Изобретение относится к способу получения соединений, которые ингибируют протеазу, кодируемую вирусом иммунодефицита человека (ВИЧ), которые имеют значение в предотвращении заражения ВИЧ, лечении заражения ВИЧ и лечения полученного синдрома приобретенного иммунодефицита (СПИД), а также к промежуточным соединениям для их получения.
Ретровирус, обозначаемый как вирус иммунодефицита человека (ВИЧ), представляет собой этиологический агент комплексного заболевания, которое включает прогрессирующее разрушение иммунной системы (синдром приобретенного иммунодефицита - СПИД) и дегенерацию центральной и периферической нервной системы. Общим признаком репликации ретровируса является экстенсивный посттрансляционный процессинг предшественников полипротеинов с помощью вирусно-кодируемой протеазы с получением зрелых вирусных протеинов, необходимых для сборки вируса и его функционирования. Ингибирование этого процессинга предотвращает получение нормального инфекционною вируса. Например, Kohl N.E. et al., Proc. Nat'l Acad. Sci. 85, 4686 (1988), сообщает, что генетическая инактивация ВИЧ кодированной протеазы приводит к получению незрелых, незаразных вирусных частиц. Эти результаты показывают, что ингибирование ВИЧ протеазы представляет жизнеспособный способ лечения СПИДа и предотвращения или лечения заражения ВИЧ.
Нуклеотидная последовательность ВИЧ указывает на присутствие pol гена в одной открытой рамке считывания [Raptner L. et al., Nature, 313, 277 (1985)] . Гомология аминокислотной последовательности подтверждает то, что pol последовательность кодирует обратную транскриптазу, эндонуклеазу и ВИЧ протеазу [Toh H. et аl., EMBO J. 4, 1267, (1985); Power M.D. et al. Science, 231, 1567 (1986); Pearl L.H. et al., Nature, 329, 351 (1987)]. Ранние попытки найти ингибиторы ВИЧ протеазы включают EPA 432695; EPA 435365; J. Med. Chem. 34, 1228 (1991) и Science, 248, 358 (1990). Заявители показывают, что соединения этого изобретения являются ингибиторами ВИЧ протеазы.
Краткое описание изобретения
Раскрывается способ получения охарактеризованных далее соединений формулы I. Эти соединения являются полезными при ингибировании ВИЧ протеазы, предотвращении заражения ВИЧ, при лечении инфекции ВИЧ и при лечении СПИДа либо сами по себе, либо в виде фармацевтически приемлемых солей, ингредиентов композиции, либо в сочетании с другими антивирусами, иммуномодуляторами, антибиотиками или вакцинами, либо без них. Также раскрываются способы лечения СПИДа, способы предотвращения заражения ВИЧ и способы лечения заражения ВИЧ с помощью указанных соединений.
В табл. A указаны некоторые аббревиатуры, которые встречаются в данной заявке.
Подробное описание изобретения и предпочтительные варианты воплощения изобретения
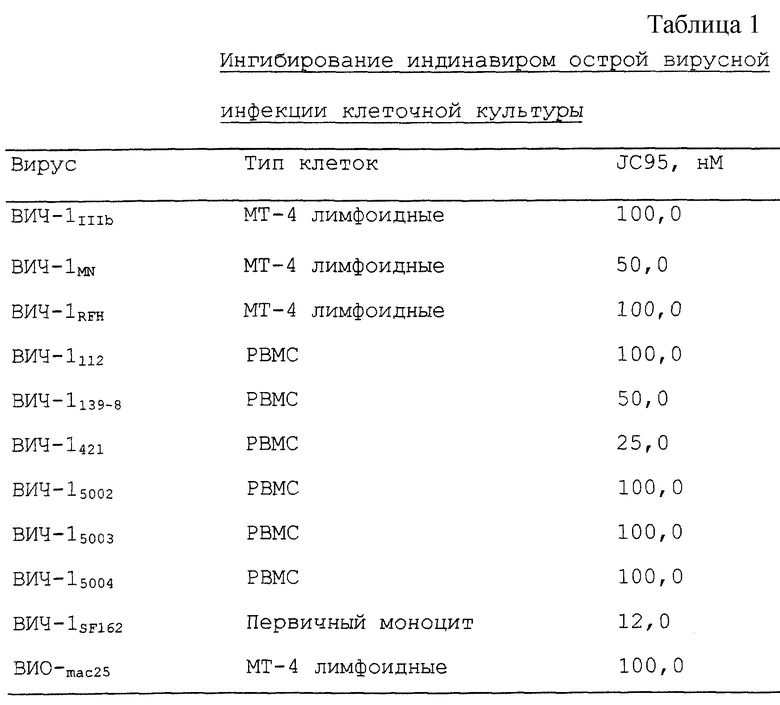
Данное изобретение относится к способу получения производных пиперазинилпентанамида общей формулы I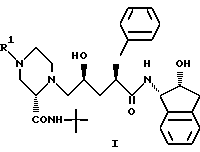
где R1 - C1-C4-алкил, незамещенный или замещенный одной или более группами, выбранными из:
(1) арила, незамещенного или замещенного одним или более из C1-C4-алкила, аминогруппы, гидроксигруппы или арила;
(2) 5-7-членного циклоалкила, незамещенного или замещенного одной или более из групп галогена, C1-C3-алкокси или арила; либо
(3) гетероцикла, незамещенного или замещенного одной или более из групп: галогена, оксо-, C1-C4-алкокси, C1-C4-алкила, -C(O)OC1-C3-алкила, -NHC(O)C1-C3-алкила или бутилоксикарбонила,
включающему взаимодействие соединения формулы (i)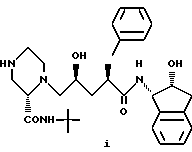
с соединением формулы R1-X, где X выбран из Cl, Br или I.
В частности, изобретение относится к получению соединений I, где R1 представляет собой C1-C4-алкил, незамещенный или замещенный гетероциклом, который может быть незамещен или замещен одной или более группами из галогена, оксогруппы, C1-C4-алкокси, C1-C4-алкила, -C(O)OC1-C3-алкила, -NHC(O)C1-C3-алкила или бутилоксикарбонила.
Предпочтительно указанный гетероцикл выбран из пиперидинила, пиридила, тиенила, пирролила, тиазолила, имидазолила, фурила, бензимидазолила, пиразинила, изоксозолила, пиридазинила или хинолинила, каждый из которых может быть замещен или незамещен одной или более группами из галогена, оксогруппы, C1-C4-алкокси, C1-C4-алкила, -C(O)OC1-C3-алкила, -NHC(O)C1-C3-алкила или бутилоксикарбонила.
В предпочтительном варианте способ получения соединения I заключается в том, что соединение (i) подвергают взаимодействию с соединением формулы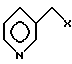
где X определен выше, с получением соединения формулы I, где предпочтительно X представляет собой атом хлора.
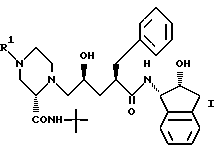
Еще один аспект изобретения заключается в том, что предложено производное пиперазинилпентанамида формулы (i)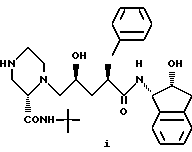
или его соль в качестве промежуточного соединения для вышеуказанного способа получения соединения I.
Наиболее предпочтительны следующие соединения.
Соединение B: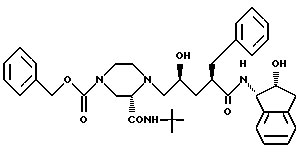
N-(2(R)-гидрокси-1(S)-инданил)-2(R)-фенилметил-4(S)-гидрокси- 5-(1-(4-карбобензилокси-2(S)-N'-(т-бутилкарбоксамидо)-пиперазинил))- пентанамид.
Соединение D: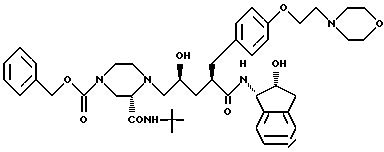
N-(2(R)-гидрокси-1(S)-инданил)-2(R)-((4-(2-(4-морфолинил)-этокси)- фенил)метил)-4(S)-гидрокси-5-(1-(4-карбобензилокси-2(S)-N'-(т- бутилкарбоксамидо)-пиперазинил))-пентанамид.
Соединение J:
(индинавир)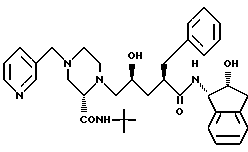
N-(2(R)-гидрокси-1(S)-инданил)-2(R)-фенилметил-4(S)-гидрокси- 5-(1-(4-(3-пиридилметил)-2(S)-N'-(т-бутилкарбоксамидо)-пиперазинил))- пентанамид.
Соединения данного изобретения могут иметь асимметрические центры и встречаться как в виде рацематов, рацематических смесей, так и в виде индивидуальных диастереоизомеров или энантиомеров. Все изомерные формы включены в данное изобретение. В тех случаях, когда какая-либо переменная встречается более чем один раз в каком-либо заместителе или в формуле I, ее значение в каждом положении не зависит от ее значения в любом другом положении. Кроме того, комбинации заместителей и/или переменных допустимы только если такие комбинации приводят к стабильным соединениям.
Предполагается, если не оговорено особо, что используемый здесь термин "алкил" включает как разветвленные, так и неразветвленные насыщенные алифатические углеводородные группы, имеющие определенное число углеродных атомов (Me - метил, Et - этил, Pr - пропил, Bu - бутил); "алкокси" представляет собой алкильную группу с указанным числом углеродных атомов, присоединенную через кислородный мостик; и предполагается, что "циклоалкил" включает насыщенные кольцевые группы, такие как циклопропил, циклобутил, циклопентил, циклогексил (Cyh) и циклогептил. Имеется в виду, что "алкенил" включает углеродные группы либо неразветвленной, либо разветвленной конфигурации с одной или более углерод-углеродными двойными связями, которые могут встречаться в любой стабильной точке вдоль цепи, такой как этенил, пропенил, бутенил, пентенил и т.п. Имеется в виду, что "алкинил" включает углеводородные группы либо неразветвленной, либо разветвленной конфигурации с одной или более углерод-углеродными тройными связями, которые могут встречаться в любой стабильной точке вдоль цепи, такой как этинил, пропинил, бутинил, пентинил и т.п. "Галоид", используемый здесь, означает фтор, хлор, бром или йод, и "противоион" используют для обозначения единичного отрицательно заряженного иона, такого как хлорид, бромид, гидроксид, ацетат, трифторацетат, перхлорат, нитрат, бензоат, малеат, тартрат, гемитартрат, бензосульфонат и т. п.
Имеется в виду, если специально не оговорено, что используемый здесь термин "арил" означает фенил (Ph) или нафтил. Имеется в виду, что термин "карбоциклический" означает любое стабильное 5- до 7-членное углеродное кольцо или 7- до 10-членное бициклическое углеродное кольцо, любое кольцо которого может быть насыщенным или ненасыщенным.
Используемый здесь, если не оговорено особо, термин "гетероцикл" или "гетероциклический" означает стабильную 5- до 7-членную моно- или бициклическую или стабильную 7- до 10-членную бициклическую гетероциклическую кольцевую систему, любое кольцо которой может быть насыщенным или ненасыщенным и которая состоит из углеродных атомов и из от одного до трех гетероатомов, набранных из группы, состоящей из N, O и S, и в которой гетероатомы азота и серы могут быть окислены, и гетероатом азота может быть кватернизован и включает любую бициклическую группу, в которой любое из вышеопределенных гетероциклических колец конденсировано с бензольным кольцом. Гетероциклическое кольцо может быть присоединено к любому гетероатому или углеродному атому, что приводит к образованию стабильной структуры. Примерами таких гетероциклических элементов являются пиперидинил, пиперазинил, 2-оксопиперазинил, 2-оксопиперидинил, 2-оксопирролодинил, 2-оксоазепинил, азепинил, пирролил, 4-пиперидонил, пирролидинил, пиразолил, пиразолидинил, имидазолил, имидазолинил, имидазолидинил, пиридил, пиразинил, пиримидинил, пиридазинил, оксазолил, оксазолидинил, изоксазолил, изоксазолидинил, морфолинил, тиазолил, тиазолидинил, изотиазолил, хинуклидинил, изотиазолидинил, индолил, хинолинил, изохинолинил, бензимидазолил, тиадиазолил, бензопиранил, бензотиазолил, бензоксазолил, фурил, тетрагидрофурил, тетрагидропиранил, тиенил, бензотиенил, тиаморфолинил, тиамоpфолинил сульфоксид, тиаморфолинил сульфон и оксадиазолил. Морфолино означает то же, что и морфолинил.
Фармацевтически приемлемые соли соединений формулы I (в форме водо- или маслорастворимых продуктов или способных к диспергированию продуктов) включают обычные нетоксичные соли или четвертичные аммониевые соли, которые образуются, например, из неорганических или органических кислот или оснований. Примерами таких кислых солей присоединения являются ацетат, адипат, альгинат, аспартат, бензоат, бензолсульфонат, бисульфат, бутират, цитрат, камфорат, камфорсульфонат, циклопентанпропионат, диглюконат, додецилсульфат, этансульфонат, фумарат, глюкогептаноат, глицерофосфат, гемисульфат, гептаноат, гексаноат, гидрохлорид, гидробромид, гидройодид, 2-гидроксиэтансульфонат, лактат, малеат, метансульфонат, 2-нафталинсульфонат, никотинат, оксалат, памоат, пектинат, персульфат, 3-фенилпропионат, пикрат, пивалат, пропионат, сукцинат, тартрат, тиоцианат, тозилат и ундеканоат. К основным солям относятся соли аммония, соли щелочных металлов, такие как соли натрия и калия, соли щелочноземельных металлов, такие как соли кальция и магния, соли с органическими основаниями, такие как соли дициклогексиламина, N-метил-D-глюкамин, и соли с аминокислотами, такими как аргинин, лизин, и так далее. Кроме того, основные азотсодержащие группы могут быть кватернизованы такими агентами, как низшие галоидалкилы, такие как метил-, этил-, пропил- и бутилхлорид, бромиды и йодиды; диалкилсульфаты, такие как диметил, диэтил, дибутил; и диамилсульфаты, длинноцепочечные галогениды, такие как децил-, лаурил-, миристил- и стеарилхлориды, бромиды и йодиды, аралкил галогениды, такие как бензил и фенетил бромиды и другие.
