Изобретение описывает до сих пор неизвестный и поэтому новый класс соединений, которые являются аналогами 1 α, 25-(OH)2D3 и проявляют селективную активность в отношении клеточных функций.
Уровень техники
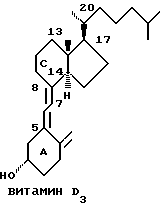
Витамин D, либо поставляемый с пищей (витамин D2 или D3) либо вырабатываемый в коже под воздействием ультрафиолетового света, подвергается метаболизму в некоторых тканях, давая вначале 25-гидроксивитамин D3[25-OHD3] и позднее 1 α, 25-дигидроксивитамин D3[1 α, 25-(OH)D3] и многочисленные другие метаболиты (1-6) витамина D. Некоторые гидроксилазы присутствующие в различных тканях (например, печень, почки, плацента, кератиноциты, фибробласты, моноциты, лимфоциты, костные клетки.) ответственны за активацию и инактивацию метаболических путей родительских молекул витамина D. 1 α, 25-(OH)2D3 ведет себя как классический стероидный гормон, так как его синтез имеет обратную связь с регулированием некоторыми гормонами, ионами и гуморальными факторами, поддерживая нормальный гомеостаз тела плазмы и костных минералов. Кроме того, гормон(ы) витамина D действуют, связывая и активируя специфические рецепторы витамина D, присутствующие в большинстве тканей и клеток. Затем, стероидно-рецепторный комплекс действует как трансактивирующий фактор посредством связывания со специфическими ДНК последовательностями, известными как витамин D чувствительные элементы, т.о., что транскрипция многочисленных генов либо активируется, либо инактивируется (7,8). Эта генная (ин)активация проявляется совместно с другими ядерными вспомогательными факторами, частью которых является рецептор витамина A (R X R) (9, 10). Кроме того, существует некоторое доказательство того, что витамин D, его метаболиты и аналоги действуют по негеномному механизму, либо путем активации кальциевых каналов или другой мембраны, либо вторичных сигналов предвестников (11-13). Витамин D, его метаболиты и аналоги оказывают сильные воздействия на кальциевый и фосфатный метаболизм. Кроме того, они могут быть использованы для профилактики и терапии дефицита витамина D и других нарушений плазменного и костно-минерального гомеостаза (например, остеомаляция, остеопороз, почечная остеодистрофия, нарушения паратироидной функции). Кроме того, найдены рецепторы витамина D в многочисленных тканях и клетках, которые не принадлежат "ткани- мишени" ответственной за только что упомянутый кальциевый гомеостаз. Такие клетки включают больше всего клетки, относящиеся к эндокринной системе, и витамин D, его метаболиты и аналоги способны влиять на гормональную секрецию этих желез или тканей (например, инсусил, паратироид, кальцитонин, гипофизарные гормоны). Зафиксирована также активность рецепторов витамина D и витамина D в переносящих кальций тканях, кроме кишки и костей (например, плацента и молочные железы). Вдобавок, наблюдается, что рецепторы витамина D и витамин D действуют на большинство других клеток (например, клетки, относящиеся к иммунной системе, клетки кожи). Эти клетки или ткани могут быть доброкачественного, аденоматозного и злокачественного типа. Эти так называемые некальцемические эффекты витамина D, его метаболитов и аналогов создают возможность применения таких соединений в различных терапевтических целях, таких как модификация иммунной системы, модификация секреции гормонов, изменение транспорта кальция в различных тканях, модификация внутриклеточной концентрации кальция, индукция дифференциации клеток и ингибирование пролиферации клеток 14, 15). В частности, такие соединения могут быть полезны в терапии нарушений, характеризуемых пролиферацией клеток (например, псориаз, карцинома) (16-18).
Для увеличения терапевтической возможности природного витамин D гормона(ов), могут быть синтезированы аналоги с повышенной эффективностью для специфического воздействия и снижения воздействия другого типа. Например, для получения лекарственного средства против псориаза, могут быть синтезированы аналоги с повышенным воздействием на кератиноциты и лимфоциты, присутствующие в пораженной кожной поверхности, но с пониженными воздействиями на кальций сыворотки, мочи или кости (19-23). Подобные аналоги могут обладать повышенной эффективностью к ингибированию пролиферации клеток карциномы (например, лейкоз или раковые клетки молочной железы) и/или повышению дифференциации таких клеток, либо сами по себе, в результате присущей им эффективности, либо в результате возрастания таких воздействий в комбинации с другими лекарственными средствами (например, факторы роста или цитокины, другие стероидные или антистероидные гормоны или ретиновые кислоты, или родственные соединения) и в то же самое время, обладать пониженной эффективностью в отношении воздействия на кальций сыворотки, мочи и кости или фосфатный гомеостаз.
Другим таким примером могут служить аналоги с повышенной активностью в отношении специфической секреции гормона (например, паратироидный гормон, инсулин) без подобной соответствующей эффективности в отношении других деятельностей природных витамин D гормонов(а). Аналоги с повышенной активностью в отношении доброкачественных клеток, относящихся к иммунной системе, могут быть использованы для лечения иммунных расстройств (например, аутоиммунные расстройства, СПИД, предупреждение отторжения трансплантанта или реакции "трансплантант против хозяина") особенно если их воздействие на другие системы (например, кальциевый и фосфатный метаболизм) должно быть сравнительно ослабленным. Кроме того, могут быть разработаны аналоги с повышенной активностью в отношении образующих кости клеток без одновременного воздействия на костные поглощающие клетки или наоборот и такие аналоги могут быть полезны в лечении костных нарушений.
Ряд аналогов витамина D с видоизмененным специфическим воздействием на различные ткани (особенно, соотношение дифференциации клеток и кальцемических эффектов) описан ранее с переменным успехом в такой дифференциации.
Особенно, окса аналоги в боковой цепи (патент WO 90/09992; EP 0385 446A 2), модификации или гомологизация боковой цепи (WO 87/00834, международная патентная классификация C 07 C 172/00), изменения в стехиометрии при углероде 20 (WO 90/09991, международная патентная классификация C 07 C 401/00, A 61 K 31/59), модификации по C11 C-кольца (EP 89/401, 262-4) и эпокси аналоги (PCT/EP 92/0126) боковой цепи, проявившие интересующие характеристики.
Описание изобретения
Данное изобретение относится к синтезам и биологической оценке оригинальных соединений, которые все же сохраняют некоторые основные характеристики воздействия витамина D, но с более селективным паттерном, (т.е., не все функции физиологического витамин гормона сохраняются в той же относительной степени) и со структурой, которая может быть основательно изменена в центральной части. Действительно, в структуре витамина D можно различить три различные части: (i) центральная часть, состоящая из бициклической C-кольцевой системы: (ii) верхняя часть, состоящая из боковой цепи, которая соединена с положением 17 D-кольца; (iii) нижняя часть, состоящая из A-цикла и Δ (5,7)-диена (так называемого seco (вторичного) B-цикла), который связан с положением 8 c-кольца. Первая цель данного изобретения состоит во внесении значительных структурных изменений в центральной части витамина D.

В частности, данное изобретение относится к аналогам витамина D, в которых отсутствуют объединенные трансконденсированные шестичленный C-цикл и пятичленный D-цикл, но все же существует центральная часть, состоящая из замещенной цепи из пяти атомов, атомы которой соответствуют положениям 8, 14, 13, 17 и 20 витамина D, и концы которой связаны в положении 20 со структурной составляющей, являющейся частью боковой цепи витамина или аналога витамина D, и в положении B с Δ (5,7)-диен составляющей, связанной с A-циклом активного 1-альфа-гидрокси метаболита или установленного аналога витамина D.
Соединения данного изобретения представлены общей формулой I, и в этой формуле: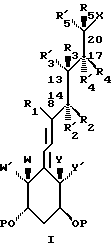
- P обозначает водород, алкил или ацил;
- X обозначает часть боковой цепи витамина D или один из его установленных аналогов;
- Y и Y', которые могут быть одинаковыми или различными, представляют водород или алкил, или, взятые вместе, представляют алкилиден группу, или образуют карбоцикл;
- W и W', которые могут быть одинаковыми, или различными, представляют водород или алкил, или, взятые вместе, представляют алкилиден группу или образуют карбоцикл;
- один из атомов углерода центральной части, соответствующий положениям 14, 13, 17 или 20,
вместе с присоединенными к нему R и R' заместителями, может быть заменен кислородом (O), серой (S) или несущим азот заместителем (NR).
R и R' (т.е. R, R1, R2, R'2, R3, R'3, R'4, R4, R5, R'5):
° когда локализованы в отношении положения 1, 3 центральной цепи, таком как R1 и R3 или R'3, R2 или R'2, и R4 или R'4. R3 или R3 и R5 или R'5, взятые вместе с тремя примыкающими атомами центральной цепи, которые соответствуют положениям 8, 14, 13 или 14, 13, 17 или 13, 17, 20, соответственно, могут образовывать насыщенный или ненасыщенный карбоциклический или гетероциклический 3-, 4-, 5-, 6- или 7- членный цикл, тем самым также включается случай, когда геминально замещенные R и R', взятые вместе, образуют циклическую ненасыщенную связь, при условии, что когда R1 и R3, образуют 6-членный карбоцикл следующей природы
(1) незамещенный и насыщенный, (2) монозамещенный при C-11 или (3) имеющий двойную связь между C-9 и C-11, R2 и R4 не образуют пятичленный карбоцикл, если R3 представляет метил, этил или этенил когда локализованы в отношении положения 1,2 (т.е. вицинальное) центральной цепи, так как R1 и R2 или R'2, R2 или R'2 и R3 или R'3, R3 или R'3 и R4 или R'4, R4 или R'4 и R5 или R'5 и когда не являются частью цикла описанного выше, взятые вместе с двумя прилежащими атомами центральной цепи, которые отвечают положениям 8, 14 или 14, 13 или 13, 17 или 17, 20 соответственно, могут образовывать насыщенный или ненасыщенный карбоциклический или гетероциклический 3-,4-, 5-, 6- или 7-членный цикл, тем самым включается также случай, когда геминально замещенный R и R', взятые вместе, образуют циклическую ненасыщенную связь.
когда локализованы в отношении положения 1,2 (т.е. вицинальное) центральной цепи, так как R1 и R2 или R'2, R2 или R'2 и R3 или R'3, R3 или R'3 и R4 или R'4, R4 или R'4 и R5 или R'5 и когда не являются частью цикла описанного выше, взятые вместе с двумя прилежащими атомами центральной цепи, которые отвечают положениям 8, 14 или 14, 13 или 13, 17 или 17, 20 соответственно, могут образовывать насыщенный или ненасыщенный карбоциклический или гетероциклический 3-,4-, 5-, 6- или 7-членный цикл, тем самым включается также случай, когда геминально замещенный R и R', взятые вместе, образуют циклическую ненасыщенную связь.
когда локализованы в отношении 1,1-положения (т.е. геминальные) центральной цепи, таком как R2 или R'2, или R3 или R'3, или R4 или R'4 или R5 или R'5, и когда не являются частью описанного выше цикла, взятые вместе с атомом углерода несущим R и R' заместители, могут образовывать либо насыщенный, либо ненасыщенный карбоциклический или гетероциклический 3-, 4-, 5-, или 6-членный цикл.
которые могут быть одинаковыми или различными, и когда они не образуют цикл или связь как описано выше, представляют водород или низшую алкил группу, или, взятые вместе в случае геминального замещения, обозначают низшую алкилиден группу.
В контексте изобретения выражение "низшая алкил группа" означает линейную или разветвленную насыщенную или ненасыщенную углеродную цепь, содержащую от 1 до 7 углеродных атома, и "низшая алкилиден группа" означает линейную или разветвленную насыщенную или ненасыщенную углеродную цепь, содержащую от 1 до 7 углеродных атомов, которая связана с одним из атомов 14, 13, 17 и/или 20 основной цепи через двойную связь.
В контексте изобретения часть боковой цепи витамина D или одного из его установленных аналогов означает замещенную алкильную цепь с 2-15 углеродными атомами, особенно как присутствует в витамине D2(C-22 - C-28) или D3(C-22 - C-27) или частично видоизмененную, как показано ниже, с нумерацией витамина D.
Главным образом:
- гидроксил заместитель в одном или более положениях, например 24, 25 и/или 26 и/или
- метил или этил заместитель в одном или более положениях, например 24, 26 и/или 27 и/или
- галоген заместитель(и) в одном или более положениях, например перфторированные в положениях 26 и/или 27 или дифторированные в положении 24 и/или
- дополнительный углеродный атом(ы), главным образом C24 между положениями 24 и 25, с таким же паттерном замещения, как упомянуто выше и/или
- сложноэфирные производные одного или более гидроксильных заместителей упомянутых выше и/или
- замена одного или более углеродных атомов на атомы кислорода, азота или серы, например в положениях 22, 23 или 24 и/или
- цикл, образованный атомами углерода 26 и 27 с помощью одной связи (циклопропан) или путем промежуточного звена из 1-4 углеродных атомов, цикл может быть насыщенным, ненасыщенным или ароматическим и может необязательно быть замещенным в любом возможном положении (иях) вышеупомянутым заместителем и/или
- цикл, образованный между углеродными атомами 26 и 27 посредством 1-4 атомов с получением гетероциклического кольца, включая ароматическое, которое может необязательно быть замещенным в любом возможном положении вышеупомянутым заместителем и/или
- ненасыщенный с одной или более двойной или тройной C-C связями (связью), эти ненасыщенные цепи могут быть замещены в любом возможном положении вышеупомянутыми заместителями и/или
- может присутствовать эпоксидная функциональная группа между углеродными атомами 22, 23 или 23, 24 или 24, 25 или 25, 26: эти эпоксидированные цепи могут быть насыщенными или ненасыщенными и могут быть замещенными в любых возможных положениях вышеупомянутыми заместителями и/или
- два или более углеродных атома боковой цепи могут быть соединены простой связью или через промежуточное звено из 1-5 углеродных атомов, атомов кислорода, азота или серы, образуя 3-7-членный насыщенный или ненасыщенный карбоциклический или гетероциклический, включая ароматический, цикл, который может оптимально быть замещен в любом возможном положении вышеупомянутыми заместителями и/или замещенный в одном или более положениях насыщенным, ненасыщенным карбоциклическим, гетероциклическим или ароматическим циклом, который может быть замещен в любом возможном положении(иях) вышеупомянутыми заместителями
- изомерные формы замещенной цепи.
Следовательно, изобретение относится к сериям аналогов с широко варьируемыми структурами, как приведено в качестве примеров в Таблице 1, где показаны некоторые специфические примеры соединений формулы 1, и которые приводятся в многочисленных препаративных методиках и примерах.
Наиболее часто соединения изобретения представлены одной из формул IIa (тип C), IIb (тип D), IIc (тип E), IId (тип CD), IIe (тип CE), IIf (тип DE) и IIg (ациклический тип):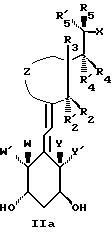

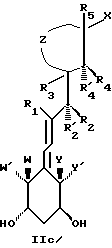




где X, Y, Y', W и W' имеют вышеуказанные значения;
- Z представляет насыщенную или ненасыщенную углеводородную цепь, содержащую ноль (следовательно, Z представляет связь между 1,3-смежными углеродными атомами центральной цепи), один, два, три или четыре атома, которые все могут быть замещенными и/или замененными гетероатомом, таким как кислород, сера и азот.
- R1, R2, R'2, R3, R'3, R4, R'4, R5, R'5, которые могут быть одинаковыми или различными, представляют водород или низший алкил, такой как метил, этил или н-пропил.
Среди этих соединений предпочтительны циклические производные типа C, D, E, CD, CE и DE, которые соответствуют структурам IIIa, IIIb, IIIc, IIId, IIIe и IIIf, соответственно.
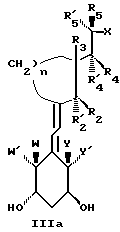

