Изобретение относится к области аналитической химии, а именно исследованию и анализу материалов путем выделения их из сложных матриц.
К настоящему времени известен ограниченный ряд методов, позволяющих достоверно извлекать β-хлорвинилдихлорарсин (люизит) из проб сложных матриц, в том числе и из воды. Особенно это необходимо при проведении эколого-аналитического контроля при определении содержания люизита в воде на уровне предельно допустимой концентрации (ПДК=2•10-4 мг/л). Наиболее чувствительный метод, основанный на конверсии люизита до ацетилена с последующим газохроматографическим определением его с применением детектора ионизации в пламени (ДИП), позволяет определять 2.5•10-5 мг люизита. Следовательно, для анализа на уровне ПДК необходимо концентрирование из 100 мл воды, а с учетом неизбежных потерь и из большего объема пробы.
Известен способ извлечения люизита из проб грунта путем жидкостной экстракции его различными растворителями с последующим определением фотометрическим методом по количеству выделяющегося ацетилена. (Руководство по работе в автомобильной радиометрической и химической лаборатории АЛ-4М. М.: Воениздат, 1988 г. , стр. 54-57). Основными недостатками данного метода является то, что коэффициент экстракции составляет 40-60%. Более того, данный способ совершенно не пригоден для извлечения люизита из воды и особенно на уровне микроконцентраций.
Существует способ извлечения люизита из растворов экстрагированием гексаном с последующим упариванием растворителя (Станьков И.Н. и др. Химико-хроматографическое определение β-хлорвинилдихлорарсина в воздухе и реакционных смесях его переработки. Журн. аналит. химии, 1996 г., Т 51, N 5, стр. 528-532). К недостаткам этого метода следует отнести то, что, во-первых, определение низких концентраций вещества сопряжено с большим расходом растворителя (экстрагента) и, во-вторых, при упаривании раствора испаряется существенное количество люизита вследствие его высокой летучести, что в свою очередь приводит к большой погрешности анализа.
Разработан способ предварительного концентрирования люизита на адсорбенты: тенакс, хромосорб-1, хромосорб-106 и др. (Fowler N.K., sRi-Арс-1023-6840-8, Souther Research Institute, 1990). Недостатком данного способа является то, что при последующей термодесорбции или элюировании невозможно определение микроколичеств анализируемого вещества. Однако данный способ по своему техническому решению наиболее близок к заявляемому объекту и поэтому выбран в качестве прототипа.
Задачей настоящего изобретения являлось увеличение степени извлечения люизита из водных растворов для последующего определения на уровне предельно допустимых концентраций.
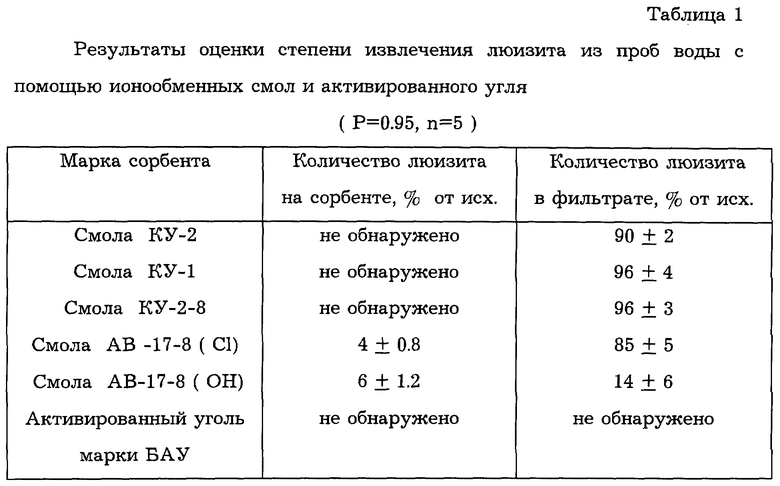
Чтобы исключить нежелательную стадию десорбции, представлялось целесообразным определять люизит по количеству выделяющегося ацетилена при воздействии щелочи непосредственно на сорбент с веществом. Предварительно исследовали возможность применения для этих целей различных типов ионообменных смол и сорбентов. Для этого раствор люизита (10 мл) с концентрацией 0,01 мг/мл пропускали через колонку с соответствующим сорбентом. Степень извлечения люизита оценивали по его остаточному содержанию в фильтрате фотометрическим методом. Сорбент с люизитом обрабатывали раствором щелочи и по количеству выделяющегося ацетилена оценивали возможность определения люизита непосредственно на поверхности сорбента. Было показано, что практически ни один из исследованных сорбентов не приводит к достижению поставленной цели (см. табл. 1). При этом невозможность прямого определения на активированном угле, возможно, обусловлена его развитой поверхностью, а на анионите АВ-17-8 (OH-форма) - химической агрессивностью последнего по отношению к люизиту.
