Изобретение относится к технологии получения полимера олефинов и их применения в полимерных сплавах и формованных изделиях, в частности к металлоценовому катализатору и способу получения сополимера циклоолефинов, а также к полимерному сплаву и формованному изделию, включающим по меньшей мере один сополимер циклоолефинов.
Известен металлоценовый катализатор для получения сополимера циклоолефинов, включающий по меньшей мере одно металлоценовое соединение с (замещенными) индениловыми остатками в качестве лигандов, в особенности изопропилен(циклопентадиенил)(1- инденил)дихлорцирконий (см. заявку EP N 0407870, МКИ C 08 F 32/08, 1991 г.).
Недостаток получаемых при помощи известного металлоценового катализатора сополимеров циклоолефинов, которые могут применяться для получения полимерных сплавов и формованных изделий, состоит в том, что их физико-механические свойства являются не совсем удовлетворительными. То же самое относится и к их активности полимеризации.
Задачей настоящего изобретения является предоставление катализатора, с помощью которого возможно получение сополимера циклоолефинов с улучшенными физико-механическими свойствами, в особенности, полиолефинов с повышенной прочностью на разрыв и повышенной прозрачностью, и, кроме того, более высокой активности полимеризации.
Поставленная задача решается предлагаемым металлоценовым катализатором для получения сополимера циклоолефинов, представляющим собой стерически жесткое металлоценовое соединение, содержащее в качестве лигандов по меньшей мере две замещенные или незамещенные циклопентадиенильные группы, связанные между собой через моноциклическую кольцевую систему, причем одна циклопентадиенильная группа анеллирована к моноциклической системе, и эта система лигандов отлична от 4-( η5- -3-алкил-циклопентадиенил)- 4,6,6-триметил-( η5- 2-алкил-4,5-тетрагидропенталена) и представляет собой соединение формулы (I)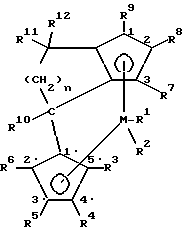
где M - металл группы IVб Периодической системы,
R1 и R2 одинаковы или различны и означают атом галогена, алкил с 1-10 атомами углерода, или группу NR2 13, где R13 означает алкил с 1-10 атомами углерода,
R3, R4, R5, R6, R7, R8 и R9 одинаковы или различны и означают атом водорода, алкил с 1-10 атомами углерода, или остаток -R13-SiR3 13-, где R13 имеет вышеуказанное значение,
R10 - алкил с 1-20 атомами углерода,
R11 и R12 одинаковы или различны и означают алкил с 1-20 атомами углерода, арил с 6-20 атомами углерода, алкенил с 2-12 атомами углерода,
n - целое число от 1 до 10.
Моноциклическая система предпочтительно имеет 6 кольцевых атомов.
Предпочитается металлоценовый катализатор вышеприведенной формулы (I), в которой
M означает цирконий,
R1 и R2 одинаковы и означают атом галогена,
R3 R4 R5, R6, R7, R8 и R9 одинаковы или различны и представляют собой водород, алкил с 1-4 атомами углерода,
R10 - алкил с 1-6 атомами углерода,
R11 и R12 одинаковы или различны и означают метил или фенил,
n - 2.
Предлагаемый металлоценовый катализатор может быть нанесен на носитель и/или форполимеризован.
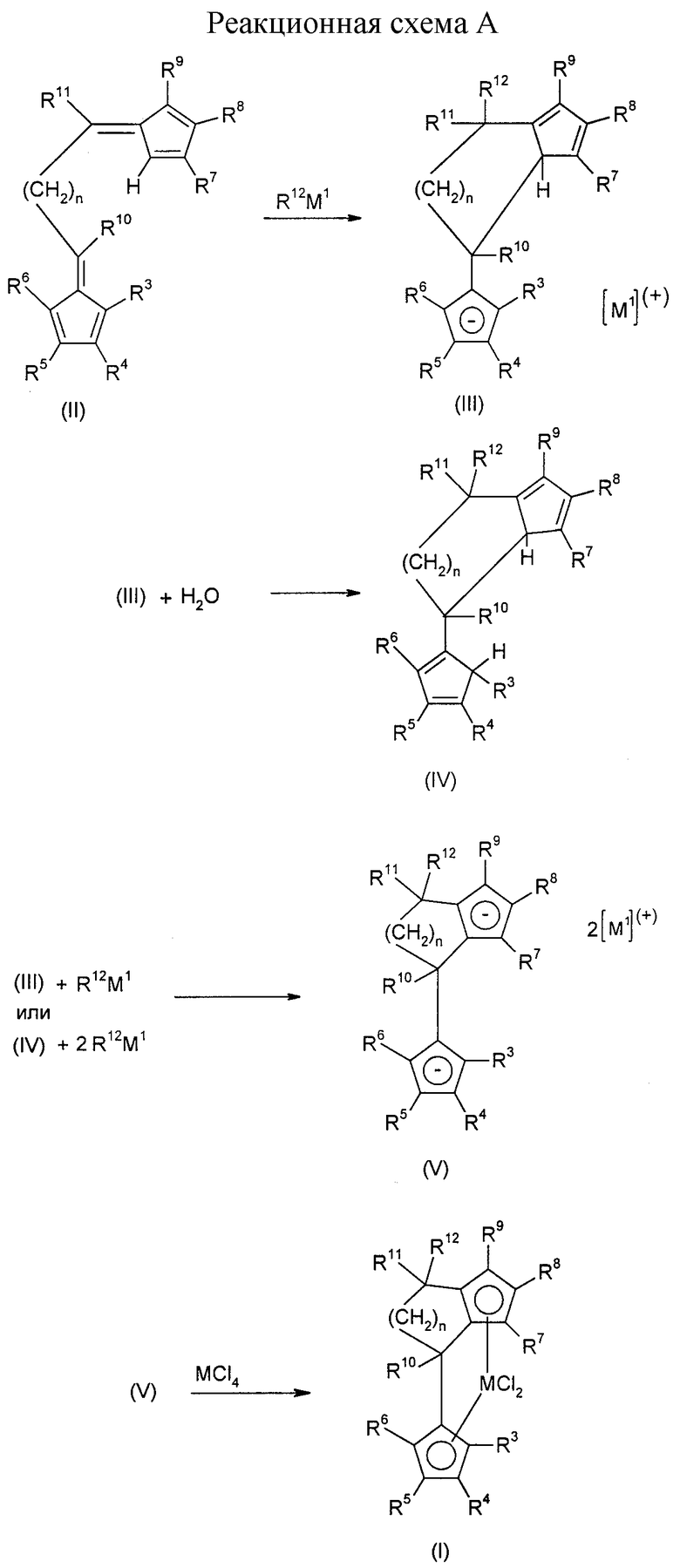
Получение предлагаемого металлоценового катализатора вышеприведенной формулы (I) поясняется реакционной схемой A (см. в конце описания). При этом M1 означает металл основной группы Iа, IIа или IIIa.
Дифульвены формулы (II) получаются из дикетонов (см. Chem. Ber. 114, 1226 (1981); там же 109, 3426 (1976); там же 107, 2453 (1974) или кетоальдегидов по известным из литературы способам (см. J. Org. Chem. 57 (1992) 2504; там же, 49 (1984) 1849: журнал Химия, 46 (1992) 377).
Превращение дифульвена (II) до лигандной системы формулы (III) осуществляют путем взаимодействия с металлоорганическим соединением (таким, как, например, метиллитий, бутиллитий, фениллитий) или реагентом Гриньяра.
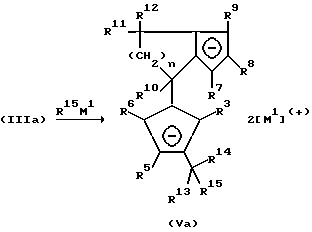
Соли формулы (III) могут подвергаться непосредственному превращению до соответствующих дианионных соединений формулы (V) путем депротонирования, например, бутиллитием. Гидролиз соединения (III) приводит к образованию бис-циклопентадиенового соединения (IV), которое получается как смесь структурных изомеров и может очищаться хроматографией. Путем двухкратного депротонирования соединения (IV), например, бутиллитием, образуется дианионное соединение формулы (V).
Превращение до имеющих мостик металлоценов формулы (I), а также выделение желаемых комплексов в принципе известны. Для этого дианион формулы (V) подвергается взаимодействию с соответствующим галогенидом металла, таким, как, например, тетрахлорид циркония, в инертном растворителе. Металлоцены формулы (I) можно также синтезировать непосредственно из дифульвенов структуры (II) без выделения промежуточных продуктов.
Пригодными растворителями являются алифатические или ароматические растворители, такие, как, например, гексан или толуол, эфирные растворители, такие, как, например, тетрагидрофуран или диэтиловый эфир или галогенированные углеводороды, такие, как, например, хлористый метилен или галогенированные ароматические углеводороды, такие, как, например, о-дихлорбензол.
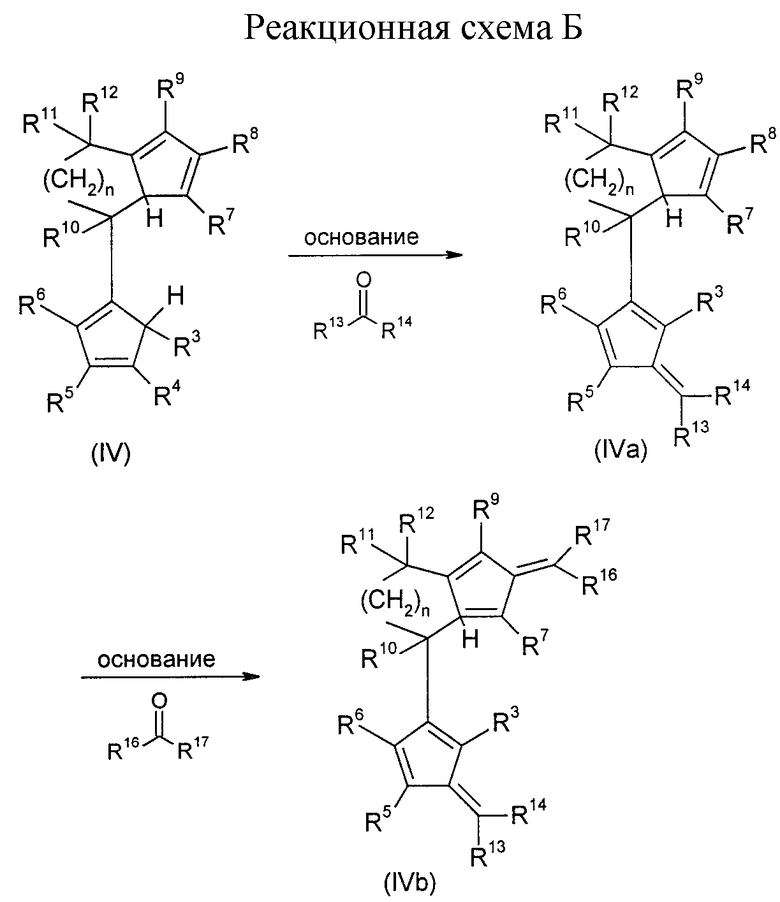
Бис-циклопентадиенильные соединения формулы (IV), при которых по меньшей мере один из остатков R3 до R6 а также по меньшей мере один из остатков R7 до R9 является водородом, и по меньшей мере один из остатков R3 до R9 отличен от водорода, могут превращаться по известным из литературы способам до фульвенов формулы (IVa) или (IVb). Это поясняется реакционной схемой Б (см. в конце описания), где R13, R14, R16 и R17 одинаковы или различны и имеют значение радикала R10.