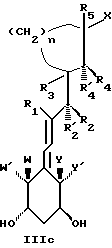

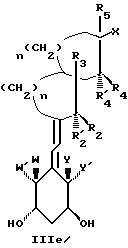

где: n - целое число, равное 2 или 3;
- X представляет одну из следующих частей боковой цепи витамина D: (4-гидрокси-4-метил)фенил, (R)- или (S)-(3-гидрокси-4-метил) фенил, (3'-гидрокси-3'-метил)бутилокси, (4-гидрокси-4-этил)гексил, (4-гидрокси-4-метил)-2-пентинил, (4'-гидрокси-4'-этил)гексилокси; 4,5-эпокси. 4-метил-2-пентинил; 4-гидрокси-4-этил-2-гексииил; (3-метил-2,3-эпокси)-бутилокси; (3-гидрокси-3-этил)пентилокси; (4-гидрокси-4-этил)-гексилокси.
Y, Y', W и W' - одинаковые и представляют водород, или, взятые вместе, представляют метилен группу =CH2;
R1, R2, R'2, R3, R'3, R4, R'4, R5 и R'5, которые могут быть одинаковыми или различными, представляют водород или метил.
Все соединения изобретения могут быть получены использованием реакций, которые хорошо известны в технике синтетической органической химии. В частности, во всех случаях, нижняя часть структуры может быть введена в соответствии со способом Lythgoe (24), по которому защищенный фосфин оксид IV взаимодействует с соответствующим карбонил производным VII, в котором различные реакционноспособные функциональные группы предпочтительно защищены и в котором группы X, Y, Y', W, W', Z, R, R1, R2...R'5, имеют те же значения, что определены выше, после чего с реакционноспособных функциональных групп снимают защиту. О синтезе таких производных, как IV сообщается также в литературе (25).
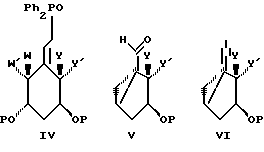
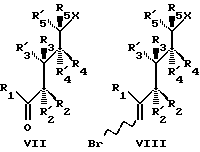
Альтернативные способы включают
a) сочетание соответствующего винильного карбаниона (из VIII) с V с последующим катализируемым кислотой сольволизом и
b) взаимодействие алкинил аниона VI с соответствующим карбонил производным VII с последующим частичным восстановлением тройной связи и катализируемым кислотой сольволизом (26). Можно изменить способ так, чтобы мог быть использован альтернативный способ сочетания, такой как сульфоновый способ (27a) или Okamura's сочетание (27b).
Как будет показано в ряде экспериментов, соединения структуры VII могут быть получены разнообразными способами. Важно отметить, что эти производные в большинстве случаев получают синтетическими способами, которые короче и более эффективны, чем способы, обычно используемые для получения аналогов витамина D.
Схема 1 (см. в конце описания).
a) AlC3 изопрен, толуол, 6 ч, -78oC, - комн. темп. (72%); b) MeONa, MeOH, 1ч, комн.темп. (94%); c) NaBH4, MeOH, 12 ч, 0oC - комн. темп. (86%); d) MEMCl, DIPEA, ТГФ (THF), 3 ч, комн. темп. (98%); e) (i) OSO4, NMMO, Me2CO: H2O (3:1), 12 ч, комн. темп. (86%); (ii) NaIO4, Me2IO4, Me2CO:H2O (3: 1), 12 ч, комн.темп. (98%); f) KOH, 12ч. 60oC (53%); g) (i) 10% Pd/C, 1 атм H2, гексан, 1,5 ч, 0oC (93%); (ii) MeONa, MeOH, 3 ч. 0oC - комн.темп. (97%); h) Ph3P= CH2, HMPA: ТГФ (1:1), 2ч, - 20oC (100%); i) (i) 9-BBN, ТГФ, 4 ч, комн. темп. ; (ii) EtOH, NaOH 6 н., H2O2 30%, 1 ч, 60oC (74%-91%); j) TsCl, DMAP, Et3N, CH2Cl2, 12 ч, комн. темп. (91%-97%); k) NaH, DMSO 2-(1-этокси)-этокси-2-метил-3-бутин, 1,5 ч, 60oC-комн. темп. (70%); l) 10% Pd/C, 4 бар, H2, EtOAc, 1ч, комн.темп. (34%); m) Me2BBr, CH2Cl2, 1 ч, -78oC (73%); n) PDC, CH2Cl2, 4 ч, комн. темп. (86%-99%); o) TSIM, ТГФ, 1 ч, комн. темп. (94%-98%); p) TBAF, ТГФ, 5 дней, 30oC (99%); q) (i) NaH, CS2, 24 ч, комн. темп. ; (ii) MeI, ТГФ, 2 ч, комн. темп. (98%); r) Bu3SnH, AIBN, толуол, 9 ч, 110oC (92%); s) K1, DMSO, 4 ч, 60oC (95%); t) метилвинилкетон, CuI, Zn, EtOH: H2O (7:3), 3,15 ч, 15oC (83%); u) MeMgCl, ТГФ, 1 ч, комн. темп. (98%); v) Amberlyst 15, MeOH, 1 неделя, 30oC (96%).
Схема 1
18-нор-витамина D скелет отражает аналоги типа IIId. Основными стадиями синтеза являются:
а) синтез транс-конденсированного декалона, (b) сужение одного кольца до транс-конденсированного гидриндана, с) построение боковой цепи.
Известно, что описанный в литературе диенофил 1.1 дает син продукт присоединения Дильса- Альдера к силокси группе (28). Таким образом, ригоселективное взаимодействие с изопреном дает 1.2a; эпимеризация индуцированная основанием приводит к 1,2b. Селективное восстановление карбонильной функциональной группы и последующая спиртовая защита приводят к промежуточному продукту 1.3b. Расщепление двойной связи и альдольная реакция образования диальдегида дают транс-гидриндан 1.4. Гидрирование 1.4 приводит к смеси С-17 эпимеров, которая, после индуцируемой основанием эпимеризации, превращается термодинамически более стабильный продукт 1,5a. Реакция Виттига и гидроборация приводят к 1.6a, затем приблизительно к 20% C-20 эпимера. После разделения, боковую цепь вводят посредством тозилата 1,6b. Наконец, каталитическое гидрирование, восстановление C-8 карбонильной функции и 25-гидроксил защита приводят к заданному предшественнику 1.8d. промежуточный продукт 1.5b также позволяет удалить C-12 окси-функциональную группу путем известной методики, включающей радикальную реакцию (29).
Гидроборация 1.9c и последующая трансформация гидроксил группы в иод-соединение 1.10c (4:1, 20S:20R). Боковая цепь вводится при воздействии ультразвука, что дает 1.10 (30). Этот кетон дает при взаимодействии с метилмагнийхлоридом третичный спирт 1.11a. Окисление до C-8 кетона 1.11.C и защита третичного спирта приводят к заданному предшественнику 1.11d.
Аналоги с шестичленной структурой IIIa могут быть синтезированы способом, который включает в качестве ключевой стадии перегруппировку Ireland-Claisen'a субстрата полученного из сложного эфира, спиртовую часть которого составляет (R) - 3-метил-2-циклогексенол (31). Два примера такого подхода приведены в схеме 2 (см. в конце описания).
Взаимодействие (R) -3-метил-2-циклогексенола с гомохиральной кислотой 2.1, получаемой из (-)-ментона (32), дает сложный эфир 2.2. После депротонации сложного эфира, анион этанола взаимодействует (на месте) с третбутилдиметисилил хлоридом; последующий термолиз приводит к циклогексену 2.3 (67% от превращения исходного материала) (33). Карбоксигруппу в 2.3 в дальнейшем превращают в метил группу, следуя стандартным условиям, что в результате дает производное 2.4. Гидроборация 2.4 дает вторичный спирт, который окисляют до циклогексанона 2.5. Последний представляет требуемый карбонильный субстрат для синтеза аналогов 4, обладающих (24S)-конфигурацией.
a) DCC, DMAP, CH2Cl2 (91%); b) LiCA, THF, HMPA; tBuMe2SiCl; c) Δ (67%); d) CH2N2, диэтиловый эфир (86%); e) LAH, THF (89%); f) TsCl, пиридин (96%); g) LAH, THF (91%); (h) 9-BBN, THF: NaOH, H2O2 (80%); i) PDC, CH2Cl2 (90%); j) TBAF, THF, 30oC (88%); k) PPh3, DEAD, pNO2PhCOOH (68%); l) K2CO3, KOH; m) TBSCl, имидазол, DMF, DMAP (97%); n) 9-BBN, THF (92%); o) PDC, CH2Cl2 (92%); p) DCC (96%); q) LDA, TBSCl; r) LAH, THF, Δ (86%); s) TsCl, py (100%); t) LAN, THF (100%); u) Hg(OAc)2, NaOH, NaBH4; v) TESCl, DMAP, DMF, имидазол; w) 9-BBN, H2O2 (95%); x) PDC (80%).
Схема 2
Синтез его (24R)-эпимера выполняют аналогичным способом после инверсии при C-24. Поэтому, исходя из промежуточного продукта 2.4. удаляют защитную группу и образовавшийся спирт обращают по методике (34). Воспроизведение приведенной выше последовательности дает циклогексанон 2.7. Обычная методика сочетания приводит затем в конечном итоге к аналогам 5 и 6, которые содержат (24R)-гидрокси группу.
Использованием аналогичного подхода может быть осуществлен синтез 25-гидрокси аналога. Поэтому (R)-3-метил-2-циклогексенол этерифицируют (R)-(+)-цитронелловой кислотой (2.8), получая сложный эфир 2.9. Следующая затем перегруппировка Ireland-Claisen'a дает кислоту 2.10. После превращения карбоксигруппы в метил группу (2.11), тризамещенную двойную связь преимущественно окисляют до третичного спирта, используя ацетат ртути, NaOH и боргидрид натрия. Последующая спиртовая защита и региоселективное окисление циклической двойной связи приводят к циклогексанону 2.12, из которого получают, используя обычную реакцию сочетания, аналоги 7 и 8.
Аналоги типа IIIa с обращенной конфигурацией при C-13 могут также быть получены способом Ireland-Claisen'a Это иллюстрируется схемой 3 (см. в конце описания). С этой целью ацетат (S)-3- метил-2-циклогексенола (3.1:≈86%) может быть непосредственно депротонирован, и соответствующий энол силилэфир перегруппировывают до кислоты 3.2. Дальнейшее обогащение заданного энантиомера достигается посредством разделения в R-(+)- α -метилбензиламине. Дальнейшая последовательность включает восстановление кислоты 3.2 и защиту образовавшегося первичного спирта до 3.3. Последний может быть окислен использованием 9-BBN и перекисью водорода до спирта 3.4. После защиты-снятия защиты, первичный спирт используют для построения окса боковой цепи. Это осуществляется взаимодействием аниона с 1-хлор-3-метил-2-бутеном. После гидролиза и окисления получают циклогексанон 3,6. Окончательное введение 25-гидрокси группы выполняют по методике восстановления с помощью меркурацетатно-гидридного восстановления. Полученное карбонилпроизводное 3.7 служит предшественником аналога 9, характеризуемого 22-окса боковой цепью и эпимерной конфигурации при C-13. Следует далее отметить, что обычное сочетание Horner-Wittig'a. также приводит в этом случае к образованию изомера с (Z)-7,8-двойной связью (соотношение 4:1).
P=SiPh2tBu
a) PGL, фосфатный буфер (≈86%); b) LDA, tBuMe2SiCl, ТГФ; HCl; разделение в R-(+)- α -метил бензиламине (48%); c) LAH, диэтиловый эфир (95%); d) tBuPh2SiCl, DMF имидазол (98%); e) 9-BBN, H2O2 (96%); f) ДНР, CH2Cl2 (93%); g) (м-Bu)4NF, ТГФ (91%); h) ClCH2CH=C(CH3)2, NaH, DMF (81%); i) TSOH, MeOH, комн. темп. (98%); j) PDC, CH2Cl2, комн. темп. (84%); k) Hg(OAc)2, NaBH4 (68%).
Схема 3
Другой подход к синтезу аналогов типа IIIa состоит в присоединении к сопряженным связям части боковой цепи, включающей 3-метил-2-циклогексанон в качестве субстрата. Пример приведен в схеме 4 (см. в конце описания).
a) tBuPh2SiCl, имидазол, DMF, 36 ч, комн. темп. (100%); b) DIBALH, гексан, 0,5 ч, - 78oC; c) t-BuOK, (MeO)2Р(O)CHN2 THF, 20ч, - 78oC, комн. темп. (90% всего из 4.2); d) В-ВГ-9-BBN, CH2Cl2, 4 ч, 0oC, затем CH3COOH, 0,5 ч, 0oC, NaOH; H2O2 0,5 ч, комн. темп. (90%); e) tBuLi, CuI/HMPT, BF3-OEt2, 3-метил циклогексенон, диэтиловый эфир, 16 ч, -120o-20oC (40%); f) TBAF, THP, 3 ч, комн. темп. (90%); g) ВДЖХ, элюент: гексан:этилацетат 6:4; h) Ph3P, имидазол, l2, THF, 6 ч, -20oC - комн.темп. (88%);
Схема 4
Необходимый гомохиральный купрат-реагент получают приведенной далее последовательностью превращений, исходя из метил (S)-3-гидрокси-2-метилпропаноата (4.1). После защиты спирта, сложный эфир восстанавливают и полученный альдегид 4.3 обрабатывают анионом, полученным из метил диазометил фосфоната (35). Образовавшийся алкин 4.4, полученный с 90% выходом из 4.2, превращают в дальнейшем в винил бромид производное 4.5. Из последнего требуемый купрат реагент получают обработкой трет- бутиллитием и CuJ при -120oC. 1,4-Присоединение к 3-метил-2- циклогексенону выполняют в диэтиловом эфире в присутствии борнитрофторида (36). После обычной обработки и очистки получают циклогексанон 4.6 вместе с C13-эпимером.
После гидролиза заданный спирт 4.7 может быть отделен от его C13-эпимера (конфигурационное отнесение соответствует CD), и далее превращают в иодид 4.8. Это карбонильное производное служит субстратом для присоединения A-цикла.
Для синтеза соединений типа IIc приведен пример в схеме 5 (см. в конце описания). Исходное соединение 5.1 получают из R-лимонно-яблочной кислоты (37).
a) TSOH, THF, 20 ч, комн.темп. (90%); b) DDQ, 3 ч, комн.темп.; c) PDC, DMF, 20 ч, комн.темп.; d) CH2N2, Et2O (94%); e) EtMgBr, 2 ч, комн. темп.; f) Pd/c, H2 (50%); g) TPAP, NMMO, 2 ч, комн.темп. (70%);
Схема 5
Построение гетероциклических ядер из 5.1 и 5.2 предусматривает сборку предшествующего скелета путем конвергенции. Оба эпимера 5.3 с соответственно и ориентированной боковой цепью получают в соотношении 1:1. Дальнейшие превращения выполняются на этой эпимерной смеси. Разделение возможно на стадии конечных аналогов. Трансформация п-метоксибензилового эфира в 5.3 (α+β) в сложном эфире 5.4 (α+β) и последующая реакция Гриньяра приводят к боковой цепи. Наконец вводят альдегидную функциональную группу и получают предшественник 5.6 (α+β) .
Группа аналогов с пятичленным циклом, как примеры общей формулы IIIc, легко может быть получена исходя из известного 6.1 (38). Расщепление простой эфирной связи в 6.1 с помощью иодида натрия приводит к ключевому промежуточному соединению, иодиду 6.2. Основные синтетические возможности при введении (a) боковой цепи использованием иод-функциональной группы через (1) прямое сочетание или (2) после превращения иодметильного заместителя в гидроксильный заместитель или (3) после обращения ориентации иодметильного заместителя или (4) после превращения иодид группы в формил группу и (b) A-циклической части после гомологизации по гидроксиметил заместителю. Примеры таких подходов приведены ниже и иллюстрированы схемой 6 (см. в конце описания).
Иод-соединение 6.2 может быть подвергнуто реакции сочетания в условиях обработки ультразвуком с метил-винил кетоном и этил-винил кетоном, что дает соответственно 6.8 и 6.9. Кетон 6.8 при взаимодействии с метилмагнийбромидом дает соответствующий третичный спирт. Окисление первичного спирта и 1-C гомологизация образующегося альдегида 6.10 с метокситрифенилфосфоний метилидом и последующий гидролиз приводят к альдегиду 6.12, требуемому для сочетания с A-циклом. Аналогично, реакция 6.9 с этилмагнийбромидом и последующая трансформация дают 6.13.
a) Cl3SiCH3, NaI, CH3CN (90%); b) DIPEA, CH3OCH2Cl, CH2Cl2 (86%); c) TBAF, THF (88%); d) OSO4, NaIO4, THP:H2O (65%); e) LiAlH4, THF, комн.темп., (95%); f) (1) 9-BBN, TMF, 60oC;
2) H2O2, NaOH (87%); g) Ph3P имидазол, I2, диэтиловый эфир: CH3CN 3:1 (93%); h) Amberlyst-15, MeOH, THF (86%); i) CuJ, Zn., MVK, EVK или T-2,4-пентадионовой кислоты этиловый эфир, EtOH:H2O 7:3 (45%); j) Mg, EtI, Et2O, 0oC (73%); j') MeLi, Et2O, - 78oC (85%); k) TPAP, NMMO, молекулярные сита  CH2Cl2 (66%); k') (CrO3)Py2 ("Collins"). CH2Cl2 (35%); l)(1) [Ph3PCH2OCH3] +Cl-, н-BuLi, диэтиловый эфир, -30oC, 2) HCl 2 н., THF (48%); m) KOH, изопренилхлорид, 18-Крон-6, толуол, ультразвук (40%); n) KOH, аллибромид, 18-Крон-6, THF (75%); o) (1) Hg (OAc)2, H2O, TH; 2) NaBH4, NaOH (94%); p) SO3P,. Et3N, CH2Cl2:DMSO 1:1 (71%); q) (1) 9-BBN, THF, 60oC, (2) H2O2, NaOH (95%); r) (1) PDC, DMF, 40oC; CH2N2, Et3O, 0oC (36%); s) Mg, Et1 (2 экв. ), Et2O, 0oC (92%); t) MEMCl, DIPEA, CH2Cl2; (80%); u) (1) NaNO2, DMF, мочевина, 25oC (45!%); (2) NaOMe (1,3 экв.), MeOH; (3) O3, Na2S - 78oC (70%); (v) (EeO)2P(O)CH2CH=CHCOOEt. LDA, THF(91%); (w) H2/Pd (4 атм.), 3 ч (80%); x) Me2BBr, ClCH2CH2Cl:CH2Cl2 1:6 (93%); y) Mg, MeBr, THF; z) TBAF, THF.
CH2Cl2 (66%); k') (CrO3)Py2 ("Collins"). CH2Cl2 (35%); l)(1) [Ph3PCH2OCH3] +Cl-, н-BuLi, диэтиловый эфир, -30oC, 2) HCl 2 н., THF (48%); m) KOH, изопренилхлорид, 18-Крон-6, толуол, ультразвук (40%); n) KOH, аллибромид, 18-Крон-6, THF (75%); o) (1) Hg (OAc)2, H2O, TH; 2) NaBH4, NaOH (94%); p) SO3P,. Et3N, CH2Cl2:DMSO 1:1 (71%); q) (1) 9-BBN, THF, 60oC, (2) H2O2, NaOH (95%); r) (1) PDC, DMF, 40oC; CH2N2, Et3O, 0oC (36%); s) Mg, Et1 (2 экв. ), Et2O, 0oC (92%); t) MEMCl, DIPEA, CH2Cl2; (80%); u) (1) NaNO2, DMF, мочевина, 25oC (45!%); (2) NaOMe (1,3 экв.), MeOH; (3) O3, Na2S - 78oC (70%); (v) (EeO)2P(O)CH2CH=CHCOOEt. LDA, THF(91%); (w) H2/Pd (4 атм.), 3 ч (80%); x) Me2BBr, ClCH2CH2Cl:CH2Cl2 1:6 (93%); y) Mg, MeBr, THF; z) TBAF, THF.
Схема 6
С другой стороны, основание полученное отщеплением иодида 6.2 после защиты гидроксил группы, дает олефин 6.3. Гидроборация 6.3 приводит к двум диастереомерам в соотношении 1:1. После выделения, изомер 6.6 превращают в иодид 6.7. Как описано для эпимера 6.2, 6.7 используют для синтеза ключевого промежуточного соединения 6.16.
Окислительное расщепление двойной связи в 6.3 и восстановление образовавшегося кетона приводит к эпимерным спиртам 6.4 и 6.5. Смесь подвергают реакции Вильямсона (получение простых эфиров), что приводит к аллиловым простым эфирам 6.17 α и 6.17 β Присоединение воды по двойной связи, гидролиз МОМ-диэтилового эфира и окисление образовавшегося первичного спирта дает эпимерные альдегиды 6.19 α и 6.19 β , которые могут быть разделены ВДЖХ (гексан-ацетон 9:1). Соответствующие структуры обоих эпимеров устанавливают nOe измерениями. 1-C гомологизация, как уже описано для 6.10, приводит к промежуточным продуктам 6.21 α и 6.21 β .
Также взаимодействие смеси анионов 6.4 и 6.5 с аллилбромидом дает смесь 6.18 (α+β) . Последовательное применение гидроборации концевой двойной связи, окисление и обработка диазометаном проводят к соответствующему сложному метиловому эфиру карбоновой кислоты, который реагирует с этилмагнийбромидом. Последующий гидролиз простого МОМ эфира и окисление первичного спирта дают эпимерные альдегиды, которые разделяют ВДЖХ. Соответствующие структуры 6.20 α и 6.20 β устанавливают nOe измерениями. Затем 1-C гомологизации дает соответственно 6.22 α и 6.22 β Сочетание альдегидов 6.12, 6.13, 6.16 6.21  , 6.21 β , 6.22 α и 6.22 β с A-циклом описано ниже.
, 6.21 β , 6.22 α и 6.22 β с A-циклом описано ниже.
Кроме того, превращение иодида 6.2, через соответствующее нитросоединение (39), в альдегид 6.24 дает возможность введения боковой цепи. Это может быть выполнено по реакции типа Horner-Wittig'a с применением фосфонокротоната с последующим каталитическим гидрированием. Затем выполняется 1-C гомологизация как описано для 6.12. Сочетание (24) образующегося 6.26 с анионом 13.1 приводит к промежуточному продукту 6.27. После чего сложноэфирная функциональная группа может быть превращена в третичные спирты. Эта последовательность служит примером построения аналогов, в которых заданная боковая цепь образуется в результате Lythgoe's сочетания.
в другом примере из этой серии иод-соединение 6.2 сочетается в условиях обработки ультразвуком со сложным этиловым эфиром транс-2,4- пентадионовой кислоты. Вслед за гидрированием 6.28, образующийся спирт 6.29 подвергают, как уже описано, гомологизации до предшественника 6.30.
Другой пример аналогов типа IIIc имеет ароматическое кольцо и легко может быть построен из 3-гидроксифенетилового спирта 7.1 (см. схему 7 в конце описания) и включает создание боковой цепи через фенольную гидроксильную группу и окисление первичного спирта до альдегидной функциональной группы, подходящей для сочетания с А циклической частью. Образование простого эфира с тозилатом 7.2 дает 7.3.
a) KOH, DMSO, 4 ч, комн.темп. (85%); b) Et3N, SO3•C5H5N, 15 мин (48%); c) CH3I, KO-t, Bu (55%).
Схема 7
После окисления первичного спирта в 7.3, образующийся альдегид бисметилируют, получая предшественник 7.4.
Кроме того, возможно несколько способов для синтеза аналогов с общей структурой IIIc. Несколько возможных вариантов показано в схеме 8 (см. в конце описания).
В первом способе описанное ранее соединение 3.4 (схема 3) этерифицируют как указано выше: после снятия защиты могут быть выделены два спиртовых диастереомера 8.1. Оба выделенных спирта 8.1 α и 8.1 β обрабатывают ацетатом ртути/боргидридом натрия, и впоследствии окисляют, получая альдегиды 8.2 и 8.3, которые после выполнения в обычном порядке сочетания дают аналоги 22 и 23, соответственно.
β -Эпимер 8.1 β может также быть превращен в диастереомерную смесь эпоксидов, которые после окисления приводят к альдегиду 8.4. Полученное соединение представляет собой субстрат для сочетания с аналогом 24. Окончательно 8.4 может также привести к эпимерной смеси первичных спиртов через окисление до соответствующего кетона, реакцию Виттига с метилентрифенилфосфораном и 9-BBN окислением. После тозилирования первичного спирта вводят боковую цепь через вытеснение анионом 3-этоксиэтил-3-метил-1-бутина; снятие защиты дает 8,5 в виде смеси эпимеров, которые теперь могут быть разделены. Окисление α -эпимера 8,5 α с помощью PDC приводит к альдегиду 8.6, предшественнику аналога 25.
a) ClCH2CH= C(CH3)2, NaH (89%); b) (нВ)4NF (81%); c) Hg(OAc)2; NaOH, NaBH4 (76%) 2: 1 смесь; d) PDC, CH2Cl2, комн.темп. (80%); e) мCPBA, CH2C2, 0oC (86%); f) PDC, CH2Cl2 (73%); g) PDC, (96%); h) Ph3P+CH3Br-, нBuLi, THF (83%); i) 9-BBN (90%); j) TSCl, пиридин (95%); k) HC=CC(Me)2OEE, NaH, DMSO (62%); l) (нBu)4NF, THF (92%); m) PDC, CH2C2 (71%).
aa) т-бутилдиметилсилил этил кетен ацеталь, HgI2, CH2Cl2; ab) LiAlH4, Et2O; ac) TBAF, THF (61% из 8.1); ad) TBD MSCl, имидазол, DMF (99%); ae) O3, MeOH, -30oC, FeSO4, Cu(OAc)2; af) Pd, H2 (4 атм) (61% из 8.2); ag) TBAF, THF (100%); ah) MEMCl, EtiPr2N, CH2Cl2 (99%); ai) NaBH4 MeOH (70%); aj) KOH, 18-крон-6, хлор-3-метил-2-бутен, толуол, ультразвук (43%); ak) Hg (OAc)2 NaOH, NaBH4 (78%); al) Amberlyst-15, MeOH:THF1:1 (100%); am) CH2Cl2; DMSO 1: 2, пиридинсульфуртриоксидный комплекс, Et3N (69%).
ba) K2CO3, MeOH, 1 ч, комн.темп. (55%); bb) BnO-Cl= (=NH)Cll3, CF3SO, CH2Cl2 /холоди.гексан, 90 мин, 0oC (60%); bc) (i) FOSMIC, BuLi, Et2O 2 часа, 0oC; (ii) HCl (37%) раствор), 12 час. комн.темп. (67%); bd) ⊘3P =CH-CH2COO-, THF, 2 ч, комн. темп.; be) CH2N2, Et2 (28% общий выход); bf) MeLi, LiBr, диэтиловый эфир; 2 час. 0oC; bg) Pd/C 10%, EtOAc, H2, 6 ч, комн.т. (53%); bh) NMMO, TPAP, CH2Cl2, 2 ч, комн.темп. (85%).
Схема 8
Пример синтеза аналогов общей формулы IIIc, исходя из R-карвона (8.7) также показан в схеме 8. Ключевые моменты: а) диастереоселективное 1,4-присоединение
b) удаление изопропилиденовой группы (40) (с) введение окса-боковой цепи. Этот путь приводит к разделению диастереоизомеров.
1,4-Присоединение включающее силилированный кетен ацеталь на 8.7 приводит к простому энол силил эфиру. Перед гидролизом сложноэфирную функциональную группу в этом соединении обычно восстанавливают до гидроксильной группы. Озонолиз 8.8 и последующая обработка солями железа и меди позволяет расщеплять изопропилиденовый заместитель. Каталитическое гидрирование образовавшейся двойной связи и изменение защитной группы дают простой MEM эфир 8.10. Впоследствии восстановление боргидридом натрия приводит к изомерному спирту 8.11. Эта смесь подвергается воздействию изопренилхлорида с образованием простого эфира. Простые эфиры 8.12, 8.13 и 8.14 могут быть разделены. Каждый индивидуально превращают в соответствующие третичные спирты 8.16, 8.17 и 8.18.
Может быть представлен еще один способ получения аналогов формулы IIIc, исходящий из соединения 8.19, кетона, описанного в литературе (41). Он включает построение боковой цепи использованием карбонильной функциональной группы.
Реакция с диэтил(изоцианометил)фосфонатом с последующим кислотным гидролизом дает альдегид 8.21. Боковая цепь вводится гомологизацией Виттига. Воздействие метилалюминия на 8.22 приводит к третичному спирту. Двойную связь гидрируют с сопутствующим расщеплением простого бензил эфира. Окончательно окисление первичной гидроксильной группы в 8.23 дает предшествующий альдегид 8.24.
Пример синтеза аналога общей формулы IIIe показан в схеме 9 (см. в конце описания). При использовании в качестве исходного соединения известного гомохирального энона 9.1 (42) разлагающее металл аммоний восстановление приводит к транс-конденсированному декалону 9.2. Введение боковой цепи включает реакцию с натриевой солью защищенного 2-метил-3-бутин-2-ола, с последующей дегидратацией до 9.3. Каталитическое гидрирование в конечном счете приводит к декалону 9.4., предшественнику аналога 31.
a) Li, INH3 (56%); b) NaC=C-C(Me)2OEE DMSO (74%); с) Tf2O CH2Cl2, пиридин, DMAP (25%); d) H2, Pd, EtOA2 (65%); e) TMS, имидазол;
aa) HgT2, CH2(OTBAS)(OEt), Et3N, CH2Cl2, 3 ч, -78oC комн.темп. (97%); ab) толуол, гликоль, H2O4, молекулярные сита  10 ч нагревание до кипения с обратным холодильником (75%); ac) DIBAN, толуол, 4 ч, -78oC (93%); ad) триэтилфосфонацетат, BuLi, THF, 17 ч, -78oC - комн. темп. (88%); ae) Pd/C, гексан, 1 атм H2, 1,5 ч, 0oC (99%); af) MeMgI, диэтиловый эфир, 5 ч, комн. темп. (85%); ag) Amberlyst-15, THF:H2O 2:1, 12 ч, комн.темп. (99%); ah) TSIM, THF, 2 ч, комн. темп. (97%); ai) EtMgI, диэтиловый эфир, 2 ч, комн. темп. (89%); aj) Ph3 + (CH2)3COOBzBr-, LDA ДА, HMPA: THF 1:1,2 ч. -20oC (21%);
10 ч нагревание до кипения с обратным холодильником (75%); ac) DIBAN, толуол, 4 ч, -78oC (93%); ad) триэтилфосфонацетат, BuLi, THF, 17 ч, -78oC - комн. темп. (88%); ae) Pd/C, гексан, 1 атм H2, 1,5 ч, 0oC (99%); af) MeMgI, диэтиловый эфир, 5 ч, комн. темп. (85%); ag) Amberlyst-15, THF:H2O 2:1, 12 ч, комн.темп. (99%); ah) TSIM, THF, 2 ч, комн. темп. (97%); ai) EtMgI, диэтиловый эфир, 2 ч, комн. темп. (89%); aj) Ph3 + (CH2)3COOBzBr-, LDA ДА, HMPA: THF 1:1,2 ч. -20oC (21%);
Схема 9
Дальнейшие примеры аналогов общей формулы IIIe, в которых один из циклов бициклической системы представляет гетеро-цикл, также показаны в схеме 9. Синтез начинается от известного энона 9.5 (28) и протекает через присоединение к сопряженным связям, образование гетероцикла и конденсацию Виттига, как показано в схеме. Получают различные карбонил производные, которые конденсируют с A-циклом обычным способом.
Примеры предшественников аналогов типа IIIb, с циклогексановым D-циклом, описаны в схеме 10 (см. в конце описания). Исходным материалом для этих частных примеров служит известный 10.1 (43); для построения боковой цепи используют сложноэфирную функцию, хотя карбонильная функция также может быть трансформирована в формил группу. Алкилирование 10.2 приводит к 10.3 как основному (95%) эпимеру в соответствии с приведенными литературными данными (44). После превращения сложноэфирной функции в метил группу, следуя классической методике, концевую двойную связь в 10.6 расщепляют озонолизом. Окончательное снятие защиты приводит к кетону 10.7.
a) PPTS. ацетон, 2 ч, кипение (86%); b) LDA, THF, 1 ч. - 30oC; 5-Br-1-пентен, HMPA, 3 ч. -78oC (93%); c) LiAlH4, Et2O (99,8%); d) TosCl, TEA, DMAP, DCM, 20 ч, комн. темп. (95%); e) LiAlH4, Et2O, 5 ч. кипение (88%); f) O3, DCM: 2,5 М NaOH в MeOH 4:1 (об./об.). 45 мин. -78oC (64%); g) PPTS, ацетон, вода (кат.), 3 ч, кипение (75%); h) FOSMlC, BuLi, Et2O, 15 мин., -60oC, HCl 37%, 12 ч, комн.т. (64%), i) Me2S=CH2, THF, 2 ч; комн.т. (33%), j) BF3•OEt2Et2O 12 ч, комн.темп. (65%).
Схема 10
Образование формил заместителя из кетона хорошо известно. Здесь используется два способа: один из которых включает взаимодействие с диэтил (изоцианометил)фосфонатом (45). Эпимерные альдегиды 10.8 и 10.9 могут быть разделены. Кроме того, катализируемая основанием эпимеризация 10.9 дает термодинамически более стабильный 10.8. Оба предшественника 10.8 и 10.9 могут быть превращены в аналоги через сочетание с 13.1 и органометаллические реакции при условиях, аналогичных условиям синтеза 19 из 6.27. Другой способ включает в качестве промежуточного соединения эпоксид 10.10, который затем превращают в смесь 10.8 и 10.9.
Примеры предшествующих соединений типа IIb с 5-членным D-циклом описаны в схеме 11 (см. в конце описания).
В одном случае синтез начинается с простого т-бутилдиметилсилилового эфира 11.1 коммерчески доступного 5-(гидроксиметил)фурфурала. Реакция Виттига с илидом 11.2 дает простой эфир 11.3, который легко превращают в третичный спирт 11.4. Окончательно снятие защитной группы и окисление первичной гидроксильной группы приводит к предшественнику 11.5.
Предшественник 11.11 может быть получен из известного 11.6 (46) и синтез включает гидроборацию двойной связи после восстановительного отщепления атома брома и образования тозилата. Эпимеры 11.9 подвергают затем реакции сочетания с боковой цепью. Окисление дает эпимерические альдегиды 11.11 α + 11.11 β
Близкородственный предшественник может быть получен из(-)-камфарной кислоты (11.12). Вслед за восстановлением, образование простого моно-эфира, катализируемое SAM 11 липазой. предусматривает требуемое различие двух гидроксильных функциональных групп.
После окисления соответствующего альдегида, может быть введена боковая цепь из 11.13. Это приводит к промежуточному продукту 11.14.
С одной стороны, реакция Гриньяра и окисление первичного спирта приводят к предшественнику 11.21. С другой стороны, 11.14 может легко быть превращен в предшественник 11.19 и 11.20; теперь включается дополнительная стадия каталитического гидрирования.
Другой D-циклический аналог типа IIb, называемый 8,9-втор-1 α , 25-(OH) у витамин D3, получают из 11.22 (из 12.1). Образование энольного производного (например, трифлат) через кинетически индуцированный энолят анион и последующий озонолиз дают 11.24. Восстановление соответствующего тозилата 11.25 и последующее окисление первичной гидроксильной группы в 11.26 дают 8,9-втор C/D циклический предшественник 11.27.
a) THF, HMPA, 2ч, -20oC (62%); b) EtMgBr Et2O, 5 ч. -10oC (86% 11.14, 75% 11.17); c) TBAF, THF, 1ч, комн. темп.; d) SO3 - пиридин, CH2Cl2, DMSO, 3 ч, -10oC (40%, 11.4; 63% 11.10; 80% 11.13); e) нBu3SnH, 100oC,; f) TsCl, Et3N, CH2Cl2, DMAP 71%; i) 9-BBN, THF, 60oC; ii) H2O2, (85%); (h)≡-C(Et)2OEE, NaH, DMSO, 90 мин, 65oC (63%); i) LiAlH4 THF, Et2O, 4 ч. (88%); j) винилацетат, SAM 11, 66 ч. 37oC (60%); k) три-этил-4-фосфонацетат, LDA, THF, 24 ч, 0o--->25oC; l) K2CO3, EtOH, комн.темп. (65% всего); m) 5% Rh/Al2O3, EtOAc, H2 (90%); n) MeMgBr, Et2O, 90 мин, комн. темп. (86% 11.18, 94% 11.15); o) TPAP, NMNO, CH2Cl2, 2 ч, комн.темп. (80-78%); p) LDA, THF, 15 мин, -78oC, 2 ч, комн.темп., затем PhNTf2, 18ч 0oC (65%); q) O3, NaHCO3, MeOH, -78oC, затем NaBH4, MeOH, 18 ч. -78oC до комн. темп. (91% всего); r) LiAlH4, THF, Δ 36 ч (61%); s) TPAP, NMNO, CH2Cl2, 1 ч, комн. темп. (50%);
Схема 11
Примеры предшественников для синтеза аналогов типа II d, которые характеризуются цис-конденсированной бициклической системой, показаны в схеме 12 (см. в конце описания). Эти предшественники могут быть получены через a) озонолиз витамина D2, (b) введение боковой цепи и (c) эпимеризацию при C-13. Эпимеризация известного кетона 12.1 (47) приводит к приблизительно 3:1 соотношению в пользу цис-конденсированного изомера, 25-гидроксил группу предварительно защищают перед сочетанием с A-циклом. Можно исходить также из известного диола Lythgoe'a (48), который легко может быть превращен в монотозилат 12.3. Реакция 12.3 с анионом 3-этоксиэтил-3-метил-1-бутина приводят к 12.4 промежуточного продукта для двух предшественников. Окисление и эпимеризация дают кетон 12.5.
С другой стороны, отделение 25-оксигруппы ведет к 12.6, в котором двойная связь может быть селективно эпоксидирована. Окисление гидроксильной группы и последующая ДВИ промежуточная эпителизация дают цис-конденсированный кетон 12.7.
a) NaOMe, MeOH, 24 часа, комн. темп. (73% для 12,2; 65% для 12,5); b) TMS имидазол, CH2Cl2, 3 часа, комн. темп. (79%); c) NaH, DMSO, HC=C-C(CH3)2OEE, (67% для 12,4; 56% для 12,12); d) PDC:CH2Cl2, 2 часа (84% для 12.5; 69% для 12,7; 70% для 12,12); e) TSOH, толуол. 60oC (74%); f) мCPBA, Na2HPO4, THF (81%); g) DBU, CH2Cl2, 3 дня, комн. темп.; (h) (i) O3, CH2Cl2: MeOH (1: 1), -78oC; ii) Me2S, комн. темп.; i) 5% HCl, THF (1:3), 30oC, 36 час; j) NaBH4 MeOH. комн. темп. (99%); k) TSCl, пиридин, 0oC, 12 час (56%); l) триэтил-4- фосфонокротонат, DLA, THF, -78oC комн. темп., 3 ч (85%); m) NaOEt, EtOH, комн.темп., 21 ч (62%); n) H2, Rh/Al2O3, EtOAc комн. темп., 1,5 ч (89-93%)
Схема 12
Важным является тот факт, что одновременно может быть выполнена эффективная эпимеризация при C-20 и C-13. Озонолиз витамина D2 без восстановительной обработки дает кето-альдегид 12.8, который при катализируемой кислотой эпимеризации приводит к смеси четырех возможных изомеров, из которых основной компонент 12.9 может быть выделен ВДЖХ. Легче выделять два цис-конденсированных изомера вместе и восстанавливать карбонильные функциональные группы перед разделением C-20 эпимеров. Первичная гидроксильная группа в 12.10 может быть тозилирована с достаточной селективностью. Сочетание тозилата 12.11 с анионом 3-этоксиэтил-3-метил-1-бутина и последующее окисление дают предшественник 12.12. Аналогичное сочетание приводит к предшественнику 12.13. Эти кетоны и тетрагидропроизводное 12.14 могут сочетаться с анионом 13.2, давая соответственно аналоги 46.48 и 47.
Селективное взаимодействие Horner-Wittig'a альдегидной группы в 12.9 с анионом триэтил 4-фосфонкротоната представляет альтернативный путь построения боковой цепи. Он приводит к 12.15 и в дальнейшем к 12.16. Сочетание с 13.2 с последующим взаимодействием с подходящим органометаллом приводит к аналогам 49 и 52. Та же последовательность, но исходящая из S-эпимера 12.8 приводит к 12.17 и 12.18 предшественникам аналогов 53 и 55.
Предшествующие альдегиды или кетоны, описанные в схемах 1, 2, 3, 5, 6, 7, 8, 9, 10, 11 и 12, сочетаются с A-циклом фосфин оксидов 13.1 и 13.2 использованием методики Lythgoe'a (схема 13 в конце описания). Этим способом получены аналоги витамина D3 с 1 по 55, показанные в таблице 1. Что касается 5- и 6-членных циклов типа C, и E, и комбинацией CD, CE и DE (см. таблицу 1), следует отметить, что циклы могут быть насыщенными, такие как циклопентан или циклогексан, ненасыщенными, такие как циклопентен или циклогексен.
a) n.BuLi, THF, -78oC; b) n.Bu4NF, THF; c) Amberlyst-15, MeOH; d) PPTS, CH2Cl2; e) MeMgX, THF, r.t. (комн.т.); f) EtMgX, THF, r.t. (комн.темп.).
Схема 13
Циклы могут также быть замещены одним или более заместителями состоящими из группы, включающей алкил, алкенил, алкинил, арил, галоген, гидрокси и их производные функциональные группы, такие как простые эфиры и сложные эфиры, и амины и их функциональные группы, такие как N-алкилированные амины и амиды.
Сочетание по Horner-Wittig с использованием классического A-циклического фосфиноксида и транс-конденсированного CD-циклического кетона приводит исключительно к E-стереохимии при 7,8-двойной связи. Глубокое изменение центральной CD-циклической системы в новых аналогах, описанных выше, может приводить к изменению в стереоселективности для этой трансформации. Это особенно справедливо в случаях, когда конденсация Виттига выполняется на циклоалканах для которых α - положения могут быть менее дифференцированными по сравнению с классическим примером. Следовательно, эту трудность следует ожидать, особенно в случае синтеза аналогов типа IIIa, III и IIIe. В качестве примера, конденсация Виттига на декалоне 9.4 приводит к смеси 2:1 E- и Z-производных 14.1 и 14.2, которые затем гидролизуют до аналога 31, выделяемого в виде смеси 2: 1 изомеров. Аналогичным примером является реакция на 3.6, которая приводит к разделяемой 4:1 смеси E: Z-изомеров 14.3 и 14.4 (см. схему 14 в конце описания).
Однако также в других случаях, может возникать эта проблема стереоселективности. К примеру, конденсация Виттига на альдегиде 11.27 приводит к E: Z смеси 14.5 и 14,6, которая может быть разделена, один из них приводит после гидролиза к аналогу 40.
Известно, что при повышенных температурах производные витамина D, обладающие природной триеновой системой, легко перегруппировываются в так называемые провитамин D производные (схема 15 в конце описания). В природных сериях структура витамина D преобладает в равновесии (приблизительное соотношение при 25oC = 9:1). Существенное изменение в CD-циклической части молекулы может, однако, существенно сказываться на этой равновесной композиции. Кроме того, конверсия формы витамина в форму провитамина может происходить легче, чем в природных производных.
a) nBuLi, 13.2, THF, -78oC; b) nBuLi, 13.1, THF, -78oC.
Схема 14
К примеру, было найдено, что карбонильное производное 2,7 приводит, после обычного сочетания Wittig-Horner'a и отчасти затрудненного гидролиза силилзащитных групп ( 40oC, 40 ч, TBAF в THF) к смеси аналога 5 и его соответствующей формы провитамина, соединения 58.
Схема 15
Для некоторых типов аналогов присутствие 19-нор А цикла обязательно. Катоны типа VII, когда используются как предшественники 19-нор аналогов, могут сочетаться, с использованием методики Lyjhgoe'a, с 13.2, например фосфин оксидом IV, или, напротив, с алкинами типа VI. Возможно также превращение кетонов VII в виниловый бромид VIII, который может взаимодействовать с карбонильной функциональной группой в V. 19-нор-A циклические предшественники V и VI, альтернативные для 13.2, могут быть получены из (-)-1, 3,4, 5-тетраоксициклогексанкарбоновой кислоты 16.1. Способ основан на стратегии "цикловитамин", для которой существуют примеры в случае природных серий (19 метилен). Две существенные особенности состоят в одновременном удалении 1- и 4-гидроксильных групп в 16.1 и в образовании бицикло [3.1.0]гексанового скелета. 5-Гидроксил группу в лактоне 16.2 защищают, например, в виде простого т-бутил-диметилсилилового эфира; 16.3 может быть отделен от меньшего региоизомера. Две гидроксильных группы удаляют, в качестве одного из нескольких потенциальных способов (29), используя обескислороживание Barton-Mc Combie через бис-тиокарбонилимидазолид 16.4. Сольволиз образовавшегося 16.5 дает 16,6. Превращение гидроксильной группы в подходящую отщепляемую группу и индуцируемое основанием образование циклопропана дают сложный эфир 16.8. Два предшественника 16.10 и 16.11 теперь легко доступны; один из возможных способов образования алкина состоит во взаимодействии альдегида 16.10 с диметил диазометилфосфонатом (35). Сочетание 16.11 с подходящим кетоном типа VII (таким как 12.2b) может быть выполнено, как описано в природных сериях, и включает взаимодействие аниона 16.11, LiAlH4 восстановление образовавшей пропаргиловой спиртовой группы и катализируемый кислотой сольволиз, с образованием 19-нор- витамин аналога 43. Альдегид 16.10 может также непосредственно быть использован в реакции с подходящим виниловым анионом, полученным из винилового галогенида типа VIII (такой как 16.12). Виниловый галогенид может быть получен из кетона, например, по типу олефинизации Виттига.
a) TSOH, толуол, Δ, 15ч (79%); b) TBDMSCl, имид, DMAP, DMF, комн.темп., 12 ч (66%); c) (имид)2C=S, DMAP, Δ, 3d (87%); d) Bu3SnH, AIBN, толуол, Δ, 5ч (55%); e) NaOMe, MeOH, 0oC, 1 ч (100%); f) п-BrC6M4SO2Cl, CHCl3, пиридин, 0oC, комн. темп. , 13,5 ч (100%); g) т-BuOK, т-BuOH, Δ, 1ч (71%); h) DOBAH, толуол, -78oC, 2 ч (98%); i) PCC, CH2Cl2, комн.темп., 2ч (90%); j) (MeO)2P(O)CHN2, т-BuOK, -78oC---> комн. темп., 18 ч (89%); k) 16.12, т-BuLi, Et2O, -78oC; 50 мин, 16.10, 1 ч (46%); l) п-TSOH, H2O-диоксан (1:3), 63oC, 6 ч (78%); m) Ph3P+ CH2Br, Br-, NaN (TMS)2, THF, -68oC, 1 ч; 12.2, -68oC, 1 ч, комн. темп., в течение ночи (56%); n) 16.11, н-BuLi, THF, - 50oC, 1 ч, 12.2, комн. темп. , 30 мин (55%); o) Li AlH4, THF, кипение, 2 ч. (50%); p) п-TOH; H2O-диоксан (1:3), 63oC; 6 ч (40%).
Схема 16
Некоторые аналоги витамина D, относящиеся к данному изобретению, характеризуются центральной частью, структура которой основательно изменена, но все же сохраняют подобную витамину D биологическую активность. Особенно те производные, в которых недостает совместного присутствие шести- и пятичленных циклов, характерных для скелета витамина D, и которые могут рассматриваться как не стероидные аналоги витамина D, составляют первые примеры совершенно новых серий аналогов витамина D.
В частности, это показывает, что классическая транс-конденсированная пергидриндан CD-циклическая система не является необходимой для биологической активности. В этом отношении, было также сделано открытие, что стероидные аналоги, несущие неприродную CD-циклическую систему, в действительности активны; в этих случаях, однако, структура A-цикла не дает возможности предпочтительной перегруппировки для образования провитамина D.
Наконец, также очевидно, что необходимо наличие некоторых конформационно ограниченных структурных особенностей, таких как циклы и/или алкил заместители в центральной части, поскольку производное (1) с линейной незамещенной центральной цепью не активно.
Нами найдено, что соединения, описанные выше, и принадлежащие к новому классу лекарственных средств, включающие аналоги витамина D с модификациями CD циклической структуры, обладают селективным воздействием на функцию клеток, таким как ингибирование пролиферации клеток (доброкачественные клетки, такие как кератиноциты, равно как злокачественные клетки, такие как карцинома молочной железы, остео-саркома и лейкозные клетки) и также обладают высокой активностью в отношении дифференциации клеток (например, только что упомянутые типы клеток), но с другой стороны, обладают замечательно понижающим воздействием на кальциевый и костный гомеостаз, определяемый у рахитичных цыплят (путем измерения кальция сыворотки и кости, измерением двух витамин D зависящих белков, остеокальцина сыворотки и дуоденального калбиндина) равно как у витамин D насыщенной здоровой мыши (использование аналогичных конечных точек). Таким образом, не являясь классическими соединениями витамина D, новые лекарственные средства не проявляют такого же токсического воздействия на кальциевый и костный гомеостаз. В свете предшествующей техники и изучений, неожиданным и удивительным оказалось то, что центральная часть классической структуры витамина D известная как CD цикл, не является существенной для всех воздействий гормона витамина D и что наоборот, изменения в этой части выражают селективную активность спектра активности витамина D, это может применяться терапевтически при различных нарушениях. Особенно новые лекарственные средства могут быть использованы в терапии или профилактике
- иммунных нарушений, таких как аутоиммунные нарушения (такие как, но не ограничиваемые ими, сахарный диабет типа 1, множественный (рассеянный) склероз, волчанка и нарушения типа волчанки, астма, гломерулонефрит, и т.д.), селективных дисфункций иммунной системы (например, СПИД) и предупреждение иммунного отторжения такое как отторжение трансплантанта (например, почки, сердце, костный мозг, печень, панкриатический островок или вся поджелудочная железа, кожа и т.д.) или предупреждение гомологичной болезни. Вновь разработанные лекарственные средства могут быть использованы сами по себе, либо в комбинации с другими лекарственными средствами, способными интерферировать иммунную систему (например, циклоспорин, FK 506, глюкокортикоиды, моноклональные антитела, цитокины или факторы роста...). По аналогии с иммунной активностью новых соединений, можно ожидать аналогичные эффекты при других воспалительных заболеваниях (например, ревматоидный артрит).
- кожные заболевания, характеризуемые либо гиперпролиферацией и/или воспалительной и/или иммунной реакцией (например, псориаз, дискератоз, акне). Кроме того, поскольку эти лекарственные средства могут стимулировать дифференциацию кожных клеток, они могут быть использованы для лечения или профилактики алопеции различного происхождения, включая алопецию, вызванную химиотерапией или облучением.
- гиперпролиферативные нарушения и карцинома, такие как гиперпролиферативные кожные заболевания (например, псориаз) и некоторые типы карцином и их метастаз (все типы карцином, которые были или могут быть вызваны наличием витамин D рецепторов, такие как, но не ограничивающиеся ими, карцинома молочной железы, лейкоз, миелодиспластические синдромы и лимфомы, сквамозные клеточные карциномы и желудочно-кишечные карциномы, меланомы, остеосаркома.. . ). Вновь изобретенные лекарственные средства, опять как и в случае других показаний, могут быть использованы сами по себе в подходящей форме и подходящим способом введения, или в комбинации с другими лекарственными средствами, про которые известно, что они обладают терапевтическим значением при таких заболеваниях. Эти новые лекарственные средства могут быть в частности полезны для таких заболеваний, в которых, в противоположность классическим химиотерапевтическим средствам, они могут также стимулировать дифференциацию клеток.
- эндокринные расстройства, поскольку витамин D аналоги могут модулировать секрецию гормонов, такие как повышенная инсулиновая секреция или селективное подавление секреции паратироидного гормона (например, при хронической почечной недостаточности или при вторичном гиперпаратиреозе).