В конце описания представлены cхемы I-III получения исходных соединений и новых соединений данного изобретения. Схемы I-III не ограничиваются какими-либо конкретными заместителями, применяемыми в схемах с иллюстративными целями. Примеры иллюстрируют применение следующих схем для конкретных соединений.
Амидная конденсация, используемая для получения соединений этого изобретения, обычно проводится карбодиимидным способом с помощью реагентов, таких как дициклогексилкарбодиимид или 1-этил-3-(3-диметиламинопропил)карбодиимид. Другие способы образования амидной или пептидной связи включают (но не ограничиваются) синтетические пути через хлорангидрид, азид, смешанный ангидрид или активированный сложный эфир. Обычно амидное связывание проводят в растворе, но может быть применен твердофазный синтез согласно классическим методам Merrifield. Практикуется также присоединение и удаление одной или более защищающих групп.
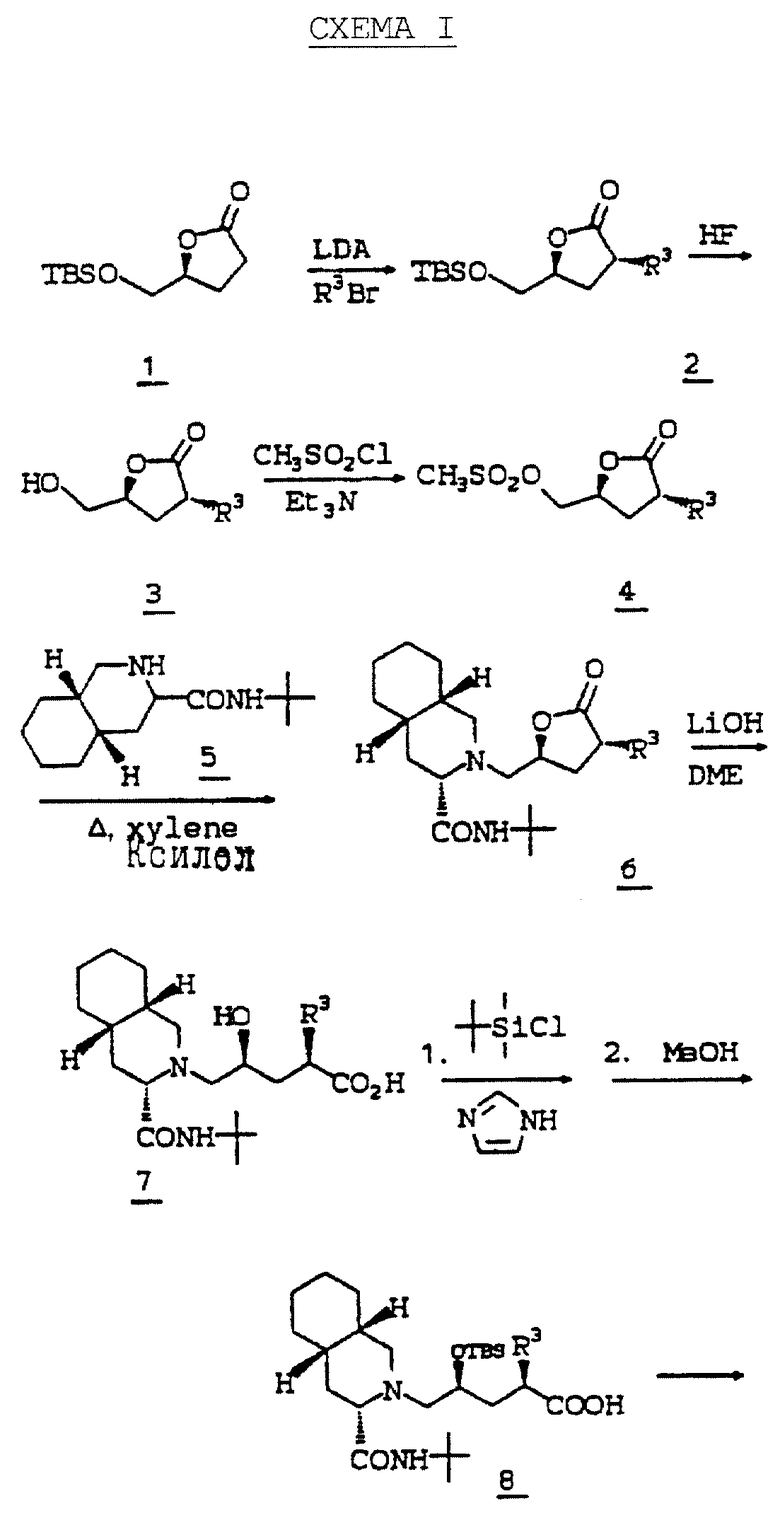
Дигидро-5(S)-(трет-бутилдиметилсилилоксиметил)-3(2H)-фуранон (соединение 1) получают стандартными методами, известными в данной области, из коммерчески доступного дигидро-5(S)-(гидроксиметил)-2(3H)-фуранона. После алкилирования соединения 1 с получением соединения 2 защищающую группу лактона 2 удаляют водным HF, получая соединение 3.
Спиртовую группу 3 активируют путем превращения в отщепляемую группу, такую как мезилат, тозилат или трифилат, путем обработки спирта сульфонилхлоридом или ангидридом сульфокислоты, таким как ангидрид трифторметансульфокислоты, в присутствии объемного аминового основания, такого как триэтиламин, диэтилизопропиламин или 2,6 лутидин, получая соединение, такое как соединение 4. Отщепляемую группу соединения 4 замещают амином 5, таким как N'-т-бутил-(4aS,8aS)-(декагидроизохинолин)-3(S)-карбоксамид, в высококипящем растворителе, таком как ДМФ или ксилол, получая соединение, такое как 6. Трифторметансульфонилоксигруппа может быть замещена амином при комнатной температуре в растворителе, таком как изопропанол, обработкой N,N-диизопропилэтиламином.
Соединение 6 гидролизуют водным гидроксидом лития или натрия, и образовавшуюся гидроксикислоту 7 превращают в защищенную гидроксикислоту 8. Гидроксильную группу обычно защищают стандартной силильной защищающей группой, такой как т-бутилдиметилсилил или т-бутилдифенилсилил.
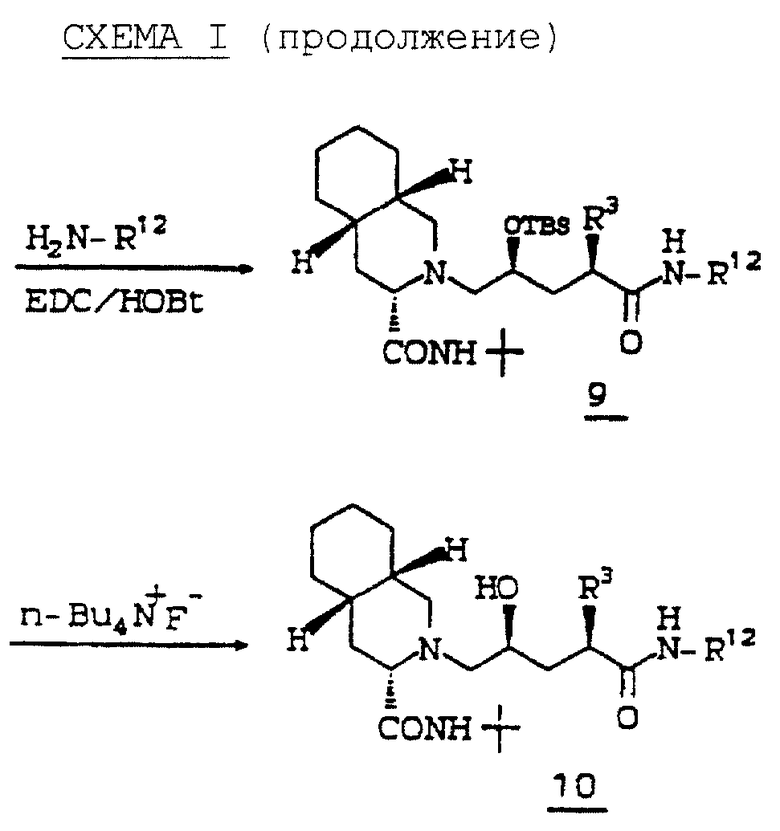
Защищенную гидроксикислоту 8 затем связывают с требуемым R12 амином, получая соединение 9, и силильную защищающую группу удаляют с помощью фтор-иона, получая соединение 10 (схема I).
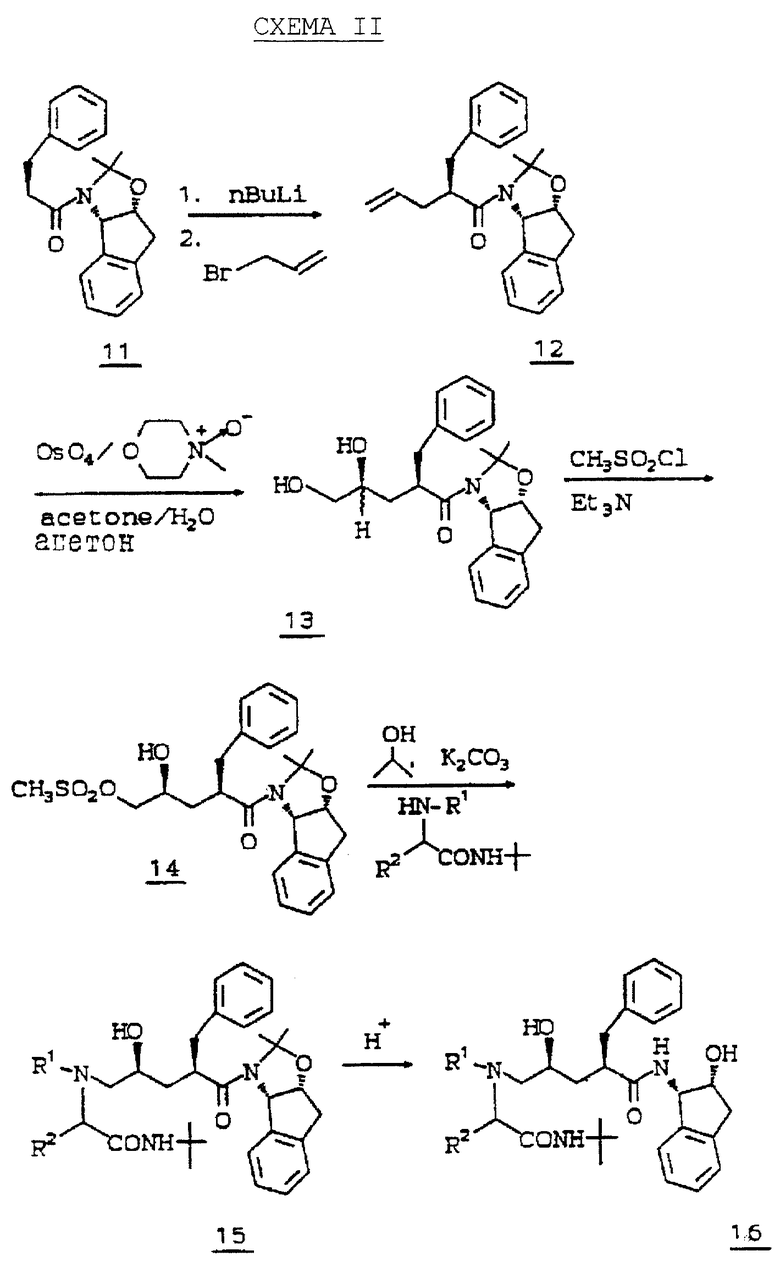
Второй способ получения продуктов общей формулы I представлен cхемой II. В cхеме II алкилирование 11 проводят при помощи первой стадии депротонирования 11 н-бутиллитием или диизопропиламидом лития (LDA) и следующей за ней второй стадии добавления галоидалкенила (такого как аллилбромид), получая 12.
Дигидроксилированием олефина 12 тетроксидом осмия и N-метилморфолин-N-оксидом (NMO) получают диастереомерную смесь диолов 13. Селективное мезилирование первичного спирта 13 метансульфонилхлоридом и либо триэтиламином, либо пиридином дает мезилат 14.
Нагревание мезилата 14 с амином при кипячении с обратным холодильником в спиртовом растворителе, таком как метанол или изопропанол, содержащем избыток карбоната калия, приводит к получению амино-спирта, такого как соединение 15. Диастереомеры можно разделить на этой стадии стандартными методами, хорошо известными специалистам в данной области. Альтернативно разделение можно осуществить после удаления кеталя.
Удаление кеталя в соединении 15 проводят путем обработки кислотой в присутствии метанола или водной кислоты с помощью 1н HCl в ТГФ, получая соединение 16.
