Настоящее изобретение относится к способу преобразования в условиях изомеризации линейных олефинов в соответствующие им разветвленные метил-изоолефины, к способу получения усовершенствованных катализаторов изомеризации олефина и способу регенерации этих катализаторов для поддержания высокой каталитической характеристики.
Предшествующий уровень техники.
Увеличивающаяся потребность в высокооктановом бензине, смешанном с низшими алифатическими алкиловыми эфирами, такими как октановые присадки и дополнительные топлива, создает значительную потребность в изоалкилэфирах, в особенности с C5 до C7 метил-, этил- и изопропил-t-алкиловых эфирах, таких как метил-t-бутиловый эфир, этил-t-бутиловый эфир, t-амил-метиловый эфир и t-амил-этиловый эфир. Следовательно, увеличивается потребность в соответствующих исходных изоалкеновых веществах, таких как изобутен, изоамилены и изогексены.
Для получения изоолефинов желательно преобразовать алкен, например нормальный бутен, в разветвленный метил-алкен, например изобутилен, посредством такого преобразования, как структурная изомеризация. Эти преобразованные изоалкены затем могут быть далее обработаны, например посредством полимеризации, этерификации или окисления для получения полезных продуктов. Нормальные алкены, содержащие четыре атома углерода (1-бутен, транс-2-бутен и цис-2-бутен) и пять атомов углерода (1-пентен, транс-2-пентен и цис-2-пентен), являются относительно недорогими исходными компонентами (сырьем). Традиционно бутены и амилены, включая в меньшей степени изобутилен и изоамилен, получаются в качестве побочных продуктов от нефтепереработки и нефтехимических процессов, например, в каталитических и термических устройствах крекинга. Обычно бутены получаются также из бутадиена с помощью выборочной гидрогенизации.
Известно, что цеолиты, как натуральные, так и синтетические, обладают каталитическими свойствами для многих процессов обработки углеводородов. Цеолитами обычно являются пористые кристаллические алюминосиликаты, имеющие определенную структуру с полостями, соединенными каналами. Эти полости и каналы во всем кристаллическом веществе обычно могут быть такого размера, который позволяет выборочное разделение углеводородов. Такое разделение углеводородов с помощью кристаллических алюминосиликатов существенно зависит от различия в молекулярных размерах. Поэтому эти вещества известны как "молекулярные сита" и их используют не только как катализаторы, но и для некоторых селективных адсорбционных процессов. Цеолитовые молекулярные сита более подробно рассматриваются в D.W.Breck, "Zeolite Molecular Sieves", Robert E. Krieger Publishing Company, Malabar, Florida (1984).
Вообще термин "цеолит" включает в себя широкое разнообразие и натуральных и синтетических содержащих положительные ионы кристаллических алюминосиликатных веществ, включая молекулярные сита. Они обычно характеризуются как кристаллические алюминосиликаты, которые содержат каркасы из SiO4 и AlO4 тетраэдров, в которых атомы алюминия и кремния соединяются в трехмерную решетку, разделенную атомами кислорода. Эта решетчатая (каркасная) структура содержит каналы или межсоединительные пустоты, которые заполнены такими катионами, как натрий, калий, аммоний, водород, магний, кальций, и молекулами воды. Вода может обратимо удаляться, например при нагревании, что делает кристаллическую решетку пригодной для использования в качестве каталитического вещества. Термин "цеолит", используемый в настоящем описании, не ограничивается кристаллическими алюминосиликатами. Используемый здесь указанный термин включает в себя силикоалюминофосфаты (SAPO), металлосодержащие алюминофосфаты (MeAPO и ELAPO) и металлосодержащие силикоалюминофосфаты (MeAPSO и ELAPSO). Группы MeAPO, MeAPSO, ELAPO и ELAPSO имеют дополнительные элементы, включенные в их кристаллическую решетку. Например, Me представляет собой Со, Fe, Mg, Mn или Zn, a El представляет собой Li, Be, Ga, Ge, As или Ti. Другим определением для обозначения веществ, применяемых в данном изобретении, может быть "молекулярное сито типа цеолита".
Развитие техники привело к появлению множества синтетических цеолитических кристаллических веществ. Кристаллические алюминосиликаты являются наиболее преобладающими и обозначаются буквами или другими подходящими символами. Различные цеолиты имеют специфическое обозначение, например Zeolite A (US-A-2,882,243), Zeolite X (US-A-2,882,244), Zeolite Y (US-A-3,130,007), Zeolite ZSM-5 (US-A-3,702,886), Zeolite ZSM-11 (US-A-3,709,979), Zeolite ZSM-12 (US-A-3,832,449), Zeolite ZSM-23 (US-A-4,076,842), Zeolite ZSM-35 (US-A-4,016,245 и 5,190,736), Zeolite ZSM-48 (US-A-4,375,573) и Zeolite NU-1 (US-A-4,060,590) и другие. Различные ферриэритовые цеолиты, включая водородную форму ферриэрита, описаны в патентах США US-A-3,933,974, 4,000,248 и 4,942,027 и патентах, упоминаемых в них. Катализаторы типа SAPO раскрыты в патенте США US-A-4,440,871. Катализаторы типа MeAPO раскрыты в патентах США US-A-4,544,143 и 4,567,029; катализаторы типа ELAPO - в патенте США US-A-4,500,651, а катализаторы типа ELAPSO - в патенте EP-A-159,624.
Раскрываются два общих класса катализаторов как особенно полезных для изомеризации линейного олефина в соответствующий разветвленный метил-изоолефин. Они включают в себя пористые некристаллические тугоплавкие катализаторы на основе оксидов и катализаторы на основе цеолита.
Примерами пористых некристаллических тугоплавких катализаторов на основе оксидов являются оксиды алюминия, например гамма- или эта- Al2O3, галогенизированные оксиды алюминия, оксиды алюминия, провзаимодействовавшие с кремнием, бором или цирконием, различные фосфаты и твердые фосфорные кислоты. Примеры таких катализаторов описаны в патентах США US-A-5,043,523, 3,531,542, 3,381,052, 3,444,096, 4,038,337, 3,663,453, Великобритании GB-A-2,060,424, и V.R.Choudhary and L.K.Doraswamy "lzomerization of n-Butene to Isobutene, I. Selection of Catalist by Group Screening", Journal of Catalysis, volume 23, pages 54-60, 1971. Примеры пористых некристаллических тугоплавких катализаторов описаны в патенте США US-A-4,434,315, который раскрывает в качестве катализатора пористый алюминий, ацилированный (подкисленный) критическим количеством оксидом кремния и содержащий от 5 частей на миллион до 2 весовых процентов палладия, хрома, никеля, марганца или серебра посредством пропитки. Использование перечисленных металлов приводит к большей гибкости каталитической регенерации. Все эти катализаторы быстро дезактивируются. В соответствии с примерами в GB-A-2,060,424 срок службы составляет не более 1 - 2 часов. Часто необходимо добавить водяной пар и галогенные компоненты для продления срока службы катализаторов. DE-A-3,000,650 устанавливает, что срок службы такими способами может быть увеличен примерно до 50 часов, хотя это все еще меньше желаемого срока.
С точки зрения катализаторов на основе цеолитов, наиболее широкое применение имеют те, которые содержат цеолиты с большими порами или те, которые имеют двух- или более мерные межсоединительные каналы. Примеры катализаторов на основе цеолитов, имеющих двух- или более мерные межсоединительные каналы и используемых совместно с каталитическими металлами, приведены в патентах США US-A-4,435,311 (с платиной и палладием), US-A-4,503,282 и 5,227,569 (пропитанный или ионно-обменный с металлами, содержащимися в 7 группе). Примеры катализаторов на основе цеолитов с большими порами, используемые совместно с каталитическими металлами, приведены в патенте США US-A-5,227,569 (пропитанный или ионно-обменный с металлами, содержащимися в 7 группе) и US-A-4,392,003 (с галлием).
Более новый патент EP-A-523,838 раскрывает способ для структурной изомеризации линейного олефина в соответствующий ему разветвленный метил-изоолефин, используя в качестве катализатора цеолит с одно- или более одномерной пористой структурой, имеющей размер пор, достаточно малый для замедления димеризации побочного продукта и образования коксовых отложений в пористой структуре и достаточно большой для входа линейного олефина и образования разветвленного метил-изоолефина. EP-A-5396015 раскрывает способ изомеризации скелетной конструкции н-алкенов, используя алюминофосфатное молекулярное сито с порами от 0,4 до 0,6 нм. Установлено, что при применении этих пористых катализаторов в них образуются коксовые отложения, которые снижают их эффективность. Для восстановления их эффективности катализаторы могут быть регенерированы при повышенных температурах посредством контакта с кислородом. Такой процесс регенерации при многократном повторении может иметь неблагоприятное воздействие на срок службы катализатора и избирательность.
Типовая температура регенерации цеолитического катализатора раскрыта в "Chemystry Of Catalitic Processes" B.C.Gates, J.R.Katzer and G.C.A.Schuit, McGraw-Hill Book Company, New-York (1979) pp. 1-5 и составляет 650oC-750oC. Сейчас имеются тенденции к повышению температур регенерации. Например, температура регенерации до 850oC используется при промышленной регенерации цеолитовых катализаторов, используемых в жидком каталитическом крекинге (FCC). J.Biaswas and I.E.Maxwell "Applied Catalisis", 63 (1990), 197-258.
