УРОВЕНЬ ТЕХНИКИ
1. Область техники
[0001] Настоящее изобретение относится к новым соединениям, проявляющим антагонизм по отношению к прогестерону и не имеющим каких-либо признаков частичной агонистической активности. Указанные соединения можно применять для регуляции фертильности и лечения гормонально-зависимого рака груди. Настоящее изобретение также относится к способам получения и применению указанных новых соединений в терапии.
2. Описание уровня техники
[0002] В прошлом было сделано предположение, что антагонисты прогестерона могут оказывать благоприятное действие при лечении ряда заболеваний, включая рак груди, и при различных формах регуляции фертильности.
[0003] В настоящее время только два соединения, принадлежащие к указанному классу, одобрены для клинического применения. Являющийся прототипом антагонист мифепристон (см. фигуру 1) показан для индуцирования аборта, а улипристал одобрен для посткоитальной регуляции фертильности.
[0004] Оба соединения характеризуются тем, что они являются модуляторами рецепторов прогестерона, и это означает, что в общую активность указанных соединений может вносить вклад и частичная агонистическая активность.
[0005] Соединения, не имеющие какой-либо частичной агонистической активности, должны проявлять повышенную активность при одобренных показаниях, а также, возможно, при лечении рака груди.
Фигура 1
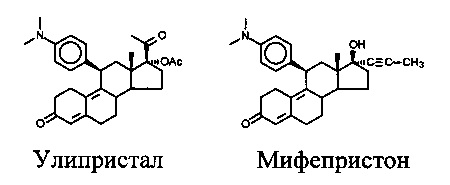
[0006] Общеизвестно, что заместитель в положении 11' определяет свойства в отношении рецептора прогестерона (Nickisch et al Steroids Vol 78,255-267,2013). В то время как диметиламиногруппа, присутствующая в мифепристоне и улипристале, обеспечивает преимущественно антагонистическую активность у соединений, ароматические заместители, такие как фураны или пиридины, обеспечивают соединения с высокой активностью частичного агониста (ЕР 000002417148), которые можно применять для лечения гинекологических показаний, таких как эндометриоз, но не для посткоитальной регуляции фертильности и лечения рака груди.
[0007] Заместители в положении 17 влияют на селективность связывания прогестерона и глюкокортикоидного рецептора. Сообщалось, что фрагментом в положении 17, который обеспечивает высокую селективность в отношении рецептора прогестерона, является, например, перфторалкильная группа, что первоначально описано в DE 19706061.
[0008] Позднее были описаны различные заместители в положении 11, содержащие перфторалкильную группу в положении 17, см., например, WO 2008058767, WO 2011005929, WO 2011009530, WO 2011009531, WO 2011009534, WO 2011098436 и WO 2011098437.
[0009] Другие заместители в положении 17, обладающие хорошей специфичностью в отношении рецептора прогестерона, включают 17-спирофуран-3'-илиден, описанный в ЕР 569041, 17-спиролактоны, описанные в ЕР 558416, и дифтор-17-спирофуран-3'-илиден, описанный в WO 20100118025.
[0010] В указанных патентах описаны разнообразные заместители в положении 11', включая различные гетероциклы, но неожиданно отсутствует описание 11'-N-имидазолов, что вероятно связано с тем фактом, что для синтеза таких молекул требуются специальные способы, даже несмотря на то, что общая структура была предложена Куком (Cook) с соавторами в WO 99/45022.
[0011] Поэтому еще более неожиданным оказалось то, что описанные 11'-N-имидазолы обладают очень высокой антипрогестагенной активностью в отсутствие каких-либо агонистических компонентов, что делает их идеальными кандидатами для стимуляции родов, посткоитальной регуляции фертильности, прерывания беременности и лечения рака груди.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
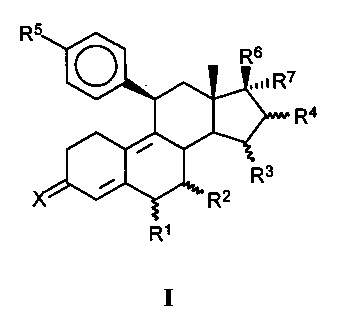
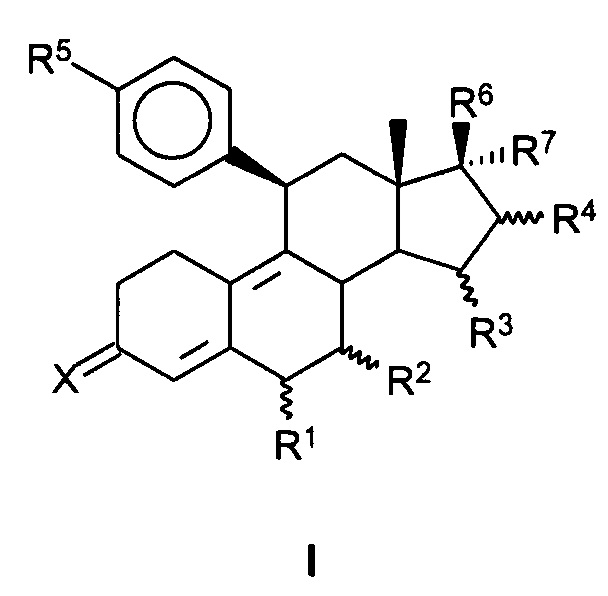
[0012] В одном из вариантов реализации антагонист прогестерона имеет структуру формулы (I):
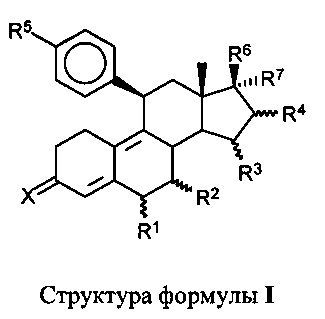
где:
X представляет собой O или H2;
R1 представляет собой атом водорода, линейную C1-C5 алкильную группу, разветвленную C1-C5 алкильную группу, C3-C5 циклоалкильную группу или атом галогена;
R2 представляет собой атом водорода, линейную C1-C5 алкильную группу, разветвленную C1-C5 алкильную группу, C3-C5 циклоалкильную группу или атом галогена; или
R1 и R2 совместно представляют собой метиленовую группу,
R3 представляет собой атом водорода, линейную C1-C5 алкильную группу, разветвленную C1-C5 алкильную группу, C3-C5 циклоалкильную группу или атом галогена;
R4 представляет собой атом водорода, линейную C1-C5 алкильную группу, разветвленную C1-C5 алкильную группу, C3-C5 циклоалкильную группу или атом галогена; или
R3 и R4 совместно представляют собой дополнительную связь или метиленовую группу,
R5 представляет собой N-имидазолильную группу, которая может быть необязательно замещена одной или более алкильными группами,
R6 представляет собой свободную, превращенную в простой или сложный эфир, гидроксильную группу,
R7 представляет собой радикал формулы CnFmHo, где n равен 2, 3, 4, 5 или 6, причем m≥1 и m+о=2n+1 или 2n-1 или 2n-3.
[0013] В одном из вариантов реализации антагонист прогестерона имеет структуру формулы (I),
где
X представляет собой O;
R1 и R2 представляют собой атомы водорода;
R3 и R4 представляют собой атомы водорода;
R5 представляет собой N-имидазолильную группу;
R6 представляет собой гидроксильную группу;
R7 представляет собой радикал формулы CnFmHo, где n равен 2, 3, 4, 5 или 6, причем m≥1 и m+o=2n+1 или 2n-1 или 2n-3.
[0014] В одном из вариантов реализации антагонист прогестерона имеет структуру формулы (I),
где
X представляет собой O;
R1 и R2 совместно представляют собой метиленовую группу;
R3 и R4 совместно представляют собой дополнительную связь или метиленовую группу;
R5 представляет собой N-имидазолильную группу;
R6 представляет собой гидроксильную группу;
R7 представляет собой радикал формулы CnFmHo, где n равен 2, 3, 4, 5 или 6, причем m≥1 и m+о=2n+1 или 2n-1 или 2n-3.
[0015] В одном из вариантов реализации антагонист прогестерона имеет структуру формулы (I),
где
X представляет собой O;
R1 и R2 представляют собой атомы водорода;
R3 и R4 представляют собой атомы водорода;
R5 представляет собой N-имидазолильную группу;
R6 представляет собой гидроксильную группу;
R7 представляет собой радикал формулы CnFmHo, где n равен 2, 3, 4, 5 или 6, причем m≥1 и m+о=2n+1.