Взаимодействие фульвена (IVa) с металлоорганическими соединениями формулы R15M1 (причем R13, R14, R15, R16 и R17 одинаковы или различны и имеют значение радикала R10; M1 имеет вышеуказанное значение) приводит к образованию моноанионного соединения (IIIa).
Применение двух эквивалентов соединения формулы R15M1 приводит непосредственно к образованию дианионного соединения (Va):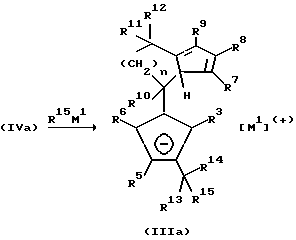
Превращение фульвена (VIb) приводит, соответственно превращению (IVa), к образованию дианионного соединения (Vb).
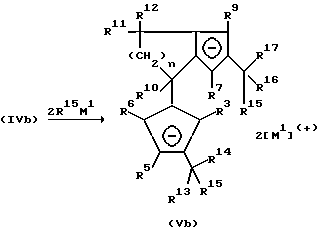
Бис-циклопентадиенильные анионы формулы (Vb) могут подвергаться взаимодействию с соединениями Rp 18MX,
где M - имеет вышеуказанное значение, X - удаляемая группа, такая, как галоген, тозилат, трифторметансульфонат, R18 имеет значение радикала R10, и p - целое число от 1 до 5.
Это поясняется следующей реакционной схемой: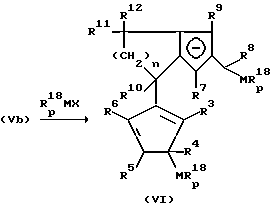
Соединения формулы (VI), при которых по меньшей мере один из остатков R3 до R6, а также по меньшей мере один из остатков R7 до R9 является водородом, могут превращаться до металлоценов согласно изобретению.
Соли соединений формулы (IIIa) могут непосредственно превращаться до соответствующих дианионовых соединений формулы (Va) путем депротонирования, например, бутиллитием. Превращение до имеющих мостик металлоценов формулы (I) осуществляется соответственно превращению соединения формулы (V) до целевого соединения формулы (I).
Дальнейшая возможность получения металлоценовых соединений согласно изобретению состоит в том, что моноциклические системы, к которым анеллирована циклопентадиенильная группа, причем эти моноциклические системы имеют функциональные группы, которые могут служить уделяемыми группами в реакциях замещения (такими, как, например, бромид или тозилат), подвергают взаимодействию, например, с циклопентадиенильными или инденил-литиевыми соединениями.
Металлоцены согласно изобретению представляют собой высокоактивные катализаторы для полимеризации циклоолефинов. В зависимости от структуры замещения лигандов металлоцены могут получаться как смесь изомеров. Металлоцены преимущественно применяются в виде чистых изомеров. Но применение рацемата является, в большинстве случаев, достаточным.
Можно, однако, применять и чистый энантиомер в (+)- или (-)- форме. С помощью чистых энантиомеров возможно получение оптически активного полимера. Рекомендуется, однако, разделение конфигурационных изомерных форм металлоценов, так как ответственный за полимеризацию центр (атом металла) в этих соединениях создает полимер, имеющий другие свойства. Для определенных областей применения, например, мягких формованных изделий, это может быть вполне желательным.
Металлоценовый катализатор согласно изобретению может включать также еще сокатализатор.
В принципе, в качестве сокатализатора пригодно любое соединение, которое благодаря своей кислотности по Льюсу может переводить нейтральный металлоцен в катион и стабилизировать последний ("лабильная координация"). Сверх этого, сокатализатор или образовавшийся из него анион не должен вступать в дальнейшую реакцию с образовавшимся металлоценовым катионом. Как сокатализатор предпочтительно применяется алюминиевое соединение и/или соединение бора, в частности применяют алюмоксан. При этом предпочитают линейный алюмоксан формулы (VII) и/или циклический алюмоксан формулы (VIII),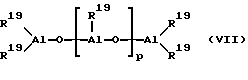
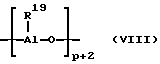
где остатки R19 одинаковы или различны и означают водород или углеводородную группу с 1-20 атомами углерода, такую, как алкил с 1 - 18 атомами углерода, арил с 6-18 атомами углерода или бензил, а p - целое число от 2 до 50, предпочтительно 10 до 35.
Предпочтительно остатки R19 одинаковы и означают водород, метил, изобутил, фенил или бензил, особенно предпочтительно метил.
В случае если остатки R19 различны, то они предпочтительно означают метил и водород или альтернативно метил и изобутил, причем содержание водорода или изобутила в остатках R19 предпочтительно составляет от 0,01 до 40%.
Способы получения алюмоксанов известны. Точная пространственная структура алюмоксанов неизвестна (см. J. Am. Chim. Soc. (1993) 115, 4971). Является, например, мыслимым, что цепи и кольца объединяются с образованием двухмерных или трехмерных структур.
Независимо от способа получения, всем алюмоксановым растворам свойственно меняющееся содержание непрореагировавшегося исходного алюминиевого соединения, имеющегося в свободном виде или в качестве аддукта.
Перед применением для осуществления реакции полимеризации металлоценовое соединение можно предварительно активировать сокатализатором, в особенности, алюмоксаном, что позволяет заметно повысить его активность. Предварительная активация металлоценового соединения осуществляется предпочтительно в растворе. При этом металлоценовое соединение предпочтительно растворяется в растворе алюмоксана в инертном углеводороде. Как инертный углеводород годится алифатический или ароматический углеводород. Предпочтительно используется толуол.
Концентрация алюмоксана в растворе составляет примерно от 1 вес.% до предела насыщения, предпочтительно от 5 до 30 вес.%, в пересчете на общее количество раствора. Металлоцен может применяться в такой же концентрации, предпочтительно, однако, он применяется в количестве от 10-4 до 1 моль на моль алюмоксана. Продолжительность предварительной активации составляет от 5 минут до 60 часов, предпочтительно 5 - 60 минут. Рабочая температура составляет от -78 до +100oC, предпочтительно 0 - 70oC.
С помощью металлоцена можно осуществлять форполимеризацию, для чего предпочтительно используют используемый для полимеризации циклоолефин, или один из используемых для полимеризации циклоолефинов.
Металлоцен можно нанести на носитель, такой, как, например, силикагели, окиси алюминия, твердый алюминоксан, другие неорганические носители или также полиолефин в виде мелкодисперсного порошка.
В том случае если к реакционной смеси добавляется растворитель, то в качестве растворителя используют известный инертный растворитель, как, например, алифатические или циклоалифатические углеводороды, фракции бензина или гидрированные фракции дизельного масла, или толуол.
Металлоцены предпочтительно используют в виде их рацематов. Металлоценовое соединение применяется предпочтительно в концентрации, в пересчете на переходный металл, 10-3 - 10-8, предпочтительно 10-4 - 10-7 моль переходного металла на дм3 растворителя или на дм3 объема реактора. Алюмоксан используется предпочтительно в концентрации 10-4 - 10-1 моль, предпочтительно 10-3 - 10-2 моль на дм3 объема реактора, в пересчете на содержание алюминия. В принципе, однако, возможны также более высокие концентрации.
Дальнейшим объектом изобретения является способ получения сополимера циклоолефинов путем полимеризации по меньшей мере одного циклоолефина, который заключается в том, что в качестве металлоценового катализатора используют стерически жесткое металлоценовое соединение вышеприведенной общей формулы (I).
При осуществлении предлагаемого способа предпочтительно используют один или несколько полициклических олефинов, в частности, нижеприведенных формул (IX) - (XIV),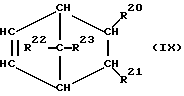
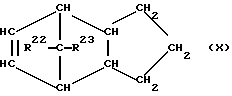
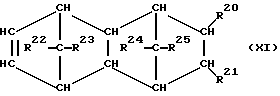
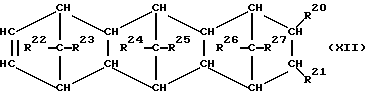
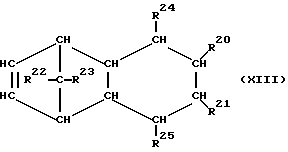
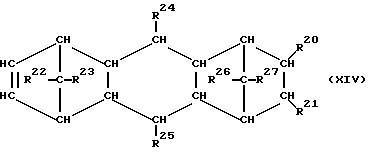
в которых R20 - R27 одинаковы или различны и означают атом водорода или углеводородный остаток с 1-20 атомами углерода, такой, как алкил с 1-8 атомами углерода или арил с 6-10 атомами углерода, или два или больше из остатков R20 - R27 вместе образуют циклическую систему с 4-40 атомами углерода, причем одинаковые остатки R20 - R27 в различных формулах могут иметь различное значение. Особенно предпочтительными являются циклоолефины формул (X) или (XII), где R20 - R27 одинаковы или различны и означают атом водорода или углеводородный остаток с 1- 20 атомами углерода, в частности, арильный остаток с 6-10 атомами углерода, или алкильный остаток с 1-8 атомами углерода, причем одинаковые остатки R20 - R27 в различных формулах могут иметь различное значение.
В случае необходимости, при получении сополимеров циклоолефинов применяется также моноциклический олефин формулы (XV)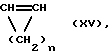
где n означает число 2-10.
Кроме того, при получении сополимеров циклоолефинов применяют один или несколько ациклических 1-олефинов, предпочтительно имеющих формулу (IV)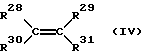
где R28 - R31 одинаковы или различны и означают атом водорода или углеводородный остаток с 1-20 атомами углерода, предпочтительно арильный остаток с 6-10 атомами углерода, и алкил с 1-8 атомами углерода. Предпочитается этилен.
В частности сополимеры полициклических олефинов, предпочтительно формул (IX) и (XI), получают с применением этилена.
Особенно предпочтительными полициклическими олефинами являются норборнен и тетрациклододецен, которые могут быть замещены алкилом с 1-6 атомами углерода. Сополимеризуют их предпочтительно этиленом, причем особое значение имеют сополимеры этилена и норборнена.
Полициклический олефин применяют в количестве от 0,1 до 99,9 вес.%, моноциклический олефин - в количестве от 0 до 99,9 вес.%, а ациклический олефин - в количестве от 0,1 до 99,9 вес.%, каждый раз в пересчете на общее количество мономеров.
Концентрация применяемого ациклического олефина зависит от его растворимости в реакционной среде при конкретных давлении и температуре.
Под полициклическими олефинами, моноциклическими олефинами и ациклическими олефинами следует понимать также смеси двух или нескольких олефинов соответствующего типа. То есть, согласно предлагаемому способу кроме полициклических сополимеров можно также получать тройные полимеры или полимеры на основе по меньшей мере четырех мономеров. Кроме того, по вышеописанному способу можно получать также сополимеры моноциклических и ациклических олефинов.
Среди моноциклических олефинов предпочитают циклопентен, который может быть замещенным.
Предлагаемый способ осуществляют предпочтительно при температурах -78 - 150oC, в особенности, 0-100oC, и давлении 0,01 - 64 бар.
Полимеризацию проводят в среде самого жидкого циклоолефина или в растворе циклоолефина, причем целесообразно давление превышает 1 бар.
При получении сополимеров молярное соотношение полициклического олефина к применяемому олефину с открытой цепью можно варьировать в широких пределах. Предпочтительно молярное соотношение циклоолефина и олефина с открытой цепью составляет от 3: 1 до 100:1. Количество втроенного сомономера можно почти в любой мере регулировать путем выбора соответствующих температуры полимеризации, концентрации компонентов катализатора, молярного соотношения и давления газообразного олефина с открытой цепью. Предпочитают содержание циклических компонентов в пределах 20 - 80 моль.%, в частности 40 - 60 моль. %.
