Настоящее изобретение относится к новым контрастным агентам, более конкретно к новым газосодержащим или газовыделяющим контрастным агентам, предназначенным для использования в изобразительной диагностике.
Хорошо известно, что получение ультразвуковых диагностических изображений сопряжено с необходимостью располагать потенциально ценным диагностическим оборудованием, например при исследовании сосудистой системы, в особенности в кардиографии, и капиллярной системы тканей. С целью улучшить получаемые акустическим путем изображения, были предложены разнообразные контрастные агенты, включая сюда суспензии твердых частиц, эмульгированные капли жидкости, газовые пузырьки, а также инкапсулированные газы и жидкости. Общеизвестно, что особенно эффективны в смысле акустического обратного рассеяния, которое они обеспечивают, контрастные агенты низкой плотности, которые являются легкосжимаемыми, поэтому интересны газосодержащие и газогенерирующие системы.
Первоначальные исследования, включающие в себя использование пузырьков свободного газа, которые генерировались ин виво вследствие внутрисердечной инъекции физиологически приемлемых веществ, продемонстрировали потенциальную эффективность таких пузырьков в качестве контрастных агентов в эхокардиографии; однако такая техника на практике весьма ограничена из-за краткой продолжительности жизни свободных пузырьков. Известен способ стабилизации газовых пузырьков для эхокардиографии и других ультразвуковых исследований, например, с использованием эмульгаторов, масел, загустителей или сахаров, или же путем удержания или инкапсулирования газа или его предшественника в различных полимерных системах, в частности в форме пористых газосодержащих полимерных микрочастиц или газовых "микрошариков", инкапсулированных посредством полимерных покрытий.
Известно (патентная заявка WO 80/02365) использование газовых микропузырьков, инкапсулированных в желатину, что позволяет улучшить получаемые ультразвуковые изображения. Однако, из-за крайне малой толщины инкапсулирующего покрытия не обеспечивается необходимая стойкость таких микропузырьков в диапазоне их предпочтительных размеров для использования в эхокардиографии /1-10 мкм/.
В описании US A-4774958 предлагается использовать дисперсии микропузырьков, стабилизированные инкапсулированием в денатурированный белок, например в альбумин сыворотки человека. Такие системы позволяют готовить системы микропузырьков, размеры которых составляют, например, 2-5 мкм, но, тем не менее, не обеспечивают эффективной визуализации левой половины сердца и миокарда. Кроме того, применение этих получаемых из белка агентов может создать проблемы, связанные с потенциальными аллергическими реакциями.
В описании к европейской заявке на патент EP-A-0327490 помимо прочего предлагаются ультразвуковые агенты, которые включают в себя синтетический биоразлагаемый полимер в форме части, содержащих газ или летучую текучую среду (т.е. текучую среду с температурой кипения ниже 60oC) в свободном или связанном состоянии. Типичные синтетические биоразлагаемые полимеры охватывают полиэфиры оксикарбоновых кислот, полиалкилцианоарилаты, полиаминокислоты, полиамиды, полиарилированные сахариды и полиортоэфиры.
Аналогичные биоразлагаемые полимеры в виде микрочастиц на основе полимеризованных альдегидов предлагаются и в описании к европейской заявке на патент EP-A-0441468, тогда как системы на основе микрочастиц из производных полиаминокислоты-полициклического имида предлагаются в описании к европейской заявке на патент EP-A-0458079.
В описании к европейской заявке на патент EP-A-0458745 предлагаются воздухо- или газонаполненные микропузырьки, в которых инкапсулированный материал представляет собой деформируемый и эластичный межфазно осажденный полимер, предпочтительно являющийся биоразлагаемым. Его примеры охватывают полисахариды, полиаминокислоты, полиактиды, полигликозиды, лактидо-лактоновые сополимеры, полипептиды, белки, сложные полиортоэфиры, полидиоксанон, полибета-аминокетоны, полифосфазены, полиангидриды и полиалкилцианакрилаты. Такие микропузырьки получают по обычной эмульсионной технологии, осуществление которой приводит к осаждению полимера вокруг капелек летучей жидкости, которая в дальнейшем испаряется.
В описании к международной заявке на патент WO 91/12823 предлагаются ультразвуковые контрастные агенты, представляющие собой газо- или паронаполненные полимерные микрокапсулы; к предпочтительным полимерам относятся несолюбилизированные белки, в частности денатурированный альбумин. Такие микрокапсулы могут быть получены формированием вокруг твердой или жидкой сердцевины белковой оболочки (например, согласно методам с использованием простой или сложной коацервации, двойной эмульсии или сведения к минимальному растворимости вблизи изоэлектрической точки), отверждением оболочки (например, химической или тепловой обработкой) и удалением сердцевины (например, сублимацией или за счет испарения). Применение технологии двойной эмульсии позволяет получать микрокапсулы с сотовой структурой, в которой предусмотрены многочисленные газо- или паронаполненные камеры.
Известно, что при получении магнитного резонансного (МР) изображения эффективна также газосодержащая контрастная среда, например в виде чувствительных контрастных агентов, действие которых ослабляет интенсивность МР - сигнала. В качестве парамагнитных МР - контрастных агентов может также служить кислородосодержащая контрастная среда.
Более того в технике получения X-лучевого изображения было установлено, что в качестве негативных пероральных контрастных агентов могут быть использованы такие газы, как двуокись углерода.
Общеизвестно, что желательным свойством контрастных агентов на полимерной основе является биоразлагаемость, что позволяет упростить их последующее удаление из обследуемого субъекта или абсорбцию его организмом. Однако для максимизации в достижении этой цели мало внимание было уделено конкретной структуре полимеров, поскольку основные надежды при этом обычно связывают с полимерами, которым характерна хотя и медленная, но биоразлагаемость, в частности с такими, как полиэфиры, полиангидриды, поликарбонаты, и полиамиды и полиуретаны, что в основном обусловлено чувствительностью сложноэфирных, амидных и уретановых групп к энзимному гидролизу.
В EP-A-0458745, в качестве контрастных агентов для ультразвукового исследования, помимо прочих, предлагаются этерифицированные полипептидные производные особой категории, проявляющих, регулируемую биоразлагаемость. Эти полимеры, о которых в описании к заявке на европейский патент EP-A- 0130935 сказано как о постепенно выделяющихся носителях для лекарств, включает в себя соединения общей формулы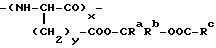
где
Ra и Rb - алкильные группы или водородные атомы;
Rc - возможно замещенная алифатическая или ароматическая группа.
или же Rb - водородный атом или алкильная группа, а Ra и Rc совместно образуют двухвалентную группу, в частности диметиленовую, виниленовую или фениленовую группу,
Y - 1 или 2;
значения символа x таковы, что молекулярный вес полимера составляет по меньшей мере 5000,
и их сополимеры с другими полиаминокислотами. Первая стадия при биоразложении таких полимеров состоит в отщеплении в качестве боковых цепей метилендиэфирных групп с образованием полимеров, молекулы которых содержат звенья общей формулы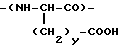
Было установлено, что такие полимеры затем подвергаются дальнейшему разложению пептидазами до их составляющих аминокислот, которые могут абсорбироваться организмом субъекта, которому ввели сочетание полимер/лекарство, в ходе протекания характерного медленного процесса. Кроме того, такие пептидные структуры могут оказаться способными вызывать аллергические реакции.
Таким образом, необходимость в контактных агентах на полимерной основе, которые сочетают в себе свойства хорошей стойкости при хранении, стабильность ин виво при введении в организм, предпочтительнее в течение по меньшей мере нескольких циклов циркулирования в случае внутрисердечных инъекций и быстрого последующего биологического разложения, продолжает сохраняться.
Настоящим изобретением установлено, что вышеуказанные цели могут быть достигнуты созданием контрастных агентов, основанных на полимерах, молекулы которых содержат метиленовые диэфирные группы формулы I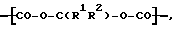
где каждым из R1 и R2 обозначен водородный атом или связанная с углеродным атомом одновалентная органическая группа или же R1 и R2 совместно способны образовывать двухвалентную органическую группу, связанную с углеродными атомами,
что и легло в основу настоящего изобретения. Такие молекулярные звенья особенно быстро разлагаются обычными эстеразными энзимами, но в отсутствии энзимов они стойки. Они могут быть соединены не только с органическими группами, связанными с углеродными атомами, как это имеет место в случае обычных сложных карбоксилатных эфиров, на также с кислородными атомами -O-, как это имеет место в случае сложных карбонатных эфиров.
Полимеры этого типа и разнообразные способы их получения изложены и заявлены в совместно рассматриваемой заявке на международный патент WO 92/04392, поданной авторами настоящего изобретения, содержание которой здесь упомянуто в качестве ссылки. Звенья формулы 1 в молекулах таких полимеров могут, например, входить в состав полимерной основной цепи либо в форме повторяющихся звеньев, либо в форме соединительных звеньев между полимерными звеньями, либо они могут содержаться в поперечных сшивающих группах между основными полимерными цепями.
Еще один класс полимеров этого типа и способы их получения предлагаются и заявлены в одновременно поданной и рассматриваемой заявке на патент. К ним относятся неструктурированные полимеры с низкой или нулевой растворимостью в воде, молекулы которых включают в себя неполипептидную полимерную основную цепь, к которой присоединены боковые цепи, причем по меньшей мере часть указанных боковых цепей содержит липофильные остатки, связанные с полимерной основной цепью посредством сложных метилендиэфирных звеньев формулы I, благодаря чему упомянутые липофильные остатки способны отщепляться в результате биологического разложения с образованием водорастворимого полимера.
Так, например, в соответствии с одним из аспектов настоящего изобретения предлагаются контрастные агенты, включающие в себя газосодержащие или газогенерирующие полимерные микрочастицы и/или микропузырьки, характеризующиеся тем, что такой полимер представляет собой биоразлагаемый полимер, молекулы которого содержат звенья формулы II.

где
значения символов R1 и R2 определены выше;
каждым из m и n, которые могут быть как идентичными, так и различными, обозначает 0 или 1.
Полимеры, молекулы которых содержат звенья формулы II, где один из или каждый из n и m служит для обозначения 1, то есть содержащие карбонатные сложноэфирные группы, ранее предлагались только в вышеупомянутой международной патентной публикации WO 92/04392; в некоторых случаях они могут быть особенно легко биоразлагаемыми.
Полимеры, молекулы, которых содержат полипептидную основную цепь, могут вызвать нежелательную аллергическую реакцию, поэтому в общем предпочтительнее использовать неполипептидные полимеры.
Молекулы полимеров, которые могут быть использованы в соответствии с настоящим изобретением, могут включать в себя звенья формулы III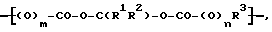
где значения каждого из символов R1, R2, m и n определены выше;
R3 - двухвалентная органическая группа, например двухвалентная органическая группа, связанная с углеродным атомом.
Молекулы таких полимеров могут включать в себя множество звеньев формулы III, характеризующихся различными значениями символов m, n, R1, R2 и R3, например в форме блок- или графт-сополимеров. Через некоторые интервалы по всей молекуле полимера могут встречаться сложные диэфирные связи, например в форме сшивающих групп; они могут встречаться также и между сополимерными секциями, причем в этом последнем варианте символом R3 обозначена полимерная группа. В другом варианте такие связи или мостики могут встречаться повсеместно практически по всей молекуле полимера, причем в этом варианте символ R3 предпочтительно является группой с низким молекулярным весом.
Особенно интересные звенья III представляют собой, те в которых m = 0, n = 0 или 1, т.е. дикарбоксилатные звенья формулы IV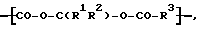
или карбоксилатные звенья формулы V
каждый из R1 и R2 может обозначать, например, связанную с водородным или углеродным атомом гидрокарбильную или гетероциклическую группу, содержащую, в частности, 1 - 20 углеродных атомов, такую как алифатическая группа, например, алкильная или алкенильная группа (предпочтительнее содержащая до 10 углеродных атомов), циклоалкильная группа (предпочтительнее содержащая до 10 углеродных атомов), аралифатическая группа, в частности аралкильная группа (предпочтительнее содержащая до 20 углеродных атомов), арильная группа (предпочтительнее содержащая до 20 углеродных атомов) или гетероциклическая группа, включающая в себя до 20 углеродных атомов и один или несколько гетероатомов, выбираемых из кислорода, серы и азота. Такая гидрокарбильная или гетероциклическая группа может включать в себя одну или несколько функциональных групп, в частности атомы галогена или группы формулы -NR4R5, -CONR4R5, -CR6, R6 и COOR7, где каждый из R4 и R5, которые могут быть как идентичными, так и различными, обозначает водородный атом, ацильную группу или гидрокарбильную группу такого типа, как описанная выше в определении значения символов R1 и R2; R6 - водородный атом или ацильная группа, или же группа такого типа, как описанная выше в определении значений символов R1 и R2, R7 - водородный атом или группа, аналогичная группе R1 или R2. В этом случае, когда R1 и R2 обозначают двухвалентные группы, они могут представлять собой, например, алкилиденовые, алкенилиденовые, алкиленовые или алкениленовые группы (предпочтительнее содержащие до 10 углеродных атомов в каждой), у которых могут быть по одной или несколько функциональных групп, как это определено выше.
Как указано выше, сложные диэфирные группы формулы I могут быть разделены группами широкого диапазона. В том случае, когда желательно, чтобы молекула полимера была разделена на относительно короткие секции, что способствует биоразлагаемости, группа R3, которая разделяет сложные диэфирные звенья формулы II, может представлять собой, например, алкиленовую или алкениленовую группу, (содержащую, в частности, до 20, более предпочтительно до 10 углеродных атомов), циклоалкиленовую группу (предпочтительнее содержащую до 10 углеродных атомов), аралкиленовую группу (предпочтительнее содержащую до 20 углеродных, атомов и возможно связанную посредством арильных и/или алкильных остатков, причем такие аралкильные остатки охватывают, например, две арильные группы, соединенные алкиленовой цепью) или гетероциклическую группу, включающую в себя один или несколько гетероатомов, выбираемых из кислорода, серы и азота (предпочтительнее содержащую до 20 углеродных атомов). Группа R3 может включать в себя функциональные группы, например, такие, которые перечислены в определении значений символов R1 и R2 и/или заместители, в частности такие, как оксогруппы; углеродные цепи групп R3 могут прерываться и/или заканчиваться гетероатомами, в частности атомами кислорода, азота или серы, например в связанном с оксозаместителями состояния с образованием таких связей или мостиков, как сложноэфирные, сложные тиоэфирные или амидные группы. Для улучшения гидрофильности полимеров в радикал R3 можно включить, например, один или несколько рядов оксиэтиленовых или полиоксиэтиленовых звеньев и/или гидроксилзамещенных углеродных цепочек (например в форме оксиалкильных групп или сахарных групп). Такие ряды звеньев могут быть, например, связаны посредством оксикарбонильных групп, в частности короткоцепочечных остатков двухосновных кислот, таких как оксалил, малонил, сукцинил, глутарил или адипоил.
В таком случае, когда группа R3 представляет собой полимерную группу, оно может быть, например, полиамидной, полиоксикислотной, полиэфирной, поликарбонатной, полисахаридной, полиоксиэтиленовой, полиоксиэтилен-полиоксипропиленовой блок-сополимерной группой, поливиниловой спиртовой или поливинилэфирной/ спиртовой группой.
Большой ряд возможных групп R1, R2 и R3 позволяет регулировать гидрофобность или гидрофильность полимера в соответствии с любой требуемой целью применения. Так, например, можно с успехом придать полимерам нерастворимость в воде, но при этом продукты разложения в результате энзимного гидролиза могут быть водорастворимыми.
Алифатические группы, обозначаемые, например, символами R1 и R2, могут быть прямыми или разветвленными, насыщенными или ненасыщенными, охватывая, в частности, алкильные и алкенильные группы, такие как метильная, этильная, пропильная, изопропильная, бутильная, децильная и аллильная группы. К аралифатическим группам относятся (монокарбоциклические арил)-алкильные группы, например бензильные группы. Арильные группы включают в себя моно- и бициклические арильные группы, например, фенильная, толильная и нафтильная группы. Гетероциклические группы охватывают 5- или 6-членные гетероциклические группы, предпочтительнее, содержащие по одному гетероатому, например фурильная, тиенильная и пиридильная группы. Атомы галогена в качестве заместителей могут представлять собой, например, хлор, бром и иод.
Биоразложение полимеров, молекулы которых включают в себя звенья формулы III, обычно протекает в результате энзимного гидролитического разветвления связей, которыми группы формулы -O-C(R1R2)-O- соединяются со смежными карбонильными группами, вследствие чего как правило образуются альдегиды или кетоны формулы R1-CO-R2. Находящиеся в промежутках секции образуют различные продукты в зависимости от того, равно или 0 или 1. В случае, когда m или n = 0, в результате гидролитического расщепления обычно образуется карбоксильная группа, а в том случае, когда m или n = 1, образуется гипотетический остаток карбоновой кислоты -R3-O-COOH, который обычно выделяет углекислый газ с образованием группы формулы -R3-OH. Это явление может быть использовано в тех случаях, когда высвобождение двуокиси углерода желательно физиологически или функционально.
Как указано выше, звенья формулы III внутри молекулы одного и того же полимера могут быть различными, то есть полимеры могут представлять собой сополимеры, в частности блок- или графт-сополимеры. Такие полимеры могут быть сополимерами, образованными из небиоразлагаемых мономеров, причем небиоразлагаемые секции, которые остаются после энзимного или другого расщепления, по предпочтительному варианту должны характеризоваться приемлемыми размерами, которые обеспечивают их водорастворимость или диспергируемость в воде, что обуславливает, таким образом, легкость их рассредоточения или удаления. Такие небиоразлагаемые секции можно рассматривать как часть групп R3 в формуле III, которые в действительности связывают между собой биоразлагаемые группы формулы II.
Полимеры могут быть линейными, разветвленными или структурированными. Разветвленные и структурированные полимеры обычно образуются благодаря наличию функциональных групп или двойных связей в соответствующих группах R2, R2 или R3 их мономеров. Таким образом, молекулы получаемых структурированных или разветвленных полимеров содержат некоторые звенья формулы III, где группы R1, R2 и/или R3 замещены структурирующими или боковыми цепями.
Обычно в тех случаях, когда углеродные атомы, которые связывают группы R3 с группами формулы II, являются хиральными, предпочтительная хиральность обнаружена у природных продуктов, поскольку такие структуры обычно более эффективно разлагаются под действием деструктирующих энзимов.
Согласно наблюдениям, в структурированных биоразлагаемых полимерах структурирующих секции чаще всего разрушаются первыми, вследствие чего энзимному гидролизу становятся доступными остатки структуры. Таким образом, в качестве структурирующих цепей особенно полезными являются группы формулы II в молекуле полимера. Таким образом, одна из возможностей состоит в конверсии водорастворимых длинноцепочечных природных или синтетических небиоразлагаемых или медленно биоразлагаемых веществ, например, полисахаридов или олигосахаридов, или короткоцепочечных полиакриламидов в водорастворимую форму путем структурирования с использованием в качестве структурирующих звеньев такие радикалы, которые содержат группы формулы II. Это позволяет свести к минимуму стоимость готового продукта за счет уменьшения количества относительно дорогостоящих биоразлагаемых звеньев формулы II.
Блок-сополимеры могут, например, отвечать нижеследующей общей формуле: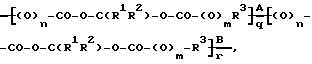
где значения соответствующих символов R, R, R, m и n таковы, что повторяющиеся звенья в блоках A и B различны, а каждым из q и r обозначено целое положительное число, например 10-207. К тем блокам, что представлены выше, может присоединяться один или несколько дополнительных блоков.
Полимеры, молекулы которых включают в себя звенья формулы III, которые могут быть использованы в соответствии с настоящим изобретением, можно, например, получать так, как это изложено в описании к вышеупомянутой заявке на международный патент WO 92/04392.
Молекулы полимеров другого класса, которые также могут быть использованы в соответствии с настоящим изобретением, включают в себя звенья формулы VI.
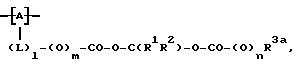
где символом A обозначено повторяющее звено неполипептидной полимерной основной цепи:
L обозначает связующую группу;
l=0 или 1;
значение символов, m, n, R1 и R2 определены выше;
R3a обозначает липофильную органическую группу, например органическую группу, которая определена выше для значений символов R1 и R2.
Группа A и группа L (если она имеется) должны быть такими, чтобы водорастворимостью обладали полимерные продукты разложения, которые образуются в результате биологической деструкции с отщеплением сложной метилендиэфирной группы, которая обычно содержит звенья формулы VII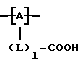
где значения символов A, L и l определены выше,
когда m в формуле VI обозначает 0, и звенья формулы VIII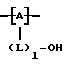
где значения символа A, L и l определены выше,
когда m в формуле VI обозначает 1.
Факторы, которые влияют на водорастворимость таких продуктов разложения полимеров, охватывают природу повторяющихся звеньев A любых сомономерных звеньев, которые могут входить в их состав, длину любой из связывающих групп L и общую длину полимерной цепи.
Предпочтительные повторяющиеся звенья A и все сомономерные звенья должны быть относительно короткими, то есть содержащими, например, до 10, в частности 1-6, углеродных атомов, и перемежаться одним или несколькими гетероатомами, выбираемыми из кислорода, азота и серы, и/или должны быть замещенными одним или несколькими заместителями, включающими в себя такие гетероатомы [например, оксо-, гидроксильными и аминогруппами]. В том случае, когда повторяющиеся звенья A и/или любые сомономерные звенья включают в себя гидрофильные группы, размеры таких звеньев не обязательно ограничены, причем к возможным звенья относятся, таким образом, полиоксиэтилен (остатки полиэтиленоксидных сложных эфиров метакриловой кислоты).
Длина предпочтительных связующих групп должна быть небольшой, причем к таким группам относятся, например, алкиленовые группы C1 - C3, в частности метиленовые, этиленовые и пропиленовые, возможно соединенные с полимерной основной цепью и/или (когда это уместно) перемещающиеся, в частности, с окси-, карбонильными, оксикарбонильными, имино, или иминокарбонильными группами. В том числе, когда содержатся полярные группы, в частности кислородные атомы или иминогруппы, связующие группы могут обладать большей длиной, что не оказывает нежелательного подавления водорастворимости. Таким образом, приемлемые продукты разложения полимеров включают в себя, например, поливиниловый спирт, полиакриловую кислоту, полиметакриловую кислоту, полиоксиалкилакрилаты и -метакрилаты, в частности поли-2-оксиэтилакрилат, полисахариды, в частности крахмал и декстран, сложные полиэфиры, простые полиэфиры, в частности полиоксиэтилены и полиоксипропилены, полиакриламиды и полиметакриламиды, такие как поли-N-оксиалкилакриламиды и -метакриламиды (например, поли-N-(2-оксипропил)-метакриламид, полиамиды, полиуретаны и эпоксидные полимеры.
