Настоящее изобретение относится к способу получения производного 2-аминотиазолкарбоксамида, представленного следующей формулой (I):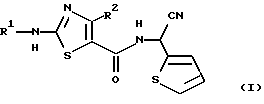
в которой R1 представляет C1-5 алкил с прямой или разветвленной цепью, C1-5 галогеналкил или С3-6 циклоалкил, и R2 представляет С1-3 алкил или C1-3 галогеналкил.
Соединения формулы (I) используются в качестве микробицидов при лечении болезней растений, вызываемых Pythiaceae или Peronosporaceae. Соединения формулы (I) уже были описаны в корейской выложенной заявке 94-19960 и в соответствующих иностранных заявках, например патентной заявке США 08/287917, патентной заявке Японии 192529 и патентной заявке ЕР 94112652.6, которые были поданы заявителем настоящей заявки.
Кроме того, способ получения производных 2-аминотиазолкарбоксамида, включая соединения формулы (I), использующий 2-аминотиазолкарбоновую кислоту в качестве промежуточного соединения, описан в выложенной корейской заявке 97-24120. Однако недостатком этого способа является то, что он оказывается неэкономичным при промышленном производстве из-за многостадийности получения промежуточного соединения и низкого выхода.
Соответственно, заявители провели исследования по усовершенствованию ранее известного способа путем решения вышеупомянутых проблем, и в результате пришли к настоящему изобретению.
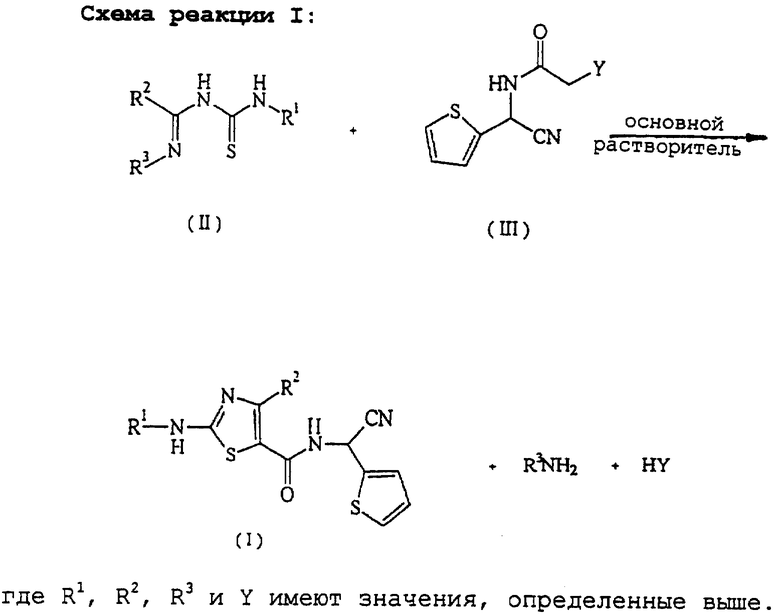
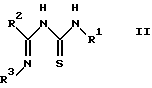
Настоящее изобретение относится к способу получения производного 2-аминотиазолкарбоксамида, представленного вышеприведенной формулой (I), отличающемуся тем, что соединение иминотиомочевины, представленное следующей формулой (II):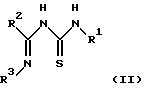
в которой R1 и R2 имеют значения, определенные выше, и R3 представляет фенил, который может быть необязательно моно- - пентазамещенным независимо хлором, метокси, этокси, фенокси или нитро,
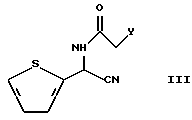
подвергают реакции с соединением тиофенацетамида, представленным следующей формулой (III):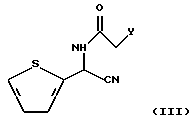
в которой Y представляет уходящую группу, такую как хлорид, бромид и т. п.
Соединение формулы (I) может быть получено путем реакции соединения формулы (II) с соединением формулы (III) в растворителе и в присутствии основания, как описывается схемой реакции I (см. в конце описания).
Примеры основания, используемого в вышеописанной реакции, включают органическое основание, такое как триэтиламин, трибутиламин, диизопропилэтиламин, N,N-диметиланилин, пиридин, 4-диметиламинопиридин и т.п., и неорганическое основание, такое как гидроокись натрия, гидроокись калия, карбонат калия, бикарбонат натрия, бикарбонат калия, гидрид натрия, гидрид калия и т. п. Органическое основание является предпочтительным, и более предпочтительным является алкиламин, такой как триэтиламин, трибутиламин, диизопропилэтиламин и т.п. Основание может использоваться в количестве от 1 до 5 эквивалентов, предпочтительно в количестве от 1 до 2 эквивалентов.
Вышеописанная реакция может осуществляться при температуре между 20oС и 120oС, предпочтительно между 40oС и 80oС, и требуемое время реакции составляет примерно от 8 до 12 часов.
