Изобретение относится к технологии производства полиамидов, более конкретно к способу непрерывного получения полиамидов, способу получения высокомолекулярного поликапролактама и поликапролактаму с молекулярным весом 3000 - 14000 г/моль.
В заявке DE N 2443566 описан способ непрерывного получения полиамидов путем непрерывного перемещения смеси по меньшей мере одного лактама и 1 - 15 вес.% воды, а также, в случае необходимости, других полиамидобразующих компонентов, в котором смесь исходных веществ на первой стадии нагревают до температур от 210 до 330oC, на второй стадии смесь поликонденсации подвергают адиабатическому сбросу давления напряжения, после чего на дальнейших стадиях смесь полимеризуют до высокомолекулярных полиамидов, при этом
а) смесь исходных веществ на первой стадии при давлениях, находящихся свыше соответствующих давлений пара исходных веществ и предотвращающих образования паровой фазы, нагревают в течение 5 минут до 2 часов, предпочтительно 10 до 60 минут, до достижения конверсии, равной 70%, предпочтительно 80%;
б) давление смеси поликонденсации на второй стадии сбрасывают до давления, равного 1 - 11 бар, предпочтительно 1 - 6 бар, и непосредственно после этого на третьей стадии, предпочтительно вместе с образовавшимся во время сброса давления водяным паром при подводе тепла и испарении основного количества воды при указанном давлении или более низком давлении в течение меньше 10 минут, предпочтительно меньше 5 минут, реакционную смесь нагревают до температур от 250 до 350oC, предпочтительно от 260 до 280oC, и
в) смесь поликонденсации на четвертой стадии отделяют от водяного пара и на дальнейших стадиях полимеризуют до высокомолекулярных полиамидов.
Недостаток известного способа заключается в том, что вязкость расплава получаемого таким образом поликапролактама слишком высока. Высокая вязкость расплава, как правило, вызывает проблемы при подаче расплава и при отводе теплоты реакции. В связи с этими проблемами образуются, как правило, отложения на внутренних стенках производственного оборудования, что, между прочим, приводит к ухудшению качества получаемого продукта. Кроме того, обеспечиваемый известным способом выход на объем/время низок, а низкомолекулярный поликапролактам со степенью конверсии свыше 85% не доступный.
Задачей настоящего изобретения является разработка способа получения полиамидов, не обладающего вышеуказанными недостатками известного способа.
Поставленная задача решается предлагаемым способом непрерывного получения полиамидов из смеси по меньшей мере одного лактама и воды в полиамидобразующих условиях, при этом на первой стадии указанную смесь нагревают при давлении, при котором реакционная смесь является однофазной и жидкой, а на второй стадии подвергают адиабатическому сбросу давления и дальнейшей полимеризации, за счет того, что на первой стадии используют смесь, содержащую 0,5 - 7 вес.% воды, указанную смесь нагревают до температуры 220 - 310oC и полимеризуют до достижения конверсии, равной по меньшей мере 85%, после чего на второй стадии после адиабатического сброса давления при температуре 215 - 300oC продолжают полимеризацию.
Применяемая в предлагаемом способе смесь может дополнительно содержать дикарбоновые кислоты, диамины или их смеси и/или целевые добавки.
В качестве лактама можно применять, например, капролактам, энантлактам, каприллактам и лауриллактам и их смеси, предпочтительно капролактам.
В качестве дикарбоновых кислот пригодны, например, алкандикарбоновые кислоты с 6-12 атомами углерода, в частности с 6-10 атомами углерода, такие, как, например, адипиновая кислота, пимелиновая кислота, пробковая кислота, азелаиновая кислота или себациновая кислота, а также терефталевая кислота и изофталевая кислота, в качестве диаминов, например, алкилдиамины с 4-12 атомами углерода, в частности с 4-8 атомами углерода, такие, как, например, гексаметилендиамин, тетраметилендиамин или октаметилендиамин, кроме того м-ксилилендиамин, бис-(4- аминофенил)метан, бис-(4-амино-фенил)-пропан-2,2 или бис-(4- аминоциклогексил)метан, смеси дикарбоновых кислот любого состава, смеси диаминов любого состава и смеси дикарбоновых кислот и диаминов, предпочтительно в эквивалентном соотношении, например, в случае смесей, содержащих гексаметилендиаммонийадипат, гексаметилен-диаммонийтерефталат или тетраметилендиаммонийадипат, предпочтительно гексаметилендиаммонийадипат и гексаметилендиаммонийтерефталат, в количествах от 0 до 60%, предпочтительно от 10 до 50%, от веса исходных компонентов. Особенное техническое значение имеют поликапролактам и полиамиды, состоящие из капролактама, гексаметилендиамина, а также адипиновой кислоты, изофталевой кислоты и/или терефталевой кислоты.