Полимеризацию можно также осуществлять многоступенчатой, причем могут получаться также блок-сополимеры.
Средний молекулярный вес образовавшегося полимера можно регулировать известным образом также путем дозировки водорода, варьирования концентрации катализатора или варьирования температуры.
Полидисперсность Mw/Mn сополимеров циклоолефинов имеет величину от 1,9 до 3,5, то есть, колеблется лишь в узких пределах. Благодаря этому эти сополимеры в особой степени пригодны к литью под давлением.
Путем предлагаемого способа можно получить аморфные сополимеры циклоолефинов, не включающие частично кристаллических полимеров этилена. Эти сополимеры являются прозрачными и твердыми, и они поддаются термопластичной переработке. Удлинение при разрыве (согласно промышленному стандарту Германии DIN 53457) находится в пределах от 50 до 100 МПа, предпочтительно между 55 и 70 МПа. Как при экструзии, так и при литье под давлением при температуре 300oC не обнаруживается ни реакции разложения, ни уменьшение вязкости.
Получаемые согласно изобретению сополимеры циклоолифенов особенно годятся для получения формованных изделий, таких, как полученные путем экструзии изделия (например, пленки, рукава, трубы, прутки и волокна) или полученные путем литья под давлением изделия любого вида и размера. Пленки могут быть экструдированными пленками, каландированными листами, поливными пленками, пленками с моно- и двухаксиальной ориентацией или многослойными пленками, пригодными, в частности, для упаковки пищевых продуктов или в качестве черновых упаковок. Они обладают высоким гидроизоляционным действием и низкой газопроницаемостью. Получаемые согласно изобретению сополимеры циклоолефинов годятся также как добавки в других полимерных пленках (в частности, полиолефиновых пленках, таких, как полипропиленовые пленки или полиэтиленовые пленки), например, с целью улучшения текучести, улучшения лакируемости, влияния на модуль упругости и получения непрозрачных пленок.
Важным свойством получаемых по способу согласно изобретению сополимеров циклоолефинов является их прозрачность. Поэтому особое значение имеет применение экструдированных или полученных путем литья под давлением изделий из сополимера циклоолефинов в области оптики. Определяемый при помощи рефрактометра Аббе и смешанного света показатель преломления описываемых нижеследующих примерах продуктов реакции находится в пределах от 1,520 до 1,555. Следовательно, показатель преломления лишь в незначительной мере отличается от показателя преломления крона (n = 1,51), благодаря чему продукты согласно изобретению можно применять вместо стекла для разных целей, например, как линзы, призмы, несущие пластинки и пленочные подложки для оптических ЗУ, видеопластинок, компакт-дисков, как покровные и фокусирующие пластинки для солнечных элементов, как покровные и рассеивающие стекла в области мощной оптики, как лучевые волноводы в виде волокна или пленок.
В модифицированном в отношении ударной вязкости виде полученные по способу согласно изобретению сополимеры циклоолефинов могут использоваться в качестве структурных материалов в различных технических областях.
Полученные по способу согласно изобретению сополимеры циклоолефинов могут использоваться также для получения полимерных сплавов. Сплавы можно получать в расплаве или в растворе. Каждый сплав имеет выгодную для определенной области применения комбинацию свойств компонентов. Для сплавов, включающих сополимеры циклоолефинов, полученные по способу согласно изобретению, применяются предпочтительно следующие полимеры:
полиэтилен, полипропилен, сополимеры этилена и пропилена, полибутилен, поли-(4-метил-1-пентен), полиизопрен, полиизобутилен, натуральный каучук, поли-(метил-метакрилат), дальнейшие полиметакрилаты, полиакрилаты, сополимеры акрилата и метакрилата, полистирол, сополимеры стирола и акрил-нитрила, бис-фенол-A- поликарбонат, дальнейшие поликарбонаты, ароматические сложные полиэфиркарбонаты, полиэтилентерефталат, полибутилен-терефталат, аморфные полиакрилаты, полиамид-6, полиамид-66, дальнейшие полиамиды, полиарамиды, полиэфиркетоны, полиоксиметилен, полиокси-этилен, полиуретаны, полисульфоны, полиэфирсульфоны, поливинилиден-фторид.
Способ согласно изобретению дает возможность получения, при высокой активности, в частности, прозрачных сополимеров циклоолефинов, обладающих высокой прочностью на разрыв.
Указанные в нижеследующих примерах температуры стеклования Tg были определены путем дифференциально-сканирующей калометрии при скорости нагревания 20oC/мин. Указанные коэффициенты вязкости были определены согласно промышленному стандарту Германии DIN 53728. Механические свойства были измерены путем опыта удлинения при растяжении (согласно промышленному стандарту Германии DIN 50457, 4302).
Критерием активности катализатора служит выход полимера на единицу времени и ммоль металлоцена: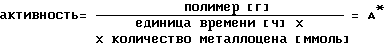
Нижеследующие примеры служат для более подробного пояснения изобретения.
Получение металлоорганических соединений и обращение с ними проводят с исключением воздуха и влаги в атмосфере аргона (техника Шленка). Все требуемые растворители до использования абсолютируют путем многочасового кипячения над пригодным осушителем и последующей дистилляции в атмосфере аргона.
Получение исходных дикетонов и кетоальдегидов осуществляют по известным в литературе методам. Циклопентадиен и метилциклопентадиен получают путем крекинга димеров и хранят при -35oC.
Определение соотношения Al/CH3 в алюмоксане осуществляют путем разложения пробы серной кислотой и определения объема образующихся газов в нормальных условиях, а также путем комплексометрического титрования алюминия в растворенной пробе по Шварценбаху.
Целевые соединения идентифицируют путем 1H-ЯМР, 13C-ЯМР и ИК-спектроскопии.
Нижеследующие примеры поясняют не только получение целевых продуктов формулы (I), но и их исходных и промежуточных продуктов. Для названия промежуточных и целевых продуктов употреблена номенклатура МСТПХ (Междунар. союз по теоретической и прикладной химии).
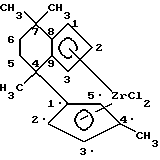
Данная номенклатура поясняется на примере соединения [4-( η5- 4'-метил-циклопентадиенил)-4,7,7-триметил-( η5- 4,5,6,7- тетрагидроинденил]-дихлорциркония. Циклическая система, которая является мостиком обоих циклопентадиенильных лигандов этого соединения, имеет шесть кольцевых атомов углерода (C4, C5, C6, C7, C8, C9) и три метиловых заместителя. Одна циклопентадиениловая группа однократно анеллирована к циклической системе, вторая находится как заместитель на циклической системе.
А. Получение бис-фульвенов формулы (II)
Пример 1а
Синтез 2,5-бис-(2,4-циклопентадиен-1-илиден)гексана
11,0 г (96,3 ммоль) 2,5-гександиона и 12,7 г (193 ммоль) свежеполученного путем крекинга циклопентадиена растворяют в 60 мл метанола, охлаждают до 0oC и смешивают с 8,60 г (121 ммоль) пирролидина. После 90-минутного перемешивания при 0oC реакционный раствор гидролизуют 5 мл ледяной уксусной кислоты и 50 мл воды, подвергают двухкратному экстрагированию, каждый раз 70 мл диэтилового эфира, а объединенные органические фазы промывают насыщенным раствором хлористого натрия, сушат над сульфатом магния и растворитель удаляют в вакууме. Получают 18,0 г (89%) дифульвена в качестве маслянистого остатка оранжево-красного цвета.
Пример 1б
Синтез 2,5-бис-(циклопента-2,4-диен-1-илиден)ундекана
Раствор 3,50 г (19,9 ммоль) 2,5-ундекандиона в 100 мл метанола и 10 мл тетрагидрофурана охлаждают до 0oC и смешивают с 3,92 мл (3,14 г, 47,5 ммоль) свежеполученного путем крекинга циклопентадиена. К оранжево-красному прозрачному реакционному раствору в течение 10 мин добавляют каплями 6,28 мл (5,40 г, 76,0 ммоль) свежедистиллированного пирролидина. При этом реакционный раствор в течение 10 мин окрашивается в темно-красный цвет. После этого реакционному раствору дают нагреваться до комнатной температуры и для завершения реакции перемешивают в течение дальнейших 3 дней. В целях переработки пирролидин нейтрализуют 4 мл ледяной уксусной кислоты и гидролизуют 100 мл воды. Экстрагируют два раза, каждый раз 100 мл пентана, промывают объединенные органические фазы несколько раз насыщенным водным раствором хлористого натрия и сушат над сульфатом магния. После удаления растворителя в вакууме получают 4,16 г (78%) сырого циклопентадиенилиден в качестве темно-красного масла.
В результате хроматографии на колонке, содержащей силикагель, дезактивированной триэтиламином, с применением в качестве элюента смеси пентана и диэтилового эфира в соотношении 100:1, получают дифульвен в качестве оранжево-красного масла.
Б. Синтез имеющих мостик бис-циклопентадиенильных анионов формулы (V)
Пример 2
Синтез 4-(η5- циклопентадиенил)-4,7,7-триметил- ( η5- 4,5,6,7-тетрагидроилиденил)-дилития
К раствору 10,0 г (47,5 ммоль) 2,5-бис-(2,4-циклопентадиен-1- илиден)гексана в 150 мл диэтилового эфира при интенсивном перемешивании медленно добавляют каплями 62,4 мл (99,8 ммоль) 1,60 М эфирного раствора метиллития при 0oC. Реакционному раствору дают нагреваться до комнатной температуры.
После 24-часового перемешивания получают осадок бежевого цвета. После фильтрации и многократной промывки пентаном получают 13,2 г (89%) дилитиевой соли в качестве порошка бежевого цвета, которая координирована с молярным эквивалентом диэтилового эфира.
Пример 3
Синтез 4-( η5- циклопентадиенил)-4,7-диметил-7-фенил-( η5- 4,5,6,7-тетрагидроилиденил)-дилития
83,4 мл (74,3 моль) 0,89 М раствора фениллития в диэтиловом эфире добавляют каплями при 0oC к раствору 7,10 г (33,7 ммоль) дифульвена по примеру 1 в 100 мл диэтилового эфира. При этом приблизительно через 5 минут начинается выпадение осадка бежевого цвета. Реакционному раствору дают нагреваться до комнатной температуры и перемешивают в течение дальнейших 12 часов при 25oC. После фильтрации, многократной промывки пентаном и сушки в вакууме, создаваемом при помощи масляного насоса, получают 10,3 г (82%) дилитиевой соли в качестве порошка бежевого цвета, имеющей высокую чувствительность к гидролизу.
Пример 4
Синтез 4-( η5- циклопентадиенил)-4,7-диметил-7-бутил-( η5- 4,5,6,7-тетрагидроилиденил)-дилития
Раствор 15,0 г (71,3 ммоль) дифульвена по примеру 1, растворенного в 100 мл диэтилового эфира, охлаждают до -30oC и, при интенсивном перемешивании, медленно смешивают с 94 мл (150 ммоль) 1,60 М раствора н-бутиллития в гексане. При этом образуется осадок лимонно-желтого цвета. Реакционному раствору дают нагреваться до комнатной температуры и перемешивают для завершения реакции еще в течение дальнейших 24 часов. После этого выпавший осадок отфильтровывают, многократно промывают пентаном и сушат в вакууме, создаваемом при помощи масляного насоса. Получают 29,0 г (91%) дилитиевой соли в качестве имеющего высокую чувствительность к гидролизу порошка бежевого цвета, с которым еще координирован молярный эквивалент диэтилового эфира.