Обычно нет необходимости в том, чтобы полимерные продукты разложения биоразлагаемых полимеров, молекулы которых содержат звенья формулы VI, были сами по себе биоразлагаемыми, поскольку они являются водорастворимыми. Так, например, к этим продуктам относятся поливиниловые и полиакриловые материалы. Таким образом, рамками настоящего изобретения охватывается применение полимеров, молекулы которых содержат звенья формулы VI, где A - повторяющиеся звень полиолефина, например этилена или пропилена. Необходимо иметь в виду, что полимеры этого типа могут быть получены согласно технологии свободнорадикальной полимеризации, которая сравнительно проста и экономична в осуществлении в противоположность, например, более сложной технологии полипептидного синтеза, которую необходимо осуществлять для получения таких полимеров, как те, что предлагается в описании к заявке на европейский патент EP-A-0130935.
Природа и размеры групп R1, R2 и R3a в звеньях формулы VI оказывают влияние как на степень придания полимерам, молекулы которых содержат такие звенья, липофильных свойств, и следовательно, способности несолюбилизироваться в отношении воды, так и на скорость, с которой при биологическом разложении отщепляются эти боковые цепи. Так, например, большие и/или громоздкие группы сообщают тенденцию к снижению скорости биологического разложения из-за пространственных затруднений, одновременно повышая липофильность полимера. Одна из полезных с вышеописанной цепью категорий боковых цепей включает в себя группы R1 и R2, каждую из которых выбирают из водородного атома и алкильных групп C1 - C4, в частности метильной группы, а также группы R3a, которая представляет собой низшую алкильную группу, например содержащую по меньшей мере 3 углеродных атома, в частности пропильную или бутильную группу. Такие боковые цепи сочетают в себе существенную степень липофильности и биоразлагаемость.
Необходимо иметь в виду, что, например, линейные полимеры, молекулы которых содержат звенья формулы VI, могут проявлять параметры улучшенной перерабатываемости (например, растворимости в органических растворителях и перерабатываемости в расплаве) в сравнении с аналогичными свойствами структурированных полимеров, молекулы которых содержат, в частности, введенные в них звенья с поперечно сшивающими группами формулы II. В этом аспекте они могут представлять собой противоположность полимерам, предлагаемым в описании к заявке не европейский патент EP-A-0130935, потенциальный недостаток которых состоит в том, что высокий уровень водородной связи, проявляемый полипептидами, проявляет тенденцию сообщать им относительно высокую температуру плавления, вследствие чего они могут не обладать способностью перерабатываться в расплаве без нежелательной деструкции.
Полимеры, молекулы которых содержат звенья формулы VI, могут быть получены любым удобным путем, например либо (A) реакцией предварительно полученного водорастворимого полимера с реагентом, который обеспечивает возможность введения в молекулу желательной липофильной сложной метилендиэфирной боковой цепи, либо (B) полимеризацией функционального мономера, молекула которого содержит желаемую липофильную сложную метилендиэфирную боковую цепь.
Способ (A) можно осуществлять, например, реакцией полимера, молекулы которого содержат боковые спиртовые гидроксильные группы (например, поливинилового спирта, полиоксиалкилакрилата или -метакрилата, или полисахарида), с соединением формулы IX
X-CO-O-C-(R1R2)-O-CO-(O)n-R3a,
где значения R1, R2, R3a и n определены выше;
символом X обозначена отделяемая группа, в частности, атом галогена, например, фтора, хлора, брома или иода.
Реагенты формулы IX могут быть, например, получены так, как это изложено Фолкманом и Лундом в журнале Synthesis 1990, 1159. Реакции, в результате которых получают полимеры, молекулы которых содержат звенья формулы VI, где m = 1, обычно проводят в растворе, например, в таком растворителе, где тетрагидрофуран, в присутствии слабо нуклеофильного основания, в частности пиридина. При этом можно использовать каталитическое количество четвертичного амина, такого же как 4-диметиламинопиридина. Число гидроксильных групп молекулы полимера, которые поступают в реакцию с образованием желательных липофильных сложных метилендиэфирных групп, можно регулировать путем соответствующего выбора таких факторов, как количество реагентов, продолжительность и температура реакции, что позволяет влиять на конечный гидрофильно-липофильный баланс липофилизированного полимера. Полученный продукт можно очищать по стандартной технологии, в частности экстракцией растворителем и/или растворением/ осаждением, и/или тонкослойной хроматографией.
По другому варианту способ (A) можно осуществлять реакцией полимера, молекулы которого содержат боковые карбоксильные группы (например, полиакриловой кислоты, полиметакриловой кислоты или водорастворимого пептида) с соединением формулы X
X-CR1R2-O-CO-(O)n-R3a,
где значения символов R1, R2, R3a, X и n определены выше.
Такие реакции, в результате которых получают полимеры, молекулы которых содержат звенья формулы VI, где m = 0, обычно проводят в растворе, например, в таком растворителе, как N,N'-диметилформамид, в присутствии сильного основания, в частности трет-бутоксида калия. Можно также использовать каталитическое количество краун-эфира, например 18 - краун-эфира - 6. Как и в предыдущем случае, гидрофильно-липофильный баланс полимерного продукта можно регулировать соответствующим выбором реакционных параметров, что позволяет определять число карбоксильных группы, которые поступают в реакцию, и полученный продукт можно очищать по обычной технологии.
Реагенты формулы X можно получать, например, реакцией альдегида или кетона формулы R1-CO-R2 с галоидангидридом кислоты или галоидформиатным сложным эфиром формулы R3a-(O)n-CO-X, например в присутствии такого катализатора, как хлористый цинк или пиридин.
Способ (A) можно также осуществлять, например, реакцией полимера, молекулы которого содержат такие функциональные группы, как эпоксигруппы, с реагентом, молекулы которого содержат желаемые липофильные сложные метилендиэфирные группы и концевые группы, способные вступать в реакцию с упомянутыми функциональными группами. К концевым группам, вступающим в реакцию с эпоксидными группами, относятся амино-, гидроксильные и карбоксильные группы. Подобным же образом эти последние группы могут содержаться в молекулах исходного полимера, а концевые эпоксидные группы могут содержаться в молекулах реагента.
В том случае, когда эти продукты предназначены для внутривенного применения, молекулярная масса предпочтительных полимерных исходных материалов, используемых при осуществлении способа (A), обычно не должна превышать приблизительно 40000. В тех же случаях, когда такие продукты применяют для других целей, их молекулярная масса не имеет решающего значения.
Способ (B) можно осуществлять с использованием любых мономеров, которые можно полимеризовать или сополимеризовать с получением неструктурированных полимеров и у которых молекулы содержат по одному или несколько заместителей, которые во время полимеризации не осаждаются и которые могут быть подвергнуты обработке перед полимеризацией с введением в молекулы желаемых липофильных сложных метилендиэфирных групп. С этой целью можно осуществлять технологию свободнорадикальной, конденсационной или ионной полимеризации.
Свободнорадикальную полимеризацию можно, например, проводить с использованием мономеров, молекулы которых содержат карбоксильные группы, в частности, акриловой кислоты или метакриловой кислоты, обработанной для получения производного реакцией с соединением формулы X, или с использованием мономеров, молекулы которых содержат гидроксильные группы, в частности 2-оксиэтилакрилата или N-(2-оксипропил)-метакриламида, обработанного с получением производного реакцией с соединением формулы IX. По другому варианту можно провести реакцию мономеров, молекулы которых содержат гидроксильные группы, с соединением формулы XI
X-CO-O-C(R1R2)-X,
где значения символов R1, R2 и X определены выше,
с последующей реакцией полученного продукта с соответствующей солью карбоновой кислоты формулы R3a-COOH.
Свободнорадикальную полимеризацию можно также проводить с использованием сложных винилкарбонатных эфиров формулы XII
CH2=CH-O-CO-O-C(R1R2)-O-CO(O)m- R3a,
где значения символов n, R1, R2 и R3a определены выше.
Такие мономеры, у которых, например, n = 0, могут быть получены реакцией винилхлорформиата с альдегидом или кетоном формулы R1R2 C=0 в присутствии каталитического количества основания, в частности пиридина или кислот в результате чего получают возможно замещенный хлорметилвинилкарбанат формулы XIII
CH2=CH-O-CO-O-C(R1R2)-CI
где значения символов R1 и R2 определены выше,
после чего проводят реакцию, например, с соответствующей солью карбоновой кислоты формулы R3a-COOH, предпочтительнее в присутствии каталитического количества соответствующего краун-эфира. Необходимо принять во внимание, что формально соединения XII можно рассматривать как "виниловый спирт", обработанный с получением производного соединения формулы IX. Полученные из них полимеры соответственно должны быть энзимно биоразлагаемыми до поливинилового спирта.
Можно осуществлять обычную технологию блочной в растворе, эмульсионной или суспензионной полимеризации. Молекулярную массу полимерного продукта, которая по предпочтительному варианту воплощения настоящего изобретения не должна превышать приблизительно 40000, можно регулировать с помощью регуляторов степени полимеризации, в частности меркаптонов, из которых растущая полимерная цепь способна извлекать протон, что приводит к завершению цепи и образованию серного радикала, который способен инициировать новую полимерную цепь; таким образом, рост молекулярной массы полимера управляется типом и концентрацией регулятора степени полимеризации.
Соответствующие виниловые мономеры, молекулы которых содержат, например, карбонильную группу вблизи виниловой группы, как это имеет место в молекулах акриловых или метакриловых сложных эфиров, например полученные согласно вышеизложенному, можно также подвергать полимеризации согласно ионной технологии (как анионной, так и катионной); такая технология особенно приемлема для получения полимера со строго определенной молекулярной массой, в особенности в сравнении с низкомолекулярными материалами.
Конденсационную полимеризацию можно проводить с использованием широкого диапазона мономеров с соответствующими функциональными группами, в частности таких, которые отвечают общим формулам XIV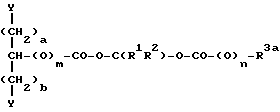
и XV
где значения символов R1, R2, R3a, m и n определены выше;
- реакционноспособная группа, в частности карбокси-, гидроксильная или эпоксигруппа, например 2,3-эпоксипропилоксигруппа;
каждым из символов a, b и c обозначен 0 или небольшое целое положительное число, равное 1, 2 или 3.
В формуле XV группы R1, R2 и R3, а также значения m и n в двух боковых цепях могут быть как идентичными, так и различными.
Такие мономеры могут быть использованы в ходе проведения обычных реакций конденсации с соответствующими реагентами, в частности с дикарбоновыми кислотами, двухатомными спиртами, диаминами, дихлорангидридами кислот, диизоцианатами и диэпоксисоединениями, с получением таких полимеров, как сложные полиэфиры, полиамиды, полиуретаны и эпоксидные полимеры. Молекулярную массу получаемого полимерного продукта можно регулировать выбором соответствующей продолжительности реакций, температуры и/или с использованием монофункциональных агентов обрыва цепи.
В тех случаях, когда это приемлемо, полимеры могут быть получены с осуществлением технологии эмульсионной полимеризации. Она может оказаться особенно ценной тогда, например, когда желательно получить полимеры в форме монодисперсных частиц. Способы эмульсионной полимеризации с получением частиц, в особенности монодисперсных частиц, изложены в описаниях к европейским заявкам на патент EP-A-0003905, EP-A-0091453, EP-A-0010986 и EP-A-0106873.
Желательно, когда молекулярная масса полимеров, используемых в контрастных агентах в соответствии с настоящим изобретением, является относительно малой, например не превышает 40000, поскольку это может способствовать как биоразложению, так и диспергированию продуктов разложения. Таким образом, термин "полимер", который в данном описании использован в связи с настоящим изобретением, следует понимать как охватывающий низкомолекулярные материалы, в частности олигомеры.
Необходимо учитывать, что поскольку полимеры предназначены для использования в медицинских целях, они должны образовывать нетоксичные, физиологически приемлемые продукты разложения. Таким образом, группы R1, R2, R3 и R3a в звеньях формул III и VI следует выбирать с учетом этого требования и необходимости в том, чтобы продукты разложения были легко диспергируемыми. Углекислый газ, который выделяется за счет отщепления карбонатных сложноэфирных групп, обычно является физиологически и приемлемым.
Контрастные агенты настоящего изобретения могут быть использованы в соответствии с различными технологиями получения диагностических изобретений, включая сюда ультразвуковое, MP- и X- лучевое изображение. Их применение при получении диагностических ультразвуковых и MP- изображений, например, в качестве чувствительных контрастных агентов, составляет предпочтительные варианты воплощения настоящего изобретения.
В системе контрастных агентов настоящего изобретения может быть использован любой биологически совместимый газ, в частности воздух, азот, кислород, водород, окись азота, двуокись углерода, гелий, аргон, гексафторид серы и низкомолекулярные, возможно фторсодержащие углеводороды, в частности метан, ацетилен и четырехфтористый углерод. Этот газ может находится в свободном состоянии внутри микропузырьков или же может быть связан или захвачен содержащимися в них веществами. Используемый в данном описании термин "газ" служит для обозначения любых веществ, которые при температуре 37oC находятся в газообразном состоянии.
Газовые предшественники охватывают карбонаты и бикарбонаты, например, бикарбонат натрия или алюминия и аминомалонатные сложные эфиры.
В случае применения в ультразвуковой технике, в частности, в эхокардиографии, с целью обеспечить свободное прохождение через легочную систему и достичь резонанса с предпочтительной частотой получения изображения, равной приблизительно 0,1-15,0 мГц, может оказаться удобным применение микропузырьков, средний размер которых составляет 0,1-10,0 мкм, например 1-7 мкм. Контрастные агенты настоящего изобретения могут быть получены целым рядом путей, обычной эмульсионной технологией, которая известна в полимерной области техники. Так, например, техника микроинкапсулирования для приготовления микрокапсул, стенки или оболочки, которых состоит из полимерного материала, описана в литературе, в частности в работе "Microencapsuecition and Related Drug Processes" П.Д. Дизи, Марсел Деккер инк., Нью-Йорк, 1984 г.
Один полезный способ соответствует технологии межфазного осаждения, которая изложена в описании к вышеупомянутой заявке на европейский патент EP-A-0458745 и включает в себя растворение или суспендирование полимерного материала, образующего стенки, в не смешивающимся с водой низкокипящем органическом растворителе (например, в алифатическом или циклоалифатическом углеводороде или перфторуглероде, в частности, в таком, молекулы которого содержат до 10 углеродных атомов, или же в соответствующем простом эфире, сложном эфире или в другом липофильном растворителе), эмульгирование (например, путем перемешивания с высокой степенью сдвига) приготовленного раствора или суспензии в водной фазе (предпочтительнее содержащей поверхностно-активное вещество для стабилизации образующейся эмульсии типа масло - в - воде) и последующее удаление органической фазы (например, выпариванием или лиофилизацией, предпочтительнее в атмосфере того газа, который желательно ввести), благодаря чему на межфазной границе между водной и органической фазами полимер образует оболочку.
Размер образующихся таким образом микрочастиц (микропузырьков регулируют регулированием скорости перемешивания во время эмульгирования, причем повышение скорости перемешивания проявляет тенденцию к образованию частиц меньшего размера. На размер влияние оказывает также природа поверхностно-активного вещества, которое можно выбрать, например, из жирных кислот (например, из прямоцепочечных насыщенных или ненасыщенных кислот, молекулы которых содержат по 10-20 углеродных атомов) и углеводов, а также их триглицеридных сложных эфиров, фосфолипидов (например, из лецитина), белков (например, из сывороточного альбумина человека), полиоксиэтилена и блоксополимеров, молекулы которых содержат гидрофильные и гидрофобные блоки (например, из полиоксиэтилен-полиоксипропиленовых блок-сополимеров, таких как продукты "Pluronies").
Такие эмульгаторы обычно используют в количествах 1-10% (вес/объем) относительно водной фазы. В полимер можно также вводить обычные добавки. Так, например, для модификации эластичности и/или полярности оболочки можно вводить такие материалы, как полиэтиленгликоли. По другому варианту полимерные частицы можно покрывать, например, полиэтиленгликолевыми звеньями, белками или полисахаридами с целью модификации их тенденции к агрегированию и/или их биологических свойств.
Пористость оболочки и, следовательно, ее проницаемость для растворителей, растворенных веществ и газов, также можно регулировать, причем она зависит от разности точек кипения у летучей органической фазы и окружающей водной фазы. Так, например, пористость мембраны с увеличением разницы между указанными точками кипения повышается.
По другому варианту полимер можно растворять в соответствующем органическом растворителе (например, в дихлориде метилена, диметил сульфоксиде, тетрагидрофуране или диметилформамиде), а затем диспергировать (например, с помощью высокоскоростной мешалки) в водной фазе (предпочтительнее содержащей такой полимерный материал, как поливиниловый спирт или полоксамер) таким образом, чтобы вызвать осаждение частиц полимерного материала, который можно собрать и липофилизировать с получением пористого полимера в форме частиц, соответствующего настоящему изобретению. Такая технология предлагается в вышеупомянутом описании к заявке на европейский патент EP-A-0458079. Варианты приготовления микрочастиц охватывают инжекцию органического раствора полимера, предпочтительнее совместно с физиологически приемлемым стабилизатором, в частности с оксипропилцеллюлозой, в жидкий азот. По другому варианту такой полимер можно растворять в соответствующем органическом растворителе (например, в хлористом метилене или тетрагидрофуране) с последующей распылительной сушкой раствора или эмульсии типа масло - в - воде или вода - в - масле этого органического раствора полимера в водной фазе.
Микрочастицы в соответствии с настоящим изобретением можно также приготовить с применением технологии коацервации или двойной эмульсии, как это изложено, например, в описании к вышеупомянутой заявке на международный патент WO 91/12823. Так, например, водную фазу, содержащую водорастворимый полимер (в дальнейшем называемый "форполимером"), можно эмульгировать с использованием летучего органического растворителя (например, алифатического или циклоалифатического углеводорода или перфторуглерода, молекулы которого содержат до 10 углеродных атомов каждая) до образования эмульсии типа масло - в - воде. Добавление коацервационного агента (например, дегидратационного агента, в частности изопропанола или такой соли, как сульфат натрия) инициирует концентрирование "формолимера" вокруг масляных капелек, вследствие чего с целью подавить агломерирование микрочастиц, которые образуются за счет структурирования формолимера при введении биоразлагаемых звеньев формулы II, желательно добавлять поверхностно-активное вещество, в результате чего образуются микрочастицы водонерастворимого пористого полимера, которые можно высушить лиофилизацией. Соответствующие технология структурирования и реагенты для структурирования водорастворимых "формолимеров", в частности полиакриламидов, подробно представлены в описании к вышеупомянутой заявке на международный патент WO 92/04392, поданной авторами настоящего изобретения.
Контрастные агенты настоящего изобретения можно хранить и транспортировать в сухом виде, в котором они обычно сохраняют стойкость в течение неопределенного промежутка времени, и смешивать с соответствующим жидким носителем (например, стерилизованной водой для инъекций, физиологическим солевым раствором или фосфатным буфером) перед введением в организм. Таким путем по желанию можно варьировать концентрацию вводимого инъекцией или каким-либо другим путем контрастного агента в зависимости от конкретной природы области применения. Их можно также хранить в форме суспензии в таких же носителях, в особенности в случае микропузырьков, когда пористость инкапсулирующей полимерной оболочки относительно низка, а сама она практически совершенно стабильна в водной среде в отсутствии эстеразных энзимов.
Существо настоящего изобретения проиллюстрировано далее с помощью нижеследующих неорганических примеров.
Общая информация.
Для удаления стабилизатора метакриловую кислоту перегоняли под очень глубоким вакуумом.
Тепловой инициатор 2,2'-азобисизобутиронитрил (АИБН) очищали перекристаллизацией из метаноле.
Все реакции проводили в атмосфере азота.
Хроматография с исключением размеров (ХИР):
Насос: насос Кнойера 64 для высокоэффективной жидкостной хроматографии
Детектор: последовательно смонтированные PL-гелевые колонки фирмы "Полимер лабораториз"; размером пор - 104 500
500  и 100
и 100 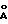 , размеры частиц - 5 мкм, длина соответственно 30, 30 и 60 см.
, размеры частиц - 5 мкм, длина соответственно 30, 30 и 60 см.
Растворитель: ТГФ;
Калибровка: полистирольные стандарты (фирмы "Полимер лабораториз");
Внутренний стандарт степени истечения: толуол;
Программное обеспечение: программное обеспечение ГПХ/ХИР фирмы "Полимер лабораториз", версия 5.10;
Mw - средневесовая молекулярная масса;
Mn - среднечисленная молекулярная масса;
Mw/Mn: полидисперсность;
Mp: молекулярная масса при максимальной высоте детектора.
Список аббревиатур:
Tc: температура стеклования;
ТБА-ОН: тетрабутиламмонийгидроксид;
ТБА: тетрабутиламмоний;
АИБН: 2,2-азобисизобутиронитрил;
SO2Cl2: хлористый сульфирил;
EtSCl этансульфенилхлорид;
ДДУ: 1,8 - диазадицикло(5.4.0)унден-7-ен-(1;5:-5);
MgSO4: сульфат магния;
ТГФ: тетрагидрофуран;
ДМФ: N,N-диметилформамид;
САЧ: сывороточный альбумин человека.
Пример 1. Получение полупродуктов.
a) Метилендиметакрилат.
40,00 мл 1,00 М раствора гидрата окиси калия при температуре 0oC добавили к 3,44 г (40,00 ммоль) метакриловой кислоты и раствор высушили вымораживанием в течение 16 ч. Затем добавили 230 мл, сухого диметилформамида и суспензию нагрели до температуры 60oC в атмосфере сухого азота. В течение 10 мин двумя порциями добавили 1,61 мл (20,00 ммоль) дииодметана и реакционную смесь оставили стоять на 4 дня при температуре 60oC. Под пониженным давлением (0,05 мм рт.ст.) удалили растворитель, после чего добавляли 140 мл диэтилового эфира, 50 мл насыщенного водного раствора бикарбоната натрия и 50 мл воды. Водный слой подвергли экстракционной обработке 6 порциями по 60 мл диэтилового эфира и объединенные эфирные экстракты промыли 4 порциями по 50 мл воды, высушили над сульфатом магния и выпаривали с получением 2,63 г. /72%-ный выход/ соединения, указанного в заголовке.
1H-ЯМР - спектрограмма (60 мгГц, CDCl3 δ ): 1,97 (2х метил, м.,), 5,63 (2•H-C; м.), 5,88 (CH2, с.), 6,18 (2•H-C; м.).
ИК - спектрограмма (пленки, см-1): 2987 (сл.,), 2962 (сл.,), 2930 (сл., ), 1732 (сн.), 1638 (сл.), 1454 (сл.), 1315 (сл.), 1295 (см.,), 1158 (сл.), 1100 (сн.), 1012 (ср.), 989 (ср.).
b) Метилен-бис-(16-оксигексадеканоат).
I. 16-трифенилметоксигексадекановая кислота.
Раствор 1,36 г (5,00 ммоль) 16-оксигексадекановой кислоты, 1,53 г (5,50 ммоль) трифенилметилхлорида, 1,25 мл триэтиламина и 10,03 г (0,25 ммол). 4-диметиламинопиридина перемешивали при комнатной температуре в атмосфере азота в течение ночи в сухом диметилформамиде. По истечении 16 ч пемешивания в смесь воды со льдом вылили коричневый мутный раствор и подвергали смесь экстракционной обработке 5 порциями по 50 мл дихлорметана. Органическую фазу промыли 2 порциями по 100 мл насыщенного раствора хлористого аммония, 2 порциями по 100 мл воды и высушили над сульфатом магния. Под пониженным давлением удалили растворитель и продукт очистили тонкослойной хроматографической обработкой в колонке с двуокисью кремния с использованием смеси дихлорметана с метанолом в соотношении 20:1 в качестве элюента, в результате чего в виде маслоподобного продукта получили 0,41 г соединения, указанного в заголовке.
