Изобретение направлено на контрастные средства, в частности на частицы контрастного средства в виде оксида металла, покрытого полиэлектролитом, для применения в МР, рентгеновских, ЭИТ и магнитометрических исследованиях, особенно там, где такие частицы оксида металла проявляют суперпарамагнитные свойства.
Применение контрастных средств в медицинских диагностических технологиях для улучшения контрастности тканей или для облегчения исследования процессов в организме хорошо устоялось. Вид улучшения контрастности варьирует в зависимости от способа получения изображения, но в магнитно-резонансной томографии большинство традиционных контрастных средств получают свою способность улучшать контрастность от их воздействия на время выбора ткани.
Одним из значительных преимуществ МР томографии является высокое качество внутритканевого контрастирования, возникающего в зависимости от времени релаксации ткани. Исходно считалось, что даже без добавления контрастных средств параметры релаксации могут быть использованы для различения нормальной и патологической тканях (смотри, Damadian, Science 171:1151-1153 (1971)). Однако, как только первые МР томограммы были получены Lauterbur (смотри Nature 242: 190-191 (1973)), стало ясно, что дифференцировать аномальную ткань от нормальной наверняка было невозможно. Таким образом, теперь в течение некоторого времени представляется весьма интересным применение материалов, которые улучшают контрастность путем воздействия на ключевые параметры контрастирования. Впервые описала применение МР контрастных средств у животных группа Lauterbur (смотри Lauterbur et al., Frontiers of Biological Energetics, New York, Academic Press 1978, с. 752). Потенциал внутривенно вводимого контрастного средства для диагностики в клинике был показан Carr et al. в 1984 году (смотри Carr et al., AJR 143:215-224 (1984)), а первое МР контрастное средство, GdDTPA, получило одобрение для клинического применения в 1988 году.
На сегодняшний день хорошо документально подтверждено, что GdDTPA и сходные вещества, такие как GdDTPA-BMA, GdHPFO3A и GdDOTA, являются безопасными и выгодными для усиленной МР томографии центральной нервной системы. Благодаря своей низкой молекулярной массе и гидрофильным свойствам, данные хелатные соединения металлов распределяются экстрацеллюлярно и быстро выводятся почками. В последнее время были разработаны другие контрастные средства с улучшенными фармакокинетическими свойствами, разрешающими более специфическое распределение по органам и в зависимости от заболевания.
Вообще говоря, существуют два подхода, которые могут быть использованы для улучшения доставки МР контрастного средства в область-мишень. В соответствии с традиционным подходом усилия направлены на применение меченых парамагнетиком природных или синтетических молекул или макромолекул со специфичным накоплением или локализацией (например, гепатобилиарные средства, средства, накапливающиеся в кровяном русле, порфирины). Альтернативным подходом является применение более сильных магнитных меток, таких как частицы суперпарамагнетика, которые накапливаются в желаемой области благодаря своей особой природе или посредством их связывания со специфическими молекулами-мишенями. В целом, диагностически выгодные отношения мишень/фон суперпарамагнитных средств значительно выше, чем таковые парамагнитных средств, и, таким образом, суперпарамагнитные средства могут определяться при очень низких тканевых концентрациях (смотри Weissleder et al., Magn. Reson. Quart 8: 55-63 (1992)).
Применение суперпарамагнитных средств в качестве МР контрастных средств возникло по причине их идеального сочетания сильного воздействия на интенсивность сигнала ткани, что приводит к мощному усилению контраста и их высокоспецифичного выбора мишени. Существует множество потенциальных мишеней для дисперсных средств в зависимости от пути введения и физикохимических признаков материала частиц, в частности размера частиц и поверхностных характеристик. Их два важнейших применения представляют собой энтеральное введение для исследования желудочно-кишечного тракта или парентеральное введение для исследований части кровяного русла и/или ретикулоэндотелиальной системы и областей ее анатомического распределения, например печени, селезенки, костного мозга и лимфатических узлов. Сверхмалые частицы оксида железа с диаметром менее чем приблизительно 30 нм обладают относительно продолжительным внутрисосудистым временем полужизни по сравнению с более крупными традиционными частицами оксида железа. В дополнение к сокращению T2, обычно связанному с частицами оксида железа, сверхмалые частицы также дают сокращение T1, увеличивая таким образом сигнал внутри сосудов. Последние достижения в области дисперсных средств также сделали возможным мечение рецепторными лигандами или фрагментами антител/антитела. Краткое описание описанных применений различных суперпарамагнитных средств приведено в Fahlvik et al., JMRI 3: 187-194 (1993).
До настоящего момента большая часть работы, проводимая в области суперпарамагнитных средств, была сосредоточена на оптимизации их эффективности контрастирования и биокинетики. Публикациям, относящимся к фармацевтическим составам или аспектам безопасности препаратов частиц, уделялось мало внимания.
Однако, что касается парентеральных дисперсных препаратов, адекватная эффективность контрастирования и биокинетика не являются сами по себе достаточными и приходится сталкиваться с определенными проблемами. Так, например, для традиционного препарата оксида железа - декстрана, который был опробован в клинических испытаниях, было показано, что он обладает низкой коллоидной стабильностью. Частицы следует редиспергировать и/или разбавлять и отфильтровывать непосредственно перед применением, а препарат вводят медленным вливанием сквозь встроенный фильтр, чтобы избежать тяжелых токсических эффектов.
Несмотря на то что частицы могут быть снабжены покрытием для адекватной стабильности и поверхностная область частицы из парентерального дисперсного средства прочна, мы обнаружили, что покрывающие агенты, которые традиционно считались совершенно безопасными, как например полисахариды накопления крахмал и декстран и их производные, могут сами по себе оказывать вредное воздействие на сердечно-сосудистые показатели, истощение тромбоцитарного звена, время свертывания крови и на систему комплемента.
Однако заявители обнаружили, что этих проблем можно избежать или уменьшить путем использования меньших, чем традиционные, плотностей покрытия определенных полиэлектролитных покрывающих материалов, таких как синтетические полиаминокислоты, синтетические полимеры и в особенности структурные полисахариды.
Таким образом, с одной точки зрения изобретение описывает диагностическое средство, содержащее особый композитный материал, причем частицы, сделанные из него, содержат диагностически эффективный, в значительной мере нерастворимый в воде металлооксидный кристаллический материал и полиионный покрывающий агент, где указанные частицы имеют размеры менее 300 нм, указанный кристаллический материал имеет размер кристалла от 1 до 100 нм, массовое отношение указанного кристаллического материала к указанному покрывающему агенту лежит в интервале от 1000:1 до 11:1, и указанный покрывающий агент выбран из группы, содержащей природные и синтетические структурные полисахариды, синтетические полиаминокислоты, физиологически переносимые синтетические полимеры и их производные.
Полисахариды широко распространены в природе и могут быть в целом разбиты на две категории полисахаридов накопления (таких как крахмал, гликоген, декстран и их производные) и структурных полисахаридов, таких как пектины и пектиновые фрагменты, такие как полигалактуроновая кислота, гликозаминогликаны и гепариноиды (например, гепарин, гепаран, кератан, дерматан, хондроитин и гиалуроновая кислота), целлюлозы и полисахариды морского происхождения, такие как альгинаты, каррагинаны и хитозаны и их производные.
Настоящее изобретение касается второй категории как природных, так и синтетических форм данных полисахаридов, включая такие полисахариды, которые были фрагментированы или химически модифицированы, например, для получения производных с введенными участками прикрепления для связывания с металлооксидными кристаллами.
Особенно предпочтительными в качестве полиионных полисахаридных покрывающих агентов являются природные и синтетические гепариноподобные полисахариды, такие как гепарины, хондроитины (например, хондроитин-4-сульфат) и полисахариды морского происхождения альгинаты, каррагинаны и хитозаны.
Менее предпочтительно в качестве покрывающих агентов могут применяться синтетические полиионные полимеры, например полиаминокислоты, полиакрилаты и полистиролсульфонаты (и другие синтетические полимеры, как указано в EP-A-580818). Среди полиаминокислот предпочтительными являются гомо- и сополимеры лизина, глютаминовой кислоты и аспарагиновой кислоты и их эфиров (например, метиловых и этиловых эфиров).
Вообще покрывающий агент должен содержать множество ионных групп, например аминовых, карбоксильных, сульфатных, сульфонатных, фосфонатных или фосфатных групп, расположенных вдоль цепи полимера для обеспечения множественных точек прикрепления к поверхности металлооксидного кристалла, так же, как и для обеспечения композитной частицы как единого целого с сетевым электрическим зарядом, предпочтительно отрицательным, который измеряется как дзета-потенциал. Множественное прикрепление гарантирует прочное устойчивое к автоклавированию связывание и устойчивость при хранении, тогда как сетевой заряд способствует улучшению биологической переносимости частицы после введения в сосудистую систему.
В общем случае полиионный покрывающий агент будет иметь молекулярную массу в интервале от 500 до 2000000 Да, более особенно от 1000 до 500000, конкретно от 1500 до 250000, более конкретно от 2000 до 150000 Да.
Поверхностно-связанный покрывающий агент составляет небольшую часть всей композитной частицы, причем массовое отношение кристаллического материала к покрывающему агенту предпочтительно находится в интервале от 1000:1 до 15:1, особенно от 500: 1 до 20:1, конкретно от 100:1 до 25:1, более конкретно по крайней мере 20:1 в случае гепаринового или хондроитинового покрытий.
Производство композитной частицы в общем случае будет осуществляться по одно- или двухстадийной методике покрытия. По одностадийной методике кристаллический материал получают путем осаждения при высоких значениях pH (например, выше 9, предпочтительно выше 11) с помощью основания в присутствии покрывающего агента, тогда как по двухстадийной методике сначала получают кристаллический материал, а затем формируют покрытие.
Таким образом, с дальнейшей точки зрения, изобретение описывает способ производства контрастного средства по изобретению, причем указанный способ включает в себя:
(а) при значениях pH выше 9 соосаждение кристаллов диагностически эффективного в значительной мере нерастворимого в воде оксида металла размером от 1 до 100 нм и покрывающего агента или
(б) покрытие кристаллов диагностически эффективного в значительной мере нерастворимого оксида металла покрывающим агентом,
посредством чего получают композитную частицу, имеющую размер менее 300 нм и массовое отношение кристалла к поверхностно-связанному покрывающему агенту от 1000:1 до 11:1, причем указанный покрывающий агент выбран из группы, содержащей природные и синтетические структурные полисахариды и их производные, синтетические полиаминокислоты и физиологически переносимые синтетические полимеры, однако предпочтительно, чтобы покрывающий агент представлял собой структурный полисахарид.
Методики соосадительного и послеосадительного формирования покрытия хорошо известны и широко описаны в литературе, как описано ниже (смотри, например, US-A-4795698 и US-A-4452773).
Поскольку осаждается не весь покрывающий агент, может оказаться необходимым применять от 1 1/2- до 7-, обычно приблизительно 2-кратный избыток (по отношению к количеству, необходимому, если бы связывалось 100% покрывающего агента), чтобы получить желаемую плотность покрытия.
Природа кристаллического материала в композициях по изобретению будет, разумеется, зависеть от ее предназначения. Несмотря на то что в общем случае изобретение применимо ко всем в значительной мере нерастворимым кристаллическим материалам, для которых желательным является парентеральное введение с последующей прицельной специфической доставкой или удлиненным временем пребывания в кровяном русле, однако оно особенно применимо в случае металлооксидных диагностических контрастных средств и, в частности, оксидов металлов, которые проявляют суперпарамагнитные свойства. В данном случае оксид может играть роль диагностического контрастного средства в ЭИТ и магнитометрических исследованиях и особенно в МР томографии.
Широкий спектр оксидов металлов, которые проявляют суперпарамагнитные свойства и у которых размер кристалла меньше размера одного домена, хорошо известен и описан, например, в US-A-4827945 (Groman), EP-A-525199 (Meito Sangyo) и EP-A-580878 (BASF). Смешанные ферриты, содержащие более чем один элемент металла, например, как тот, на который ссылается BASF, могут обеспечить особенно эффективные кристаллы с точки зрения релаксации. Данные различные оксиды металлов могут применяться в соответствии с настоящим изобретением, но особенно предпочтительными являются железооксидные суперпарамагнитные кристаллы, например соединения формулы (FeO)nFe2O3, где n находится в интервале от 0 до 1, примером которых служат магнемит (γ-Fe2O3) и магнетит (Fe3O4) так же, как и их композиты. С применением таких железооксидных кристаллов при их метаболическом усвоении, главным образом, осуществляемом ретикулоэндотелиальной системой, не высвобождается аномального токсического металла, а железо просто вносится в депо железа в организме.
Размер кристалла для суперпарамагнитных кристаллов предпочтительно будет находиться в интервале от 2 до 50 нм, особенно от 3 до 35 нм и конкретно от 4 до 20 нм. Частицы композитного средства строения кристалл/покрытия могут содержать единственные кристаллы или по желанию кластеры из множества кристаллов. В этом последнем случае кластерное "ядро" композитной частицы составляет желательно менее чем 100 нм по размеру.