Однако, установлено, что использование таких высоких температур регенерации, которые применяются в FCC, приводят к низкой характеристике изомеризации олефины (низкой избирательности) для такого катализатора на основе цеолита со средним размером пор, который описан в EP-A-523,838. В соответствии с патентом US-A-5,043,523 для катализаторов модифицированного оксида алюминия описанного выше типа рекомендуется температура регенерации от 550oC до 600oC. Сообщалось, что катализатор из модифицированного оксида алюминия не показал снижения активности после 10 циклов регенерации при 575oC по способу A в примере 29. Однако, установлено, что цеолитические катализаторы с одномерной или более, чем одномерная, пористой структурой, имеющие размер пор малый, но достаточным для замедления димеризации побочного продукта и образования коксовых отложений в пористой структуре и достаточно большой для входа линейного олефина и образования разветвленного метил-изоолефина, например ферриэрит, ZSM-22 и ZSM-23, имеют тенденцию к потере избирательности для образования изоолефинов, когда подвергаются температурам выше 565oC в течение периода, используемого в упомянутых выше процессах регенерации.
Внедрение в промышленность способа изомеризации для образования изоолефинов из линейных олефинов будет далее затрудняться из-за более длинных времен регенерации, сравнимых со сроком службы.
Поэтому целью настоящего изобретения является создание способа с катализатором на основе цеолита со средним размером пор для структурной изомеризации линейного олефина в соответствующий ему разветвленный метил-изоолефин с увеличенным сроком службы и/или сокращенным временем регенерации и с увеличенным общим выходом продукта.
Раскрытие изобретения.
В способе согласно настоящему изобретению один или более линейных олефинов преобразуются при условиях изомеризации в соответствующий им разветвленный метил-изоолефин посредством контакта при температуре от 200oC до 650oC с катализатором изомеризации, содержащим (i) по меньшей мере один цеолит, имеющим только одномерную пористую структуру с размером пор достаточно малый для замедления димеризации побочного продукта и образования коксовых отложений и достаточно большой для входа линейного олефина и образования разветвленного метил-изоолефина, (ii) связующего и (iii) до 15% по весу металла, способствующего окислению коксовых отложений. После рабочего периода, когда достаточное количество коксовых отложений находится в катализаторе, которое сокращает активность и/или избирательность катализатора, этот катализатор выделяется из подачи олефина и контактирует при температуре менее чем приблизительно 565oC с кислородсодержащим газом, с парциальным давлением кислорода приблизительно от 0,001 до приблизительно 40 атмосфер в течение достаточного времени для выгорания в значительной степени коксовых отложений, т.е. для регенерации катализатора. После регенерации способ изомеризации продолжается.
Улучшенная рабочая характеристика катализатора достигается тогда, когда катализатор готовится с помощью смешивания до пастообразного состояния вместе с порошком цеолита, порошком оксида алюминия, водой, пептизирующим количеством кислоты и металлом, способствующим окислению коксовых отложений, формирования смеси в виде гранулы, и спекания гранулы при температуре от 300oC до 700oC.
Особенно желательно использование палладия и/или платины в качестве металла, способствующего окислению коксовых отложений.
Краткое описание чертежей.
В дальнейшем изобретение поясняется конкретными вариантами его воплощения со ссылками на прилагаемый чертеж, который является графиком избирательности к изобутилену, полученным за ряд регенераций с катализаторами A (без палладия) и B (с палладием).
Описание предпочтительных вариантов воплощения.
Установлено, что способ структурной изомеризации линейного олефина в соответствующий ему разветвленный метил-изоолефин со сроком службы более длинным, чем время регенерации, может быть осуществлен с помощью внедрения в катализаторы некоторого металла, способствующего окислению коксовых отложений в эффективном количестве для содействия выгоранию коксовых отложений из катализатора, и выполнения регенерации при температуре, меньшей чем примерно 565oC, пока коксовые отложения не выгорят в значительной степени. С помощью введения металла, способствующего окислению коксовых отложений, и использования специального парциального давления кислорода, было установлено, что регенерация с цеолитом с порами среднего размера может быть эффективна при более низких температурах, которые в основном не оказывают отрицательного воздействия на производительность катализатора. К тому же было установлено, что приготовление катализатора с помощью скрепления и спекания смешанных до пастообразного состояния порошка цеолита, алюминиевого порошка, воды, пептизирующего вещества и металла, способствующего окислению коксовых отложений, обеспечивает получение катализатора изомеризации олефина с повышенной производительностью.
Катализаторы для изомеризации. Изомеризующие катализаторы, применяемые в способе, содержат цеолит, определенный далее, связующее вещество и металл, способствующий окислению коксовых отложений.
Цеолит, используемый в катализаторе для изомеризации, согласно настоящему изобретению содержит цеолит, имеющий одномерную пористую структуру с размером пор, значительно большим, чем 0,42 нм и меньше, чем примерно 0,7 нм. Цеолиты с таким специфическим размером пор обычно относятся к цеолитам со средним или промежуточным размером пор и имеют обычно 10-элементную (или сложенную 12-элементную) кольцевую канальную структуру в одном направлении и 9-элементную или менее (малая пористость) в других направлениях. Для целей настоящего изобретения рассматривается одномерная пористая структура, в которой каналы, имеющие желаемый размер пор, не связаны с другими каналами такой же или большей размерности, она может также рассматриваться с другой стороны как канальная, пористая структура (см. US-A-3,864,283) или одномерное сито.
Цеолитовый катализатор предпочтительно содержит в основном только цеолиты со специфическим размером пор в одном измерении. Цеолиты, имеющие размер пор более 0,7 нм, являются чувствительными к нежелательным ароматизации, олигомеризации, алкилированию, коксообразованию и образованию побочных продуктов. Далее, двух и трехмерные цеолиты, имеющие размер пор более 0,42 нм в двух или более направлениях, разрешают димеризацию и тримеризацию алкена. Следовательно, цеолиты, имеющие диаметр пор более 0,7 нм в любом направлении, или имеющие двух- или трехмерную пористую структуру, в которой любые два направления имеют размер пор более 0,42 нм, обычно исключаются. Цеолиты, которые содержат только малые поры (менее 0,42 нм), не позволяют диффундировать разветвленному метил-изоолефиновому продукту.
Примеры цеолитов, которые могут быть применены в процессах согласно изобретению, имеющие одномерные пористые структуры с размерами пор от примерно 0,42 нм и 0,7 нм, включают в себя водородную форму ферриэрита, AlPO-31, SAPO-11, SAPO-41, FU-9, NU-10, NU-23, ZSM-12, ZSM-22, ZSM-23, ZSM-35, ZSM-48, ZSM-50, ZSM-57, MeAPO-11, MeAPO-31, MeAPO-41, MeAPSO-11, MeAPSO-31 и MeAPSO-41, MeAPSO-46, ELAPO-11, ELAPO-31, ELAPO-41, ELAPSO-11, ELAPSO-31 и ELAPSO-41, лаумонтита, канкринита, офретита, водородной формы стилбита, магниевой или кальциевой формы морденита и партеита. Другие структуры этих решеток, известные под иными названиями, рассматриваются как эквивалентные. Обзор, описывающий состав решеток многих цеолитов, приводится в New Developments in Zeolite Science Technology, "Aluminophosphate Molecular Sieves and the Periodic Table" Flanigen et al. (Kodansha Ltd., Tokyo, Japan 1986).
Многим натуральным цеолитам, таким как ферриэрит, геоландит и стилбит, свойственна одномерная пористая структура с диаметром пор, равным или немногим меньше 0,42 нм. Эти же цеолиты могут быть преобразованы в цеолиты с желаемым большим размером пор с помощью удаления связанного щелочного или щелочно-земельного металла известными способами, такими как замещением ионом аммония, необязательным спеканием, для получения цеолита в его водородной форме, см. например патенты США US-A-4,795,623 и 4,942,027. Замена связанного щелочного или щелочно-земельного металла водородной формой соответственно увеличивает диаметр пор. Понятно, что диаметр пор или упоминаемый здесь термин "размер" должен означать эффективный диаметр пор или размер для диффузии. С другой стороны, натуральные цеолиты со слишком большим размером пор, например мерденит, могут быть изменены заменой щелочного металла на больший ион, например большего щелочно-земельного металла для уменьшения размера пор.