[0016] В одном из вариантов реализации антагонист прогестерона имеет структуру формулы (I),
где
X представляет собой O;
R1 и R2 представляют собой атомы водорода;
R3 и R4 представляют собой атомы водорода;
R5 представляет собой N-имидазолильную группу;
R6 представляет собой гидроксильную группу;
R7 представляет собой радикал формулы CnFmHo, где n равен 2, 3, 4, 5 или 6, причем m≥1 и m+o=2n-1.
[0017] В одном из вариантов реализации антагонист прогестерона имеет структуру формулы (I),
где
X представляет собой O;
R1 и R2 представляют собой атомы водорода;
R3 и R4 представляют собой атомы водорода;
R5 представляет собой N-имидазолильную группу;
R6 представляет собой гидроксильную группу;
R7 представляет собой радикал формулы CnFmHo, где n равен 2, 3,4, 5 или 6, причем m≥1 и m+o=2n-3.
[0018] В одном из вариантов реализации антагонист прогестерона имеет структуру:
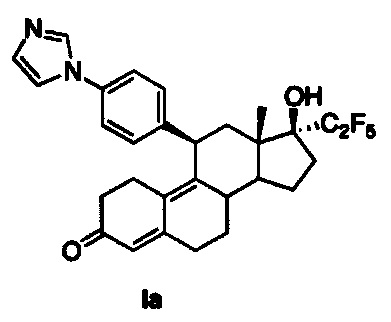 .
.
[0019] В одном из вариантов реализации антагонист прогестерона имеет структуру:
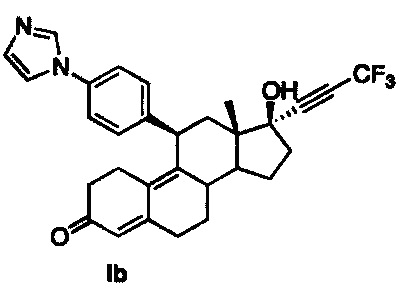 .
.
[0020] В одном из вариантов реализации антагонист прогестерона имеет структуру:
 .
.
[0021] В одном из вариантов реализации антагонист прогестерона имеет структуру:
 .
.
[0022] В одном из вариантов реализации антагонист прогестерона имеет структуру формулы (I):
где:
X представляет собой O или H2;
R1 представляет собой атом водорода, линейную C1-C5 алкильную группу, разветвленную C1-C5 алкильную группу, C3-C5 циклоалкильную группу или атом галогена;
R2 представляет собой атом водорода, линейную C1-C5 алкильную группу, разветвленную C1-C5 алкильную группу, C3-C5 циклоалкильную группу или атом галогена; или
R1 и R2 совместно представляют собой метиленовую группу;
R3 представляет собой атом водорода, линейную C1-C5 алкильную группу, разветвленную C1-C5 алкильную группу, C3-C5 циклоалкильную группу или атом галогена;
R4 представляет собой атом водорода, линейную C1-C5 алкильную группу, разветвленную C1-C5 алкильную группу, C3-C5 циклоалкильную группу или атом галогена; или
R3 и R4 совместно представляют собой дополнительную связь или метиленовую группу;
R5 представляет собой N-имидазолильную группу, необязательно замещенную одной или более алкильными группами; и
R6 и R7 представляют собой
 или
или 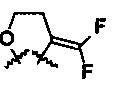 .
.
Волнистые линии обозначают, что данный заместитель может находиться в α- или β-положении.
[0023] В одном из вариантов реализации антагонист прогестерона имеет структуру формулы (I):
X представляет собой O;
R1 и R2 представляют собой атомы водорода;
R3 и R4 представляют собой атомы водорода;
R5 представляет собой N-имидазолильную группу, необязательно замещенную одной или более алкильными группами; и
R6 и R7 представляют собой
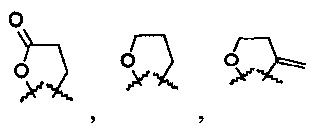 или
или 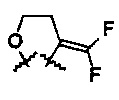 .
.
[0024] В одном из вариантов реализации антагонист прогестерона имеет структуру формулы (I):
где
X представляет собой O;
R1 и R2 совместно представляют собой метиленовую группу;
R3 и R4 совместно представляют собой дополнительную связь или метиленовую группу;
R5 представляет собой N-имидазолильную группу, необязательно замещенную одной или более алкильными группами; и
R6 и R7 представляют собой
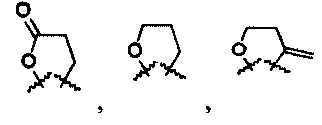 или
или 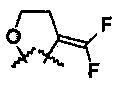 .
.
[0025] В одном из вариантов реализации антагонист прогестерона имеет структуру формулы (I):
где
X представляет собой O;
R1 и R2 представляют собой атомы водорода;
R3 и R4 представляют собой атомы водорода;
R5 представляет собой N-имидазолильную группу, необязательно замещенную одной или более алкильными группами; и
R6 и R7 представляют собой 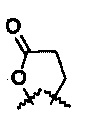 .
.
[0026] В одном из вариантов реализации антагонист прогестерона имеет структуру формулы (I):
где
X представляет собой O;
R1 и R2 представляют собой атомы водорода;
R3 и R4 представляют собой атомы водорода;
R5 представляет собой N-имидазолильную группу, необязательно замещенную одной или более алкильными группами; и
R6 и R7 представляют собой 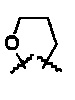 .
.
[0027] В одном из вариантов реализации антагонист прогестерона имеет структуру формулы (I):
где
X представляет собой O;
R1 и R2 представляют собой атомы водорода;
R3 и R4 представляют собой атомы водорода;
R5 представляет собой N-имидазолильную группу, необязательно замещенную одной или более алкильными группами; и
R6 и R7 представляют собой 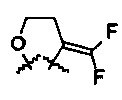 .
.
[0028] В одном из вариантов реализации антагонист прогестерона имеет структуру:
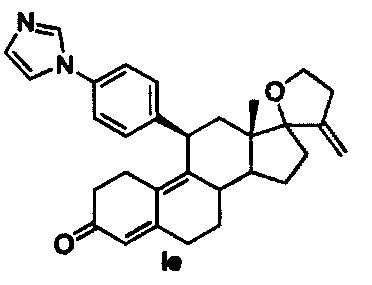 .
.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
[0029] Преимущества настоящего изобретения станут понятны специалистам в данной области техники после изучения следующего подробного описания вариантов реализации и прилагаемых чертежей, где:
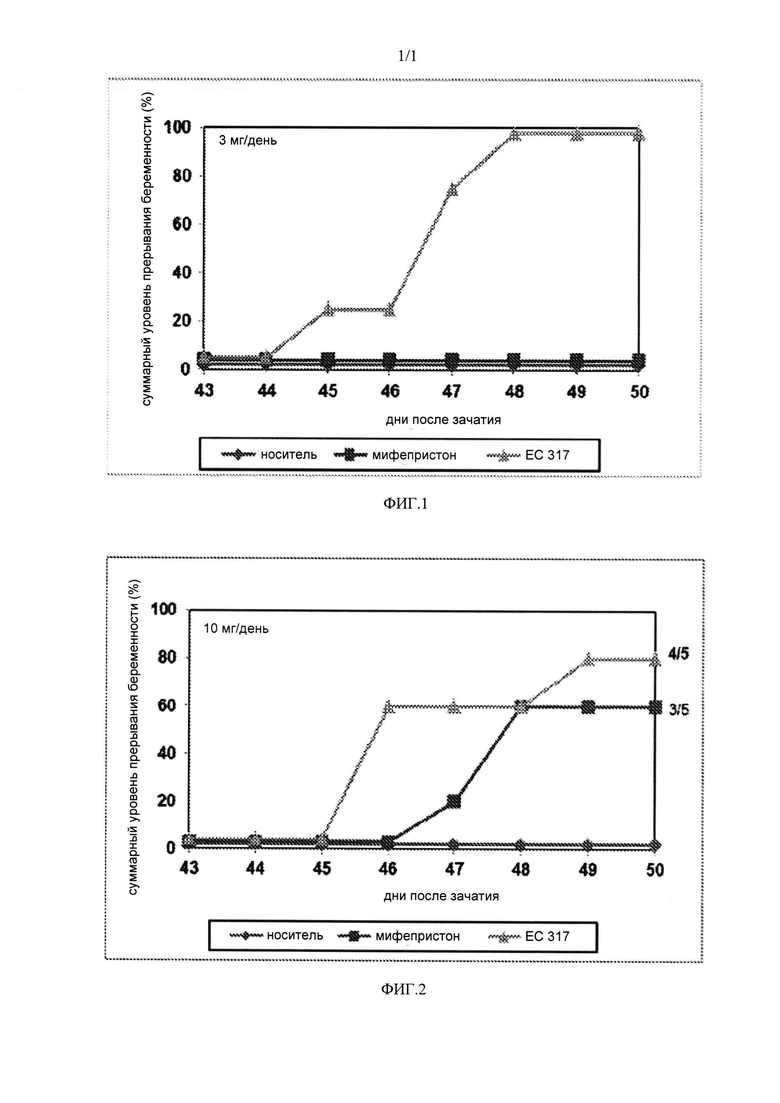
На ФИГ. 1 приведен график суммарного уровня прерывания беременности при исследовании 3 мг/день соединения, имеющего структуру 1, на модели беременных морских свинок; и
На ФИГ. 2 приведен график суммарного уровня прерывания беременности при исследовании 10 мг/день соединения, имеющего структуру 1, на модели беременных морских свинок.