Размеры кристалла, кластера и композитной частицы могут быть легко определены с помощью стандартных методов, таких как электронная микроскопия, рассеяние лазерного луча или гидродинамическая хроматография, например, как обсуждается ниже до примеров.
Массовое отношение металлооксидного кристалла к покрывающему агенту может быть легко определено с помощью элементного анализа, такого как индуктивный парный плазменный анализ, например сравнение сигналов металла из оксида металла и серы из сульфатных групп прикрепления покрывающего агента (или, аналогично, сигнала других характеристических атомов или групп покрывающего агента, где они не являются сульфатом). Подобным образом отношение может быть определено с помощью гравиметрического анализа.
Полиионная природа соединений покрытия позволяет им связываться с поверхностью кристалла во множестве участков на одну молекулу полимера. Это дает возможность образования прочной связи покрытие - кристалл, которая способна выдерживать условия, применяющиеся обычно для автоклавирования диагностических средств (121oC в течение 15 минут), и дает продукт, для которого не существует проблемы низкой коллоидной стабильности, как упоминалось выше, для продуктов декстран : оксид железа.
Как следствие своей полиионной природы, покрывающий агент придает композитным частицам суммарный электрический заряд, определяемый как ненулевой дзета-потенциал. При особенно низких плотностях покрытия заряд покрывающего агента может вывести из равновесия заряд металлооксидных кристаллов, и обнаруживается, что в изоэлектрической точке устойчивость частиц низка и может происходить агрегация, что дает агрегаты свыше 1000 нм по размеру. Это является нежелательным, но воздействие уровня покрытия на размер частиц может легко отслеживаться, а возникновение таких агрегатов - избегаться. В общем случае предпочтительными являются дзета-потенциалы, имеющие абсолютные значения по крайней мере 10 мВ (т.е. ≤ -10 мВ или ≥ +10 мВ), особенно от 20 до 100 мВ и конкретно от 30 до 70 мВ.
Таким образом, в предпочтительном осуществлении контрастное средство по данному изобретению содержит железооксидное ядро, которое покрыто и стабилизировано полиионными полисахаридами или полиаминокислотами. Железооксидные частицы с покрытием проявляют хорошую устойчивость, будучи подвергнуты воздействию высоких температур, разбавлению и во времени. По сравнению с традиционными препаратами оксида железа, покрытыми и стабилизированными полисахаридами накопления декстраном (или его производными) и крахмалом, новые средства обладают низкой токсичностью и высокой биологической совместимостью.
Для получения препарата оксида железа со специфической химической композицией и строением требуются тщательно контролируемые условия получения. Множество промышленных применений коллоидных суспензий оксида железа (феррожидкостей) было описано для записывающей технологии, в качестве ингредиентов красителей и красок и для электромеханических устройств. Существует большое количество методов синтеза и стабилизации технических чистых оксидов железа, однако они неприменимы для фармацевтических оксидов железа, главным образом, по причине своей неводной природы и/или благодаря токсичности добавляемых покрытий и поверхностно-активных веществ.
Наиболее часто применяемый способ синтеза суперпарамагнитных железооксидных контрастных средств происходит из методики, впервые описанной Molday (смотри Molday and Mackinzie, J. Immunol. Meth. 52: 353-367 (1982) и US-A-4452773 (Molday)), для мечения и разделения биологических молекул и клеток. Так называемый Molday-метод, или метод одностадийного соосаждения, основан на осаждении оксидов железа в щелочном растворе, содержащем водорастворимый полисахарид, предпочтительно декстран. Коллоидные отобранные по размеру композитные частицы содержат единственный или множественные железооксидные кристаллы, внедренные в декстран или покрытые декстраном. Кристаллы обычно построены из магнетита или магнетита/магнемита размером 3-30 нм. Общий диаметр частиц, однако, может колебаться от менее чем приблизительно 10 нм до свыше нескольких сотен нм. Как правило, препараты оксида железа, полученные по Molday-методу, являются неоднородными, и в данных препаратах определяется множество фракций частиц, отличающихся по размеру.
Таким образом, в целях выделения частиц в пределах более однородного интервала размеров применяли методы центрифугирования, фильтрования или гель-фильтрации (смотри Weissleder et al., Radiology 175: 489-493 (1990)).
Традиционные железооксидные частицы с покрытием с общим диаметром в интервале 50-1000 нм экспериментально испытывали в качестве контрастных средств для исследований печени и селезенки. Более мелкие выделенные частицы показали удлиненное время полужизни в кровяном русле и также способность проникать сквозь стенку капилляров. Существует множество потенциальных применений для данных средств, как например для получения изображений лимфатических узлов и костного мозга, для визуализации кровообращения/кровяного русла или для активного нацеливания. Однако было показано, что средства на основе декстрана являются относительно неустойчивыми и давали значительные побочные эффекты. Например, средство, недавно применявшееся в клинических испытаниях, не может быть приготовлено в виде готового к применению продукта. Непосредственно перед введением дозу разбавляют и вводят по типу медленного вливания через встроенный фильтр в целях снижения частоты и тяжести побочных явлений, таких как повышение кровяного давления, боль в нижней части спины и гематологические сдвиги.
Заявители обнаружили, что частицы по настоящему изобретению обладают повышенной устойчивостью и пониженной токсичностью по сравнению с традиционными частицами. Данные частицы, применяемые в соответствии с изобретением, могут, например, быть синтезированы по простой двухстадийной методике, где стадия 1 представляет собой осаждение металлооксидных кристаллов из щелочного раствора, а стадия 2 представляет собой процедуру покрытия кристаллов полимером полиэлектролитной природы, либо, альтернативно, они могут быть синтезированы путем соосаждения металлооксидных кристаллов и полиэлектролитного покрывающего полимера.
Металлооксидные кристаллы в общем случае могут быть осаждены из водных растворов растворимых солей металла(ов) путем добавления основания. Суперпарамагнитные железооксидные кристаллы могут быть осаждены из водного раствора смеси солей железа путем быстрого добавления щелочи до значений pH выше 10 при энергичном перемешивании или во время облучения ультразвуком. Может применяться широкий спектр солей железа, таких как FeCl2•nH2O, FeCl3•nH2O, цитрат Fe(III), глюконат Fe(III), FeSO4•nH2O, Fe2(SO4)3, оксалат Fe(II), Fe(NO3)3, ацетилацетат Fe(II), сульфат Fe(II) этилендиаммония, фумарат Fe(II), фосфат Fe(III), пирофосфат Fe(III), цитрат аммония Fe(III), сульфат аммония Fe(II), сульфат аммония Fe(III) и оксалат аммония Fe(II). Отношение между двухвалентным и трехвалентным железом должно предпочтительно находиться в интервале от 1:5 до 5:1. Такие осажденные железооксидные кристаллы могут быть представлены следующей формулой: (FeO)x•Fe2O3, где x может представлять собой число в интервале от 0≤x≤1. Магнемит, γ-Fe2O3, представляет низкое значение x, тогда как магнетит, Fe3O4, представляет высокое значение x.
Применяемые основания могут быть выбраны из широкого спектра сильных неорганических или органических оснований, таких как NaOH, NH4OH, LiOH, KOH, триэтиламин и гуанидин. В общем случае противоионы для металла и основания должны представлять собой физиологически переносимые ионы с тем, чтобы свести к минимуму необходимость очистки осажденных кристаллов от потенциально токсичных побочных продуктов.
Осаждение оксида железа или, альтернативно, соосаждение оксида железа и полимера может происходить в воде, в смеси воды и органического(их) растворителя(ей) или в вязкой среде. Например, могут применяться такие органические растворители, как метанол, этанол, ацетон, эфиры и гексан. Вязкая основа может содержать гидрогели полисахаридов или полиаминов, трийодзамещенные ароматические соединения, глицерин или полиэтилен- и полипропиленгликоли. Разумеется осаждение из водного раствора, свободного от физиологически непереносимых сорастворителей является предпочтительным, поскольку, опять же, сокращается необходимость в очистке после получения.
Покрывающий материал как один из компонентов нового препарата оксида железа обладает полиэлектролитной структурой вследствие преимуществ с точки зрения устойчивости и токсичности. Полиэлектролиты включают полианионные и поликатионные соединения или их смесь, которые прочно связаны с поверхностью оксида железа посредством множества точек прикрепления.
Покрывающие материалы могут быть разбиты на группы в зависимости от их заряда и функциональных групп, как например отрицательно заряженные полимеры с функциональными группами, содержащими атомы фосфора или серы или карбоксильные группы, и положительно заряженные полимеры с функциональными группами, содержащими атомы азота. Примерами отрицательно заряженных полимеров являются определенные модификации карбоксицеллюлозы, альгинаты, каррагинаны, полигалактуронат, гепарины и гепариноподобные соединения, такие как хондроитин-4-сульфат, дерматансульфат, кератинсульфат и гиалуронат, синтетические полимеры, такие как полистиролсульфонат, и аминокислоты, такие как полиглютамат и полиаспартат. Примеры положительно заряженных полимеров включают хитозан и полилизин.
Как обсуждалось ранее, степень замещенности и плотность заряда полиэлектролитных полимеров не должны быть слишком низкими, когда токсичность и устойчивость частиц становится критической. Таким образом, полимеры должны содержать множественные (более чем одну) функциональные группы, чтобы гарантировать множественные точки прикрепления к металлооксидной поверхности и придать частицам заряженную поверхность.
Диаметр суперпарамагнитного железооксидного ядра будет обычно находиться в интервале от приблизительно 4 нм до приблизительно 100 нм. Более мелкие ядра будут содержать только один субдоменный суперпарамагнитный кристалл, тогда как более крупные ядра могут содержать кластер кристаллов. Отдельные ядра кристаллов небольшого диаметра могут быть стабилизированы малыми количествами полимеров низкой или высокой молекулярной массы, тогда как для того, чтобы покрыть и стабилизировать кластеры, требуются большие количества полимеров вследствие их (кластеров - прим. перев.) плотности, а также большей магнитной нагрузки на одну частицу. Суммарный диаметр композитной частицы, включая железооксидное ядро и слой полимерного покрытия, будет обычно лежать в интервале приблизительно от 5 до 300 нм в зависимости от условий получения и молекулярной массы, структуры и количества полимера.
Релаксация частиц, содержащих суперпарамагнитный железооксидный кристалл, будет варьировать в зависимости от размера и состава ядра и покрытой частицы. Релаксация T1(r1) может составлять минимум 5 и максимум 200, тогда как релаксация T2(r2) может колебаться от 5 до 500 при 0,5 T (значения релаксации приведены в с-1мМ-1 Fe). Отношение r2/r1 может колебаться от 1 до свыше 100 или предпочтительно от 2 до 10. Небольшие частицы с единственным кристаллом будут иметь значения отношений r2/r1 в более низком интервале, тогда как более крупные частицы и многокристалльные частицы будут показывать более высокие значения отношений. Магнитный момент частиц будет относительно независим от размера частиц и кристаллов до тех пор, пока железооксидные кристаллы проявляют суперпарамагнитные свойства. При 1 T магнитный момент равен приблизительно 20-100 или предпочтительно 30-90 электромагнитных единиц/г оксида железа.
С дальнейшей точки зрения изобретение описывает диагностические композиции, содержащие диагностические средства по изобретению совместно по крайней мере с одним физиологически приемлемым носителем или разбавителем, например с водой для инъекций.
Композиции по изобретению могут быть представлены в любой удобной фармацевтической форме, например, суспензии, дисперсии, порошка и так далее и могут входить в состав с водными носителями (такими как вода для инъекций) и/или ингредиентами регуляции осмоляльности, pH, вязкости и устойчивости. В идеале композиция находится в форме суспензии, причем суспензия изотонична и изогидрична с кровью. Например, изотоничная суспензия может быть получена путем добавления солей, таких как хлорид натрия, низкомолекулярных сахаров, таких как глюкоза (декстроза), лактоза, мальтоза или маннитол или растворимая фракция полимерного покрывающего агента или их смеси. Изогидричность может быть достигнута путем добавления кислот, таких как соляная кислота, или оснований, таких как гидроксид натрия, если требуется лишь немного сдвинуть pH. Также могут быть применены буферы, такие как фосфатный, цитратный, ацетатный, боратный, тратратный и глюконатный. Химическая устойчивость суспензии частиц может быть модифицирована путем добавления антиоксидантов, таких как аскорбиновая кислота или пиросульфит натрия, и хелатообразователей, таких как лимонная кислота, натриевая соль ЭДТУ и натрийкальциевая соль ЭДТУ. Для улучшения физической устойчивости препарата также могут быть добавлены наполнители. Наиболее часто применяемые наполнители для парентерально вводимых суспензий представляют поверхностно-активные вещества, такие как полисорбаты, лецитин или сложные эфиры сорбита, модификаторы вязкости, такие как глицерин, пропиленгликоль и полиэтиленгликоли (макроголи), или модификаторы критической точки мицеллообразования, предпочтительно неионные поверхностно-активные вещества.