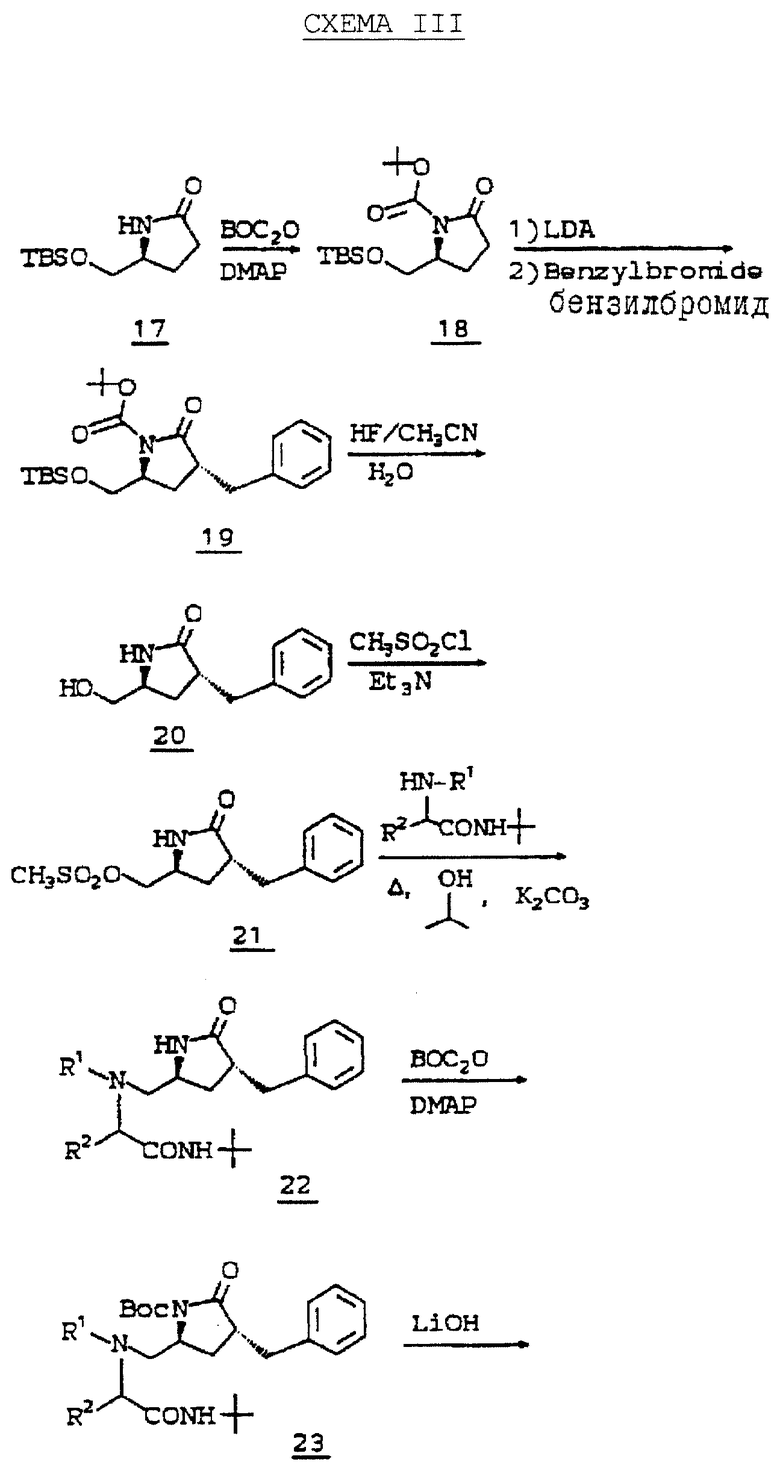
Третий способ получения продуктов общей формулы I иллюстрируется схемой III. Защиту пирролидиновой-NH-группы соединения 17 проводят бутилоксикарбонил (BOC)-ангидридом и диметиламинопиридином, получая защищаемое соединение 16. Алкилирование 18 осуществляют с помощью первой стадии депротонирования 18 сильным основанием, таким как гексаметилдисилиламид лития (LHMDS) или диизопропиламидом лития (LDA) и следующей за ней второй стадии добавления галоидалкила (такого как бензилбромид), получая соединение 19.
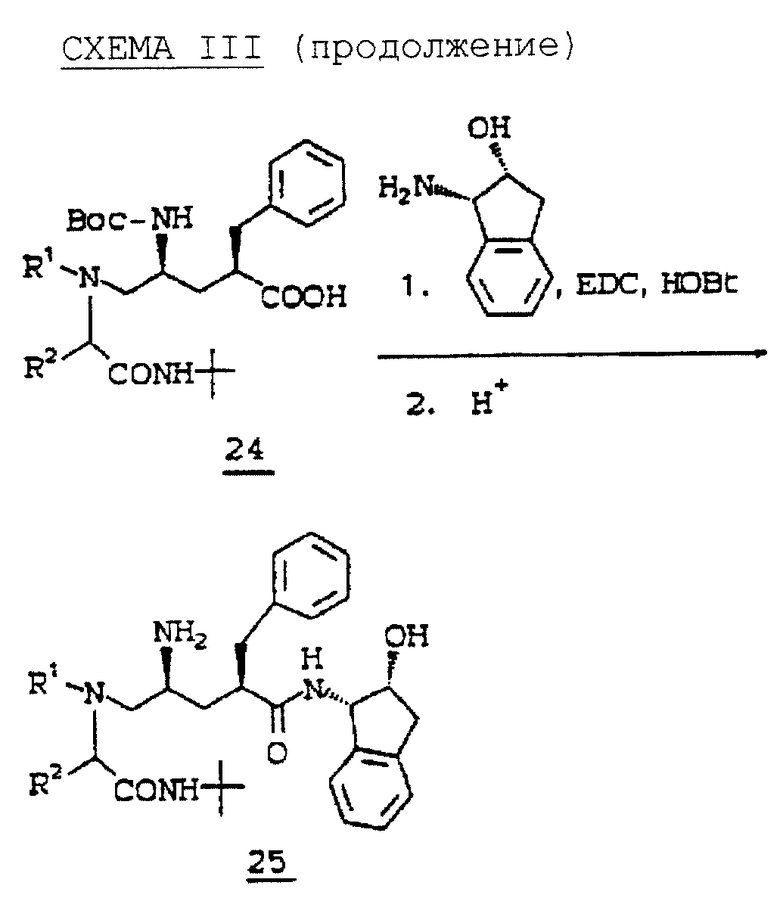
TBS защищающую и BOC защищающую группу 19 удаляют обработкой водным HF в ацетонитриде, получая спирт 20. Мезилирование первичного спирта 20 метансульфонилхлоридом и либо триэтиламином, либо пиридином дает мезилат 21, который кипятят с амином с обратным холодильником в спиртовом растворителе, таком как метанол или изопропанол, содержащем избыток карбоната калия, получая аминопирролидинон, такой как соединение 22. Пирролидиновую -NH-группу 22 повторно защищают с помощью BOC группы, как описано выше, и образовавшееся соединение 23 гидролизуют с раскрытием основанием, таким как гидроксид лития или натрия, получая кислоту 24. Затем соединение 24 связывают с NH2R12 амином стандартным способом, и BOC удаляют газообразным HCl или трифторуксусной кислотой, получая требуемый продукт, иллюстрируемый соединением 25.
Соединение формулы 26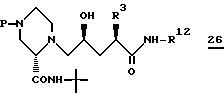
где P есть группа, защищающая азот, такая как -BOC или -CBZ, предпочтительно получают согласно способу, описанному cхемой I, предпочтительно применяя в нем 5-трифторметансульфонилоксиметильный аналог лактона 4 (см. пример 15, стадия 1).
Целевое cоединение I формулы 27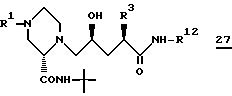
где R12 группа формулы
можно получить различными путями из соединения (i) формулы 28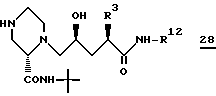
которое получают после удаления группы, защищающей азот, в соединении 26, используя способы, хорошо известные в данной области (например, каталитическое гидрирование, чтобы удалить CBZ группу, или обработка триметилсилилтрифлатом и 2,6-лутидином при около 0oC в растворителе, таком как CH2Cl2, чтобы удалить BOC группу).
Пиперазильный азот в 4-положении соединения 28 алкилируют соединением формулы R1-X в растворителе, таком как DMF в присутствии Et3N при комнатной температуре, где X есть -Cl, Br или -I. Методики выполнения таких процедур хорошо известны специалистам в данной области. R1 группа в R1-X определена выше при определении соединений формулы I.
Соединения данного изобретения используют для ингибирования ВИЧ протеазы, предотвращения или лечения заражения вирусом иммунодефицита человека (ВИЧ) и лечении последующих патологических изменений, таких как СПИД. Считают, что лечение СПИДа или предотвращение или лечение заражения ВИЧ включает, но не ограничивает, лечение широкого диапазона состояний инфекции ВИЧ, СПИД, АРС (СПИД-ассоциированный комплекс) как симптоматическое, так и асимптоматическое, действительное или потенциальное воздействие ВИЧ. Например, соединения данного изобретения используют при лечении заражения ВИЧ у подозреваемых после воздействия ВИЧ, например, при переливании крови, трансплантации органа, обмене общей жидкости организма, укусах, травматическом повреждении иглой или при воздействии на пациента крови во время операционного вмешательства.
Для этих целей соединения данного изобретения могут вводиться орально, парентерально (включая подкожные инъекции, внутривенные, внутримышечные, внутригрудинные инъекции или методику инфузий) путем ингаляционного разбрызгивания или ректально в составах с единичными дозами, содержащими обычные нетоксичные фармацевтически приемлемые носители, адъюванты и наполнители.
Таким образом, согласно данному изобретению разработан также способ лечения и фармацевтическая композиция для лечения ВИЧ инфекции и СПИДа. Лечение включает введение пациенту при необходимости такого лечения фармацевтической композиции, включающей фармацевтический носитель и терапевтически эффективное количество соединения данного изобретения или фармацевтически приемлемую его соль.
Эти фармацевтические композиции могут быть в форме орально вводимых суспензий или таблеток; назальных аэрозолей; стерильных препаратов для инъекций, например в виде стерильных инъецируемых водных или маслянистых суспензий, или суппозиториев.
Уровень доз порядка от 0,02 до 5,0 или 10,0 г в день используют при лечении или профилактике и вышеуказанных условиях, причем оральные дозы в два-пять раз выше. Например, заражение ВИЧ эффективно лечат путем введения от 10 до 50 мг соединения на 1 кг веса тела при приеме от одного до трех раз в день. Следует иметь в виду, однако, что характерный уровень дозы и частота приема дозы для конкретного пациента может варьировать и будет зависеть от ряда факторов, включая активность конкретного применяемого соединения, метаболическую стабильность и продолжительность действия этого соединения, возраст, вес тела, общее состояние здоровья, пол, диету, способ и время приема, скорость введения, комбинации лекарственных средств, серьезности конкретного состояния, и пациента, подвергающегося лечению.
Испытание на ингибирование микробной экспрессированной вирусной протеазы
Исследования ингибирования реакции протеазы, экспрессируемой в Eschericia coli, пептидным субстратом [Val-Ser-Gln-Asn-(бетанафил)Ala-Pro-Ile-Val 0,5 мг/мл во время инициирования реакции] проводят в 50 мМ Na ацетате, pH 5,5, при 30oC в течение 1 ч. К 25 мкл раствора пептида в воде добавляют различные концентрации ингибитора в 1,0 мкл ДМСО. Реакцию инициируют добавлением 15 мкл 0,33 нМ протеазы (0,11 нг) в растворе 0,133 М Na ацетата, pH 5,5 и 0,1% сыворотки бычьего альбумина. Реакцию гасят 160 мкл 5% фосфорной кислоты. Продукты реакции разделяют с помощью ЖХВР (VYDAC широкопористая 5 см C-18 обращенная фаза, градиент ацетонитрила, 0,1% фосфорная кислота). Степень ингибирования реакции определяли по высоте пиков продуктов. ЖВХР продуктов, независимо синтезированных, дает количественное содержание и подтверждение состава продукта. Продукты синтеза в примерах 1-7 включительно дают IC50 значения в диапазоне 1-100 нМ. Соединения A, B и J демонстрируют IC50 значения между около 0,3 и около 6 нМ.
ПРИМЕР 1
Получение N-(2(R)-гидрокси-1(S)-инданил)-2(R)-фенилметил-4(S)- гидрокси-5-(1-(N'-(т-бутил)-4(S)-феноксипролинамид)-ил)-пентанамида
Стадия 1: Получение N-(2(R)-гидрокси-1(S)-инданил)-3-фенилпропанамида
К холодному (0oC) раствору метиленхлорида (30 мл), содержащего 2(R)-гидрокси-1(S)-аминоиндана (750 мг, 5,0 ммоль) и триэтиламин (606 мг, 6,0 ммоль), добавляют раствор гидроциннамоилхлорида (843 мг, 5,0 ммоль) в 5 мл метиленхлорида. Через 2 ч реакционную смесь выливают в делительную воронку, содержащую 50 мл метиленхлорида, и промывают 10%-ным раствором лимонной кислоты (2 х 30 мл). Органический слой сушат, фильтруют и концентрируют, получая белое твердое вещество.
Стадия 2: Получение N-(2(R)-гидрокси-1(S)-индан-N, O- изопропилиден-ил)-3-фенил-пропанамида
Неочищенное белое твердое вещество, полученное по вышеупомянутой стадии 1, растворяют в 50 мл метиленхлорида, добавляют 5 мл диметоксипропана, и затем добавляют 100 мг п-толуолсульфокислоты. Реакционную смесь перемешивают при комнатной температуре в течение 18 ч и затем выливают в делительную воронку и промывают насыщенным раствором NaHCO3 (2 х 30 мл). Органический слой сушат, фильтруют и концентрируют, получая масло, которое разделяют путем хроматографирования (SiO2, 40% ЭА/гексан) с получением масла, в конечном счете кристаллизуемом.
Стадия 3: Получение N-(2(R)-гидрокси-1(S)-индан-N, O- изопропилиден-ил)-2(S)-фенилметил-пент-4-енамида
К раствору N-(2(R)-гидрокси-1(S)-индан-N,O-изопропилиден-ил)-3- фенил-пропанамида (1,03 г, 2,9 ммоль) в 20 мл ТГФ, охлажденному до -78oC, добавляют H-BuLi (2,5 М, 1,40 мл, 3,5 ммоль). Через 20 мин добавляют аллилбромид (0,48 мг, 3,9 ммоль), реакционную смесь перемешивают при -78oC в течение 1 ч и затем добавляют 10 мл насыщенного раствора NH4Cl, чтобы погасить реакцию. Реакционную смесь разбавляют 50 мл воды, экстрагируют этилацетатом (2 х 50 мл), органический слой промывают насыщенным раствором NaCl (50 мл), сушат, фильтруют и концентрируют, получая сырой продукт. Сырой продукт очищают на силикагеле, получая названное соединение.