Особенно предпочтительными цеолитами являются те, которые имеют ферриэритовую изотипическую структуру (или гомеотипическую). См. "Atlas of Zeolite Structure Types" by W.M.Meier and D.H.OIson, Butterworth-Heinemann, 1992, p. 98. Выдающиеся структурные свойства ферриэрита, обнаруженные с помощью рентгеновской кристаллографии, заключаются в параллельных каналах в алюминосиликатной решетке почти эллиптической формы в поперечном сечении. Примеры таких цеолитов, имеющих ферриэритовую изотипическую каркасную структуру, включают в себя натуральные и синтетический ферриэриты (может быть орторомбическим или моноклиническим), Sr-D, FU-9 (EP-B-55,529), ISI-6 (US-A-4,578,259), NU-23 (EP-A-103,981), ZSM-35 (US-A-4,016,245) и ZSM-38 (US-A-4,375,573). ZSM-22 и ZSM-23 также являются полезными цеолитами для изготовления катализаторов. Водородная форма ферриэрита (H-ферриэрит) является наиболее предпочтительным цеолитом и рассматривается как имеющий в основном одномерную структуру, эллиптическую форму пор (< 0,54 нм и > 0,42 нм), достаточно большие, чтобы разрешить вход линейного олефина и диффузии разветвленного метил-изоолефина и достаточно малого для замедления образования коксовых отложений. Способы изготовления различных Н-ферриэритов раскрыты в US-A-4,251,499, 4,795,623 и 4,942,027.
Виды цеолитов, которые неприемлемы для процессов согласно настоящему изобретению, включают в себя ZSM-5, ZSM-10, Бета, эрионит, цеолит Y, водородная форма морденита и фаугасит.
Цеолиты, используемые в настоящем изобретении, объединяются с тугоплавкими оксидами, которые служат в качестве связующего вещества. Подходящие тугоплавкие оксиды включают в себя натуральные отбеливающие глины, такие как бентонит, монтмориллонит, аттапульгит и каолин; оксид алюминия; оксид кремния; алюмино-силикат; гидратированный алюминий; двуокись титана, двуокись циркония и их смеси. Весовое соотношение цеолита и связующего вещества в нужных пределах изменяются приблизительно от 60:40 до приблизительно 99,5: 0,5, предпочтительно - приблизительно от 75:25 до приблизительно 99:1, более предпочтительно - приблизительно от 80:20 до приблизительно 98:2, наиболее предпочтительно - приблизительно от 85:15 до приблизительно 95:5 измеренном как оксид в окончательном катализаторе. Предпочтительным связующим веществом является оксид алюминия.
Связующие, полезные при изготовлении катализаторов, могут быть любыми из обычных алюминийсодержащих связующих, применяемых для изготовления катализаторов и включают в себя, например, оксиды алюминия, алюминосиликаты и отбеливающие глины. Термин "связующее, содержащее оксид алюминия", используемое в настоящем изобретении, включает в себя любой из предшествующих оксидов алюминия, в том числе гидратированные формы оксида алюминия, такие как байерит, боемин и гиббсит, который при спекании преобразуется в оксид алюминия (Al2O3). Предпочтительными алюминосиликатами являются аморфные, например алюминосиликатные гели и коллоидные растворы. Примерами, не ограничивающими изобретение, являются соответствующие отбеливающие глины, включая бентонит, гекторит, каолин и аттапульгит. Связующие вещества поставляются в любой подходящей форме - порошках, суспензиях, гелях или коллоидных растворах. Когда связующие вещества поставляются в виде суспензий, гелей или коллоидных растворов, по меньшей мере часть воды, используемая на стадии образования пасты, будет составлять часть суспензии, геля или коллоидного раствора.
Предпочтительными связующими веществами являются оксиды алюминия, например псевдобоэмит, гамма- и байерит оксиды алюминия. Эти связующие уже являются коммерчески доступными. Фирмы LaRoche Chemicals через свою группу VERSAL® оксидов алюминия и Vista Chemical Company через свою CATAPAL® группу оксидов алюминия поставляют подходящие порошки оксида алюминия, которые могут быть использованы для изготовления натуральных катализаторов. Предпочтительными связующими оксидами алюминия, используемыми в изготовлении катализатора, в особенности когда применяется экструзия, являются порошки оксида алюминия с высокой дисперсностью. Такие высокодисперсные оксиды алюминия, например обычный CATAPAL®D, имеет дисперсность более 50% в водно-кислотной дисперсии, имеющей содержание кислоты 0,4 мг эквивалента кислоты (уксусной) на грамм Al2O3.
Металлы, входящие в состав катализатора, являются металлом, который способствует окислению коксовых отложений в присутствии кислорода при температуре выше, чем, скажем 250oC. В то время как термин "металл" используется в данном случае как катализатор окисления, специалистам понятно, что эти металлы не обязательно будут находиться в состоянии с валентностью, равной нулю, а во многих случаях будут находится в состоянии с более высокой степенью окисления. Таким образом, "металл" может охватывать оксиды металла, также как и металл.
Предпочтительным используемым металлом, способствующим окислению коксовых отложений, является переходный или редкоземельный металл. Более предпочтительный металл, способствующий окислению коксовых отложений, выбирается из группы IB, VB, VIB, VIIB и VIII рядов переходных металлов Периодической таблицы (CAS версия). Особенно предпочтительными являются Pd, Pt, Ni, Co, Mn, Ag и Cr. Наиболее предпочтительными являются благородные металлы, такие как палладий и/или платина.
Количество введенного металла(ов), способствующего окислению коксовых отложений, обычно варьируется до 15% по весу, предпочтительно с нижней границей, равной 5 частям на миллион, а верхней границей - приблизительно до 15% по весу, предпочтительно до 18% по весу металла от общего веса катализатора. Когда используются благородные металлы, такие как палладий и/или платина, предпочтительно введение меньшего, а не большего количества металлов в цеолит/связующее. Предпочтительно, чтобы благородные металлы присутствовали в количестве от 5 частей на миллион до приблизительно 2 %, предпочтительно - около 1%, более предпочтительно - около 3000 частей на миллион, наиболее предпочтительно - около 2000 частей на миллион по весу основного металла в конечном полученном катализаторе. В наиболее предпочтительном варианте воплощения предпочтительно использовать благородный металл в количестве, достаточном для того, чтобы способствовать регенерации без ухудшения качественной характеристики катализатора, обычно приблизительно от 30 частей на миллион до 100 частей на миллион. Большие количества платины и/или палладия, скажем, больше 2% по весу, могут иметь неблагоприятное воздействие на срок службы, активность изомеризации олефина и/или избирательность катализатора.
Катализаторы могут быть приготовлены различными способами. В одном варианте воплощения цеолит объединяется со связующим и формируется в виде гранул с помощью, например, сжатия или экструзии, а каталитический металл добавляется с помощью пропитывания гранулы металлсодержащим раствором. После пропитки катализатор может быть спечен при температуре приблизительно от 200oC до 700oC, предпочтительно приблизительно от 200oC до 650oC, более предпочтительно - от 300oC до 600oC.
В предпочтительном варианте воплощения порошок цеолита и порошок оксида алюминия смешиваются, скажем, посредством перемешивания пасты с водой и одним или более компонентов каталитического металла, а полученная смесь формируется в виде гранулы. Обнаружено, что катализаторы, приготовленные посредством перемешивания пасты, имеют большую производительность изомеризации олефина, чем катализатор, приготовленный посредством пропитывания. Термин "паста" используется для обозначения смешивания порошков, к которому добавляется достаточное количество воды для образования густой пасты, и в которой смешивание сопровождается сопутствующим разделением пасты. Для образования пасты могут быть использованы коммерчески доступные смесители, например Lancaster Mix Muller и Simpson Mix Muller.
Предпочтительно, чтобы гранула формировалась в процессе экструзии. При использовании экструзии к смеси могут быть добавлены пептизирующая кислота (кислоты), например, азотная, уксусная, лимонная или их смеси; необязательно, но могут быть использованы целлюлозные ускорители экструзии, например гидроксипропил метилцеллюлоза METHOCEL® F4M. Количества используемых пептизирующих кислот могут быть определены экспериментально, эти количества будут обеспечивать пластичный, пригодный к экстудированию материал. Термин "гранула" обозначает в данном случае любую форму до тех пор, пока вещества объединены.
Эти гранулы спекаются при температуре от нижнего предела 200oC, предпочтительно от 300oC, более предпочтительно от 450oC до верхнего предела 700oC, предпочтительно до 600oC, более предпочтительно до 525oC.
Поток подачи углеводорода. Подаваемый углеводород, применяемый в настоящем изобретении, содержит один или более линейных алкенов, содержащих по меньшей мере 4, обычно от 4 до 10, атомов углерода. Предполагается также, что для целей настоящего изобретения линейными алкенами являются те, которые содержат линейный сегмент алкена с 4-10 атомами углерода, которые могут проникать через цеолитовый катализатор на расстояние, эффективное для разрешения изомеризации. Таким образом, целая молекула не должна быть достаточно малой, чтобы целиком заполнить пористую структуру катализатора. Предпочтительный поток содержит бутилен и/или амилен.
