируют на силикагеле со смесью этилацетата/гексана. 38 г искомого соединения получают в виде желтой пены.
[α]
д) 11β , 19-[4-(4-цианофенил)-о-фенилен] 17β -гидрокси- 17α -(3-гидроксипроп-1-инил)-4-андростен-3-он
37 г кетонацеталя, полученного на стадии в), растворяют в 950 мл ацетона и смешивают под защитным газом с 95 мл водного раствора соляной кислоты (4 n). После перемешивания в течение 2 часов при 50oC реакционную смесь выливают на холодный насыщенный раствор бикарбоната натрия (с основным pH) и перегоняют большую часть ацетона. После добавления хлористого метилена водную фазу отделяют и несколько раз экстрагируют с хлористым метиленом. Комбинированные органические фазы высушивают на сульфате натрия и концентрируют испарением в вакууме. Осадок хроматографируют на силикагеле со смесью этилацетат/гексана. 23,8 г искомого соединения получают в виде желтоватой пены.
е) 11β , 19-[4-(4-цианофенил)-о-фенилен] 17β -гидрокси 17α -(3-гидроксипроп-1(Z)-енил-4-андростен-3-он
23 г пропаргилового спирта, полученного на этапе д), растворяют под защитным газом в 825 мл тетрагидрофурана, смешивают с 23 мл пиридина и гидрируют с использованием 2,3 г палладия (10%) на сульфате бария, взятом в качестве катализатора, при стандартном давлении. После поглощения эквивалента водорода (в дополнение к тонкослойной хроматографии) реакционную смесь фильтруют на целите, осадок повторно промывают тетрагидрофураном и фильтрат концентрируют испарением в вакууме. Пиридин удаляют азеотропной дистилляцией с толуолом. Осадок рекристаллизуют из ацетон/тетрагидрофурана и полученный таким образом кристаллизуют
из хлористого метилена/метанола. 14,3 г искомого соединения получают в виде белого кристаллизата.
Точка плавления: 265 - 266oC (при разложении);
[α]
Пример приготовления 2
11β, 19-[4-(3-пиридинил)-о-фенилен] 17β -гидрокси- 17α -(3-гидроксипроп-1(Z)-енил-4-андростен-3-он
а) 11β, 19-[4-(3-пиридинил)-о-фенилен]-3,3-диметил-триметилендиокси- 17α -[3-тетрагидропиран-2-илокси)-проп-1-инил]-андростан 5α , 17β -диол
13,7 г нонафлата, полученного на стадии 1с), растворяют в смеси из 140 мл толуола и 70 мл этанола и последовательно смешивают под защитным газом с 877 мг тетракиса (трифенилфосфин) палладия (O), 1,29 г хлористого лития, 19 мл 2 М раствора карбоната натрия и 2,46 г диэтил(3-пиридинил)борана. Реакционную смесь затем перемешивают в течение 2 часов при температуре масляной бани 95oC, охлаждают до комнатной температуры и смешивают с водой и этилацетатом. Водную фазу отделяют и экстрагируют этилацетатом. Комбинированные органические фазы высушивают на сульфате натрия и концентрируют испарением в вакууме. Осадок хроматографируют на силикагеле со смесью этилацетат/гексана. 8,8 г искомого продукта получают в виде желтоватой пены.
б) 11β, 19-[4-(3-пиридинил)-о-фенилен] 17β -гидрокси 17α -(3-гидроксипроп-1-инил)-4-андростен-3-он
8,8 г кетонацеталя, полученного на стадии а), растворяют в 250 мл ацетона и смешивают под защитным газом с 5 мл водного раствора соляной кислоты (4 n). После 2 часов перемешивания при температуре 50oC реакционную смесь выливают на холодный насыщенный раствор бикарбоната натрия (с основным pH) и перегоняют большую часть ацетона. После добавления хлористого метилена водную фазу отделяют и несколько раз экстрагируют хлористым метиленом. Комбинированные органические фазы высушивают на сульфате натрия и концентрируют испарением в вакууме. Осадок хроматографируют на силикагеле смесью этилацетат/гексан. 5,0 г искомого соединения получают в виде желтоватой пены.