[0030] Несмотря на то, что изобретение допускает различные модификации и альтернативные формы, конкретные варианты реализации показаны в качестве примеров на чертежах и подробно описаны в настоящей заявке. Чертежи необязательно являются масштабируемыми. Следует понимать, тем не менее, что чертежи и подробное описание не ограничивают изобретение конкретной описанной формой, и, напротив, охватывают все модификации, эквиваленты и альтернативные формы, не выходящие за рамки сущности и объема настоящего изобретения, определенного прилагаемой формулой изобретения.
ПОДРОБНОЕ ОПИСАНИЕ ПРЕДПОЧТИТЕЛЬНЫХ ВАРИАНТОВ РЕАЛИЗАЦИИ
[0031] Следует понимать, что настоящее изобретение не ограничено конкретными устройствами или биологическими системами, которые, безусловно, могут быть различными. Также следует понимать, что терминология, используемая в настоящем описании, приведена исключительно для описания конкретных вариантов реализации и не является ограничивающей. При использовании в настоящем описании и прилагаемой формуле изобретения формы единственного числа включают ссылки на отдельные объекты и множество объектов, если в контексте явно не указано иное. Таким образом, например, ссылка на «линкерную группу» включает одну или более линкерных групп.
[0032] Если отсутствуют иные определения, все технические и научные термины, используемые в настоящем описании, имеют значения, общепринятые специалистами в данной области техники.
[0033] Термин «алкил», используемый в настоящем описании, в общем случае относится к химическому заместителю, содержащему одновалентную группу CnH2n, где n представляет собой положительное целое число. В некоторых вариантах реализации n равен от 1 до 12. Термин «алкил» включает разветвленные или неразветвленные одновалентные углеводородные радикалы. Примеры алкильных радикалов включают, но не ограничиваются ими: метил, этил, пропил, изопропил, бутил, изобутил, втор-бутил, пентил, 3-пентил, гексил, гептил, октил, нонил, децил, ундецил, додецил. Алкильную группу, которая содержит 1-6 атомов углерода, называют «низшим алкилом». Подходящие низшие алкильные радикалы включают, но не ограничиваются ими, метил, этил, н-пропил, изопропил, 2-пропенил (или аллил), н-бутил, трет-бутил и изобутил (или 2-метилпропил).
[0034] Термин «замещенные алкилы», используемый в настоящем описании, в общем случае относится к алкильным радикалам, содержащим одну или более функциональных группу, присоединенных к любому атому углерода алкильного радикала. Функциональные группы включают, но не ограничиваются ими, арил, аралкил, ацил, галогены, гидроксил, амино, алкиламино, ациламино, ацилокси, алкокси и меркапто. Согласно настоящему описанию термин «замещенный низший алкил» относится к алкильному остатку, содержащему 1-6 атомов углерода и одну или более функциональных групп, присоединенных к любому атому углерода алкильного радикала.
[0035] Термин «алкокси» в общем случае относится к группе -OR, где R представляет собой низший алкил, замещенный низший алкил, арил, замещенный арил, аралкил или замещенный аралкил. Подходящие алкоксирадикалы включают, но не ограничиваются ими, метокси, этокси, фенокси, трет-бутокси, метоксиэтокси и метоксиметокси.
[0036] Термин «ацилокси» используют 6 настоящей заявке для описания органического радикала, полученного из органической кислоты в результате удаления атома водорода. Органический радикал может быть дополнительно замещен одной или более функциональными группами, включая, но не ограничиваясь ими, алкил, арил, аралкил, ацил, галоген, амино, тиол, гидроксил, алкокси и т.д. Подходящие ацилоксигруппы включают, например, ацетокси, т.е. CH3COO-, который является производным уксусной кислоты.
[0037] Термин «галоген» используют в настоящей заявке для описания атомов фтора, брома, хлора и йода.
[0038] Термин «гидроксил» используют в настоящей заявке для описания группы -OH.
[0039] Термин «алкилацил» обозначает группы -C(O)R, где R представляет собой алкил или замещенный алкил, арил или замещенный арил, такие как определено в настоящем описании.
[0040] Термин «циклоалкилацил» обозначает группы -C(O)R, где R представляет собой циклоалкил или замещенный циклоалкил, такие как, например, циклопропилацил-, циклопентилацил и циклогексилацил.
[0041] Термин «арил» используют для описания ароматического заместителя, который может представлять собой отдельное кольцо или несколько конденсированных колец, соединенных посредством ковалентных связей или общей группы, такой как этиленовый фрагмент. Ароматическое(-ие) кольцо(-а) включает(-ют), но не ограничивается(-ются) ими, фенил, нафтил, бифенил, дифенилметил и 2,2-дифенил-1-этил. Арильная группа также может быть замещена заместителями, включая, но не ограничиваясь ими, алкильные группы, атомы галогенов, нитрогруппы, карбоксильные группы, алкокси и фенокси, с образованием «замещенной арильной группы». Заместители могут быть присоединены через любое положение арильного радикала, которое в иных случаях занято атомом водорода.
[0042] Термин «фторированный алкинил», используемый в настоящем описании, в общем случае относится к алкинильным радикалам, содержащим один или более атомов фтора, присоединенных к любому атому углерода алкинильного радикала вместо атома водорода.
[0043] Термин «фармацевтически приемлемые соли» включает соли, полученные в результате взаимодействия фармацевтически приемлемых нетоксичных оснований или кислот, включая неорганические и органические основания, с неорганическими или органическими кислотами. Фармацевтически приемлемые соли могут включать соли, полученные из неорганических оснований, включая алюминий, аммоний, кальций, медь, железо (III), железо (II), литий, магний, соли марганца (III), марганца (II), калия, натрия, цинка и т.д. Примеры включают соли аммония, кальция, магния, калия и натрия. Соли, полученные из фармацевтически приемлемых нетоксичных оснований, включают соли первичных, вторичных и третичных аминов, замещенных аминов, включая природные замещенные амины, циклических аминов и основных ионообменных смол, таких как аргинин, бетаин, кофеин, холин, N,N'-дибензилэтилендиамин, диэтиламин, 2-дибензилэтилендиамин, 2-диэтиламиноэтанол, 2-диметиламиноэтанол, этаноламин, этилендиамин, N-этилморфолин, N-этилпиперидин, глюкамин, глюкозамин, гистидин, гидрабамин, изопропиламин, лизин, метилглюкамин, морфолин, пиперазин, пиперидин, полиаминовые смолы, прокаин, пурины, теобромин, триэтиламин, триметиламин, трипропиламин, трометамин и т.д.
Следующие примеры включены для демонстрации предпочтительных вариантов реализации изобретения. Специалистам в данной области техники должно быть понятно, что способы, описанные в последующих примерах, представляют собой способы, предложенные авторами настоящего изобретения для реализации изобретения, и, таким образом, их можно рассматривать как предпочтительные способы реализации изобретения. Тем не менее, специалистам в данной области техники с учетом настоящего описания должно быть понятно, что можно проводить многочисленные изменения конкретных раскрытых вариантов реализации и при этом добиваться схожих или аналогичных результатов, не выходя за рамки сущности и объема изобретения.