13C-ЯМР-спектрограмма (75 мгГц, CDCl3 δ ): 24,9, 25,7, 26,3, 29,2, 29,5, 29,6, 29,7, 30, 32,8; 34,1; 62,9; 63,7; 86,2, 144,5; 177,2.
Масс-спектрограмма (Cl): 515/M+H/.
II. Цезиевая соль 16-трифенилметоксигексадекановой кислоты.
0,16 мл ГМ водного раствора карбоната цезия по каплям добавляли в раствор 0,16 г (0,31 ммоль). 16-трифенилметоксигексадекановой кислоты из вышеприведенной части Ib(I) примера в 10 мл тетрагидрофурана до момента достижения величины pH приблизительно 8, после чего под пониженным давлением удалили растворитель и остаток высушили в вакууме в течение 2 ч. Маслоподобный полукристаллический остаток диспергировали в 10 мл диметилформамида и выпарили досуха в вакууме.
III. Метилен-бис-(16-трифенилметоксигексадеканоат).
0,04 г (0,16 ммоль) диодметана добавляли в суспензию цезиевой соли 16-трифенилметоксигексадекановой кислоты (0,31 ммоль) из вышеприведенной части IB(II) примера в 10 мл сухого диметилформамида. Реакционную смесь выдерживали при температуре 60oC в течение 2 дней в токе азота. Растворитель удалили в вакууме и продукт очистили тонкослойной хроматографической обработкой 2 порциями по 5 см в колонке с двуокисью кремния с использованием хлороформа в качестве элюента, в результате чего в виде коричневого маслоподобного продукта получили 0,10 г соединения, указанного в заголовке.
13C-ЯМР-спектрограмма (75 мгГц, CDCl3, δ\): 24,6; 26,3; 29,0; 29,2, 29,4, 29,5, 29,6, 29,7, 30,0, 34,0, 63,7, 79,0, 86,2, 126,7, 127,2, 127,6, 127,9, 128,7, 144,5, 172,5
IV. Метилен-бис-(16-оксигексадеканоат).
0,07 г (0,07 ммоль) метилен-бис-(16-трифенилметоксигексадеканоата) из вышеприведенной части Ib(III) примера растворили в 8 мл ледяной уксусной кислоты и нагрели до температуры 55oC. За ходом реакции следили тонкослойной хроматографией. По истечении 10 ч реакционную смесь вылили в лед и сырой продукт отфильтровали, промыли водным раствором бикарбоната натрия и водой и высушили под пониженным давлением. Далее этот продукт очистили тонкослойной хроматографической обработкой в колонке с двуокисью кремния, используя смесь хлороформа с метанолом в соотношении 20:1 в качестве элюента, в результате чего в виде белого твердого вещества получили соединение, указанное в заголовке.
1H-ЯМР-спектрограмма (300 мгГц, CDCl3, δ/ ) : 1,2 - 1,4 (м., 44H), 1,5 - 1,6 (м., 8H), 2,35 (т., 4H), 3,65 (т., 4H), 5,75 (с., 2H).
aa) Метилен-бис-(12-оксидодеканоат).
2,0 ммоль ДБУ добавили в раствор 12-оксидодекановой кислоты в 2 мл ДМФ. Раствор перемешивали в течение 5 мин с последующим добавлением в него 1,0 ммоль иодистого метила и раствор перемешивали в течение 12 ч при температуре 60oC. Затем под пониженным давлением удалили ДМФ и остаточный материал растворили в 50 мл хлороформа, промыли 3 порциями по 20 мл 10%-ного раствора карбоната калия, высушили над сульфатом магния и выпарили. Сырой продукт очистили тонкослойной хроматографической обработкой на силикагеле, используя для элюирования смесь хлороформа с метанолом в соотношении 95:5 (выход - 75%).
1H-ЯМР-спектрограмма (CDCl3 δ ): 1,20 - 1,40 (м., 28H), 1,50 - 1,68 (м., 10H), 2,35 (т.н. 7,5 Гц, 4H), 3,63 (т., 6,6 Гц, 4H), 5,74 (с., 2H).
13C-ЯМР-спектрограмма (CDCl3, δ ): 24,62, 25,75; 28,98; 29,19; 29,40; 29,42; 29,49; 29,56; 32,80; 33,99; 63,01; 79,06; 172,53.
Масс-спектрограмма "E1/: 445 (M+1, 100).
d) Метилен-бис-(10-оксидеканоат).
4,24 г (0,027 моль) ДДУ добавили в 5,0 г (0,027 моль) 10-оксидодекановой кислоты в 100 мл ДМФ. По истечении 5 мин перемешивания добавили 4,09 г (0,014 моль) диидметана и смесь оставили перемешиваться при комнатной температуре в течение 3 дней. Под пониженным давлением выпарили ДМФ и остаток растворили добавлением 100 мл хлороформа и 50 мл воды. После разделения на фазы водный слой подвергли экстракционной обработке 3 порциями по 75 мл хлороформа и объединенную органическую фазу высушили над сульфатом магния. Растворитель удалили под пониженным давлением и тонкослойной хроматографической обработкой получили 2,98 г (выход - 54,9%) продукта, указанного в заголовке.
1H-ЯМР (60 мгГц, CDCI3, δ ): 1,30 - 1,80 (м., 28H., CH2), 2,35 (м., 4H, CH2CO), 3,65 (м., 6H, 2•CH2O + 2•OH), 5,75 (с., 2H, OCH2O-).
e) Бис-(хлоркарбонилоксиметил)терефталат.
I. Бис-(этилтиокарбонилоксиметил)-терефталат.
3,24 г (0,029 моль) трет-бутоксида калия добавили в раствор 2,40 г (0,014 моль) терефталевой кислоты в 100 мл ДМФ. В приготовленную суспензию добавили 4,50 г (0,028 моль) о-хлорметил-этилкарбонотиоата1. Затем добавили 0,23 г (0,87 ммоль) продукт 18 - краун-6 и реакционную смесь оставили перемешиваться при комнатной температуре в течение 4 дней. Далее реакционную смесь отфильтровали и под пониженным давлением удалили растворитель. Остаток очистили тонкослойной хроматографической обработкой (двуокись кремния/хлороформ), в результате чего получили 3,38 г (62%-ный выход) продукта, указанного в заголовке.
1H-ЯМР-спектрограмма (60 мгГц, CDCl3 δ ): 1,30 (т., 6H, CH3CH3), 2,95 (к., 4H, CH3CH2), 5,80 (с., 4H, OCH2O), 8,20 (с., 4H, Ph).
II. Бис-(хлоркарбонилоксиметил)-терефталат.
При температуре 0-5oC с перемешиванием в течение 15 мин 0,73 г (0,0054 моль) хлористого тионила добавили к 1,02 г (0,0054 моль) бис-(этилтиокарбонилоксиметил)-терефталата из вышеприведенной части Ie(I) примера, после чего перемешивание продолжали в течение 45 мин при комнатной температуре. В результате выпаривания EtSCl при комнатной температуре под остаточным давлением 0,1 мм рт.ст. получили светло-желтые кристаллы. Выход - 0,80 г (90%-ный).
1H-ЯМР-спектрограмма (60 мгГц, CDCl3, δ ): 5,76 (с., 4H, OCH2O), 8,20 (с., 4H, Ph).
f) Метилен-бис-(4-оксиметилбензоат).
9,90 г (0,065 моль) ДДУ добавили в 9,89 г (0,065 моль) 4-оксиметилбензойной кислоты в 325 мл ДМФ. По истечении 5 мин перемешивания добавили 8,705 г (0,035 моль) диидметана и смесь оставили перемешиваться при комнатной температуре в течение 3 дней. Под пониженным давлением выпарили ДМФ и остаток растворили добавлением 100 мл хлороформа и 50 мл воды. После разделения на фазы водный слой подвергали экстракционной обработке 3 порциями по 75 мл хлороформа и объединенную органическую фазу высушили над сульфатом магния. Под пониженным давлением испарили растворитель, получив 3,0 г (27%-ный выход) продукта, указанного в заголовке.
1H-ЯМР-спектрограмма (60 мгГц, CDCl3, δ ): 4,7 (с., 4H., HO-CH2-Ph), 6,62 (с., 2H, O-CH2-O), 7,4-8,2 (м., 8H, Ph).
k) Общая процедура для хлорметилкарбонатов.
В раствор хлорметилхлорформиата и указанного спирта в 200 мл хлористого метилена при температуре 0oC добавили пиридин. По истечении 20 мин выдержки при температуре 0oC и 21 ч при температуре 25oC реакционную смесь промыли водным раствором соляной кислоты (10 мл, 1 М), 10 мл водного насыщенного раствора бикарбоната натрия и 10 мл воды. Под пониженным давлением после сушки над сульфатом магния удалили растворитель, получив сырой хлорметилкарбонат.
g) Метилхлорметилкарбонат.
Указанное соединение получили из хлорметилхлорформиата и метанола.
1H-ЯМР-спектрограмма (60 мгГц, CDCl3, δ ): 3,98 (с., 3H, OCH3), 5,85 (с. , 2H, CH2Cl).
h) Этилхлорметилкарбонат.
Это соединение получили из хлорметилхлорформиата и этанола.
1H-ЯМР-спектрограмма (60 мгГц, CDCl3, δ ): 1,25 (т., 3H, CH3), 4,25 (к., 2H, CH2), 5,70 (с., 2H, OCH2Cl).
i) Бутилхлорметилкарбонат.
Соединение получили из хлорметилхлорформиата и бутанола.
1H-ЯМР-спектрограмма (60 мгГц, CDCl3, δ ): 0,86 (м., 3H, CH3CH), 1,40 (м., 4H, CH2CH2), 4,15 (т., 2H, CH2-O), 5,63 (с., 2H, OCH2Cl).
j) Децилхлорметилкарбонат.
Это соединение получили из хлорметилхлорформиата и децилового спирта.
1H-ЯМР-спектрограмма (60 мгГц, CDCl3, δ ): 0,90-1,50 (м., 19H, CH3- и CH2), 4,20 (м., 2H, CH2O), 5,75 (с., 2H, OCH2Cl).
k) Бензилхлорметилкарбонат.
Это соединение получили из хлорметилхлорформиата и бензильного спирта.
1H-ЯМР-спектрограмма (60 мгГц, CDCl3, δ ): 5,20 (с., 2H, PhCH2O), 5,70 (с., 2H, ClCH2O), 7,32 (с., 5H, Ph).
I)-p) Общая процедура для метакрилоилоксиметилкарбонатов.
В раствор метакриловой кислоты в 200 мл ДМФ добавили трет.бутоксид калия. В образовавшуюся суспензию добавили хлорметилкарбонат из вышеприведенной части Igk примера. Затем добавили продукт 18-краун-6 и реакционную смесь оставили перемешиваться при комнатной температуре в течение 24 ч. Далее реакционную смесь профильтровали и под пониженным давлением удалили растворитель. Остаток растворили в 30 мл хлороформа и промыли 10 мл насыщенного водного раствора бикарбоната натрия и 20 мл воды. Органическую фазу высушили над сульфатом магния и под пониженным давлением удалили растворитель (см. табл.2).
l) Метилметакрилоилоксиметилкарбонат.
Это соединение получили из метилхлорметилкарбоната и метакрилата калия.
ИК-спектрограмма (бромид калия): 1772 (C=O, сильн.), 1737 (C=O, сильн.), 1635 (C=C, сильн.) см-1.
1H-ЯМР-спектрограмма (300 мгГц, CDCl3, δ ): 1,91 (с., 3H, CH3C=), 3,79 (с. , 3H, CH3O), 5,64 (м., 1H, CH2=), 5,80 (с., 2H, -OCH2O-), 6,16 (м., 1H, CH2O).
13C-ЯМР-спектрограмма (75 мгГц, CDCl3, δ ): 17,95 (CH3C), 55,13 (CH3O), 82,18 (-OCH2O-), 127,52 (CH2), 135,02 (CO), 154,44 (C=O), 165,46 (C=O).
m) Эталметакрилоилоксиметилкарбонат.
Это соединение получили из этилхлорметилкарбоната и метакрилата калия.
ИК-спектрограмма (бромид калия): 1772 (C:O, сильн.), 1736 (C:O, сильн.), 1635 (C:C, сильн.) см-1.
1H-ЯМР-спектрограмма (300 мгГц, CDCl3, δ): 1,27 (т., 3H, CH3), 1,92 (с., 3H, CH3C: ), 4,23 (кг., 2H, CH2), 5,66 (1H, CH2:), 5,80 (с., 2H, -OCH2O-), 6,20 (м., 1H, CH2:).
13C-ЯМР-спектрограмма (75 мгГц, CDCl3, δ): 15,70 (CH3CH2), 19,60 (CH3C: ), 65,72 (CH2), 83,05 - (-OCH2O-), 127,76 (CH2:), 135,40 (C:),
153,82 (C/O), 165,42 (C:O).
n) Бутилметакрилоилоксиметилкарбонат.
Данное соединение получили из бутилхлорметилкарбоната и метакрилата калия.
ИК-спектрограмма (бромид калия): 1772 (C:O, сильн.), 1736 (C:O, сильн.), 1635 (C:C, сильн.) см-1.
1H-ЯМР-спектрограмма (300 мгГц, CDCl3, δ) : 0,99 (т., 3H, CH3CH2), 1,47 (с. , 2H, CH2CH2), 1,72 (м., 2H, CH2CH2), 2,01 (c., 3H, CH3=), 4,25 (т., 2H, CH2-O), 5,74 (м., 1H, CH2=), 5,89 (c., 2H., -OCH2O), 6,27 (м., 1H, CH2=).
13C-ЯМР-спектрограмма (75 мгГц, CDCl3, δ) : 13,47 (CH3CH2), 17,97 (CH3CH2), 18,71 (CH3C=), 30,36 (CH2), 68,46 (CH2O), 82,07 (-OCH2O), 127,46 (CH2=), 135,05 (C=), 153,89 (C=O), 165,50 (C=O).
o) Децилметакрилоилоксиметилкарбонат.
Данное соединение получили из дециклохлорметилкарбоната и метакрилата калия.
ИК-спектрограмма (бромид калия): 1772 (C:O, сильн.), 1763 (C:O, сильн.), 1635 (C:C, сильн.) см-1.
1H-ЯМР - спектрограмма (300 мгГц, CDCl3, δ ): 0,90 (т., 3H, CH3), 1,28 (м. , 14H, CH2), 1,72 (м., 2H, CH2), 1,90 (с., 3H., CH3C-), 4,21 (т., 2H, CH2O), 5,70 (м., 1H, CH2=), 5,86 (с., 3H., - OCH2O-), 6,24 (м., 1H, CH2=);
13С-ЯМР - спектрограмма (75 мгГц, CDCl3, δ ): 13,78 (CH3), 17,76 (CH3C= ), 22,76-31,55 (CH2), 68,60 (CH2O), 81,90 (-OCH2O-), 127,28 (CH2:), 134,86 (C=-), 153,73 (C:O), 165,33 (C:O).
p) Бензилметакрилоилоксиметилкарбонат.
Указанное соединение получили из бензилхлорметилкарбоната и метакрилата калия.
ИК-спектрограмма (бромид калия): 3077 (фенил), 1772 (C:O, сильн.), 1763 (C:O, сильн.), 1635 (C:C, сильн.) см-1.
1H-ЯМР - спектрограмма (300 мгГц, CDCl3, δ ): 1,96 (с., 3H, CH3C), 5,22 (с. , 2H, CH2O), 5,70 (м., 1H, CH2-), 5,87 (с., 3H., -OCH2O-), 6,22 (м., 1H, CH2=), 7,39 (с., 5H, Ph).
13С-ЯМР - спектрограмма (75 мгГц, CDCl3, δ ): 17,96 (CH3C), 69,91 (CH2O), 82,03 (-OCH2O-), 127,41 (CH2=), 128,32 (Ph), 134,68 (C:), 153,58 (C: O), 165,28 (C:O).
g) Этил-1-метакрилоилоксиэтилкарбонат.
I. Этилхлорэтилкарбонат.
В раствор 23,16 г (0,162 моль) хлорэтилхлорформиата и 7,45 г (0,162 моль) этанола в 200 мл хлористого метилена при температуре 0oC добавили 12,82 г (0,162 моль) пиридина. По истечении 10 мин выдержки при температуре 0oC и 21 ч при температуре 25oC реакционную смесь промыли 100 мл водного раствора соляной кислоты, 100 мл насыщенного водного раствора бикарбоната натрия и 100 мл воды. После сушки над сульфатом магния под пониженным давлением удалили растворитель, получив в виде сырого промежуточного продукта 18,5 г (74%-ный выход) этилхлорэтилкарбоната.
1H-ЯМР - спектрограмма (60 мгГц, CDCl3 δ ) : 1,30 (т., 3H, CH3), 1,85 (д., 3H, CH3CH), 4,25 (к., 2H, CH2), 6,45 (к., 1H, CH).
II. Этил-1-метакрилоилоксилоксиэтилкарбонат.
3,70 г (0,033 моль) трет-бутоксида калия добавили в раствор 2,84 г. (0,033 моль) метакриловой кислоты в 100 мл ДМФ. В образовавшуюся суспензию добавили 5,08 г. (0,033 моль) этилхлориэтилкарбоната из вышеприведенной части 1 (I) примера. Затем добавили 0,61 г (2,3 ммоль) продукта 18-краун-6 и реакционную смесь оставили перемешиваться при комнатной температуре в течение 3 дней. Далее реакционную смесь профильтровали и под пониженным давлением удалили из нее растворитель. Остаток растворили в 100 мл хлороформа и промыли 50 мл насыщенного водного раствора бикарбоната натрия и 50 мл воды. Органическую фазу высушили над сульфатом магния и под пониженным давлением удалили растворитель. В результате тонкослойно хроматографической обработки получили 2,50 г (38%-ный выход) продукта, указанного в заготовке (в пересчете на выделенный исходный материал выход составил 75%).
1H-ЯМР - спектрограмма (60 мгГц, CDCl3, δ ): 1,30 (т., 3H, CH3 CH), 1,60 (д. , 3H, CH3CH), 2,00 (с., 3H, CH3C=) 4,20, (к., 2H, CH2), 5,70 (м., 1H, CH2=), 6,25 (к., 1H, - OCH(CH3)O-), 6,90 (м., 1H, CH2=).
r) Метакрилоилоксиметилбензоат
10,0 г (0,090 моль) трет-бутоксида калия добавили в раствор 7,75 г (0,090 моль) метакриловой кислоты в 300 мл ДМФ. В образовавшуюся суспензию добавили 15,0 г (0,088 моль) хлорметилбензоата2. Затем добавили 1,8 г (6,9 ммоль) продукта 18-крауз-6- и реакционную смесь оставили перемешиваться при комнатной температуре в течение 2 дней. Реакционную смесь профильтровали и растворитель удалили под пониженным давлением. Остаток растворили в 100 мл хлороформа и промыли 50 мл насыщенного водного раствора бикарбоната натрия и 50 мл воды. Органическую фазу высушили над сульфатом магния и под пониженным давлением удалили растворитель. В результате тонкослойной хроматографической обработки получили 15,9 г (82%-ный выход) продукта, указанного в заголовке продукта.
1H-ЯМР - спектрограмма (60 мгГц, CDCl3, δ ): 2,00 (с., 3H, CH3C:), 5,65 (м. , 1H, CH2:), 6,15 (с., 2H, OCH2O-), 6,35, (м., 1H, CH2:), 7,50 (м., 3H, Ph), 8,05 (м., 2H, Ph).
S) N-(2-ацетоксиметоксикарбонилоксипропил)-метакриламид.
I. N-(2-хлорметоксикарбонилоксипропил)-метакриламид
В раствор 2,86 г (20 ммоль) N-(2-оксипропил)-метакриламида3 и 1,90 г (25 ммоль) пиридина в 100 мл хлористого метилена при температуре 0oC добавили 3,87 г (30 ммоль) хлорметилхлороформиата в 120 мл хлористого метилена. По стечении 15 мин выдержки при температуре 0oC и 24 ч при температуре 25oC реакционную смесь промыли 5 порциями по 25 мл воды. После сушки над сульфатом магния под пониженным давлением отогнали растворитель. В результате тонкослойной хроматографической обработки (силикагель, хлороформ) получили 3,30 г (70%-ный выход) продукта, указанного в заготовке.
1H-ЯМР - спектрограмма (60 мгГЦ, CDCl3, δ ): 1,42 (д., 3H, 3H3-CH-O), 2,0 (м. , 3H, CH3C: ), 3,2 - 4,0 (м., 2H, NH-CH2-CH), 4,8 - 5,3 (м., 1H, CH3-CH-O), 5,3 (м., 1H, CH2:), 5,70 (м., 1H, CH2:), 5,7 (с., 2H, CH2Cl), 6,6 - 6,7 (широк. с., 1H, H).
II. N-(2-ацетоксиметоксикарбонилоксипропил)-метакриламид
Способ 1.
В перемешиваемый раствор 0,943 г (4 ммоль) N-(2-хлор-метоксикарбонилоксипропил)-метакриламида из вышеприведенной части IS (1) примера в 10 мл ТГФ при комнатной температуре добавили 30 мл. ТГФ-раствора 1,21 г (4 ммоль) ТБА-ацетата, приготовленного сушкой вымораживанием водного раствора эквимолярных количеств ТБА-ОН и уксусной кислоты. После перемешивания в течение 5 дней под пониженным давлением удалили растворитель. Остаток растворили в 50 мл хлороформа и промыли 5 порциями по 10 мл воды. Органическую фазу высушили над сульфатом магния и под пониженным давлением удалили растворитель. В результате тонкослойной хроматографической обработки (силикагель) этилацетат в соотношении 3: 4 получили 0,486 г (47%-ный выход) продукта, указанного в заголовке примера.
1H-ЯМР - спектрограмма (60 мгГц, CDCL3, δ ): 1,4 (д., 3H, CH3-CH-O), 2,0 (c. , 3H, CH3C-), 2,2 - (c., 3H, CH3C:O), 3,2 - 4,0 (м., 2H, NH-CH2-CH), 4,8 - 5,3 (м., 1H, CH3-CH-O), 5,3 (м., 1H, CH2:), 5,70 (м., 1H, CH2-), 5,8 (с., OCH2O), 6,1 - 6,7 (широк. с., 1H, NH).
Способ 2.
В раствор 0,430 г (3,0 ммоль) N-(2-оксипропил)-метакриламида3 и 0,285 г (3,6 ммоль) пиридина в 30 мл хлористого метилена при температуре 0oC добавили 0,500 г (3,3 ммоль) ацетоксиметилхлороформиата в 6 мл хлористого метилена. По истечении 10 мин выдержки при температуре 0oC и 3 дней при комнатной температуре реакционную смесь промыли 100 мл воды. После сушки над сульфатом магния под пониженным давлением удалили растворитель. В результате тонкослойной хроматографической обработки (силикагель, гексан/этилацетат в соотношении 3:4) получили 0,41 г (51%-ный выход) продукта, указанного в заголовке.