В. Синтез имеющих мостик циклопентадиенов формулы (IV)
Пример 5 Синтез 4-циклопентадиенил-4,7,7-триметил-4,5,6,7- тетрагидро-1H-индена
К суспензии 7,35 г (23,5 ммоль) дилитиевой соли по примеру 2 в 50 мл диэтилового эфира добавляют каплями при 0oC 50 мл дегазированной воды. При этом суспензия бежевого цвета сразу исчезает и получают прозрачную оранжевого цвета фазу диэтилового эфира. Затем фазы разделяют в делительной воронке, водную фазу экстрагируют еще два раза, каждый раз 25 мл диэтилового эфира и объединенные органические фазы промывают 20 мл насыщенного раствора хлористого натрия. После сушки над сульфатом магния и удаления растворителя в вакууме получают 5,1 г (96%) гидролизованного продукта в качестве масла оранжево-красного цвета.
Пример 6 Получение 4-циклопентадиенил-4,7-диметил-4-фенил- 4,5,6,7-тетрагидро-1H-индена
Охлажденную до 0oC желтую суспензию 3,64 г (9,72 ммоль) дилитиевой соли по примеру 3 в 50 мл диэтилового эфира гидролизуют путем медленного добавления 20 мл дегазированной воды. При этом суспензия исчезает, и получают прозрачный раствор оранжевого цвета. После двухкратной экстракции, каждый раз 20 мл диэтилового эфира, объединенные органические фазы многократно промывают насыщенным водным раствором хлористого натрия и сушат над сульфатом магния. Путем последующего удаления растворителя в вакууме получают 2,62 г (94%) гидролизованного продукта в качестве оранжевого масла.
Пример 7 Получение 4-циклопентадиенил-4,7-диметил-4-бутил- 4,5,6,7-тетрагидро-1H-индена
Охлажденную до 0oC желтую суспензию 5,00 г (17,33 ммоль) дилитиевой соли по примеру 4 в 50 мл диэтилового эфира гидролизуют путем медленного добавления 20 мл дегазированной воды. При этом суспензия исчезает, и получают прозрачный раствор оранжевого цвета. После двухкратной экстракции, каждый раз 20 мл диэтилового эфира, объединенные органические фазы многократно промывают насыщенным водным раствором хлористого натрия и сушат над сульфатом магния. Путем последующего удаления растворителя в вакууме получают 4,59 г (96%) гидролизованного продукта в качестве оранжевого масла.
Г. Синтез имеющих мостик циклопентадиеново-фульвеновых лигандов формулы (IVa) путем дополнительного введения заместителей (введение различных остатков R12, R13, R14, R16, R17)
Пример 8a
Синтез 4-(3'-изопропилиден-циклопента-1,4-диенил)-4,7,7- триметил-4,5,6,7-тетрагидро-1H-индена
7,70 г (34,0 ммоль) циклопентадиенилтетрагидроинденила по примеру 5 растворяют в 70 мл метанола и охлаждают до 0oC. Затем к оранжево-красному реакционному раствору добавляют последовательно 2,96 г (51,0 ммоль) ацетона и 4,83 г (68,0 ммоль) пирролидина. После 5-часового перемешивания при 0oC для завершения реакции продолжают перемешивать в течение 2 часов при комнатной температуре, после чего реакцию прекращают путем добавления 4 мл ледяной уксусной кислоты. Красный прозрачный реакционный раствор гидролизуют 200 мл воды и получаемую при этом желтую суспензию подвергают трехкратной экстракции диэтиловым эфиром, взятым в количестве по 50 мл. После многократной промывки объединенных органических фаз насыщенным водным раствором хлористого натрия и сушки над сульфатом магния
получают 8,00 г (88%) целевого фульвена в качестве воскообразного осадка оранжево-красного цвета.
Пример 8б
Получение 4-циклопентадиенил-4.7-диметил-7-аллил-4,5,6,7- тетрагидро-1H-индена
К раствору 16,8 г (79,8 ммоль) 2,5-бис-(2,4-циклопентадиен-1- илиден)гексана по примеру 1, растворенного в 100 мл диэтилового эфира, и 50 мл тетрагидрофурана добавляют каплями, интенсивно перемешивая, при 0oC в течение часа 0,60 М раствора аллилового реагента Гриньяра (175 ммоль) в диэтиловом эфире. После окончания добавления перемешивают при комнатной температуре в течение ночи, получаемую суспензию желто-оранжевого цвета охлаждают до 0oC и осторожно гидролизуют водным насыщенным раствором хлористого аммония. Органическую фазу отделяют, три раза промывают, каждый раз 50 мл насыщенного водного раствора хлористого натрия и сушат над сульфатом магния. После удаления растворителя в вакууме масляного насоса получают 17,5 г (87%) продукта в качестве масла оранжевого цвета.
Д. Синтез дианионных комплексов формулы (Va)
Пример 9а
Синтез 4-[3'-трет-бутил-( -η5- циклопентадиенил)]- 4,7,7-триметил-( η5- 4,5,6,7-тетрагидро-инденил)дилития
При взаимодействии тетрагидроинденилфульвена по примеру 8 с 2 эквивалентами эфирного раствора метиллития при 0oC получают уже через несколько секунд осадок интенсивного желтого цвета. Перемешивают еще в течение 12 часов при комнатной температуре. После фильтрации, промывки пентаном и сушки в вакууме, создаваемом при помощи масляного насоса, получают дилитиевую соль, которая без предварительной идентификации далее превращается.
Пример 9б
Синтез 4-( -η5- циклопентадиенил)]-4,7-диметил-7- аллил-( η5- 4,5,6,7-тетрагидро-инденил)дилития
10,5 г продукта по примеру 8б растворяют в 100 мл диэтилового эфира, охлаждают до 0oC, затем добавляют каплями 57,6 мл раствора н-бутиллития (1,60-молярного в гексане, 92,0 ммоль). После перемешивания в течение 18 часов при комнатной температуре получают осадок желто-бежевого цвета, который отфильтровывают, многократно промывают пентаном и сушат в вакууме, создаваемом при помощи масляного насоса. Дилитиевую соль получают в качестве твердого вещества бежевого цвета, с которым координирован еще один молярный эквивалент диэтилового эфира. Выход: количественный.
Е. Синтез металлоценов формулы (I)
Пример 10
Синтез 4-( η5- циклопентадиенил)-4,7,7-триметил-( η5- 4,5,6,7- тетрагидроинденил)-дихлорциркония
К охлажденной до -78oC суспензии 9,58 г (30,7 ммоль) дилитиевого соединения по примеру 2 в 200 мл толуола прибавляют порциями в течение 10 мин 7,50 г (32,2 ммоль) тетрахлорида циркония. После 50-часового перемешивания при комнатной температуре осадок отфильтровывают и фильтрат оранжевого цвета сгущают досуха в вакууме. После многократной промывки пентаном получают 4,38 г (37%) дихлорида цирконоцена в качестве порошка оранжево-желтого цвета.
Для очистки порошок оранжево-желтого цвета в течение нескольких дней экстрагируют пентаном в циркуляционной фритте, причем после удаления растворителя в вакууме, создаваемом при помощи масляного насоса, получают 1,70 г (14%) дихлорида цирконоцена в качестве желтого порошка с точкой плавления 223oC (разл., определенной путем дифференциально-сканирующей калометрии).
Пример 11
Синтез 4-( η5- циклопентадиенил)-4,7,7-триметил- ( η5- 4,5,6,7-тетрагидро-инденил)дихлортитана
Суспензию 5,46 г (17,5 ммоль) дилитиевого эфирата по примеру 2 в 200 мл толуола охлаждают до -78oC и смешивают с 3,3 г (17,5 ммоль) тетрахлорида титана. При этом реакционный раствор сразу же окрашивается в темно-красный цвет. Перемешивают в течение 30 часов при комнатной температуре, отфильтровывают от нерастворимого вещества и темно-красную толуольную фазу сгущают досуха в вакууме, создаваемом при помощи масляного насоса. После многократной промывки пентаном получают 1,85 г дихлорида титаноцена в качестве коричнево-бежевого порошка. Сырой продукт экстрагируют пентаном в течение нескольких дней в циркуляционной фритте, причем после удаления растворителя получают 780 мг (13%) дихлорида титаноцена в качестве коричневого твердого вещества с точкой плавления 259oC (разл., определенной путем дифференциально-сканирующей калометрии).
Пример 12
Синтез [4-( -η5- циклопентадиенил)]-4,7,7-триметил- ( η5- 4,5,6,7-тетрагидроинденил)] -дихлорциркония из 2,5- бис-(2,4-циклопентадиен-1-илиден)гексана
К раствору 10,0 г (47,5 ммоль) 2,5-бис-(2,4-циклопентадиен-1- илиден)гексана по примеру 1 в 150 мл толуола прибавляют медленно при интенсивном перемешивании 62,4 мл (99,8 ммоль) 1,60 М эфирного раствора метиллития при 0oC. После окончания добавления перемешивают в течение 24 часов при комнатной температуре, затем охлаждают до -30oC и добавляют 9,32 г (40 ммоль) тетрахлорида циркония. После 30- часового перемешивания при комнатной температуре отфильтровывают от хлористого лития и фильтрат сгущают досуха в вакууме. После многократной промывки пентаном получают 4,02 г (26%) дихлорида циркония.
Пример 13
Синтез обоих диастереомеров 4-( η5- циклопентадиенил)-4,7- диметил-7-фенил-( η5- 4,5,6,7-тетрагидроинденил)-дихлорциркония
К охлажденной до -78oC суспензии 4,37 г (11,7 ммоль) дилитиевой соли по примеру 3 в 200 мл толуола добавляют порциями 2,72 г (11,7 ммоль) тетрахлорида циркония. Реакционной смеси дают нагреваться до комнатной температуры и суспензию оранжевого цвета перемешивают еще в течение 20 часов при 20oC. После фильтрации из фильтрата удаляют растворитель в вакууме, создаваемом при помощи масляного насоса, и получаемый при этом маслянистый остаток оранжево-красного цвета переводят в порошок путем интенсивного перемешивания в 20 мл пентана. После удаления пентана в вакууме получают 2,64 г (50%) дихлорида цирконоцена в качестве порошка желто-оранжевого цвета. На основе данных спектра 1H-ЯМР сырого продукта соотношение диастереомеров составляет примерно 8:1.
1H-ЯМР (200.1 МГц, CDCl3): δ = 7.24-6.89 (m, 5H, Ph-H); 6.78- 6.73, 6.54-6.50, 6.22-6.20, 5.97-5.94 (4xm, 7H, 1-H до 3-H и 2'-H до 5'-H); 2.30-2.22, 1,92-1.84 (2 x m, 4H,5-H,6-H); 1.76, 1.71 млн.д. (2xs,6H, 10-H и 11-H).
