Область техники, к которой относится изобретение
Настоящее изобретение обеспечивает пиразолпиримидиновые производные и их применения в способах лечения злокачественных заболеваний и расстройств и способах для лечения воспалительных заболеваний и расстройств.
Предпосылки создания изобретения
Семейство казеинкиназ 1 (CK1 или CKI) представляет собой серин/треонинкиназы с шестью членами (изоформы) у людей: α, γ1, γ2, γ3, δ и ε. Они различаются длиной и последовательностью N-концевого (9-76 аминокислот) и особенно C-концевого (24-200 аминокислот) некаталитического домена (Schittek and Sinnberg, Molecular Cancer 2014, 13:231).
CK1δ и CK1ε на 98% идентичны в их киназном домене и на 53% идентичны в их C-концевом регуляторном домене (Fish KJ et al. J Biol Chem 1995, 270:14875-14883). Принимая во внимание, что существует некоторая избыточность в отношении фосфорилирования субстрата CK1, большинство изоформ CK1 имеют разные биологические роли. Широкий диапазон субстратов CK1 показывает, что члены семейства CK1 участвуют во множестве клеточных процессов: от регуляции переноса через мембраны, цитокинеза, везикулярного транспорта, биогенеза рибосом, репарации ДНК, пути передачи сигналов, апоптоза и до циркадного ритма (Knippschild U et al. Cell Signal 2005, 17:675-689; Cheong JK and Virshup DM. Int J Biochem Cell Biol 2011, 43:465-469; Zemp I, et al. J Cell Sci 2014, 127:1242-1253).
CK1α играет роль в формировании митотического веретена в процессе деления клеток и в механизмах репарации ДНК и участвует в метаболизме РНК (Knippschild U et al. Cell Signal 2005, 17:675-689). Это способствует активации mTOR посредством поддерживаемой деградации эндогенного ингибитора mTOR DEPTOR (Duan S et al. Mol Cell 2011, 44:317-324).
CK1α играет главную роль в регуляции сигнального пути Wnt/β-катенина. Авторы настоящего изобретения показали, что CK1α является ключевым компонентом комплекса деструкции β-катенина. Когда Wnt рецепторы не задействованы, CK1α фосфорилирует β-катенин на сериновом остатке S45, что необходимо для инициирования фосфорилирования другой киназы, GSK3 (Amit et al. Genes Dev. 2002 16: 1066-1076).
Фосфорилирование β-катенина посредством GSK3 на остатках T41, S37 и S33 генерирует дегрон убиквитинирования, рекрутируя E3 SCF-β-TrCP, приводя к убиквитинированию и разрушению β-катенина (Clevers H and Nusse R Cell 2012, 149: 1192-1205). Авторы настоящего изобретения также показали, что индуцируемая абляция CK1α в эпителии кишечника мыши запускает массированный ответ эпителиального Wnt, что, к удивлению, не изменяет гомеостаз желудочно-кишечного тракта, с очень незначительным усилением пролиферации и отсутствием онкогенеза (Elyada et al. Nature 2011, 470: 409-413). Это непохоже на последствия острой абляции других компонентов комплекста деструкции β-катенина, таких как APC, что приводит к потере гомеостаза и онкогенезу (O.J. Sansom, O.J. et al. Genes Dev. 2004, 18:1385-1390).
Авторы настоящего изобретения обнаружили, что причиной для поддержания гомеостаза после CK1α абляции является то, что наряду с Wnt активацией абляция CK1α индуцирует некоторые опухоль-супрессорные пути, среди которых ответ на повреждение ДНК (DDR), клеточное старение и активация p53 пути (Elyada E et al. Nature 2011, 470: 409-413, Pribluda A et al. Cancer Cell 2013, 24: 1-5).
В то время как молекулярные механизмы, лежащие в основе активации этих антинеопластических путей, все еще остаются неясными, авторы настоящего изобретения обнаружили, что CK1α абляция индуцирует диспропорционально минимальное повреждение ДНК, без каких-либо признаков ATM активации, что указывает на то, что CK1α-индуцированая активация DDR и p53 по-видимому связана с необычными молекулярными механизмами (Burstain I et al., неопубликованный). Кроме того, авторы настоящего изобретения обнаружили, что CK1α абляция приводит к индукции нового типа воспалительного ответа, называемого паравоспалением, который затрагивает только эпителий, без каких-либо обычных признаков воспалительного ответа (инфильтрация клетками воспалительного происхождения, жар, покраснение, опухоль и воспаление) (Pribluda A et al. Cancer Cell 2013, 24: 1-5, Lasry A and Ben-Neriah Y 2015, Trends in Immunology, Vol. 36: 217-228). Паравоспаление содействует WT p53 активации в супрессии онкогенеза, но при этом переключается на провоцирующий опухоль механизм в отсутствие функционального p53 (Pribluda A et al. Cancer Cell 2013, 24: 1-5, Aran et al., Genome Biol. 2016 Jul 8;17(1):145).
Хотя уже было установлено, что CK1α является основным регулятором p53, авторы настоящего изобретения также обнаружили, что совместная абляция CKIδ и CK1ε в эпителии кишечника также приводит к активации p53, что может синергически действовать с CK1α-индуцированой p53 активацией.
IRAK1 был идентифицирован как терапевтическая мишень для MDS и некоторых разновидностей AML и тройного негативного рака молочной железы (Garrett W. Rhyasen et al., 2013, Cancer Cell 24, 90-104, Rhyasen GW, Bolanos L, Starczynowski DT, 2013, Exp Hematol. 41:1005-7, Zhen Ning Wee et al., 2015, NATURE COMMUNICATIONS, 6:8746). IRAK1 мРНК сверхэкспрессируется у ~20-30% MDS пациентов, и IRAK1 белок существенно сверхэкспрессирован и гиперактивирован в большинстве исследованных образцов костного мозга при MDS. IRAK1 представляет собой серин/треонинкиназу, которая опосредует сигналы, индуцируемые Toll-подобным рецептором (TLR) и Интерлейкин-1 Рецептором (IL1R). После активации рецептора IRAK1 становится фосфорилированным, что затем приводит к рекрутменту TRAF6, приводя к TRAF6 активации NF-κB и JNK путей. Молекулярный источник сверхэкспрессии и/или гиперактиваци IRAK1 при MDS (или AML) не является убедительным. Считают, что сверхэкспрессия TLR или необходимых кофакторов в MDS клонах может приводить к хронической IRAK1 активации даже в отсутствие инфекции. Низкомолекулярные ингибиторы, таргетирующие IRAK1 (Ингибитор IRAK1/4, Amgen Inc.), изначально были разработаны для аутоиммунных и воспалительных заболеваний. Учитывая, что IRAK1 гиперактивирован (то есть фосфорилирован) в MDS, но не нормальных клетках костного мозга, Starczynowski с коллегами показали, что лечение IRAK-Ингибитором (IRAK1/4, Amgen) и выключение IRAK1 приводило к существенному ухудшению пролиферации, прогениторной функции и жизнеспособности MDS клеток in vitro и in vivo. Yu с коллегами показали, что сверхэкспрессия IRAK1 придает клеткам тройного негативного рака молочной железы (TNBC) преимущества роста через NF-κB-связанную секрецию цитокинов, и метастатические TNBC клетки демонстрируют приобретенную IRAK1 зависимость, что приводит к высокой чувствительности к генетическому и фармакологическому ингибированию IRAK1. Лечение паклитакселом TNBC клеток индуцирует сильное IRAK1 фосфорилирование, повышение экспрессии воспалительных цитокинов, обогащение раковых стволовых клеток и приобретенную резистентность к лечению паклитакселом. Фармакологическое ингибирование IRAK1 способно реверсировать резистентность к паклитакселу, запуская массовый апоптоз. Также было обнаружено, что IRAK1 является транскрипционной мишенью DEK и имеет важное значение для выживания клеток рака головы и шеи (Adams AK et al. Oncotarget. 2015, 22; 6(41): 43395-43407) и также в качестве потенциальной мишени в лечении воспалительных и связанных с нарушением иммунной системы расстройств (Bahia MS et al. Cell Signal. 2015 Jun;27(6):1039-55).
Таким образом, авторы настоящего изобретения обнаружили, что соединения по настоящему изобретению способны ингибировать IRAK1, важный апстрим регулятор NF-κB пути, который играет важную роль в гематологических злокачественных опухолях.
Общее описание
Настоящее изобретение обеспечивает соединение, имеющее общую формулу (I), включая любой его стереоизомер или соль:
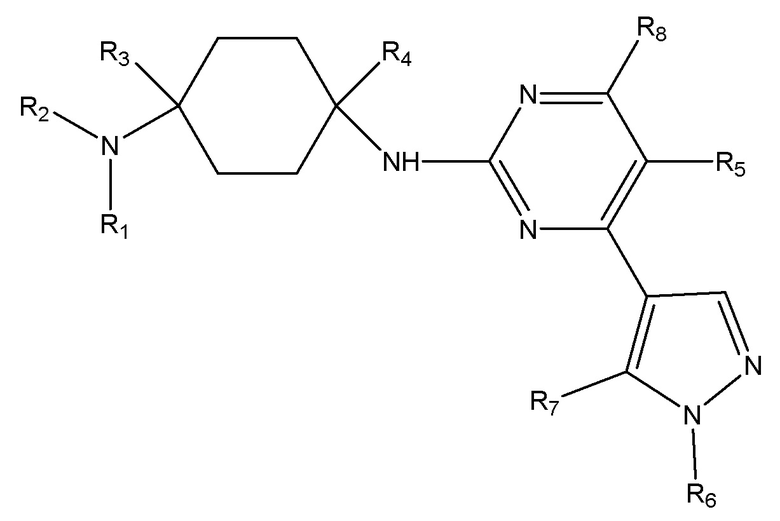
(I)
где
R1 и R2 каждый независимо выбран из H, линейного или разветвленного C1 - C8 алкила, линейного или разветвленного C1 - C5 алкокси, линейного или разветвленного C1 - C5 ацила, C5 - C15 арила, C3 - C7 гетероарила, каждый из которых необязательно замещен по меньшей мере одним из галогенида, гидроксила, сложного эфира, простого эфира, C5 - C15 арила, C3 - C7 гетероарила и амида; или
R1 и R2 вместе с атомом азота, с которым они связаны, образуют 4-7-членное насыщенное, ненасыщенное или ароматическое кольцо, которое необязательно может включать по меньшей мере один из N, O, NH, C=N, C=O или SO2 и необязательно может быть замещенно по меньшей мере одним из линейного или разветвленного C1 - C5 алкила, C5 - C15 арила, C3 - C7 гетероарила, гидроксила, галогенида и циано;
R3 и R4 каждый независимо выбран из H, линейного или разветвленного C1 - C8 алкила, необязательно замещенного по меньшей мере одним из галогенида, гидроксила, алкокси, C5 - C15 арила, C3 - C7 гетероарила, сложного эфира и амида; или
R1 или R2 вместе с R3 и атомом углерода и атомом азота, с которыми каждый из них связан, образуют 4-7-членное насыщенное, ненасыщенное или ароматическое кольцо, которое необязательно может включать по меньшей мере один из N, NH, O, C=N, C=O, SO2 и необязательно может быть замещено по меньшей мере одним из линейного или разветвленного C1 - C5 алкила, C5 - C15 арила, C3 - C7 гетероарила, гидроксила, карбонила и галогенида;
R5 и R8 каждый независимо выбран из H, галогенида, линейного или разветвленного C1 - C8 алкила, линейного или разветвленного C2 - C8 алкенила, линейного или разветвленного C2 - C8 алкинила, необязательно замещенных по меньшей мере одним галогенидом;
R6 выбран из линейного или разветвленного C1 - C8 алкила, линейного или разветвленного C2 - C8 алкенила, линейного или разветвленного C2 - C8 алкинила, C5 - C10 циклоалкила, насыщенного или ненасыщенного 4-6-членного гетероциклила, необязательно замещенных по меньшей мере одним из линейного или разветвленного C1 - C8 алкила, C3 - C7 циклоалкила, 4-6-членного гетероциклила, C5 - C15 арила, C3 - C7 гетероарила, галогенида, гидроксила, C1 - C5 алкилгалогенида;
R7 выбран из линейного или разветвленного C1 - C8 алкила, линейного или разветвленного C2 - C8 алкенила, линейного или разветвленного C2 - C8 алкинила, замещенных по меньшей мере одним C3 - C7 циклоалкилом, 4-6-членным гетероциклилом, C5 - C15 арилом, C3 - C7 гетероарилом, галогенидом, гидроксилом, C1 - C5 алкилгалогенидом.
Настоящее изобретение обеспечивает соединение, имеющее общую формулу (I), включая любой его стереоизомер или соль, где:
R1 и R2 каждый независимо выбран из H, линейного или разветвленного C1 - C8 алкила, линейного или разветвленного C2 - C8 алкенила, линейного или разветвленного C2 - C8 алкинила, линейного или разветвленного C1 - C5 алкокси, линейного или разветвленного C1 - C5 ацила, C5 - C15 арила, C3 - C7 гетероарила, каждый из которых необязательно замещен по меньшей мере одним из галогенида, гидроксила, сложного эфира, простого эфира, C5 - C15 арила, C3 - C7 гетероарила и амида; или
R1 и R2 вместе с атомом азота, с которым они связаны, образуют 4-7-членное насыщенное, ненасыщенное или ароматическое кольцо, которое необязательно может включать по меньшей мере один из N, O, NH, C=N, C=O или SO2 и необязательно может быть замещено по меньшей мере одним из линейного или разветвленного C1 - C5 алкила, линейного или разветвленного C2 - C5 алкенила, линейного или разветвленного C2 - C5 алкинила, C5 - C15 арила, C3 - C7 гетероарила, гидроксила, галогенида и циано;
R3 и R4 каждый независимо выбран из H, линейного или разветвленного C1 - C8 алкила, линейного или разветвленного C2 - C8 алкенила, линейного или разветвленного C2 - C8 алкинила, необязательно замещенных по меньшей мере одним из галогенида, гидроксила, алкокси, сложного эфира, C5 - C15 арила, C3 - C7 гетероарила и амида; или
R1 или R2 вместе с R3 и атомом углерода и атомом азота, с которыми они связаны, образуют 4-7-членное насыщенное, ненасыщенное или ароматическое кольцо, которое необязательно может включать по меньшей мере один из N, NH, O, C=N, C=O, SO2 и необязательно может быть замещено по меньшей мере одним из линейного или разветвленного C1 - C5 алкила, линейного или разветвленного C2 - C5 алкенила, линейного или разветвленного C2 - C5 алкинила, C5 - C15 арила, C3 - C7 гетероарила, гидроксила, карбонила и галогенида;
R5 и R8 каждый независимо выбран из H, галогенида, линейного или разветвленного C1 - C8 алкила, линейного или разветвленного C2 - C8 алкенила, линейного или разветвленного C2 - C8 алкинила, необязательно замещенных по меньшей мере одним галогенидом (в некоторых вариантах осуществления CF3);
R6 выбран из линейного или разветвленного C1 - C8 алкила, линейного или разветвленного C2 - C8 алкенила, линейного или разветвленного C2 - C8 алкинила, C5 - C10 циклоалкила, насыщенного или ненасыщенного 4-6-членного гетероциклила, необязательно замещенных по меньшей мере одним из линейного или разветвленного C1 - C8 алкила, C3 - C7 циклоалкила, 4-6-членного гетероциклила, C5 - C15 арила, C3 - C7 гетероарила, галогенида, гидроксила, C1 - C5 алкилгалогенида;
R7 выбран из линейного или разветвленного C1 - C8 алкила, линейного или разветвленного C2 - C8 алкенила, линейного или разветвленного C2 - C8 алкинила, замещенных по меньшей мере одним C3 - C7 циклоалкилом, 4-6-членным гетероциклилом, C5 - C15 арилом, C3 - C7 гетероарилом, галогенидом, гидроксилом, C1 - C5 алкилгалогенидом.
В некоторых вариантах осуществления R1 и R2 каждый независимо выбран из H, линейного или разветвленного C1 - C8 алкила, необязательно замещенного по меньшей мере одним из галогенида, гидроксила, сложного эфира и амида.
В некоторых вариантах осуществления R1 и R2 каждый независимо выбран из H, линейного или разветвленного C1 - C5 алкокси, необязательно замещенного по меньшей мере одним из галогенида, гидроксила, сложного эфира и амида.
В некоторых вариантах осуществления R1 и R2 каждый независимо выбран из H, C1 - C5 ацила, необязательно замещенного по меньшей мере одним из галогенида, гидроксила, сложного эфира, простого эфира и амида.
В других вариантах осуществления R1 и R2 каждый независимо выбран из H, C5 - C15 арила, необязательно замещенного по меньшей мере одним из галогенида, гидроксила, сложного эфира, простого эфира и амида.
В некоторых вариантах осуществления по меньшей мере один из R1 и R2 представляет собой H.
В некоторых вариантах осуществления R4 представляет собой H. В некоторых вариантах осуществления R3 и R4 представляют собой H.
В некоторых вариантах осуществления R5 выбран из H, Cl и линейного или разветвленного C1 - C4 алкила. В некоторых вариантах осуществления R5 представляет собой H. В некоторых вариантах осуществления R8 выбран из H, Cl и линейного или разветвленного C1 - C4 алкила. В некоторых вариантах осуществления R8 представляет собой H. В некоторых других вариантах осуществления один из R5 или R8 представляет собой H (то есть, только один из R5 или R8 представляет собой H, другими словами, один из R5 или R8 отличен от H).
В некоторых вариантах осуществления R6 выбран из линейного или разветвленного C1 - C8 алкила, C5 - C10 циклоалкила, насыщенного или ненасыщенного 4-6-членного гетероциклила; и R7 выбран из линейного или разветвленного C1 - C8 алкила, замещенного по меньшей мере одним C3 - C7 циклоалкилом, 4-6-членным гетероциклилом, C5 - C15 арилом, C3 - C7 гетероарилом, галогенидом, гидроксилом, C1 - C5 алкилгалогенидом.
В некоторых вариантах осуществления R6 выбран из линейного или разветвленного C1 - C8 алкила, C5 - C10 циклоалкила, 4-6-членного насыщенного гетероциклила.
В некоторых вариантах осуществления R7 представляет собой линейный или разветвленный C1 - C8 алкил, замещенный по меньшей мере одним из C3 - C7 циклоалкила и гидроксила.
В некоторых вариантах осуществления R6 выбран из линейного или разветвленного C1 - C8 алкила, насыщенного или ненасыщенного 4-6-членного гетероциклила, каждый из которых необязательно замещен по меньшей мере одним из линейного или разветвленного C1 - C8 алкила, C3 - C7 циклоалкила, галогенида, гидроксила, CF3.
В некоторых вариантах осуществления R7 представляет собой линейный или разветвленный C1 - C8 алкил, замещенный по меньшей мере одним C3 - C7 циклоалкилом.
В некоторых вариантах осуществления R1 и R2 вместе с атомом азота, с которым они связаны, образуют 4-7-членное насыщенное кольцо, необязательно содержащее по меньшей мере один из N или O, NH, C=N, C=O или SO2 (то есть в дополнение к атому N, с которым R1 и R2 связаны), и которое необязательно может быть замещено по меньшей мере одним из линейного или разветвленного C1 - C5 алкила, гидроксила, галогенида и циано.
В некоторых вариантах осуществления R1 и R2 вместе с атомом азота, с которым они связаны, образуют 4-7-членное насыщенное кольцо.
В некоторых вариантах осуществления R1 и R2 вместе с атомом азота, с которым они связаны, образуют 4-7-членное насыщенное кольцо, содержащее по меньшей мере один из N или O (в дополнение к атому N, с которым R1 и R2 связаны).
В других вариантах осуществления R1 и R2 вместе с атомом азота, с которым они связаны, образуют 4-7-членное ароматическое кольцо, необязательно содержащее по меньшей мере один из N или O (в дополнение к атому N, с которым R1 и R2 связаны).
В некоторых вариантах осуществления R1 или R2 вместе с R3 и атомом углерода и атомом азота, с которыми они связаны, образуют 4-7-членное насыщенное кольцо, которое необязательно содержит по меньшей мере один из N, NH, O, C=O, SO2 и необязательно может быть замещено по меньшей мере одним из линейного или разветвленного C1 - C5 алкила, гидроксила, карбонила и галогенида.
В некоторых вариантах осуществления R1 или R2 вместе с R3 и атомом углерода и атомом азота, с которыми ни связаны, образуют 4-7-членное насыщенное кольцо, которое содержит по меньшей мере один из NH, O или C=O.
В некоторых вариантах осуществления соединение по настоящему изобретению выбрано из следующих:
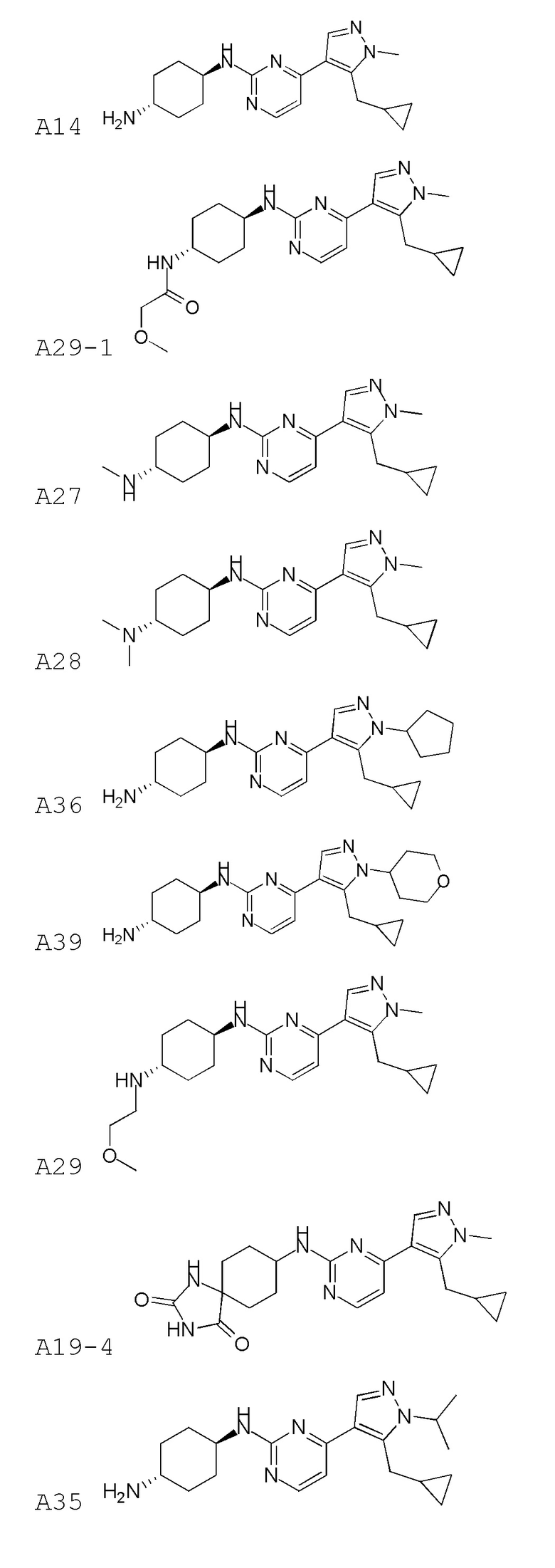
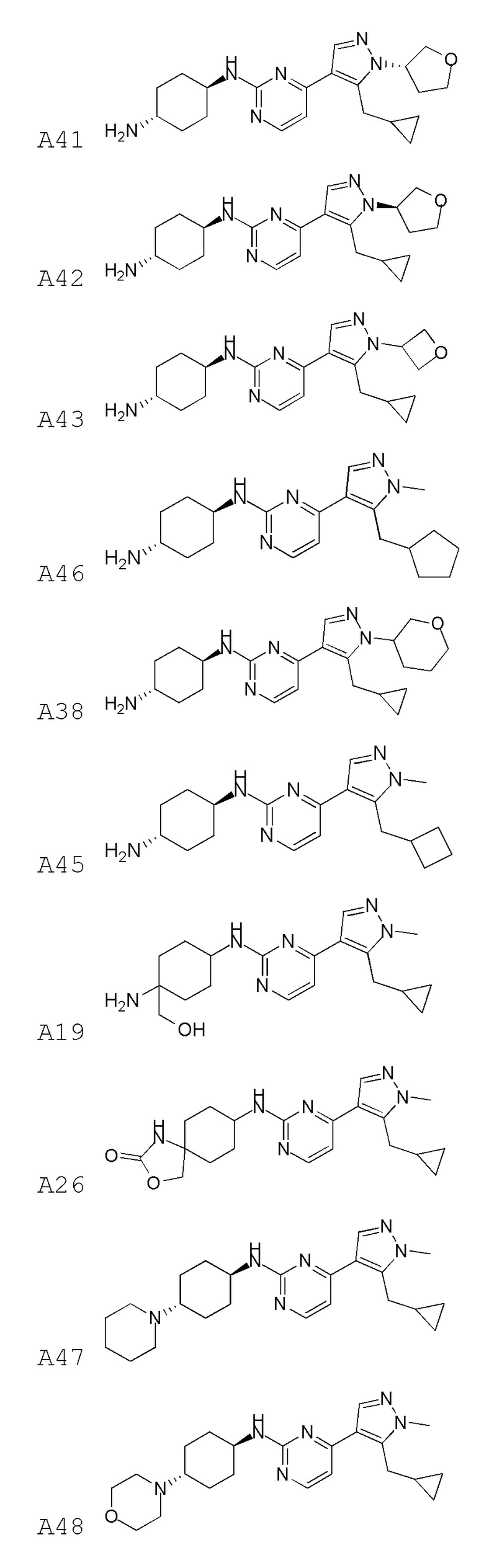
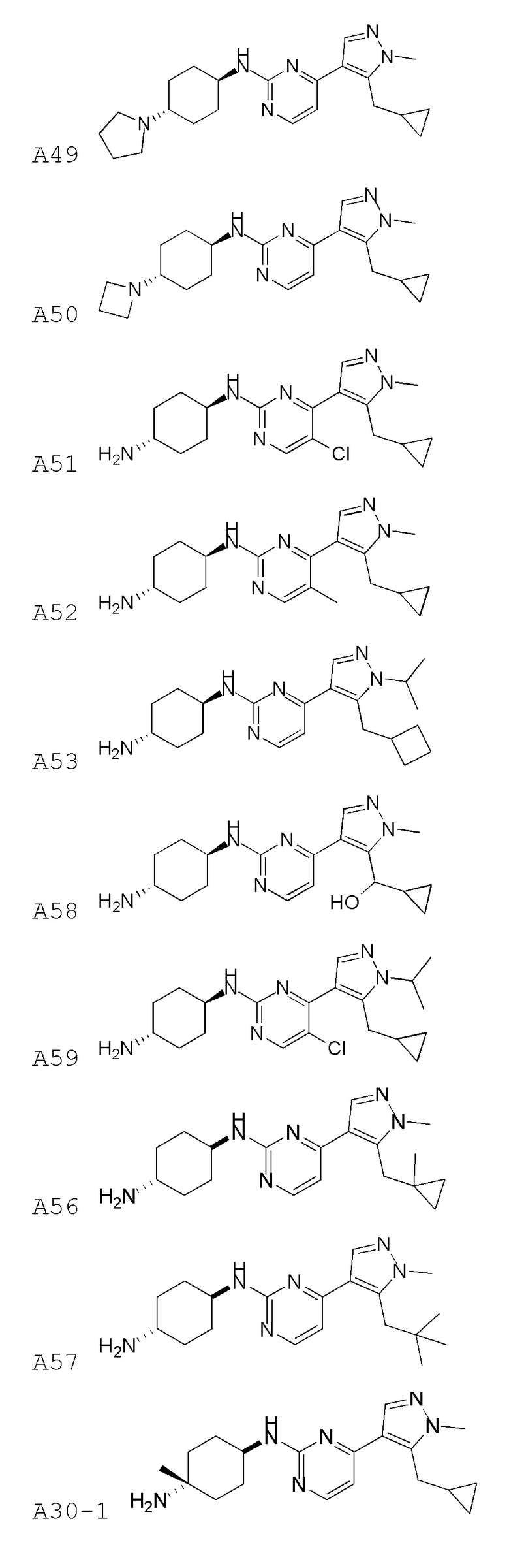
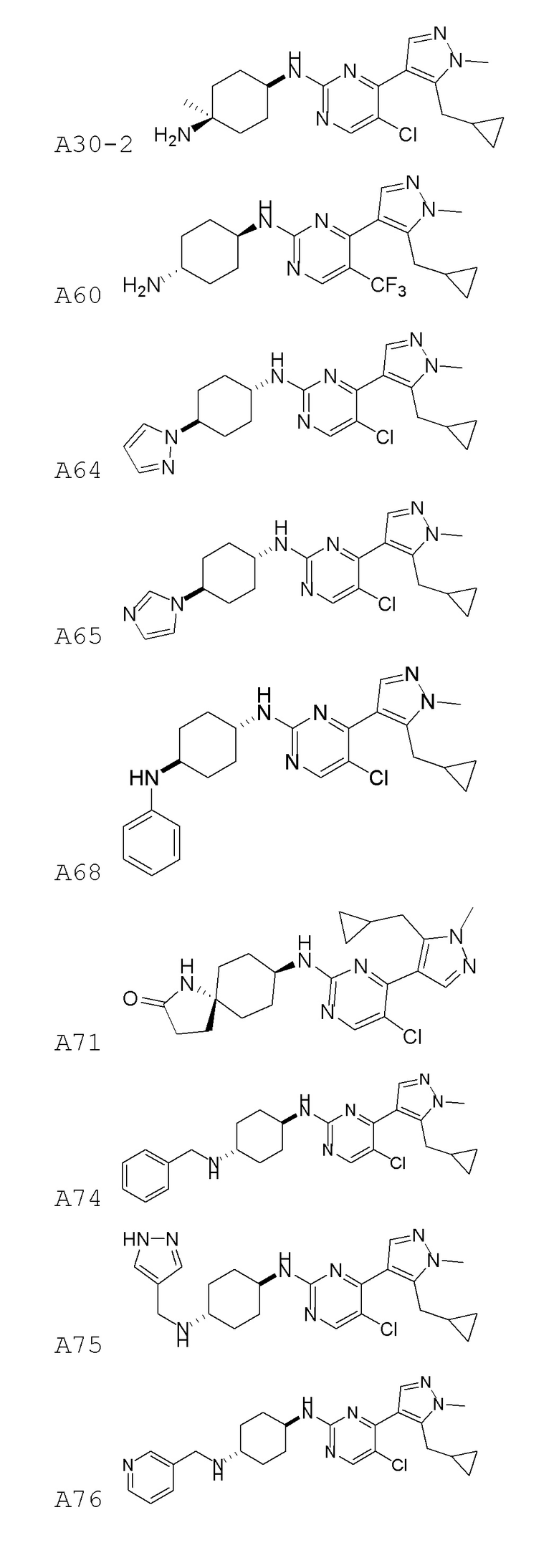
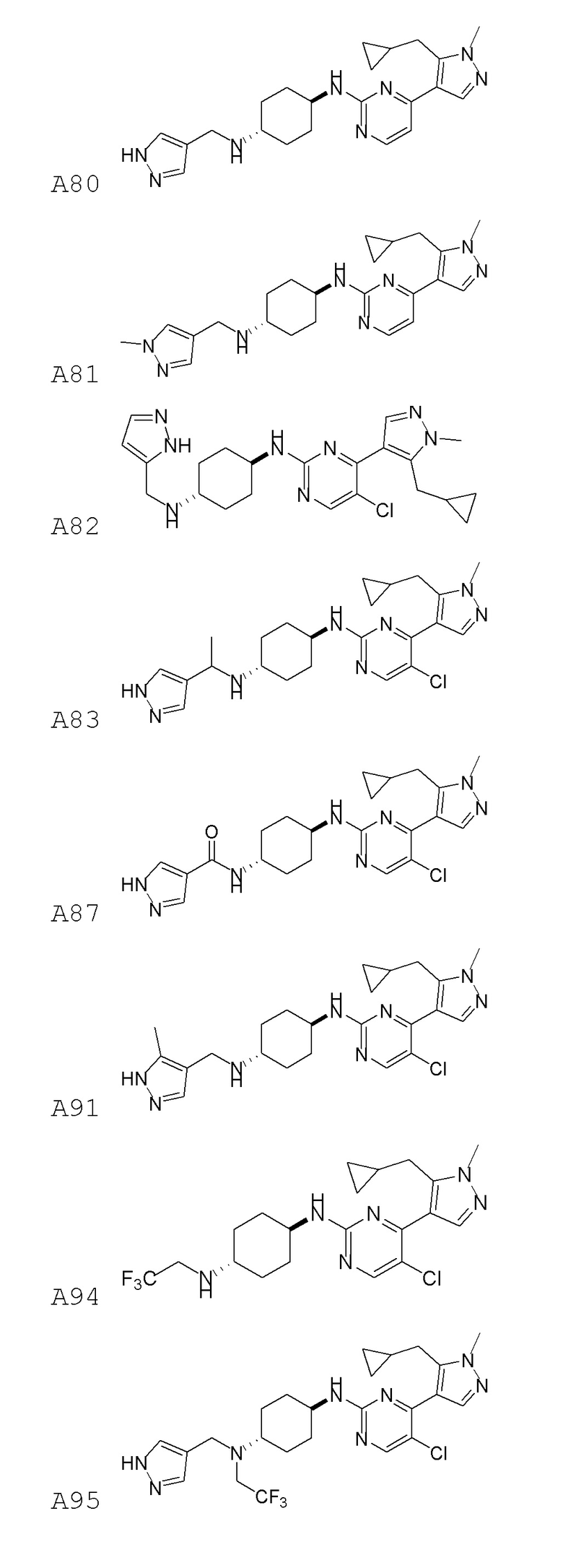
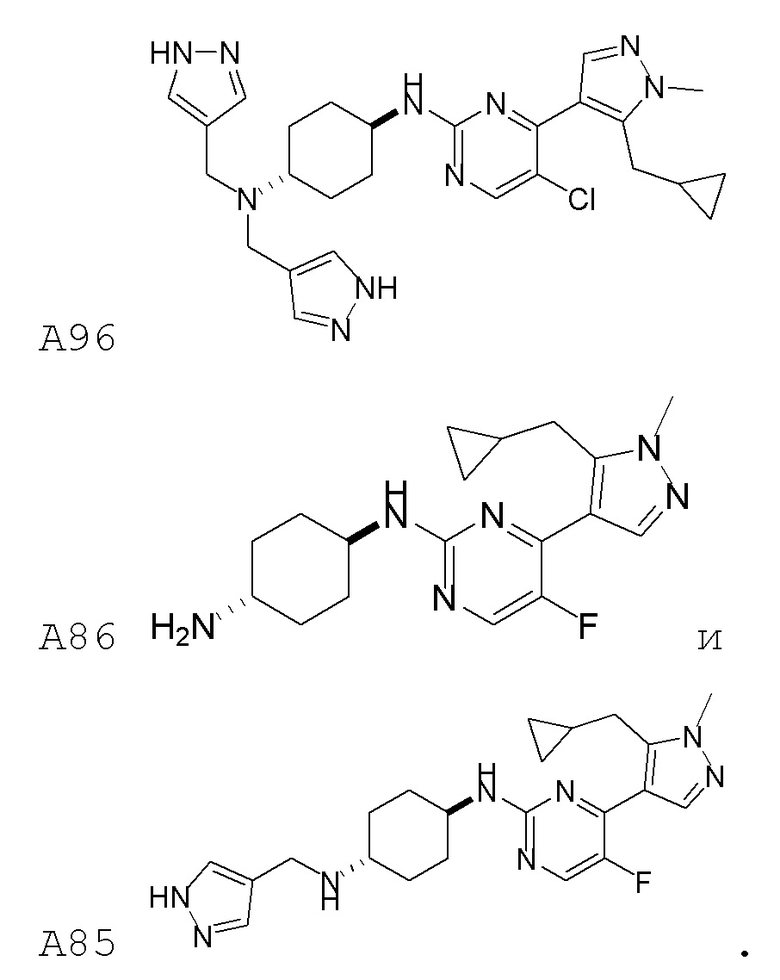
В некоторых вариантах осуществления соединение по настоящему изобретению представляет собой:
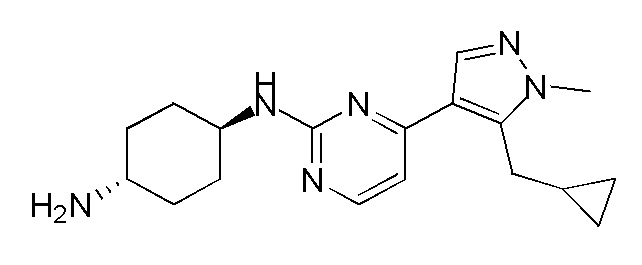 A14.
A14.
В некоторых других вариантах осуществления соединение по настоящему изобретению представляет собой:
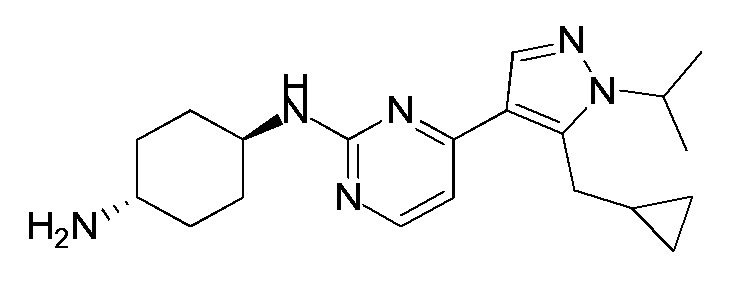 A35.
A35.
В других вариантах осуществления соединение по настоящему изобретению представляет собой:
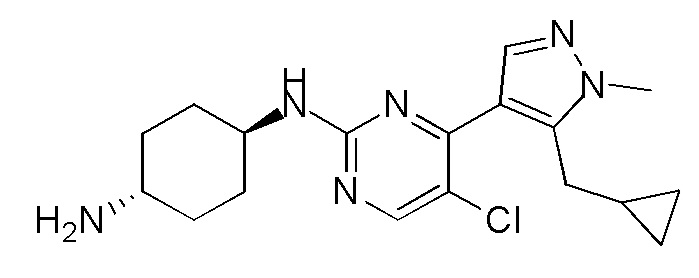 A51.
A51.
Термин "линейный или разветвленный C1 - C8 алкил" следует понимать как охватывающий углеводородную насыщенную цепь, которая может быть прямой или разветвленной, включающей 1, 2, 3, 4, 5, 6, 7 или 8 атомов углерода.
Термин "линейный или разветвленный C2 - C8 алкенил" или "линейный или разветвленный C2 - C5 алкенил" следует понимать как охватывающий углеводородную цепь, содержащую по меньшей мере одну двойную связь между любыми двумя атомами углерода в цепи, которая может быть прямой или разветвленной, включающей 2, 3, 4, 5, 6, 7 или 8 атомов углерода или 2, 3, 4, 5 атомов углерода, соответственно.
Термин "линейный или разветвленный C2 - C8 алкинил" следует понимать как охватывающий углеводородную цепь, содержащую по меньшей мере одну тройную связь между любыми двумя атомами углерода в цепи, которая может быть прямой или разветвленной, включающей 2, 3, 4, 5, 6, 7 или 8 атомов углерода.
Термин "линейный или разветвленный C1 - C5 алкокси" следует понимать как охватывающий группу -OR9, где R9 представляет собой линейный или разветвленный C1 - C5 алкил.
Термин "галогенид" следует понимать как охватывающий любой галогеновый радикал, выбранный из -F, -Br, -Cl, -I.
Термин "C1 - C5 алкилгалогенид" следует понимать как охватывающий любую прямую или разветвленную алкильную цепь, содержащую от 1 до 5 атомов углерода, замещенную по меньшей мере одним галогеновым радикалом, выбранным из -F, -Br, -Cl, -I, в любом месте прямой или разветвленной цепи. В некоторых вариантах осуществления алкилгалогенид содержит один галоген; в других вариантах осуществления алкилгалогенид содержит два атома галогена (одинаковых или разных); в других вариантах осуществления, алкилгалогенид содержит три атома галогена (одинаковых или разных) и так далее.
Термин "гидроксил" следует понимать как охватывающий -OH.
Термин "сложный эфир" следует понимать как охватывающий любой из -C(=O)OR10 или -OC(=O)R10, где R10 представляет собой линейный или разветвленный C1 - C8 алкил.
Термин "амид" следует понимать как охватывающий любой из -C(=O)NR11R12', -NR11C(=O)R12', где R11 и R12' каждый независимо представляет собой H или линейный или разветвленный C1 - C8 алкил.
Термин "простой эфир" следует понимать как охватывающий любой из -R13OR14' или -OR15', где R13 выбран из линейного или разветвленного C1 - C8 алкилена, и R14' и R15' каждый независимо выбран из линейного или разветвленного C1 - C8 алкила.
Термин "линейный или разветвленный C1 - C5 ацил" следует понимать как охватывающий любой -C(=O)R16, где R16 представляет собой C1 - C5 линейный или разветвленный алкил.
Термин "C5 - C15 арил" следует понимать как охватывающий любую одиночную или конденсированную ароматическую кольцевую систему, содержащую от 5 до 7 атомов углерода. Примеры включают, но не ограничиваются этим, фенил, пенталенил, нафталенил, антраценил и любые их комбинации.
Термин "C3 - C7 гетероарил" следует понимать как охватывающий любую простую или конденсированную ароматическую кольцевую систему, содержащую от 5 до 7 атомов углерода и по меньшей мере один гетероатом, выбранный из N, O и S. Примеры включают, но не ограничиваются этим, фуранил, бензофуранил, изобензофуранил, пирролинил, индолинил, изоиндолинил, тиофенил, бензотиофенил, бензо[c]тиофенил, имидазолил, бензимидазолил, пуринил, пиразолил, индазолил, оксазолил, бензоксазолил, изоксазолил, бензизоксазолил, тиазолил, бензотиазолил, пиридинил, хинолинил, изохинолинил, пиримидинил, хиназолинил, пиридазинил, циннолинил и любые их комбинации.
Когда идет речь о варианте осуществления, где R1 и R2 вместе с атомом азота, с которым они связаны, образуют 4-7-членное насыщенное, ненасыщенное или ароматическое кольцо, следует понимать, что это относится к любому кольцу, которое может быть образовано, включающему 4, 5, 6 или 7 членов, в том числе указанный атом азота. Указанное кольцо может быть насыщенным, то есть содержащим все сигма-связи, ненасыщенным, то есть содержащим по меньшей мере одну двойную или по меньшей мере одну тройную связь, или любые их комбинации, или ароматическим, то есть кольцевой системой, которая обладает ароматическим характером, циклически сопряженной молекулярной кольцевой системой со стабильностью (из-за делокализации) значительно большей, чем у гипотетической локализованной структуры (например, структуры Кекуле).
Например, указанное кольцо может быть выбрано из пиперидинила, пирролидинила, азетидинила и так далее.