Данные ЯМР-спектрограммы хорошо согласовались с вышеприведенными.
t) N-[2-(1-ацетоксиэтоксикарбонилокси)-пропил]- метакриламид.
I N-[2-(1-хлорэтоксикарбонилокси)-пропил]-метакриламид
В раствор 3,15 г (22 ммоль) N-(2-оксипропил)-метакриламида3 и 2,088 г (26,4 ммоль) пиридина в 100 мл хлористого метилена при температуре 0oC добавили 4,718 г (33 ммоль) 1-хлорэтилхлорформиата в 20 мл хлористого метилена. По истечении 10 мин выдержки при температуре 0oC и 5,5 ч при температуре 25oC реакционную смесь промыли 5 порциями по 40 мл воды. После сушки над сульфатом магния под пониженным давлением удалили растворитель, получив 4,84 г (88%-ный выход) продукта, указанного в заголовке.
1H-ЯМР - спектрограмма (60 мгГц, CDCl3 δ ): 1,37 (д., 3H, CH2- CH 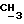 O-), 1,83 (д., 3H,
O-), 1,83 (д., 3H, 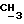 - CH-Cl), 1,97 (м., 3H,
- CH-Cl), 1,97 (м., 3H, 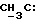 ), 3,3-3,6 (м., 2H, H - CH2-CH), 4,7 - 5,3 (м., 1H, CH2-
), 3,3-3,6 (м., 2H, H - CH2-CH), 4,7 - 5,3 (м., 1H, CH2- 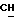 (CH3)-O), 5,3 (м., 1H, CH2:) 5,70 (м., 1H, CH2:) 6,0 - 6,6 (м., 2H, NH + -Cl
(CH3)-O), 5,3 (м., 1H, CH2:) 5,70 (м., 1H, CH2:) 6,0 - 6,6 (м., 2H, NH + -Cl 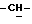 CH3).
CH3).
II. 2-(1-ацетоксиэтоксикарбонилокси)-пропил-метакриламид.
100 мл ТГФ-раствора 6,93 г (23 ммол). ТБА-ацетата, приготовленного сушкой вымораживанием водного раствора эквимолярных количеств ТБА-ОН и уксусной кислоты, добавили при комнатной температуре в перемешиваемый раствор 4,736 г (19 ммоль) N-[2-(1-хлорэтоксикарбонилокси)-пропил]-метакриламида из вышеприведенной части 1t(I) примера в 100 мл ТГФ. После перемешивания в течение 4 дней под пониженным давлением удалили растворитель. Остаток растворили в 100 мл хлороформа и промыли 5 порциями по 20 мл воды. Органическую фазу высушили над сульфатом магния и под пониженным давлением удалили растворитель. В результате тонкослойной хроматографической обработки (силикагель, гексан-этилацетат в соотношении 3:4) получили 1,29 г (25%-ный выход) продукт, указанного в заголовке.
1H-ЯМР-спектрограмма (60 мгГц, CDCl3 δ ) : 1,3 (д., 3H, CH2-CH(CH3)O), 1,5 (д., 3H, O-CH(CH3)O), 2,0 (м., 3H, CH3C:), 2,1 (с., 3H, CH3C:O), 3,3-3,6 (м. , 2H, NH-CH2-CH), 4,7 - 5,3 (м., 1H, CH2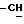 (CH3)-O), 5,4 (м., 1H, CH2: ), 5,7 (м. , 1H, CH2:), 6,1 - 6,6 (широк.с., 1H, NH), 6,6 - 6,9 (м., 1H, O
(CH3)-O), 5,4 (м., 1H, CH2: ), 5,7 (м. , 1H, CH2:), 6,1 - 6,6 (широк.с., 1H, NH), 6,6 - 6,9 (м., 1H, O 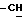 (CH3)O).
(CH3)O).
u) Метил-1-матакрилоилоксиэтилкарбонат.
I. Метил-1-хлорэтилкарбонат.
В раствор 35,74 г (0,25 моль) 1-хлорэтилхлорформиата и 8,00 г (0,25 моль) метанола в 300 мл хлористого метилена при температуре 0oC добавили 19,78 г (0,25 моль) пиридина. По истечение 10 мин выдержки при температуре 0oC и 2 дней при температуре 25oC реакционную смесь промыли 100 мл водного раствора соляной кислоты, 100 мл насыщенного водного раствора бикарбоната натрия и 100 мл воды. После сушки над сульфатом магния под пониженным давлением удалили растворитель, получив 25,5 г (74%-ный выход) метил-1-хлорэтилкарбоната в форме сырого промежуточного продукта.
1H-ЯМР - спектрограмма (60 мгГц, CDCl3, δ ): 1,85 (д., 3H, CH3CH), 3,80 (с., 3H, CH3O), 6,50 (к., 1H, CH).
II. Метил-1-метакрилоилоксиэтилкарбонат.
3,70 г (0,033 моль) трет-бутоксида калия добавили в раствор 2,84 г (0,033 моль) метакриловой кислоты в 100 мл ДМФ. В образовавшуюся суспензию добавили 4,55 г (0,033 моль) метил-1-хлорэтилкарбоната из эксперимента вышеприведенной части 1 и (I) примера. Затем добавили 0,61 г. (2,3 ммоль) продукта 18-крон-6 и реакционную смесь оставили перемешиваться при комнатной температуре в течение 3 дней. Далее реакционную смесь профильтровали и под пониженным давлением удалили растворитель. Остаток растворили в 100 мл хлороформа и промыли 50 мл насыщенного водного раствора бикарбоната натрия и 50 мл воды. Органическую фазу высушили над сульфатом магния и под пониженным давлением удалили растворитель. В результате тонкослойно хроматографической обработки получили 4,46 г (72%-ный выход) продукта, указанного в заголовке примера.
1H-ЯМР - спектрограмма (60 мгГц, CDCl3, δ ): 1,65 (д., 3H, CH3CH), 2,00 (с. , 3H, CH3C: ), 3,90 (с., 3H, CH3O), 5,65 (м., 1H, CH2:), 6,26 (м., 1H, CH2:), 6,90 (к., 1H, CHCH3).
v) Этиленди-(хлорметилкарбонат).
19,12 г (148,4 ммоль) хлорметилхлорформиата добавили в охлаждаемый льдом (0oC) раствор 2,8 мл (50 ммоль) этиленгликоля в 200 мл хлористого метилена. Затем добавили 8,70 г (110 ммоль) пиридина и реакционную смесь оставили перемешиваться в течение 15 мин при 0oC и в течение 6 ч при комнатной температуре. Затем реакционную смесь промыли 100 мл ГМ раствора соляной кислоты, 100 мл насыщенного водного раствора бикарбоната натрия и 100 мл воды и высушили над сульфатом магния. После этого растворитель выпарили, получив 11,88 г (выход 96,2%) продукта, указанного в заголовке.
1H-ЯМР - спектрограмма (60 мгГц, CDCl3, δ ): 4,48 (м., 4H, OCH2CH2O), 5,75 (с., 4H, OCH2Cl).
w) Ацетоксиметилхлорформиат.
I. O-ацетоксиметил-этилкарбонтиоат.
4,50 г (0,028 моль) O-хлорметил-S-этилкарбонтиоата1 в 20 мл ДМФ добавили в раствор 2,74 г (0,028 моль) ацетата калия в 100 мл ТГФ. Затем добавили 0,22 г (0,84 ммоль) продукта 18-краун-6 и реакционную смесь оставили перемешиваться при комнатной температуре в течение 3 дней. Далее реакционную смесь профильтровали и под пониженным давлением удалили растворитель. Остаток очистили тонкослойной хроматографической обработкой (силикагель, хлороформ), получив 4,23 г (85%-ный выход) продукта, указанного в заголовке.
1H-ЯМР - спектрограмма (60 мгГц, CDCl3, δ ): 1,30 (т., 3H, 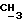 CH2), 2,20 (с., 3H, CH3C:O), 2,95 (к., 2H,
CH2), 2,20 (с., 3H, CH3C:O), 2,95 (к., 2H, 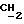 CH3), 5,80 (с., 2H, OCH2O).
CH3), 5,80 (с., 2H, OCH2O).
II. Ацетоксиметилхлорфомиат.
При температуре 0-5oC и перемешивании в течение 15 мин 2,43 г (0,018 моль) SO2Cl2 добавили к 3,15 г (0,019 моль) -о-ацетоксиметил-S-этилкарбонтиоата из эксперимента вышеприведенной части Iw(I) примера и последующим перемешиванием в течение 45 мин при комнатной температуре. В результате выпаривания EtSCl при комнатной температуре под остаточным давлением 11 мм рт. ст. получили бесцветную жидкость. Выход - 2,44 г (89%).
1H-ЯМР - спектрограмма (60 мгГц, CDCl3, δ ): 2,20 (с., 3H, CH3C:O), 5,76 (с., 2H, OCH2O).
x) Гексаметиленди-(хлорметилкарбонат).
19,12 г (148,5 ммоль) хлорметилхлорформиата при температуре 0oC 19,12 г (148,5 ммоль) хлорметилхлорформиата добавили в охлаждаемый льдом (0oC) раствор 5,90 г (50 ммоль) 1,6-гександиола в 200 мл хлористого метилена. Затем добавили 8,70 г (110 ммоль) пиридина и реакционную смесь оставили перемешиваться в течение 15 мин при температуре 0oC и 5 ч при комнатной температуре. Далее реакционную смесь промыли 100 мл ГМ раствора соляной кислоты 100 мл насыщенного водного раствора бикарбоната натрия, 100 мл воды и высушили над сульфатом магния. Удалили растворитель, получив 13,25 г (95%-ный выход) продукта, указанного в заголовке.
1H-ЯМР (60 мгГц, CDCl3 δ ): 1,20 - 2,00 (м., 8H,: (CH2)4), 4,22 (т., 4H, 2 • (CH2O)), 5,73 (с., 4H, 2 • OCH2Cl).
y) Метакрилоилоксиметилацетат.
5,0 г (0,045 моль) трет-бутоксида калия добавили в раствор 3,87 г (0,045 моль) метакриловой кислоты в 150 мл ДМФ. В образовавшуюся суспензию добавили 4,86 г (0,045 моль) хлорметилацетата3. Затем добавили 0,9 г (3,45 ммоль) продукта 18-крауон-6- и реакционную смесь оставили перемешиваться при комнатной температуре в течение 4 дней. Далее реакционную смесь профильтровали и под пониженным давлением удалили растворитель. Остаток растворили в 100 мл хлороформа и промыли 50 мл насыщенного водного раствора бикарбоната натрия и 50 мл воды. Органическую фазу высушили над сульфатом магния и под пониженным давлением удалили растворитель. В результате тонкослойной хроматографической обработки получили 5,19 г (75%-ный выход) продукта, указанного в заголовке примера.
1H-ЯМР - спектрограмма (60 мгГц, CDCl3 δ ) : 2,00 (с., 3H, CH3C=), 2,18 (с., 3H, CH3O=O), 5,70 (м., 1H, CH2:), 5,85 (с., 2H, -OCH2O-), 6,25 (м., 1H, CH2=).
z) Бутилакрилоилоксиметилкарбонат.
5,84 г (0,052 моль) трет. бутоксида калия добавили в раствор 4,47 г (0,045 моль) акриловой кислоты в 220 мл ДМФ. В образовавшуюся суспензию добавили 6,5 г (0,052 моль) бутилхлорметилкарбоната из эксперимента вышеприведенной части Ii примера в 150 мл ДМФ. Затем добавили 0,6 г продукта 18-краун-6 и реакционную смесь оставили перемешиваться при комнатной температуре в течение 2 дней. Далее реакционную смесь профильтровывали и под пониженным давлением удалили растворитель. Остаток растворили в 100 мл хлороформа и промыли 50 мл насыщенного водного раствора бикарбоната натрия и 50 мл воды. Органическую фазу высушили над сульфатом магния и под пониженным давлением удалили растворитель. В результате тонкослойной хроматографической обработки получили 4,57 г продукта, указанного в заголовке примера.
1H-ЯМР - спектрограмма (60 мгГц, CDCl3, δ ): 0,80 (т., 3H, 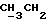 1,28 (м. , 2H, CH2), 1,60 (м., 2H, CH2), 4,15 (т., CH2O), 5,78 (с., 2H, OCH2O), 5,88 (дд., 1H, CH:), 6,1 (дд., 1H, CH2:), 6,45 (дд., 1H, CH2:
1,28 (м. , 2H, CH2), 1,60 (м., 2H, CH2), 4,15 (т., CH2O), 5,78 (с., 2H, OCH2O), 5,88 (дд., 1H, CH:), 6,1 (дд., 1H, CH2:), 6,45 (дд., 1H, CH2: 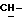 ).
).
aa) Дихлорангидрид 3,6,9-триоксаундекандикислоты.
2,0 ммоль 3,6,9-триоксаундександикислоты прокипятили с обратным холодильником в 1 мл хлористого тионила в течение 6 ч, после чего под пониженным давлением выпарили избыток хлористого тионила. Сырой продукт использовали на последующей стадии без очистки.
1H-ЯМР - спектрограмма (CDCl3, δ ): 3,64-3,68 (м., 4H), 3,76-3,82 (м., 4H), 4,49 - 4,51 (м., 4H).
13C-ЯМР - спектрограмма (CDCl3, δ ): 70,70; 71,29; 76,65; 172,03.
ab) 1-(7-бензилоксикарбонилгептансилокси)-этилдецилкарбонат
I. 1-бензилнонандикислота.
Раствор 25,0 г (0,13 моль) нонандикислоты в 550 мл бензола добавили к 0,71 г (3,72 ммоль) п-толуолсульфокислоты и нагрели до температуры 80oC. По каплям добавили 14,3 г (0,13 моль) бензилового спирта в 50 мл бензола. Затем реакционную смесь прокипятили с обратным холодильником в течение ночи. Из реакционной смеси удалили воду и ее собрали в ловушку Дина-Старка. По истечении 24 ч тонкослойный хроматографический анализ не позволял обнаружить никакого бензилового спирта. Реакционную смесь охладили до комнатной температуры, а затем в бане со льдом. Фильтрованием удалили выпавшую в осадок непрореагировавшую нонандикислоту. Фильтрат сконцентрировали досуха и остаток очистили в хроматографической колонке с использованием в качестве элюента смеси дихлорметана с метанолом в соотношении 10:1. Выход - 28%.
1H-ЯМР - спектрограмма (300 мгГц, CDCl3, δ ): 7,35 - 7,31 (м., Ar), 5,10 (c., ArCH2), 2,33 (т., CH2CO), 1,62 (м., 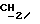 CH2CO), 1,29 (м., (CH2)3).
CH2CO), 1,29 (м., (CH2)3).
II. Цезий-1-бензилнонандиоат.
6,3 г (21,6 ммоль) 1-бензилнонандикислоты (из эксперимента выше приведенной части 1ac(I) примера суспендировали в 100 мл дистиллированной воды и нагревали массу до температуры 50oC. По каплям до величины pH, равной 7, добавили 3,5 г (10,8 ммоль) карбоната цезия. Лиофилизацией в течение 2 дней удалили воду. Выход - 95%.
III. 1-хлорэтилдецилкарбонат.
В перемешиваемый раствор 6,0 г (7,23 ммоль) деканола в 150 мл дихлорметана добавили 3,66 мл (45,6 ммоль) сухого пиридина. Этот раствор охладили в ледяной бане, а затем в него по каплям добавили 6,5 г (45,6 ммоль) 1-хлорэтилхлорформиата. Реакционную смесь для завершения реакции выдержали в течение ночи, затем разбавили дихлорэтаном и промыли 0,5 н. раствором соляной кислоты, дважды насыщенным раствором бикарбоната натрия и наконец дистиллированной водой. Раствор высушили над сульфатом магния, профильтровали через двуокись кремния и сконцентрировали досуха. Выход - 93%.
1H-ЯМР - спектрограмма (300 мгГц, CDCl3, δ ): 6,43 (к., CHCl), 4,19 (т., CH2O), 1,83 (д.,  ); 1,69 (
); 1,69 ( 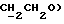 ); 1,40 - 1,22 (м., (CH2)8), 0,88 (т.,
); 1,40 - 1,22 (м., (CH2)8), 0,88 (т., 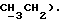 ).
).
IV. 1-(7-бензилоксикарбонилгептаноилокси)-этилдецилкарбонат.
5,0 г (12,2 ммоль) цезий-1-бензилнонандиоата (из эксперимента вышеприведенной части 1ас(II) примера) растворили в 150 мл ДМФ. В этот раствор добавили 3,25 г 1-хлорэтилдецилкарбоната (из эксперимента вышеприведенной части Iac(III) примера), а затем 125 мг (0,75 ммоль) иодистого калия. Реакцию провели при температуре 50oC в течение 3 дней. Под пониженным давлением удалили растворитель. Остаток суспендировали в дихлорметане и трижды промыли насыщенным раствором бикарбоната натрия и наконец дважды - водой. После сушки над сульфатом магния раствор выпаривали досуха. Продукт очистили хроматографической обработкой в колонке с использованием в качестве элюента смеси петролейный эфир) этилацетат в соотношении 12:1. Выход - 65%.
1H-ЯМР - спектрограмма (300 мгГц, CDCl3, δ ): 7,35 - 7,31 (м., Ar), 6,78 (к., 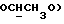 5,10 (с., ArCH2), 4,19 (т., CH2O), 2,33 (т., CH2CO).
5,10 (с., ArCH2), 4,19 (т., CH2O), 2,33 (т., CH2CO).
V. 1- (7-карбоксигептаноилокси)-этилдецилкарбонат.
В раствор 4,0 г (7,9 ммоль) 1-(7-бензилоксикарбонилгептаноилокси)-этилдецилкарбоната (из эксперимента вышеприведенной части 1ac(IV) примера) в 15 мл уксусной кислоты добавили каталитическое количество (150 мг) палладия на древесном угле. Смесь подвергли гидрогенизации водородом при комнатной температуре в течение 5 ч. Под пониженным давлением удалили уксусную кислоту. Остаток очистили хроматографической обработкой в колонке с использованием смеси н-гептан/этилацетат в соотношении 4:1 в качестве элюента. Выход - 52%.
1H-ЯМР - спектрограмма (300 мгГц, CDCl3, δ ): 6,76 (к.,  ), 4,19 (т., CH2O); 2,32 (т., CH2CO).
), 4,19 (т., CH2O); 2,32 (т., CH2CO).
13C-ЯМР - спектрограмма (300 мгГц, DMCO-d6, δ ): 174,67 (COOH); 171,30 (CH2COO), 152,68 (OCOO), 91,09 (OCHCH3).
ac) Нонилкарбонилоксиметилхлорфомиат.
(2) Деканоат калия.
Раствор 2,6 г (46,4 ммоль) гидрата окиси калия в 50 мл воды по каплям добавили в суспензию 8,0 г (46,4 ммоль) декановой кислоты в 300 мл воды при температуре 60oC до момента достижения величины pH, равной 7. Затем лиофилизацией удалили воду. Выход - 9,28 г (95%).
II. O-Нонилкарбонилоксиметил-s-этилкарбонтиоат.
4,79 г (0,031 моль) o-хлорметил-s-этилкарбонтиоата1 в 20 мл ДМА добавили в суспензию деканоата калия (из эксперимента вышеприведенной части 1ad (I) примера 6,5 г (0,031 моль) в 500 мл ДМА. Затем добавили 0,25 г (0,93 ммоль) продукта 18-краун-6 и реакционную смесь оставили перемешиваться при комнатной температуре в течение 22 ч. Далее реакционную смесь профильтровали и под пониженным давлением удалили растворитель. Остаток очистили тонкослойной хроматографической обработкой (силикагель, гексан/этилацетат в соотношении 30:1). Выход - 5,96 г (67%).
1H-ЯМР - спектрограмма (300 мгГц, CDCl3 δ : 0,88 (т., 3H, CH3) (CH2)8, 1,27 (м. , 12H, (CH2)6), 1,33 (т., 3H, 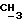 CH2S), 1,63 (м., 2H,
CH2S), 1,63 (м., 2H, 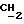 CH2=0), 2,37 (т., 2H, CH2C:O), 2,89 (к., 2H, CH2S), 5,81 (с., 2H, OCH2O).
CH2=0), 2,37 (т., 2H, CH2C:O), 2,89 (к., 2H, CH2S), 5,81 (с., 2H, OCH2O).
13C-ЯМР - спектрограмма (300 мгГц, CDCl3, δ ) 14,1; 14,8; 22,7; 24,6; 25,4; 29,0; 29,2; 29,3; 29,4; 31,9; 33,9; 80,2 (OCH2O), 170,7 (C:O), 172,2 (C:O).
III. Нонилкарбонилоксиметилхлорформиат.
1,17 г (8,65 ммоль) SO2Cl2 при температуре 0oC и с перемешиванием добавили к 2,10 г (7,22 ммоль) o-нонилкарбонилоксиметил-s-этил-карбонтиоата (из эксперимента вышеприведенной части 1ad (II) примера) с последующим перемешиванием при комнатной температуре в течение 17 ч. В результате выпаривания EtSCI при температуре 30oC под остаточным давлением 20 мм рт.ст. получили желтую жидкость. Выход - 1,62 г (85%).
1H-ЯМР - спектрограмма (300 мгГц, CDCl3, δ ): 0,88 (т., 3H, CH3), 1,27 (м., 12H, (CH2)6), 1,66 (м., 2H, CH2CH2C:O), 2,41 (т., 2H, CH2C O), 5,32 (с. , 2H, OCH2O).
13C-ЯМР - спектрограмма (300 мгГц, CDCl3 δ ) : 14,1; 22,7; 24,5; 29,0; 29,19; 29,24; 29,4; 31,9; 33,8; 83,3 (OCH2O), 150,1 (CIC:O), 171,7 (C:O).
dd) 1 - ацетокси-1-фенилметилвинилкарбонат.
I. 1-хлор-1-фенилметилвинилкарбонат.
3,0 г (0,028 моль) винилхлорформиата и 4,14 г (0,039 моль) растворили в 30 мл 1,2 - дихлорэтана и по каплям в перемешиваемый раствор добавили 0,1 г (1,28 моль) пиридина. Этот раствор перемешивали в течение 1 дня при температуре 80oC, промыли 25 воды и водную фазу подвергли обратной экстракционной обработке 25 мл хлористого метилена. Объединенные органические фазы высушили над сульфатом магния и сконцентрировали, получив 3,0 г (50%-ный выход) продукта, указанного в заголовке.
1H-ЯМР - спектрограмма (60 мгГц CDCl3 δ ) : 4,55 (дд, 1H, CH2:) 4,95 (дд, 1H, CH2:), 7,05 (дд., 1H, CH2: 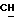 ), 7,25 (с., 1H, CH-Ph), 7,40 (м., 5H, ph).
), 7,25 (с., 1H, CH-Ph), 7,40 (м., 5H, ph).
II. 1 - ацетокси-1-фенилметилвинилкарбонат.
2,0 г (0,012 моль) ацетата серебра добавили в раствор 2,50 г (0,012 моль) 1-хлор-1-фенилметилвинилкарбоната в 60 мл ДМФ. Реакционную смесь оставили перемешиваться при комнатной температуре в течение 12 ч. Далее реакционную смесь профильтровали и под пониженным давлением удалили растворитель. Остаток очистили тонкослойной хроматографической обработкой (силикагель, хлористый метилен), получив 0,56 г (20%-ный выход) продукта, указанного в заголовке.