Композиции по изобретению будут преимущественно содержать оксид металла в диагностически эффективной концентрации металла, обычно от 0,1 до 250 мг Fe/мл, предпочтительно от 1 до 100 мг Fe/мл и особенно предпочтительно от 5 до 75 мг Fe/мл.
Изобретение далее описывает способ получения усиленного контрастом изображения организма человека или другого животного, предпочтительно млекопитающего, причем указанный способ включает в себя введение в указанный организм, предпочтительно парентерально и особенно предпочтительно внутрисосудисто, суспензии контрастного средства по изобретению и получение изображения по крайней мере части указанного организма, в котором распределяется указанное средство, например, по методу МР, ЭТ или магнитометрии.
Что касается способа по изобретению, применяемая дозировка будет представлять собой дозировку, обеспечивающую эффективное контрастирование для применяемого способа получения изображения. Обычно она будет лежать в области от 1 до 500 мкмоль Fe/кг, предпочтительно от 2 до 250 мкмоль Fe/кг и особенно предпочтительно от 5 до 50 мкмоль Fe/кг.
Могут быть применены дозировки и концентрации, традиционно применяемые в данной области.
Известно, что различные препараты оксида железа, полученные по методам данной области, оказывают значительные побочные эффекты, будучи введены внутрисосудисто. Наиболее часто встречающимися изменениями показателей по сообщениям являются снижения системного кровяного давления и острое истощение тромбоцитарного звена. Заявители обнаружили, что данные побочные эффекты представляют собой физиологическую и гематологическую реакции на индуцированную частицами активацию системы комплемента. Тогда как традиционные железооксидные частицы сильно активируют каскад комплемента, композитные частицы по настоящему изобретению не оказывают или оказывают минимальное влияние на количество циркулирующих тромбоцитов, тогда как традиционные препараты вызывают островыраженную и транзиторную тромбоцитопению. Чужеродные поверхности углеводной природы, такие как немодифицированный декстран и определенные модификации декстрана, могут являться мощными активаторами комплемента подобно многим штаммам грамположительных и грамотрицательных бактерий. Поверхности активируют альтернативный путь комплемента, поскольку нуклеофильные поверхностные группы, такие как OH, образуют ковалентную связь с белком комплемента C3b (смотри Immunology (второе издание), Gower Medical Publishing, New York, 1989; 13.3). В условиях сепсиса последствия активации комплемента могут быть полезными, поскольку система комплемента является важной частью реакции организма на повреждение, такое как инвазия инфекционного агента. Однако активация комплемента после инъекции дисперсного контрастного средства является скорее вредной, чем полезной.
К удивлению, было показано, что покрытые частицы по настоящему изобретению не воздействуют на систему комплемента или на параметры, связанные с комплементом, такие как кровяное давление и количество тромбоцитов. Подобранный покрывающий материал дает возможность получения поверхности частицы, которая не вызывала бы активации комплемента сходным с традиционными частицами образом. Аналогично, малое количество полиэлектролитных полимеров, применяемых для стабилизации частиц, также имеет супрессирующий эффект на активацию комплемента, возможно вследствие изменений в опсонизации (уровне и типе опсонинов) по сравнению с традиционными частицами.
Настоящее изобретение будет теперь описано еще подробнее со ссылкой на следующие неограничивающие примеры.
В следующих примерах концентрации железа определяли с помощью плавки железооксидных частиц с последующим анализом методом ICP. pH измеряли, применяя pH-метр Beckman ⊘ 10, оснащенный pH-электродом Orion Sureflow Ross. Распределение частиц по размеру измеряли методом гидродинамической хроматографии (ГДХ) (смотри Small and Langhorst, Analytical Chem. 54:892A (1982)) или методом рассеяния лазерного луча (PCS), применяя Malvern Zetasizer 4. Поверхностный заряд частицы, выраженный как дзета-потенциал или электрофоретическая подвижность, также определяли с помощью Malvern Zetasizer 4. Релаксацию T1 и T2, r1 и r2, измеряли в водных образцах при 37oC и 0,47 T (Minispec PC-20). Для T1 и последовательности CPMG (TE = 4 мс) для T2 применяли последовательность ИК-импульсов. Кривые магнитного момента получали при температуре окружающей среды на магнитометре с вибрирующими образцами (Molspin), работающем в области магнитных полей от +1 до -1 T.
Пример сравнения 1.
Декстран (5 г, Sigma), имеющий среднюю молекулярную массу 9000 Да растворяли в воде (10 мл). FeCl3•6H2O (1,35 г) и FeCl2•4H2O (0,81 г) растворяли при температуре 60oC в растворе углеводов, после чего смесь медленно вносили в 0,18 М NaOH (100 мл) при 60oC при облучении ультразвуком. Облучение ультразвуком продолжали в течение еще 10 минут с последующим центрифугированием при 4000 об/мин в течение 5 минут. Супернатант отделяли и фракцию подвергали диализу против 0,9% NaCl (5х1 л). Для декстрановых частиц был выявлен большой разброс частиц по размеру, причем присутствовали фракция с размером менее 12 нм и фракция с размером более 300 нм, что было определено с помощью ГДХ.
Пример сравнения 2.
(В соответствии с примером 7.3 из US-A-5314679).
К водному раствору (8,5 мл) FeCl3•6H2O (1,17 г) и FeCl2•4H2O (0,53 г) добавляли 1 М карбонат натрия до pH 2,3, а затем добавляли декстран, имеющий среднюю молекулярную массу 9000 Да (5,00 г). Раствор нагревали до 60-70oC и затем охлаждали приблизительно до 40oC. Добавляли 7,5% NaOH до pH около 9,5, прежде чем суспензию нагревали до 95oC в течение 15 мин. Дисперсию подвергали диализу против воды (5х1 л) (отсечка по 15000 Дальтон).
Пример сравнения 3.
(В соответствии с примером 6.1 из US-A-54770183).
К раствору 50 мл 0,28 М FeCl3, 0,16 М FeCl2 и 6,25 г декстрана, имеющего среднюю молекулярную массу 70000 Да (Pharmacia, Uppsala, Sweden) в течение трех минут добавляли 50 мл 7,5% NH4OH. Суспензию перемешивали в течение 5 минут и затем нагревали до 700oC в течение 30 минут. Раствор центрифугировали при 5000 об/мин в течение 15 минут и супернатант подвергали диализу против воды (5х1 л).
Пример сравнения 4.
Крахмал (3 г, Reppe Glucose, Sweden), имеющий среднюю молекулярную массу 7000 Да, растворяли в воде (10 мл). FeCl3•6H2O (2,7 г) и FeCl2•4H2O (4,5 г) растворяли при температуре 60oC в растворе углеводов, после чего смесь медленно вносили в 1,2 М NaOH (50 мл) при 60oC при облучении ультразвуком. Облучение ультразвуком продолжали в течение еще 10 минут с последующим центрифугированием при 5000 об/мин в течение 5 минут. Супернатант отбирали и подвергали диализу против водного раствора 0,9% NaCl. Кривая магнитного момента показала, что крахмальные частицы проявляли суперпарамагнитные свойства. Размер частиц составлял 450 нм, что было определено с помощью PCS. Было определено, что размер кристаллов магнетита составлял около 10 нм.
Пример сравнения 5.
а) Дисперсию магнетитовых частиц, полученных как в примере 1а (равную 0,3 г магнетитовых частиц), разбавляли водой (50 мл) и к ней добавляли карбоксидекстран со средней молекулярной массой 65000 Да (30 мг, Pharmacia Ab, Uppsala, Sweden), растворенный в воде (30 мл). Дисперсию облучали ультразвуком, центрифугировали при 5000 об/мин в течение 13 минут и супернатант фильтровали (фильтр 0,22 мкм). Для карбоксидекстрановых частиц был показан средний диаметр 88 нм, что было определено с помощью ГДХ. Было измерено, что дзета-потенциал составлял -26 мВ.
б) Дисперсию магнетитовых частиц, полученных как в примере 1а (равную 0,3 г магнетитовых частиц), разбавляли водой (50 мл) и к ней добавляли карбоксидекстран со средней молекулярной массой 65000 Да (50 мг, Pharmacia Ab, Uppsala, Sweden), растворенный в воде (5 мл). Дисперсию облучали ультразвуком, центрифугировали при 5000 об/мин в течение 13 минут и супернатант фильтровали (фильтр 0,22 мкм). Для карбоксидекстрановых частиц был показан средний диаметр 74 нм, что было определено с помощью ГДХ. Было измерено, что дзета-потенциал составлял -32 мВ. Значение r1 составляло 35,2 (мM•с)-1, а значение r2 составляло 358 (мМ•с)-1.
в) Дисперсию магнетитовых частиц, полученных как в примере 1а (равную 0,3 г магнетитовых частиц), разбавляли водой (50 мл) и к ней добавляли карбоксидекстран с молекулярной массой 3000-4000 Да (15 мг), растворенный в воде (1,5 мл). Дисперсию облучали ультразвуком, центрифугировали при 5000 об/мин в течение 13 минут и супернатант фильтровали.
Пример сравнения 6.
а) Дисперсию магнетитовых частиц, полученных как в примере 1а (равную 0,5 г магнетитовых частиц), разбавляли водой (85 мл) и к ней добавляли декстранфосфат со средней молекулярной массой 74000 Да (50 мг, Pharmacia Ab, Uppsala, Sweden), растворенный в воде (5 мл). Дисперсию облучали ультразвуком, центрифугировали и супернатант фильтровали (фильтр 0,22 мкм). Кривая магнитного момента показала, что декстранфосфатные частицы проявляли суперпарамагнитные свойства. Размер частиц составлял 74 нм, что было определено с помощью PCS.
б) Дисперсию магнетитовых частиц, полученных как в примере 1а (равную 0,3 г магнетитовых частиц), разбавляли водой (50 мл) и к ней добавляли декстранфосфат со средней молекулярной массой 71800 Да (50 мг, TdB Consultancy AS, Uppsala, Sweden), растворенный в воде (5 мл). Дисперсию облучали ультразвуком, центрифугировали при 5000 об/мин в течение 13 минут и супернатант фильтровали (фильтр 0,22 мкм). Для декстранфосфатных частиц был показан средний диаметр 48 нм, что было определено с помощью ГДХ. Было измерено, что дзета-потенциал составлял -51 мВ. Значение r1 составляло 37 (мМ•с)-1, а значение r2 составляло 342 (мM•c)-1.
в) Дисперсию магнетитовых частиц, полученных как в примере 1а (равную 0,3 г магнетитовых частиц), разбавляли водой (50 мл) и к ней добавляли декстранфосфат со средней молекулярной массой 71800 Да (15 мг, TdB Consultancy AS, Uppsala, Sweden), растворенный в воде (1,5 мл). Дисперсию облучали ультразвуком, центрифугировали при 5000 об/мин в течение 13 минут и супернатант фильтровали (фильтр 0,22 мкм). Для декстранфосфатных частиц был показан средний диаметр 48 нм, что было определено с помощью ГДХ. Было измерено, что дзета-потенциал составлял -36 мВ.
Пример сравнения 7.
Дисперсию магнетитовых частиц из примера 1а (равную 0,5 г магнетитовых частиц) разбавляли водой (50 мл) и к ней добавляли декстрансульфат со средней молекулярной массой 500000 Да (30 мг, Sigma, D-6001), растворенный в воде. Дисперсию облучали ультразвуком, центрифугировали при 5000 об/мин в течение 13 минут и супернатант фильтровали (фильтр 0,22 мкм). Кривая магнитного момента показала, что декстрансульфатные частицы проявляли суперпарамагнитные свойства, и для них был показан средний диаметр 42 нм, что было определено с помощью ГДХ. Было измерено, что дзета-потенциал составлял -57 мВ. На поверхность частиц адсорбировалось пятьдесят шесть процентов декстрансульфата. Значение r1 составляло 37,7 (мМ•с)-1, а значение релаксации r2 составляло 307 (мМ•с)-1.
Пример 1.
а) Магнетитовые частицы осаждали из водного раствора (500 мл) FeCl2•4H2O (12,50 г, 6,29•10-2 моль) и FeCl3•6H2O (33,99 г, 1,26•10-1 моль) путем быстрого добавления NH4OH (28-30%, 72 мл) до значения pH свыше 10 при энергичном перемешивании. Частицы собирали с помощью магнита и промывали водой до значения pH 6-7. Частицы диспергировали в около 200 мл воды. Реакционную смесь выдерживали в безазотных условиях для декантирования и редиспергирования. Для непокрытых магнетитовых частиц, стабилизированных HCl, был показан гидродинамический диаметр 97 нм. Было измерено, что дзета-потенциал составлял +36 мВ. Значение r1 составляло 27,8 (мМ•с)-1, а значение r2 составляло 324 (мМ•с)-1.