В соответствии с этим было высказано предположение о необходимости модификации сорбента (желательно с неразвитой поверхностью) веществом, специфично связывающим люизит. Известно, что 1,2-диолы образуют с люизитом устойчивые циклические соединения, именно на этом основано применение некоторых из них для лечения и профилактики поражений люизитом (Александров, В. Емельянов. Отравляющие вещества. М., Воениздат, 1990 г., стр. 149). Необходимым условием связывания соединения на поверхности ионообменной смолы является наличие в его структуре анионной или катионной группы. Наиболее доступным веществом, сочетающим в себе необходимые свойства, является натриевая соль 2,3-димеркаптопропансульфокислоты (унитиол). Наличие в структуре унитиола сульфоанионной группы позволяет привить его на поверхности анионита.
Таким образом, решение поставленной задачи достигается тем, что в качестве адсорбента используется анионная смола, модифицированная натриевой солью 2,3-димеркаптопропансульфокислоты.
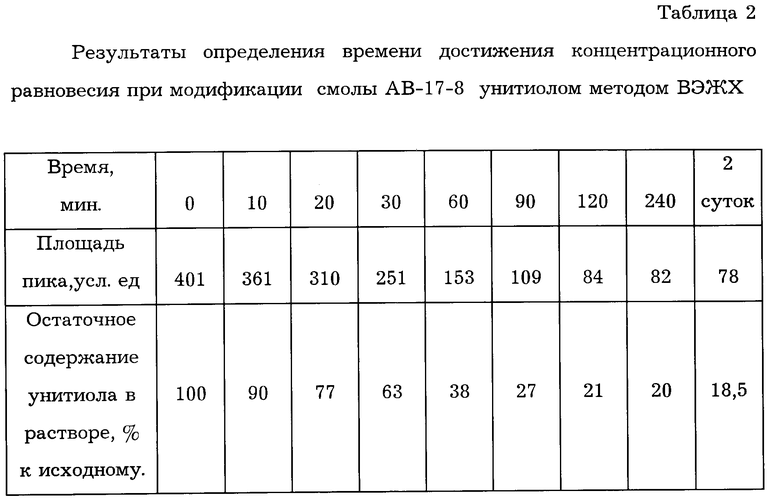
В качестве примера для модификации был выбран анионит АВ-17-8. Для модификации анионита сухую смолу АВ-17-8 (Cl-форма) заливали унитиолом из расчета 5±0,2 мл 5% водного раствора унитиола на 1±0,1 г смолы. Несмотря на то, что ионообменные процессы идут довольно быстро, в случае унитиола концентрационное равновесие наступает не сразу. Определение времени достижения концентрационного равновесия при модификации поверхности смолы проводили методом высокоэффективной жидкостной хроматографии (ВЭЖХ) по оценке остаточного содержания унитиола в растворе. Результаты исследований приведены в табл.2.
Из данных, представленных в табл.2, следует, что концентрационное равновесие при модификации смолы унитиолом наступает через 1,5 -2,0 часа.