Стадия 4: Получение N-(2(R)-гидрокси-1(S)-индан-N,O- изопропилиденил)-2(S)-фенилметил-(4(RS),5-дигидрокси)-пентанамида
К 800 мг (2,2 ммоль) N-2(R)-гидрокси-1(S)-индан-N,O- изопропилиден-ил)-2(S)-фенилметил-пент-4-енамида, растворенного в 40 мл 9:1 смеси ацетон/вода, добавляют 0,8 мл 60%-ного раствора N-метилморфолин-N-оксида в воде, а затем 4 мл 2,5%-ного раствора тетроксида осмия в т-BuOH. Через 18 ч добавляют избыток твердого бисульфата натрия, реакционную смесь перемешивают в течение 2 ч и затем фильтруют через прокладку целита. Фильтрат концентрируют, разбавляют 50 мл воды, экстрагируют метиленхлоридом (2 х 50 мл), органический слой сушат, фильтруют и концентрируют, получая продукт в виде пены.
Стадия 5: Получение N-(2(R)-гидрокси-1(S)-индан-N, O- изопропилиден-ил)-2(S)-фенилметил-4(RS)-гидрокси-5- метансульфонилоксипентанамида
К 200 мг (0,527 ммоль) N-(2(R)-гидрокси-1(S)-индан-N,O- изопропилиден-ил)-2(S)-фенилметил-(4(RS), 5-дигидрокси)-пентанамида, растворенного в 7 мл метиленхлорида, при 0oC добавляют триэтиламин (59 мг, 0,58 ммоль), а затем метансульфонилхлорид (66 мг, 0,579 ммоль). Через 4 ч реакционную смесь обрабатывают путем промывания 10%-ным раствором лимонной кислоты (2 х 50 мл), и органический слой сушат, фильтруют и концентрируют, получая мономезилат в виде смеси спиртов.
Стадия 6: Получение N'-т-бутил-N-BOC-4(R)-гидрокси-L-пролинамида
К раствору N-BOC-4(R)-гидроксипролина (2,00 г) в ДМФ (20 мл), охлажденному до 0oC, добавляют ЭДК (1,987 г) HOBt (1,401 г), трет-бутиламин (1,09 мл) и триэтиламин (2,41 мл). Через 18 ч реакционную смесь разбавляют этилацетатом (150 мл) и промывают 10%-ной HCl, насыщенным NaHCO3, водой и соляным раствором. Затем раствор сушат над MgSO4 и концентрируют, получая белое твердое вещество.
Стадия 7: Получение N'-т-бутил-N-BOC-4(S)-фенокси-L-пролинамида
К раствору N'-т-бутил-N-BOC-4(R)-гидрокси-L-пролинамида (0,6 г) в ТГФ (5 мл) добавляют фенол (0,295 г), трифенилфосфин (0,824 г) и затем по каплям диэтилазодикарбоксилат (0,495 мл). Реакционную смесь перемешивают при комнатной температуре в течение 24 ч и разбавляя этилацетатом (200 мл), и промывают насыщенным NaHCO3, водой, соляным раствором и сушат над MgSO4 Концентрированием в вакууме получают желтое масло, которое очищают с помощью флеш-хроматографии (элюирование гексан:ЭА - 1:1, колонка 30 мм).
Стадия 8: Получение соли трифторуксусной кислоты N'-т-бутил-4(S)-фенокси-L-пролинамида
К раствору N'-т-бутил-N-BOC-4(S)-фенокси-L-пролинамида (0,596 г) в метиленхлориде (4 мл) при 0oC добавляют трифторуксусную кислоту (2 мл). После 30 мин реакционную смесь нагревают до комнатной температуры и перемешивают в течение двух часов. Растворитель удаляют в вакууме и получают светло-желтое масло.
Стадия 9: Получение N-(2(R)-гидрокси-1(S)-индан-N, O- изопропилиден-ил)-2-(R)-фенилметил-4(S)-гидрокси-5-(1-(N'-(т- бутил)-4(S)-фенокси-пролинамид)ил)-пентанамида
К раствору соли трифторуксусной кислоты N'-т-бутил-4(S)-фенокси-L-пролинамида (0,36 г) и N-(2(R)-гидрокси-1(S)-индан-N, O-изопропилиден-ил)-2(S)-фенилметил-4(RS)- гидрокси-5-метансульфонилокси-пентанамида (0,226 г) в 3 мл изопропанола добавляют карбонат калия (0,441 г), и реакционную смесь нагревают до 80oC. После 18 ч реакционную смесь охлаждают до комнатной температуры, фильтруют через целит, который промывают далее порциями ЭА. Фильтрат концентрируют, осадок растворяют в ЭА (100 мл) и промывают водой, соляным раствором и сушат над MgSO4. Растворитель удаляют в вакууме, и получающееся масло очищают с помощью флеш-хроматографии, получая продукт в виде смеси диастереомеров.
Стадия 10: Получение N-(2(R)-гидрокси-1(S)-инданил)-2-(R)- фенилметил-4-(S)-гидрокси-5-(1-(N'-т-бутил-4(S)-феноксипролинамид)-ил)- пентанамида
К раствору N-(2(R)-гидрокси-1(S)-индан-N,O-изопропилиден-ил)-2-(R)- фенилметил-4-(S)-гидрокси-5-(1-N'-(т-бутил)-4(S)-феноксипролинамид)-ил)- пентанамида (0,13 г) в MeOH (5 мл) добавляют камфорсульфокислоту (КСК) (0,070 г) при комнатной температуре. После 5 ч добавляют еще КСК (0,025 г), и реакционную смесь перемешивают суммарно 18 ч. Реакцию гасят насыщенным NaHCO3 (5 мл), и растворитель удаляют до объема 4 мл. Водный слой тщательно экстрагируют ЭА, и органический слой промывают водой, соляным раствором и сушат. После удаления растворителя в вакууме полученное масло очищают с помощью флеш-хроматографии, получая названное соединение в виде белой пены. Пену растворяют в ЭА: гексаны, и маточный раствор декантируют с масла. Масло затем сушат в эксикаторе под высоким вакуумом, получая белую пену.
ПРИМЕР 2
Получение N-(2(R)-гидрокси-1(S)-инданил)-2 (R)-фенилметил-4(S)- гидрокси-5-(1-(N'-т-бутил-4(S)-2-нафтилокси-пролинамид)ил)-пентанамида
Стадия 1: Получение соли трифторуксусной кислоты N-т-бутил-4(S)-2-нафтилокси-L-пролинамида
Следуя в основном той же самой методике синтеза соли трифторуксусной кислоты N-т-бутил-4(S)-фенокси-L-пролинамида, описанной в общих чертах в примере 1, стадии с 6 по 8, но замещая фенол, используемый в ней, на 2-нафтол, получают 2-нафтилокси-пролинамид.
Стадия 2: Получение N-(2(R)-гидрокси-1(S)-инданил)-2(R)- фенилметил-4(S)-гидрокси-5-(1-(N'-т-бутил-4(S)-2-нафтилокси- пролинамид)ил)-пентанамида
Названное соединение получают, следуя в основном той же самой методике, описанной в общих чертах в примере 1, стадии 9 и 10, но заменяя соль трифторуксусной кислоты N-т-бутил-4(S)-фенокси-L-пролинамида, используемую в ней на стадии 9, на соль трифторуксусной кислоты N-т-бутил-4(S)-2-нафтилокси-L-пролинамида.
ПРИМЕР 3
Получение N-(2(R)-гидрокси-1(S)-инданил)-2-(R)-фенилметил-4(S)- гидрокси-5-(1-(N'-т-бутил-4(S)-1-нафтилокси-пролинамид)ил)-пентанамида
Стадия 1: Получение соли трифторуксусной кислоты N-т-бутил-4(S)-1-нафтилокси-L-пролинамида
Следуя в основном той же самой методике синтеза соли трифторуксусной кислоты N-т-бутил-4(S)-фенокси-L-пролинамида, описанной в общих чертах в примере 1, стадии с 6 по 8, но заменяя фенол, используемый в ней, на 1-нафтол, получают 1-нафтилокси-пролинамид.
Стадия 2: Получение N-(2(R)-гидрокси-1(S)-инданил)-2(R)- фенилметил-4(S)-гидрокси-5-(1-(N'-т-бутил-4(S)-2-нафтилокси- пролинамид)ил)-пентанамида
Названное соединение получают, следуя методике, описанной в общих чертах в примере 1, стадии 9 и 10, но заменяя соль трифторуксусной кислоты N-т-бутил-4(S)-фенокси-L-пролинамида, используемую в ней на стадии 9, на соль трифторуксусной кислоты N-т-бутил-4(S)-1-нафтилокси-L-пролинамида.
ПРИМЕР 4
Получение N-(2(R)-гидрокси-1(S)-инданил)-2(R)-фенилметил-4(S)- гидрокси-5-(2-(3(S)-N'-(т-бутил-карбоксамидо)-(4aS, 8aS)- декагидроизохинолин)ил)-пентанамида
Стадия 1: Получение дигидро-5(S)-(т-бутилдифенилсилил)-оксиметил)- 3(R)фенилметил-3(2H)-фуранона
Раствор диизопропиламида лития (ЛДА) генерируют путем добавления 1,55 мл н-BuLi (2,5 М в гексане) к 0,55 мл (3,9 ммоль) диизопропиламина в 10 мл ТГФ при -78oC. Через 30 мин добавляют раствор дигидро-5-(S)-((т-бетилдифенилсилил)-оксиметил)-3(2H)-фуранона (1,38 г, 3,89 ммоль) в 5 мл ТГФ. После перемешивания в течение 30 мин, добавляют бензилбромид (0,68 г, 3,9 ммоль), и продолжают перемешивание в течение 3 ч, и после этого времени реакцию останавливают добавлением 10%-ного водного раствора лимонной кислоты. Раствор экстрагируют этилацетатом (2 х 50 мл), который промывают обратной струей соляным раствором, сушат, фильтруют и концентрируют, получая масло. Продукт очищают с помощью хроматографии (SiO2, 20% ЭА/гексан), получая названное соединение.
Стадия 2: Получение дигидро-5(S)-(гидроксиметил)-3(R)фенилметил-3(2H)-фуранона
К 5,26 г дигидро-5(S)-((т-бутилдифенилсилил)оксиметил)-3(R)- фенилметил-3(2H)-фуранона в 40 мл ацетонитрила добавляют 1,34 мл 49%-ного водного раствора HF. Через 18 ч при комнатной температуре реакционную смесь концентрируют до сухого остатка, и остаток распределяют между водой (50 мл) и этилацетатом (50 мл). Органический слой промывают соляным раствором, сушат, фильтруют и концентрируют, получая продукт в виде желтовато-коричневого твердого вещества (т.пл. 69-72oC).
Стадия 3: Получение дигидро-5(S)-((метансульфонил)оксиметил)- 3(R)фенилметил-3(2H)-фуранона
К раствору 2,93 г (14 ммоль) дигидро-5-(S)-(гидроксиметил)-3(R)-фенилметил-3(2H)-фуранона в метиленхлориде, охлажденному до 0oC, добавляют триэтиламин (1,98 мл, 15,6 ммоль), а затем метансульфонилхлорид (1,20 мл, 15,6 ммоль). Через 1 ч при 0oC реакционную смесь выливают в 10%-ный водный раствор лимонной кислоты, промывают этилацетатом (2 х 100 мл), обратной струей воды (100 мл), соляным раствором (100 мл), сушат, фильтруют и концентрируют, получая продукт в виде воскообразного коричневого твердого вещества.
Стадия 4: Получение дигидро-5(S)-(2-(3(S)-N-(т- бутилкарбоксамидо)-(4aS, 8aS)-(декагидроизохинолин)ил)метил)-3(R)- фенилметил-3(2H)-фуранона
К 70 мг дигидро-5(S)-((метансульфонил)-оксиметил)-3(R)- фенилметил-3(2H)-фуранона (0,25 ммоль) в 10 мл ксилола, содержащего 100 мг, карбоната калия, добавляют 65 мг (0,27 ммоль) N-т-бутил-(4aS,8aS)-(декагидроизохинолин)-3(S)-карбоксамида, и реакционную смесь нагревают до 140oC. Через 6 ч реакционную смесь охлаждают, выливают в 30 мл воды и промывают этилацетатом
(2 x 30 мл). Органическую фазу сушат, фильтруют и концентрируют, получая осадок, который хроматографируют (50/50 ЭА/гексан), получая указанный продукт.
Стадия 5: Получение 2(R)-фенилметил-4(S)-т-бутилдиметилсилилокси)- 5-(2-(3(S)-N-(т-бутилкарбоксамидо)-(4aS, 8aS)-декагидроизохинолин)ил)- пентановой кислоты
К 130 мг (0,305 ммоль) дигидро-5(S)-(2-3(S)-N-(т-бутил-карбоксамидо)- (4aS, 8aS)-(декагидроизохинолин)ил)метил)-3(R)-фенилметил-3-(2H)фуранона в 2 мл ДМЭ добавляют 1 мл раствора гидроксида лития. Через 4 ч при комнатной температуре реакционную смесь концентрируют до сухого остатка и сушат путем азаотропной перегонки с толуолом (3Х) для удаления избытка воды. Остаток растворяют в 5 мл ДМФ и добавляют 414 мг (6,10 ммоль) имидазола и 465 мг (3,05 ммоль) т-бутилметилсилилхлорида. Через два дня при комнатной температуре к реакционной смеси добавляют 1 мл метанола и через 1 ч раствор выпаривают досуха. Остаток распределяют между насыщенным раствором NH4Cl (водн.) и промывают этилацетатом, который сушат, фильтруют и концентрируют, получая масло, которое является смесью продукта и фуранона исходного вещества. Это вещество в неочищенном виде используют в следующей реакции.
Стадия 6: Получение N-(2(R)-гидрокси-1(S)-инданил)-2(R)- фенилметил-4(S)-(т-бутилдиметилсилилокси-5-(2-(3(S)-N'-(т- бутилкарбоксамидо)-(4aS, 8aS)-декагидроизохинолин)ил)-пентанамида
Вышеупомянутый неочищенный продукт стадии 5 растворяют в 3 мл ДМФ вместе с 47 мг (0,246 ммоль) ЭДК, 33 мг (0,246 ммоль) HOBT и 37 мг 2(R)-гидрокси-1(S)-аминоиндана; pH раствора доводят до 8,5-9,0 триэтиламином, и через 18 ч его концентрируют до сухого остатка, остаток растворяют в 10%-ном водном растворе лимонной кислоты и промывают водный слой этилацетатом. Органический слой сушат, фильтруют и концентрируют, и образовавшееся масло хроматографируют (SiO2, 30% ЭА/гексан), получая названное соединение.