В некоторых вариантах осуществления указанное кольцо необязательно может включать (в качестве членов кольца) по меньшей мере один из N, O, NH, C=N, C=O или SO2. В некоторых других вариантах осуществления указанное кольцо может быть необязательно замещенным (на кольцевой системе путем замещения -H атома на указанном кольце) по меньшей мере одним из линейного или разветвленного C1 - C5 алкила, гидроксила, галогенида и циано (-CN).
Когда речь идет о вариантах осуществления, где R1 или R2 вместе с R3 и атомом углерода и атомом азота, с которыми они связаны, образуют 4-7-членное насыщенное, ненасыщенное или ароматическое кольцо, следует понимать, что это относится к любому кольцу, которое может быть образовано, содержащему 4, 5, 6 или 7 членов, включая указанный атом азота. Это кольцо образует спиро-бикольцевую систему с циклогексильным кольцом в каркасе соединения формулы I. Указанное кольцо может быть насыщенным, то есть содержащим все сигма-связи, или ненасыщенным, то есть содержащим по меньшей мере одну двойную или по меньшей мере одну тройную связь или любые их комбинации. В некоторых вариантах осуществления кольцо представляет собой ароматическое кольцо.
В некоторых вариантах осуществления указанное кольцо необязательно содержит по меньшей мере один из N, NH, O, C=N, C=O, SO2 как образующие кольцо. В некоторых других вариантах осуществления указанное кольцо необязательно замещено (на кольцевой системе путем замещения -H атома на указанном кольце) по меньшей мере одним из линейного или разветвленного C1 - C5 алкила, гидроксила, карбонила (-C(=O)R, где R представляет собой H или C1 - C5 линейный или разветвленный алкил) и галогенида.
Термин "C5 - C10 циклоалкил" или термин "C3 - C7 циклоалкил" следует понимать как охватывающий насыщенное (то есть, кольцо, содержащее только сигма-связи между его членами) углеводородное кольцо, которое содержит 5, 6, 7, 8, 9 или 10 атомов углерода или 3, 4, 5, 6 или 7 атомов углерода, соответственно.
Термин "насыщенный, ненасыщенный или ароматический 4-6-членный гетероциклил" следует понимать как охватывающий насыщенное (то есть, кольцо, содержащее только сигма-связи между его членами), ненасыщенное или ароматическое (то есть, кольцо, содержащее по меньшей мере одну двойную связь или по меньшей мере одну тройную связь или любые их комбинации) кольцо, содержащее 4, 5 или 6 членов, по меньшей мере один из которых представляет собой гетероатом, выбранный из N, O, S, P.
Термин "необязательно замещенный" при использовании в настоящей заявке, означает, что рассматриваемые группы либо незамещенные, либо замещены одним или несколькими указанными заместителями. Когда рассматриваемые группы замещены более чем одним заместителем, заместители могут быть одинаковыми или отличными друг от друга.
Некоторые из соединений, описанных в настоящей заявке, могут содержать один или несколько хиральных центров или, иначе говоря, могут быть способны существовать в виде двух энантиомеров или нескольких диастереомеров. Соответственно, соединения по настоящему изобретению включают также смеси энантиомеров, а также очищенные энантиомеры или энантиомерно обогащенные смеси. Соединения по настоящему изобретению включают также смеси диастереомеров, а также очищенные диастереомеры или диастереомерно обогащенные смеси.
Настоящее изобретение также включает любую соль соединения формулы (I), включая любую фармацевтически приемлемую соль, где соединение по настоящему изобретению имеет полный заряд (положительный или отрицательный), и к нему для образования указанной соли добавляют по меньшей мере один противоион (имеющий отрицательный или положительный заряд). Фраза "фармацевтически приемлемая соль(и)", при использовании в настоящей заявке, означает те соли соединений по настоящему изобретению, которые являются безопасными и эффективными для фармацевтического применения для млекопитающих и которые обладают желаемой биологической активностью. Фармацевтически приемлемые соли включают соли кислотных или основных групп, присутствующих в соединениях по настоящему изобретению. Фармацевтически приемлемые кислотно-аддитивные соли включают, но не ограничиваются этим, гидрохлорид, гидробромид, гидройодид, нитрат, сульфат, бисульфат, фосфат, кислотный фосфат, изоникотинат, ацетат, лактат, салицилат, цитрат, тартрат, пантотенат, битартрат, аскорбат, сукцинат, малеат, гентизинат, фумарат, глюконат, глюкаронат, сахарат, формиат, бензоат, глутамат, метансульфонат, этансульфонат, бензолсульфонат, пара-толуолсульфонат и памоат (то есть, 1,1'-метилен-бис-(2-гидрокси-3-нафтоат)). Некоторые соединения по настоящему изобретению могут образовывать фармацевтически приемлемые соли с различными аминокислотами. Подходящие основные соли включают, но не ограничиваются этим, соли алюминия, кальция, лития, магния, калия, натрия, цинка и диэтаноламина. Для обзора фармацевтически приемлемых солей см. BERGE ET AL., 66 J. PHARM. SCI. 1-19 (1977), включенный в настоящую заявку посредством ссылки.
Настоящее изобретение также обеспечивает композицию, содержащую по меньшей мере одно соединение, определенное в настоящей заявке выше и ниже (в соответствии с общей формулой I).
Настоящее изобретение также относится к фармацевтическим композициям, содержащим соединение заявленного изобретения в смеси с фармацевтически приемлемыми вспомогательными веществами и необязательно другими терапевтическими средствами. Вспомогательные вещества должны быть ʺприемлемымиʺ в смысле совместимости с другими ингредиентами композиции и не вредными для их реципиентов.
Фармацевтические композиции включают композиции, подходящие для перорального, ректального, назального, местного (включая трансдермальное, буккальное и сублингвальное), вагинального или парентерального (включая подкожное, внутримышечное, внутривенное и внутрикожное) введения или введения через имплантат. Композиции можно получить любым способом, хорошо известным в области фармацевтики.
Такие способы включают стадию приведения в ассоциацию соединений по настоящему изобретению или их комбинаций с любым вспомогательным агентом. Вспомогательный агент(ы), также называемый вспомогательным ингредиентом(ами), включают такие, которые известны в данной области техники, такие как носители, наполнители, связующие вещества, разбавители, разрыхлители, смазывающие вещества, красители, ароматизаторы, антиоксиданты и смачивающие агенты.
Фармацевтические композиции, подходящие для перорального введения, могут быть представлены в виде дискретных дозированных единиц, таких как пилюли, таблетки, драже или капсулы, или в виде порошка или гранул, или в виде раствора или суспензии. Активный ингредиент также может быть представлен в виде болюса или пасты. Композиции могут быть затем сформулированы в виде суппозитория или клизмы для ректального введения.
Настоящее изобретение дополнительно включает фармацевтическую композицию, как описано выше, в сочетании с упаковочным материалом, включающим инструкции по применению композиции для использования, как описано выше.
Для парентерального введения, подходящие композиции включают водную и неводную стерильную инъекцию. Композиции могут быть представлены в однодозовых или многодозовых контейнерах, например, герметизированных флаконах и ампулах, и могут храниться в высушенном замораживанием (лиофилизированном) состоянии, требующем только добавления стерильного жидкого носителя, например, воды, перед использованием. Для трансдермального введения могут быть предусмотрены, например, гели, пластыри или спреи. Композиции или составы, подходящие для внутрилегочного введения, например, путем назальной ингаляции, включают порошкообразные или пылевидные составы, которые могут быть образованы при помощи дозированных аэрозолей под давлением, небулайзеров или инсуффляторов.
Точная доза и режим введения композиции обязательно будут зависеть от терапевтического или полезного для здоровья эффекта, который должен быть достигнут, и могут варьироваться в зависимости от конкретного состава, пути введения и возраста и состояния индивидуума, которому вводят композицию.
Настоящее изобретение также обеспечивает соединение, как определено в настоящей заявке выше и ниже (в соответствии с общей формулой I), для применения в терапии.
Термин "лечение" или "терапия" при использовании в настоящей заявке означает ведение и уход за пациентом с целью борьбы с заболеванием, расстройством или состоянием. Этот термин предназначен для включения задержки прогрессирования заболевания, расстройства или состояния, смягчение или облегчение симптомов и осложнений и/или лечение или устранение заболевания, расстройства или состояния. Подлежащий лечению пациент предпочтительно представляет собой млекопитающее, в частности, человека.
Следует понимать, что диапазоны доз, указанные выше, являются только иллюстративными и не предназначены для ограничения объема настоящего изобретения. Терапевтически эффективное количество для каждого активного соединения может варьироваться в зависимости от факторов, включая, но не ограничиваясь этим, активность используемого соединения, стабильность активного соединения в организме пациента, тяжесть состояний, требующих облегчения, общую массу тела пациента, подлежащего лечению, путь введения, характер абсорбции, дистрибуции и экскреции активного соединения организмом, возраст и чувствительность подлежащего лечению пациента и т.п., как должно быть очевидно для квалифицированного специалиста в данной области. Вводимое количество может корректироваться, поскольку различные факторы меняются со временем.
Для пероральной доставки, активные соединения могут быть включены в композицию, которая содержит фармацевтически приемлемые носители, такие как связующие (например, желатин, целлюлоза, камедь трагаканта), эксципиенты (например, крахмал, лактоза), смазывающие вещества (например, стеарат магния, диоксид кремния), разрыхлители (например, альгинат, Primogel и кукурузный крахмал) и подсластители или ароматизаторы (например, глюкоза, сахароза, сахарин, метилсалицилат и перечная мята). Композиция может доставляться перорально в виде желатиновых капсул или прессованных таблеток. Капсулы и таблетки можно получить любым обычным методом. Капсулы и таблетки также могут быть покрыты различными покрытиями, известными в данной области, для модификации ароматов, вкусов, цвета и формы капсул и таблеток. Кроме того, в капсулы также могут быть включены жидкие носители, такие как жирные масла.
Подходящие пероральные составы также могут быть в форме суспензии, сиропа, жевательной резинки, облатки, эликсира и подобного. При желании, также могут быть включены обычные агенты для модификации ароматов, вкусов, цвета и формы отдельных форм. Кроме того, для удобного введения при помощи зонда пациентам, неспособным глотать, активные соединения могут быть растворены в приемлемом липофильном носителе, таком как растительное масло, такое как оливковое масло, кукурузное масло и сафлоровое масло.
Активные соединения также можно вводить в форме раствора или суспензии или в лиофилизированной форме, которую можно преобразовать в форму раствора или суспензии перед использованием. В таких составах можно использовать разбавители или фармацевтически приемлемые носители, такие как стерильная вода и физиологический солевой буфер. Могут быть включены другие обычные растворители, рН-буферы, стабилизаторы, антибактериальные агенты, поверхностно-активные вещества и антиоксиданты. Например, полезные компоненты включают хлорид натрия, ацетаты, цитраты или фосфатные буферы, глицерин, декстрозу, жирные масла, парабены, полиэтиленгликоль, пропиленгликоль, бисульфат натрия, бензиловый спирт, аскорбиновую кислоту и т.п. Парентеральные составы могут храниться в любых удобных контейнерах, таких как сосуды и ампулы.
Пути для местного введения включают назальное, буккальное, мукозальное, ректальное или вагинальное применения. Для местного введения, активные соединения может быть сформулированы в лосьоны, кремы, мази, гели, порошки, пасты, спреи, суспензии, капли и аэрозоли. Таким образом, один или несколько загустителей, увлажнителей и стабилизаторов могут быть включены в композиции. Примеры таких агентов включают, но не ограничиваются этим, полиэтиленгликоль, сорбит, ксантановую камедь, вазелин, пчелиный воск или минеральное масло, ланолин, сквален и т.п. Специальной формой местного введения является доставка при помощи трансдермального пластыря. Способы получения трансдермальных пластырей раскрыты, например, в Brown, et al. (1988) Ann. Rev. Med. 39:221-229, который включен в настоящую заявку посредством ссылки.
Подкожная имплантация для замедленного высвобождения активных соединений также может быть подходящим путем введения. Это предполагает хирургические процедуры для имплантации активного соединения в любой подходящей композиции в подкожное пространство, например, под переднюю брюшную стенку. См., например, Wilson et al. (1984) J. Clin. Psych. 45:242-247. Гидрогели можно использовать в качестве носителя для замедленного высвобождения активных соединений. Гидрогели в целом известны в данной области техники. Их обычно получают путем сшивания высокомолекулярных биосовместимых полимеров в сеть, которая набухает в воде с образованием гелеобразного материала. В некоторых случаях гидрогели являются биодеградируемыми или биоразрушаемыми. Для целей настоящего изобретения могут быть полезны гидрогели, полученные из полиэтиленгликолей, коллагена или сополимеров L-молочной-гликолевой кислот. См., например, Phillips et al. (1984) J. Pharmaceut. Sci., 73: 1718-1720.
Настоящее изобретение также обеспечивает соединение, как определено в настоящей заявке выше и ниже (в соответствии с общей формулой I), для применения в ингибировании по меньшей мере одной из Казеинкиназы I (CKI) и Киназы 1, ассоциированной с рецептором Интерлейкина-1 (IRAK1). В некоторых вариантах осуществления соединение, определенное в настоящей заявке выше и ниже (в соответствии с общей формулой I), используют в ингибировании CKI и IRAK1. Согласно вышеприведенным вариантам осуществления, применение соединения по настоящему изобретению, определенного в настоящей заявке выше и ниже (в соответствии с формулой I), обеспечивает возможность лечения заболеваний, расстройств, симптомов и состояний, связанных с по меньшей мере одной из CKI и IRAK1 (или, в некоторых вариантах осуществления, с обеими CKI и IRAK1). Согласно таким аспектам настоящее изобретение обеспечивает лечение заболеваний, расстройств, симптомов и состояний, связанных с ингибированием по меньшей мере одной из CKI и IRAK1 (или, в некоторых вариантах осуществления, обеих CKI и IRAK1).
В другом из его аспектов настоящее изобретение обеспечивает соединение, определенное в настоящей заявке выше и ниже (в соответствии с общей формулой I), для применения в ингибировании киназы 1, ассоциированной с рецептором интерлейкина-1 (IRAK1).
Настоящее изобретение также обеспечивает соединение, определенное в настоящей заявке выше и ниже (в соответствии с общей формулой I), для применения в ингибировании казеинкиназы I (CKI).
Термин "Казеинкиназа I" следует понимать как охватывающий семейство протеинкиназ, которые являются серина/треонин-селективными ферментами, которые функционируют как регуляторы путей сигнальной трансдукции в большинстве типов эукариотических клеток. CK1 изоформы участвуют в передаче сигналов Wnt, циркадных ритмах, ядерно-цитоплазматическом транслировании транскрипционных факторов, репарации ДНК, активации р53 и транскрипции ДНК.
Термин "киназа 1, ассоциированная с рецептором интерлейкина-1" следует понимать как охватывающий фермент, кодируемый геном IRAK1, который был признан важным апстрим регулятором NF-kB пути, вовлеченным в пути развития гематологических злокачественных опухолей, таких как множественная миелома, MDS, лейкоз и лимфома, рак молочной железы, рак головы и шеи, воспалительные и иммунные заболевания и другие.
Когда речь идет об "ингибировании" указанного фермента, следует понимать этот термин как охватывающий любое качественное или количественное снижение активности указанного фермента из-за прямого или опосредованного связывания по меньшей мере одного соединения по настоящему изобретению с указанным ферментом.
Настоящее изобретение также обеспечивает соединение, определенное в настоящей заявке выше и ниже (в соответствии с общей формулой I), для применения в лечении состояния, симптома или заболевания, ассоциированного с злокачественным состоянием.
В некоторых вариантах осуществления указанное злокачественное состояние представляет собой рак. В других вариантах осуществления указанный рак имеет либо WT, либо мутантный p53 (мутации, которые дезактивируют р53, типичные для онкологических состояний). В других вариантах осуществления указанный рак выбран из лейкоза, злокачественной меланомы, рака молочной железы, рака предстательной железы и колоректального рака. В некоторых вариантах осуществления указанный рак имеет WT p53.
Настоящее изобретение также обеспечивает соединение, определенное в настоящей заявке выше и ниже, для применения в лечении рака, имеющего WT p53, где указанный WT p53 является биомаркером для эффективности указанного соединения. Таким образом, в этом аспекте WT p53 служит в качестве измеряемого индикатора эффективности лечения рака соединением или композицией, содержащей соединение по изобретению. Настоящее изобретение также обеспечивает способ лечения рака, имеющего WT p53, у нуждающегося в этом субъекта, где указанный WT p53 является биомаркером для эффективности указанного соединения.
В некоторых вариантах осуществления указанное применение, кроме того, включает индукцию ответа на иммунотерапию рака. Таким образом, в некоторых вариантах осуществления настоящего изобретения соединение или композиция, содержащая соединение по настоящему изобретению, обеспечивает как лечение рака (противоопухолевая, противозлокачественная активность), так и ответ на иммунотерапию рака.
В некоторых вариантах осуществления указанное злокачественное состояние выбрано из гематологических злокачественных опухолей (множественная миелома, миелодиспластический синдром (MDS), острый миелоидный лейкоз (AML), меланома и ER-негативный рак молочной железы, диффузная крупноклеточная В-клеточная лимфома (DLBCL), хронический миелоидный лейкоз (CML), хронический лимфоцитарный лейкоз (CLL), рак головы и шеи и любые их комбинации.
Еще в одном из его аспектов настоящее изобретение обеспечивает соединение, определенное в настоящей заявке выше и ниже, для применения в индуцировании противоопухолевого ответа. В некоторых вариантах осуществления указанный противоопухолевый ответ включает ответ на иммунотерапию рака.
Термин "индуцированный противоопухолевый ответ" следует понимать как охватывающий любую качественную или количественную химиотерапию раковых опухолей.
Термин "ответ на иммунотерапию рака" следует понимать как охватывающий любую качественную или количественную индукцию иммунотерапии рака иммунной системой самого субъекта для борьбы с раковыми клетками. Как правило, иммунотерапия может быть классифицирована как активная, пассивная или гибридная (активная и пассивная) и предназначена для использования того факта, что раковые клетки часто имеют молекулы на их поверхности, которые могут быть обнаружены иммунной системой субъекта, известные как опухоль-ассоциированные антигены (TAAs); они часто представляют собой белки или другие макромолекулы (например, углеводы). Активная иммунотерапия направляет иммунную систему на атаку опухолевых клеток путем таргетирования TAAs. Пассивные иммунотерапии усиливают существующие противоопухолевые ответы.
В некоторых вариантах осуществления указанный ответ на иммунотерапию рака относится к изменению экспрессии контрольных точек иммунного ответа на опухолевой клетке, на антиген-презентирующей клетке, на Т-клетке или на природной киллерной (NK) клетке.
В некоторых вариантах осуществления указанный ответ на иммунотерапию рака относится к снижению экспрессии молекулы иммунной контрольной точки на опухолевой клетке, которая индуцирует подавление противоопухолевой активности Т-клетки.
В некоторых вариантах осуществления указанный ответ на иммунотерапию рака относится к снижению экспрессии белка контрольной точки PD-L1. В некоторых других вариантах осуществления указанная реакция на иммунотерапию относится к ингибированию PD-L1. В некоторых других аспектах соединение по настоящему изобретению используют в способе ингибирования PD-L1.
Настоящее изобретение также обеспечивает соединение, определенное в настоящей заявке выше и ниже (в соответствии с общей формулой I), для применения в лечении воспалительного и иммунного заболевания, включая ассоциированное с ним состояние, симптом или заболевание.
Когда речь идет о "воспалительных и иммунных заболеваниях", должно быть понятно, что это относится к любому типу расстройства (включая ассоциированные с ним состояния, симптомы и заболевания), которые можно лечить ингибиторами киназы, ассоциированной с рецептором интерлейкина-1. Было показано, например, что IRAK1 является незаменимым элементом IL-Rs и TLR путей, которые могут регулировать аномальные уровни цитокинов, и поэтому может использоваться для лечения связанных с иммунной системой и воспалением расстройств, таких как, например, ревматоидный артрит, воспалительное заболевание кишечника, псориаз, подагра, астма и рак (Bahia MS et al. Cell Signal. 2015 Jun;27(6):1039-55).
Настоящее изобретение также обеспечивает способ лечения состояния, симптома или заболевания, ассоциированного с злокачественным состоянием у нуждающегося в этом субъекта, при этом указанный способ включает стадию введения указанному субъекту по меньшей мере одного соединения, определенного в настоящей заявке выше и ниже (в соответствии с общей формулой I).
Настоящее изобретение также обеспечивает способ ингибирования по меньшей мере одной из казеинкиназы I (CKI) и киназа 1, ассоциированной с рецептором интерлейкина-1 (IRAK1), у нуждающегося в этом субъекта, включающий стадию введения указанному субъекту по меньшей мере одного соединения, определенного в настоящей заявке выше и ниже (в соответствии с общей формулой I).
Еще в одном из его аспектов настоящее изобретение обеспечивает способ ингибирования киназы 1, ассоциированная с рецептором интерлейкина-1 (IRAK1), у нуждающегося в этом субъекта, включающий стадию введения указанному субъекту по меньшей мере одного соединения, определенного в настоящей заявке выше и ниже (в соответствии с общей формулой I).
В некоторых вариантах осуществления способ по настоящему изобретению дополнительно включает индукцию ответа на иммунотерапию рака у указанного субъекта.
Еще в одном из его аспектов настоящее изобретение обеспечивает способ индукции ответа на иммунотерапию рака у нуждающегося в этом субъекта, где указанный способ включает стадию введения указанному субъекту по меньшей мере одного соединения, определенного в настоящей заявке выше и ниже.
Настоящее изобретение также обеспечивает способ лечения воспалительного и иммунного расстройства, включая ассоциированное с ним состояние, симптом или заболевание; указанный способ включает введения субъекту, нуждающемуся в этом, соединения, определенного в настоящей заявке выше и ниже (в соответствии с общей формулой I).
Настоящее изобретение также обеспечивает способ ингибирования казеинкиназы I у нуждающегося в этом субъекта, включающий стадию введения указанному субъекту по меньшей мере одного соединения, определенного в настоящей заявке выше и ниже (в соответствии с общей формулой I).
Краткое описание чертежей
Для лучшего понимания изобретения, раскрытого в настоящей заявке, и для иллюстрации того, как его можно осуществить на практике, варианты осуществления будут описаны при помощи неограничивающих примеров только со ссылкой на прилагаемые чертежи, в которых:
Фиг. 1A и 1B показывают доза-ответ для указанных соединений по настоящему изобретению (Фиг. 1А, Соединения A36, A39, A6, A14, A35, A51, A29, A19-4, A41, A28, A42, A43, A46; Фиг. 1B соединения A30-1, A30-2, A51, A60, A64, A65)-тестированные в RKO колоректальной клеточной линии. RKO клетки инкубировали в течение 16 часов при 37°C с указанными концентрациями соединений и анализировали методом вестерн-блоттинга. Показаны стабилизация β-катенина и p53 и фосфорилирование H2AX (γH2AX), маркера повреждения ДНК, указывающие на ингибирование CKIα киназы. Следует отметить, что, хотя стабилизация β-катенина и p53 и фосфорилирование H2AX являются общим эффектом для большинства соединений, некоторые соединения не стабилизируют β-катенин, тогда как близкие аналоги (например, A19-4) только стабилизируют β-катенин. Уровни CKIα белка служат в качестве нагрузочного контроля.
Фиг. 2A-2D показывают, что ингибитор CKIα A14 индуцирует апоптоз клеток костного мозга, выделенных у мышей с бластным кризом CML, дозозависимым образом (ex vivo). Фиг. 2A схематически представляет экспериментальную процедуру; Инкубация с A14 или DMSO в течение 8 часов. Фиг. 2B показывает существенное уменьшение количества лейкозных клеток после обработки соединением A14 - кривая доза-ответ. Фиг. 2C показывает увеличение процента апоптозных клеток (Аннексин5+/7AAD-) дозозависимым образом. Фиг. 2D показывает увеличение процента общей гибели (7AAD+) клеток дозозависимым образом.
Фиг. 3A-3C демонстрирует, что A14 существенно снижает лейкозную клеточную нагрузку в периферической крови и костном мозге in vivo у мышей с бластным кризом CML. Фиг. 3A схематически представляет экспериментальную процедуру инокуляции BM клеток от мыши с бластным кризом CML (GFP+ клетки) нормальным C57Bl/6 мышам и ежедневную обработку (пероральное введение) соединением A14 или только носителем. Фиг. 3B показывает процент GFP+ лейкозных клеток в периферической крови на день 9 после обработки; A14 (N=6) по сравнению с мышами, которых обрабатывали носителем (N=6). Фиг. 3C показывает процент GFP+ лейкозных клеток в костном мозге на день 9 после обработки: A14 (N=6) по сравнению с мышами, которых обрабатывали носителем (N=6).
Фиг. 4A-4F показывают общий анализ крови на день 9 после A14 обработки мышей с бластным кризом CML: Фиг. 4A показывает абсолютное число лейкоцитов (WBC) в периферической крови (109/л) (N=5). Фиг. 4B показывает абсолютное число лимфоцитов (Lymph) в периферической крови (109/л) (N=5). Фиг. 4C показывает абсолютное число моноцитов (Mono) в периферической крови (109/л) (N=5). Фиг. 4D показывает абсолютное число гранулоцитов (Gran) в периферической крови (109/л). Фиг. 4E показывает число эритроцитов (RBC, 1012/л). Фиг. 4F показывает уровень гемоглобина (HGB, г/л).
Фиг. 5 показывает репрезентативные фотографии мазков крови от A14-обработанной по сравнению с обработанной носителем мыши с бластным кризом CML на день 9 после трансплантации костного мозга.
Фиг. 6 схематически представляет подготовку мышиной модели AML.
Фиг. 7A-7D показывают ингибиторный эффект A14 на прогрессирование AML. Фиг. 7A схематически представляет экспериментальные процедуры. Фиг. 7B демонстрирует процент GFP+ лейкоцитов в периферической крови (PB) A14-обработанных мышей по сравнению с мышами, которых обрабатывали носителем. Фиг. 7C и 7D показывают полученные методом проточной цитометрии репрезентативные графики GFP+ лейкоцитов в PB за один день до (Фиг. 7C) и три дня (Фиг. 7D) после A14-обработки.
Фиг. 8A-8G показывают общий анализ крови на день 9 после обработки AML мышей соединением A14 по настоящему изобретению. Фиг. 8A показывает абсолютное число лейкоцитов (WBC) в периферической крови (109/л). Фиг. 8B показывает абсолютное число лимфоцитов (Lymph) в периферической крови (109/л). Фиг. 8C показывает абсолютное число моноцитов (Mono) в периферической крови (109/л). Фиг. 8D показывает абсолютное число гранулоцитов (Gran) в периферической крови (109/л). Фиг. 8E показывает число эритроцитов (RBC, 1012/л). Фиг. 8F показывает гемоглобин (г/л). Фиг. 8G показывает тромбоциты (PLT) в периферической крови (109/л).
Фиг. 9 показывает репрезентативные фотографии мазков крови от A14-обработанных AML мышей по сравнению с обработанными носителем на день 9 после первой A14 обработки.
Фиг. 10 показывает ингибирование IRAK1 ингибиторами, представляющими собой соединения по настоящему изобретению A51 и A14. Клетки собирали и анализировали методом вестерн-блоттинга. Блоты инкубировали со следующими антитела: Фосфо-IRAK1 (Thr209), (A1074, AssayBiothechnology; 1/1000), Фосфо-IKKα/β (Ser176/180) (16A6, Cell Signaling; 1/1000), IKKα (2682, Cell Signaling; 1/11000), IKK β (2370, Cell Signaling; 1/11000), Фосфо-c-Jun (Ser 63) (9261, Cell Signaling; 1/11000), p53 (DO-1&1801 смесь гибридом; разведение 1:20 супернатантов от каждой), CKIα (C-19; 1/1000; Santa Cruz Biotechnology) и фосфо-гистон H2AX (S139; 1/1000; Millipore). Вторичные антитела представляли собой HRP-связанные козлиные антимышиные, козлиные антикроличьи и кроличьи антикозлиные антитела (все 1/10000; Jackson). Ингибирование Фосфо-IRAK1, Фосфо-IKKα/β и Фосфо-c-Jun указывает на ингибирование IRAK1. Стабилизация p53 и фосфорилирование H2AX (γH2AX), маркера повреждения ДНК, указывают на ингибирование CKIα киназы. Уровни CKIα белка служат в качестве нагрузочного контроля.
Фиг. 11A-11E относятся к экспериментальным результатам, достигаемым при однодозовой обработке AML мышей ингибитором CKI A51. Фиг. 11A схематически показывает подготовку AML мышей и их обработку соединением A51 (20мг/кг) в день 30 после индукции заболевания (заражение лейкозом). Фиг. 11B - 11E показывают эффект A51 через 16 часов после обработки (по сравнению с обработкой только носителем) снижения общего количества лейкозных клеток в крови: Фиг. 11B показывает уменьшение WBC в периферической крови (PB), Фиг. 11C показывает сокращение лейкозной селезенки, и Фиг. 11D и 11E показывают уменьшение пропорции лейкозных бластных клеток (GFP+ клетки) в периферической крови (PB) и костном мозге (BM), соответственно.
Фиг. 12A и 12B показывают снимки селезенки и кости обработанных AML мышей. Реальное уменьшение размера селезенки (спленомегалия) после обработки соединением A51, как раскрыто выше (Фиг. 12A), и бледные кости возвращают нормальный цвет после однодозовой обработки (Фиг. 12B).
Фиг. 13A-13E показывают эффекты на AML клетки, выделенные из костного мозга лейкозных мышей при обработке ингибиторами CKI in vitro. Показаны процент мертвых клеток (7AAD+) и эффекты ингибиторов на экспрессию PD-L1 белка главной иммунной контрольной точки в лейкозных клетках; обработка ингибитором 10 или 100 нМ для нескольких временных точек (5 часов - Фиг. 13A, 6 часов - Фиг. 13B и 13C и 9 часов Фиг. 13D и 13E).
Подробное описание вариантов осуществления
Получение (E)-1-циклопропил-4-(диметиламино)-3-(2-(метилтио)пиримидин-4-ил)бут-3-ен-2-она (Ядро A) и 4-(5-(циклопропилметил)-1H-пиразол-4-ил)-2-(метилтио)пиримидина (Ядро B)
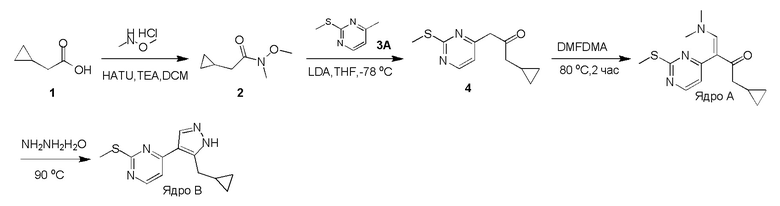
Стадия 1: N,O-Диметилгидроксиламингидрохлорид (25,14 г, 257,69 ммоль, 1,72 экв.), HATU (56,97 г, 149,82 ммоль, 1,00 экв.) и TEA (45,48 г, 449,46 ммоль, 3,00 экв.) добавляли к раствору 2-циклопропилуксусной кислоты (15,00 г, 149,82 ммоль, 1,00 экв.) в DCM (500 мл) при 0°C и затем смесь перемешивали при 30°C в течение 3 часов. Полученную смесь выливали в воду (500 мл). Водную промывочную фазу экстрагировали при помощи DCM (3×250 мл). Объединенные органические слои сушили над безводным Na2SO4, фильтровали и концентрировали. Остаток очищали при помощи колоночной хроматографии (SiO2, петролейный эфир(PE): этилацетат (EA)=50:1-10:1) с получением желаемого соединения 2 (13,20 г, 82,97 ммоль, 55,4% выход) в виде бесцветной жидкости.
1H ЯМР (CDCl3, 400 МГц) δ 3,65 (с, 3H), 3,17 (с, 3H), 2,33 (д, J=6,8 Гц, 2H), 1,09-1,06 (м, 1H), 0,54-0,50 (м, 2H), 0,16-0,14 (м, 2H).
Стадия 2: К раствору 4-метил-2-метилсульфанил-пиримидина (9,00 г, 64,19 ммоль, 1,00 экв.) в THF (500 мл) добавляли LDA (2 M, 48,46 мл, 1,51 экв.) при -78°C. После перемешивания в течение 1 часа добавляли по каплям раствор соединения 2 (13,79 г, 96,29 ммоль, 1,50 экв.) в THF (500 мл) при -78°C и затем реакционную смесь перемешивали при -78°C в течение 4 часов. Гасили насыщенным водным раствором NH4Cl (100 мл), водную фазу экстрагировали этилацетатом (3 × 50 мл). Объединенные органические слои промывали насыщенным солевым раствором, сушили над безводным Na2SO4, фильтровали и концентрировали. Остаток кристаллизовали из смеси петролейный эфир/этилацетат с получением желаемого соединения 4 (13,60 г, 55,06 ммоль, 85,8% выход) в виде желтого твердого вещества.
ЖХМС: RT=0,629 мин, m/z 223,0 [M+H]+.
Стадия 3: Раствор соединения 4 (13,60 г, 61,18 ммоль, 1,00 экв.) в DMF-DMA (51,42 г, 2,45 моль, 40 экв.) перемешивали при 90°C в течение 2 часов. Растворитель удаляли в вакууме. Остаток очищали при помощи колоночной хроматографии на силикагеле с получением Ядра A (10,60 г, 36,30 ммоль, 59,3% выход) в виде желтого твердого вещества.
ЖХМС: RT=0,634 мин, m/z 278,2 [M+H]+
1H ЯМР (CDCl3, 400 МГц) δ 8,38 (д, J=6,8 Гц, 1H), 7,62 (с, 1H), 6,96 (с, 1H), 2,96-2,87 (м, 6H), 2,56 (с, 3H), 2,38 (д, J=8,8 Гц, 2H), 1,04-1,02 (м, 1H), 0,52-0,46 (м, 2H), 0,09-0,04 (м, 2H).
Стадия 4: Раствор Ядра A (10,60 г, 38,21 ммоль, 1,00 экв.) и гидразингидрата (6,75 г, 114,63 ммоль, 3,00 экв.) в этаноле (200 мл) перемешивали при 90°C в течение 3 часов. Растворитель удаляли в вакууме. Остаток очищали при помощи колоночной хроматографии на силикагеле с получением Ядра B (9,00 г, 35,81 ммоль, 93,7% выход) в виде светло-желтого твердого вещества.
ЖХМС: RT=2,018 мин, m/z 247,1 [M+H]+
1H ЯМР (CDCl3, 400 МГц) δ 8,43 (д, J=5,2 Гц, 1H), 8,04 (с, 1H), 7,10 (д, J=5,2 Гц, 1H), 3,12 (д, J=7,2 Гц, 2H), 2,60 (с, 3H), 1,19-1,14 (м, 1H), 0,66 (кв., J=5,2 Гц, 2H), 0,32 (кв., J=5,2 Гц, 2H).
Получение 4-(5-(циклопропилметил)-1-метил-1H-пиразол-4-ил)-2-(метилсульфонил)пиримидина (Ядро C)
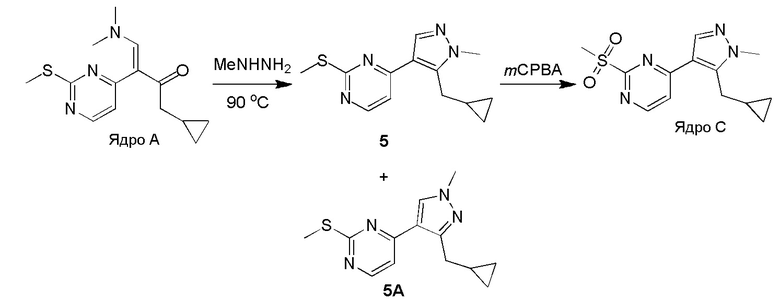
Стадия 1: Раствор Ядра A (6,20 г, 22,35 ммоль, 1,00 экв.) и метилгидразина (8,00 г, 69,46 ммоль, 3,11 экв.) в этаноле (100 мл) перемешивали при 90°C в течение 16 часов. Растворитель удаляли в вакууме. Остаток очищали при помощи препаративной ВЭЖХ (щелочные условия) с получением соединения 5 (1,80 г, 6,84 ммоль, 30,6% выход) в виде желтого твердого вещества и изомера 5A (2,00 г, 7,30 ммоль, 32,6% выход) в виде желтого масла.
Соединение 5:
ЖХМС: RT=2,551 мин, m/z 261,1 [M+H]+
1H ЯМР (CDCl3, 400 МГц) δ 8,38 (д, J=5,2 Гц, 1H), 7,90 (с, 1H), 7,11 (д, J=5,2 Гц, 1H), 3,93 (с, 3H), 3,24 (д, J=6,4 Гц, 2H), 2,62 (с, 3H), 1,12-1,09 (м, 1H), 0,54-0,49 (м, 2H), 0,32-0,28 (м, 2H).
Региоизомер 5A:
ЖХМС: RT=2,486 мин, m/z 261,1 [M+H]+
1H ЯМР (CDCl3, 400 МГц) δ 8,41 (д, J=5,2 Гц, 1H), 7,90 (с, 1H), 7,04 (д, J=5,2 Гц, 1H), 3,92 (с, 3H), 2,96 (д, J=6,4 Гц, 2H), 2,61 (с, 3H), 1,20-1,17 (м, 1H), 0,50-0,45 (м, 2H), 0,26-0,22 (м, 2H).
Стадия 2: К раствору соединения 1 (1,50 г, 5,76 ммоль, 1,00 экв.) в DCM (20 мл) добавляли m-CPBA (2,98 г, 17,28 ммоль, 3,00 экв.) при 0°C и перемешивали при 30°C в течение 2 часов. Полученную смесь промывали NaHSO3 (2 × 100 мл), насыщенным водным раствором NaHCO3 (100 мл) и насыщенным солевым раствором, сушили над безводным Na2SO4, фильтровали и концентрировали. Остаток очищали при помощи колоночной хроматографии на силикагеле (петролейный эфир: этилацетат=10:1-1:1) с получением Ядра C соединения (1,50 г, 5,08 ммоль, 88,2% выход) в виде желтого твердого вещества.
ЖХМС: RT=1,891 мин, m/z 293,0 [M+H]+
1H ЯМР (CDCl3, 400 МГц) δ 8,74 (д, J=5,6 Гц, 1H), 7,97 (с, 1H), 7,58 (д, J=5,6 Гц, 1H), 3,94 (с, 3H), 3,37 (с, 3H), 3,25 (д, J=6,8 Гц, 2H), 1,14-1,11 (м, 1H), 0,52-0,49 (м, 2H), 0,37-0,35 (м, 2H).
Общие процедуры для получения соединения A-n:
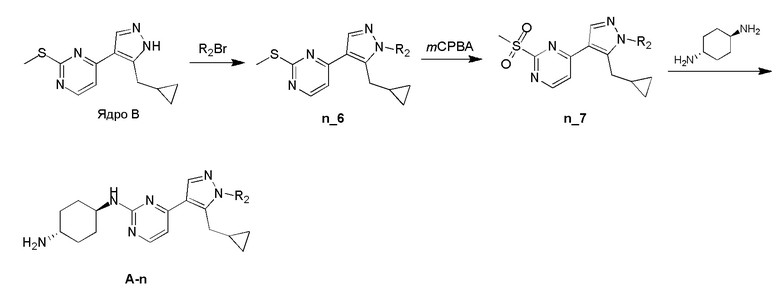
Способ A: получение соединения n_6
Раствор Ядра B (25 ммоль, 1,00 экв.) в DMF (50 мл) охлаждали до 0°C и добавляли NaH (1,50 экв.). После перемешивания при 0°C в течение 1 часа добавляли RBr (1,60 экв.) и реакционную смесь перемешивали при 20°C в течение 15 часов. Добавляли воду (10,00 мл) и экстрагировали этилацетатом (3×10 мл). Объединенные органические слои промывали водой (10 мл) и насыщенным солевым раствором (10 мл), сушили над Na2SO4, фильтровали и концентрировали с получением неочищенного продукта, который очищали с получением соединения n_6.
Способ B: получение соединения n_7
К раствору соединения n_6 (10 ммоль, 1,00 экв.) в дихлорметане (30 мл) добавляли m-CPBA (3,00 экв.) при 0°C и реакционную смесь перемешивали при 20-30°C в течение 5 часов. Полученную смесь промывали NaHSO3 (2 × 50,00 мл), насыщенным водным раствором NaHCO3 (50 мл) и насыщенным солевым раствором (10 мл), сушили над безводным Na2SO4, фильтровали и концентрировали с получением соединения n_7.
Способ C: получение соединения A-n
Смесь соединения n_7 (10 ммоль, 1,0 экв.), DIEA (10,00 экв.) и транс-4-амино-циклогексанола (1,0~3,0 экв.) в DMSO (80 мл) нагревали при 160°C в течение 3 часов. Реакционную смесь охлаждали до температуры окружающей среды и выливали в воду (100 мл) и затем экстрагировали дихлорметаном (3×50 мл). Объединенные слои промывали насыщенным солевым раствором, сушили над безводным Na2SO4, фильтровали и концентрировали. Остаток очищали при помощи препаративной ВЭЖХ (щелочные условия) с получением соединения A-n.
Способ D: получение соединения A-n
Раствор соединения n_7 (10 ммоль, 1,00 экв.) и транс-циклогексан-1,4-диамина (2,0~4,0 экв.) в диоксане (40 мл) перемешивали при 130°C в течение 2 часов с микроволновым нагревом. Смесь фильтровали и фильтрат очищали при помощи препаративной ВЭЖХ (щелочные условия) с получением соединения A-n.
Получение (1R,4R)-N1-(4-(5-(циклопропилметил)-1-изопропил-1H-пиразол-4-ил)пиримидин-2-ил)циклогексан-1,4-диамина (A-35)
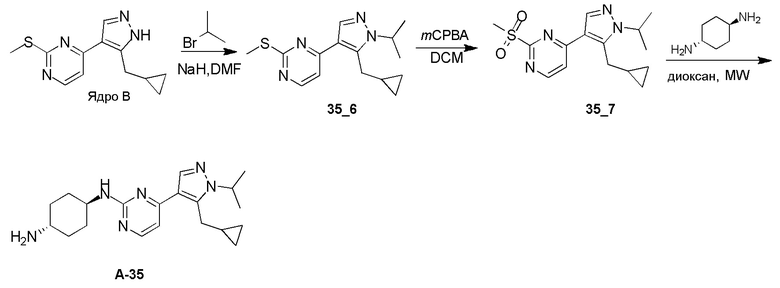
Стадия 1: Соединение 35_6 синтезировали в соответствии с процедурой, описанной в способе A, и его получали в виде желтого твердого вещества после очистки при помощи ВЭЖХ (TFA условия). Выход: 17,1%;
ЖХМС: RT=1,959 мин, m/z 289,1 [M+H]+
1H ЯМР (CDCl3, 400 МГц) δ 8,49-8,47 (д, J=2,8 Гц, 1H), 8,06 (с, 1H), 7,21-7,20 (д, J=5,6 Гц, 1H), 4,63-4,60 (т, J=6,6 Гц, 1H), 3,26-3,24 (д, J=6,4 Гц, 2H), 2,65 (с, 3H), 1,56 (с, 3H), 1,55 (с, 3H), 1,07-1,04 (м, 1H), 0,54-0,51 (м, 2H), 0,30-0,28 (м, 2H).
