И:К)бретение относится к способу получения новых солей нафтилиденовых и хииолиновых соединений общей формулы
где X - атом азота или группа СН; Z - галактуроновая, аспарагиновая, глюконовая или глутами новая кислота при условии, что когда Z - D-гапактуроновая кислота, X не может ознчать атома азота. Цель изобретения - получение новых нафтилиденовых и хинолиновых соединений , обладающих лучшими характеристиками, необходимыми-для парэнтерального ввода в организм, чем из вестные структурные аналоги подобного действия.
Изобретение иллюстрируется нижеследующими примерами.
Пример 1. L-Аспарагат 1-зтил-6-фтор-1 ,4-дигидро-4-оксо-7-(1-пиперазинил)-1,8-нафтиридин-З-карбоновой кислоты.
Смесь 199 мг (1,5 ммоль) L-acnaрагиновой кислоты, 480 мг (1,5 ммоль 1-зтил-6 фтор-1,4-дигидро-4 оксо-7-(1 -пиперазинил)-- , 8-нафтиридин-З-карбоновой кислоты и 10 мл воды нагревают до 60 С. Полученный раствор высушивают при температуре ниже , в результате чего получают 730 мг твердого вещества. 600 мг этого вещества кристаллизуют из раствора 2 мл воды и 4 мл абсолютного этилового спирта. Кристаллы фильтруют, промывают абсолютным этиловым спиртом и высушивают, в результате чего получают 310 мг целевого продукта: Т.Ш1. (с разложением) 214-215 С. (Ы) - 4,3 (с 2, ). Анализ показывает, что продукт содержит 0,8 моль воды на моль соединения.
Пример 2. L-Глутамат 1 этил 6-фтор-1,4-дигидро-4-оксо-7-(1-пиперазинил)-1,8-нафтиридин-З-карбоновой кислоты.
Смесь 220 мг (1,5 ммоль) L-глутаминовой кислоты, 480 мг (1,5 ммоль) 1-этил-6-фтор-1,4-дигидро-4-оксо-7926242
-(1-пиперазинил)-1,8-пафтиридин-З-карбоновой кислоты и 10 мл воды нагревают до 60 С. Полученный раствор охлаждают при температуре ниже ОС, 5 в результате чего получают 690 мг
твердого вещества. 620 мг этого твер дого вещества кристаллиг уют из раствора 2 мл воды и 6 мл абсолютного этилового спирта. Кристаллы фильтру-
10 ют, промывают абсолютным этиловым спиртом и высушивают, в результате чего получают 484 мг целевого продукта с т.пл. (разложения) 196-200°С. W - :0,9° (с 2, HgO).
15 Анализ показывает наличие 0,6 моль воды на моль соединения.
Пример 3. L-Аспарагат I-этил-6-фтор- 1 ,4-дигидро-4-оксо-7г-(1-пиперазинил)-3-хинолинкарбоновой
20 кислоты.
Смесь 1,28 г (4 ммоль 1-этил-6-фтор-1,4-дигидро 4-оксо-7-(1-пиперазинил )-3-хинолинкарбоновой кислоты, 532 мг (4 ммоль) L-аспарагиновой
25 кислоты и 30 мл воды перемешивают при 40-50 С в течение 20 мин и при комнатной температуре в течение 2,5 ч. Раствор осветляют фильтрацией и фильтрат высушивают при температуре
0 ниже О С, в результате чего получают 1,77 г твердого вещества. Это твердое вещество кристаллизуют из 6 мп воды и 20 мл абсолютного этилового спирта. Кристаллы фильтруют, промывают абсолютным этиловым спиртом и простым эфиром и высушивают, в результате чего получают 1,11 г целевого продукта с т.пл. (с одновременным разложением ) 208°С.
0 (ot)p - 5,1 (с 2, ).
Анализ показывает содержание 0,8 моль воды на моль соединения.