Экспериментальная часть:
Конкретные примеры соединений, имеющих формулу (I), включают следующие соединения:
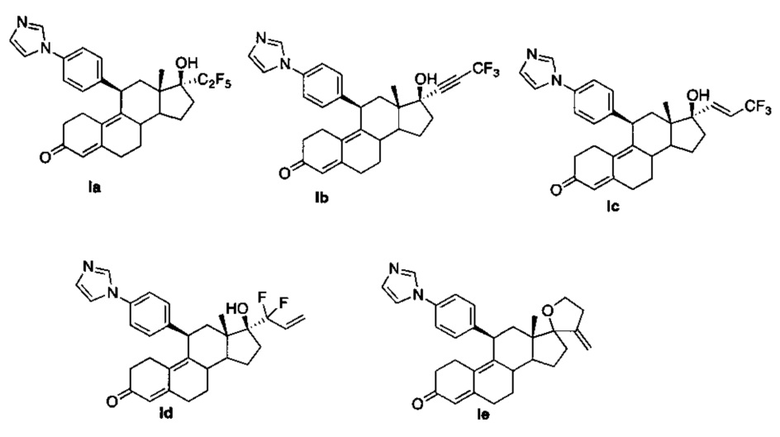
Синтез соединений Ia, Ib, Ic, Id, Ie можно проводить согласно следующим схемам.
Соединение I можно синтезировать согласно схеме, указанной ниже.
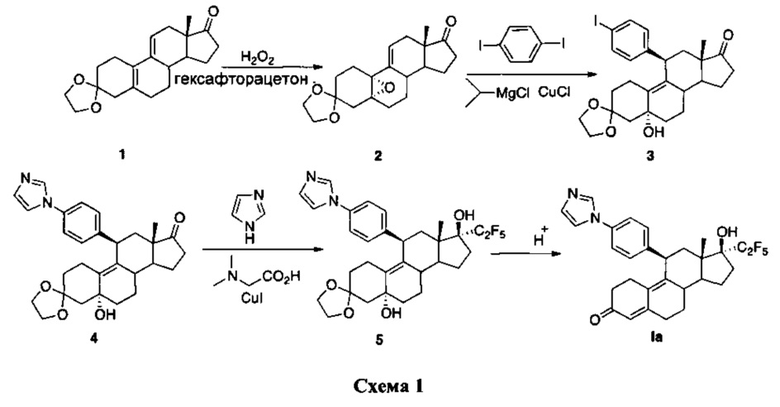
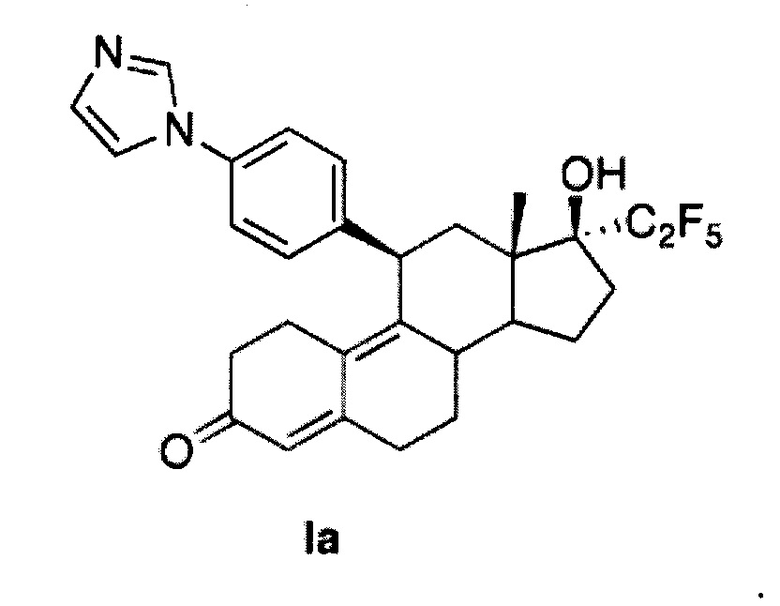
Промежуточное соединение 2 можно синтезировать согласно способу, описанному в Steroids, 1998, 63, 253. Промежуточное соединение 3 получают путем добавления арилкупрата, полученного в результате взаимодействия 1,4-дииодбензола, хлорида изопропилмагния и каталитических количеств хлорида меди (I), к промежуточному соединению 2. Проводят сочетание полученного таким образом йодарильного производного 3 с имидазолом в условиях реакции Улльмана с использованием йодида меди (I) в качестве медного катализатора и N,N-диметилглицина в качестве лиганда. Добавление пентафторлития к 17-кетогруппе соединения 4 и последующий гидролиз приводят к получению соединения Iа.
Соединения Iа и Ib можно синтезировать согласно следующей схеме.
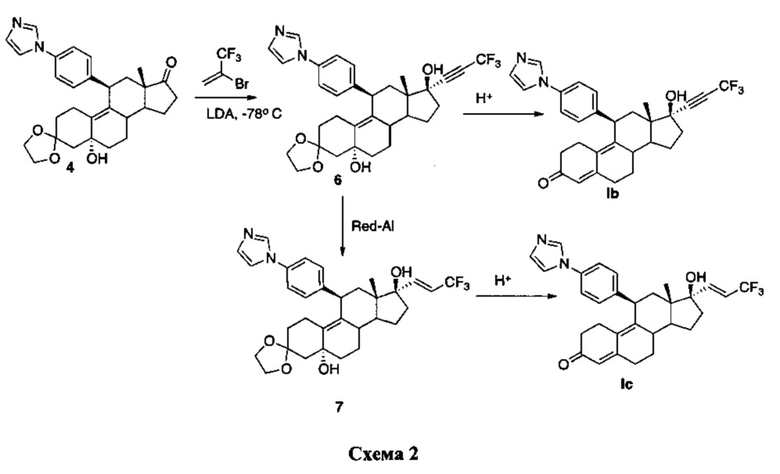
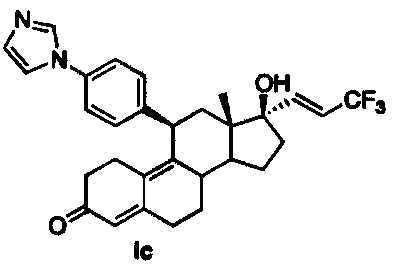
3,3,3-трифторпропиниллитий, полученный в результате обработки 2-бром-3,3,3-трифторпропена с использованием LDA при -78°C, добавляли к 17-кетогруппе промежуточного соединения 3 для получения соединения 6, из которого в результате кислотного гидролиза получали соединение Ib. Восстановление с использованием Red-Al промежуточного соединения 6 приводило к получению соединения 7, из которого путем гидролиза с использованием 4н. хлороводородной кислоты получали соединение Iс.
Соединение Id можно получать согласно схеме, указанной ниже.
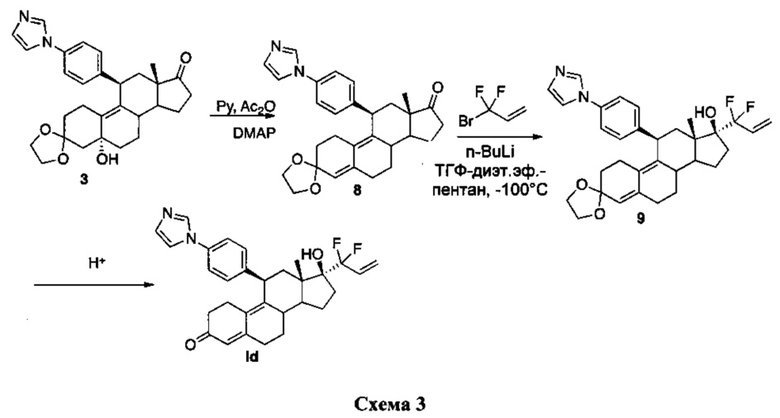
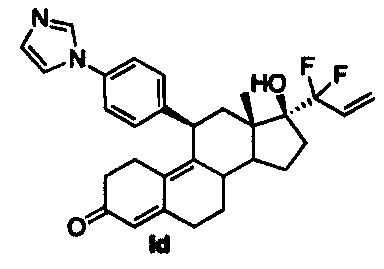
Промежуточное соединение 3 дегидрировали по положению 5 с использованием избытка ангидрида уксусной кислоты и пиридина. Полученное неочищенное вещество использовали без очистки и добавляли дифтораллиллитий при -100°C для получения соединения 9, из которого путем кислотного гидролиза получали соединение Id.