Пример 14
Синтез обоих диастереомеров 4-( η5- циклопентадиенил)-4,7- диметил-7-бутил-( η5- 4,5,6,7-тетрагидроинденил)-дихлорциркония
Суспензию 7,80 г (22,0 ммоль) дилитиевой соли по примеру 4 в 200 мл толуола охлаждают до -78oC, а затем порциями добавляют 5,10 г (22,0 ммоль) тетрахлорида циркония. Реакционной смеси дают нагреваться до комнатной температуры и суспензию желто-оранжевого цвета перемешивают еще в течение 48 часов, после чего отфильтровывают от нерастворимого вещества и удаляют растворитель в вакууме, создаваемом при помощи масляного насоса. Получаемое масло красно-оранжевого цвета переводят в порошок путем интенсивного перемешивания с пентаном. Получают 2,72 г (30%) дихлорида цирконоцена, который очищают путем экстракции пентаном в течение нескольких дней в циркуляционной фритте. Спектр 1H-ЯМР тонкого желтого осадка показывает два набора сигналов в соотношении 15: 1. Из получаемого желтого фильтрата можно после хранения при -30oC получать небольшое количество кристаллов. Эти кристаллы чистых диастереомеров дихлорида цирконоцена позволяют идентификацию отдельных сигналов спектра 1H-ЯМР. При этом выкристаллизовавшиеся из пентанового раствора кристаллы соответствуют тому диастереомеру, который образуется меньше всего.
Кристаллы можно получать также из 1,35 г (14%) желтого тонкого порошка тем, что приблизительно 100 мг порошка растворяют в небольшом количестве хлористого метилена и путем диффузии пентана в этот раствор осуществляют очень медленную кристаллизацию. Главным продуктом является другой диастереомер.
1H-ЯМР (200.1 МГц, CDCl3): δ = 6.73-6.67, 6.52-6.47, 6,41-6.38, 6.08-6.04, 5.87-5.83, 5.76-5.73 (6 x m, 7H, 2'-H до 5'-H и 1-H до 3-H); 2.48-2.16 (м, 3H, 5eq-H, 5ax-H, 6ax-H): 1.91-1.84 (m, 1H, 6eq-H); 1.83 (s, 3H, 10-H): 1.35-0.85 (3 x m, 6H, nBu-CH2); 1.25 (s, 3H, 11-H); 0.82-0.77 млн.д. (pt, 3H, nBu-CH3).
Пример 15
Синтез 4-( η5- циклопентадиенил)-4,7-диметил-7-бутил-( η5- 4,5,6,7-тетрагидро-инденил)-дихлоргафния
К охлажденной до -78oC суспензии 2,00 г (5,64 ммоль) дилитиевой соли по примеру 4 в 150 мл толуола прибавляют 1,81 г (5,65 ммоль) тетрахлорида гафния. Оранжевой суспензии дают нагреваться до комнатной температуры и для завершения реакции перемешивают еще в течение двух дней. Затем отфильтровывают от нерастворимого вещества и оранжево-красный фильтрат сгущают досуха в вакууме масляного насоса. К оранжево-красному остатку добавляют 30 мл пентана и интенсивно перемешивают в течение ночи. После удаления растворителя в вакууме получают 700 мг (24%) дихлорида гафноцена в качестве порошка бежевого цвета. В спектре 1H-ЯМР неочищенного продукта можно найти лишь один диастереомер.
1H-ЯМР (200.1 МГц, CDCl3): δ = 6.63-6.58, 6.42-6.39, 6.33- 6.30, 5.99-5.96, 5.81-5.78, 5.66-5.63 (6 x m, 7H, 2'-H до 5'-H и 1-H до 3-H); 2.38 (ddd, 1H, 5eq-H, 2J(H5ax/H5eq)=14.0 Гц, 3J(H6ax/H5eq)=5.7 Гц, 3J(H6eq/H5eq)= 3.2 Гц); 2.23 (ddd, 1H, 5ax-H, 2J(H5eq/H5ax)=14.0 Гц, 3J(H6ax/H5ax)=14.4 Гц, 3J(H6eq/H5ax)= 6.8 Гц); 1.98 (ddd, 1H, 6ax-H, 2J(H6eq/H6ax)= 14,4 Гц, 3J(H5ax/H6ax)=14,4 Гц, 3J(H5eq/H6ax)=5.7 Гц; 1,85 (ddd, 1H, 6eq-H, 2J(H6ax/H6eq)=14.4 Гц, 3J(H5ax/H6eq)=6.8 Гц, 3J(H5eq/H6eq)=3.2 Гц); 1.90 (s, 3H, 10-H); 1.27 (s, 3H, 11-H); 1.35-1.07 (3 x m, 6H, nBu-CH2); 0.84-0.76 млн.д. (pt, 3H, nBu-CH3).
Точка плавления: 151oC (определенная путем дифференциально-сканирующей калометрии).
Пример 16
Синтез обоих диастереомеров 4-( η5- циклопентадиенил)-4,7- диметил-7-бутил-( η5- 4,5,6,7-тетрагидроинденил)-дихлортитана
Если 5,95 г (16,8 ммоль) дилитиевой соли по примеру 4 суспендируют в 120 мл толуола, то при добавлении 3,18 г (16,8 ммоль) тетрахлорида титана при -78oC суспензия бежевого цвета сразу же окрашивается в темно-красный цвет. Суспензию перемешивают еще 36 часов при комнатной температуре, после чего отделяют от осадка и темно-красный фильтрат сгущают досуха в вакууме, создаваемом при помощи масляного насоса. При этом получают 1,54 г (24%) обоих диастереомеров дихлорида титаноцена в качестве коричнево-красного порошка. Согласно спектру 1H-ЯМР неочищенного продукта соотношение обоих диастереомеров составляет 8:1. Если коричнево-красный порошок экстрагируют пентаном в течение нескольких дней в циркуляционной фритте, то из фильтрата выпадает коричневый осадок. При этом в спектре 1H-ЯМР можно установить, что пентановый раствор включает оба изомера в соотношении 1:1 (150 мг, 2,3%), в то время как коричневый порошок (720 мг, 11%) представляет собой почти чистый диастереомер.
1H-ЯМР (200.1 МГц, CDCl3): δ = 7.06-6.96, 6.95-6.92, 6.82- 6.74, 5,89-5.85, 5.83-5.79, 5.64-5.61 (6 x m, 7H, 2'-H до 5'-H и 1-H до 3-H); 2.40-2.27, 1.89-1.75 (2х m, 4H, 5eq-H. 5ax-H, 6ax-H, 6eq-H); 1.86 (s, 3H, 10-H): 1.35-0.89 (m, 6H, nBu-CH2); 1.21 (s, 3H, 11-H); 0.84-0.77 млн.д. (pt, 3H, nBu-CH3).
Точка плавления: 134oC (определенная путем дифференциально-сканирующей калометрии).
Пример 17
Синтез {4-(3'-[трет-бутил-( η5- циклопентадиенил)]-4,7,7- триметил-( η5- 4,5,6,7-тетрагидроинденил)}дихлорциркония
Суспензию 2,84 г (7,71 ммоль) дилитиевой соли по примеру 9а в 150 мл толуола охлаждают до -78oC. После добавления порциями 1,79 г (7,71 ммоль) тетрахлорида циркония реакционной смеси дают нагреваться до комнатной температуры и перемешивают еще 48 часов. Затем отделяют от нерастворимого вещества, оранжевую толуольную фазу сгущают в вакууме, создаваемом при помощи масляного насоса, и получаемое оранжево-красное масло переводят в порошок путем интенсивного перемешивания в пентане. При этом получают 787 мг (23%) региоизомерных дихлоридов цирконоцена в качестве оранжево- желтого порошка. Согласно спектру 1H-ЯМР неочищенный продукт включает оба диастереомера в соотношении 1: 1. Путем экстракции оранжево-желтого порошка пентаном в циркуляционной фритте получают 370 мг (11%) дихлоридов цирконоцена в соотношении 1:1.
1H-ЯМР (200.1 МГц, CDCl3): δ = 6.76-6.72, 6.69-6.65, 6.53- 6.50, 6.43-6.32, 5.99-5.97, 5.88-5.82, 5.72-5.61 (7 x m, 12H, 1-H, 2-H, 3-H, 2'-H, 4'-H, 5'-H); 2.37-2.33, 2.10-1.85, 1.75-1.62 (3 x m, 8H, 5-H и 6-H); 1.82, 1.81 (2 x s, 6H, 10-H); 1,33 (bs, 6H, 11-H или 12-H); 1.31, 1.25 (2 x s, 18H, tBu-CH3); 1,12, 1,09 млн.д. (2 x s, 6H, 11-H или 12-H).
Ж. Синтез металлоценово-диалкильных комплексов
Пример 18
Синтез 4-( η5- циклопентадиенил)-4,7,7-триметил- ( η5- 4,5,6,7-тетрагидроинденил)-цирконийдиметила
К суспензии 1,03 г (2,66 ммоль) дихлорида цирконоцена по примеру 10 в 50 мл диэтилового эфира медленно добавляют каплями 3,30 мл (5,33 ммоль) 1,60 М эфирного раствора метиллития при -78oC. Реакционной смеси дают медленно нагреваться до комнатной температуры в ванне охлаждающей смеси и дополнительно перемешивают еще 5 часов при комнатной температуре. Растворитель удаляют в вакууме и бесцветный остаток экстрагируют три раза пентаном, взятым в количестве по 50 мл. Объединенные пентановые растворы сгущают и для кристаллизации хранят при -25oC. После отделения растворителя и сушки в вакууме, создаваемом при помощи масляного насоса, получают 700 мг (76%) цирконоцендиметила в качестве бесцветного кристаллического порошка.
1H-ЯМР (200.1 МГц, CDCl3): δ = 6.58-6.50, 6.38-6.34, 5.91-5.87, 5.47-5.53, 5.34-5.31 (5 x m, 7H, 1-H до 3-H и 2'-H до 5'-H); 2.18 (ddd, 1H, 5eq-H, 2J(H5ax/H5eq)= 14.0 Гц, 3J(H6ax/H5eq)=5.9 Гц, 3J(H6eq/H5eq)=3.1 Гц; 2,09 (ddd, 1H, 5ax-H, 2J(H5eq/H5ax)= 14,0 Гц, 3J(H6eq/H5ax)=14.0 Гц, 3J(H6eq/H5ax)= 5.3 Гц); 1.80 (ddd, 1H, 6ax-H, 2J(H6eq/H6ax) =14.0 Гц 3J(H5ax/H6ax)=14.0 Гц, 3J(H5eq/H6ax)=5.9 Гц; 1.57 (ddd, 1H, 6eq-H, 2J(H6ax/H6eq)= 14.0 Гц, 3J(H5ax/H6eq)=5.3 Гц, 3J(H5eq/H6eq)=3.1 Гц; 1,53 (s, 3H, 10-H); 1,19, 1.12(2xs, 6H, 11-H и 12-H), 0.26 и 0.34 млн.д. (2хs,6H, 13-CH3 и 14-CH3).
Т. пл.: 114oC (определенная путем дифференциально-сканирующей калометрии).
Пример 19
Синтез обоих диастереомеров 4-( η5- циклопентадиенил)-4,7- диметил-7-(2-пропен-1-ил)-( η5- 4,5,6,7-тетрагидроинденил)- дихлортитана
2,45 г (7,24 ммоль) дилитиевого соединения по примеру 9б растворяют в 80 мл тетрагидрофурана, получаемый прозрачный раствор оранжевого цвета охлаждают до -78oC и смешивают с 2,42 г (7,24 ммоль) аддукта тетрахлорида титана и бис-тетрагидрофурана. При этом реакционная смесь сразу же окрашивается в темно-красный цвет. Реакционной смеси дают нагреваться до комнатной температуры и перемешивают еще 2 дня. После удаления растворителя в вакууме получают порошок коричневого цвета. Путем экстракции неочищенного продукта пентаном в циркуляционной фритте получают 0,22 г (9%) обоих аллил-титаноценов в качестве коричневого порошка. Согласно данным спектра 1H-ЯМР продукт содержит оба диастереомера в соотношении 2:1.