Пример 4. L-Глутамат 1-этил-6-фтор- 1,4-диги. ,ро-4-оксо-7- (t -пи-
5 пер зинил)-3-хинолинкарбоновой кислоты. Смесь 1,28 г (4 ммоль) 1-этил-ь-фтор-1,4-дигидро-4-оксо-7-(1-пиперазинил) -3-хинолинкарбоновой кислоты, 538 мг (4 ммоль) L-глутаминовой кислоты и 50 мл воды нагревают над паровой баней. Полученный раствор осветляют путем фильтрации, и фильтрат лиофилизуют, в результате чего
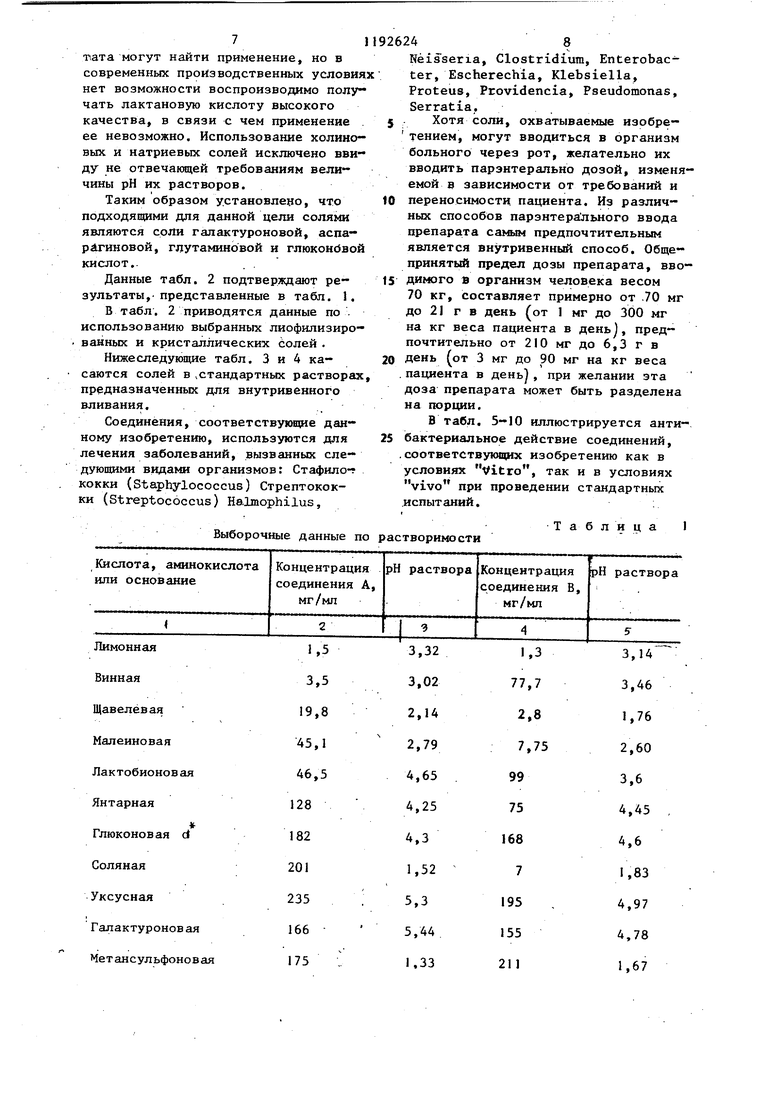
5 получают 1,31 г твердого вещества. Этот продукт кристаллизуют из 6 мл воды и 45 мл абсолютного этилового спирта. Кристаллы фильтруют про3мывают абсолютлым этиловым спиртом и эфиром и высушивают, в результате чего получают 1,39 г целевого проду та с т.гш. (с одновременным разложе нием) 184-185°С.()1 + 1,3° (с 2, ). Анализ показывает содержание 0,1 моль этанола и 0,25 моль воды на моль соединения. Пример 5. D-Галактуронат 1-этш1-6 фтор 1,4 дигидрог4-оксо-7 - (1--пиперазинил)-3--хинолинкарбоново кислоты Смесь 1,28 г (4 ммоль) 1-этил 6-фтор-,4 дигидро 4-оксо-7-(1-пипер зинил)-З-хинолинкарбоновой кислоты 817 мг (4 ммоль) гидрата В галак туроновой кислоты и 30 мл воды перемешивают в течение 1,5 ч при комнатной температуре. Раствор осветляют путем фильтрации, и фильт рат высушивают лри температуре ниже , в результате чего получают 1,98 г твердого продукта. Этот про дукт кристаллизуют из раствора 7,5 воды и 14 мл абсолютного этанола. Кристаллы фильтруют,промывают абсолютным этиловым спиртом и простым эфиром и высушивают, в результате чего получают 1,36 г целевого продукта с т.пл. (с одновременным разложением) 142 С. (Di)+ 8,4 (с 2, НгО). Анализ показывает содержание 0,2 моль этилового спирта и 0,15 мо воды на моль соединения. П р и м р 6. D-Глюконат 1-этил-6-фтор-1,4-дигидро-4-оксо-7- -(1-пиперазинил)-1,8-нафтиридин 3-карбоновой кислоты. Суспензию 2,78 г (8 ммоль) полуторагидрата 1-этил-6-фтор-1,4-дигид- ро-4-оксо-7- (1 -пиперазинил ) -1,8-наф тиридин-3-карбоновой кислоты, 1,21 мл (2,8 ммоль) 2,34 нормального промышленного раствора D-глюконовой кислоты и 60 мл воды перемешивают при комнатной температуре. По истечении 12 ч добавляют дополнительно 2,7 мл (6,3 ммоль) промьшшенного раствора D-глюконовой кислоты в воде, и в течение пяти минут образуется желтый раствор. Этот раствор осветляют путем фильтрации, и фильтрат высушивают при температуре ниже О С. Твердый продукт кристаллизуют из раствора примерно 10 мл воды и 40 мл абсолютного этилового спирта. Крис- 4 галлы перемешивают в течение 4 ч при комнатной температуре, фильтруют, промывают абсолютным этиловым спиртом и простым эфиром и высушивают, в результате чего получают 3,97 г целевого продукта с т.пл. (с разложением) 164-165°С. (о)+ 4,9 (с 2, ). Анализ показывает содержание примерно 1 моль воды на моль соединения. Пример 7. D-Глюконат 1- -этил-6-фтор-1,4-дигидро-4-оксо-7-(1-пиперазинил)-3-хинолинкарбоновой кислоты. A.Суспензию 2,7 г (8 ммоль) моногидрата 1-этил-6-фтор-1,4-дигидро- -4-ОКСО-7-(1-пиперазинил)-3-хинолинкарбоновой кислоты. 