Согласно предпочтительной форме осуществления предлагаемого способа применяют капролактам и гексаметилендиаммонийадипат (так называемую соль АГ), при этом соль АГ применяют в виде водного раствора. Обычно применяют капролактам и гексаметилендиаммонийадипат в молярном соотношении в диапазоне от 99,95 : 0,05 до 80 : 20, предпочтительно от 95 : 5 до 85 : 15.
В качестве целевых добавок можно назвать, например, пигменты, такие, как, например, двуокись титана, двуокись кремния или тальк, регуляторы роста цепи, такие, как, например, алифатические и ароматические карбоновые и дикарбоновые кислоты, такие, как, например, пропионовая кислота или терефталевая кислота, стабилизаторы, такие, как, например, галогениды меди (I) и галогениды щелочного металла, затравочные вещества, такие, как, например, силикат магния или нитрид бора, катализаторы, такие, как, например, фосфористая кислота, а также антиокислители в количестве от 0 до 5%, предпочтительно от 0,05 до 1%, от веса исходных компонентов. Целевые добавки добавляют, как правило, перед гранулированием и перед, во время или после, предпочтительно после, полимеризации.
Предлагаемый способ осуществляют, как правило, таким образом, что смесь лактама и 0,5-7 вес.%, предпочтительно 1 - 4,5 вес.%, особенно предпочтительно 2 - 3 вес.%, воды, целесообразно предварительно нагретой до температуры 75 - 90oC, подают в реактор, при этом реакционную смесь нагревают до температуры 220 - 310oC, предпочтительно 240 - 290oC.
Реактор преимущественно оснащен встроенными элементами, такими, как, например, упорядоченные смесительные элементы (так называемые насадки Зульцера) или же неупорядоченные смесительные элементы, такие, как, например, насадки (например, кольца Рашига, шарики или кольца Паля). Эти элементы должны обеспечивать минимальную продолжительность пребывания мономеров в расплаве (далее: время реакции), обеспечивающую высокую конверсию. Кроме того, они позволяют в основном предотвращать образование зон, в которых подача расплава не происходит или лишь в незначительной степени (так называемые "мертвые зоны"), и эффект обратного смешивания.
Как уже указывалось выше, давление реакции выбирают согласно изобретению так, что реакционная смесь является однофазной и жидкой. Это позволяет предотвратить образование газовых "подушек", вызывающих пульсацию потока, что обуславливало бы эффект обратного смешивания и неравномерную полимеризацию. При этом давление, как правило, составляет 5 - 30 бар, предпочтительно 8 - 18 бар (абсолютно).
Время реакции, в основном зависящее от температуры, давления и содержания воды реакционной смеси, согласно изобретению составляет от 2 до 4 часов, предпочтительно от 2 до 2,5 часов. При времени реакции меньше 2 часов и содержании воды меньше 1 вес.% достигаемая конверсия в общем составляет только меньше 86%. Время реакции свыше 4 часов в общем приводит к плохим выходам на объем/время, что, к тому же, требует применения больших по размеру и технически более сложных реакторов.
При применении капролактама после первой стадии предлагаемого способа обычно получают поликапролактам с молекулярным весом в диапазоне от 3000 до 9000, предпочтительно от 5000 до 6700 г/моль. При этом общая концентрация концевых групп в общем составляет 220 - 670, предпочтительно 300 - 400 ммоль/кг, вязкость расплава равна 100 - 10000, предпочтительно 200 - 4000 мПа•сек (при 270oC). Данный поликапролактам является дополнительным объектом изобретения.
Конверсия (рассчитанная по содержанию экстракта, причем
конверсия = 100 - содержание экстракта)
согласно изобретению составляет по меньшей мере 85%, предпочтительно ≥ 87%, особенно предпочтительно ≥ 89%.
Согласно изобретению находящуюся под давлением реакционную смесь на второй стадии подвергают адиабатическому сбросу давления, при котором необходимое для испарения тепло подводят не снаружи, причем давление на второй стадии составляет, как правило, от 0,1 мбар до 1,1 бар, предпочтительно от 500 до 1050 мбар. При этом реакционная смесь с первой стадии в общем охлаждается до температур в пределах от 215 до 300oC, предпочтительно от 235 до 265oC.