Стадия 7: Получение N-(2(R)-гидрокси-1(S)-инданил)-2(R)- фенилметил-4(S)-гидрокси-5-(2-(3(S)-N'-(т-бутилкарбоксамидо)-(4aS, 8aS)- декагидроизохинолин)ил)-пентанамида
Продукт вышеописанной стадии 6 растворяют в 1 мл ТГФ и добавляют 1 мл 1 М раствора тетрабутиламмония фторида в ТГФ. Через 18 ч при комнатной температуре реакционную смесь разбавляют 20 мл насыщенного раствора NaHCO3 (водн. ), и продукт экстрагируют этилацетатом, который сушат, фильтруют и концентрируют, получая пену. Полученное вещество хроматографируют на препаративной пластинке (0,5 мм, 5% MeOH/CHCl3), и названный продукт выделяют обычным способом в виде твердого вещества с т.пл. 105-107oC.
ПРИМЕР 5
Получение N-2(R)-гидрокси-1(S)-инданил)-2(R)-фенилметил-4(S)- амино-5-(2-(3(S)-N'-(т-бутилкарбоксамидо)-(4aS, 8aS)- декагидроизохинолин)ил)-пентанамида
Стадия 1: Получение 5(S)-((т-бутил-диметилсилилокси)метил)-3(R)- фенилметил-N-BOC-2-пирролидинона
Раствор 5(S)-((т-бутил-диметилсилилокси)метил)-N-BOC-2-пирролидинона (400 мг, 1,26 ммоль) в 2 мл ТГФ добавляют к предварительно охлажденному (-78oC) 1 М раствору гексаметилдисилазида лития (1,3 мл) в 5 мл ТГФ. Через 45 мин добавляют 0,15 мл бензилбромида (1,3 ммоль), и продолжают перемешивание. Через 5 ч реакционную смесь выливают в разделительную воронку, содержащую 30 мл 10%-ного водного раствора лимонной кислоты. Водный слой экстрагируют (2 х 30 мл ЭА), и это промывают обратной струей соляного раствора (50 мл), сушат, фильтруют и концентрируют до масла. Остаток хроматографируют (SiO2, 20% ЭА/гексан), получая продукт в виде масла.
Стадия 2: Получение 5(S)-гидроксиметил-3(R)-фенилметил-2-пирролидинона
К 130 мг (0,34 ммоль) 5(S)-((т-бутил-диметилсилилокси)-метил)-3(R)- фенилметил-N-BOC-2-пирролидинона в 5 мл ацетонитрила добавляют 0,1 мл раствора 48% HF в воде. Через 3 ч при комнатной температуре реакционную смесь концентрируют до сухого состояния и разбавляют 30 мл водного 10%-ного раствора NaHCO3. Смесь экстрагируют ЭА (2 х 30 мл), сушат, фильтруют и концентрируют, получая неочищенный продукт.
Стадия 3: Получение 5(S)-(метансульфонилокси)-метил-3(R)- фенилметил-2-пирролидинона
К раствору неочищенного продукта из стадии 2 в 5 мл метиленхлорида, охлажденного до 0oC, добавляют триэтиламин (42 мг, 0,41 ммоль) и метансульфонилхлорид (47 мг, 0,41 ммоль). Реакционной смеси дают возможность медленно нагреться до комнатной температуры и перемешивают в течение 18 ч, и после этого ее разбавляют 30 мл метиленхлорида, промывают 30 мл 10%-ным раствором лимонной кислоты, сушат, фильтруют и концентрируют, получая продукт в виде масла.
Стадия 4: Получение 5(S)-(2-(3(S)-N-(т-бутилкарбоксамидо)- (4aS,8aS)-(декагидроизохинолин)ил)-метил)-3(R)-фенилметил-2-пирролидинона
К раствору 380 мг (1,34 ммоль) 5(S)-(метансульфонилокси)-метил-3(R)-фенилметил-2-пирролидинона в 20 мл изопропанола добавляют 350 мг карбоната калия и 360 мг N-т-бутил-(4aS,8aS)-(декагидроизохинолин)-3(S)-карбоксамида, и реакционную смесь нагревают до 85oC. Через 18 ч охлажденную реакционную смесь фильтруют через целит, выпаривают досуха и остаток растворяют в воде, что и экстрагируют ЭА (2 х 50 мл). Органический слой сушат, фильтруют и концентрируют, и остаток хроматографируют (SiO2, 50/50 ЭА/гексан), получая продукт в виде масла.
Стадия 5: Получение 5(S)-(2-(3(S)-N'-(т-бутилкарбоксамидо)-(4aS,8aS)-(декагидроизохинолин)ил)-метил)-3(R)-фенилметил-N-BOC-2-пирролидинона
К раствору вышеупомянутого продукта стадии 4 (260 мг, 0,611 ммоль) в 10 мл метиленхлорида добавляют диметиламинопиридин (74 мг, 0,6 ммоль) и 133 мг (0,61 ммоль) BOC-ангидрида. Через 18 ч при комнатной температуре реакционную смесь обрабатывают путем разбавления 30 мл метиленхдорида, и органический слой промывают 30 мл 10%-ного раствора лимонной кислоты, соляным раствором (30 мл), сушат, фильтруют и концентрируют, получая масло. Хроматографирование (SiO2, 40% ЭА/гексан) дает названное соединение.
Стадия 6: Получение 5-(2-(3(S)-N'-(т-бутилкарбоксамидо)- (4aS,8aS)-(декагидроизохинолин)ил)-4(S)-[(1', 1')- (диметилэтоксикарбонил)-амино] -2(R)-фенилметилпентановой кислоты
К раствору вышеуказанного продукта стадии 5 (260 мг, 0,495 ммоль), растворенному в 3 мл диметоксиэтана, добавляют 1,5 мл 1 М водного раствора гидроксида лития (1,5 ммоль). Реакционную смесь обрабатывают спустя 2 ч путем концентрирования досуха, растворения остатка в насыщенном водном растворе хлорида аммония, и водную фазу промывают этилацетатом (2 х 50 мл), который сушат, фильтруют и концентрируют, получая неочищенную кислоту.
Стадия 7: Получение N-(2(R)-гидрокси-1(S)-инданил)-2(R)- фенилметил-4(S)-[(1', 1')-(диметилэтоксикарбонил)-амино] - 5-(2-3(S)- N'-(т-бутилкарбоксамидо)-(4aS,8aS)-декагидроизохинолин)ил)-пентанамида
К раствору вышеупомянутого продукта стадии 6 (260 мг, 0,49 ммоль) в метиленхлориде добавляют EDS (94 мг, 0,49 ммоль), HOBT (66 мг, 0,49 ммоль), 2(R)-гидрокси-1(S)-аминоиндан (73 мг, 0,49 ммоль) и pH реакционной смеси доводят до 8,5-9,0, используя триэтиламин. Через 5 ч при комнатной температуре реакционную смесь обрабатывают путем разбавления 50 мл метиленхлорида и промывания органики насыщенным водным раствором хлорида аммония. Органическую фазу сушат, фильтруют и концентрируют, и остаток хроматографируют, получая названное соединение в виде пены.
Стадия 8: Получение N-(2(R)-гидрокси-1(S)-инданил)-2(R)- фенилметил-4(S)-гидрокси-5-(2-(3(S)-N'-(т-бутилкарбоксамидо)- (4aS, 8aS)-декагидроизохинолин)-пентанамид
К раствору вышеупомянутого продукта стадии 7 (180 мг, 0,28 ммоль) в 5 мл метиленхлорида, охлажденного до 0oC, добавляют 1 мл трифторуксусной кислоты. Через 4 ч реакционную смесь концентрируют досуха, и остаток растворяют в 50 мл метиленхлорида и промывают 10%-ным водным раствором NaHCO3. Органический слой сушат, фильтруют и концентрируют, получая продукт в виде твердого вещества, которое хроматографируют (SiO2, 7% MeOH/CH2Cl2), получая названное соединение, т.пл. = 92-95oC.
ПРИМЕР 6
Получение N-(2(R)-гидрокси-1(S)-инданил)-2(R)-фенилметил-4(S)- гидрокси-5-(1-(4-карбобензилокси-2-(S)-N'-(т-бутилкарбоксамидо)- пиперазинил))-пентанамида
Используя в основном ту же самую методику, что в примере 1, но заменяя N-т-бутил-4(S)-фенокси-L-пролинамид, используемый в ней на стадии 9, на N-т-бутил-4-CBZ-пиперазин-2(S)-карбоксамид, получают названное соединение.
ПРИМЕР 7
Получение N''-(N-(2-пиридил)-валил)-2(R)-фенилметил-4(S)- гидрокси-5-(2-(3(S)-(N'-т-бутилкарбоксамидо)-(4aS, 8aS)- декагидроизохинолин)-ил)-пентанамида
Используя в основном ту же самую методику, что в примере 4, но заменяя 2(R)-гидрокси-1(S)аминоиндан, применяемый в ней на стадии 6, на N-2-пиридил-валин, получают названное соединение.
ПРИМЕР 8
Получение N-(2(R)-гидрокси-1(S)-инданил)-2-(R)-фенилметил-4(S)- гидрокси-5-(2(S)-(N'-т-бутил-3-фенил-пропионамид)амино)-пентанамида
Используя в основном ту же самую методику, что и в примере 1, но заменяя N'-т-бутил-4(S)-фенокси-L-пролинамид, применяемый в ней на стадии 9, на N-т-бутилфенилаланинамид, получают названное соединение.
ПРИМЕР 9
Получение N-(4(S)-3,4-дигидро-1H-2,2-диоксо-бензотиопиранил)- 2(R)-фенилметил-4(S)-гидрокси-5-(2-(3(S)-N'-(т-бутилкарбоксамидо)- (4aS,8aS)-декагидроизохинолин)ил)-пентанамида
Стадия 1: Получение N-(4(S)-3,4-дигидро-1H-бензотиопиранил)-2(R)- фенилметил-4(S)-гидрокси-5-(2-(3(S)-т-бутилкарбоксамидо)-(4aS,8aS)- декагидроизохинолин)ил)-пентанамида
Используя в основном ту же самую методику, что в Примере 4, но заменяя 2(R)-гидрокси-1(S)-аминоиндан, используемый в стадии 6, на 4(S)-амино-3,4-дигидро-1H-бензотиопиран, получают вышеназванное соединение.
Стадия 2: Получение N-(4(S)-3,4-дигидро-1H-2,2- диоксобензотиопиранил)-2(R)-фенилметил-4(S)-гидрокси-5-(2-(3(S)- т-бутилкарбоксамидо)-(4aS, 8aS)-декагидрозохинолин)ил)-пентанамида
Соединение из вышеупомянутой стадии 1 растворяют в смеси 1:1 метанола и воды. К ней добавляют 10 экв. OXONE, и реакционную смесь перемешивают при комнатной температуре. По завершении реакции реакционную смесь концентрируют до сухого состояния, добавляют воду и экстрагируют этилацетатом, экстракт сушат, фильтруют и концентрируют, получая названное соединение.
ПРИМЕР 10
Получение N-(4(S)-3,4-дигидро-1H-2,2-диоксобензотиопиранил-2(R)- фенилметил-4(S)-гидрокси-5-(1-(4-карбобензилокси-2(S)-N'-(т- бутилкарбоксамидо)-пиперазинил))-пентанамида
Стадия 1: Получение дигидро-5(S)-(1-(4-карбобензилокси-2-(S)-N'-(т- бутилкарбоксамидо)-пиперазинил)метил)-3(R)-фенилметил-3(2H)-фуранона
Используя в основном ту же самую методику, что в примере 4, стадия 4, но замещая N'-т-бутил-(4aS, 8aS)-(декагидроизохинолин)-3(S)-карбоксамид, используемый в ней, на 4-карбобензилокси-2(S)-N'-(т-бутилкарбоксамидо)-пиперазин, получают названное соединение.
Стадия 2: Получение 2(R)-фенилметил-4(S)-(т-бутилдиметилсилилокси)- 5-(1-(4-карбобензилокси-2(S)-N'-(т-бутилкарбоксамидо)-пиперазинил))- пентановой кислоты
Используя в основном ту же самую методику, как в примере 4, стадия 5, но заменяя дигидро-5(S)-(2-(3(S)-N'-(т-бутилкарбоксамидо)- (4aS,8aS)-декагидроизохинолин)ил)метил)-3(R)-фенилметил-3(2H)-фуранон, используемый в ней, на дигидро-5(S)-(1-(4-карбобензилокси-2(S)-N'-(т- бутилкарбоксамидо)-пиперазинил)метил)-3(R)-фенилметил-3(2H)-фуранон, получают названное соединение.
Стадия 3: Получение N-(4(S)-3,4-дигидро-1H-бензотиопиринил)-2(R)- фенилметил-(4(S)-(т-бутилдиметилсилилокси)-5-(1-(4-карбобензилокси- 2(S)-N'-(т-бутилкарбоксамидо)-пиперазинил))-пентанамида
Неочищенную 2(R)-фенилметил-4(S)-(т-бутилдиметилсилилокси)- 5-(1-(4-карбобензилокси-2(S)-N'-(т-бутилкарбоксамидо)-пиперазинил))- пентановую кислоту растворяют в 3 мл ДМФ вместе с 1 экв. ЭДК, 1 экв. HOBT и 1 экв. 4(S)-амино-3,4-дигидро-1H-бензотиопирана; pH раствора доводят до 8,5-9,0 триэтиламином, и через 18 ч реакционную смесь подвергают обработке путем концентрирования досуха, растворения остатка в 10%-ном водном растворе лимонной кислоты и промывки водного слоя этилацетатом. Органический слой сушат, фильтруют и концентрируют, и получающийся остаток хроматографируют, получая названное соединение.