1,43 г (8 ммоль)8 -лактона D-глюконовой кислоты и 60 мл воды перемешивают при комнатной температуре в течение 19,5 ч. Раствор осветляют путем фильтрации, и фильтрат высушивают при температуре ниже О С. Твердый продукт кристаллизуют из раствора примерно 10 мл воды и 50 мл абсолютного этилового спирта. Кристаллы фильтруют, промывают абсолютным этиловым спир- том и простьЕМ эфиром и высушивают, в результате чего получают 3,56 г целевого продукта с т.пл. 171-172 С (плавление с разложением) ()i + 5,1° (с 2, ). Анализ показывает содержание О,1 моль воды на моль соединения и слабую гигроскопичность продукта. B.Суспензию 0,98 г (2,9 ммоль) моногидрата 1-этил-6-фтор-1,4-дигидрр-4-оксо-7- (l-пиперазинил) нолинкарбоновой кислоты, 2,48 мл (2,9 ммоль) 1,17-нормальной D-глюконовой кислоты в воде, полученной путем разбавления промышленного раствора, и 20 мл воды перемешивают при комнатной температуре в течение 2 ч. Полученный желтый раствор осветляют путем фильтрации, и фильтрат высушивают при температуре ниже ОС. Твердый продукт перекристаллизовывается из раствора примерно 4 мл воды и 17 мл абсолютного этилового спирта. Кристаллы фильтруют, промывают абсолютным этиловым спиртом простым эфиром и высушивают, в результате чего получают 1,35 г цеевого продукта в виде кристаллов: . пл. (с одновременным разложением; 165-168 С. 5,7 (с 2, I О). (()i)g - t - 2 Анализ показывает содержание примерно 0,2 моль на моль соединения. D-глюконатные соли, полученные из промышленного раствора глюконовой кислоты (примеры 6 В и 7 BJ, имеют более цизкие температуры разложения, чем соответствукяцие соли, полученные из 8 -лактона D-глюконовой кислоты. Глюконатные соли, полученные из промьшшенного раствора D-глюконовой кислоты, имеют также более сильное окрашивание (получаются более темные растворы, чем соответствующие соли, полученные из 8 -лактона D-глю коновой кислоты, в связи с этим получение глюконатной соли с использованием о -лактона D-глюконовой кислоты является предпочтительным способом осуществления изобретения. В табл. 1 представлены данные растворимости и величины рН, которые демонстрируют, что большинство фарма цевтически пригодных солей, полученных из соединений А и В П-этил-6-фтор-1J4 дигидро-4-оксо-7- 1-пипера зинил)-3-хинолинкарбоновая кислота и 1-ЭТИЛ-6-ФТОР-1,4-дигидро-4-оксо-8- (1-пиперазонил -1,8-нафтипиридин- -3 карбоновая кислота соответственн не могут использоваться для приготов ления ценных парэйтерально вводимых в организм дозированных форм, содержащих эти соединения. I Данные, приведенные в табл. 1, получены при осуществлении процедуры выбора растворимости. . В мерные колбы вводят отвешенное количество соединения А и соединения В, рассчитанное по выбранным концентрациям. Равномолярные количества присоединенной кислоты, аминокислоты или основания вводят в мерные колбы либо в виде раствора, либо в