Кроме того, с помощью водяного пара целесообразно удаляют летучие компоненты, как, например, применяемый лактам и дополнительные полиамидофазующие компоненты, а также их улетучивающиеся вместе с водяным паром олигомеры на второй стадии. Согласно дальнейшей предпочтительной форме осуществления изобретения летучие компоненты непрерывно количественно возвращают в процесс, то есть предпочтительно на первую стадию.
Время реакции на второй стадии в общем составляет 2 - 60 минут, предпочтительно 3 - 30 минут.
При применении капролактама после второй стадии предлагаемого способа обычно получают поликапролактам с молекулярным весом в диапазоне от 3000 до 14000, предпочтительно от 6000 до 12000 г/моль. При этом общая концентрация концевых групп в общем составляет 140 - 670, предпочтительно 170 - 330 ммоль/кг, вязкость расплава равна 100 - 10000, предпочтительно 200 - 4000 мПа•сек (при 270oC). Данный поликапролактам является дальнейшим объектом изобретения.
Как правило, после второй стадии получают поликапролактам, который можно превращать в куски по стандартным методам, например, путем выпуска полимера в профилированном виде с последующим охлаждением за счет пропускания через водяную ванну и гранулированием.
Согласно изобретению на обеих стадиях процесса осуществляется оптимальное использование теплоты реакции. В качестве примера можно назвать использование высвобождающейся теплоты реакции при нагревании смеси на первой стадии и отказ от обычно требуемого подвода тепла на второй стадии процесса. К тому же, из-за относительно низкого молекулярного веса и одновременно высокой общей концентрации концевых групп получаемые согласно изобретению полимеры обладают высокой реакционноспособностью, имеющейся в распоряжении при дальнейшей переработке. Примерами такой переработки являются связывание предлагаемого полиамида с наполнителями, как, например, стекловолокнами, и другими добавками и получение блок- сополимеров путем смешивания в расплавленном состоянии с дальнейшими полимерами.
Получаемый предлагаемым способом низкомолекулярный поликапролактам можно переводить в высокомолекулярный поликапролактам за счет того, что поликапролактам с молекулярным весом 3000 - 9000 г/моль или 3000 - 14000 г/моль либо подвергают газофазной экстракции и одновременно в твердой фазе дополнительной конденсации при термообработке либо экстрагируют водой или метанолом и в твердой фазе подвергают дополнительной конденсации при нагревании.
В случае обоих вышеуказанных вариантов получения высокомолекулярного поликапролактама получают целевой продукт с коэффициентом вязкости, составляющей, как правило, от 140 до 350 мл/г.
Благодаря тому, что экстракции подвергают низкомолекулярный полиамид - а не высокомолекулярный полиамид, как согласно прототипу - достигается значительное сокращение времени экстракции, а также общего времени получения полиамидов и, таким образом, высокий выход на объем/время.
Примеры
Определение концевых карбоксильных групп осуществляли путем ацидиметрического титрования (двойное определение). Для этого сначала определяли слепое значение и фактор, потом повторяли определение с применением испытуемого полиамида, на основе которого определяли содержание концевых групп.
Для определения слепого значения 30 мл дистиллированного бензилового спирта нагревали с обратным холодильником в течение 15 минут на нагревательной плите с добавкой нескольких стеклянных выварочных шариков, после чего добавляли 6 капель индикатора (50 мг крезолового красного, растворенного в 50 мл чистого для анализа н-пропанола) и в горячем состоянии титровали с помощью титрованного раствора (смесь 80 мл 0,5-м. метанольного раствора гидроокиси калия и 860 мл н-пропанола, доведенная гексанолом до 2000 мл) до изменения цвета (от желтого до серого).
Для определения фактора испытание повторяли с той лишь разницей, что к бензиловому спирту добавляли 0,015 г гексаметилендиаммонийадипата. Фактор вычисляли по навеске гексаметилендиаммонийадипата: [расход - слепое значение: 131.2].
Для определения проб испытание повторяли с применением 0,5 г испытуемого полиамида.
По уравнению [расход - слепое значение]•фактор: навеска вычислялось содержание концевых карбоксильных групп в ммоль/кг.