Стадия 4: Получение N-(4(S)-3,4-дигидро-1H-бензотиопиранил)-2(R)- фенилметил-(4(S)-гидрокси)-5-(1-(4-карбобензилокси-2(S)-(т- бутилкарбоксамидо)-пиперазинил))-пентанамида
Продукт из вышеописанной стадии 3 растворяют в 1 мл ТГФ и добавляют 1 мл 1 М раствора фторида тетрабутиламмония в ТГФ. Через 18 ч при комнатной температуре реакционную смесь разбавляют 20 мл насыщенного (водного) раствора NaHCO3, и продукт экстрагируют этилацетатом, экстракт сушат, фильтруют и концентрируют, получая остаток. Остаток хроматографируют, получая продукт.
Стадия 5: Получение N-(4(S)-3,4-дигидро-1H-2,2- диоксобензотиопиранил)-2(R)-фенилметил-4(S)-гидрокси-5-(1-(4- карбобензилокси-2(S)-N'-(т-бутилкарбоксамидо)-пиперазинил))-пентанамида
Соединение из вышеприведенной стадии 4 растворяют в смеси 1:1 метанола и воды. К ней добавляют 10 экв. OXONE, и реакционную смесь перемешивают при комнатной температуре. По завершении реакции реакционную смесь концентрируют досуха, добавляют воду и экстрагируют этилацетатом, экстракт сушат, фильтруют и концентрируют, получая названное соединение.
ПРИМЕР 11
Получение N-(2(R)гидрокси-1(S)-инданил)-2(R)-((4-((2- гидрокси)этокси)фенил)метил-4(S)-гидрокси-5-(2-(3(S)-N'-(т- бутилкарбоксамидо)-(4aS,8aS)-декагидроизохинолин)ил)-пентанамида
Стадия 1: Получение N-(2(R)-гидрокси-1(S)-инданил)-2(R)-((4-(2- аллилокси)фенил)метил)-4(S)-гидрокси-5-(2-(3(S)-т-бутилкарбоксамидо)- (4aS, 8aS)-декагидроизохинолин)ил)-пентанамида
К раствору N-(2(R)-гидрокси-1(S)-инданил)-2(R)-((4- гидроксифенил)метил)-4(S)-гидрокси-5-(2-(3(S)-т-бутилкарбоксамидо)- (4aS, 8aS)-декагидроизохинолин)ил)пентанамида в диоксане добавляют 6 экв. аллилбромида в 6 экв. карбоната цезия. Реакционную смесь нагревают до 90oC. По завершении реакции осадок отфильтровывают, диоксан отгоняют досуха, и остаток разбавляют водой, и эту смесь промывают этилацетатом. Органическую фазу сушат, фильтруют и концентрируют, получая продукт.
Стадия 2: Получение N-(2(R)-гидрокси-1(S)-инданил)-2(R)-((4-((2- гидрокси)этокси)фенил)метил)-4(S)-гидрокси-5-(2-(3(S)-N'-(т- бутилкарбоксамидо)-(4aS,8aS)-декагидроизохинолин)ил)-пентанамида
Продукт стадии 1 растворяют в метаноле, добавляют 1 экв. п-толуолсульфокислоты, и реакционную смесь охлаждают до -78oC. Через реакционную смесь пропускают озон до тех пор, пока не появится устойчивый синий цвет. Сосуд продувают азотом для удаления следов озона и добавляют избыток раствора боргидрида натрия. Реакционную смесь нагревают до комнатной температуры и затем добавляют насыщенный раствор NaHCO3. Метанол удаляют на роторном испарителе, и водный остаток промывают этилацетатом, экстракт сушат, фильтруют и концентрируют, получая названное соединение.
ПРИМЕР 12
Получение N-(2(R)-гидрокси-1(S)-инданил)-2(R)-((4-((2- гидрокси)этокси)фенил)метил)-4(S)-гидрокси-5-(1-(4-карбобензилокси- 2(S)-N'-(т-бутилкарбоксамидо)-пиперазинил)-пентанамида
Используя в основном ту же самую методику, как в примере 11, но заменяя N-(2(R)-гидрокси-1(S)-инданил)-2(R)-((4-гидроксифенил)метил)- 4(S)-гидрокси-5-(2-(3(S)-т-бутилкарбоксамидо)-(4aS, 8aS)- декагидроизохинолин)ил)-пентанамид, используемый в ней, на N-(2(R)-гидрокси-1(S)-инданил)-2(R)-((4-гидроксифенил)метил)- 4(S)-гидрокси-5-(1-(4-карбобензилокси-2(S)-(т- бутилкарбоксамидо)пиперазинил)-пентанамид, получают названное соединение.
ПРИМЕР 13
Получение N-(2(R)-гидрокси-1(S)-инданил)-2(R)-((4-(2-(4- морфолинил)этокси)фенил)метил)-4(S)-гидрокси-5-(2-(3(S)- N'-(т- бутилкарбоксамидо)-(4aS,8aS)-декагидроизохинолин)ил)-пентанамида
К раствору N-(2(R)-гидрокси-1(S)-инданил)-2(R)-((4- гидроксифенил)метил)-4(S)-гидрокси-5-(2-(3(S)-N'-(т- бутилкарбоксамидо)-(4aS,8aS)-декагидроизохинолин)ил)-пентанамида в диоксане добавляют 6 экв. хлорэтилморфолина и 6 экв. карбоната цезия. Реакционную смесь нагревают до 90oC. По завершении реакции осадок отфильтровывают, отгоняют диоксан, и остаток разбавляют водой, затем промывают этилацетатом. Органическую фазу сушат, фильтруют и концентрируют, получая названное соединение.
ПРИМЕР 14
Получение N-(2(R)-гидрокси-1(S)-инданил)-2(R)-((4-(2-(4- морфолинил)этокси)фенил)метил)-4(S)-гидрокси-5-(1-(4- карбобензилокси-2(S)-N'-(т-бутилкарбоксамидо)-пиперазинил))-пентанамида
К раствору N-(2(R)-гидрокси-1(S)-инданил)-2(R)-((4- гидроксифенил)метил)-4(S)-гидрокси-5-(1-(4-карбобензилокси-2(S)-(т- бутилкарбоксамидо)-пиперазинил)-пентанамида в диоксане добавляют 6 экв. хлорэтилморфолина и 6 экв. карбоната цезия. Реакционную смесь нагревают до 90oC. По завершении реакции осадок отфильтровывают, отгоняют диоксан, и остаток разбавляют водой, затем промывают этилацетатом. Органическую фазу сушат, фильтруют и концентрируют, получая названное соединение.
ПРИМЕР 15
Получение N-(2(R)-гидрокси-1(S)-инданил)-2(R)-фенилметил-4(S)- гидрокси-5-(1-(4-(3-пиридилметил)-2-(S)-N'-(т-бутилкарбоксамидо)- пиперазинил))-пентанамида
Стадия 1: Получение дигидро-5(S)-((трифторметансульфонил)оксиметил)-3(R)-фенилметил-3(2H)-фуранона
К раствору 18,4 г (89,2 ммоль) дигидро-5(S)-(гидроксиметил)-3(R)-фенилметил-3(2H)-фуранона в 350 мл метиленхлорида, охлажденному до 0oC, добавляют 13,51 мл 2,6-литидина (115,98 ммоль), а затем по каплям добавляют 16,51 мл ангидрида трифторметансульфокислоты (98,1 ммоль). Через 1,5 ч при 0oC, реакционную смесь выливают в смесь 300 мл лед/соляной раствор и перемешивают в течение 0,5 ч. Затем водный слой экстрагируют метиленхлоридом (3 х 150 мл), органические слои промывают 10%-ной HCl (2 х 75 мл), насыщенным NaHCO3 (100 мл), водой (100 мл), сушат над MgSO4 фильтруют и концентрируют, получая твердое вещество. Очистка с помощью флеш-хроматографии (колонка 120 х 150 мм, градиент элюирования смесью гексаны:ЭА, 4:1 до 3:1) приводит к получению названного продукта; т.пл. 53-54oC.
Стадия 2: Получение 4-(1,1-диметилэтил)-1-(фенилметил)-1,2(S),4- пиперазинтрикарбоксилата
Названное соединение получают, следуя способу Bigge C.F.; Hays S.J.; Novak P.M.; Drummond J.T.; Johnson G.; Bobovski T.P.; Tetrahedron Lett. 1989, 30, 5193; исходя из 2(S)-пиперазинкарбоновой кислоты (см. Felder E.; Maffei S.; Pietra S.; Pitre D.; Helv. Chim. Acta. 1960, 117, 888.
Стадия 3: Получение N-т-бутил-4-(1,1-диметилэтоксикарбониламино)- 1-(фенилметилкарбониламино)пиперазин-2(S)-карбоксамида
К 9,90 г (27,16 ммоль) 4-(1,1-диметилэтил)-1-((фенилметил)- 1,2(S),4-пиперазинтрикарбоксилату, растворенному в 75 мл ДМФ и охлажденному до 0oC, добавляют 5,73 г (29,88 ммоль) ЭДК, 4,03 г (29,88 ммоль) HOBt, 3,14 мл (29,88 ммоль) т-бутиламина, и, наконец, 4,16 мл (29,88 ммоль) триэтиламина. Реакционную смесь перемешивают в течение 18 ч, и реакционный объем концентрируют наполовину. Затем смесь разбавляют 600 мл ЭА и промывают 10%-ной HCl (2 х 75 мл), насыщенным NaHCO3 (1 х 75 мл), водой (3 х 75 мл) и соляным раствором (1 х 50 мл), сушат над MgSO4 и концентрируют до твердого вещества. Это твердое вещество растирают в порошок в смеси ЭА:гексан (1:2) и фильтруют, получая названный продукт в виде твердого белого вещества; т.пл. 134-135oC.
Стадия 4: Получение N-т-бутил-4-(1,1-диметилэтоксикарбониламино) пиперазин-2(S)-карбоксамида
К 1,20 г (2,86 ммоль) N-т-бутил-4-(1,1-диметилэтоксикарбониламино)- 1-(фенилметилкарбониламино)пиперазин-2(S)-карбоксамида и 1,1 г (0,086 ммоль) 10% Pd/C добавляют 15 мл метанола. Сосуд продувают водородом, и реакционную смесь перемешивают в течение 2 ч, фильтруют через целит и промывают этанолом. Растворители удаляют в вакууме, получая названный продукт в виде пены.
1H ЯМР (300 МГц, CDCl3) δ 6,65 (ш, 1H), 4,10 (м, 1H), 3,81 (ш, 1H), 3,21 (дд, J = 18 и 7 Гц, 1H), 3,02-2,70 (м, 4H), 2,10-2,0 (ш, 1H), 1,50 (с, 9H), 1,41 (с, 9H).
Стадия 5: Получение дигидро-5(S)-(4-(1,1- диметилэтоксикарбониламино))-2(S)-N-(т- бутилкарбоксамидо)пиперазинил)метил)-3(R)-фенилметил-3(2H)-фуранона
К раствору 22,40 г (0,0662 моль) дигидро-5(S)- ((трифторметансульфонил)оксиметил)-3(R)-фенилметил-3(2H)-фуранона (получен на стадии 1) и 18,0 г (0,063 моль) н-т-бутил-4-(1,1-диметилэтоксикарбониламино)пиперазин-2(S)-карбоксамида, растворенных в 10 мл изопропанола, добавляют 11,53 мл (0,0662 моль) N,N-диизопропилэтиламина. Через 2,5 ч добавляют еще 1,2 г дигидро-5(S)-((трифторметансульфонил)оксиметил)-3(R)-фенилметил- 3(2H)-фуранона. Реакция завершается через 3,5 ч контролем с помощью тонкослойной хроматографии (тсх) и реакционную смесь концентрируют до получения густого масла. Растирание в смеси ЭА:гексаны (1:2, 200 мл) приводит к получению белого твердого вещества, которое фильтруют и выгружают. Масло очищают с помощью флеш-хроматографии (колонка 120 х 150 мм, ЭА:гексаны, градиент элюирования 1:1, 2:1, 3:1, до чистого ЭА), получая названное соединение.
1H ЯМР (400 МГц, CDCl3) δ 7,34-7,17 (м, 5H), 6,31 (ш.с, 3H), 4,38 (ш.м, 1H), 3,96-3,92 (м, 1H), 3,79 (ш.м, 1H), 3,16 (дд, J = 13,6 и 4,4 Гц, 1H), 3,08-2,99 (м, 3H), 2,90-2,82 (м, 1H), 2,80 (дд, J = 13,5 и 8,9 Гц, 1H), 2,78 (м, 1H), 2,32-2,04 (м, 1H), 1,99-1,92 (м, 1H), 1,45 (с, 9H), 1,29 (с, 9H).
Стадия 6: Получение 2(R)-фенилметил-4(S)-(т- бутилдиметилсилилокси)-5-(1-(4-(1,1-диметилэтоксикарбониламино)))- 2(S)-N-(т-бутилкарбоксамидо)-пиперазинил))-пентанамида
К 25,50 г (52,50 ммоль) дигидро-5(S)-(4-(1,1- диметилэтоксикарбониламино))-2(S)-N-(т-бутилкарбоксамидо)-пиперазинил)- метил)-3(R)-фенилметил-3(2H)-фуранона, растворенного в 120 мл ДМЭ, охлажденном до 0oC, добавляют раствор 60 мл воды и 1,512 г (63,01 ммоль) гидроксида лития. Через 0,5 ч реакцию останавливают путем добавления 10%-ной HCl до pH 6, и раствор концентрируют в вакууме. Остаток растворяют в 50 мл воды и экстрагируют ЭА (4 х 75 мл), и органические соли промывают водой (1 х 20 мл), соляным раствором (1 х 20 мл). Водный слой снова экстрагируют ЭА (2 х 75 мл), и объединенные вместе органические слои сушат над MgSO4 и концентрируют, получая желтое твердое вещество. Этот неочищенный продукт растворяют в 100 мл ДМФ и добавляют 17,87 г (0,262 моль) имидазола, охлаждают до 0oC и затем добавляют 31,50 г (0,21 моль) т-бутилдиметилсилилхлорида. Реакционную смесь перемешивают 1 ч при 0oC и затем нагревают до комнатной температуры. Через 20 ч реакцию останавливают 10 мл метанола, и реакционную смесь концентрируют до половины объема. Добавляют 100 мл водного буфера с pH 7, и водный слой экстрагируют ЭА (4 х 100 мл), объединенные органические слои промывают 10%-ной HCl (2 х 50 мл), водой (3 х 75 мл) и соляным раствором (1 х 50 мл), сушат над MgSO4 и концентрируют, получая названное соединение. Это вещество непосредственно используют в следующей стадии.