Стадия 2: Соединение 35_7 синтезировали в соответствии с процедурой, описанной в способе B, и его получали в виде желтого твердого вещества после очистки при помощи ТСХ. Выход: 57,8%;
ЖХМС: RT=0,711 мин, m/z 321,1 [M+H]+
Стадия 3: Соединение A-35 синтезировали в соответствии с процедурой, описанной в способе D, с использованием транс-циклогексан-1,4-диамина (4,0 экв.), и его получали в виде белого твердого вещества. Выход: 19,7%
ЖХМС: RT=1,223 мин, m/z 355,3 [M+H]+
1H ЯМР (CDCl3, 400 МГц) δ 8,17-8,16 (д, J=5,2 Гц, 1H), 7,89 (с, 1H), 6,72-6,71 (д, J=5,2 Гц, 1H), 4,85-4,83 (д, J=8,8 Гц, 1H), 4,60-4,53 (м, 1H), 3,23-3,22 (д, J=6,4 Гц, 2H), 2,20-2,18 (д, J=10 Гц, 2H), 2,02-1,99 (м, 2 H), 1,54 (с, 3H),1,52 (с, 3H), 1,40-1,27 (м, 4H), 1,08-1,06 (м, 1H), 0,52-0,48 (м, 2H), 0,28-0,26 (м, 2H).
Получение (1R,4R)-N1-(4-(1-циклопентил-5-(циклопропилметил)-1H-пиразол-4-ил)пиримидин-2-ил)циклогексан-1,4-диамина (A-36)
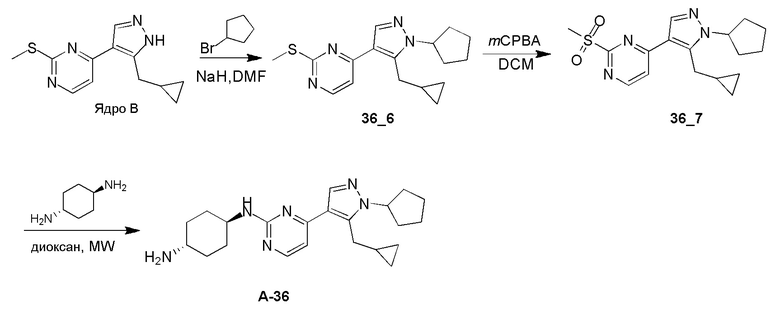
Стадия 1: Соединение 36_6 синтезировали в соответствии с процедурой, описанной в способе A, и его получали в виде желтого масла после очистки при помощи препаративной ВЭЖХ (TFA условия). Выход: 14,7%
ЖХМС: RT=2,228 мин, m/z 315,2 [M+H]+
1H ЯМР (CDCl3, 400 МГц) δ 8,31-8,30 (д, J=5,6 Гц, 1H), 7,84 (с, 1H), 7,03-7,01 (д, J=5,2 Гц, 1H), 4,67-4,63 (м, 1H), 3,20-3,18 (д, J=6,4 Гц, 2H), 2,52 (с, 3H), 2,05-2,02 (м, 7H), 1,65-1,62 (м, 2H), 0,98 (м, 1H), 0,43-0,39 (м, 2H), 0,23-0,20 (м, 2H).
Стадия 2: Соединение 36_7 получали в соответствии с процедурой, описанной в способе B, и его получали в виде желтого твердого вещества после очистки при помощи колоночной хроматографии. Выход: 66,6%
ЖХМС: RT=0,707 мин, m/z 347,2 [M+H]+
Стадия 3: Соединение A-36 получали в соответствии с процедурой, описанной в способе D, с использованием транс-циклогексан-1,4-диамина (2,0 экв.), и его получали в виде белого твердого вещества после очистки при помощи препаративной ВЭЖХ (TFA условия). Выход: 41,35%
ЖХМС: RT=0,591 мин, m/z 381,4 [M+H]+
1H ЯМР (CD3OD, 400 МГц) δ 8,26 (с, 1H), 8,08-8,06 (д, J=6,4 Гц, 1H), 7,27-7,25 (д, J=6,8 Гц, 1H), 4,15 (с, 1H), 3,33 (с, 3H), 3,20 (с, 1H), 2,25-1,99 (м, 11H), 1,75 (м, 2H), 1,64-1,58 (м, 4H), 1,16 (м, 1H), 0,57-0,55 (м, 2H), 0,36-0,35 (м, 1H).
Получение (1R,4R)-N1-(4-(5-(циклопропилметил)-1-(тетрагидро-2H-пиран-4-ил)-1H-пиразол-4-ил)пиримидин-2-ил)циклогексан-1,4-диамина (A-39)
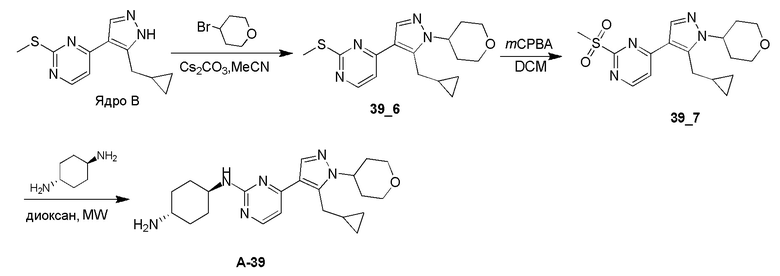
Стадия 1: К раствору Ядра B (700,00 мг, 2,84 ммоль, 1,00 экв.) в MeCN (14 мл) добавляли CS2CO3 (1,85 г, 5,68 ммоль, 2,00 экв.) при 0°C. Через 30 минут добавляли 4-бромтетрагидропиран (703,03 мг, 4,26 ммоль, 1,50 экв.). Смесь перемешивали при 100°C в течение 16 часов в плотно закрытой пробирке. Реакционную смесь фильтровали и фильтрат концентрировали. Неочищенный продукт очищали при помощи препаративной ВЭЖХ (TFA) с получением соединения 39_6 (45,0 мг, 136,18 мкмоль, 4,8% выход) в виде желтого твердого вещества.
ЖХМС: RT=0,702 мин, m/z 331,1 [M+H]+
1H ЯМР (CDCl3, 400 МГц) δ 8,42-8,40 (д, J=5,6 Гц, 1H), 7,94 (с, 1H), 7,11-7,10 (д, J=5,2 Гц, 1H), 4,37-4,33 (м, 1H), 4,19-4,15 (м, 1H), 3,59-3,54 (м, 2H), 3,28-3,26 (д, J=6,4 Гц, 2H), 2,6 (с, 3H), 2,44-2,40 (м, 2H), 1,87-1,84 (м, 2H), 1,05-1,02 (м, 1H), 0,55-0,50 (м, 2H), 0,33-0,30 (м, 2H).
Стадия 2: Соединение 39_7 получали в соответствии с процедурой, описанной в способе B, и его получали в виде желтого твердого вещества после очистки при помощи препаративной ТСХ. Выход: 72,6%
ЖХМС: RT=0,607 мин, m/z 363,1 [M+H]+
Стадия 3: Соединение A-39 получали в соответствии с процедурой, описанной в способе C, с использованием транс-циклогексан-1,4-диамина (3,0 экв.) без DIEA, и его получали в виде желтого твердого вещества после очистки при помощи препаративной ВЭЖХ (щелочные условия). Выход: 40,2%
ЖХМС: RT=1,104 мин, m/z 397,4 [M+H]+
1H ЯМР (CDCl3, 400 МГц) δ 8,17-8,16 (д, J=4,4 Гц, 1H), 7,89 (с, 1H), 6,72-6,70 (д, J=5,2 Гц, 1H), 5,12-5,07 (м, 1H), 4,37-4,34 (м, 1H), 4,18-4,15 (м, 2H), 3,84 (с, 1H), 3,59-3,53 (м, 2H), 3,26-3,25 (д, J=6,4 Гц, 2H), 2,77 (с, 1H), 2,44-2,40 (м, 2H), 2,16 (с, 2H), 1,93 (с, 2H), 1,87-1,83 (м, 2H), 1,32-1,27 (м, 4H), 1,05 (с, 1H), 0,54-0,49 (м, 2H), 0,29-0,27 (м, 2H).
Получение (1R,4R)-N1-(4-(5-(циклопропилметил)-1-(оксетан-3-ил)-1H-пиразол-4-ил)пиримидин-2-ил)циклогексан-1,4-диамина (A-43)
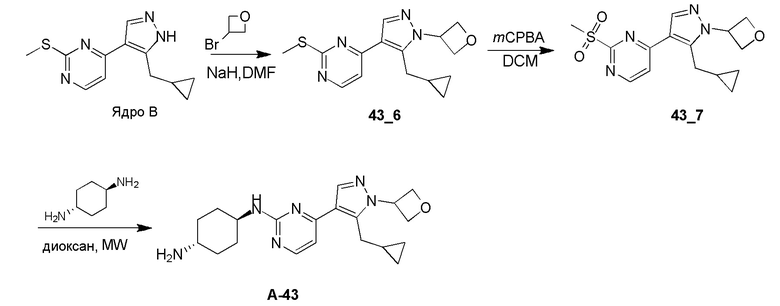
Стадия 1: Соединение 43_6 синтезировали в соответствии с процедурой, описанной в способе A, и его получали в виде желтого твердого вещества после очистки при помощи препаративной ВЭЖХ (TFA условия). Выход: 17,9%
ЖХМС: RT=0,664 мин, m/z 303,1 [M+H]+
1H ЯМР (CDCl3, 400 МГц) δ 8,44-8,42 (д, J=5,2 Гц, 1H), 8,04 (с, 1H), 7,13-7,12 (д, J=5,6 Гц, 1H), 5,89-5,34 (м, 1H), 5,28-5,25 (т, J=6,6 Гц, 2H), 5,03-5,00 (т, J=7,0 Гц, 2H), 3,21-3,20 (д, J=6,4 Гц, 2H), 2,6 (с, 3H), 0,97-0,94 (м, 1H), 0,51-0,46 (м, 2H), 0,25-0,22 (м, 2H).
Стадия 2: Соединение 43_7 синтезировали в соответствии с процедурой, описанной в способе B, и его получали в виде желтого твердого вещества после очистки при помощи препаративной ТСХ. Выход: 79,6%
ЖХМС: RT=0,566 мин, m/z 335,1 [M+H]+
Стадия 3: Соединение A-43 синтезировали в соответствии с процедурой, описанной в способе D, с использованием транс-циклогексан-1,4-диамина (3,0 экв.), и его получали в виде белой смолы после очистки при помощи препаративной ВЭЖХ (TFA условия). Выход: 24,7%;
ЖХМС: RT=0,987 мин, m/z 369,3 [M+H]+
1H ЯМР (CD3OD, 400 МГц) δ 8,40 (с, 1H), 8,13-8,11 (д, J=6,8 Гц, 1H), 7,31-7,29 (д, J=7,2 Гц, 1H), 5,83-5,80 (т, J=7,0 Гц, 1H), 5,15-5,12 (т, J=6,2 Гц, 2H), 5,06-5,03 (т, J=7,0 Гц, 2H), 4,12 (с, 1H), 3,28-3,26 (д, J=6,4 Гц, 2H), 3,19 (с, 1H), 2,23-2,17 (м, 4H), 1,62-1,60 (м, 4H), 1,06 (с, 1H), 0,53-0,52 (м, 2H), 0,28-0,27 (м, 2H).
Получение (R)-тетрагидрофуран-3-илметансульфоната (41_4)
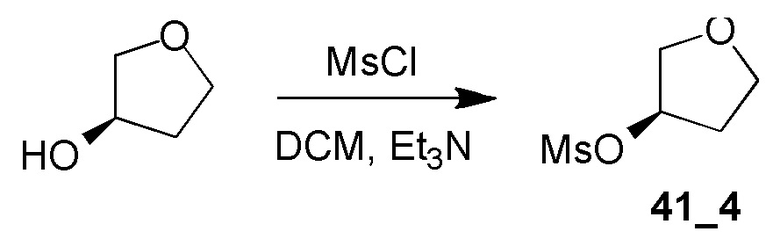
К раствору (R)-тетрагидрофуран-3-ола (500,00 мг, 5,68 ммоль, 1,00 экв.) в DCM (5 мл) добавляли TEA (1,15 г, 11,36 ммоль, 2,00 экв.) и метансульфонилхлорид (650,11 мг, 5,68 ммоль, 1,00 экв.) при 0°C. Смесь перемешивали при 20°C в течение 1 часа. Смесь разбавляли водой (20 мл) и экстрагировали при помощи DCM (10 мл × 2). Объединенные органические слои концентрировали с получением соединения 41_4 (900,00 мг, 5,42 ммоль, 95,3% выход) в виде бесцветной жидкости.
1H ЯМР (CDCl3, 400 МГц) δ 5,33-5,30 (м, 1H), 4,06-3,90 (м, 4H), 3,05 (с, 3H), 2,28-2,23 (м, 2H).
Получение (1R,4S)-N1-(4-(5-(циклопропилметил)-1-((S)-тетрагидрофуран-3-ил)-1H-пиразол-4-ил)пиримидин-2-ил)циклогексан-1,4-диамина (A-41)
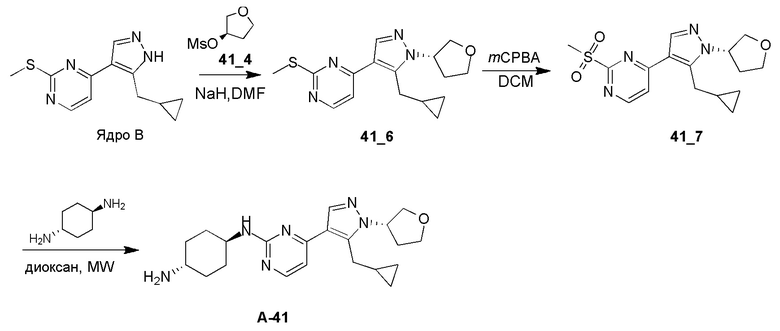
Стадия 1: Соединение 41_6 синтезировали в соответствии с процедурой, описанной в способе A, и его получали в виде желтого твердого вещества после очистки при помощи препаративной ВЭЖХ (TFA условия). Выход: 13,1%
1H ЯМР (CDCl3, 400 МГц) δ 8,42-8,40 (д, J=4,4 Гц, 1H), 7,94 (с, 1H), 7,11-7,10 (д, J=7,2 Гц, 1H), 5,04-5,00 (м, 1H), 4,27-4,03 (м, 4H), 3,29-3,27 (д, J=8,4 Гц, 2H), 2,6 (с, 3H), 2,48-2,42 (м, 2H), 1,07-1,02 (м, 1H), 0,53-0,49 (м, 1H), 0,30-0,28 (м, 2H).
Стадия 2: Соединение 41_7 синтезировали в соответствии с процедурой, описанной в способе B, и его получали в виде желтого твердого вещества после очистки при помощи препаративной ТСХ. Выход: 57,5%
ЖХМС: RT=0,585 мин, m/z 349,2 [M+H]+
Стадия 3: Соединение A-41 синтезировали в соответствии с процедурой, описанной в способе D, с использованием транс-циклогексан-1,4-диамина (4,0 экв.), и его получали в виде белого смолообразного твердого вещества после очистки при помощи препаративной ВЭЖХ (TFA условия). Выход: 17,2%
ЖХМС: RT=0,969 мин, m/z 383,3 [M+H]+
1H ЯМР (CD3OD, 400 МГц) δ 8,26 (с, 1H), 8,10-8,09 (д, J=6,8 Гц, 1H), 7,25-7,23 (д, J=6,8 Гц, 1 H), 5,28-5,23 (м, 1H), 4,22-4,12 (м, 3H), 3,99-3,95 (м, 2H), 3,35-3,33 (д, J=6,8 Гц, 2H), 3,19 (с, 1H), 2,47-2,44 (м, 2H), 2,35-2,17 (м, 4H), 1,62-1,57 (м, 4H), 1,14 (с, 1H), 0,58-0,56 (м, 2H), 0,36 (м, 2H).
Получение (S)-тетрагидрофуран-3-илметансульфоната (42_4)
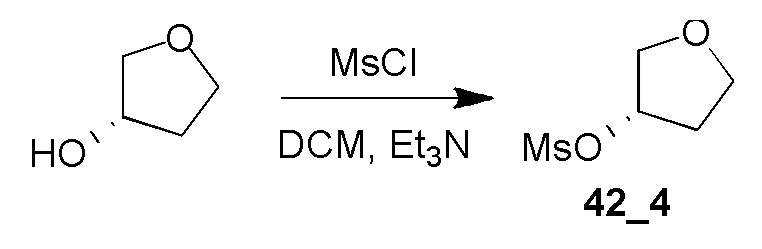
Соединение 42_4 получали таким же способом, как соединение 41_4, из (S)-тетрагидрофуран-3-ола, и оно представляло собой бесцветную жидкость. Выход: 90,0%
1H ЯМР (CDCl3, 400 МГц) δ 5,33-5,30 (м, 1H), 4,04-3,86 (м, 4H), 3,05 (с, 3H), 2,27-2,22 (м, 2H).
Получение (1R,4R)-N1-(4-(5-(циклопропилметил)-1-((R)-тетрагидрофуран-3-ил)-1H-пиразол-4-ил)пиримидин-2-ил)циклогексан-1,4-диамина (A-42)
Стадия 1: Соединение 42_6 синтезировали в соответствии с процедурой, описанной в способе A, и его получали в виде желтого твердого вещества после очистки при помощи препаративной ВЭЖХ (TFA условия). Выход: 21,4%
ЖХМС: RT=0,692 мин, m/z 317,1 [M+H]+
1H ЯМР (CDCl3, 400 МГц) δ 8,42-8,40 (д, J=4,4 Гц, 1H), 7,95 (с, 1H), 7,12-7,10 (д, J=7,2 Гц, 1H), 5,04-5,00 (м, 1H), 4,27-4,03 (м, 4H), 3,29-3,27 (д, J=8,4 Гц, 2H), 2,6 (с, 3H), 2,47-2,42 (м, 2H), 1,06-1,03 (м, 1H), 0,55-0,50 (м, 1H), 0,31-0,29 (м, 2H).
Стадия 2: Соединение 42_7 синтезировали в соответствии с процедурой, описанной в способе B, и его получали в виде желтого твердого вещества после очистки при помощи препаративной ТСХ. Выход: 83,2%
ЖХМС: RT=0,587 мин, m/z 349,1 [M+H]+
Стадия 3: Соединение A-42 синтезировали в соответствии с процедурой, описанной в способе D, и его получали в виде белого смолообразного твердого вещества после очистки при помощи препаративной ВЭЖХ (TFA условия). Выход: 51,5%
ЖХМС: RT=1,078 мин, m/z 383,3 [M+H]+
1H ЯМР (CD3OD, 400 МГц) δ 8,26 (с, 1H), 8,10-8,08 (д, J=6,8 Гц, 1H), 7,28-7,26 (д, J=6,8 Гц, 1H), 5,27-5,24 (м, 1H), 4,22-4,13 (м, 3H), 3,99-3,96 (м, 2H), 3,35-3,31 (т, J=6,8 Гц, 2H), 3,19 (с, 1H), 2,46-2,44 (м, 2H), 2,36-2,17 (м, 4H), 1,63-1,58 (м, 4H), 1,15 (с, 1H), 0,58-0,56 (м, 2H), 0,36 (м, 2H).
Получение (1R,4R)-N1-(4-(5-(циклопропилметил)-1-(тетрагидро-2H-пиран-3-ил)-1H-пиразол-4-ил)пиримидин-2-ил)циклогексан-1,4-диамина (A-38)
Стадия 1: Смесь соединения 38_1 (1,20 г, 11,99 ммоль, 1,40 экв.) и бензил N-аминокарбамата (1,42 г, 8,56 ммоль, 1,00 экв.) в MeOH (15 мл) перемешивали при 30°C в течение 2 часов. Добавляли NaBH3CN (2,69 г, 42,82 ммоль, 5,00 экв.). Полученную смесь перемешивали при 30°C в течение 16 часов. Смесь концентрировали и разбавляли водой (50 мл) и EA (50 мл). Органический слой концентрировали и очищали при помощи колоночной хроматографии (PE:EA=10:1~2:1) с получением соединения 38_2 (1,50 г, 5,99 ммоль, 70% выход) в виде бесцветного масла.
ЖХМС: RT=0,568 мин, m/z 273,2 [M+H]+
1H ЯМР (CDCl3, 400 МГц) δ 7,40-7,35 (м, 5H), 6,25 (с, 1H), 5,26-5,14 (м, 2H), 3,86-3,73 (м, 3H), 3,48-3,30 (м, 2H), 1,91-1,85 (м, 2H), 1,59-1,46(м, 2H).
Стадия 2: К раствору соединения 38_2 (1,60 г, 6,39 ммоль, 1,00 экв.) в MeOH (15 мл) добавляли Pd(OH)2 (179,54 мг, 1,28 ммоль, 0,20 экв.). Смесь перемешивали при 20°C в атмосфере H2 (1,055 кг/см2) в течение 16 часов. Смесь фильтровали и фильтрат концентрировали с получением соединения 38_3 (450 мг, неочищенное) в виде бесцветного масла, которое непосредственно использовали на следующей стадии без очистки.
Стадия 3: Соединение 38_6 получали из Ядра A и соединения 38_4 в соответствии с процедурой, аналогичной описанной для синтеза Ядра B, и его получали в виде желтого масла после очистки при помощи препаративной ВЭЖХ (TFA условия). Выход: 40,3%
ЖХМС: RT=0,851 мин, m/z 331,1 [M+H]+
1H ЯМР (CDCl3, 400 МГц) δ 8,41-8,40 (д, J=5,2 Гц, 1H), 7,92 (с, 1H), 7,10-7,09 (д, J=5,2 Гц, 1H), 4,34-4,33 (м, 1H), 4,03-4,00 (м, 2H), 3,87-3,81 (т, J=10,8 Гц, 1H), 3,51-3,50 (м, 1H), 3,29-3,19 (м, 2H), 2,60 (с, 3H), 2,35 (м, 1H), 2,13 (м, 1H), 1,89-1,85 (м, 2H), 1,06-1,03 (м, 1H), 0,53-0,51 (м, 2H), 0,31 (м, 2H).
Стадия 4: Соединение 38_7 синтезировали в соответствии с процедурой, описанной в способе B, и его получали в виде желтого твердого вещества после очистки при помощи препаративной ТСХ. Выход: 56,1%
ЖХМС: RT=0,739 мин, m/z 363,1 [M+H]+
Стадия 5: Соединение A-38 синтезировали в соответствии с процедурой, описанной в способе D, с использованием транс-циклогексан-1,4-диамина (4,0 экв.), и его получали в виде желтого твердого вещества после очистки при помощи препаративной ВЭЖХ (щелочные условия). Выход: 40%
ЖХМС: RT=1,154 мин, m/z 397,3 [M+H]+
1H ЯМР (CDCl3, 400 МГц) δ 8,18-8,17 (д, J=5,2 Гц, 1H), 7,86 (с, 1H), 6,70-6,69 (д, J=5,2 Гц, 1H), 4,88-4,86 (м, 1H), 4,32-4,29 (м, 1H), 4,02-4,00 (т, J=5,6 Гц, 2H), 3,86-3,81 (м, 2H), 3,51-3,49 (т, J=3,00 Гц, 1H), 3,27-3,19 (м, 2H), 2,81 (с, 1H), 2,34-2,31 (м, 1H), 2,19-2,16 (м, 3H), 2,00-1,97 (м, 4H),1,38-1,27 (м, 4H), 1,06 (с, 1H), 0,52-0,50 (м, 2H), 0,28 (м, 2H)
Общая процедура для синтеза соединения A-n
Способ E
Реакционную смесь Ядра C (1,00 экв.) и амина n (4,00 экв.) в DMSO (8 мл) перемешивали при 160°C в течение 3 часов. Реакционную смесь охлаждали до комнатной температуры и выливали на смесь лед-H2O (20 мл). Водный слой экстрагировали при помощи EA (50 мл × 3). Объединенные органические слои промывали насыщенным солевым раствором (50 мл × 3), сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали при помощи колоночной хроматографии с получением A-n.
Способ F
Реакционную смесь Ядра C (1,00 экв.), TBAF (2,00 экв.), K2CO3 (4,00 экв.) и амина n (4,00 экв.) в DMSO (10 мл) перемешивали при 160°C в течение 3 часов. Реакционную смесь охлаждали до 15°C и выливали в H2O (20 мл). Водный слой экстрагировали при помощи EA (20 мл × 3). Объединенные органические слои промывали насыщенным солевым раствором (20 мл × 3), сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали при помощи препаративной ВЭЖХ (HCl условия) с получением A-n.
Способ G
Реакционную смесь амина n (1,20 экв.) и Ядра C (1,00 экв.) в диоксане (3 мл) перемешивали при 130°C в условиях микроволнового нагрева в течение 1,5 часов. Реакционную смесь выливали в H2O (20 мл). Водный слой экстрагировали при помощи EA (20 мл × 3). Объединенные органические слои промывали насыщенным солевым раствором (20 мл × 3), сушили над Na2SO4, фильтровали и концентрировали. Остаток очищали при помощи препаративной ВЭЖХ (щелочные условия) с получением A-n.
Получение (1R,4R)-N1-(4-(5-(циклопропилметил)-1-метил-1H-пиразол-4-ил)пиримидин-2-ил)циклогексан-1,4-диамина (A-14) и (1R,4R)-N1-(4-(5-(циклопропилметил)-1-метил-1H-пиразол-4-ил)пиримидин-2-ил)-N4,N4-диметилциклогексан-1,4-диамина (A-28)
Стадия 1: Соединение A-14 получали в соответствии с процедурой, описанной в способе G, и его получали в виде желтого твердого вещества. Выход: 38,4%
ЖХМС: RT=2,043 мин, m/z 327,2 [M+H]+.
1H ЯМР (CDCl3, 400 МГц) δ 8,17 (д, J=5,3 Гц, 1H), 7,83 (с, 1H), 6,70 (д, J=5,3 Гц, 1H), 4,91 (с, 1H), 3,93-3,85 (м, 4H), 3,21 (д, J=6,3 Гц, 2H), 2,74 (с, 1H), 2,16 (д, J=4,0 Гц, 2H), 1,94 (д, J=6,7 Гц, 2H), 1,30-1,25 (м, 4H), 1,11-1,10 (м, 1H), 0,50-0,46 (м, 1H), 0,27-0,24 (м, 1H)
Стадия 2: К смеси соединения A-14 (18,52 мг, 122,53 мкмоль, 1,00 экв.) в EtOH (500 мкл) добавляли 2,3-дигидробензотриазол-1-илметанол (18,52 мг, 122,53 мкмоль, 1,00 экв.) одной порцией при 0°C. Смесь перемешивали при 15°C в течение 1 часа и добавляли NaBH4 (9,27 мг, 245,06 мкмоль, 2,00 экв.). Полученную смесь перемешивали при 15°C в течение 1 часа и выливали в H2O (10 мл). Водный слой экстрагировали при помощи DCM (20 мл × 3). Объединенный органический слой промывали насыщенным солевым раствором, сушили над NaSO4 и концентрировали. Остаток очищали при помощи препаративной ВЭЖХ (щелочные условия) с получением A-28 (5,00 мг, 14,10 мкмоль, 11,5% выход, 100% чистота) в виде белого твердого вещества.
ЖХМС: RT=2,535 мин, m/z 355,2 [M+H]+
1H ЯМР (CDCl3, 400 МГц) δ 8,10 (д, J=5,2 Гц, 1H), 7,76 (с, 1H), 6,63 (д, J=5,2 Гц, 1H), 4,78 (д, J=3,6 Гц, 1H), 3,81 (с, 3H), 3,77-3,71 (м, 1H), 3,13 (д, J=6,4 Гц, 2H), 2,27 (с, 6H), 2,17 (д, J=12,4 Гц, 2H), 1,93 (д, J=11,6 Гц, 2H), 1,36-1,31 (м, 2H), 1,23-1,17 (м, 2H), 1,04-1,02 (м, 1H), 0,44-0,39 (м, 2H), 0,20-0,18 (м, 2H).
Получение 8-амино-1,3-диазаспиро[4,5]декан-2,4-дионгидрохлорида (амин-19)
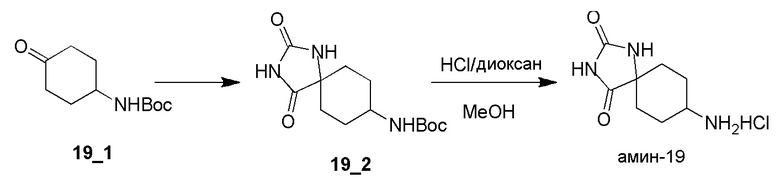
Стадия 1: Раствор NaCN (2,75 г, 56,02 ммоль, 2,39 экв.) в H2O (10 мл) добавляли к смеси соединения 19_1 (5,00 г, 23,44 ммоль, 1,00 экв.) и (NH4)2CO3 (4,96 г, 51,57 ммоль, 2,20 экв.) в EtOH (25 мл) и H2O (25 мл). Реакционную смесь перемешивали при 10°C в течение 16 часов и затем при 70°C еще в течение 24 часов. ТСХ (PE:EA=3:1) показала, что исходное вещество израсходовано (Rf=0,55) и сформировалось крупное пятно (Rf=0,25). Реакционной смеси давали остыть и фильтровали. Фильтровальную лепешку промывали при помощи H2O (100 мл) и сушили. Соединение 19_2 (4,00 г, 14,12 ммоль, 60,2% выход) получали в виде белого твердого вещества.
1H ЯМР (DMSO-d6, 400 МГц) δ 10,51 (с, 1H), 8,48 (с, 1H), 6,70-6,72 (д, J=8,0 Гц, 1H), 3,16 (с, 1H), 1,79-1,76 (м, 2H), 1,65-1,64 (д, J=4,0 Гц, 2H), 1,62-1,52 (м, 2H), 1,60-1,59 (м, 1H), 1,37 (с, 11H).
Стадия 2: К смеси соединения 19_2 (1,00 г, 3,53 ммоль, 1,00 экв.) в MeOH (10 мл) добавляли HCl/диоксан (4 M, 10 мл, 11,33 экв.) при 0°C и реакционную смесь перемешивали при 10°C в течение 16 часов. ЖХМС показала, что реагент израсходован. Реакционную смесь концентрировали с получением Амина-19 (700,00 мг, 3,19 ммоль, 90,3% выход, HCl) в виде белого твердого вещества.
1H ЯМР (DMSO-d6, 400 МГц) δ 8,52 (с, 1H), 8,21 (с, 3H),3,06 (с, 1H), 1,96-1,89 (м, 1H), 1,76-1,59 (м, 6H).
Получение 8-((4-(5-(циклопропилметил)-1-метил-1H-пиразол-4-ил)пиримидин-2-ил)амино)-1,3-диазаспиро[4,5]декан-2,4-диона (A19_4) и (1-амино-4-((4-(5-(циклопропилметил)-1-метил-1H-пиразол-4-ил)пиримидин-2-ил)амино)циклогексил)метанола (A-19)
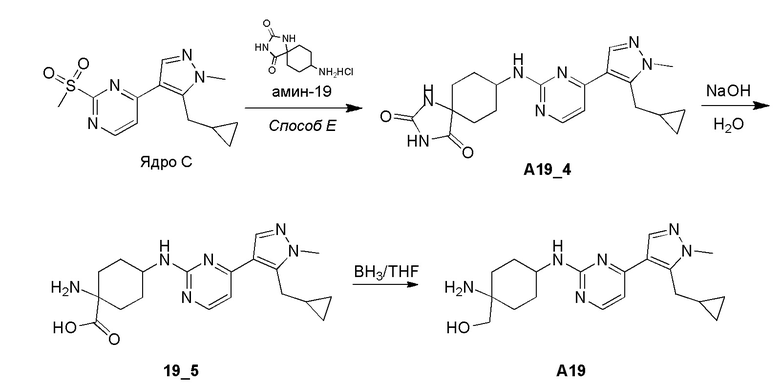
Стадия 1: Соединение A19_4 получали в соответствии с процедурой, описанной в способе E, с использованием амина-19 (4 экв.) и DIEA (4 экв.), и его получали в виде белого твердого вещества после очистки при помощи препаративной ВЭЖХ (щелочные условия). Выход: 72,2%
ЖХМС: RT =0,573 мин, m/z 397,2 [M+H]+
1H ЯМР (CD3OD, 400 МГц) δ 8,22 (с, 1H), 8,14-8,09 (м, 1H), 7,24 (д, J=5,2 Гц, 1H), 5,19 (д, J=6,8 Гц, 1H), 4,21 (с, 1H), 3,94 (с, 3H), 3,30 (д, J=6,8 Гц, 2H), 2,20 (д, J=6,0 Гц, 2H), 2,09-2,05 (т, 2H), 1,86 (д, J=14,8 Гц, 2H),1,69-1,65 (м, 2H), 1,25-1,21 (м, 1H), 0,60 (д, J=7,6 Гц, 2H), 0,39-0,35 (м, 2H),
Стадия 2: Соединение A19_4 (95,00 мг, 240,23 мкмоль, 1,00 экв.) добавляли к смеси NaOH/H2O (3 M, 640,60 мкл, 8,00 экв.) и реакционную смесь перемешивали при 120°C в течение 16 часов. Реакционную смесь охлаждали до 15°C и выливали в H2O (20 мл). Водный слой доводили до pH=7 2N раствором HCl. Смесь концентрировали. Неочищенный продукт растирали в порошок с MeOH (50 мл × 3). Фильтрат концентрировали с получением соединения 19_5 (300,00 мг, неочищенное) в виде белого твердого вещества.
ЖХМС: RT =1,148 мин, m/z 371,2 [M+H]+
1H ЯМР (CD3OD, 400 МГц) δ 8,15 (д, J=5,2 Гц, 1H), 7,95-7,93 (м, 1H), 6,89-6,85 (м, 1H), 4,03 (с, 1H), 3,91 (с, 3H), 3,28 (с, 2H), 2,32-2,25 (м, 2H), 2,16-2,13 (д, J=9,6 Гц, 2H), 1,99-1,95 (д, J=9,6 Гц, 2H),1,57-1,54 (д, J=14Гц, 2H), 1,37 (с, 1H), 0,51-0,48 (м, 2H), 0,31-0,27 (м, 2H),
Стадия 3: К смеси 19_5 (330,00 мг, 890,81 мкмоль, 1,00 экв.) в THF (500 мкл) добавляли BH3 (1M, 7,13 мл, 8,00 экв.) и реакционную смесь перемешивали при 100°C в течение 16 часов. Реакционную смесь охлаждали до 15°C и выливали в MeOH (20 мл). Смесь концентрировали. Неочищенный продукт очищали при помощи препаративной ВЭЖХ (щелочные условия) с получением соединения A-19 (23,00 мг, 64,52 мкмоль, 7,2% выход, 100% чистота) в виде белого твердого вещества.
ЖХМС: RT =1,060 мин, m/z 357,2 [M+H]+
1H ЯМР (CD3OD, 400 МГц) δ 8,12 (д, J=5,6 Гц, 1H), 7,93 (с, 1H), 6,85-6,82 (м, 1H), 3,88-3,80 (м, 4H), 3,28 (с, 2H), 3,39 (с, 2H), 3,27 (д, J=6,4 Гц, 2H), 2,03-1,94 (м, 3H),1,68-1,58 (м, 6H), 1,13 (д, J=5,2 Гц, 1H), 0,50-0,47 (м, 2H), 0,30-0,28 (м, 2H).
Получение 8-((4-(5-(циклопропилметил)-1-метил-1H-пиразол-4-ил)пиримидин-2-ил)амино)-3-окса-1-азаспиро[4,5]декан-2-она (A-26)
К смеси соединения A-19 (20,00 мг, 56,11 мкмоль, 1,00 экв.) в THF (500 мкл) добавляли CDI (9,1 мг, 56,11 мкмоль, 1,00 экв.) и реакционную смесь перемешивали при 25°C в течение 16 часов. Реакционную смесь концентрировали. Неочищенный продукт очищали при помощи препаративной ВЭЖХ (щелочные условия) с получением соединения A-26 (4,00 мг, 10,46 мкмоль, 18,6% выход, 100% чистота) в виде белого твердого вещества.
ЖХМС: RT =2,210 мин, m/z 383,2 [M+H]+
1H ЯМР (CDCl3, 400 МГц) δ 8,19 (с, 1H), 7.,84 (с, 1H), 6,74 (с, 1H), 5,97 (с, 1H), 5,02 (с, 1H), 4,15 (с, 1H), 3,89 (с, 4H), 3,17 (с, 2H), 2,16-2,13 (д, J=12,4 Гц, 2H), 1,98-1,95 (д, J=13,2 Гц, 2H), 1,72-1,68 (д, J=12,8 Гц, 2H), 1,48-1,45 (д, J=10,08Гц, 2H), 1,10 (с, 1H), 0,49 (с, 2H), 0,24 (с, 2H).
Получение (1R,4R)-N1-(4-(5-(циклопропилметил)-1-метил-1H-пиразол-4-ил)пиримидин-2-ил)-N4-метилциклогексан-1,4-диамина (A-27)
Стадия 1: Соединение 27_2 получали в соответствии с процедурой, описанной в способе E, с использованием амина-27 (1,0 экв.) и DIEA (10 экв.) при 160°C в течение 6 часов, и его получали в виде желтого твердого вещества после очистки при помощи препаративной ВЭЖХ (TFA). Выход: 41,1%
1H ЯМР (CDCl3, 400 МГц) δ 8,18 (д, J=5,2 Гц, 1H), 7,83 (с, 1H), 6,71 (д, J=5,6 Гц, 1H), 4,87 (с, 1H), 4,41 (с, 1H), 3,89-3,82 (м, 4H), 3,49 (с, 1H), 3,19 (д, J=6,3 Гц, 2H), 2,19-2,05 (м, 4H), 1,46 (с, 9H), 1,35-1,29 (м, 7H), 1,26 (м, 2H), 1,23 (м, 2H), 0,50-0,46 (м, 2H), 0,26-0,24 (м, 2H)
Стадия 2: К раствору соединения 27_2 (100,00 мг, 234,44 мкмоль, 1,00 экв.) в THF (4,00 мл) добавляли LiAlH4 (26,69 мг, 703,32 мкмоль, 3,00 экв.) при 0°C. Смесь перемешивали при 70°C в течение 3 часов. Смесь охлаждали до 0°C и затем гасили водным раствором NH4Cl (3 капли). Полученный раствор сушили над Na2SO4 и затем концентрировали. Остаток не удалось очистить при помощи препаративной ВЭЖХ. Таким образом, соединение A-27 (70 мг, неочищенное) нужно было очистить после защиты при помощи Boc группы. К раствору соединения A-27 (неочищенное) в THF (1 мл) добавляли (Boc2)2 (200 мг, 2 экв.). После перемешивания в течение 1 часа при 20°C реакционную смесь очищали при помощи препаративной ТСХ (EA) с получением соединения 27_Boc (30 мг, 29,13% общий выход), которое использовали непосредственно на следующей стадии.
Стадия 3: К раствору 27_Boc (30,00 мг, 68,09 мкмоль, 1,00 экв.) в DCM (100 мкл) добавляли HCl/диоксан (4 M, 85 мкл, 5,00 экв.) одной порцией при 0°C и смесь перемешивали при 15°C в течение 5 часов. Реакционную смесь концентрировали с получением соединения A-27 (12,00 мг, 30,88 мкмоль, 45,35% выход, 97% чистота, HCl) в виде светло-желтого твердого вещества. Выход: 45,4%
ЖХМС: RT =2,558 мин, m/z 341,2 [M+H]+
1H ЯМР (CD3OD, 400МГц) δ 8,26 (с, 1H), 8,14 (д, J=6,4 Гц, 1H), 7,31 (д, J=6,4Гц, 1H), 4,19-4,12 (м, 1H), 3,95 (с, 3H), 3,71 (с, 2H), 3,17 (с, 1H),2,76 (с, 3H), 2,31-2,20 (м, 4H), 1,65 (м, 4H), 1,21 (с, 1H), 0,51 (с, 2H), 0,37 (д, J=3,2 Гц, 2H).
Получение N-((1R,4R)-4-аминоциклогексил)-2-метоксиацетамидгидрохлорида (амин-29)
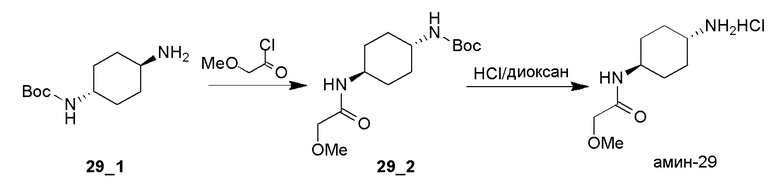
Стадия 1: К смеси соединения 29_1 (658,25 мг, 3,07 ммоль, 1,0 экв.) и 2-метоксиацетилхлорида (500 мг, 4,61 ммоль, 1,5 экв.) в DCM (5 мл) добавляли DIEA (310,82 мг, 3,07 ммоль, 1,00 экв.) при 0°C. Затем реакционную смесь перемешивали при 15°C в течение 2 часов. Реакционную смесь концентрировали под вакуумом. Остаток очищали при помощи колоночной хроматографии с получением соединения 29_2 (490,00 мг, 1,71 ммоль, 55,7% выход) в виде белого твердого вещества.