Синтез соединения Iе можно проводить согласно способу, указанному на схеме 4.
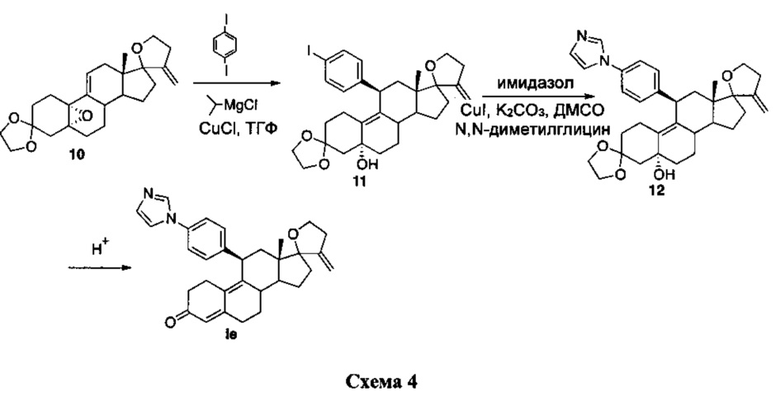
Промежуточное соединение 10 получали согласно способу, описанному в Jiang, et al., Bioorg Med Chem (2006) 14: 6726-6732. Добавление арилкупрата, полученного с использованием 1,4-дииодбензола, хлорида изопропилмагния и хлорида меди (I), к эпоксиду 10 приводило к получению соединения 11.
Сочетание по Улльману промежуточного соединения 11 с имидазолом с использованием йодида меди (I) в качестве катализатора, N,N-диметилглицина в качестве лиганда и карбоната калия в качестве основания приводило к получению промежуточного соединения 12, из которого путем кислотного гидролиза получали соединение Iе.
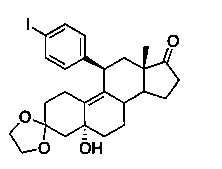
3,3-этилендиокси-5α-гидрокси-11β-[4'-йодфенил]эстр-9-ен-17-он (3)
Раствор 1,4-дииодбензола (13,2 г, 40 ммоль) в безводном ТГФ (80 мл) охлаждали до -10°C, после чего по каплям в течение 15 минут добавляли 2 М раствор хлорида изопропилмагния (20 мл, 40 ммоль). После перемешивания в течение 20 минут добавляли твердый хлорид меди (I) (898 мг, 9,07 ммоль) и реакционную смесь перемешивали в течение 30 минут. По каплям добавляли раствор эпоксида 2 (6 г, 18 ммоль) в 60 мл ТГФ и перемешивали в течение 2 часов, медленно нагревая до 10°C. Реакцию гасили насыщенным водным раствором хлорида аммония (50 мл) и смесь экстрагировали этилацетатом (3×50 мл). Объединенные органические слои дополнительно промывали водой и солевым раствором, сушили над сульфатом натрия и выпаривали в вакууме с получением неочищенного продукта. Неочищенный продукт растирали с диизопропиловым эфиром (120 мл) для осаждения чистого продукта, который отфильтровывали, промывали ледяным диизопропиловым эфиром (30 мл) и сушили в вакууме с получением 6,9 г (72%) соединения 3 в виде беловатого твердого вещества.
1Н ЯМР (δ, CDCl3, 300 МГц): 0,49 (s, 3Н), 3,88-4,04 (m, 4Н), 4,26 (d, J=7,1 Гц), 4,39 (s, 1Н), 6,98 (d, J=8,1 Гц, 2Н), 7,57 (d, J=8,4 Гц, 2Н).
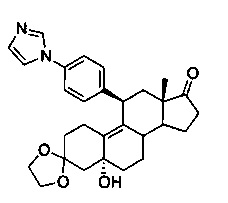
3,3-этилендиокси-5α-гидрокси-11β-[4'-(1-имидазолил)фенил]эстр-9-ен-17-он(4)
Смесь соединения 3 (9,7 г, 18 ммоль), имидазола (1,4 г, 20 ммоль), йодида меди (I) (346 мг, 1,8 ммоль), N,N-диметилглицина (374 мг, 3,6 ммоль) и карбоната калия (5 г, 36 ммоль) в безводном ДМСО (10 мл) трижды дегазировали путем вакуумирования и добавления азота и помещали на предварительно нагретую до 110°C масляную баню. Реакционную смесь грели в течение 60 часов. После охлаждения до комнатной температуры реакционную смесь разбавляли этилацетатом (150 мл) и фильтровали через подложку Celite. Переносили фильтрат в делительную воронку и промывали водой, солевым раствором и сушили над безводным сульфатом натрия. Удаляли растворитель в вакууме с получением неочищенного продукта, из которого при помощи очистки путем хроматографии на колонке с SiO2, элюируя 30% ацетоном в дихлорметане, получали 7,6 г (91%) целевого продукта 4 в виде бледно-желтого твердого вещества.
1Н ЯМР (δ, CDCl3, 300 МГц): 0,51 (s, 3Н), 3,92-4,04 (m, 4Н), 4,37-4,39 (m, 2Н), 7,19 (s, 1Н), 7,27-7,35 (m, 5Н), 7,84 (s, 1Н).
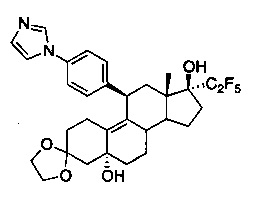
3,3-этилендиокси-5α,17β-дигидрокси-17-(1,1,2,2,2-пентафторэтил)-11β-(4'-(1-имидазолил)фенил)эстр-9-ен (5)
Пентафторйодэтан (3,9 г, 16 ммоль) конденсировали в растворе соединения 4 (1,3 г, 2,7 ммоль) в толуоле (45 мл), выдерживаемом при -78°C. По каплям в течение 15 минут добавляли 1,5 М раствор комплекса метиллитий-бромид лития (8,9 мл, 13,5 ммоль). Полученную реакционную смесь перемешивали при -78°C в течение часа и оставляли перемешиваться при 0°C еще на 1 час. Реакцию гасили путем добавления насыщенного раствора бикарбоната натрия (30 мл). Смесь экстрагировали этилацетатом (2×50 мл) и объединенные органические слои однократно промывали водой, солевым раствором и сушили над сульфатом натрия. Удаляли растворитель в вакууме с получением неочищенного продукта, из которого при помощи очистки путем хроматографии на колонке с SiO2, элюируя 10% ацетоном в дихлорметане, получали 1,28 г (80%) целевого продукта 5 в виде бледно-желтого твердого вещества.
1Н ЯМР (δ, CDCl3, 300 МГц): 0,60 (s, 3Н), 3,89-4,04 (m, 4Н), 4,37 (s, 2Н), 7,18 (s, 1Н), 7,27-7,32 (m, 5Н), 7,67 (s, 1Н).
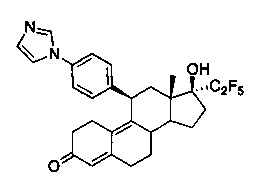
11β-(4'-(1-имидазолил)фенил)-17β-гидрокси-17-(1,1,2,2,2-пентафторэтил)эстра-4,9-диен-3-он (Iа)
Раствор соединения 5 (1 г, 1,68 ммоль) в метаноле (10 мл) охлаждали до 0°C, после чего по каплям добавляли 5н. хлороводородную кислоту (1,6 мл, 8,4 ммоль). Реакционную смесь перемешивали в течение часа при нагревании до комнатной температуры. Реакцию гасили, осторожно добавляя насыщенный раствор бикарбоната натрия, и смесь экстрагировали этилацетатом (2×25 мл). Объединенные органические слои промывали водой, солевым раствором и сушили над безводным сульфатом натрия. Удаляли растворитель в вакууме с получением неочищенного продукта, из которого при помощи очистки путем хроматографии на колонке с SiO2, элюируя 10% ацетоном в дихлорметане, получали 0,8 г (90%) целевого соединения Iа в виде беловатого твердого вещества.