- заболевания, характеризуемые ненормальным внутриклеточным кальциевым регулированием, поскольку новые лекарственные средства обладают благоприятными воздействиями на клетки, чьи функции в значительной степени зависят от внутриклеточного движения кальция (например, эндокринные клетки, мышцы... ).
Новые соединения могут находить применение как при заболеваниях людей, так и в ветеринарной медицине.
Количество новых соединений, необходимое для их терапевтического действия может изменяться в соответствии с показанием, способом введения и вида обрабатываемой особи (животное/человек). Соединения могут вводиться энтеральным, парентеральным или местным локальным путем. При лечении дерматологических нарушений предпочтительно местное нанесение в виде мази, крема или лосьона для системной обработки, предпочтительно в дозе от 0,1 до 500 мкг/г. Системное введение в виде таблеток, капсул, жидкого или стерильного препарата в подходящем носителе, разбавителе и/или растворителе для парентеральной инъекции требует применения микрограммовых количеств соединений в день в зависимости от показания и клинического/ветеринарного состояния.
Преимущество новых соединений перед природными или существующими метаболитами витамина состоит в свойственной им активности в индуцировании клеточной дифференциации, ингибировании пролиферации клеток и модуляции клеточной активности в целом, хотя, тем не менее, они проявляют понижающий кальцемический эффект in vivo (в живом организме). Действительно, такие кальцемические эффекты, присутствующие в других метаболитах витамина D или аналогах, считались бы за нежелательные побочные эффекты, поскольку доза, требуемая для вышеупомянутых состояний в несколько раз превышает физиологическую и будет выражаться в серьезных кальцемических нарушениях при использовании других метаболитов витамина D или аналогов.
БИОЛОГИЧЕСКАЯ ОЦЕНКА НОВЫХ АНАЛОГОВ ВИТАМИНА
1. Связывающие свойства новых неизвестных аналогов витамина D.
Способы, используемые для оценки связывающих свойств новых аналогов служат примерами описанных ранее способов испытаний по связыванию стероидных гормонов (включая витамин D).
Сродство аналогов 1 α, 25-(OH)2D3 витамин D рецепторам оценивают по их способности конкурировать с [3H]1 α 25-(OH)2D3 (специфическая активность 180 кюри/ммоль, Amersham, Buckinghamshire, ИК) в связывании с высокой скоростью супернатанта из кишечных мукозных гомогенатов, получаемых от здоровых свиней (22, 23). Инкубацию выполняют при 4oC в течение 20 ч и фазовое разделение осуществляют добавлением смеси декстран/слой активированного угля. Сродство для 1 α , 25-(OH)2D3 составляет 1.06±0.38• 1010 М-1 (M±SD, n=10). Относительное сродство аналогов рассчитывают из их концентрации, необходимой для вытеснения 50% [3H] 1 α , 25-(OH)2D3 из этого рецептора в сравнении с 1 α , 25-(OH)2D3 (принятый за 100%) (Табл. 2 в конце описания).
Относительное сродство для hDBP измеряют инкубацией [3H] 1 α , 25-(OH)2D3 и увеличением концентрации 1,25-(OH)2 или его аналогов с очищенным hDBP (0,2 мкМ) в 1 мл (0,01 М Трис-HCl, 0,154 М NaCl, pH 7,4) в течение 3 ч при 4oC с последующим фазовым разделением путем добавления смеси холодной декстран-слой активированного угля (22,23).
Результаты, полученные с несколькими примерами новых аналогов, приведены в Таблице 2. Эти данные ясно показывают связывание с витамин D рецептором, необходимое для их биологической активности, хотя их связывание с витамин D связывающим белком, известным как DBP, понижено по сравнению с 1 α , 25-(OH)2D3. Нами, а также другими, ранее показывалось, что для других аналогов витамина D, которые так ослабляют связывание с DBP, повышается отношение клеточной дифференциации к кальцемическим эффектам (23,37).
2. Воздействия новых аналогов витамина D на пролиферацию клеток и дифференциацию клеток.
Системы клеточных культур используют в соответствии с современным уровнем развития техники:
- для оценки воздействия на пролиферацию доброкачественных клеток и особенно для оценки их эффективности при использовании для лечения дерматологических нарушений, новые соединения испытывают в культурах человеческих нормальных кератиноцитов.
Человеческие кожные кератиноциты выделяют и культивируют, используя модификацию способа,- Ketano и Okado (38).
Кратко, взятую на биопсию кожу пациентки с опухолью молочной железы, режут на кусочки размером 3-5 и пропитывают в течение ночи при 4oC раствором диспазы (dispase (20 Boehringer единиц/мл). Эпидермис чистят от дермы, промывают свободным от кальция и магния раствором соли с фосфатным буфером и инкубируют, встряхивают в 0,25% растворе трипсина в течение 10 мин при комнатной температуре. Затем реакцию прекращают добавлением PBS, содержащего 10% FCS. Клетки собирают после центрифугирования при 4oC в течение 10 мин при 800 об/мин. После дополнительной промывки PBS, осадок суспендируют в культуральной среде в 25 см2 первичных сосудах Becton Dickinson'a Кератиноциты культивируют при 37oC в атмосфере 5% CO2 в воздухе. Спустя несколько часов среду заменяют на новую. Среду [Кератиноцитовая среда от Gibco содержащая эпидермальный ростовый фактор (5 нг, мл), бычий гипофизарный экстракт (35-50 мг/мл) и антибиотики] обновляют через день до сливания.
Кератиноциты культивируют на пластине с 96 ячейками, и через 24 часа обрабатывают различными концентрациями аналогов витамина D, с последующим импульсным введением метки с 1 мккюри [3H]тимидина за 3 часа. Культуры промывают 3 раза РВ и дважды 10% (об/об) охлажденной льдом трихлоруксусной кислотой. Клетки солюбилизуют с 1 М NaOH и считают радиоактивность с помощью сцинтилляционного счетчика.
Для оценки действия на пролиферацию клеток и индукции дифференциации клеток, малигнизированные клетки выращивают in vitro (в лабор. сосуде) и их пролиферацию оценивают измерением числа клеток, протеинового содержания и вошедшего радиоактивного тимидина. В качестве примеров малигнизированной клетки используют человеческие лейкозные клетки (HL 60), человеческие остесаркомные клетки (MG 63 клетки) и как муреина так и человеческие раковые клетки молочной железы (MCF 7, MF M223 и GR клетки). Вдобавок, действие новых лекарственных средств проявляет дополнительные эффекты при испытании в комбинации с другими противораковыми лекарственными средствами (например, ретиновые кислоты анти-эстрогены...).
HL-60 клетки высевают при 1,2•105 клеток/мл и добавляют 1 α , 25-(OH)2D3 или его аналоги в этаноле (конечная концентрация <0,2%) в RPMI 1640 среду пополненную 10% инактивированной нагреванием сыворотки плода коровы (FCS) на 4 дня при 37oC. Затем клетки испытывают на злокачественность NBT восстанавливающим испытанием, как описано в (22), используя гемоцитомер, или на пролиферацию, подсчитывая клетки и [3H] тимидин инкорпорирование. MG63 клетки, высеянные при 5•103 клетки/мл в плоскодонные культуральные чашки с 96 ячейками (Falcon, Becton Dickinson, NJ) в объеме 0,2 мл DMEM и 2% FCS, инкубируют с 1 α , 25-(OH)2D3 или его аналогами в течение 72 ч. Затем измеряют остеокальцин в культуральной среде, используя R1A (радиоиммуноанализ) гомологического человеческого остеокальцина (39). Клетки карциномы молочной железы (MCF-7 или GR) выращивают в DMEM/nut смеси F-12 (HAM) среде дополненной 10% FCS. Клетки (5000/ячейка) инкубируют в течение 24 ч в чашках для культуральных тканей с 96 ячейками (Falcon 3072) и впоследствии 72 часа инкубируют с/без 1 α, 25-(OH)2D3 или аналогами.
Затем клетки инкубируют с [3H] тимидином (1 мккюри/ячейка) в течение 4 ч и собирают после этого в NaOH (0,1 М) и подсчитывают радиоактивность. Содержание белка в клетках измеряют BCA анализом протеинов Pierce (Rockford, 1L).
Полученные с некоторыми из новых аналогов результаты представлены в Таблице 2 и на фиг. 1-5.
Для оценки иммунного потенциала новых лекарственных средств их биологическую активность исследуют по реакции смешанных лимфоцитов in vitro на современном техническом уровне; вдобавок исследуют воздействия аналогов на индукцию дифференциации HL 60 клеток в созревших моноцитах in vitro. Кроме того, их иммунный потенциал дмонстрируется путем воздействия на снижение реакции "трансплантант против хозяина" на мышах и для предотвращения неврологических явлений на модели мыши с экспериментальным аллергическим энцифалитом.
Способность новых аналогов активировать геномный путь метаболизма, обычно используемый метаболитами природного витамина D, демонстрируют трансфекционным изучением, используя конструкцию нескольких прямых дупликаций реактивных элементов витамина D (используя мышиную остеопонтин или крысиную остеокальцин DRE последовательность сочетаемую с геном по данным CAT (компьютерная аксиальная томография) или hGH (конструкции изготовлены (I. White and G.V.Hendy, Montreal, Canada and M.R.Haussler, Tucson, Arizona).
Таблица 2
Суммарные биологические свойства некоторых отдельных аналогов витамина D: сродство к рецептор витамина D/ витамин D - связывающий белок плазмы, их способность к индуцированию клеточной дифференциации /ингибированию пролиферации клеток в человеческих лейкозных (HL-60), остеосаркомных (MG-63), человеческих раковых молочной железы (MGF-7) клетках и человеческих кератиноцитах. Биологические значения представлены в % от активности 1 α, 25-(OH)2D3 при 50% его активности (B50). Подробное описание методики смотри в тексте. Нумерация соединений идентична нумерации, используемой для описания их химической структуры в Таблице 1 (см. в конце описания).
3. Оценка иммунного потенциала in vivo.
Для оценки иммунного потенциала аналогов используют хорошо известную модель предупреждения рецидива аутоиммунного заболевания в спонтанно диабетической NOD мыши. Когда изогенные NOD островки трансплантируют под почечную капсулу спонтанно диабетической NOD мыши, диабет вылечивается всего за несколько дней, поскольку в отсутствии иммуномодуляторной обработки, вновь трансплантированные островки разрушаются в течение 14 дней. Циклоспорин A, хорошо известный иммуносупрессант, может только замедлять рецидив при величине дозы около токсической (15 мг/кг/день). Комбинация субтерапевтической дозы CyA (7,5 мг/кг/день) с низкой, некальцемической дозой одного из новых аналогов (номер 46, из Таблицы 1: 10 мкг/кг/2дня) дает эффективное увеличение срока жизнеспособности трансплантанта, с жизнеспособностью трансплантанта даже после прекращения терапии (день 60). (Таблица 3 в конце описания).
4. Кальцемические эффекты новых аналогов витамина
Для оценки кальцемических эффектов осуществляют испытания in vivo, используя цыплят и мышей с недостаточностью витамина D в возрасте 3 недель, проводя последовательно в течение 10 дней инъекции 1 α, 25-(OH)2D3 или его аналогов (22,23). Измеряют содержание кальция в сыворотке (путем атомной абсорбциометрии) и остеокальцин (путем специфического RIA- радиоиммунноанализа), дуоденальный калбиндин D-28K (путем RIA) и костного кальция. Исследовали также гиперкальцемический эффект наиболее интересующих аналогов на витамин-D насыщенных нормальных NMRI мышах, путем ежедневной подкожной инъекции 1 α, 25-(OH)2D3 его аналогов или растворителя в течение 7 последовательных дней, используя сыворотку, кости и мочевыделение и сывороточный остеокальцин (путем специфического RIA на мышах) в качестве параметров (40).
Характерные данные, полученные с некоторыми из новых аналогов представлены на фиг. 6.
1. Haussler MR. McCain TA. N Engl J Med 1977:297:974-983.
2. Hausslcr MR. McCain TA. N Engl J Med 1977:297:1041.1050.
3. Kanis JA. J Bone J Surg 1982;64B:542-560.
4. Henry HL, Norman AW. Disorders of Bone and Mineral Metabolism 1992: 149-162.
5. Bouillon R, Van Baclen H. Saudi Med J 1989:10:260-266.
6. DcLuca HF. Endocrinology 1992:130:1763-1763.
7. Hausslcr MR, Mangelsdorf DJ, Komm BS, Terpenning CM. Yamaoka K, Allegretto EA, Baker AR, Shine J, McDonnell DP, Hughes M, Weigel NL, O'Malley BW, Pike JW. Recent Prog Horm R 1988,44:263-305.
8. Ozono К, Sone T, Pike JW. J Bone Miner Res 1991:6:1021-1027.
9. Carlberg C, Bendik I, Wyss A, Meier E, Sturzenbecker J, Grippo JF, Hunziker W. Nature 1993:361:657-660.
10. Ross TK, Moss VE, Prahl JM, DeLuca HF. Proc Natl Acad.Sci USA 1992: 89:256-260.
11. Zhou L-X, Nemere I, Norman AW. J Bone Min Res 1992;7:457-463.
12. Baran DT, Sorensen AM, Honeyman RW, Ray R.Holick MF. J Bone Min Res 1990:5:517-524.
13. Lieberherr M, Grosse B. Duchambon P, Drueke T. J Biol Chem 1989, 264:20404-20406.
14. Binderup L. Biochem Pharmacol 1992:43:1885-1892.
15. Manolagas SC, Hustmyer FG, Yu X-P.Kidney lntl 1990; 38, Supp.29: S9-S16.
16. Kragballe K. Arch Dermatol Res 1992;284:S30-S36.
17. Colston KW, Mackay AG. James SY, Binderup L, Chander S. Coombex RC. Biochem Pharmacol 1992:44:2273-2280.
18. Zhou JY, Norman AW, Chen D, Sun G. Uskokovic MR, Koeffler HP. Proc Natl Acad Sci USA 1990:87(10):3929-3932.
19. Binderup L, Iatini S, Binderup E, Bretting C. Calverley M, Hansen K. Biochem Pharmacol 1991:42:1569-1575.
20. Okamura WH, Palenzuela JA, Plumet J, Midland MM. J Cell Biochem 1992:49:10-18.
21. Ikekawa N. Mod Res Rev 1987:7 110.3:333-366.
22. Bouillon R, Allewaert K, Vanleeuwen JPTM, Tan B-K, Xiang D-7, Declereq P, Vandewalle M, Pols HAP, Bos MP, Van Baelen H, Birkenhager JC. J Biol Chem 1992:267:3044-3051.
23. Bouillon R. Allewaert K. Xiang D-Z, Tan B-K, Van Baelen H. J Bone Min Res 1991:6:1051-1057.
24. a. Lythgoe B, Moran TA, Nambudiry MEN. Fideswell J, Wright PW. J Chem Soc Perkin Trans I 1978:590.
b. Lythgoe B. Chem Soc Rev 1981:449-475.
25. a. Baggiolini E, Lacobelli J, Henessy B, Batcho A, Gereno I, Uskokovic M. J Org chem 1986:51:3098-3108.
b. Perlman KL, Swenson RE, Paaren HE, Schnoes HK, DeLuca HP. Tetrahedron Lett 1991;32:7663-7666.
26. a. Nemoto H, Kimura Т. Kuroba H, Fukumoto К. J Chcm Soc Perkin trans I 1986:1777-1780.
b. Wilson SR, Venkatesan AM, Augelli-Szafran CE, Yasmin A. Tetrahedron Lett 1991;32:2339-2342.
27. a. Kocienski PJ, Lythgoe B, Ruston S. J Chem Soc Perkin Trans I 1979:1290.
b. Okamura WH. Acc Chem Res 1983:16:81.
28. Audia JE, Boisvert I., Danishefsky SJ. Patten AD, Villalobos A, J Org Chem 1989, 54:3738-3740.
29. Barton DH, McCombie SW. J Chem Soc Perkin Trans I 1975; 16:1574-1585.
30. Castedo L, Mascarenas JC, Mourino A, Perez-Sestelo J. Tetrahedron Lett 1991:32:2813-2816.
31. Lam L, Hui R, Jones JP: J Org Chem 1986:51:2047.
32. Grieco P, Yokoyama Y, Gilman S, Ohfune Y. J Chem Soc. 1977:870.
33. Ireland RE, Mueller RH, Willard AK. J Am Chem Soc 1976;98:1868.
34. Mitsunobu O. Synthesis 1981:1.
35. Gilbert JC. Weerasooriya U. J Org Chem 1982:47:1837-1845.
36. Rossiter BE, Swingle NM. Chem Rev 1992:771-806.
37. Yamada S, Nakayama K, Takayama H. Tetrahedron Lett 1981:22:2591.
38. Erickson GW, Fry JL. J Org Chem 1980;45:970-972.
39. McMurry JE, Melton J, Padgett H. J Org Chem 1974; 39:259-260.
40. Solladie G, Hutt J. J Org Chem 1987152:3560.
41. Chapuis C, Brauchlo R. Helv Chim Acta 1992:75:1527.
42. Pfan M, Jabin I, Revial G. J Chem Soc Perkin Trans I 1993:1935.
43. Liu H-J. Ralitsch M. J Chem Soc Chem Commun 1990:997-998.
44. Wicha J, Bal K. J Chem Soc Perkin Trans I 1978:1282-1288.
45. Moskal J, Van Leusen A. Recl Trav Chim Pays-Bas 1987, 106:137-141.
46. Hutchinson JH, Money T. Can J Chem 1985,63:3182.
47, Bovicelli P, Lupattelli P, Mincione E. J Org Chem 1992; 57:5052-554.
48. Dusso AS, Negrea L, Gunawardhana S, Lopez-Hilker S, Finch J, Mori T, Nishii Y, Slatopolsky E, Brown AJ. Endocrinology 1991:128:1687-1692.
В приведенных далее ПМР спектрах:
S синглет,
m мультиплет,
t триплет,
d дублет,
dd двойной дублет
br, brs уширенный сигнал
q - квартет
Пример 1. Синтез цис-декалона 1.2b
К суспензии AlCl3 (2 г, 14,99 ммоль) в толуоле (250 мл) при -78oC добавляют 1.1. Раствор перемешивают в течение 1 ч в атмосфере АГ до тех пор, пока температура не достигнет комнатной. Добавляют с помощью механического шприца изопрен (11 мл: 0,11 моль) в толуоле (80 мл) со скоростью 2 мл/15 мин. Через 6 ч смесь выливают в охлажденный льдом насыщенный раствор NaHCO3. Раствор экстрагируют Et2O и сушат (NaSO4) объединенные органические слои. Упаривание использованного для экстракции растворителя, фильтрация через короткую колонку с силикагелем при элюировании Et2O и ВДЖХ (силикагель: E OAc: изооктан 4:96) дают 1.2a (3.27 г, 74%).
К раствору 1.2а (1.3 г, 4.5 ммоль) в MeOH (94 мл), добавляют по каплям 2М NaOH в MeOH (67 мл, 139.14 ммоль). Через 2 ч добавляют твердый CO2 и раствор концентрируют. Остаток выливают в воду и экстрагируют Et2O. Объединенные органические слои промывают раствором соли, сушат (Na2SO4) и после упаривания фильтруют через короткую силикагелевую колонку, элюируя Et2O. ВДЖХ очистка (силикагель: EtOAc: изооктан 4:96) дает 1.2b (1,25 г, 94%).
Rf: 0.52 (изооктан:ацетон 90:10).
ИК (KBr): 1712, 1469, 1443, 1366, 1254 см-1.
ПМР: (500 MHz, CDCl3): δ : 5.34 (1H, bs): 3.91(1H, s); 2.83(1HtdЯ, J= 5.81, 14.1); 2.58(1H, dt, J=19.2, 26,45); 2.33 (1H, tm, J=15.07); 2.22 (1H, ddd J= 13.9, 4.25, 2.4); 2.17 (1H, m); 2.09(1H), dddd, J=13.9, 5.9, 3.4, 2.5); 1.84 (1H, ddd, J=1.18, 4.28, 14.1); 1.8(1H, m); 1.74(1H, dd, J=16.6, 5.0); 0.94(9H, s); 0.12(6H, s,); 0.65(3H, s) м.д.
Пример 2. Синтез 1.3b
К раствору 1.2b (1,1 г, 3.74 ммоль) в MeOH (60 мл) добавляют небольшими порциями при 0oC NaBH4 (0,7 г, 18,67 ммоль). Раствор перемешивают в течение ночи при комнатной температуре. Раствор концентрируют и остаток растворяют в воде. Экстракция CH2Cl2, промывание объединенных органических слоев раствором соли, сушка (MgSO4) упаривание растворителя и ВДЖХ очистка (силикагель; ацетон: гексан 8:2) дают 1.3а (954 г. 86% c Rf 0.36; ацетон:гексан 1:9).
К охлажденному (0oC) раствору 1.3а (800 мг, 2.7 ммоль) и DIPEA (6 мл, 65.61 ммоль) в CH2Cl2 (30 мл), добавляют по каплям MEMCl (2.75 мл, 24.08 ммоль). Раствор перемешивают 3 ч при комнатной температуре и затем разбавляют Et2O (100 мл). Смесь промывают 0.1 н HCl раствором (30 мл), насыщенным NaHCO3 (30 мл) раствором соли. Органический слой сушат (MgSO4) и концентрируют. Колоночная хроматография (силикагель: ацетон:гексан 1:9) дает 1.3b (1,02 г, 98%).
Rf: 0.72 (ацетон:гексан 1:9).
ПМР: (360 MHz, CDCl3): δ : 5.3 (1H, m): 4.86 (1H, d, J=7.12); 4.69 (1H, d, J= 7.12); 3.67-3.79 (3H, m); 3.55(2H, t, J=7.3); 3.38 (3H, s); 3.22 (1H, dt, J=4.75, 10.1); 2.4-2.48 (1H, m); 2.16(1H, t,J=13.5); 1.84- 1.60(5H, m); 1.63(3H, s); 1.47-1.37(3H, m); 0.9 (9H, s); 0.05(6H, s) м.д.
Пример 3. Синтез 1.4
К раствору 1.3b (1 г, 2.6 ммоль) в смеси ацетон: вода 3:1 (20 мл) добавляют NMMO (335 мг, 2.9 мл) и OSO4 (100 мг, 0,39 ммоль) и раствор перемешивают в течение ночи при комнатной температуре. Добавляют твердый Na2S2O3 и смесь экстрагируют CH2Cl2. Органические слои сушат (MgSO4), концентрируют и фильтруют через короткую силикагелевую колонку (элюируют Et2O). α -диол (86%) растворяют в смеси ацетон:вода 3:1 (24 мл) и раствор охлаждают (0oC). К раствору небольшими порциями добавляют NaJO4 (1.132 г, 5.294 ммоль). Смесь перемешивают в течение ночи в атмосфере Ar при комнатной температуре. Затем раствор фильтруют, концентрируют и остаток растворяют в воде. Раствор экстрагируют CH2Cl2 (3х), органические слои промывают раствором соли и сушат (MgSO4). После упаривания растворителя получают бесцветное масло (98%). Его (650 мг, 1.56 ммоль) растворяют в дегазированном растворе KOH (2 г) в воде (100 мл). Смесь перемешивают в течение ночи при 60-80oC в токе Ar. Затем раствор экстрагируют CH2Cl2 и органические слои сушат (MgSO4). Фильтрация через короткую силикагелевую колонку (элюирование Et2O) и ВДЖХ очистка (силикагель: ацетон:гексан 5:95), дают 1.4 (436 мг, 53 %).
Rf: 0.81 (EtOAc).
ИК (пленка): 1669; 1560; 1458; 1376; 1235; 1035 см-1.
ПМР: (500 MHz, CDCl3): δ ; 6.70 (1H, dd, J=5.25, 2.40); 4.83(1H, d, J= 7.07); 4.73(1H, d, J=7.07); 4.72(1H, m); 3.76-3.67(2H, m); 3.64(1H, dt, J= 10.36, 4.35); 3.55(2H, dt,J=4.7); 3.38(3H, s); 2.64(1H, ddd, J=16,8, 6.6, 3.2); 2.45 (1H, ddd, J=22.4, 6.6); 2.35(1H, dm, J=12); 2.24(3H, s); 2.02(1H, dddd, J= 2.02, 3.98, 11.3, 16.8); 1.87(1H, m); 1.73(1H, ddd, J=13.3, 6.03, 2.9); 1.60-1.43(2H, m); 0.80(9H, s); 0.01 (3H, s); -0.1(3H, s) м.д.
Пример 4. Синтез 1.5а
Охлажденный (0oC) раствор 1.4 (320 мг, 0,80 ммоль) в гексане (3 мл) перемешивают 1,5 часа при 1 атм H2 в присутствии 10% Pd/C (20 мг) и затем фильтруют через короткую колонку с силикагелем (элюируют Et2O). Раствор упаривают и остаток растворяют в MeOH (10 мл). Добавляют NaOMe (40 мг) при 0oC и раствор перемешивают 3 ч, пока температура не достигнет комнатной. Смесь концентрируют и остаток растворяют в насыщенном NH4Cl растворе. Экстракция Et2O, сушка (MgSO4) и ВДЖХ очистка (силикагель:ацетон;гексан 1:9), дают 1.5а(311 мг, 97%).
Rf: 0.15 (ацетон: петролейный эфир 1:9).
ИК (пленка): 1709; 1471; 1357; 1253; 1201 см-1.
ПМР: (500 MHz, CDCl3): δ :4.77(1H, d, J =7.08); 4.64(1H, d, J=7.09); 3.88(1H, ppd, J= 2.46); 3.68-3.59(2H, m); 4.49(2H, t, J=4.7); 3.32(3H, s); 3.31(1H, dt, J= 4.3, 10.5); 2.77(1H, dt, J=10.6, 8.0); 2.06(3H, s); 1.99-1.89(2H, m); 1.88- 1.80(1H, m); 1.76(1H, ddd, J=11.4, 3.7, 7.1); 1.69(1H, ddd, J= 13.7, 6.1, 3.0); 1.62-1.52(2H, m), 1,51- 1.43(1H, m); 1,38(1H, ddt, J=13.7, 3.7, 2.3); 1.24-1.17(1H, m); 0.82(9H, s); -0.05(3H, s); -0.10(3H, s) м.д.
Пример 5. Синтез 1.6а
К охлажденному (0oC) раствору Ph3P+CH3Br (112 мг, 0,31 ммоль) в ТГФ (1 мл) и HMPA (1 мл) в атмосфере Ar добавляют BuLi в ТГФ (0,116 мл, 0,289 ммоль) и затем спустя 1 ч 1.5а (48 мг, 0,12 ммоль) в ТГФ (1 мл). Раствор перемешивают 3 ч в атмосфере пока температура не повысится до комнатной. Концентрация и очистка колоночной хроматографией (силикагель: ацетон; гексан 1:9) дают 1.5 (48 мг, 100% с Rf 0.47; ацетон: гексан 1:9).
К перемешиваемому раствору 1.5b (48 мг, 0,12 ммоль) в ТГФ (0.5 мл) в атмосфере Ar при комнатной температуре добавляют 0.5 М раствор 9-BBN в ТГФ (1 мл, 0,5 ммоль). Через 4 ч добавляют EtOH (0,1 мл), NaOH 6 н (0,125 мл) и 30% H2O2 (0.25 мл) и раствор перемешивают в атмосфере Ar в течение 1 ч при 60oC. Смесь выливают в раствор соли и экстрагируют Et2O (3х). Сушка (MgSO4), упаривание растворителя и ВДЖХ очистка (силикагель: ацетон: гексан 15:85) дают 1.6а (37 мг,74%).
Rf: 0.24 (ацетон:гексан 2:8).
ИК(пленка): 3445; 2930; 2878; 1469; 1364; 1252 см-1.
ПМР: (500 MNZ, CDCl3): δ : 4.82(1H, d, J=7.1); 4.71(1H, d, J=7.1); 3.99(1H, s); 3.75-3.63(3H, m); 3.54(2H, t, J= 4.64); 3.4-3.32(2H, m); 3.38(3H, s); 1.95- 1.15(12H, m); 1.10(1H, t,J=9.81); 0.97(3H, d, J=6.85); 0.88(9H, s); 0.04 (3H, s); 0.02 (3H, s) м.д.
Пример 6. Синтез 1.8а
При 0oC к перемешанному раствору спирта 1.6а (35 мг, 0.083 ммоль) в CH2Cl2 (2 мл) и Et3N (0.5 мл) добавляют TsCl (32 мг, 0.168 ммоль) при 0oC и раствор перемешивают 12 ч при комнатной температуре. Смесь фильтруют через короткую силикагелевую колонку (элюируя смесью ацетон:гексан 15:85). ВДЖХ очистка (силикагель: ацетон:гексан 15:85) дает 1.6b (43 мг, 91% с Rf 0.26; ацетон:гексан 15:85).
NaH (15 мг, 0.38 ммоль) в ДМСО (1,5 мл) перемешивают 2 часа при 60oC в атмосфере Ar, затем раствор перемешивают, пока не остынет до комнатной температуры. После чего добавляют по каплям при комнатной температуре 2-(1-этокси)-этилокси-2-метил-3-бутин (547 мг, 3,5 ммоль). Через 30 мин. добавляют 1.6 (200 мг, 0,35 ммоль) в ДМСО(1.3 мл) и смесь перемешивают 1,5 ч при комнатной температуре. Затем смесь выливают в насыщенный раствор NaHCO3 и экстрагируют Et2O. Сушка (MgSO4), упаривание растворителя и ВДЖХ очистка (силикагель: ацетон: гексан 1:9) дают 1,7 (136 мг, 70% с Rf 0.38; ацетон:гексан 1:9).
1.7 (40 мг, 0,0721 ммоль) растворяют в EtOAc (3 мл) и добавляют 10% Pd/C (3 мг). Суспензию трясут при давлении H2 4 бар и комнатной температуре в течение 1 часа. Затем смесь фильтруют через короткую силикагелевую колонку (элюируют EtOAc), получая 1.8а (12 мг, 34%).
Rf: 0.20 (ацетон:гексан 15:85)
ИК (пленка): 3456; 2932; 2861; 1368; 1251 см-1.
ПМР: (500 MHz, CDCl3): δ : 4.83(1H, d, J=7.1); 4.71(1H, d, J=71); 3.92(1H, s); 3.76- 3.68(2H, m); 3.56(2H, t, J=4.7); 3.39(3H, s); 3.35(1H, dt J= 10.3, 4.0); 1.96-1.73(5H, m); 1.61-1.22(10H, m); 1.21(6H, s); 1.14-0.98(4H, m); 0.88(12H, m); 0.03 (3H, s); 0.01(3H, s) м.д.
Пример 7. Синтез 1.8d
К охлажденному (-78oC) и перемешанному раствору 1.8а (10 мг, 0,02054 ммоль) в CH2Cl2 (1 мл) добавляют 1,5 M Me2BBГ в CH2Cl2 (0,034 мл 0.05 ммоль). Смесь перемешивают 3 ч при -78oC и затем добавляют по каплям к энергично перемешиваемому раствору насыщенного NaHCO3 и ТГФ. Смесь экстрагируют Et2O. Сушка (MgSO4), упаривание растворителя и ВДЖХ очистка (силикагель, ацетон:гексан 2:8), дают 1.8b (6 мг, 73%).
К раствору диола 1.8b (5 мг, 0,0125 ммоль) в CH2Cl2 (2 мл) при комнатной температуре добавляют PDC (14 мг, 0,0376 ммоль). Раствор перемешивают 4 ч и затем фильтруют через короткую силикагелевую колонку (ацетон:гексан 3:7), получая 1.8c (5 мг, 99%).
Rf: 0.21 (ацетон: гексан 2:8).
ПМР: (500 MHz, CDCl3): δ :3.93(1H,brs); 3.35 (1H, dt, J=4.0, 10.1); 1.96-1.15(17H, m); 1.21(6H, s); 1.12- 0,99(3H, m); 0.9-0.88 (12H, m); 0.05(3H, s); 0,02 (3H, s) м.д.
Кетон 1.8с (5 мг, 0.0126 ммоль) растворяют в ТГФ (1 мл) и добавляют TSIM (0,5 мл, 3.41 ммоль). Смесь перемешивают 1 ч при комнатной температуре и затем чистят хроматографически (силикагель: ацетон:гексан 1:9), получая 1,8d (5,8 мг, 98% с Rf 0.55; ацетон:гексан 1:9).
Пример 8. Синтез 1.9с
Алкен 1.5 с (105 мг, 0,264 ммоль) перемешивают с 1 М TBAF (1 мл, 1 ммоль) в ТГФ (1,5 мл) в течение 10 дней при 30oC. Смесь концентрируют и чистят хроматографически (силикагель; ацетон:гексан 3:7), получая 1.9а (5,8 мг, 99%). К суспензии NaH (70 мг, 2.92 ммоль) в ТГФ (5 мл) добавляют по каплям 1.9а (32 мг, 0,113 ммоль) в ТГФ (2 мл). После перемешивания в течение 0,5 ч и охлаждении (0oC) добавляют по каплям CS2 (0,349 мл, 5.8 ммоль) и перемешивание продолжают 24 ч, пока температура не снизится до комнатной. Добавляют по каплям Mel (0,375 мл, 6 ммоль) и раствор перемешивают 2 ч. Смесь выливают в 0,1 н. HCl раствор. Экстракция Et2O, сушка (MgSO4), упаривание и ВДЖХ очистка (силикагель, ацетон:гексан 2:8), дают 1,9b (41 мг, 97%). К раствору Bu3SnH (1 мл, 1,08 ммоль) и AIBN (2 мг) в толуоле (5 мл) при 110oC добавляют по каплям (0,5 ч) 1,90 (41 мг, 0,11 ммоль) в толуоле (2 мл). Смесь перемешивают 8 ч при 110oC. Колоночная хроматография (силикагель, ацетон:гексан 1:9) с последующей ВДЖХ (силикагель; ацетон; гексан 3:7), дают 1.9с (27 мг, 92%).
Rf: 0.3 (ацетон:гексан 5:95).
ИК (пленка): 2927; 2872; 1447; 1113; 1043 см-1.
ПМР: (500 MHz, CDCl3): δ : 4.83(1H, d, J=7.1); 4.71(1H, d, J=7.1); 4.7-4.67(2H, m); 3.77-3.66 (2H, m); 3.57(2H, t, J=4.8); 3.4(3H, s); 3.38(1H, dt, J= 4.2, 9.9); 2.16(1H, m); 2.08(1H, m); 1,99(1H, m; 1,88-1.76(2H, m); 1.67(3H, s); 1.57-1.46(2H, m); 1.36-1.1 (6H, m); 0.90-0.80(2H, m) м.д.
Пример 9. Синтез 1.10b
Из 1.9с как описано для 1.6b из 1.5b. Общий выход эпимерной смеси 20-S, 20-R (соотношение 8:2) составляет 82%.
Rf: 0.35 (ацетон: гексан 15:85).
ИК (пленка): 2931; 2876; 1458; 1362; 1189 см-1.
ПМР: (500 MHz, CDCl3): δ : 7.78(2H, d, J=8.3); 7.35(2H, d, J=8.3); 4.80(1H, d, J= 7.1); 4.68 (1H, d, J=7.1); 3.97 (1H, dd, J=9.5, 5.0); 3.83 (1H, m);, 3.73- 3.64(2H, m); 3.54(3H, t, J=4.7); 3.39 (3H, s); 3.38(3H, s); 3.28(1H, dt, J= 10.1, 4.3); 2.54(3H, s); 2.03(1H, m); 1.91- 1.81(2H, m); 1.78-1.47(4H, m); 1.3-1.03(5H, m); 0.95-0.79(2H, m); 0.91(3H, d, J=6.9); 0.78(3H, d, J=6.9) м.д.
Пример 10. Синтез 1.10d
К раствору 1.10 (40 мг, 0.091 ммоль) в ДМСО (1,5 мл) добавляют Kl(150 мг, 0,91 ммоль). Смесь перемешивают 4 ч при 60oC и затем выливают в раствор соли. Раствор экстрагируют. Экстракция с помощью Et2O, сушка (MgSO4), упаривание растворителя и флэш хроматография дают 1.10c (34,3 мг, 95%).
1.10c (5 мг, 0,0126 ммоль) растворяют в EtOH: вода 7:3 (0.5 мл). Добавляют CuI (20 мг, 0.105 мл), Zn порошок (30 мг, 0.458 ммоль) и метилвинилкетон (0.150 мл, 1,81 ммоль) и раствор перемешивают 35 минут при 15oC в сонофикаторе (Banson. 220). Процесс сонофикации (обработка ультразвуком прерывают для того, чтобы охладить жидкость в сонофикаторе, и повторяют его в течение 40 минут. Добавляют дополнительно CuI (10 мг, 0.105 ммоль), Zn (14 мг, 0.23 ммоль) и метилвинилкетон (0,075 мл, 0.95 ммоль) и сонофикацию продолжают 2 ч. Затем смесь фильтруют через короткую силикагелевую колонку (элюируют Et2O) и сушат (MgSO4). ВДЖХ очистка (ацетон: гексан 15:85) что дает 1.10d (3.6 мг, 83%).
Rf: 0.18 (ацетон:гексан 5:95).
ИК(пленка): 2928; 2871; 1716; 1459 см-1.
ПМР; (500 MHz, CDCl3): δ : 4,82(1H, d, J=7.1); 4.7(1H, d, J=7.1); 3.96-3.66(2H, m); 3.56(2H, t, J= 4.7); 3.39(3H, s); 3.33(1H, dt, J=10.4, 4.3); 2.44-2.34(2H, m); 2.12(3H, s); 2.05(1H, dm, J=12.4); 1.91(1H, m); 1.79(1H, dm, J= 11.0); 1.7- 1.4(5H, m); 1.36-1.2(4H, m); 1.19-0.94(4H, m); 0.88(3H, d, J=8.5); 0.87(1H, m); 0.77(3H, d, J=6,6) м.д.
Пример 11. Синтез 1.11
Кетон 1.10d (10 мг, 0,0294 ммоль) растворяют в ТГФ (1.5 мл) и добавляют 2М MeMgCl (0,4 мл. 1.2 ммоль). Смесь перемешивают 1 ч при комнатной температуре и затем добавляют 0,1 н. HCl до прекращения образования газа. Раствор фильтруют через короткую силикагелевую колонку и сушат безводным MgSO4, получая 1.11a (10,3 мг, 98%).
Раствор 1.11a (10 мг, 0,028 ммоль) в MeOH (2 мл) перемешивают с амберлитом, покрытым силикагелем 15(200 мг) в течение 1 недели при 30oC. Затем смесь фильтруют через короткую силикагелевую колонку (промывают Et2O). ВДЖХ очистка (силикагель: ацетон:гексан 3:7) дает 1.11b (7.2 мг, 96%).
Затем спирт 1.11b превращают в 1.11b (Rf 0.58; ацетон:гексан 15:85) как описано для 1.8d из 1.8b (80% выход).
Rf: 0.2 (ацетон:гексан 15:85).
ИК (пленка): 3422; 2958; 2872; 1713; 1464; 1377 см-1.
ПМР: (500 MHz, CDCl3): δ : 2.37-2.22(3H, m); 2.13(1H, m): 2.07(1H, dm, J= 12.4); 1.74-1.20(15H, m); 1.21(6H, s); 1.09(1H, m); 0.90(3H, d, J=6.83); 0.78 (3H, d, J =6. 78) м.д.
Пример 12. Синтез кислоты 2.1
Суспензию бензолселеновой кислоты (9.6 г, 0.05 моль) в смеси ТГФ (50 мл) и фосфатный буфер (0,1 М, pH=7.25 мл) обрабатывают приблизительно 30% перекисью водорода (88 г, 0,4 моль) при комнатной температуре. Добавляют раствор ментона (6,16 г, 0,04 моль) в ТГФ (25 мл) и реакционную смесь перемешивают при комнатной температуре 17 ч. Добавляют насыщенный водный раствор NaHCO3 до достижения реакционной смесью pH 9. После удаления H2O2 и ТГФ при пониженном давлении, реакционную смесь подкисляют до pH 5. После насыщения солью реакционную смесь экстрагируют диэтиловым эфиром (250 мл, 3 минуты) и объединенные эфирные фазы сушат над безводным MgSO4. После фильтрации, фильтрат концентрируют в вакууме. Оставшуюся бесцветную жидкость (14 г) растворяют в 150 мл метанола и 37% HCl (3,75 мл) добавляют. Эту смесь кипятят с обратным холодильником в течение 3 часов. После охлаждения, реакционную смесь обрабатывают насыщенным водным раствором NaHCO3 до pH 8. Органический растворитель удаляют упариванием при пониженном давлении. Образовавшийся остаток экстрагируют диэтиловым эфиром (3 раза). Объединенный эфирный раствор сушат над безводным MgSO4. После фильтрации и концентрации сырой продукт чистят колоночной хроматографией (EtOAc/гексан 1:4), получая чистый метиловый эфир (7,32 г, 91%).
Rf: 0,4: (EtOAC: гексан 1:2).
ИК(пленка): 3434 (ГП); 2957, 2873(s); 1736 (s); 1461, 1437(m); 1287, 1261, 1205, 1164(5); 734 (s) см-1.
ПМР: (360 MHz, CDCl3): δ : 3,68 (3H, s); 2.34(1H, dd, J=6.1, 14.8); 2.14(1H, dd, J=8.0, 14.8); 1.95(1H, m); 1.65(1H, m); 1.52(2H, m); 1.35(2H, m); 1.22(1H, m); 0,96(3H, d, J=6.8); 0,90(6H, dd, J=7.1, 7.5) м.д.
MS (m/z): 202(2%); 187(1%); 184(1%); 184(2%); 159 (2%); 43(100%).
К раствору полученного ранее сложного эфира (5.17 г, 25.5 ммоль) в ДМФ, добавляют третбутилдиметилсилил хлорид (RBDMS-Cl, 5.79 г, 38.4 ммоль), DМАР (50 мг) и имидазол (3.92 г, 57.6 ммоль). Раствор перемешивают в течение ночи при комнатной температуре в атмосфере азота. Разбавленную диэтиловым эфиром реакционную смесь промывают водой. Органическую фазу сушат над безводным MgSO4. После фильтрации и концентрации полученный сырой продукт чистят колоночной хроматографией (силикагель, EtOAc: гексан 1:50), что дает 7,85 г продукта (98 % выход).
Rf: 0.58 (EtOAc: гексан 1:2).
ИК(пленка): 2896(s); 2857(s)); 1743(s); 1471, 1462, 1436, 1385 (m); 1253(); 1210, 1165, 1101(m); 1057, 837,773 см-1.
ПМР: (360 MHz, CDCl3): δ : 3.67(3H, s); 3.40 (1H, m); 2.30(1H, dd, J = 6.4, 14.8); 2.12(1H, dd, J=8.0, 14.8); 1.90 (1H, m); 1.68(1H, m); 1.40(3H, m); 1.15(1H, m); 0.95(3H, d, J= 6.8); 0.88(9H, s); 0,84(6H, dd, J =6.8, 10.5); 0,02(6H, s) м.д.
M (m/z): 316(1%); 301(2%); 249(3%); 191(5%); 115(80%).
К перемешиваемой суспензии трет-бутилата калия (16,85 г, 165 ммоль) в сухом диэтиловом эфире (150 мл) добавляют 0.752 мл воды через шприц при 0oC. Образующуюся суспензию перемешивают 10 минут при той же температуре и затем обрабатывают предыдущим продуктом (6 г, 19 ммоль). Убирают ледяную баню и реакционную смесь перемешивают при комнатной температуре 50 часов. К реакционной смеси добавляют лед до образования двух прозрачных слоев. Эту реакционную смесь подкисляют 10% HCl водным раствором до pH 1. После экстракции диэтиловым эфиром объединенные эфирные фазы сушат над MgSO4. После фильтрации и концентрации сырой продукт чистят колоночной хроматографией на силикагеле 1:20 и 1:4 EtOAc: гексан), получая 5,34 г кислоты 2.1 (выход: 93%).
Rf: 0.54 (EtOAc: гексан 1: '').
ИК(пленка): 3500 (m); 2958, 2857 (s); 1708 (s);
1471, 1462, 1410(s); 1294, 1252, 1226 (s) 836.773 (s) см-1
ПМР: (360 MHz, CDCl3): δ : 3.40(1H, m); 2.35(1H, dd, J=6.0, 15.0 Hz); 2.15(1H, dd, J= 8.0, 15.0, Hz); 1.92 (1H, m); 1.70(1H, m); 1.42(4H, m); 1.00(3H, d,J=6.8); 0.88(9H, m); 0.87 (6H, dd, J=6.6, 10.5); 0,03(6H, s)м.д.
MS (m/z): 302(1%); 287(1%); 258(10%); 245(5%); 187(50%); 115(80%).
Пример 13. Синтез сложного эфира 2.2
К перемешиваемому раствору (R)-3-метил-2-циклогексен-1- ола (0,70 г, 6.25 ммоль) в метиленхлориде (50 мл) добавляют кислоту 2.1 (1.51 г, 5 ммоль) при 0oC. После добавления СС (3,25 г, 15,8 ммоль) и DMAP (0,732 г, 6 ммоль) при той же температуре, смесь выдерживают 5 мин при 0oC и затем нагревают до комнатной температуры и оставляют перемешиваться в течение ночи. Добавляют по 2 мл этанола и уксусной кислоты и смесь дополнительно перемешивают в течение 2 ч. После фильтрации реакционную смесь концентрируют до объема 20 мл. После разбавления диэтиловым эфиром (200 мл) реакционную смесь промывают водой. Эфирный раствор сушат над безводным MgSO4 и концентрируют в вакууме. Оставшуюся жидкость разделяют колоночной хроматографией на силикагеле, получая 1,8 г сложного эфира 2.2 (выход: 91%).
Rf: 0.6 (EtOAc:гексан 1:20).
ИК(пленка): 2950 (s); 2857 (s); 1730(s); 1462, 1380(m); 1251(s); 1162, 1055(m.) см-1.
ПМР: (360 MHz, CDCl3): δ : 5.45(1H, m); 5.25 (1H, m); 3.40(1H, m); 2.30(1H, dd,J=8,14); 2.10(1H, dd,J=8.15); 1.95(2H, m); 1.75(3H, m); 1.70(3H, s); 1.65(2H, m); 1.62(2H, m); 1.40(4H, m); 1.10(1H, m); 0.95(3H, d, J=6.4); 0.88(9H, s); 0.85(6H,q, J=7.14); 0.02(6H, s) м.д.
MS(m/z): 396(1%); 339(1%); 267(1%); 167(30%); 109(50%); 95(80%); 75(100%).
Пример 14. Синтез кислоты 2.3
К перемешиваемому раствору N-изопропил-N-циклогекси- ламина (158 мг, 1,12 ммоль) в 1 мл сухого гексана добавляют по каплям н-бутиллитий (2.40 М раствор в гексан, 0,467 мл, 1.12 ммоль) при 5oC за несколько минут. После добавления, бесцветный раствор перемешивают при -5oC 20 минут. После чего гексан и избыток амина удаляют в вакууме при 0oC. В среде аргона оставшийся белый твердый продукт растворяют в ТГФ (2 мл) и HМРА (0,7 мл). Затем смесь охлаждают до -78oC и добавляют по каплям кислоту 2.2 за 2 минуты. Спустя 10 минут после добавления, реакционной смеси дают нагреться до -30oC и выдерживают при этой температуре 1 ч. Реакционную смесь охлаждают до -78oC и TBDMS-Cl (168 мг, 1.12 ммоль) добавляют. Реакционную смесь перемешивают при -78oC 10 мин. затем очень медленно в течение 1 часа нагревают до комнатной температуры. Наконец, реакционную смесь кипятят с обратным холодильником в атмосфере аргона 17 часов и затем охлаждают до комнатной температуры. После разбавления диэтиловым эфиром, реакционную смесь промывают 2,5% водным раствором HCl и водой. Органическую фазу сушат над безводным MgSO4 и концентрируют в вакууме. Остаток разделяют колоночной хроматографией на силикагеле (EtOAc: гексан), получая 117 мг кислоты 2.3 и 220 мг исходного материала 2.2 (выход: 67% в расчете на израсходованный исходный материал).
Rf: 0.56 (EtOAc: гексан 1:5).
ИК(пленка): 3400 (m); 2980(s); 1704(s); 1462, 1381 (m); 1253, 1202(m.) см-1.
ПМР: (360 MHZ,CDCl3): δ : 5.26(1H, m); 5.40(1H, d, J=10.3); 3.36(1H, m); 2.21(1H, d, J= 5.5); 1.95(2H, m); 1.60-2.80(6H, m); 1.42(1H, s); 1.25 (3H, m); 1.11(3H, d); 1.02(3H, d, J=6.8 Hz); 0.88(9H, s); 0.83(3H, d, J=6.8); 0.82(3H, d, J=6.8); 0,03(6H, s) м.д.
MS (m/z): 396(1%); 352(1%); 381(1): 339(1%); 281(2%): 237(5%); 115(30%); 95(80%).
Пример 15. Синтез алкена 2.4
К раствору кислоты 2.3 (474 мг, 1.2 ммоль) в сухом диэтиловом эфире (10 мл) добавляют 30 мл диазометана (0,5 М раствор в диэтиловом эфире) при 0oC. Реакционную смесь перемешивают при 0oC 1 час. Диэтиловый эфир и избыток диазометана удаляют упариванием при пониженном давлении. Остаток (425 мг, 86% выход) растворяют в ТГФ и добавляют к суспензии литийалюмогидрида (114 мг, / ммоль) в ТГФ (20 мл) через шприц. Реакционную смесь перемешивают 3 часа при комнатной температуре и затем кипятят 1 час. Избыток литийалюмогидрида разлагают осторожным добавлением этанола и затем обрабатывают разбавленным водным раствором HCl. Спирт экстрагируют диэтиловым эфиром и объединенные эфирные фазы сушат над безводным MgSO4. После фильтрации и концентрации, сырой продукт выделяют колоночной хроматографией на силикагеле и чистят ВДЖХ, получая 382 мг спирта (89% выход).
Rf: 0.4 (EtOAc: гексан 1:10).
ИК(пленка): 3355 (m); 2956(s): 1462, 1385, 1251, 1048(s) 836.773 (s); 941,732, 664 (m) см-1.
ПМР: (360 MHZ, CDCl3): δ : 5.67(1H, m); 5.53 1H, d,br,J=10): 3.81(1H,q, J= 6.0, 11.4): 3.72(1H, dd,J=5.5, 11.3); 3.38(1H, m); 1.95(2H, m); 1.70(2H, m); 1,60(4H, m); 1.40(1H, m); 1.35-1.15(4H, m); 1.03(3H, s); 1.00(3H, s); 0,90(9H, s); 0.85 (6H, dd, J=6.9, 9.9); 0.05(6H, s) мм.рт.ст.
MS (m/z); 339(M+-iPr 1%); 341(1%); 325(1%); 251(1%).
К раствору спирта (250 мг, 0.65 ммоль) в пиридине (10 мл) добавляют п-толуолсульфонилхлорид (420 мг, 2,2 ммоль) при комнатной температуре. Светло-желтый раствор перемешивают при комнатной температуре в течение 18 часов, затем выливают на лед. Смесь экстрагируют диэтиловым эфиром и объединенные эфирные фазы промывают 5% водным раствором HCl до pH 3. После высушивания над безводным MgSO4 органическую фазу концентрируют в вакууме. Остаток фильтруют через короткую силикагелевую колонку и чистят ВДЖХ, получая 336 мг тозилата (выход: 96%).
Rf: 0.37 (EtOAc: гексан 1:20).
ИК(пленка): 2958, 2857(s); 1741, 1599 (m); 1462, 1367 (s); 1250, 1178 (s): 1047, 953(s): 837, 773 (s) см-1.
ПМР: (360 MHZ, CDCl3): δ : 7.80(2H, d,J=8.3); 7.33(2H, d,J=8.4Hz); 5.59(1H, m); 5.32(1H, d,br=10.2); 4.20(1H, dd, J=4.7, 10.0); 4.10(1H, dd, J= 7.4, 10.0); 3.30(1H, m); 2.46(3H, s); 1.90 (2H, m); 1.62(2H, m); 1.53(3H, m, ); 1.45(3H, m); 1.26(2H, m); 1.15(1H, m); 0.95(3H, s); 0.90(3H, d, J=7.1); 0.88 (9H, s); 0.82(6H, dd,J=6,9,8.8); 0,01(3H, s); -0.01 (3H, s) м.д.
MS (m/z); 512(1%); 486(1%); 455(1%); 426(1); 364(2%); 321(2%); 307(5%); 229(20%); 9.55 (100%).
К суспензии литийалюмогидрида (71 мг, 1,88 ммоль) в ТГФ (12 мл) добавляют тозилат (336 мг, 0.627 ммоль) в виде раствора в ТГФ при комнатной температуре. Реакционную смесь кипятят 2 часа. Избыток LiAlH4 разлагают добавлением этанола. Затем смесь обрабатывают 5% водным раствором HCl. Эту смесь экстрагируют диэтиловым эфиром и объединенные эфирные фазы сушат над безводным MgSO4. Чистый алкен 2.4(234 мг) выделяют колоночной хроматографией (тонкоизмельченный силикагель) с выходом 91%.
Rf: 0.54 (чистый гексан).
ИК(пленка); 3011(w); 2957, 2858(s); 1462, 1383, 1386(s); 1253, 1082, 1054 (s); 836, 772 (s); 941 731 (w) см-1.
ПМР: (360 MHz, CDCl3): δ : 5.58(1H, m): 5.45 (1H, m); 3.37(1H, m); 1.92(2H, m); 1.70(1H, m); 1.60 (4H, m); 1.52(2H, m); 1.36(1H, m); 1.28(1H, m); 1.20(1H, m); 0,93(3H, s): 0,89(9H, s): 0.87(3H, d, J =6.8): 0.85(3H, d, J = 0 7.3); 0.82(6H, q, J=6.9); 0.02(3H, s); 0,01(3H, s) м.д.
MS (m/z); 366(1%); 364(1%); 309(10%); 287(1%); 233(5%), 75(100%).
Пример 16. Синтез циклогексанона 2.5
К раствору алкена 2.4 (60 мг, 0.164 ммоль) в ТГФ (6 мл) добавляют 9-BBN (0.5 М раствор в ТГФ, 3,3 мл, 1,04 ммоль) при комнатной температуре в атмосфере азота. Раствор перемешивают при комнатной температуре 1 час и затем кипятят с обратным холодильником 20 часов. Органоборан окисляют, добавляя последовательно этанол (0,5 мл), 6 н. NaOH (0,4 мл) и 30% перекись водорода (0,8 мл). Эту смесь нагревают при 50oC 1 час. Реакционную смесь экстрагируют диэтиловым эфиром и объединенные эфирные фазы промывают 5% водным раствором HCl. Органическую фазу сушат над безводным MgSO4, фильтруют и фильтрат концентрируют в вакууме. Остаток чистят колоночной хроматографией (тонкоизмельченный силикагель), получая соответствующий спирт (51 мг) с 80% выходом в виде смеси диастереомеров. Смесь этого спирта (51 мг, 0.133 ммоль) и PDC (175 мг, 0,442 ммоль) в метиленхлориде (4 мл) перемешивают при комнатной температуре 15 часов и непосредственно чистят на силикагеле. Конечная очистка методом ВДЖХ приводит к кетону 2.5 46 мг) с выходом 90%.
Rf: 0.4 (EtOAc: гексан 1:10).
ИК(пленка): 2931, 2857(s); 1715(s); 1472, 1385(s), 1250, 1081, 1058 (s): 941,667 (m); 837, 773(s) см-1.
ПМР: (360 MHz, CDCl3): δ : 3.36(1H, m); 2.26 (3H, m); 2.05(1H, d, J= 13.3); 1.90(1H, m); 1.82(1H, m); 1.65(3H, m): 1,58(1H, m): 1.50(3H, m): 1.25(1H, m): 0.92(3H, d, J =6.9); 0.88(9H, s); 0,85 (3H, s); 0.84(6H, q); 0.79 (3H, d, J =7.3): 0.02(6H, s) м.д., MS (m/z) 382(2%); 368(10%); 340(1%); 326(60%); 185(60%); 95(70%); 75(100%).
Пример 17. Синтез алкена 2.6
Раствор 2.4 (160 мг, 0,437 ммоль) и TBAF (1M раствор в ТГФ, 2,18 мл, 2,18 ммоль) в ТГФ (10 мл) нагревают при 30oC при перемешивании в течение 3 дней. Реакционную смесь разбавляют гексаном и сразу же хроматографируют. Сырой продукт чистят затем ВДЖХ (1:12 EtOAc/гексан), получая незащищенный 24S-спирт (106 мг, 88%).
ИК(пленка): 3378(m); 2959, 2870(s); 1646(w); 1462, 1380(s); 1060, 989(m); 732(m) см-1.
ПМР(500 MHz, CDCl3): δ :5.59(1H, dt, J=10.2, 3.5); 5.46(1H, dq, J =10.2, 2.0); 3.31(1H, m); 1,92(2H, m); 1.65(1H, m); 1.58(4H, m); 1.37(2H, m); 1.30-1.20(4H, m); 0,94(3H, s): 0.92(3H, d, J=6.8); 0.90(3H, d, J=6.8); 0.88(3H, d, J=6.8); 0.82 (3H, d, J=7.3).
MS (m/z); 252(M+, 3); 234(5); 149(20), 122(20); 95(100) м.д.
Раствор вышеприведенного спирта (100 мг, 0.397 ммоль) в ТГФ (4 мл) обрабатывают трифенилфосфином (260 мг, 0,99 ммоль) и 4-нитробензойной кислотой (166 мг, 0,99 ммоль) в атмосфере азота при комнатной температуре. В дальнейшем, медленно добавляют диэтил азодикарбоксилат. Реакционную смесь перемешивают при комнатной температуре 15 часов. После разбавления гексаном смесь фильтруют через силикагелевую колонку. Дальнейшая очистка ВДЖХ дает соответствующий обращенный п-нитробензоат сложный эфир (100 мг, 63%).
Смесь последнего (100 мг, 0,25 ммоль) и K2CO3 (173 мг, 1.25 ммоль) в метаноле перемешивают при комнатной температуре в течение 0,5 ч. Реакционный процесс не наблюдается. К реакционной смеси добавляют затем KOH (745 мг) и смесь перемешивают при комнатной температуре 1,5 ч. Добавляют воду и смесь экстрагируют диэтиловым эфиром. Объединенный эфирный раствор промывают водой, сушат над безводным MgSO4 и концентрируют в вакууме. Сырой продукт чистят ВДЖХ (1:1 EtOAc /гексан), получая 24 R-спирт (59 мг, 94%).
ИК (пленка): 3379(m): 2959, 2870(s): 1644 (w); 1462, 1380 (s):, 1060, 989(m); 732(m) см-1.