1H ЯМР (CDCl3, 400 МГц) δ 3,80 (с, 1H), 3,74-3,67 (м, 1H), 3,40 (с, 1H), 3,33 (с, 3H), 1,97-1,87 (м, 4H), 1,37 (с, 9H), 1,22-1,12 (м, 3H)
Стадия 2: Соединение 29-2 подвергали процедуре снятия защиты аналогично тому, как показано на стадии 3 синтеза соединения A27, и Амин-29 получали в виде белого твердого вещества. Выход: 91,9%
1H ЯМР (CD3OD, 400МГц) 4,05 (с, 1H), 374 (с, 2H), 3,70-3,69 (м, 1H), 3,36 (с, 3H), 3,28-3,27 (м, 1H), 3,09-3,07 (м, 1H), 2,07-2,04 (м, 2H), 1,94-1,92 (м, 2H), 1,56-1,40 (м, 4H)
Получение N-((1R,4R)-4-((4-(5-(циклопропилметил)-1-метил-1H-пиразол-4-ил)пиримидин-2-ил)амино)циклогексил)-2-метоксиацетамида (A29_1)
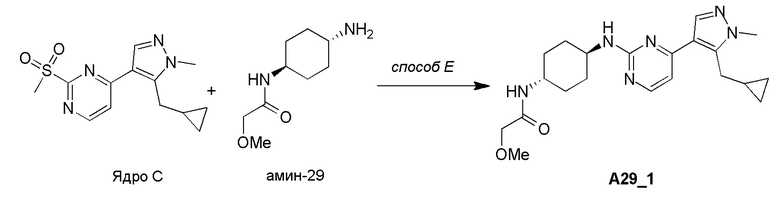
Соединение A29_1 получали в соответствии с процедурой, описанной в способе E, с использованием амина-29 (1,0 экв.) и DIEA (4 экв.), и его получали в виде светло-желтого твердого вещества после очистки при помощи препаративной ВЭЖХ (щелочные условия). Выход: 27,4%
ЖХМС: RT=2,535 мин, m/z 355,2 [M+H]+
1H ЯМР (CDCl3, 400 МГц) δ 8,20 (д, J=5,2 Гц, 1H), 7,85 (с, 1H), 6,73 (д, J=5,2 Гц, 1H), 6,40 (д, J=8,0 Гц, 1H), 4,92 (с, 1H), 3,91 (м, 7H), 3,45 (м, 3H), 3,27- 3,20 (м, 2H), 2,21 (с, 2H), 2,10 (с, 2H), 1,45-1,36 (м, 4H), 1,13-1,12 (м, 1H), 0,53-0,48 (м, 2H), 0,28-0,26 (кв., J=4,9 Гц, 2H)
Получение (1R,4R)-N1-(4-(5-(циклопропилметил)-1-метил-1H-пиразол-4-ил)пиримидин-2-ил)-N4-(2-метоксиэтил)циклогексан-1,4-диамина (A-29)
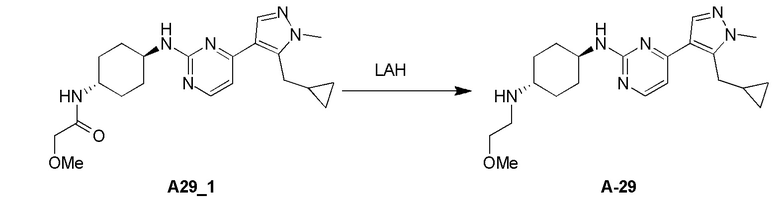
К смеси LAH (40,00 мг, 1,05 ммоль, 10,50 экв.) в THF (4 мл) добавляли раствор соединения A29_1 (40,00 мг, 100,38 мкмоль, 1,00 экв.) в THF (1 мл) при 15°C. Смесь перемешивали при 70°C в течение 36 часов. Реакционную смесь охлаждали и выливали в H2O (0,1 мл). Добавляли 1N раствор NaOH (0,1 мл) и фильтровали. Фильтрат концентрировали. Неочищенный продукт очищали при помощи препаративной ВЭЖХ (щелочные условия) с получением A-29 (4,00 мг, 9,88 мкмоль, 9,8% выход, 95,0% чистота) в виде желтого твердого вещества.
ЖХМС: RT=2,535 мин, m/z 355,2 [M+H]+
1H ЯМР (CDCl3, 300 МГц) δ 8,17 (д, J=6,8 Гц, 1 H), 7,82 (с, 1 H), 6,76-6,69 (м, 1 H), 4,88 (д, J=10,4 Гц, 1 H), 3,89 (с, 4 H), 3,54-3,50 (м, 2 H), 3,38 (с, 3 H), 3,22 (д, J=8,40 Гц, 2 H) 2,84-2,81 (м, 2H), 2,49 (с, 1H) 2,19 (с, 2H), 2,01 (с, 1H) 1,38 (с, 4H),1,07(с, 1H) 0,49-0,43 (м, 2H),0,25-0,20 (м, 2 H)
Получение (1R,4R)-4-(пиперидин-1-ил)циклогексанамин гидрохлорида (амин-47)
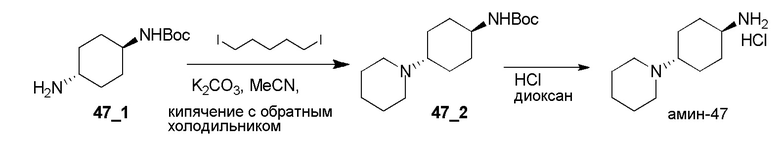
Стадия 1: Смесь соединения 47_1 (200,00 мг, 933,27 мкмоль, 1,00 экв.), 1,5-дийодопентана (302,32 мг, 933,27 мкмоль, 1,00 экв.) и K2CO3 (515,95 мг, 3,73 ммоль, 4,00 экв.) в MeCN (10 мл) перемешивали при 90°C в течение 16 часов. Смесь фильтровали и фильтрат концентрировали с получением 47_2 (300 мг, неочищенный) в виде белого твердого вещества и его использовали на следующей стадии непосредственно.
Стадия 2: Неочищенное соединение 47_2 подвергали процедуре снятия защиты аналогично тому, как показано на стадии 3 синтеза соединения A27, и амин-47 получали в виде белого твердого вещества.
1H ЯМР (D2O, 400 МГц) δ 3,55-3,10 (м, 6H), 2,16-2,08 (м, 4H), 1,59-1,41 (м, 10H).
Получение 4-(5-(циклопропилметил)-1-метил-1H-пиразол-4-ил)-N-((1R,4R)-4-(пиперидин-1-ил)циклогексил)пиримидин-2-амина (A-47)
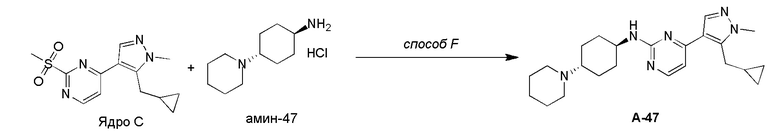
Соединение A-47 получали в соответствии с процедурой, описанной в способе F с амин-47, и его получали в виде белого твердого вещества после очистки при помощи препаративной ВЭЖХ (HCl условия). Выход: 49,3%
ЖХМС: RT=1,530 мин, m/z 395,2 [M+H]+
1H ЯМР (CD3OD, 400 МГц) δ 8,21 (с, 1H), 8,06 (д, J=6,8 Гц, 1H), 7,24-7,21 (м, 1H), 4,10 (с, 1H), 3,88 (с, 3H), 3,49 (д, J=11,2 Гц, 2H), 3,23 (с, 2H), 3,06-3,00 (м, 4H), 2,26 (д, J=10 Гц, 4H), 1,96-1,57 (м, 10H), 1,15 (с, 1H), 0,50 (с, 2H), 0,31-0,30 (м, 2H)
Получение (1R,4R)-4-морфолиноциклогексанамин гидрохлорида (амин-48)
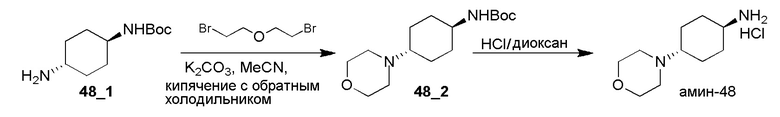
Неочищенный амин-48 синтезировали способом, аналогичным описанному для синтеза амина-47, и его получали в виде желтого твердого вещества.
1H ЯМР (D2O, 400 МГц) δ 3,87 (с, 4H), 3,24-3,11 (м, 6H), 2,20-2,12 (м, 4H), 1,59-1,39 (м, 5H).
Получение 4-(5-(циклопропилметил)-1-метил-1H-пиразол-4-ил)-N-((1R,4R)-4-морфолиноциклогексил)пиримидин-2-амина (A-48)
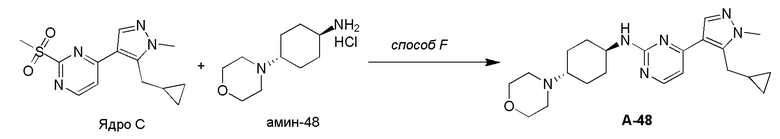
Соединение A-48 получали в соответствии с процедурой, описанной в способе F, с использованием амина-48 с TBAF (1,0 экв.), и его получали в виде белого твердого вещества после очистки при помощи препаративной ВЭЖХ (HCl условия). Выход: 51,2%
ЖХМС: RT=2,095 мин, m/z 397,3 [M+H]+
1H ЯМР (CD3OD, 400 МГц) δ 8,16 (с, 1H), 8,01 (с, 1H), 7,19 (с, 1H), 4,06-3,97 (м, 3H), 3,83-3,78 (м, 5H), 3,51-3,42 (м, 2H), 3,24 (с, 1H), 3,14-3,12 (м, 1H), 2,29-2,19 (м, 4H), 1,69-1,63 (м, 2H), 1,54-1,48 (м, 2H), 1,02(с, 1H),0,45 (с, 2H), 0,25 (с, 2H).
Получение (1R,4R)-4-(пирролидин-1-ил)циклогексанамин гидрохлорида (амин-49)
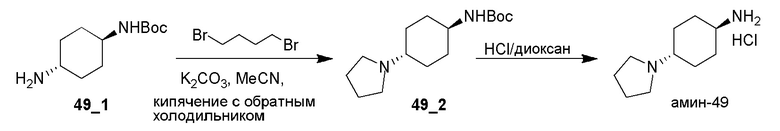
Неочищенный амин-49 синтезировали способом, аналогичным описанному для синтеза амина-47, и его получали в виде белого твердого вещества.
1H ЯМР (D2O, 400 МГц) δ 3,55-3,53 (м, 3H), 3,13-2,99 (м, 4H), 2,21-2,09 (м, 2H), 2,06-1,99 (м, 7H), 1,49-1,40 (м, 4H).
Получение 4-(5-(циклопропилметил)-1-метил-1H-пиразол-4-ил)-N-((1R,4R)-4-(пирролидин-1-ил)циклогексил)пиримидин-2-амина (A-49)
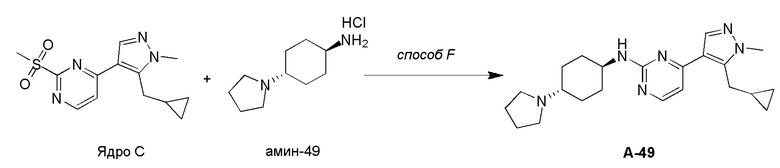
Соединение A-49 получали в соответствии с процедурой, описанной в способе F, с использованием амина-49, и его получали в виде белого твердого вещества после очистки при помощи препаративной ВЭЖХ (HCl условия). Выход: 89,3%
ЖХМС: RT=2,607 мин, m/z 381,3 [M+H]+
1H ЯМР (CDCl3, 400 МГц) δ 8,16 (с, 1H), 8,01 (с, 1H), 7,19 (с, 1H), 4,06 (с, 1H), 3,83 (с, 3H), 3,08 (с, 2H), 2,26-1,48 (м, 13H), 1,09 (с, 1H), 0,45 (с, 2H), 0,25 (с, 2H).
Получение (1R,4R)-4-(азетидин-1-ил)циклогексанамин гидрохлорида (амин-50)
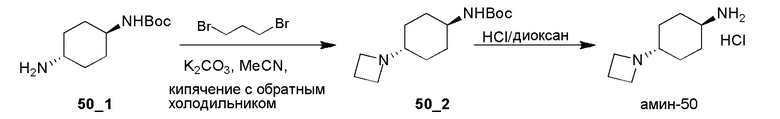
Неочищенный амин-50 синтезировали способом, аналогичным описанному для синтеза амина-47, и его получали в виде белого твердого вещества
1H ЯМР (CDCl3, 400 МГц) δ 4,15-4,08 (м, 2H), 3,65-3,12 (м, 1H), 2,28-2,03 (м, 6H), 1,50-1,42 (м, 4H), 1,26-1,20 (м, 1H).
Получение N-((1R,4R)-4-(азетидин-1-ил)циклогексил)-4-(5-(циклопропилметил)-1-метил-1H-пиразол-4-ил)пиримидин-2-амина (A-50)
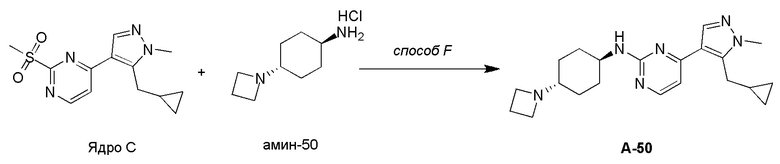
Соединение A-50 получали в соответствии с процедурой, описанной в способе F с амином-50, и его получали в виде белого твердого вещества после очистки при помощи препаративной ВЭЖХ (щелочные условия). Выход: 16%
ЖХМС: RT=2,494 мин, m/z 367,3 [M+H]+
1H ЯМР (CDCl3, 400 МГц) δ 8,17 (д, J=6,8 Гц, 1H), 7,88 (с, 1H), 6,70 (д, J=6,8 Гц, 1H), 4,86 (д, J=11,6 Гц, 1H), 3,91 (м, 4H), 3,25-3,20 (м, 5H), 2,31-2,07 (м, 6H), 2,05-2,02 (м, 2H), 1,24-1,10 (м, 5H), 0,47-0,43 (м, 2H), 0,25-0,22 (с, 2H).
Получение (1R,4R)-N1-(4-(5-(циклобутилметил)-1-метил-1H-пиразол-4-ил)пиримидин-2-ил)циклогексан-1,4-диамина (A-45)
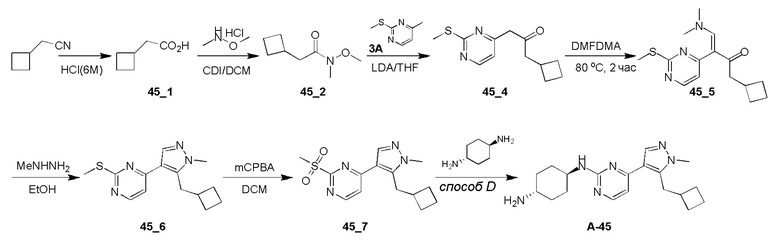
Стадия 1: Раствор 2-циклобутилацетонитрила (1,00 г, 10,51 ммоль, 1,00 экв.) в HCl (6 M, 10,00 мл, 5,71 экв.) перемешивали при 120°C в течение 16 часов. Смесь разбавляли водой (50 мл) и экстрагировали при помощи EA (20 мл × 2). Объединенный органический слой промывали водой (40 мл), сушили и концентрировали с получением соединения 45_1 (750 мг, 6,57 ммоль, 62,5% выход) в виде бесцветной жидкости
1H ЯМР (CDCl3, 400 МГц) δ 2,72-2,68 (м, 1H), 2,47-2,45 (д, J=8,0 Гц, 2H), 2,17-2,15 (м, 2H), 1,90-1,88 (м, 2H), 1,75-1,70 (м, 2H).
Стадия 2-4: Соединение 45_5 синтезировали способом, аналогичным описанному для синтеза Ядра A на схеме 1.1.
Соединение 45_2 получали в виде бесцветной жидкости.
1H ЯМР (CDCl3, 400 МГц) δ 3,68 (с, 3H), 3,16 (с, 3H), 3,75-3,71 (м, 1H), 2,55-2,53 (м, 2H), 2,16-2,13 (м, 3H), 1,89-1,87 (м, 2H), 1,73-1,68 (м, 2H).
Соединение 45_4 получали в виде желтого масла
ЖХМС: RT=0,795 мин, m/z 237,1 [M+H]+
Соединение 45_5 получали в виде темно-коричневого твердого вещества:
1H ЯМР (CDCl3, 400 МГц) δ 8,39-8,38 (д, J=4,8 Гц, 1H), 8,02 (с, 1H), 7,59 (с, 1H), 6,96-6,92(м, 1H), 2,96 (с, 3H), 2,89 (м, 5H), 2,72-2,68 (м, 1H), 2,57 (с, 3H), 2,10-2,05 (м, 2H), 1,87-1,82(м, 4H).
Стадия 5-6: Соединение 45_7 синтезировали способом, аналогичным описанному для синтеза Ядра C на схеме 1.2.
Соединение 45_6 получали в виде желтого масла после очистки при помощи препаративной ВЭЖХ (TFA условия). Выход: 21,2%;
1H ЯМР (CDCl3, 400 МГц) δ 8,41-8,39 (д, J=7,2 Гц, 1H), 7,88 (с, 1H), 7,09-7,07 (д, J=6,8 Гц, 1H), 3,89 (с, 3H), 3,35-3,33 (д, J=9,6 Гц, 2H), 2,72-2,67 (м, 1H), 2,6 (с, 3H), 2,02-1,98 (м, 2H), 1,84-1,76 (м, 4H).
Соединение 45_7 получали в виде желтого масла после очистки при помощи препаративной ТСХ. Выход: 86,6%;
ЖХМС: RT=0,706 мин, m/z 307,2 [M+H]+
Стадия 7: Соединение A-45 получали в соответствии с процедурой, описанной в способе D с транс-циклогексан-1,4-диамином (4,0 экв.), и его получали в виде желтого твердого вещества после очистки при помощи препаративной ВЭЖХ (щелочные условия). Выход: 21,9%;
ЖХМС: RT=0,539 мин, m/z 341,3 [M+H]+;
1H ЯМР (CDCl3, 400 МГц) δ 8,18-8,17 (д, J=5,2 Гц, 1H), 7,82 (с, 1H), 6,69-6,68 (д, J=5,2 Гц, 1H), 4,85-4,83 (д, J=8,0 Гц, 1H), 3,91-3,84 (м, 4H), 3,33-3,31 (д, J=7,6 Гц, 2H), 2,75-2,68 (м, 2H), 2,18-2,17 (м, 2H), 2,00-1,93 (м, 4H), 1,80-1,77 (м, 4H), 1,31-1,26 (м, 4H).
Получение (1R,4R)-N1-(4-(5-(циклопентилметил)-1-метил-1H-пиразол-4-ил)пиримидин-2-ил)циклогексан-1,4-диамина (A-46)
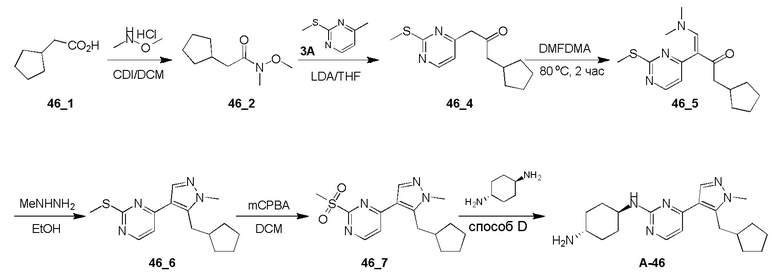
Соединение A-46 синтезировали способом, аналогичным описанному для синтеза соединения A-45.
Соединение 46_2 получали в виде бесцветного масла.
1H ЯМР (CDCl3, 400 МГц) δ 3,64 (с, 3H), 3,14 (с, 3H), 2,41-2,40 (т, J=7,2 Гц, 2H), 2,26-2,25 (м, 1H), 1,82-1,80 (м, 2H), 1,59-1,51 (м, 4H), 1,15-1,13 (м, 2H).
Соединение 46_5 получали в виде желтого масла.
ЖХМС: RT=1,294 мин, m/z 306,2 [M+H]+
Соединение 46_6 получали в виде желтого масла после очистки при помощи препаративной ТСХ. Выход: 26,5%
ЖХМС: RT=0,866 мин, m/z 289,1 [M+H]+
1H ЯМР (CDCl3, 400 МГц) δ 8,42-8,41 (д, J=5,2 Гц, 1H), 7,90 (с, 1H), 7,10-7,09 (д, J=5,2 Гц, 1H), 3,90 (с, 3H), 3,26-3,24 (д, J=7,6 Гц, 2H), 2,61-2,60 (д, J=4,8 Гц, 3H), 2,21 (м, 1H), 1,72-1,67 (м, 4H), 1,27-1,26 (м, 2H), 1,25 (м, 3H).
Соединение 46_7 получали в виде белого твердого вещества после очистки при помощи препаративной ТСХ. Выход: 44,0%
ЖХМС: RT=1,219 мин, m/z 321,2 [M+H]+
Соединение A-46 получали в виде желтого твердого вещества после очистки при помощи препаративной ВЭЖХ (щелочные условия). Выход: 52,7%
ЖХМС: RT=2,381 мин, m/z 355,2 [M+H]+
1H ЯМР (CD3OD, 400 МГц) δ 8,16 (с, 1H), 8,07-8,03 (т, J=8,0 Гц, 1H), 7,20-7,18 (д, J=6,8 Гц, 1H), 4,06-4,02 (м, 1H), 3,88 (с, 3H), 3,31-3,30 (м, 2H), 3,16 (с, 1H), 2,17-2,15 (м, 5H), 1,67 (м, 4H), 1,56 (м, 6H), 1,30-1,25 (м, 2H)
Получение (1R,4R)-N1-(5-хлор-4-(5-(циклопропилметил)-1-метил-1H-пиразол-4-ил)пиримидин-2-ил)циклогексан-1,4-диамина (A-51)
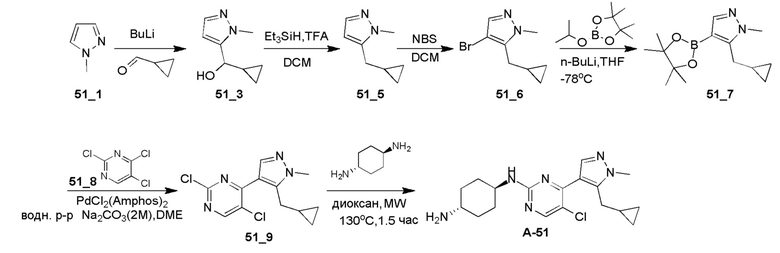
Стадия 1: К раствору соединения N-метилпиразола (51_1, 8,00 г, 97,44 ммоль, 1,00 экв.) в THF (160 мл) добавляли по каплям n-BuLi (2,5 M, 46,77 мл, 1,20 экв.) при -78°C. Затем полученную смесь перемешивали при этой температуре в течение 1 часа, добавляли по каплям раствор циклопропанкарбальдегида (8,20 г, 116,93 ммоль, 1,20 экв.) в THF (80 мл), и затем реакционную смесь перемешивали при 20°C в течение 16 часов. ТСХ (PE:EA=2:1) показала расход реагента 1 (Rf=0,3) и образование продукта (Rf=0,05). Смесь выливали в водный раствор NH4Cl (300 мл) и перемешивали в течение 10 минут. Водную фазу экстрагировали этилацетатом (100 мл×2). Объединенную органическую фазу промывали насыщенным солевым раствором (100 мл), сушили над безводным Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали при помощи колоночной хроматографии (SiO2, PE:EA=8:1~0:1) с получением соединения 51_3 (12,00 г, 78,85 ммоль, 80,9% выход, 100% чистота) в виде бесцветного масла.
ЖХМС: RT=0,118 мин, m/z 153,1 [M+H]+
Стадия 2: Реакционную смесь соединения 51_3 (9,00 г, 59,14 ммоль, 1,00 экв.), TFA (40,46 г, 354,84 ммоль, 26,27 мл, 6,00 экв.) и Et3SiH (41,26 г, 354,84 ммоль, 56,52 мл, 6,00 экв.) в DCM (900 мл) перемешивали при 40°C в течение 36 часов. Смесь доводили до уровня pH=8 водным раствором NaHCO3 и разделяли. Органический слой концентрировали и очищали при помощи препаративной ВЭЖХ (щелочные условия) с получением соединения 51_5 (2,10 г, 15,42 ммоль, 26,1% выход) в виде темно-коричневого масла.
ЖХМС: RT=0,565 мин, m/z 137,1 [M+H]+
Стадия 3: К раствору соединения 51_5 (2,10 г, 15,42 ммоль, 1,00 экв.) в DCM (21 мл) добавляли NBS (3,02 г, 16,96 ммоль, 1,10 экв.) при 0°C. Смесь перемешивали при 20°C в течение 2 часов. Смесь концентрировали и очищали при помощи колоночной хроматографии (SiO2, PE:EA=20:1) с получением соединения 51_6 (3,00 г, 13,95 ммоль, 90,5% выход) в виде желтого масла.
ЖХМС: RT=0,784 мин, m/z 217,1 [M+H]+
1H ЯМР (CDCl3, 400 МГц) δ 7,39 (с, 1 H), 3,87 (с, 3 H), 2,65-2,63 (д, J=8,8 Гц, 2 H), 0,98-0,94 (м, 1H), 0,55-0,51 (м, 2H), 0,29-0,25 (м, 2H).
Стадия 4: К раствору соединения 51_6 (3,00 г, 13,95 ммоль, 1,00 экв.) в THF (60 мл) добавляли n-BuLi (2 M, 10,46 мл, 1,50 экв.) по каплям при -78°C. После перемешивания в течение 0,5 часа при этой температуре, добавляли раствор 2-изопропокси-4,4,5,5-тетраметил-1,3,2-диоксаборолана (5,19 г, 27,90 ммоль, 2,00 экв.) в THF (6 мл). Полученную смесь нагревали до 20°C и перемешивали в течение 0,5 часа. ТСХ (PE:EA=5:1) показала расход реагента (Rf=0,6) и образование продукта (Rf=0,5). Смесь гасили насыщенным раствором NH4Cl (50 мл) и экстрагировали при помощи EA (100 мл). Органический слой концентрировали и очищали при помощи колоночной хроматографии (SiO2, PE:EA=20:1~10:1) с получением соединения 51_ 7 (3,30 г, 11,40 ммоль, 81,7% выход, 90,5% чистота) в виде бесцветного масла.
ЖХМС: RT=0,801 мин, m/z 263,2 [M+H]+
1H ЯМР (CDCl3, 400 МГц) δ 7,67 (с, 1 H), 3,85 (с, 3 H), 2,82-2,81 (д, J=6,8 Гц, 2 H),1,30 (с, 12H), 0,92-0,90 (м, 1H), 0,45-0,42 (м, 2H), 0,29-0,27 (м, 2H).
Стадия 5: К раствору соединения 51_7 (500,00 мг, 1,91 ммоль, 1,00 экв.) в DME (10 мл) добавляли 2,4,5-трихлорпиримидин (51_8, 420,40 мг, 2,29 ммоль, 1,20 экв.), Na2CO3 (2 M, 2,10 мл, 2,20 экв.) и катализатор PdCl2(Amphos)2 (67,62 мг, 95,50 мкмоль, 0,05 экв.) в атмосфере азота. Полученную смесь перемешивали при 85°C в течение 2 часов в атмосфере азота. ТСХ (PE:EA=5:1) показала расход реагента (Rf=0,4) и образование продукта (Rf=0,5). Смесь разбавляли водой (50 мл) и экстрагировали при помощи EA (50 мл). Органический слой концентрировали и очищали на колонке с силикагелем (PE:EA=5:1) с получением соединения 51_9 (350,0 мг, 1,05 ммоль, 55% выход, 85% чистота) в виде желтого масла.
ЖХМС: RT=0,819 мин, m/z 283,1 [M+H]+
Стадия 6: Реакционную смесь соединения 51_9 (300,00 мг, 1,06 ммоль, 1,00 экв.) и транс-циклогексан-1,4-диамина (484,17 мг, 4,24 ммоль, 4,00 экв.) в диоксане (4,5 мл) перемешивали при 130°C в течение 2 часов в условиях микроволнового нагрева. Смесь фильтровали и концентрировали. Неочищенное вещество очищали при помощи препаративной ВЭЖХ (HCl условия) с получением соединения A-51 (80,00 мг, 200,04 мкмоль, 18,9% выход, 99,3% чистота, HCl) в виде желтого твердого вещества.
ЖХМС: RT=2,817 мин, m/z 361,1 [M+H]+
1H ЯМР (DMSO-d6, 400 МГц) δ 8,37 (с, 1 H), 8,29 (с, 3 H), 8,08 (с, 1 H), 3,86 (с, 3 H), 3,70-3,68 (м, 2 H), 3,06-3,05 (д, J=6,4 Гц, 2H), 2,96 (с, 1H), 2,03-1,95 (м, 4H), 1,49-1,32 (м, 4H), 0,98 (с, 1H), 0,42-0,40 (м, 2H), 0,16 (м, 2H).
Получение (1R,4R)-N1-(4-(5-(циклопропилметил)-1-метил-1H-пиразол-4-ил)-5-метилпиримидин-2-ил)циклогексан-1,4-диамина (A-52)
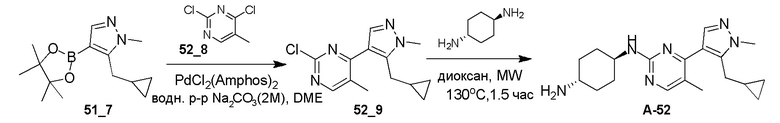
Стадия 1: К раствору соединения 51_7 (500,00 мг, 1,91 ммоль, 1,00 экв.) в DME (10 мл) добавляли соединение 52_8 (373,60 мг, 2,29 ммоль, 1,20 экв.), Na2CO3 (водн.) (2 M, 2,1 мл, 2,20 экв.) и катализатор PdCl2(Amphos)2 (67,62 мг, 95,50 мкмоль, 0,05 экв.) в атмосфере азота. Полученную смесь перемешивали при 85°C в течение 2 часов в атмосфере азота. ТСХ (PE:EA=5:1) показала расход реагента (Rf=0,4) и образование продукта (Rf=0,25). Смесь разбавляли водой (50 мл) и экстрагировали при помощи EA (50 мл). Органический слой концентрировали и очищали при помощи колоночной хроматографии (SiO2, PE:EA=20:1~5:1) с получением соединения 52_9 (350,00 мг, 1,15 ммоль, 60,3% выход, 86,5% чистота) в виде желтого масла.
ЖХМС: RT=0,761 мин, m/z 263,2 [M+H]+
Стадия 2: Реакционную смесь соединения 52_9 (350,00 мг, 1,33 ммоль, 1,00 экв.) и транс-циклогексан-1,4-диамина (607,49 мг, 5,32 ммоль, 4,00 экв.) в диоксане (5 мл) перемешивали при 130°C в течение 2 часов в условиях микроволнового нагрева. ТСХ (PE:EA=1:1) показала расход реагента (Rf=0,6) и образование продукта (Rf=0,05). Смесь фильтровали и концентрировали. Неочищенное вещество очищали при помощи препаративной ВЭЖХ (HCl условия) и затем препаративной ВЭЖХ (щелочные условия) с получением соединения A-52 (30,0 мг, 88,00 мкмоль, 6,6% выход, 99,8% чистота) в виде желтого твердого вещества.
ЖХМС: RT=2,429 мин, m/z 341,2 [M+H]+
1H ЯМР (CDCl3, 400 МГц) 8,09 (с, 1H), 7,67 (с, 1H), 4,72-4,70 (д, J=8,4Гц, 1H), 3,91(с, 3H), 3,82-3,80 (м, 1H), 3,02-3,00 (д, J=6,4Гц, 2H), 2,79-2,76 (м, 1H), 2,22 (с, 3H), 2,14-1,93 (м, 4H), 1,32-1,23 (м, 4H), 0,97 (м, 1H),0,45-0,42 (м, 2H), 0,12-0,10 (м, 2H).
Получение (1R,4R)-N1-(5-хлор-4-(5-(циклопропилметил)-1-метил-1H-пиразол-4-ил)пиримидин-2-ил)-4-метилциклогексан-1,4-диамина (A-30_1) и (1S,4S)-N1-(5-хлор-4-(5-(циклопропилметил)-1-метил-1H-пиразол-4-ил)пиримидин-2-ил)-4-метилциклогексан-1,4-диамина (A-30_2)
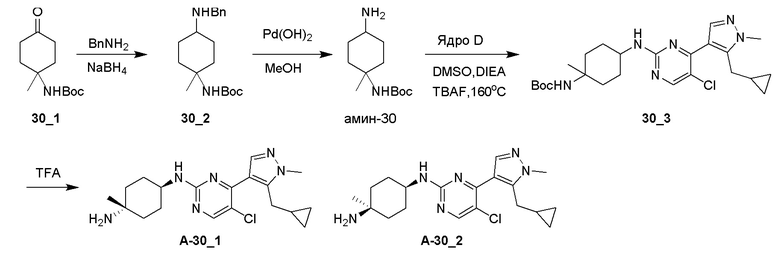
Стадия 1: К смеси соединения 30_1 (900,00 мг, 3,96 ммоль, 1,00 экв.) и BnNH2 (424,26 мг, 3,96 ммоль, 432,92 мкл, 1,00 экв.) в DCM (18,00 мл) добавляли AcOH (237,77 мг, 3,96 ммоль, 226,45 мкл, 1,00 экв.) и NaBH(OAc)3 (1,68 г, 7,92 ммоль, 2,00 экв.). Полученную смесь перемешивали при 20°C в течение 2 часов. Смесь гасили при помощи H2O (50 мл) и разделяли. Органический слой концентрировали. Остаток очищали при помощи колоночной хроматографии на Al2O3 (DCM:MeOH=20:1) с получением соединения 30_2 (650,00 мг, 2,03 ммоль, 51,3% выход, 99,6% чистота) в виде красного масла.
ЖХМС: RT=1,520 мин, m/z 319,2 [M+H]+
1H ЯМР (CDCl3, 400 МГц) δ 7,33-7,31 (м, 5 H), 4,43-4,35 (м, 1 H), 3,81 (д, J=10,8 Гц, 2 H), 2,62-2,50 (м, 1H), 2,12-2,09 (м, 1H), 1,82-1,79 (м, 4H), 1,43(с, 9H), 1,38-1,27 (м, 7H).
Стадия 2: К раствору соединения 30_2 (350,00 мг, 1,10 ммоль, 1,00 экв.) в MeOH (3,5 мл) добавляли Pd(OH)2 (35,00 мг). Смесь перемешивали при 20°C в течение 16 часов в атмосфере H2 (1,125 кг/см2). ТСХ (DCM:MeOH=10:1) показала расход реагента (Rf=0,6) и образование продукта (Rf=0,3). Смесь фильтровали и маточный раствор концентрировали с получением амина-30 (220,00 мг, 963,5 мкмоль, 87,6% выход) в виде красного масла.
MS: m/z 229,2 [M+H]+
1H ЯМР (CDCl3, 400 МГц) δ 4,43-4,33 (м, 1 H), 3,82-2,66 (м, 1 H), 2,08 (с, 1H), 1,79-1,66 (м, 4H), 1,43 (с, 9H), 1,33-1,27 (м, 7H).
Стадия 3: К раствору Амина-30 (300,00 мг, 917,99 мкмоль, 1,00 экв.) и Ядра D (230,57 мг, 1,01 ммоль, 1,10 экв.) в DMSO (4 мл) добавляли DIEA (480,97 мкл, 2,75 ммоль, 3,00 экв.) и TBAF (48,00 мг, 2,75 ммоль, 0,20 экв.). Смесь перемешивали при 160°C в течение 3 часов. Смесь разделяли между EA (50 мл) и H2O (50 мл). Органический слой концентрировали с получением соединения 30_3 (400,00 мг, неочищенный) в виде красного масла.
ЖХМС: RT=1,081 мин, m/z 475,2 [M+H]+
Стадия 4: К раствору соединения 30_3 (400,00 мг, 842,05 мкмоль, 1,00 экв.) в DCM (4 мл) добавляли TFA (800 мкл) при 0°C. Полученную смесь перемешивали при 20°C в течение 1 часа. ТСХ (PE:EA=2:1) показала расход реагента (Rf=0,6) и образование продукта (Rf=0,05). Смесь концентрировали. Остаток очищали при помощи препаративной ВЭЖХ (HCl условия) с получением соединения A-30_1 (55,00 мг, 127,60 мкмоль, 15,1% выход, 95,4% чистота, HCl) и A-30_2 (40,00 мг, 95,31 мкмоль, 11,3% выход, 98,0% чистота, HCl) в виде желтого твердого вещества.
ЖХМС(A-30_1): RT=2,495 мин, m/z 375,2 [M+H]+
1H ЯМР_Пик-1 (MeOD, 400 МГц) δ 8,49 (с, 1 H), 8,45 (с, 1 H), 4,10 (с, 1 H), 3,96 (с, 3H), 3,23 (д, J=6,4 Гц, 2H), 2,11-2,10 (м, 2H), 2,07-1,94 (м, 2H), 1,84-1,74 (м, 4H), 1,47 (с, 3H), 1,09-1,08(м, 1H), 0,55-0,51 (м, 2H), 0,28-0,27 (м, 2H).
ЖХМС(A-30_2): RT=2,527 мин, m/z 375,2 [M+H]+
1H ЯМР_Пик-2 (MeOD, 400 МГц) δ 8,47 (с, 2 H), 4,19 (с, 1 H), 3,96 (с, 3H), 3,19 (д, J=6,4 Гц, 2H), 2,05-2,1,99 (м, 2H), 1,87-1,78 (м, 2H), 1,42 (с, H), 1,08-1,07 (м, 1H), 0,54-0,52 (м, 2H), 0,30-0,28 (м, 2H).
Получение (1R,4R)-N1-(4-(5-(циклобутилметил)-1-изопропил-1H-пиразол-4-ил)пиримидин-2-ил)циклогексан-1,4-диамина (A53)
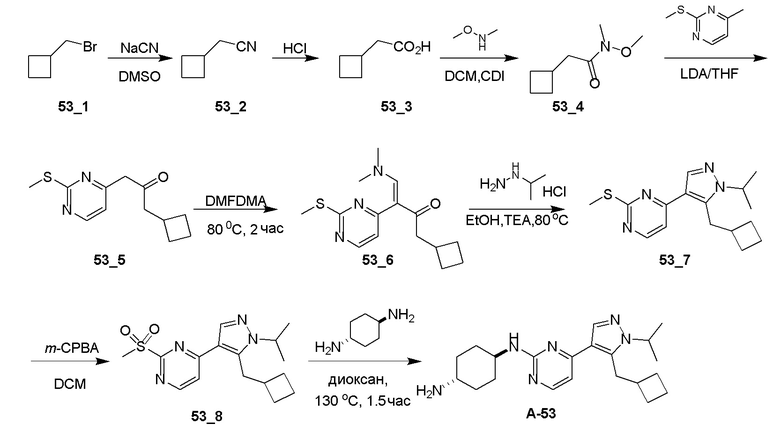
Стадия 1: К смеси NaCN (13,81 г, 281,82 ммоль, 1,40 экв.) в DMSO (240 мл) добавляли соединение 53_1 (30,00 г, 201,30 ммоль, 22,56 мл, 1,00 экв.) по каплям при 60°C. Смесь хранили при 75°C в течение 16 часов. Смесь охлаждали и разбавляли водой (500 мл). Раствор экстрагировали при помощи EtOAc (200 мл × 3). Объединенные органические слои промывали насыщенным солевым раствором (100 мл × 3) и сушили. Органический слой концентрировали с получением соединения 53_2 (15,00 г, 157,66 ммоль, 78,3% выход) в виде светло-желтой жидкости.
1H ЯМР (CDCl3, 400 МГц) δ 2,64-2,63 (м, 1 H), 2,42-2,41 (д, J=6,4 Гц, 2 H), 2,20-2,17 (м, 2 H), 1,90-1,83 (м, 4H).
Стадия 2: Раствор соединения 53_2 (15,00 г, 157,66 ммоль, 1,00 экв.) в HCl (6 M, 150 мл, 5,71 экв.) перемешивали при 120°C в течение 16 часов. Смесь разбавляли водой (500 мл) и экстрагировали при помощи EtOAc (200 мл × 2). Объединенные органические слои промывали водой (400 мл). Органический слой сушили и концентрировали с получением соединения 53_3 (16,00 г, 140,18 ммоль, 88,9% выход) в виде бесцветной жидкости.
1H ЯМР (CDCl3, 400 МГц) δ 2,69-2,68 (м, 1 H), 2,47-2,45 (м, 2 H), 2,17-2,15 (м, 2H), 1,89-1,87 (м, 2H), 1,75-1,70 (м, 2H).
Стадия 3: К раствору соединения 53_3 (16,00 г, 140,18 ммоль, 1,00 экв.) и N-метоксиметанамина (20,51 г, 210,27 ммоль, 1,50 экв., HCl) в DCM (160 мл) добавляли CDI (45,46 г, 280,36 ммоль, 2,00 экв.) при 0°C порциями. Смесь перемешивали при 20°C в течение 16 часов. Смесь разбавляли водой (50 мл) и экстрагировали при помощи DCM (30 мл × 3). Объединенный органический слой сушили и концентрировали с получением неочищенного продукта. Неочищенный продукт очищали на колонке с силикагелем (PE:EA=50:1~10:1) с получением соединения 53_4 (12,00 г, 76,33 ммоль, 54,4% выход) в виде бесцветной жидкости.
1H ЯМР (CDCl3, 400 МГц) δ 3,68-3,67 (д, J=2,4 Гц, 3 H), 3,16 (с, 3 H), 2,77-2,73 (м, 1H), 2,54-2,53 (м, 2H), 2,16-2,13 (м, 2H), 1,88-1,86 (м, 2H), 1,73-1,72 (м, 2H), 1,70-1,68 (м, 2H).
Стадия 4: К раствору соединения 53_4 (4,50 г, 32,09 ммоль, 1,00 экв.) в THF (225 мл) добавляли LDA (2 M, 24,07 мл, 1,50 экв.) при -78°C. После перемешивания в течение 1 часа в эту смесь добавляли раствор 2-циклобутил-N-метокси-N-метил-ацетамида (6,05 г, 38,51 ммоль, 1,20 экв.) в THF (120 мл) при -78°C. Полученную смесь перемешивали при -78°C в течение 4 часов, гасили насыщенным NH4Cl (200 мл) и водную фазу экстрагировали этилацетатом (200 мл × 3). Объединенные слои промывали насыщенным солевым раствором (200 мл), сушили над безводным Na2SO4, фильтровали и концентрировали с получением соединения 53_5 (10,00 г, неочищенный) в виде желтого масла.
ЖХМС: RT=0,788 мин, m/z 237,1 [M+H]+
Стадия 5: Раствор соединения 53_5 (10,00 г, 42,31 ммоль, 1,00 экв.) в DMF-DMA (201,68 г, 1,69 моль, 224,09 мл, 40,00 экв.) перемешивали при 90°C в течение 2 часов. Смесь концентрировали с получением неочищенного продукта. Неочищенный продукт очищали на колонке с силикагелем (DCM:MeOH=1:0~10:1) с получением соединения 53_6 (8,80 г, неочищенный) в виде черно-коричневого твердого вещества.
Стадия 6: К раствору соединения 53_6 (800,00 мг, 2,75 ммоль, 1,00 экв.) в EtOH (12 мл) добавляли изопропилгидразин (364,95 мг, 3,30 ммоль, 1,20 экв., HCl) и TEA (333,93 мг, 3,30 ммоль, 457,43 мкл, 1,20 экв.). Смесь перемешивали при 90°C в течение 1 часа. Смесь концентрировали. Неочищенное вещество очищали при помощи препаративной ВЭЖХ (TFA) с получением соединения 53_7 (600,00 мг, 1,88 ммоль, 68,5% выход, 95% чистота) в виде желтого масла.