Определение концевых аминогрупп осуществляли путем ацидиметрического титрования (двойное определение). Для этого сначала определяли слепое значение и фактор, потом повторяли определение с применением испытуемого полиамида, на основе которого определяли содержание концевых групп.
Для определения слепого значения 25 мл смеси растворителей (1000 г чистого для анализа фенола, 540 г чистого для анализа метанола и 1 мл 0,1-м. метанольного раствора гидроокиси калия) нагревали с обратным холодильником при температуре 150 - 160oC на магнитной смесительной плите в течение 25 минут. После охлаждения смеси до температуры, которую терпит рука, к смеси растворителей добавляли 2 капли индикатора (смесь 0,1 г бензилового оранжевого и 10 мл чистого для анализа метанола, доведенная этиленгликолем до 100 мл, + смесь 500 мг метиленового синего и 5 мл чистого для анализа метанола, доведенная этиленгликолем до 50 мл) и с помощью титрованного раствора (смесь 3,44 мл 70 вес.%-ной хлорной кислоты и 200 мл чистого для анализа метанола, доведенная этиленгликолем до 2000 мл) титровали до изменения цвета (от зеленого до серого).
Для определения фактора испытание повторяли с той лишь разницей, что вместо чистой смеси растворителей применяли 25 мл раствора 0,16 г высушенного гексаметилендиаммонийадипата в 500 мл смеси растворителей. Фактор вычисляли по навеске гексаметилендиаммонийадипата: [расход - слепое значение: 131.2].
Для определения проб испытание повторяли с применением 0,5 г испытуемого полиамида, растворенного в 25 мл смеси указанных растворителей.
По уравнению [расход - слепое значение]•фактор: навеска вычислялось содержание концевых аминогрупп в ммоль/кг.
Вязкость расплава определяли в ротационном вискозиметре (типа Haake RV2) при 270oC.
Молекулярный вес (МВ) определяли на основе содержания концевых групп (в ммоль/кг) по уравнению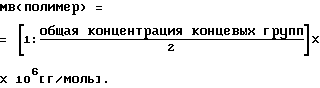
Вязкость раствора обозначается коэффициентом вязкости (KB), указывающим на относительное повышение вязкости растворителя добавлением 0,1 до 1,0 г/100 мл растворенного полимера, деленное на концентрацию в г/100 мл. Коэффициент вязкости увеличивается по мере повышения степени полимеризации.

где 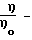 соотношение вязкости,
соотношение вязкости,
η - вязкость раствора полимера определенной концентрации;
ηo - вязкость растворителя.
Вязкость раствора определяли при температуре 25oC.
Для определения содержания экстракта 10 г соответствующего полиамида в 150 мл метанола нагревали с обратным холодильником в течение 16 часов.
Затем еще горячую пробу (около 50 - 60oC) с помощью складчатого фильтра освобождали от твердых веществ, при этом остаток на фильтре три раза промывали метанолом, взятым в количестве по 25 мл. Фильтрат подавали в имеющую несколько стеклянных выварочных шариков, аналитически взвешенную плоскодонную колбу и упаривали на масляной бане при максимальной температуре бани, равной 110oC. После интенсивной внешней очистки колбы оставшийся в колбе экстракт освобождали от еще прилипшего метанола в вакуумном сушильном шкафу в течение 2 часов при 60oC в вакууме водоструйного насоса (20 - 30 торр). Затем экстракт охлаждали в эксикаторе и подвергали аналитическому взвешиванию.
Пример 1
При давлении 1050 мбар 20,4 л/час имеющего температуру 80oC расплава капролактама с водосодержанием 2 вес.% с помощью насоса подавали в атмосфере азота из обогреваемого сборника в обогреваемый теплообменник с рабочей площадью 6 м2 и температурой на входе, равной 270oC, в котором его нагревали до 260oC в течение 2 минут. Давление на напорной стороне насоса составляло 15 бар; приток являлся однофазным и жидким.
Исходный раствор непрерывно с помощью насоса пропускали через цилиндрическую трубу длиной 5000 мм и внутренним диаметром 130 мм, наполненную кольцами Рашига диаметром 5 мм с перемычкой, при этом среднее время пребывания раствора в трубе составляло 2,5 часа.