ПМР (500 MHz, CDCl3): δ 5.59(1H, ddd, J=3.4, 4.2, 10.2 Hz); 5.46(1H, dq, J=10.1, 2.8); 3.3(1H, m); 1.92(2H, m); 1.70(1H, m); 1.63(1H, m); 1.58(3H, m, ); 1.46(2H, m); 1.35(1H, m); 1.28(2H, m); 1.10(1H, m); 0,94(3H, s); 0.92(3H, d, J =6.7 Hz); 0.90(3H, d, J=6.7); 0,87 (3H, d, J =0,87); 0.82(3H, d, J=7.3) м.д. MS (m/z); 252(M+, 3); 234(5); 149(20), 122(20); 95(100).
Раствор этого спирта (59 мг, 0,234 ммоль), имидазола (32 мг, 0,468 ммоль), TBDMS-Cl (71 мг, 0,468 ммоль) и DМАР (10 мг) в ДМФ (3 мл) перемешивают при комнатной температуре 16 часов, и затем обрабатывают TBDMS-Cl (71 мг, 0,468 ммоль), имидазолом (32 мг) и DMAP (10 мг). После 5 часов перемешивания при комнатной температуре добавление повторяют еще раз. Реакционную смесь перемешивают 10 часов и затем обрабатывают 10% HCl (1 мл). После перемешивания в течение 10 мин реакционную смесь экстрагируют диэтиловым эфиром. Объединенный эфирный раствор промывают 5% HCl и водой, сушат MgSO4 и концентрируют в вакууме. Оставшийся продукт разделяют колоночной хроматографией и чистят ВДЖХ (чистый гексан), получая 2.6 (83 мг, 97%).
ИК(пленка): 2857(s); 1645(w); 1408; 1375(m); 1289, 1156 (m): 945 см-1.
ПМР(500 MHz, CDCl3): δ 5.58(1H, dt, J=10.2, 3.4); 5.4:(1H, dq, J=10,1, 1.8); 3,36 (1H,q, J=5.0); 1.91 (2H, m); 1.68(1H, m); 1,58(4H, m); 1.48(1H, m); 1.36(2H, m); 1.27(2H, m), 1.22(1H, m); 0.93(3H, s); 0.89(9H, s); 0.87(3H, d, J=6.8); 0.84((3H, d, J =6.8); 0.83(3H, d, J=6,8); 0.80(3H, d, J= 7.3); 0.03(3H, s);0.02(3H, s) м.д.
MS (m/z): 366 (M,1%).
Пример 18. Синтез циклогексанона 2.7
К раствору 2.6 (80 мг, 0,219 ммоль) в ТГФ (8 мл) добавляют 9-BBN (0,5 М раствор в ТГФ, 4.37 мл, 2.19 ммоль) при комнатной температуре в атмосфере азота. Реакционную смесь нагревают при температуре кипения с обратным холодильником в течение 20 часов. Органобораны окисляют, добавляя последовательно EtOH (0.66 мл), 6 н. NaOH (0,53 мл) и 30% H2O2 (1,06 мл). Эту смесь нагревают при 50oC в течение 1 часа. После разбавления диэтиловым эфиром реакционную смесь промывают 5% водным раствором HCl, водой и сушат над MgSO4. После концентрации оставшееся масло хроматографируют и чистят ВДЖХ, получая спирт (77, 3 мг, 92%).
Раствор последнего (77,3 мг, 0,2 ммоль) и PDC (396 мг, 1 ммоль) в CH2Cl2 (10 мл) перемешивают при комнатной температуре в течение 24 часов. Непосредственная колоночная хроматография реакционной смеси и затем ВДЖХ очистка дают заданный кетон 2.7 (71 мг, 92%).
ПМР (500 MHz, CDCl3) δ : 3.36 (1H, dt,J=9.7, 4.8); 2.28(1H, d, J=13.7); 2.26(2H, m); 2.05(1H, dt, J=13.4, 1.4); 1.90(1H, m); 1.82 (1H, m); 1.70-1.60 (4H, m); 1.50(1H, m); 1.34-1.20(3H, m); 0,97(1H, m); 0.89(33, d, J= 6.8); 0.88(9H, s); 0.86(3H, s); 0.84(3H, d, J=6.8); 0,83(3H, d, J=6,8); 0.79(3H, d, J=7.2); 0,02(3H, s); 0,01(3H, s) м.д.
Пример 19. Синтез сложного эфира 2,9
К перемешиваемому раствору (R)-(+)-цитронеловой кислоты (2.8; 0,98 г, 5.76 ммоль) в метиленхлориде (50 мл) добавляют (R)- 3-метилциклогексен-1-ола (84% е. е. (исключая ошибки) 0,64 г, 5,76 ммоль) при 0oC в атмосфере азота. Реакцию инициируют добавлением DCC (2.96 г, 14,4 ммоль) и DMAP (,732 г, 6 ммоль). После 5 минут при 0oC, реакционную смесь нагревают до комнатной температуры и оставляют перемешиваться при комнатной температуре в течение ночи (18 ч). Добавляют этанол (4 мл) и уксусную кислоту (4 мл) к реакционной смеси при 0oC. Смесь перемешивают при 0oC 20 мин и при комнатной температуре 1 час. Добавляют диэтиловый эфир и образовавшийся белый твердый продукт удаляют фильтрацией. Фильтрат упаривают при пониженном давлении и оставшуюся жидкость растворяют в диэтиловом эфире. Эфирный раствор промывают водой, сушат над безводным MgSO4. Колоночная хроматография сырого продукта дает сложный эфир 2.9 (1,521 г) с 96% выходом.
Rf: 0.52 (EtOAc: гексан 1:20).
ИК (пленка); 2931(s); 2360 (w); 1732(s), 1456, 1378(m): 1150, 1071(s): 921(m) см-1.
ПМР (360 MHz, CDCl3): δ 5.45 (1H, m)-, 5.25(1H, m); 5.07(1H, t, J=7.1 H); 2.28(1H, dd, J= 6.0, 14.4 Hz), 2.10(1H, dd,J=8.2, 14.4); 1.95(3H, m); 1.71(3H, s); 1.68(3H, s); 1.58(3H, s); 1.70(4H, m); 1.20(4H, m7); 0.92(3H, d, J =6.6) м.д.
MS (m/z): 264(M+, 1%); 249(1%); 227(1%); 191(1); 169(30); 109(30); 95(100).
Пример 20. Синтез кислоты 2.10
К перемешиваемому раствору диизопропиламина (456 мкл, 3.27 ммоль) в ТГФ(10 мл) добавляют н-бутиллитий (2.45 М раствор в гексане. 1.33 мл, 3.27 ммоль) при -15oC в атмосфере азота. Реакционную смесь перемешивают при той же температуре 20 мин и затем добавляют HМРА (3 мл). Реакционную смесь охлаждают до -78oC. Раствор 2.9 (0,77 г, 2,92 ммоль) в ТГФ (2 мл) добавляют 2.9 очень медленно к реакционной смеси при -78oC. Через 10 минут после добавления образовавшемуся энолу дают нагреться до -50oC за 20 мин. TBOMS-Cl (491 мг, 3,27 ммоль) в виде твердого продукта добавляют при -50oC и реакционную смесь перемешивают при той же температуре 20 мин и затем нагревают до комнатной температуры. Реакционную смесь перемешивают при комнатной температуре 3 часа и затем нагревают до кипения с обратным холодильником 16 часов. К смеси добавляют 5% водный раствор HCl (15 мл) и перемешивают при комнатной температуре 60 мин. Смесь экстрагируют диэтиловым эфиром. Объединенный эфирный раствор промывают водой, сушат над безводным MgSO4 и концентрируют в вакууме. Полученное масло разделяют колоночной хроматографией, получая 2.10(448 мг, 58%).
Rf: 0.35 (EtOAc/гексан 1:5).
ИК(пленка): 2930 (s); 1704(s); 1462 1381 (m); 1285, 1253, 1202 (m); 836 (m) см-1.
ПМР(360 MHz, CDCl3): δ 5.62(1H, m); 5.41(1H, d, J=11.0); 5.09(1H, t, J= 6.9 HZ); 2.22(1H, d, J=5.9); 2.08(1H, m); 1.90(3H, m); 1.68(3H, s); 1.58 (3H, s); 1.62(7H, m); 1.10(3H, s); 1.02(3H, d, J=6.8) м.д.
MS (m/z): 264(M+. , 5%); . 249(1%); 221(1); 208(5); 154(15); 109(15); 96(100).
Пример 21. Синтез алкена 2.11
К суспензии LiAlH4 (302 мг, 7.95 ммоль) в ТГФ (10 мл) добавляют раствор 2.10 (420 мг, 1.59 ммоль) в ТГФ (5 мл). Реакционную смесь кипятят с обратным холодильником в течение 48 часов. Избыток LiAlH4 разрушают, добавляя 5% водного раствора HCl. Смесь экстрагируют диэтиловым эфиром. Объединенные эфирные фазы промывают водой, сушат над MgSO4 и концентрируют в вакууме. Образовавшийся продукт хроматографируют, получая первичный спирт (344 мг, 88%). Диастереоизомер может быть удален дальнейшей ВДЖХ очисткой (EtOAc/гексан 1: 6).
Rf: 0.35 (EtOAc/гексан 1:5).
ИК(пленка): 3339 (m); 2928, 2871(s); 1454, 1376(m); 1028(m); 732(m) см-1.
ПМР (500 MHz, CDCl3): δ 5.67(1H, dt,J=10.1,3.5 Hz); 5.53 (1H, dt, J= 10.2, 1.8 Hz); 5.12(1H, tt,J=6.8,1.6); 3.78(1H, m); 3.72(1H, m); 2.06(1H, m); 1.93(2H, m); 1.88(1H, tt,J=7.8, 7.8); 1.70(2H, m); 1.68(3H, s); 1,61(2H, m); 1.59(3H, s); 1.50(1H, m); 1.40(1H, ddd, J=l. 2, 4.5, 10.5); 1.33(1H, m); 1.28(2H, m); 1.13(1H, m); 1.13(3H, d, J= 7.0); 1.01(3H, s) м.д. MS (m/z): 250(М+, 5); 232 3); 219(10); 137(40); 95(100).
К раствору спирта (250 мг, 1 ммоль) в пиридине (15 мл) добавляют п-толуолсульфонил хлорид (572 мг, 3 ммоль). Светло-желтый раствор перемешивают при комнатной температуре в течение 17 часов и затем выливают в ледяную воду. Смесь экстрагируют диэтиловым эфиром и объединенный эфирный раствор промывают 5% водным раствором HCl и водой. После сушки над безводным MgSO4 эфирный раствор концентрируют при пониженном давлении. Остаток разделяют колоночной хроматографией, получая соответствующий тозилат.
К суспензии LiAlH4 (342 мг, 9 ммоль) в ТГФ (30 мл) добавляют раствор тозилат в ТГФ (5 мл). Эту реакционную смесь нагревают при температуре кипения с обратным холодильником в течение 2 часов. Избыток LiAlH4 разлагают, добавляя этанол. Смесь экстрагируют диэтиловым эфиром. Объединенный эфирный раствор промывают 2% HCl водным раствором и водой. После высушивания над безводным MgSO4 диэтиловый раствор концентрируют в вакууме. Колоночная хроматография оставшегося продукта дает 211 (245 мг, 100% от двух стадий).
ИК(пленка): 2925, 2865(s); 1647(w); 1453, 1378 (s); 731 (s) см-1.
ПМР (500 MHz, CDCl3): δ 5.58(1H, dt, J=10.2, 3.5); 5.45(1H, dm, J=10.2); 5.12(1H, tt, J= 7,0,1.3); 2.02(1H, m); 1.92(2H, m); 1.85(1H, tt, J=7.9, 15.4); 1.69(1H, m); 1.68(3H, s); 1,59(3H, s); 1.56(2H, m); 1.46(1H, m); 1.35(1H, m); 1.25(3H, m); 0.93(3H, s); 0.88(3H, d, J=6.8); 0.78(3H, d, J= 7.1) м.д.
MS (m/z): 234(1%); 57(100%).
Пример 22. Синтез кетона 2.12
К бесцветному раствору Hg(OAc)2 в H2O (1.25 мл) добавляют ТГФ (1.25 мл). Реакционная смесь становится желтой и образуется некоторое количество осадка. К этой смеси добавляют раствор 2.11 (212 мг, 0,906 ммоль) в ТГФ (2.5 мл) и реакционную смесь перемешивают при комнатной температуре 2 часа. Добавляют 3М NaOH (1.09 мл) и впоследствии добавляют 1М раствор NaBH4 в 3 М NaOH (1.09 мл). Эту смесь перемешивают 10 мин. Экстракция диэтиловым эфиром с последующей колоночной хроматографией дает 25-гидрокси производное (154 мг) с 67% выходом.
ИК(пленка): 3364(m); 2963, 2867(s); 1462, 1379 (m); 1153(m); 732 (m) см-1.
ПМР (500 MHz, CDCl3): δ 5.58(1H, dt, J=10.3,3.5); 5.46(1H, dm, J=10.3); 1.93(2H, m); 1.70(1H, m); 1.58(4H, m); 1.50-1.35(5H, m); 1.26(2H, m); 1.21(6H, s); 0.94(3H, s); 0.88(3H, d, J =7,0); 0.80(3H, d, J=7.1) м.д.
MS(m/z): 252(M+, 1%); 95(100).
К раствору последнего (50 мг, 0.20 ммоль) в ДМФ (3 мл) добавляют последовательно хлортриэтилсилан (90 мг, 0.60 ммоль), имидазол (54 мг, 0.80 ммоль) и DMAP (10 мг). Этот раствор перемешивают при комнатной температуре 20 часов и затем разбавляют диэтиловым эфиром. Эфирный раствор промывают 5% водным раствором HCl и водой, соответственно. После сушки над MgSO4 растворители удаляют в вакууме. Оставшийся продукт делят колоночной хроматографией (2% EtOAc в гексане), что дает защищенный силил простой эфир (67 мг, 92%).
ИК(пленка): 2958(s): 1460, 1380(m); 1235, 1156 (m); 1042(s); 730(s) см-1.
ПМР(360 MHz, CDCl3); δ 5.59(1H, dt, J=10.2, 3.6); 5.46(1H, dm, J=10.2); 1.92(2H, m); 1.70(1H, m); 1.79(3H, m); 1.45-1.33(6H, m); 1.25(2H, m); 1.19(6H, s); 0.94(9H, t, J=8.0); 0.93(3H, s); 0.88(3H, d, J=6.8); 0.80 (3H, d, J=7.3); 0.56(6H, q,J=8.0) м.д. MS (m/z); 366(М+, 1%); 337(10%); 337(10%); 233(20%); 173(30%); 103(100%).
К раствору силил простого эфира (121 мг, 0.33 ммоль) в ТГФ (8 мл) добавляют 9-BBN (0.5 М раствора в ТГФ, 6,6 мл, 3,3 ммоль). Этот раствор нагревают при температуре кипения с обратным холодильником в течение 30 часов. Органобораны окисляют, добавляя последовательно EtOH (1 мл), 6 н. NaOH (0,8 мл) и 30% H2O2 (1,6 мл). Эту реакционную смесь нагревают при 50oC 1 час и затем экстрагируют диэтиловым эфиром. Объединенный эфирный раствор промывают 5% водным раствором HCl, водой и сушат над MgSO4. После удаления растворителей оставшийся продукт делят колоночной хроматографией, получая циклогексанол (121 мг, 95%).
Смесь последнего (121 мг, 0,34 ммоль) и PDC (480 мг, 1,2 ммоль) в CH2Cl2 перемешивают при комнатной температуре 20 часов и сразу же фильтруют через короткую колонку. Очистка сырого продукта ВДЖХ (1:20 EtOAc/гексан) дает циклогексанон 2.12 (97 мг, 80%).
ИК(пленка): 2958(s); 1722(s); 1461, 1381(m, ); 1282, 1234, 1042(s); 742(m) см-1.
ПМР (500 MHz, CDCl3): δ 2.28(1H, d, J=13.2); 2.26 (2H, m); 2.06(1H, dt, J= 13.3, 1.6), 1.90(1H, m); 1.83 1H, m); 1.67(3H, m); 1.18(6H, s); 0,93(9H, t, J =8.0); 0.90(3H, d, J=6.9 Hz); 0.86(3H, s); 0.79(3H, d, J=7.2); 0.56(6H, q, J=8.0) м.д.
MS (m/z): 354(M+, 10%); 353(5%); 173(30); 111(60); 55(100).
Пример 23. Синтез алкина 4.4
Раствор 4.1 (48 г, 0.04 ммоль), имидазола (6,6 г, 0,68 моль) и т-бутилдифенилхлорсилана (13,2 г, 0,048 моль) в сухом ДМФ (16 мл) перемешивают 36 ч при комнатной температуре в атмосфере азота, затем добавляют диэтиловый эфир (100 мл) к раствору и органический слой промывают водой (20 мл) три раза, сушат над безводным MgSO4 и упаривают, получая 15,58 г 4.2. Очистка колоночной хроматографией (гексан: этилацетат 90:1) дает 14.2 г 4.2 со 100% выходом.
Rf: 0.48 (гексан: этилацетат 5:1).
ИК(пленка): 2932(m); 1741(s); 1428(s); 1199 (s); 1111(s), 739 (s), 702 (s) см-1.
ПМР (500 MHz, CDCl3): δ 7.65 (4H, m); 7.4(6H, m); 3.82(1H, dd, J=6.9, 9.7); 3.72(1H, dd, J= 5.8, 9.7): 3.68 (3H, s); 2.72(1H, секстет, J=6.9); 1.15(3H, d,J=6.9); 1.03(9H, s) м.д.
К раствору 4.2 (1,5 г, 4,2 ммоль) в сухом гексане (9 мл) добавляют далее по каплям при -78oC в атмосфере азота диизобутилалюминий гидрид (10 M/гексан, 4.2 мл, 4,2 ммоль). Обработка реакционной смеси 2 н. раствором калий натрий тартрата в воде при перемешивании и последующая экстракция водного слоя диэтиловым эфиром (200 мл), сушка органического слоя (MgSO4, безводный) и удаление растворителя дают 1,32 г альдегида 4.3, содержащего небольшое количество 4.2.
Rf: 0.32 (гексан: этилацетат 4:1).
ПМР(500 MHz, CDCl3): δ 9.76(1H, d, J= 2); 7.65(4H, m), 7.4(6H, m); 3.87(2H, m), 2.57(1H, m); 1.11(3H, d, J=6.9); 1.03(9H, s) м.д.
К суспензии калий b-бутилата (0,68 г, 6.05 ммоль) в сухом ТГФ (14 мл) добавляют по каплям за одну минуту в атмосфере азота при -78oC метил (диазометил)фосфонат (0,59 г, 6.0 ммоль). Полученный красный раствор оставляют перемешиваться в течение пяти минут при -78oC и, впоследствии, по каплям за одну минуту добавляют раствор альдегида 4.3(1,78 г, 5,5 ммоль) в сухом ТГФ (13 мл). Реакционную смесь перемешивают 18 ч при -78oC и 2 ч при комнатной температуре, и затем добавляют воду (200 мл), образовавшийся раствор экстрагируют трижды дихлорметаном (400 мл) и диэтиловым эфиром (200 мл). Органические слои промывают раствором соли, сушат над безводным MgSO4 концентрируют и чистят колоночной хроматографией (гексан: этилацетат), получая 1,68 г 4.4 с 90% выходом (из 4.2).
Rf: 0.67 (гексан, этилацетат 4:1).
ИК(пленка): 3307 (s); 2959(m); 2116(s); 1428 (s), 1112(s); 702(s); 739(s) см-1
ПМР(500 MHz, CDCl3): δ 7.69(4H, m): 7.4(6H, m); 3.74(1H, dd, J=5,7, 9.6); 3.55(1H, dd,J=7.6, 9.6); 2.66(1H, ddf, J= 2,3, 5.6, 7.6); 2.03(1H, d, J=2); 1.23(3H, d, J=6.8); 1.07(9H, s) м.д.
Пример 24. Синтез 4.5
К хорошо перемешиваемому раствору B-Br-9-BBN ( 1M CHCl 8,04 мл, 8.04 ммоль) в дихлорметане (12 мл) добавляют по каплям 4.4 (2.16 г, 6.6 ммоль) в дихлорметане (24 мл) при 0oC в атмосфере азота. Реакционную смесь перемешивают 4 ч при 0oC. Затем добавляют уксусную кислоту (4.26 мл) и смесь перемешивают дополнительно один час при 0oC, после чего добавляют 51 мл 3 М NaOH в воде и 8.52 мл 30% перекиси водорода. После перемешивания в течение 30 мин при комнатной температуре (25oC), продукт экстрагируют трижды гексаном и органический слой промывают водой, водным NaHCO3 и снова водой и в конце сушат MgSO4 (безв. ). Остаток, полученный после концентрации чистят колоночной хроматографией (гексан: этилацетат 300:1), получая 2,4 г 4.5 с 90% выходом.
Rf: 0.6 (гексан:этилацетат 10:1).
ИК (пленка): 2931(m); 1625(s); 1427(s); 1112(s); 887(s); 823(s); 739(s); 701(s) см-1.
ПМР (500 MHz,; CDCl3): δ 7.65(4H, m); 7.42 (6H, m); 5.69(1H, d, J=1.6); 5.49(1H, d, J=1.6); 3.71(1H, dd, J=6,9,10); 3.56(1H, dd,J=5,8,10); 2.61(1H, перекрывание, J=6,9, 6,85, 5.87); 1.09(3H, d,J=6.85); 1.05(9H,.s) м.д.
Пример 25. Синтез 4.8
К раствору бромида 4.5(520 мг, 1,29 ммоль) в сухом диэтиловом эфире (2,5 мл) добавляют трет-бутиллитий 2,6 ммоль) быстро, по порциям, при -120oC (избыток жидкого H2, в MeOH). Далее к этому раствору добавляют свежеприготовленный раствор CuI (250 мг, 1,29 ммоль)/HMPT (484 мг, 2,96 ммоль) в диэтиловом эфире (4.5 мл) при -120oC. Реакционной смеси дают постепенно нагреться до -78oC, затем перемешивают 1 час и после этого обрабатывают свежеперегнанным BF3•OEt2) (310 мг, 2,2 ммоль), после чего добавляют по каплям 3-метил-циклогексенон (116 мг, 1 ммоль) в сухом диэтиловом эфире (2,5 мл). Реакционную смесь нагревают до -20oC и оставляют при этой температуре на 10 ч. Приведенный выше раствор выливают в водный NH4Cl/6 н. HCl (4:1 по объему) и экстрагируют диэтиловый эфир (2х50 мл). Объединенные экстракты промывают 20% водным H4OH (2х30 мл), 2% водной HCl (30 мл) и водой (30 мл), сушат над безводным MgSO4 и упаривают. Остаток чистят колоночной хроматографией (гексан: этилацетат 10:1) и ВДЖХ (гексан: этилацетат 4:1), что дает 174 мг 4.6 и его C13-эпимер с 40% выходом.
Rf:0.32 (гексан:этилацетат 5:1).
К раствору этой смеси (35 мг, 0,08 ммоль) в сухом ТГФ (1,5 мл) добавляют TBAF (1.1M/THF, 0,3 мл, 0,32 ммоль) при комнатной температуре. После перемешивания в течение 2 ч при комнатной температуре растворитель упаривают и остаток чистят колоночной хроматографией (H:Е 1:1), получая 14.1 мг 4.7 с его C13-эпимером. Тщательное разделение ВДЖХ (гексан:этилацетат 6:4; дважды) дает 4.7 перед его C13-эпимером (1:1).
Rf: 0,27 (гексан-этилацетат 1:1).
ИК(пленка): 3386 (s, br); 2932 (m), 2253 (s), 1704 (s); 1590(m); 1468(s); 1384(m); 1073(s) см-1.
ПМР (360 MHz, CDCl3): δ 5.02(1H, dd, J=1.3); 4.94(1H, s); 3.56(1H, dd, J=6.3,10.6); 3.45(1H:, dd,J= 7.5, 10.6); 2.58(1H, AB, d, J=14); 2.45(1H, m); 2.30(2H, m); 2.22(1H, AB, d, J=14); 1.82(2H, m); 1,61(2H, m); 1.10(3H, s); 1.08(3H, d, J =6,8) м.д.
Пример 26. Синтез 6.2
К перемешиваемому раствору 6.1 (1 г, 6.50 ммоль) и иодиду натрия (2,34 г, 15,60 ммоль) в ацетонитриле (12 мл) добавляют по каплям при 0oC метилтрихлорсилан (1,84 мл, 15,60 ммоль). После двух часов нагревания до кипения с обратным холодильником, смесь охлаждают и добавляют воду. Экстракция диэтиловым эфиром с последующим промыванием органической фазы водным раствором тиосульфата, водой и раствором соли, сушка (Na2SO4) и концентрация в вакууме дают сырой иодид, который чистят на силикагелевой колонке (пентан: этилацетат 8:2), что дает 6.2(1,58 г, 86,4%).
Rf: 0,21 (пентан:этилацетат 85:15).
УФ: λmex =254
ИК(пленка): 3302 (s); 2954(s); 2866(s); 1453(m); 1365 (m); 1262(m); 1183(m); 1035(s) см-1.
ПМР: (360 MHz, CDCl3): δ 3.58(1H, d, J=10.8); 3.43(1H, d); 3.30(1H, dd, J= 9, J= 3,5); 2.95(1H, dd, J= 11.5); 2.35-2.10(2H, m); 1.61-1.05(3H, m); 1.01(3H, s); 0.98(3H, s) м.д.
Пример 27. Синтез 6.3
К охлажденному раствору (0oC) 6.2 (5 г, 17,73 ммоль) в дихлорметане (50 мл) добавляют по каплям N,N-диизопропилэтиламин (DIPEA, 6.96 мл, 3.99 ммоль) и простой хлорметил метиловый эфир (MOMCl, 1,98 мл, 26,65 ммоль). После перемешивания при комнатной температуре 3 часа, смесь доводят до pH 1-1 и экстрагируют диэтиловым эфиром (3х). Объединенные органические фракции промывают раствором соли и насыщенным бикарбонатом натрия, сушат (Na2SO4 безводн. ) и концентрируют в вакууме. Очистка колоночной хроматографией (двуокись кремния; гексан:ацетон 95:5) дает 4.95 г (86%) МОМ диэтилового эфира 6.2. К перемешиваемому раствору этого промежуточного продукта (870 мг, 2,67 ммоль) в тетрагидрофуране (6 мл) добавляют раствор тетрабутиламмоний трифторида (TBAF, 1М в THF, 21,33 ммоль, 21,33 мл). После перемешивания в течение 4 ч при комнатной температуре добавляют воду. Экстракция диэтиловым эфиром, сушка органической фазы (MgSO4) и очистка на колонке с двуокисью кремния (гексан:ацетон 95:5) дают 465 мг чистого 6.3(88%).
Rf:0.76 (гексан:ацетон 9:1).
ИК(пленка): 2966(s); 2877(s); 1651(m); 1464(m); 1369(m); 1213(w); 1151(s); 1108(s), 1049(s) см-1.
ПМР: (360 MHz, CDCl3): δ 4.76(2H, dd, J=2.2 Hz); 4.60(2H, dd,J=6.4 Hz); 3.35(3H, s); 3.33(1H, d); 3.25 (1H, d, J=9.3 Hz); 2.43-2.37(2H, m); 1.83-1.75(1H, m); 1.48-1.38(1H, m); 0.97(3H, s); 0.94(3H, s) м.д.
Пример 28. Синтез 6.4 и 6.5
Раствор осмий тетроксида (0,67% в т-бутаноле, 0,184 ммоль, 7 мл) добавляют по каплям к смеси 6.3 (372 мг, 1.88 ммоль) и периодата натрия (998 мг, 4,7 ммоль) в ТH: вода 1:1 (4 мл). После перемешивания в течение 30 часов при комнатной температуре, добавляют насыщенный раствор тиосульфата натрия в воде (1 мл) и полученную смесь экстрагируют дихлорметаном. Очистка колоночной хроматографией (двуокись кремния: гексан:этилацетат 93:7) дает 244 мг (65%) кетона. Раствор этого кетона (244 мг, 1,22 ммоль) в тетрагидрофуране (1 мл) добавляют по каплям к суспензии литийалюмогидрида (47 мг, 1,22 ммоль) в тетрагидрофура (2 мл). После перемешивания при комнатной температуре в течение 1 часа, добавляют декагидрат сульфата натрия и полученную смесь перемешивают дополнительно 2 часа и впоследствии фильтруют для удаления солей металлов. Фильтрат концентрируют в вакууме и чистят колоночной хроматографией (двуокись кремния; гексан; этилацетат 85:15), что дает 222 мг (90%) смеси диастереомеров 6.4 и 6.5.
Rf: 0.16(гексан:этилацетат 8:2).
ИК(пленка): 3426 (s, br); 2958(s); 2876(s); 1467(m); 1369(m); 1216(m); 1151(m); 1108(s); 1046(s) см-1.
ПМР: 360 MHz, CDCl3): δ : 4,63(2H: 2, s); 4.59(2H;2, s): 3.79(1H: 2, dd) 4.01(1H: 2, dd); 3.41(1H:2, d); 3.30(1H:2, d); 3.29(1H:2, d); 3.25(1H: 2, d); 3.39(3H:2, s); 3.36(3H:2, s); 2.20-1.40(4H, m); 1.00-0.35(9H, 3xs) м.д.
Пример 29. Синтез 6.6
К раствору 6.3 (1.552 г, 7.84 ммоль) в тетрагидрофуране (35 мл) добавляют раствор 9 борабицикло 3.3.1 нонана (0,5М в тетрагидрофуране, 15,7 мл, 7,85 ммоль) и полученный раствор перемешивают 5 часов при 55oC. После охлаждения до комнатной температуры добавляют этанол (4,71 мл) и 6М водный раствор гидроокиси натрия (1,57 мл, 9,42 ммоль), с последующим добавлением по каплям при 0oC 35% водного раствора перекиси водорода (3,68 мл). Перемешивание в течение 1 ч при температуре кипения, последующая экстракция водного слоя диэтиловым эфиром, сушка органической фазы Na2SO4 безводн.) и удаление растворителя дают 2,8 г сырого масла. Очистка колоночной хроматографией (гексан: ацетон 8: 2) и ВДЖХ (силикагель:гексан:этилацетат 75:25) дает 6.6 (617 мг, 36%) перед заданным эпимером (864 мг, 51%).
Rf:0.25(дихлорметан: метанол 9:1).
ИК(пленка): 3418(s, br); 2946(s); 2875(s); 1464(s); 1368(m); 1215(m); 1150(s); 1108 (s); 1047(s) см-1.
ПМР: (500 MHz, CDCl3): δ :4,61(2H, 2xd,J=6.5); 3.72(1H, dd,J=5.6, 10.2); 3.52(1H, dd, J=10.2, 9.3); 3.35(3H, s); 3.34(1H, d, J =9.15); 3.25(1H, d); 2.10(1H, ddd); 1.88(2H, m); 1.65(1H, m); 1.43(1H, m); 1.35(1H, m); 0,91(3H, s); 0.90(3H, s); 0.83(3H, s) м.д.
Пример 30. Синтез 6,7
К смеси трифенилфосфина (1,46 г, 5.56 ммоль), имидазола (378 мг, 3,56 ммоль) и 6.6 (600 мг, 2,78 ммоль) в смеси диэтиловый эфир: ацетонитрил 3:1 (12 мл) добавляют при 0oC, порциями, иод (1,41 г, 5.56 ммоль) и полученную смесь перемешивают в темноте в течение 3 ч при комнатной температуре. Экстракция смесью диэтиловый эфир: гексан 1:1, промывка собранных органических фракций раствором соли, сушка (Na2SO4 безводн.) и удаление растворителя дают бледно-желтое масло, которое чистят колоночной хроматографией (силикагель: гексан:этилацетат 95:5), что дает 825 мг(91%) иодида. Раствор этого простого эфира (460 мг, 1,41 ммоль) в смеси метанол:тетрагидрофуран 3:1 (70 мл) перемешивают в присутствии Amberlyst-15 в течение 72 час при комнатной температуре в темноте. После этого Amberlyst-15 отфильтровывают и фильтрат концентрируют в вакууме и чистят колоночной хроматографией (силикагель:этилацетат 85:15 до 70:30), что дает 6.7(342 мг, 86%).
Rf: 0,20(гексан:этилацетат 8:2).
ИК(пленка); 3380 (s); 2953(s); 2872 (m); 1452 (m); 1368(m); 1264(m); 1183(m); 1028(s) см-1.
ПМР: (500 MHz, CDCl3): δ : 3.41(2H, s,br); 3.30(1H, dd, J =9, 3.6); 2.98(1H, dd, J=11.5); 2.35-2.27(1H, m); 2.13-2.05(1H, m); 1.81-1.73(1H, m); 1.44-1.30(3H, m); 0,89(3H, s); 0.88(3H, s); 0,72(3H, s) м.д.
Пример 31. Синтез 6.8
К суспензии иодида меди (1) (676 мг, 3,55 ммоль) и цинковой пыли (928 мг, 14,18 ммоль) в смеси этанол:вода 7:3 (10 мл) добавляют метилвинилкетон (380 мкл, 4,62 ммоль) и 2 (1 г, 3.55 ммоль). Реакционную смесь обрабатывают ультразвуком в течение 1 часа в атмосфере аргона с последующим добавлением дополнительного количества иодида меди (1) (338, 1,775 ммоль) и цинковой пыли (464 мг, 7,09 ммоль). После других минут обработки ультразвуком, смесь фильтруют через целит и медные и цинковые соли промывают этилацетатом. Фильтрат экстрагируют этилацетатом, сушат (безв. магнийсульфат) и концентрируют. Очистка на колонке с двуокисью кремния (пентан: этилацетат 7:3) дает 510 мг 6.8(64%).
Rf: 0,29 (пентан:этилацетат 85:15).
ИК(пленка): 3420(s); 2946 (s); 2869(s); 1714 (s); 1454(s); 1367(s); 1231(m); 1163(s); 1024(s) см-1.
ПМР: (360 MHz, CDCl3): δ : 3.58(1H, d, J = 10.7) 3.45(1H, d); 2.50-2.35(2H, m); 2.14(3H, s); 1.96-1.84(1H, m); 1.80-1.13(8H, m); 0.98(3H, s); 0,88 (3H, s); 0.70(3H, s) м.д.
Пример 32. Синтез 6.10
К раствору метиллития (1,5 М в диэтиловом эфире, 11,6 мл, 17,34 ммоль) добавляют по каплям раствор 6.8 (490 мг, 2,17 ммоль) в диэтиловом эфире (5 мл) при -78oC. После перемешивания в атмосфере аргона в течение 2 часов, добавляют насыщенный хлорид аммония и полученную смесь экстрагируют диэтиловым эфиром, органическую фазу сушат (MgSO4) и растворитель удаляют. Очистка сырого продукта колоночной хроматографией (двуокись кремния; пентан: этилацетат 8: 2) дает 455 мг белого кристаллического диола (85%). Раствор этого диола (200 мг, 0,826 ммоль) в дихлорметане (0,8 мл) добавляют к смеси дипиридин хром (Y) оксида (1,066 г, 4,13 ммоль) и дихлорметана. Смесь перемешивают при комнатной температуре; через два часа добавляют диэтиловый эфир (5 мл) и целит. Фильтрация через силикагель-целит, промывка диэтиловым эфиром и удаление растворителя дают остаток, который чистят на колонке с двуокисью кремния (пентан:этилацетат 8:2) и ВДЖХ (пентан:этилацетат 75:25). Получают 70 мг чистого 6.10 (35%).
Rf:0.24(пентан:этилацетат 85:15).
ИК(пленка): 3480(s); 2964(s); 1715(m); 1470(m); 1372 (m) см-1.
ПМР: (360 MHz, CDCl3): δ : 9,64(1H, s); 2.39- 2.28(1H, m); 2.04-1.92(1H, m); 1.83-1.72(1H, m); 1.53- 1.23(8H, m); 1.21(6H, s); 1.03(3H, s); 0.96(3H, s); 0,75(3H, s) м.д.
Пример 33. Синтез 6.12
Раствор н-бутиллития (2.5М в гексане, 467 мкл, 1.17 ммоль) добавляют по каплям к раствору (метоксиметил)-три-фенилфосфоний хлорида (400 мг, 1,17 ммоль) в тетрагидрофуране (4 мл) при -78oC. Полученную смесь перемешивают 20 минут и впоследствии добавляют по каплям раствор 6.10 (70 мг, 291 ммоль) в тетрагидрофуране (700 мкл). После перемешивания в течение 10 мин, при -78oC, смеси дают нагреться до комнатной температуры и перемешивают дополнительно 21 час. Добавление воды, экстракция диэтиловым эфиром, сушка (Na2SO4) и концентрация дают сырой простой энолэфир. К раствору энолэфира (65 мг, 243 мкмоль) в тетрагидрофуране (700 мкл) добавляют раствор хлористоводородной кислоты (2М в THF, 66 мкл). Через 30 мин смесь экстрагируют диэтиловым эфиром; органические фазы промывают насыщенным бикарбонатом натрия и раствором соли и сушат (Na2SO4). После удаления растворителя на роторном испарителе оставшееся масло чистят колоночной хроматографией (двуокись кремния: пентан:этилацетат 8:2); получая 25 мг чистого 6.12(45%).
Rf:0.17(пентан:этилацетат 8:2).
ИК(пленка): 3440(s); 2928 (m); 1715 (m); 1470(m); 1380(m) см-1.
ПМР: (500 MHz, CDCl3): δ 9.84(1H, dd,J=4.1, 2.4); 2.31(2H, dd); 1.99-1.90(1H, m); 1.87-1.23(10H, m); 1.22(6H, s); 0.98(3H, s); 0.82(3H, s); 0.68(3H, s) м.д.
Пример 34. Ситез 6.9
К суспензии иодида меди(I)(676 мг, 3,55 ммоль) и цинковой пыли (928 мг, 14,18 ммоль) в смеси этанол:вода 7:3 (10 мл) добавляют этилвинилкетон (458 мкл, 4,61 ммоль) и 6.2 (1 г, 3.55 ммоль). Реакционную смесь обрабатывают ультразвуком в течение 1 часа в атмосфере аргона с последующим добавлением дополнительного количества иодида меди (I) (338 мг, 1,775 ммоль) и цинковой пыли (464 мг, 7,09 ммоль). После других 35 минут обработки ультразвуком, смесь фильтруют через целит и медную и цинковую соли промывают диэтиловым эфиром в звуковой бане. После промывания сульфатом натрия, фильтрат концентрируют и чистят на колонке с двуокисью кремния (пентан: этилацетат 85: 15), получая 433 мг 6.9(51%).
Rf:0,29 (пентан:этилацетат 85:15).
ИК(пленка): 3473(s, br); 2940(s); 2871 (s); 1712(s); 1460(s); 1376(s); 1113(s); 1028(s) см-1.
ПМР: (200 MHz, CDCl3): δ 3.52 (2H, 2хd, J=10.6); 2.42(2H,q, J=7.3); 1.98-1.14(11H, m); 1.06(3H, t,J= 7.3); 0.98(3H, s); 0.89(3H, s); 0.70(3H, s) м.д.
Пример 35. Синтез 6.11
К раствору этилодида (305 мкл, 3,75 ммоль) в диэтиловом эфире (3,75 мл) добавляют трет-бутиллитий (3,21 мл 2,34 М раствор в пентане, 7,5 ммоль) при -78oC и полученный раствор перемешивают в течение 1 часа. Впоследствии добавляют по каплям раствор 6.9 (300 мг, 1,25 ммоль) в сухом диэтиловом эфире (3 мл). Смесь перемешивают 2 часа при -73oC в атмосфере аргона и затем приводят к комнатной температуре. Добавляют насыщенный хлористый аммоний и полученную смесь экстрагируют диэтиловым эфиром и дихлорметаном. Органическую фазу сушат (MgSO4), фильтруют, концентрируют и чистят на колонке с двуокисью кремния (пентан:этилацетат 8:2), получая 251 мг (74%) диола.
К смеси 4-метилморфолин N-оксида (158 мг, 1,35 ммоль) с активированными порошкообразными молекулярными ситами  (450 мг) и диода (243 мг, 0,9 ммоль) в дихлорметан (1,8 мл) добавляют при 0oC тетра(н-пропил) аммоний перрутенат (15,8 мг, 45 мкмоль), порциями. После 2 ч перемешивания при комнатной температуре, реакционную смесь фильтруют через силикагель, промывают дихлорметаном и концентрируют в вакууме. Очистка колоночной хроматографией (двуокись кремния; пентан: этилацетат 85: 15) дает 193 мг 6.11 (60%).
(450 мг) и диода (243 мг, 0,9 ммоль) в дихлорметан (1,8 мл) добавляют при 0oC тетра(н-пропил) аммоний перрутенат (15,8 мг, 45 мкмоль), порциями. После 2 ч перемешивания при комнатной температуре, реакционную смесь фильтруют через силикагель, промывают дихлорметаном и концентрируют в вакууме. Очистка колоночной хроматографией (двуокись кремния; пентан: этилацетат 85: 15) дает 193 мг 6.11 (60%).
Rf:0.20(пентан:этилацетат 9:1),
ИК(пленка): 3436 (s,br); 2965 (s); 2938 (s); 2877 (s); 1721(s); 1460(s); 1370(m); 1266(m); 1186(m) см-1.
ПМР: (200 MHz., CDCl3): δ : 9.65(1H, s); 2.48-2.25(1H, m); 2.09-1.90(1H, m); 1.88-1.70(1H, m); 1.65-1.20(13H, m); 1.02(3H, s); 0.97(3H, s); 0.87(6H, t, J=7.4H); 0.76(3H, s); м.д.
Пример 36. Синтез 6.13
К раствору (метоксиметил)трифенилфосфоний хлорида (330 мг, 963 мкмоль) в диэтиловом эфире (2,5 мл) добавляют н-бутиллитий (2,5 М раствор в гексане, 347 мкл, 866 мкмоль) при 0oC. После перемешивания в течение 10 мин красную суспензию доводят до комнатной температуры, перемешивают 10 минут и затем охлаждают снова до -30oC. Добавляют по каплям раствор 6.11 (86 мг, 321 мкмоль) в диэтиловом эфире (860 мкл) и после 1/2 часа охлаждающую баню убирают и смесь перемешивают при комнатной температуре 18 часов. Добавление воды, последующая экстракция диэтиловым эфиром, сушка (Na2SO4) и концентрация в вакууме дают 200 мг сырого винилэфира. После фильтрации через силикагель и упаривание растворителя, фильтрат растворяют в тетрагидрофуране (1 мл) и обрабатывают водной соляной кислотой (2 н. раствор в тетрагидрофуране). Через 30 мин добавляют воду и смесь экстрагируют диэтиловым эфиром, сушат (Na2SO4) и фильтруют. Фильтрат концентрируют в вакууме и чистят на колонке с двуокисью кремния (пентан:этилацетат 9:1) и ВДЖХ(гексан: этилацетат 8:2), что дает 30 мг (33%) 6.13.
Rf: 0.26 (пентан: этилацетат 9:1).
ИК(пленка): 3455(m, br); 2965(s); 2938(s); 2876(m); 1720(s); 1460(m); 1144(m) см-1.
ПМР: (200 MHz, CDCl3): δ : 9.8(1H, dd,J=3, 3.5 Hz); 2.15(2H, 2хd, J<1,3,3.5 Hz); 2.05-1.52 (2H, m); 1,52- 1.4(4H, q, J=7.5 H); 1.4-1.0(6H, m); 0,99(3H, s); 0,86(6H, t, J=7.5 Hz); 0.81(3H, s); 0,69 (3H, s) м.д.
Пример 37. Синтез 6.17 (α+β) (R=Me)
Смесь 6.4 и 6.5 (730 мг, 3,61 ммоль), гидроокиси калия (порошок, 400 мг, 7.22 ммоль) и 1-хлор-3-метил-2-бутена (610 мкл, 5,42 ммоль) в толуоле (8 мл) обрабатывают ультразвуком в течение 30 минут. После добавления следов 18-крон-6 и дополнительного количества гидроокиси калия (200 мг, 3.61 ммоль) смесь обрабатывают ультразвуком дополнительно 1 час. После чего смесь фильтруют через небольшой слой силикагеля, осадок промывают диэтиловым эфиром, фильтрат концентрируют и чистят колоночной хроматографией (гексан:этилацетат 95: 5 8: 2), что дает 286 мг 6.17 (α+β) (R=Me) (29%) и 450 мг непрореагировавшего вещества.
Rf: 0.63(гексан:этилацетата 8:2).
ИК(пленка): 3015 (s); 1617(m); 1420(m); 1215(s); 1015(m); 923(s) см-1.
ПМР: в соответствии со структурами обоих эпимеров.
Пример 38. Синтез 6.18 (α+β) (R-H)
Смесь свежерастертой гидроокиси калия (700 мг, 12,5 ммоль), 18-крон-6 простого эфира (50 мг, 193 мкмоль), 6.4 и 6.5 (700 мг, 3,47 ммоль) и аллил бромида (644 мкл, 7.62 ммоль) в тетрагидрофуране (7 мл) перемешивают 48 часов при комнатной температуре. Смесь фильтруют через силикагель и фильтрат концентрируют в вакууме. Очистка колоночной хроматографией (двуокись кремния; гексан:этилацетат 95:5) дает 630 мг 6.18 (α+β) (R=H) (75%).
Rf:0.67(гексан:этилацетат 8:2).
ИК(пленка): 3079(m); 2956(s); 2875(s)); 1646(m); 1465(m); 1371(m); 1150(s); 1106(s); 1049(s); 918(s) см-1.
ПМР: в соответствии со структурами обоих эпимеров.
Пример 39. Синтез 6.19 α (R=Me) и 6.19 β (R=Ме)
К суспензии ацетата ртути (432 мг, 1,36 ммоль) в вод (1,35 мл) и тетрагидрофуране (1, 35 мл) добавляют раствор 6.17 (α+β) (R=Me) (307 мг, 1.138 ммоль) в тетрагидрофуране (2,7 мл); через несколько минут окраска смеси становится бледно- желтой. Смесь перемешивают 1ч при комнатной температуре и в дальнейшем добавляют 3М водного раствора гидроокиси натрия (1,35 мл), сразу же с последующим добавлением раствора боргидрида натрия (1М в 3М гидроокиси натрия, 0,68 мл). Это дает темно-серую суспензию, которую фильтруют через небольшой слой силикагеля. Концентрированный фильтрат чистят колоночной хроматографией (двуокись кремния; гексан:этилацетат 8:2), получая 308 мг 19 (α+β) (R= Me) (94%) третичного спирта. К раствору этого спирта (288 мг, 1,0 ммоль) в метанолтетрагидрофуране 2:1 (90 мл) добавляют Amberlyst-15 (32 г). Полученную смесь перемешивают 55 ч при комнатной температуре и впоследствии фильтруют через силикагель. Фильтрат концентрируют в вакууме и чистят колоночной хроматографией (двуокись кремния; гексан:этилацетат 6:4), получая диол (240 мг; 97%). К смеси 4-метилморфолин N-оксида (NMMO, 157 мг, 1,348 ммоль), активированных порошкообразных молекулярных сит  (449 мг) и диола (230 мг, 0,94 ммоль) в сухом дихлорметане (3 мл) добавляют порциями при -10oC твердый тетpa (н-пропил)аммоний перрутенат (TPAP, 15,8 мг, 45 мкмоль). После перемешивания в течение 1,5 часа при комнатной температуре смесь фильтруют через целит и остаток промывают этилацетатом. Окрашенный в темный цвет фильтрат концентрируют на роторном испарителе и чистят колоночной хроматографией (двуокись кремния; гексан: этилацетат 7:3). Два C-20 диастереомера 6.19 α (R=Me; 90 мг, 40%) и 6.19 β (R=Me; 60 мг, 26%) разделяют ВДЖХ (гексан: ацетон 92:8) и относительную конфигурацию обоих устанавливают NOE-экспериментами.
(449 мг) и диола (230 мг, 0,94 ммоль) в сухом дихлорметане (3 мл) добавляют порциями при -10oC твердый тетpa (н-пропил)аммоний перрутенат (TPAP, 15,8 мг, 45 мкмоль). После перемешивания в течение 1,5 часа при комнатной температуре смесь фильтруют через целит и остаток промывают этилацетатом. Окрашенный в темный цвет фильтрат концентрируют на роторном испарителе и чистят колоночной хроматографией (двуокись кремния; гексан: этилацетат 7:3). Два C-20 диастереомера 6.19 α (R=Me; 90 мг, 40%) и 6.19 β (R=Me; 60 мг, 26%) разделяют ВДЖХ (гексан: ацетон 92:8) и относительную конфигурацию обоих устанавливают NOE-экспериментами.
19 α:Rf:0,36(гексан:ацетон 75:25).
ИК(пленка): 3444(s, br); 2969 (s); 2875(5); 1718(s), 1466(s); 1367(s); 1161(s); 1087(s) см-1.
ПМР: (500 MHz, CDCl3): δ : 9,65(1H, s); 3.76-3.71 (1H, ddd); 3.55-3.49(2H, m); 2.38-2.32(1H, ddd); 2.12-2.04 (1H, m); 1.75-1.72(2H, t); 1.72-1.68(1H, m); 1.45-1.38 (1H, ddd); 1.23(6H, s); 1.01(3H, s); 0.99(3H, s); 0,95 (3H, s) м.д.
19 β : Rf: 0,41 (гексан:ацетон 75:25).
ИК(пленка); 3446(s, br); 2964(s); 2872(s); 1718(s): 1466(s); 1384(s); 1367(s); 1154(5); 1098(s) см-1.
ПМР: (500 MHz, CDCl3): δ :9,61(1H, s); 3.79-3.73 (1H, ddd, J=5,6,5.7 H); 3.60-3.54(1H, ddd); 3.50- 3.45(1H, t, J =7.3 H); 2,18-2.03(2H, m); 1.77-1.74(2H, t); 1.65- 1.57(2H, m); 1.52-1.45(1H, m); 1.23(6H, s); 1.09(3H, s); 1.00(3H, s); 0.90(3H, s) м.д.
Пример 40. Синтез 6.20 α (R=Et) и 6.20 β (R=Et)
Раствор 6.18 α,β (R=H (600 мг, 2.48 ммоль) и 9-бора-бицикло 3.3.1 нонана (0,5 М в ТГФ, 19,8 мл, 9,92 ммоль) в тетрагидрофуране перемешивают 5 ч при 55oC. Смесь приводят к комнатной температуре, добавляют этанол (5,26 мл) и 6М водный раствор гидроокиси натрия (1,75 мл, 9,92 ммоль) и смесь в дальнейшем охлаждают до 0oC. Медленно добавляют 35% водный раствор перекиси водорода (4,2 мл), после чего нагревают с обратным холодильником при температуре кипения 1 ч. Экстракция диэтиловым эфиром, сушка (MgSO4) и упаривание растворителя дают остаток, который чистят колоночно хроматографией (двуокись кремния: гексан:этилацетат 6:4), что дает 615 мг спирта (95%).
Смесь этого спирта (220 мг, 0,846 ммоль) и пиридиний дихромата (143 г, 5,08 ммоль) в N,N-диметилформамиде (6 мл) перемешивают 12 часов при 40oC. Добавляют воду и смесь экстрагируют диэтиловым эфиром. Сушка органической фазы (MgSO4) и концентрация в вакууме дают желтое масло, которое разбавляют диэтиловым эфиром. Раствор охлаждают до 0oC и добавляют по каплям раствор диазометана в диэтиловом эфире до тех пор, пока тонкослойная хроматография не покажет полного завершения метилирования. После чего добавляют равный объем гексана и органическую фазу промывают водой, сушат (MgSO4) и концентрируют в вакууме. Очистка колоночной хроматографией (двуокись кремния: гексан/этилацетат 8: 2) в ВДЖХ (гексан: этилацетат 9:1) дают 85 мг (35%) сложного метилового эфира.
Раствор этого эфира (90 мг, 0,31 ммоль) в диэтиловом эфире добавляют к этилмагний иодиду (1,3 ммоль). Полученную смесь перемешивают 2 часа при комнатной температуре и в дальнейшем реакцию прерывают добавлением насыщенного аммоний хлорида. Экстракция диэтиловым эфиром, сушка органической фракции (MgSO4), удаление растворителя и очистка на двуокиси кремния (гексан:этилацетат 7:3) дают 90 мг (92%) третичного спирта.
К раствору этого спирта (90 мг, 0,285 ммоль) в метаноле: тетрагидрофуране 3: 1 (20 мл) добавляют Amberlyst-15(7г). После 2 ч перемешивания при комнатной температуре Amberlyst фильтруют, фильтрат концентрируют в вакууме и чистят на колонке с двуокисью кремния (гексан: этилацетат 6:4), получая 65 мг (84%) диола.
К раствору этого диола (50 мг, 0,184 ммоль) и триэтиламина (211 мкл, 1,84 ммоль) в смеси диметилсульфоксид: дихлорметан 1:1(2 мл) добавляют порциями сера триаоксид пиридин комплекс (179 мг, 1,104 ммоль). После 2 ч перемешивания в атмосфере азота при комнатной температуре, смесь фильтруют через силикагель и фильтрат, после удаления растворителя, чистят колоночной хроматографией (двуокись кремния: гексан: ацетон 9:1). ВДЖХ (гексан:ацетон 92: 8) разделение дает два эпимерных спирта 6.20 α (R=Et, 13 мг, 26%) и 6.20 β (R=Et, 20 мг, 40%). Относительная стереохимия установлена ОЕ экспериментами.
6.20 α :Rf:0,32 (гексан: этилацетат 8:2).
ИК(пленка): 3519(s, br); 2966 (s); 2878 (s); 2728(w), 1718(s); 1462(s); 1371(m); 1264(w); 1138(s); 1089(s) см-1.
ПМР: (500 MHz, CDCl3): δ : 9,65 (1H, s); 3.71 (1H, m); 3,50(2H, m); 2.39-2.31(1H, m); 2.12-2.04(1H, m); 1.75-1.67(3H, m); 1.55-1.38(6H, m); 1.01(3H, s); 0,99(3H, s); 0.95(3H, s); 0.86(6H, t) м.д.
6.20 β :Rf:0.35(гексан: этилацетат 8:2).
ИК(пленка): 3516(s, br); 2962 (s); 2877 (s); 2716(m); 1721(s); 1463(s); 1368(m); 1139(s); 1100(s) см-1.
ПМР: (500 MHz, CDCl3): δ : 9,61(1H, s); 3,75(1H, m); 3.54(1H, m); 3.47(1H, t); 2.17-2.03(2H, m); 1.72(2H, t); 1.65-1.42(7H, m); 1.10(3H, s); 1.00(3H, s); 0.90(3H, s); 0,86(6H, t) м.д.
Пример 41. Синтез 6.21 α (R=Ме),
Раствор н-бутиллития (2,5М в гексане, 195 мкл, 0,486 ммоль) добавляют по каплям при -10oC к суспензии (метоксиметил)трифенил фосфоний хлорида (233 мг, 0,680 ммоль) в диэтиловом эфире (1,8 мл). Через 20 минут полученную красную суспензию приводят к комнатной температуре, перемешивают 10 минут и затем снова охлаждают до -30oC. Раствор 6.19 α (R=Ме) (47 мг, 194 мкмоль) в диэтиловом эфире (0,5 мл) добавляют по каплям, после перемешивания в течение 1/2 ч при -30oC смесь приводят к комнатной температуре и перемешивают 15 часов. Обработка фильтрацией через силикагель, промывка остатка диэтиловым эфиром и концентрация фильтрата дают 64 мг бледно-желтого масла, которое растворяют в тетрагидрофуране (1 мл). Раствор соляной кислоты (2 н. в тетрагидрофуране, 120 мкл) добавляют и полученный раствор перемешивают 2 ч при комнатной температуре. Фильтрация через силикагель, концентрация фильтрата и очистка ВДЖХ (гексан: ацетон 9:1) дает 6.21 (R =Me; 24 мг; 48%).,
Rf:0,21(гексан: этилацетат 85:15).
ПМР: (360 MHz., CDCl3): δ : 9.82(1H, dd, J=2,2, 4 Hz); 3.79-3.72(1H, dt, J= 5,7, 9.5); 3.64-3.56(1H, dd, J=5.6 Hz); 3.61-3.54(1H, dt); 2.42-2.37(1H, dd, J=2.2, 14.5 Hz); 2.33-2.27(1H, dd, J=4,14.5 Hz); 2.19-2.09(1H, m); 1.99- 1.89(1H, m); 1.75(2H, t); 1.66-1.54(3H, m); 1.23(6H, s); 1.00(3H, s); 0,90(3H, s); 0,83(3H, s) м.д.
Пример 42. Синтез 6.21 β (R=Ме)
Как описано для 6.21 α (R= Me, выход 36%).
Rf:0.15 (гексан: этилацетат 85:15).
ИК(пленка): 3452(s, br); 2968(s); 2877(m); 1720(s); 1468(m), 1366(m); 1155(m), 1094() см-1.
ПМР: (200 MHz, CDCl3): δ : 9,84(1H, dd, J=3.6 Hz); 3,81-3.42(3H, m); 2.38-2.01(4H, m); 1.80-1.55(5H, m); 1.22 (6H, 2xs); 1.10(3H, s); 0,88(6H, 2xs) м.д.
Пример 43. Синтез 6.22 α (R=Et)
Раствор н-бутиллития (2,5 М в гексане, 57 мкл. 0.142 ммоль) добавляют по каплям при -10oC к суспензии (метокси-метил) трифенил фосфонийхлорида (56 мг, 0,163 ммоль) в диэтиловом эфире (0,8 мл). Через 10 минут полученную красную суспензию приводят к комнатной температуре, перемешивают 10 минут и затем снова охлаждают до -30oC. Раствор 6.20 α - (11 мг, 40,7 мкмоль) в диэтиловом эфире (0,2 мл) добавляют по каплям, через 1/2 часа при -30oC смесь приводят к комнатной температуре и перемешивают 15 часов. Обработка фильтрацией через силикагель, промывка остатка диэтиловым эфиром и концентрация фильтрата дает 35 мг бледно-желтого масла, которое растворяют в тетрагидрофуране (1 мл). Добавляют раствор хлористоводородной кислоты (2 н. в тетрагидрофуране, 150 мкл) и полученный раствор перемешивают 2 ч при комнатной температуре). Фильтрация через силикагель, концентрация фильтрата и очистка ВДЖХ (гексан:ацетон 9:1) дают 3 мг (26%) 6.22 α
Rf: 0,27 (гексан:этилацетат 8:2).
ПМР:(500 MHz, CDCl3): δ :9.81(1H, t); 3.72(1H, m); 3.52(2H, m); 2.40(1H, dd); 2.31(1H, dd); 2.18-2.08(1H, m); 1,99-1.90(1H, m); 1.75-1.38(9H, m); 1.01(3H, s); 0,91 (3H, s); 0,87(3H, s); 0,85(6H, t) м.д.
Пример 44. Синтез 6.22 β (R=Et)
Как описано для 6.2- α (R=Et); выход 36%.
Rf: 0,29 (гексан:этилацетат 8:2).
ИК(пленка): 3513(s, br); 2963(s); 2879(m); 2732 (w); 1720(s); 1463(m); 1387(m); 1094(s) см-1.
ПМР: (500 MHz, CDCl3): δ : 9.80(1H, dd): 3.72(1H, m); 3.49(2H, m); 2.30(1H, dd, J= 14,3, 3.5 Hz); 2.21(1H, dd, J= 2.6 Hz), 2.15- 2.08(1H, m), 1.80-1.42 (10H, m); 1.10(3H, s); 0.89 (3H, s); 0.87 (3H, s); 0.86 (6H, t) м. д.
Пример 45. Синтез 6.16
Исходя из 6.7 как описано дл синтеза 6.13 из 6.3
6.14: Rf :0,26(гексан:этилацетат 8:2).
ПМР: (360 MHz, CDCl3): δ : 3,42 (2H, s, br); 2.42(4H, m); 1.90-1.24 (9H, m); 1,05(4H, t, J =7.3 Hz); 0.89 (3H, s); 0.80(3H, s); 0.67(3H, s) м.д.
6.15: Rf:0,21(гексан, этилацетат 8:2).
ПМР: (500 MHz, CDCl3): δ : 9.65(1H, s); 2.00(2H, m); 1.65(2H, m); 1.45(4H, q); 1.40-1.05(8H, m); 1.01 (3H, s); 0,.93(3H, s); 0.85(6H, t); 0.71(3H, s) м.д.
6.16: Rf:0.26(гексан:этилацетат 8:2).
ИК(пленка):3426(s,br); 2935(s); 1714(m); 1650(s); 1390(m); 1112(m) см-1
ПМР: (500 MHz, CDCl3): δ :9.86(1H, t, J=3.1 Hz); 2.29 (1H, dd, J=14,5 Hz); 2.24(1H, dd); 1.91(1H, m); 1,75 (1H, m); 1.66(1H, m); 1,59(1H, m); 1.45 (4H, q, J=7.6 Hz); 1.42- 1.07(8H, m); 1.05(3H, s); 0.86(6H, t); 0.80(3H, s): 0.69(3H, s) м.д.
Пример 46. Синтез 6.24
К раствору 6.2 (1,9 г, 6.7 ммоль) в CH2Cl2 (120 мл) при 0oC добавляют DIPEA (20 экв. , 20 мл). После перемешивания в течение 40 мин при 0oC добавляют MEMCl (8 экв., 6 мл) и перемешивают в течение 2 ч. Смесь выливают в смесь вода-диэтиловый эфир. Сушат органическую фазу (MgSO4). После фильтрации и упаривания остаток чистят колоночной хроматографией (силикагель, диэтиловый эфир: гексан 1:3), получая 6.23 (2.02 г, 80%).
Rf: 0.49(диэтиловый эфир: гексан 1:1).