Растворитель включает спирт, такой как метанол, этанол, изопропиловый спирт и т.д.; ароматический углеводород, такой как бензол, толуол, ксилол и т. д. ; простой эфир, такой как диэтиловый эфир, диоксан, 1,2-диметоксиэтан, тетрагидрофуран и т.д.; кетон, такой как ацетон, метилэтилкетон, циклогексанон и т.п.; нитрил, такой как ацетонитрил, пропионитрил и т.д.; галогенированный углеводород, такой как дихлорметан, 1,2-дихлорэтан, хлороформ и т. д.; сложный эфир, такой как метилацетат, этилацетат и т.д.; и полярный растворитель, такой как N,N-диметилформамид, N,N-диметилацетамид, диметилсульфоксид и т.д.; и спирт является предпочтительным.
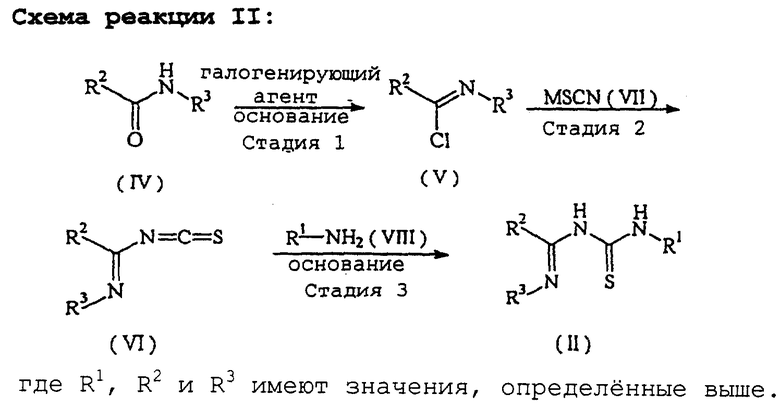
Соединение формулы (II), используемое как исходное вещество в схеме реакции I, является новым и может быть получено в соответствии со схемой реакции II (см. в конце описания).
Иначе говоря, соединение формулы (II) может быть получено по способу, отличающемуся тем, что:
на стадии 1 амидное соединение, представленное следующей формулой (IV):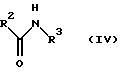
в которой R2 и R3 имеют значения, определенные выше, подвергается реакции с галогенирующим агентом в растворителе в присутствии основания с получением имидоилхлоридного соединения, представленного следующей формулой (V):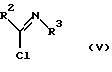
в которой R2 и R3 имеют значения, определенные выше;
на стадии 2 полученное имидоилхлоридное соединение формулы (V) подвергают реакции с тиоцианатным соединением, представленным следующей формулой (VII):
MSCN (VII),
в которой М представляет щелочной металл, такой как натрий, калий, и т. д., или NH4,
по которой хлоридная группа заменяется изотиоцианидной группой с получением имидоилизотиоцианатного соединения, представленного формулой (VI):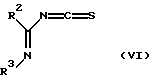
где R2 и R3 определены, как описано выше;
на стадии 3 полученное имидоилизотиоцианатное соединение формулы (VI) подвергают реакции с первичным аминовым соединением, представленным следующей формулой (VIII):
R1-NН2 (VIII),
где R1 определен, как описано выше, в присутствии основания.
На стадии 1 для получения имидоилхлоридного соединения формулы (V) в качестве галогенирующего агента могут быть использованы тионилхлорид (SОСl2), фосген (COCl2), оксихлорид фосфора (РОС13) и т.п. Галогенирующий агент подходящим образом используется в количестве от 1 до 4 эквивалентов. Эту реакцию проводят при температуре между -20oС и 80oС, предпочтительно между -10oС и 20oС. Время реакции составляет обычно от 2 до 5 часов. В качестве основания может быть использовано органическое основание, такое как пиридин, 4-диметиламинопиридин, триэтиламин, N,N-диметиланилин, трибутиламин и т. п. Предпочтительными являются слабые основания, такие как пиридин. Основание подходящим образом используется в количестве от 1 до 4 эквивалентов.
В качестве растворителя может быть использован ароматический углеводород, такой как бензол, толуол, ксилол и т.п.; галогенированный углеводород, такой как дихлорметан, 1,2-дихлорэтан, хлороформ, и т.п.; простой эфир, такой как диэтиловый эфир, диоксан, 1,2-диметоксиэтан, тетрагидрофуран, и т.п. ; кетон, такой как ацетон, метилэтилкетон, циклогексанон и т.п.; нитрил, такой как ацетонитрил, пропионитрил и т.п.; сложный эфир, такой как метилацетат, этилацетат и т.п.; или полярный растворитель, такой как N,N-диметилформамид, N, N-диметилацетамид, диметилсульфоксид и т.п.; и, предпочтительно, галогенированный углеводород, такой как дихлорэтан, хлороформ и т.п. Кроме того, N,N-диметилформамид может быть использован в качестве катализатора.