Цилиндрическую трубу обогревали до 270oC с помощью масла в качестве теплоносителя. Температура продукта на конце трубы составляла 270oC. Давление, при котором реакционная смесь еще являлась однофазной и жидкой, составляло 10 бар. Продукт, который под давлением отбирался в конце цилиндрической трубы, имел следующую характеристику:
Коэффициент вязкости (определенный в качестве 0,55 вес.%-ного раствора в 96 вес.%-ной серной кислоте) = 57 мл/г; содержание концевых кислотных групп = 157 ммоль/кг; содержание концевых аминогрупп = 155 ммоль/кг; содержание экстракта = 10,5%; вязкость расплава (однофазного и жидкого под давлением при 270oC, определенная с помощью ротационного вискозиметра) = 280 мПа•сек.
Через регулировочный клапан реакционную смесь непрерывно подавали в обогреваемый сепаратор с одновременным сбросом давления до атмосферного. При этом реакционная смесь становилась двухфазной и температура снизилась на 8oC до 262oC путем адиабатического испарения воды.
В кубе сепаратора получали плавкий преполимер со следующей характеристикой:
Коэффициент вязкости (определенный в качестве 0,55 вес.%-ного раствора в 96 вес.%-ной серной кислоте) = 81 мл/г; содержание концевых кислотных групп = 99 ммоль/кг; содержание концевых аминогрупп = 102 ммоль/кг; содержание экстракта = 9,7%; вязкость расплава (270oC) = 350 мПа•сек.
Газообразные вторичные пары состояли из 70 вес.% воды и 30 вес.% улетучивающихся вместе с водяным паром компонентов (определение состава осуществляли путем определения показателя преломления лактама в конденсате вторичного пара при 25oC, при этом за основу взяли градуировочную кривую, составленную с учетом различных соотношений капролактама и воды) и отводились из головной части сепаратора, сжижались в конденсаторе, после чего их возвращали в процесс.
После 5-минутного пребывания в сепараторе преполимер с помощью насоса непрерывно отводили из сепаратора и через сопло подавали в профилированном виде в водяную ванну, где его затвердевали и гранулировали. Полученный таким образом преполимер аналогично уровню техники (см. заявку ГДР N 206999) подвергали противоточной экстракции водой и подвергали термообработке до достижения молекулярного веса, равного 28500 г/моль.
Пример 2
При давлении 1050 мбар 20,4 л/час имеющего температуру 80oC расплава капролактама с водосодержанием 2 вес.% с помощью насоса подавали в атмосфере азота из обогреваемого сборника в обогреваемый теплообменник с рабочей площадью 6 м2 и температурой на входе, равной 270oC, в котором его нагревали до 260oC в течение 2 минут. Давление на напорной стороне насоса составляло 15 бар; приток являлся однофазным и жидким.
Исходный раствор непрерывно с помощью насоса пропускали через цилиндрическую трубу длиной 5000 мм и внутренним диаметром 130 мм, наполненную кольцами Рашига диаметром 5 мм с перемычкой, при этом среднее время пребывания раствора в трубе составляло 2,5 часа.
Цилиндрическую трубу обогревали до 270oC с помощью масла в качестве теплоносителя. Температура продукта на конце трубы составляла 270oC. Давление, при котором реакционная смесь еще являлась однофазной и жидкой, составляло 10 бар. Продукт, который под давлением отбирался в конце цилиндрической трубы, имел следующую характеристику:
Коэффициент вязкости (определенный в качестве 0,55 вес.%-ного раствора в 96 вес.%-ной серной кислоте) = 53 мл/г; содержание концевых кислотных групп = 166 ммоль/кг; содержание концевых аминогрупп = 166 ммоль/кг; содержание экстракта = 10,3%; вязкость расплава (однофазного и жидкого под давлением при 270oC) = 260 мПа•сек.
Через регулировочный клапан реакционную смесь непрерывно подают в обогреваемый сепаратор с одновременным сбросом давления до атмосферного. При этом реакционная смесь становилась двухфазной. В этот момент через трубу изотермически (270oC) подавали перегретый водяной пар в расплав, при этом капролактам и дальнейшие улетучивающиеся вместе с водяным паром компоненты, как часть олигомеров капролактама, уносились вместе с паром.
В кубе сепаратора получали плавкий преполимер со следующей характеристикой:
Коэффициент вязкости (определенный в качестве 0,55 вес.%-ного раствора в 96 вес.%-ной серной кислоте) = 91 мл/г; содержание концевых аминогрупп = 95 ммоль/кг; содержание экстракта = 4,8%.