ЖХМС: RT=0,901 мин, m/z 303,2 [M+H]+
Стадия 7: К раствору соединения 53_7 (600,00 мг, 1,98 ммоль, 1,00 экв.) в DCM (9 мл) добавляли m-CPBA (1,01 г, 4,96 ммоль, 85% чистота, 2,50 экв.) при 0°C. Смесь перемешивали при 20°C в течение 3 часов. Реакцию гасили насыщенным водным раствором NaS2O3 (50 мл) и экстрагировали при помощи DCM (20 мл × 2). Объединенные органические слои промывали водным раствором NaHCO3 (40 мл). Органический слой сушили и концентрировали. Неочищенное вещество очищали при помощи препаративной ТСХ (DCM: MeOH=10:1)(Rf=0,6) с получением соединения 53_8 (500,00 мг, 1,50 ммоль, 75,5% выход) в виде желтого масла.
ЖХМС: RT=0,803 мин, m/z 335,1 [M+H]+
Стадия 8: К раствору соединения 53_8 (500,00 мг, 1,50 ммоль, 1,00 экв.) в диоксане (7,5 мл) добавляли транс-циклогексан-1,4-диамин (685,14 мг, 6,00 ммоль, 4,00 экв.). Смесь перемешивали при 130°C в течение 2 часов в микроволновой печи. Смесь фильтровали и концентрировали. Неочищенное вещество очищали при помощи препаративной ВЭЖХ (основание) и препаративной ВЭЖХ (HCl) с получением соединения A-53 (250,00 мг, 615,9 мкмоль, 41% выход, 99,8% чистота, HCl) в виде желтого твердого вещества.
ЖХМС: RT=2,57 мин, m/z 369,2 [M+H]+
1H ЯМР (DMSO, 400 МГц) δ 8,88-8,87 (м, 1 H), 8,38-8,25 (м, 5 H), 7,30-7,28 (м, 1H), 4,78-4,76 (м, 1H), 3,36-3,34 (м, 2H), 3,05 (с, 1H), 2,61 (с, 1H), 2,05(м, 4H), 1,92-1,91 (м, 2H),1,78 (м, 4H), 1,47-1,39 (м, 10H).
Получение (1R,4R)-N1-(4-(1-метил-5-((1-метилциклопропил)метил)-1H-пиразол-4-ил)пиримидин-2-ил)циклогексан-1,4-диамина (A56)
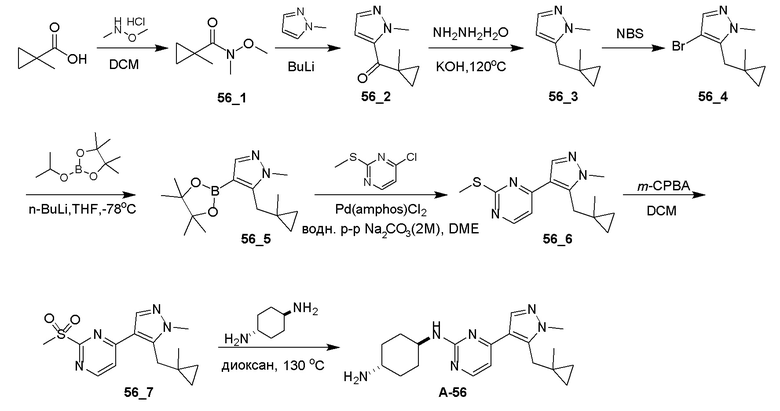
Стадия 1: К раствору 1-метилциклопропан-1-карбоновой кислоты (4,00 г, 39,95 ммоль, 1,00 экв.) и CDI (7,13 г, 43,95 ммоль, 1,10 экв.) в DCM (40 мл) при 0°C порциями добавляли N-метоксиметанамин (4,68 г, 47,94 ммоль, 1,20 экв., HCl). Смесь перемешивали при 20°C в течение 16 часов. Смесь разбавляли водой (50 мл) и экстрагировали при помощи DCM (20 мл × 3). Объединенные органические слои сушили и концентрировали с получением неочищенного продукта. Неочищенный продукт очищали на колонке с силикагелем (PE:EA=30:1~10:1) с получением соединения 56_1 (3,30 г, 23,05 ммоль, 57,7% выход) в виде бесцветной жидкости.
1H ЯМР (CDCl3, 400 МГц) δ 3,73 (с, 3 H), 3,23 (с, 3 H), 1,37 (с, 3H), 1,05-1,03 (м, 2H), 0,58-0,55 (м, 2H).
Стадия 2: К раствору 1-метилпиразола (1,70 г, 20,71 ммоль, 1,72 мл, 1,00 экв.) в THF (35,00 мл) добавляли n-BuLi (2,5 M, 9,94 мл, 1,20 экв.) при -78°C. Перемешивали в течение 1 часа. К этой смеси добавляли соединение 56_1 (3,26 г, 22,78 ммоль, 1,10 экв.) в THF (35 мл) при -78°C. Полученную смесь перемешивали при 20°C в течение 1 часа. Смесь гасили насыщенным NH4Cl (20 мл) и экстрагировали при помощи EtOAc (20 мл × 2). Органический слой концентрировали и очищали на колонке с силикагелем (PE:EA=1:0~20:1) с получением соединения 56_2 (2,50 г, 13,41 ммоль, 64,8% выход, 88,1% чистота) в виде желтого масла.
ЖХМС: RT=0,609 мин, m/z 165,1 [M+H]+
Стадия 3: Реакционную смесь соединения 56_2 (2,00 г, 12,18 ммоль, 1,00 экв.) и KOH (2,73 г, 48,72 ммоль, 4,00 экв.) в NH2NH2.H2O (2,57 г, 48,72 ммоль, 2,49 мл, 95% чистота, 4,00 экв.) и дигликоле (40 мл) нагревали до 110°C в течение 1,5 часов, затем при 200°C еще в течение 1 часа с использованием аппарата Дина-Старка. Смесь разбавляли водой (50 мл) и экстрагировали при помощи MTBE (50 мл × 2). Объединенные органические слои концентрировали с получением соединения 56_3 (1,10 г, неочищенный) в виде бесцветного масла.
ЖХМС: RT=0,639 мин, m/z 151,1 [M+H]+
Стадия 4: К раствору соединения 56_3 (1,10 г, 7,32 ммоль, 1,00 экв.) в DCM (11 мл) добавляли NBS (1,30 г, 7,32 ммоль, 1,00 экв.) при 0°C. Смесь перемешивали при 20°C в течение 1 часа. Смесь концентрировали. Неочищенное вещество очищали на колонке с силикагелем (PE:EA=1:0~50:1) с получением соединения 56_4 (1,45 г, 5,65 ммоль, 77,1% выход, 89,2% чистота) в виде бесцветного масла.
ЖХМС: RT=0,835 мин, m/z 229,0 [M+H]+
1H ЯМР (CDCl3, 400 МГц) δ 7,39 (с, 1 H), 3,85 (с, 3 H), 2,73 (с, 2H), 1,05 (с, 3H), 0,44-0,31 (м, 4H).
Стадия 5: К раствору соединения 56_4 (1,45 г, 6,33 ммоль, 1,00 экв.) в THF (29 мл) добавляли n-BuLi (2 M, 4,75 мл, 1,50 экв.) при -78°C. Через 30 минут к этой смеси добавляли 2-изопропокси-4,4,5,5-тетраметил-1,3,2-диоксаборолан (2,36 г, 12,66 ммоль, 2,59 мл, 2,00 экв.) в THF (2,5 мл). Полученную смесь нагревали до 20°C и перемешивали в течение 0,5 часа. Смесь гасили насыщенным NH4Cl (50 мл) и экстрагировали при помощи EtOAc (100 мл). Органический слой концентрировали и очищали на колонке с силикагелем (PE:EA=20:1~10:1) с получением соединения 56_5 (1,25 г, 4,11 ммоль, 64,9% выход, 90,7% чистота) в виде бесцветного масла.
ЖХМС: RT=0,928 мин, m/z 277,1 [M+H]+
Стадия 6: К раствору соединения 56_5 (500,00 мг, 1,81 ммоль, 1,00 экв.) в DME (10 мл) добавляли 4-хлор-2-метилсульфанил-пиримидин (290,79 мг, 1,81 ммоль, 210,72 мкл, 1,00 экв.), NA2CO3 (2 M, 1,99 мл, 2,20 экв.) и 4-дитрет-бутилфосфанил-N,N-диметил-анилин;дихлорпалладий (64,09 мг, 90,50 мкмоль, 64,09 мкл, 0,05 экв.) в атмосфере азота. Полученную смесь перемешивали при 85°C в течение 2 часов в атмосфере азота. Смесь разбавляли водой (50 мл) и экстрагировали при помощи EtOAc (50 мл). Органический слой концентрировали и очищали на колонке с силикагелем (PE:EA=5:1) с получением соединения 56_6 (350,00 мг, 1,14 ммоль, 63% выход, 89% чистота) в виде желтого масла.
ЖХМС: RT=0,829 мин, m/z 275,1 [M+H]+
Стадия 7: К раствору соединения 56_6 (350,00 мг, 1,28 ммоль, 1,00 экв.) в DCM (5,5 мл) добавляли MCPBA (690,28 мг, 3,20 ммоль, 80% чистота, 2,50 экв.) при 0°C. Смесь перемешивали при 20°C в течение 2 часов. Смесь гасили водным раствором Na2S2O3 (100 мл) и экстрагировали при помощи DCM (50 мл × 2). Объединенные органические слои концентрировали и очищали на колонке с силикагелем (PE:EA=1:1) с получением соединения 56_7 (200,00 мг, 617,72 мкмоль, 48,6% выход, 94,6% чистота) в виде желтого твердого вещества.
ЖХМС: RT=0,653 мин, m/z 307,1 [M+H]+
Стадия 8: Смесь соединения 56_7 (200,00 мг, 652,78 мкмоль, 1,00 экв.) и транс-циклогексан-1,4-диамина (298,17 мг, 2,61 ммоль, 4,00 экв.) в диоксане (3 мл) перемешивали при 130°C в течение 2 часов в микроволновой печи. Смесь фильтровали и концентрировали. Неочищенное вещество очищали при помощи препаративной ВЭЖХ (основание) с получением соединения A-56 (50,00 мг, 144,85 мкмоль, 22,2% выход, 98,6% чистота) в виде желтого твердого вещества.
ЖХМС: RT=1,999 мин, m/z 341,2 [M+H]+
1H ЯМР (CDCl3, 400 МГц) δ 8,18-8,17 (д, J=5,2 Гц, 1 H), 7,82 (с, 1 H), 6,69-6,67 (д, J=5,2 Гц, 1H), 4,87-4,85 (д, J=5,6 Гц, 1H), 3,88 (с, 3H), 3,84 (с, 1H), 3,38 (с, 2H), 2,75-2,73 (м, 1H), 2,17-2,16 (м, 2H), 1,94-1,92 (м, 2H), 1,31-1,27 (м, 4H), 1,08 (с, 3 H), 0,31-0,25 (м, 4H).
Получение (1R,4R)-N1-(4-(1-метил-5-неoпентил-1H-пиразол-4-ил)пиримидин-2-ил)циклогексан-1,4-диамина (A57)
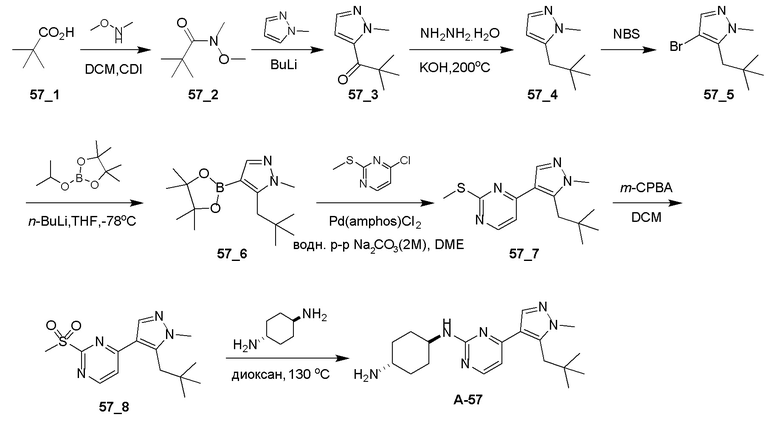
Соединение A-57 синтезировали способом, аналогичным описанному для синтеза соединения A-56.
ЖХМС: RT=1,558 мин, m/z 343,2 [M+H]+
1H ЯМР (MeOD, 400 МГц) δ 8,20-8,13 (м, 2 H), 7,29-7,27 (д, J=5,2 Гц, 1H), 4,12-4,09 (м, 1H), 3,92 (с, 3H), 3,34 (с, 1H), 3,21 (с, 1H), 2,20 (м, 4H), 1,63 (м, 4H), 0,98 (с, 9 H).
Получение (4-(2-(((1R,4R)-4-аминоциклогексил)амино)пиримидин-4-ил)-1-метил-1H-пиразол-5-ил)(циклопропил)метанола (A58)
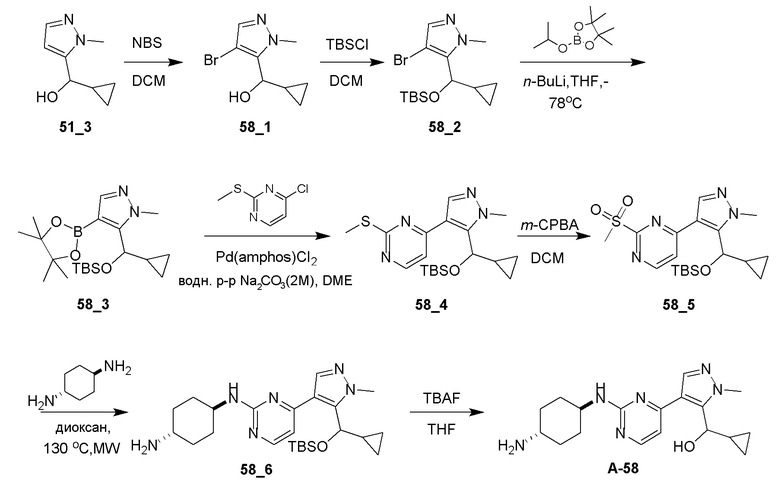
Стадия 1 - Стадия 6: Промежуточное соединение 58_6 синтезировали из соединения 51_3 способом, аналогичным описанному для синтеза соединения A-56.
ЖХМС: RT=0,676 мин, m/z 457,4 [M+H]+
Стадия 7: К раствору соединения 58_6 (300,00 мг, 656,89 μмоль, 1,00 экв.) в THF (300 мкл) добавляли TBAF.3H2O (414,51 мг, 1,31 ммоль, 2,00 экв.) при 20°C. Смесь перемешивали в течение 30 минут. Смесь фильтровали и концентрировали. Неочищенный продукт очищали при помощи препаративной ВЭЖХ (основание) с получением соединения A-58 (27,00 мг, 76,89 мкмоль, 11,7% выход, 97,5% чистота) в виде желтого твердого вещества.
ЖХМС: RT=2,29 мин, m/z 343,2 [M+H]+
1H ЯМР (CDCl3, 400 МГц) δ 8,23-8,22 (д, J=5,2 Гц, 1 H), 7,86 (с, 1 H), 6,81-6,80 (д, J=5,2 Гц, 1H), 5,09 (с, 1H), 4,32-4,30 (м, 1H), 3,89 (с, 3H), 3,81-3,79 (м, 1H), 2,93-2,88 (м, 1H), 2,20-2,06 (м, 4H), 1,52-1,31 (м, 5H), 0,59-0,28 (м, 4H).
Получение (1R,4R)-N1-(5-хлор-4-(5-(циклопропилметил)-1-изопропил-1H-пиразол-4-ил)пиримидин-2-ил)циклогексан-1,4-диамина (A59)
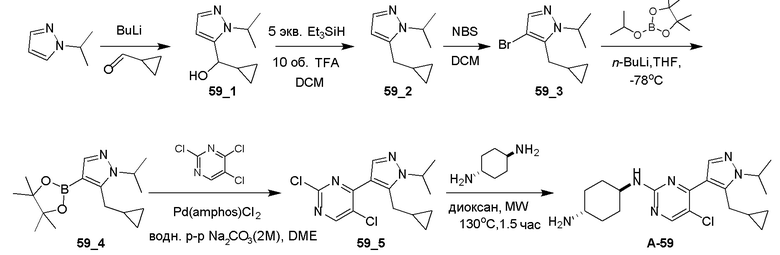
Соединение A-59 синтезировали способом, аналогичным описанному для синтеза соединения A-51.
ЖХМС: RT=2,478 мин, m/z 389,2 [M+H]+
1H ЯМР (MeOD, 400 МГц) δ 8,42-8,36 (м, 2 H), 4,82-4,75 (м, 1H), 4,00 (с, 1H), 3,21-3,20 (с, 3H), 2,20-2,15 (м, 4H), 1,60-1,56 (м, 4H), 1,51 (с, 6H), 1,05 (с, 1H), 0,55-0,53 (м, 2H), 0,28 (м, 2H).
Получение (1R,4R)-N1-(4-(5-(циклопропилметил)-1-метил-1H-пиразол-4-ил)-5-(трифторметил)пиримидин-2-ил)циклогексан-1,4-диамина(A60)
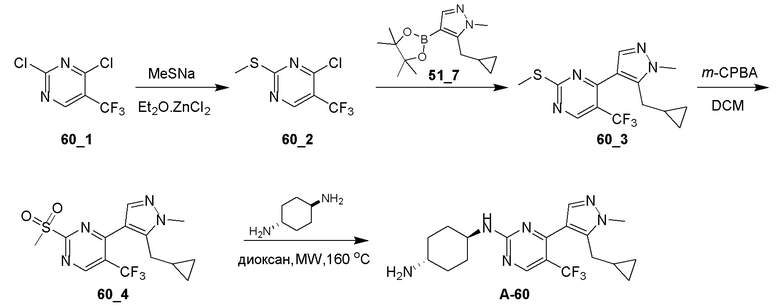
Стадия 1: К раствору соединения 60_1 (2,00 г, 9,22 ммоль, 1,00 экв.) в THF (40 мл) добавляли ZnCl2-Et2O (1 M, 11,06 мл, 1,20 экв.) при 0°C под защитой азота. Смесь перемешивали в течение 2 часов при 0°C. Добавляли метилсульфанилнатрий (646,23 мг, 9,22 ммоль, 587,48 мкл, 1,00 экв.). Полученную смесь перемешивали при 20°C в течение 16 часов. ТСХ (чистый PE) показала расход реагента 1 (Rf=0,5) и образование продукта (Rf=0,3). Смесь гасили раствором 1M HCl (20 мл) и концентрировали. Водный слой экстрагировали при помощи DCM (20 мл × 3). Объединенный органический слой концентрировали и очищали на колонке с силикагелем (PE: EA=1:0~50:1) с получением соединения 60_2 (1,00 г, 1,97 ммоль, 21,4% выход, 45,1% чистота) в виде бесцветного масла.
ЖХМС: RT=0,794 мин, m/z 228,9 [M+H]+
1H ЯМР (CDCl3, 400 МГц) δ 8,67 (с, 1H), 2,62 (с, 3H).
Стадия 2: К раствору соединения 51_7 (574,11 мг, 2,19 ммоль, 1,00 экв.) в THF (10 мл) добавляли 60_2 (500,00 мг, 2,19 ммоль, 1,00 экв.), K3PO4 (1 M, 4,38 мл, 2,00 экв.) и Ad2nBuP.Бифенил (50,00 мг) в атмосфере азота. Полученную смесь перемешивали при 85°C в течение 2 часов в атмосфере азота. ТСХ (PE:EA=1:1) показала расход реагента (Rf=0,6) и образование продукта (R=0,5). Смесь концентрировали и разбавляли при помощи H2O (50 мл). Водный слой экстрагировали при помощи EA (20 мл × 2). Органический слой концентрировали. Остаток очищали на колонке с силикагелем (PE:EA=20:1~5:1) с получением соединения 60_3 (300,00 мг, 611,96 мкмоль, 27,9% выход, 66,9% чистота) в виде желтого твердого вещества.
ЖХМС: RT=0,927 мин, m/z 329,0 [M+H]+
1H ЯМР (CDCl3, 400 МГц) δ 8,75 (с, 1H), 7,75 (с, 1H), 3,94 (с, 3H), 2,95 (д, J=6,8Гц, 2H), 2,57 (с, 3H), 0,98-0,94 (м, 1H), 0,48-0,44 (м, 2H), 0,17-0,13 (м, 2H).
Стадия 3: К раствору соединения 60_3 (400,00 мг, 1,22 ммоль, 1,00 экв.) в DCM (7 мл) добавляли m-CPBA (657,92 мг, 3,05 ммоль, 80% чистота, 2,50 экв.) при 0°C. Смесь перемешивали при 20°C в течение 1 часа. ТСХ (DCM:MeOH=20:1) показала расход реагента (Rf=0,6) и образование продукта (Rf=0,5). Смесь гасили водным раствором Na2S2O3 (100 мл) и экстрагировали при помощи DCM (50 мл × 2). Органический слой концентрировали и очищали на колонке с силикагелем (PE:EA=1:1) с получением соединения 60_4 (300,00 мг, 738,23 мкмоль, 60,5% выход, 88,7% чистота) в виде желтого твердого вещества.
ЖХМС: RT=0,72 мин, m/z 361,0 [M+H]+
Стадия 4: Смесь соединения 60_4 (200,00 мг, 555,02 мкмоль, 1,00 экв.) и транс-циклогексан-1,4-диамина (253,51 мг, 2,22 ммоль, 4,00 экв.) в диоксане (3 мл) перемешивали при 130°C в течение 2 часов в условиях микроволнового нагрева. Смесь фильтровали и фильтрат концентрировали. Остаток очищали при помощи препаративной ВЭЖХ (HCl условия) с получением соединения A-60 (80,00 мг, 185,66 мкмоль, 33,5% выход, 100% чистота, HCl) в виде желтого твердого вещества.
ЖХМС: RT=2,417 мин, m/z 395,2 [M+H]+
1H ЯМР (MeOH, 400 МГц) δ 8,66 (с, 1H), 7,90 (с, 1H), 4,15 (с, 1H), 3,97 (с, 3H), 3,20 (с, 1H), 3,10 (с, 2H), 2,18 (с, 4H), 1,62 (с, 4H), 1,05 (с, 1H), 0,53 (с, 2H), 0,25 (с, 2H).
Получение N-((1R,4R)-4-(1H-пиразол-1-ил)циклогексил)-5-хлор-4-(5-(циклопропилметил)-1-метил-1H-пиразол-4-ил)пиримидин-2-амина (A64)
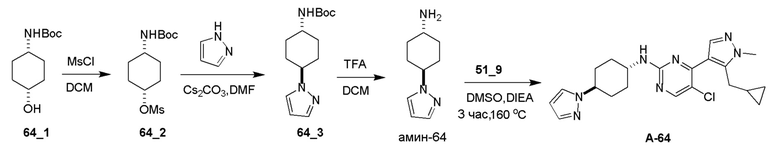
Стадия 1: К раствору соединения 64_1 (2,00 г, 9,29 ммоль, 1,00 экв.) в DCM (20 мл) добавляли TEA (1,88 г, 18,58 ммоль, 2,58 мл, 2,00 экв.) и метансульфонилхлорид (1,06 г, 9,29 ммоль, 719,03 мкл, 1,00 экв.) при 0°C. Смесь перемешивали при 20°C в течение 2 часов. Смесь разбавляли при помощи H2O (30 мл) и экстрагировали при помощи DCM (20 мл × 2). Объединенные органические слои сушили и концентрировали с получением соединения 64_2 (2,50 г, 8,52 ммоль, 91,7% выход) в виде белого твердого вещества.
1H ЯМР (CDCl3, 400 МГц) δ 4,88 (с, 1 H), 4,48 (с, 1 H), 3,52 (с, 1 H), 3,01 (с, 3H), 2,07-2,03 (м, 2H), 1,83-1,82 (м, 2H), 1,73-1,70(м, 2H), 1,61-1,55 (м, 7H), 1,448 (с, 9H).
Стадия 2: К раствору 1H-пиразола (195,25 мг, 2,87 ммоль, 1,20 экв.) в MeCN (7,00 мл) добавляли Cs2CO3 (1,56 г, 4,78 ммоль, 2,00 экв.) и соединение 64_2 (700,00 мг, 2,39 ммоль, 1,00 экв.). Смесь перемешивали при 100°C в течение 2 часов. Смесь разбавляли при помощи H2O (20 мл) и экстрагировали при помощи EtOAc (10 мл × 3). Объединенные органические слои промывали насыщенным солевым раствором (20 мл) и концентрировали. Остаток очищали при помощи препаративной ВЭЖХ (щелочные условия) с получением соединения 64_ 3 (200,00 мг, 732,25 мкмоль, 30,6% выход, 97,1% чистота) в виде желтого твердого вещества.
MS: m/z RT=0,888 мин, 266,0 [M+H]+
Стадия 3: К раствору соединения 64_3 (180,00 мг, 678,35 μмоль, 1,00 экв.) в DCM (2 мл) добавляли TFA (400,00 мкл) при 0°C. Смесь перемешивали при 20°C в течение 1 часа. Смесь разбавляли при помощи H2O (30 мл) и экстрагировали при помощи DCM (10 мл × 2). Водный слой лиофилизировали с получением амина-64 (180,00 мг, 633,91 мкмоль, 93. 5% выход, 98,3% чистота, TFA) в виде бесцветного масла.
ЖХМС: RT=0,272 мин, m/z 166,2 [M+H]+
Стадия 4: К раствору соединения 51_9 (100,00 мг, 353,16 мкмоль, 1,00 экв.) в DMSO (1,5 мл) добавляли амин-64 (95,48 мг, 529,74 мкмоль, 1,50 экв.) и DIEA (182,57 мг, 1,41 ммоль, 246,71 мкл, 4,00 экв.). Полученную смесь перемешивали при 160°C в течение 3 часов, фильтровали и маточную жидкость концентрировали. Остаток очищали при помощи препаративной ВЭЖХ (щелочные условия) и затем препаративной ВЭЖХ (HCl условия) с получением соединения A-64 (10,00 мг, 23,42 мкмоль, 6,6% выход, 100% чистота) в виде желтого твердого вещества.
ЖХМС: RT=1,981 m/z 412,2[M+H]+
1H ЯМР (MeOD, 400 МГц) δ 8,41-8,37 (м, 2 H), 7,83 (с, 1 H), 7,67 (с, 1 H),6,39 (с, 1H), 4,36 (с, 1H), 4,12-4,08 (м, 1H), 3,95 (с, 3H), 3,25-3,23 (д, J=6,4Гц, 2H), 2,26-2,23 (м, 4H), 2,05-2,02(м, 2H), 1,99-1,96 (м, 2H), 1,73-1,67(м, 2H), 1,09 (с, 1H), 0,55-0,50 (м, 2H), 0,27-0,26 (м, 2H).
Получение N-((1R,4R)-4-(1H-имидазол-1-ил)циклогексил)-5-хлор-4-(5-(циклопропилметил)-1-метил-1H-пиразол-4-ил)пиримидин-2-амина (A-65)
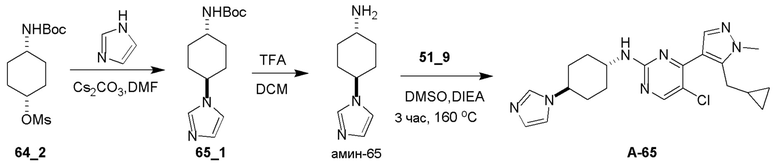
Соединение A-65 синтезировали способом, аналогичным описанному для синтеза соединения A-64, в виде желтого твердого вещества.
ЖХМС: RT=1,461 мин, m/z 412,2 [M+H]+
1H ЯМР (MeOD, 400 МГц) δ 9,10 (с, 1 H), 8,42 (с, 1 H), 7,81 (с, 1 H), 7,62 (с, 1 H), 4,55-4,49 (м, 1H), 4,13 (с, 1H), 3,96 (с, 3H), 3,25-3,24 (д, J=6,4Гц, 2H), 2,36-2,28 (м, 4H), 2,08-1,98 (м, 2H), 1,80-1,74 (м, 2H), 1,09 (с, 1H), 0,53-0,51 (м, 2H), 0,27-0,26 (м, 2H).
Получение (1R,4R)-N1-(5-хлор-4-(5-(циклопропилметил)-1-метил-1H-пиразол-4-ил)пиримидин-2-ил)-N4-фенилциклогексан-1,4-диамина (A-68)
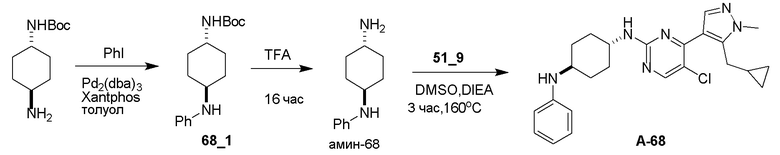
Стадия 1: К раствору трет-бутил ((1R,4R)-4-аминоциклогексил)карбамата (500,00 мг, 2,33 ммоль, 1,00 экв.) и йодбензола (713,01 мг, 3,49 ммоль, 389,63 мкл, 1,50 экв.) в THF (15 мл) добавляли t-BuOK (784,35 мг, 6,99 ммоль, 3,00 экв.) и RuPhos Индолин (100,00 мг). Полученную смесь перемешивали при 80°C в течение 12 часов в атмосфере азота. Смесь фильтровали и маточную жидкость концентрировали. Остаток очищали на колонке с силикагелем (PE:EA=10:1~4:1) с получением соединения 68_1 (300,00 мг, 869,39 мкмоль, 37,3% выход, 84,1% чистота) в виде белого твердого вещества.
ЖХМС: RT=1,024 мин, m/z 291,0 [M+H]+
Стадия 2 и стадия 3: Соединение A-68 синтезировали способом, аналогичным описанному для синтеза соединения A-64, в виде желтого твердого вещества.
ЖХМС: RT=1,194 мин, m/z 437,2 [M+H]+
1H ЯМР (MeOD, 400 МГц) δ 8,29 (с, 1 H), 8,03 (с, 1 H), 7,75-7,34 (м, 5 H), 3,86 (с, 3H), 3,05 (д, J=6,4Гц, 2H), 2,00-1,97 (м, 4H), 1,55 (с, 2H), 1,37-1,29 (м, 2H), 0,97 (с, 1H), 0,39 (с, 2H), 0,14 (с, 2H).
Получение (5R,8R)-8-((5-хлор-4-(5-(циклопропилметил)-1-метил-1H-пиразол-4-ил)пиримидин-2-ил)амино)-1-азаспиро[4,5]декан-2-она (A-71)
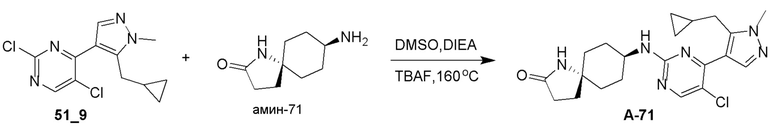
К раствору амина-71 (180,00 мг, 879,34 мкмоль, 1,00 экв., HCl) и соединения 51_9 (248,99 мг, 879,34 мкмоль, 1,00 экв.) в DMSO (2,7 мл) добавляли DIEA (614,30 мкл, 3,52 ммоль, 4,00 экв.) и TBAF (1 M, 175,87 мкл, 0,20 экв.). Смесь перемешивали при 160°C в течение 3 часов. Смесь охлаждали и фильтровали. Твердое вещество промывали при помощи MeOH (10 мл) при комнатной температуре и затем EtOAc (3 мл) при 50°C с получением соединения A-71 (40,00 мг, 90,39 мкмоль, 11,4% выход, 93,7% чистота) в виде белого твердого вещества.
ЖХМС: RT=2,836 мин, m/z 415,2 [M+H]+
1H ЯМР (DMSO, 400 МГц) δ 8,26 (с, 1 H), 8,00 (с, 1 H), 7,72 (с, 1 H), 7,20 (с, 1 H), 3,85 (с, 3H), 3,70-3,68 (м, 1H), 3,06 (д, J=6,4Гц, 2H), 2,19-2,15 (м, 2H), 1,86-1,80 (м, 5H), 1,64-1,60 (м, 3H), 1,49-1,41 (м, 5H), 0,97 (с, 1H), 0,40 (с, 2H), 0,13 (с, 2H).
Получение (1R,4R)-N1-бензил-N4-(5-хлор-4-(5-(циклопропилметил)-1-метил-1H-пиразол-4-ил)пиримидин-2-ил)циклогексан-1,4-диамина (A-74)
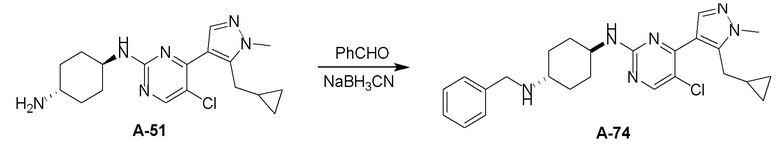
К раствору соединения A-51 (200,00 мг, 554,20 мкмоль, 1,00 экв.) и бензальдегида (58,81 мг, 554,20 мкмоль, 56,01 мкл, 1,00 экв.) добавляли AcOH (33,28 мг, 554,20 мкмоль, 31,70 мкл, 1,00 экв.) и NaBH3CN (69,65 мг, 1,11 ммоль, 2,00 экв.). Смесь перемешивали при 15°C в течение 16 часов. Смесь гасили водным раствором NaHCO3 (1 мл) и концентрировали. Остаток очищали при помощи препаративной ВЭЖХ (колонка: Phenomenex Gemini 150×25мм×10мкм; подвижная фаза: [вода (0,05% гидроксида аммония об/об)-ACN]; B%: 55%-85%, 12 мин) с получением соединения A-74 (100,00 мг, 217,80 мкмоль, 39,3% выход, 98,2% чистота) в виде розового твердого вещества.
ЖХМС: RT=2,74 мин, m/z 451,2 [M+H]+
1H ЯМР (CDCl3, 400 МГц) δ 8,20-8,13 (м, 2 H), 7,34-7,33 (м, 5 H), 4,87 (д, J=8,4Гц, 1H), 3,91 (с, 3H), 3,84 (с, 3H), 3,07-3,06 (м, 2H), 2,58-2,52 (м, 1H), 2,16-2,02 (м, 4H), 1,32-1,22 (м, 4H), 1,01-0,99 (м, 1H), 0,49-0,45 (м, 2H), 0,18-0,16 (м, 2H).
Получение (1R,4R)-N1-((1H-пиразол-4-ил)метил)-N4-(5-хлор-4-(5-(циклопропилметил)-1-метил-1H-пиразол-4-ил)пиримидин-2-ил)циклогексан-1,4-диамина (A-75)
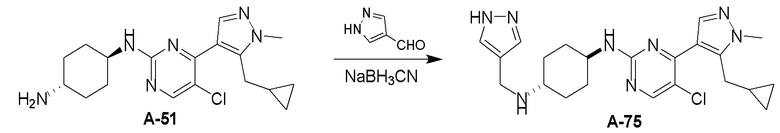
К раствору соединения A-51 (200,00 мг, 554,20 мкмоль, 1,00 экв.) и 1H-пиразол-4-карбальдегида (53,25 мг, 554,20 мкмоль, 1,00 экв.) добавляли AcOH (34,86 мкл, 609,62 мкмоль, 1,10 экв.) и NaBH3CN (69,65 мг, 1,11 ммоль, 2,00 экв.). Смесь перемешивали при 15°C в течение 16 часов. Смесь гасили водным раствором NaHCO3 (1 мл) и концентрировали. Остаток очищали при помощи препаративной ВЭЖХ (колонка: Phenomenex Gemini 150×25мм×10мкм; подвижная фаза: [вода (0,05% гидроксид аммония об/об)-ACN]; B%: 39%-39%,12 мин) с получением соединения A-75 (4,00 мг, 9,04 мкмоль, 1,6% выход, 99,7% чистота) в виде желтого твердого вещества.
ЖХМС: RT=2,95 мин, m/z 441,2 [M+H]+
1H ЯМР (CDCl3, 400 МГц) δ 8,21-8,12 (м, 2 H), 7,61 (с, 2 H), 4,90 (д, J=8,4Гц, 1H), 3,91 (с, 3H), 3,85-3,80 (м, 3H), 3,06-3,05 (м, 2H), 2,70 (м, 1H), 2,19-2,07 (м, 4H), 1,42-1,23 (м, 4H), 1,09 (м, 1H), 0,50-0,45 (м, 2H), 0,18-0,14 (м, 2H).
Получение (1R,4R)-N1-(5-хлор-4-(5-(циклопропилметил)-1-метил-1H-пиразол-4-ил)пиримидин-2-ил)-N4-(пиридин-3-илметил)циклогексан-1,4-диамина (A-76)
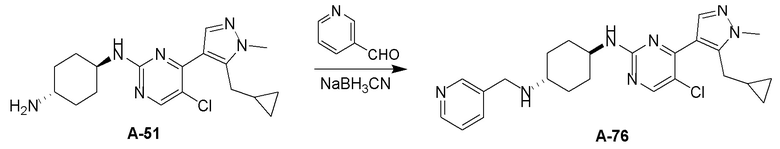
К раствору соединения A-51 (200,00 мг, 554,20 мкмоль, 1,00 экв.) и пиридин-3-карбальдегида (59,36 мг, 554,20 мкмоль, 52,07 мкл, 1,00 экв.) добавляли AcOH (33,28 мг, 554,20 мкмоль, 31,70 мкл, 1,00 экв.) и NaBH3CN (69,65 мг, 1,11 ммоль, 2,00 экв.). Смесь перемешивали при 15°C в течение 16 часов. Смесь гасили водным раствором NaHCO3 (1 мл) и концентрировали. Остаток очищали при помощи препаративной ВЭЖХ (колонка: Phenomenex Gemini 150×25мм×10мкм; подвижная фаза: [вода (0,05% гидроксида аммония об/об)-ACN]; B%: 40%-58%,12 мин.) с получением соединения A-76 (100,00 мг, 219,09 мкмоль, 39,5% выход, 98,8% чистота) в виде желтого твердого вещества.
ЖХМС: RT=2,381 мин, m/z 452,2 [M+H]+
1H ЯМР (CDCl3, 400 МГц) δ 8,58-8,51 (м, 2 H), 7,69 (д, J=8,0Гц, 1H), 7,27 (с, 1 H), 4,87 (д, J=8,0Гц, 1H), 3,91 (с, 3H), 3,85-3,81 (м, 3H), 3,07-3,06 (м, 2H), 2,56-2,51 (м, 1H), 2,16-2,01 (м, 4H), 1,30-1,22 (м, 4H), 1,09 (м, 1H), 0,50-0,46 (м, 2H), 0,19-0,16 (м, 2H).
Получение (1R,4R)-N1-((1H-пиразол-4-ил)метил)-N4-(4-(5-(циклопропилметил)-1-метил-1H-пиразол-4-ил)пиримидин-2-ил)циклогексан-1,4-диамина (A80)
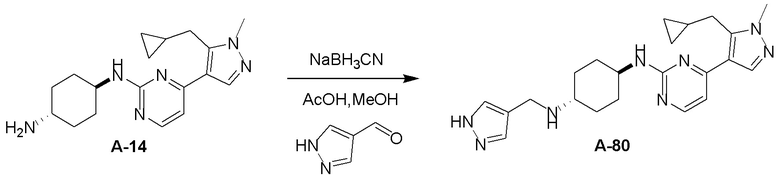
К раствору соединения A-14 (200,00 мг, 612,67 мкмоль, 1,00 экв.) и 1H-пиразол-4-карбальдегида (58,87 мг, 612,67 мкмоль, 1,00 экв.) добавляли AcOH (36,79 мг, 612,67 мкмоль, 35,04 мкл, 1,00 экв.) и NaBH3CN (77,00 мг, 1,23 ммоль, 2,00 экв.). Смесь перемешивали при 15°C в течение 16 часов. Смесь гасили водным раствором NaHCO3 (1 мл) и концентрировали. Остаток очищали при помощи препаративной ВЭЖХ (щелочные условия) с получением соединения A-80 (20,00 мг, 48,90 мкмоль, 8% выход, 99,4% чистота) в виде желтого твердого вещества.
ЖХМС: RT=2,418 мин, m/z 407,2 [M+H]+
1H ЯМР (CDCl3, 400 МГц) δ 8,17 (д, J=5,2Гц, 1H), 7,83 (с, 1H), 7,58 (с, 1H), 6,71 (д, J=6,2Гц, 1H), 4,88 (д, J=7,6Гц, 1H), 3,89 (с, 4H), 3,81 (с, 2H), 3,21 (д, J=6,0Гц, 2H), 2,61 (с, 1H), 2,21-2,03 (м, 4H), 1,36-1,25 (м, 4H), 1,09 (с, 1H), 0,48-0,46 (м, 2H), 0,25-0,24 (м, 2H).
Получение (1R,4R)-N1-(4-(5-(циклопропилметил)-1-метил-1H-пиразол-4-ил)пиримидин-2-ил)-N4-((1-метил-1H-пиразол-4-ил)метил)циклогексан-1,4-диамина (A81)
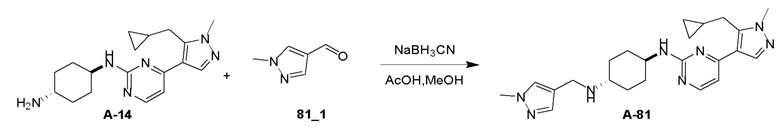
К раствору соединения A-14 (200,00 мг, 612,67 мкмоль, 1,00 экв.) и соединения 81_1 (67,46 мг, 612,67 мкмоль, 1,00 экв.) добавляли AcOH (36,79 мг, 612,67 мкмоль, 35,04 мкл, 1,00 экв.) и NaBH3CN (77,00 мг, 1,23 ммоль, 2,00 экв.). Смесь перемешивали при 15°C в течение 16 часов. Смесь гасили водным раствором NaHCO3 (1 мл) и концентрировали. Остаток очищали при помощи препаративной ВЭЖХ (щелочные условия) с получением соединения A-81 (40,00 мг, 87,81 мкмоль, 14,3% выход, 92% чистота) в виде желтого твердого вещества.
ЖХМС: RT=2,469 мин, m/z 421,2 [M+H]+
1H ЯМР (CDCl3, 400 МГц) δ 8,17 (д, J=5,2Гц, 1H), 7,82 (с, 1 H), 7,57 (с, 1 H), 7,51 (с, 1 H), 6,7 (д, J=6,2Гц, 1H), 4,84 (д, J=7,6Гц, 1H), 3,89-3,85 (м, 8H), 3,19 (д, J=6,0Гц, 2H), 2,73 (с, 1H), 2,25-2,12 (м, 4H), 1,54 (м, 1 H), 1,29-1,23 (м, 2H), 1,07 (с, 1H), 0,50-0,46 (м, 2H), 0,26-0,24 (м, 2H).