б) Дисперсию магнетитовых частиц из примера 1а (равную 0,5 г магнетитовых частиц) разбавляли водой (70 мл) и к ней добавляли гепарин (2 мл, Гепарин 5000 МЕ/мл, Прод. N F1 NA, Nycomed Pharma, Oslo, Noway). Дисперсию облучали ультразвуком, центрифугировали при 4000 об/мин в течение 13 минут и супернатант фильтровали (фильтр 0,22 мкм). Кривая магнитного момента показала, что гепариновые частицы проявляли суперпарамагнитные свойства, и для них был показан
средний диаметр 48 нм, что было определено с помощью ГДХ. Размер магнетитовых кристаллов составлял около 10 нм, что было определено с помощью электронной микроскопии. Было измерено, что дзета-потенциал составлял -61 мВ. На поверхность частиц адсорбировалось пятьдесят четыре процента гепарина, что соответствует 10 мкг серы на мг железа. Значение r1 составляло 40,5 (мМ•с)-1, а значение r2 составляло 304 (мМ•с)-1.
в) Дисперсию магнетитовых частиц из примера 1а (равную 0,5 г магнетитовых частиц) разбавляли водой (90 мл) и к ней добавляли низкомолекулярный гепарин, молекулярная масса 4000-6000 Да (0,8 мл Fragmin 10000 МЕ/мл, Kabi Pharmacia AB, Sweden). Дисперсию облучали ультразвуком, центрифугировали при 4000 об/мин в течение 13 минут и супернатант фильтровали (фильтр 0,22 мкм). Кривая магнитного момента показала, что гепариновые частицы проявляли суперпарамагнитные свойства. Было измерено, что магнитный момент насыщения составлял 78 электромагнитных единиц/г оксида железа. Размер частиц составлял 85 нм, в что было определено с помощью PCS. Было измерено, что дзета-потенциал составлял - 40 мВ. На поверхности частиц адсорбировалось шестнадцать процентов добавленного полиэлектролита.
г) Дисперсию магнетитовых частиц из примера 1а (равную 0,5 г магнетитовых частиц) разбавляли водой (70 мл) и к ней добавляли гепарин (1 мл, Гепарин 5000 МЕ/мл, Прод. N F1 NA, Nycomed Pharma, Oslo, Norway). Дисперсию облучали ультразвуком, центрифугировали при 4000 об/мин в течение 13 минут и супернатант фильтровали (фильтр 0,22 мкм). Кривая магнитного момента показала, что гепариновые частицы проявляли суперпарамагнитные свойства, и для них был показан средний диаметр 64 нм, что было определено с помощью PCS. На поверхность частиц адсорбировалось шестьдесят девять процентов гепарина, что соответствует 7 мкг серы на мг железа. Значение r1 составляло 38 (мМ•с)-1, а значение r2 составляло 273 (мМ•с)-1.
Пример 2.
Дисперсию магнетитовых частиц из примера 1а (равную 0,3 г магнетитовых частиц) разбавляли водой (50 мл) и к ней добавляли дерматансульфат (36 мг, Sigma C-241 3), растворенный в воде (5 мл). Дисперсию облучали ультразвуком, центрифугировали при 5000 об/мин в течение 13 минут и супернатант фильтровали (фильтр 0,22 мкм). Для дерматансульфатных частиц был показан средний диаметр 49 нм, что было определено с помощью ГДХ. На поверхность частиц адсорбировалось сорок процентов дерматансульфата. Было измерено, что дзета-потенциал составлял -58 мВ.
Пример 3.
Дисперсию магнетитовых частиц из примера 1а (равную 0,3 г магнетитовых частиц) разбавляли водой (50 мл) и к ней добавляли гиалуроновую кислоту (60 мг, Sigma H-401 5), растворенную в воде (6 мл). Дисперсию облучали ультразвуком, центрифугировали при 5000 об/мин в течение 13 минут и супернатант фильтровали (фильтр 0,22 мкм). Кривая магнитного момента показала, что гиалуроновые частицы проявляли суперпарамагнитные свойства, и для них был показан средний диаметр 123 нм, что было определено с помощью ГДХ. Было измерено, что дзета-потенциал составлял -55 мВ. Значение r1 составляло 33,7 (мМ•с)-1, а значение r2 составляло 318 (мМ•с)-1.
Пример 4.
Дисперсию магнетитовых частиц из примера 1а (равную 0,3 г магнетитовых частиц) разбавляли водой (50 мл) и к ней добавляли хондроитин-4-сульфат (60 мг, Sigma C-8529), растворенный в воде (5 мл). Дисперсию облучали ультразвуком, центрифугировали при 5000 об/мин в течение 13 минут и супернатант фильтровали (фильтр 0,22 мкм). Кривая магнитного момента показала, что хондроитин-4-сульфатные частицы проявляли суперпарамагнитные свойства, и для них был показан средний диаметр 54 нм, что было определено с помощью ГДХ. Было измерено, что дзета-потенциал составлял -52 мВ. На поверхность частиц адсорбировалось двадцать девять процентов хондроитин-4-сульфата. Значение r1 составляло 40,4 (мМ•с)-1, а значение r2 составляло 314 (мМ•с)-1.
Пример 5.
Составы оксида железа из примера сравнения 7 (декстрансульфатный оксид железа) и из примеров 1б и г (гепариновый оксид железа) инкубировали с человеческой плазмой in vitro в концентрации, эквивалентной дозировке 1 мг Fe/кг и изучали их эффект на параметр свертывания активированное частичное тромбопластиновое время (АЧТВ) с помощью CephotestTM (Nycomed Pharma AS). Составы гепаринового оксида железа в примерах 1б и г увеличивали АЧТВ в зависимости от дозы гепарина по факторам 4,5 и 2,5 соответственно. Это ясно указывает на желательность минимизации используемой плотности покрытия. Состав в примере сравнения 7 увеличивал АЧТВ по фактору 2,7.
Пример 6.
Составы оксида железа из примеров 1б и г (гепариновый оксид железа) вводили внутривенно крысам (n= 3) в дозировках 1 мг Fe/кг (только 1б) и 2 мг Fe/кг и отбирали образцы крови до и через 10, 30 и 60 минут после введения. Эффект составов на параметр свертывания активированное частичное тромбопластиновое время (АЧТВ) изучали in vitro с помощью CephotestTM (Nycomed Pharma AS). Составы увеличивали АЧТВ в зависимости от дозы и времени. Через 10 и 30 минут после введения дозировки 2 мг Fe/кг составов из примеров 1б и г увеличивали АЧТВ по факторам 4 и 1,5 соответственно. Дозировка 1 мг Fe/кг состава из примера 1б увеличивала АЧТВ по фактору 1,3 через 10 минут.
Пример 7.
Дисперсию магнетитовых частиц из примера 1а (равную 0,1 г магнетитовых частиц) разбавляют водой (15 мл) и к ней добавляют гепарансульфат (20 мг, Sigma H-7641), растворенный в воде. Дисперсию облучают ультразвуком и центрифугируют. Супернатант отбирают.
Пример 8.
Дисперсию магнетитовых частиц из примера 1а (равную 0,1 г магнетитовых частиц) разбавляют водой (15 мл) и к ней добавляют кератансульфат (15 мг, Sigma K-3001), растворенный в воде. Дисперсию облучают ультразвуком и центрифугируют. Супернатант отбирают.
Пример 9.
а) Дисперсию магнетитовых частиц из примера 1а (равную 0,3 г магнетитовых частиц) разбавляют водой (50 мл) и к ней добавляют лямбда-каррагинан (30 мг, Sigma C-3889), растворенный в воде (3 мл). Дисперсию облучали ультразвуком, центрифугировали при 4000 об/мин в течение 13 минут и супернатант фильтровали (фильтр 0,22 мкм). Для лямбда-каррагинановых частиц был показан средний диаметр 53 нм, что было определено с помощью ГДХ. Было измерено, что дзета-потенциал составлял -56 мВ.
б) Дисперсию магнетитовых частиц из примера 1а (равную 0,3 г магнетитовых частиц) разбавляют водой (50 мл) и к ней добавляют лямбда-каррагинан (50 мг, Sigma C-3889), растворенный в воде (5 мл). Дисперсию облучали ультразвуком, центрифугировали при 4000 об/мин в течение 13 минут и супернатант фильтровали (фильтр 0,22 мкм). Для лямбда-каррагинановых частиц был показан средний диаметр 61 нм, что было определено с помощью ГДХ. Было измерено, что дзета-потенциал составлял -61 мВ. Значение r1 составляло 38,6 (мМ•с)-1, а значение r2 составляло 309 (мМ•с)-1.
в) Дисперсию магнетитовых частиц из примера 1а (равную 0,3 г магнетитовых частиц) разбавляют водой (50 мл) и к ней добавляют лямбда-каррагинан (15 мг, Sigma C-3889), растворенный в воде (1,5 мл). Дисперсию облучали ультразвуком, центрифугировали при 4000 об/мин в течение 13 минут и супернатант фильтровали (фильтр 0,22 мкм). Для лямбда-каррагинановых частиц был показан средний диаметр 52 нм, что было определено с помощью ГДХ. Было измерено, что дзета-потенциал составлял -50 мВ.
Пример 10.
а) Дисперсию магнетитовых частиц из примера 1а (равную 0,3 г магнетитовых частиц) разбавляют водой (50 мл) и к ней добавляют йота-каррагинан (30 мг, Fluka Prod. 22045), растворенный в воде (3 мл). Дисперсию облучали ультразвуком, центрифугировали при 5000 об/мин в течение 13 минут и супернатант фильтровали (фильтр 0,22 мкм). Для йота-каррагинановых частиц был показан средний диаметр 63 нм, что было определено с помощью ГДХ. Было измерено, что дзета-потенциал составлял -47 мВ.
б) Дисперсию магнетитовых частиц из примера 1а (равную 0,3 г магнетитовых частиц) разбавляют водой (50 мл) и к ней добавляют йота-каррагинан (15 мг, Fluka Prod. 22045), растворенный в воде (1,5 мл). Дисперсию облучали ультразвуком, центрифугировали при 5000 об/мин в течение 13 минут и супернатант фильтровали (фильтр 0,22 мкм). Для йота-каррагинановых частиц был показан средний диаметр 54 нм, что было определено с помощью ГДХ. Было измерено, что дзета-потенциал составлял -39 мВ.
Пример 11.
а) Дисперсию магнетитовых частиц из примера 1а (равную 0,5 г магнетитовых частиц) разбавляли водой (80 мл) и к ней добавляли альгинат Protanal LF 10/60, имеющий среднюю молекулярную массу приблизительно 180000 Да (50 мг, Pronova, Drammen, Norwey), растворенный в воде (10 мл). Дисперсию облучали ультразвуком, центрифугировали при 5000 об/мин в течение 13 минут и супернатант фильтровали (фильтр 0,22 мкм). Для альгинатных частиц был показан средний диаметр 57 нм, что было определено с помощью ГДХ. Было измерено, что дзета-потенциал составлял -63 мВ. Значение r1 составляло 39,9 (мМ•с)-1, а значение r2 составляло 305 (мМ•с)-1.
б) Дисперсию магнетитовых частиц из примера 1а (равную 0,5 г магнетитовых частиц) разбавляли водой (80 мл) и к ней добавляли альгинат Protanal LF 60, имеющий среднюю молекулярную массу приблизительно 325000 Да (25 мг, Pronova, Drammen, Norwey), растворенный в воде (10 мл). Дисперсию облучали ультразвуком, центрифугировали при 5000 об/мин в течение 13 минут и супернатант фильтровали (фильтр 0,22 мкм). Для альгинатных частиц был показан средний диаметр 67 нм, что было определено с помощью ГДХ. Было измерено, что дзета-потенциал составлял -58 мВ.
в) Дисперсию магнетитовых частиц из примера 1а (равную 0,3 г магнетитовых частиц) разбавляли водой (50 мл) и к ней добавляли альгинат Protanal LFR 5/60, имеющий среднюю молекулярную массу приблизительно 380000 Да (15 мг, Pronova, Drammen, Norwey), растворенный в воде (15 мл). Дисперсию облучали ультразвуком, центрифугировали при 5000 об/мин в течение 13 минут и супернатант фильтровали (фильтр 0,22 мкм). Для альгинатных частиц был показан средний диаметр 62 нм, что было определено с помощью ГДХ. Было измерено, что дзета-потенциал составлял -53 мВ.
Пример 12.
а) Дисперсию магнетитовых частиц из примера 1а (равную 0,3 г магнетитовых частиц) разбавляли водой (50 мл) и к ней добавляли натрий-карбоксицеллюлозу (30 мг), растворенную в воде (3 мл). Дисперсию облучали ультразвуком, центрифугировали при 5000 об/мин в течение 13 минут и супернатант фильтровали (фильтр 0,22 мкм). Для карбоксицеллюлозных частиц был показан средний диаметр 56 нм, что было определено с помощью ГДХ. Было измерено, что дзета-потенциал составлял -57 мВ. Значение r1 составляло 40,1 (мМ•с)-1, а значение r2 составляло 303 (мМ•с)-1.