ИК(пленка): 3480, 3308, 1782, 1150, 1085 см-1.
ПМР: (500 MHz, CDCl3): δ 4.688 и 4.671 (2х1H, J=6.7): 3.68 и 3.56(2х2H, 2хm); 3.47(1H, d, J=9.3); 3.39(3H, s); 3.297(1H, dd, J=3.3, 9.0); 3.281(1H, d, J= 9.4); 2.94(1H, dd, J=9.0, 11.7); 2.28(2H, dtd, J=3,10, 12); 2.24(1H, m); 0,99(3H, s); 0.96(3H, s); 0.72(3H, s) м.д.
К раствору 6.23 (1 г, 2,7 ммоль) в DMF (160 мл), добавляют нитрит натрия (400 мг, 2 экв.) и каталитическое количество мочевины. После перемешивания в течение 2 дней при комн. температуре раствор выливают в смесь лед-вода. Эфирную фазу сушат (MgSO4). После фильтрации и упаривания, остаток чистят колоночной хроматографией (силикагель, диэтиловый эфир: гексан 1:6--->1:3), получая нитросоединение (350 мг; 45%) с Rf=0.36 (диэтиловый эфир:гексан 1: 1).
К раствору нитросоединения (345 мг, 1,2 ммоль) в безводном метаноле (25 мл) добавляют NaOMe (98 мг, 1,3 экв.). После перемешивания в течение 30 мин, раствор охлаждают до -78oC и пропускают через него ток озона (20 ммоль/ч) до появления синей окраски (30 мин), затем раствор продувают азотом в течение 30 мин при -78oC с последующим добавлением диметилсульфида (3,5 мл). Смесь нагревают до комнатной температуры и после упаривания растворителя добавляют смесь диэтиловый эфир-раствор соли. Эфирную фазу сушат (MgSO4). После фильтрации растворитель упаривают и колоночной хроматографией (силикагель, нитрометан: бензол 1:14) получают 6.24 (215 мг, 70%).
Rf: 0.14 (нитрометан:бензол 1:14).
ИК (пленка): 1717 см-1.
ПМР: (500 MHz, CDCl3): δ :9,76(1H, d, J=2.24); 4.692 и 4.675(2х1H, J= 6.7); 3.69 и 3.56(4H); 3.49(1H, d, J=9.4); 3.39(3H, s); 3.32(1H, d, J=9.4); 2.69(1H, td, J=2.2, 9.1); 2.11(1H, m)); 1.20(3H, s); 1.02(3H, s); 0.91(3H, s) м.д.
Пример 47. Синтез 6.25
К раствору н-бутиллития (500 мкл, 2.4М, 1.5 экв., в гексане) в THF (6 мл) при -78oC в атмосфере аргона добавляют диизопропиламин (1,5 экв., 168 мкл). После перемешивания в течение 20 мин при -78oC добавляют по каплям три-этил-4-фосфонкротонат (1,5 экв. , 333 мг, 90%, 300 мкл). После перемешивания в течение 2 ч при -78oC, добавляют по каплям раствор 6.24(215 мг, 833 мкмоль, 1 экв.) в ТH (5 мл), и перемешивание продолжают в течение 2 ч при -78oC. Затем смесь медленно нагревают до комнатной температуры и выливают в смесь диэтиловый эфир-раствор соли. Эфирную фазу сушат (MgSO4). Фильтрация, упаривание и колоночная хроматография (диэтиловый эфир:гексан 1:4) дает диеновый сложный эфир (267 мг, 91% с Rf=0.39(диэтиловый эфир:гексан 1: 1). К раствору этого продукта (267 мг, 754 мкмоль) в EtOAc (10 мл) добавляют каталитическое количество палладия на углероде (10%), после чего смесь гидрируют в течение 3 ч (4 атм.). Фильтрация через целит, добавление Et3N (200 мкл), упаривание и колоночная хроматография (диэтиловый эфир:гексан 1: 14--->1: 6) дают насыщенный продукт (215 мг, 80%, с Rf =0.53 (диэтиловый эфир:гексан 1:1)).
К раствору этого продукта (60 мг, 169 мкмоль) в CH2Cl2 (12000 мкл), при -78oC добавляют раствор диметилборбромида (10 экв., 1 мл, 1,5М в CH2Cl2:Cl2: ClCH2CH2Cl 2:1). После перемешивания в течение 1ч при -78oC, смесь переносят к энергично перемешиваемой смеси THF (8 мл) и насыщенного раствора NaHCO3 (4 мл). Реакционный сосуд промывают дихлор-метаном (2х2) мл), с последующим добавлением диэтилового эфира и раствора соли. Органическую фазу сушат (MgSO4). После фильтрации и упаривания, колоночной хроматографией (диэтиловый эфир: гексан 1:3) получают 6.25(42 мг, 93%).
Rf: 0.38 (диэтиловый эфир: гексан 1:1).
ИК(пленка:): 1717 см-1.
ПМР: (500 NHZ, CDCl3): δ : 4.12(2H, q, J=7.13); 3.57 (1H, br, d, J=10) , 3.45(1H, br, d, J= 10); 2.29 (2H, m); 1.87(1H, m); 1.73(1H,qd, J=2,10); 1.25(3H, t, J=7.13); 0.98(3H, s); 0.89(3H, s); 0.71(3H, s) м.д.
Пример 48. Синтез 6.26
К раствору 6.25 (140 мг, 520 мкмоль) в CH2Cl2 -DMSO (2.5 мл: 5 мл), при -15oC добавляют по каплям раствор Et3N (3 экв., 220 мкл) и триоксид серы-пиридин-комплекс (25 экв., 205 мг) в CH2Cl2 - DMSO (1 мл:2 мл). После перемешивания в течение 3 ч между -10oC и -4oC, смесь выливают в диэтиловый эфир - раствор соли. Органическую фазу сушат (MgSO4). Фильтрация, упаривание и колоночная хроматография (силикагель, диэтиловый эфир:гексан 1:9) дают альдегид (100 мг, 72% с Rf=0.60 (диэтиловый эфир: гексан 1:1)).
Этот альдегид превращают в 6.26 как описано для 6.12 из 6.10 (выход 57%).
Rf: 0.50 (Et2O: гексан 1:1)
ПМР: (500 MHz, CDCl3): δ : 9.83(1H, dd, J=2.48, 4.12); 4.13(2H, q, J= 7.13); 2.30(4H, m); 1.93(1H, m); 1.27(3H, t, J=7.1); 0.95(3H, s); 0.80(3H, s); 0.68(3H, s); м.д.
Пример 49. Синтез 6.27
Альдегид 6.26 подвергают сочетанию с 13.1 как описано для аналога 11 из 6.12 (выход 91% с Rf 0.73, Et2) гексан: 1:1).
ПМР (360 MHz, CDCl3): δ : 6.34(1H, dd, J=11,15); 5.92(1H, d, J=11); 5.66(dt, J= 8.15); 5.20(1H, br, s); 4.87(1H, br, s); 4.39(1H, t, J=5.5); 4.185(1H, m); 4.13(2H, q, J= 7.14); 2.40(1H, dd, J=3.13); 2.30(2H, m); 2,18(1H, dd J=7,13); 1.26(3H, t, J=7.14); 0.882(9H, s); 0.866(9H, s); 0.80 (3H, s); 0.78(3H, s); 0.66(3H, s); 0.07 (12H, s) м.д.
Пример 50. Синтез 6.29
К суспензии иодида меди (I) (420 мг, 2,2 ммоль) и цинковой пыли (600 мг, 9,2 ммоль) в смеси этанол-вода 7:3(27 мл) добавляют транс-2,4-пентадиеновой кислоты этиловый эфир (270 мкл, 1.93 ммоль) и иодид 6.2(420 мг, 1,5 ммоль). Смесь обрабатывают ультразвуком в течение 1ч в атмосфере аргона при 0oC. Смесь фильтруют через целит и промывают EtOAc. Фильтрат экстрагуруют EtOAc, сушат (MgSO4) и концентрируют. Колоночная хроматография (силикагель; диэтиловый эфир: гексан 1:9--->1:5) дает 6.28 (145 мг, 35%) и выделяется 6.2(145 мг, 35%).
Rf: 0.38(диэтиловый эфир: гексан 1:1).
ПМР (500 MHz, CDCl3): δ :5.55 (2H, 2xdt, J=6,15); 4.14(2H, q, J=7.13); 3.58(1H, brd, J= 11); 3.45(1H, brd, J=11); 3.02(2H, d, J=6); 2.10(1H, m); 1.90(2H, m); 1.75 (1H, qd, J= 3,10); 1.26(3H, t, J=7.12); 0.98(3H, s); 0.89(3H, s); 0.72(3H, s) м.д.
К раствору 6.28 (40 мг, 142 мкмоль) в сухом EtOAc (8 мл), добавляют каталитическое количество Pd/C(10%), после чего смесь гидрируют в течение 3 ч (4 атм). Фильтрация через целит, добавление Et3N (200 мкл), упаривание и колоночная хроматография (диэтиловый эфир: гексан 1:4) дают 6.29 32 мг, 80%, с Rf=0.36 (диэтиловый эфир:гексан 1:1).
ПМР: (500 MHz, CDCl3): δ : 4.13(2H, q, J=7); 3.58 (1H, d, J=10); 3.46(1H, d, J= 10); 2.30(2H, t, J =7); 1.87(1H, m); 1.72(1H, qd, J=3,10); 1.26(3H, t, J=7); 0,99 (3H, s) 0,89(3H, s); 0,71(3H, s) м.д.
Пример 51. Синтез 10.2
Раствор 10.1 (3,44 г, 17,36 ммоль), этилен гликоля (5,3 мл, 95 ммоль) и пиридин п-толуолсульфонат (500 мг, 1,99 ммоль) в циклогексане (190 мл) нагревают до кипения с обратным холодильником в течение 3 ч при непрерывном отделении воды. После охлаждения до комнатной температуры, растворитель упаривают, и остаток растворяют в диэтиловом эфире (300 мл). Промывка насыщенным NaHCO3 - раствором и раствором соли, сушка (Na2SO4), упаривание растворителя и очистка колоночной хроматографией (силикагель, гексан: ацетон 9:1) и ВДЖХ (изооктан:ацетон 95:5) дают 10.2 (3,6 г, 86%).
Rf: 0.20 (гексан:ацетон 95:5).
ИК(пленка): 2950, 2881, 1740, 1436, 1280, 1189 см-1.
ПМР: (500 MHz CDCl3): δ :3.93(4H, m); 3.65(3H, s); 2.47(1H, dd, J=14.45, 3.13); 2.06(1H, tt, J= 11.15, 3.31); 2.02(1H, dd, J=14.42, 10.72); 1.63-1.52(5H, m); 1.22(1H, m); 0.911(, s), 0.898(3H, s) м.д.
Пример 52. Синтез 10.3
К перемешиваемому раствору ДА (2М гексана, 4.67 мл, 9.348 ммоль) в ТГФ (5.45 мл) при температуре +30oC добавляют раствор 10.2 (1.510 г, 6.232 ммоль) в ТГФ (21.8 мл) и продолжают перемешивать в течение 1 часа. После охлождения при температуре - 78oC добавляют смесь 5-бром-1-пентена (2.34 мл, 19.76 ммоль) и гегсаметилфосфорамида (5.5 мл, 31.16 ммоль); перемешивание продолжают в течение 3 часов. Смесь продолжают медленно перемешивать еще в течение ночи, а затем растворяют в воде и диэтиловом эфире. Экстракция водой в диэтиловом эфире, сушка (Na2SO4), упаривание раствора и очистка колоночной хроматографией (силикагель: ацетон 9:1) и ВДЖХ (изооктан:ацетон 97:3) дают 10.3 (1.81 г, 93%).
Rf: 0.24 (изооктан:ацетон 97:3).
ИК(пленка):3076; 2950; 2881; 1732; 1641; 1435; 1186 см-1.
ПМР (500 MHz, CDCl3): δ : 5.76(1H, ddt, J=17.10, 10.17, 6.65(t)); 4.99 (1H, ddt, J= 17.13, 1.82, 1.59); 4.94(1H, m); 3.92(4H, m); 3.65(3H, s); 2.49(1H, dt, J=11.61, 3.39(t)); 2.03(2H, m): 1.95(1H, dt, J=12.87, 3.52(t)); 1.65-1.59 (3H, m); 1.57(1H, m); 1.53(1H, m); 1.51-1.40(3H, m); 1. 35-1. 19(3H, m); 0.961(3H, s); 0.924(3H, s) м.д.
Пример 53. Синтез 10.6
К суспензии LiAlH4 (332,5 мг, 8.762 ммоль) в диэтиловом эфире (165 мл) добавляют по каплям при 0oC раствор 10.3(1,600 г, 5.154 ммоль) в диэтиловом эфире (62 мл); смесь перемешивают в течение 1 ч при 0oC в течение 3 ч при комнатной температуре. К энергично перемешиваемой смеси затем добавляют, очень медленно, насыщенный раствор до образования белого хлопьевидного осадка. Суспензию перемешивают в течение 1 ч, осадок фильтруют через целит и растворитель упаривают, получая 10.4 (1,453 г, 99,8%).
Rf: 0,22 (гексан: ацетон 8:2).
ПМР: (500 MHz, CDCl3): δ : 5.79(1H, ddt, J=17.10, 10.17, 6.64 (t)); 4.99(1H, ddd, J=17.14, 1.94, 1.61); 4.94 (1H, m); 3.92(4H, m); 3.55(1H, m); 3.40(1H, m); 2.03(2H, m); 1.77 (1H, dd, J= 12.82, 3.55); 1.63(1H, m); 1.57(1H, dd, J=12.72, 3.67); 1.54-1.28(12H, m); 0.978(3H, s); 0.910(3H, s) м.д.
К раствору 10.4(1.453 г, 5.145 ммоль) в дихлорметане 25.7 мл) и триэтиламине (3.9 мл, 20,58 ммоль), добавляют раствор TsCl (1,962 г, 10.29 ммоль) в дихлорметане (15,4 мл) и небольшое количество 4-диметиламинопиридина при 0oC. После перемешивания в течение 20 ч при комнатной температуре объем уменьшают до 50%, с последующей фильтрацией осадка. Полное упаривание растворителя и ВДЖХ очистка (гексан: ацетон 85:15) дает 10.5(2.126 г, 95%).
Rf:0.25(гексан:ацетон 85:15).
К раствору 10.5 (2,126 г, 4.870 ммоль) в диэтиловом эфире (250 мл) добавляют LiAlH4 (3,69 г, 97,40 ммоль) и нагреваемую до кипения суспензию перемешивают в течение 5 ч. После охлаждения до 0oC осторожно добавляют насыщенный водный раствор Na2SO4 до появления серого осадка. Небольшое количество Na2SO4 - раствора добавляют и перемешивание продолжают 3 ч. Осадок фильтруют через целит и промывают дважды, суспендируя в диэтиловом эфире, с последующей новой фильтрацией. После упаривания растворителя, остаток чистят ВДЖХ (изооктан: этилацетат 98:2), давая 10.6(1.14 г, 88%).
Rf:0.20 (изооктан:этилацетат 98:2).
ИК(пленка): 3076, 2949, 2869, 1641, 1464, 1090 см-1.
ПМР: (500 MHz. , CDCl3): δ : 5.81(1H, d, J=17,17, 10.21, 6,63 (t)); 4.99(1H, ddd, J=17.02, 2.08, 1.58); 4.93(1H, m); 3.92(4H, m); 2.02(2H, m); 1.64(2H, m); 1.54 (2H, m); 1.52-1.33(7H, m); 1.26(2H, m); 0.971 (3H, s); 0.911(3H, s); 0.907(3H, d, J=6.83); 0.895(1H, m) м.д.
Пример 54. Синтез 10.7
При -78oC озон пропускают через раствор 10.5 (565 мг, 2.121 ммоль) в дихлорметане (16,8 мл) и 2,5М раствор гидроокиси натрия в метаноле (4,24 мл) до установления устойчивого синего цвета. Реакционную смесь разбавляют диэтиловым эфиром и водой. После того как температура достигает комнатной, органическую фазу промывают раствором соли, сушат (Na2SO4) и растворитель упаривают. Очистка остатка колоночной хроматографией (гексан: ацетон 9:1) и ВДЖХ (гексан: ацетон 97:3) дают сложный эфир (405 мг, 64%). Раствор этого сложного эфира (400 мг, 1,340 ммоль) и пиридин паратолуолсульфоната (101 мг, ммоль) в ацетоне 13,4 мл) и несколько капель воды нагревают при температуре кипения с обратным холодильником в течение 3 ч. После охлаждения до комнатной температуры, растворитель упаривают и остаток растворяют в диэтиловом эфире, после чего промывают насыщенным NaHCO3 - раствором и раствором соли. Сушка (Na2SO4), упаривание растворителя и очистка колоночной хроматографией (силикагель; гексан:ацетон 9:1), и ВДЖХ (гексан:ацетон 96:4), дают 10.7 (256 мг, 75%) перед 10.6(83 мг, 21%).
Rf: 0.19(гексан:ацетон 93:7).
ИК(пленка): 1739, 1705(s); 1454; 1436; 1249 см-1.
ПМР: (500 MHz, CDCl3): δ :3.67(3H, s); 2.49(1H, dt, J=13.62(t), 6.39); 2.33-2.24(3H, m); 2.03(1H, ddd, J= 12.90; 6.24; 3.29); 1.80-1.58(4H, m); 1.56-1.47(3H, m); 1.33(1H, m); 1.11(3H, s); 1.07(3H, s); 1.02(1H, m); 0.943(3H, d, J=6.90) м.д.
Пример 55. Синтез альдегида 10.8 и 10.9
A. Раствор FORMIC (19,6 мкл, 113,6 мкмоль) в диэтиловом эфире (475 мкл) добавляют при -60oC 2,5М раствор бутиллития в гексане (52 мкл, 130,5 мкмоль), и полученный раствор перемешивают 15 минут. Раствор 10.7 (28,9 мг, 113,6 мкмоль) в диэтиловом эфире (119 мкл) добавляют затем и дают смеси нагреться до 0oC и перемешивание продолжают 1,5 ч. После перемешивания, осторожно, (вводят) 37%-водный HCl-раствор (200 мкл) смесь энергично перемешивают в течение ночи. После разбавления диэтиловым эфиром, водный слой экстрагируют диэтиловым эфиром и этилацетатом, с последующим промыванием органической фазы раствором соли и сушкой (Na2SO4). Раствор обрабатывают диазометаном, избыток разлагают добавлением силикагеля. Фильтрация, упаривание растворителя и очистка колоночной хроматографией (силикагель: гексан:ацетон 9:1) дают 10.8 и 10.9 (7.4:1 соотношение: 17 мг, 64%).
Rf: 0.35 (гексан:ацетон 9:1).
ИК(пленка): 2934; 2863; 1739; 1719; 1438; 1374; 1248; 1171 см-1.
ПМР: (500 MHz, CDCl3): δ : 9.80(1H, d, J=2.89); 3.66(3H, s); 2.29(2H, m); 1.99(1H, dt, J=12.53, 3.29 (t)); 1.86(1H, m); 1.74(2H, m); 1.63-1.44(4H, m); 1.39 (1H, m); 1.23(2H, m)); 1.17(3H, s); 1.02(1H, m) м.д.
B. Суспензию триметилсульфоний иодида (107,0 мг, 0,514 ммоль) и 2,5М раствор бутиллития (в гексане 132 мкл, 0,29 ммоль) в ТH (6,2 мл) перемешивают в течение 1ч при комнатной температуре. После охлаждения до 0oC, раствор 10.7 (52,3 мг, 0,206 ммоль) в ТH (4,1 мл) добавляют и перемешивание продолжают 2 ч при комнатной температуре. Смесь разбавляют дихлорметаном, экстрагируют водой и раствором соли, сушат (Na2SO4) и растворитель упаривают. Остаток чистят колоночной хроматографией (силикагель:гексан: ацетон 9:1) и ВДЖХ (гексан: ацетон 96:4), получая два диастереомера 10.10 (17 мг, 33% соотношение 6:4) и исходный продукт 10.7(18 мг, 3/%).
Rf: 0.17 (гексан:ацетон 96.5:3,5).
К раствору 10.7 (17 мг, 63,34 мкмоль) в диэтиловом эфире (3,2 мл) добавляют при 0oC бор трифторид диэтил эферат (40 мкл, 324,6 мкмоль). Раствор перемешивают в течение 1 ч при 0oC и 12 ч при комнатной температуре. Смесь выливают в диэтиловый эфир и промывают насыщенным NaHCO3-раствором и раствором соли. После упаривания растворителя остаток чистят колоночной хроматографией (силикагель: гексан: ацетон 9:1), получая смесь 10.8 и 10.9 (11 мг, 65%; соотношение 1,6:1). Смесь может быть разделена ВДЖХ (гексан:ацетон 96:4).
Rf: 0.38 (гексан:ацетон 9:1).
ИК(пленка): 2950; 2867; 1739; 1713; 1437 см-1
ПМР: (500 MHz, CDCl3): δ : 9,98(1H, d, J=2.60); 3.67(3H, s); 2.30(2H, m); 2.01(1H, m); 1.84(1H, m); 1.77-1.64(4H, m); 1.55-1.28 (6H, m); 1.18(3H, s); 1.01 (3H, s); 0.941(1H, m); 0.927(3H, d, J=6.88) м.д.
Пример 56. Синтез 11.13
Раствор (-)-камфорной кислоты 11.12 (2 г, 15 ммоль) в сухом ТH (45 мл) медленно добавляют к перемешиваемой суспензии LiAlH4 (1.9г, 50 ммоль) в сухом Et2O (40 мл); смесь нагревают при температуре кипения с обратным холодильником в течение 4 ч. После охлаждения до комнатной температуры, добавляют Na2SO4•10H2O. Фильтрация, упаривание растворителя и кристаллизация из EtOAc дают диол (2,28 г, 88%).
Раствор диола 0,54 г, 3,14 ммоль) в винилацетате (10 мл) обрабатывают 66 ч SAM 11 липазой (300 мг) при 37oC.
Упаривание растворителя и колоночная хроматография (силикагель, пентан: EtOAc 8:2) дают моноацетат 11.13 (0,4 г, 60%).
Rf: 0.28 (пентан: EtOAc 8:2).
ИК(пленка): 3440 (с), уширен. 2962 (s); 2874 (m); 1739(s); 1463(m); 1369(m); 1246(s); 1144(w); 1033(s); 971(w) см-1.
ПМР: (360 MHz, CDCl3): δ : 4.08(1H, dd, J=10.80); 3.98(1H, dd, J=6.10, 8.20); 3.58(1H, d, J=10.75); 3.46 (1H, d); 2.20(1H, ddt, J=8.9); 2.03(3H, s); 1.89(1H, ddd), 1.58(1H, dt); 1.35(2H, 2); 1.01(6H, s); 0,81(3H, s) м.д.
Пример 57. Синтез 11.14
К 11.13 (317 мг, 1,48 ммоль) и Et3N (1,71 мл, 14.8 ммоль) в CH2Cl2:DMSO (1: 1; 8 мл) добавляют SO3 пиридин комплекс (1,42 г, 8,88 ммоль). После перемешивания в течение 3 ч при комнатной температуре смесь выливают в H2O и экстрагируют с Et2O. Органический слой промывают 1 н. HCl и раствором соли, сушат (Na2SO4) и концентрируют. Колоночная хроматография (силикагель, пентан: EtOAc 9: 1) дают альдегид (250 мг, 80%). Этот альдегид (190 мг, 0.90 ммоль) в сухом ТHF (2 мл) добавляют к литий триэтил-4-фосфонацетату (2.83 ммоль; из фосфонацетата и LDA) в сухом THF (8 мл) при 0oC. После перемешивания в течение 12 ч при 25oC, смесь промывают раствором соли и сушат (MgSO4). Упаривание растворителя дает сырой ацетат, который подвергают сольволизу с K2CO3 в EtOH при комнатной температуре. Фильтрация, упаривание растворителя и колоночная хроматография (силикагель: пентан: EtOAc 75:25) дают 11.14 (155 мг, 65%).
Rf: 0.31 (пентан: EtOAc 8:2)
ИК (пленка): 3436 (s, уширен. ; 2965 (s); 2870(m); 1712(s); 1636(s); 1462(s); 1369(s); 1330(m); 1253(s); 1140(s); 1007 (s); 882 (m) 832 (w) см-1.
ПМР: (500 MHz, CDCl3): δ : 7.28(1H, dd. J=10.5,15.4); 6.18(1H, d, J= 15.5); 6.10(1H, dd); 5.80(1H, d); 4.19(2H, q J=7.2); 3.71(1H, dd, J=10.3, 5.8); 3.53(1H, dd, J=8.2); 2.13(1H, m); 2.00(1H, m); 1.94(1H, m); 1.48 (1H, m); 1.41(1H, m); 1.28(3H, t); 1.01(3H, s); 0.94(3H, s); 0.68(3H, s) м.д.
Пример 58. Синтез 11.16
Раствор 11.14(17 мг, 0.064 ммоль) в EtOAc (1 мл) и 5% Rh на Al2O3 (20 мг) перемешивают при комнатной температуре 2 часа в атмосфере H2. Фильтрация на силикагеле, упаривание растворителя и ВДЖХ (пентан: EtOAc 7:3) очистка дают 11.16(16 мг 90%).
Rf: 0.29 (пентан: EtOAc 75:25).
ИК(пленка): 3385 (s, уширен. ); 2941 (s); 2860(m); 1735(s); 1455 (s); 1371 (s); 1248 (s); 1152 (s); 1022 (s); 945(m); 870 (w) см-1.
ПМР: (500 MHz, CDCl3): δ :4.12(2H, q, J=7.1); 3.72(1H, dd, J=10,2, 5.4); 3.50(1H, dd, 3=8.7); 2.30 (2H, t, J=7.4); 2.07(1H), 1.88(1H); 1.60(2H, m); 1.53(1H); 1.40(1H); 1.38- 1.18(5H, m); 1.25(3H, t); 0.89(3H, s); 0.84(3H, s); 0.68(3H, s) м.д.
Пример 59. Синтез 11.18
Раствор 11.6 (10 мг, 0,037 ммоль) в сухом Et2O (1 мл) и EtMgCl (2М раствор в Et2O, 144 мкл, 289 мкмоль) перемешивают 90 мин при комнатной температуре. Затем добавляют одну каплю насыщен. раствора NH4Cl. Фильтрация на силикагеле, с пентан: EtOAc (6: 4), концентрация растворителя и ВДЖХ (пентан: EtOAc 6:4) очистка дают 11.18 (7.8 мг, 75%).
Rf: 0.20(пентан: EtOAc 8:2).
ИК(CH2Cl2): 3349 (s, уширен); 2966 (s); 2936 (s); 2874(m); 1457(m); 1388(m); 1374(m); 1264(w); 1094(m); 1034(m); 973(w); 946(w); 878(w) см-1.
ПМР: (360 MHz, CDCl3): δ : 3.72(1H, dd, J=5.4, 10.1); 3.51(1H, dd, J= 8.7); 2.09(1H, dddd, J= 9.6); 1.90(1H, dddd, J=13); 1.46(4H, q, J=7.4); 1.65-1.15(11H, m); 0.90 (3H, s); 0.86(6H, t); 0.84(3H, s); 0.69(3H, s) м.д.
Пример 60. Синтез 11.17
Из 11.16 и MeMgBr как описано для 11.18(выход 86%).
Rf: 0.37 (пентан: EtOAc 5:5).
ИК(CH2Cl2): 3354(s, уширен. ); 2937(s); 2868(m); 1466(s); 1375(s); 1204(w); 1150(w); 1090(w); 1040(m); 1008(m); 905 (w) см-1.);
ПМР: (500 MHz, CDCl3):  : 3,72(1H, dd, J=5.15, 9.85); 3.51(1H, dd, J= 9.3); 2.08(1H, m); 1.89(1H, m); 1.53(1H, m); 1,50-1.15(10H, m); 1.21(6H, s); 0.90(3H, s) 0.85(3H, s); 0.69(3H, s) м.д.
: 3,72(1H, dd, J=5.15, 9.85); 3.51(1H, dd, J= 9.3); 2.08(1H, m); 1.89(1H, m); 1.53(1H, m); 1,50-1.15(10H, m); 1.21(6H, s); 0.90(3H, s) 0.85(3H, s); 0.69(3H, s) м.д.
Пример 61. Синтез 11.19
К раствору 11.17 (12 мг, 47 мкмоль), N-метил морфолин оксида (8,5 мг, 72 мкмоль) и активированных молекулярных сит  24 мг) в CH2Cl2 (400 мкл) добавляют тетра-н-пропил аммоний перрутенат (0.8 мг, 2.35 мкмоль). После перемешивания в течение 2 ч при комн. темп. смесь фильтруют через силикагель. Остаток промывают пентан: EtOAc 5:5. Упаривание растворителя и ВДЖХ (пентан: EtOAc 85:15) очистка дают 11.19 (8; 3 мг, 70%).
24 мг) в CH2Cl2 (400 мкл) добавляют тетра-н-пропил аммоний перрутенат (0.8 мг, 2.35 мкмоль). После перемешивания в течение 2 ч при комн. темп. смесь фильтруют через силикагель. Остаток промывают пентан: EtOAc 5:5. Упаривание растворителя и ВДЖХ (пентан: EtOAc 85:15) очистка дают 11.19 (8; 3 мг, 70%).
Rf: 0.38 (пентан:EtOAc 8:2).
ИК(CH2Cl2): 3421(s, уширен. ); 2966 (s); 2862 (m); 2718(w); 1713(s); 1466(s); 1377(s); 1133(w); 905(w) см-1.
ПМР: (500 MHz, CDCl3): δ : 9.76(1H, d, J=2.3); 2.70(1H); 2.04(1H), 1.75-1.21(1H, m); 1.21(6H, s); 1.09 (3H, s); 0.87(3H, s); 0.84(3H,.s) м.д.
Пример 62. Синтез 11.20
Из 11.18 как описано для 11.19 из 11.17 (выход 78%).
Rf: 0.15 (гексан: этилацетат 85:15).
ИК(пленка): 3442(s, уширен. ); 2938(s); 2871(w); 2720(w); 1717(s); 1457(s); 1377(s); 1262(m); 1092(m), 1031(s); 947(w); 877(w) см-1.
ПМР: (500 MHz, CDCl3): δ : 9.76(1H, d, J=2.2); 2.70(1H, ddd); 2.06(1H, m); 1.74-1.59(2H, m); 1.51(1H, m); 1.46(4H, q, J=7.5); 1.41(1H, m); 1.39-1.20(1H, m); 1.09(3H, s); 0.87(3H, s); 0.86(6H, t); 0.84(3H, s) м.д.
Пример 63. Синтез 11.23
К перемешиваемому раствору LDA (2.11 ммоль) в THF (2 мл) добавляют при -78oC раствор 11.22 (0,58 г, 1.85 ммоль) в THF (0,5 мл) за 10 мин. Полученный раствор медленно нагревают до комнатной темп. После перемешивания при комн. темп. 2 ч реакционную смесь охлаждают до -78oC и добавляют по каплям PhNTf2 (0,71 г, 2.0 ммоль) в ТH (2,5 мл). Раствор нагревают медленно до 0oC и перемешивают в течение ночи. Добавляют воду. Экстракция пентаном, сушка (MgO4) и упаривание растворителя и очистка колоночной хроматографией (силикагель, гексан: EtOAc 10:1) дают 11.23(0.47 г, 65%).
Rf: 0.30(гексан: EtOAc 10:1).
ИК(пленка): 3750; 2973; 1417; 1209; 114 см-1.
ПМР: (500 MHz, CDCl3): δ : 5.60(1H, dd, J=6.86, 3.50); 4.90(1H, q, J= 5.3); 3.54(1H, m); 3.48(1H, m); 2.5(1H, m); 2.3(2H, m); 2.0(2H, m); 1.8(1H, m); 1.5-1.4(10H, m); 1.33 (3H, d, J= 5.28); 1.21(3H, s); 1.19(3H, s); 1.18(3H, t, 3=7.05); 1.05(1H, m); 0.94(3H, d, J=6.52); 0.76(3H, s) м.д.
Пример 64. Синтез 11.24
Через раствор 11.23 (400 мг, 0.83 ммоль) и твердого NaHCO (112 мг, 1,3 ммоль) в MeOH (200 мл) пропускают ток озона (18 ммоль/ч), вырабатываемого WELSBACH газогенератором, при -78oC за период 30 мин, до приобретения раствором темно-синего цвета. Затем через раствор пропускают обильный ток азота, пока раствор не обесцветится. Добавляют NaBH4 (1,0 г, 26 ммоль) к смеси при - 78oC, через 15 мин добавляют другую порцию (1,0 г, 26 ммоль). Смеси дают медленно нагреться до комн. темп. и перемешивают 18 ч. Добавляют боргидрид натрия (2.0 г, 52 ммоль) при -20oC и реак. смесь перемешивают 2 ч и затем медленно нагревают до комн. темп. MeOH упаривают и добавляют насыщенный раствор NH4Cl. Экстракция CH2Cl2, сушка (MgSO4) и упаривание растворителя дают 11.24(300 мг, 91%).
Rf: 0.23(гексан: EtOAc 2:1).
ИК(пленка): 3397, 2970; 2348; 1713; 1416; 1209; 114, 904 см-1.
ПМР: (500 MHz, CDCl3: δ : 4.90(1H, q, J=5.3); 3.65(3H, s); 3.64(2H, m); 3.54(1H, m); 3.48(1H, m); 2.70(1H, t, J=9.36); 2.0(1H, m); 1.80(1H, m); 1.70 (2H, m); 1.5- 1.4(7H, m); 1.30(6H, m); 1.20(10H, m); 1.10(1H, m); 1.0(3H, t, J=6.43); 0.80(3H, s) м.д.
Пример 65. Синтез 11.26
К раствору 11.24 (90 мг, 0.2 ммоль) добавляют раствор TSCl (167 мг, 0.87 ммоль) в пиридине (2,5 мл). Смесь перемешивают при - 4oC 18 ч. Добавляют раствор ацетата аммония, экстрагируют CH2Cl2, сушат (MgSO4) и растворитель упаривают, получая неочищенное масло, которое используют в следующей реакции. К LiAlH4 (160 мг, 4,2 ммоль) в сухом THF (5 мл) добавляют 11.25 (460 мг, 0,11 ммоль) в сухом THF (5 мл) при 0oC. Смесь нагревают при кипении 36 ч, затем осторожно добавляют 10% HCl раствор до нейтрализации. Упаривание растворителя и ВДЖХ очистка(гексан: EtOAc 1:1) дают 11.26(31 мг, 61%).
Rf: 0,29 (гексан: Et OAc 1:1).
ИК(пленка): 3855; 2957 см-1.
ПМР 5 (500 MHz, CDCl3): δ : 3.72(1H, dd, J=10.20, 4.70); 3.41(1H, dd, J= 10,20, 9.07); 1.92(1H, m); 1.70(2H, m); 1.60(1H, sbr); 1.56 (1H, m); 1.5-1.3(12H, m); 1.25(1H, m); 1.20(6H, s); 1.04(1H, m); 0.95(3H, d, J=6.71); 0,85(3H, t, J=7.15); 0,67(3H, s) м.д.
Пример 66. Синтез 11.27
К смеси 11.26(30 мг, 100 мкмоль), N-метилморфолин оксид (1, 6 6 экв., 166 мкмоль, 19 мг) и молекулярных сит (54 мг, тип  2а 3 мк) в CH2Cl2 (1,5 мл), добавляют тетрапропиламмоний перрутенат (5 мкмоль, 1,8 мг). После перемешивания в течение 1 ч черносерую суспензию чистят непосредственно колоночной хроматографией (Et2O: гексан 1: 4--->1:1), получая 11.27(15 мг, 50%).
2а 3 мк) в CH2Cl2 (1,5 мл), добавляют тетрапропиламмоний перрутенат (5 мкмоль, 1,8 мг). После перемешивания в течение 1 ч черносерую суспензию чистят непосредственно колоночной хроматографией (Et2O: гексан 1: 4--->1:1), получая 11.27(15 мг, 50%).
Rf: 0.33 (Et2O: гексан 1:1);
ПМР: (500 MHz, CDCl3): δ :9,69(1H, d, J=3.31); 2.58(td, J=3,3, 9.1); 1.98(1H, m); 1.87(1H, m); 1.22(6H, s); 0.96(3H, d, J =6.68); 0.92(3H, t, J= 7.2); 0.87(3H, s) м.д.
Пример 67. Синтез 12.2
Смесь 12.1 (75 мг, 0,268 ммоль) и метилат натрия (каталитическое количество) в супер сухом метаноле (1,5 мл) перемешивают 24 ч при комн. темп. в атмосфере Ar. Затем смесь фильтруют через силикагель, фильтрат концентрируют в вакууме и разделяют ВДЖХ (силикагель; гексан; этилацетат 75:25), получая цис-конденсированный кетон (55 мг, 73%) перед транс-изомером (18 мг).
Цис-кетон (50 мг, 0,179 ммоль) и 1-(триметилсилил)-имидазол (104 мкл, 0,716 ммоль) в дихлорметане (1,8 мл) перемешивают 3 ч при комнатной температуре.
После удаления растворителя в вакууме, добавляют диэтиловый эфир и полученный осадок через небольшой слой силикагеля. Концентрация фильтрата дает 73 мг сырого продукта, который чистят ВДЖХ (силикагель; гексан: этилацетат 95:5), получая защищенный спирт 12.2 (50 мг, 79%).
Rf: 0.58(гексан: этилацетат 85:15).
ИК(пленка): см-1, 2958(s); 1710(s); 1464(m), 1379(m) 1320(w), 1249(s); 1156(m) чс-1.
ПМP: (500 MHz, CDCl3): δ :2.31 (3H, m); 2.15(1H, m); 1.95-1.70(5H, m); 1.57(1H, m); 1.43-1.25(8H, m); 1.18 (6(+1)H, s); 1.04(3H, s); P,91(3H, d); 0.10(9H, s) м.д.
Пример 68. Синтез 12.5
Суспензию NaH (956 мг, 23,9 ммоль) в безводн. диметилсульфоксиде (30 мл) перемешивают при 65oC в атмосфере азота, 1,5 ч, после чего медленно добавляют 3-этоксиэтил-3-метил-1-бутил (3,68 г,: 23,9 ммоль). Раствор 12.3 (1,77 г, 4,84 ммоль) в сухом диметилсульфоксиде (10 мл) добавляют затем и перемешивание продолжают 0.5 ч при комн. темп. Реакционную смесь выливают в охлажденный льдом насыщенный раствор NH4Cl. Водную фазу экстрагируют диэтиловым эфиром и объединенные экстракты промывают раствором соли, сушат (MgSO4) и упаривают при пониженном давлении. Остаток чистят колоночной хроматографией (силикагель; гексан: этилацетат 8:2), получая 12.4 (1,24 г, 67% выход) продукта. Смесь этого спирта и пиридиндихромата (1,8 г: 4,8 ммоль) в дихлорметане (10 мл) перемешивают 2 ч при комн. темп. Полученный кетон непосредственно чистят колоночной хроматографией (гексан: этилацетат 8:2); 467 мг (84%) получают.
Раствор этого кетона (369 мг; 1,06 ммоль) и каталитическое количество метоксида натрия в сухом метаноле (10 мл) перемешивают в атмосфере азота при комн. темпер. 12 ч. Реакционную смесь фильтруют через силикагель, используя метанол в качестве элюента. Упаривание при пониженном давлении дает остаток, который чистят колоночной хроматографией (этилацетат: гексан 2:8). Чистый 12.5 (149 мг; 65%) получают разделением ВДЖХ (этилацет: гексан 2:8).
Rf: 0.48 (гексан:этилацетат 8:2).
ИК(пленка): 3398, 2979, 2934, 2875, 2291, 2226, 1708, 1464, 1443, 1378, 1360, 1334, 1253, 1160, 1124, 1081, 1053 см-1.
ПМР: (200 MHz, CDCl3): δ : 1.18 (3H,); 3.67(1H, dq, J=9.11, 7.05 Hz); 3.49(1H, dq, J=9.11, 7.10 Hz); 5.68(1H, q, 5.24 Hz); 1.32 (3H, d, 5.24 Hz); 1.49 (3H, s); 1.43 (3H, s); 2.23(2H, m); 1.06 (3H, d, 6.48 Hz); 1.06(3H, s), 2.44(1H, m) м.д.
Пример 69. Синтез 12.6
Раствор 12.4(1.035 г; 3,1 ммоль) и п-толуол сульфоновой кислоты (295 мг, 1,55 ммоль) в толуоле (50 мл) перемешивают при 60oC 1 ч. Затем реакционную смесь выливают в насыщенный раствор NaHCO3, экстрагируют диэтиловым эфиром, промывают водой и сушат (MgSO4). Колоночная хроматография (силикагель; гексан- этилацетат 85:15) остатка, полученного после фильтрации и упаривание растворителя дают 12.6 (567 мг, 70%).
Rf: 0.45(гексан: этилацетат 8:2).
ИК(пленка): 3426, 2932, 2868, 2223, 1615, 1457, 1372, 1165 см-1.
ПМР: (500 MHz, CDCl3): δ : 5.14 (1H, s, J=28.78 Hz); 5.20 (1H, s); 1.88 (3H, s); 2.34 (1H, d, J=3.41 Hz); 2.38(1H, d, Jdd=3.45 Hz, Jcc' =16.76 Hz); 1.06(3H, d, J=6.56 Hz); 0.93 (3H, s); 4.08 (1H, m) м.д.
Пример 70. Синтез 12.7
Смесь 126 (300 мг,: 1,15 ммоль), 3-хлор-пероксибензойной кислоты (497 мг, 80%; 2.31 ммоль и Na2HPO4 (163 мг; 1.15 ммоль) в сухом ТH (30 мл) перемешивают при 0oC в атмосфере азота. После 3 дней перемешивания, смесь разбавляют этилацетатом: гексаном (1:1), промывают 10% Na2SO3 раствором, насыщенным Na2CO3 раствором и раствором соли и сушат (MgSO4). Фильтрация, удаление растворителей при пониженном давлении и колоночная хроматография (силикагель:этилацетат: гексан 2:8) дают эпоксид (90 мг; 66%).
Смесь этого продукта (50 мг: 0.181 ммоль) и пиридин дихромата (203 мг; 0,54 ммоль) в дихлорметане (4 мл) перемешивают 2 ч при комн. темп. Реакционную смесь чистят колоночной хроматографией (гексан: этилацетат 8:2), получая транс-конденсированный кетон (36 мг.: 69%).
Раствор кетона (80 мг: 0,328 ммоль) и 1,8-диазабицикло[5.4.0] ундек-7-ена (76 мг: 0.500 ммоль) в дихлорметане (2 мл) перемешивают при комнатной температуре 3 дня. Реакционную смесь выливают в насыщенный NH4Cl раствор, экстрагируют диэтиловым эфиром, промывают насыщенным NaHCO3 и раствором соли и сушат (MgSO4). Остаток, после фильтрации и удаления растворителя, чистят ВДЖХ (этилацетат: гексан 15: 85) и получают 12.7 (8 мг, 27%) перед транс-конденсированным изомером (30 мг).
Rf: 0.24 (этилацетат: гексан 15:85).
ИК (пленка): 3410, 2952, 2239, 1712, 1460, 1379, 1139, 1307, 1271, 1232, 1152, 1068 см-1.
ПМР: (200 MHz, CDCl3): δ : 1.52(3H, s): 2.95 (1H, d, 4.47 Hz); 2.71(1H, d, 5.65 Hz); 1.05(3H, s); 1.05(3H, d, 6.5 Hz) м.д.
Пример 71. Синтез 12.10
Озонолиз витамина D2 (2г, 5.05 ммоль) в дихлорметан: метанол 50 мл, (1: 1) выполняют при -78oC. Последующая обработка диметилсульфидом (8 мл) при -78oC в течение 30 мин и упаривание растворителя дают сырой 12.8. Его растворяют в тетрагидрофуране (30 мл) и добавляют при перемешивании 5% HCl (10 мл). Перемешивание продолжают при 30oC в атмосфере азота в течение 36 ч. Упаривание растворителя, добавление диэтилового эфира, промывка насыщенным раствором NaHCO3, сушка (MgSO4) и концентрация в вакууме дают остаток. Флэш хроматография (силикагель; гексан-этилацетат 8:2) дает белый кристаллический 12.9 вместе с 20-S - изомером 736 мг. 70%; 2,5:1 соотношение). Эту смесь (200 мг; 0,96 ммоль) в метаноле (25 мл) обрабатывают NaBH4 (73 мг. 1,92 ммоль) при комн. темп., в атмосфере N2 20 мин. Добавляют HCl раствор (10%, 8 мл), после перемешивания в течение 10 мин метанол упаривают. Добавляют диэтиловый эфир, промывают насыщенным NaHCO3, сушат (MgSO4), упаривают и разделяют C-20 эпимеры (ВДЖХ; гексан:этилацетат:метанол; 100:100:1,5) получают 12.10(140 мг, 69%).
Rf: 0.45(гексан:этилацетат:метанол 5:4:1).
ИК(пленка):2795, 2703, 1719, 1700, 1380 см-1.
ПМР: (360 MHz, CDCl3): δ : 0,86(3H, d, J=6.75 Hz) 0.89(s) и 1.02(s) (3H), 3.30-3.94(3H, m) м.д.
Пример 72. Синтез 12.12
Раствор 12.10 (170 мг, 0,8 ммоль) и TsCl (229 мг, 1.20 ммоль) в сухом пиридине (10 мл) выдерживают при 0oC 13 ч. Затем смесь выливают в ледяную воду, экстракция, промывка (NaHCO3 насыщен. раствор), сушка (MgSO4), упаривание растворителя и флэш хроматография (силикагель: гексан: этилацетат 6: 4) дают 12.11 (163 мг, 56%). Реакция 12.11 (160 мг, 0,44 ммоль) с анионом 3-этокси-этил-1-бутина (2,2 мл) как описано для 12.4, в примере 61, дает после обработки и флэш хроматографии (силикагель; гексан: этилацетат 8:2) 86 мг (56%) продукта. Смесь этого спирта (56 мг, 0,16 ммоль) и пиридин дихромата (241 мг, 0,64 ммоль) в сухом дихлорметане (7 мл) перемешивают при комн. темп. в атмосфере азота в течение 1 ч. Прямая флэш хроматография (силикагель; гексан: этилацетат 8:2) дает 12.12 (39 мг, 70%).
Rf:0.21 (гексан: этилацетат 9:1).
ИК(пленка: 2231, 1708 см-1.
ПМР:(500 MHz, CDCl3): δ : 0,95 (d, J=6,56 Hz) и 0,96(d, J=6,67 Hz) (3H), 1.03(3H, s), 1.18(3H, m), 1.31 (3H, d, J=5,31 Hz), 1.42(3H, s), 1.478(s) и 1.482(s) (3H), 3.43-3.69(2H, m), 5.08(1H, m) м.д.
Пример 73. Синтез 12.13
Из 12.10 и 3-(этокси)-этокси-этил-1-пентина как описано для 12.12.
Rf: 0.35 (гексан: EtOAc - 9:1).
ИК(пленка): 2234; 1708 см-1.
ПМР: (500 MHz, CDCl3): δ : 5.10(1H,q, J=5.21); 3,68(1H, m); 3.48(1H, m); 1.31(3H, d, J=5.21); 1.17 (3H, dd, J=7.03, 7.03); 1.04(3H, s); 0.97(3H, d, J=6.63); 0.94(6H, m) м.д.
Пример 74. Синтез 12.14
Смесь 12.12(19 г, 0,055 ммоль), 5% Rh/Al2O3 (8 мг) и EtOAc (2,5 мл) перемешивают при комн. темп. в атмосфере H2 (атмосферное давление) в течение 1 ч. Смесь фильтруют через короткую силикагелевую колонку (гексан: EtOAc 7:3). ВДЖХ очистка (гексан: 9:1) дает 12.14 (17 мг, 89%).
Rf: 0.50 (гексан: EtOAc 8:2).
ИК (пленка): 1708 см-1.
ПМР: (500 MHz, CDCl3): δ : 4.86(1H,q, J=5.33); 3.50(2H, m); 1.26(3H, d, J= 5.33); 1.19(3H, dd, J=6.90, 6.90); 1.17(3H, s); 1.02(3H, s); 0.82(3H, d, J=6.67) м.д.
Пример 75. Синтез 12.15
К раствору LDA (0,50 ммоль) в сухом THF (3,5 мл) при -78oC в атмосфере N2 добавляют по каплям триэтил 4-фосфоно-кротонат (90%, 124 мкл, 0,50 ммоль). Перемешивание продолжают при -78oC 30 мин. Добавляют по каплям раствор 12.9(88 мг, 0,42 ммоль) в сухом THF (1,5 мл). Реакционную смесь перемешивают при -78oC в течение 2 ч. и затем дают нагреться до комнатной температуры за 1 час. Смесь разбавляют диэтиловым эфиром, промывают раствором соли, сушат (MgSO4) и упаривают. ВДЖХ очистка (гексан: EtOAc 88:12) дает 12.15 (110 мг, 85%).
Rf: 0.41 (гексан: EtOAc 8:2).
ИК(пленка): 1708, 1639, 1616, 1004 см-1.
ПМР: (500 MHz, CDCl3): δ : 7.23(1H, dd, J=15.39, 11.00); 6.12(1H, dd, J= 15.19, 10.99); 5.93(1H, dd, J=15.19, 9.66); 5.80(1H, d, J=15.39); 4.20(2H, m); 1.29(3H, dd, J=7.08, 7.08); 0.96(3H, d, J=6.65); 1.01(3H, s) м.д.
Пример 76. Синтез 12.14
Смесь 12.13 (67 мг, 0,23 ммоль), 5% Rh/Al2O3 (30 мг и EtOAc (4 мл) перемешивают в атмосфере H2 (атмосферное давление) при комн. темп. в течение 1,5 ч. Затем смесь фильтруют через короткую силикагелевую колонку (гексан: EtOAc 1:1). ВДЖХ очистка (гексан: EtOAc 88:12) дает 12.14 (63 мг, 93%).
Rf: 0.49 (гексан: EtOAc -8:2).
ИК(пленка): 1735, 1707 см-1.
ПМР: (500 MHz, CDCl3): δ : 4,12(2H, q, J=7.20 Hz); 1.25(3H, t, J=7.20 Hz), 1.02(3H, s); 0,82 (3H, d, J=6,65) м.д.
Пример 77. Синтез 12.15
Сочетание по Horner-Wittig 12.14(60 мг, 0.19 ммоль) с 13.2 (162 мг, 0,28 ммоль), с использованием н-BuLi (1,6 М раствор в гексане, 175 мкл, 0,28 ммоль) как основания, выполняют как описано для 10. Флэш хроматография (гексан: EtOAc 1: 1) и ВДЖХ разделение (гексан: EtOAc 18:1) дают (80 мг, 62%).
Rf: 0.64 (гексан: EtOAc:1).
Пример 78. Синтез 12.17
Из 12.8 по реакции Horner-Wittig как описано для 12.15 из 12.9 с последующей индуцируемой NaOEt-EtOH эпимеризацией при комн. темп. в течение 21 ч (общий выход 48%).
Rf: 0.28(н-пентан:ацетон 94:6).
ИК(пленка): 2957(s); 1713(s); 1641(s); 1463(m); 1137 (s) см-1
ПМР: (500 MHz, CDCl3): δ : 7.21 (1H, dd, J=10.8, 15.3); 6.10(1H, dd, J= 10.8, 15.1); 5.99(1H, dd, J=8,8, 15.1); 5.77(1H, d, J=15.3); 4.19(2H, q, J= 7.1); 2.32 (4H, m); 2.15(1H, m); 1.92(1H, m); 1.84(1H, m); 1.75(3H, m): 1.60(2H, m); 1,44(1H, m); 1.35(1H, m); 1.29(3H, t, J=7.1), 1.05(3H, d, J= 6,5); 1.04(3H, s) м.д.
Пример 79. Синтез 12.18
Из 12.17 как описано для 12.16 из 12.15 (выход: 88%).
Rf:0.35(H-пентан:ацетон 96:4).
ИК(пленка): 2954(s); 1733(s); 1713(s); 1380(s); 1159(s); 1097 (s) см-1.
ПМР: (500 MHz, CDCl3): δ : 4,12(2H, q, J=7.1); 2.31(5H, m); 2.15(1H, m); 1.91(3H, m); 1.75(2H, m); 1.56(3H, m); 1.43-1.24(6H, m); 1.25(3H, t, J=7.1); 1.19(1H, m); 1.03(3H, s); 0.89(3H, d, J=6.6) м.д.
Пример 80. Синтез аналога 1
Как описано для 11, исходя из 13.1 и 10-гидрокси-10-метилдеканала.
Rf: 0.40 (дихлорметан: метанол 9:1).
ИК(пленка): 3374(s); 3025(w), 2969 (s), 2929 (s); 1634(m); 1466(w); 1432(w); 1366 (w), 1306(w); 1266(w); 1218(w); 1149(w); 1054(w); 975(w); 958(w), 907(w); 800(w); 737(w) см-1.
ПМР: (360 MHz, CDCl3): δ : 6.37(1H, dd J=11.15); 6.03(1H, d, J=11 Hz); 5.71(1H, dt, J=15 Hz); 5.31 (1H, d, J= 7 Hz); 4.99(1H, d); 4.42(1H, t, J=5.5 Hz); 4.20(1H, m); 2.58(1H, dd, J=13 Hz); 2.54(1H, dd, J=4 Hz); 2.06(2H, dd, J=7Hz); 1,96 (2H, t, J=5,5Hz); 1,85-1,65 (3H, m); 1.50-1.15(18H, m).
Пример 81. Синтез аналога 2
Из 1,8d как описано для 13.
Rf: 0.37 (дихлорметан: метанол 88:12).
ИК(пленка): 3368; 1610; 1374; 1049 см-1.
ПМР: (500 MHz, CDCl3): δ : 6.31 (1H, d, J=11.2); 6.06(1H, d, J=1172); 4.15(1H, ds); 4.13-4.04(2H, m); 2.75-2.64(2H, m); 2.49(1H, dd, J=13.1, 3.8); 2.40(1H, m); 2.28(1H, dd, J=13,8, 7.9); 2.21(1H, dd, J=13.5, 7.1); 2.15-0.70(22H,); 1,21(6H, s), 0.9(3H, dd, J=6.73) м.д.
Пример 82. Синтез аналога 3
Из 1.11d как описано для 11.
Rf: 0.32(ацетон:гексан 4:6).
ИК(пленка): 3356; 1441; 1378; 1215; 1144 см-1.
ПМР: (500 MHz, CDCl3): δ 6.37 (1H, d, J=11.4); 6.18(1H, d, J=11,4); 5.32(1H, bs); 5.02(1H, s); 4.43(1H, m); 4.21(1H, m); 2.87(1H, dm, J=13.6); 2.61(1H, dd, J=3.4 Hz, J=13.2 Hz); 2.30(1H, dd, J=7.3, 13.2); 2.01-1.93(2H, m); 1.25(6H, s, 20% 20-эпи), 1.21(6H, s), 0.87(3H, d, J= 6.7); 0.76(3H, d, J=6,6, 20% 20-эпи) м.д.
Пример 83. Синтез аналога 4
К раствору 13.1 (76 мг, 0,13 ммоль) в THF (2 мл) добавляют по каплям н-бутиллитий (52 мкл, 0.13 ммоль, 2,5М раствор в гексане) при -78oC в атмосфере азота. Образовавшийся темно-красный раствор перемешивают в течение 1 ч при -78oC, после чего раствор 2.5 (25 мг, 0,065 ммоль) в THF (1 мл) добавляют. Красный раствор перемешивают при -78oC в течение 1 ч и затем нагревают до комнатной температуры. Реакционную смесь сразу же фильтруют через силикагелевую колонку (EtOAc: гексан 1:30) и сырой продукт (74 мг) чистят затем ВДЖХ (EtOAc:гексан 1:200), получая 45,0 мг (92%) продукта сочетания.
Раствор продукта сочетания (45,0 мг, 0,06 ммоль) и TBAF (1,27 мл, 1,27 ммоль, 1М раствор в THF) в THF (3 мл) перемешивают при комнатной температуре (25-30oC) 39 ч. Реакционную смесь сразу же фильтруют через колонку с силикагелем (MeOH: CH2Cl2 1: 20) и сырой продукт (59 мг) разделяют ВДЖХ (MeOH: CH2Cl2 1: 16), получая 4(19,1 мг, 78%). Получают также продукт (8,3 мг), который не идентифицирован.
Rf: 0,21(дизлорметан: метанол 1:20).
ИК(пленка): 3378(s); 2954(s); 1643(w); 1453; 1383(s); 1264(s); 1142(w); 1057(s); 742(s) см-1.
ПМР: (500 MHz, CDCl3): δ : 6.32(1H, d, J=11.2 Hz); 6.05(1H, d, J=11.2 Hz); 5.32(1H, m); 4.98(1H, m); 4.45 (1H, m); 4.20(1H, m); 3.30(1H, m); 2.60(1H, dd, J= 3.9, 13.2 Hz); 2.42(1H, m); 2.30(1H, dd, J=7.4, 13.2 Hz); 2.24(2H, m); 1.96(2H, m); 1.79(1H, d, J=13.1, Hz); 1.65(6H, m), 1.47(2H, m); 1.25(1H, m), 0.90(3H, d, J=6.6 Hz); 0.89(6H, dd, J=6,8 Hz); 0,75(3H, s); 0.74(3H, d, J= 7.2 Hz).
Пример 84. Синтез аналога 5 и провитамина 56
К раствору 13.1 (110 мг, 0,188 ммоль) в THF(3 мл) добавляют по каплям н-бутиллитий (76 мкл, 0,188 ммоль, 2,5 М раствор в гексане) при -78oC в атмосфере азота. Полученный темно-красный раствор перемешивают 1ч при -78oC, после чего добавляют раствор 2.7 (36 мг, 0,094 ммоль) в THF(1 мл). Красный раствор перемешивают при -78oC в течение 1 ч и затем медленно нагревают до комнатной температуры. Реакционную смесь сразу же фильтруют через колонку с силикагелем (EtOAc: гексан 1:20) и сырой продукт (117 мг) чистят далее ВДЖХ (EtOAc: гексан 1:200), получая 66,0 мг (93%) продукта сочетания.
Раствор продукта сочетания (65,0 мг, 0,087 ммоль) и TBAF (2,61 мл, 2,61 ммоль, 1М раствор THF) в THF (8 мл) перемешивают при 30-40oC в течение 40 часов. Реакционную смесь сразу же фильтруют через колонку с силикагелем (MeOH: CH2Cl2 1: 20) и сырой продукт (82 мг) снова разделяют ВДЖХ (MeOH: CH2Cl2 1:20), что дает 5 (23,3 мг, 66%) и 56(4,2 мг, 12%).
5 : Rf: 0.15 (дихлорметан:метанол 1:20).
IR (пленка): 3385 (s). 2956 (s): 1642 (w): 1450, 1383 (s); 1056. 909 (s); 734 (m) cm-1.
1H NMR: (500 MHz, CDCl3): δ : 6.32 (1H, d, J =11.2 Hz); 6.06 (1H, d, J = 11.2 Hz); 5.31 (1H, d, J = 2.2 Hz); 4.99 (1H, d, J = 2.2 Hz); 4.42 (1H, m); 4.21 (1H, m); 3.31 (1H, m); 2.60 (1H, dd, J = 3.9, 13.2 Hz); 2.42 (1H, m); 2.29 (1H, dd, J = 7.4. 13.1 Hz), 2.02 (4H, m); 1.80 (1H, d, J = 13.0 Hz); 1.65 (5H, m); 1.45 (4H, m); 0.90 (3H, d, J = 6.6 Hz); 0.89 (6H, dd, J = 6.6 Hz); 0.75 (3H, s); 0.74 (3H, d, J = 7.2 Hz).
56: Rf: 0.15 (дихлорметан:метанол 20:1),
IR (пленка): 3384 (s), 2956, 2863 (s), 1640 (w), 1453, 1383 (s), 1056, 90B(s) cm-1.
ПМР: (500 MHz, CDCl3): δ :5.89(1H, d, J=12.2 Hz); 5.68 (1H, d, J =13.0 Hz) , 5.65(1H, br s); 4.18(1H, br s); 4.10(1H, m); 3.33(1H, m); 2.43(1H, dd, J= 4,5, 16.8 Hz); 2.10(2H, m); 2.00(2H, m); 1,72(3H, s); 1.71 (12H, m); 1.60(4H, m); 1.45(3H, m); 1.33(2H, m); 0.92(3H, d, J=6.8 Hz); 0.90(3H, d, J= 6,6 Hz); 0,88(3H, d, J =5,9H); 0,80 (3H, s); 0,77 (3H, d, J=7,2 Hz).
MS (m/z): 404(5), 387(6), 386(4), 229(30), 211(30), 95(40).
Пример 85. Синтез 6
К раствору 13.2 (57 мг, 0,099 мноль) в THF (2 мл) добавляют по каплям н-бутиллитий (40 мкл, 0,099 ммоль, 2,48 М раствор в гексане) при -78oC в атмосфере азота. Образовавшийся темно-красный раствор перемешивают 1 час при -78oC, после чего добавляют раствор 2.7(19 мг, 0,049 ммоль) в THF (1,5 мл). Красный раствор перемешивают при -78oC в течение 1 часа и затем медленно нагревают до комнатной температуры. Реакционную смесь немедленно фильтруют через колонку с силикагелем (EtOAc: гексан 1:20) и сырой продукт (44 мг чистят далее ВДЖХ (EtOAc: гексан 1:140), получая 32,0 мг (88%) продукта сочетания.
Раствор продукта сочетания (32,0 мг, 0,043 ммоль) и TBAF (1,96 мл, 1,96 ммоль, 1М раствор в THF) в THF (4 мл) перемешивают при 30 - 45oC 40 часов. Реакционную смесь немедленно фильтруют через колонку с силикагелем (MeOH: CH2Cl2 1:20) и сырой продукт (17 мг) снова разделяют ВДЖХ(MeOH:CH2Cl2 1:20), получая 4/1 смесь E- и Z-изомеров (15,5 мг, 91%).
Эту смесь снова разделяют на специальной ВДЖХ колонке (RSiLCN 10 микрон, 5,0 мг/500 мкл/доза) с элюентом гексан: и -PrOH:CH3CN 89:10:1 что дает 13.2 мг аналога 6 (E-изомер) и 2 мг Z-изомера. Разделение затруднено и оба продукта разделяют несколько раз.
ИК(пленка): 3380(s); 2957(s); 1616(w); 1452; 1381(m);1047(s); 736(m) см-1.
ПМР (CDCl3): δ (6.26(1H, d, J=11.2), 5.94(1H, d, J= 11.2); 4.08(2H, m); 3.33(1H, m); 2.64(1H, dd, J=3.8,13.3); 2.48(1H, dd, J=3.7, 13.3); 2.43(1H, m); 2.29(1H, dd, J= 7.7, 13.3); 2.18(1H, dd, J=6.7, 13.3), 2.07(1H, d, J= 13.0); 2.00(1H, m), 1.88(2H, m), 1.86(1H, d, J=13.1); 0.93(3H, d, J=6.3); 0.91(3H, d, J =6.4); 0.89 (3H, d, J=6.5); 0.79 (3H, d, J=7,8); 0,87 (3H, s) м.д.
MS (m/z); 392 M+, 5); 374(8); 308(10); 235(50); 217(40), 55(100).
Пример 86. Синтез аналога 7
Как описано для 11.
ИК(пленка): 3380(s); 2939(s); 1625(w); 1452; 1383 (m); 909 (m) см-1.
ПМР (CDCl3): δ 6.32(1H, d, J=11.2); 6.04(1H, d, J=11.2); 5.32(1H, t, J= 1.4); 4.99(1H, m); 4.43(1H, m); 4.22(1H, m); 2.60(1, dd, J=3,9, 13.2); 2.43(1H, dt, J= 13.7, 5.1); 2.29(1H, dd, J=7.4, 13.2); 2.03(1H, m); 2.02(1H, d, J= 13.1); 1.96(2H, m); 1.80(1H, d, J=13.0); 1.21(6H, s); 0.89(3H, d, J= 6.8); 0.75(3H, s); 0,73(3H, s, J =6.3) м,д.
MS (m/z): 404 (M+, 1%).
Пример 87. Синтез аналога 8
Как описано для 13.
УФ: λmax =249, 5 нм.
ИК(пленка): 3382(s), 2935(s), 1615(w), 1454, 1380(m), 1048(m), 909, 734(s) см-1.
ПМР (CDCl3): δ 6.26(1H, d, J=11.2); 5.94(1H, d, J=11.2); 4.09(2H, m), 2. 67(1H, dd, J= 3,8, 13.3); 2,49(1H, dd, J=3,8, 13.3); 2.43(1H, m); 2.29(1H, dd, J= 7.6, 13.3); 2.19(1H, dd, J= 6,7, 13.2); 2.04(1H, d, J=13.0 Hz); 2.00(1H, m); 1.90(1H, m); 1.86(1H, m); 1.84(1H, d, J=13.0 Hz), 1.70(1H, m); 1.56(3H, m),:1.21(6H, s); 0,89(3H, d, J =6.9), 0.77(3H, d, J=7.3), 0.76(3H, s) м.д.
Пример 88. Синтез аналога 9
Как описано для 11.
Rf: 0.30 (дихлорметан: метанол 1:20)
ИК(пленка): 3386(s); 2932; 2874(s); 1640(w); 1456, 1475(s); 1141; 1053(s), 816(m) см-1.