1H-ЯМР - спектрограмма (60 мгГц, CDCl3, δ ) : 2,24 (с., 3H, CH3C:C), 4,60 (дд. , 1H, CH2:), 4,95 (дд., 1H, CH2:) 7,00 (дд., 1H, CH:), 7,50 (м., 5H, Ph), 8,00 (с., 1H, - CH-Ph).
Бензосилоксиметилхлорформиат.
I. O-бензоилоксиметил-s-этилкарбонтиоат.
5,73 г (0,037 моль) o-хлорметил-s-этилкарбонтиоата1 в 20 мл ДМФ добавили в раствор 5,94 г (0,037 моль) бензоата калия, а затем добавили 0,485 г (1,85 ммоль) продукта 18-карун-6- в 130 мл ДМФ и реакционную смесь оставили перемешиваться при комнатной температуре в течение 24 ч. Под пониженным давлением удалили растворитель. Остаток растворили в 150 мл хлороформа и промыли 5 порциями по 20 мл воды, а затем высушили над сульфатом магния. Под пониженным давлением удалили растворитель и остаток очистили тонкослойной хроматографической обработкой (силикагель, хлороформ), получив 7,16 г (91%-ный выход) продукта, указанного в заголовке.
1H-ЯМР - спектрограмма (60 мгГц, CDCl3, δ ): 1,3 (т., 3H, CH3), 2,9 (к., 2H, CH2CH3), 6,1 (с., 2H, OCH2O), 7,3 - 7,7 (м., 3H, Ph), 8,0 - 8,2 (м., 2H, Ph).
II. Бензоилоксиметилхлорформиат.
4,03 г (0,030 моль) SO2Cl2 при температуре 0-5oC с перемешиванием в течение 15 мин добавили к 7,16 г (0,030 моль) o-бензоилоксиметил-s-этилкарбонтиоата, после чего перемешивание при комнатной температуре продолжали в течение 2 ч. В результате выпаривания EtSCI при комнатной температуре и остаточном давлении 11 мм рт.ст. получили желтую жидкость. Выход - 5,30 г (83%).
1H-ЯМР - спектрограмма (60 мгГц, CDCL3, δ ): 6,1 (с., 2H, OCH2O), 7,3 - 7,7 (м., 3H, Ph), 8,0 - 8,2 (м., 2H, Ph).
Пример 2. Получение полимеров.
а) Эмульсионная сополимеризация метилендиметакрилата и стирола.
50 мл раствора концентрацией 1% (вес/объем) додецилсульфата натрия в воде предварительно нагрели в токе азота до температуры 60oC. При интенсивном перемешивании добавили 0,20 г (1,09 ммоль) диметакрилата метилена из эксперимента вышеприведенного примера Ia и 9,80 г (0,094 мл) стирольного мономера. Полимеризацию инициировали метабисульфит / персульфатной окислительно-восстановительной системой, которая включала в себя 1,6 мг (7,2 ммоль) метабисульфита калия и 0,08 мг (0,3 ммоль) персульфата калия. Процесс полимеризации проводили в течение 8 ч с последующим охлаждением до комнатной температуры. Содержание сухого вещества в приготовленной эмульсии составляло 11,2%, что соответствовало степени конверсии 68%. Полученный полимер не растворялся в ТГФ, хорошем растворителе для полистирола, что указывало на структурированность полимера.
b) Полимер из метилен-бис-(16-оксигексадеканоата) и адипоилхлорида.
Раствор 0,657 г (3,59 ммоль) адипоихлорида в 5 мл смеси ксилола с трихлорэтиленом в весовом соотношении 80:20 при температуре 60oC по каплям добавили в раствор 2,000 г (3,59 ммоль) метилен-бис (16-оксигексадеконсата) из эксперимента вышеприведенного примера 1b (IV) в 160 мл смеси ксилола с трихлорэтиленом в объемном соотношении 80:20. По истечении 44 ч выдержки при температуре 60oC под пониженным давлением реакционную смесь охладили до температуры 20oC и под пониженным давлением выпарили растворитель, получив 0,227 белого твердого вещества.
ИК - спектрограмма (чистый), см-1, см-1 : 2915 (сильн.), 1759, 1732 (сильн. ), 1466, 1417 (слаб.) 1380, 1263, 1175 (слаб.), 1105 (слаб.), 991 (слаб.), 798 (слаб.), 726.
1Н-ЯМР - спектрограмма (300 мгГц, CDCl3 δ): 1,58 (м., 44H, CH2), 1,63 (м, 12H, CH2), 2,29 (м., 8H, CH2CO), 4,04 (м., 4H, 2•CH2O), 5,73 (м., 2H, -OCH2O-).
Хроматография с исключением размеров (ХИР) : Mw - 11100, Mn - 6500, Mp - 14100, Mw/Mn - 1,7.
c) Полимер из метилен-бис(12-оксидодеканоата) и адипоилхлорида.
Раствор 1,22 г (6,7 ммоль) адипоилхлорида к 5 мл смеси ксилола с трихлорэтиленом в весомо соотношении 80:20 при температуре 60oC по каплям добавили в раствор 3,00 г (6,7 ммоль) метилен-бис (12-оксидодеканоата) из эксперимента вышеприведенного примера 1с в 100 мл смеси ксилола с трихлорэтиленом в объемном соотношении 80: 20. По истечении 4 дней выдержки при температуре 60oC под пониженным давлением реакционную смесь охладили до температуры 20oC и под пониженным давлением выпарили растворитель, получив желтое твердое соединение. Это соединение очистили тонкослойной хроматографической обработкой (двуокись кремния, постепенный переход от хлороформа к этилацетату). Хроматография с исключением размеров (ХИР) : Mw - 18276, Mn - 12840, Mw - 1,423.
(d) Полимер из метилен-бис(10-оксидеканоата) и сукцинилхлорида.
0,200 г (1,29 ммоль) сукцинилхлорида добавили при температуре 70oC в раствор 0,500 г (1,29 ммоль) метилен-бис (10-оксиденаноата) из эксперимента вышеприведенного примера 1 в 60 мл толуола. По истечении 100 ч выдержки при температуре 70oC под пониженным давлением реакционную смесь охладили до температуры 20oC и под пониженным давлением выпарили растворитель, получив 0,436 г желтого твердого вещества.
ИК - спектрограмма (чистый) : 2933 (сильн.), 1787, 1738 (сильн.), 1650, 1465 (слаб.), 1413 (слаб.), 1357 (слаб.), 1262 (слаб.), 1164, 1099 (слаб.), 1049 (слаб.), 988, 906, 802 см-1).
1Н-ЯМР - спектрограмма (300 мгГц, CDCl3, δ): 1,25 (м., 20H, CH2), 1,57 (м. , 8H, CH2), 2,32 (м., 4H, CH2CO), 2,61 (м., 4H, CH2CO), 4,04 (м., 4H, 2•CH2O), 5,70 (c. , 2H, -OCH2O-). Хроматография с исключением размеров (ХИР)'' Mw - 1870, Mn - 1580, Mp - 1310, Mw/Mn - 1,18
e) Олигомер из метилен-бис(10-оксидеканоата) и янтарной кислоты.
0,152 г (1,29 ммоль) янтарной кислоты при температуре 130oC добавки в раствор 0,500 г (1,29 ммоль) метилен-бис (10-оксидеканоата) из эксперимента вышеприведенного примера 1d и 0,007 г (0,004 ммоль) п-толуолсульфокислоты в 12 мл толуола. По истечении 84 ч выдержки при температуре 140oC с непрерывным удалением образовавшейся воды за счет отгонки реакционную смесь охлаждали до температуры 20oC. Под пониженным давлением выпарили растворитель, получив 0,425 г желтого твердого продукта.
ИК - спектрограмма (чистый): 2933 (сильн.), 1739 (сильн.), 1550, 1467 (слаб.), 1415, 1360 (слаб.) 1261, 1168, 1100, 995, 803, 724 см-1.
1Н-ЯМР - спектрограмма (300 мгГц, CDCl3, δ ): 1,27 (м., 20H, CH2), 1,59 (м. , 8H, CH2), 2,33 (м., 4H, CH2CO), 2,64 (м., 4H, CH2CO), 4,05 (м., 4H, 2•CH2O), 5,72 (с., 2H, -6CH2C-), 10,00 (широк, с., 2H).
Хроматография с исключением размеров (ХИР): не показала образования каких-либо полимеров (только олигомеры).
f) Полимер из метилен-бис(10-оксидеканоата) и адипоилхлорида.
Раствор 0,943 г (5,15 ммоль) адипоилхлорида в 7 мл смеси ксилола с трихлорэтиленом в весовом соотношении 80:20 добавили при температуре 60oC в раствор 2,000 г (5,15 ммоль) метилен-бис(10-оксидеканоата) из эксперимента вышеприведенного примера 1 в 120 мл смеси ксилола с трихлорэтиленом в объемном соотношении 80:20. По истечении 48 ч выдержки при температуре 60oC под пониженным давлением реакционную смесь охладили до температуры 20oC и под пониженным давлением выпарили растворитель, получив белое твердое вещество. В результате тонкослойной хроматографической обработки (двуокись кремния, этилацетат) получили 0,44 г полимерной фракции.
1Н-ЯМР - спектрограмма (300 мгГц, CDCl3, δ ): 1,32 (м., 20H, CH2), 1,57 (м. , 12H, CH2), 2,34 (м., 8H, CH2CO), 4,03 (м., 4H, 2•CH2O), 5,71 (с., 2H, -OCH2O-).
Хроматография с исключением размеров (ХИР): Mw - 20964, Mn - 12382, Mp - 22843, Mw/Mn - 1,693.
g) Полимер из бис(хлоркарбонилоксиметил)-терефталата и 1,6-диаминогексена.
0,23 г (0,002 моль) 1,6-диаминогексана и 0,40 г (0,004 моль) триэтиламина в 5 мл ТГФ добавили в раствор 0,70 г (0,002 моль) бис(хлоркарбонилоксиметил)-терефталата из эксперимента вышеприведенного примера 1e (II) в 20 мл ГТФ. Затем реакционную смесь оставили перемешиваться в течение 6 дней при комнатной температуре. Далее реакционную смесь профильтровали и под пониженным давлением удалили растворитель, в результате чего получили полимер, который не растворялся в хлороформе.
1Н-ЯМР - спектрограмма (60 мгГц, CDCl3: δ ): 1,20 (м., 8H, 4•CH2), 2,85 - 3,20 (м., 6H, 2•NH и 2•CH2H), 5,80 (с., 2H, OCH2O), 8,00 (с., 4H, Ar).
h) Полимер из метилен-бис(4-оксиметилбензоата) и адипоилхлорида
Раствор 1,26 г (6,89 ммоль) адипоилхлорида в 5 мл смеси 1,1,2,2- тетрахлорэтана с трихлорэтиленом в весовом соотношении 80:20 по каплям добавили при температуре 60oC в раствор 2,18 г (6,89 ммоль) метилен-бис(4-оксиметилбензоата) из эксперимента вышеприведенного примера 1f в 90 мл смеси 1,1,2,2-тетрахлорэтана с трихлорэтиленом в весовом соотношении 80:20. По истечении 4 дней выдержки при температуре 60oC под пониженным давлением реакционную смесь охладили до 20oC и под пониженным давлением выпарили растворитель, получив 2,82 г коричневого вязкого маслоподобного продукта. В результате осаждения в метаноле получили 0,80 г желтого соединения. Хроматография с исключением размеров (ХИР): Mm - 3793, Mn - 2715, Mp - 2845, Mw/Mn - 1,724.
1H-ЯМР-спектрограмма (200 мгГц, CD3COCD3, δ ) : 1,65 (широк.с., 4H, CH2), 2,40 (широк. с. , 4H, CH2CO), 5,18 (с., 4H, O-CH2-Ph), 6,25 (с., 2H, OCH2), 7,4 - 7,6 (м., 4H, Ph), 7,9 - 8,1 (м., 4H, Ph).
i)-m) Общая процедура полимеризации метакрилоилоксиметилкарбонатов.
Раствор 1,0 г метакрилоилоксиметилкарбоната из эксперимента вышеприведенного примера 1l - p в 8,0 г ДМФ нагрели до температуры 60oC и добавили 0,005 г (0,03 ммоль) АИБН. По истечении 24 ч реакционную смесь охладили и полимерный раствор по каплям добавили в перемешиваемое избыточное количество метаноал(нерастворитель). Полимер отфильтровали и промыли метанолом и водой, а затем высушили под пониженным давлением.
i) Полимер из метилметакрилоилоксиметилкарбоната.
ИК - спектрограмма (бромид калия) : 1763 (C:O, сильн.) см-1.
1H-ЯМР (300 мгГц, CDC3, δ ): 1,00 (м., 2H, CH2), 1,90 (м., 3H), 3,85 (с. , 3H, CH3O), 5,70 (c., 2H, OCH2O).
13C-ЯМР - спектрограмма (75 мгГц, CDCl3, δ ): 46,35 ( 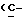 -CH3), 56,55 (CH3O), 83,59 (-OCH2O-), 154,41 (C:O), 175,50 (C:O).
-CH3), 56,55 (CH3O), 83,59 (-OCH2O-), 154,41 (C:O), 175,50 (C:O).
Дифференциальная сканирующая калориметрия (ДСК) показала, что Tc = 59,8oC, а начальная температура деструкции была равной 242,2oC.
Термомеханический анализ показал, что температура стеклования составляла 59:9oC.
Хроматография с исключением размеров (ХИР): Mw - 100000, Mn - 59000, Mw/Mn - 1,7.
j) Полимер из этилметакрилоилоксиметилкарбоната.
ИК - спектрограмма (бромид калия): 1763 (C:O, сильн.) см-1.
1H-ЯМР - спектрограмма (300 мгГц, CDCl3, δ ) :1,00 (м, 2H, CH2), 1,32 (т. , 3H, CH3), 1,90 (м. , 3H, CH3), 4,25 (м., 2H, CH2O), 5,70 (c., 2H, OCH2O).
13C-ЯМР - спектрограмма (75 мгГц, CDCl3, δ ): 15,77 (-OCH2O-), 46,35 ( 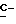 -CH3), 65,90 (CH2O), 83,50 (-OCH2O-), 153,69 (C:O), 175,80 (C:O).
-CH3), 65,90 (CH2O), 83,50 (-OCH2O-), 153,69 (C:O), 175,80 (C:O).
Дифференциальная сканирующая калориметрия (ДСК) показала, что Tc = 35,9oC, а начальная температура деструкции была равной 260,9oC. Термомеханический анализ показал, что температура стеклования составляла 31,2oC.
Хроматография с исключением размеров (ХИР): Mw - 34000, Mn - 20000, Mw/Mn - 1,7.
k) Полимер из бутилметакрилоилоксиметилкарбоната.
ИК - спектрограмма (бромид калия) : 1763 (C:O) см-1.
1H-ЯМР - спектрограмма (300 мгГц, CDCl3, δ ): 0,90 (т. 3H, CH3), 1,00 (м. , 2H, CH2), 1,39 (м., 2H, CH2), 1,70 (м., 2H, CH2), 1,90 (м., 3H, CH3), 4,20 (т., 2H, CH2O), 5,68 (с., 2H, OCH2O).
13C-ЯМР - спектрограмма (75 мгГц, CDCl3, δ ): 13,54 (CH3CH2), 18,73 (CH2), 30,39 (CH2), 46,26 ( 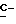 -CH3), 69,72 (CH2O), 83,67 (-OCH2O-), 153,86 (C:O), 175,80 (C:O).
-CH3), 69,72 (CH2O), 83,67 (-OCH2O-), 153,86 (C:O), 175,80 (C:O).
Дифференциальная сканирующая калориметрия (ДСК) показала, что начальная температура деструкции составляла 239,9oC (Tc не установили). Термомеханический анализ показал, что температура стеклования составляла 24,7oC. Хроматография с исключением размеров (ХИР): Mw - 60000, Mn - 29000, Mw/Mn - 2,1.
l) Полимер из децилметакрилоилоксиметилкарбоната.
ИК - спектрограмма (бромид калия): 1763 (C:O, сильн.) см-1.
1H-ЯМР - спектрограмма (300 мгГц, CDCl3, δ ): 0,90 (т., 3H, CH3), 0,90 (м. , 3H, CH2), 1,30 (м., 14H, CH2), 1,70 (м., 2H, CH2), 1,90 (м., 2H), 4,19 (т., 2H, CH2O), 5,66 (с., 2H, OCH2O).
13C-ЯМР - спектрограмма (75 мгГц, CDCl3, δ ): 13,78 (CH3), 22,34 - 31,57 (CH2), 46,26 ( 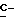 -CH3), 68,70 (CH2O), 33,67 (-OCH2O-), 153,55 (C:O), 175,80 (C:O).
-CH3), 68,70 (CH2O), 33,67 (-OCH2O-), 153,55 (C:O), 175,80 (C:O).
Дифференциальная сканирующая калориметрия (ДСК) показала, что начальная температура деструкции была равной 232,9oC (Tc не была установлена). Термомеханический анализ показал, что температура стеклования составляла - 3,3oC.
Хроматография с исключением размеров (ХИР): Mw - 160000, Mn - 90000, Mw/Mn - 1,7.
m) Полимер из бензилметакрилоилоксиметилкарбоната.
ИК - спектрограмма (бромид калия) : 3077 (Ph), 1763 (C:O, сильн.) см-1.
1H-ЯМР - спектрограмма (300 мгГц, CDCl3, δ ), 0,95 (м., 3H, CH3), 1,90 (м., 2H), 5,25 (с., 2H, CH2O), 5,75 (c., 2H, OCH2O), 6,70 (c., 5H, P).
13C-ЯМР - спектрограмма (75 мгГц, CDCl3, δ ): 46,26 ( 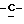 -CH3), 68,03 (
-CH3), 68,03 (  PR), 82,02 (-OCH2O-), 129,45 (Ph), 153,67 (C:O), 175,80 (C:O).
PR), 82,02 (-OCH2O-), 129,45 (Ph), 153,67 (C:O), 175,80 (C:O).
Дифференциальная сканирующая калориметрия (ДСК) показала, что Tc = 31,6oC, а начальная температура деструкции была равной 197,1oC. Термомеханический анализ показал, что температура стеклования составляла 32,8oC.
Хроматография с исключением размеров (ХИР) : Mw - 92000, Mn - 44000, Mw/Mn - 2,1.
n) Свободнорадикальная полимеризация в растворе бензилметакрилоилоксиметилкарбоната, дающая низкомолекулярный полимер.
Раствор 0,5 г (2,0 ммоль) бензилметакрилоилоксиметилкарбоната из эксперимента вышеприведенного примера 1p в 7,5 г ДМФ нагрели до температуры 60oC и добавили в него 0,0015 г (0,02 ммоль) аллилмеркаптана совместно с 0,0025 г (0,015 ммоль) АИБН. По истечении 24 ч реакционную смесь охладили и по каплям полимерный раствор добавили в избыточное количество метанола /нерастворитель/ Полимер отфильтровали и промыли метанолом и водой, после чего высушили под пониженным давлением.
Хроматография с исключением размеров (ХИР): Mw - 22000, Mn - 14000.
o) Свободнорадикальная полимеризация метил-1-метакрилоилоксиэтилкарбоната.
0,005 г (0,03 ммоль) АИБН добавили в атмосфере сухого азота при температуре 60oC в раствор 1,0 г (5,9 ммоль) метил-1-метакрилоилоксиэтилкарбоната из эксперимента вышеприведенного примера 1 (II) и в 8 г сухого ТГФ. По истечении 24 ч реакционную смесь охладили до температуры 20oC и под пониженным давлением удалили растворитель. Полученный полимер растворили в CH2Cl2 и осадили в метаноле. Метанол отделили от полимера фильтрованием, получив белый порошок.
1H-ЯМР - спектрограмма (200 мгГц, CDCl3 δ : 0,90 (м., 3H, CH3), 1,45 (с. , 3H, CH3CH), 1,87 (м., 2H, CH2), 3,80 (с., 3H, CH3O), 5,65 (широк с., 1H, 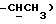 ).
).
Хроматография с исключением размеров (ХИР) : Mw-16033, Mn-6641, Mp-16192, Mw/Mn-2,41. Дифференциальная сканирующая калориметрия (ДСК) показала, что Tc=57,62oC.
p) Свободнорадикальная полимеризация этил-1-метакрилоилоксиэтилкарбоната.
0,033 г (0,02 ммоль) АИБН в атмосфере сухого азота добавили при температуре 50oC в раствор 0,504 г (2,49 ммоль) этил-1-метакрилоилоксиэтилкарбоната из эксперимента вышеприведенного примера 1g (II) в 9 мл сухого ТГФ. По истечении 7 г реакционную смесь охладили до температуры 20oC и полимер осадили в 50 мл метанола с последующим фильтрованием раствора. Выделенный полимер растворили в ТГФ, осадили в метаноле (70 мл) и отфильтровали, получив 0,138 г белого порошка.
1H-ЯМР - спектрограмма (300 мгГц, CDCl3, δ): 0,90 (м., 3H, CH3), 1,25 (с., 3H, CH3), 1,45 (с., 3H, CH3) , 1,87 (м., 2H, CH2), 4,15 (широк. с., 2H, CH2O), 6,62 (широк.с., 1H, 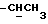 ).
).
Хроматография с исключением размеров (ХИР) : Mw-26500, Mn-18600, Mp-22000, Mw/Mn-1,43.
q) Полимер из этилметакрилоилоксиметилкарбоната, эмульсионная полимеризация.
Раствор 0,056 г (0,19 ммоль) додецилсульфоната натрия в 20,5 мл воды нагрели до температуры 60o в токе азота, после чего добавили 5,266 г (28,00 ммоль) этилметакрилоилоксиметилкарбоната из эксперимента вышеприведенного примера 1m. Полимеризацию инициировали 53,4 мг (0,24 ммоль) метабисульфита и 4,38 мг (0,02 ммоль) персульфата калия в качестве окислительно-восстановительной системы. По истечении 16 ч выдержки при температуре 60oC добавили 4,38 мг (0,02 ммоль) персульфата калия и полимеризацию продолжали в течение дополнительных 3 ч при температуре 60oC и в атмосфере азота с последующим охлаждением до температуры 20oC.
r) Полимер из метакрилоилоксиметилбензоата.
При температуре 60oC в атмосфере сухого азота 0,005 г (0,03 ммоль) АИБН добавили в раствор 1,00 г (4,55 ммоль) метакрилоилоксиметилбензоата из эксперимента вышеприведенного примера 1 г. в 8 г. ТГФ. По истечении 24 ч реакционную смесь охладили до температуры 20oC и под пониженным давлением удаляли растворитель. Полученный полимер растворили в хлористом метилене и пересадили в метаноле. Метанол отделили от полимера фильтрованием, в результате чего получили белый порошок.
1H-ЯМР - спектрограмма (200 мгГц, CDCl3, δ) : 0,85 (м., 3H, CH3), 1,87 (м., 2H, CH2), 5,70 (м., 2H -OCH2O-), 7,45 (м., 3H, Ph), 8,05 (м., 2H, Ph).