Пример оценки степени извлечения и возможности последующего определения люизита из проб воды
Модифицированной смолой набивали колонку диаметром 4 мм и высотой столба смолы 60±5 мм, что соответствует 0,5±0,1 г сорбента.
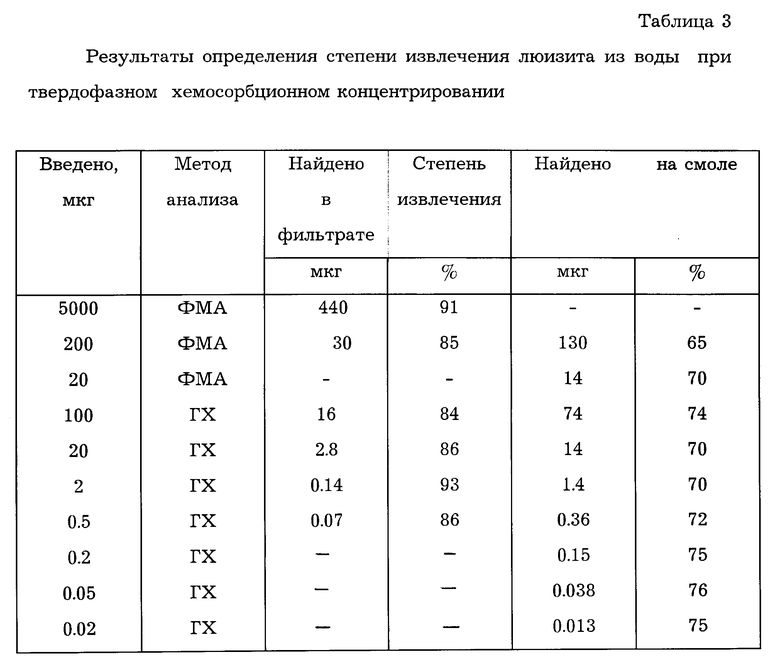
Исходный раствор готовили на этиловом спирте из a-люизита с действующим началом 95±2% концентрацией 1 мг/мл. Из него путем соответствующего разведения в воде готовили серии стандартных растворов с концентрациями от 0,1 до 1•10-7 мг/мл. Полученные растворы пропускали через колонку с хемосорбентом со скоростью 3±0,5 мл/мин. Полноту сорбции оценивали по остаточному содержанию люизита в фильтрате. После этого сорбент переносили в дрексель или пенициллиновый пузырек в зависимости от последующего вида анализа и обрабатывали 7 мл водного раствора 30% щелочи. По количеству выделяющегося ацетилена определяли содержание люизита и оценивали принципиальную возможность непосредственного определения хемосорбированного люизита на поверхности анионита предлагаемым способом. Результаты исследований представлены в табл.3.
Как следует из результатов, приведенных в табл.3, степень извлечения люизита из проб воды составляет 87±6%. Установлено, что химически сорбированный люизит выделяет в 1,18 раза меньшее количество ацетилена при воздействии щелочи в условиях опыта по сравнению с аналогичным количеством люизита в стандартных растворах. В то же время значительно удобнее пользоваться калибровками на стандартных растворах, поэтому при определении люизита на смоле необходимо учитывать экспериментально найденный поправочный коэффициент.
Количество сорбированного унитиола, находящегося в колонке в условиях опыта, составляет примерно 0,6 ммоль, что является заведомо избыточным и по расчетам позволяет связывать более 100 мг люизита. В то же время, снижение количества унитиола в 10 раз приводит к уменьшению полноты извлечения до 36-43%. Поэтому уменьшение содержания унитиола является нецелесообразным.