На стадии 2 имидоилизотиоцианатное соединение формулы (VI) получают путем реакции имидоилхлоридного соединения формулы (V), полученного на стадии 1, с тиоцианатным соединением формулы (VII). Тиоцианатное соединение формулы (VII) предпочтительно используют в количестве от 1 до 2 эквивалентов. Температура реакции может быть между -20oС и 50oС, предпочтительно между 0oС и 20oС, и время реакции предпочтительно варьирует обычно от 2 до 5 часов.
На стадии 3 иминотиомочевиновое соединение формулы (II) получают из имидоилизотиоцианатного соединения формулы (VI). На данной стадии атом углерода изотиоцианата атакуется аминовым соединением формулы (VIII) в присутствии основания, благодаря чему получается тиомочевиновое соединение формулы (II). Аминовое соединение формулы (VIII) может использоваться в количестве от 1 до 4 эквивалентов, предпочтительно от 2 до 3 эквивалентов. Данная реакция может проводиться при температуре между -20oС и 80oС, предпочтительно между 0oС и 30oС. Время реакции составляет, как правило, от 2 до 4 часов.
Вышеописанные способы по настоящему изобретению будут более подробно объяснены нижеследующими примерами. В качестве типичных примеров соединений формулы (II) по настоящему изобретению могут быть упомянуты соединения, которые описаны в таблице (см. в конце описания).
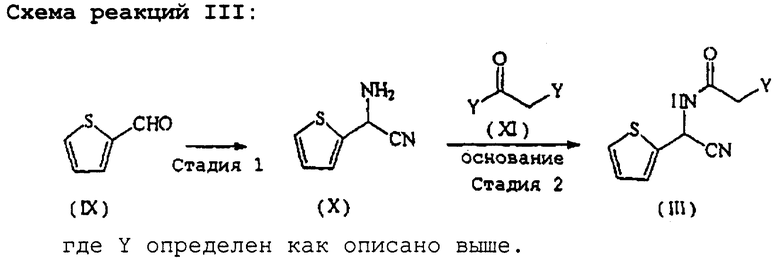
Соединение формулы (III), используемое в качестве исходного вещества в схеме реакций (I), также является новым и может быть получено в соответствии со схемой реакций III (см. в конце описания).
Иными словами, соединение формулы (III) может быть получено по способу, отличающемуся тем, что на стадии 1 альдегидное соединение, представленное следующей формулой (IX):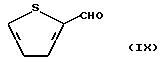
превращают в аминонитрильное соединение, представленное следующей формулой (X):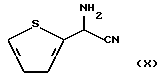
по известному синтезу Стрекера;
на стадии 2 полученное аминонитрильное соединение формулы (X) вводят в реакцию с соединением, представленным следующей формулой (XI):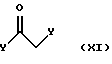
где Y определен, как описано выше, в присутствии основания.
На стадии 1 вышеописанной реакции альдегидное соединение формулы (IX) может быть легко превращено в аминонитрильное соединение формулы (X) путем известного синтеза Стрекера, как указано выше.
На стадии 2 тиофенацетамидное соединение формулы (III) может быть получено путем реакции аминонитрильного соединения формулы (X) с 1-3 эквивалентами, предпочтительно с 1-1,5 эквивалентами хлорацетилхлорида или бромацетилбромида формулы (XI) в присутствии основания. Данная реакция может осуществляться при температуре между -20oС и 80oС, предпочтительно между 0oС и 20oС. Время реакции подходящим образом составляет от 30 минут до 2 часов.
В качестве основания может использоваться органическое основание, такое как пиридин, 4-диметиламинопиридин, триэтиламин, N,N-диметиланилин, трибутиламин, диизопропилэтиламин и т.п., предпочтительно пиридин или 4-диметиламинопиридин. Основание может предпочтительно использоваться в количестве от 1 до 3 эквивалентов.
В качестве растворителя может использоваться галогенированный углеводород, такой как дихлорметан, 1,2-дихлорэтан, хлороформ и т.п.; ароматический углеводород, такой как бензол, толуол, ксилол и т.п.; простой эфир, такой как диэтиловый эфир, диоксан, 1,2-диметоксиэтан, тетрагидрофуран и т.п.; кетон, такой как ацетон, метилэтилкетон, циклогексанон и т.п.; нитрил, такой как ацетонитрил, пропионитрил и т.п.; сложный эфир, такой как метилацетат, этилацетат и т.п.; или полярный растворитель, такой как N,N-диметилформамид, N,N-диметилацетамид, диметилсульфоксид и т.п.; и, предпочтительно, галогенированный углеводород или ароматический углеводород.
Настоящее изобретение более подробно объясняется с помощью нижеследующих препаративных примеров и примеров. Однако должно быть ясно, что настоящее изобретение никоим образом не ограничивается этими примерами.
Препаративный пример 1: Синтез N-фенилпропионамида
В реакционный сосуд помещают анилин (279,4 г, 3,0 моль) и дихлорметан (2400 г), реакционный сосуд охлаждают до 0oС и медленно добавляют в него по каплям гидроокись натрия (132,0 г, 3,3 моль), растворенную в воде (660 г).