Хроматография с исключением размеров (ХИР) : Mw-30281, Mn-11580, Mp-32286, Mw/Mn - 2615.
Дифференциальная сканирующая калориметрия (ДСК) показала, что Tc= 60,98oC.
s) Свободнорадикальная полимеризация N-(2-ацетоксиметоксикарбонилоксипропил) - метакриламида.
0,0138 г (0,084 ммоль) АИБН при температуре 50oC в атмосфере сухого азота добавили в раствор 0,519 г (2 ммоль) N-(2-ацетоксиметоксикарбонилоксипропил) - метакриламида из эксперимента вышеприведенного примера 1s(II) в 8 мл сухого ТГФ. По истечении 3 дней под пониженным давлением удалили растворитель, получив 0,439 г белого порошка.
1H-ЯМР - спектрограмма (200 мгГц, CDCl3, δ/ ): 0,8 - 1,2 (м., 3H, CH3), 1,2 - 1,4 (м. , 3H, CH2-CH 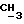 O), 1,6 - 2,0 (м., 2H, CH2), 2,1 (с., 3H, CH3CO), 2,9 - 3,9 (м., 2H, NH
O), 1,6 - 2,0 (м., 2H, CH2), 2,1 (с., 3H, CH3CO), 2,9 - 3,9 (м., 2H, NH 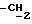 ), 4,7 - 5,0 (м., 1H, CH2
), 4,7 - 5,0 (м., 1H, CH2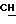 (CH3)-O), 5,8 (с., 2H, O-OH2-O), 6,2 - 7,0 (м., 1H, NH).
(CH3)-O), 5,8 (с., 2H, O-OH2-O), 6,2 - 7,0 (м., 1H, NH).
Хроматография с исключением размеров (ХИР) : Mw-5411, Mn-2857, Mw/Mn- 1,894.
Дифференциальная сканирующая калориметрия (ДСК) показала, что Tc= 52,91oC.
t) Свободнорадикальная полимеризация N-[2-(1-ацетоксиэтоксикарбонилокси) - пропил] - метакриламид.
0,0031 г (0,18 ммоль) АИБН при температуре 50oC в атмосфере сухого азота добавили в раствор 1,23 г (4,5 ммоль) N-[2(1-ацетоилоксикарбонилокси) - пропил] - метакриламида из эксперимента вышеприведенного примера 1t(II) в 18 мл сухого ТГФ. По истечении 3, под пониженным давлением удалили растворитель. В результате тонкослойной хроматографической обработки (ступенчатый градиент перехода от смеси гексана с этилацетатом в соотношении 3:4 к метанолу) получили 0,96 г белого порошка.
1H-ЯМР - спектрограмма (200 мгГц, CDCl3, δ : 0,8 - 1,2 (м., 3H, CH3), 1,2 - 1,4 (м., 3H, CH2-CH 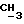 O), 1,5 (д., 3H, O-CH
O), 1,5 (д., 3H, O-CH 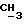 -O), 1,6 - 2,0 (м., 2H, CH2), 2,0 - 2,2 (с., 3H, CH3CO), 2,9 - 3,9 (м., 2H, NH -
-O), 1,6 - 2,0 (м., 2H, CH2), 2,0 - 2,2 (с., 3H, CH3CO), 2,9 - 3,9 (м., 2H, NH - 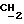 ), 4,7 - 5,0 (м., 1H, CH2
), 4,7 - 5,0 (м., 1H, CH2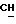 (CH3)-O), 6,2 - 7,0 (м., 2H, NH -O-
(CH3)-O), 6,2 - 7,0 (м., 2H, NH -O- 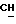 (CH3)-O).
(CH3)-O).
Хроматография с исключением размеров (ХИР) : Mw - 1991, Mn - 1268, Mp - 2105, Mw/Mn - 1,548.
Дифференциальная сканирующая калориметрия (ДСК) показала, что Tc= 51,53oC.
u) Олигомер из этиленди-(хлорметилкарбоната)- и дикалийтерефталата.
1,62 г (0,014 моль) трет-бутоксида калия добавили в раствор 1,20 г (0,0072 моль) терефталевой кислоты в 40 мл ДМФ. В образовавшуюся суспензию добавили 1,78 г (0,0072 моль) этиленди-(хлорметилкарбоната) из эксперимента вышеприведенного примера 1. Затем добавили 0,056 г (0,21 ммоль) продукта 18-краун-6 и реакционную смесь оставили перемешиваться при комнатной температуре в течение 2 дней, а затем при температуре 60oC в течение 11 дней. Реакционную смесь профильтровали и под пониженным давлением удалили растворитель. Остаток растворили в 50 мл этилацетата и промыли 30 мл насыщенного водного раствора бикарбоната натрия и 30 мл воды. Органическую фазу высушили над сульфатом магния и под пониженным давлением удалили растворитель, получив продукт, указанный в заголовке.
1H-ЯМР - спектрограмма (60 мгГц, CDCl3, δ ): 4,48 (м., 4H, OCH2 CH2O), 6,02 (с., 4H, OCH2O), 8,12 (с., 4H, Ar).
Хроматография с исключением размеров (ХИР): Mw - 1938, Mn - 1511, Mp - 2137, Mw/Mn - 1,283
V) Свободнорадикальная эмульсионная гомополимеризация бензилметакрилоилоксиметилкарбоната.
Раствор 1,6 • 10-2 г (5,5 • 10-2 ммоль) додецилсульфата натрия в 6,0 мл воды, из которой удалили кислород, ввели в 50-миллилитровую двугорлую круглодонную колбу, снабженную магнитной мешалкой и холодильником. В этот раствор добавили 0,015 г (6,7 • 10-2 ммоль) метабисульфита калия, растворенного в 1,0 мл воды, из которой был удален кислород, и 2,0 (8,0 ммоль) бензилметакрилоилоксиметилкарбоната из эксперимента вышеприведенного примера 1p. Реакционную смесь нагрели до температуры 60oC. В нагретую реакционную смесь добавили 1,25 • 10-3 г (4,6 • 10-3 ммоль) персульфата калия и провели реакцию. По истечении приблизительно 5 ч полимеризацию прекратили и полимерную эмульсию по каплям добавили в большое избыточное количество метанола (нерастворитель). Далее полимер отфильтровали и промыли метанолом и водой. Эту процедуру полностью повторили трижды с целью очистки полимера. Затем полимер собрали и высушили в вакууме с целью удалить из него все примеси растворителя. Некоторое количество стойкой эмульсии не подвергали экстракционной обработке по вышеизложенному, но сохранили для анализа размеров частиц с помощью оптического микроскопа. Размеры частиц эмульсии вычисляли с помощью оптической микроскопии, в результате чего было установлено, что их диаметр составляет чуть меньше 1 мкм.
W) - Z) Свободнорадикальная растворная сополимеризация этилметакрилоилоксиметилкарбоната и метакриловой кислоты.
Исходную мономерную смесь, которая состояла из этилметакрилоилоксиметилкарбоната из эксперимента вышеприведенного примера 1m и метакриловой кислоты в 8,0 г. ДМФ нагрели до температуры 60oC и добавили в нее 0,005 г (0,03 моль) АИБН. По истечении 24 ч по каплям в перемешиваемое избыточное количество хлороформа (нерастворитель) добавили полученный полимерный раствор, массу профильтровали и промыли дополнительным количеством хлороформа, после чего высушили под пониженным давлением.
1H-ЯМР (200 мгГц, CDCl3, δ ): 10 (с., 6H, 2 • CH3), 1,27 (т., 3H, 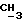 CH2), 1,90 (с., 4H, 2 • CH2), 3,52 (широк.с., 1H, OH), 4,2 (м., 2H, CH3CH2), 5,72 (с., -OCH2O-).
CH2), 1,90 (с., 4H, 2 • CH2), 3,52 (широк.с., 1H, OH), 4,2 (м., 2H, CH3CH2), 5,72 (с., -OCH2O-).
aa) Олигомер из гексаметиленди-(хлорметилкарбоната) и дикалийтерефталата.
7,87 г (0,068 моль) трет-бутоксида калия добавили в раствор 5,66 г (0,034 моль) терефталевой кислоты в 200 мл ДМФ. В образовавшуюся суспензию добавили 9,50 г (0,034 моль) гексаметилендихлорметилкарбоната из эксперимента вышеприведенного примера IX. Затем добавили 0,24 г (0,82 ммоль) продукта 18-краун-6 и реакционную смесь оставили перемешиваться при комнатной температуре в течение 5 ч при температуре 60oC в течение 14 дней.
Далее реакционную смесь профильтровали и под пониженным давлением удалили растворитель. Остаток растворили в 100 мл хлороформа и промыли 50 мл насыщенного водного раствора бикарбоната натрия и 50 мл воды. Органическую фазу высушили над сульфатом магния и под пониженным давлением удалили растворитель, получив желтый продукт.
1H-ЯМР - спектрограмма (60 мгГц, CDCl3, δ ): 1,25 - 1,90 (м., 8H, 4 • CH2), 4,20 (т., 4H, -OCH2CH2), 6,00 (с., 4H, OCH2O), 8,10 (с., 4H, Ar).
Хроматография с исключением размеров (ХИР) : Mw - 2987, Mn - 1754, Mp - 3014, Mw/Mn - 1,703.
Дифференциальная сканирующая калориметрия (ДСК): показала, что Tc < 20oC.
ab) Полимер из метакрилоилоксиметилацетата.
При температуре 60oC в атмосфере сухого азота в раствор 1,00 г (4,00 ммоль) метакрилоилоксиметилацетата из эксперимента вышеприведенного примера 1y в 8 г сухого ТГФ добавили 0,005 г (0,03 ммоль) АИБН. ПО истечении 24 ч реакционную смесь охладили до температуры 20oC и под пониженным давлением удалили растворитель. Образовавшийся полимер растворили в CH2Cl2 и пересадили в метаноле. Метанол отделили от полимера фильтрованием, получив белый порошок.
Дифференциальная сканирующая калориметрия (ДСК) показала, что Tc = 54,99oC.
Хроматография с исключением размеров (ХИР) : Mw - 184678, Mn - 2446, Mp - 54732, Mw/Mn - 7,66.
ac) Олигомер из метилен-бис(10-оксидеканоата) и мелонилхлорида.
0,254 г (1,80 ммоль) мелонилхлорида при температуре 60oC добавили в раствор 0,700 г (1,800 ммоль) метилен-бис(10-оксидеканоата) из эксперимента вышеприведенного примера 1 в 50 мл смеси ксилола с трихлорэтиленом в объемном соотношении 80 : 20. По истечении 77 ч выдержки при температуре 60oC под пониженным давлением реакционную смесь охладили до температуры 20oC и выпарили растворитель, получив 0,666 г коричневой вязкой жидкости. Хроматография с исключением размеров (ХИР) : Mw - 2700, Mn - 2100, Mp - 1600, Mw/Mn - 1,28.
ad) Полимер из этил-1-метакрилоилоксиэтилкарбоната, эмульсионная полимеризация.
При температуре 60oC в атмосфере азота смесь 6,5 мг (0,023 ммоль) додецилсульфата натрия в 2,40 мл воды и 6,3 мг (0,028 ммоль) в 0,82 мл воды нагрели до температуры 60oC с последующим добавлением 0,617 г (3,10 ммол) этил-1-метакрилоилоксиэтилкарбоната из эксперимента вышеприведенного примера 1q (II). Полимеризацию инициировали добавлением 0,54 мг (0,002 ммоль) персульфата калия в 0,25 мл воды. Далее процесс полимеризации провели в течение 20 ч при температуре 60oC в атмосфере азота с последующим охлаждением до температуры 20oC.
ae) Полимер из метилен-бис(12 оксидодеканоата) и трифосгена.
Раствор 2,0 ммоль метилен-бис(12-оксидодеканоата) из эксперимента вышеприведенного примера 1c и 0,67 ммоль трифосгена в 2 мл смеси ксилола с трихлорэтиленом в соотношении 95:5 выдержали при температуре 60oC в течение 36 ч под остаточным давлением 50 мм рт.ст. с последующим выпариванием, в результате чего получили полимерный материал.
af) Полимер из метилен-бис(12-оксидодеканоата) и дихлорангидрида 3,6,9 - триоксаундекандикислоты.
Раствор 2,0 ммоль метилен-бис (12-оксидодеканоата) из эксперимента вышеприведенного примера 1aa и 2,0 ммол, дихлорангидрида 3,6,9 - триоксаундекандикислоты в 2 мл смеси ксилола с трихлорэтиленом в соотношении 95:5 выдерживали при температуре 60oC в течение 36 ч под остаточным давлением 50 мм рт.ст., а затем выпарили с получением полимерного материала.
ag) Декстран-10-1-(7-карбоксигептаноилокси)-этилдецилкарбонат.
В раствор 0,65 г продукта декстран 10 в 40 мл ДМСО добавили 1,5 г (36 ммоль) 1-(7-карбоксигептаноилокси)-этилдецилкарбоната из эксперимента примера 1ac, 0,83 г (4,3 ммоль) N-этил-N'- -(3-диметиламинопропил)-карбодиимида и 42 мг (0,28 ммоль)-4-пирролидинопиридина, растворенных в 30 мл ДМСО. После перемешивания при комнатной температуре в течение 2 дней реакционную смесь разбавили 250 мл воды и подвергли диализу против воды в течение 30 ч. В результате лиофилизации раствора получили 1,3 г окрашенного в светло-желтый цвет порошка. На 13С-ЯМР - спектрограмме при 171,28 ч. /1000000 ч. появился новый карбонильный сигнал. Это имело место внутри ожидаемой области для сложноэфирного карбонильного сигнала продукта. Остальные сигналы находились в соответствии со структурой продукта.
ah) Полимер из продукта Плуроник F68 и бензоилоксиметилхлороформиата.
9,889 г (1,191 ммоль) продукта Плуроник F68 растворили в 30 мл сухого толуола. После нагревания до температуры 45oC при постоянном перемешивании добавили 0,70 мл триэтиламина. По капле добавили 1,072 г (5,00 ммоль) бензилоксиметилхлорформиата из эксперимента вышеприведенного примера 1ah(II), растворенного в 4 мл толуола, после чего добавили дополнительно 0,25 мл триэтиламина и 2,5 мл толуола. Реакционную смесь выдержали при температуре 45oC в течение 8 ч, а затем при температуре 55oC в течение 16 ч после чего охладили и профильтровали. Под пониженным давлением удалили растворитель и выделенное соединение растворили в толуоле, после чего пересадили в 500 мл н-гептана с перемешиванием, получив 8,45 г белого порошка. ИК - спектрограмма (бромид калия): 1722 (C:O) см-1.
ai) Свободнорадикальная полимеризация 1-ацетокси-1-фенилметилвинилкарбоната
0,005 г (0,03 моль) АИБН добавляют при температуре 60oC в атмосфере сухого азота в раствор 1,0 г. 1-ацетокси-1-фенилметилвинилкарбоната из эксперимента вышеприведенного примера ad (II) в 8 мл сухого ТГФ. По истечении 12 ч под пониженным давлением удаляют растворитель. Полученный полимер растворяют в CH2Cl2 и переосаждают в подходящем растворителе. Этот растворитель отделяют от продукта фильтрованием, получая белый порошок.
a) Свободнорадикальная растворная сополимеризация N-(2-оксипропил)-метакриламида с N-(2-ацетоксиметоксикарбонилоксипропил)-метакриламида
0,430 г (3,0 ммоль) N-(2-оксипропил)-метакриламида3 и 0,778 г (3,0 ммоль) - (2-ацетоксиметоксикарбонилоксипропил)-метакриламида из эксперимента вышеприведенного примера 1s растворили в 10 мл тетрагидрофурана и раствор нагрели до температуры 55oC. Добавили 0,0207 г (0,126 ммоль) АИБН и смесь перемешивали при температуре 55oC в течение 3 дней, получив прозрачную желеобразную массу. Эту последнюю растворили в тетрагидрофуране и под пониженным давлением выпарили растворитель, получив 1,33 г белого порошка.
Хроматография с исключением размеров (ХИР) показала образование полимера.
Пример 3. Получение полимерных частиц.
a) Частицы из полимера, полученного из метилендиметакрилата и стирола.
13 мл образца полимерной эмульсии из эксперимента вышеприведенного примера 2а при комнатной температуре смешали с 13 мл гептана. По истечении 40 мин этот образец лиофилизировали, получив продукт в форме белого порошка.
b) Частицы из полимера, полученного из метилен-бис(16-оксигексадеканоата) и адипоилхлорида.
6,204 г раствора концентрацией 4,1 вес.% полимера из эксперимента вышеприведенного примера 2b в смеси ксилола с трихлорэтиленом в соотношении 90: 10 добавили в 25 мл раствора продукта Плуроник®F68 в воде концентрацией 0,5% (вес/объем). Эту смесь подвергали интенсивному встряхиванию (вручную) в течение 1 мин и высушили вымораживанием в течение 16 ч. Оптическое микроскопическое исследование показало образование микрочастиц.
c) Частицы из полимера, полученного из метилен-бис(16-оксигексадеканоата) и адипоилхлорида.
6,204 г раствора концентрацией 4,1 вес.% полимера из эксперимента вышеприведенного примера 2b в смеси ксилола с трихлорэтиленом в соотношении 90: 10 добавили к 25 мл раствора концентрацией 0,5% (вес/объем) продукта Плуроник®F68 в воде. Эту смесь перемешивали в смесителе Юлтра Тьюракс T25 при скорости вращения мешалки 20500 об/мин в течение 40 сек и высушили вымораживанием в течение 16 ч. Оптическое микроскопическое исследование показало образование микрочастиц.
d) Частицы полимера, полученного из метилен-бис(16-оксигенсадеканоата) и адипоилхлорида
12,408 г раствора концентрацией 4,1 вес.% полимера из эксперимента вышеприведенного примера 2b в смеси ксилола с трихлорэтиленом в соотношении 90: 10 добавили в 50 мл 0,5%-ного (вес/объем) раствора продукта Плуроник®F68 в воде. Эту смесь подвергли перемешиванию в смесителе Юлтра Тьюракс Т25 при скорости вращения мешалки 24000 об/мин в течение 40 сек, а затем сушки вымораживанием в течение 16 ч. Оптическое микроскопическое исследование показано образование микрочастиц.
e) Частицы полимера, полученного из метилен-бис(12-оксидодеканоата) и адипоилхлорида.
0,40 г полимера метилен-бис(12-оксидодеканоата) из эксперимента вышеприведенного примера 2c и адипоилхлорида в 4 мл смеси ксилола с трихлорэтиленом в соотношении 10: 1 добавили в 20 мл 0,5%-ного [вес/объем] раствора продукта Плуроник®F68 в воде. Эту смесь подвергли перемешиванию в смесителе Юлтра Тьюракс Т25 при скорости вращения мешалки 20500 об/мин в течение 30 сек с последующей сушкой вымораживания в течение 16 ч под остаточным давлением 0,5 мм рт.ст. Оптическое микроскопическое изучение показало образование микрочастиц.
f) Частицы олигомера, полученного из этилендихлорметилкарбоната и дикалийтерефталата.
22,5 мл раствор концентрацией 4% (вес/объем) олигомера этилендихлорметилкарбоната и дикалийтерефтарата из эксперимента вышеприведенного примера 2u в хлороформе, приготовленного растворением полимера и с осторожным подогревом, добавили в 30 мл 0,5%-ного раствора (вес/объем) продукта Плуроник®F68 в воде. Эту смесь подвергли перемешиванию в смесителе Юлтра Тьюракс Т25 при скорости вращения мешалки 24000 об/мин в течение 40 сек с последующей сушкой вымораживанием в течение 16 ч. Оптическое микроскопическое изучение показано образованием микрочастиц.
g) Частицы полимера, полученного из этилметакрилоилоксиметилкарбоната
9 мл 10%-ного [вес/объем] раствора полимера, полученного из этилметакрилоилоксиметилкарбоната из эксперимента вышеприведенного примера 2j в хлороформе добавили в 30 мл 0,5%-ного [вес/объем] раствора продукта Плуроник®F68 в воде. Эту смесь подвергли перемешиванию в смесителе Юлтра Тьюракс T25 при скорости вращения мешалки 24000 об/мин в течение 40 сек и сушке вымораживанием в течение 16 ч. Оптическое микроскопическое изучение показало образование микрочастиц.
h) Частицы полимера, полученного из метил-1-метакрилоилоксиэтилкарбоната.
0,462 г полимера метил-1-метакрилоилоксиэтилкарбоната из эксперимента вышеприведенного примера 2с в 5 мл толуола добавили в 20 мл 1,0%-ного (вес/объем) раствора продукта Плуроник®F68 в воде. Эту смесь подвергли перемешиванию в смесителе Юлтра Тьюракс T25 при скорости вращения мешалки 20500 об/мин в течение 30 сек и сушке вымораживанием под остаточным давлением 0,05 мм рт. ст. в течение 16 ч. Оптическое микроскопическое изучение показало образование микрочастиц.
i) Частицы полимера, полученного из метакрилоилоксиметилбензоата.
0,45 г полимера из метакрилоилоксиметилбензоата из эксперимента вышеприведенного примера 2 r в 2 мл смеси толуола с трихлорэтиленом в соотношении 10:1 добавили в 20 мл 1,0%-ного (вес/объем) раствора продукта Плуроник®F68 в воде. Эту смесь подвергли перемешиванию в смесителе Юлтра Тьюракс T25 при скорости вращения мешалки 20500 об/мин в течение 30 сек с последующей сушкой вымораживанием в течение 4 ч под остаточным давлением 0,05 мм рт.ст. Оптическое микроскопическое изучение показало образование микрочастиц.
j) Частицы полимера, полученного из этилметакрилоилоксиметилкарбоната.
В соответствии с вышеприведенным примером 2g приготовили эмульсию полимера. 14,783 г эмульсии добавили в 47,305 г толуола. Эту смесь подвергли интенсивному перемешиванию в течение 20 ч и сушке вымораживанием в течение 16 ч получив 1,813 г белого порошка. Оптическое микроскопическое изучение и сканирующая электронная микроскопия показали образование микрочастиц.
k) Частицы полимера, полученного из этилметакрилоилоксиметилкарбоната.
Полимерную эмульсию приготовили в соответствии с вышеизложенным в примере 2g 12,7612 г этой эмульсии добавили в 40,836 г хлороформа. Такую смесь подвергли интенсивному перемешиванию в течение 20 ч и сушке вымораживанием в течение 16 ч, получив 1,496 г белого порошка. Оптическое микроскопическое изучение и сканирующая электронная микроскопия показали образование микрочастиц.
l) Частицы олигомера, полученного из этилендихлорметилкарбоната и дикалийтерефталата.