Газообразные вторичные пары состояли из 80 вес.% воды и 20 вес.% улетучивающихся вместе с водяным паром компонентов, отводились из головной части сепаратора и разделялись в колонне. Продукт куба колонны использовали для приготовления исходной смеси, а головной продукт после нагревания до 270oC возвращался на вторую стадию процесса.
Преполимер с помощью насоса непрерывно отводили из сепаратора и через сопло подавали в профилированном виде в водяную ванну, где его затвердевали и гранулировали. Полученный таким образом преполимер аналогично уровню техники (см. заявку ГДР N 206 999) подвергали противоточной экстракции водой и подвергали термообработке до достижения молекулярного веса, равного 33500 г/моль (коэффициент вязкости (определенный в качестве 0,55 вес.%-ного раствора в 96 вес.%-ной серной кислоте) = 250 мл/г, общая концентрация концевых групп = 60 ммоль/кг).
Пример 3
При давлении 1050 мбар 20,4 л/час имеющего температуру 80oC расплава капролактама с водосодержанием 2 вес.% с помощью насоса подавали в атмосфере азота из обогреваемого сборника в обогреваемый теплообменник с рабочей площадью 6 м2 и температурой на входе, равной 270oC, в котором его нагревают до 260oC в течение 2 минут. Давление на напорной стороне насоса составляло 15 бар; приток являлся однофазным и жидким.
Исходный раствор непрерывно с помощью насоса пропускали через цилиндрическую трубу длиной 5000 мм и внутренним диаметром 130 мм, наполненную кольцами Рашига диаметром 5 мм с перемычкой, при этом среднее время пребывания раствора в трубе составляло 2,5 часа.
Цилиндрическую трубу обогревали до 270oC с помощью масла в качестве теплоносителя. Температура продукта на конце трубы составляла 270oC. Давление, при котором реакционная смесь еще являлась однофазной и жидкой, составляло 10 бар. Продукт, который под давлением отбирался в конце цилиндрической трубы, имел следующую характеристику:
Коэффициент вязкости (определенный в качестве 0,55 вес.%-ного раствора в 96 вес.%-ной серной кислоте) = 55 мл/г; содержание концевых кислотных групп = 162 ммоль/кг; содержание концевых аминогрупп = 158 ммоль/кг; содержание экстракта = 10,4%; вязкость расплава (однофазного и жидкого под давлением при 270oC) = 280 мПа•сек.
Через регулировочный клапан реакционную смесь непрерывно подают в обогреваемый сепаратор с одновременным сбросом давления до 90 мбар. При этом реакционная смесь становилась двухфазной и температура снизилась на 12oC до 258oC путем адиабатического испарения воды.
В кубе сепаратора получали плавкий преполимер со следующей характеристикой:
Коэффициент вязкости (определенный в качестве 0,55 вес.%-ного раствора в 96 вес.%-ной серной кислоте) = 75 мл/г; содержание концевых кислотных групп = 117 ммоль/кг; содержание концевых аминогрупп = 121 ммоль/кг; содержание экстракта = 2,5%.
Газообразные вторичные пары состояли из 42 вес.% воды и 58 вес.% улетучивающихся вместе с водяным паром компонентов, отводились из головной части сепаратора, сжижались в конденсаторе и использовали для приготовления исходной смеси.
Преполимер с помощью насоса непрерывно отводили из сепаратора и через сопло подавали в профилированном виде в водяную ванну, где его затвердевали и гранулировали. Полученный таким образом преполимер аналогично уровню техники (см. заявку ГДР N 206 999) подвергали противоточной экстракции водой и подвергали термообработке до достижения коэффициента вязкости, равного 192 мл/г.