После подтверждения того, что температура сосуда составляет 0oС, в него по каплям в течение 2 часов добавляют пропионилхлорид (291,5 г, 3,2 моль). Затем смесь перемешивают при комнатной температуре (20oС) в течение 2 часов и реакция заканчивается. После завершения реакции слои разделяют и дихлорметан удаляют отгонкой под пониженным давлением, получая коричневое твердое вещество. Твердое вещество перекристаллизовывают из толуола, получая указанное в заголовке соединение (434,7 г, 2,9 моль) с выходом 97%.
1H ЯМР (СDСl3): δ 7,75 (1Н, с, шир.), 7,52 (2Н,д), 7,29 (2Н, д), 7,08 (1Н, т), 2,37 (2Н, кв.), 1,22 (3Н, т).
Пример 1: Синтез N-этил-N'-(1-фенилиминопропил)мочевины
N-фенилпропионамид (149,2 г, 1,0 моль) и пиридин (261,0 г, 3,3 моль) растворяют в дихлорметане (300 г) и смесь охлаждают до 0oС. К ней по каплям в течение двух часов добавляют оксихлорид фосфора (168,7 г, 1,1 моль) и затем смесь перемешивают при комнатной температуре (20oС) в течение 2 часов для получения N-фенилпропионимидохлорида.
Впоследствии реагент в течение 2 часов медленно добавляют по каплям в реакционный сосуд, содержащий смесь тиоцианата калия (145,8 г, 1,5 моль) и карбоната калия (318,0 г, 3,0 моль) в ацетоне (1000 мл) при температуре 10oС или ниже и затем смесь перемешивают в течение 1 часа с получением изотиоцианата N-фенилпропионимидоила. В реакционный сосуд в течение 2 часов по каплям добавляют этиламин (128,8 г, 2,0 моль), поддерживая температуру 10oС или ниже, и затем смесь перемешивают в течение 1 часа.
После завершения реакции растворитель удаляют отгонкой при пониженном давлении. Продукт экстрагируют толуолом и промывают раствором гидроокиси натрия. Затем толуол удаляют отгонкой под пониженным давлением и остаток перекристаллизовывают из изопропилового спирта, получая указанное в заголовке соединение (157,2 г, 0,7 моль) с выходом 67%.
1H ЯМР (СDСl3): δ 11,84 (1Н, с, шир.), 8,18 (1Н, с, шир.), 7,32 (2Н, м), 7,12 (1Н, т), 6,79 (2Н, д), 3,69 (2Н, м), 2,23 (2Н, кв.), 1,26 (3Н, т), 1,15 (3Н, т).
Пример 2: Синтез N-(1-(2,4-дихлорфенил)иминопропил)) -N' -этилтиомочевины
N-(2,4-дихлорбензол)пропионамид (21,8 г, 0,1 моль) и пиридин (27,7 г, 0,35 моль) растворяют в дихлорметане (30 г) и смесь охлаждают до 0oС. К ней по каплям в течение двух часов добавляют оксихлорид фосфора (16,9 г, 0,11 моль) и затем смесь перемешивают при комнатной температуре (20oС) в течение 2 часов с получением N-(2,4-дихлорбензол) пропионимидоилхлорида.
После этого реагент в течение 2 часов медленно добавляют по каплям в реакционный сосуд, содержащий смесь тиоцианата калия (14,6 г, 0,15 моль) и карбоната калия (31,8 г, 0,3 моль) в ацетоне (100 мл) при температуре 10oС или ниже и затем смесь перемешивают в течение 1 часа с получением изотиоцианата N-(2,6-дихлорбензол)пропионимидохлорида. В реакционный сосуд в течение 2 часов по каплям добавляют этиламин (12,9 г, 0,2 моль), при поддержании температуры 10oС или ниже, и затем смесь перемешивают в течение 1 часа.
После завершения реакции по такой же методике, как в примере 1, получают указанное в заголовке соединение (21,9 г, 72 ммоль) с выходом 72%.
1H ЯМР (СDСl3): δ 11,69 (1Н, с, шир.), 8,65 (1Н, с, шир.), 7,43 (1Н, д), 7,21 (1Н, м), 6,78 (1Н, д), 3,70 (2Н, м), 2,23 (2Н, кв.), 1,27 (3Н, т), 1,14 (3Н, т).
Пример 3: Синтез N-изопропил-N'- (1-фенилиминопропил) тиомочевины
N-фенилпропионамид (7,46 г, 0,05 моль) и пиридин (13,8 г, 0,18 моль) растворяют в дихлорметане (300 г) и смесь охлаждают до 0oС. К ней по каплям в течение двух часов добавляют оксихлорид фосфора (8,43 г, 0,05 моль) и затем смесь перемешивают при комнатной температуре (20oС) в течение 2 часов с получением N-фенилпропионимидохлорида.
Впоследствии реагент в течение 2 часов медленно добавляют по каплям в реакционный сосуд, содержащий смесь тиоцианата калия (7,3 г, 0,08 моль) и карбоната калия (15,9 г, 0,15 моль) в ацетоне (50 мл) при температуре 10oС или ниже и затем смесь перемешивают в течение 1 часа с получением изотиоцианата N-фенилпропионимидоилхлорида. В реакционный сосуд в течение 2 часов по каплям добавляют изопропиламин (5,9 г, 0,1 моль), при поддержании температуры 10oС или ниже, и затем смесь перемешивают в течение 1 часа.