Используемый здесь термин "н-бутилен" включает в себя все формы н-бутилена, например 1-бутен и 2-бутен, или транс-2-бутен или цис-2-бутен и их смеси. Используемые здесь термины "н-амилен" или "н-пентен" включают в себя 1-пентен, цис- или транс-2-пентен или их смеси. Н-бутилен или н-амилен, используемый в способе настоящего изобретения, обычно присутствует в других веществах, например в других углеводородах. Таким образом, подаваемый поток, используемый в способе согласно настоящему изобретению, содержащий н-бутилен или н-амиилен, также может содержать другие углеводороды, например, алканы, другие олефины, ди-олефины, такие как бутадиен, ароматические соединения, водород и инертные газы. Обычно подаваемый поток н-бутена, применяемый в настоящем изобретении, содержит от 10 до 100% по весу н-бутена. Например, фракционированный поток углеводорода после жидкокаталитического крекинга обычно содержит приблизительно от 20 до 60 весовых процентов нормального бутена, а углеводородный поток из устройства обработки эфиров, например метил-трет-бутилового эфира (МТВЕ), обычно содержит от 40 до 100 весовых процента н-бутилена. Потоки от крекинг-установок и установок каталитического крекинга также в основном содержат алканы, скажем до 80% по весу. Эти подаваемые потоки также могут быть образованы выборочно гидрогенизирующим бутадиеном для образования линейных бутенов.
Используемый здесь термин "алкен", с другой стороны, может быть заменен на "олефин"; термин "линейный" - на "нормальный"; а термин "изоолефин" - на "разветвленный метил-изоолефин" Аналогично, бутен и бутилен относятся к одному и тому же алкену с 4 атомами углерода; пентен и амилен - к одному и тому же алкену с 5 атомами углерода.
Условия изомеризации. В процессах согласно настоящему изобретению поток углеводородов, содержащий по меньшей мере один линейный олефин, контактирует с каталитическим цеолитом при условиях изомеризации. Обычно поток углеводородов контактирует в описанным выше цеолитическим катализатором в газообразной фазе при соответствующих температуре, давлении и объемной скорости реакции. Обычно соответствующие условия реакции включают в себя температуру приблизительно от 200oC до 650oC, предпочтительно от 340oC до 600oC, парциальное давление олефина выше примерно 0,5 атмосферы и суммарное давление от 0,5 до 10,0 атмосфер или выше, молярное отношение водород/углеводород от 0 до 30 или выше (т.е. присутствие водорода необязательно), в значительной степени свободным от воды (т.е. менее примерно 2% по весу потока) и количества углеводорода на единицу веса катализатора в час (WHSV-объемная скорость) приблизительно от 0,5 до 100 час-1. Эти реагирующие потоки могут содержать нереагирующие растворители, такие как алканы. Водород может быть добавлен прямо в подаваемый поток перед введением его в зону изомеризации или прямо в зону изомеризации.
Предпочтительная температура реакции будет зависеть от ряда факторов, таких как давление, объемная скорость (WHSV) и состав потока. Олефины с более низким молекулярным весом, такие как бутены, изомеризуются лучше при температуре приблизительно от 200oC до 650oC, в то время как олефины с более высоким молекулярным весом лучше изомеризуются при более низких температурах. Пентены лучше изомеризуются при температуре приблизительно от 200oC до 550oC, а гексены лучше изомеризуются при температуре приблизительно от 200oC до 500oC. Смешанные бутены и пентены лучше изомеризуются при температуре приблизительно от 200oC до 600oC, а смешанные пентены и гексены приблизительно от 200oC до 525oC. Использование низкой температуры может быть преимуществом, когда олефин легко расщепляется на легкие нежелательные вещества при высоких температурах. Возможно также получить более высокую концентрацию желаемых продуктов при более низких температурах вследствие того факта, что более высокие равновесные концентрации разветвленных олефинов возможны при более низких температурах.
В типовой схеме способа изомеризации бутена пары бутена контактируют с таким катализатором в реакторе при температуре примерно от 320oC до 650oC, парциальном давлении олефина примерно от 351,62 г/см2 до 3516,2 г/см2, а общем давлении от 1054,86 г/см2 до 7032,4 г/см2, объемной скорости (WHSV) олефина приблизительно от 0,5 до 50 час-1. Предпочтительные условия изомеризации выполняются при температуре приблизительно от 320oC до 450oC при атмосферном давлении и объемной скорости (WHSV) олефина приблизительно от 2 до 25 час-1, более предпочтительно - приблизительно от 2 до 15 час-1.
В типовой схеме способа изомеризации пентена пары пентена контактируют с таким катализатором в реакторе при температуре примерно от 250oC до 550oC, парциальном давлении олефина примерно от 210,97 г/см2 до 7032,4 г/см2, общем давлении от 1054,86 г/см2 до 7032,4 г/см2, объемной скорости (WHSV) олефина приблизительно от 1 до 100 час-1. Предпочтительные условия изомеризации выполняются при температуре приблизительно от 300oC до 425oC при атмосферном давлении и объемной скорости олефина (WHSV) приблизительно от 2 до 25 час-1 более предпочтительно - приблизительно от 2 до 40 час-1.
Для смешанных потоков могут быть использованы промежуточные условия между процессами изомеризации пентена и бутена в зависимости от желаемой смеси продуктов.
Способ согласно настоящему изобретению может использовать комбинацию цеолитов с одно- или более мерной пористой структурой, имеющей размер пор, достаточно малый для затруднения димеризации побочных продуктов и образования коксовых отложений с пористой структурой, достаточно большой для входа линейного(ых) олефина(ов) и диффузии изоолефинового(ых) продукта(ов). Эти комбинации могут включать в себя гранулы смешанных цеолитов и образование сложенных слоев катализаторов, таких как ZSM-22 и/или ZSM-23 на ферриэрите, ферриэрит на ZSM-22 и/или ZSM-23 и ZSM-22 на ZSM-23. Сложенные слоями катализаторы могут быть одинаковой формы и/или размера или различной формы и/или размера, например 0,318-сантиметровые трехлопастной формы на 0,08-сантиметровые цилиндрической формы.
Условия регенерации. Во время процесса на поверхности катализатора будут образовываться коксовые отложения. Коксовые отложения могут расти на внешней поверхности катализатора и/или на поверхности внутренних каналов и/или пор катализатора. Поэтому полезно регенерировать катализатор, когда по меньшей мере, скажем 2%, предпочтительно - по меньшей мере 5%, более предпочтительно - по меньшей мере 10%, но менее 30%, предпочтительно до 25%, наиболее предпочтительно - до 20% по весу образовалось коксовых отложений (в пересчете на катализатор без коксовых отложений).
Когда рост коксовых отложений на катализаторе достигает точки, при которой необходима регенерация, подача углеводородов к катализатору прекращается, любые отгоняемые (десорбируемые) углеводороды на поверхности катализатора удаляются горячим газом (например, азотом и/или водородом), а катализатор затем регенерируется посредством горячей обработки его кислородсодержащим газом. Отгонка (десорбция) может быть выполнена при высоком давлении, в вакууме или посредством циклического увеличения и снижения давления в реакторе. Десорбция может быть объединена с регенерацией. Например, в процессе изомеризации бутена подача бутена может быть остановлена и заменена на подачу водорода для десорбции отложений, а затем заменена на поток кислородсодержащего газа для регенерации.
Регенерация предпочтительно выполняется при температуре по меньшей мере 250oC. Важно, чтобы температура во время регенерации оставалась менее приблизительно 565oC, предпочтительно менее или равной 530oC, более предпочтительно - менее или равной приблизительно 500oC, наиболее предпочтительно - менее или равной приблизительно 490oC в течение времени, эффективного для существенного выгорания коксовых отложений на поверхности катализатора. Считается, что коксовые отложения в значительной степени выгорели, если более 80% по весу коксовых отложений удалено по сравнению с их начальным уровнем, когда изомеризация олефина или подача линейного олефина (далее "весовой процент от начальных коксовых отложений") остановлены. Предпочтительно, чтобы регенерация выполнялась до выгорания всех коксовых отложений. Считается, что все коксовые отложения выгорели, когда более 95 весовых процентов от начальных коксовых отложений удалены. Температуры регенерации измеряются как средняя температура среды реактора (т.е. температуры объемной газовой фазы) и случающихся время от времени пиков в течение короткого времени или внутри части окружающей реактор среды в процессе согласно настоящему изобретению. Коксовые отложения рассматриваются здесь как любые углеродсодержащие окисляемые вещества. Уровень коксовых отложений может быть измерен удобным тестом, описываемым ниже.
Предпочтительные условия регенерации включают в себя давление системы, изменяющиеся в пределах от более 1 атмосферы, предпочтительно от 1406.5 г/см2 избыточного давления до 105486.04 г/см2 избыточного давления, более предпочтительно - до 70324.03 г/см2 избыточного давления. Такое высокое давление в системе приводит к большему парциальному давлению кислорода, в то же время поддерживая соотношение кислорода к инертному газу, используемому для поглощения теплоты.
Отношение парциального давления кислорода к общему давлению системы обычно составляет приблизительно от 0,001 атмосферы, предпочтительно от 0,01 атмосферы до приблизительно 40 атмосфер, предпочтительно до 10 атмосфер. Предпочтительным кислородсодержащим газом является воздух, хотя воздух может быть разбавлен дополнительно азотом, диоксидом углерода или продуктами сгорания углеводорода.