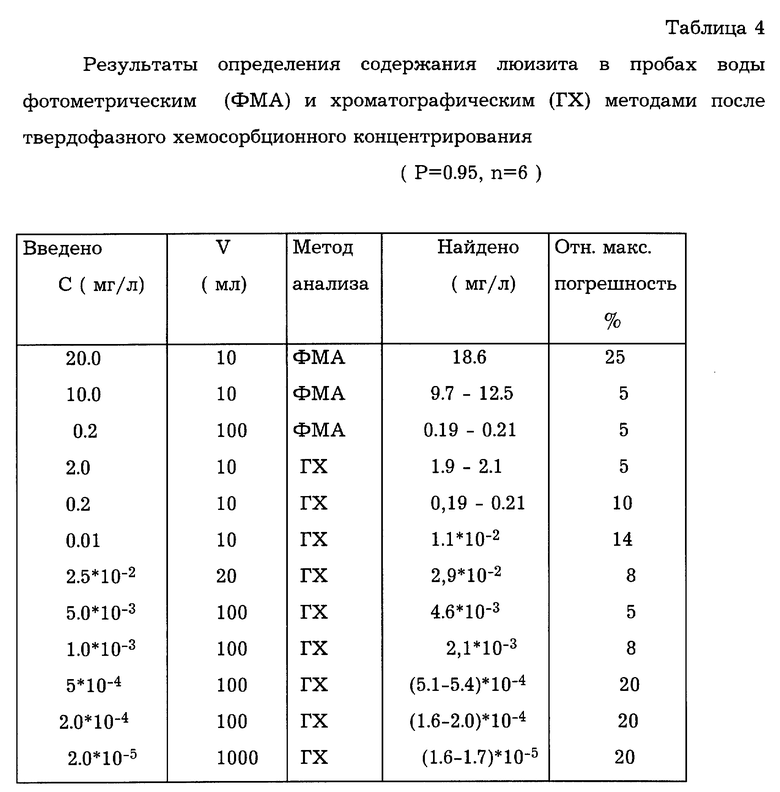
Для оценки правильности определения люизита в воде растворы с известной концентрацией пропускали через колонки с сорбентом. Объем раствора брали с учетом чувствительности последующего метода анализа. Расчет результатов анализа проводили с учетом степени извлечения люизита. Результаты оценки правильности определения люизита в пробах воды фотометрическим и газохроматографическим методами, после твердофазного хемосорбционного концентрирования, приведены в табл.4.
Таким образом, использование предлагаемого способа твердофазного хемосорбционного концентрирования позволяет достоверно извлекать люизит из проб воды для последующего определения его различными методами анализа в широком диапазоне концентраций, в том числе и ниже уровня ПДК.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ РАЗДЕЛЬНОГО ОПРЕДЕЛЕНИЯ α-, β-ЛЮИЗИТА, ИХ ОКСИДОВ И ХЛОРВИНИЛАРСОНОВЫХ КИСЛОТ ПРИ СОВМЕСТНОМ ПРИСУТСТВИИ В ПОЧВЕ | 2000 |
|
RU2201780C2 |
| СПОСОБ РАЗДЕЛЬНОГО ОПРЕДЕЛЕНИЯ В ВОДЕ β-ХЛОРВИНИЛДИХЛОРАРСИНА НА УРОВНЕ ПДК И β-ХЛОРВИНИЛАРСОНОВОЙ КИСЛОТЫ | 1998 |
|
RU2163016C2 |
| СПОСОБ ОПРЕДЕЛЕНИЯ β-ХЛОРВИНИЛДИХЛОРАРСИНА В ВОЗДУХЕ НА УРОВНЕ ПДК | 1998 |
|
RU2146364C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ β-ХЛОРВИНИЛАРСИНОКСИДА В ВОЗДУХЕ НА УРОВНЕ ПДК | 2000 |
|
RU2202782C2 |
| СОСТАВ ЭКСТРАГЕНТА ДЛЯ ИЗВЛЕЧЕНИЯ β-ХЛОРВИНИЛАРСИНОКСИДА ИЗ ПРОБ ПОЧВЫ | 1996 |
|
RU2129454C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ЛЮИЗИТА В ВОДНО-СПИРТОВЫХ ЭКСТРАКТАХ | 1998 |
|
RU2141108C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ МАССОВОЙ КОНЦЕНТРАЦИИ ЛЮИЗИТА В ВОДЕ, СОДЕРЖАЩЕЙ ИПРИТ, ГАЗОХРОМАТОГРАФИЧЕСКИМ МЕТОДОМ С ПРИМЕНЕНИЕМ ПЛАМЕННО-ИОНИЗАЦИОННОГО ДЕТЕКТОРА | 2011 |
|
RU2472149C1 |
| СПОСОБ УТИЛИЗАЦИИ ОТРАВЛЯЮЩЕГО ВЕЩЕСТВА КОЖНО-НАРЫВНОГО ДЕЙСТВИЯ ТИПА ЛЮИЗИТ | 1999 |
|
RU2172196C2 |
| СПОСОБ ДЕТОКСИКАЦИИ ОТХОДОВ СТРОИТЕЛЬНЫХ МАТЕРИАЛОВ, ЗАГРЯЗНЕННЫХ ЛЮИЗИТОМ И ПРОДУКТАМИ ЕГО ПРЕВРАЩЕНИЙ | 2009 |
|
RU2460596C2 |
| СПОСОБ ОЦЕНКИ МИКРОСОМАЛЬНОЙ СИСТЕМЫ ПЕЧЕНИ ПРИ ДЕЙСТВИИ КСЕНОБИОТИКОВ АЛКИЛИРУЮЩЕГО ТИПА | 1995 |
|
RU2104539C1 |