После завершения реакции по такой же методике, как в примере 1, получают указанное в заголовке соединение (8,1 г, 0,03 моль) с выходом 65%.
1H ЯМР (СDСl3): δ 11,80 (1Н, с, шир.), 7,92 (1Н, с, шир.), 7,35 (2Н, м), 7,13 (1Н, т), 6,79 (2Н, д), 4,50 (1Н, м), 2,25 (2Н, кв.), 1,28 (3Н, с), 1,22 (3Н, с), 1,17 (3Н, т).
Пример 4: Синтез 2-хлор-N-(α-циано-2-тенил) ацетамида
Гидрохлорид аминотиофен-2-ил-ацетонитрила (17,5 г, 0,1 моль) растворяют в дихлорметане (100 мл) и затем добавляют к нему пиридин (16,6 г, 0,21 моль). Смесь охлаждают до 10oС и затем в течение 1 часа добавляют к ней по каплям хлорацетилхлорид (12,4 г, 0,11 моль).
После завершения реакции смесь трижды промывают водой (по 60 мл на каждую промывку), растворитель удаляют отгонкой под пониженным давлением и остаток перекристаллизовывают из толуола, получая указанное в заголовке соединение (19,8 г, 0,09 моль) с выходом 92%.
1H ЯМР (СDСl3): δ 7,42 (1Н, д), 7,32 (1Н, д), 7,23 (1Н, с, шир.), 7,05 (1Н, т), 6,28 (1Н, д), 4,15 (2Н, с.).
Пример 5: Синтез 2-6ром-N-(α-циано-2-тенил)ацетамида
Гидрохлорид аминотиофен-2-ил-ацетонитрила (8,8 г, 0,05 моль) растворяют в дихлорметане (50 мл) и затем добавляют к нему пиридин (8,7 г, 0,11 моль). Смесь охлаждают до 0oС и затем в течение 1 часа добавляют к ней по каплям бромацетилбромид (10,1 г, 0,05 моль).
После завершения реакции по такой же методике, как в примере 4, получают указанное в заголовке соединение (11,4 г, 0,04 моль) с выходом 88%.
1H ЯМР (СDСl3): δ 7,41 (1Н, м), 7,32 (1Н, м), 7,11 (1Н, д, шир.), 7,05 (1Н, м), 6,25 (1Н, д), 3,94 (2Н, с.).
Пример 6: Синтез N-(α-циано-2-тенил)-4-этил-2- (этиламино) -5-тиазолкарбоксамида
Способ 1
N-этил-N'-(1-фенилиминопропил) тиомочевину (23,5 г, 0,1 моль) и 2-хлор-N-(α-циано-2-тенил)ацетамид (21,4 г, 0,1 моль) растворяют в метаноле (200 мл). Затем в смесь вводят триэтиламин (15,2 г, 0,15 моль) и смесь кипятят с обратным холодильником в течение 8 часов.
После завершения реакции смесь охлаждают и фильтруют. Остаток промывают холодным метанолом и сушат, получая указанное в заголовке соединение (24,0 г, 0,08 моль) с выходом 75%.
Способ 2
N-(1-(2,6-дихлорфенил) иминопропил)-N'- этилтиомочевину (31 г, 0,1 моль) и 2-xnop-N-(α-циано-2-тенил)ацетамид (21,4 г, 0,1 моль) растворяют в метаноле (200 мл). Затем в смесь вводят триэтиламин (15,2 г, 0,15 моль) и смесь кипятят с обратным холодильником в течение 8 часов.
После завершения реакции смесь охлаждают с использованием холодного метанола и сушат с получением указанного в заголовке соединения (24,0 г, 0,08 моль) с выходом 75%.
1H ЯМР (СDСl3): δ 7,38 (1Н, д), 7,33 (1Н, д), 7,04 (1Н, т), 6,43 (1Н, д), 5,94 (1Н, д, шир.), 5,59 (1Н, с, шир.), 3,26 (2Н, кв.), 2,93 (2Н, кв.), 1,26 (6Н, м).
Пример 7: Синтез N-(α-циано-2-тенил)-2-(этиламино)-4-метил -5-тиазолкарбоксамида
N-этил-N'-(1-фенилиминоэтил) тиомочевину (22,1 г, 0,1 моль) и 2-хлор-N-(α-циано-2-тенил) ацетамид (21,4 г, 0,1 моль) растворяют в этаноле (200 мл). Затем в смесь вводят диизопропилэтиламин (15,5 г, 0,12 моль) и смесь перемешивают при температуре 60oС в течение 10 часов.
После завершения реакции растворитель удаляют отгонкой при пониженном давлении, получая коричневое твердое вещество. Твердое вещество перекристаллизовывают из смеси толуола и воды (объемное соотношение 10/1) с получением указанного в заголовке соединения (22,1 г, 0,07 моль) с выходом 72%.