б) Дисперсию магнетитовых частиц из примера 1а (равную 0,3 г магнетитовых частиц) разбавляли водой (50 мл) и к ней добавляли натрий-карбоксицеллюлозу (15 мг), растворенную в воде (1,5 мл). Дисперсию облучали ультразвуком, центрифугировали при 5000 об/мин в течение 13 минут и супернатант фильтровали (фильтр 0,22 мкм). Для карбоксицеллюлозных частиц был показан средний диаметр 65 нм, что было определено с помощью ГДХ. Было измерено, что дзета-потенциал составлял -53 мВ.
Пример 13.
а) Дисперсию магнетитовых частиц из примера 1а (равную 0,3 г магнетитовых частиц) разбавляли водой (50 мл) и к ней добавляли хитозан, имеющий среднюю молекулярную массу приблизительно 2000000 Да (30 мг, Fluka 22743), растворенный в 1%-ной уксусной кислоте (4,5 мл). Дисперсию облучали ультразвуком, центрифугировали при 5000 об/мин в течение 13 минут и супернатант фильтровали (фильтр 0,22 мкм). Кривая магнитного момента показала, что хитозановые частицы проявляли суперпарамагнитные свойства. Размер частиц составлял 64 нм, что было определено с помощью PCS. Было измерено, что дзета-потенциал составлял +48 мВ. Значение r1 составляло 35,1 (мМ•с)-1, а значение r2 составляло 281 (мМ•с)-1.
б) Дисперсию магнетитовых частиц из примера 1а (равную 0,3 г магнетитовых частиц) разбавляли водой (50 мл) и к ней добавляли хитозан, имеющий среднюю молекулярную массу приблизительно 2000000 Да (50 мг, Fluka 22743), растворенный в 1%-ной уксусной кислоте (7,5 мл). Дисперсию облучали ультразвуком, центрифугировали при 5000 об/мин в течение 13 минут и супернатант фильтровали (фильтр 0,22 мкм). Кривая магнитного момента показала, что хитозановые частицы проявляли суперпарамагнитные свойства. Размер частиц составлял 64 нм, что было определено с помощью PCS. Было измерено, что дзета-потенциал составлял +47 мВ.
в) Дисперсию магнетитовых частиц из примера 1а (равную 0,3 г магнетитовых частиц) разбавляли водой (50 мл) и к ней добавляли хитозан, имеющий среднюю молекулярную массу приблизительно 2000000 Да (15 мг, Fluka 22743), растворенный в 1%-ной уксусной кислоте (2,25 мл). Дисперсию облучали ультразвуком, центрифугировали при 5000 об/мин в течение 13 минут и супернатант фильтровали (фильтр 0,22 мкм). Кривая магнитного момента показала, что хитозановые частицы проявляли суперпарамагнитные свойства. Размер частиц составлял 64 нм, что было определено с помощью PCS. Было измерено, что дзета-потенциал составлял +47 мВ.
Пример 14.
а) Дисперсию магнетитовых частиц из примера 1а (равную 0,3 г магнетитовых частиц) разбавляли водой (50 мл) и к ней добавляли хитозан, имеющий среднюю молекулярную массу приблизительно 750000 Да (50 мг, Fluka 22742), растворенный в 1%-ной уксусной кислоте (7,5 мл). Дисперсию облучали ультразвуком, центрифугировали при 5000 об/мин в течение 13 минут и супернатант фильтровали (фильтр 0,22 мкм). Кривая магнитного момента показала, что хитозановые частицы проявляли суперпарамагнитные свойства. Размер частиц составлял 62 нм, что было определено с помощью PCS. Было измерено, что дзета-потенциал составлял +48 мВ. Значение r1 составляло 33,4 (мМ•с)-1, а значение r2 составляло 279 (мМ•с)-1.
б) Дисперсию магнетитовых частиц из примера 1а (равную 0,3 г магнетитовых частиц) разбавляли водой (50 мл) и к ней добавляли хитозан, имеющий среднюю молекулярную массу приблизительно 750000 Да (50 мг, Fluka 22742), растворенный в 1%-ной уксусной кислоте (2,25 мл). Дисперсию облучали ультразвуком, центрифугировали при 5000 об/мин в течение 13 минут и супернатант фильтровали (фильтр 0,22 мкм). Кривая магнитного момента показала, что хитозановые частицы проявляли суперпарамагнитные свойства. Размер частиц составлял 64 нм, что было определено с помощью PCS. Было измерено, что дзета-потенциал составлял +49 мВ.
Пример 15.
а) Дисперсию магнетитовых частиц из примера 1а (равную 0,3 г магнетитовых частиц) разбавляли водой (50 мл) и к ней добавляли хитозан, имеющий среднюю молекулярную массу приблизительно 70000 Да (30 мг, Fluka 22741), растворенный в 1%-ной уксусной кислоте (4,5 мл). Дисперсию облучали ультразвуком, центрифугировали при 5000 об/мин в течение 13 минут и супернатант фильтровали (фильтр 0,22 мкм). Кривая магнитного момента показала, что хитозановые частицы проявляли суперпарамагнитные свойства. Размер частиц составлял 62 нм, что было определено с помощью PCS. Было измерено, что дзета-потенциал составлял +49 мВ. Значение r1 составляло 34,3 (мМ•с)-1, а значение r2 составляло 327 (мМ•с)-1.
б) Дисперсию магнетитовых частиц из примера 1а (равную 0,3 г магнетитовых частиц) разбавляли водой (50 мл) и к ней добавляли хитозан, имеющий среднюю молекулярную массу приблизительно 70000 Да (50 мг, Fluka 22741), растворенный в 1%-ной уксусной кислоте (2,25 мл). Дисперсию облучали ультразвуком, центрифугировали при 5000 об/мин в течение 13 минут и супернатант фильтровали (фильтр 0,22 мкм). Кривая магнитного момента показала, что хитозановые частицы проявляли суперпарамагнитные свойства. Размер частиц составлял 64 нм, что было определено с помощью PCS. Было измерено, что дзета-потенциал составлял +48 мВ.
Пример 16.
а) Дисперсию магнетитовых частиц из примера 1а (равную 0,3 г магнетитовых частиц) разбавляли водой (50 мл) и к ней добавляли поли(4-стиролсульфонат натрия) (30 мг, Janssen 22.227.14), растворенный в воде (3 мл). Дисперсию облучали ультразвуком, центрифугировали при 5000 об/мин в течение 13 минут и супернатант фильтровали (фильтр 0,22 мкм). Для частиц из поли(4-стиролсульфоната натрия) был показан средний диаметр 43 нм, что было определено с помощью ГДХ. Было измерено, что дзета-потенциал составлял -53 мВ.
б) Дисперсию магнетитовых частиц из примера 1а (равную 0,3 г магнетитовых частиц) разбавляли водой (50 мл) и к ней добавляли поли(4-стиролсульфонат натрия) (15 мг, Janssen 22.227.14), растворенный в воде (1,5 мл). Дисперсию облучали ультразвуком, центрифугировали при 5000 об/мин в течение 13 минут и супернатант фильтровали (фильтр 0,22 мкм). Для частиц из поли(4-стиролсульфоната натрия) был показан средний диаметр 36 нм, что было определено с помощью ГДХ. Было измерено, что дзета-потенциал составлял -49 мВ.
Пример 17.
а) Дисперсию магнетитовых частиц из примера 1а (равную 0,3 г магнетитовых частиц) разбавляли водой (50 мл) и к ней добавляли поли-L-глютаминову кислоту, имеющую молекулярную массу 2000-15000 Да (30 мг, Sigma P-4636), растворенную в воде (3 мл). Дисперсию облучали ультразвуком, центрифугировали при 4000 об/мин в течение 13 минут и супернатант фильтровали (фильтр 0,22 мкм). Кривая магнитного момента показала, что частицы из поли-L-глютаминовой кислоты проявляли суперпарамагнитные свойства и для них был показан средний диаметр 37 нм, что было определено с помощью ГДХ. Было измерено, что дзета-потенциал составлял -68 мВ.
б) Дисперсию магнетитовых частиц из примера 1а (равную 0,3 г магнетитовых частиц) разбавляли водой (50 мл) и к ней добавляли поли-L-глютаминову кислоту, имеющую молекулярную массу 2000-15000 Да (50 мг, Sigma P-4636), растворенную в воде (5 мл). Дисперсию облучали ультразвуком, центрифугировали при 4000 об/мин в течение 13 минут и супернатант фильтровали (фильтр 0,22 мкм). Для частиц из поли-L-глютаминовой кислоты был показан средний диаметр 37 нм, что было определено с помощью ГДХ. Было измерено, что дзета-потенциал составлял -66 мВ. Значение r1 составляло 40,4 (мМ•с)-1, а значение r2 составляло 281 (мМ•с)-1.
в) Дисперсию магнетитовых частиц из примера 1а (равную 0,3 г магнетитовых частиц) разбавляли водой (50 мл) и к ней добавляли поли-L-глютаминовую кислоту, имеющую молекулярную массу 2000-15000 Да (15 мг, Sigma P-4636), растворенную в воде (1,5 мл). Дисперсию облучали ультразвуком, центрифугировали при 4000 об/мин в течение 13 минут и супернатант фильтровали (фильтр 0,22 мкм). Для частиц из поли-L-глютаминовой кислоты был показан средний диаметр 38 нм, что было определено с помощью ГДХ. Было измерено, что дзета-потенциал составлял -65 мВ.
Пример 18.
а) Дисперсию магнетитовых частиц из примера 1а (равную 0,3 г магнетитовых частиц) разбавляли водой (50 мл) и к ней добавляли поли-L-глютаминовую кислоту, имеющую молекулярную массу 15000-50000 Да (30 мг, Sigma P-4761), растворенную в воде (3 мл). Дисперсию облучали ультразвуком, центрифугировали при 4000 об/мин в течение 13 минут и супернатант фильтровали (фильтр 0,22 мкм). Для частиц из поли-L-глютаминовой кислоты был показан средний диаметр 37 нм, что было определено с помощью ГДХ. Было измерено, что дзета-потенциал составлял -66 мВ.
б) Дисперсию магнетитовых частиц из примера 1а (равную 0,3 г магнетитовых частиц) разбавляли водой (50 мл) и к ней добавляли поли-L-глютаминовую кислоту, имеющую молекулярную массу 15000-50000 Да (50 мг, Sigma P-4761), растворенную в воде (5 мл). Дисперсию облучали ультразвуком, центрифугировали при 4000 об/мин в течение 13 минут и супернатант фильтровали (фильтр 0,22 мкм). Для частиц из поли-L-глютаминовой кислоты был показан средний диаметр 36 нм, что было определено с помощью ГДХ. Было измерено, что дзета-потенциал составлял -66 мВ. Значение r1 составляло 41,7 (мМ•с)-1, а значение r2 составляло 286 (мМ•с)-1.
в) Дисперсию магнетитовых частиц из примера 1а (равную 0,3 г магнетитовых частиц) разбавляли водой (50 мл) и к ней добавляли поли-L-глютаминовую кислоту, имеющую молекулярную массу 15000-50000 Да (15 мг, Sigma P-4761), растворенную в воде (1,5 мл). Дисперсию облучали ультразвуком, центрифугировали при 4000 об/мин в течение 13 минут и супернатант фильтровали (фильтр 0,22 мкм). Для частиц из поли-L-глютаминовой кислоты был показан средний диаметр 36 нм, что было определено с помощью ГДХ. Было измерено, что дзета-потенциал составлял -63 мВ.
Пример 19.
а) Дисперсию магнетитовых частиц из примера 1а (равную 0,3 г магнетитовых частиц) разбавляли водой (50 мл) и к ней добавляли поли-L-глютаминовую кислоту, имеющую молекулярную массу 50000-100000 Да (30 мг, Sigma P-4886), растворенную в воде (3 мл). Дисперсию облучали ультразвуком, центрифугировали при 4000 об/мин в течение 13 минут и супернатант фильтровали (фильтр 0,22 мкм). Кривая магнитного момента показала, что частицы из поли-L-глютаминовой кислоты проявляли суперпарамагнитные свойства. Был показан средний диаметр 40 нм, что было определено с помощью ГДХ. Было измерено, что дзета-потенциал составлял -70 мВ. Значение r1 составляло 39,6 (мМ•с)-1, а значение r2 составляло 289 (мМ•с)-1.
б) Дисперсию магнетитовых частиц из примера 1а (равную 0,3 г магнетитовых частиц) разбавляли водой (50 мл) и к ней добавляли поли-L-глютаминовую кислоту, имеющую молекулярную массу 50000-100000 Да (15 мг, Sigma P-4886), растворенную в воде (1,5 мл). Дисперсию облучали ультразвуком, центрифугировали при 4000 об/мин в течение 13 минут и супернатант фильтровали (фильтр 0,22 мкм). Был показан средний диаметр 39 нм, что было определено с помощью ГДХ. Было измерено, что дзета-потенциал составлял -66 мВ.