[α]
в) 11β , 19-[4-(3-пиридинил)-о-фенилен] 17β -гидрокси- 17α -(3-гидроксипроп-1(Z)-енил)-4-андростен-3-он
5 г пропаргилового спирта, полученного на стадии б), растворяют под защитным газом в 200 мл тетрагидрофурана, смешивают с 5 мл пиридина и гидрируют с использованием 500 мг палладия (10%) на сульфате бария, взятом в качестве катализатора, при стандартном давлении. После поглощения эквивалента водорода (в дополнение к тонкослойной хроматографии) реакционную смесь фильтруют на целите, осадок повторно промывают тетрагидрофураном, а фильтрат концентрируют испарением в вакууме. Пиридин удаляют азеотропной дистилляцией с толуолом. Осадок хроматографируют на силикагеле со смесью этилацетата/гексана. 3,4 г искомого продукта получают в виде желтоватой пены. В результате кристаллизации этилацетата получают 3,12 г белых кристаллов.
Точка плавления: 219 - 221oC; [α]
Пример 3.Технология изготовления препарата
10,0 мг 11β -[(4-N,N-диметиламино)-фенил]- 17α- гидрокси- 17β -(3-гидроксипропил)- 13α- метил-4,9(10)-гонадиен-3-он
140,5 мг лактозы
69,5 мг кукурузного крахмала
2,5 мг поливинилпирролидона 25
2,0 мг аэрозила
0,5 мг стеарата магния
225,0 мг общей массы.
Из этой композиции обычным способом изготавливают таблетки по 10 мг для перорального применения.
Для применения фармацевтического агента, изготовленного в соответствии с данным изобретением, существенно, чтобы, по меньшей мере, одна лечебная доза вводилась в фолликулярной фазе менструального цикла (до овуляции), и желательно, чтобы, по меньшей мере, одна лечебная доза вводилась в лютеальной фазе менструального цикла (после овуляции).
Предпочтительно, чтобы фармацевтический агент, изготовленный в соответствии с данным изобретением, вводился в индивидуальных лечебных дозах через каждые 4 - 10 дней, желательно еженедельно или в один и тот же день, начиная с любого дня перед овуляцией во время первого в курсе лечения менструального цикла. Желательно также, чтобы временные интервалы между введением индивидуальных лечебных доз были постоянными.
Предпочтительно, чтобы фармацевтический агент в соответствии с данным изобретением вводился еженедельно, в один и тот же день недели, например, по понедельникам ("пилюля понедельника"). При еженедельном ритме введения препарата в один и тот же день недели достигается высокая степень надежности лечения. Тем не менее, представляется возможным вводить лечебную дозу ежедневно, через каждые два или три дня, либо только в фолликулярной фазе, либо дополнительно и в лютеальной фазе менструального цикла. Кроме того, имеется возможность варьировать интервалы между введением индивидуальных лечебных доз фармацевтического агента по данному изобретению или вводить его постоянно при помощи имплантированного носителя медленного действия.
С целью определения доз конкурентных антагонистов прогестерона, ингибирующих овуляцию, было проведено описанное ниже испытание на крысах; дозы, вызывающие аборт, были определены в испытании на крысах, уже известном из опубликованных источников (например, патент США 5 095 129).
Коэффициенты диссоциации для соединений I, II и для RU 486;
Соединение - Коэффициент диссоциации
I - > 100
II - > 30
RU 486 - < 10
Было обнаружено, что соединения I и II, при необычайно сильной антиимплантационной эффективности (соединение I способно полностью ингибировать овуляцию у крысы при суточной дозе в 0,1 мг, а соединение II - при суточной дозе в 0,3 мг), не обладают антиглюкокортикоидной эффективностью. Это подтверждено в испытании на антиглюкокортикоидный эффект при инволюции вилочковой железы (EP-A-0 283 428).