1H ЯМР (СDСl3): δ 7,36 (1Н, д), 7,30 (1Н, д), 7,04 (1Н, т), 6,10 (1Н, д), 5,99 (1Н, с, шир.), 3,28 (2Н, кв.), 2,53 (2Н, с), 1,30 (3Н, т).
Пример 8: Синтез N-(α-циано-2-тенил)-4-этил-2-(изопропиламино) -5-тиазолкарбоксамида
N-изопропил-N'-(1-фенилиминопропил)тиомочевину (2,5 г, 0,01, моль) и 2-бpом-N-(α-циано-2-тенил)ацетамид (2,6 г, 0,01 моль) растворяют в метаноле (20 мл). Затем в смесь вводят триэтиламин (1,5 г, 0,02 моль) и смесь перемешивают при температуре 60oС в течение 7 часов.
После завершения реакции растворитель удаляют отгонкой под пониженным давлением, получая коричневое твердое вещество. Твердое вещество перекристаллизовывают из смешанного растворителя этанол-вода (объемное соотношение 1/1), получая указанное в заголовке соединение (2,0 г, 0,01 моль) с выходом 60%.
1H ЯМР (СDСl3): δ 7,38 (1Н, д), 7,30 (1Н, д), 7,01 (1Н, т), 6,44 (1Н, д), 6,00 (1Н, д), 5,49 (1Н, с, шир.), 3,61 (1Н, м), 2,91 (2Н, кв.), 1,27 (9Н, м).
Пример 9: Синтез N-этил-N'-(1-(4-феноксифенил)иминопропил)-тиомочевины
N-(4-феноксибензол)пропионамид (120,6 г, 0,5 моль) и пиридин (130,6 г, 1,65 моль) растворяют в дихлорметане (200 г), и смесь охлаждают до 0oС. К смеси по каплям в течение двух часов добавляют оксихлорид фосфора (84,3 г, 0,55 моль), и затем смесь перемешивают при комнатной температуре (20oС) в течение 2 часов с получением N-(4-феноксибензол) пропионимидоилхлорида.
Впоследствии реагент медленно добавляют по каплям в реакционный сосуд, содержащий смесь тиоцианата калия (72,9 г, 0,75 моль) и карбоната калия (159,0 г, 1,5 моль) в ацетоне (500 мл), при температуре 10oС или ниже в течение 2 часов и затем смесь перемешивают в течение 1 часа с получением изотиоцианата N-(4-феноксибензол)пропионимидоила. В реакционный сосуд по каплям добавляют этиламин (64,4 г, 1,0 моль), при поддержании температуры 10oС или менее в течение 2 часов, и затем смесь перемешивают в течение 1 часа.
После завершения реакции растворитель удаляют отгонкой при пониженном давлении. Продукт экстрагируют толуолом и промывают раствором гидроокиси натрия. Затем толуол удаляют перегонкой при пониженном давлении и остаток перекристаллизовывают из изопропилового спирта с получением указанного в заголовке соединения (98,2 г, 0,3 моль) с выходом 60%.
1H ЯМР (СDСl3): δ 11,97 (1Н, с.), 8,90 (1H, с), 7,33 (3Н, м), 7,01 (4Н, м), 6,75 (2Н, м), 3,68 (2Н, кв.), 1,25 (3Н, т.), 1,16 (3Н, т).
Пример 10: Синтез N-этил-N'-(1-(4-метоксифекил)иминопропил)-тиомочевины
N-(4-метоксибензол)пропионамид (89,6 г, 0,5 моль) и пиридин (130,6 г, 1,65 моль) растворяют в дихлорметане (200 г), и смесь охлаждают до 0oС. К смеси по каплям в течение двух часов добавляют оксихлорид фосфора (84,3 г, 0,55 моль), и затем смесь перемешивают при комнатной температуре (20oС) в течение 2 часов с получением N-(4-метоксибензол) пропионимидоилхлорида.
Впоследствии реагент медленно добавляют по каплям в реакционный сосуд, содержащий смесь тиоцианата калия (72,9 г, 0,75 моль) и карбоната калия (159,0 г, 1,5 моль) в ацетоне (500 мл), при температуре 10oС или менее в течение 2 часов, и смесь перемешивают в течение 1 часа с получением N-(4-метоксибензол)пропионимидоилизотиоцианата. В реакционный сосуд по каплям добавляют этиламин (64,4 г, 1,0 моль) при поддержании температуры 10oС или менее в течение 2 часов, и затем смесь перемешивают в течение 1 часа.
После завершения реакции растворитель удаляют перегонкой при пониженном давлении. Продукт экстрагируют толуолом и промывают раствором гидроокиси натрия. Затем толуол удаляют перегонкой при пониженном давлении, и остаток перекристаллизовывают из изопропилового спирта с получением указанного в заголовке соединения (84,9 г, 0,32 моль) с выходом 64%.