Получение (1R,4R)-N1-((1H-пиразол-3-ил)метил)-N4-(5-хлор-4-(5-(циклопропилметил)-1-метил-1H-пиразол-4-ил)пиримидин-2-ил)циклогексан-1,4-диамина (A-82)
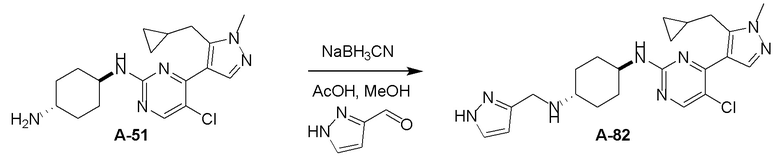
К раствору соединения A-51 (200,00 мг, 554,20 мкмоль, 1,00 экв.) и 1H-пиразол-3-карбальдегида (53,25 мг, 554,20 мкмоль, 1,00 экв.) добавляли AcOH (31,7 мкл, 554,20 мкмоль, 1,00 экв.) и NaBH3CN (69,65 мг, 1,11 ммоль, 2,00 экв.). Смесь перемешивали при 15°C в течение 16 часов. Смесь гасили водным раствором NaHCO3 (1 мл) и концентрировали. Остаток очищали при помощи препаративной ВЭЖХ (щелочные условия) с получением соединения A-82 (35,00 мг, 79,28 мкмоль, 14,3% выход, 99,9% чистота) в виде желтого твердого вещества.
ЖХМС: RT=2,867 мин, m/z 441,2 [M+H]+
1H ЯМР (CDCl3, 400 МГц) δ 8,20 (с, 1H), 8,13 (с, 1H), 7,53 (с, 1H), 6,22 (с, 1H), 4,91 (д, J=7,6Гц, 1H), 3,93 (с, 2H), 3,91 (с, 3H), 3,81 (с, 1H), 3,49 (с, 1H), 3,07 (д, J=5,6Гц, 2H), 2,56-2,53 (м, 1H), 2,16-2,02 (м, 4H), 1,33-1,32 (м, 4H), 1,05 (с, 1H), 0,48-0,45 (м, 2H), 0,18-0,16 (м, 2H).
Получение (1R,4R)-N1-(1-(1H-пиразол-4-ил)этил)-N4-(5-хлор-4-(5-(циклопропилметил)-1-метил-1H-пиразол-4-ил)пиримидин-2-ил)циклогексан-1,4-диамина (A83)
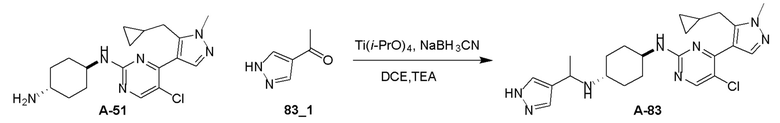
К раствору соединения A-51 (100,00 мг, 251,67 мкмоль, 1,00 экв., HCl), соединения 83_1 (27,71 мг, 251,67 мкмоль, 1,00 экв.) добавляли TEA (503,34 мкмоль, 69,77 мкл, 2,00 экв.) и Ti(i-PrO)4 (149 мкл, 503,34 мкмоль, 2,00 экв.). Смесь перемешивали при 80°C в течение 12 часов. Затем добавляли NaBH3CN (39,54 мг, 629,18 мкмоль, 2,50 экв.). Полученную смесь перемешивали при 15°C в течение 4 часов. Смесь гасили водным раствором NaHCO3 (50 мл) и экстрагировали при помощи DCM (50 мл × 2). Органический слой концентрировали и очищали при помощи препаративной ВЭЖХ (щелочные условия) с получением соединения A-83 (20,00 мг, 43,96 мкмоль, 17,5% выход) в виде желтого твердого вещества.
ЖХМС: RT=1,897 m/z 455,2[M+H]+
1H ЯМР (CDCl3, 400 МГц) δ 8,19 (с, 1H), 8,12 (с, 1H), 7,52 (с, 2H), 4,90 (д, J=7,6Гц, 1H), 4,05-4,00 (м, 1H), 3,91 (с, 3H), 3,78-3,76 (м, 1H), 3,06 (д, J=5,6Гц, 2H), 2,49-2,46 (м, 1H), 2,11-2,08 (м, 4H), 1,40-1,38 (м, 3H), 1,25-1,19(м, 4H), 1,05 (с, 1H), 0,47-0,44 (м, 2H), 0,16-0,15 (м, 2H).
Получение N-((1R,4R)-4-((5-хлор-4-(5-(циклопропилметил)-1-метил-1H-пиразол-4-ил)пиримидин-2-ил)амино)циклогексил)-1H-пиразол-4-карбоксамида (A87)
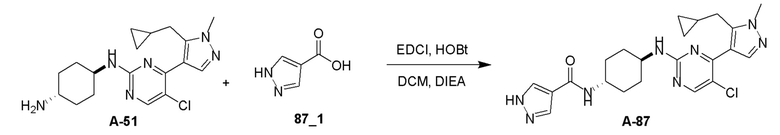
К раствору соединения 87_1 (46,59 мг, 415,65 мкмоль, 1,00 экв.) в DCM (4 мл) добавляли DIEA (181,5 мкл, 1,04 ммоль, 2,50 экв.), EDCI (95,62 мг, 498,78 мкмоль, 1,20 экв.) и HOBt (11,23 мг, 83,13 мкмоль, 0,20 экв.) при 0°C. Реакционную смесь перемешивали при 0°C в течение 0,5 часа. Добавляли раствор соединения A-51 (150,00 мг, 415,65 мкмоль, 1,00 экв.) в DCM (4,5 мл). Полученную смесь перемешивали при 20°C в течение 16 часов. Смесь разбавляли при помощи H2O (50 мл) и экстрагировали при помощи DCM (40 мл × 3). Органический слой концентрировали и очищали при помощи препаративной ВЭЖХ (HCl условия) с получением соединения A-87 (80,00 мг, 160,61 мкмоль, 38,6% выход, 98,6% чистота, HCl) в виде желтого твердого вещества.
ЖХМС: RT=2,391 m/z 455,2[M+H]+
1H ЯМР (MeOD, 400 МГц) δ 8,48 (с, 1H), 8,39 (с, 1H), 8,16 (с, 2H), 4,04 (с, 1H), 3,96 (с, 3H), 3,93-3,90 (м, 1H), 3,24 (д, J=6,4Гц, 2H), 2,18-2,04 (м, 4H), 1,67-1,53(м, 4H), 1,18 (с, 1H), 0,57-0,55 (м, 2H), 0,30-0,29 (м, 2H).
Получение (1R,4R)-N1-(5-хлор-4-(5-(циклопропилметил)-1-метил-1H-пиразол-4-ил)пиримидин-2-ил)-N4-((5-метил-1H-пиразол-4-ил)метил)циклогексан-1,4-диамина (A-91)
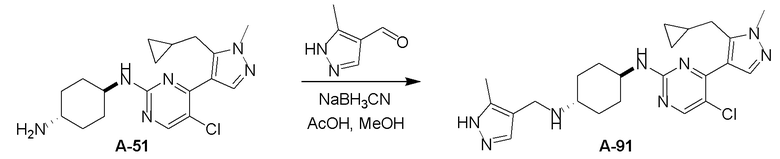
К раствору соединения A-51 (150,00 мг, 415,65 мкмоль, 1,00 экв.) и 5-метил-1H-пиразол-4-карбальдегида (54,92 мг, 498,78 мкмоль, 1,20 экв.) в MeOH (1,5 мл) добавляли AcOH (23,77 мкл, 415,65 мкмоль, 1,00 экв.) и NaBH3CN (78,36 мг, 1,25 ммоль, 3,00 экв.). Смесь перемешивали при 15°C в течение 16 часов. Смесь гасили водным раствором NaHCO3 (10 мл) и экстрагировали при помощи EA (20 мл × 2). Объединенные органические слои концентрировали и очищали при помощи препаративной ВЭЖХ (HCl условия) с получением соединения A-91 (20,00 мг, 36,22 мкмоль, 8,7% выход, 89% чистота, HCl) в виде белого твердого вещества.
ЖХМС: RT=2,425 m/z 455,2[M+H]+
1HЯМР (MeOD, 400 МГц) δ 8,44-8,41 (м, 2H), 8,23 (с, 1H), 4,26 (с, 2H), 4,13-4,06 (м, 1H), 3,95 (с, 3H), 3,24 (д, J=6,4Гц, 2H), 2,51 (с, 3H), 2,40-2,24 (м, 4H), 1,76-1,61 (м, 4H), 1,09 (с, 1H), 0,54-0,53 (м, 2H), 0,28-0,27 (м, 2H).
Получение (1R,4R)-N1-(5-хлор-4-(5-(циклопропилметил)-1-метил-1H-пиразол-4-ил)пиримидин-2-ил)-N4-(2,2,2-трифторэтил)циклогексан-1,4-диамина (A94)
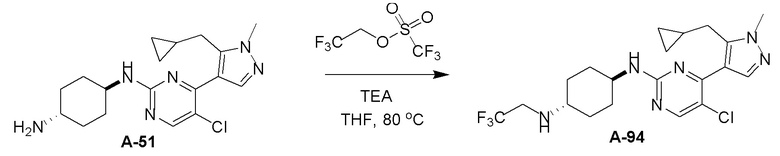
К раствору соединения A-51 (150,00 мг, 415,65 мкмоль, 1,00 экв.) в THF (4,5 мл) добавляли TEA (144,04 мкл, 1,04 ммоль, 2,50 экв.) и 2,2,2-трифторэтилтрифторметансульфонат (115,77 мг, 498,78 мкмоль, 1,20 экв.). Смесь перемешивали при 50°C в течение 16 часов. Смесь концентрировали и очищали при помощи препаративной ВЭЖХ (HCl условия) с получением соединения A-94 (80,00 мг, 163,38 мкмоль, 39,3% выход, 97,9% чистота, HCl) в виде желтого твердого вещества.
ЖХМС: RT=2,137 мин, m/z 443,2 [M+H]+
1H ЯМР (MeOD, 400 МГц) δ 8,45 (м, 2H), 4,15-4,09 (м, 2H), 4,09 (с, 1H), 3,95 (с, 3H), 3,39-3,36 (м, 1H), 3,22-3,21 (м, 2H), 2,33-2,25 (м, 4H), 1,71-1,56 (м, 4H), 1,09 (с, 1H), 0,54-0,52 (м, 2H), 0,28-0,27 (м, 2H).
Получение Получение (1R,4R)-N1-((1H-пиразол-4-ил)метил)-N4-(5-хлор-4-(5-(циклопропилметил)-1-метил-1H-пиразол-4-ил)пиримидин-2-ил)-N1-(2,2,2-трифторэтил)циклогексан-1,4-диамина (A-95)
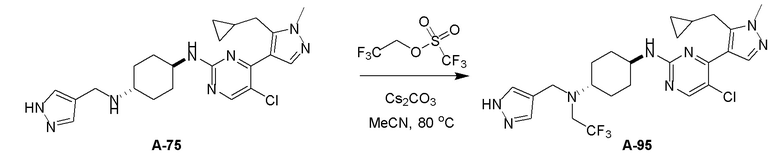
К раствору соединения A-75 (150,00 мг, 340,16 мкмоль, 1,00 экв.) в MeCN (4,5 мл) добавляли Cs2CO3 (221,66 мг, 680,32 мкмоль, 2,00 экв.) и 2,2,2-трифторэтилтрифторметансульфонат (118,43 мг, 510,24 мкмоль, 1,50 экв.). Смесь перемешивали при 70°C в течение 16 часов. Смесь фильтровали, фильтрат концентрировали и очищали при помощи препаративной ВЭЖХ (HCl условия) с получением соединения A-95 (20,00 мг, 35,7 мкмоль, 10,5% выход, 100% чистота, HCl) в виде белого твердого вещества.
ЖХМС: RT=2,285 m/z 523,2[M+H]+
1H ЯМР (MeOD, 400 МГц) δ 8,41-8,39 (м, 2H), 8,02 (с, 1H), 7,76 (с, 1H), 5,03-4,97 (м, 2H), 4,23 (с, 2H), 4,12-4,04 (м, 1H), 3,95 (с, 3H), 3,21 (д, J=6,4Гц, 3H), 2,34-2,23 (м, 4H), 1,65-1,53 (м, 4H), 1,09 (с, 1H), 0,54-0,52 (м, 2H), 0,27-0,26 (м, 2H).
Получение (1R,4R)-N1,N1-бис((1H-пиразол-4-ил)метил)-N4-(5-хлор-4-(5-(циклопропилметил)-1-метил-1H-пиразол-4-ил)пиримидин-2-ил)циклогексан-1,4-диамина (A-96)
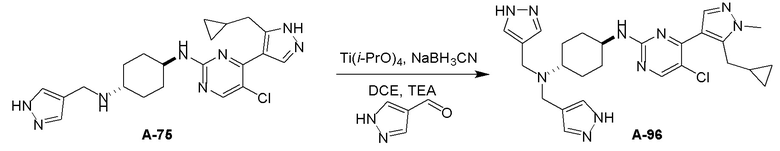
К раствору соединения A-75 (50,00 мг, 113,4 мкмоль, 1,00 экв.) и соединения 1H-пиразол-4-карбальдегида (32,69 мг, 340,17 мкмоль, 3,00 экв.) добавляли TEA (31,44 мкл, 226,78 мкмоль, 2,00 экв.) и Ti(Oi-Pr)4 (67,1 мкл, 226,78 мкмоль, 2,00 экв.). Смесь перемешивали при 80°C в течение 12 часов. Добавляли NaBH3CN (21,38 мг, 340,17 мкмоль, 3,00 экв.) и полученную смесь перемешивали при 15°C в течение 4 часов. Реакцию гасили водным раствором NaHCO3 (1 мл) и фильтровали. Фильтрат очищали при помощи препаративной ВЭЖХ (TFA и затем щелочные условия) с получением соединения A-96 (7,00 мг, 13,10 мкмоль, 11,6% выход, 97,5% чистота) в виде белого твердого вещества.
ЖХМС: RT=2,382 m/z 521,3[M+H]+
1H ЯМР (MeOD, 400 МГц) δ 8,23 (м, 2H), 8,03 (с, 1H), 7,78 (с, 4H), 4,47-4,29 (м, 4H), 3,91-3,87 (м, 4H), 3,12 (д, J=6,4Гц, 2H), 2,27-2,22 (м, 4H), 1,94-1,85 (м, 2H), 1,46-1,36 (м, 2H), 1,01-1,00 (м, 1H), 0,45-0,44 (м, 2H), 0,14-0,13 (м, 2H).
Получение (1R,4R)-N1-(4-(5-(циклопропилметил)-1-метил-1H-пиразол-4-ил)-5-фторпиримидин-2-ил)циклогексан-1,4-диамина (A-86)
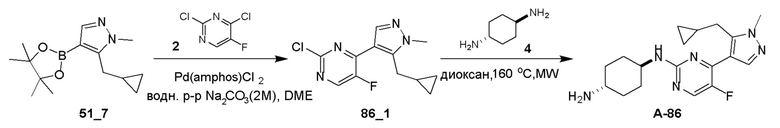
Стадия 1: К раствору соединения 51_7 (800 мг, 3,05 ммоль, 1,00 экв.) в DME (16 мл) добавляли соединение 2 (509,54 мг, 3,05 ммоль, 1,00 экв.), Na2CO3 (2 M, 4,6 мл, 3,00 экв.) и 4-дитрет-бутилфосфанил-N,N-диметил-анилин дихлорпалладий (108,04 мг, 152,50 мкмоль, 108 мкл, 0,05 экв.) в атмосфере азота. Полученную смесь перемешивали при 85°C в течение 2 часов в атмосфере азота. Смесь разбавляли при помощи H2O (50 мл) и экстрагировали при помощи EA (50 мл). Органический слой концентрировали с получением соединения 86_1 (1,20 г, неочищенный) в виде желтого масла.
ЖХМС: RT=1,819 мин, m/z 267,1 [M+H]+
Стадия 2: Смесь соединения 86_1 (1,10 г, 4,12 ммоль, 1,00 экв.) и соединения 4 (1,88 г, 16,48 ммоль, 4,00 экв.) в диоксане (16 мл) перемешивали при 130°C в течение 8 часов в условиях микроволнового нагрева. Смесь фильтровали и концентрировали. Остаток очищали при помощи препаративной ВЭЖХ (HCl условия) с получением соединения A-86 (1,30 г, неочищенный) в виде желтого твердого вещества. Соединение A-86 (300 мг) очищали снова при помощи препаративной ВЭЖХ (HCl условия) с получением соединения A-86 (20,00 мг, 52,51 мкмоль, 12,1% выход, 100% чистота, HCl) в виде желтого твердого вещества.
ЖХМС: RT=2,321 мин, m/z 345,2 [M+H]+
1H ЯМР (MeOD, 400 МГц) δ 8,37-8,36 (м, 1 H), 8,23-8,22 (м, 1 H), 4,07-4,03 (м, 1H), 3,96 (с, 3H), 3,21 (д, J=6,4Гц, 2H), 2,24-2,18 (м, 4H), 1,68-1,57 (м, 2H), 1,19 (с, 1H), 0,57-0,55 (м, 2H), 0,38-0,34 (м, 2H).
Получение (1R,4R)-N1-((1H-пиразол-4-ил)метил)-N4-(4-(5-(циклопропилметил)-1-метил-1H-пиразол-4-ил)-5-фторпиримидин-2-ил)циклогексан-1,4-диамина (A-85)
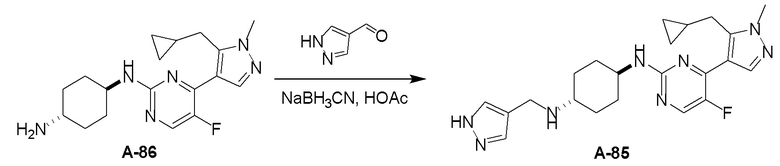
К раствору соединения A-86 (300,00 мг, 783,90 мкмоль, 1,00 экв.) и 1H-пиразол-4-карбальдегида (82,86 мг, 862,29 мкмоль, 1,10 экв.) в MeOH (3 мл) добавляли AcOH (49,31 мкл, 862,29 мкмоль, 1,10 экв.) и NaBH3CN (98,52 мг, 1,57 ммоль, 2,00 экв.). Смесь перемешивали при 150°C в течение 16 часов. Смесь гасили водным раствором NaHCO3 (1 мл) и концентрировали. Оставшийся продукт очищали при помощи препаративной ВЭЖХ (щелочные условия) с получением соединения A-85 (50,00 мг, 111,53 мкмоль, 14,2% выход, 94,7% чистота) в виде желтого твердого вещества.
ЖХМС: RT=2,472 мин, m/z 425,3 [M+H]+
1H ЯМР (CDCl3, 400 МГц) δ 8,10-8,09 (м, 1 H), 8,02-8,01 (м, 1 H), 7,60 (с, 1H), 4,80 (д, J=8,4Гц, 1H), 3,91 (с, 3H), 3,84 (м, 3H), 3,22-3,21 (м, 2H), 2,68 (с, 1H), 2,21-2,06 (м, 4H), 1,41-1,35 (м, 2H), 1,30-1,24 (м, 3H), 1,10-1,09 (м, 1H), 0,49-0,45 (м, 2H), 0,24-0,22 (м, 2H).
Биологические экспериментальные процедуры
Первичный скрининг в клетках RKO для определения соединений, обладающих типичной ингибирующей CKIα активностью (стабилизация β-катенина и фосфорилирование p53 и гистона H2AX; см. Elyada et al, Nature 2011 Feb 17; 470(7334):409-13; Pribluda et al, Cancer Cell 2013 Aug 12; 24(2):242-56). Колоректальные клетки RKO инкубировали с 10 мкM каждого из соединений в течение 16 часов при 37°C. Клетки промывали при помощи PBS и клеточные осадки инкубировали с ледяным белковым лизисным буфером, содержащим коктейль ингибиторов протеаз (1/200; Calbiochem) и ингибиторы фосфатаз (20мМ пара-нитрофенилфосфата (PNPP), 20мМ β-глицерофосфата и 300 нМ окадаиковой кислоты). Вестерн-блот-анализ осуществляли стандартными методами. Блоты инкубировали с антителами для детекции β-катенина (1/2,500; BD Transduction), p53 (DO-1&1801 смесь гибридом; разбавление супернатантов 1:20 от каждой), CKIα (C-19; 1/1,000; Santa Cruz Biotechnology) и фосфо-гистон H2AX (S139; 1/1000; Millipore). Вторичные антитела представляли собой HRP-связанные козлиные антимышиные, козлиные антикроличьи и кроличьи антикозлиные антитела (все 1/10000; Jackson). Блоты проявляли с использованием ECL (GE Healthcare).
Доза-ответа анализ наиболее активных соединений. Активные соединения дополнительно анализировали в эксперименте доза-ответ (Фиг. 1A и 1B). По аналогии с первичным скринингом, RKO клетки инкубировали с понижением концентраций каждого из активных соединений в течение 16 часов при 37°C. Выделение клеточного экстракта и Вестерн-блот-анализ осуществляли способом, аналогичным описанному для первичного скрининга.
Создание мышиной модели бластного криза CML и исследование ингибитора в этой модели. Для создания модели BCR-ABL-индуцируемого хронического миелоидного лейкоза (CML) клетки костного мозга (BM) от 10-недельной мыши дикого типа выделяли и добавляли к cKit экспрессирующим клеткам (EasySep #18757) и инкубировали в течение ночи при 37°C в питательной среде RPMI, дополненной 15% FCS, L-глутамином, пенициллином/стрептомицином (Biological Industries, Israel) и фактором стволовых клеток (SCF), IL-3, IL-6 и TPO (Peprotech). Культуру затем инфицировали p210BCR-ABL-IRES-GFP ретровирусным конструктом, содержащим супернатантную среду, в течение 4 часов, затем добавляли питательную среду и инфицированные клетки инкубировали при 37°C еще в течение 24 часов. Культуру затем инъецировали внутривенно сублетально облученным мышам (500 рад). При обнаружении быстрого стабильного увеличения числа GFP-экспрессирующих клеток в периферической крови инокулированных мышей (методом FACS) и увеличения числа лейкоцитов и незрелых клеток (определяли при помощи Wright-Giemsa окрашенных мазков крови) мышей умерщвляли и их BM клетки переносили к новым сублетально облученным WT хозяевам; каждый такой перенос завершался возникновением заболевания. К четвертому переносу хозяева больше не были сублетально облученными перед переносом заболевания. Развитие бластного криза легко можно было определить по аномально высокому количеству бластных клеток, более чем 30% лейкоцитов (WBC) в периферической крови (PB), и коротким временным интервалам между переносами. Исследования ингибитора CKI осуществляли на последних генерированных заболеваниях, в которых PB бласты были легко определяемыми, без облучения хозяина и коротким временем генерирования (до 12 дней). Мышей контролировали ежедневно для определения кахексии, потери массы тела, выраженной вялости и покрытия шерстью и агонизирующих мышей умерщвляли при признаках агонии.
Для оценки эффекта ингибирования CKIα на CML селективный ингибитор CKIα (A14) вводили через желудочный зонд один раз в день при дозе 10 мг/кг, начиная через 24 часа после трансплантации костного мозга (BMT) (Фиг. 3A). Ингибитор растворяли в 1% растворе метилцеллюлозы с 0,1% Tween 80 и 0,2% поли-этиленгликоля (Носитель). Контрольных мышей обрабатывали только носителем.
Ex vivo эффекты ингибитора (Фиг. 2A и 13A-E). Свежевыделенный BM у мышей с AML (13A-E) или с бластным кризом CML (2A) выращивали в RPMI, дополненной 15% FCS, L-глутамином, пенициллином/стрептомицином, Hepes, пируватом натрия и не относящимися к незаменимым аминокислотами (Biological Industries, Israel). Ингибиторы CKI (A14, A51, A75 или A86) растворяли в DMSO и добавляли к тканевой культуральной среде при указанной концентрации; контрольные культуры обрабатывали только DMSO. Через несколько часов после обработки (как указано в каждом эксперименте) клетки собирали и подсчитывали вручную с использованием камеры и стандартного инвертационного светового микроскопа. Мертвые клетки исключали с использованием трипанового синего (Sigma). Аннексин V-PE (MBL), 7AAD (Tonbo) и PD-L1 (BioLegend) окрашивание оценивали при помощи FACS в соответствии с рекомендациями изготовителя.
Создание мышиной модели острого миелоидного лейкоза (AML) (Фиг. 6) и обработка ингибитором CKIα, таким как A14 (Фиг. 7, 8 и 9) или A51 (Фиг. 11 и 12). Для создания модели MLL-AF9 острого миелоидного лейкоза (AML) клетки костного мозга (BM) от мыши дикого типа возраста 10 недель выделяли и добавляли к cKit-экспрессирующим клеткам (EasySep #18757) и инкубировали в течение ночи в RPMI, дополненной 15% FCS L-глутамина, пенициллином/стрептомицином (Biological Industries, Israel) и фактором стволовых клеток (SCF), IL-3, IL-6 и TPO (Peprotech). Культуру инфицировали MSCV-MLL-AF9-IRES-GFP ретровирусным конструктом, содержащим супернатантную среду, в течение 4 часов, затем добавляли питательную среду и инфицированные клетки инкубировали при 37°C еще в течение 24 часов. Культуру затем инъецировали внутривенно сублетально облученным мышам (500 рад). При обнаружении быстрого стабильного увеличения числа GFP-экспрессирующих клеток в периферической крови мышей (методом FACS) и увеличения числа лейкоцитов и незрелых клеток (определяли при помощи Wright-Giemsa окрашенных мазков крови) мышей умерщвляли и их BM клетки переносили (1й BMT) к сублетально облученным WT хозяевам. При возникновении AML заболевания мышей умерщвляли и 50000 BM клеток трансплантировали (2й BMT) WT мышам-хозяевам. GFP-экспрессирующие клетки отслеживали в периферической крови и при обнаружении >10% GFP+ в PB (день 11 после BMT) мышей обрабатывали A14 ингибитором (Фиг. 7A). A14 вводили через желудочный зонд один раз в день при дозе 20 мг/кг в течение 3 дней, затем при 10мг/кг/день еще в течение 6 дней. Ингибитор растворяли в 1% растворе метилцеллюлозы с 0,1% Tween 80 и 0,2% полиэтиленгликоля (носитель). Контрольных мышей обрабатывали только носителем. Мышей контролировали ежедневно для определения кахексии, потери массы тела, выраженной вялости и покрытия шерстью и агонизирующих мышей умерщвляли. Для однодозового эксперимента (Фиг. 11A) A51 вводили через желудочный зонд при одной дозе 20 мг/кг и мышей умерщвляли через 16 часов после обработки.
FACS анализ. Все анализы осуществляли на BD's оборудовании: измерительное устройство для FACS, FACS ARIA сортер или LSR II аппараты. Для иммуноокрашивания клетки суспендировали в 1% BSA/PBS буфере с 5мкМ EDTA. Клетки затем анализировали с использованием набора для детекции апоптоза Annexin V PE Apoptosis Detection Kit (eBioscience), 7-AAD (TONBO biosciences) и PE антимышиного CD274 (B7-H1, PD-L1) антитела (клон 10F.9G2, BioLegend); Анализы осуществляли в соответствии с инструкциями изготовителя. Моноклональные антитела, специфические в отношении CD16 и CD32 (Miltenyi Biotec), использовали для блокады Fc рецепторов перед окрашиванием.
Общий анализ крови. Периферическую венозную кровь получали из лицевой вены мыши с использованием стандартных процедур и анализировали с использованием автоматического гематологического анализатора BC-2800 (Mindray) в соответствии с инструкциями изготовителя.
Таблица 1 представляет квантифицированные данные для соединений по настоящему изобретению в активации p53 и ответе на повреждение ДНК (DDR) и стабилизации β-катенина как индикатора активации Wnt пути. Активацию p53 определяли в соответствии со степенью стабилизации белка в некоторых вестерн-блот анализах (примеры вестерн-блоттинга показаны на Фиг. 1A и 1B). Например, A43-стабилизированный p53 существенно выше необработанного контроля при 6 мкМ, с отсутствием активности при 2 мкМ (Фиг. 1A, нижняя правая панель), и, таким образом, получил среднюю оценку+для активации p53. В отличие от этого, A35 начал стабилизировать p53 при 0,2 мкМ, с максимальной стабилизацией при 1 мкМ (Фиг. 1A, верхняя правая панель), и, таким образом, получил среднюю оценку +++ для активации p53. A19-4 не показал ни стабилизации p53 ни индукции γH2AX (индикатор ответа на повреждение ДНК [DDR]), но стабилизировал β-катенин при 2 мкМ подобно лучшим β-катенин-стабилизирующим соединениям (Фиг. 1A, нижняя левая панель) и, таким образом, получил оценку +++ для активации β-катенина/Wnt.
Стабилизация p53, ответ на повреждение ДНК и активация Wnt/β-катенина, вызываемые соединениями по настоящему изобретению
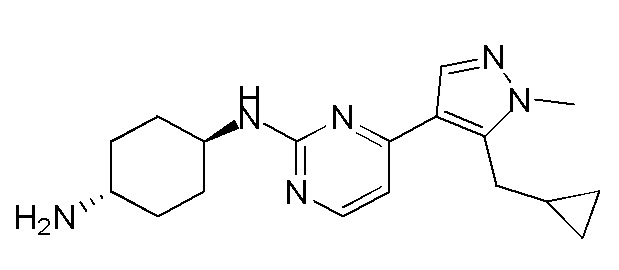
Найдено: 327,2
Найдено:399,2
Найдено:341,2
Найдено:355,2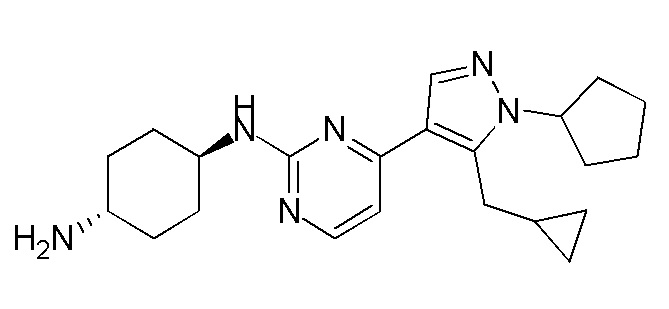
Найдено:381,4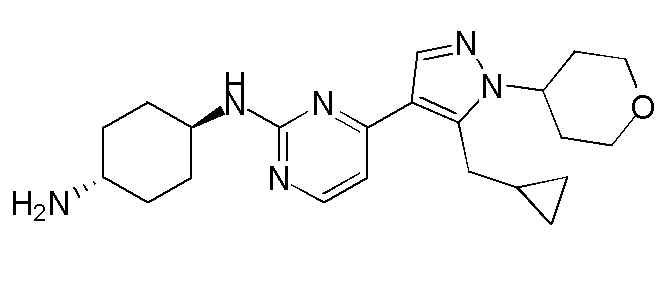
Найдено:397,4
Найдено:385,2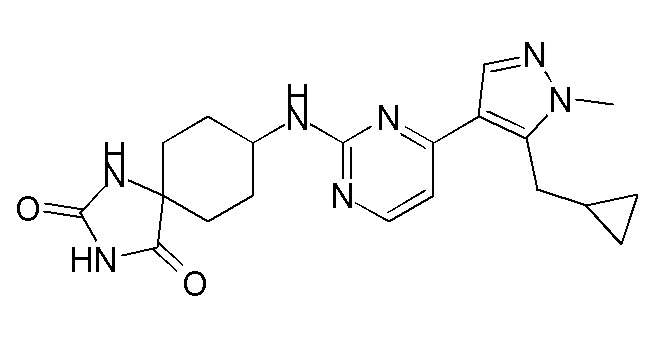
Найдено:396,2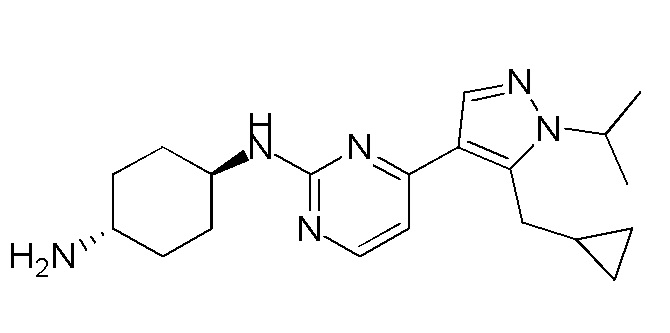
Найдено:355,3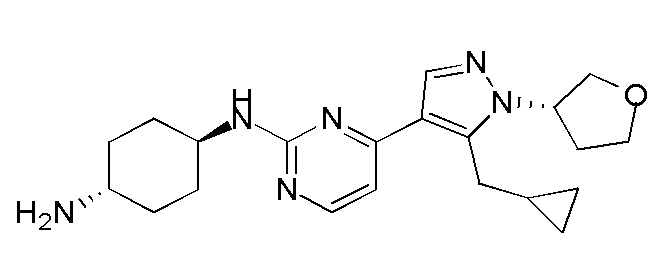
Найдено:383,3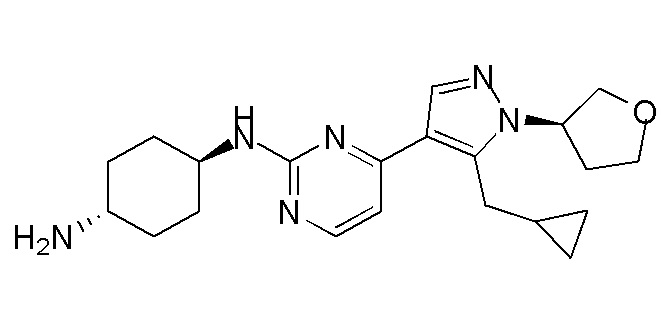
Найдено:383,2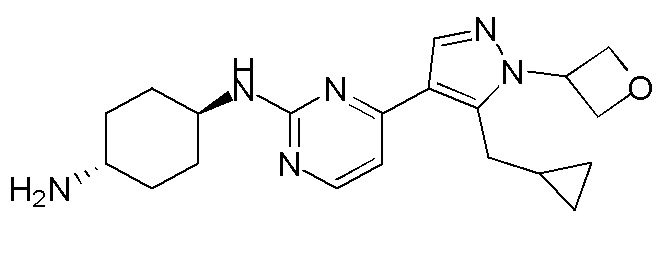
Найдено:369,3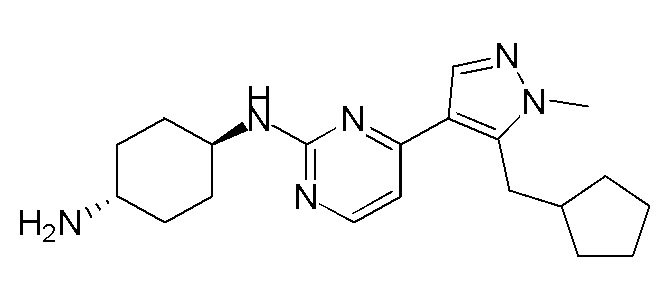
Найдено:355,2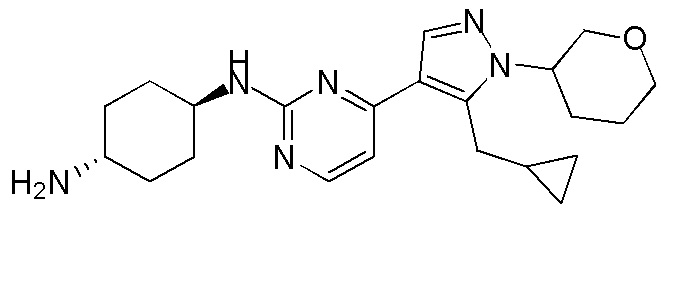
Найдено:397,3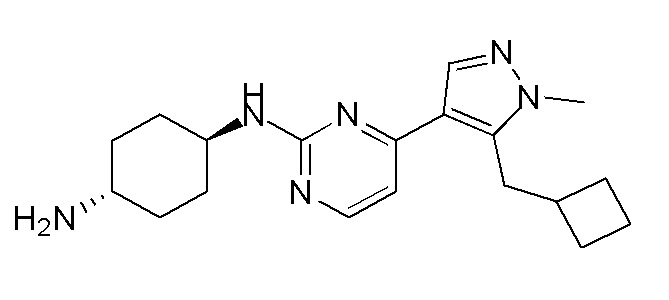
Найдено:341,3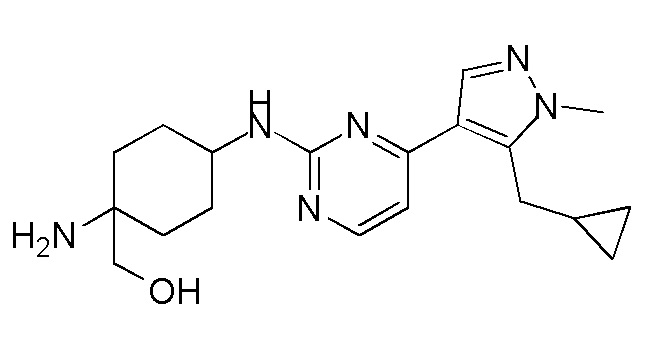
357,2;
Найдено:357,2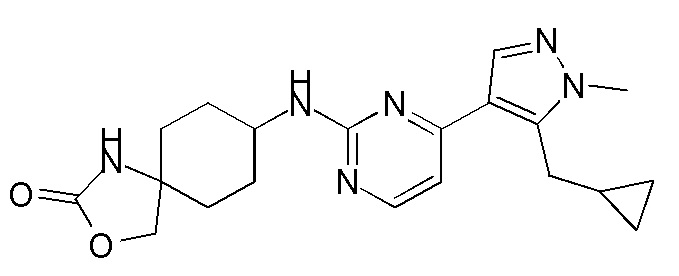
383,2;
Найдено:383,2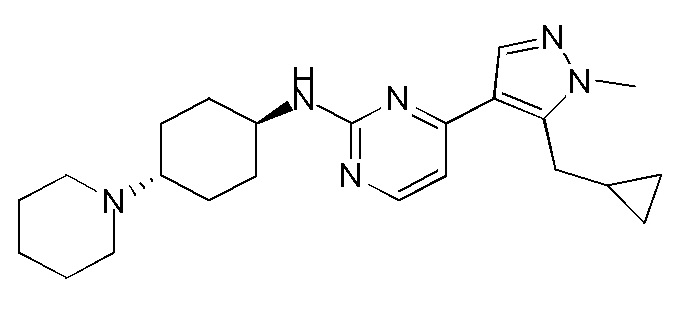
395,3;
Найдено:395,2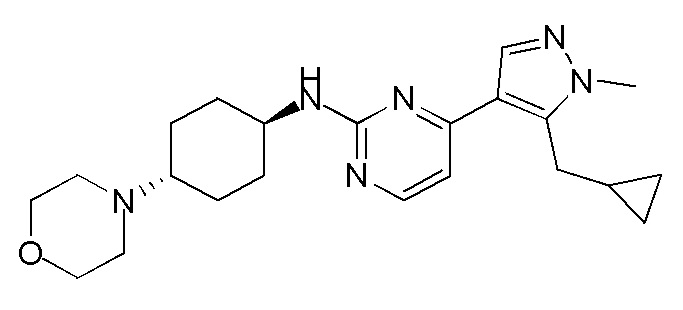
397,3;
Найдено:397,3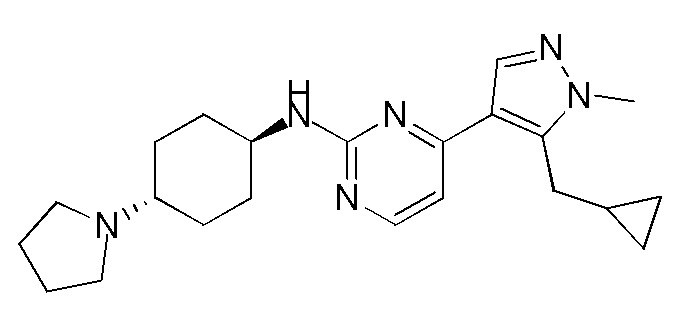
381,3;
Найдено:381,3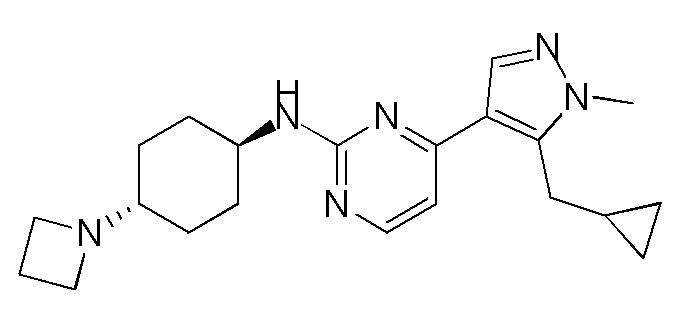
367,3;
Найдено:367,3
361,2;
Найдено: 361,1
Найдено:341,2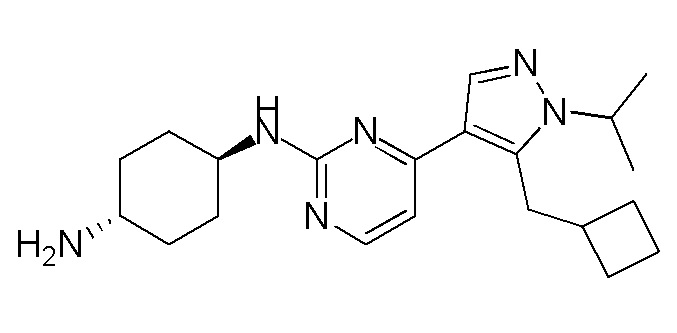
369,3;
Найдено:369,2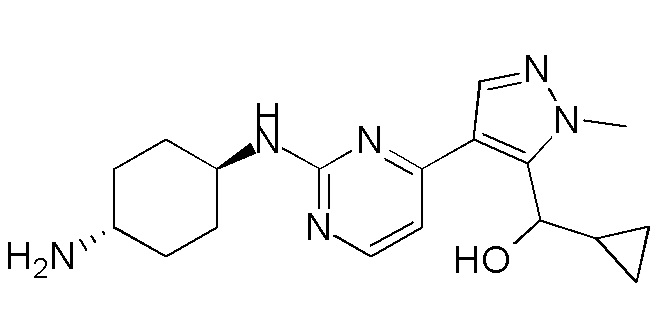
343,2;
Найдено:343,2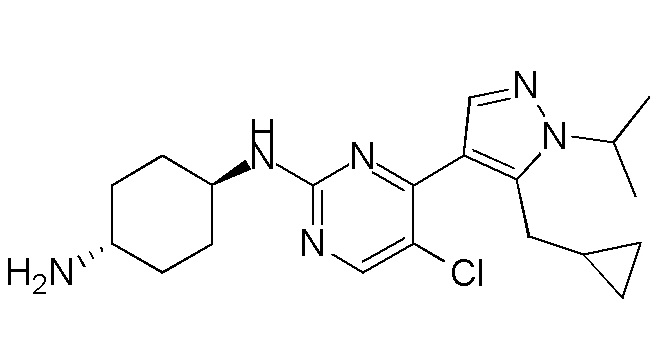
389,2;
Найдено: 389,2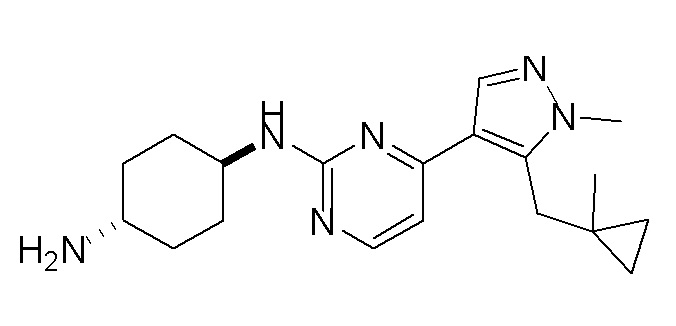
4-ил)пиримидин-2-ил)циклогексан-1,4-диамин
341,2;
Найдено:341,2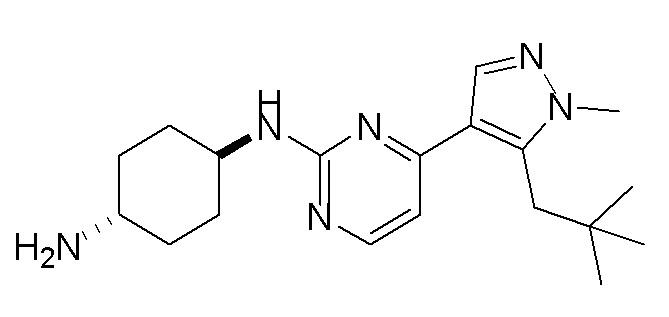
343,3;
Найдено:343,2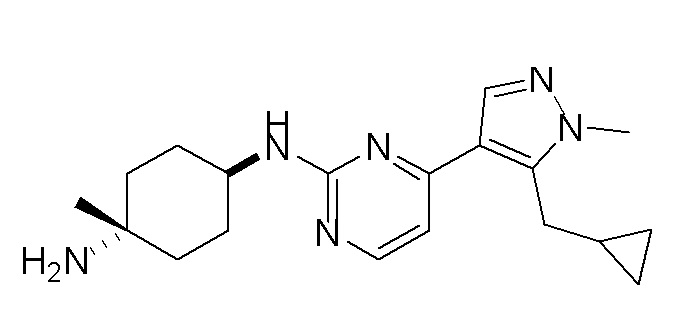
375,2;
Найдено: 375,2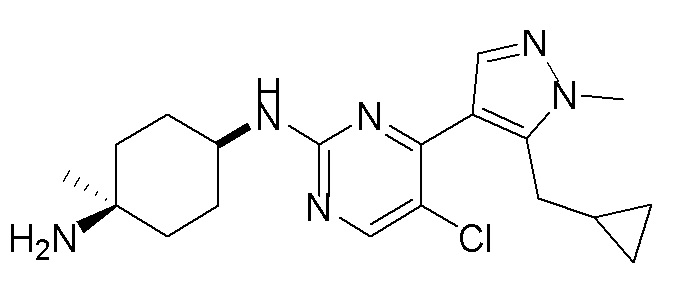
375,2;
Найдено: 375,2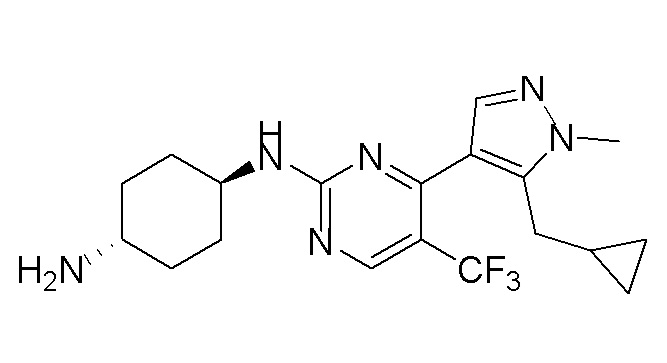
395,2;
Найдено:395,2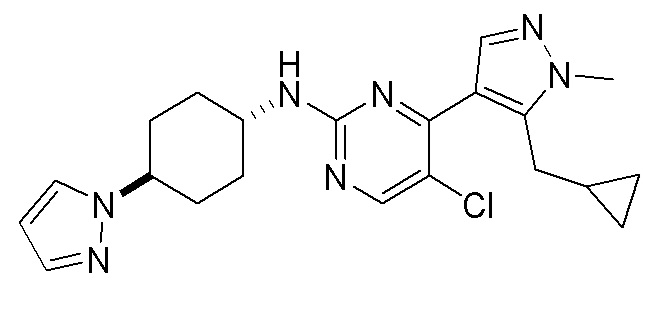
412,2;
Найдено: 412,2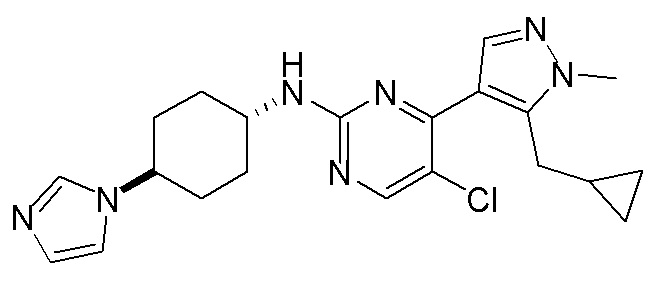
412,2;
Найдено: 412,2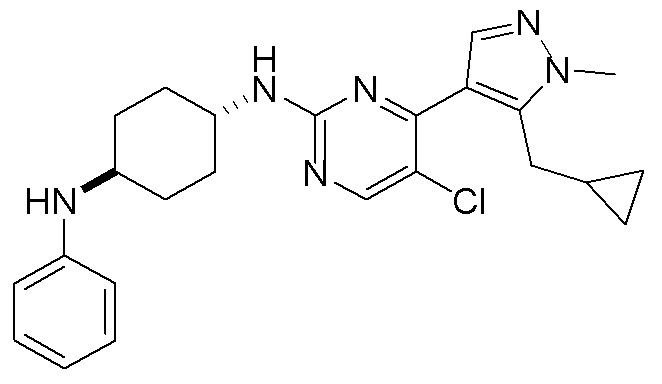
437,2;
Найдено: 437,2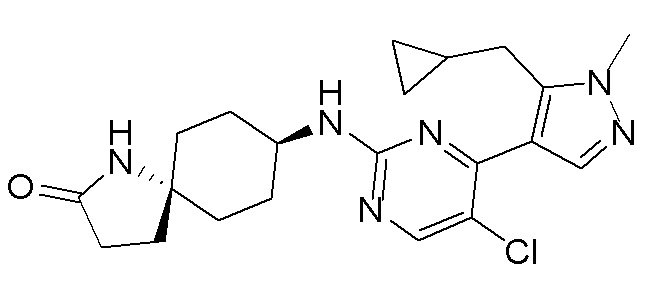
415,2;
Найдено: 415,2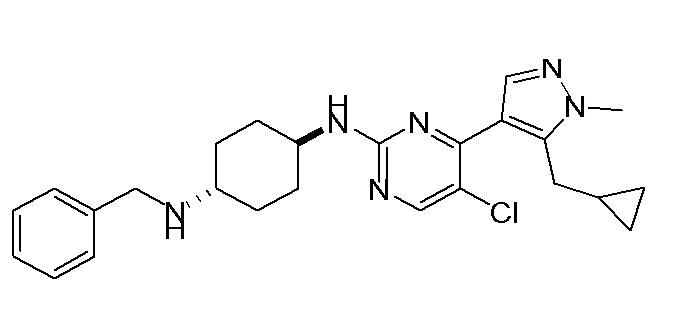
451,2;
Найдено: 451,2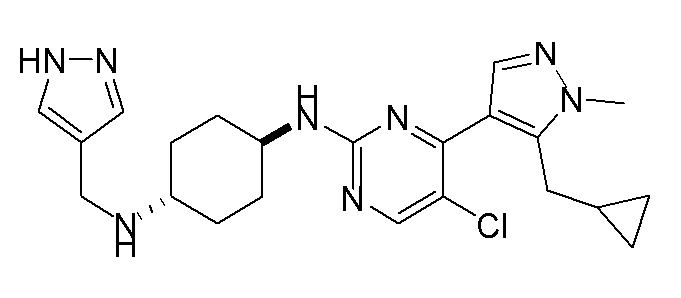
441,2;
Найдено: 441,2
452,2;
Найдено: 452,2
407,3;
Найдено: 407,2
421,3;
Найдено: 421,2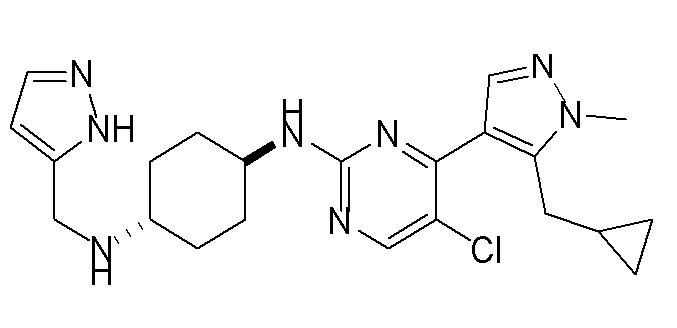
441,2;
Найдено: 441,2
455,2;
Найдено: 455,2
455,2;
Найдено: 455,2
455,2;
Найдено: 455,2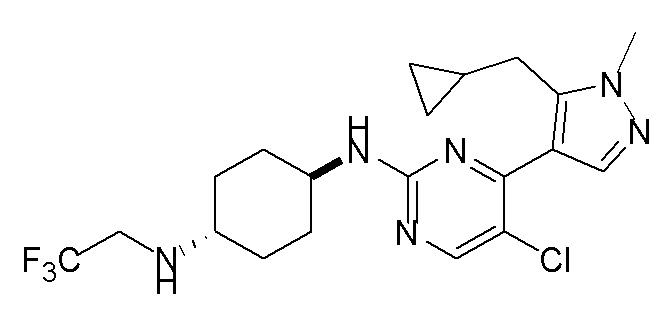
443,2;
Найдено:443,2
523,2;
Найдено:523,2
521,3;
Найдено:521,3
345,2;
Найдено: 345,2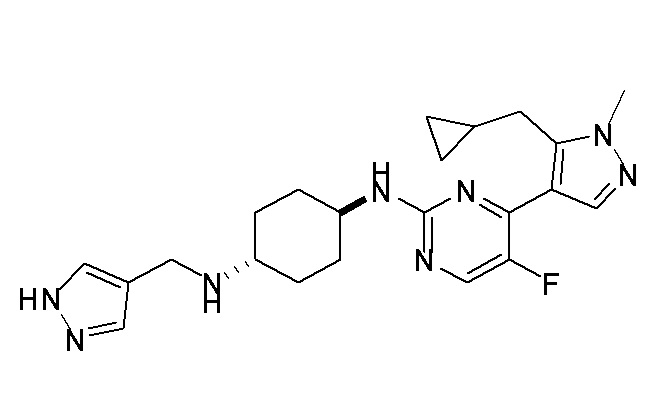
425,3;
Найдено: 425,3
+ означает низкую, но значимую активацию при концентрации соединения 6 мкМ; +++ означает максимальную стабилизацию β-катенина или p53 при >2 мкМ; ++++ означает максимальную активацию p53 и DDR при >0,5мкМ, +++++ означает максимальную активацию p53 и DDR при >0,1мкМ.