ПМР: (500 MHz, CDCl3): δ 6.32(1H, d, J=11.3 Hz); 6.10(1H, d, J=11.3 Hz); 5.25(1H, d, J= 1.7 Hz); 5.05(1H, d, J=2.2 Hz); 4.40(1H, m); 4.24(1H, m); 3.65(2H, m); 3.46(2H, m); 2.62(1H, dd, J= 4.1, 12.8 Hz), 2.47(1H, m); 2.25(1H, dd, J= 10.8, 12.3 Hz), 2.12(2H, m); 2.02(1H, m); 1.80(3H, m); 1.70(3H, m); 1.55(4H, m); 1.35(2H, m); 1.23(6H, s); 0.8(3H, s).
Пример 89. Синтез аналога 10
Как описано для 11. Оба эпимера могут быть разделены ВДЖХ (двуокись кремния: этилацетат: пентан 15: 85) на стадии TBMB простых эфиров bij NOE измерениями,
Rf: 0,36(дихлорметан: метанол 7:1).
10: Rf:0.20 дихлорметан:метанол 92:8).
ИК(пленка): 3324, 2986, 2880, 1455, 1414, 1378, 1260, 1130 см-1.
ЯМР: (MHz, CDCl3): δ : 6,45(1H, dd, J=11,15 Hz); 6.05(1H, d, J=11 Hz); 5.71(1H, dt, J=7.5 и 15 Hz); 5.30 (1H, m); 5.00(1H, d, J=4.5 Hz); 4.98 (1H, m); 4.42(1H, m); 4.21(1H, m); 3.80 (1H, d, J=8.2 Hz); 3.50(1H, d, J=8.2 Hz); 3.48(1H, s); 2.58(1H, dd, J=4, 13.2 Hz); 2.35(2H, d, J =7.5 Hz), 2.28(1H, dd, J=7.2, 13.2 Hz), 1.95(2H, t, J=5,5 Hz), 1.7-1.5 (4H, m), 1.48(4H, q, J = 7.5 Hz), 1.43(2H, m), 1.25(3H, s), 0,85(6H, t, 3 =7:5 Hz),
10 β: ПМР: (500 MHz, CDCl3): δ : 6.46(1H, dd, J =11,15 Hz), 6.05(1H, d, J=11H), 5.69(1H, dt, J=7.5, 15 Hz); 5.31(1H, t, J=1.5 Hz); 5.00(1H, t, J=4,7 Hz); 4.98(1H, m), 4.45(1H, m), 4.21(1H, m), 3.67(1H, d, J=8 Hz), 3.62(1H, d, J= 8 Hz), 2.58(1H, dd, J=3.4, 13.3 Hz), 2.40(1H, dd, J=7.5, 14 Hz), 2.35(1H, dd, J= 7.5, 14 Hz), 2.28 (1H, dd, J=7,13.3 Hz), 1.97(2H, t, J=5,5 Hz), 1.75-1.55 (4H, m); 1.47(4H, q, J=7.5 Hz): 1.43(2H, m), 1.27(3H, s), 0,85(6H, t, J=7,5 Hz).
Пример 90. Синтез аналога 11
К раствору сухого A-циклического фосфин оксида 13.1 (87 мг, 150 мкмоль) в тетрагидрофуране (1.4 мл) добавляют раствор - бутиллития (2,5 М в гексане, 57 мкл, 142,5 мкмоль) при -78oC. После перемешивания полученной красной суспензии в течение 1 ч добавляют по каплям раствор 6.12 (12 мг, 47,2 мкмоль) в тетрагидрофуране (0,5 мл). Реакционную смесь перемешивают 1 час при -78oC и затем убирают охлаждающую баню. Медленно добавляют воду до полного исчезновения оранжевой окраски и тетрагидрофуран удаляют. После добавления диэтилового эфира и насыщенного бикарбоната натрия, водный слой экстрагируют несколько раз диэтиловым эфиром. Объединенные органические фазы фильтруют через силикагель, фильтрат концентрируют и оставшееся масло чистят ВДЖХ (пентан:этилацетат 8:2), получая 24 мг (82%) продукта сочетания.
К раствору этого продукта (24 мг, 38,8 мкмоль) в тетрагидрофуране (0,5 мл) добавляют раствор тетра (н-бутил)-аммонийфторида (1М в THF, 311 мкл, 311 мкмоль) и образовавшуюся смесь перемешивают при комнатной температуре в атмосфере аргона в темноте 87 часов. После упаривания THF, остаток чистят на колонке с двуокисью кремния (дихлорметан:метанол 9:1) и ВДЖХ (CH2Cl2:MeOH 94:6), что дает 15 мг (98%) 11.
Rf: 0.16 (дихлорметан:метанол 9:1).
IR (пленка):3354 (s); 2966 (s); 2936 (о); 2869 (m); 1635 (w); 1468 (m); 1377 (s); 1366 (s); 1265 (m); 1215 (m); 1152 (s); 1056 (s); 976 (m) см-1.
1H NMR: (360 MHz, CDCl3): δ : 6.36 (1H, dd, J = 11 Hz, J = 15 Hz): 6.15(1H, d, J= 11 Hz); 5.77 (1H, dt, J= 15 Hz, J = 7.5 Hz): 5.31 (1H, d, J<1): 5.00 (1H, d); 4.43 (1H, t, J = 5.5): 4.21 (1H, m): 2.57 (1H, dd, J = 13, 3.7 Hz): 2.27 (1H, dd, J = 7); 2.10-1.71 (7H, m): 1.60-1.25 (10H, m): 1.21 (6H, s); 0.82 (3H, s); 0.78 (3H, s); 0.66 (3H, s).
Пример 91. Синтез аналога 12
Как описано для 11.
Rf: 0.25 (дихлорметан:метанол 95:5)
IR (пленка): 3374 (s, br); 2965 (s); 2938 (s); 2876 (m); 1631 (m); 1460 (m); 1057 (s); 976 (m); 935 (s); 909 (s) cм-1.
1H NMR: (200 MHz. CDCl3): δ : 6.36 (1H, dd, J = 10.7, 15 Hz): 6.05 (1H. d, J = 10.7 Hz); 5.76 (1H, dt, J = 15, 7.5 Hz); 5.31 (1H, d); 4.99 (1H, d): 4.43 (1H, t, J= 5.6 Hz): 4.21 (1H, m); 2.58 (1 H, dd, J = 7, 13.2 Hz): 2.25 (1H, dd, J = 3.7, 13.2 Hz); 2.06-2.00 (1H, m): 2.00-1.92 (2H, t); 1.84-1.54 (5H, m); 1.53-1.38 (6H, m); 1.38-1.00 (8H, m): 0.87 (6H, t): 0.83 (3H, s); 0.79 (3H, s); 0.66 (3H, s).
Пример 92. Синтез аналога 13
К раствору 13.2 (76 мг, 133 мкмоль) в тетрагидрофуране (1,3 мл) добавляют раствор н-бутиллития (2,5 М в гексане, 51 мкл, 127 мкмоль) при -75oC. После перемешивания полученной суспензии в течение 1 ч, добавляют по каплям раствор 6.13 (13 мг, 56 мкмоль) в тетрагидрофуране (0,5 мл). Реакционную смесь перемешивают 1 ч при -75oC, после чего охлаждающую баню убирают. После добавления этилацетата (2 мл) и насыщенного бикарбоната натрия (2 мл) водный слой экстрагируют несколько раз этилацетатом. Объединенные органические фазы фильтруют через силикагель, фильтрат концентрируют в вакууме и полученное масло чистят ВДЖХ (пентан:этилацетат 95:5), получая 11 мг (38%) продукта сочетания.
К раствору этого продукта (11 мг, 17 мкмоль) в метаноле (2,5 мл) и тетрагидрофуране (2,5 мл) добавляют Amberlyst-15 (1,6 г) и полученную смесь перемешивают при комнатной температуре в атмосфере аргона, в темноте 9 часов. Смесь фильтруют через силикагель. Amberlyst-15 промывают несколько раз метанолом и фильтруют через силикагель. Фильтрат концентрируют в вакууме и полученное масло чистят ВДЖХ (дихлорметан: метанол 95:5), получая 13 (6 мг, 86%).
Rf: 0.19 (дихлорметан: метанол 95:5).
УФ: λmax =240,9 им; (ε = 32.535,7).
ИК(пленка): 3443(s, br): , 2913(w), 1518(m); 1433(s), 1256(w); 1087(w), 1024(w) см-1.
ПМР: (500 MHz,CDCl3): δ : 6,26(1H, dd, J=10.8, 14.9 Hz); 6.05(1H, d, J= 10.8, Hz); 5.72(1H, dt, J=15, 7.7 Hz); 4.12-4.08(2H, m); 2.54(1H, dd, J= 13.5,3.9 Hz); 2.47(1H, dd, J=13.1, 3.5 Hz); 2.3(1H, dd, J=13.3, 7.6 Hz); 2.15(1H, dd, J=13.3, 6.9 Hz); 2.1 1H, dd, J=13.3, 7.1 Hz); 2.03(1H, dd, J= 13,6, 8.4 Hz); 1.8-1.9 (3H, m); 1.75(1H, m); 1.64-1.5(3H, m); 1.48-1.4 4+2, q+m), 1.4- 1.7(7H, m); 0,87(6H, y, J=7.4 Hz), 0.84(3H, s); 0.79(3H, s); 0.67(3H, s).
Пример 93. Ситез аналога 14
Сочетание 6.21а с 13.1 выполняют как описано для 6.12. Однако расщепление простого силил эфира выполняют при перемешивании метанольного раствора в присутствии Amberlyst-15 в течение 4 ч при комнатной температуре. После фильтрации соединение 14 чистят ВДЖХ (дихлорметан: метанол 95:5).
Rf: 0.40 (дихлорметан: метанол 9:1).
ИК(пленка): 3386; 2969(s); 1641(w); 1468(w); 1366 (s); 1154(m); 1058(s); 978(m) см-1
ПМР: (500 MHz, CDCl3): δ :6.37(1H, dd, J=10,8, 15.1 Hz); 6.04(1H, d, J= 10,8 Hz); 5.71(1H, dt, J=15.1 Hz, J= 7.5 Hz), 5.30(1H, d, J<1); 4.98(1H, d); 4.42(1H, m); 4.21(1H, m); 3.75(1H, dt); 3.61- 3.55(2H, m); 2.56(1H, dd, J= 13.2, 3.9 Hz); 2.25(1H, dd, J=7.3 Hz); 2.11-1.90(4H, m); 1.81-1.30(9H, m); 1.24(6H, s); 0.89(3H, s); 0.83(3H, s); 0,81 (3H, s).
Пример 94. Синтез аналога 15
Как описано для 14 исходя из 6.22d.
Rf: 0.59 (дихлорметан:метанол 9:1).
ИК(пленка): 3380 (s, br): 2964(s), 1632(w); 1462(s), 1376(s), 1262(m), 1067(s) см-1.
ПМР: (500 MHz, CDCl3): δ : 6.37(1H, dd, J=10.8, 15.1 Hz): 6.04(1H, d); 5.71(1H, dt, J=7.5 Hz); 5.30(1H, d); 4.98(1H, d); 4.42(1H, m); 4.21(1H, m); 3.71(1H, m); 3.60-3.46(2H, m); 2.57(1H, dd, J=13,4, 3.7 Hz); 2.25 (1H, dd, J= 7.4 Hz); 2.11-1.9(4H, m); 1.73-1.65(3H, m); 1.60-1.20 (10H, m); 0.89(3H, s), 0,87(6H, t); 0.83(3H, s); 0.81(3H, s).
Пример 95. Синтез аналога 16
Как описано для 14 исходя из 6.16.
Rf: 0.27 (дихлорметан:метанол 95:5).
ИК(пленка): 3380 (s,br), 2966(s), 1616(w), 1551 (m), 1422(s), 1278(s), 1156(w) см-1.
ПМР: (360 MHz, CDCl3): δ : 6.35(1H, dd, J=10.9, J= 15.2 Hz); 6.07 (1H, d, J=10.9Hz); 5.76(1H, dt); 5.31 (1H, d, J<1); 5.00 (1H, d); 4.44 (1H, t, J= 5.8 Hz); 4.22(1H, m)); 2.57 (1H, dd, J=13.3, 3.6 Hz), 2.27 (1H, dd, J= 6.8 Hz); 2.10-1.50 (11H, m); 1.45(4H, q, J=7.5 Hz); 1.42 (7H, m); 0,86(6H, t, J= 7.5 Hz); 0.80 (3H, s), 0.78 (3H, s), 0.67 (3H, s).
Пример 96. Синтез аналога 17.
Как описано для 11.
Rf: 0.31 (дихлорметан:метанол 9:1).
ИК(пленка): 3384 (s); 2968(s); 1631 (m); 1467 (s). 1365(s);1265(m); 1154(m); 1090(s); 1056(s) см-1
ПМР: (500 MHz, CDCl3): δ : 6.35, dd, J=10.6, 15 Hz); 6,05(1H, d, J= 10.6); 5.73(1H, dt, J=15, 7.5 Hz); 5.31 (1H, d); 4.99(1H, d, J≈1); 4.43(1H, t, J=5,5 Hz); 4.22(1H, m); 3.75(1H, m); 3.55(1H, m); 3.48(1H, m); 2.57 (1H, dd): , 2.27(1H, dd); 2.00(4H, m); 1.75(2H, t, J= 5.6 Hz); 1.83-1.20(7H, m); 1.25(6H, s); 0,88(3H, s),
Пример 97. Синтез аналога 18
Как описано для 14 исходя из 6.2 β.
Rf: 0.54 (дихлорметан:метанол 9:1).
ИК(пленка): 3380 (s,br); 2964(s); 2875(s); 1632 (w); 1462(s); 1364(m); 1266 (m); 1091(s); 1065(s) см-1
ПМР: (500 MHz, CDCl3): δ : 6.35(1H, dd, J=10.7, 15.1 Hz).; 6.05(1H, d, J= 10.7 Hz); 5.72(1H, dt, J=7.5, Hz); 5.31(1H, d); 4.99(1H, d); 4.43(1H, m); 4.22(1H, m); 3.71 (1H, dt); 3.55-3.45(2H, m); 2.57(1H; dd, J=13,3, 3.7 Hz); 2,28(1H, dd, J= 7,0 Hz); 2.02-1.90(4H, m);1.72(2H, t); 1.61-1.43 (11H, m); 0.87(3H, s), 0,85(3H, s); 0.84(6H, t); 0.83(3H, s).
Пример 98. Синтез аналога 19
К раствору 6.27 (12 мг, 19 мкмоль) в THF (1 мл) при -5oC добавляют по каплям раствор MeMB (50 мкл, 3М в Et2O, 8 экв). После нагревания в течение ночи при комнатной температуре, смесь выливают в смесь лед-раствор аммонийхлорида - диэтиловый эфир. Органическую фазу сушат (MgSO4). Фильтрация, упаривание и колоночная хроматография (диэтиловый эфир:гексан 1:9--->1;4) дают бис- силилированный аналог (10 мг, 82%). Снятие защиты TBAF как описано для аналога 11 дает 19 (5 мг, 80%).
Rf: 0.27 (MeOH:CH2Cl2 1:19).
ПМР: (500 MHz, CDCl3): δ : 5.36(1H, dd, J=10.8, 15.2); 6.05(1H, 10.8); 5.76(1H, dt, J=7,7, 15.1);. 5.31(1H, dd, J=1,2); 5.00(1H,brs); 4.43(1H, t, J= 5.7); 4.22(1H, m); 2.57(1H, dd, J=3.8, 13.3); 2.26(1H, dd, J=7.5, 13.3); 1.21(6H, s); 0.82(3H, s); 0,78(3H, s); 0.66 (3H, s) м.д.
Пример 99. Синтез аналога 20
Из 6.27 с EtMgBr как описано для 19(выход 50%).
Rf: 0.29 (MeOH: CH2Cl2 1:19).
ПМР: (500 MHz, CDCl3): δ : 6.36(1H, dd, J=10.8, 15.1); 6.06(1H, d, J= 10.8); 5.76(1H, dt, J= 7.5, 15.1); 5.31 (1H, dd, J=1.2); 5.00(1H, brs); 4.43(1H, t, J=5.5); 4.22(1H, m); 2.57(1H, dd;J=3.6, 13.2); 2.26(1H, dd, J= 7.2, 13.3); 1.46(4H, q, 7.5); 0.86(6H, t, J=7.5); 0.82(3H, s); 0.78(3H, s); 0.66(3H, s) м.д.
Пример 100. Синтез 21
Из 6.30 как описано для 19 из 6.26 (выход 48%).
Rf: 0,16 CH2Cl2 (MeOH 1:19).
ПМР: (500 MHz, CDCl3): δ : 6.36(1H, dd, J=10.8, 15.1); 6.06(1H, d, J= 10.8); 5.76(1H, dt, J= 7.5, 15.1); 5.31(1H, dd, J=1,2); 5.00 (1H, brs); 4.43(1H, t, J=5,5); 4.22(1H, m); 2.57(1H, dd, J=3.6, 13.2); 2.26(1H, dd, J= 7.2, 13.3); 1.46(4H, q, 7.5); 0.86(6H, t, J=7.5); 0.82(3H, s); 0.78(3H, s); 0.66(3H, s) м.д.
Пример 101. Синтез аналога 22
Как описано для 11.
Rf: 0.30 (дихлорметан:метанол 1:20).
ИК(пленка): 3389(s); 2932 (s); 1632 (w); 1462, 1366 (w); 1089, 1057(s); 736(m) см-1.
ПМР: (500 MHz, CDCl3): δ : 6.38(1H, dd, J=10.7, 15.1 Hz); 6.04(1H, d, J= 10.7 Hz); 5.72(1H, m); 5.30(1H, s); 4.98(1H, s); 4.41(1H, m); 4.21(1H, m); 3.73(1H, m); 3.66(1H, m); 3.38(1H, m); 2.58(1H, m); 2.27 (1H, m); 1.97(4H, m); 1.73(2H, m); 1.60(4H, m); 1.40(2H, m); 1.22(6H, s); 0.87(3H, s).
Пример 102. Синтез аналога 23
Как описано для 11.
Rf:0.21 (дихлорметан: метанол 1:17).
ИК(пленка): 3384, 2932(s), 1630(w); 1455, 1365(m), 1265, 1152(m), 1089, 1054(s); 909, 734(s) см-1.
ПМР: (500 MHz, CDCl3): δ : 6.38(1H, m); 6.05(1H, d, J=10,8 Hz); 5.70(1H, m); 5.28(1H, s); 4.95(1H, s); 4.41 (1H, m); 4.21(1H, m); 3.70(2H, m); 3.41(1H, m), 2.55(1H, m); 2.25(1H, m); 2.15(2H, m); 2.00(4H, m); 1.72(4H, m); 1.60(2H, m); 1,40(2H, m); 1,21(6H, s); 1.10(2H, m); 0.89(3H, s).
Пример 103. Синтез аналога 24
Как описано для 11.
Rf: 0.29 (дихлорметан:метанол 1:20)
IR (пленка): 3421 (m); 2931 (s); 1637 (w); 1458, 1379 (m); 1085 (s); 911 (s); 935 (s) cм-1.
1H NMR: (500 MHz, CDCl3): δ : 6-38 (1H, m); 6.04 (1H, d, J = 10.8 Hz); 5.70 (1H, m); 5.30 (1H, m); 4.98 (1H, s); 4.40 (1H, m); 4.20 (1H, m); 3.58 (1H, m); 3.53 (1H, m); 3.43 (1H, m); 2.92 (1H, m); 2.55 (1H, m); 2.25 (1H, m); 2.08 (1H, m); 1.98 (4H, m); 1.80 (3H, m), 1.60 (1H, m); 1.40 (3H, m); 1.32 (3H, s); 1.28 (3H, s); 0.90 (3H, s).
Пример 104. Синтез аналога 25
Как описано для 11.
Rf: 0.30 (дихлорметан:метанол 1:20).
IR (пленка): 3401 (s); 2924 (s); 1633 (w); 1453, 1374 (m); 1164, 1054 (s); 738 (s) cм-1.
1H NMR: (500 MHz, CDCl3): δ : 6.39 (1H, m), 6.06 (1H, d, J = 10.8 Hz); 5.70 (1H, m); 5.32 (1H, t, J = 1.6 Hz); 5.00 (1 H, m); 4.43 (1H, m); 4.21 (1H, m); 2.55 (1H, m); 2.25 (1H, m); 2.15 (1H, dd, J = 8.2, 14.0 Hz); 2.00 (5H, m); 1.90 (2H, m); 1.60 (4H, m); 1.49 (6H, s); 1.40 (2H, m); 1.10 (1H, m); 0.87 (3H, s).
Пример 105. Синтез аналога 26
Как описано для 11.
Rf: 0.36 (дихлорметан:метанол 7:1).
1H NMR: (500 MHz, CDCl3): δ : 6.39 (1H, dd, 10.8, 15.2 Hz), 6.04 (1H, d; 10.8 Hz); 5.68 (1H, dt, 7, 15 Hz); 5.31 (1H, dd, 1,2 Hz); 4.99 (1H, d, 1Hz); 4.43 (1H, m); 4.22 (1H, m); 3.72 (1H, ddd, 5, 7, 9.5 Hz); 3.66 (1H, ddd, 5, 7, 9.5 Hz); 3.27 (1H, dt, 4, 11 Hz); 2.57 (1 H, dd, 3.7, 13.3 Hz); 2.27 (1H, dd, 7.0, 13.4 Hz); 1.24 (6H, s); 0.76 (3H, d, 7.02 Hz).
Пример 106. Синтез аналога 27
Как описано для 11.
Rf: 0.23 (дихлорметан:метанол 9:1)
IR (пленка): 3382 (s); 2930 (s); 1632, 1445, 1359, 1261, 1153, 1091 cм-1.
1H NMR: (360 MHz, CDCl3):δ : 6.37 (1H, dd, 10.8, 15.1 Hz); 6.03 (1H, d, 10.8 Hz); 5.66 (1H, dt, 7.5, 15.1 Hz); 5.30 (1H,brs): 4.98 (1H,brs); 4.43 (1H, m); 4.21 (1H, m); 3.87 (1H, ddd, 4.5, 7, 9 Hz); 3.53 (1H, ddd, 5, 7,9 Hz); 2.90 (1H, m); 2.80 (1 H, td, 4, 10 Hz); 2.57 (1H, dd, 3.6, 13.3 Hz); 2.31 (1H, m); 2.25 (1H, dd, 7.4. 13.3 Hz); 2.10 (1H, m); 1.24 (6H, s); 1.01(3H, d,6.03Hz).
Пример 107. Синтез аналога 28.
Как описано для 11.
Rf: 0.26 (дихлорматан:метанол 9:1).
IR (пленка): 3384 (s); 2929 (s); 3026, 1631, 1443, 1363, 1261, 1218 cм-1.
1H NMR: (360 MHz, CDCl3): δ : 6.35 (1H, dd, 10.8, 15.2 Hz); 6.02 (1H, d, 10.8 Hz); 5.67 (1H, dt, 7, 15 Hz); 5.29 (1H, d,1 Hz), 4.97 (1H, d, 1 Hz), 4.42 (1H, m); 4.21 (1H, m); 3.84 (1H, ddd, 4, 7, 9 Hz); 3.49 (1H, ddd, 4, 7,9 Hz); 3.36 (1H. W1/2, 8 Hz, m); 2.56 (1H, dd, 4, 13 Hz); 2.25 (1H, dd, 7, 13 Hz); 2.19 (1H, m); 1.25 (6H, s); 0.98 (3H, d, 6.7 Hz).
Пример 108. Синтез аналога 29
Как описано для 11.
Rf: 0.38 (дихлорматан:метанол 9:1).
IR (пленка): 3357, 2926, 2857, 1366, 1056, 975, 908, 801, 734 cм-1.
1H NMR: (500 MHz, CDCl3): δ : 6.34 (1H, dd, J =15.2, 11.0 Hz); 6.04 (1H, d, J = 10.9 Hz); 5.67 (1H, ddd, J = 15.1, 8.5, 6.0 Hz); 5.31 (1H, bs); 4.99 (1H, bs); 4.43 (1H, m); 4.21 (1H, m); 2.57 (1H, dd, J = 13.2, 3.62 Hz); 2.36 (1H, dd, J = 13.8, 5.8 Hz): 2.26 (1 H, dd, J = 13.4, 7.2 Hz); 1.95 (2H, m); 1.70-1.44 (12H, m); 1.42-1.36 (2H, m); 1.27-1.14 (2H, m); 1.20 (6H, s); 1.05-0.87 (2H, m); 0.23 (3H, s); 0.62 (3H, s).
Пример 109. Синтез аналога 30
Как описано для 11.
Rf: 0.29 (дихлорметан:метанол 13:1).
IR (пленка): 3370 (s); 3082, 3045, 2964 (s), 1602, 1581, 1460, 1291 cм-1.
1H NMR: (500 MHz, CDCl3): δ : 7.20 (1H, t, 7.96 Hz); 6,90 (1H, ddd, 0.8, 1.5, 8 Hz); 6.86 (1 H, dd, 1.6, 2.2 Hz); 6.71 (1H, ddd, 0.7, 2.1, 8 Hz): 6.43 (1H, dd, 10.7, 15.5 Hz); 6.08 (1H, d, 10.7 Hz); 5.89 (1H, d, 15.5 Hz); 5.31 (1H, dd, 1.4, 1.8 Hz); 5.00 (1H,br s); 4.44 (1H, dd, 5, 7 Hz); 4.23 (1H, m); 3.96 (2H, t, 6.4 Hz); 2.57 (1H, dd, 3.73, 13.4 Hz); 2.28 (1H, dd, 6.9, 13.4 Hz); 1.978 (1 H, dd, 5, 7 Hz); 1.962 (1 H, ddd, 0.6, 4, 8 Hz); 1.50 (4H, q, 7.52 Hz); 1.39 (6H, s); 0.88 (6H, t, 7.52 Hz).
Пример 110. Синтез аналога 31
После защиты третичного спирта в виде триметилсилилового эфира построение боковой цепи нор A-цикла выполняют обычным образом. После удаления силил эфирных защитных групп (TBAF, THF) смесь чистят колоночной хроматографией (силикагель: дихлорметан: метанол 24:1), что приводит к смеси E-аналога 31 и Z-изомера 7,8 (соотношение 2:1).
Rf : 0,20(CH3OH:CH2Cl2 1:19)
ПМР: E-изомер (CDCl3): δ 6.25(1H, d, J=11.2); 5.94(1H, d, J=11.3); 4.10(1H, m); 4.05(1H, m); 2.81 (1H, m); 2.69(1H, dd, J=3.8, 13.2); 2.25(1H, dd, J=7,8, 13.3); 1.20(6H, s); 0.67(3H, s).
1H NMR: Z-изомер (CDCl3): δ 6.22 (1H, d, J = 11.1); 6.08 (1H, d, J = 11.1); 4.10 (1H, m): 4.05 (1H, m); 2.39 (1H, dd, J=6.7, 13.4); 1.20 (6H, s); 0.67(3H, s).
Пример 111. Синтез аналога 32
Как описано для 11. Получен вместе с 7-Z-изомером (1:1).
Rf: 0.43 (дихлорметан:метанол 94:6).
IR (пленка): 3356-2924; 1436; 1374, 1205; 1144; 1054 cм-1.
1H NMR: (500 MHz, CDCl3): δ : 6.30-6.29 (1H, 2xd, J = 11.35, 11.11); 6.17-6.14 (1H, 2xd, J= 11.51-11.46); 5.48 (1H, m); 5.33 (1H, m); 5.00 (1H, dpp s); 4.43 (1H, m): 4.22 (1H, m); 3.94 (1H, m); 2.60 (1H, dm, J = 13); 2.35-2.15 (4H, m); 2.50-2.36 (2H, m); 1.23 (6H, 2xs); 2.12-1.91 (5H, m); 1.88-1.79 (2H, m); 1.71-1.45 (7H, m) ppm.
Пример 112. Синтез аналога 33
Как описано для 11. Получен вместе с 7-Z-изомером (6:4).
Rf: 0.31 (дихлорметан:метанол 94:6).
IR (пленка): 3367, 2936, 2865, 1433, 1363, 1308, 1217, 1152 cм-1.
1H NMR: (500 MHz, CDCl3): δ : 6.25 (1H, 2xd, J = 11.55, 11.65); 6.18 (1H, 2xd, J = 11.37, 11.36); 5.32 (1H, bs); 4.99 (1H, bs); 4.43 (1H, m: 4.21 (1H, m); 3.99 (1H, ddd, J = 7.2, 5.1. 5.1); 3.93 (1 H, ddd, J = 5.7, 4.9, 4.9); 3.85 (1H, m); 3.77 (1H, m); 2.60 (1H, dd, J = 13.18, 3.79); 2,49 (1H, dd, J = 6.0, 14.1); 2.45-1.23 (20H, m); 1.20 (6H, 2xs) ppm.
Пример 113. Синтез аналога 34
Как описано для 11. Получен вместе с 7-Z-изомером (6:4).
Rf: 0.3 (дихлорметан:метанол 94:6).
IR (пленка): 3371, 2929, 2865, 1428, 1360, 1298, 1152, 1049, 974 cм-1.
1H NMR: (500 MHz, CDCl3): δ :6.18 (1H, 2xd, J = 11.36, 11.46), 6.08 (1H, 2xd, J= 11.35, 11.5); 5.65 (2H, m); 4.10(2H, m); 4.05 (H.m); 3.95 (1H, m); 3.90 (1H, m); 3.83 (1H, m); 2.71 (1H, dd, J = 13.25, 3.75); 2.59 (1H, dd, J = 13.6, 3.6); 2.52-1.56 (17H, m); 1.31 (3H, s); 1.29 (3H, s); 1.21 (1H, m) ppm.
Пример 114. Синтез аналога 35
Как описано для 11. Получен вместе с 7-Z-изомером (1:1)
Rf: 0.30 (дихлорметан:метанол 94:6).
1H NMR: (500 MHz, CDCl3): δ : 6.21 (1H, 2xd, J = 9.32, 9.7); 6.08 (1H, 2xd, J= 11.1, 11.71); 4.2-4.0 (4H, m); 2.62 (1H, dm, J= 11.7); 2.49 (1H, dm, J =16.2) ppm.
Пример 115. Синтез аналога 36
Как описано для 11. Получен вместе с 7-Z-изомером (1:1).
Rf: 0.26 (дихлорметан:метанол 94:6).
IR (пленка): 3390, 2925, 2855, 1458, 1361, 1172, 1051 cм-1.
1H NMR: (500 MHz, CDCl3): δ : 6.28 (1 H, 2xd, J = 11.22, 11.38 Hz); 6.18 (1H, 2xd, J= 11.95, 11.17 Hz); 5.33 (1H, appd, J= 1.45 Hz); 5.0 (1H, m); 4.44 (1H, m); 4.21 (1H, m); 4.13 (1H, m); 4.04 (1H, m); 1.60 (1H, dm, J= 12.80 Hz); 2.40 (5H, m); 2.20 (5H, m); 2.10 (4H, m); 1.92 (4H, m); 1.85 (12H, m); 1.40 (12H, m); 1.25 (6H,2xs); 1.21 (6H,2xs).
Пример 116. Синтез аналога 37
Из 11.19 как описано для аналога 11.
Rf: 0.48 (CH2Cl2:MeOH 9:1).
IR (CH2Cl2): 3343 (br, s); 2962 (s); 2861 (m); 1640 (w); 1558 (w); 1456 (s); 1375 (s); 1261 (m); 1057 (s) cм-1.
1H NMR: (500 MHz, CDCl3): δ : 6.34 (1H, dd, J 10.8, 15.1): 6.08 (1H, d, J= 10.8); 5.65 (1H, dd, J= 15.1, 8.6); 5.32 (1H, d, J=1): 5.01 (1H, d); 4.42(1H, m); 4.21 (12H, m); 2.58 (1H, dd, J=13.1, 3.9); 2.47 (1H); 2.27(1H, dd. J= 7.6); 1.99(1H, m); 1.95(1H, m)1.76(1H, m); 1.60-1.20 (14H, m); 1.21 (6H, s); 0.84 (3H, s); 0.76 (3H, s); 0.67 (3H, s) ppm
Пример 117. Синтез аналога 38
Из 11.20 как описано для аналога 11.
Rf: 0.19 (CH2Cl2:MeOH 95:5).
IR (CH2Cl2): 3380 (s); 2960 (s); 2939 (s); 2872 (m); 1633 (m); 1454 (s); 1374 (s); 1253 (m); 1092 (s); 1054 (s) cm-1.
1H NMR: (500 MHz, CDCl3):  : 6.34 (1H, dd, J = 10.8, 15.1); 6.08 (1H, d); 5.65 (1H, dd, J = 8.5); 5.32 (1H, d, J= 1); 5.01 (1H, d); 4.42 (1H, m); 4.22 (1H, m); 2.58 (1H, dd, J = 13.2, 4.0); 2.47 (1H, dd); 2.27 (1H, dd, J = 7.6); 2.23-1,90 (2H, m); 1.76 (1H, m); 1.60-1.50 (5H, m); 1.46 (4H, q, J = 7.5); 1.50-1.38 (4H, m); 1.35-1.15 (5H, m); 0.86 (6H, t); 0.84 (3H, s); 0.76 (3H, s); 0.67 (3H, s) ppm.
: 6.34 (1H, dd, J = 10.8, 15.1); 6.08 (1H, d); 5.65 (1H, dd, J = 8.5); 5.32 (1H, d, J= 1); 5.01 (1H, d); 4.42 (1H, m); 4.22 (1H, m); 2.58 (1H, dd, J = 13.2, 4.0); 2.47 (1H, dd); 2.27 (1H, dd, J = 7.6); 2.23-1,90 (2H, m); 1.76 (1H, m); 1.60-1.50 (5H, m); 1.46 (4H, q, J = 7.5); 1.50-1.38 (4H, m); 1.35-1.15 (5H, m); 0.86 (6H, t); 0.84 (3H, s); 0.76 (3H, s); 0.67 (3H, s) ppm.
Пример 118. Синтез аналога 39
Из 11.21 как описано для аналога 11.
Rf: 0.20 (CH2Cl2:MeOH 95:5).
IR (CH2Cl2): 3402 (s); 2967 (s) 2872 (m); 1634 (w); 1422 (m); 1373 (m); 1265 (s); 1138 (w) cм-1.
1H NMR: (500 MHz, CDCl3): δ : 6.35 (1H, dd, J = 10.8, 15.1); 6.21 (1H, dd, J = 10.8, 15.5); 6.07 (1H, d); 5.96 (1H, dd, J = 15.5); 5.75 (2H, 2xd); 5.63 (1H, dd, J= 8.6); 5.31 (1H, d, J=<1); 5.01 (1H, d); 4.44 (1H, m); 4.22 (1H, m); 2.57 (1H, dd); 2.50 (1H, dd, J = 8.9); 2.26 (1H, dd); 2.02-1.83 (4H, m); 1.55 (m); 1.45 (m); 1.34 (6H, s); 0.98 (3H, s); 0.77 (3H, s); 0.65 (3H, s) ppm.
Пример 119. Синтез аналога 40
Из 11.27 как описано для аналога 11. Образуется также 7,8- Z-изомер 40Z (соотношение 34:34' 4:1). Они могут быть разделены колоночной хроматографией на силикагеле, пропитанном нитратом серебра (элюент MeOH: CH2Cl2 1:24--->1: 6).
40: Rf: 0.14 (MeOH CH2Cl2 1:14 on AgNO3-силикагель)
1H NMR; (500 MHz, CDCl3): δ : 6.30 (1H, dd, J = 10.8, 15.2); 6.08 (1H, d, J = 10.8); 5.60 (1H, dd, J = 8.8, 15.2); 5.30 (1H, brs); 4.98 (1H, d, J = 1.8); 4.43 (1H, m); 4.20 (1H, m); 2.58 (1H, dd, J = 4.0, 13.0); 2.33 (1H, q, J = 9); 2.25 (1H, J = 8.3,13.0); 2.05 (1H, m); 1.88 (1H, ddd, J = 3.8, 8.3, 13); 1.78 (1H, m); 1.21 (6H, s); 0.93 (2H, t, J = 7); 0.94 (3H, d, J = 7.0); 0.85 (3H, t, J = 6.6); 0.65 (3H, s) ppm.
40Z: Rf:0.10(MeOH:CH2Cl2 1:14 on AgNO3- силикагель).
1H NMR: (500 MHz, CDCl3): δ : 6.35 (1 H, t, J = 11); 6.26 (1H, d, J = 12.5); 5.33 (1H, dd, J = 1,2); 5.28 (1H, t, J= 11); 5.01 (1H, brs); 4.43 (1H, m); 4.22 (1H, m); 2.84 (1H, q, J = 9); 2.59 (1H, dd, J =4.1, 13.2); 2.31 (1H, dd, J = 6.9, 13.4); 1.97 (1H, ddd, J =4, 8, 1.2); 1.82 (1H, m); 1.22 (6H, s); 0.96 (3H, d, J = 6.7); 0.94 (3H, 1, J = 7.3); 0.84 (2H, t, J = 6.5); 0.70 (3H, s) ppm.
Пример 120. Синтез аналога 41
Как описано для 11.
Rf: 0.37 (дихлорметан:метанол 9:1).
IR (пленка): 3376 (s, br), 2934 (s), 2242 (w), 1631 (w), 1461 (s), 1381 (m), 1056 (s), 958 (m), 911 (s) cм-1.
1H NMR: (360 MHz, CDCl3): δ : 6.41-6.30 (1H, m); 6.10-6.00 (1H, m); 5.70-5.59 (1H, m); 5.31 (1H, d); 5.00 (1H, s, br); 4.44 (1H, m); 4.22 (1H, m);2.60-2.52 (1H, m); 2.30-2.00 (4H, m); 1.96 (2H, t); 1.90-1.10 4 (14H, m); 1.05 (6H, t); 0.95-0.80 (6H, m).
Пример 121. Синтез аналога 42
Как описано для 11.
Rf: 0.28 (дихлорметан:метанол 9:1).
IR (пленка): 3382 (s): 2925, 1660, 1455, 1261, 1055 cм-1.
1H NMR: (360 MHz, CDCl3): δ : 7.12 (1H, dd, 11.2, 15.5 Hz); 6.30 (1H, d, 15.5 Hz); 6.19 (1H, d, 3.3Hz); 6.18 (1H, d, 11 Hz); 6.17 (1H, d, 3.3 Hz); 6.06 (1H, dt, 1, 11 Hz); 5.50 (1H, ddd, 7, 8, 11 Hz); 5.27 (1H, d, 2 Hz); 5.05 (1H, d, 2 Hz); 4.42 (1H, m, W1/2 11 Hz); 4.25 (1H, m, W1/2 19 Hz); 2.91 (1H, m); 2.64 (1H, dm, 13 Hz), 2.44 (2H, m); 2.31 (1H, dd, 8.5, 13 Hz); 1.83 (1H, ddd, 4, 9, 13 Hz); 0.86 (3H, t, 7.5 Hz); 0.86 (3H, t, 8 Hz).
Пример 122. Синтез аналога 43
Как описано для 13.
Rf: 0.39 (дихлорметан:метанол 9:1).
IR (пленка): 3377 (s, br), 2931 (s), 1610 (w), 1454 (s), 1376 (s), 1265 (s), 1214 (w), 1152 (w), 1049 (s), 976 (m) cм-1.
1H NMR: (500 MHz, CDCl3): δ : 6.26 (1H, d, J = 11.2); 6.04 (1H, d); 4.09 (2H, m); 2.69 (1H, dd, J= 3.8, 13.3 Hz); 2.47 (2H, m); 2.29 (1H, dd, J= 13.3, 7.8 Hz); 2.21-2.07 (3H, m); 1.91 (1H, m); 1.85 (2H, m); 1.73-1.30 (17H, m); 1.23 (6H, s); 1.05 (1H, m); 0.93 (3H, s); 0.88 (3H, d, J = 6.6 Hz);
Пример 123. Синтез аналога 44
Как описано для 13.
Rf: 0,062 (этилацетат: гексан 5:95).
ИК(пленка): 3379, 2927, 2291, 3224, 1608, 1452, 1374, 1261, 1125, 1087, 1044 см-1.
ПМР: (360 MHz, CDCl3): δ : 4.10 (2H, dddd); 6.25(1H, d, J=11.3 Hz); 6.05(1H, d, J= 11.4Hz); 0,95 (3H, s); 1.03(3H, d, J=6.49 Hz); 1.50(6H, s); 2.49 (1H, dd, J=13.4, 3.55 Hz); 2.69 (1H, dd, J=13.3, 3.83 Hz)
Пример 124. Синтез аналога 45
Как описано для 13.
Rf:0,24 (дихлорметан:метанол 4:96)
ИК(пленка): 3422, 2976, 1642, 1451, 1267, 1088, 1048, 880 см-1.
ПМР: (500 MHz, CDCl3): δ : 1.53(3H, s); 2.97(1H, d, J, =5.55 Hz); 2.73(1H, d, J=5.54 Hz); 1.04 (3H, d, J = 5.17 Hz); 0.95(3H, s); 6.25(1H, d, J=11.38 Hz); 6.04(1H, d, J=11.30 Hz); 4.00 (2H, m).
Пример 125. Синтез аналога 46
Как описано для 13.
Rf: 0.17 (дихлорметан:метанол 95:5).
ИК(пленка): 3360, 3040, 2233, 1647, 1611, 811 см-1.
ПМР: (500 MHz, CDCl3): δ : 0,93(3H, s); 0.94(3H, d, J=7.81 Hz); 1.49(6H, s); 4.09(2H, m); 6.04(1H, d, J=11.11 Hz); 6.25(1H, d, 7=11,11Hz).
Пример 126. Синтез аналога 47
Из 12.14 как описано для аналога 13.
Rf:0.24 (CH2Cl2: MeOH 95:5).
УФ (MeOH) λmax =249 нм.
ИК(пленка): 3360, 3036, 1649, 1610; 811 см-1.
ПМР: (500 MHz, CDCl3): δ : 6.26(1H, d, J=11,31); 6.03(1H, d, J =11.31); 4.09(2H, m); 1.21(6H, s); 0.92 (3H, s); 0.84(3H, d, J=6.61) м.д.
MS: m/z 386(3); 353(1); 303(1); 45(100).
Пример 127. Синтез аналога 48
Из 12.13 как описано для 13.
Rf: 0.31 (CH2Cl2: MeOH 95:5).
ИК(пленка): 3361; 3036; 2236; 1612; 811 см-1
ПМР: (500 MHz, CDCl3): δ :6.25(1H, d, J =11.04); 6.04 (1H, d, J=11.04 Hz); 4.10(2H, m); 1.02 (6H, t, J=7.37); 0.95(3H, d, J=6.71); 0,93(3H, s) м. д.
Пример 128. Синтез аналога 49
Из 12.15 и 13.2 как описано для 19 из 6.26.
Rf 0.17(CH2Cl2: MeOH 95:5).
ИК(пленка): 3364; 3026; 1599; 990; 810 см-1
ПМР (500 MHz, CDCl3): δ : 6.26(1H, d. J=11.20); 6.16(1H, dd, J=15.44, 30); 6.04(1H, d, J= 11.20); 5.95(1H, dd, J=15.29, 10.30); 5.70(1H, d, J= 15.44); 5.58 (1H, dd, J=15.28, 8.22); 4.08(2H, m); 1.34((6H, s); 0.96(3H, d, J=6.70); 0,93(3H, s) м.д.
Пример 129. Синтез аналога 50
Из 12.15 как описано для 20 из 6.26.
Rf: 0.26(CH2Cl2: MeOH 95:5).
ПМР: (500 MHz, CDCl3): δ : 56.26(1H, d, J=11.16); 6.16(1H, dd, J=15,46, 10.32); 6.04(1H, d,7=11.16); 5.97 (1H, dd, J=15.24, 10.32); 5.57(1H, dd, J= 15.24, 8.07); 5.52(1H, d, J= 15.46); 4.09(2H, m); 0.98(3H, d, J=6.71); 0.94(3H, s); 0.87(6H, dd, J=7,44, 7.44) м.д.
Пример 130. Синтез аналога 51
Из 12.16 как описано для 19 из 6.26.
Rf: 0.17 (CH2Cl2: MeOH≈95:5).
ИК(пленка): 3360, 3036, 1610; 811 см-1.
ПМР: (360 MHz, CDCl3): δ : 6.26(1H, d, J= 11.09); 6.03(1H, d, J=11.09); 4.08(2H, m); 1.20(6H, s); 0.92(3H, s); 0.82(3H, d, J=6.48) м.д.
MS: m/z 400(29); 382(10); 303(4); 275(11); 257 (12); 59(100).
Пример 131. Синтез аналога 52
Из 12.16 как описано для 20 из 6.26.
Rf: 0.28 (CH2Cl2: MeOH 95:5).
ИК(пленка): 3364, 3037; 1611; 811 см-1.
ПМР: (360 MHz, CDCl3): δ 6.26(1H, d, J=10.98); 6.03(1H, d, J=10,98); 4.08(2H, m); 1.45(4H, q, J= 7.40); 0.92 (3H, s); 0,85(6H, t, J =7.40); 0.82(3H, d, J =6.55) м.д.
MS: m/z 428(34); 381(11); 299(6); 45(100).
Пример 132. Синтез аналога 53
Из 12.17 как описано для 19 из 6.26.
Rf: 0.20 (CH2Cl2 MeOH 95:5).
ИК(пленка): 3370(s, br); 3051(w); 2970(w); 1607 (m); 1263(s); 1095(s) cm-1.
ПМР: (500 MHz, CDCl3): δ : 6.23(2H, m); 5.99(2H, m); 5.73(1H, d, J= 15.5); 5.65(1H, dd, 7=8.5, 15.35); 4.10(1H, m); 4.06(1H, m); 2.69 (1H, m); 2.48 (3H, m); 2.26 (1H, dd, J = 8.0, 13.0); 2.15 (2H, m); 1.91 (1H, m); 1.83 (3H, m); 1.72 (1H, m); 1.62-1.44 (4H, m): 1.35 (6H, 2s); 1.25 (3H, m); 0.99 (3H, d, J= 6.8); 0.94 (3H, s)ppm.
Пример 133. Синтез аналога 54
Из 12.18 как описано для 19 из 6.26.
Rf: 0.19 (CH2Cl2:MeOH≈95:5)
IR (CH2Cl2): 3372 (s); 2928 (s); 1620 (w); 1462 (m); 1374 (s); 1019 (s); 974 (m) cм-1.
1H NMR: (500 MHz, CDCl3): δ : 6.26 (1H, d, J= 11.3); 6.04 (1H, d, J = 11.3); 4.09 (2H, m); 2.69 (1H, dd, J = 3.8, 13.2); 2.47 (3H, m); 2.30 (1H, dd, J = 7.7, 13.3); 2.19 (1H, dd, J = 6.4, 13.2); 2.10 (1H, m); 1.90 (1H, m); 1.84 (2H, m); 1.70 (1H, m); 1.62-1.23 (19H, m); 1.22 (6H, s); 0.93 (3H, s); 0.87 (3H, d, J = 6.6) ppm.
MS: m/z
Пример 134. Синтез аналога 55
Из 12.18 как описано для 20 из 6.26.
Rf: 0.19 (CH2Cl2:MeOH≈95:5)
IR (пленка): 3383 (s, br); 2927 (s); 1610 (m); 1046 (s); 974 (m) cм-1.
1H NMR: (500 MHz, CDCl3): δ : 6.26 (1H, d, J = 11.2); 6.04 (1H, d, J = 11.2); 2.46 (2H, m); 2.29 (1H, dd, J = 7.7, 13.2); 2.19 (1H, dd, J=6.5, 13.3); 2.10 (2H, m); 1.91 (1H, m); 1.84 (2H, m); 1.70 (1H, m); 1.62-1.50 (6H, m); 1.47 (4H, q, J = 7.5); 1.50-1.17 (13H, m); 0.92 (3H, s); 0.86 (6H, t, J= 7.5); 0.86 (3H, d, J= 6.5) ppm.
Пример 135. Синтез аналога 56
Из 10.8 как описано для 19 из 6.26.
Rf: 0.26 (hexane:aceton 6:4)(гексан:ацетон 6:4).
IR (пленка): 3388, 2927, 1634, 1464, 1367, 1058, 909, 734 cм-1.
1H NMR (500 MHz, CDCl3): δ :6.31 (1H, dd, J = 15.16. 10.81); 6.05 (1H, d, J = 10.84); 5.65 (1H. dd, J = 15.15, 8.97); 5.30 (1H, m); 4.99 (1H, m); 4.43 (1H, m); 4.22 (1H, m); 2.58 (1H, dd, J = 13.32, 3.88); 2.16 (1H, dd, J= 13.15, 7.11); 2.00 (1H, m); 1.94 (1H, m); 1.77-1.71 (3H, m); 1.54-1.24 (m); 1.21 (6H, s); 0.896 (3H, d, J = 7.58); 0.889 (3H, s); 0.740 (3H, s) ppm.
Пример 136. Синтез аналога 57
Из 10.9 как описано для 19 из 6.26.
Rf: 0.26 (hexane:aceton 6:4)(гексан:ацетон 6:4).
IR (пленка): 3387, 2934, 2865, 1634, 1454, 1366, 1057, 736 cм-1.
1H NMR (500 MHz, CDCl3): δ :6.34 (1H, dd, J = 14.98, 10.65); 6.07 (1H, d, J = 10.86); 6.05 (1H, dd, J = 15.11, 9.62); 5.31 (1H. m); 4.99 (1H. m); 4.44 (1H. m); 4.23 (1H, m); 2.57 (1H, d, J= 13.14, 3.78); 2.26 (1H. dd, J = 13.18. 6.69); 1.97 (2H, m); 1.88 (1H, m); 1.78 (1H. m); 1.65 (1H, m); 1.53-1.24 (m); 1.21 (6H. s); 0.958 (3H, s); 0.906 (3H, d); 0.830 (3H, s) ppm.
Пример 137. Синтез 16.3
Суспензия (-)-1,3,4,5-тетраоксициклогексанкарбоновой кислоты (16.1:47,5 г, 0,24 моль) и TSOH (200 мг) в толуоле (400 мл) нагревают до кипения с обратным холодильником и образующуюся H2O удаляют с помощью насадки Дина-Старка. Через 12 ч смесь фильтруют и сушат (Na2SO4). Упаривание растворителя дает сырой продукт 16.2 (42 г, 99%), который используют как таковой в следующей стадии.
Смесь 16.2 (1,1 г, 6.3 ммоль), т-бутилдиметилсилилхлорида (1,09 г, 7.24 ммоль), DMAP (13 мг, 0,11 ммоль) и имидазола (549 мг, 8.08 ммоль) в DMF (5.8 мл) перемешивают 12 ч при комнатной температуре в атмосфере азота. Смесь разбавляют Et2O, реакцию прерывают H2O и экстрагируют Et2O. Органический слой промывают раствором соли, сушат (Na2SO4), фильтруют и концентрируют. Колоночная хроматография (силикагель: гексан: EtOAc 2:1) и ВДЖХ разделение (CH2Cl2: MeOH, 97:3) дают 16.3 (1.16 г, 66%). Т.пл. 94-96oC.
Rf: 0.29 (гексан: EtOAc 2:1).
ИК(пленка): 3480, 3308, 1782, 1150, 1085 см-1.
ПМР: (500 MHz, CDCl3): δ : 4.87(1H, dd, J=4.9, 6.0); 3.97(1H, dd, J=4.4, 4.9); 3.89(1H, ddd, J= 4,4, 7.0, 10.8); 2.97 (1H, s, D2O обмениваемый), 2.79(1H, s, D2O обмениваемый); 2.62(1H, d, J=11,6); 2.29(1H, ddd, J=2.8, 6.0, 11.6); 2.02(1H, ddd, J=2,8, 1.0, 12.1); 1.97(1H, dd, J =10.8, 12.1); 0.91(9H, s): 0.10(6H, s) м.д.
Пример 138. Синтез 16.5
Смесь 16.3(8, 43 г, 29.2 ммоль), 1,1-тиокарбонилдиимидазола (28,3 г, 0,154 моль) и DMAP (203 мг, 1,67 ммоль) в дихлорэтане (80 мл) нагревают при температуре кипения с обратным холодильником 3 дня. Раствор декантируют и остаток промывают теплым CH2Cl2. Упаривание объединенных органических фаз и хроматография (силикагель: гексан: EtOAc 1:4) дают 16.4 (12,9 г, 87%).
Трибутилин гидрид (0,42 мл, 1,58 ммоль) добавляют по каплям к раствору 26.4 (200 мг, 0,395 ммоль) и AIBN (8 мг) в дегазированном сухом толуоле (5 мл). После кипячения в течение 5 ч растворитель упаривают. Колоночная хроматография (силикагель; гексан: EtOAc 5:1) дает 16.5 (56 мг, 55%). Т.пл. 52-54oC.
Rf: 0.60 (гексан: EtOAc 2:1).
ИК(пленка): 1777, 1259, 1124, 838, 776 см-1.
ПМР: (500 MHz, CDCl3): δ : 4.84(1H, dd, J=5.7, 5.0); 4.03(1H, ddd, J= 6.5, 6.4, 9.6, 9.6); 2.68(1H, m); 2.41(1H, ddddd, J=1,9, 2.0, 5.0, 6.5, 13.4); 2.35(1H, dddd, J=1.9, 2.0, 5.7, 11.5); 2.24(1H, ddddd, J=2.0, 2.0, 4.9, 6.4, 12.7); 1.81(1H, d, J= 11.5); 1.58(1H, m); 1,52(1H, dd, J=9.6, 13.4); 0.88 (9H, s); 0.05(6H, s) м.д.
Пример 139. Синтез 16.6
30% раствор NaOMe в сухом MeOH (3,7 мл, 19,47 ммоль) добавляют к 16.5 (2,49 г, 9.73 ммоль) в сухом MeOH (40 мл) при 0oC в атмосфере азота. После перемешивания в течение 1 ч при 0oC, добавляют насыщенный раствор (40 мл) и раствор нейтрализуют 2 н. HCl. Смесь экстрагируют CH2Cl2, объединенный органический слой промывают раствором соли, сушат (MgSO4). Фильтрация, упаривание растворителя и фильтрация через небольшой слой силикагеля (гексан: EtOAc 2:1) дают чистый 16,6 (2,8 г, 100%).
Rf: 0.28 (гексан: EtOAc 2:1).
ИК(пленка): 3385, 1739, 1257, 1039, 837, 778 см-1.
ПМР: (500 MHz, CDCl3): δ : 4,24(1H, s); 4.04(1H, m); 3.70(3H, s); 2.85(1H, dddd, J= 3.67, 3.67, 11.95, 11,95); 2.22(1H, m); 1.98(1H, m); 1.85(1H, m); 1.32-1.57 (3H, m; 0.90(9H, s); 0.08 (6H, s) м.д.
Пример 140. Синтез 16.7
Смесь 16.6 (2.77 г, 9.63 ммоль), п-бромфенилсульфонилхлорида (4.00 г, 15.6 ммоль), DMAP (30 мг, 0,25 ммоль) в безводном пиридине (4,6 мл) и хлороформ (1,8 мл) перемешивают 1,5 ч при 0oC и 12 ч при комнатной температуре. Добавляют воду и диэтиловый эфир. Объединенную органическую фазу промывают последовательно 2% HCl раствором, насыщенным NaHCO3 раствором и водой и сушат (MgSO4). Фильтрация, концентрация и хроматография (силикагель:гексан, EtOAc 5:1) дают 16.7 (4.88 г, 100%).
Т.пл. 62-64oC.
Rf: 0.57 (гексан: EtOAc 2:1).
ИК(пленка): 1737, 1577, 1369, 1188, 1049, 967, 822 см-1
ПМР: (500 MHz CDCl3): δ :7.73(4H, m); 4.72 (1H, dddd, J=4.52, 4.52, 11.29, 11.29); 4.19(1H, m); 3.69 (3H, s); 2.81(1H, dddd, J=3.64, 3.64, 12.47, 12.47); 2.31 1H, m); 1.86(1H, m); 1. 60(1H, m); 1.43-1.52(3H, m); 0,83(9H, s); 0.02(3H, s); 0.02(3H, s) м.д.
Пример 141. Синтез 16.8
К перемешиваемому раствору 16.7 (4.64 г, 9.15 ммоль) в безводном т-BuOH (30 мл) добавляют по каплям 1 М раствор т-BuOK в t-Buoh (10,6 мл, 10,6 ммоль) при 50oC в атмосфере N2. Полученную смесь нагревают при температуре кипения с обратным холодильником в течение 1 ч, добавляют насыщенный раствор NH4Cl (20 мл) и воду (5 мл). Смесь экстрагируют диэтиловым эфиром. Объединенную органическую фазу сушат (MgSO4), фильтруют и растворитель упаривают ниже 18oC. Хроматография (силикагель; диэтиловый эфир: пентан 5:95) дает 16.8 (1,63 г, 71%).
Rf: 0.48(гексан: EtOAc 5:1).
ИК(пленка): 1727, 1371, 1256, 1114, 1097, 838 см-1.
ПМР: (500 MHz, CDCl3: δ : 3.93 (1H, m): 3.66(3H, s); 2.20(1H, dd, J = 7.2, 12.9); 2.14(1H, dd, J=8.2, 12.9); 2.07(1H, dd, J=7.1, 12.0); 1.81(1H, m); 1.77(1H, m); 1.29(1H, dd, J=5.0, 8.5); 0.87 (9H, s); 0.67(1H, dd, J=5.0, 5.0); 0.02(6H, s).
MS: m/z 239(10), 213(100), 199(9), 167(35), 149(39), 125(20), 111(18), 89(96), 45(98) м.д.
Пример 142. Синтез 16.9
К перемешиваемому раствору 16.8(570 мг, 2.11 ммоль) в безводном толуоле (25 мл) добавляют по каплям раствор диизобутилалюминийгидрида (5.28 мл, 5.28 ммоль) 1М в гексане при -78oC в атмосфере N2. Перемешивание продолжают 2 ч при -78oC. Реакцию прерывают 2 н. раствором калий натрий тартрата (25 мл). Перемешивание продолжают в течение ночи, пока температура постепенно не станет комнатной. Смесь экстрагируют CH2Cl2, сушат (MgSO4) и упаривают. Хроматографическая (силикагель: гексан: EtOAc 4:1) очистка дает 16.9(500 мг, 98%).
Rf: 0.30(гексан: EtOAc 4:1).
ИК(пленка): 3328, 1256, 1115, 1094, 1032, 904, 775 см-1
ПМР: (500 MHz, CDCl3): δ : 4.03(1H, m); 3.62(1H, dd, J=5.1, 11.1); 3.51(1H, dd, J= 5.1, 11.1); 2.05(1H, dd, J =6.4, 12.6 Hz); 1.92(1H, dd, J= 6.4, 12.6); 1.75-1.84 (2H, m); 1.18(1H, ddd, J=4.2, 4.2, 8.4); 0.89(9H, s); 0.51(1H, dd, J=5.1, 8.4); 0.02(6H, s); 0.38(1H, dd, J=4.2, 4.2) м.д.
Пример 143. Синтез 16.10
К перемешиваемому раствору 16.9(480 мг, 1.98 ммоль) в дихлорметане (20 мл) добавляют PCC (750 мг, 3.49 ммоль) при комнатной температуре в атмосфере азота. После 2 ч перемешивания, смесь фильтруют через целит, который промывают дихлорметаном. Объединенный фильтрат промывают последовательно раствором соли, NaHCO3 раствором и раствором соли. Сушка (Na2SO4), фильтрация и хроматография (силикагель, диэтиловый эфир: пентан 1:9) дают 16.10 (430 мг, 90%).
Rf: 0.40 (гексан: EtOAc 9:1).
ИК(пленка): 1706, 1256, 1121, 1072, 838, 778 см-1.
ПМР: (500 MHZ, CDCl3): δ : 8.90(1H, s); 4.04(1H, m); 2.17(1H, ddd, J= 1.1, 8,0, 13.0); 2.13(1H, dd, J= 7.2, 13.0); 2.10(1H, dd, J=7.2, 13.0); 1.93(1H, ddd, J= 5.1, 5.3, 8.8); 1.80(1H, ddd, J=5.1, 8.0, 13.0); 1.35(1H, dd, J=5.6, 8.8); 0.97(1H, dd, J=5.3, 5.6); 0.88(9H, s); 0,04(6H, s) м.д.
Пример 144. Синтез 16.11
К суспензии т-BuOK (352 мг, 3.14 ммоль) в сухом THF (2 мл) добавляют по каплям раствор диметил диазометил фосфоната (219 мг, 1,45 ммоль) в сухом THF (2 мл) при -78oC в атмосфере азота. Через 10 мин, добавляют по каплям раствор 1610 (290 мг, 1.21 ммоль) в сухом THF (2 мл) при -78oC. Перемешивание продолжают при -78oC 4 часа, при -15oC -8 часов, при комнатной температуре - 5 часов. Добавляют воду, после чего экстрагируют дихлорметаном и сушат (MgSO4). Фильтрация, упаривание растворителя ниже 18oC и хроматография (силикагель, пентан, затем диэтиловый эфир; пентан 1:9) дают 16.11 (254 мг, 89%) в виде бесцветного масла.
Rf: 0.69(гексан:EtOAc 9:1),
ИК(пленка): 3467, 3315, 2113, 1111, 1095, 836, 776 см-1
ПМР: (500 MHz, CDCl3): δ : 3,84(1H, m); 2.28(1H, J=7.1, 12.5); 2.05(1H, dd, J= 7.1, 12.7); 1.92(1H, s); 1.90(1H, ddd, J=1.0, 8.3, 12.5); 1.83(1H, ddd, J= 4.9, 8.1, 12.7); 1.60 (1H, ddd, J=4.9, 4.9,8.3); 0,88 (9H, s); 0.81(1H, dd, J=4.9, 8.1 Hz); 0.55(1H, dd, J=4.9, 4.9); 0,01(6H, s) м.д.
Пример 145. Синтез 16.12
Гексаметилдисилазид натрия (1М в THF, 5.2 мл, 5.2 ммоль) добавляют к (бромометилен) трифенил фосфоний бромиду (2.35 мг, 5.4 ммоль) в сухом THF (7 мл) при -68oC. Через 1ч добавляют раствор 12.2 (357 мг, 0,91 ммоль) в 2 мл THF. После перемешивания в течение 1 ч смеси дают нагреться до комнатной температуры и перемешивают в течение ночи. Фильтрация через небольшой слой целита, промывка гексаном и концентрация дают масляный остаток, который хроматографируют (силикагель, гексан), получая (E) - и (Z) - 16.12 (в 3:1 соотношении) с общим выходом 56% (237 мг).
Rf: 0.41 (гексан).
ИК(пленка): 2955, 2874, 1622, 1462, 1380, 1235, 1043, 743 см-1.
ПМР: (500 MHz, CDCl3): δ : 5.87(1H, brs; 2.50(1H, ddd, J=4,6, 4.8, 14.5); 2.22(1H, dd, J= 8.1, 9.7); 2.10(1H, m):, 1.86(1H, m); 1.28(3H, s); 1.20(6H, s); 0,94(9H, t, J=8.0); 0.87(3H, d, J=5.5); 0.56(6H, q, J=8.0) м.д.
MS m/z: 115(17); 103(89).
Пример 146. Синтез 16.15
К перемешиваемому раствору 16.12 (51 мг, 0.11 ммоль) в диэтиловом эфире (0.6 мл) добавляют по каплям раствор т-бутиллития 1,7 м в н-пентане, 0.16 мл, 0.27 ммоль) при -78oC в атмосфере аргона и перемешивание продолжают 50 мин. Затем добавляют по каплям раствор 16.10(12 мг, 0,05 ммоль) в диэтиловом эфире (0.2 мл). Смесь перемешивают 1 ч при -78oC и реакцию прерывают добавлением насыщенного водного NH4Cl (2 мл) и экстрагируют Et2O и EtOAc. Объединенную органическую фазу сушат (MgSO4), концентрируют, фильтруют через небольшой слой силикагеля (EtOAc: гексан 1:6) и чистят ВДЖХ (силикагель: EtOAc: гексан 1:9), получая эпимерную смесь (в соотношении 6.4) (E) - 16.15(11 мг) и (Z)-16.15 (3,5 мг) с общим выходом 46%.
Rf: 0.59 (EtOAc: гексан 1:6).
ИК(пленка): 3394, 2954, 2876, 1465, 1390, 1383, 1092, 1043 см-1.
ПМР: (500 MHz, CDCl3): δ : 5.14(1H, 2хd, J=8,5, 8.7); 4.42(1H, d, J=7,7 min); 4.23(1H, d, J=8.5, основн.); 4.02(1H, m); 1.20(3H, s); 0.94(12H, t, J= 8.0, перекрывание с s): 0.87(12H, s+d, перекрывание): 0.56 (6H, q, J=8.0); 0.28(1H, dd, J= 4.4, 4.6, min); 0.23 (1H, dd, 3=4,4, 4.6 основн.) 0.02(6H, 2xs); м.д.
Пример 147. Синтез 16.13 г
К перемешиваемому раствору 16.11(22 мг. 0, 093 ммоль) в сухом THF (4 мл) при -50oC, добавляют н-бутиллитий (1,6 М раствор в н-гексане, 0.14 мл, 0.23 ммоль). После перемешивания в течение 1 ч 12.2 (40 мг, 0,10 ммоль) в сухом THF (1 мл) добавляют. Температуре дают подняться до комнатной и перемешивание продолжают 30 мин. Резкое охлаждение водой, экстракция Et2O, обычная обработка и ВДЖХ очистка (силикагель: EtOAc: гексан 1:20) дают 16.13 (22 мг, 55% в расчете на превращенный 12.2, 15 мг) в виде отдельного диастереомера.
Rf: 0.52 (EtOAc: гексан 1:9).
ИК(пленка): 3477, 2953, 2875, 2226, 1463, 1380, 1253, 1093 см-1.
ПМР: (500 MHz, CDCl3): δ : 3.83(1H, J=7,7, 7.8, 15.2); 2.23(1H, dd, J= 7.1, 12.5); 2.03(1H, dd, J=7.1, 12.6); 1.20(6H, s); 0.99(3H, s); 0,94(9H, t, J=8.0); 0,88(3H, d, J=6,6); 0.85(9H, s); 0.73(1H, dd, 5.0, 8.3); 0.56(6H, q, J=8.0); 0.52 (1H, dd, J=4.9, 4.9); 0.0 (6H, s) м.д.
Пример 148. Синтез 16.14
Спирт 16.13 (18 мг, 29 мкмоль) нагревают до кипения в THF (5 мл) в присутствии LiAlH4 (4 мг) и метилата натрия (4 мг). Через 2 ч смесь охлаждают, реакцию прерывают насыщенным NH4Cl и экстрагируют Et2O. Обычная обработка с последующей хроматографической очисткой (силикагель, EtOAc гексан: 1:25) дают 16.14 (9 мг, 50%).
Rf: 0.43(EtOAc: гексан 1:9).
ИК (пленка): 3508, 2930, 2872, 2463, 1380, 1256, 1093, 837 см-1.
ПМР: (500 MHz, CDCl3): δ : 5.34(2H, s); 3.95(1H, m); 2.06(2H, m); 1.27(3H, s); 1.19(6H, s); 0.94 (9H, t, J=8.0; 0,93(3H, d, J=6.0); 0.87(9H, s); 0.55 (6H, q, J=8.0); 0.07(1H, dd, J=3.2, 3.4); 0.02(6H, 2xs) м.д.
Пример 149. Синтез аналога 43
a) Из 16.15:
Смесь (E) -16.15(10 мг, 0.016 ммоль); PTSA (0,9 мг), воды (0.4 мл) и 1,4-диоксана (165 мл) перемешивают 6 ч при 63oC. Смесь обрабатывают насыщен. NaHCO3 (1,5 мл) и экстрагируют CH2Cl2. Объединенную органическую фазу сушат (MgSO4), концентрируют, фильтруют через короткую колонку с силикагелем (ацетон: гексан 4:6) и чистят ВДЖХ (силикагель: MeOH:CH2Cl2 5:95), получая 43 (5.0 мг, 78%).
b) Из 16.14;
Как описано из 16.15, выход 40% перед 7-изомером (соотношение 3:1).
Пример 147
1H NMR: (500 MHz, CDCl3) δ : 6.31 (1H, d, J = 11.2 Hz); 5.96 (1H, d, J = 11.2 Hz): 4.15 (1H, bs); 4.13-4.04 (2H, m); 2.72-2.64 (2H, m); 2.49 (1H, dd, J= 13.1, 3.8 Hz); 2.40 (1H, m); 2.28 (1H, dd, 13.8, 7.9 Hz); 2.21 (1H, dd, 13.5, 7.1 Hz); 1.21 (6H, s); 0.9 (3H, d, J = 6.73 Hz), 2.15-0.70 (22H, m) ppm.
Пример 148
1H NMR: (500 MHz, CDCl3):  : 6.38 (1H, d, J = 14.3 Hz); 6.1 (1H, d, J = 13.3 Hz); 5.32 (1H, s); 5.02 (1H, s); 4.43 (1H, m); 4.29 (1H, dd, J = 8.1, 1.6 Hz); 4.24-4.15 (2H, m); 4.02 (1H, d, 8.1 Hz); 2.68 (1H, m); 2.61 (1H, m); 2.41 (1H, dd, J = 13.4, 3.5 Hz); 2.31 (1H, m); 2.31 (1H, dd, 13.4, 7.2 Hz), 1.99 (3H, s) ppm.
: 6.38 (1H, d, J = 14.3 Hz); 6.1 (1H, d, J = 13.3 Hz); 5.32 (1H, s); 5.02 (1H, s); 4.43 (1H, m); 4.29 (1H, dd, J = 8.1, 1.6 Hz); 4.24-4.15 (2H, m); 4.02 (1H, d, 8.1 Hz); 2.68 (1H, m); 2.61 (1H, m); 2.41 (1H, dd, J = 13.4, 3.5 Hz); 2.31 (1H, m); 2.31 (1H, dd, 13.4, 7.2 Hz), 1.99 (3H, s) ppm.
Пример 149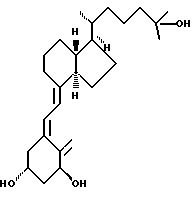
1H NMR: (500 MHz, CDCl3): δ : 6.37 (1H, d, J = 11.4 Hz); 6.08 (1H, d, J = 11.4 Hz); 5.32 (1H, bs); 5.02 (1H, s); 4.43 (1H, m);4.21 (1H, m); 2.87 (1H, dm, J = 13.6 Hz); 2.61 (1H, dd. J = 13.2, 3.4 Hz); 2.30 (1H, dd, J = 13.2, 7.3 Hz); 2.01-1.93 (2H, m); 1.21 (6H, s); 0.87 (3H, s, J = 6.7 Hz) ppm.
Пример 150
1H NMR: (500 MHz, CDCl3): δ : 6.31 (1H, d, J = 11.3 Hz); 5.94 (1H, d, J = 11.3 Hz); 4.09 (2H, m); 2.86 (1H, dm, J = 14.1 Hz); 2.71 (1H, dd, J = 13.3, 4.2 Hz); 2.49 (1H, dd. J = 13.3, 3.4 Hz); 2.27 (1H, dd, 13.3, 7.8 Hz); 2.23-2.11 (3H, m); 1.21 (6H, s); 0.77 (3H, d, J = 6.6 Hz) ppm.
Пример 151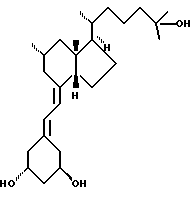
1H NMR: (500 MHz, CDCl3): δ : 6.29 (1H, d, J =11.3 Hz); 6.05 (1H, d, J = 11.3 Hz); 4.10 (2H, m); 2.70 (1H, dd, J = 13.3, 3.8 Hz); 2.57 (1H, bd, J = 9.5 Hz); 2.49 (1H, dd, J = 13.3, 3.8 Hz); 2.29 (1H, dd, J = 13.3, 3.8 Hz); 2.20 (1H, dd, J = 13.2, 6.4 Hz); 2.15 (1H, dd, J = 11.0, 7.6 Hz); 1.92 (1H, m); 1.85 (2H, m), 1.71 (1H, m); 1.23 (6H, s), 0.93 (3H, s); 0.90 (3H, d, J = 6.0 Hz); 0.88 (3H, d, J= 6.9 Hz) ppm.
Пример 152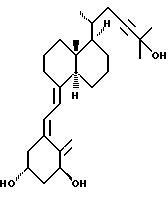
1H NMR: (500 MHz, CDCl3):  : 6.36 (1H, d, J = 11 Hz); 5.98 (1H, d, J = 11 Hz); 5.33 (1H, s); 4.99 (1H, s); 4.43 (1H, m); 4.23 (1H, m); 2.87 (1H, m); 2.60 (1H, m); 2.31 (1H, dd, J = 6.8, 13.5 Hz); 2.25 (1H, dd, J = 16,3 Hz); 1.49 (6H, s), 1.05 (3H, d, J = 6.8 Hz); 0.69 (3H, s) ppm.
: 6.36 (1H, d, J = 11 Hz); 5.98 (1H, d, J = 11 Hz); 5.33 (1H, s); 4.99 (1H, s); 4.43 (1H, m); 4.23 (1H, m); 2.87 (1H, m); 2.60 (1H, m); 2.31 (1H, dd, J = 6.8, 13.5 Hz); 2.25 (1H, dd, J = 16,3 Hz); 1.49 (6H, s), 1.05 (3H, d, J = 6.8 Hz); 0.69 (3H, s) ppm.
Пример 153
1H NMR: (500 MHz, CDCl3): δ : 6.29 (1H, d. J = 11 Hz); 5.83 (1H, d, J = 11 Hz); 4.12 (1H, m); 4.04 (1H, m); 2.84 (1H, m); 2.74 (1H, dd, J = 13, 3.5 Hz); 2.48 (1H, dd, J = 13, 3 Hz); 1.49 (6H, s); 1.05 (3H, d, J = 7 Hz); 0.69 (3H, s) ppm.
Пример 154.