1H ЯМР (СDСl3): δ 11,90 (1Н, с.), 8,12 (1H, с.), 6,87 (2Н, д.), 3,80 (3Н, с.), 3,69 (2Н, кв.), 2,24 (2Н, кв.), 1,26 (3Н, т.), 1,14 (3Н, т).
Пример 11: Синтез N-этил-N'-(1- (4-нитрофенил) иминопропил) -тиомочевины
N-(4-ниробензол)пропионамид (19,4 г, 0,1 моль) и пиридин (26,1 г, 0,33 моль) растворяют в дихлорметане (30 г), и смесь охлаждают до 0oС. К смеси по каплям в течение двух часов добавляют оксихлорид фосфора (16,9 г, 0,11 моль), и затем смесь перемешивают при комнатной температуре (20oС) в течение 2 часов с получением N-(4-нитробензол)пропионимидоилхлорида.
Впоследствии реагент медленно добавляют по каплям в реакционный сосуд, содержащий смесь тиоцианата калия (14,6 г, 0,15 моль) и карбоната калия (31,8 г, 0,3 моль) в ацетоне (100 мл), при температуре 10oС или менее в течение 2 часов, и смесь перемешивают в течение 1 часа с получением N-(4-нитробензол)пропионимидоилизотиоцианата. В реакционный сосуд по каплям добавляют этиламин (12,9 г, 0,2 моль), при поддержании температуры 10oС или менее в течение 2 часов, и затем смесь перемешивают в течение 1 часа.
После завершения реакции растворитель удаляют перегонкой при пониженном давлении. Продукт экстрагируют толуолом и промывают раствором гидроокиси натрия. Затем толуол удаляют перегонкой при пониженном давлении, и остаток перекристаллизовывают из изопропилового спирта с получением указанного в заголовке соединения (17,4г, 0,062 моль) с выходом 62%.
1H ЯМР (СDСl3): δ 11,20 (1Н, с.), 8,23 (2Н, д.), 7,88 (1Н, с.), 6,92 (2Н, д.), 3,71 (2Н, кв.), 2,22 (2Н, кв.), 1,28 (3Н, т.), 1,17 (3Н, т).
Пример 12: Синтез N-(α-циано-2-этил)-4-этил-2-(этиламино)-5-тиазолкарбоксамида
Способ А)
Получение целевого соединения с использованием исходного материала с фенокси группой
N-этил-N'-(1-(4-феноксифенил)иминопропил)тиомочевину (32,7 г, 0,1 моль) и 2-хлор-N-(α-циано-2-тенил)ацетамид (21,4 г, 0,1 моль) растворяют в метаноле (200 мл). Затем в смесь вводят триэтиламин (15.2 г, 0.15 моль), и смесь нагревают с обратным холодильником в течение 8 часов. После завершения реакции смесь охлаждают и фильтруют. Остаток промывают холодным метанолом и сушат с получением указанного в заголовке соединения (22,5 г, 0.07 моль) с выходом 70,2%.
Способ В)
Получение целевого соединения с использованием исходного материала с метокси группой
N-этил-N'-(1-(4-метоксифенил)иминопропил)тиомочевину (26,5 г, 0,1 моль) и 2-хлор-N-(α-циано-2-тенил)ацетамид (21,4 г, 0,1 моль) растворяют в метаноле (200 мл). Затем в смесь вводят триэтиламин (15.2 г, 0.15 моль), и смесь нагревают с обратным холодильником в течение 8 часов. После завершения реакции смесь охлаждают и фильтруют. Остаток промывают холодным метанолом и сушат с получением указанного в заголовке соединения (20,0 г, 0,06 моль) с выходом 62,4%.
Способ С)
Получение целевого соединения с использованием исходного материала с нитро группой
N-этил-N-(1-(4-нитрофенил)иминопропил)тиомочевину (28,0 г, 0,1 моль) и 2-хлор-N-(α-циано-2-тенил)ацетамид (21,4 г, 0,1 моль) растворяют в метаноле (200 мл). Затем в смесь вводят триэтиламин (15.2 г, 0.15 моль), и смесь нагревают с обратным холодильником в течение 8 часов. После завершения реакции смесь охлаждают и фильтруют. Остаток промывают холодным метанолом и сушат с получением указанного в заголовке соединения (21,5 г, 0.067 моль) с выходом 67,1%. Данные ЯМР целевого соединения
1H ЯМР (СDСl3): δ 7,38 (1Н, д.), 7,33 (1H, д.), 7,04 (1Н, т.), 6,43 (1H, д. ), 5,94 (1H, д. шир.), 5,59 (1H, с., шир.), 3,26 (2Н, кв.), 2,93 (2Н, кв. ), 1,26 (6Н, м.).