1H-ЯМР (200.1 МГц, CDCl3): δ = 7.05-7.01, 6.94-6.91, 6.85- 6.80, 6.75-6.73, 5.87-5.79, 5.63-5.61 (6 x m, 2'-H до 5'-H и 1-H до 3-H); 5.63-5.40, 5.15-4.80 (2 x m, винил-CH и винил-CH2); 2.38-2.17, 2.10-2.06, 1.86-1.77 (3 x m, 5-H, 6-H, аллил-CH2); 1,85 (s, 10-H); 1.24, 1.10 млн.д. (2хs, 11-H).
Пример 20
Синтез обоих диастереомеров [4-( η5- циклопентадиенил)-4,7- диметил-7-(2-пропен-1-ил)-( η5- 4,5,6,7-тетрагидроинденил)- дихлорциркония
7,56 г (22,3 ммоль) дилитиевого соединения по примеру 96 суспендируют в 200 мл толуола и охлаждают до -78oC. Добавляют порциями 5.21 г (22,3 ммоль) тетрахлорида циркония. Реакционную смесь оставляют стоять в течение 30 минут при -78oC, после чего ей дают нагреваться в течение 4 часов до комнатной температуры и перемешивают еще в течение дальнейших 12 часов. Получаемую суспензию оранжевого цвета фильтруют на фритте типа Г4, остаток два раза промывают толуолом, взятым в количестве по 30 мл, и фильтрат сгущают досуха в вакууме, создаваемом при помощи масляного насоса. При этом получают масло оранжево-красного цвета, которое путем добавления 50 мл пентана и последующего интенсивного перемешивания переводят в порошок. После удаления растворителя в вакууме получают 5,04 г (55%) порошковых аллилцирконоценов желто- оранжевого цвета. Путем многократной экстракции сырого продукта 100 мл пентана в циркуляционной фритте получают 2,34 г (26%) аллилцирконоценов в качестве желтого порошка. Точка плавления: 99oC (определенная путем дифференциально-сканирующей калометрии). Согласно данным спектра 1H-ЯМР продукт содержит оба диастереомера в соотношении 1,5:1.
1H-ЯМР (200.1 МГц, CDCl3): δ = 6.73-6.67, 6.55-6.47, 6.41-6.38, 6.08-6.04, 5.94-5.80, 5.76-5.72 (6 x m, 2'-H до 5'-H и 1-H до 3-H); 5.62-5.41, 5.20-4.83 (2 x m, винил-CH и винил-CH2); 2,55-2.17, 2.09-2.05, 1.97-1.69 (3 x m, 5-H, 6-H, аллил-CH2): 1.83 (s, 3H, 10-H); 1.27, 1.09 млн.д. (2xs, 11-H (цис-изомер) и 11-H (трансизомер).
Пример 21
Синтез обоих диастереомеров 4-( η5- циклопентадиенил)- 4,7-диметил-7-(3-(9-борабицикло}нонил-B)пропил-( η5- 4,5,6,7- тетрагидроинденил)]-дихлорциркония
210 г (0,51 ммоль) дихлоридов аллилцирконоцена по примеру 20 растворяют в 50 мл толуола и смешивают при комнатной температуре с 62 мг (0,51 ммоль) 9-борабициклононила. Перемешивают в течение 36 часов при комнатной температуре, удаляют растворитель в вакууме и получаемое масло оранжево-желтого цвета смешивают с 30 мл диэтилового эфира. Прозрачный раствор сгущают до 10 мл и охлаждают в течение нескольких часов до -30oC, в результате чего получают 208 мг (78%) диастереомеров в качестве оранжево-желтого порошка с точкой плавления 74oC (определенной путем дифференциально- сканирующей калометрии).
Пример 22
Синтез { 4-[3'-изопропил-( η5- циклопентадиенил)]-4,7,7-триметил- ( η5- 4,5,6,7-тетрагидроинденил)}дихлорциркония
а) Синтез 4-(3'-изопропил-циклопентадиенил)-4,7,7-триметил- 4,5,6,7-тетрагидро-1H-индена
К суспензии 2,17 г (57,3 ммоль) гидрида литийалюминия в 100 мл диэтилового эфира добавляют каплями при комнатной температуре раствор 6,11 г (22,9 ммоль) тетрагидроинденилфульвена по примеру 8а в 20 мл диэтилового эфира. После сильной, но не очень экзотермической реакции суспензию оранжевого цвета нагревают с обратным холодильником еще в течение трех часов. Затем охлаждают в ледяной ванне до 0oC и гидролизуют осторожно ледяной водой. Получаемый при этом белый объемистый осадок два раза экстрагируют диэтиловым эфиром, взятым в количестве по 50 мл, и объединенные органические фазы промывают водным раствором хлористого натрия. После сушки над сульфатом магния и после удаления растворителя в вакууме получают 5,63 г (92%) замещенного изопропилом анса- лиганда в качестве масла оранжевого цвета. Также в этом случае продукт состоит из большого количества обусловленных двойной связью изомеров, так что возможна лишь грубая классификация сигнальных групп в спектре 1H-ЯМР.
б) Синтез { 4-[3'-изопропил-( η5- циклопентадиенил)]-4,7,7- триметил-( η5- 4,5,6,7-тетрагидроинденил)}дилития
4,21 г (15,7 ммоль) замещенного изопропилом лиганда растворяют в 70 мл диэтилового эфира и добавляют каплями при 0oC 21,6 мл (34,5 ммоль) 1,60 М раствора метиллития. Раствор быстро обесцвечивается с образованием белого осадка. После окончания прикапывания перемешивают еще в течение дальнейших 15 часов при комнатной температуре. Затем осадок отфильтровывают и два раза промывают диэтиловым эфиром, взятым в количестве по 15 мл. Получают 5,20 г (93%) крайне чувствительной к воздействию воздуха дилитиевой соли в качестве порошка бежевого цвета, включающей один молярный эквивалент диэтилового эфира.
в) Синтез{4-[3'-изопропил-( η5- циклопентадиенил)]-4,7,7- триметил-( η5- 4,5,6,7-тетрагидроинденил)}дихлорциркония
К охлажденной до -78oC суспензии 5,20 г (14,7 ммоль) дилитиевой соли в 200 мл толуола медленно добавляют 3,40 г (14,6 ммоль) тетрахлорида циркония. Образовавшуюся суспензию бежевого цвета перемешивают в течение 24 часов при комнатной температуре, нерастворимое вещество отделяют и прозрачный остаток оранжевого цвета сгущают приблизительно до 50 мл в вакууме, создаваемом при помощи масляного насоса. Спектроскопическое исследование 1H-ЯМР толуольной фазы показывает, что в этой фазе находятся оба диастереомера в соотношении 1:1. Путем добавления 2 мл пентана и хранения в холодильнике при -20oC осаждается твердое вещество желтого цвета (1,42 г), в котором обогащен один из диастереомеров (соотношение 8:1). В толуольной фазе в обратном соотношении обогащен другой диастереомер (1,62 г); общий выход составляет 49%.
Если растворять приблизительно 100 мг выпавшего желтого порошка в хлористом метилене, то после последующей медленной диффузии пентана в этот раствор получают кристаллы, которые представляют собой диастереомер 4R*-{4-[3'-изопропил- ( η5- циклопентадиенил)] -4,7,7-триметил-( η5- 4,5,6,7-pR*-тетрагидроинденил)}дихлорцирконий.
1H-ЯМР (200.1 МГц, CDCl3): δ = 6.79-6.75, 6.48-6.38, 6.24- 6.21, 5.99-5.95, 5.56-5.54, 5.44-5.41 (6 x m, 6H, 1-H, 2-H, 3- H, 2'-H, 4'-H, 5'-H); 2,91 (s, 1H, iPr-CH, 3J(iPr-CH3/iPr/CH)= 6.9 Гц); 2.41 (ddd, 1H, 5eq-H, 2J(H5ax/H5eq)= 14.5 Гц, 3J(H6ax/H5eq) =6.2 Гц, 3J(H6eq/H5eq)=2.8 Гц); 2.28 (ddd, 1H, 5ax-H, 2J(H5eq/H5ax)=14.5 Гц, 3J(H6ax/H5ax)=14.5 Гц, 3J(H6eq/H5ax)= 5.6 Гц); 2.01 (ddd, 1H, 6ax-H, 2J(H6eq/H6ax)= 14.5 Гц, 3J(H5ax/H6ax)=14.5 Гц, 3J(H5eq/H6ax)=6.2 Гц; 1.72 (ddd, 1H, 6eq-H, 2J(H6ax/H6eq) =14.5 Гц, 3J(H5ax/H6eq)=5.6 Гц, 3J(H5eq/H6eq)=2.8 Гц; 1.78 (s, 3H, 10-H); 1.32, 1.11 (2 x s, 6H, 11-H и 12-H); 1.24, 1.09 млн.д. (2 xd, 6H, iPr-CH3, 3J(iPr-CH/iPr-CH3)=6.9 Гц.
Точка плавления: 193oC (определенная путем дифференциально-сканирующей калометрии).
Пример 23
Синтез { 4-[3'-изопропил-( η5- циклопентадиенил)]-4,7,7-триметил- [2-изопропил-( η5- 4,5,6,7-тетрагидроинденил)]}дихлорциркония
а) Синтез 4-(3'-изопропил-циклопентадиенил)-4,7,7-триметил- (2-изопропил-4,5,6,7-тетрагидро-1H-индена)
Вариант I
Стадия А
2-изопропилиден-4-(3'-изопропилиден-циклопента-1', 4'- диенил)-4,7,7-триметил-(4,5,6,7-тетрагидро-2H-инден)
8,32 г (34,2 ммоль) монофульвена по примеру 8а растворяют в смеси 50 мл метанола и 20 мл пентана и получаемый прозрачный раствор оранжево-красного цвета охлаждают до 0oC. Путем последовательного добавления 2,61 г (3,31 мл, 45,0 ммоль) ацетона и 6,08 г (7,10 мл, 85,5 ммоль) пирролидина реакционный раствор становится темно-красным через 30 минут. После перемешивания в течение 7 дней при комнатной температуре к реакционной смеси добавляют последовательно 5 мл ледяной уксусной кислоты, 150 мл воды и 50 мл пентана.
После двухкратной экстракции водной фазы пентаном объединенные органические фазы несколько раз промывают насыщенным водным раствором хлористого натрия и сушат над сульфатом магния. После удаления растворителя в вакууме, создаваемом при помощи масляного насоса, получают 9,04 г (86%) указанного дифульвена в качестве красного масла. Часть красного масла смешивают с пентаном и подвергают хроматографии на содержащей силикагель (величиной зерен 60 меш; продукт фирмы Мерк, DE) колонке, предварительно дезактивированной триэтиламином. В качестве растворителя применяют смесь пентана и триэтилового эфира в соотношении 100:5 (общий выход < 10%).
Стадия Б
4-(3'-изопропил-циклопентадиенил)-4,7,7-триметил-(2- изопропил-4,5,6,7-тетрагидро-1H-инден)
В трехгорлую колбу с холодильником интенсивного охлаждения и капельной воронкой подают 3,03 г (80,0 ммоль) гидрида литийалюминия в 100 мл диэтилового эфира, и при интенсивном перемешивании при комнатной температуре добавляют 6,47 г (21,1 ммоль) полученного на стадии А дифульвена, растворенного в 50 мл диэтилового эфира. По окончании добавления реакционную смесь нагревают с обратным холодильником в течение 5 часов и затем осторожно гидролизуют 100 мл воды. При этом получают серый осадок окиси алюминия и желтую фазу диэтилового эфира. Последнюю декантируют, серый осадок экстрагируют несколько раз диэтиловым эфиром, и объединенные фазы диэтилового эфира промывают насыщенным водным раствором хлористого натрия. После сушки над сульфатом магния и удаления растворителя в вакууме получают 6,25 г (96%) восстановленного дифульвена в качестве масла оранжево-красного цвета, которое можно перерабатывать без дальнейшей очистки.