Стадия 7: Получение N-(2(R)-гидрокси-1(S)-инданил)-2(R)- фенилметил-4(S)-(т-бутилдиметилсилилокси)-5-(1-(4-(1,1- диметилэтоксикарбониламино)))-2(S)-N-(т-бутилкабоксамидо)- пиперазинил))-пентанамида
К 27,0 г (0,0446 моль) неочищенного вещества из стадии 6, растворенного в 180 мл ДМФ и охлажденного до 0oC, добавляют 8,98 г (0,0468 моль) ЭДК, 6,32 г (0,0468 моль) HOBt и 7,31 г (0,049 моль) аминогидрокси-индана. Добавляют триэтиламин (6,52 мл, 0,0468 моль), и реакционную смесь перемешивают при 0oC в течение 2 ч, при комнатной температуре в течение 16 ч и гасят путем разбавления 500 мл ЭА. Органический слой промывают 10%-ной HCl (2 х 100 мл), насыщенным NaHCO3 (1 х 100 мл), водой (3 х 150 мл), соляным раствором (1 х 75 мл), сушат над MgSO4 и концентрируют, получая названное соединение в виде белой пены.
1H ЯМР (400 МГц, CDCl3) δ 7,4-7,17 (м, 9H), 6,51 (ш.с, 1H), 5,79 (ш.с, 1H), 5,23 (м, 1H), 4,23 (ш.с, 1H), 4,06 (м, 1H), 3,96-3,84 (м, 2H), 3,07-2,78 (м, 8H), 3,65 (дд, J = 9,6 и 4,1 Гц, 1H), 2,56-2,44 (м, 2H), 2,29 (дд, J = 12,0 и 4,5 Гц, 1H), 2,17-2,09 (м, 1H), 1,79 (ш.с, 1H), 1,44 (с, 9H), 1,35 (с, 9H), 1,10 (с, 1H), 0,84 (с, 9H), 0,12 (с, 3H), 0,08 (с, 3H).
Стадия 8: Получение N-(2(R)-гидрокси-1(S)-инданил)-2(R)- фенилметил-4(S)-(гидрокси)-5-(1-(4-(1,1-диметилэтоксикарбониламино)))- 2(S)-N-(т-бутилкарбоксамидо)-пиперазинил))-пентанамида
К 32,20 г (0,0437 моль) N-(2(R)-гидрокси-1(S)-инданил-(2(R)- фенилметил-4(S)-(т-бутилдиметилсилилокси)-5-(1-(4-(1,1- диметилэтоксикарбониламино)))-2(S)-N-(т-бутилкарбоксамидо)- пиперазинил))-пентанамида добавляют 437 мл (0,437 моль) фторида тетрабутиламмония (1,0 М раствор в ТГФ, Aldrich). Реакционную смесь перемешивают в течение 18 ч, и затем концентрируют до 200 мл и разбавляют 700 мл ЭА. Смесь промывают водой (2 х 100 мл), соляным раствором (1 х 50 мл), и водные слои снова экстрагируют ЭА (2 х 200 мл). Объединенные органические слои сушат над MgSO4 и концентрируют до масла. Очистка флеш-хроматографией (120 х 150 мм колонка, градиентное элюирование CH2Cl2: CHCl3/насыщенный NH3:метанол, при увеличении метанола от 1%, 5%, 2%) приводит к получению названного соединения в виде белой пены.
1H ЯМР (400 МГц, CDCl3) δ 7,31-7,11 (м, 9H), 6,41 (ш.с, 1H), 6,23 (д, J = 8,6 Гц, 1H), 5,25 (дд, J = 8,6 и 4,7 Гц, 1H), 4,21 (м, 1H), 3,83-3,82 (м, 2H), 3,78-3,61 (м, 2H), 3,22-3,19 (м, 2H), 3,03-2,78 (м, 8H), 2,62-2,58 (м, 1H), 2,41-2,35 (м, 2H), 2,04-2,02 (м, 1H), 1,57-1,50 (м, 1H), 1,45 (с, 9H), 1,32 (с, 9H).
Стадия 9: Получение N-(2(R)-гидрокси-1(S)-инданил)-2(R)- фенилметил-4(S)-(гидрокси)-5-(1-(2(S)-N-(т-бутилкарбоксамидо)- пиперазинил)-пентанамида
К 21,15 г (0,034 моль) N-(2(R)-гидрокси-1(S)-инданил)-2(R)- фенилметил-4(S)-(гидрокси)-5-(1-(4-(1,1-диметилэтоксикарбониламино)))- 2(S)-N-(т-бутилкарбоксамидо)-пиперазинил))-пентанамида, растворенного в 350 мл метиленхлорида и охлажденного до 0oC, добавляют 22,43 мл (0,204 моль) 2,6-лутидина и затем 32,85 мл (0,170 моль) триметилсилилтрифлата на протяжении 5 мин. Через 0,5 ч реакционную смесь гасят 10%-ной HCl (80 мл), и смесь перемешивают в течение 0,5 ч. К смеси добавляют 100 мл насыщенного NaHCO3, затем твердый NaHCO3 до pH 8. Затем водный слой экстрагируют ЭА (4 х 100 мл), и объединенные органические слои промывают водой (1 х 50 мл), соляным раствором (1 х 75 мл), сушат над MgSO4 и концентрируют. Остаток очищают при помощи колоночной хроматографии (колонка 120 х 150 мм, градиентное элюирование CH2Cl2: CHCl3, насыщенный NH3:MeOH, при медленном увеличении метанола от 2%, 3%, 4%, 5%, 6% до 10%). При этом получают названный продукт в виде белой пены.
1H ЯМР (400 МГц, CDCl3) δ 7,53 (с, 1H), 7,29-7,09 (м, 9H), 6,52 (д, J = 8,3 Гц, 1H), 5,24 (дд, J = 8,2 и 4,9 Гц, 1H), 4,23 (дд, J = 4,7 и 4,03 Гц, 1H), 4,25-4,00 (ш. с, 1H), 3,83-3,81 (м, 1H), 3,03-2,88 (м, 4H), 2,82-2,73 (м, 7H), 2,50-1,60 (ш.с, 2H), 2,45 (д, J = 6,2 Гц, 2H), 2,32-2,29 (м, 1H), 1,98 (м, 1H), 1,51 (м, 1H), 1,33 (с, 9H).
Стадия 10: Получение N-(2(R)-гидрокси-1(S)-инданил)-2(R)- фенилметил-4(S)-гидрокси-5-(1-(4-(3-пиридилметил)-2(S)-N'-(т- бутилкарбоксамидо)-пиперазинил))-пентанамида
К 10 г (0,019 моль) N-(2(R)-гидрокси-1(S)-инданил)-2(R)- фенилметил-4(S)-гидрокси)-5-(1-(2-(S)-N-(т-бутилкарбоксамидо)- пиперазинил))-пентанамида и 3,45 г (0,021 моль) 3-пиколилхлорила, растворенным в 40 мл ДМФ, добавляют 5,85 мл (0,042 моль) триэтиламина. Через 3 ч добавляют дополнительно 0,313 г 3-пиколилхлорида. После последующих 2 ч реакционную смесь разбавляют 400 мл ЭА и промывают водой (3 х 75 мл), соляным раствором (1 х 100 мл), сушат над MgSO4 и концентрируют. Остаток растирают в порошок в 30 мл ЭА, и собирают образующийся белый осадок. Последующей перекристаллизацией из ЭА получают названный продукт (т.пл. 167,5-168oC).
ПРИМЕР 16
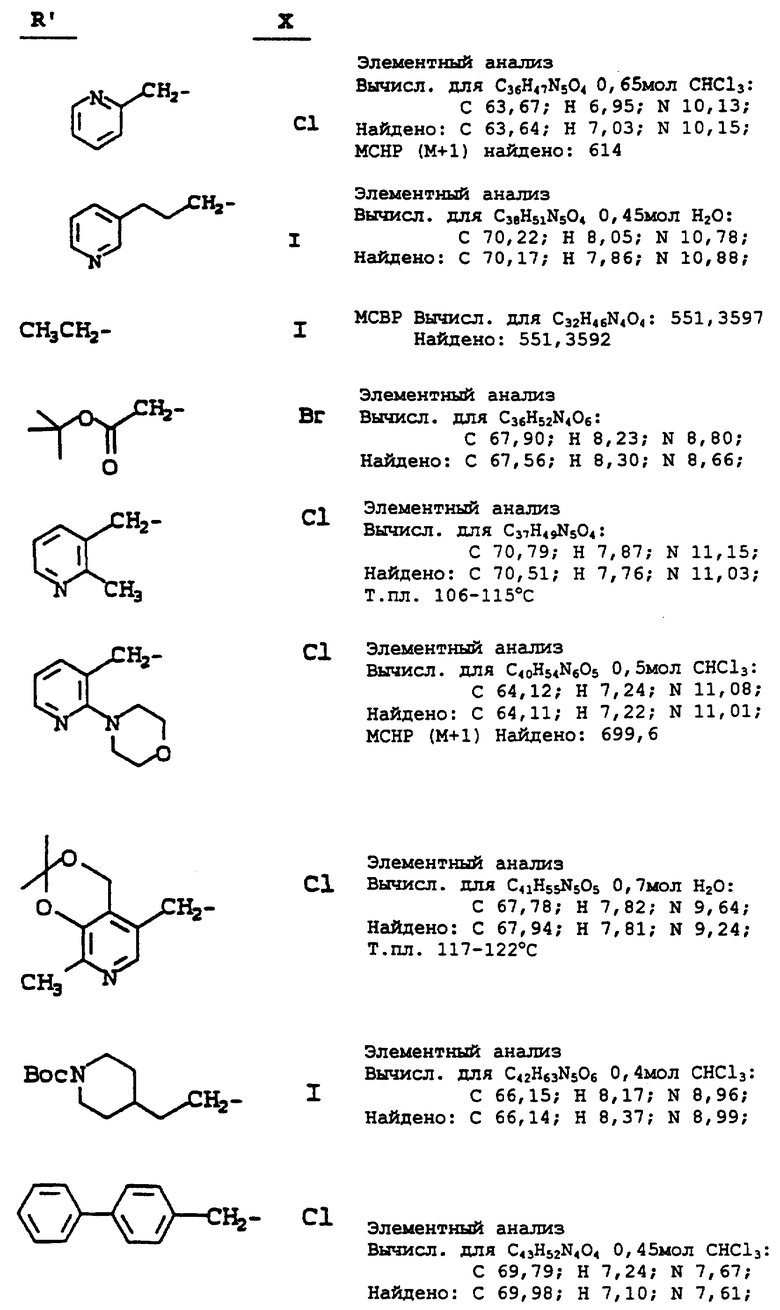
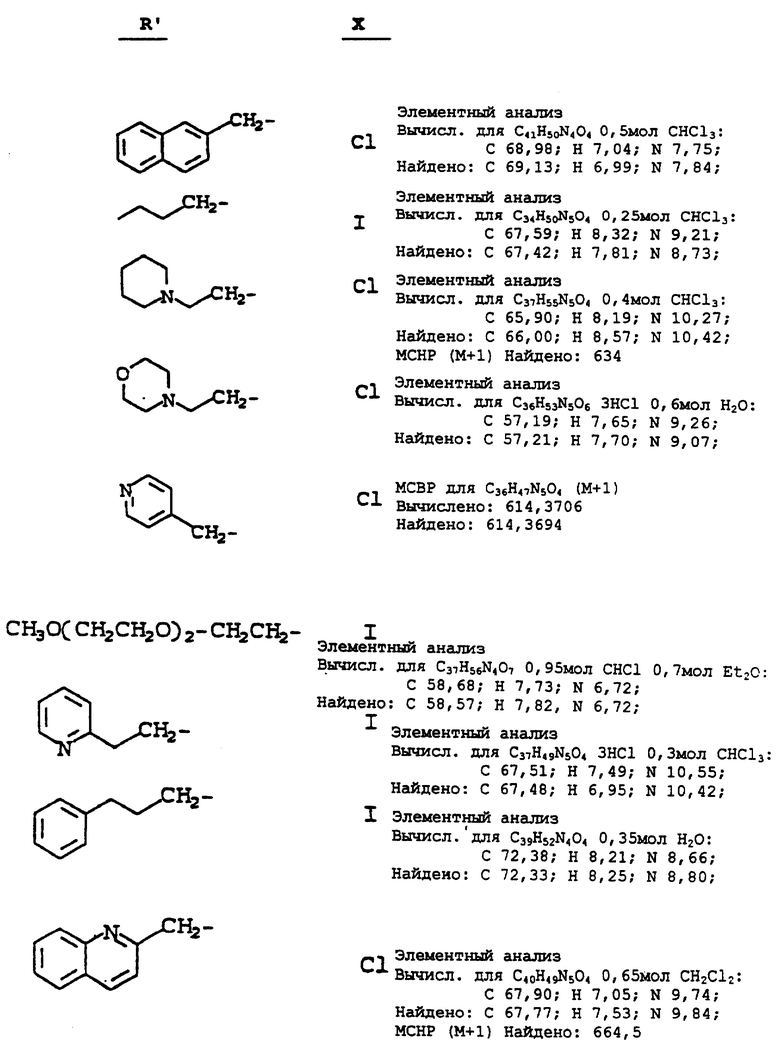
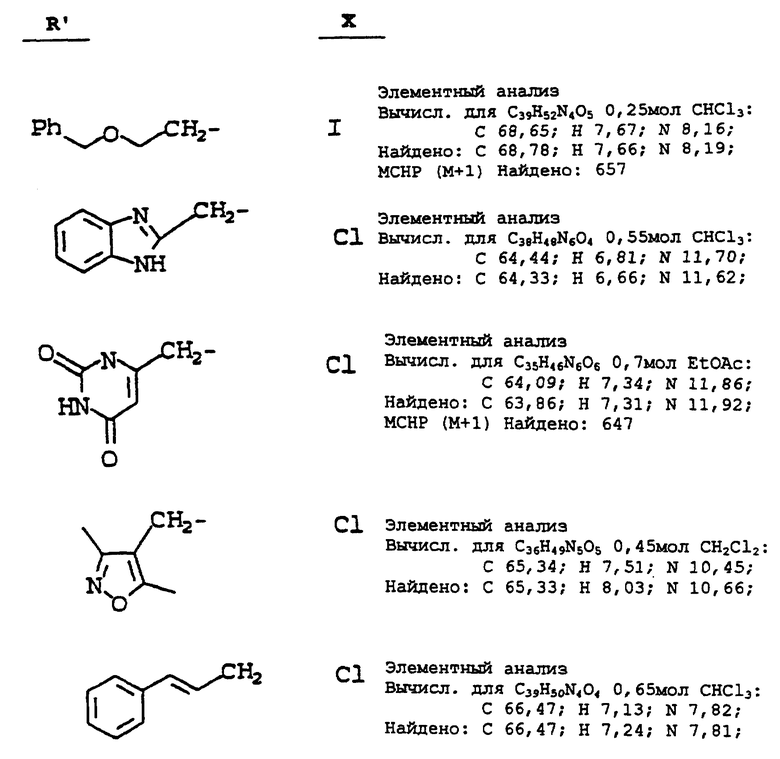
Используя в основном ту же самую методику, как описано в примере 15, но обрабатывая N-(2(R)-гидрокси-1(S)-инданил)-2(R)- фенилметил-4(S)-гидрокси-5-(1-(2-(S)-N'-(т-бутилкарбоксамидо)- пиперазинил))-пентанамид, используемый в ней (соединение i), алкилирующим агентом (ii) вместо 3-пиколилхлорида, используемого на стадии 10 в ней, получают продукты, определяемые формулой (iii) (см. схему A, представленную в конце описания).