Ингибирование IRAK1 соединениями по настоящему изобретению. RKO клетки инкубировали в течение 16 часов при 37°C с указанными концентрациями соединений по настоящему изобретению A51 (1мкМ) и A14 (2мкМ) (Фиг. 10). В указанных точках времени RKO обрабатывали при помощи TNFα (100 единиц/мл). Клетки собирали и анализировали методом вестерн-блоттинга. Блоты инкубировали со следующими антителами: Фосфо-IRAK1 (Thr209), (A1074, AssayBiothechnology; 1/1000), Фосфо-IKKα/β (Ser176/180) (16A6, Cell Signaling; 1/1000), IKKα (2682, Cell Signaling; 1/11000), IKK β (2370, Cell Signaling; 1/11000), Фосфо-c-Jun (Ser 63) (9261, Cell Signaling; 1/11000), p53 (DO-1&1801 смесь гибридом; разбавление супернатантов 1:20 от каждой), CKIα (C-19; 1/1000; Santa Cruz Biotechnology) и фосфо-гистон H2AX (S139; 1/1000; Millipore). Вторичные антитела представляли собой HRP-связанные козлиные антимышиные, козлиные антикроличьи и кроличьи антикозлиные антитела (all 1/10000; Jackson). Блоты проявляли с использованием ECL (GE Healthcare). Фиг. 10 показывает ингибирование фосфорилирования IRAK1, а также Фосфо-IKKα/β и Фосфо-c-Jun, указывающее на ингибирование IRAK1 киназы. Также показана стабилизация p53 и фосфорилирование H2AX (γH2AX), маркера повреждения ДНК, что свидетельствует об ингибировании CKIα киназы. Уровни CKIα белка служат в качестве нагрузочного контроля.
Скан, показывающий аффинность к киному для A51 (WXL5846, см. Таблицу 2 ниже), показывает, что ключевыми мишенями A51 являются все члены CKI семейства и IRAK1, с некоторыми контрольными киназами. KINOMEscan™ основан на анализе конкурентного связывания, который количественно определяет способность соединения конкурировать с иммобилизованным активным сайт-направленным лигандом. Анализ осуществляли путем объединения трех компонентов: ДНК-меченной киназы; иммобилизованного лиганда; и испытываемого соединения. Способность испытываемого соединения конкурировать с иммобилизованным лигандом измеряли при помощи количественной ПЦР ДНК метки; % Контр.=0 (ноль) указывает на полное ингибирование испытываемой киназы ингибитором при концентрации 1мкМ (Fabian, M.A. et al. A small molecule-kinase interaction map for clinical kinase inhibitors. Nat. Biotechnol. 23, 329-336 (2005) and Karaman, M.W. et al. A quantitative analysis of kinase inhibitor selectivity. Nat. Biotechnol. 26, 127-132 (2008)).
Скан, показывающий аффинность к киному для A51 A51 (WXL5846)
A51
Таблица 3 показывает Kd измерения для определения взаимодействия A14 (WXL-4085) и A51 (WXL-5846) с IRAK1
Kd A14 (WXL-4085) и A51 (WXL-5846) с IRAK1
A14
A51
Заключение. IRAK1 как главная мишень в скане кинома показывает нулевое связывание IRAK1 с ее мишенью в присутствии ингибиторов. Пиразолпиримидиновые соединения по настоящему изобретению A51 и A14 показали отличную Kd связывания с IRAK1. Соединения также показали ингибирование активации IRAK1 и ингибирование активации IRAK1 мишени IKK (I каппа B киназа) в клетках RKO (вестерн-блот анализ). Следует отметить, что соединение A51 показало полное (100%) ингибирование фосфо- (активной) IRAK1 при концентрации 1 мкМ в RKO клеточной линии. В качестве сравнения, Garrett W. Rhyasen et al. показали, что ингибитор IRAK1-4 от Amgen, используемый в лечении MDS и рака молочной железы, ингибировал только 70% IRAK1/4 в клеточных линиях при 10 мкМ (Garrett W. Rhyasen et al, 2013, Cancer Cell 24, 90-104, см., в частности, Фиг. 2 в этом документе). Таким образом, было обнаружено, что соединения по настоящему изобретению, такие как A51, являются отличными ингибиторами IRAK1, важного апстрим-регулятора NF-kB пути, который играет важную роль в гематологических злокачественных опухолях (включая, среди прочих, множественную миелому, MDS, лейкоз и лимфому, рак головы и шеи и рак молочной железы).
Эффект однодозовой обработки ингибитором CKI мышей с AML и экспрессия PD-L1. AML мышей подготавливали путем инокуляции MLL-AF9 онкоген-трансдуцированных клеток костного мозга мышам C57/BL6. MLL-AF9 слияние представляет собой один из имеющих плохой прогноз AML человека, индуцированный хромосомной транслокацией. Через 30 дней после инокуляции лейкоза мыши-реципиенты имели высокое число лейкоцитов (WBC) (×10 больше чем у нормальной мыши) и имели >95% лейкозных бластов в костном мозге, и 50% периферических WBC у этих мышей представляли собой AML бласты. Эти мышей имели спленомегалию, и их кости были бледными и хрупкими из-за острого лейкоза (Фиг. 12). Пероральное введение A51 (20мг/кг) в течение 16 часов приводило к существенному уменьшению общего количества лейкозных клеток в крови (Фиг. 11B), сокращению лейкозной селезенки (Фиг. 11C и Фиг. 12A), 50% и >90% уменьшению пропорции лейкозных бластных клеток (GFP+ клетки) в костном мозге и крови, соответственно (Фиг. 11D и Фиг. 11E). Бледные кости возвращали свой нормальный цвет после однодозовой обработки (Фиг. 12B).
In vitro эффекты CKI ингибиторов на AML клетки, выделенные из костного мозга лейкозных мышей. Показаны процент мертвых клеток (7AAD+) после 10 или 100 нМ обработки ингибитором через 6 и 9 часов после обработки (Фиг. 13B и 13D). DMSO обработка приводила к <10% мертвых клеток через 9 часов. Также, показаны эффекты ингибиторов на экспрессию PD-L1 белка главной иммунной контрольной точки в лейкозных клетках с использованием метода проточной цитометрии: уменьшение средней интенсивности флуоресценции (MFI) через 5 часов и уменьшение фракции PD-L1-положительных лейкозных клеток после обработки ингибитором по сравнению с DMSO-обработанными клетками через 6 и 9 часов (уменьшение выражали в % от DMSO контроля) (Фиг. 13A, 13C и 13E).
| название | год | авторы | номер документа |
|---|---|---|---|
| N1-(4-(5-(ЦИКЛОПРОПИЛМЕТИЛ)-1-МЕТИЛ-1H-ПИРАЗОЛ-4-ИЛ)ПИРИДИН-2-ИЛ)ЦИКЛОГЕКСАН-1,4-ДИАМИНОВЫЕ ПРОИЗВОДНЫЕ И РОДСТВЕННЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ СK1 И/ИЛИ IRAK1 ДЛЯ ЛЕЧЕНИЯ РАКА | 2018 |
|
RU2761457C2 |
| СОЕДИНЕНИЯ И КОМПОЗИЦИИ, ПРИМЕНЯЕМЫЕ ДЛЯ ЛЕЧЕНИЯ РАССТРОЙСТВ, СВЯЗАННЫХ С NTRK | 2016 |
|
RU2744974C2 |
| ИНГИБИТОРЫ ВЗАИМОДЕЙСТВИЯ МЕНИН-MLL | 2017 |
|
RU2799820C2 |
| СОЕДИНЕНИЯ И СПОСОБЫ ЛЕЧЕНИЯ ПАРАЗИТАРНЫХ ЗАБОЛЕВАНИЙ | 2018 |
|
RU2793122C2 |
| ИНГИБИТОРНЫЕ СОЕДИНЕНИЯ | 2013 |
|
RU2673079C2 |
| ГЕТЕРОАРИЛЬНЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ IRAK II ТИПА И ИХ ПРИМЕНЕНИЯ | 2019 |
|
RU2810338C2 |
| АРИЛ-ИЛИ ГЕТЕРОАРИЛЗАМЕЩЕННЫЕ БЕНЗОЛЬНЫЕ СОЕДИНЕНИЯ | 2012 |
|
RU2632193C2 |
| ПРОИЗВОДНЫЕ ХИНОЛИНА И СОДЕРЖАЩИЕ ИХ ИНГИБИТОРЫ MELK | 2011 |
|
RU2582610C2 |
| БИЦИКЛИЧЕСКИЕ ЛАКТАМЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2716136C2 |
| ПИРИДИНЫ, ПИРИМИДИНЫ И ПИРАЗИНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ТИРОЗИНКИНАЗЫ БРУТОНА И ИХ ПРИМЕНЕНИЕ | 2014 |
|
RU2712220C2 |

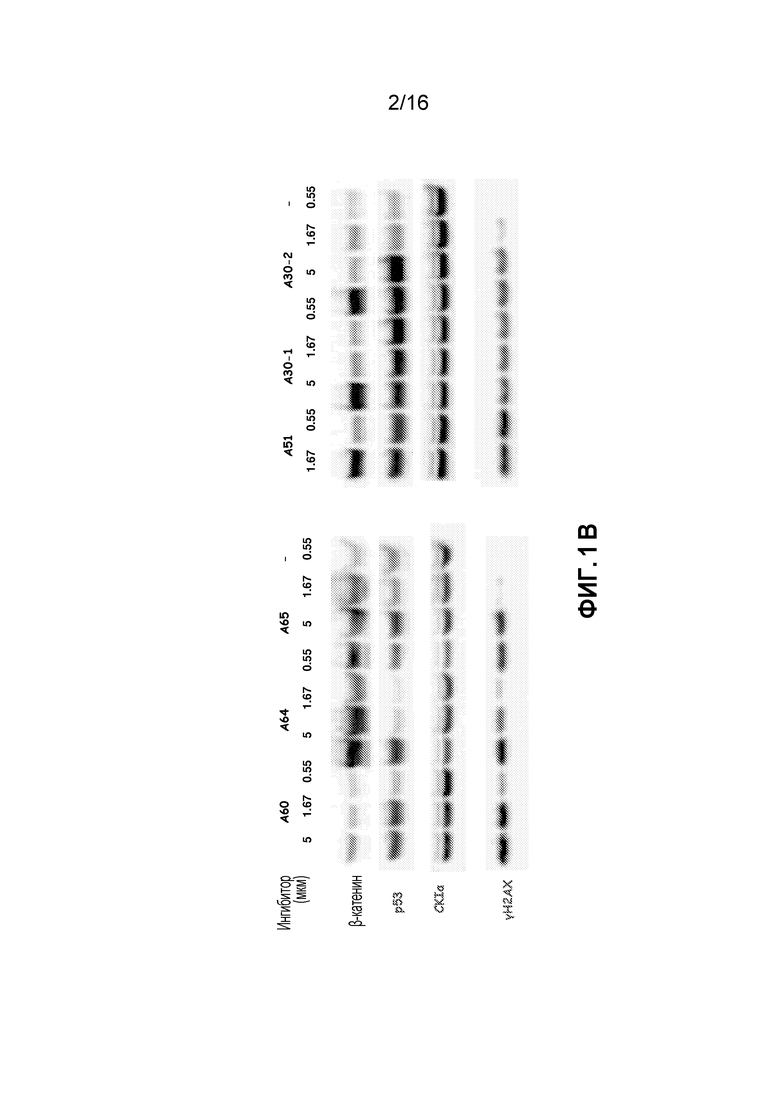
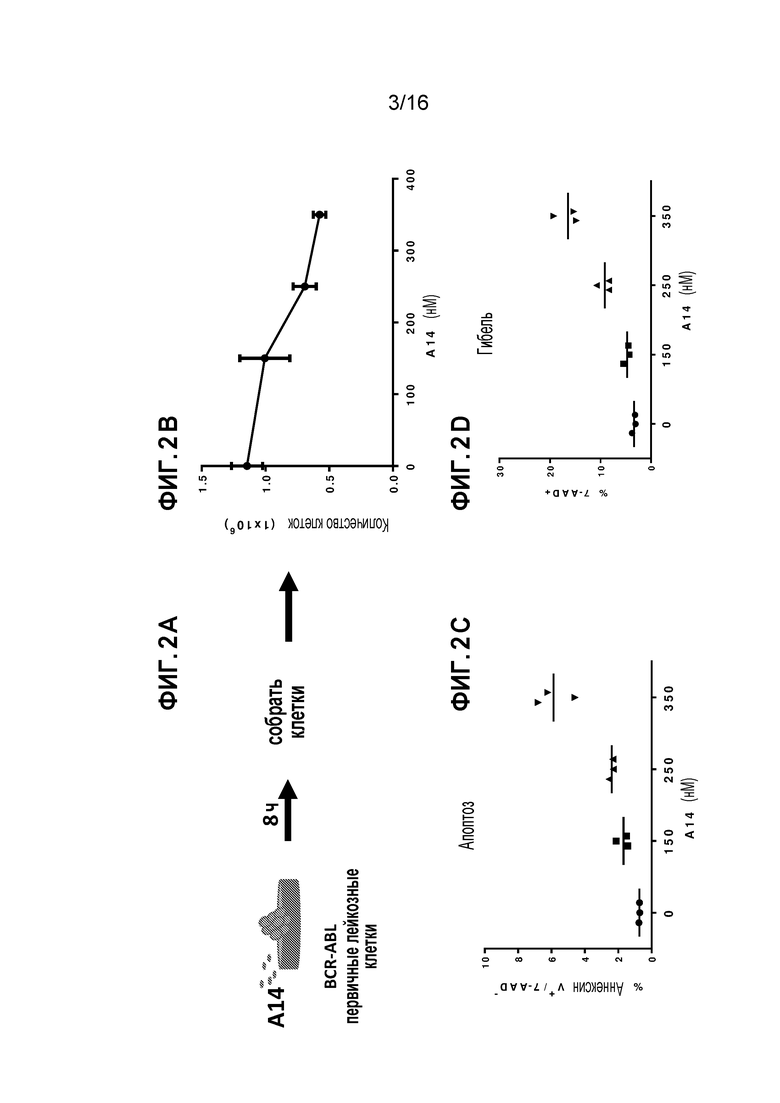
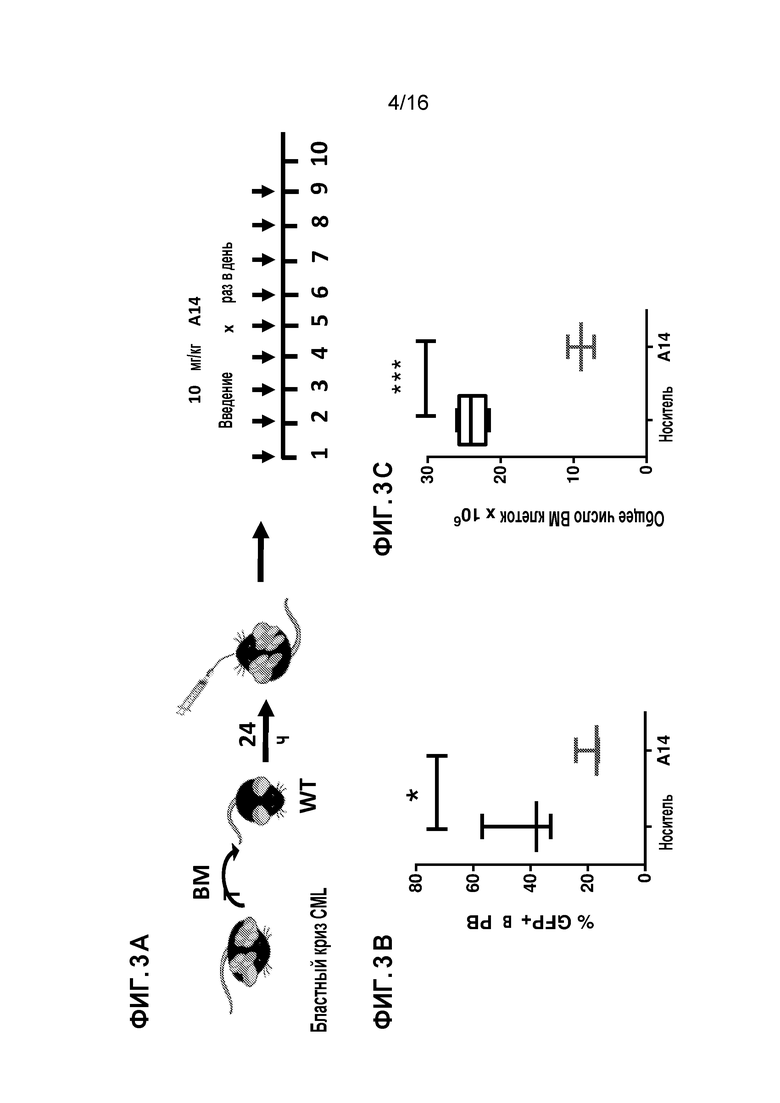
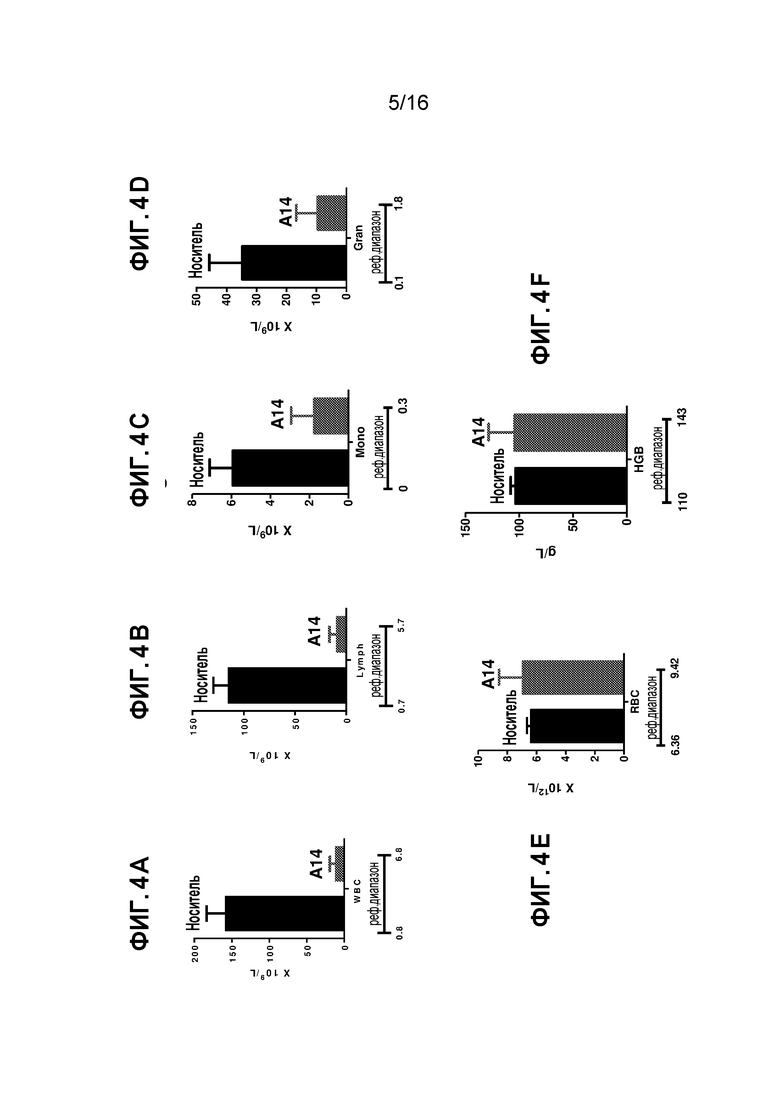
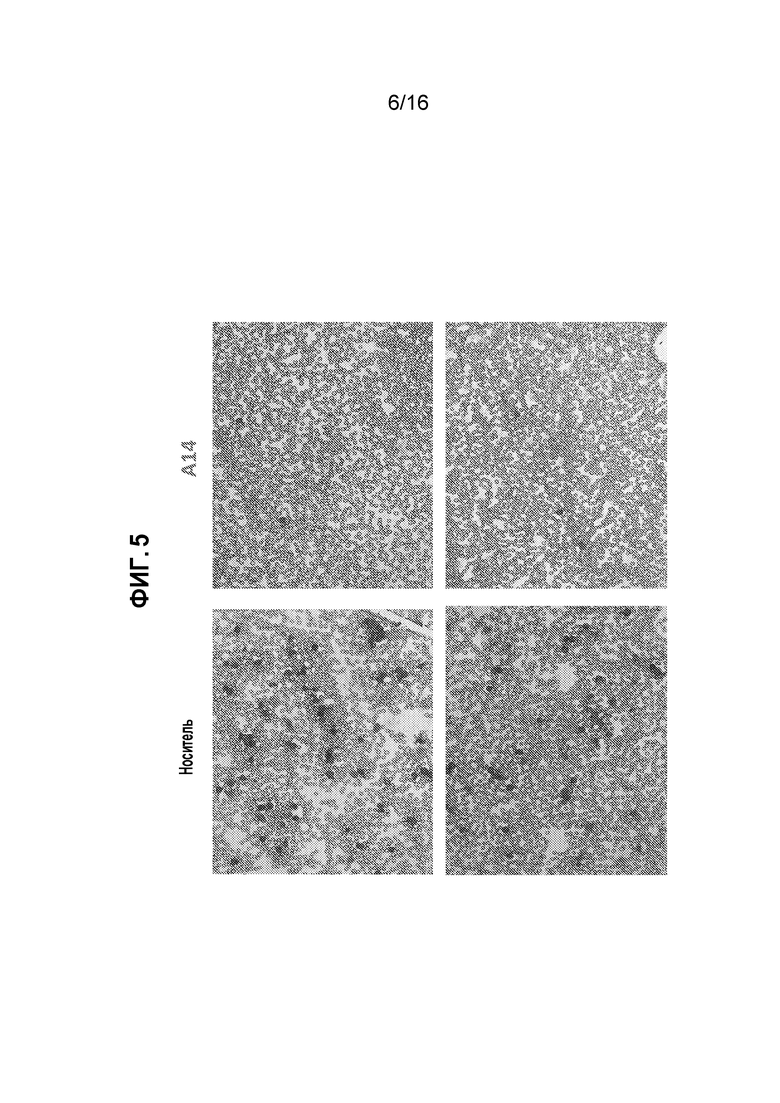
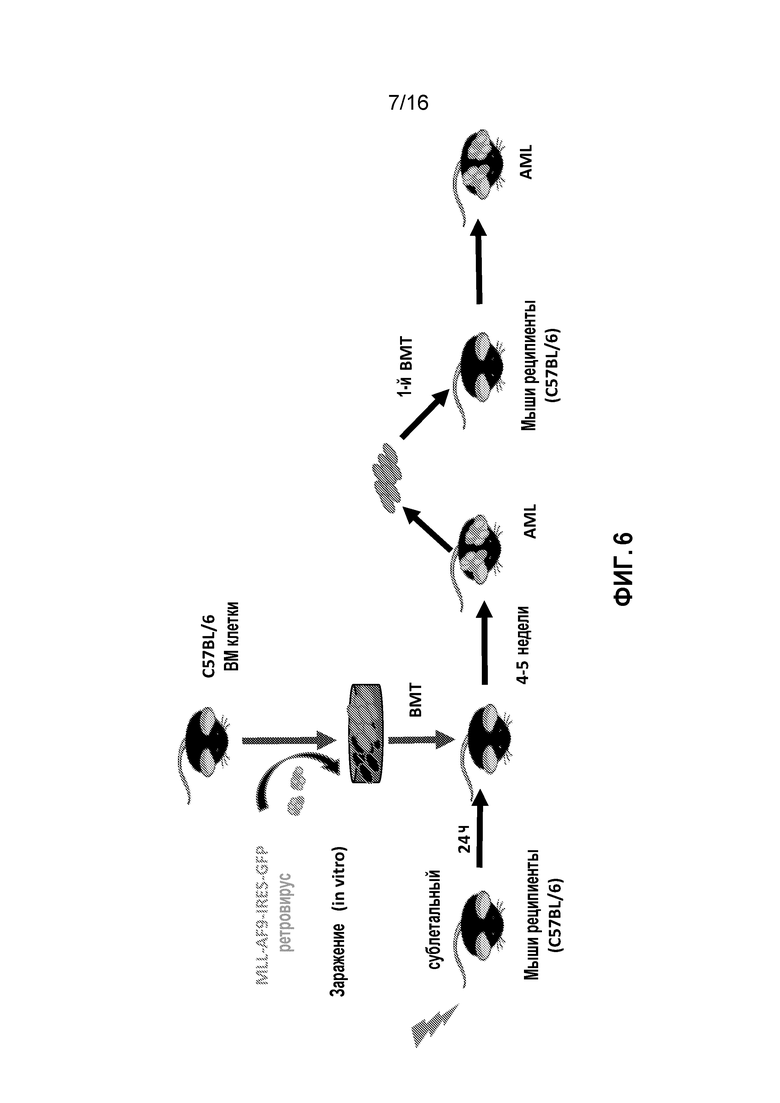
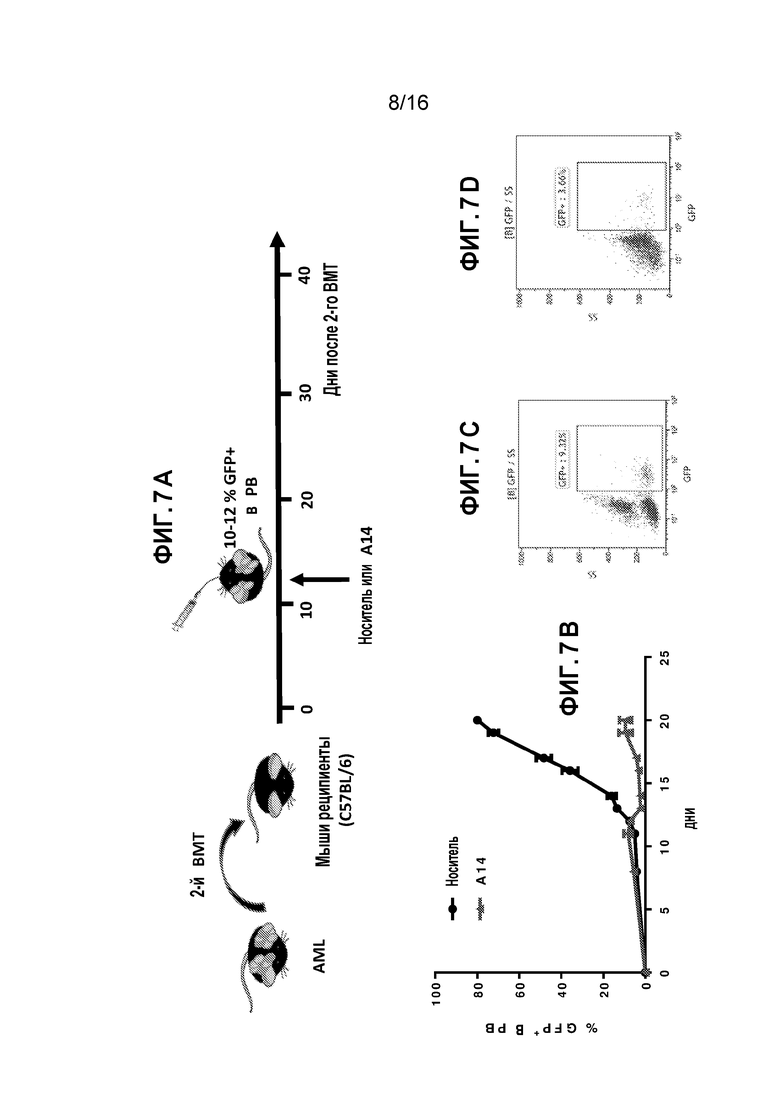
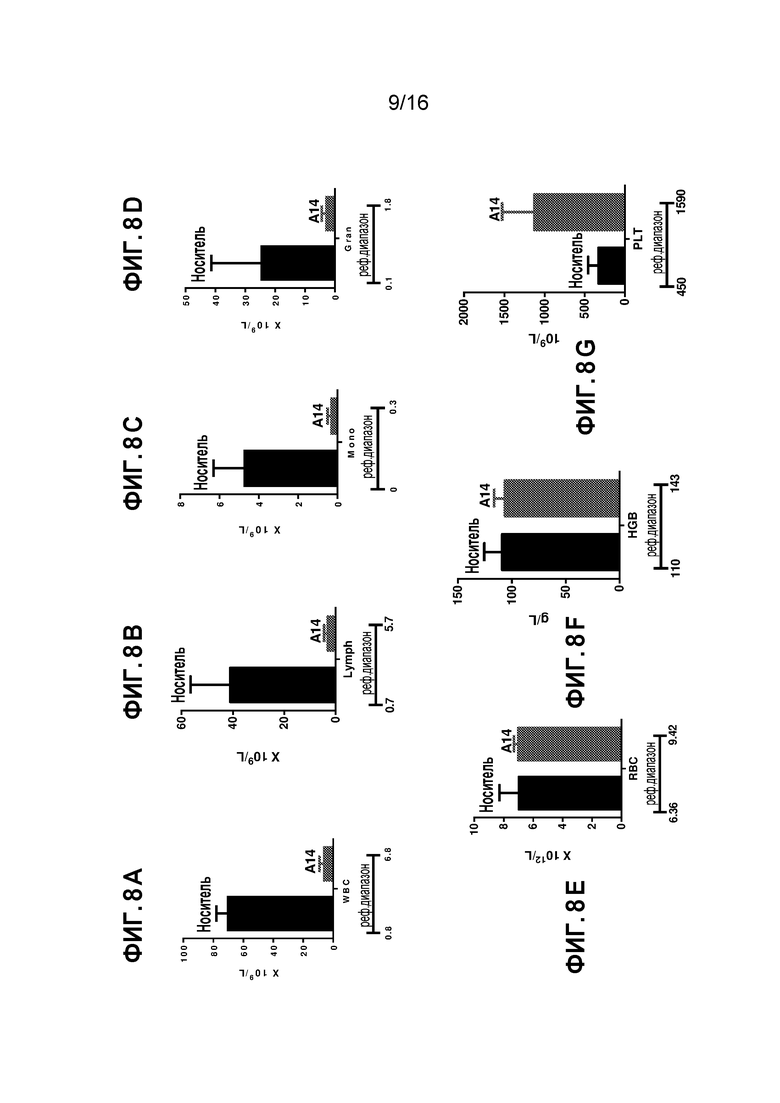


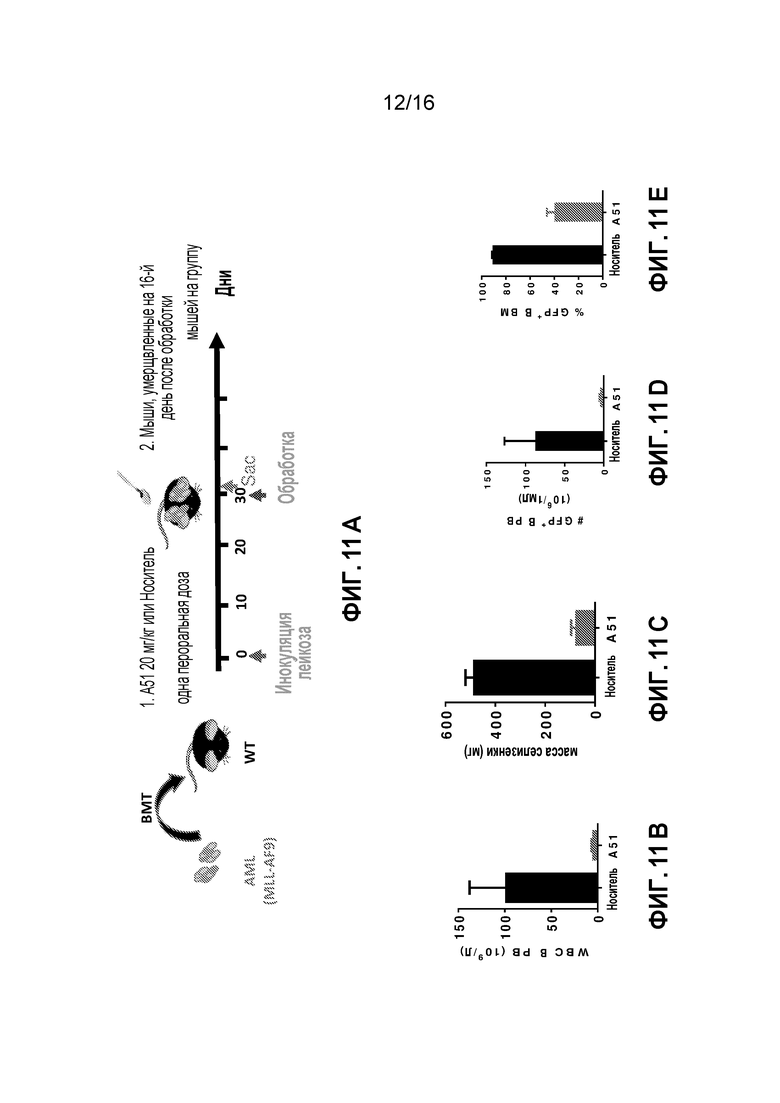
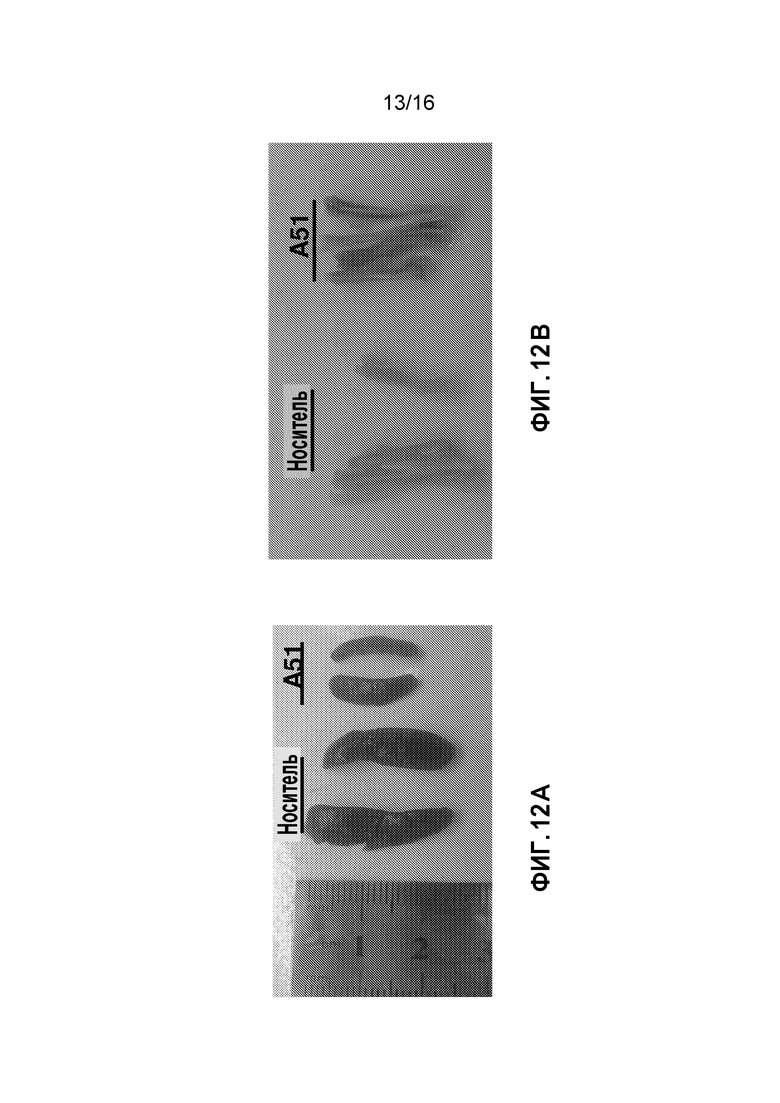
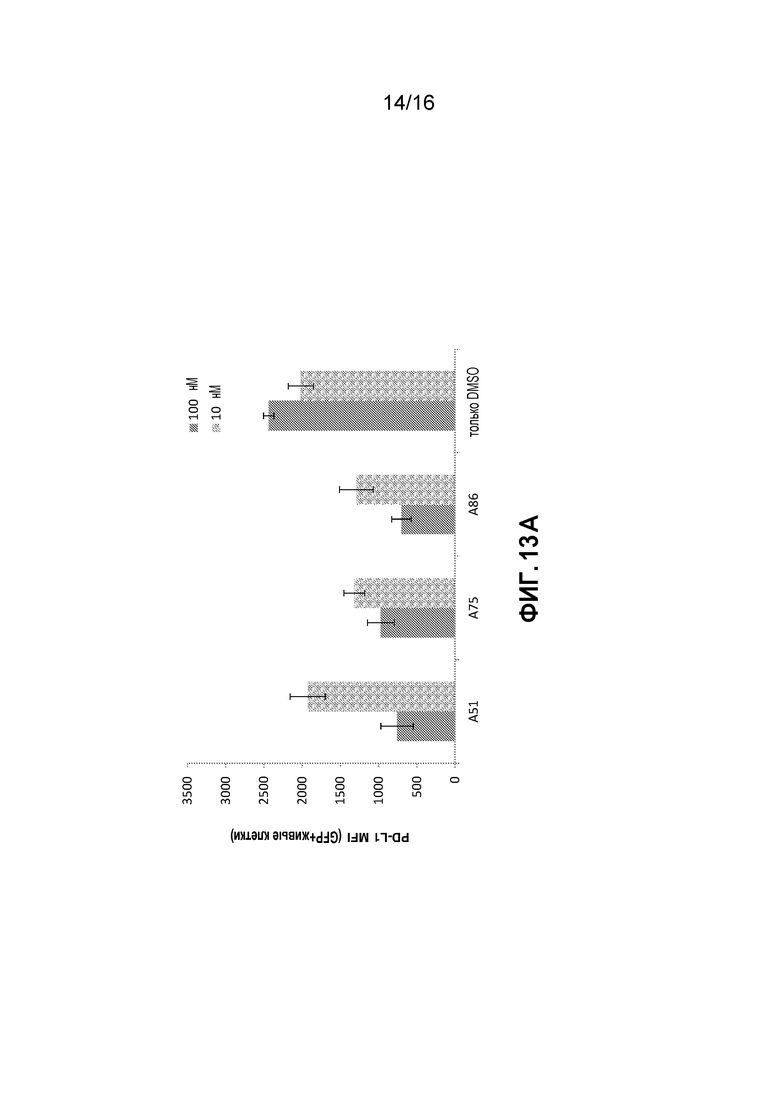
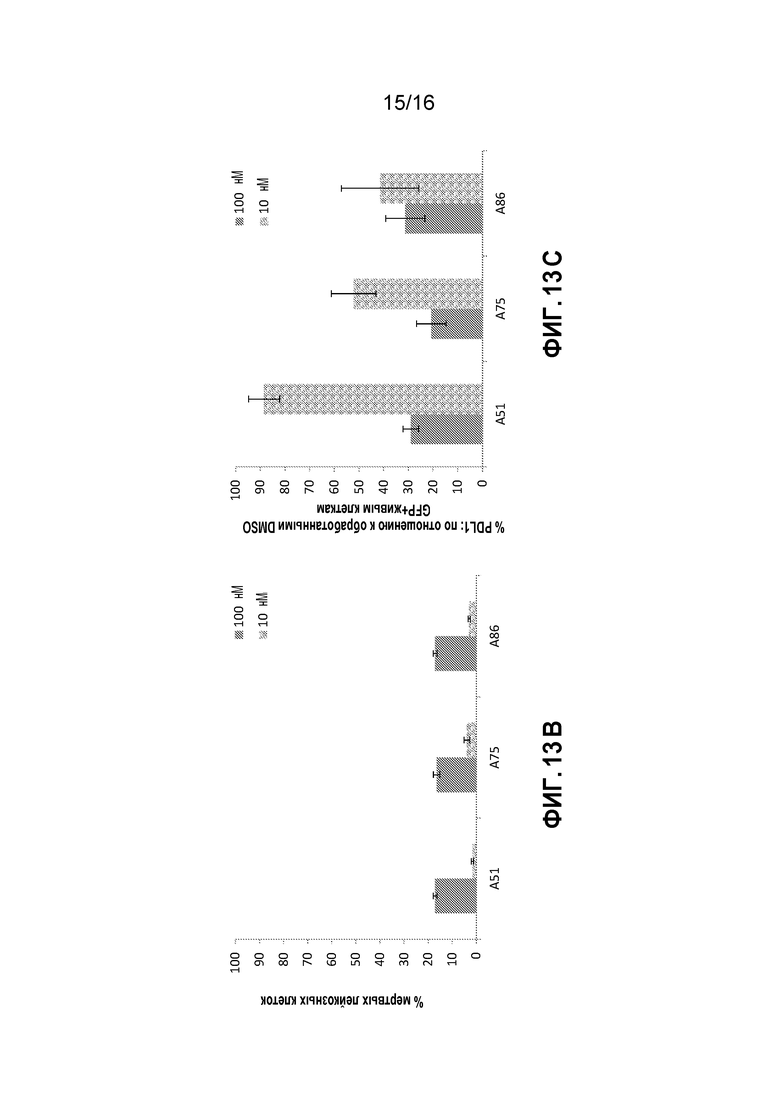
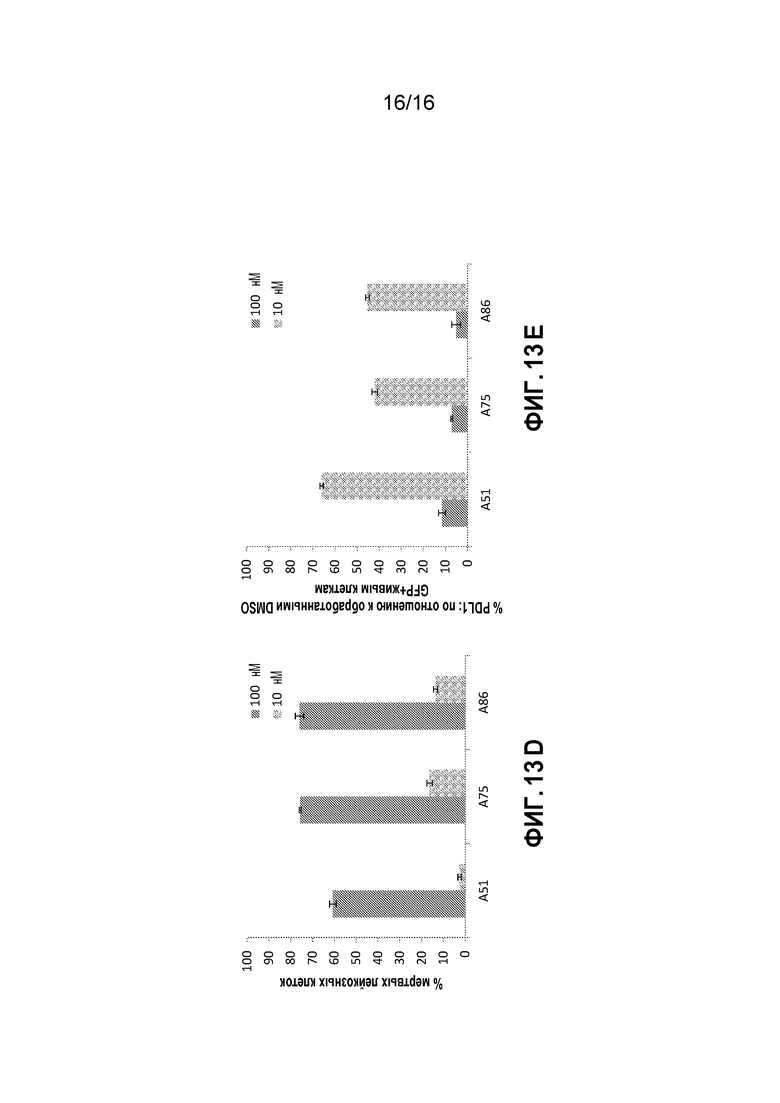
Изобретение относится к соединениям общей формулы (I), их стереоизомерам или фармацевтически приемлемым солям, где в формуле (I) R1 и R2 каждый независимо означает H, C1-C8 алкил, C1-C5 ацил, каждый из которых необязательно замещен по меньшей мере одним из галогенида, гидроксила, C1–C5 алкокси и C3–C6 гетероарила, содержащего один или два атома азота в цикле, необязательно замещенного метилом; или R1 и R2 вместе с атомом азота, с которым они связаны, образуют 4-7-членное насыщенное кольцо, которое необязательно включает O, или 5-членное ароматическое кольцо, которое содержит N; R3 означает H или линейный C1-C8 алкил, необязательно замещенный гидроксилом; или R1 или R2 вместе с R3 и атомом углерода и атомом азота, с которым каждый из них связан, образуют 5-членное насыщенное кольцо, которое необязательно включает по меньшей мере один из NH, O и C=O; R4 и R8 означают Н; R5 означает галогенид или C1-C8 алкил, необязательно замещенный по меньшей мере одним галогенидом; R6 означает C1-C8 алкил, C5–С6 циклоалкил или 4-6-членный насыщенный гетероциклил, содержащий атом кислорода в цикле; R7 означает C1-C8 алкил, замещенный C3-C7 циклоалкилом. Также изобретение относится к конкретным соединениям, указанным в формуле изобретения, к способам ингибирования и способу лечения злокачественных заболеваний. Технический результат – пиразолпиримидиновые производные формулы (I) в качестве ингибиторов казеинкиназы I (CKI) и киназы 1, ассоциированной с рецептором интерлейкина-1 (IRAK1). 6 н. и 34 з.п. ф-лы, 3 табл., 42 ил.
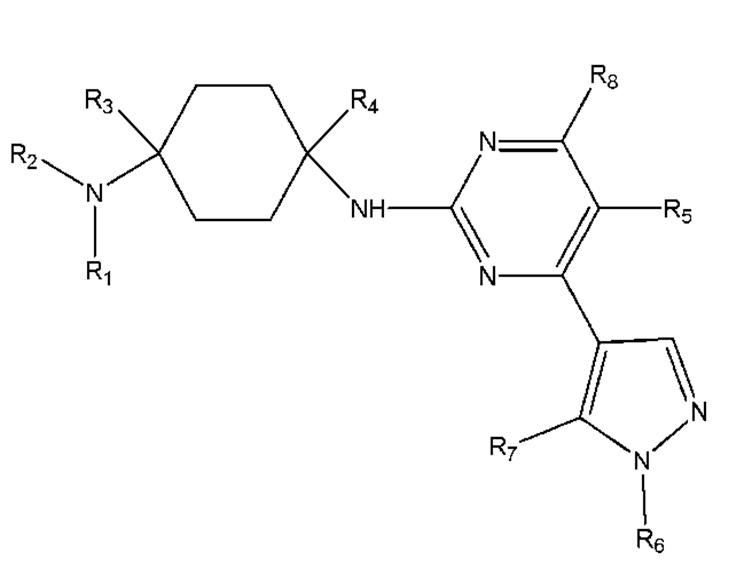 (I)
(I)
1. Соединение формулы (I) или его стереоизомер или фармацевтически приемлемая соль:
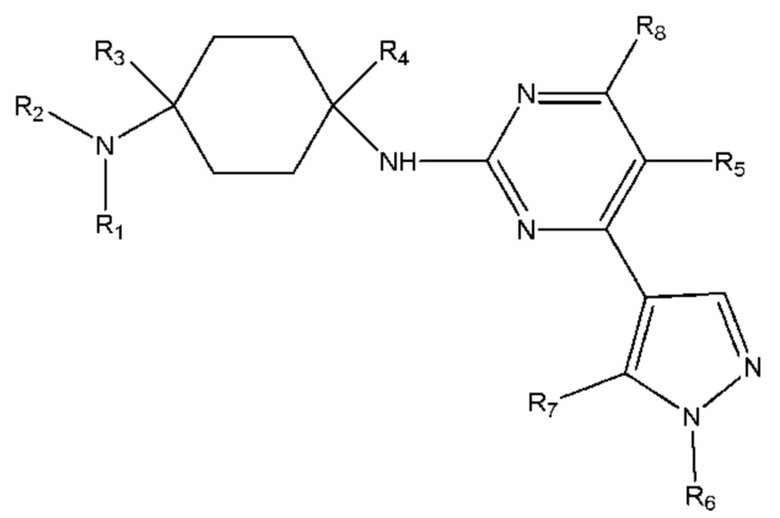
(I)
где
R1 и R2 каждый независимо означает (i) H; или (ii) линейный или разветвленный C1 - C8 алкил, линейный или разветвленный C1 - C5 ацил, каждый из которых необязательно замещен по меньшей мере одним из галогенида, гидроксила, C1 – C5 алкокси и C3 – C6 гетероарила, содержащего один или два атома азота в цикле, необязательно замещенного метилом; или