Вариант II
Стадия А
2,5-бис-[(изопропил)циклопента-2,4-диен-1-илиден]-гексан
К раствору 2,78 мл (2,71 г, 23,8 ммоль) 2,5-гександиона и 4,00 г (47,6 ммоль) изопропилциклопентадиена в 50 мл метанола добавляют каплями при 0oC 5,90 мл (5,07 г, 71,3 ммоль) свежедистиллированного пирролидина. При этом реакционный раствор сразу же становится темно-красным. После перемешивания в течение дальнейших 15 часов при 0oC избыточный пирролидин нейтрализуют путем добавления раствора 2 мл ледяной уксусной кислоты в 100 мл воды. Экстрагируют два раза диэтиловым эфиром, взятым в количестве по 100 мл, объединенные органические фазы промывают несколько раз насыщенным водным раствором хлористого натрия и сушат над сульфатом магния. После удаления растворителя в вакууме получают 5,20 г (75%) дифульвена в качестве темно-красного масла.
Очистка дифульвена осуществляется путем хроматографии на колонке, содержащей силикагель, дезактивированный триэтиламином, используемым в качестве смеси с пентаном в соотношении 1:100). В качестве элюента применяют смесь пентана и диэтилового эфира в соотношении 1:1. Получают 1,72 г (25%) дифульвена в качестве красного масла.
Стадия Б
а) 4-(3'-изопропил-циклопентадиенил)-4,7,7-триметил-(2- изопропил-4,5,6,7-тетрагидро-1H-инден)
600 мг (2,04 ммоль) замещенного бис-изопропилом дифульвена со стадии А растворяют в 10 мл диэтилового эфира и медленно смешивают при 0oC с 2,55 мл 1,60 М эфирного раствора метиллития. Реакционной смеси дают нагреваться до комнатной температуры. Через 24 часа получают суспензию оранжевого цвета, которую охлаждают до 0oC и затем гидролизуют 10 мл воды. После экстракции диэтиловым эфиром в количестве 20 мл и сушки над сульфатом магния получают 520 мл (82%) целевого продукта в качестве оранжевого масла.
д) {4-[3'-изопропил-( η5- циклопентадиенил)]-4,7,7-триметил-[2- изопропил-( η5- 4,5,6,7-тетрагидроинденил)}дихлорцирконий
К раствору 500 г (1,61 ммоль) замещенного бис-изопропилом соединения со стадии Б варианта I или II в 20 мл пентана добавляют каплями 2,00 мл (3,22 ммоль) 1,60 М эфирного раствора метиллития при 0oC. Реакционной смеси дают нагреваться до комнатной температуры, и через 12 часов получают мутную суспензию оранжевого цвета, которую охлаждают до -78oC и смешивают с 373 мг (1,61 ммоль) тетрахлорида циркония. После 24-часового перемешивания при комнатной температуре отфильтровывают от нерастворимого вещества и удаляют растворитель в вакууме. Получают 300 мг (40%) обоих диастереомеров анса-цирконоцена в качестве порошка оранжевого цвета. В спектре 1H-ЯМР обнаруживают резонансные сигналы обоих диастереомеров в соотношении 1:1 (определены при помощи изопропиловых групп).
1H-ЯМР (200.1 МГц, CDCl3): δ = 6.44-6.41, 6.24-6.15, 5.95-5.91, 5.68-5.60, 5.33-5.29, 5.18-5.17 (6 x m, 10H, 2'-H, 4'-H, 5'-H, 1-H и 3-H); 3.13-2.83, 2.46-2.16, 2.08-1.93, 1.78-1.72 (4 x m, 12H, 2xiPr-CH и 5-H и 6-H); 1.76, 1.72 (2 x s, 6H, 10-H), 1.28-1.07 мнл.д. (m, 36H, 11-H, 12-H, 4хiPr-CH3).
Пример 24
Синтез { 4-[3'-триметилсилил-( η5- циклопентадиенил)]-4,7,7- триметил-[2-триметил-силил-( η5- 4,5,6,7-тетрагидроинденил)]} дихлорциркония
а) Синтез 4-(3'-триметилсилил-циклопентадиенил)-4,7,7- триметил-(2-триметилсилил-4,5,6,7-тетрагидро-1H-индена)
Раствор 6,81 г (21,8 ммоль) дилитиевого эфирата по примеру 9 в 50 мл тетрагидрофурана охлаждают до 0oC и при перемешивании прикапывают 5,50 мл (4,74 г, 43,6 ммоль) триметилсилилхлорида. Реакционной смеси дают нагреваться в течение ночи до комнатной температуры, получаемую мутную суспензию оранжевого цвета гидролизуют путем добавления 50 мл дегазированной воды, и затем экстрагируют петролейным эфиром. После сушки над сульфатом магния и удаления растворителя в вакууме получают 6,54 г (81%) масла красно-оранжевого цвета.
б) Синтез {4-[3'-триметилсилил-( η5- циклопентадиенил)]-4,7,7- триметил-[2-триметил-силил( η5- 4,5,6,7-тетрагидроинденил)]}дилития
К охлажденному до 0oC раствору 3,30 г (8,90 ммоль) замещенного бис-триметилсилилом соединения со стадии а) в 40 мл пентана прикапывают при перемешивании 11,1 мл (17,8 ммоль) 1,60 М эфирного раствора метиллития. При этом получают, при выделении газа, осадок белого цвета. Для завершения реакции продолжают перемешивать еще в течение 24 часов при комнатной температуре, после чего белый осадок отфильтровывают и промывают пентаном. После сушки в вакууме, создаваемом при помощи масляного насоса, получают 2,60 г (76%) дилитиевой соли в качестве белого пирофорного остатка.
в) Синтез { 4-[3'- триметилсилил-( η5- циклопентадиенил)]-4,7,7-триметил-[2-триметил- силил( η5- 4,5,6,7-тетрагидроинденил)]}дихлорциркония
К охлажденной до -78oC суспензии 2,60 г (6,79 ммоль) замещенной бис-триметил-силилом дилитиевой соли со стадии б) в 100 мл толуола добавляют порциями 1,58 г (6,79 ммоль) тетрахлорида циркония. Реакционной смеси дают нагреваться до комнатной температуры, и после 24-часового перемешивания получают суспензию оранжевого цвета. После отделения нерастворимого вещества и сгущения досуха получают красное масло. Путем добавления 20 мл пентана и последующей переработки получают 1,54 г (43%) обоих диастереомеров анса-цирконоцена в качестве порошка оранжевого цвета. Точка плавления: 151oC (разл., определенная путем дифферециально-сканирующей калометрии).
Hижеследующие примеры более подробно поясняют способ согласно изобретению. В этих примерах физико-механические свойства продуктов определяют по вышеуказанным промышленным стандартным терминам.
Пример 25
В автоклав емкостью 1,5 дм3, предварительно тщательно промытый этеном, подают 600 см3 85 вес.%-ного раствора норборнена в толуоле. Путем неоднократной напрессовки этена (18 бар) раствор насыщают этеном. После этого противотоком подают 5 см3 толуольного раствора метилалюмоксана (19,1 вес.%-ный раствор этилалюмоксана с молекулярным весом 1300 г/моль, определенным криоскопическим способом) и перемешивают в течение 30 минут при 70oC. После предварительной активации продолжительностью 15 минут добавляют раствор 1,0 мг 4-( η5- циклопентадиенил)-4,7,7- триметил-( η5- 4,5,6,7-тетрагидроинденил)дихлорциркония в 5 см3 толуольного раствора метилалюмоксана. (В случае регулирования молекулярного веса с помощью водорода можно здесь напрессовать водород).
При перемешивании (750 об/мин) полимеризуют в течение часа при 70oC, причем давление этена путем дополнительной подачи держится при 18,0 бар.
По истечении времени реакции полимеризационную смесь выпускают в сосуд и сразу подают в 5 дм3 ацетона, перемешивают в течение 10 минут, а затем осажденный продукт отфильтровывают. Фильтровальный осадок промывают по три раза попеременно 10%-ной соляной кислотой и ацетоном. В конце промывают водой до нейтральности, осадок суспендируют в ацетоне и снова фильтруют. Очищенный таким образом полимер сушат в течение 15 часов при 80oC в вакууме (0,2 бар).
После сушки получают 224 г бесцветного полимера, имеющего температуру стеклования 179oC, коэффициент вязкости 52 см3/г, прочность на разрыв 59 МПа и относительное удлинение при разрыве 3,1%. Активность A* составляет 80512 г полимера/ч•ммоль.
Пример 26 (сравнительный пример)
Повторяют пример 25 с той разницей, что в качестве металлоценового соединения используют изопропилен(циклопентадиенил)(1-инденил)дихлорцирконий. Получают 89 г полимера, имеющего температуру стеклования 150oC, коэффициент вязкости 57 см3/г, прочность на разрыв 61 МПа и относительное удлинение при разрыве 3,3%. Активность A* составляет 34000 г полимера/ч•ммоль.
Пример 27
В автоклав емкостью 1,5 дм3, предварительно тщательно промытый этеном, подают 600 см3 85 вес.%-ного раствора норборнена в толуоле. Путем неоднократной напрессовки этена (18 бар) раствор насыщают этеном. После этого противотоком подают 5 см3 толуольного раствора метилалюминоксана (10,1 вес.%-ный раствор этилалюмоксана с молекулярным весом 1300 г/моль, определенным криоскопическим способом) и перемешивают в течение 30 минут при 80oC. После предварительной активации продолжительностью 15 минут добавляют раствор 1,0 мг 4-( η5- изопропил-циклопентадиенил)-4,7,7- триметил-( η5- 4,5,6,7-тетрагидроинденил)дихлорциркония в 5 см3 толуольного раствора метилалюмоксана. (В случае регулирования молекулярного веса с помощью водорода можно здесь напрессовать водород).
При перемешивании (750 об/мин) полимеризуют в течение часа, причем давление этена путем дополнительной подачи держится при 17,8 бар, а температура - при 80oC.
По истечении времени реакции полимеризационную смесь выпускают в сосуд и сразу подают в 5 дм3 ацетона, перемешивают в течение 10 минут, а затем отфильтровывают осажденный продукт. Фильтровальный осадок промывают по три раза попеременно 10%-ной соляной кислотой и ацетоном. В конце промывают водой до нейтральности, осадок суспендируют в ацетоне и снова фильтруют. Очищенный таким образом полимер сушат в течение 15 часов при 80oC в вакууме (0,2 бар).
После сушки получают 16,0 г бесцветного полимера, имеющего температуру стеклования 145oC, коэффициент вязкости 156 см3/г, прочность на разрыв 64 МПа и относительное удлинение при разрыве 3,3%. Активность A* составляет 68300 г полимера/ч•ммоль.
Пример 28
В автоклав емкостью 1,5 дм3 предварительно тщательно промытый этеном, подают 600 см3 50 вес.%-ного раствора норборнена в толуоле. Путем неоднократной напрессовки этена (18 бар) раствор насыщают этеном. После этого противотоком подают 5 см3 толуольного раствора метилалюмоксана (10,1 вес.%-ный раствор этилалюмоксана с молекулярным весом 1300 г/моль, определенным криоскопическим способом) и перемешивают в течение 30 минут при 80oC. После предварительной активации продолжительностью 15 минут добавляют раствор 0,2 мг 4-( η5- изопропил-циклопентадиенил)-4,7,7-триметил- ( η5- 4,5,6,7-тетрагидроинденил)дихлорциркония в 5 см3 толуольного раствора метилалюмоксана. (В случае регулирования молекулярного веса с помощью водорода можно здесь напрессовать водород).