сухрм виде. Вводят деионизйрованнук) воду до такого объема, чтобы достига лись предварительно заданные концентрации. Все колбы подвергаются акустическому воздействию в течение 15 мин, вьщерживаются при комнатной температуре в течение 30 мин, затем содержимое колбы фильтруют через воронку из оплавленного матированного стекла. После установления величи ны рН фильтрата соответствующие разбавленные растворы вводят в 0,1 н. НС1 для ультрафиолетового спектроФотометрического определения концен4 , 1 рации. Десятикратно разбавленные растворы исходных фильтратов вводят в стандартные растворы (IV). Эти разбавленные растворы анализируют, определяя выпадение осадка. Результаты влияния 10-кратного разбавления растворимых форм соединений А и В стандартными растворами (IV) приведены в табл. 3 и 4. При выборе растворимости первый, эксперимент проводят для приготовления растворов, которые должны иметь конечную концентрацию соединения А или соединения В примерно 25 мг/мл. Те кислоты и основания, которые не придают достаточную растворимость материалу для достижения заданной концентрации, рассматриваются как непригодные для данной цели. Кислоты и основания, которые могут найти полезное применение, это такие кислоты и основания, которые способны образовьшать растворы, содержащие по меньшей мере 150 мг/мл соединения А или соединения В. Эти более концентрированные растворы, которые кроме того имеют величину рН в пределах примерно 4-8, рассматриваются как растворы, успешно прошедшие процедуры выбора. Не прогнозируемые ранее свойства солей, полученньк из соединений А и В, ясны из результатов представленных в табл. 1. Эти свойства рассматриваются с точки зрения требований, необходимых для получения парэнтерально вводимых в организм дозированных форм препарата. Так, например, хлоргидрат соединения А обладает удовлетворяющей требованиям растворимостью, но не удовлетворяющей требованиям величиной рН, в то время как хлоргидрат соединения В не удовлетворяет требованиям ни в отношении растворимости, ни в отношении величины рН. Метансульфонаты и изотионаты как соединения А, так и соединения В проявляют хорошую растворимость, но не удовлетворяют в отношении рН, так же как и. обе соли, полученные из L-цистеиновой кислоты. Соли ацетата как соединения А, так и соединения В могут быть применены согласно данному изобретению, но они недостаточно хорошо лиофилизируются.в связи с чем использование их ограничено. Из данных табл. 1 ясно, что соли лакпата, могут найти применение, но в современных производственных условия нет возможности воспроизводимо полу чать лактановую кислоту высокого качества, в связи с чем применение ее невозможно. Использование холиновых и натриевых солей исключено ввиду не отвечающей требованиям величины рН их растворов.
Таким образом установлено, что подходящими для данной цели солями являются соЛи галактуроновой, аспарАгиновой, глутамииовой и глюконЬвой кислот,.