1H NMR: (500 MHz, CDCl3): δ : 6.37 (1H, d, J = 11 Hz); 5.99 (1 H, d, J = 11 Hz); 5.34 (1H, bs); 4.43 (1H, m); 4.23 (1H, m); 2.87 (1H, bd, J = 16 Hz); 2.60 (1H, bd, J = 13 Hz); 2.31 (1H, dd, J = 13.5, 7 Hz); 0.68 (3H, s) ppm.
Пример 155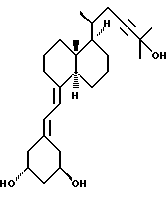
1H NMR: (500 MHz, CDCl3): δ : 6.30 (1H, d, J = 11 Hz); 5.83 (1 H, d, J = 11 Hz); 4.11 (1H, m); 4.04 (1H, m); 2.85 (1H, m); 2.74 (1H, dd, J = 13, 3.5 Hz); 2.48 (1H, dd, J = 13, 3 Hz); 1.51 (6H, s); 0.85 (3H, d, J = 6.5 Hz); 0.67 (3H, s) ppm.
Пример 156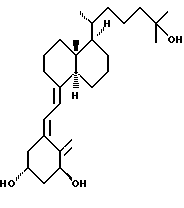
1H NMR: (500 MHz, CDCl3):  : 6.37 (1H, d, J = 11 Hz); 5.98 (1H, d, J = 11 Hz); 5.34 (1H, bs); 4.44 (1H, m); 4.25 (1H, m); 2.86 (1H, m); 2.61 (1H, dd, J= 13.5, 3 Hz); 2.31 (1H, dd, J = 13, 6.5 Hz); 0.67 (3H, s) ppm.
: 6.37 (1H, d, J = 11 Hz); 5.98 (1H, d, J = 11 Hz); 5.34 (1H, bs); 4.44 (1H, m); 4.25 (1H, m); 2.86 (1H, m); 2.61 (1H, dd, J= 13.5, 3 Hz); 2.31 (1H, dd, J = 13, 6.5 Hz); 0.67 (3H, s) ppm.
Пример 157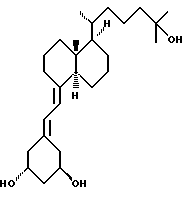
1H NMR: (500 MHz, CDCl3): δ : 6.30 (1H, d, J = 11 Hz); 5.83 (1H, d, J = 11 Hz); 4.11 (1H, m); 4.03 (1H, m); 2.84 (1H, bd, J = 14.5 Hz); 2.74 (1H, dd, J= 13.2, 4 Hz); 2.48 (1H, bd, J = 10 Hz); 1.20 (6H, s); 0.91 (3H, d, J = 7 Hz); 0.67 (3H, s) ppm.
Пример 158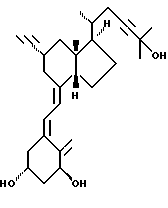
1H NMR: (500 MHz, CDCl3): δ : 6.27 (1H, d, J = 11.3 Hz); 6.10 (1H, d, J = 11.4 Hz); 4.1 (2H, m); 2.84 (1H, dd, J = 14.0, 3.3 Hz); 2.67 (1H, dd, J = 13.3, 3.9 Hz); 2.51-2.40 (2H, m); 1.56 (3H, s); 1.50 (3H, s); 1.0 (3H, d, J = 7.0 Hz); 0.98 (3H, s) ppm.
Пример 159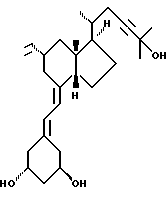
1H NMR: (500 MHz, CDCl3): δ : 6.28 (1H, d, J = 11.9 Hz); 6.09 (1H, d, J = 11.1 Hz); 5.78 (1H, ddd, J = 18, 10.4, 6.6 Hz); 5.02 (1H, dt, J = 1.5 Hz); 4.94 (1H, dt, J = 9.0, 1.4 Hz); 4.1 (1H, m); 2.68 (1H, dd, J = 14.0, 4.0 Hz); 2.61 (1H, m); 2.5 (1 H, m); 1.56 (3H, s); 1.50 (3H, s); 1.02 (3H, d, J = 7.5 Hz), 1.0 (3H, s) ppm.
Пример 160
1H NMR: (500 MHz, CDCl3):  6.24 (1H, d, J = 11.2 Hz); 6.05 (1H, d, J = 11.2 Hz); 4.13 (1H, m); 4.07 (1H, m); 2.72 (1H, dd, J = 13.5, 3.8 Hz); 2.48 (1H, dd, J = 13.5, 3.8 Hz); 1.05 (3H, d, J = 7 Hz); 0.95 (3H, s) ppm.
6.24 (1H, d, J = 11.2 Hz); 6.05 (1H, d, J = 11.2 Hz); 4.13 (1H, m); 4.07 (1H, m); 2.72 (1H, dd, J = 13.5, 3.8 Hz); 2.48 (1H, dd, J = 13.5, 3.8 Hz); 1.05 (3H, d, J = 7 Hz); 0.95 (3H, s) ppm.
Пример 161
1H NMR: (500 MHz, CDCl3): δ 6.30 (1H, d, J = 11.2 Hz); 5.83 (1H, d, J = 11 Hz); 4.12 (1H, m); 4.04 (1H, m); 2.86 (1H, bd, J = 12 Hz); 2.74 (1H, dd, J = 13, 3.8 Hz); 2.48 (1H, dd, J = 13.5, 3.5 Hz); 1.22 (6H, s); 0.76 (3H, d, J = 7 Hz); 0.67 (3H, s) ppm.
Пример 162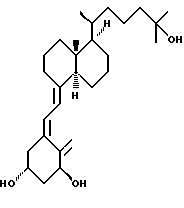
1H NMR: (500 MHz, CDCl3): δ : 6.37 (1H, d, J = 11 Hz); 5.99 (1H, d, J = 11 Hz), 5.34 (1H, bs); 5.0 (1H, bs), 4.42 (1H, m); 4.23 (1H, m); 2.87 (1H, m); 2.60 (1H, m); 2.31 (1H, m) ppm.
Пример 163
1H NMR: (500 MHz, CDCl3): δ : 6.31 (1H, d, J = 11 Hz); 6.12 (1H, d, J = 11 Hz); 4.1 (2H, bs); 2.68 (2H, dd, J = 13.5, 3.5 Hz); 2.49 (1H, dd, J = 13.5, 3.5 Hz); 2.29 (1H, dd. J = 13.5, 8 Hz); 2.21 (1H, dd, J = 13.5, 6.5 Hz); 1.19 (6H, s); 1.03 (3H, s); 0.78 (3H, d, J = 7 Hz) ppm.
Примеры на состав фармацевтической композиции.
Крем-масло в воде:
соединение формулы I - 0,0005 - 0,1%
эмульсионный агент - 5-10%
гелеобразующий агент - 0 - 0,2%
минеральное масло - 15 - 20%
пропиленгликоль - 0-5%
консервант - 0,025 - 0,05%
H2O (qs100)-вода - 65 - 80%
мазь:
соединение формулы I - 0,0005 - 0,1%
эмульсионный агент - 5-10%
минеральное масло - 5-10%
пропиленгликоль - 0-5%
H2O (вода) - 9 - 10%
консервант - 0,025 - 0,05%
вазелин - 65 - 80%
гель:
соединение формулы I - 0,0005 - 0,1%
эмульсионный агент - 0,5-1%
пропиленгликоль - 0-5%
спирт - 40-45%
хелатирующий агент - 0,05-0,1%
H2O (qs 100)-вода - 48 - 60%