ПРИМЕР 17
Получение дигидро-5(S)-(трет-бутилдиметилсилилоксиметил)-3(2H)-фуранона
К раствору 3,00 г (25,8 ммоль) дигидро-5(8)-(гидроксиметил)-2(3H)-фуранона, растворенного в 25 мл дихлорметана, добавляют 3,51 г (51,6 ммоль) имидазола и затем 4,67 г (31,0 ммоль) трет-бутилдиметилсилилхлорида. Реакционную смесь перемешивают при комнатной температуре в течение 8 ч и гасят 2 мл метанола. Смесь концентрируют до получения масла и затем разбавляют 150 мл эфира и промывают 5%-ной HCl (2 х 10 мл), насыщенным NaHCO3 (1 х 10 мл), водой (1 х 10 мл) и соляным раствором (1 х 10 мл), сушат над MgSO4 и концентрируют. Остаток очищают флеш-хроматографией (колонка 40 х 150 мм, градиентное элюирование, гексаны: этилацетат от 5:1 до 4:1), получая продукт в виде прозрачного масла.
1H ЯМР (300 МГц, CDCl3) δ 4,68-4,60 (м, 1H), 3,89 (дд, J = 3,3 и 11,3 Гц, 1H), 3,71 (дд, J = 3,2 и 11,3 Гц, 1H), 2,71-2,45 (м, 2H), 2,35-2,16 (м, 2H), 0,91 (с, 9H), 0,10 (с, 3H), 0,09 (с, 3H).
ПРИМЕР 18
Получение индинавир-сульфата
К раствору N-(2(R)-гидрокси-1(S)-инданил)-2(R)-фенилметил-4(S)- гидрокси-5-(1-(4-(3-пиридилметил)-2(S)-N'-(т-бутилкарбоксамидо)- пиперазинил-пентамида (соединение J, индинавир) (1,77 г, 2,805 ммоль) в 4 мл этанола (95%) добавляют концентрированную H2SO4 (0,149 мл, 2,805 ммоль) и смесь упаривают до пены. Остаток повторно растворяют в горячем изопропиловом спирте, медленно охлаждают до комнатной температуры, затем перемешивают при 0oC в течение 1 ч и фильтруют под потоком аргона. Белые кристаллы сушат под высоким вакуумом при 40oC, в результате получают кристаллическую соль - сульфат гидрат: т.пл. 150-153oC (размягчается при 135oC);
ПМР (400 МГц, CDCl3/CD3OD, 9/1) δ 1,19 (д, (CH3)2CHOH, J = 6,05 Гц), 1,30 (с, 9H), 1,51-1,57 (м, 1H), 1,90 (т, J = 11,2 Гц, 1H), 2,77-3,17 (м, 15H), 3,27 (дд, J = 9,1, 9,5 Гц, 1H), 3,60 (д, J = 12,1 Гц, 1H), 3,83 (д, J = 13,9 Гц, 1H), 3,90 (д, J = 13,9 Гц, 1H), 3,97-4,04 (м, (CH3)2CHOH), 4,05-4,11 (м, 2H), 4,30-4,33 (м, 1H), 5,19 (д, J = 4,9 Гц, 1H), 7,12-7,32 (м, 9H), 7,49 (дд, J = 2,3, 5,3 Гц, 1H), 8,01 (д, J = 7,7 Гц, 1H), 8,55 (д, J = 4,4 Гц, 1H), 8,76 (с, 1H). Анал. (C36H4N5O4• 1,0 H2SO4•1,0 H2O•0,2 C3H8O) C, H, N.
Испытание на ингибирование вируса
Индинавир (соединение J) испытывают на способность ингибировать распространение вируса в опыте острой инфекции. Опыт проводят с использованием MT-4 T-лимфоидных клеток человека, периферических мононуклеарных клеток крови человека (PBMC) или первичных моноцитов/макрофагов человека. MT-4 клетками (2,5 х 105 клеток на 1 мл) инфицируют при множественности заражения ≅ 0,01 и инкубируют в течение ночи в культуральной среде RPM1 1640, содержащей 10% (объем/объем) фетальной бычьей сыворотки. Инфицированные клетки затем дважды промывают в свежей культуральной среде и высевают на 96-ячеечные культуральные платы с концентрацией 5,0 х 104 клеток на ячейку. Серийные разбавления 1:2 соединения добавляют в ячейки. Испытываемые культуры дополнительно инкубируют 72 ч при 37oC, в течение которых оценивают содержание вируса.
Результаты (см. табл. 1) показывают, что соединение является эффективным ингибитором как адаптированных к T-лимфоидным клеткам вариантов ВИЧ-1, так и первичных вирусных изолятов. Чувствительность вируса к ингибитору протеазы не связана с чувствительностью к 3'-азидо-3'-деокситимидину (AZT) или ингибитору ненуклеозидной обратимой транскриптазы (RT). Индинавир эффективен также для профилактики распространения острой инфекции вируса иммунодефицита обезьяны (SIVmac251) и моноцитотропного варианта ВИЧ-1.
ВИЧ-линии: IIIb, MN и RF11, варианты, адаптированные к T-лимфоидным клеткам: 112, 139-8, 421, 5002, 5003, 5004, первичные изоляты; 139-8, высокоустойчивый к нескольким ненуклеозидным RT ингибиторам; 421, устойчивый к AZT; SF 162, моноцитотропный вариант.
95%-ную Ингибирующую концентрацию (IC95) определяют как концентрацию испытуемого соединения, при которой рост вируса подавляется по меньшей мере на 95% по сравнению с необработанными инфицированными контрольными культурами. Приведенные выше значения представляют собой результат многократных измерений.
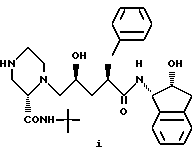
Способность индинавира предотвращать расщепление корового белка оценивают, используя линии персистентно-инфицированных T-лимфоидных клеток человека. При концентрациях ингибитора 0,5 и 12,0 мкМ количество зрелых вирионовых матриц (p17) и капсидных (p24) белков существенно снижается относительно количества, присутствующего в необработанных варионах (см. чертеж). Относительное количество p55 корового предшественника (core precursor) и промежуточных продуктов процессинга возрастает. Кроме того, ингибитор предотвращает введение в вирусные частицы зрелых RT субъединиц (p51 и p66) и зрелых вирусных ферментов интегразы (p31). Обе субъединицы образуются процессингом вирусной протеазы из полипротеинового предшественника.
На чертеже показано ингибирование ВИЧ-1 прекурсорного полипротеина посредством индинавира и влияние на процессинг удаления ингибитора. Частицы вируса анализируют сразу после выделения из клеток, обработанных ингибитором (зоны A и B), или через 6 ч после удаления ингибитора посредством диализа (зоны C и D).
Индинавир используют в концентрациях 12,0 мкМ (зоны A и C) или 0,5 мкМ (зоны B и L). Зона E представляет собой зрелый вирус из необработанных клеток. P24 - процессированный вирусный капсидный белок, Р1 представляет промежуточные продукты корового процессинга (the core processing intermediates), p66 и p51 - процессированные RT субъединицы и p31 - зрелый процессированный фермент интегразы вируса.
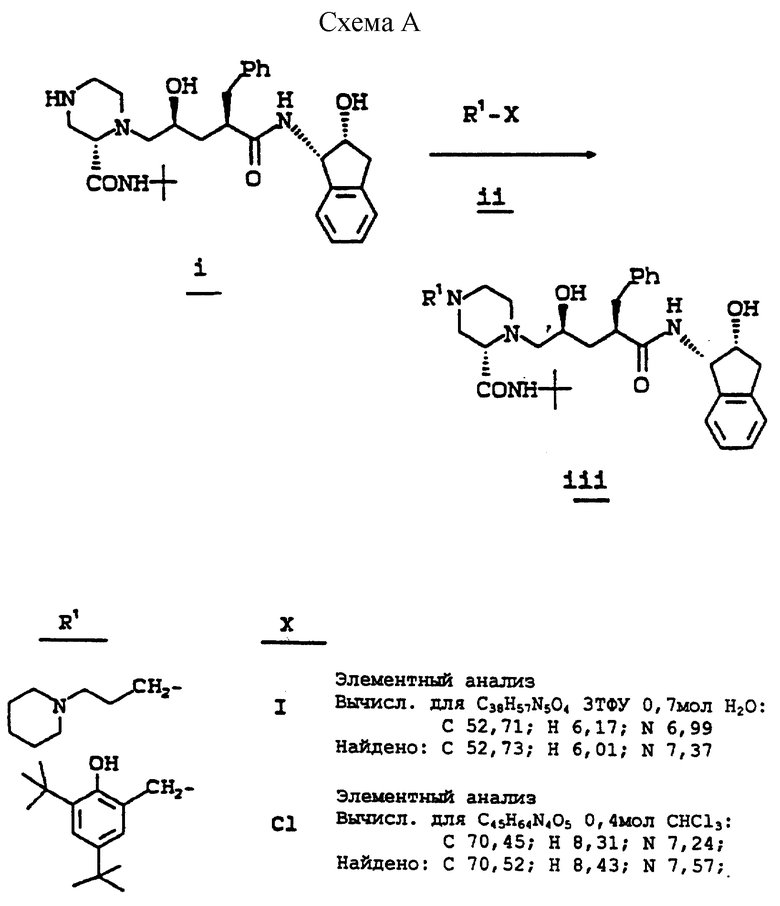


Описывается способ получения производных пиперазинилпентанамида общей формулы I, где R1 - C1-C4-алкил, незамещенный или замещенный одной или более группами, выбранными из (1) арила, незамещенного или замещенного одной или более группами, выбранными из C1-C4-алкила, гидрокси или арила, где арил - фенил или нафтил; (2) гетероцикла, незамещенного или замещенного одной или более группами, выбранными из C1-C4-алкила или бутилоксикарбонила, где гетероцикл означает 5-6-членную, насыщенную или ненасыщенную гетероциклическую кольцевую систему, возможно конденсированную с бензольным кольцом, содержащую в качестве гетероатомов один или два атома азота или азота и кислорода, заключающийся в том, что соединение формулы (i) подвергают взаимодействию с соединением формулы R1-Х, где Х выбран из Сl, Br или I. Способ позволяет получить новые соединения формулы I, которые используют для профилактики или лечения инфекции ВИЧ и при лечении СПИДа, в виде соединений, фармацевтически приемлемых солей, фармацевтических ингредиентов композиции, независимо от того, находятся они или нет в комбинации с другими противовирусными препаратами, иммуномодуляторами, антибиотиками или вакцинами. Описывается также промежуточнoе соединениe (i) для способа получения соединений I. 2 с. и 4 з.п. ф-лы, 1 ил., 2 табл.
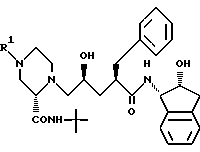
где R1 - C1-C4-алкил, незамещенный или замещенный одной или более группами, выбранными из: (1) арила, незамещенного или замещенного одной или более группами, выбранными из C1-C4-алкила, гидрокси или арила, где арил - фенил или нафтил; (2) гетероцикла, незамещенного или замещенного одной или более группами, выбранными из C1-C4-алкила или бутилоксикарбонила, где гетероцикл означает 5 - 6-членную, насыщенную или ненасыщенную, гетероциклическую кольцевую систему, возможно конденсированную с бензольным кольцом, содержащую в качестве гетероатомов один или два атома азота или азота и кислорода,
заключающийся в том, что соединение формулы i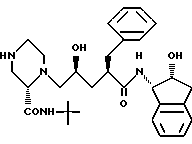
подвергают взаимодействию с соединением формулы
R1-Х,
где Х выбран из Сl, Br или I.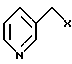
где Х определен в п.1,
с получением соединения формулы J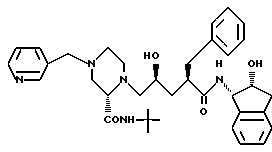
5. Способ по п.2, отличающийся тем, что Х представляет собой атом хлора.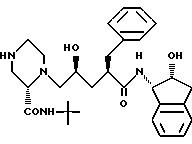
или его соль.
| Устройство для горячей вулканизации резинового низа обуви | 1944 |
|
SU68544A1 |
| Способ получения гетероциклических соединений | 1975 |
|
SU673173A3 |
| ДУГОВАЯ РТУТНАЯ ЛАМПА С ЛЮМИНЕСЦИРУЮЩИМ | 0 |
|
SU385350A1 |