Пример 4 - Экстракция метанолом
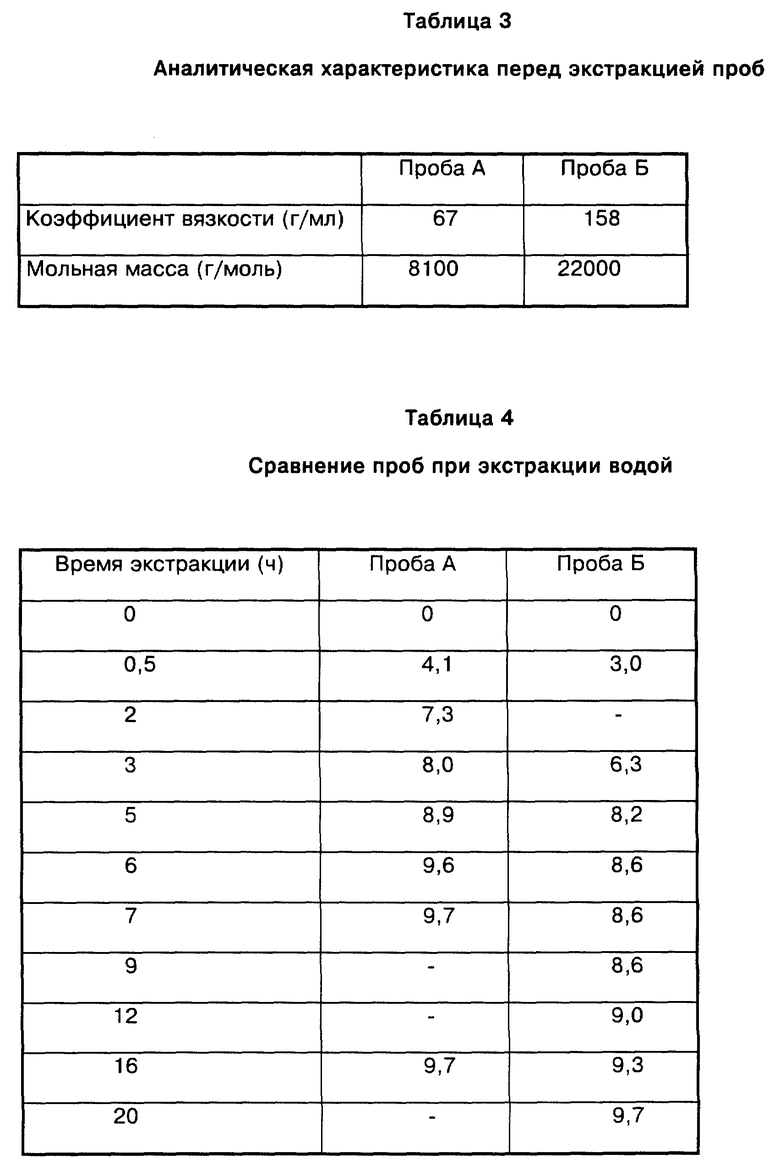
10 г низкомолекулярного поликапролактама (А) (получаемого аналогично примеру 1 с той лишь разницей, что 0,15% от веса мономеров пропионовой кислоты добавляли к смеси мономеров) и 10 г высокомолекулярного поликапролактама (Б) (получаемого в трубе в результате взаимодействия капролактама с 0,5 вес.% воды в течение 13 часов, при этом головная температура составляла 259oC, а температура трубы - 260 до 280oC) в 150 мл метанола нагревали с обратным холодильником в течение определенного периода времени (см. табл. 1 и 2).
Еще теплую пробу (около 50 - 60oC) с помощью складчатого фильтра освобождали от твердых веществ, при этом остаток на фильтре три раза промывали метанолом, взятым в количестве по 25 мл. Фильтрат подавали в аналитически взвешенную плоскодонную колбу, имеющую несколько стеклянных выварочных шариков, и упаривали на масляной бане при максимальной температуре бани 110oC. После интенсивной внешней чистки колбы оставшийся в колбе экстракт освобождали от прилипшего метанола в вакуумном сушильном шкафу в течение 2 часов при 60oC в вакууме водоструйного насоса (20 - 30 торр). Затем экстракт охлаждали в эксикаторе и подвергали аналитическому взвешиванию.
Время экстракции низкомолекулярной пробы А до достижения постоянного конечного значения составляло 6 - 7 часов, а в случае экстракции пробы Б - 20 часов.
Пример 5
Экстракция водой
Повторяли пример 4 с той лишь разницей, что вместо метанола в качестве экстрагента использовали воду.
Время экстракции низкомолекулярной пробы А до достижения постоянного конечного значения составляло 6 - 7 часов, а в случае экстракции пробы Б - 20 часов ( см. табл. 3 и 4).