1,0 г олигомера из этилендихлорметилкарбоната и дикалийтерефталата из эксперимента вышеприведенного примера 2 растворители при температуре 100oC в 19,0 г жидкого нафталина. Этот нафталиновый раствор эмульгировали при температуре 90oC в 200 мл водного раствора 8,0 г поливинилового спирта (Mw - 13000 - 23000) содержащего 0,2 г продукта Плуроник®F68. В качестве эмульгирующего прибора использовали смеситель Юлтра Тьюракс T25. Затем эту эмульсию разбавили с перемешиванием при температуре 15oC 500 мл той же самой водной фазы и подвергли перемешиванию в течение 8 мин. Нафталиновые капельки затвердевали с образованием шариков, которые профильтровали через фильтр с размером отверстий 50 мкм для отделения частиц диаметром более 50 мкм. Суспензию подвергли центрифугированию с 1000 х и шарики промыли водой и подвергли повторному центрифугированию. Эту стадию повторили дважды. Затем шарики вновь суспендировали в 100 мл воды с 0,8 г лактозы и суспензию заморозили в блок при температуре - 40oC. После этого такой блок подвергли сушке вымораживанием в течение 16 ч. Оптическое микроскопическое изучение показало образование микрочастиц.
m) Частицы полимера, полученного из метилен-бис(оксигексадеканоата) и адипоилхлорида
3 мл раствора концентрацией 3,37% (вес/объем) полимера из эксперимента вышеприведенного примера 2b в смеси ксилола с трихлорэтиленом в соотношении 90:10 добавили в 10 мл 0,5%-ного (по весу) раствора продукта Твин 80 в воде. Эту смесь подвергли перемешиванию в смесителе Юлтра Тьюракс®T25 при скорости вращения мешалки 20500 об/мин в течение 1 мин и 30 сек и высушили вымораживанием в течение 18 ч, в результате чего получили белый порошок. Оптическое микроскопическое изучение показало образование микрочастиц.
n) Частицы полимера, полученного из метилен-бис(16-оксигексадеканоата) и адипоилхлорида.
3 мл раствор концентраций 3,37% (вес/объем) полимера из эксперимента вышеприведенного примера 2b в смеси ксилола с трихлорэтиленом в соотношении 90:10 добавили в 10 мл 0,5%-ного (по весу) раствора продукта Brij®99 в воде. Эту смесь подвергли перемешиванию в смесителе Юлтра Тьюракс®T25 при скорости вращения мешалки 20500 об/мин в течение 1 мин и 30 сек и сушке вымораживанием в течение 17 ч, в результате чего получили белый порошок. Оптическое микроскопическое изучение показано образование микрочастиц.
o) Частицы полимера, полученного из метилен-бис(16-оксигексадеканоата) и адипоилхлорида
5,5 мл раствора концентрацией 1,84% (вес/объем) полимера из эксперимента вышеприведенного примера 2b в смеси ксилола с трихлорэтиленом в соотношении 90:10 добавили в 10 мл раствора концентрацией 0,5 вес.% продукта Кремофор®H40 в воде. Эту смесь подвергли перемешиванию в смесителе Юлтра Тьюракс®T25 при скорости вращения мешалки 20500 об/мин в течение 1 мин и 30 сек и сушке вымораживанием в течение 16 ч, получив белый порошок. Оптическое микроскопическое изучение показало образование микрочастиц.
p) Частицы полимера, полученного из метилен-бис(16-оксигексадеканоата) и адипоилхлорида.
5,5 мл раствора концентрацией 1,84 (вес/объем) полимера из эксперимента вышеприведенного примера 2b в смеси ксилола с трихлорэтиленом в соотношении 90:10 добавили в 10 мл раствора концентрацией 0,5 вес, % продукта Коллидон®30 в воде. Эту смесь подвергли перемешиванию в смесителе Юлтра Тьюракс®T25 при скорости вращения мешалки 20500 об/мин в течение 1 мин и 30 сек и сушке вымораживанием в течение 16 ч, получив белый порошок. Оптическое микроскопическое изучение показало образование микрочастиц.
q) Частицы полимера, полученного из метилен-бис(10-оксидеканоата) и адипоилхлорида
4 мл раствора концентрацией 2,52% (вес/объем) полимера из эксперимента вышеприведенного примера 2f в смеси ксилола с трихлорэтиленом в соотношении 90:10 добавили в 10 мл раствора концентрацией 0,5 вес.% продукта Плуроник®F68 в воде. Эту смесь подвергли перемешиванию в смесителе Юлтра Тьюракс®T25 при скорости вращения мешалки 20500 об/мин в течение 1 мин и 30 сек и сушке вымораживанием в течение 15 ч, получив белый продукт. Оптическое микроскопическое изучение показало образование микрочастиц.
r) Частицы олигомера из гексаметилендихлорметилкарбоната и дикалийтерефталата.
22,5 мл раствора концентрацией 4% (вес/объем) полимера из эксперимента вышеприведенного примера 2aa в хлороформе добавили в 30 мл раствора концентрацией 0,5 вес.% продукта Плуроник®F68 в воде. Эту смесь подвергли перемешиванию в смесителе Юлтра Тьюракс®T25 при скорости вращения мешалки 24000 об/мин в течение 40 сек и сушке вымораживанием в течение 16 ч, получив желтый каучукоподобный твердый продукт. Оптическое микроскопическое изучение показало образование микрочастиц.
s) Частицы полимера, полученного из N-[2-(1-ацетокси-этоксикарбонилокси)-пропил]-метакриламида.
8 мл раствора концентрацией 2,55% (вес/объем) полимера из эксперимента вышеприведенного примера 2t в смеси ксилола с трихлорэтиленом в соответствии 90:10 добавляли в 20 мл раствора концентрацией 0,5 вес.% продукт Плуроник®F68 в воде. Эту смесь подвергли перемешиванию в смесителе Юлтра Тьюракс®T25 при скорости вращения мешалки 20500 об/мин в течение 1 мин и 30 сек и сушке вымораживанием в течение 16 ч, получив белый порошок, оптическая микроскопия, указывает образование микрочастиц.
t) Частицы полимера, полученного из метакрилоилоксиметилацетата.
8 мл раствора концентрацией 2,48% (вес/объем) полимера из эксперимента вышеприведенного примера 2a в смеси ксилола с трихлорэтиленом в соотношении 90: 10 добавляли в 20 мл раствора концентрацией 0,5 вес.% продукта Плуроник F68 в воде. Эту смесь подвергли перемешиванию в смесителе Юлтра Тьюракс®T25 при скорости вращения мешалки 20500 об/мин в течение 1 мин и 30 сек, и сушке вымораживанием в течение 16 ч, получив белый порошок.
u) Частицы полимера, полученного из N-(2-ацетоилоксиметоксикарбонилоксипропил)-метакриламида
4 мл раствора концентрацией 2,54% (вес/объем) полимера из эксперимента приведенного примера 2s в хлороформе добавляли в 10 мл раствора концентрацией 0,5 вес.% продукта Плуроник®F68 в воде. Эту смесь подвергли перемешиванию в смесителе Юлтра Тьюракс®T25 при скорости вращения мешалки 24000 в течение 50 сек. и сушке вымораживанием в течение 16 ч, получив белый порошок.
v) Частицы полимера, полученного из этил-1-метакрилоилоксиэтилкарбоната.
Полимерную эмульсию приготовили в соответствии с методом эмульсионной полимеризации с получением полимера из этил-1-метакрилоилоксиэтилкарбоната, из эксперимента вышеприведенного примера 2ad 2,00 г такой эмульсии добавляли к 7,41 г толуола. Смесь подвергли интенсивному перемешиванию в течение 20 ч и сушке вымораживанием в течение 16 ч, получив 0,250 г белого порошка. Оптическое микроскопическое изучение показано образование микрочастиц.
w) Частицы полимера, полученного из этил-1-метакрилоилоксиэтилкарбоната.
Полимерную эмульсию приготовили в соответствии с методом эмульсионной полимеризации с получением полимера из этил-1-метакрилоилоксиэтилкарбоната, из эксперимента вышеприведенного примера 2ad 2,00 г такой эмульсии добавили к 6,40 г хлороформа. Смесь подвергли интенсивному перемешиванию в течение 20 ч и сушке вымораживанием в течение 16 ч получив 0,250 г белого порошка. Оптическое микроскопическое изучение показано образование микрочастиц.
x) Частицы полимера, полученного из бутилметакрилоилоксиметилкарбоната.
0,45 г полимера из бутилметакрилоилоксиметилкарбоната, из эксперимента вышеприведенного примера 2k растворители в 9 мл толуола. Добавляли 30 мл воды, содержащей 0,3 г продукта Плуроник®F68 = и эмульсию приготовили с использованием гомогенизатора Истрал® при скорости вращения мешалки 2000 об/мин в течение 30 сек. Эту эмульсию подвергли сушке вымораживанием в течение 19 ч. Оптическое микроскопическое изучение показано образование микрочастиц.
y) Покрытие САЧ частицы полимера, полученного из метакрилоилоксиметилбензоата.
0,9 г полимера, полученного из метакрилоилоксиметилбензоата, из эксперимента вышеприведенного примера 2r растворили в 9 г толуола. Затем добавили 30 мл водного 5%-ного раствора сывороточного альбумина человека (САЧ) и смесь гомогенизировали с помощью гомогенизатора Истрал® при скорости вращения мешалки 20000 об/мин в течение 30 сек. Приготовленную эмульсию высушили вымораживанием в течение 16 ч. Оптическое микроскопическое изучение показало формирование микрочастиц.
z) Покрытие полиоксиэтиленом частицы полимера, полученного из метакрилоилоксиметилбензоата.
0,4 г двухблочного сополимера, один из блока которого состоит из полиметилметакрилата (Mw - примерно 1000), а другой блок - из полиоксиэтилена (ПОЭ, Mw - примерно 2000), растворили в 9 мл толуола. Затем в этом толуольном растворе растворили 0,9 г полимера, полученного из метакрилоилоксиметилбензоата, из эксперимента вышеприведенного примера 2r . Добавили 30 мл воды и смесь гомогенизировали с помощью гомогенизатора Истрал® при скорости вращения мешалки 20000 об/мин в течение 30 сек. Приготовленную эмульсию высушили вымораживанием в течение 16 ч, получив покрытие ПОЭ микрочастицы.
aa) Частицы полимера, полученного из метилметакрилоилоксиметилкарбоната.
0,9 г полимера, полученного из метилметакрилоилоксиметилкарбоната, из эксперимента вышеприведенного примера 2o растворили в 9 мл толуола. Затем добавили смесь 0,3 г додецилсульфата натрия и 0,025 г продукта Плуроник®F68 в 35 мл воды и раствор гомогенизировали с помощью гомогенизатора Истрал® при скорости вращения мешалки 20000 об/мин в течение 30 сек. Эмульсию высушили вымораживанием в течение 16 ч. Оптическое микроскопическое изучение показало образование микрочастиц.
ab) Частицы полимера, полученного из метилен-бис(12-окси- додеконоата) и дихлорангидрида 3,6,9-триоксаундекандикислоты.
0,9 г полимера, полученного из метилен-бис(12-оксидодеканоата) и дихлорангидрида 3,6,9-триоксаундекандикислоты, из эксперимента вышеприведенного примера 2af растворили в 9 мл толуола. Далее добавили 30 мл воды, содержащей 0,3 г продукта Плуроник®F68 и смесь гомогенизировали с помощью гомогенизатора Истрал® при скорости вращения мешалки 20000 об/мин в течение 30 сек. Эмульсию высушили вымораживанием в течение 48 ч. Оптическое микроскопическое изучение показало образование микрочастиц.
ac) Частицы при распылительной сушке полимера из метакрилоилоксиметилбензоата.
0,72 г полимера из эксперимента вышеприведенного примера 2 r растворили в 6 г дихлорметана. Раствор высушили распылением в минисушилке Бюхи 190 для распылительной сушки. Температуру на входе задали равной 54oC, а температура на выходе, согласно измерениям, составляла 40oC. Оптическое микроскопическое изучение показало образование микрочастиц.
ad) Покрытие продуктом Плуроник®F68 частицы, полученные при распылительной сушке полимера, полученного из метилен-бис(16-оксигенксадеканоата) и адипоилхлорида.
1,79 г смеси полимера из эксперимента вышеприведенного примера 2b и продукт Плуроник®F68 в соотношении 50:50 растворили в 100 мл дихлорметана. Этот раствор подвергли распылительной сушке в минисушилке Бюхи 190 для распылительной сушки. Температуру на входе задали равной 50oC, а температуру на выходе, согласно результатам измерений, составляла 42oC. Оптическое микроскопическое изучение показало образование микрочастиц.
ae) Нанесение покрытия на частицы, изготовленные из полимера, полученного из метилен-бис(16-окси-гексадеканоата) и адипоилхлорида.
Частицы, изготовленные в соответствии с экспериментом вышеприведенного примера 3c, повторно диспергировали в несколько водных растворах различных материалов для нанесения покрытий, содержащихся в различных концентрациях, как это указано в табл. 5. Оптическое микроскопическое изучение показало, что образовалась улучшенная дисперсия с пониженной тенденцией частиц к агрегированию.
af) Покрытие продуктов Плуроник®F68 частицы полимера, полученного из метилметакрилоилоксиметилкарбоната
0,9 г полимера из метилметакрилоилоксиметилкарбоната из эксперимента вышеприведенного примера 2 растворили в 9 мл толуола. Затем добавили 30 мл воды, содержащей 0,4 г цетилтриметиламмонийхлорида, и смесь гомогенизировали с помощью гомогенизатора Истрал®. Приготовленную эмульсию высушили вымораживанием в течение 24 ч. Полученные частицы несколько раз промыли дистиллированной водой с целью удалить из них поверхностно-активное вещество. После заключительной промывки частицы высушили вымораживанием в течение 24 ч.
Пример 4. Акустические характеристики. Общая процедура:
Сухие порошки полимерных частиц, приготовленные в соответствии с вышеизложенным в примере 3, повторно диспергировали в водном растворителе путем встряхивания в лабораторном шейкере в течение 12-16 ч. Изучение оптическое микроскопией показало образование дисперсий частиц. Частицы обладали хорошей плавучестью, как и ожидалось от частиц, содержащих газ.
Акустический эффект у вышеуказанных суспензий был установлен измерением ультразвуковой проницаемости суспензий различных концентраций (мг/мл) в водном жидком носителе с помощью преобразователя на 3,5 мгГц с расширенной полосой частот согласно импульсно-отражательному методу. В качестве эталона использовали чистый жидкий носитель, причем измерения производили вдоль линии разбавления, где исходную суспензию последовательно разбавляли жидким носителем. Измерения производили до момента ослабления сигнала приблизительно до 3-5 дБ-см. Полученные акустические эффекты оказались на таком уровне, что можно было ожидать полезность продуктов в качестве ультразвуковых контрастных агентов. В соответствии с теоретическими положениями, сплошные частицы в противоположность газосодержащим того же размера при той же степени разбавления должны давать акустическое ослабление менее чем 0,1 дБ/см.
a) Характеристики частиц полимера, полученного сополимеризацией метилендиметакрилата и стирола.
Частицы были изготовлены из полимера, полученного сополимеризацией метилендиметакрилата и стирола. Продукт проявлял сильное действие на передачу акустического сигнала, которое ослаблялось с увеличением объема разбавляющей среды, как это можно видеть на фиг. 1 прилагаемых рисунков.
b-i) Характеристика различных полимерных частиц.
Результаты суммированы в табл. 6 на фиг. 2-9 прилагаемых рисунков
В таблице 6 приведены акустические измерения частиц из эксперимента вышеприведенного примера 3. Акустические измерения представлены в колонке 3 в виде концентрации, при которой в результате измерения контрастного эффекта получили 8 дБ/см, то есть половину величины посылаемого сигнала. При более высоких концентрациях интенсивность сигнала повышалась до наблюдаемого насыщения.
Пример 5. Характеристика ин виво. Общая процедура.
Сухой порошок полимерных частиц, которые были описаны в примере 3, повторно диспергировали в стерильном водном растворе хлористого натрия концентрацией 0,9 вес.% путем встряхивания в лабораторном шейкере в течение 12-16 ч. Такие дисперсии вводили инъекциями кроликам шиншилла и измерения производили с использованием доплеровской техники, согласно которой ультразвуковой зонд помещали непосредственно на сонную артерию и нижнюю квальную вену. Дисперсии частиц вводили инъекциями в ушную вену. При этом фиксировали амплитуду и длительность сигналов. Полученные амплитуды сигналов оказались весьма значительными, что указывало на сильный ультразвуковой контрастный эффект дисперсий ин виво. Большая длительность сигнала указывала на то, что стабильность ин виво была хорошей.
В таблице 7 приведена характеристика полимерных частиц ин виво. Дозы выражались в мигрограммах частиц на килограмм живого веса. Интенсивность сигнала измеряли в доплеровских единицах (ДЕ).
Пример 6. Изучение биоразлагаемости.
a) Катализируемый энзимом гидролиз полимера из метакрилоилоксиметилбензоата.
В каждую из трех реакционных склянок ввели по 50 мг полимера (пример 2 г) в виде тонкодисперсного порошка и 20 мл 0,9%-ного водного раствора хлористого натрия. В одну из этих склянок добавили также 0,1 мл эстеразы из свиной печени в 3,2 M раствора сульфата аммония (Sigma E-3128 250U). В другую склянку добавили 0,1 мл 3,2 M раствора сульфата аммония. Добавлением 0,1 M раствора гидроокиси натрия величину pH в каждой из склянок поддерживали на уровне 8,0 с помощью pH-стата (Radiometer). Отмечая расход гидроокиси натрия, рассчитывали скорость гидролиза. Было установлено, что в течение 45 ч при температуре 37oC гидролиз полимера с эстеразой протекал в 11 раз быстрее, чем в контрольном эксперименте с использованием сульфата аммония, но без эстеразы. В контрольном же эксперименте, в котором полимер находился в 0,9%-ном растворе хлористого натрия, никакого гидролиза обнаружено не было (см. фиг. 10).
В таблице 8 приведен расход 0,1 M раствора гидроокиси натрия в склянке, содержащей полимер и эстеразу с 0,1 мл 3,2 M раствора сульфата аммония в 20 мл 0,9%-ного водного раствора хлористого натрия: (см. участок (a) фигуры 10)
В таблице 9 приведен расход 0,1 M раствора гидроокиси натрия в контрольной среде, содержащей 0,1 мл 3,2 M раствора сульфата натрия в 20 мл 0,9%-ного раствора хлористого натрия (см. участок (b) фигуры 10)
В таблице 10 приведен расход 0,1 M раствора гидроокиси натрия в контрольном эксперименте в среде, содержащей полимер в 20 мл 0,9%-ного раствора хлорида натрия (см. участок (c) фиг. 10)
b) Катализируемый энзимом гидролиз полимера из метилен-бис(16-оксигексадеканоата) и адипоилхлорида.
В каждую из трех реакционных склянок добавили 50-миллиграммовые образцы полимера (пример 2b) в виде тонкодисперсного порошка и по 20 мл 0,9%-ного раствора хлорида натрия. В одну из склянок добавили также 0,1 мл эстеразы из свиной печени в 3,2 M раствора сульфата аммония (Sigma E-3128, 250U). В другую склянку добавили 0,1 мл 3,2 M раствора сульфата аммония. Добавлением 0,1 M раствора гидроокиси натрия величину pH в каждой из склянок поддерживали на уровне 8,0 с помощью pH-стата (Rad Oneer). Отмечая расход гидроокиси натрия, рассчитывали скорость гидролиза. Было установлено, что в течение 44 ч при температуре 37oC гидролиз полимера с эстеразой протекал в 10 раз быстрее, чем в контрольном эксперименте с использованием сульфата аммония, но без эстеразы. В контрольном же эксперименте, в котором полимер находился в 0,9%-ном растворе хлористого натрия, никакого гидролиза обнаружено не было (см. фиг. 11).
В таблице 11 приведен расход 0,1 M раствора гидроокиси натрия в склянке, содержащей полимер и эстеразу с 0,1 мл 3,2 M раствора сульфата аммония в 20 мл 0,9%-ного раствора хлористого натрия (см. участок (a) фиг. 11)
В таблице 12 приведен расход расход 0,1 M раствора гидроокиси натрия в среде контрольного эксперимента, содержавшей 0,1 мл 3,2 M раствора сульфата аммония в 20 мл 0,9%-ного раствора хлорида натрия (см. участок (b) фиг. 11)
В таблице 13 приведен расход 0,1 M раствора гидроокиси натрия в среде контрольного эксперимента, содержавшей полимер в 20 мл 0,9%-ного раствора хлористого натрия (см. участок (c) фиг. 11)
Источники информации
1. Фолкманн М., Ланд Ф.Дж. Synthesis. - 1990, 1159.
2. Беннехе Т., Странде П., Вигген. У., Acta chem. Scand. 43, 1988, -74.