Как описано выше, производные 2-аминотиазолкарбоксамида формулы (I) могут быть получены с высоким выходом при использовании промежуточных соединений формул (II) и (III) по способу настоящего изобретения. С промышленной точки зрения настоящий способ более экономичен, чем известный, благодаря высоким выходам.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ИМИДАЗОЛА, ОБЛАДАЮЩИЕ ИНГИБИРУЮЩЕЙ АКТИВНОСТЬЮ В ОТНОШЕНИИ ФАРНЕЗИЛТРАНСФЕРАЗЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1998 |
|
RU2179975C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНОГО ПОЛИЕНОВОГО СПИРТА (ВАРИАНТЫ), СУЛЬФОНОВОЕ СОЕДИНЕНИЕ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 1998 |
|
RU2196133C2 |
| ИЗБИРАТЕЛЬНО ДЕЙСТВУЮЩИЕ ИНГИБИТОРЫ ТРОМБИНА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1996 |
|
RU2142939C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ 2-ФЕНИЛ-3-НАФТИЛПРОПИОНОВОЙ КИСЛОТЫ | 1997 |
|
RU2193028C2 |
| РОДСТВЕННОЕ ВИТАМИНУ A СОЕДИНЕНИЕ И СПОСОБЫ ЕГО ПОЛУЧЕНИЯ | 1998 |
|
RU2188193C2 |
| ПРОИЗВОДНЫЕ ХИНОЛИН- ИЛИ НАФТИРИДИНКАРБОНОВОЙ КИСЛОТЫ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 1995 |
|
RU2120940C1 |
| ПРОИЗВОДНОЕ 5-(2-ИМИДАЗОЛИНИЛАМИНО)БЕНЗИМИДАЗОЛА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1998 |
|
RU2193562C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ 2,4-ДИОКСОПИРРОЛИДИНА И 2,4-ДИОКСОТЕТРАГИДРОФУРАНА И ЛЕКАРСТВЕННЫЕ СРЕДСТВА, СОДЕРЖАЩИЕ ИХ В КАЧЕСТВЕ АКТИВНОГО ИНГРЕДИЕНТА | 1997 |
|
RU2179976C2 |
| НОВЫЙ СПОСОБ ПОЛУЧЕНИЯ ИОПРОМИДА | 2009 |
|
RU2451667C1 |
| ПРОИЗВОДНЫЕ ПИРИДАЗИН-3-ОНА И СОДЕРЖАЩИЕ ИХ ЛЕКАРСТВЕННЫЕ СРЕДСТВА | 2000 |
|
RU2232755C2 |

Изобретение относится к способу получения производных 2-аминотиазолкарбоксамида формулы I, в которой R1 представляет C1-5 алкил с прямой или разветвленной цепью, R2 представляет С1-3 алкил, путем взаимодействия соединения формулы II, в которой R3 представляет фенил, который может быть необязательно моно-пентазамещенным независимо хлором, метокси, этокси, фенокси или нитро, с соединением формулы III, в которой Y представляет уходящую группу, в растворителе и в присутствии основания. Соединение формулы II, в которой R1 представляет C1-5 алкил с прямой или разветвленной цепью, R2 представляет C1-3 алкил, R3 представляет фенил, который может быть необязательно моно-пента-замещенным независимо хлором, метокси, этокси, фенокси или нитро, и соединение формулы III, в которой Y представляет хлор или бром. Технический результат - усовершенствование способа получения 2-аминотиазолкарбоксамида. 3 c. и 7 з.п. ф-лы, 1 табл.
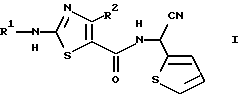
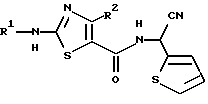
в которой R1 представляет C1-5 алкил с прямой или разветвленной цепью;
R2 представляет C1-3 алкил;
отличающийся тем, что соединение, представленное следующей формулой II: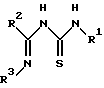
в которой R1 и R2 имеют значения, определенные выше, и R3 представляет фенил, который может быть необязательно моно-пента-замещенным независимо хлором, метокси, этокси, фенокси или нитро, подвергают реакции с соединением, представленным следующей формулой III: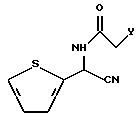
в которой Y представляет уходящую группу, в растворителе и в присутствии основания.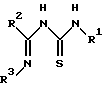
в которой R представляет C1-5 алкил с прямой или разветвленной цепью;
R2 представляет C1-3 алкил;
R3 представляет фенил, который может быть необязательно моно-пента-замещенным независимо хлором, метокси, этокси, фенокси или нитро.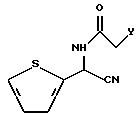
в которой Y представляет хлор или бром.
| Фильтрующий элемент | 1977 |
|
SU639574A1 |
| СПОСОБ ПОЛУЧЕНИЯ НЕПРЕДЕЛЬНЫХ УГЛЕВОДОРОДОВ | 0 |
|
SU292937A1 |
| МИКШЕР ПРОГРАММ ЦВЕТНОГО ТЕЛЕВИДЕНИЯ | 1971 |
|
SU434620A1 |
| РАСХОДОМЕР ПЫЛИ | 0 |
|
SU313091A1 |
| US 4399075 A, 16.08.1983 | |||
| СПОСОБ ПОЛУЧЕНИЯ ЗАМЕЩЕННЫХ КАРБОКСАМИДОТИАЗОЛОВ | 0 |
|
SU253685A1 |