Получение соединений I и II осуществляют в соответствии со способом синтеза, описанного в EP-A-0 283 428, как указано в вышеприведенных примерах.
Испытание способа ингибирования овуляции на крысах
Принцип, положенный в основу методики испытания
Вещества, ингибирующие овуляцию, удобнее всего выявлять в испытаниях на крысе, так как она спонтанно овулирует, и цикл легко прослеживается путем взятия вагинальных мазков. Это делает возможным контролировать ход испытания на стадии лечения.
Пример 4. Методика проведения испытания
Самки крысы весом 190 - 210 г, всего 6 животных.
Животных содержат в маролоновых клетках в комнатах с контролируемым световым режимом (10 часов в темноте и 14 часов на свету). Они получают стандартную диету (гранулированный крысиный корм) и водопроводную воду без ограничения.
Состав и введение испытуемого соединения
Испытуемые соединения растворяют в смеси бензилбензоата и касторового масла (1 + 9 по объему). Суточную дозу вводят подкожно в количестве 0,2 мл.
При пероральном введении испытуемое вещество суспендируют в жидком носителе (85 г Myri® в 100 мл раствора NaCl 0,9% по весу). Суточную дозу вводят в объеме 0,5 мл.
Экспериментальная партия животных
Перед началом испытания за животными наблюдают в течение двух циклов путем взятия вагинальных мазков. В испытании используют только животных с регулярным 4-дневным циклом. Распределение по группам проводят методом слепого (безвыборочного) отбора. Начиная с метэструса (после течки) испытуемое вещество вводят в течение 4 дней (дни 1 - 4) и продолжают контролировать цикл.
На 4-й день (после начала введения препарат) животных, у которых при взятии вагинальных мазков обнаруживают эструс (течку) или метэструс, подвергают односторонней овариэктомии под эфирным наркозом. Из труб готовят мелкоизмельченные препараты, которые исследуют под микроскопом на наличие яйцеклеток. На 5-й день всех животных (интактных и подвергнутых односторонней овариэктомии) умерщвляют газом CO2 и упомянутым выше способом готовят и исследуют препараты из труб.
Оценка результатов
В каждой группе определяют процент животных с ингибированной овуляцией.
Пример 5. Методика проведения испытания на человеке
Добровольцы:
Анамнестическое состояние пациентов: только здоровые женщины с нормальным менструальным циклом. Предпочтение отдают добровольцам, у которых перевязаны обе фаллопиевы трубы, или после хирургической экстирпации обеих фаллопиевых труб (внематочные беременности), а также женщинам, половыми партнерами которых являются мужчины, ранее подвергнутые ваээктомии.
Исследование на человеке 1: Характеристика биохимического и морфологического маркеров восприимчивости эндометрия после краткосрочного лечения онапристоном во время лютеальной фазы.
Это поисковое исследование, проводимое для выяснения, какие биохимические методы, и, в частности, методы идентификации маркерных протеинов эндометрия будут полезными при проведении дальнейших исследований эффективности лечения низкими дозами онапристона (вводимыми ежедневно и прерывистыми курсами).
Протокол эксперимента:
Группы добровольцев:
Две группы (контрольная и группа, проходящая лечение) по 8 - 10 женщин в каждой.
Длительность исследования
Оценка трех циклов: предшествующего лечению, лечения и постлечебного.
Лечение: 50 мг онапристона в сутки в дни ЛГ + 1 и ЛГ + 2 перорально.
Известно, что такая доза онапристона вызывает преждевременная менструацию у женщин, если ее дают в конце лютеальной фазы. Однако, как показывает исследование Биджмана (Swahn et al. (1990)), лечение в начале лютеальной фазы (ЛГ + 2) однократной дозой в 200 мг RU 486 не приводит к нарушениям длительности цикла.