Область использования: аналитическая химия, а именно область исследования и анализа материалов путем выделения их из сложных матриц. Данный способ может быть использован в системах войсковой и промышленной индикации, при оценке глубины распространения опасных концентраций вредных веществ, на объектах по уничтожению химического оружия, а также при решении задач по проведению экологического мониторинга. Для осуществления способа хемосорбционного концентрирования β-хлорвинилдихлорарсина (люизита) в качестве адсорбента используют анионную смолу, модифицированную водным раствором натриевой соли 2,3-димеркаптопропансульфокислоты (унитиола), а определение количества извлеченного вещества люизита проводят по количеству ацетилена, выделившегося при обработке сорбента водным раствором щелочи. Способ обеспечивает увеличение степени извлечения β-хлорвинилдихлорарсина из водных растворов путем твердофазного хемосорбционного концентрирования для последующего определения на уровне предельно допустимых концентраций. 1 з.п. ф-лы, 4 табл.
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Fowler N.K., sRi - Arc - 1023-6840-8, Souther Research Institute, 1990 | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Способ очистки сточных вод производства инсектицидных препаратов | 1985 |
|
SU1303557A1 |
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Способ извлечения пестицидов из водных растворов | 1986 |
|
SU1331832A1 |
| Очаг для массовой варки пищи, выпечки хлеба и кипячения воды | 1921 |
|
SU4A1 |
| Способ очистки воды от растворенных органических веществ | 1990 |
|
SU1799360A3 |
| Кипятильник для воды | 1921 |
|
SU5A1 |
| RU 94028249 A1, 1996 | |||
| Приспособление для точного наложения листов бумаги при снятии оттисков | 1922 |
|
SU6A1 |
| US 4648977 A, 1987 | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| US 4775475 A1, 1988 | |||
| Топка с несколькими решетками для твердого топлива | 1918 |
|
SU8A1 |
| DE 3542065 A, 1987 | |||
| Разборный с внутренней печью кипятильник | 1922 |
|
SU9A1 |
| EP 0287729 A1, 1988 | |||
| Печь-кухня, могущая работать, как самостоятельно, так и в комбинации с разного рода нагревательными приборами | 1921 |
|
SU10A1 |
| Устройство для подвода охлаждающей жидкости к полым проводникам обмотки ротора электрической машины | 1973 |
|
SU483738A2 |
| Походная разборная печь для варки пищи и печения хлеба | 1920 |
|
SU11A1 |
| Огнетушитель | 0 |
|
SU91A1 |