Пример 20.
а) Дисперсию магнетитовых частиц из примера 1а (равную 0,3 г магнетитовых частиц) разбавляли водой (50 мл) и к ней добавляли поли-L-аспарагиновую кислоту, имеющую молекулярную массу 15000-50000 Да (30 мг, Sigma P-6762), растворенную в воде (3 мл). Дисперсию облучали ультразвуком, центрифугировали при 4000 об/мин в течение 13 минут и супернатант фильтровали (фильтр 0,22 мкм). Для частиц из поли-L-аспарагиновой кислоты был показан средний диаметр 42 нм, что было определено с помощью ГДХ. Было измерено, что дзета-потенциал составлял -65 мВ.
б) Дисперсию магнетитовых частиц из примера 1а (равную 0,3 г магнетитовых частиц) разбавляли водой (50 мл) и к ней добавляли поли-L-аспарагиновую кислоту, имеющую молекулярную массу 15000-50000 Да (50 мг, Sigma P-6762), растворенную в воде (5 мл). Дисперсию облучали ультразвуком, центрифугировали при 4000 об/мин в течение 13 минут и супернатант фильтровали (фильтр 0,22 мкм). Для частиц из поли-L-аспарагиновой кислоты был показан средний диаметр 40 нм, что было определено с помощью ГДХ. Было измерено, что дзета-потенциал составлял -67 мВ. Значение r1 составляло 40,8 (мМ•с)-1, а значение r2 составляло 332 (мМ•с)-1.
в) Дисперсию магнетитовых частиц из примера 1а (равную 0,3 г магнетитовых частиц) разбавляли водой (50 мл) и к ней добавляли поли-L-аспарагиновую кислоту, имеющую молекулярную массу 15000-50000 Да (15 мг, Sigma P-6762), растворенную в воде (1,5 мл). Дисперсию облучали ультразвуком, центрифугировали при 4000 об/мин в течение 13 минут и супернатант фильтровали (фильтр 0,22 мкм). Для частиц из поли-L-аспарагиновой кислоты был показан средний диаметр 44 нм, что было определено с помощью ГДХ. Было измерено, что дзета-потенциал составлял -66 мВ.
Пример 21.
а) Дисперсию магнетитовых частиц из примера 1а (равную 0,3 г магнетитовых частиц) разбавляли водой (50 мл) и к ней добавляли поли-L-аспарагиновую кислоту, имеющую молекулярную массу 5000-15000 Да (30 мг, Sigma P-5387), растворенную в воде (3 мл). Дисперсию облучали ультразвуком, центрифугировали при 4000 об/мин в течение 13 минут и супернатант фильтровали (фильтр 0,22 мкм). Для частиц из поли-L-аспарагиновой кислоты был показан средний диаметр 38 нм, что было определено с помощью ГДХ. Было измерено, что дзета-потенциал составлял -67 мВ. Значение r1 составляло 41,1 (мМ•с)-1, а значение r2 составляло 303 (мМ•с)-1.
б) Дисперсию магнетитовых частиц из примера 1а (равную 0,3 г магнетитовых частиц) разбавляли водой (50 мл) и к ней добавляли поли-L-аспарагиновую кислоту, имеющую молекулярную массу 5000-15000 Да (50 мг, Sigma P-5387), растворенную в воде (5 мл). Дисперсию облучали ультразвуком, центрифугировали при 4000 об/мин в течение 13 минут и супернатант фильтровали (фильтр 0,22 мкм). Для частиц из поли-L-аспарагиновой кислоты был показан средний диаметр 37 нм, что было определено с помощью ГДХ. Было измерено, что дзета-потенциал составлял -70 мВ.
в) Дисперсию магнетитовых частиц из примера 1а (равную 0,3 г магнетитовых частиц) разбавляли водой (50 мл) и к ней добавляли поли-L-аспарагиновую кислоту, имеющую молекулярную массу 5000-15000 Да (15 мг, Sigma P-5387), растворенную в воде (1,5 мл). Дисперсию облучали ультразвуком, центрифугировали при 4000 об/мин в течение 13 минут и супернатант фильтровали (фильтр 0,22 мкм). Для частиц из поли-L-аспарагиновой кислоты был показан средний диаметр 37 нм, что было определено с помощью ГДХ. Было измерено, что дзета-потенциал составлял -65 мВ.
Пример 22.
а) Дисперсию магнетитовых частиц из примера 1а (равную 0,3 г магнетитовых частиц) разбавляли водой (50 мл) и к ней добавляли полиакриловую кислоту, имеющую молекулярную массу 2000 Да (30 мг, Aldrich 32366-7), растворенную в воде (3 мл). Дисперсию облучали ультразвуком, центрифугировали при 5000 об/мин в течение 13 минут и супернатант фильтровали (фильтр 0,22 мкм). Для полиакриловых частиц был показан средний диаметр 50 нм, что было определено с помощью ГДХ. Было измерено, что дзета-потенциал составлял -36 мВ. Значение r1 составляло 29,1 (мМ•с)-1, а значение r2 составляло 323 (мМ•с)-1.
б) Дисперсию магнетитовых частиц из примера 1а (равную 0,3 г магнетитовых частиц) разбавляли водой (50 мл) и к ней добавляли полиакриловую кислоту, имеющую молекулярную массу 2000 Да (50 мг, Aldrich 32366-7), растворенную в воде (5 мл). Дисперсию облучали ультразвуком, центрифугировали при 5000 об/мин в течение 13 минут и супернатант фильтровали (фильтр 0,22 мкм). Для полиакриловых частиц был показан средний диаметр 57 нм, что было определено с помощью ГДХ. Было измерено, что дзета-потенциал составлял -29 мВ.
в) Дисперсию магнетитовых частиц из примера 1а (равную 0,3 г магнетитовых частиц) разбавляли водой (50 мл) и к ней добавляли полиакриловую кислоту, имеющую молекулярную массу 90000 Да (30 мг, Aldrich 19205-8), растворенную в воде (1,5 мл). Дисперсию облучали ультразвуком, центрифугировали при 4000 об/мин в течение 13 минут и супернатант фильтровали.
Пример 23.
а) Дисперсию магнетитовых частиц из примера 1а (равную 0,3 г магнетитовых частиц) разбавляли водой (50 мл) и к ней добавляли полигалактуроновую кислоту, имеющую молекулярную массу 25000-50000 Да (30 мг, Fluka 81325), растворенную в воде (3 мл) с добавленными в нее несколькими каплями 1 М NaOH. Дисперсию облучали ультразвуком, центрифугировали при 5000 об/мин в течение 13 минут и супернатант фильтровали (фильтр 0,45 мкм). Для частиц из полигалактуроновой кислоты был показан средний диаметр 55 нм, что было определено с помощью ГДХ. Было измерено, что дзета-потенциал составлял -60 мВ.
б) Дисперсию магнетитовых частиц из примера 1а (равную 0,3 г магнетитовых частиц) разбавляли водой (50 мл) и к ней добавляли полигалактуроновую кислоту, имеющую молекулярную массу 25000-50000 Да (15 мг, Fluka 81325), растворенную в воде (1,5 мл) с добавленными в нее несколькими каплями 1 М NaOH. Дисперсию облучали ультразвуком, центрифугировали при 5000 об/мин в течение 13 минут и супернатант фильтровали (фильтр 0,45 мкм). Для частиц из полигалактуроновой кислоты был показан средний диаметр 61 нм, что было определено с помощью ГДХ. Было измерено, что дзета-потенциал составлял -55 мВ.
Пример 24.
а) Дисперсию магнетитовых частиц из примера 1а (равную 0,3 г магнетитовых частиц) разбавляли водой (50 мл) и к ней добавляли поли-L-лизин, имеющий молекулярную массу 1000-4000 Да (30 мг, Sigma P-0879), растворенный в воде (3 мл). Дисперсию облучали ультразвуком, центрифугировали при 4000 об/мин в течение 13 минут и супернатант фильтровали (фильтр 0,45 мкм). Размер частиц составлял 102 нм, что было определено с помощью PCS. Было измерено, что дзета-потенциал составлял +47 мВ.
б) Дисперсию магнетитовых частиц из примера 1а (равную 0,3 г магнетитовых частиц) разбавляли водой (50 мл) и к ней добавляли поли-L-лизин, имеющий молекулярную массу 1000-4000 Да (15 мг, Sigma P-0879), растворенный в воде (1,5 мл). Дисперсию облучали ультразвуком, центрифугировали при 4000 об/мин в течение 13 минут и супернатант фильтровали (фильтр 0,45 мкм). Размер частиц составлял 108 нм, что было определено с помощью PCS. Было измерено, что дзета-потенциал составлял +46 мВ.
Пример 25.
а) Дисперсию магнетитовых частиц из примера 1а (равную 0,3 г магнетитовых частиц) разбавляли водой (50 мл) и к ней добавляли поли-L-лизин, имеющий молекулярную массу 15000-30000 Да (50 мг, Sigma P-7890), растворенный в воде (5 мл). Дисперсию облучали ультразвуком, центрифугировали при 4000 об/мин в течение 13 минут и супернатант фильтровали (фильтр 0,22 мкм). Кривая магнитного момента показала, что поли-L-лизиновые частицы проявляли суперпарамагнитные свойства. Размер частиц составлял 78 нм, что было определено с помощью PCS. Было измерено, что дзета-потенциал составлял +56 мВ. Значение r1 составляло 38,3 (мМ•с)-1, а значение r2 составляло 295 (мМ•с)-1.
б) Дисперсию магнетитовых частиц из примера 1а (равную 0,3 г магнетитовых частиц) разбавляли водой (50 мл) и к ней добавляли поли-L-лизин, имеющий молекулярную массу 15000-30000 Да (15 мг, Sigma P-7890), растворенный в воде (1,5 мл). Дисперсию облучали ультразвуком, центрифугировали при 4000 об/мин в течение 13 минут и супернатант фильтровали (фильтр 0,22 мкм). Размер частиц составлял 89 нм, что было определено с помощью PCS. Было измерено, что дзета-потенциал составлял +57 мВ.
Пример 26.
а) Дисперсию магнетитовых частиц из примера 1а (равную 0,3 г магнетитовых частиц) разбавляли водой (50 мл) и к ней добавляли поли-L-лизин, имеющий молекулярную массу 70000-150000 Да (30 мг, Sigma P-1274), растворенный в воде (3 мл). Дисперсию облучали ультразвуком, центрифугировали при 4000 об/мин в течение 13 минут и супернатант фильтровали (фильтр 0,22 мкм). Размер частиц составлял 94 нм, что было определено с помощью PCS. Было измерено, что дзета-потенциал составлял +57 мВ.
б) Дисперсию магнетитовых частиц из примера 1а (равную 0,3 г магнетитовых частиц) разбавляли водой (50 мл) и к ней добавляли поли-L-лизин, имеющий молекулярную массу 70000-150000 Да (50 мг, Sigma P-1274), растворенный в воде (5 мл). Дисперсию облучали ультразвуком, центрифугировали при 4000 об/мин в течение 13 минут и супернатант фильтровали (фильтр 0,22 мкм). Размер частиц составлял 86 нм, что было определено с помощью PCS. Было измерено, что дзета-потенциал составлял +62 мВ. Значение r1 составляло 36,9 (мМ•с)-1, а значение r2 составляло 294 (мМ•с)-1.
в) Дисперсию магнетитовых частиц из примера 1а (равную 0,3 г магнетитовых частиц) разбавляли водой (50 мл) и к ней добавляли поли-L-лизин, имеющий молекулярную массу 70000-150000 Да (15 мг, Sigma P-1274), растворенный в воде (1,5 мл). Дисперсию облучали ультразвуком, центрифугировали при 4000 об/мин в течение 13 минут и супернатант фильтровали (фильтр 0,22 мкм). Размер частиц составлял 96 нм, что было определено с помощью PCS. Было измерено, что дзета-потенциал составлял +61 мВ.
Пример 27.
а) Дисперсию магнетитовых частиц из примера 1а (равную 0,3 г магнетитовых частиц) разбавляли водой (50 мл) и к ней добавляли поли(Asp-Na, Glu-Na) 1: 1, имеющие молекулярную массу 5000-15000 Да (30 мг, Sigma P-1408), растворенные в воде (3 мл). Дисперсию облучали ультразвуком, центрифугировали при 5000 об/мин в течение 13 минут и супернатант фильтровали (фильтр 0,22 мкм). Кривая магнитного момента показала, что частицы из поли(Asp-Na, Glu-Na) проявляли суперпарамагнитные свойства, и для них был показан средний диаметр 38 нм, что было определено с помощью ГДХ. Было измерено, что дзета-потенциал составлял -58 мВ. Значение r1 составляло 40,4 (мМ•с)-1, а значение r2 составляло 277 (мМ•с)-1.