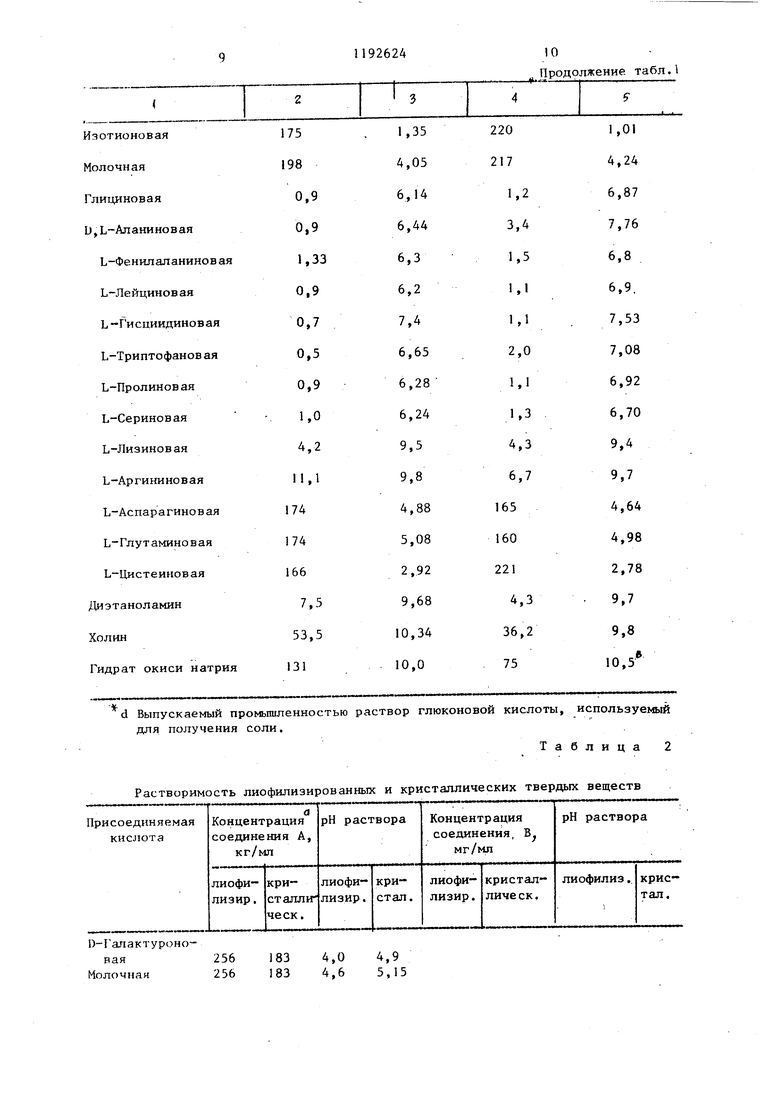
Данные табл. 2 подтверждают результаты,- представленные в табл. 1.
В табл. 2 приводятся данные по . использованию выбранных лиофилизированных и кристаллических солей .
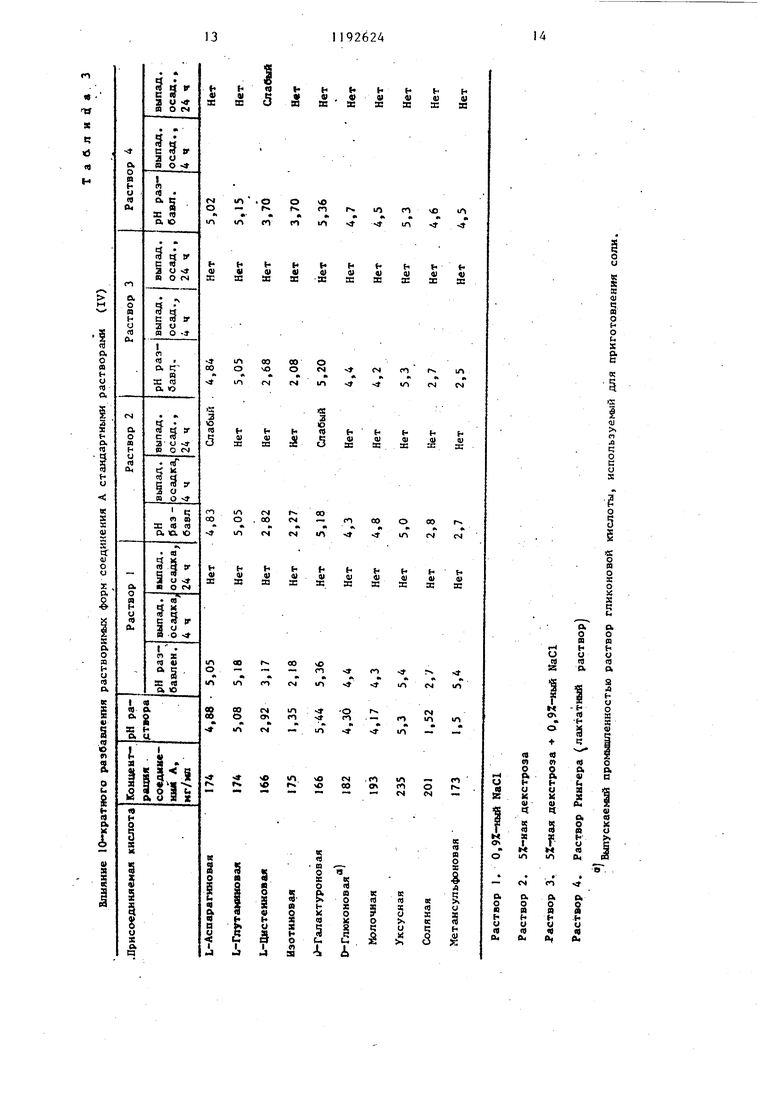
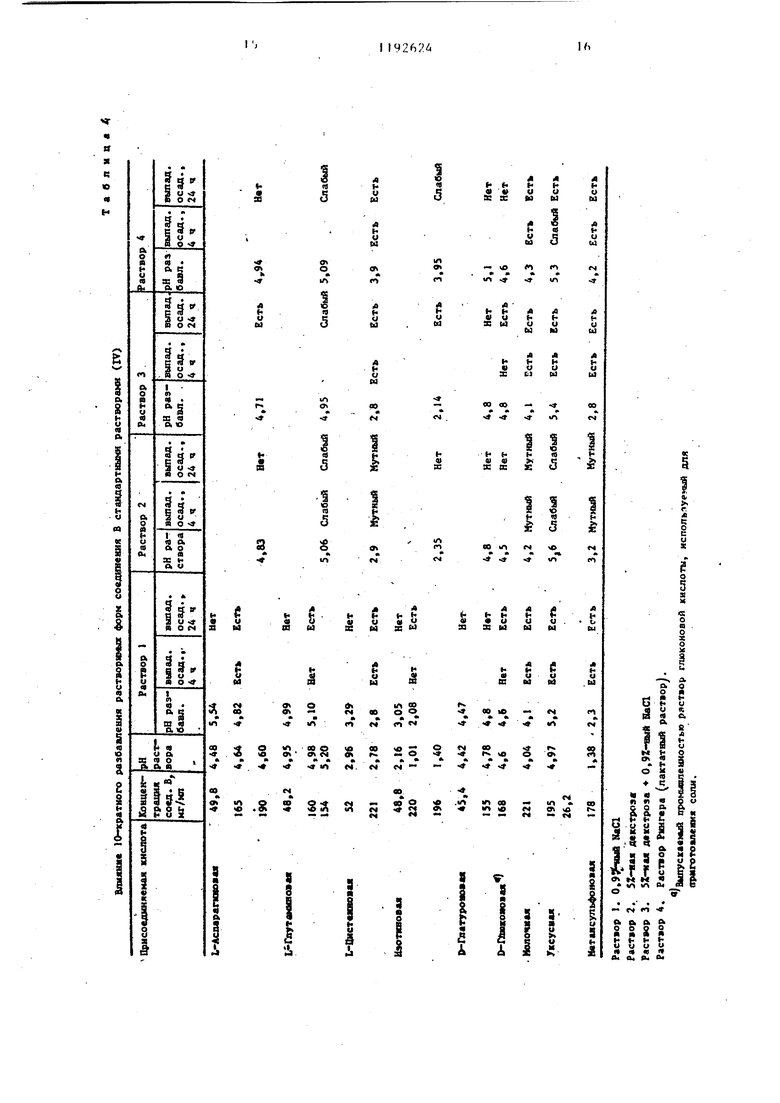
Нижеследующие табл. 3 и 4 касаются солей в стандартных растворах предназначенных для внутривенного вливания..
Соединения, соответствующие данному изобретению, используются для лечения заболеваний, вызванных следующими видами организмов: Стафило- кокки (Staphylococcus) Стрептококки (streptococcus) Halmophilus,
Выборочные данные по растворимости
Neisseria, Clostridium, Enterobac ter, Escherechia, Klebsiella, Proteus, Providencia, Pseudomonas,
Serratia. Хотя соли, охватываемые изобретением, могут вводиться в организм больного через рот, желательно их вводить парэнтерально дозой, изменяемой в зависимости от требований и
переносимости пациента. Из различных способов парзнтерального ввода препарата самьм предпочтительным является внутривенный способ. Общепринятый предел дозы препарата, вводимого в организм человека весом 70 кг, составляет примерно от .70 мг до 21 г в день (от 1 мг до 300 мг на кг веса пациента в день, предпочтительно от 210 мг до 6,3 г в
день (от 3 мг до 90 мг на кг веса .пациента в день, при желании эта доза препарата может быть разделена на порции.