1Н ЯМР (δ, CDCl3, 300 МГц): 0,68 (s, 3Н), 4,48 (d, J=6,6 Гц, 1Н), 5,79 (s, 1Н), 7,18 (s, 1Н), 7,23-7,30 (m, 5Н), 7,62 (s, 1Н).
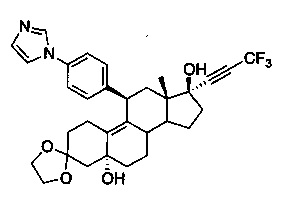
3,3-этилендиокси-5α,17β-дигидрокси-17-(3,3,3-трифтор-1-пропинил)-11β-{4'-[1'-имидазолил)фенил}эстр-9-ен (6)
Свежеприготовленный раствор диизопропиламида лития, полученный путем добавления n-BuLi (6,4 мл, 2,5 М, 16 ммоль) к диизопропиламину (1,6 г, 16 ммоль), в ТГФ (20 мл) при -78°C добавляли в раствор 2-бром-3,3,3-трифторпропена (2,4 г, 14 ммоль) в ТГФ (15 мл) при -78°C. Полученный фиолетовый раствор перемешивали при указанной температуре в течение 20 минут. В реакционную смесь вводили раствор соединения 4 (1,09 г, 2,3 ммоль) в ТГФ (10 мл) в течение 20 минут и перемешивали в течение 1 часа при -78°C и оставляли нагреваться до КТ на 16 часов. Реакцию гасили водным хлоридом аммония (50 мл) и смесь экстрагировали этилацетатом (3×100 мл). Объединенные органические слои дополнительно промывали водой и солевым раствором, сушили над безводным сульфатом натрия и выпаривали в вакууме с получением неочищенного продукта. Проводили очистку на колонке с силикагелем с использованием 10% ацетона в метиленхлориде для получения соединения 6 (1,55 г, 88%) в виде коричневого аморфного твердого вещества.
1Н ЯМР (δ, CDCl3, 300 МГц): 0,52 (s, 3Н), 3,75-4,10 (m, 4Н), 4,35-4,50 (m, 2Н), 7,16 (s, 1H), 7,27-7,36 (m, 5Н), 7,84 (s, 1Н).
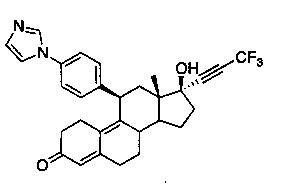
11β-(4'-(1-имидазолил)фенил)-17β-гидрокси-17-(3,3,3-трифтор-1-пропинил)эстра-4,9-диен-3-он (Ib)
В раствор соединения 6 (800 мг, 1,4 ммоль) в метаноле (10 мл) при 0°C добавляли 50% серную кислоту (0,5 мл). После перемешивания в течение 90 минут реакцию осторожно гасили путем добавления насыщенного раствора бикарбоната натрия. Смесь экстрагировали этилацетатом (2×50 мл) и объединенные органические слои промывали водой, солевым раствором и сушили над безв. сульфатом натрия. Удаляли растворитель в вакууме с получением неочищенного продукта, который очищали на колонке с оксидом кремния, элюируя 20% ацетоном в метиленхлориде, с получением соединения Ib (600 мг, 84%) в виде светло-коричневого аморфного твердого вещества.
1Н ЯМР (δ, CDCl3, 300 МГц): 0,58 (s, 3Н), 4,51 (d, J=6,5 Гц, 1Н), 5,82 (s, 1Н), 7,20 (s, 1Н), 7,27-7,34 (m, 5Н), 7,83 (s, 1Н).
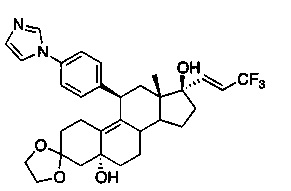
3,3-этилендиокси-5α,17β-дигидрокси-17-(3,3,3-трифторпроп-1(Е)-енил)-11β-{4'-[1'-имидазолил)фенил}эстр-9-ен (7)
Раствор соединения 6 (1,8 г, 3,1 ммоль) в безводном толуоле (30 мл) охлаждали до -78°C, после чего по каплям добавляли 65% раствор Red-Al (2,14 мл, 11 ммоль) и реакционную смесь перемешивали в течение 4 часов при -78°C. Реакцию гасили путем добавления насыщенного хлорида аммония. Отделенный органический слой промывали водой, солевым раствором и сушили над безводным сульфатом натрия. Удаляли растворитель в вакууме с получением неочищенного продукта, из которого при помощи очистки путем хроматографии на колонке с оксидом кремния, элюируя 20% ацетоном в метиленхлориде, получали соединение 7 (1,5 г, 85%) в виде коричневой пены.
1Н ЯМР (δ, CDCl3, 300 МГц): 0,57 (s, 3Н), 3,92-4,03 (m, 4Н), 4,30 (d, J=6,2 Гц, 1Н), 4,42 (s, 1H), 5,90-5,98 (m, 1H), 6,52 (dd, J1=15,4 Гц, J2=1,8 Гц, 1Н) 7,16-7,34 (m, 6Н), 7,83 (s, 1Н).
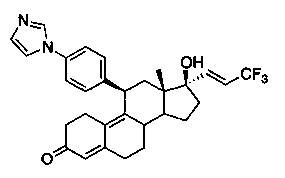
11β-(4'-(1-имидазолил)фенил)-17β-гидрокси-17-(3,3,3-трифторпроп-1(E)-енил)эстра-4,9-диен-3-он (Iс)
Раствор соединения 7 (1 г, 1,5 ммоль) в метаноле (15 мл) охлаждали до 0°C, после чего по каплям добавляли 5н. хлороводородную кислоту (1,2 мл, 6,22 ммоль). Реакционную смесь перемешивали в течение часа при нагревании до комнатной температуры. Реакцию гасили, осторожно добавляя насыщенный раствор бикарбоната натрия, и смесь экстрагировали этилацетатом (2×25 мл). Объединенные органические слои промывали водой, солевым раствором и сушили над безводным сульфатом натрия. Удаляли растворитель в вакууме с получением неочищенного продукта, из которого при помощи очистки путем хроматографии на колонке с SiO2, элюируя 10% ацетоном в дихлорметане, получали 1,06 г (67%) целевого соединения 1 с в виде бледно-коричневого твердого вещества.
1Н ЯМР (δ, CDCl3, 300 МГц): 0,64 (s, 3Н), 4,42 (d, J=6,8 Гц, 1Н), 5,80 (s, 1Н), 5,98-6,05 (m 1Н), 6,59 (dd, J1=15,5 Гц, J2=1,8 Гц, 1Н) 7,17-7,30 (m, 6Н), 7,77 (s, 1Н).
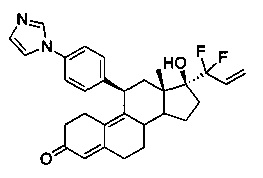
11β-(4'-(1-имидазолил)фенил)-17β-гидрокси-17-(1,1-дифторпроп-2-енил)эстра-4,9-диен-3-он (Id)
В раствор соединения 4 (1,9 г, 4 ммоль) в пиридине (15 мл) добавляли DMAP (98 мг, 0,8 ммоль), затем ангидрид уксусной кислоты (2,86 г, 28 ммоль) и полученную смесь грели при 60°C в течение 30 часов. Удаляли растворители в вакууме и неочищенный остаток быстро пропускали через короткую колонку с оксидом кремния и концентрировали с получением соединения 8 (1,82 г, 3,9 ммоль), которое растворяли в смеси ТГФ-диэтиловый эфир-пентан (4:1:1,80 мл) и охлаждали до -100°C. По каплям добавляли n-BuLi (8 мл, 2,5 М, 20 ммоль) и реакционную смесь оставляли перемешиваться на 90 минут при -95°C и нагревали до комнатной в течение 3 часов. Реакцию гасили раствором хлорида аммония (50 мл) и смесь экстрагировали этилацетатом (3×50 мл). Объединенные органические слои концентрировали в вакууме и полученный неочищенный остаток растворяли в метаноле (20 мл) и обрабатывали 5н. хлороводородной кислотой (1,7 мл) при 0°C. Реакционную смесь оставляли перемешиваться при комнатной температуре на 2 часа и реакцию осторожно гасили насыщенным раствором бикарбоната натрия (25 мл). Органические вещества экстрагировали этилацетатом (3×30 мл) и объединенные органические слои сушили над сульфатом натрия, концентрировали в вакууме. Очистка на колонке с силикагелем с использованием 10% ацетона в метиленхлориде приводила к получению Id (400 мг, 20%) в виде бледно-желтого аморфного твердого вещества.