б) Дисперсию магнетитовых частиц из примера 1а (равную 0,3 г магнетитовых частиц) разбавляли водой (50 мл) и к ней добавляли поли(Asp-Na, Glu-Na) 1: 1, имеющие молекулярную массу 5000-15000 Да (15 мг, Sigma P-1408), растворенные в воде (1,5 мл). Дисперсию облучали ультразвуком, центрифугировали при 5000 об/мин в течение 13 минут и супернатант фильтровали (фильтр 0,22 мкм). Для частиц из поли(Asp-Na, Glu-Na) был показан средний диаметр 40 нм, что было определено с помощью ГДХ. Было измерено, что дзета-потенциал составлял -60 мВ.
Пример 28.
Дисперсию магнетитовых частиц из примера 1а (равную 0,3 г магнетитовых частиц) разбавляли водой (50 мл) и к ней добавляли поли(Glu, Glu-OEt) 4:1, имеющие молекулярную массу 70000-150000 Да (30 мг, Sigma P-4910), растворенные в этаноле (1,5 мл) с добавленными в него несколькими каплями HCl. Дисперсию облучали ультразвуком, центрифугировали и супернатант отбирали.
Пример 29.
а) Дисперсию магнетитовых частиц из примера 1а (равную 0,3 г магнетитовых частиц) разбавляли водой (50 мл) и к ней добавляли поли(Glu, Lys) 1:4, имеющие молекулярную массу 150000-300000 Да (30 мг, Sigma P-0650), растворенные в воде (5,7 мл). Дисперсию облучали ультразвуком, центрифугировали при 5000 об/мин в течение 13 минут и супернатант фильтровали (фильтр 0,22 мкм). Для частиц из поли(Glu, Lys) был показан средний диаметр 79 нм, что было определено с помощью ГДХ. Было измерено, что дзета-потенциал составлял +65 мВ.
б) Дисперсию магнетитовых частиц из примера 1а (равную 0,3 г магнетитовых частиц) разбавляли водой (50 мл) и к ней добавляли поли(Glu, Lys) 1:4, имеющие молекулярную массу 150000-300000 Да (50 мг, Sigma P-0650), растворенные в воде (9,5 мл). Дисперсию облучали ультразвуком, центрифугировали при 5000 об/мин в течение 13 минут и супернатант фильтровали (фильтр 0,22 мкм). Для частиц из поли(Glu, Lys) был показан средний диаметр 77 нм, что было определено с помощью ГДХ. Было измерено, что дзета-потенциал составлял +63 мВ. Значение r1 составляло 35,5 (мМ•с)-1, а значение r2 составляло 255 (мМ•с)-1.
Пример 30.
Дисперсию магнетитовых частиц, полученных как в примере 1а (равную 0,3 г магнетитовых частиц), разбавляли водой (50 мл) и к ней добавляли дендример (полученный в соответствии с US-A-4507466 (The Dow Chemical Corporation)) (30 мг) в воде. Дисперсию облучали ультразвуком, центрифугировали при 5000 об/мин в течение 13 минут и супернатант отбирали.
Устойчивость.
Пример 31.
Устойчивость следующих железооксидных составов исследовали путем автоклавирования при 121oC в течение 15 минут: примеры сравнения 5б и 6б, примеры 1а, 1б, 2, 3, 4, 9а, 10а, 11а, б и в, 12а, 13а, б и в, 14б, 15а, 16а, 17а и б, 18б, 19а, 20а, 21б, 22а, 23а, 25а, 26а, 27б и 29а.
Непокрытые железооксидные частицы из примера 1а диспергировали до эксперимента, применяя HCl. Для других составов модификаций не делали. Составы изучали непосредственно до и после автоклавирования, так же как и одним днем и одной неделей позже. Было показано, что образец из примера 1а полностью разделялся после автоклавирования. На другие составы крайнее нагревание не влияло, и для них было показано однородное распределение по размеру до и во всех моментах времени после автоклавирования.
Пример 32.
Была изучена адсорбция одного типичного примера полиэлектролитных полимеров по данному изобретению, поли(4-сиролсульфоната натрия) (ПССNa). Проводились измерения изотермы адсорбции, равно как и электрофоретической подвижности и размера частиц как функции сокращения полимера. Непокрытые железооксидные частицы из примера 1а покрывали ПССNa, как описано в примере 16, однако дисперсии не облучали ультразвуком. Применяли такие количества ПССNa, котоыре давали отношение полимера к оксиду железа 9•10-3 к 2,5. На фиг. 1 показана изотерма адсорбции ПССNa на кристаллы оксида железа.
Непокрытые железооксидные частицы имели размер 130±5 нм, что определялось с помощью PCS. Приняв плотность частицы 5,2 г/л, можно установить площадь поверхности частиц как
площадь поверхности (м2/мл) = m•A/ρ•V,
где m представляет собой массу частиц на мл, ρ представляет собой плотность частиц, а A и V представляют собой площадь и объем отдельной частицы соответственно. Установленная площадь поверхности в суспензии была вычислена как 9,5•10-3 м2/мл. Максимальное адсорбированное количество тогда представляет 26 мг/м2, указывая на наращивание множественных слоев адсорбированного полимера или цепи/петли полимера, тянущейся с поверхности.
На фиг. 2 и 3 показаны электрофоретическая подвижность (E) частиц и их гидродинамический диаметр как функция концентрации полимера. Электрофоретическая подвижность была рассчитана как 4,2 ± 0,6 мкм•см/V•S для непокрытой железооксидной частицы и быстро падала с увеличением концентрации полимера. При высокой концентрации полимера электрофоретическая подвижность выравнивается вблизи -4 мкм•см/V•S, и создается впечатление, что размер частиц стабилизируется на 230 ± 20 нм, что определяется с помощью PCS. Результаты на фиг. 1-3 схематично объяснены в таблице.
Результаты измерений электрофоретической подвижности непокрытого, равно как и покрытого ПССNa, оксида железа в зависимости от pH суспензии показаны на фиг. 4. Непокрытая поверхность оксида железа проявляет типично амфотерную природу, меняющуюся от высоких положительных значений электрофоретической подвижности при низких значениях pH до возрастающих отрицательных значений при более высоких значениях pH. Изоэлектрическая точка расположена вблизи pH 7. Характеристики поверхности частицы полностью меняются, отражая стойкую кислую природу после покрытия ПССNa. Данная поверхность проявляет высокие отрицательные электрофоретические подвижности даже при низких значениях pH, несколько изменяясь до отчасти более отрицательной поверхности при высоких значениях pH. Результаты на фиг. 4 согласуются с фиг. 1-3, что делает очевидным полное покрытие исходной поверхности оксида молекулами кислого полимера при данной концентрации полимера.
Фиг. 1. Адсорбция ПССNa на коллоидный оксид железа. Адсорбированное количество (мг/мл) от концентрации ПССNa (мг/мл).  представляют фракционирование путем центрифугирования, а Δ представляет фракционирование путем фильтрования.
представляют фракционирование путем центрифугирования, а Δ представляет фракционирование путем фильтрования.
Фиг. 2. Электрофоретическая подвижность коллоидного оксида железа от концентрации ПССNa (мг/мл).
Фиг. 3. Размер частиц (нм) коллоидного оксида железа от концентрации ПССNa (мг/мл).
Фиг. 4. Электрофоретическая подвижность (мкм•см/V•s) чистого коллоидного оксида железа (x) и коллоидного оксида железа, покрытого ПССNA (x) от pH.
Пример 33.
Изучалась адсорбция типичного полиэлектролитного полимера гепарина на поверхность оксида железа. Электрофоретическую подвижность и размер железооксидных частиц измеряли как функцию концентрации гепарина. Непокрытые частицы из примера 1а, стабилизированные HCl, покрывали гепарином, как описано в примере 1б, однако дисперсии подвергали вихревому перемешиванию вместо облучения ультразвуком. На фиг. 5 и 6 показаны электрофоретическая подвижность и размер частиц от концентрации гепарина.
Профили электрофоретической подвижности и размера частиц от количества гепарина показывают адсорбцию полимера на поверхность частиц. Повышение количеств отрицательно заряженных молекул на положительной поверхности оксида железа сначала снижает, а затем обращает электрофоретическую подвижность. Суспензия дестабилизируется вблизи изоэлектрической точки.
Фиг. 5. Электрофоретическая подвижность (мкм•см/V•s) от концентрации добавленного гепарина (мкл/мл).
Фиг. 6. Размер частиц (мкм) от концентрации добавленного гепарина (мкл/мл).
Безопасность.
Пример 34.
Составы оксида железа из примера сравнения 4 (крахмальный оксид железа) и примера 1б (гепариновый оксид железа) вводились кроликам (n=4-5) в виде внутривенных быстрых инъекций болюса. Вводились дозы по 1, 2, 5 и 10 мг Fe/кг и до 60 минут после введения записывались среднее системное артериальное давление (САД) и среднее давление в легочной артерии (ДЛА). Для состава из примера сравнения 4 был показан зависимый от дозы эффект на ДЛА. В случае дозы 1 мг Fe/кг изменений зарегистрировано не было, тогда как небольшое увеличение с 20 ± 1 мм рт.ст. до 23 ± 1 мм рт.ст. было записано в случае дозы 2,5 мг Fe/кг. Доза 10 мг Fe/кг повышала среднее ДЛА с 22 ± 3 мм рт. ст. до 33 ± 6 мм рт.ст. Наивысшая доза также давала умеренное снижение САД с 129 ± 10 мм рт.ст. до 117 ± 8 мм рт.ст. Максимальные эффекты на ДЛА и САД были зарегистрированы через 3-4 минуты после инъекции, а реакции были кратковременны (до 10 минут). Состав из примера 1б не влиял на записываемые гемодинамические параметры.
Пример 35.
Составы оксида железа из примеров сравнения 1 (декстрановый оксид железа), 4 (крахмальный оксид железа), 5б (карбоксидекстрановый оксид железа) и 6б (декстранфосфатный оксид железа) и примеров 1б (гепариновый оксид железа), 4 (хондроитин-4-сульфатный оксид железа) и 25а (поли-L-лизиновый оксид железа) вводили внутривенно крысам (n=2-3) в дозировке 1 мг Fe/кг. Через 3, 5, 10 и 15 минут после введения записывалось количество циркулирующих тромбоцитов. Составы из примеров сравнения 1 и 4 вызывали через 3 и 5 минут после введения выраженное снижение количества тромбоцитов до 10-17% от значений до введения. Состав из примеров сравнения 5б и 6б также давал значительное снижение, примерно до 50-60% от контроля, через 3 и 5 минут. Реакция быстро компенсировалась у всех крыс и в течение 15 минут вновь достигались количества тромбоцитов, равные 75-100% от контрольных значений. Составы из примеров 1б, 4 и 25а не имели или имели минимальный эффект на количество циркулирующих тромбоцитов.
Пример 36.
Составы из примеров сравнения 1 (декстрановый оксид железа), 4 (крахмальный оксид железа), 5б (карбоксидекстрановый оксид железа) и 6б (декстранфосфатный оксид железа) и примеров 1б, 3а (гепариновый оксид железа), 4 (хондроитин-4-сульфатный оксид железа) 11а (альгинатный оксид железа), 13а (хитозановый оксид железа), 17а (поли-L-глютаматный оксид железа) и 25а (поли-L-лизиновый оксид железа) инкубировали с человеческой сывороткой in vitro в концентрациях, эквивалентных дозировкам 1 мг Fe/кг (все вещества) и 10 мг Fe/кг (примеры сравнения 4 и 5б), в течение 60 минут при 37oC. Все составы были стерилизованы путем автоклавирования и свободны от эндотоксинов. Ферментный иммуноанализ для определения человеческого комплекса комплемента SC5b-a (TCC) (Bering) применяли в целях исследования активации комплемента. В качестве положительного контроля применяли Зимозан А (Sigma), а в качестве отрицательных контролей применяли декстрозу (5%) ил воду. Все составы из примеров сравнения значительно повысили уровни TCC. Эффект был особенно выражен в случае образца из примера сравнения 5б, который дал 6-кратное увеличение в TCC, даже при более низкой дозировке 1 мг Fe/кг, по сравнению с отрицательным контролем. Зависимый от дозы эффект на TCC наблюдался в случае составов из примеров сравнения 1 и 4. Низкие дозировки давали 2-кратное, а высокие дозировки - 6-кратное увеличение измеряемых уровней TCC. Состав из примера сравнения 6б увеличивал TCC в 3 раза при низкой дозировке 1 мг Fe/кг. Другие исследованные составы не имели или имели лишь минимальные эффекты на TCC по сравнению с отрицательным контролем.