Оценка: радиоиммуноанализ: ЛГ, фолликулостимулирующий гормон, E2 и прогестерон в предлечебном, лечебном и постлечебном циклах.
- Биопсия эндометрия и маточного отделяемого в день ЛГ + 3 и в день ЛГ + 8 (лечебный цикл).
- Появление менструального кровотечения.
Исследование на человеке 2: оценка влияния ежедневного введения онапристона на овуляцию и на морфологию и биохимию эндометрия: исследование по определению оптимальной дозы.
а) Цель:
Результаты этого исследования позволяют определить дозу онапристона, которая не ведет к нарушению менструального цикла, но оказывает требуемый эффект на морфологию и функцию (маркерные протеины) эндометрия. При этом может быть установлена минимальная доза, вызывающая изменения в эндометрии.
б) Методика исследования:
Группы добровольцев: четыре группы (три группы, проходящие курс лечения, и одна контрольная группа) по 8 - 10 женщин в каждой.
Длительность исследования: оценка трех циклов - предлечебного, лечебного и постлечебного.
Схема лечения: пероральный ежедневный прем в течение 18 дней по 20 мг онапристона (Группа 1): 10 мг (группа 2) и 5 мг (группа 3), начиная с 1 дня цикла.
в) Оценка:
- РИА, ЛГ, ФСГ, E2 и прогестерон и кортизол во время предлечебного, лечебного и постлечебного циклов.
- Оценка биопсий эндометрия и взятие маточного отделяемого на 16 - и 23 -й день лечебного цикла,
- появление менструального кровотечения.
Исследование на человеке 3: влияние еженедельного введения онапристона на овуляцию, морфологию и биохимию эндометрия.
а) Цель:
Результаты этого исследования позволяют определить еженедельную дозу онапристона, не нарушающую цикл, но воздействующую на морфологию и функцию (маркерные протеины) эндометрия.
б) Методика исследования:
Группы добровольцев: шесть групп (пять групп, проходящих лечение, и одна контрольная группа) по 8 - 10 женщин с перевязанными яйцеводами.
Длительность исследования: оценка четырех циклов: предлечебного, двух лечебных и постлечебного.
Лечение:
Раз в неделю пероральное введение 50 мг (группа 1), 20 мг (группа 2), 10 мг (группа 3), 2 мг (группа 5) и 0,25 мг (группа 6) онапристона, начиная с 1 дня цикла в течение 2 циклов.
в) Оценка:
РИА, ЛГЮ ФСГ, E2 и прогестерон во время предлечебного, лечебных и постлечебного циклов.
Оценка биопсий эндометрия и взятие маточного отделяемого на 16 и 23 день обоих лечебных циклов.
Появление менструального кровотечения и нарушения цикла.
При выполнении описанной выше схемы лечения были сделаны следующие наблюдения: по сравнению с контрольной группой в лечебной группе контроль за циклом был более эффективным: были отмечены высокая степень толерантности; высокая надежность контрацепции; оральная контрацепция без эстрогенов.
Приведенные выше примеры могут быть воспроизведены с такой же степенью эффективности, если использовавшиеся в них химические соединения и/или условия их применения будут заменены на реагенты, описанные (в общих чертах или подробно) в данном изобретении.
Из приведенного выше описания специалист легко поймет существенные признаки данного изобретения и, не отходя от духа и объема последнего, может ввести в него различные изменения и модификации, чтобы приспособить его к различным условиям применения.