1Н ЯМР (δ, CDCl3, 300 МГц): 0,62 (s, 3Н), 4,44-4,46 (m, 1Н), 5,56 (5,80 (s, 1Н), 5,98-6,05 (m, 1H), 6,59 (dd, J1=15,5 Гц, J2=1,8 Гц, 1Н) 7,17-7,30 (m, 6Н), 7,77 (s, 1Н).
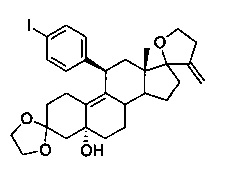
3,3-этилендиокси-5α-гидрокси-11β-(4'-(йодфенил)-17,23-эпокси-19,24-динор-17а-хола-9,20-диен (11)
Раствор 1,4-дииодбензола (5,14 г, 15,6 ммоль) в безводном ТГФ (50 мл) охлаждали до -10°C, после чего по каплям в течение 15 минут добавляли 2М раствор хлорида изопропилмагния (7,8 мл, 15,6 ммоль). После перемешивания в течение 20 минут добавляли твердый хлорид меди (I) (257 мг, 2,6 ммоль) и реакционную смесь перемешивали в течение 30 минут. По каплям добавляли раствор эпоксида 10 (2 г, 5,2 ммоль) в 20 мл ТГФ и перемешивали в течение 2 часов, медленно нагревая до 10°C. Реакцию гасили водным раствором хлорида аммония (50 мл) и смесь экстрагировали этилацетатом (2×50 мл). Объединенные органические слои дополнительно промывали водой и солевым раствором, сушили над сульфатом натрия и выпаривали в вакууме с получением неочищенного продукта. Неочищенный продукт очищали на колонке с оксидом кремния, элюируя 30% этилацетатом в гексане, с получением 2,81 г (92%) соединения 11 в виде беловатого твердого вещества.
1Н ЯМР (δ, CDCl3, 300 МГц) 0,58 (s, 3Н), 3,74 (s, 4Н), 3,81-3,94 (m, 4Н), 4,13 (d, J=6,2 Гц, 1H), 4,85 (s, 1H), 5,13 (s, 1H), 5,77 (s, 1H), 6,91 (d, J=8,5 Гц, 2H), 7,58 (d, J=8,4 Гц, 2H).
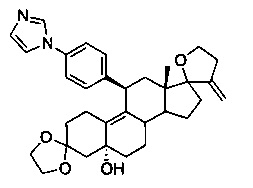
3,3-этилендиокси-5а-гидрокси-11β-(4'-[1-имидазолил)фенил)-17,23-эпокси-19,24-динор-17α-хола-9,20-диен (12)
Смесь соединения 11 (2,7 г, 4,6 ммоль), имидазола (531 мг, 4,6 ммоль), йодида меди (I) (87 мг, 0,5 ммоль), N,N-диметилглицина (94 мг, 0,9 ммоль) и карбоната калия (1,3 г, 9,2 ммоль) в безводном ДМСО (5 мл) трижды дегазировали путем вакуумирования и добавления азота и помещали на предварительно нагретую до 110°C масляную баню. Реакционную смесь грели в течение 60 часов. После охлаждения до комнатной температуры реакционную смесь разбавляли этилацетатом (100 мл) и фильтровали через подложку Celite. Переносили фильтрат в делительную воронку и промывали водой, солевым раствором и сушили над безводным сульфатом натрия. Удаляли растворитель в вакууме с получением неочищенного продукта, из которого при помощи очистки путем хроматографии на колонке с SiO2, элюируя 10% ацетоном в этилацетате, получали 2,4 г (98%) целевого продукта 12 в виде бледно-желтого аморфного твердого вещества.
1Н ЯМР (δ, CDCl3, 300 МГц) 0,54 (s, 3Н), 3,74-4,04 (m, 8Н), 4,24 (d, J=6,8 Гц, 1Н), 4,83 (s, 1Н), 5,10 (s, 1Н), 7,19 (s, 1Н), 7,27-7,36 (m, 5Н), 7,84 (s, 1Н).
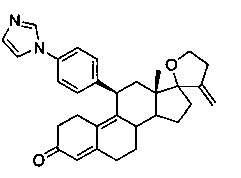
11β-(4'-[1-имидазолил]фенил)-17,23-эпокси-19,24-динон-17α-хола-4,9,20-триен-3-он (Iе)
Раствор соединения 12 (2,29 г, 4,33 ммоль) в метаноле (20 мл) охлаждали до 0°C, после чего по каплям добавляли 5н. хлороводородную кислоту (1,7 мл, 8,7 ммоль). Реакционную смесь перемешивали в течение 3 часов при нагревании до комнатной температуры. Реакцию гасили, осторожно добавляя насыщенный раствор бикарбоната натрия (30 мл), и смесь экстрагировали этилацетатом (2×50 мл). Объединенные органические слои промывали водой, солевым раствором и сушили над безводным сульфатом натрия. Удаляли растворитель в вакууме с получением неочищенного продукта, из которого при помощи очистки путем хроматографии на колонке с SiO2, элюируя 10% ацетоном в дихлорметане, получали 1,63 г (81%) целевого соединения Iе в виде белого твердого вещества.
1Н ЯМР (δ, CDCl3, 300 МГц) 0,60 (s, 3Н), 4,35 (d, J=7 Гц, 1Н), 4,86 (s,1H), 5,15 (s, 1Н), 5,78 (s,1H), 7,19 (s,1H), 7,22-7,36 (m, 5Н), 7,84 (s, 1Н).
Пример 1
Определение профиля в отношении ядерных рецепторов
Определение агонистической/антагонистической природы исследуемых соединений проводили при помощи сервиса определения профиля в отношении ядерных рецепторов в клетках SelectScreen™ производства Invitrogen, в котором применяют технологию репортера бета-лактамазы GeneBLAzer®. Вкратце, в указанном исследовании используют кДНК бета-лактамазы, транскрипцию которой регулирует вышележащая (upstream) активирующая последовательность (UAS). UAS активируется ДНК-связывающим доменом (DBD) транскрипционного фактора GAL4, который экспрессируется в виде гибридного белка лиганд-связывающего домена (LBD) рецептора-мишени. За счет связывания с лигандом GAL4(DBD)-NR(LBD) связывается с UAS, которая регулирует транскрипцию бета-лактамазы. Бета-лактамаза расщепляет специально разработанный флуоресцентный субстрат, что приводит к изменению измеряемой длины волны флуоресценции.
Обобщенный протокол, используемый для скрининга антагонистов прогестерона, активируемого контрольным агонистом R5020, приведен ниже:
Клетки НЕК 293Т, содержащие рецептор прогестерона-LBD-UAS-bla, размораживали и подготавливали так, как описано выше для скрининга агонистов. 4 мкл 10Х последовательно разбавленного контрольного антагониста RU486 (исходная концентрация 100 нМ) или исследуемых соединений добавляли в соответствующие лунки планшета для исследования, обработанного ТС. 32 мкл клеточной суспензии добавляли в лунки, затем предварительно инкубировали при 37°C/5% CO2 в инкубаторе с влажной камерой совместно с исследуемыми соединениями и контрольным титром антагониста в течение 30 минут.4 мкл 10Х контрольного агониста (см. выше) в предварительно определенной концентрации ЕС80 добавляли в лунки, содержащие контрольный антагонист или исследуемые соединения. Планшет инкубировали в течение 16-24 часов при 37°C/5% CO2 в инкубаторе с влажной камерой. 8 мкл 1 мкМ раствора, содержащего субстрат, добавляли в каждую лунку и планшет инкубировали в течение 2 часов при комнатной температуре. Затем планшет анализировали на флуоресцентном анализаторе планшетов (Тесал Safire).
Обобщенный протокол скрининга антагонистов глюкокортикоидов, активируемого контрольным агонистом дексаметазоном, соответствовал тому, что описан для скрининга антагонистов прогестерона, с тем исключением, что использовали клетки НЕК 293Т, содержащие глюкокортикоидный рецептор-LBD-UAS-bla. В качестве контрольного антагониста в исследовании глюкокортикоидов также применяли RU486.