| название | год | авторы | номер документа |
|---|---|---|---|
| БИОРАЗЛАГАЕМЫЕ НЕСШИТЫЕ ПОЛИМЕРЫ | 1993 |
|
RU2114865C1 |
| ПРИМЕНЕНИЕ ФИЗИОЛОГИЧЕСКИ ДОПУСТИМОГО, КОРПУСКУЛЯРНОГО ФЕРРИМАГНИТНОГО ИЛИ ФЕРРОМАГНИТНОГО МАТЕРИАЛА, СПОСОБ ФОРМИРОВАНИЯ МАГНИТОМЕТРИЧЕСКОГО ИЗОБРАЖЕНИЯ, ПРОЦЕСС ОБНАРУЖЕНИЯ ИЗМЕНЕНИЙ, ПРИМЕНЕНИЕ ФИЗИОЛОГИЧЕСКИ ДОПУСТИМЫХ ПАРАМАГНИТНЫХ, СВЕРХПАРАМАГНИТНЫХ, ФЕРРОМАГНИТНЫХ ИЛИ ФЕРРИМАГНИТНЫХ ЧАСТИЦ, УСТРОЙСТВО ФОРМИРОВАНИЯ ИЗОБРАЖЕНИЯ | 1992 |
|
RU2137501C1 |
| СПОСОБ МАГНИТОМЕТРИЧЕСКОГО ИССЛЕДОВАНИЯ ТЕЛА ЧЕЛОВЕКА ИЛИ ЖИВОТНОГО И СПОСОБ ОБНАРУЖЕНИЯ ИЗМЕНЕНИЙ МАГНИТНОЙ ВОСПРИИМЧИВОСТИ ЧЕЛОВЕКА ИЛИ ЖИВОТНОГО | 1991 |
|
RU2102082C1 |
| Способ получения бетулина для использования в качестве адъюванта в вакцине против коронавируса SARS-CoV-2 | 2020 |
|
RU2749193C1 |
| НАНОСТРУКТУРИРОВАННЫЕ КОМПОЗИЦИИ ДЛЯ ДОСТАВКИ СИЛИБИНИНА И ДРУГИХ АКТИВНЫХ ИНГРЕДИЕНТОВ ДЛЯ ЛЕЧЕНИЯ ГЛАЗНЫХ ЗАБОЛЕВАНИЙ | 2015 |
|
RU2726193C2 |
| КОНТРАСТНОЕ ВЕЩЕСТВО, СПОСОБ ЕГО ПОЛУЧЕНИЯ, СПОСОБ ПОЛУЧЕНИЯ ИЗОБРАЖЕНИЙ ТЕЛА ЧЕЛОВЕКА ИЛИ ЖИВОТНОГО | 1994 |
|
RU2128520C1 |
| СУСПЕНЗИЯ АГРЕГАТОВ НАНОАЛМАЗОВ И ДИСПЕРСИЯ НАНОАЛМАЗОВ ОДНОЦИФРОВОГО НАНОРАЗМЕРА | 2015 |
|
RU2700528C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПОЛИМЕРСОДЕРЖАЩЕЙ КОМПОЗИЦИИ СИЛИБИНА | 2019 |
|
RU2716706C1 |
| КОНТРАСТНЫЕ АГЕНТЫ И ИХ ПРИМЕНЕНИЕ | 1993 |
|
RU2122432C1 |
| СПОСОБ ПОЛУЧЕНИЯ ДЗЕТА-ОТРИЦАТЕЛЬНОЙ ДИСПЕРСИИ НАНОАЛМАЗОВ И ДЗЕТА-ОТРИЦАТЕЛЬНАЯ ДИСПЕРСИЯ НАНОАЛМАЗОВ | 2014 |
|
RU2681615C2 |

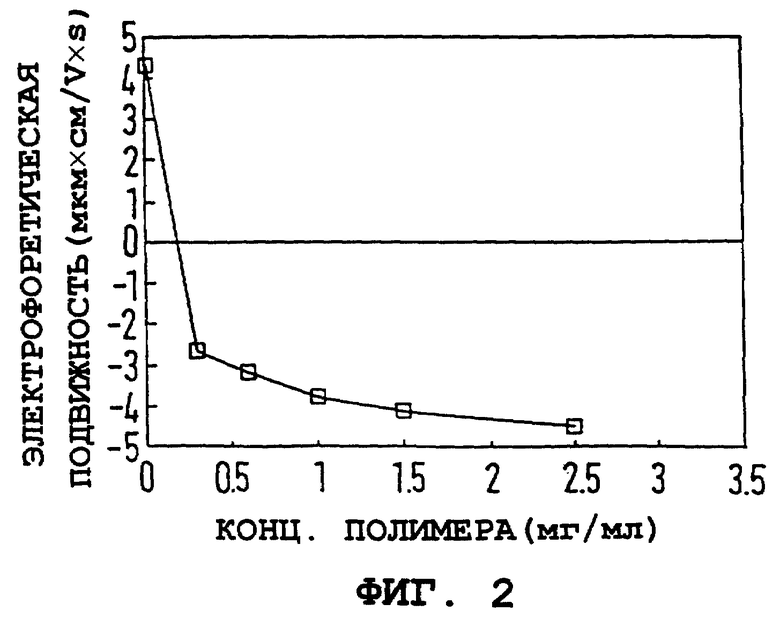
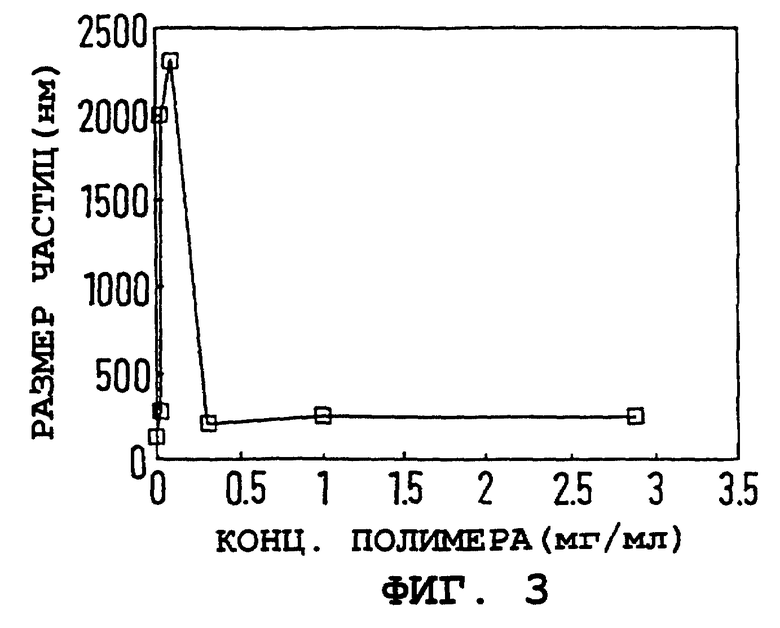
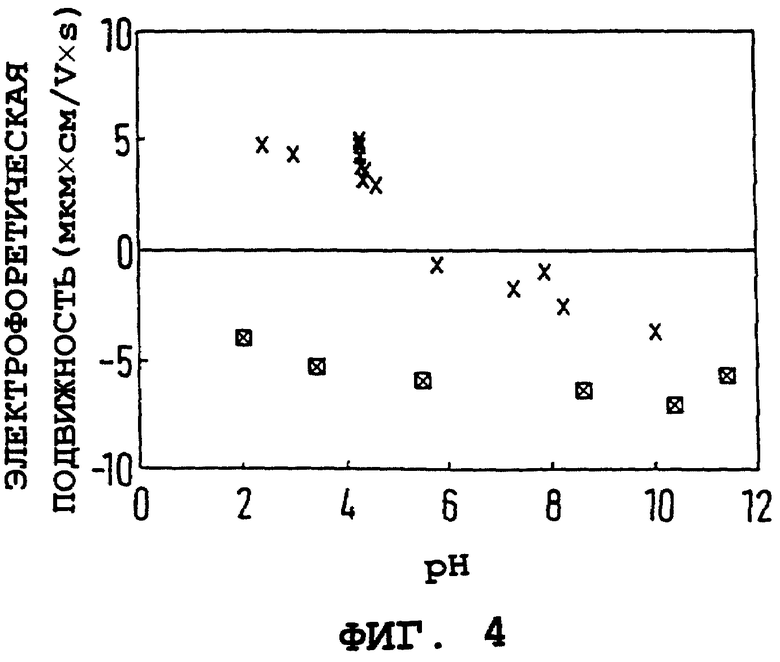
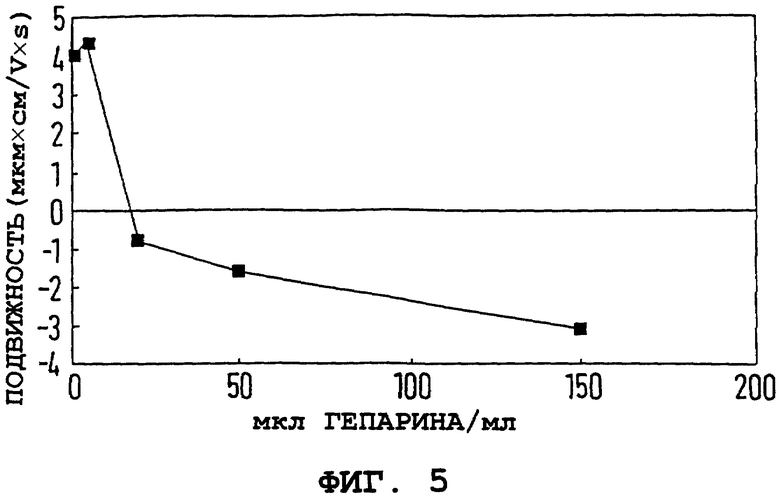
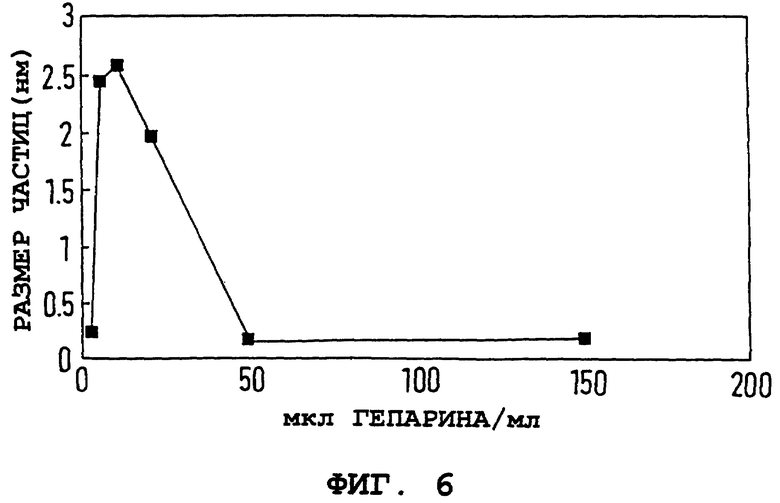
Изобретение относится к особым контрастным средствам, особенно к контрастным средствам для МР томографии, имеющим металлооксидное, предпочтительно суперпарамагнитное железооксидное, ядро, при условии низкой плотности покрытия полиэлектролитного и синтетических полимеров, особенно полиаминокислот, а также к способу его получения и применения. Изобретение обеспечивает получение низкотоксичного препарата, который оказывает сниженный или не оказывает эффект на параметры сердечно-сосудистой системы, истощение тромбоцитного звена, активацию комплемента и свертывание крови. 4 с. и 11 з.п.ф-лы, 6 ил., 1 табл.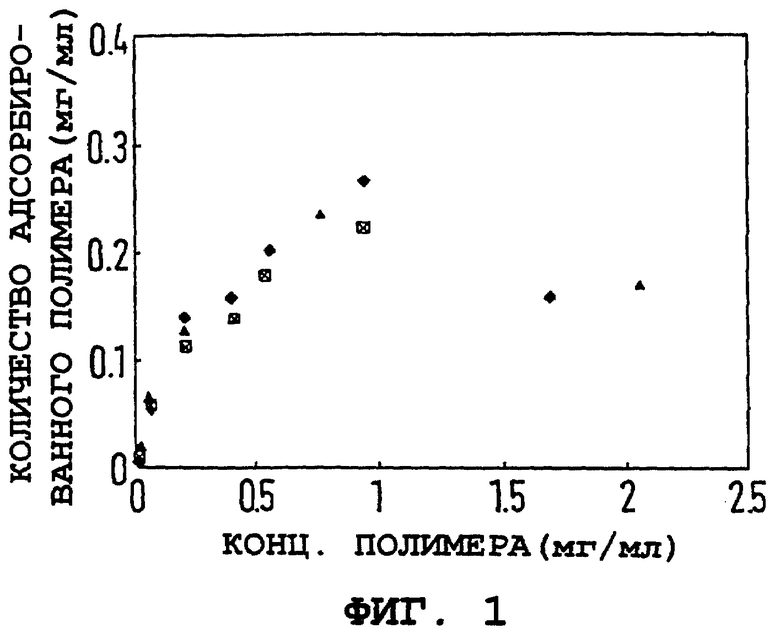
| Фильтр | 1976 |
|
SU580878A1 |
| Рентгеноконтрастное средство | 1979 |
|
SU1061821A1 |
| Способ рентгенологического исследования шейки мочевого пузыря и эластичности его стенок | 1984 |
|
SU1820838A3 |
| Станция контейнеров трубопроводов пневматической установки | 1974 |
|
SU516252A1 |