3. Строхольм И., Копечек И. Angew Macromol. Chemic 70, 1978, 109.
| название | год | авторы | номер документа |
|---|---|---|---|
| БИОРАЗЛАГАЕМЫЕ НЕСШИТЫЕ ПОЛИМЕРЫ | 1993 |
|
RU2114865C1 |
| ИОДИРОВАННЫЕ АРИЛЬНЫЕ СОЕДИНЕНИЯ И ДИАГНОСТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1995 |
|
RU2145955C1 |
| ЙОДИРОВАННЫЕ СЛОЖНЫЕ ЭФИРЫ | 1990 |
|
RU2088579C1 |
| СПОСОБ ПОЛУЧЕНИЯ 5-ЗАМЕЩЕННЫХ ПИРРОЛО (2,3-α)ПИРИМИДИНОВ | 1993 |
|
RU2127274C1 |
| Бис-аммониевые соединения на основе пиридоксина, обладающие антибактериальными и антимикотическими свойствами | 2020 |
|
RU2731999C1 |
| ПЕПТИДНЫЕ ПРОИЗВОДНЫЕ, ИХ СТЕРЕОИЗОМЕРЫ ИЛИ ФИЗИОЛОГИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, ОБЛАДАЮЩИЕ ПРОТИВОТРОМБОЗНОЙ, ПРОТИВОСВЕРТЫВАЮЩЕЙ ИЛИ ПРОТИВОВОСПАЛИТЕЛЬНОЙ АКТИВНОСТЬЮ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ПОДАВЛЕНИЯ ТРОМБИНА, СПОСОБ ПОДАВЛЕНИЯ КИНИНОГЕНАЗ, ПРИМЕНЕНИЕ СОЕДИНЕНИЙ В КАЧЕСТВЕ ИСХОДНЫХ В СИНТЕЗЕ ИНГИБИТОРА ТРОМБИНА | 1994 |
|
RU2142469C1 |
| ЗАМЕЩЕННЫЕ ПИРАЗОЛЫ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ, СПОСОБ ЛЕЧЕНИЯ, ПРОМЕЖУТОЧНЫЙ ПРОДУКТ | 1993 |
|
RU2130453C1 |
| КОНЪЮГАТ ФОТОСЕНСИБИЛИЗАТОРА И ХИТОЗАНА И ЕГО ПРИМЕНЕНИЕ | 2013 |
|
RU2675830C2 |
| СПОСОБ ПОЛУЧЕНИЯ МЕТИЛСИЛОКСАНОВЫХ СОПОЛИМЕРОВ С АЗИДОПРОПИЛЬНЫМИ И ГИДРИДСИЛИЛЬНЫМИ ФУНКЦИОНАЛЬНЫМИ ГРУППАМИ | 2021 |
|
RU2799808C2 |
| 1-АРИЛПИРРОЛО[1,2-A]ПИРАЗИН-3-КАРБОКСАМИДЫ С НЕЙРОПСИХОТРОПНОЙ АКТИВНОСТЬЮ | 2014 |
|
RU2572076C2 |
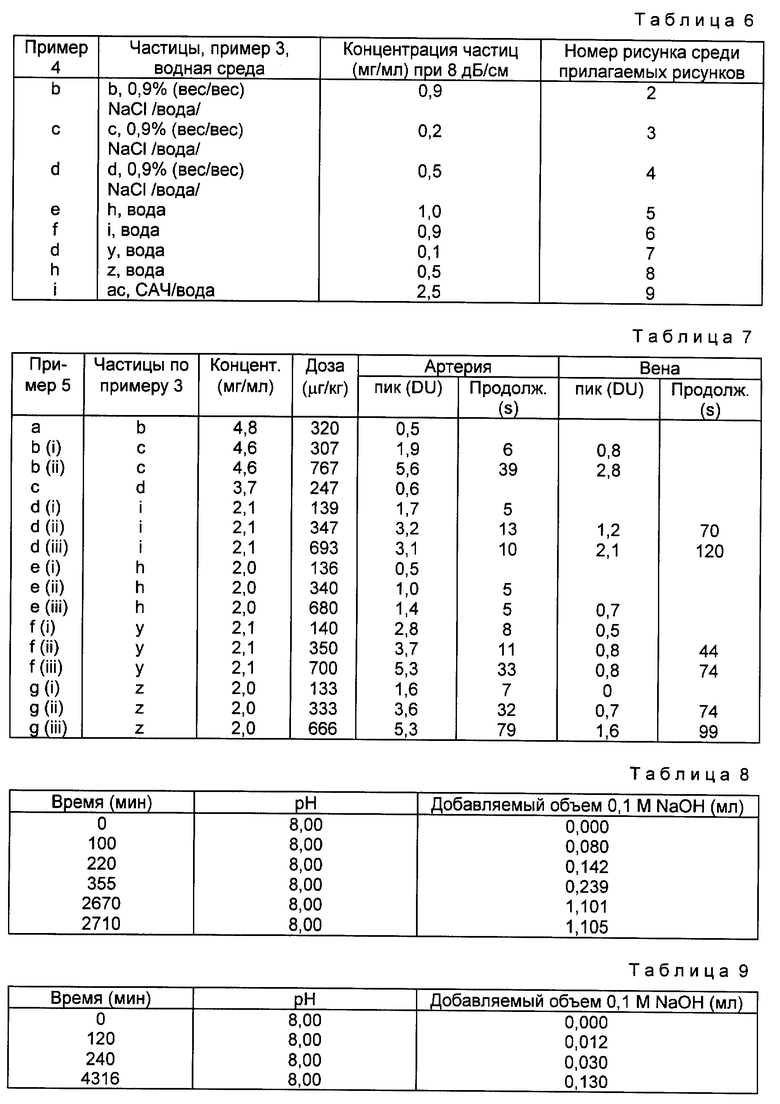
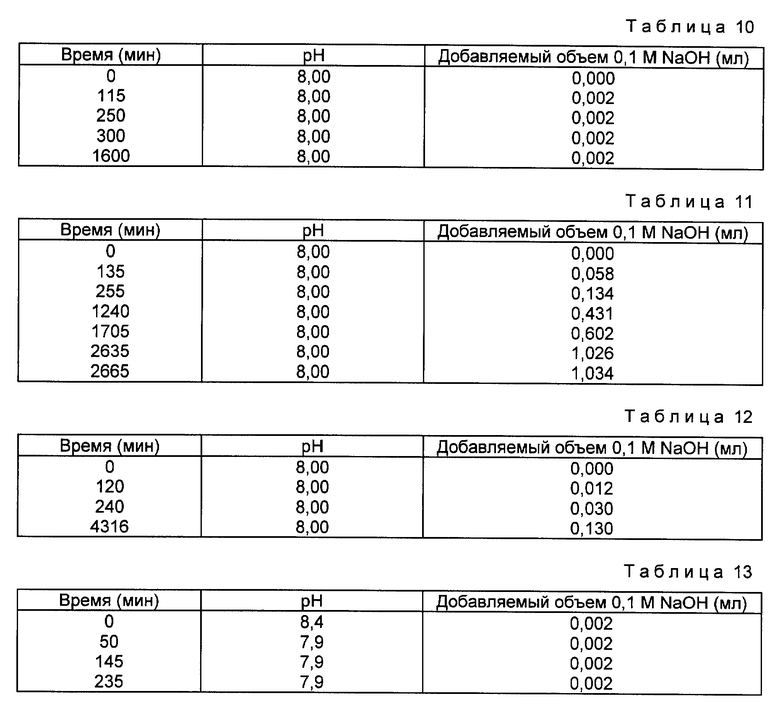

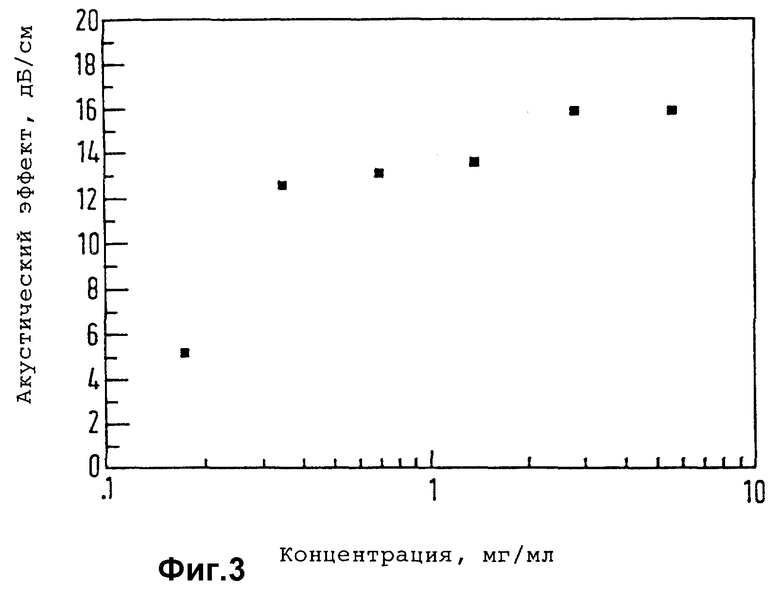
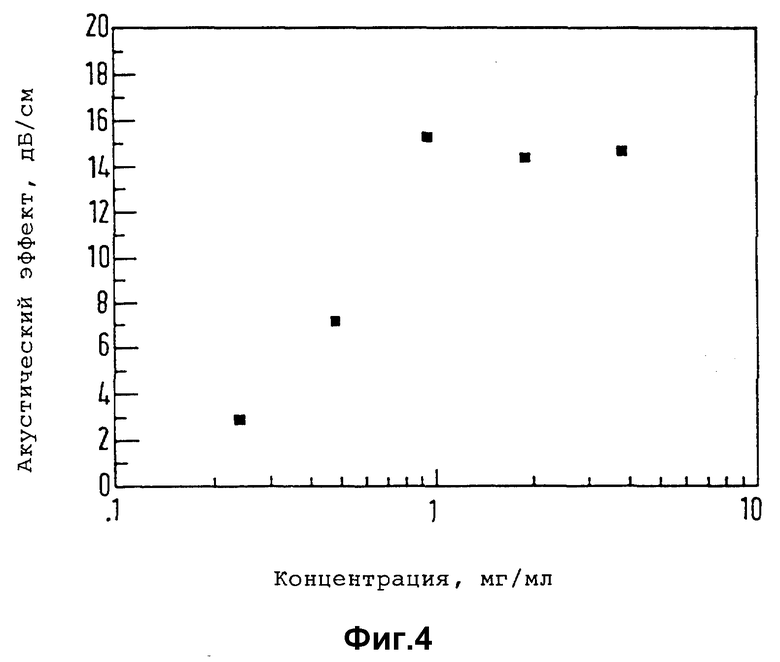
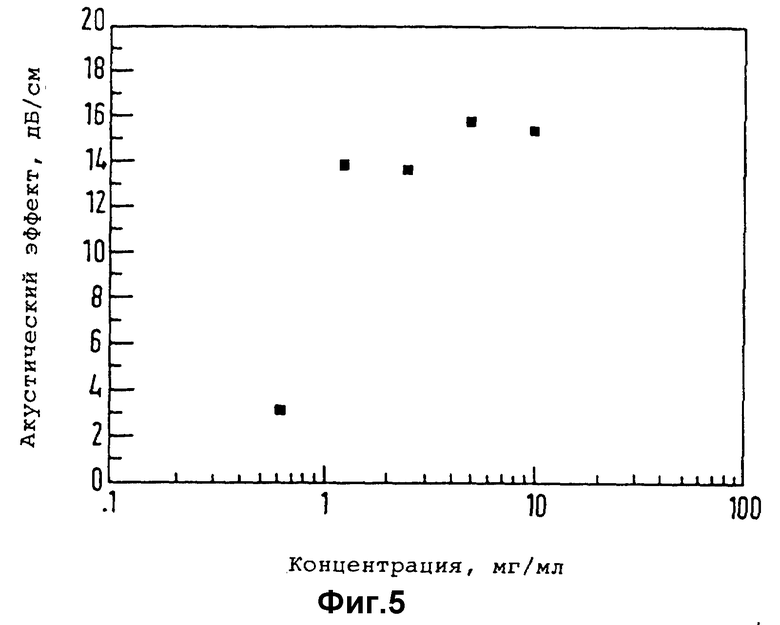
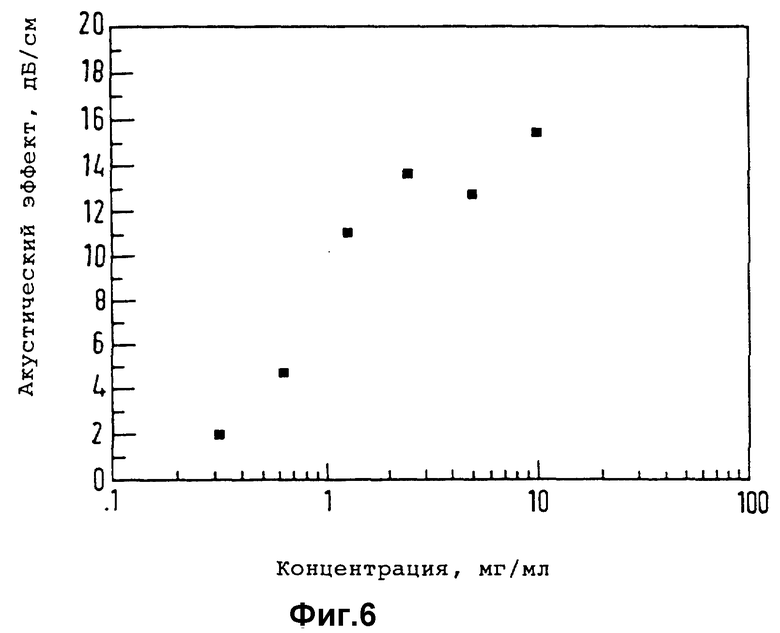
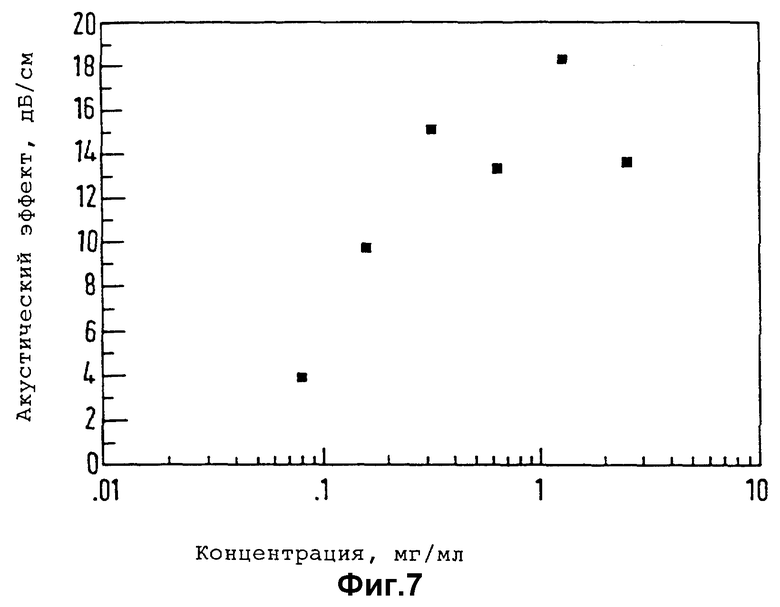
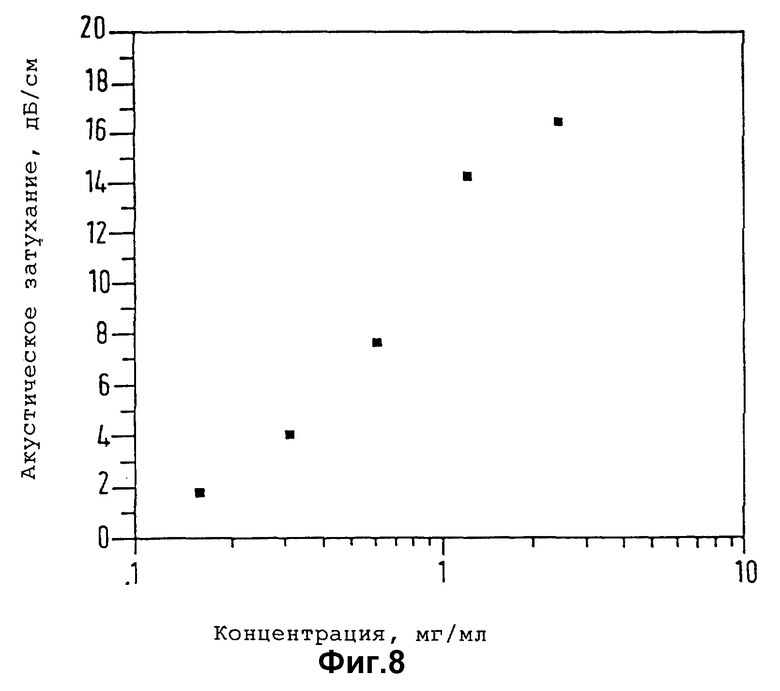
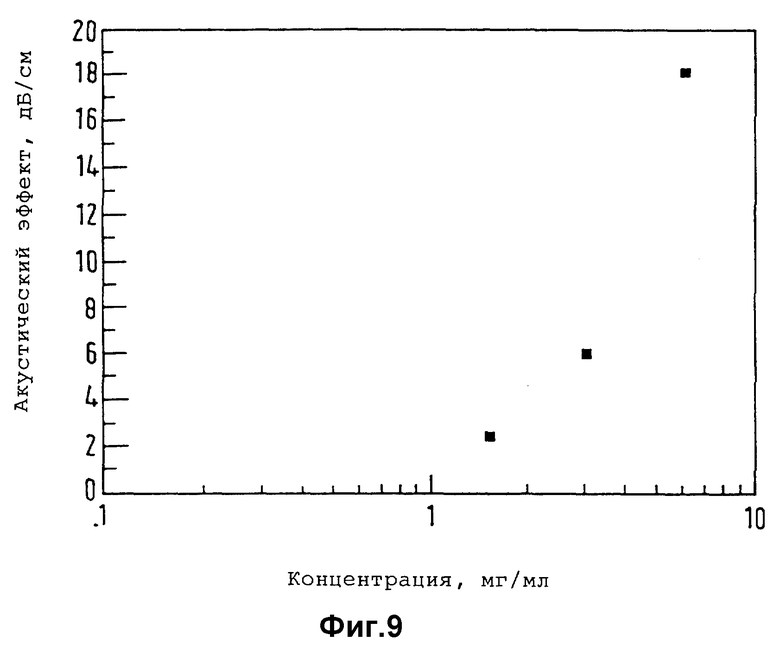
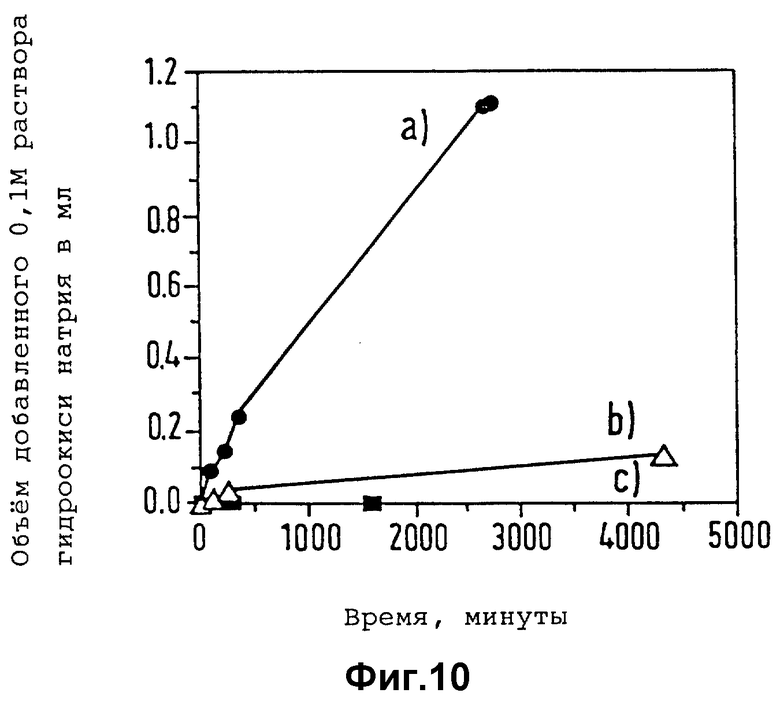
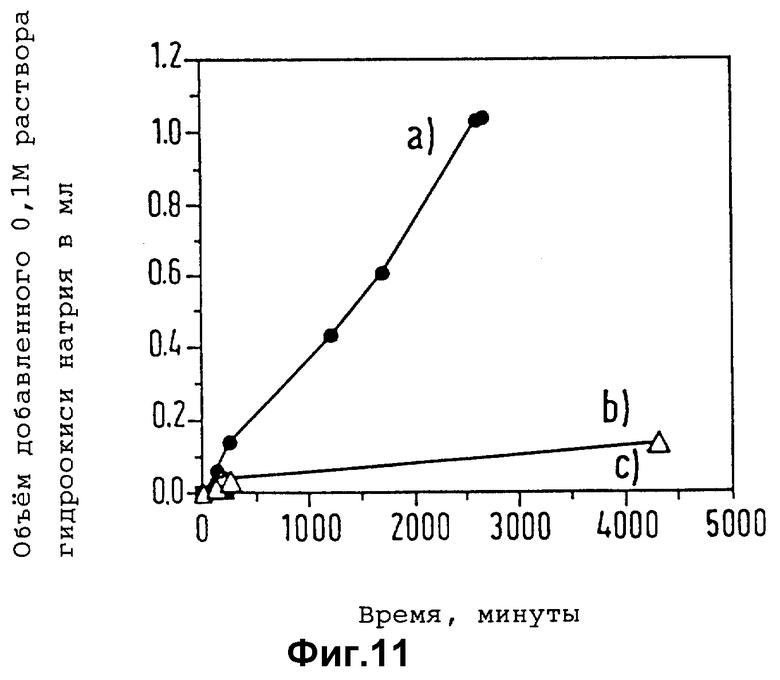
Контрастный агент может быть использован в диагностических областях техники, в частности при получении ультразвукового и магнитно-резонансного изображения. Контрастный агент представляет собой газосодержащие или газовыделяющие полимерные микрочастицы и/или микропузырьки, где полимер представляет собой биоразлагаемый полимер неполипептидной природы, молекулы которого содержат звенья формулы 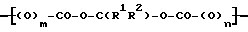 (II), где каждый из R1 и R2 - водородный атом, или связанные с углеродом одновалентные органические группы, или же совместно они образуют связанную с углеродом двухвалентную органическую группу, а каждый из m и n независимо от другого равен 0 или 1. Данный контрастный агент получают путем введения газа или газогенерирующего агента в биоразлагаемый полимер формулы II, и он обеспечивает получение улучшенного изображения тела человека или животного путем генерирования ультразвукового или МК-изображения. 3 с. и 13 з.п. ф-лы, 13 табл., 11 ил.
(II), где каждый из R1 и R2 - водородный атом, или связанные с углеродом одновалентные органические группы, или же совместно они образуют связанную с углеродом двухвалентную органическую группу, а каждый из m и n независимо от другого равен 0 или 1. Данный контрастный агент получают путем введения газа или газогенерирующего агента в биоразлагаемый полимер формулы II, и он обеспечивает получение улучшенного изображения тела человека или животного путем генерирования ультразвукового или МК-изображения. 3 с. и 13 з.п. ф-лы, 13 табл., 11 ил.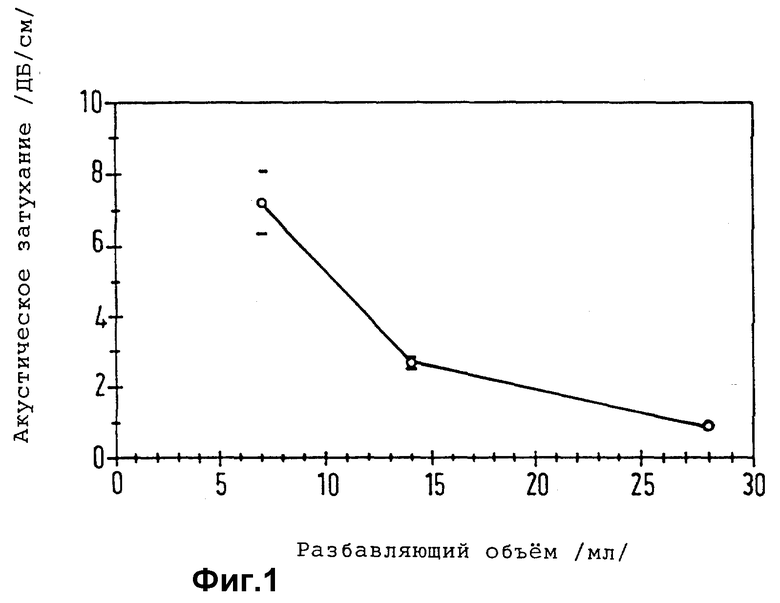
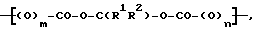
где каждый из R1 и R2 представляет собой водородный атом или связанную с углеродным атомом одновалентную органическую группу, R1 и R2 совместно образуют связанную с углеродом двухвалентную органическую группу;
m и n, которые могут быть как идентичными, так и различными, обозначают 0 или 1.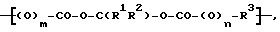
где m, n, R1 и R2 определены в п.1;
R3 - двухвалентный органический радикал.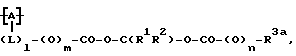
где m, n, R1 и R2 определены в п.1;
A - повторяющееся звено неполипептидной полимерной основной цепи;
L - связывающая группа;
l = 0 или 1;
R3a - липофильная органическая группа,
благодаря чему указанная липофильная группа при биологическом разложении способна отщепляться с образованием водорастворимого полимера.
где значения радикалов определены в п.1,
для образования полимерных микрочастиц и/или микропузырьков.
| EP 0458745 A1, 27.11.91 | |||
| Бликер Х | |||
| и др | |||
| По вопросу пименения констрастных агентов ультразвукового изображения для измерения скорости потока крови и определения сердечной перфузии | |||
| Ультразвуковая медицина | |||
| Способ приготовления консистентных мазей | 1919 |
|
SU1990A1 |
| Рельсовое стыковое скрепление | 1922 |
|
SU461A1 |