При перемешивании (750 об/мин) полимеризуют в течение часа, причем давление этена путем дополнительной подачи держится при 17,8 бар, а температура - при 70oC.
По истечении времени реакции полимеризационную смесь выпускают в сосуд и сразу подают в 5 дм3 ацетона, перемешивают в течение 10 минут, а затем отфильтровывают осажденный продукт. Фильтровальный осадок промывают по три раза попеременно 10%-ной соляной кислотой и ацетоном. В конце промывают водой до нейтральности, осадок суспендируют в ацетоне и снова фильтруют. Очищенный таким образом полимер сушат в течение 15 часов при 80oC в вакууме (0,2 бар).
После сушки получают 98 г бесцветного полимера, имеющего температуру стеклования 184oC, коэффициент вязкости 114 см3/г, прочность на разрыв 61 МПа и относительное удлинение при разрыве 3,1%. Активность A* составляет 104500 г полимера/ч•ммоль.
Пример 29
В автоклав емкостью 1,5 дм3, предварительно тщательно промытый этеном, подают 600 см3 50 вес.%-ного раствора норборнена в толуоле. Путем неоднократной напрессовки этена (18 бар) раствор насыщают этеном. После этого противотоком подают 5 см3 толуольного раствора метилалюмоксана (10,1 вес.%-ный раствор этилалюмоксана с молекулярным весом 1300 г/моль, определенным криоскопическим способом) и перемешивают в течение 30 минут при 80oC. После предварительной активации продолжительностью 15 минут добавляют раствор 1,0 мг 4-( η5- изопропил-циклопентадиенил)- 4,7,7-триметил-( η5- 4,5,6,7-тетрагидроинденил)дихлорциркония в 5 см3 толуольного раствора метилалюмоксана. (В случае регулирования молекулярного веса с помощью водорода можно здесь напрессовать водород.)
При перемешивании (750 об/мин) полимеризуют в течение часа, причем давление этена путем дополнительной подачи держится при 17,8 бар, а температура - при 50oC.
По истечении времени реакции полимеризационную смесь выпускают в сосуд и сразу подают в 5 дм3 ацетона, перемешивают в течение 10 минут, а затем отфильтровывают осажденный продукт. Фильтровальный осадок промывают по три раза попеременно 10%-ной соляной кислотой и ацетоном. В конце промывают водой до нейтральности, осадок суспендируют в ацетоне и снова фильтруют. Очищенный таким образом полимер сушат в течение 15 часов при 80oC в вакууме (0,2 бар).
После сушки получают 31 г бесцветного полимера, имеющего температуру стеклования 121oC, коэффициент вязкости 203 см3/г, прочность на разрыв 65 МПа и относительное удлинение при разрыве 3,3%. Активность A* составляет 13200 г полимера/ч•ммоль.
Пример 30
В автоклав емкостью 1,5 дм3 предварительно тщательно промытый этеном, подают 600 см3 50 вес.%-ного раствора норборнена в толуоле. Путем неоднократной напрессовки этена (18 бар) раствор насыщают этеном. После этого противотоком подают 5 см3 толуольного раствора метилалюмоксана (10,1 вес.%-ный раствор этилалюминоксана с молекулярным весом 1300 г/моль, определенным криоскопическим способом) и перемешивают в течение 30 минут при 80oC. После предварительной активации продолжительностью 15 минут добавляют раствор 0,83 мг 4-( η5- изопропил-циклопентадиенил)-4,7,7- триметил-( η5- 4,5,6,7-тетрагидроинденил)дихлорциркония в 5 см3 толуольного раствора метилалюмоксана. (В случае регулирования молекулярного веса с помощью водорода можно здесь напрессовать водород).
При перемешивании (750 об/мин) полимеризуют в течение часа, причем давление этена путем дополнительной подачи держится при 18,0 бар, а температура - при 90oC.
По истечении времени реакции полимеризационную смесь выпускают в сосуд и сразу подают в 5 дм3 ацетона, перемешивают в течение 10 минут, а затем отфильтровывают
осажденный продукт. Фильтровальный осадок промывают по три раза попеременно 10%-ной соляной кислотой и ацетоном. В конце промывают водой до нейтральности, осадок суспендируют в ацетоне и снова фильтруют. Очищенный таким образом полимер сушат в течение 15 часов при 80oC в вакууме (0,2 бар).
После сушки получают 45 г бесцветного полимера, имеющего температуру стеклования 130oC, коэффициент вязкости 107 см3/г, прочность на разрыв 62 МПа и относительное удлинение при разрыве 3,2%. Активность A* составляет 24200 г полимера/ч•ммоль.
Пример 31
Повторяют пример 25 с той разницей, что применяют 0,92 мг 4-( η5- бензилциклопентадиенил)-4,7,7-триметил-( η5- 4,5,6,7- тетрагидроинденил)дихлорциркония. Температура полимеризации составляет 90oC. Получают 31 г полимера, обладающего следующими свойствами: Tg = 141oC, коэффициент вязкости = 80 см3/г, прочность на разрыв = 63 МПа, относительное удлинение при разрыве = 3,6%, A* = 18900 г полимера/ч•ммоль.
Пример 32
Повторяют пример 25 с той разницей, что применяют 1,0 мг 4- ( η5- циклопентадиенил)-4,7,7-триметил-( η5- 4,5,6,7- тетрагидроинденил)бис-(диметиламино)циркония. Получают 180 г полимера, обладающего следующими свойствами: Tg = 169oC, коэффициент вязкости = 54 см3/г, прочность на разрыв = 59 МПа, относительное удлинение при разрыве = 3,2%, A* = 71900 г полимера/ч•ммоль.
Пример 33
Повторяют пример 25 с той разницей, что применяют 1,1 мг 4-( η5- трет-бутил-циклопентадиенил)-4,7,7-триметил-( η5- 4,5,6,7- тетрагидроинденил)дихлорциркония. Получают 33 г полимера, обладающего следующими свойствами: Tg = 124oC, коэффициент вязкости = 228 см3/г, прочность на разрыв = 64 МПа, относительное удлинение при разрыве = 3,8% A* = 27400 г полимера/ч•ммоль.
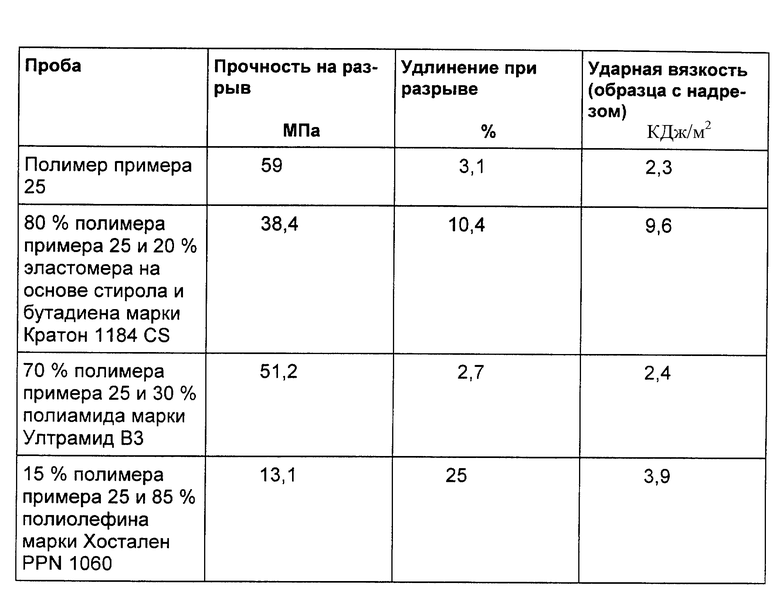
Пример 34
На двухшнековом экструдере полимер примера 25 переводят при 260oC в указанные в следующей таблице сплавы. В этой таблице приводятся и данные по механическим свойствам полимера и полимерных сплавов.
Сущность изобретения: металлоценовый катализатор для получения сополимера циклоолефинов представляет собой стерически жесткое металлоценовое соединение, содержащее в качестве лигандов по меньшей мере две замещенные или незамещенные циклопентадиенильные группы, связанные между собой через моноциклическую кольцевую систему, причем одна циклопентадиенильная группа анеллирована к моноциклической системе, и эта система лигандов отлична от 4-(η5-3-алкилциклопентадиенил)-4,6,6-триметил-(η5-2-алкил-4,5-тетрагидропенталена) и представляет собой соединение формулы (I), где М - металл группы IVб Периодической системы, R1 и R2 одинаковы или различны и означают атом галогена, алкил с 1-10 атомами углерода, или группу NR13 2, где R13 означает алкил с 1-10 атомами углерода, R3, R4, R5, R6, R7, R8 и R9 одинаковы или различны и означают атом водорода, алкил с 1-10 атомами углерода, или остаток -R13-SiR13 3-, где R13 имеет вышеуказанное значение, R10 - алкил с 1 - 20 атомами углерода, R11 и R12 одинаковы или различны и означают алкил с 1 - 20 атомами углерода, арил с 6-20 атомами углерода, алкенил с 2-12 атомами углерода, n - целое число от 1 до 10. Описывается также способ получения сополимера циклоолефинов, формованное изделие и полимерный сплав, включающие по меньшей мере один полимер циклоолефинов. Технический результат - получение катализатора, с помощью которого возможно получение сополимера циклоолефинов с улучшенными физико-механическими свойствами, в особенности полиолефинов с повышенной прочностью на разрыв и повышенной прозрачностью, и, кроме того, более высокой активности полимеризации. 5 с. и 5 з.п. ф-лы, 1 табл.
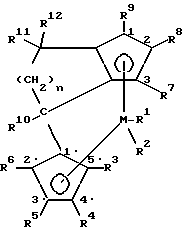
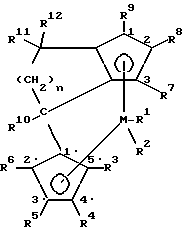
где М - металл группы IVб Периодической системы;
R1 и R2 одинаковы или различны и означают атом галогена, алкил с 1 - 10 атомами углерода, или группу NR13 2, где R13 означает алкил с 1 - 10 атомами углерода;
R3, R4, R5, R6, R7, R8 и R9 одинаковы или различны и означают атом водорода, алкил с 1 - 10 атомами углерода, или остаток -R13-SiR13 3-, где R13 имеет вышеуказанное значение;
R10 - алкил с 1 - 20 атомами углерода;
R11 и R12 одинаковы или различны и означают алкил с 1 - 20 атомами углерода, арил с 6 - 20 атомами углерода, алкенил с 2 - 12 атомами углерода;
n - целое число от 1 до 10.
Приоритет по пунктам:
21.12.1993 по пп.1 - 4;
24.12.1993 по пп.5 - 10.
| СПОСОБ ПОЛУЧЕНИЯ ВЫСОКОКИПЯЩИХ ФТОРПАРАФИНОВ | 0 |
|
SU407870A1 |
| Катализатор высокотемпературной полимеризации этилена и сополимеризации этилена с @ -олефинами | 1987 |
|
SU1641193A3 |
| Задающий генератор | 1973 |
|
SU490256A1 |
| EP 0537686 A1, 21.04.1993 | |||
| УСТРОЙСТВО ДЛЯ ОТМЫВАНИЯ КЛЕЙКОВИНЫ | 1970 |
|
SU421209A3 |