В табл. 5-10 иллюстрируется антибактериальное действие соединений, .соответствующих изобретению как в условиях vitro, так и в условиях vivo при проведении стандартных .испытаний.
Таблица





СПОСОБ ПОЛУЧЕНИЯ НАФТИЛИДЕНОВЫХ И ХИНОШШОВЫХ СОЕДИНЕНИЙ общей формулы где X - этом азота или группа СН; Z - D-галактуроновая, L-аспарагиновая, D-глюконовая или Lглутаминовая кислота при условии , что когда Z - D-ra-лактуроновая кислота, X не может означать атома азота, отличающийся тем, что соединение общей фЬрмулы О
1,5
3,5
19,8
45,1
46,5
я
128
182 d
201
235
166
ая
175 оВс1я
3,14
1,3
3,46 77,7 2,8
1,76 7,75
2,60 99 75
3,6
4,45 168 7
4,6
1,83
195
4,97
4,78 155 211
1,67
Изотионовая
Молочная
Глшдиновая
и,Ь-Аланиновая
Ь-Фенилаланиновая
L-Лейциновая
L-Гисциидиновая
L-Триптофановая
L-Пролиновая
L-Сериновая
L-Лизиновая
L-Аргининовая
L-Аспарагиновая
Ь Глутаминовая
L-Цистеиновая
/.иэтаноламин
Холин
Гидрат окиси натрия d Выпускаемый промышленностью раствор для получения соли. Растворимость лиофилизированных и
1)-Галактуроновая256 183 4,0 4,9
Молочная256183 4,6 5,15
220
1,35
1,01 4,05
217 1,2 4,24 6,14 6,87 6,44 3,4 7,76 6,3 1,5 6,8 6,2 1,1 6,9, 7,А 1,1 7,53 6,65 2,0 7,08 6,28
4,3 9,4 6,7 9,8 9,7 4,88 165 4,64 5,08 160 4,98 221 2,92 2,78 9,68
,3 9,7 36,2 ,34 9,8 10,5 75 ,0 глюконовой кислоты, используемый Таблица 2 кристаллических твердых веществ
11
Концентрации не являются необходимыми максимальными значениями, за
исключением хлоргидрата.
Для приготовления солей используетсяS -лактон глюконовой кислоты..
12
1192624 Продолжение табл.2
ft
TJ
X
e; «) «I
H
sr
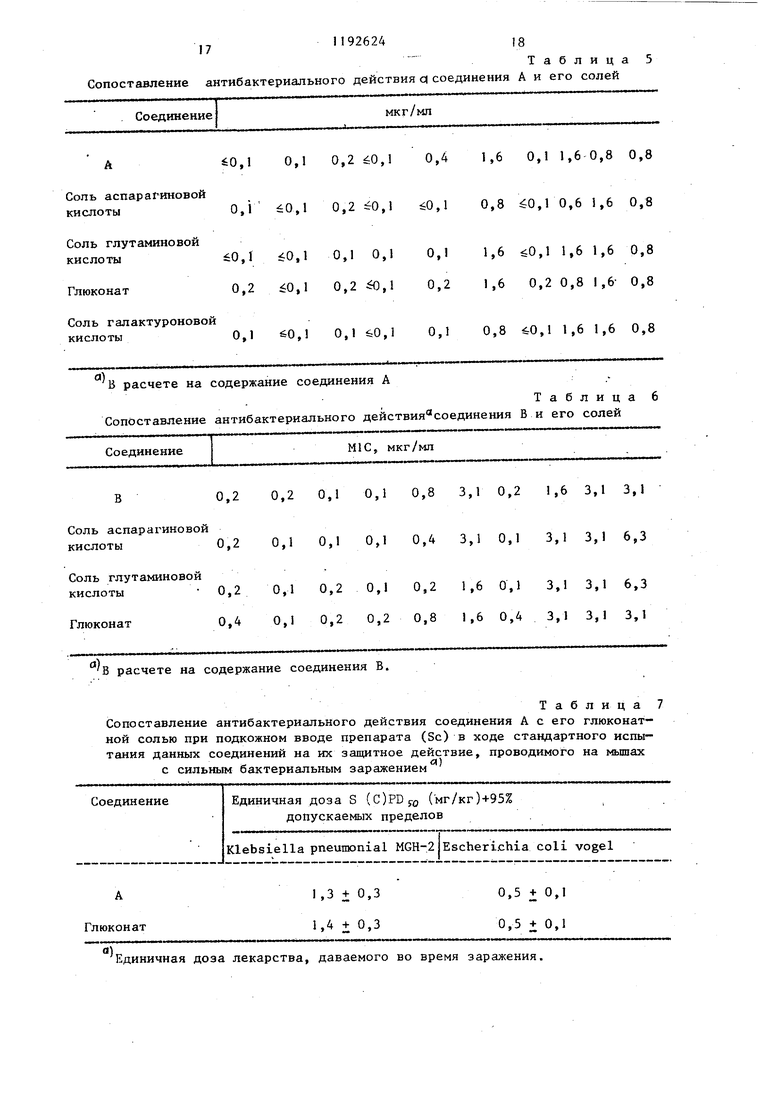
« а я к в |« 1-1 Сопоставление
Соединение А60,1 0,1 0,2 ёО,1 Соль аспарагиновой 0,1 0,1 0,,1 кислоты
Соль глутаниновой
кислоты 0,1 0,1 0,1 0,1 0,1 1,6 0,1 1,6 1,6 0,8
Глюконат0,2 0,1 0,2 ,1 0,2 1,6 0,2 0,8 1,6- 0,8
Соль галактуроновой
кислоты0,1 0,1 0,1 60,1 0,1 0,8 60,1 1,6 1,6 0,8
В расчете на содержание соединения А Сопоставление антибактериального действия - соединения В и его солей
Соединение В0,2 0,2 0,1 0,1 Соль аспарагиновой 0,2 0,1 0,1 0,1 кислоты Соль глутаминовой кислоты 0,2 0,1 0,2 0,1 Глюконат 0,4 0,1 0,2 0,2
В расчете на содержание соединения В.