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ КАПРОЛАКТАМА | 1995 |
|
RU2167862C2 |
| СПОСОБ ПОЛУЧЕНИЕ КАПРОЛАКТАМА ДЕПОЛИМЕРИЗАЦИЕЙ СМЕСЕЙ, КОТОРЫЕ СОДЕРЖАТ ПОЛИМЕРЫ ИЛИ ТЕРМОПЛАСТИЧНЫЕ ФОРМОВАННЫЕ МАТЕРИАЛЫ | 1995 |
|
RU2160253C2 |
| СПОСОБ ПОЛУЧЕНИЯ КАПРОЛАКТАМА ИЗ ПОЛИМЕРОВ, СОДЕРЖАЩИХ КАПРОЛАКТАМ, (ВАРИАНТЫ) И УСТРОЙСТВО ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ | 1995 |
|
RU2154059C2 |
| НИТИ НА ОСНОВЕ ПОЛИКАПРОЛАКТАМА С ОТНОСИТЕЛЬНОЙ ВЯЗКОСТЬЮ 2,0 - 3,0, СПОСОБ ИХ ПОЛУЧЕНИЯ, ПРЯЖА И ПЛОСКИЕ ИЗДЕЛИЯ, ПОЛУЧЕННЫЕ ИЗ НИТЕЙ | 1993 |
|
RU2114939C1 |
| СПОСОБ ВЫДЕЛЕНИЯ КАПРОЛАКТАМА ИЗ СОДЕРЖАЩИХ КАПРОЛАКТАМ ПОЛИМЕРОВ И УСТРОЙСТВО ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ | 1995 |
|
RU2159233C2 |
| ИНГЕРЕНТНО СВЕТО- И ТЕРМОСТАБИЛИЗИРОВАННЫЙ ПОЛИАМИД, СПОСОБ ЕГО ПОЛУЧЕНИЯ, НИТИ, ПОЛУЧЕННЫЕ ЕГО ВЫСОКОСКОРОСТНЫМ ФОРМОВАНИЕМ, И ПОЛОТНА, ПОЛУЧЕННЫЕ ИЗ НИТЕЙ | 1995 |
|
RU2167893C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПОЛИАМИДА ИЗ АМИНОНИТРИЛА И ПОЛИАМИД, ПОЛУЧАЕМЫЙ ИМ | 1997 |
|
RU2214425C2 |
| СПОСОБ ПОЛУЧЕНИЯ КАПРОЛАКТАМА | 1995 |
|
RU2154058C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПОЛИАМИДОВ | 1998 |
|
RU2221819C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПОЛИКАПРОЛАКТАМА | 1970 |
|
SU283942A1 |
Описывается способ непрерывного получения полиамидов из смеси по меньшей мере одного лактама и воды в полиамидобразующих условиях, при этом на первой стадии указанную смесь нагревают при давлении, при котором реакционная смесь является однофазной и жидкой, а на второй стадии подвергают адиабатическому сбросу давления и дальнейшей полимеризации, при этом на первой стадии используют смесь, содержащую 0,5 - 7 вес.% воды, указанную смесь нагревают до 220 - 310°С и полимеризуют до достижения конверсии, равной по меньшей мере 85%, после чего на второй стадии после адиабатического сброса давления при 215 - 300°С продолжают полимеризацию. Кроме того, описываются поликапролактам с молекулярным весом 3000-9000 г/моль (на основе содержания концевых групп), полученный на первой стадии вышеописанного способа при использовании в качестве лактама капролактама, а также поликапролактам с молекулярным весом 3000 - 14000 г/моль (на основе содержания концевых групп), полученный вышеописанным способом при использовании в качестве лактама капролактама. Кроме того, описывается способ получения высокомолекулярного поликапролактама путем поликонденсации низкомолекулярного поликапролактама, который заключается в том, что поликапролактам с вышеуказанным молекулярным весом подвергают газофазной экстракции и одновременно в твердой фазе дополнительной конденсации при термообработке или экстрагируют водой или метанолом и в твердой фазе подвергают дополнительной конденсации при нагревании. 5 с. и 7 з.п. ф-лы, 4 табл.
| ГОЛОВКА ДЛЯ ВЫБРАСЫВАНИЯ ЖИДКОСТИ И СПОСОБ ИЗГОТОВЛЕНИЯ ГОЛОВКИ ДЛЯ ВЫБРАСЫВАНИЯ ЖИДКОСТИ | 2009 |
|
RU2443566C1 |
| SU 686506 A, 1979 | |||
| US 3960820 A, 1976 | |||
| СКОРОСТНОЙ ЦЕНТРОБЕЖНЫЙ СЕПАРАТОР | 0 |
|
SU284968A1 |
| US 5149758 A, 1992 | |||
| US 5218080 A, 1993. | |||