В процессе регенерации важно избежать неуправляемого выделения тепла, что приведет к повышению температуры выше желаемого максимума температуры регенерации в реакторе. Это может иметь место при соответствующем повышении температуры или увеличении концентрации кислорода в кислородсодержащем газе или обоими этими причинами во время процесса регенерации для того, чтобы получить устойчивое горение коксовых отложений. Предпочтительно регенерация выполняется в течение времени, достаточного для выгорания в значительной степени всех коксовых отложений, скажем до уровня менее приблизительно 0,1 весового процента от катализатора. Время обычно будет варьироваться примерно от 5 часов до 200 часов, предпочтительно от 10 до 100 часов. Согласно изобретению предпочтительно, чтобы содержание воды не увеличивалось в процессе регенерации свыше уровня ее содержания в воздухе и/или газе для регенерации, используемом в процессе регенерации.
Способ регенерации согласно изобретению допускает плавную и управляемую каталитическую регенерацию. Температура регенерации может поддерживаться и управляться посредством регенерирующего катализатора для изомеризации, содержащего металл(ы), способствующий(е) окислению коксовых отложений при повышенных значениях давления.
Процессы изомеризации и/или регенерации могут быть выполнены совместно в реакторах с упакованным слоем, с фиксированным слоем, псевдоожиженным слоем или подвижным слоем катализатора. Слой катализатора может двигаться вверх или вниз. Способ изомеризации и процесс регенерации могут быть выполнены в одном и том же слое или в различных слоях. Для процесса регенерации может быть полезна непрерывная регенерация. Регенерация может быть выполнена также ex situ.
В предпочтительном варианте воплощения изобретение может быть определено как способ структурной изомеризации линейного олефина, содержащего по меньшей мере 4 атома углерода, в соответствующий ему разветвленный метил-изоолефин, который содержит стадии:
(а) контактирование при температуре от 200oC до 650oC потока подаваемого углеводорода, содержащего по меньшей мере один указанный олефин с изомеризующим катализатором, содержащим (i) по меньшей мере один цеолит с одномерной или более, чем одномерная, пористой структурой, имеющей размер пор, достаточно малый для затруднения димеризации побочных продуктов и образования коксовых отложений в пористой структуре и достаточно большой для входа линейного олефина и образования разветвленного метил-изоолефина (ii) связующего и (iii) металла, способствующего окислению коксовых отложений,
(b) прекращение контактирования подаваемого потока с катализатором после образования коксовых отложений на поверхности катализатора и, необязательно, отгонки любого отгоняемого углеводорода на катализаторе горячим газом,
(c) контактирования такого катализатора с коксовыми отложениями с потоком кислородсодержащего газа при температуре примерно от 250oC до самое большее 565oC в течение времени, эффективного для выгорания в значительной степени коксовых отложений, нанесенных на непокрытый коксовыми отложениями катализатор, посредством этого осуществляя регенерацию катализатора, и
(d) повторения стадии (а) с регенерированным таким образом катализатором.
В наилучшем варианте воплощения стадии с (а) до (с) могут быть повторены по меньшей мере 3 раза, более предпочтительно по меньшей мере 10 раз, прежде чем избирательность катализатора и/или производство изоолефина существенно понизятся. Полученный изоолефин может быть восстановлен или непосредственно использован в другом процессе, например, процессе для получения изоалкилэфиров, как это описано в EP-A-523,838 и US-A-5,191,146.
Для этого способа используемый катализатор готовится предпочтительно в соответствии со способом, содержащим:
(1) смешивание до пастообразного состояния порошка цеолита, порошка оксида алюминия, воды, пептизирующего количества кислоты и металла, способствующего окислению коксовых отложений,
(2) образования гранулы из смеси, изготовленной по п. (1),
(3) спекания гранулы, изготовленной по п. (2), при температуре от 300oC до 700oC.
Нижеследующие воплощения представлены для дополнительного объяснения изобретения.
Изготовление катализатора. Последующие примеры иллюстрируют способы изготовления катализаторов. Для приготовления катализаторов в примерах, описанных ниже, используются два аммониевых порошка ферриэрита ZSM-22 и ZSM-23. Эти два аммониевых ферриэрита были изготовлены одинаковым способом и обнаруживали аналогичные физические и каталитические свойства. Катализаторы A, C, E и F были приготовлены, используя аммониевые ферриэриты с молярным отношением диоксида кремния к диоксиду алюминия 53:1, площадью поверхности 391 м2/г (P/P0= 0.03), содержанием кристаллической соды 292 частей на миллион и сорбционной способностью н-гексана 7.2 г на 100 г цеолита. Катализаторы B, B' и D были изготовлены, используя аммоний-ферриэрит, имеющий молярное отношение диоксида кремния к диоксиду алюминия 62:1, с площадью поверхности 369 м2/г (P/P0= 0.03), содержанием кристаллической соды 480 частей на миллион и сорбционной способностью н-гексана 7.3 г на 100 г цеолита. Катализатор H был изготовлен, используя ZSM-22 (известный также как Тета-1 или TON), приготовленный согласно процедуре в примере TON-C в EP-A-247,802. Катализатор 1 был приготовлен, используя ZSM-23, приготовленный согласно процедуре в примере ZSM-23 в EP-A-247,802.
Компоненты катализатора были смешаны до пастообразного состояния с использованием смесителя Lancaster. Пастообразное вещество катализатора было экструдировано, используя бочковый стержневой экструдер Боннота.
Используемым связующим был оксид алюминия CATAPAL®D, а гидроксипропил метилцеллюлоза METHOCEL® (R) F4M использовалась в качестве ускорителя экструзии.
Катализатор A - без палладия. Смеситель Lancaster был загружен 944 г аммоний-ферриэрита (34.2% потерь на воспламенение ("LOI") определенных при температуре 900oC) и 93 г оксида алюминия CATAPAL®D (LOI 25.8%). Оксид алюминия был перемешан с ферриэритом в течение 5 минут, во время которых было добавлено 78 мл деионизированной воды. Смесь из 8 г ледяной уксусной кислоты и 78 мл деионизированной воды были медленно добавлены в смеситель для пептизации оксида алюминия. 10 г гидроксипропил метилцеллюлозы METHOCEL® (R) F4M были добавлены и смесь цеолит/оксид алюминия была перемешана в течение дополнительных 15 минут. Экструзионная смесь имела LOI равным 42.5%. Смесь цеолит/оксид алюминия с соотношением 90:10 была пропущена через экструдер Боннота и экструдирована, используя фильеры из нержавеющей стали с отверстиями 0.16 см. Экструдат был высушен при 120oC в течение 16 часов, а затем обожжен в воздухе при 500oC в течение 2 часов.
Катализатор B - 100 частей на миллион палладия посредством смешивания.
Смеситель Lancaster был загружен 632 г аммоний-ферриэрита (LOI 3.4%) и 92 г оксида алюминия CATAPAL®D (LOI 26.2%). Оксид алюминия был смешан с ферриэритом в течение 5 минут, во время которых было добавлено 156 мл деионизированной воды. Смесь из 6.8 г ледяной уксусной кислоты и 156 мл деионизированной воды были медленно добавлены в смеситель для пептизации оксида алюминия. Смесь была перемешана в течение 10 минут. Затем 0.20 г тетрааммиаката нитрата палладия в 156 мл деионизированной воды были медленно добавлены, когда смесь дополнительно была перемешана в течение 5 минут. 10 г гидроксипропил метилцеллюлозы METHOCEL® (R) F4M были добавлены, и смесь цеолит/оксид алюминия была перемешана в течение дополнительных 15 минут. Экструзионная смесь имела LOI равным 43.5%. Экструдат с соотношением частей 90:10 был пропущен через экструдер Боннота и экструдирован, используя фильеры из нержавеющей стали с отверстиями 0.16 см. Экструдат был высушен при 120oC в течение 16 часов, а затем обожжен в воздухе при 500oC в течение 2 часов.
Катализатор B' - 100 частей на миллион палладия посредством смешивания.
Смеситель Lancaster был загружен 645 г аммоний-ферриэрита (LOI 5.4%) и 91 г оксида алюминия CATAPAL®D (LOI 25.7%). Оксид алюминия был смешан с ферриэритом в течение 5 минут, во время которых было добавлено 152 мл деионизированной воды. Смесь из 6.8 г ледяной уксусной кислоты, 7.0 г лимонной кислоты и 152 мл деионизированной воды были медленно добавлены в смеситель для пептизации оксида алюминия. Смесь была перемешана в течение 10 минут. Затем 0,20 г тетрааммиаката нитрата палладия в 153 мл деионизированной воды были медленно добавлены, когда смесь дополнительно была перемешана в течение 5 минут. 10 г гидроксипропил метилцеллюлозы METHOCEL® (R) F4M были добавлены, и смесь цеолит/оксид алюминия была перемешана в течение дополнительных 15 минут. Экструзионная смесь имела LOI равным 43.5%. Экструдат с соотношением цеолит/оксид алюминия 90 : 10 был пропущен через экструдер Боннота и экструдирован, используя фильеры из нержавеющей стали с отверстиями 0,16 см. Экструдат был высушен при 120oC в течение 16 часов, а затем обожжен в воздухе при 500oC в течение 2 часов.
Катализатор C - 30 частей на миллион палладия посредством смешивания.
Использовался способ, аналогичный приготовлению катализатора B, с соответствующим регулированием концентраций ингредиентов для изготовления катализатора, имеющего палладия по весу 30 частей на миллион.