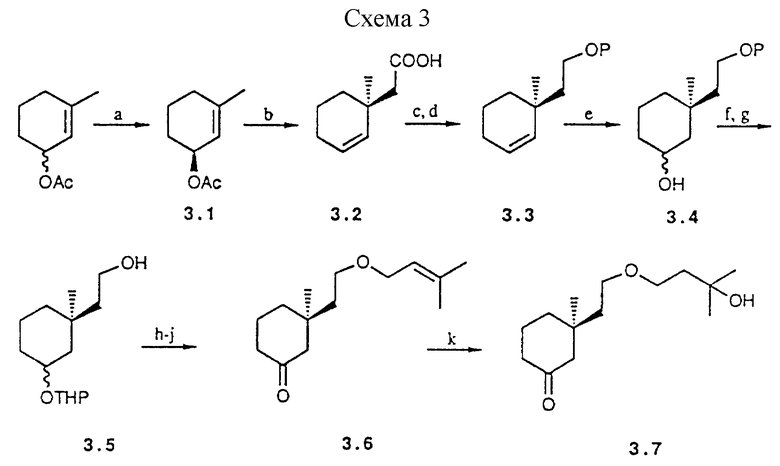

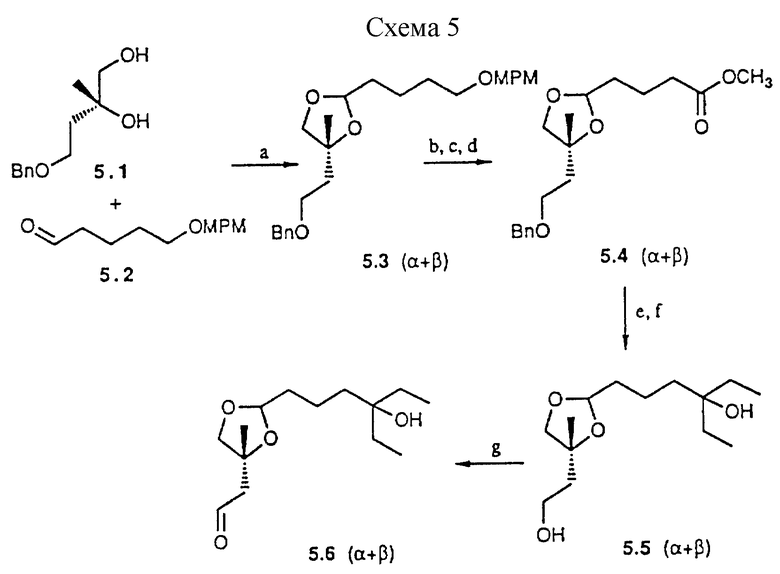
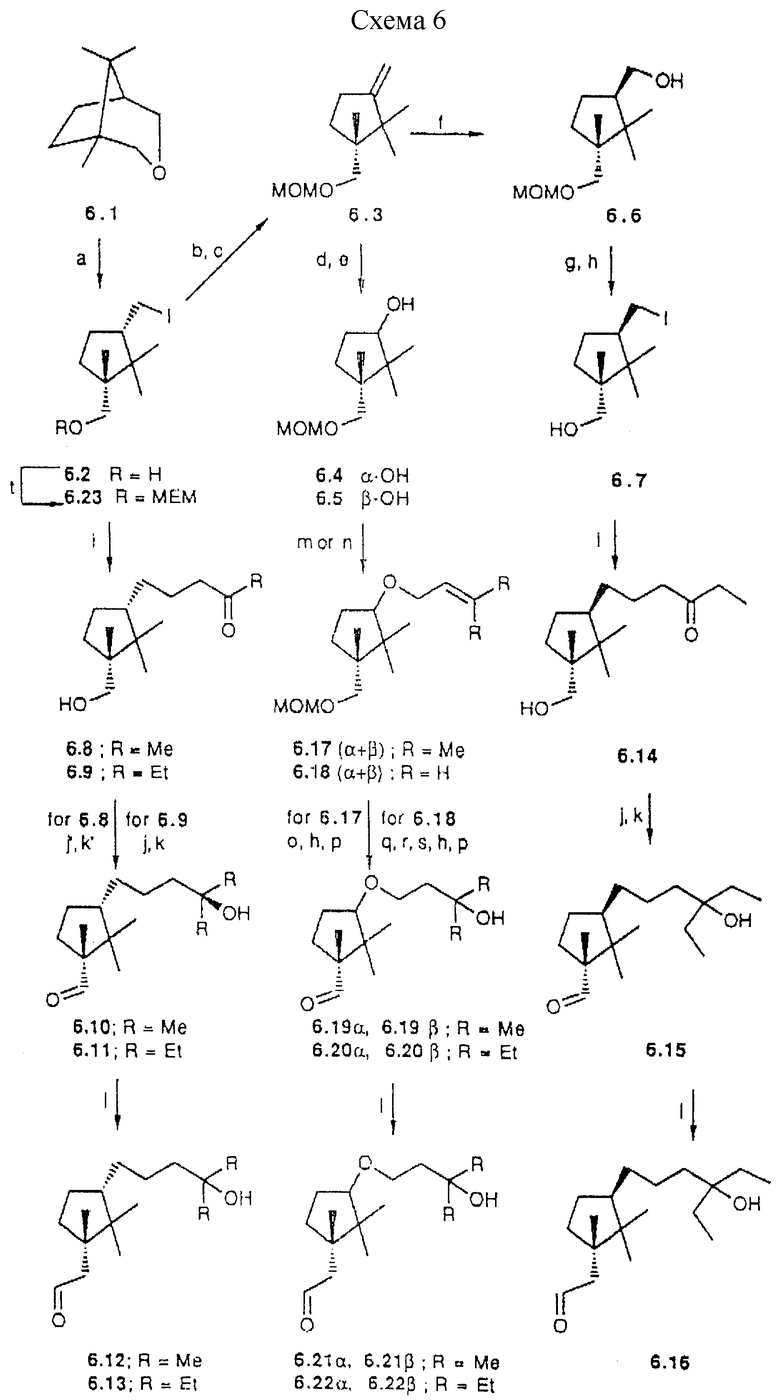
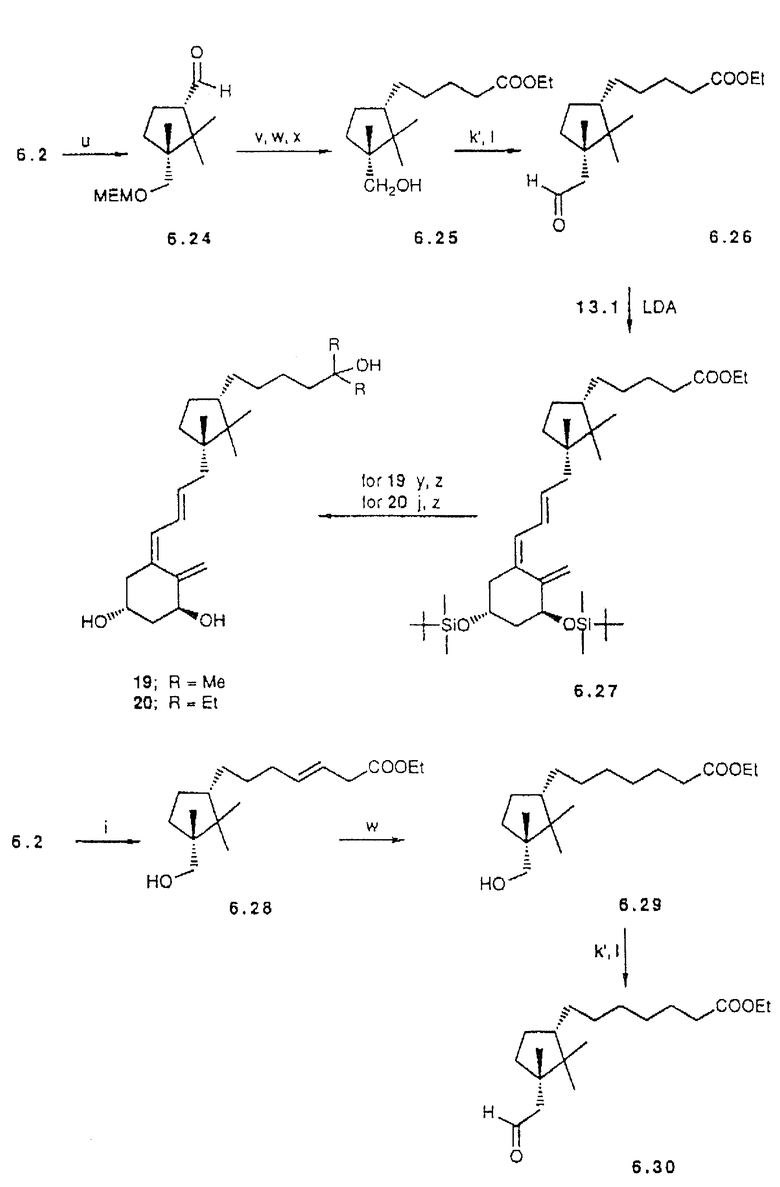



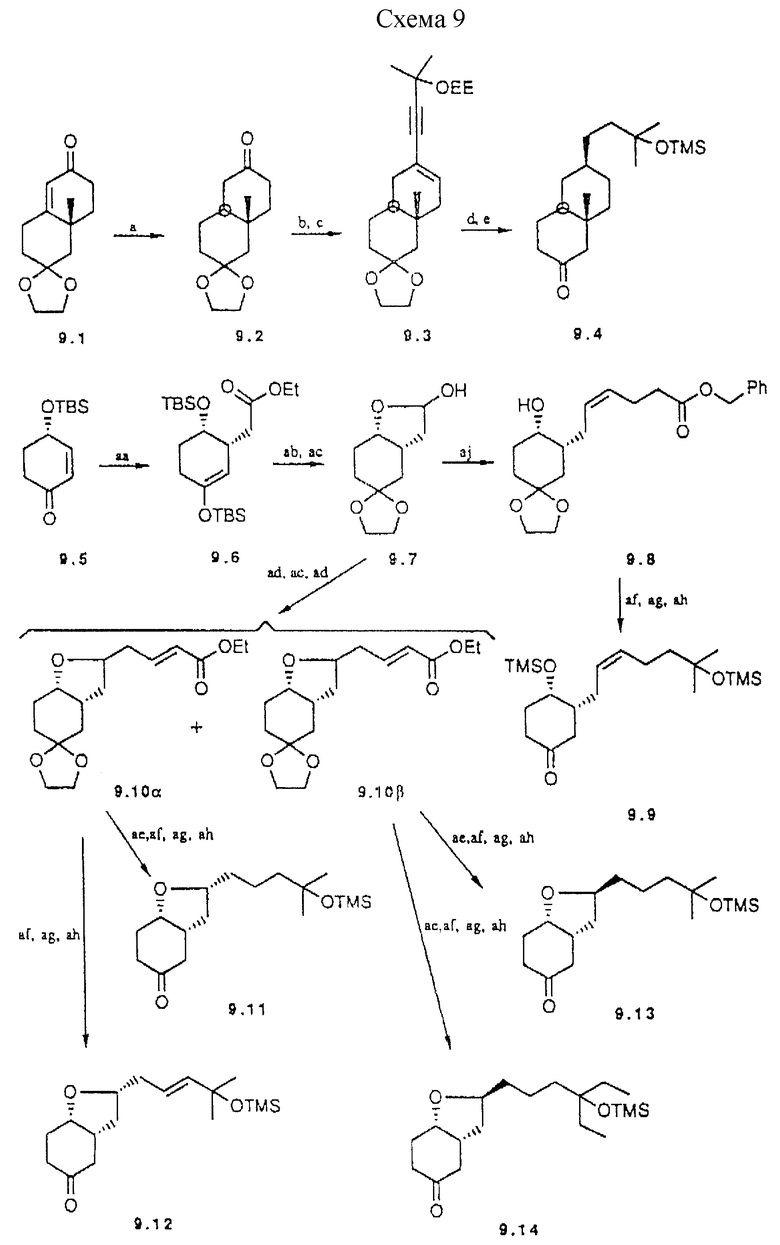
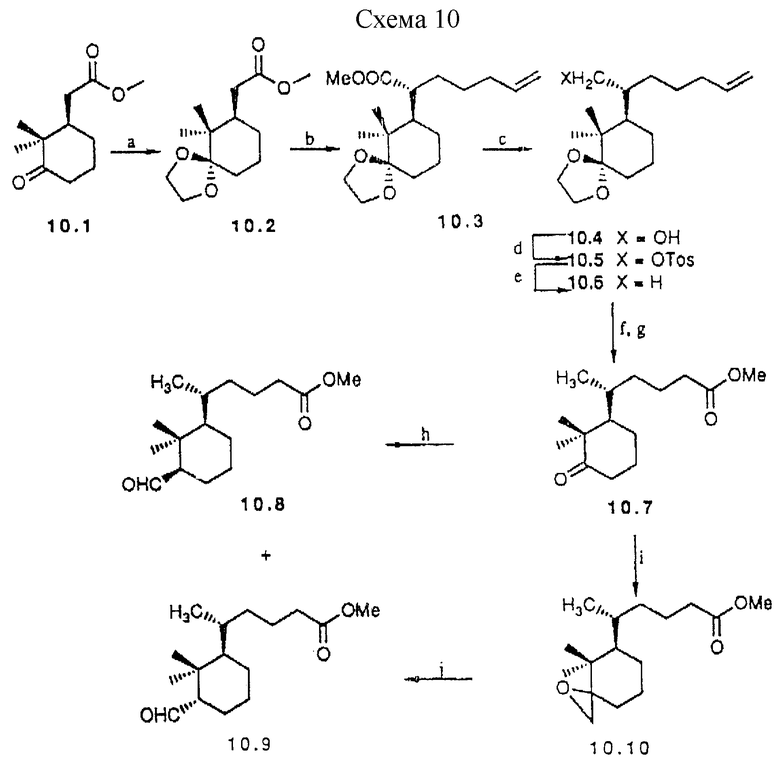

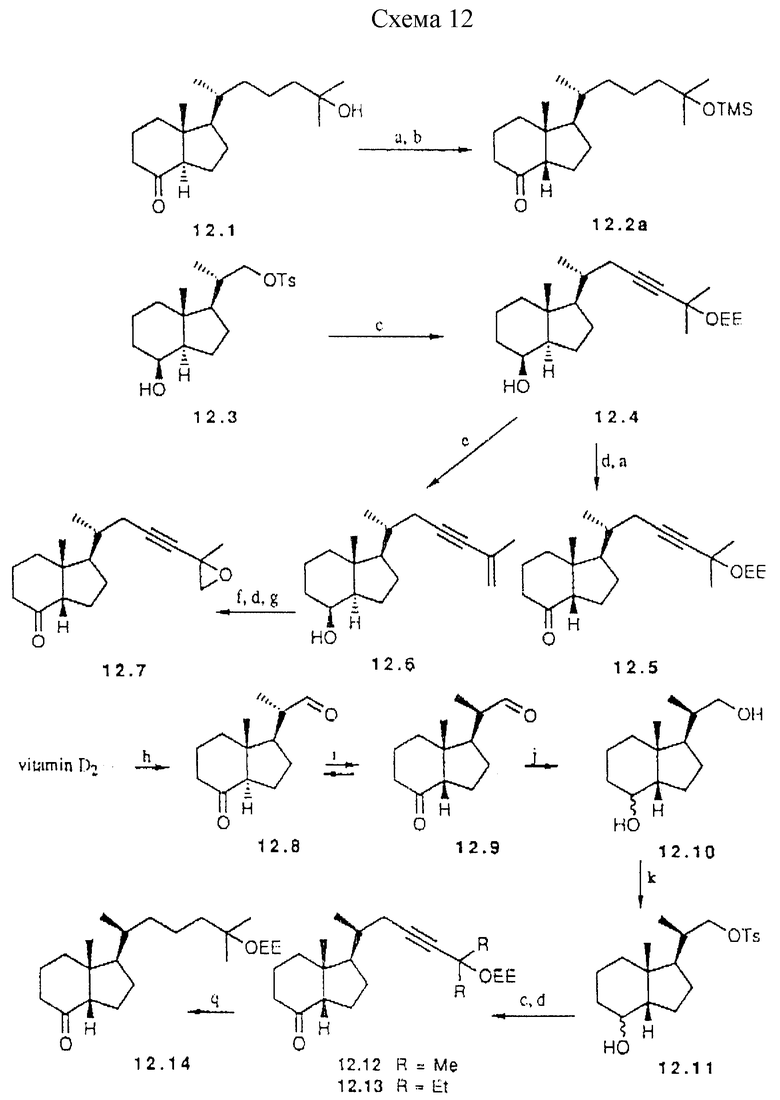

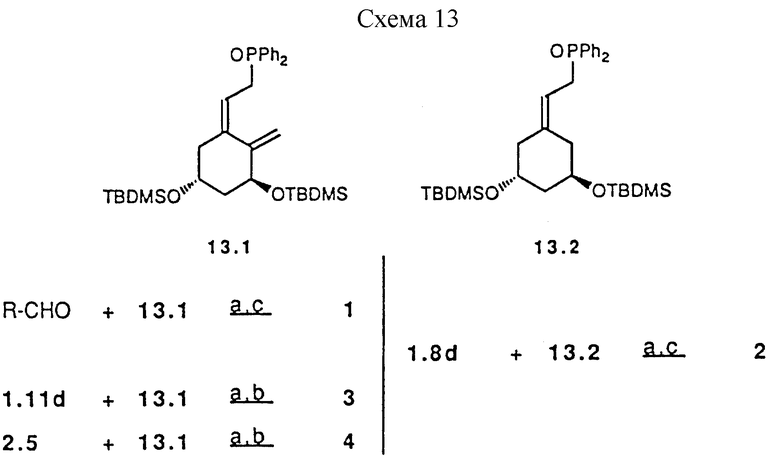
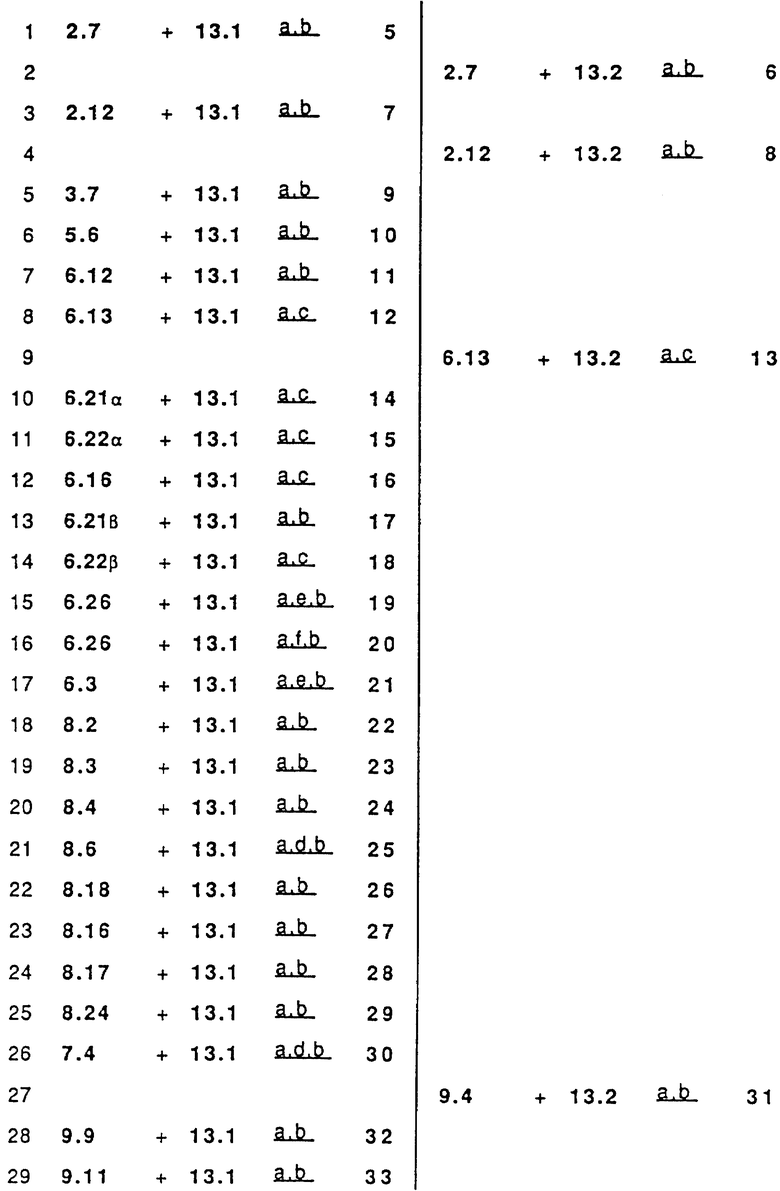


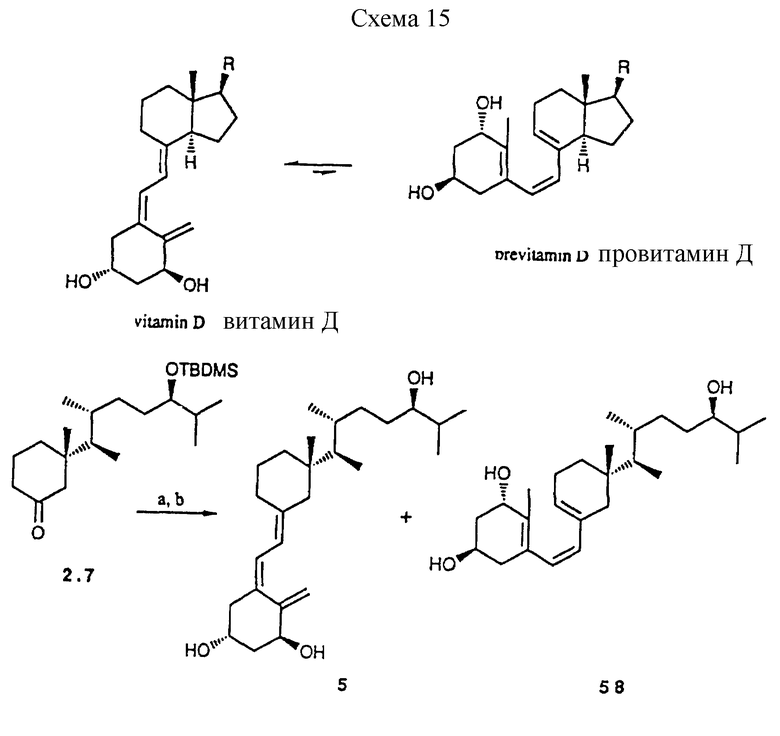




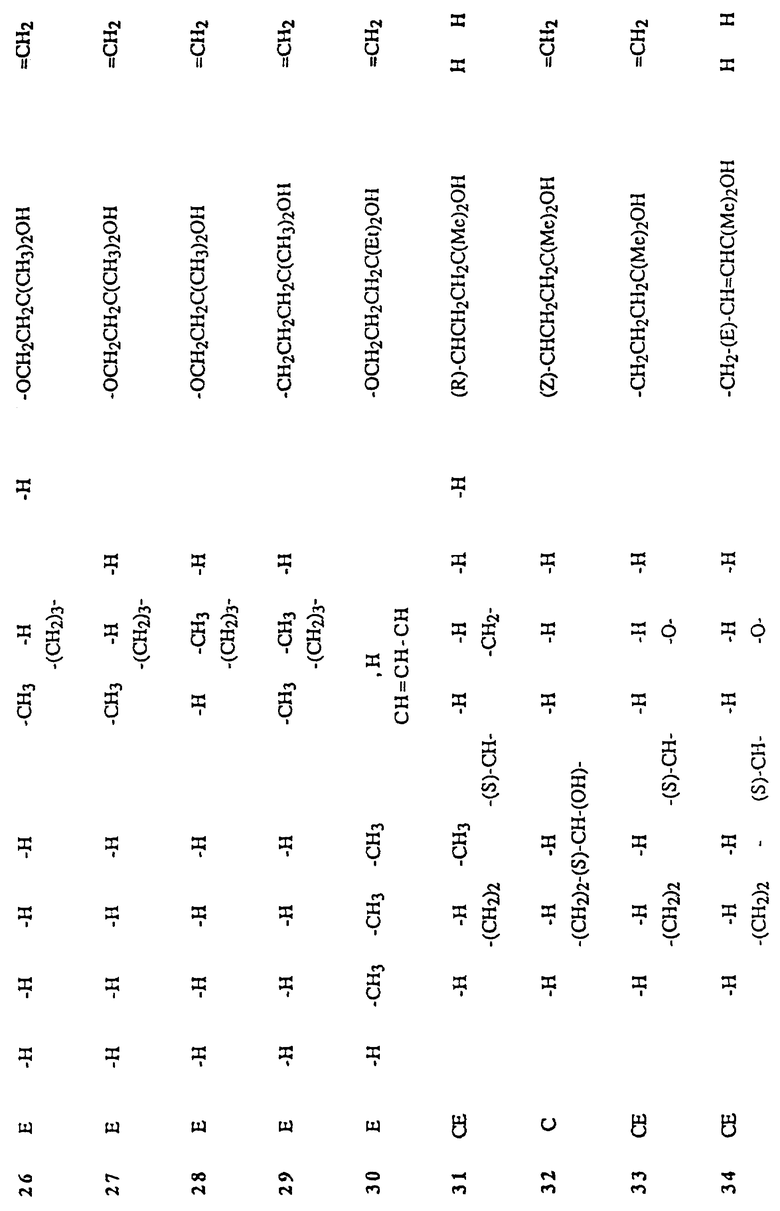




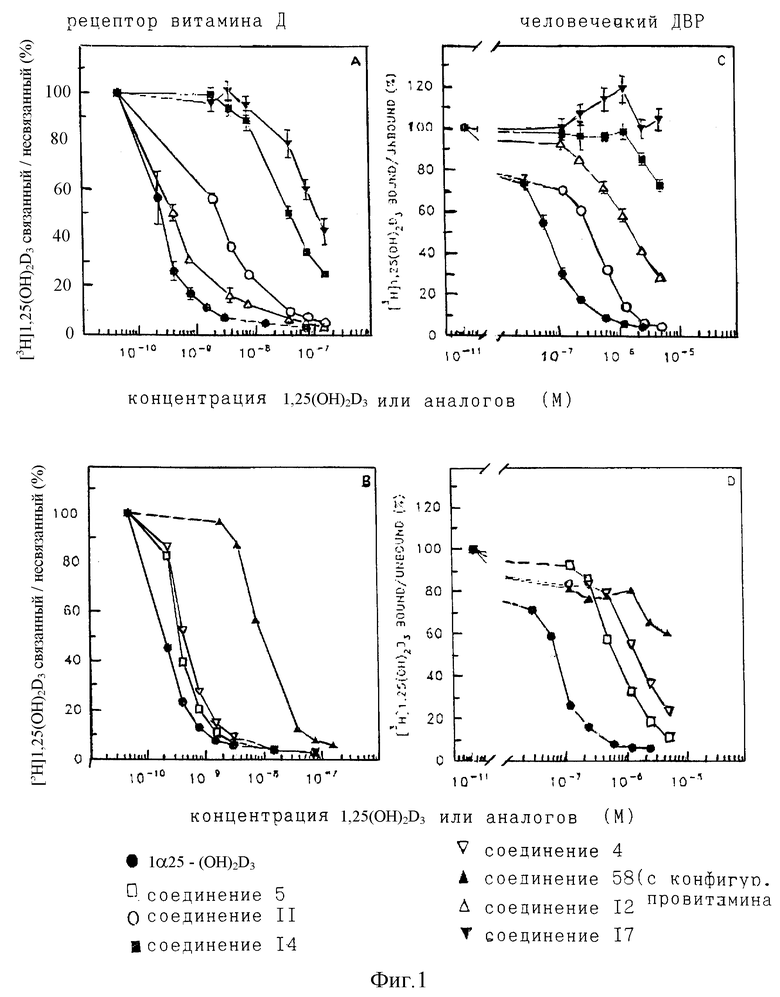


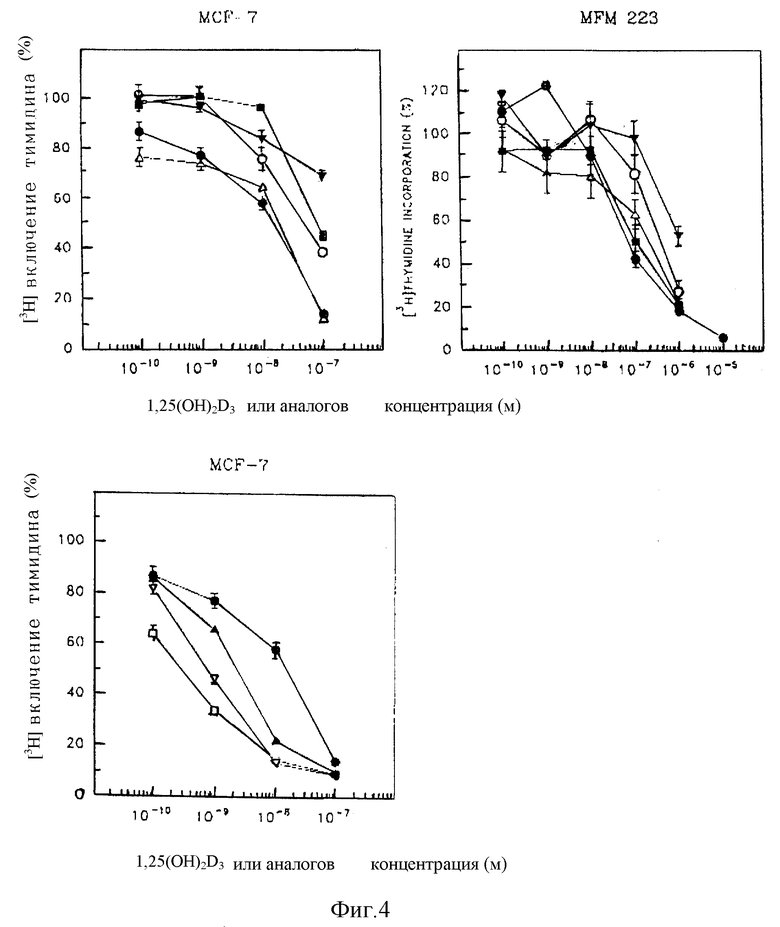


Описываются новые аналоги витамина D общей формулы I, где значения R1, R2, R3, R4, R5, R'2, R'3, R'4, R'5, X, У, У' указаны в п.1 формулы, которые проявляют селективную активность в отношении клеточных функций. Описывается способ их получения и фармацевтическая композиция на основе соединений формулы I. 10 с. и 14 з.п. ф-лы, 5 ил., 3 табл.

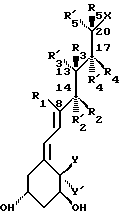
в которой Y и Y' представляют водород или взятые вместе представляют метиленовую группу =СН2;
Х представляет гидрокси(С2-С9)алкил, гидрокси(С2-С8)алкокси; гидрокси(С2-С6)алкен, гидрокси(С2-С9)алкадиен, необязательно замещенный атомом фтора; гидрокси(С2-С8)алкин, необязательно ди-замещенный трифторметилом;
R1, R2, R'2, R3, R'3, R4, R'4, R5 и R'5 принимают одно из следующих значений: а) R1, R2, R'2, R3, R'3, R4, R'4, R5 и R'5 каждый независимо представляет водород или С1-7алкил; b) R1, R3 и R'3, взятые вместе, образуют 6-членный карбоцикл, в котором один из атомов углерода необязательно замещен гидроксилом, R2, R'2, R'3 или R3, R4, R'4, R5 и R'5, как выше определено для а); с) R'3, R5 и R'5, взятые вместе, образуют 5 - 6-членный карбоцикл Е, в котором один или два атома углерода могут быть заменены атомами кислорода, R1, R2, R'2, R3, R4, R'4 и R'5 или R5, как выше определено для а); d) R2 или R'2 и R4, взятые вместе, образуют 5-членный карбоцикл D, R1, R'2, или R2, R3, R'3, R'4, или R4, R5 и R'5, имеют значения, указанные выше для а); е) R'3, взятый вместе с R1 и R'5 или R5, образуют 9- или 10-членное бициклическое СЕ-кольцо, R2, R'2, R3, R4, R'4, R5, R'5 имеют те же значения, что и для а), или f) R1 и R'3, взятые вместе, образуют 6-членный карбоцикл С, в котором один из атомов углерода необязательно замещен гидроксилом, метилом, винилом или этинилом, и R2, R'2 и R4, взятые вместе, образуют 5- или 6-членный карбоцикл D, R'2 или R2, R3, R'4, R5 и R'5 имеют значения, указанные выше для а), при условии, что, когда R1 и R'3 образуют 6-членный карбоцикл следующей природы: (1) незамещенный и насыщенный (2) монозамещенный в С-11 или (3), имеющий двойную связь между С9 и С11, R2 и R4 не образуют 5-членный карбоцикл, если R3 представляет алкил.
где R'2, R2, R3, R'4, R4, R'5 и R5 имеют указанные значения для а) в п. 1;
Х имеет то же самое значение, что в п.1;
Y, Y' имеют то же значения, что и в п.1;
3. Соединение по п.1, где R2 и R4, взятые вместе, образуют 5- или 6-членный D-цикл, как показано в IIIb1и IIIb2, соответственно или диастереоизомер IIIb1и IIIb2

в которой R1, R'2, R3, R'3, R'4, R5 и R'5 имеют указанные значения для а) в п.1;
Х имеет то же значение, что и в п.1;
Y, Y' имеют те же значения, что и в п.1.

в которой R1, R2, R'2, R3, R4, R'4 и R5 имеют те же значения, как для а) в п.1;
Х имеет то же значение, что и в п.1;
Y, Y' имеют те же значения, что и в п.1.
в которой R'2, R3, R'4, R5 и R'5 имеют те же значения, как для а) в п.1;
Х имеет те же значения, что и в п.1;
Y, Y' имеют те же значения, что и в п.1;
n равно 2;
n' равно 1 или 2 при условии, что когда n' равно 1, R3 не алкил.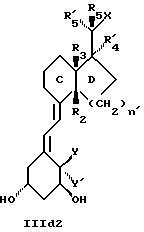
в которой R2, R3, R'4, R5 и R'5 имеют те же значения, как для а) в п.1;
Х имеет те же значения, что и по п.1;
Y, Y' имеют те же значения, что и по п.1;
n равно 1 или 2.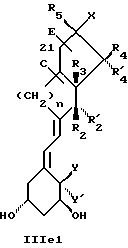
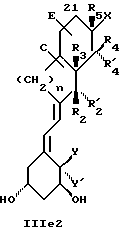
в которой n представляет собой целое число, равное 2;
R2, R'2, R3, R4, R'4, R5 имеют те же значения, что и в п.1 а);
X, Y, Y' имеют те же значения, что и в п.1.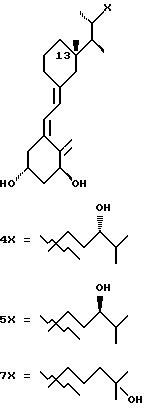
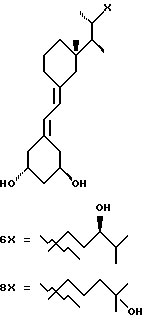
9. Соединения по 3, имеющие следующие структурные формулы: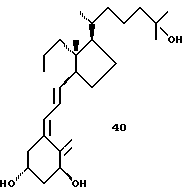
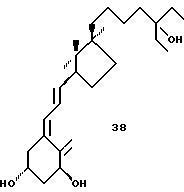
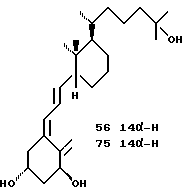
10. Соединения по п.4, имеющие структурные формулы:
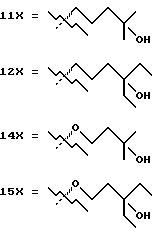

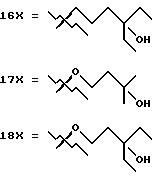

11. Соединения по п.5, имеющие следующие структурные формулы:

12. Соединения по п.6, имеющие следующие структурные формулы:
или



13. Соединение по п.12 формулы:
14. Соединения по п.12 формулы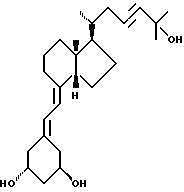
15. Соединения по п.12 формулы
16. Соединение, имеющее следующую провитамин-триеновую структуру 58:
17. Способ получения кето-альдегида 12.9 через двухстадийное последовательное превращение, отличающийся тем, что проводят кислородное расщепление витамина D2 с последующей изомеризацией катализируемой кислотой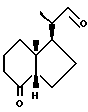
18. Способ получения аналога общей формулы I по пп.1 - 15, отличающийся тем, что соединение формулы VII, в которой R1, R'2, R2, R'3, R3, R'4, R4, R'5, R5 и Х имеют значения, как в п.1, подвергают действию соединений 13.1 или 13.2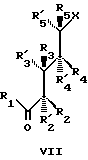


19. Соединение структуры
в котором Р представляет ацил или трет-бутилдиметилсилил;
Z представляет формил или алкоксикарбонил;
L - уходящая группа, такая, как галоген, тозил, мезил.
в котором Z и Р означает то же, что и в п.19.
в которой Р означает то же, что и по п.19.
23. Способ получения соединения по п.21, отличающийся тем, что проводят превращение формил-заместителя в алкинил-заместитель действием диметилдиазометилфосфонатом
24. Фармацевтическая композиция для ингибирования пролиферации клеток и/или индуцирования дифференциации клеток, включающая в качестве активного соединения по крайней мере одно из соединений по пп.1 - 15 и фармацевтически и/или ветеринарно приемлемый носитель или разбавитель.
| DE 3913310 A, 25.10.1990 | |||
| WO 8503299 A, 01.08.1985 | |||
| Тринус Ф.П | |||
| Фармако-терапевтический справочник | |||
| - Киев, Здоровья, 1989, с.286 | |||
| K.L.PERLMAN AND H.F.DELUCA, s' - alpha-Hydroxy-19-nor-vitamin D C-22 aldehyde | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Способ сопряжения брусьев в срубах | 1921 |
|
SU33A1 |