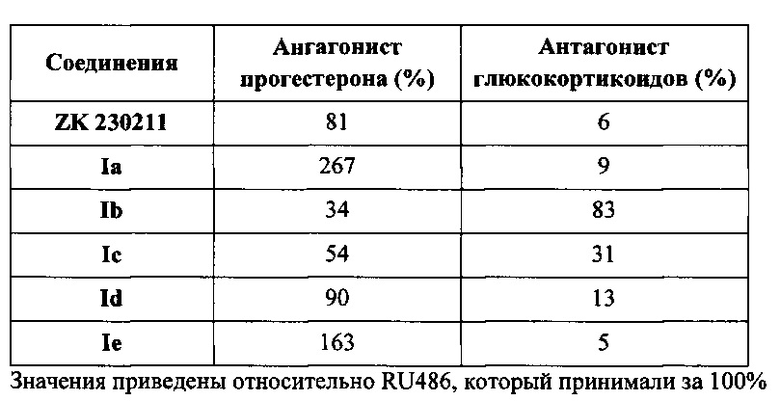
Результаты указанных исследований для приведенных исследуемых соединений показаны в таблице I.
Пример 2
Использовали модель беременных морских свинок, описанную в источнике Walter Elger et. al. J. Steroid Biochem Vol 25, No.5B, pp 835-845, 1986:
Взрослых самок морских свинок весом примерно 500 г размещали в клетках и исследовали состояние цикла путем ежедневной проверки отверстия влагалища.
Во время второго цикла трех самок размещали в клетке вместе с одним самцом на 15 день после первого наблюдения отверстия влагалища. 16 день цикла принимали за первый день беременности. Беременных животных случайным образом распределяли по различным исследуемым группам и на 43 и 44 дни беременности подкожно вводили исследуемые вещества, растворенные в 0,2 мл бензилбензоата/касторового масла. У животных проверяли вагинальное кровотечение и количество абортированных плодов и время прекращения беременности. На ФИГ. 1 показаны результаты исследования дозы 3 мг/день, тогда как на ФИГ. 2 показаны результаты исследования дозы 10 мг/день.
Дополнительные модификации и альтернативные варианты реализации различных аспектов настоящего изобретения будут очевидны специалистам в данной области техники после изучения настоящего описания. Соответственно, настоящее описание, которое следует рассматривать исключительно как иллюстративное, предназначено для описания общего способа реализации изобретения специалистам в данной области техники. Следует понимать, что формы изобретения, показанные и описанные в настоящей заявке, следует рассматривать как примеры вариантов реализации. Элементы и материалы могут быть замещены на те, что проиллюстрированы и описаны в настоящей заявке, части и способы можно применять в обратном порядке, а определенные отличительные признаки настоящего изобретения можно применять независимо, что должно быть понятно специалистам в данной области техники после изучения настоящего описания изобретения. Можно проводить изменения элементов, описанных в настоящей заявке, не выходя за рамки сущности и объема изобретения, описанного в последующей формуле изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| АНТАГОНИСТЫ ПРОГЕСТЕРОНА | 2012 |
|
RU2608521C2 |
| ПРОИЗВОДНЫЕ 11β-БЕНЗАЛЬДОКСИМ-ЭСТРА-4,9-ДИЕНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1994 |
|
RU2137777C1 |
| ГОРМОНАЛЬНЫЕ ИЛИ АНТИГОРМОНАЛЬНЫЕ СТЕРОИДНЫЕ СОЕДИНЕНИЯ, СПОСОБЫ ТЕРАПЕВТИЧЕСКОГО ВОЗДЕЙСТВИЯ, СПОСОБ ПОЛУЧЕНИЯ СОЕДИНЕНИЙ | 1999 |
|
RU2238943C2 |
| МОДУЛЯТОРЫ РЕЦЕПТОРОВ ПРОГЕСТЕРОНА | 2005 |
|
RU2381232C2 |
| 11β-ЗАМЕЩЕННЫЕ 19-НОРСТЕРОИДЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ, СОЕДИНЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1994 |
|
RU2140423C1 |
| СОЕДИНЕНИЯ С ГИДРОКСИКАРБОНИЛЬНЫМИ-ГАЛОГЕНАЛКИЛЬНЫМИ БОКОВЫМИ ЦЕПЯМИ | 2000 |
|
RU2247106C2 |
| ПРОИЗВОДНЫЕ СУЛЬФАМАТА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ СОСТАВЫ | 1996 |
|
RU2159774C2 |
| ПРОИЗВОДНЫЕ ЭСТРА-1,3,5(10)-ТРИЕНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1995 |
|
RU2139885C1 |
| ПРОИЗВОДНЫЕ 11,21-БИСФЕНИЛ-19-НОРПРЕГНАНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 1995 |
|
RU2152952C2 |
| ПРОИЗВОДНЫЕ 1Н-ИМИДАЗОЛА, ОБЛАДАЮЩИЕ CB-АГОНИСТИЧЕСКОЙ, ЧАСТИЧНОЙ CB-АГОНИСТИЧЕСКОЙ ИЛИ CB-АНТАГОНИСТИЧЕСКОЙ АКТИВНОСТЬЮ | 2002 |
|
RU2299200C2 |
Изобретение относится к соединению, имеющему структуру формулы (I), в которой X представляет собой О, R1 представляет собой атом водорода; R2 представляет собой атом водорода; R3 представляет собой атом водорода; R4 представляет собой атом водорода; R5 представляет собой N-имидазолильную группу; R6 представляет собой гидроксильную группу; R7 представляет собой радикал формулы CnFmHo, где n равен 2, 3, 4, 5 или 6, причем m≥1 и m+o=2n+1. Изобретение также относится к способу лечения субъекта с применением антагониста прогестерона. Технический результат: получены новые соединения формулы (I), проявляющие антагонизм по отношению к прогестерону. 2 н. и 1 з.п. ф-лы, 2 ил., 1 табл., 2 пр.
1. Соединение, имеющее структуру формулы (I):
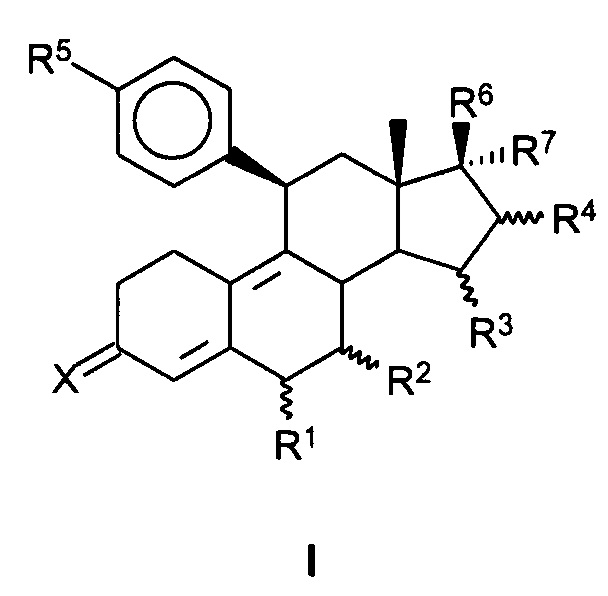 ,
,
где:
X представляет собой О,
R1 представляет собой атом водорода;
R2 представляет собой атом водорода;
R3 представляет собой атом водорода;
R4 представляет собой атом водорода;
R5 представляет собой N-имидазолильную группу;
R6 представляет собой гидроксильную группу;
R7 представляет собой радикал формулы CnFmHo, где n равен 2, 3, 4, 5 или 6, причем m≥1 и m+o=2n+1.
2. Соединение по п. 1, отличающееся тем, что указанное соединение имеет структуру:
3. Способ лечения субъекта с применением антагониста прогестерона, включающий введение субъекту эффективного количества антагониста прогестерона по любому из пп. 1-2.
| WO 2013016725 A1, 31.01.2013 | |||
| WO 2010118025 A1, 14.10.2010 | |||
| WO 2011009534 A2, 27.01.2011 | |||
| Способ получения стероидов, замещенных спирановым циклом | 1987 |
|
SU1715205A3 |
| ГОРМОНАЛЬНЫЕ ИЛИ НЕГОРМОНАЛЬНЫЕ СТЕРОИДНЫЕ СОЕДИНЕНИЯ, СПОСОБ ТЕРАПЕВТИЧЕСКОГО ЛЕЧЕНИЯ | 1999 |
|
RU2230071C2 |