Катализатор D - 2500 частей на миллион палладия посредством смешивания.
Использовался способ, аналогичный приготовлению катализатора B, с соответствующим регулированием концентраций ингредиентов для приготовления катализатора, имеющего палладия по весу 2500 частей на миллион.
Катализатор E - 100 частей на миллион палладия посредством пропитки.
Катализатор E был приготовлен посредством пропитки пористого объема катализатора A. 15 г катализатора А были пропитаны раствором, содержащим:
1) 0.015 г водного раствора нитрата палладия, содержащего 10% по весу палладия и
2) 9.6 г чистого этилового спирта.
Контакт поддерживался в течение 1 часа при комнатной температуре. Затем смесь была высушена при 120oC в течение 16 часов, а затем спечена в воздухе при 500oC в течение 2 часов.
Катализатор F - 100 частей на миллион палладия посредством пропитки.
Катализатор F был приготовлен способом, аналогичным изготовлению катализатора E за исключением того, что 0,0043 г бис-(ацетилацетонато) палладия было растворено в чистом этиловом спирте.
Катализатор G - 1000 частей на миллион палладия посредством смешивания.
Использовался способ, аналогичный получению катализатора B, с соответствующим регулированием концентраций ингредиентов для приготовления катализатора, имеющего палладия по весу 1000 частей на миллион.
Катализатор H-100 частей на миллион палладия посредством смешивания.
Использовался способ, аналогичный изготовлению катализатора B, за исключением того, что использовался ZSM-22 вместо аммоний-ферриэрита для приготовления катализатора, имеющего палладия по весу 100 частей на миллион посредством смешивания. Смесь цеолит/оксид алюминия была экструдирована через экструдер Боннота, оборудованного фильерой из нержавеющей стали с отверстиями 0.16 см. Экструдат был высушен при 120oC в течение 16 часов, а затем спечен в воздухе при 500oC в течение 2 часов.
Катализатор I - 100 частей на миллион палладия посредством смешивания.
Использовался способ, аналогичный изготовлению катализатора В, за исключением того, что использовался ZSM-23 вместо аммоний-ферриэрита для изготовления катализатора, имеющего палладия по весу 100 частей на миллион посредством смешивания. Смесь цеолит/оксид алюминия была экструдирована через экструдер Боннота, оборудованного фильерой из нержавеющей стали с отверстиями 0.16 см. Экструдат был высушен при 120oC в течение 16 часов, а затем спечен в воздухе при 500oC в течение 2 часов.
Процедура тестирования.
Тест коксовых отложений. В аналитическом тесте определяется вес коксовых отложений на катализаторе посредством измерения потерянного веса после полного выгорания коксовых отложений в кислородсодержащем потоке при повышенной температуре, обычно при 750oC в течение 1 часа. Необходима осмотрительность во избежание впитывания воды катализатором.
Изомеризация 1. Труба из нержавеющей стали с внешним диаметром 2.54 см и внутренним диаметром 1.52 см длиной 66.04 см использовалась в качестве реактора. От верха трубы вытягивался карман (гильза) термопары на 50.8 см. Для загрузки реактор сначала был перевернут, и небольшая заглушка из стекловаты соскользнула вниз трубы реактора на карман термопары и ударилась о дно трубы. Карбид кремния (после сита N 20) был добавлен на глубину около 15.2 см. На него была помещена небольшая заглушка из стекловаты. Приблизительно 4 г частиц катализатора с сеткой 6-20 меш были смешаны с 60 г свежего карбида кремния (60-80 меш) и добавлены двумя частями для равномерного распределения. Слой катализатора был толщиной около 25.4 см. Еще один кусок стекловаты был добавлен сверху на катализатор, и реактор был покрыт карбидом кремния (20 меш) и закрыт последней заглушкой из стекловаты. Многоточечная термопара была вставлена в карман и расположена так, чтобы могла регистрироваться температура выше, ниже и в трех различных местах слоя катализатора. Реактор был перевернут и установлен в печь.
Подаваемым потоком был 1-бутен, полученный от Scott Speccialty Gases с содержанием бутена более 99.2% по весу. 1-Бутен подавался в реактор в газообразном виде.
Для запуска реактора, он был сначала нагрет до желаемой рабочей температуры в течение 4 периодов по 1 часу и выдерживания рабочей температуры в течение 2 часов, в течение всего времени при протекании азота. После такой предварительной обработки подача азота была остановлена, и был добавлен 1-бутен со скоростью 36 г/час для получения желаемой объемной скорости (WHSV) 9.0 час-1. Реактор работал при выходном давлении 210,97 г/см2 и температуре 430oC.
Регенерация I. После запуска катализатора для процесса изомеризации, описанного выше, было обнаружено, что он стал черным из-за роста углеродсодержащего вещества (коксовых отложений), составляющих от 10 до 20% по весу. Каждый катализатор был удален из тестового реактора и их вес был измерен. Катализаторы были помещены обратно в тестовый реактор и регенерированы посредством следующей процедуры. В устройство было подано давление в 6329.16 г/см2 и подан воздух со скоростью приблизительно 6 литров/час. Образец нагревался с помощью следующей управляемой процедуры нагрева: подъем температуры от 25oC до 125oC в течение 10 минут, выдерживание при температуре 125oC в течение 30 минут, подъем температуры от 125oC до 350oC со скоростью 2 градуса в минуту, подъем температуры от 350oC до 470oC со скоростью 1 градус в минуту и выдерживание при 470oC в течение 24 часов. Затем реактор был охлажден и катализатор выгружен. Почти полная регенерация катализатора подтверждалась исчезновением черного цвета по сравнению с нерегенерированным катализатором. Образцы катализаторов были взвешены для измерения потерь коксовых отложений.
Изомеризация II. Фланцевая труба из нержавеющей стали с внешним диаметром 7.32 см и внутренним диаметром 6.35 см длиной 43.18 см использовалась в качестве реактора. Вторая фланцевая труба из нержавеющей стали с внешним диаметром 6.04 см и внутренним диаметром 5.08 см длиной 30.48 см использовалась для предварительного подогрева подаваемого потока. Подогреватель был расположен следующим за реактором и подсоединен U-образным коленом длиной 91.4 см к верхней части. Карман для термопары, содержащий 10 термопар длинами от 76.2 см до 203.2 см, был подсоединен к нижнему фланцу и помещен в реактор. Электронагревательные элементы простирались на всю длину реактора и подогревателя. Подогреватель был загружен поддерживающими шарами размером 0.158 см на уровне 121.9 см от верхнего края. Сначала реактор был загружен поддерживающими шарами размером 0.318 см на уровне 457.2 см от верхнего края. Затем было добавлено 15.24 см поддерживающих шаров размером 0,158 см. 241.2 г катализатора были насыпаны в реактор на верх поддерживающих шаров размером 0.158 см.
Используемым потоком был рафинат-2 коммерческого класса, содержащий приблизительно 40% по весу 1-бутена, 20% по весу транс-2-бутена, 13% по весу цис-2-бутена, 3% по весу изобутана, 23% по весу н-бутана и 1% по весу изобутилена. Рафинат-2 подавался в подогреватель в газообразном состоянии, после выпаривания в нагревателе потока при низком давлении.
Сначала при запуске реактор был нагрет до 288oC при протекающем азоте. По истечении 4 часов были взяты образцы выходящего газа на содержание кислорода. Когда содержание кислорода упало ниже уровня 0,02% по объему, этап предварительного подогрева завершился, и поток азота был прерван. Этот этап занял примерно 9 часов. Рафинат-2 был добавлен в реактор со скоростью 10888.36 г/час для обеспечения желаемой объемной скорости (WHSV) в 7.0 час-1. Как только рафинат-2 был введен в реактор, температура увеличилась до желаемого рабочего значения. Реакция изомеризации продолжалась до тех пор, пока в среднем не было достигнуто преобразования 35% нормального олефина.
Регенерация II. Выходящий из реактора поток был подсоединен к нагревателю, работающему с помощью пламени. Когда подача была прервана и азот введен в реактор, температура слоя снизилась до 343oC. Поток азота медленно увеличивался до максимум 9.8 м3/час при атмосферном давлении в течение нескольких часов до тех пор, пока вытекающий очищенный газ был свободен от углеводорода, в то же время поддерживая равномерную температуру слоя катализатора 343oC. В реактор вводился сухой воздух со скоростью 0.38 м3/час, в то же время поддерживая скорость потока азота 9.8 м3/час. Горение регистрировалось посредством наблюдения любого увеличения температуры в слое катализатора, при введении смешанного газа в реактор. Температура слоя катализатора поддерживалась так, чтобы она не превышала 471oC. Когда содержание кислорода (и в кислороде, и в диоксиде углерода) достигло 1.75 молярного процента, значение температуры в слое катализатора были записаны. Температура поддерживалась равной 471oC.