Изобретение относится к медицине, а именно к способам контрацепции. Предложено применять для оральной контрацепции конкурентные антагонисты прогестерона. Антагонисты прогестерона применяют в течение фолликулярной и необязательно лютеальной фазы менструального цикла. При этом используют дозы, которые ниже доз, ингибирующих овуляцию и вызывающих аборт. Способ позволяет ингибировать формирование желез эндометрия, делая невозможной имплантацию оплодотворенной яйцеклетки. 2 с. и 12 з.п.ф-лы, 2 табл., 1 ил.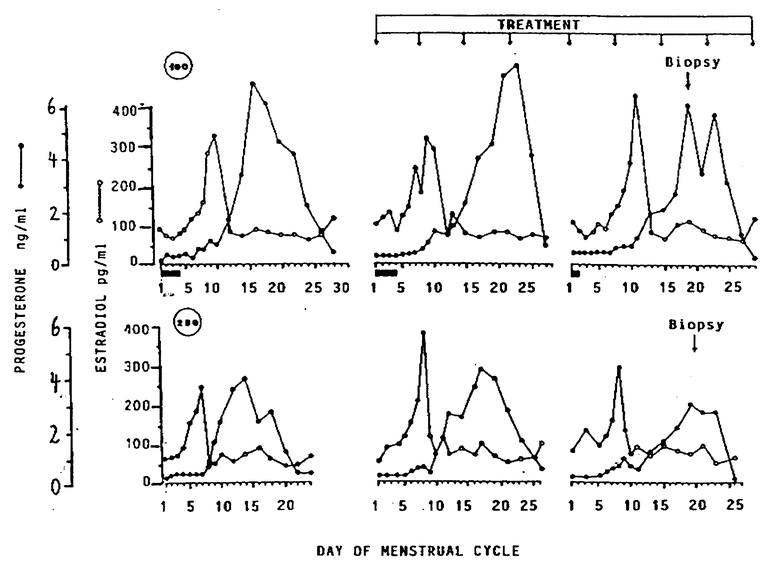
11β-[(4-N, N-диметиламино)-фенил] -17β-гидрокси-17α-пропинил-4,9(10)-эстрадиен-3-он;
11β-[(4-N, N-диметиламино)-фенил] -17β-гидрокси-18-метил-17α-пропинил-4,9(10)-эстрадиен-3-он;
11β-[(4-N, N-диметиламино)-фенил] -17аβ-гидрокси-17аα-пропинил-D-гомо-4,9(10), 16-эстрадиен-3-он;
11β- n-метоксифенил-17β-гидрокси-17α-этинил-4,9(10)-эстрадиен-3-он или
11β-(4-ацетилфенил)-17β-гидрокси-17α-(проп-1-инил)-4,9(10)-эстрадиен-3-он.
11β-(4-диметиламинофенил)-17α-гидрокси-17β-(3-гидроксипропил)-13α-метил-4,9-гонадиен-3-он или
11β- (4-ацетилфенил)-17β- гидрокси-17α-(3-гидроксипроп-1-енил)-4,9(10)-эстрадиен-3-он.
11β,19-[4-(цианофенил)-о-фенилен]-17β-гидрокси-17α-(3-гидроксипроп-1(Z)-енил)-4-андростен-3-он, или
11β,19[4-(3-пиридинил)-о-фенилен]-17β-гидрокси-17α-(3-гидроксипроп-1(Z)-енил-4-андростен-3-он).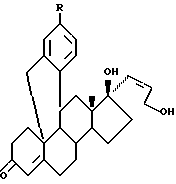
где R
или
13. Соединение по п.12, отличающееся тем, что представляет собой 11β, 19-[4-(цианофенил)-о-фенилен-17β-гидрокси-17α-(3-гидроксипроп-1(Z)-енил-4-андростен-3-он.
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| НИЖНЯЯ РЕШЕТКА ИЗМЕЛЬЧИТЕЛЯ ИЛИ БАРАБАННОЙ ДРОБИЛКИ И СПОСОБ ИЗГОТОВЛЕНИЯ НИЖНЕЙ РЕШЕТКИ | 2012 |
|
RU2598083C2 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Машковский М.Д | |||
| Лекарственные средства | |||
| - М.: Медицина, 1988, т.1, с.580-597. | |||