Сопоставление антибактериального действия соединения А с его глюконатной солью при подкожном вводе препарата (Sc) в ходе стандартного испытания данных соединений на их защитное действие, проводимого на мьшах с сильным бактериальным заражением
Единичная доза S {C)PDyo (мг/кг)+95%
Соединение допускаемых пределов
Klebsiella pneumonial MGH-2 Escherichia coli vogel A 1 ,3 + 0,3 Глюконат 1,4+0,3 ., Единичная доза лекарства, даваемогово
мкг/мп
Таблица 6
М1С, мкг/мл
Таблица 7
-2 Escherichi; ,7119262418 антибактериального действия а соединения А и его солей Таблица 5 0,4 1,6 0,1 1,6 0,8 0,8 60,1 0,8 Й0,1 0,6 1,6 0,8 0,8 3,1 0,2 1,6 3,1 3,1 0,4 3,1 0,1 3,1 3,1 6,3 0,2 1,6 0,1 3,1 3,1 6,3 0,8 1,6 0,4 3,1 3,1 3,1 0,5 + 0,1 0,5+0,1 время заражения.
Сопоставление антибактериального действия соединения А с его глюконатной солью при вводе препарата через рот (РО) в ходе стандартного испытания даргаых соединений на их защитное действие, проводимого на мьшах с сильным бактериальным заражением
15+3
А 13 + 6 Глюконат
Единичная доза лекарства у даваемого при заражении.
Сопоставление антибактериального действия соединения В с его солями при подкожном вводе препарата (si) в ходе стандартного испытания данных соединений на их . защитное действие проводимого на мьшах с сильным бактериальным заражением
Единичная доза лекарства, вводимого во время заражения.
Сопоставление антибактериального действия соединения В с его солями при вводе препарата через рот (РО) в ходе стандартного испытания на их защитное действие, проводимого на мышах с сильным бактериальным
заражением
14 + 7,0
13 + 3,0
Таблица 8
3,3 + 0,5 3,9 + 0,9
Таблица 9
Таблица 10
3,1 + 0,7
2,9 + 1,0 Соль глутаминовой кислоты12,0+3,0 Глюконат+ а), Единичная доза лекарства, даваемого при 2,9 + 1,0 2,9+0,7 заражении.
| СПОСОБ РЕЗАНИЯ ДРЕВЕСИНЫ И ИНСТРУМЕНТ ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ | 1992 |
|
RU2034698C1 |
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| Способ получения фтористых солей | 1914 |
|
SU1980A1 |
| Патент США № 4146719, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| Чугунный экономайзер с вертикально-расположенными трубами с поперечными ребрами | 1911 |
|
SU1978A1 |