Когда содержание полученного диоксида углерода упало ниже 0,05 молярного процента, температура была медленно повышена со скоростью 3-6oC в час до температуры 485oC с использованием электронагревателя. Когда температура слоя начала падать, скорость потока воздуха, поступающего в реактор, была медленно увеличена со скоростью 0,142-0,283 м3/час до достижения максимальной температуры слоя 487oC. Подача воздуха продолжалась до максимального значения 5.74 м3/час при поддержании температуры слоя 487oC до тех пор, пока температура слоя не начала падать, и в этот момент поток азота был медленно удален из системы. Регенерация продолжалась в чистом воздухе при скорости 5.74 м3/час в течение 12 часов, в это время поддерживалась температура слоя 487oC. Далее регенерация продолжалась, пока диоксида углерода в потоке газа не стало менее 0,01 молярных процентов за 1 час. Затем реактор был охлажден до температуры 288oC и наполнен азотом.
Изомеризация III. Реактор имел форму трубы из нержавеющей стали с внешним диаметром 5.08 см и внутренним диаметром 4.06 см с приваренными фланцами размером 5.08 см на каждом конце. Труба также имела трубки для подачи и приема вытекающих продуктов размером 0.63 см, приваренными на расстоянии 15.2 см от дна и верха реактора соответственно. В верхний уплотняющий фланец были установлены датчик давления и прорезанный диск. В нижний уплотняющий фланец был установлен карман термопары, приваренный прямо в центре фланца, который был присоединен и вытянут до середины трубы реактора. Термопара была в виде заваренной с одной стороны трубы из нержавеющей стали и содержащей восемь или более точек термопары. Труба реактора была помещена в 91.4 см нагревающей печью Ландберга, содержащей три зоны нагрева, но только одна нижняя зона использовалась для предварительного нагрева подаваемого потока бутилена в зону реактора. Печь управлялась тремя контроллерами. На выходном конце была расположена система трубопроводов и оборудования для подачи образцов вытекающих углеводородов прямо в газовый хроматограф.
Используемый поток был вытекающим обработанным потоком метил-трет-бутиловый эфира (MTBE) и содержал приблизительно 25-35 весовых процента бутена-2, 40-50 весовых процента бутена-1 и 20-30 весовых процента бутанов.
Сначала реактор был загружен инертным наполняющим веществом в зоне предварительного нагрева. Используемыми инертными наполняющими веществами были или корунд с частицами, размером мелкой сетки, или глиняные инертные гранулы, поддерживающие катализатор. Указанное наполняющее вещество с предварительно взвешенным количеством катализатора было добавлено для образования отчетливой зоны катализатора.
При запуске реактор был нагрет до минимальной рабочей температуры, обычно составляющей более 200oC при текущем наполняющем азоте при примерно 1054,86 - 3516,2 г/см2. Когда реактор был нагрет, поток был подан в реактор, а наполнение азотом было прекращено. Реакция изомеризации выполнялась при объемной скорости (WHSV) 7 час-1 и температуре 430oC.
Регенерация III. Предварительно муфельная печь была нагрета до 500oC. Катализатор, покрытый коксовыми отложениями, был отделен от гранул, поддерживающих катализатор. Катализатор был равномерно помещен в лоток из нержавеющей стали с примерными размерами 30.4 см на 15.24 см. Металлический лоток с катализатором был помещен в предварительно нагретую муфельную печь. Когда катализатор стал белым или почти белым, металлический лоток был удален из муфельной печи. Катализатор был перемещен в химический стакан и охлажден в эксикаторе до комнатной температуры.
Вычисления. Преобразование и избирательность были вычислены для каждого образца во время тестов. Вычисления преобразования и избирательности отражают поступающую (FD) и вытекающую (EFF) концентрации бутена-1 (B1) и изобутена (IB1). Преобразование вычислялось так: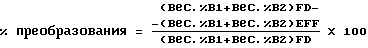
избирательность вычислялась так: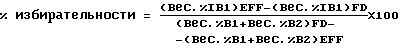
а выход вычислялся так: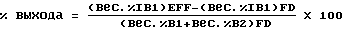
Примеры 1-9.
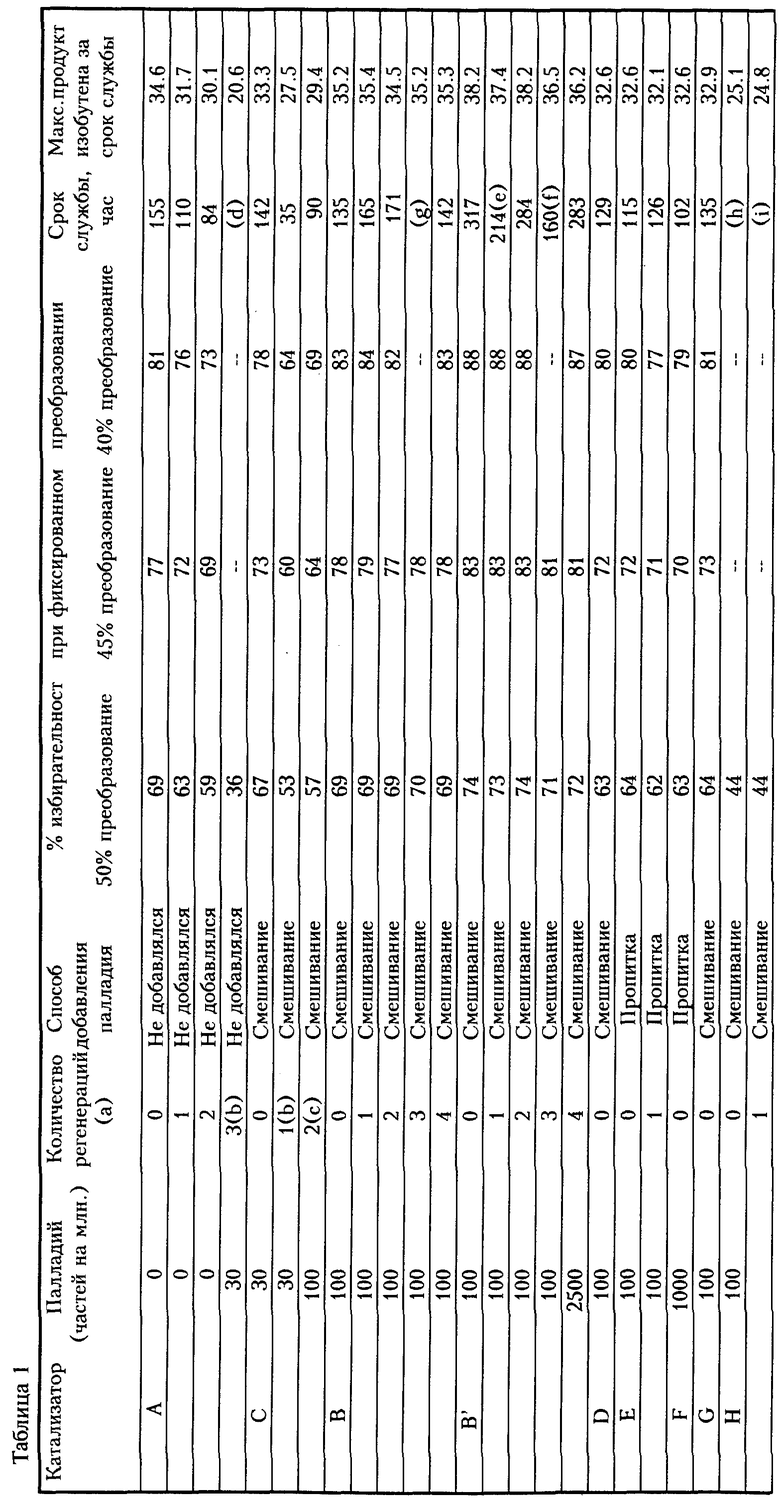
Таблица 1 представляет результаты тестирования катализаторов, изготовленных так, как указано выше. Эта таблица отражает часы срока службы катализатора в процессе изомеризации после нескольких циклов регенерации. Бутен изомеризовался согласно процессу, описанному в разделе "Изомеризация I", а катализатор был регенерирован согласно разделу "Регенерация I" для таблицы 1. Термин "срок службы" определяется здесь как время от запуска до времени, при котором концентрация разветвленного метил-изоолефина в продукте снизилась до 27% по весу после достижения своего пика. Таблица также отражает мгновенные избирательности катализаторов к изобутилену при 40% преобразовании, 45% преобразовании и 50% преобразовании и наивысшей концентрации (% по весу) разветвленного метил-изоолефина (изобутилена) в продукте, полученном во время теста. Примеры 1-9 приводятся в таблице 1 сверху вниз.
(а) Если не оговорено обратное, регенерация выполнялась при 470oC при избыточном давлении воздуха 6329.16 г/см2 при объемной скорости (WHSV) 700 час-1 в течение 24 часов.
(b) 500oC, 90 часов, 1 атмосфера в муфельной печи
(с) 490oC, 24 часа, 3 атмосферы (2109.72 г/см2 избыточного давления)
(d) Не было достигнуто производства 27% изобутена в продукте; максимум составил только 20,6%.
(е) Прервано прежде, чем в продукте достигнуто 27% изобутена; (прервано при 32.5%).
(f) Прервано прежде, чем в продукте достигнуто 27% изобутена; (прервано при 34.5%).