R1 и R2 вместе с атомом азота, с которым они связаны, образуют 4-7-членное насыщенное кольцо, которое необязательно включает O, или 5-членное ароматическое кольцо, которое содержит N;
R3 означает H или линейный C1 - C8 алкил, необязательно замещенный гидроксилом; или
R1 или R2 вместе с R3 и атомом углерода и атомом азота, с которым каждый из них связан, образуют 5-членное насыщенное кольцо, которое необязательно включает по меньшей мере один из NH, O и C=O;
R4 означает Н;
R5 означает галогенид или линейный или разветвленный C1 - C8 алкил, необязательно замещенный по меньшей мере одним галогенидом;
R6 означает линейный или разветвленный C1 - C8 алкил, C5 – С6 циклоалкил или 4-6-членный насыщенный гетероциклил, содержащий атом кислорода в цикле;
R7 означает линейный или разветвленный C1 - C8 алкил, замещенный C3 - C7 циклоалкилом; и
R8 означает Н.
2. Соединение или его стереоизомер или фармацевтически приемлемая соль по п. 1, где R1 и R2 каждый независимо означает H; или линейный или разветвленный C1 - C8 алкил, каждый необязательно замещенный по меньшей мере одним из галогенида, гидроксила и C3 – C6 гетероарила, содержащего один или два атома азота в цикле, необязательно замещенного метилом.
3. Соединение или его стереоизомер или фармацевтически приемлемая соль по п. 1, где R1 и R2 каждый независимо означает H или C1 - C5 ацил, необязательно замещенный по меньшей мере одним из галогенида или гидроксила.
4. Соединение или его стереоизомер или фармацевтически приемлемая соль по п. 1, где R1 и R2 каждый означает H.
5. Соединение или его стереоизомер или фармацевтически приемлемая соль по любому одному из пп. 1-4, где R4 представляет собой H.
6. Соединение или его стереоизомер, или фармацевтически приемлемая соль по любому одному из пп. 1-5, где каждый из R3 и R4 представляют собой H.
7. Соединение или его стереоизомер, или фармацевтически приемлемая соль по любому одному из пп. 1-6, где R5 означает F, Cl или линейный или разветвленный C1 - C4 алкил.
8. Соединение или его стереоизомер, или фармацевтически приемлемая соль по любому одному из пп. 1-7, где R5 представляет собой Cl.
9. Соединение или его стереоизомер, или фармацевтически приемлемая соль по любому одному из пп. 1-8, где R8 представляет собой H.
10. Соединение или его стереоизомер, или фармацевтически приемлемая соль по любому одному из пп. 1-9, где R6 представляет собой линейный или разветвленный C1 - C8 алкил.
11. Соединение или его стереоизомер, или фармацевтически приемлемая соль по любому одному из пп. 1-10, где R6 означает метил.
12. Соединение или его стереоизомер, или фармацевтически приемлемая соль по любому одному из пп. 1-11, где R7 представляет собой линейный или разветвленный C1 - C8 алкил, замещенный циклопропилом.
13. Соединение или его стереоизомер, или фармацевтически приемлемая соль по любому одному из пп. 1-12, где R7 означает циклопропилметил.
14. Соединение или его стереоизомер, или фармацевтически приемлемая соль по любому одному из пп. 1-13, где R1 и R2 вместе с атомом азота, с которым они связаны, образуют 4-7-членное насыщенное кольцо, необязательно содержащее атом O в цикле или 5-членное ароматическое кольцо, содержащее атом N в цикле.
15. Соединение или его стереоизомер, или фармацевтически приемлемая соль по любому одному из пп. 1-14, где R1 и R2 вместе с атомом азота, с которым они связаны, образуют 4-7-членное насыщенное кольцо.
16. Соединение или его стереоизомер, или фармацевтически приемлемая соль по любому одному из пп. 1-15, где R1 и R2 вместе с атомом азота, с которым они связаны, образуют 4-7-членное насыщенное кольцо, содержащее атом O в цикле.
17. Соединение или его стереоизомер, или фармацевтически приемлемая соль по любому одному из пп. 1-16, где R1 и R2 вместе с атомом азота, с которым они связаны, образуют 5-членное ароматическое кольцо, содержащее N в цикле.
18. Соединение или его стереоизомер, или фармацевтически приемлемая соль по любому одному из пп. 1-17, где R1 или R2 вместе с R3 и атомом углерода и атомом азота, с которыми они связаны, образуют 5-членное насыщенное кольцо, которое содержит по меньшей мере один из NH, O и C=O.
19. Соединение, выбранное из следующих:
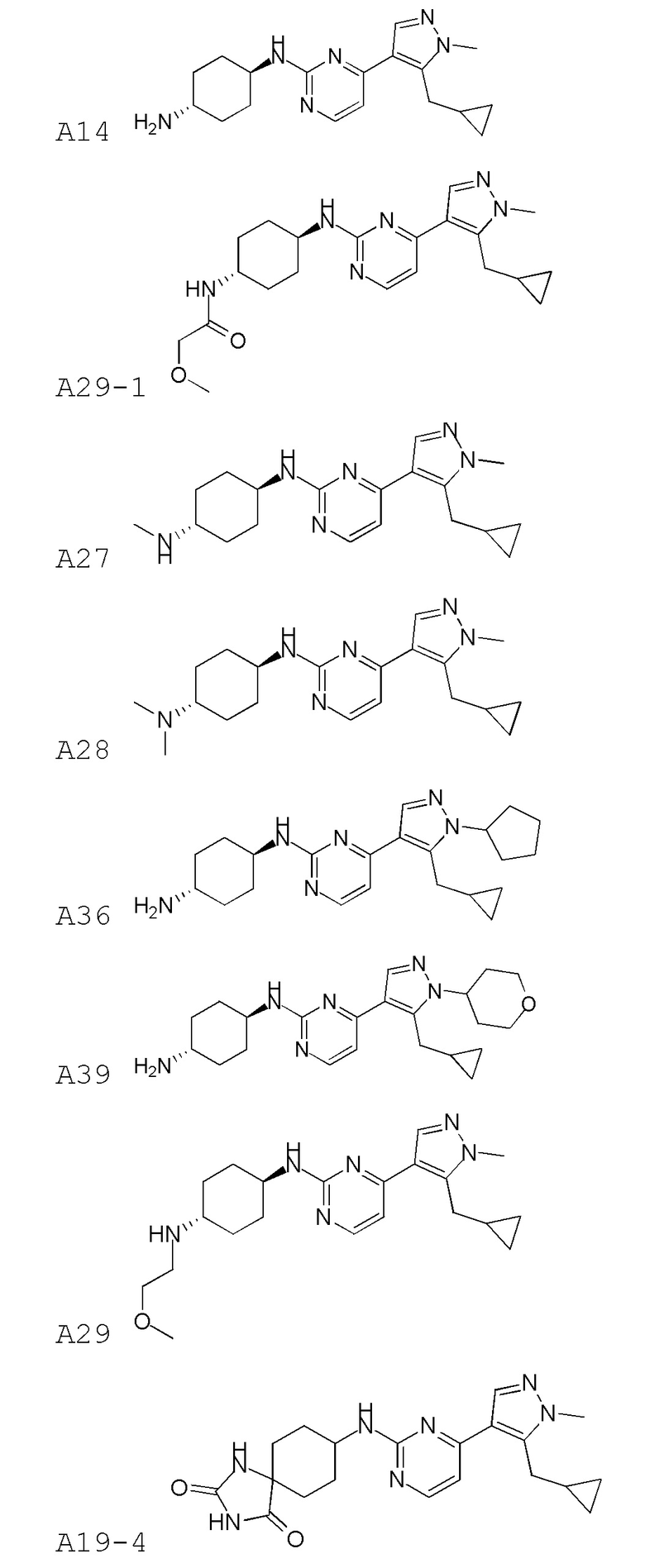
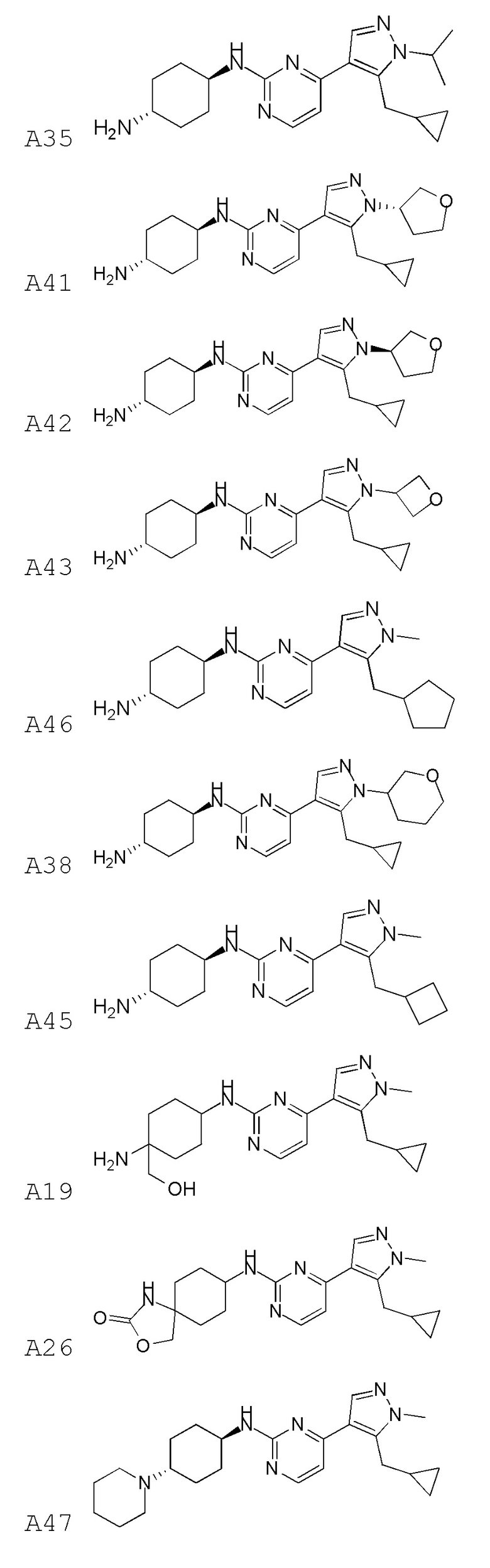
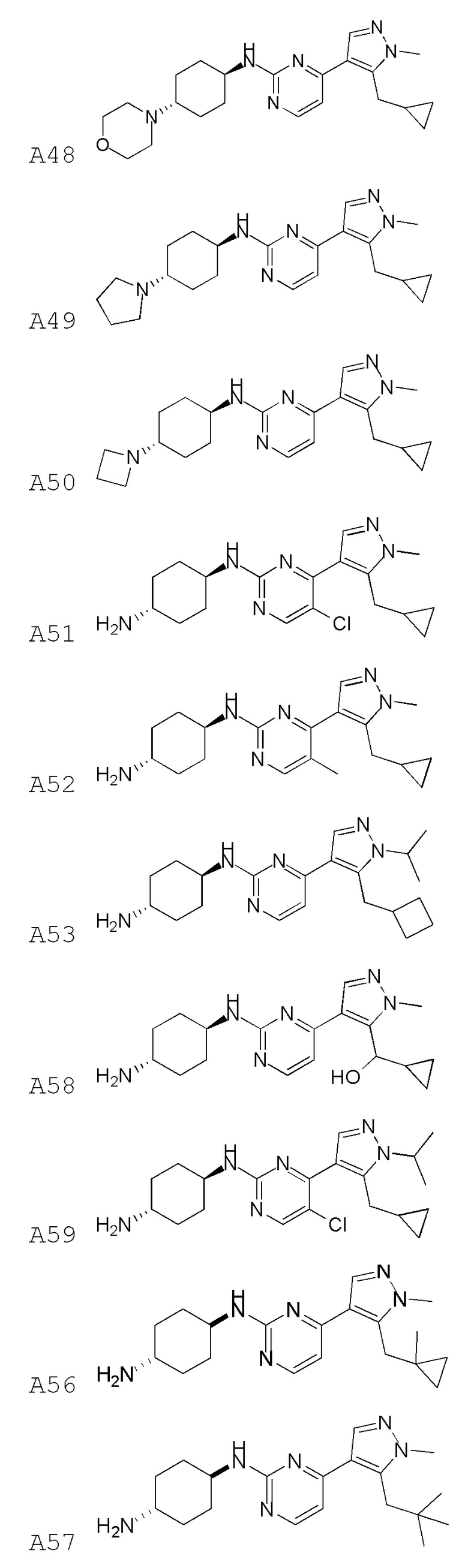
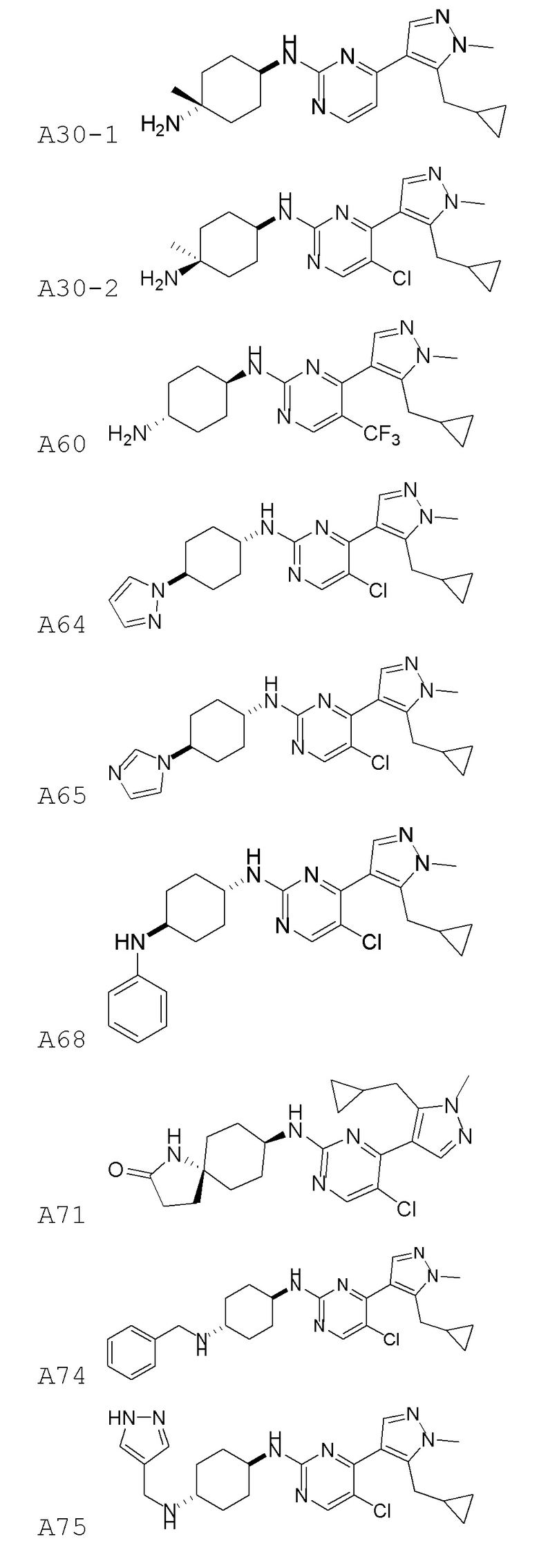
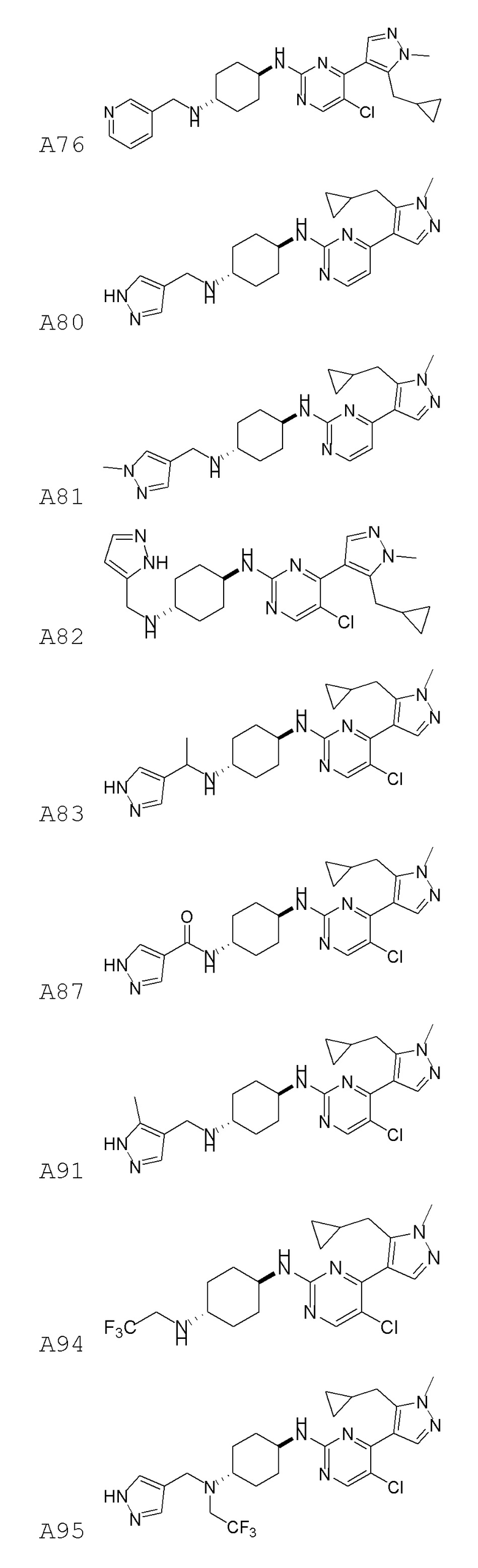
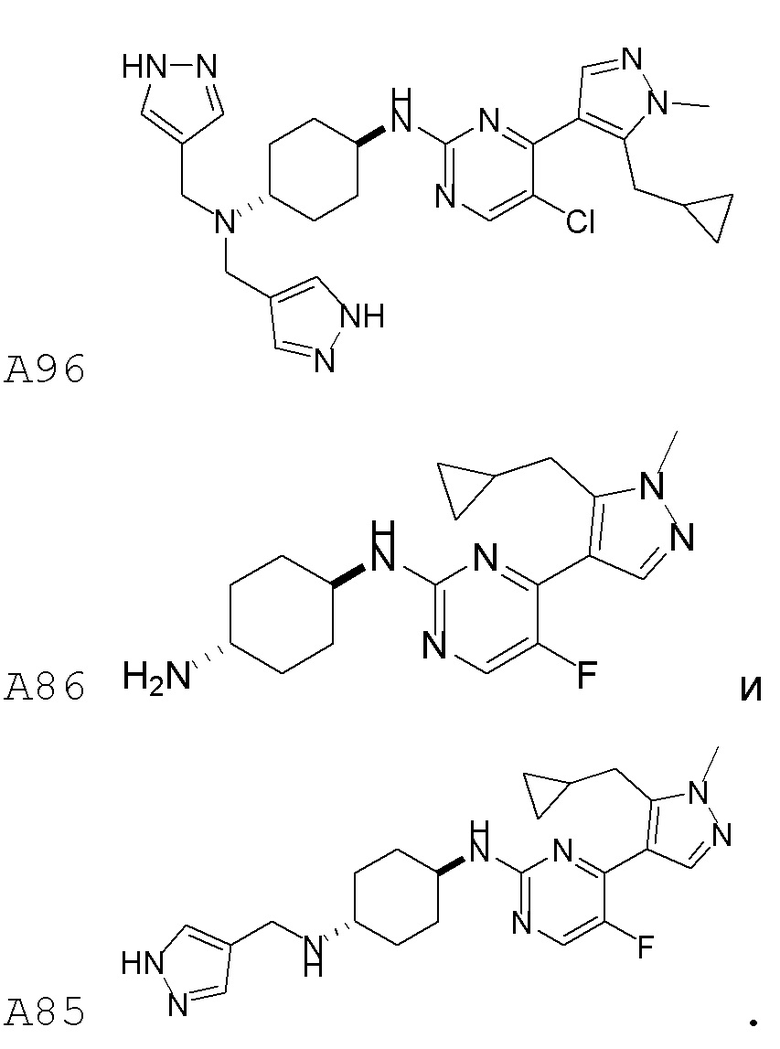
или его фармацевтически приемлемая соль.
20. Соединение по п. 19, где соединение представляет собой соединение следующей формулы
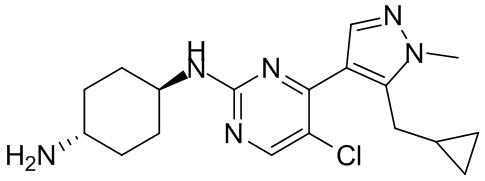
или его фармацевтически приемлемая соль.
21. Соединение или его стереоизомер, или фармацевтически приемлемая соль по любому одному из пп. 1-20, обладающее свойствами ингибитора киназы 1, ассоциированной с рецептором интерлейкина-1 (IRAK1), для применения в терапии.
22. Соединение или его стереоизомер, или фармацевтически приемлемая соль по любому одному из пп. 1-20 для применения в ингибировании по меньшей мере одной из казеинкиназы I (CKI) и киназы 1, ассоциированной с рецептором интерлейкина-1 (IRAK1).
23. Соединение или его стереоизомер, или фармацевтически приемлемая соль по любому одному из пп. 1-20 для применения в ингибировании казеинкиназы I (CKI).
24. Соединение или его стереоизомер, или фармацевтически приемлемая соль по любому одному из пп. 1-20 для применения в ингибировании киназы 1, ассоциированной с рецептором интерлейкина-1 (IRAK1).
25. Соединение или его стереоизомер, или фармацевтически приемлемая соль по любому одному из пп. 1-20 для применения в индуцировании противоопухолевого ответа.
26. Соединение или его стереоизомер, или фармацевтически приемлемая соль по любому одному из пп. 1-20, обладающее IPAK1 ингибирующей активностью, предназначенное для лечения состояния, симптома или заболевания, ассоциированного с злокачественным состоянием.
27. Соединение по п. 26, где указанное злокачественное состояние представляет собой рак.
28. Соединение по п. 26, где указанное злокачественное состояние выбрано из гематологических злокачественных опухолей (множественной миеломы, миелодиспластического синдрома (MDS), острого миелоидного лейкоза (AML), меланомы и ER-негативного рака молочной железы, диффузной крупноклеточной В-клеточной лимфомы (DLBCL), хронического миелогенного лейкоза (CML), хронического лимфоцитарного лейкоза (CLL), рака головы и шеи и любых их комбинаций.
29. Соединение по п. 27, где указанный рак имеет WT p53.
30. Соединение или его стереоизомер, или фармацевтически приемлемая соль по любому одному из пп. 1-20, обладающее IPAK1 ингибирующей активностью, предназначенное для лечения рака, имеющего WT p53, где указанный WT p53 представляет собой биомаркер для эффективности указанного соединения.
31. Соединение для применения по п. 27, где указанный рак выбран из множественной миеломы, лейкоза, злокачественной меланомы, рака молочной железы, рака предстательной железы, колоректального рака и любых их комбинаций.
32. Способ ингибирования по меньшей мере одной из казеинкиназы I (CKI) и киназы 1, ассоциированной с рецептором интерлейкина-1 (IRAK1), у нуждающегося в этом субъекта, включающий стадию введения указанному субъекту по меньшей мере одного соединения или его стереоизомера, или фармацевтически приемлемой соли по любому одному из пп. 1-20.
33. Способ ингибирования казеинкиназы I (CKI) у нуждающегося в этом субъекта, включающий введение указанному субъекту по меньшей мере одного соединения или его стереоизомера, или фармацевтически приемлемой соли по любому одному из пп. 1-20.
34. Способ ингибирования киназы 1, ассоциированной с рецептором интерлейкина-1 (IRAK1), у нуждающегося в этом субъекта, включающий введение указанному субъекту по меньшей мере одного соединения или его стереоизомера или фармацевтически приемлемой соли по любому одному из пп. 1-20.
35. Способ лечения состояния, симптома или заболевания, ассоциированного с злокачественным состоянием, у нуждающегося в этом субъекта, при этом указанный способ включает введения указанному субъекту по меньшей мере одного соединения или его стереоизомера, или фармацевтически приемлемой соли по любому одному из пп. 1-20.
36. Способ по п. 35, где указанное злокачественное состояние представляет собой рак.
37. Способ по п. 36, где указанный рак имеет WT p53.
38. Способ по п.37, где указанный WT p53 представляет собой биомаркер для эффективности указанного соединения.
39. Способ по п. 36, где указанный рак выбран из лейкоза, множественной миеломы, злокачественной меланомы, рака молочной железы, рака предстательной железы и колоректального рака.
40. Способ по п. 35, где указанное злокачественное состояние выбрано из гематологических злокачественных опухолей (множественной миеломы, миелодиспластического синдрома (MDS), острого миелоидного лейкоза (AML), меланомы и ER-негативного рака молочной железы, диффузной крупноклеточной В-клеточной лимфомы (DLBCL), хронического миелогенного лейкоза (CML) и хронического лимфоцитарного лейкоза (CLL), рака головы и шеи и любых их комбинаций.
| Устройство для закрепления лыж на раме мотоциклов и велосипедов взамен переднего колеса | 1924 |
|
SU2015A1 |
| Устройство для закрепления лыж на раме мотоциклов и велосипедов взамен переднего колеса | 1924 |
|
SU2015A1 |
| Устройство для закрепления лыж на раме мотоциклов и велосипедов взамен переднего колеса | 1924 |
|
SU2015A1 |
| ПИРИМИДИНОВЫЕ ПРОИЗВОДНЫЕ, ОБЛАДАЮЩИЕ АКТИВНОСТЬЮ В ОТНОШЕНИИ МСН | 2005 |
|
RU2373197C2 |