(g) Прервано прежде, чем в продукте достигнуто 27% изобутена; (прервано при 34.9%).
(h) Прервано прежде, чем в продукте достигнуто 27% изобутена; максимум составил только 25.1%.
(i) Прервано прежде, чем в продукте достигнуто 27% изобутена; максимум составил только 24.8%.
Как видно из таблицы 1, катализаторы, содержащие палладий (см. катализатор B) обеспечивают больший срок службы и/или достигают большей избирательности к изобутилену после ряда регенераций, в то время как катализаторы без палладия (см. катализатор A) показали существенное снижение срока службы катализатора и/или избирательность. Для катализатора B срок службы катализатора оставался выше 130 часов в течение 4 регенераций, в то время как срок службы катализатора A упал ниже 100 часов за 2 регенерации. Время, требуемое для регенерации, было значительно меньше, чем срок службы согласно настоящему изобретению. Кроме того, избирательность катализаторов с палладием оставалась существенно постоянной с минимальным уменьшением (0-2% изменения для катализаторов B, B', E и H), в то время как избирательность катализаторов без палладия значительно снижалась после каждой регенерации (3-5% изменения для катализатора A).
Регенерации при более высоких температурах, атмосферном давлении и без палладия привело к потерям избирательности катализатора при фиксированных преобразованиях. Эти потери становятся более существенными при повторяющихся регенерациях.
Далее, как можно видеть из таблицы 1, катализаторы, в которых палладий был смешан, демонстрировали увеличенный срок службы, больший выход изобутилена и большую избирательность при 40%-м, 45%-м и 50%-м уровнях преобразования по сравнению с катализатором(ами), приготовленными посредством пропитки. Больший цикл жизни и более высокая избирательность катализаторов, полученных посредством смешивания, поддерживался после многих циклов регенерации при повышенном давлении и низких температурах.
Избирательность катализатора с содержанием палладия 2500 частей на миллион ниже, чем избирательность катализатора B с содержанием 100 частей на миллион. Высокие уровни металла(ов), способствующего(их) окислению, имеющегося в катализаторе, свыше 15% по весу в пересчете на металл, от общего веса катализатора, приводят к неприемлемо сокращенным избирательности и/или сроку службы. В наилучшем варианте воплощения палладий используется в количестве, достаточном для того, чтобы способствовать регенерации, но меньшем, чем количество, которое будет несколько ограничивать срок службы катализатора.
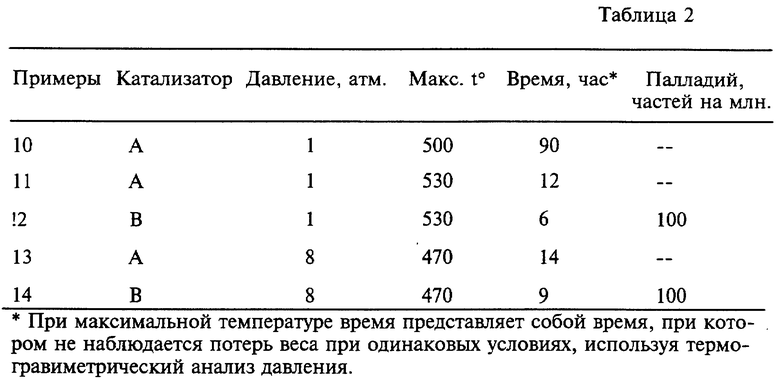
Примеры 10-14. В таблице 2 представлены времена (при максимальной температуре), требуемые для удаления коксовых отложений из катализаторов A и B при различных температурах и давлении. Эти данные получены, используя термогравиметрический анализ давления.
Использование палладия и/или более высокого парциального давления кислорода позволяет удалить коксовые отложения на катализаторе за более короткие периоды времени при более низких температурах. Как видно из таблицы 2, катализаторы с палладием (примеры 12 и 14) способны регенерировать при сокращенном времени, по сравнению с катализатором без палладия (примеры 11 и 13). Далее палладийсодержащий катализатор в примере 14, который был регенерирован при давлении 8 атмосфер (и повышенном парциальном давлении кислорода), регенерировался быстрее при более низкой температуре по сравнению с температурой в примерах 13 и 14, повышенное давление позволяет регенерировать катализатор при более низких температурах за более короткое время.
Пример 15. Использовался катализатор А (без палладия) для изомеризации исходного потока бутена при условиях, описанных в разделах "Изомеризация III" и "Регенерация III". Средняя избирательность к изобутену за ряд регенераций представлены на чертеже. Катализатор B (с палладием) использовался для изомеризации исходного потока бутена при условиях, описанных в разделах "Изомеризация II" и "Регенерация II". Средняя избирательность к изобутену за ряд регенераций представлены на фиг. 1 для этого катализатора. Линия для катализатора B представляет собой линейную регрессию, построенную по 19 точкам.
Как можно видеть из чертежа, высокая избирательность катализатора B может поддерживаться в течение по меньшей мере 19 циклов регенерации, используя способ согласно настоящему изобретению.
Все данные, представленные в таблице 1, были получены при применении коммерчески доступного бутена со степенью чистоты 99.2% или выше. Данные, показанные на чертеже, были получены при использовании потока сырья, содержащего 70-75% бутенов и 25-30% бутанов. Эти различия в потоках приводят к большей избирательности для бутаносодержащих потоков, как можно видеть на чертеже. Присутствие бутанов (или других растворителей, таких как азот) в потоке олефинов служит для использования более низкого парциального давления олефинов, что приводит к сокращению количества получаемых продуктов с количеством атомов углерода, не равным 4. Аналогично увеличиваются избирательность, когда олефин растворяется менее реактивными газами, например азотом, что показано в таблице 7 в Европейском патенте N 247,802. Полезно отметить, что катализаторы на основе ферриэрита, такие как катализатор B' очень высокой избирательности, могут быть получены и из разбавленных и из неразбавленных потоков олефина.
Описывается способ структурной изомеризации линейного олефина, имеющего по меньшей мере 4 атома углерода, в соответствующий ему разветвленный метилизоолефин, включающий стадию (а) контактирование при температуре подаваемого потока углеводородов, содержащего по меньшей мере указанный олефин с изомеризующим катализатором, содержащим по меньшей мере один цеолит с одномерной или более чем одномерная пористой структурой, имеющей размер пор более 0,42 нм и менее 0,7 нм, связующего, отличающийся тем, что на стадии (а) контактирование проводят при 200 - 650°С с катализатором, дополнительно содержащим до 15 вес. % металла, способствующего окислению коксовых отложений, способ включает дополнительно стадии: б) прекращение контактирования подаваемого потока с катализатором после образования коксовых отложений на поверхности катализатора; в) контактирование катализатора с коксовыми отложениями с потоком кислородсодержащего газа при температуре менее 565°С, давлении системы более 1 атм и парциальном давлении кислорода от 0,001 атм до 40 атм для выгорания коксовых отложений с катализатора и г) повторение стадии (а) с катализатором, полученным в соответствии со стадией (в). Описывается также каталитический состав. Технический результат - повышение выхода целевого продукта, сокращение времени регенерации катализатора и увеличение срока его службы. 2 с. и 24 з.п. ф-лы, 2 табл., 1 ил.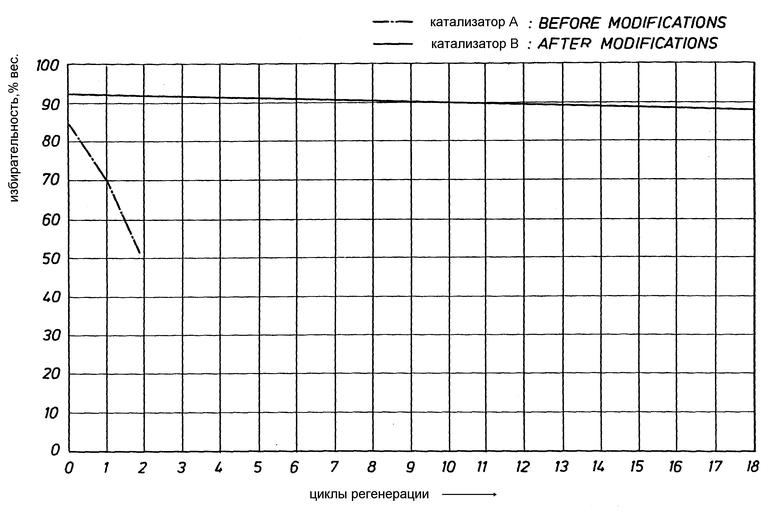
| Роликовый кантователь | 1975 |
|
SU523838A1 |
| Способ получения высокооктановых компонентов бензина | 1981 |
|
SU1015582A1 |
| Способ получения олефиновых углеводородов с -с | 1975 |
|
SU606850A1 |
| US 4855529 A, 08.08.89 | |||
| US 5227569 A, 13.07.93 | |||
| Трубчатый вертикальный колодец | 1977 |
|
SU666249A1 |
| ПОЗИЦИОННО-ЧУВСТВИТЕЛЬНЫЙ ПРОПОРЦИОНАЛЬНЫЙ СЧЕТЧИК ИЗЛУЧЕНИЯ | 1975 |
|
RU545179C |
