Настоящее изобретение относится к области химии и, более конкретно, к области химии пептидов. Точнее, предметом изобретения являются ацилированные псевдодипептиды, имеющие вспомогательное функционализированное ответвление, которые могут быть привитыми в виде конъюгатов или не привитыми, причем названное вспомогательное ответвление придает молекуле оригинальные свойства в отношении биологической активности и в физико-химическом отношении. Химическая природа вспомогательного ответвления придает ацилированному псевдодипептиду дополнительный размер, мягко модулируя его исходные свойства и сообщая ему новые свойства. Такие молекулы, имеющие вспомогательное функционализированное ответвление, могут быть конъюгированы с каким либо фармакофором, антигеном или молекулой-вектором. Биоконъюгация предполагает сочетание двух или нескольких химических структур с образованием нового молекулярного комплекса, обладающего свойствами, отличающимися от свойств его отдельных компонентов. Природные или синтетические продукты, обладающие собственными фармакологическими свойствами, могут, таким образом, быть скомбинированы между собой с целью создания новых структур с оригинальными фармакологическими свойствами или фармакологическими свойствами, улучшенными по сравнению со свойствами исходных соединений. Применение биоконъюгации представляет интерес для всех разделов медицины человека, ветеринарии и для диагностических методов.
Многочисленные агенты сочетания гомо- и гетеробифункционального типа были уже описаны и могут быть применены для сочетания столь разных молекул как аминокислоты, пептиды, белки, сахара, олигосахариды, полисахариды, нуклеиновые кислоты, олигонуклеотиды, полинуклеотиды, липиды и практически все молекулы, обладающие функциональной группой, способной образовывать связь. Последние годы значительные усилия были сосредоточены на синтезе антигенных конструкций, составленных их двух молекул, несущих различную информацию. Good et al. [(1987), Science 235, 1059-1062], в частности, синтезировали пептид, содержащий эпитопы, узнаваемые одновременно вспомогательными Т-лимфоцитами и В-лимфоцитами. Bessler и Jung [(1992) Res. Immunol. 5, 548-553] описали конъюгаты, составленные из одного пептида и одного иммуностимулятора. Hoffmann et al. [(1997) FEMS Immunol. Med. Microbiol. Microbiology 17, 225-234] описали конъюгаты между липопептидом и синтетическим пептидом мелиттина. Ulrich и Myers [(1995) Vaccine Design, Plenum Press, New York, 495-524] сделали наблюдение, что иммунный ответ был эффективен лишь тогда, когда гаптен и адъювант MPL (Monophosphoryl Lipid А) находились в одной и той же липосоме. Ими была обсуждена возможность ковалентной связи между адъювантом MPL и гаптеном. Действительно, конъюгат гаптен-адъювант мог бы оказаться более эффективным в качестве адъюванта для вакцинации. Ikeda et al. [(1999) Chem. Pharm. Bull., 47(4), 563-568] предложили синтез между структурным аналогом Липида А и опухолевым антигеном пептидной природы и доказали in vitro наличие митогенетической активности.
Такая концепция конъюгации могла бы оказаться подходящей и для белков или даже пар белок-полисахарид. Действительно, известно, что полисахариды, используемые индивидуально в качестве вакцины, вызывают у детей моложе 5 лет лишь слабый иммунологический ответ, поскольку ответ не зависит от Т-клеток [Gotschlich et al., (1977); Antibodies in Human Diagnosis and Therapy, Peltola et al., (1977), Pediatrics 60, 730-737]. Напротив, полисахариды, связанные с векторными белками (носителями), дают значительно более сильный иммунологический ответ. Этот феномен был открыт в 1931 году Avery и Goebel [(1931), J. Exp. Med. 54, 437-447]. Различные вакцины, разработанные в последние годы, свидетельствуют о прогрессе в этой области. Можно, в частности, упомянуть вакцины против Haemophilus influenzae и различных серотипов Streptococcus pneumoniae [Powell и Newman, (1995), Vaccine Design Plenum Press, New York]. Для последних была разработана многовалентная вакцина [Sood и Fattom, (1998), Exp. Opin, Invest. Drugs 7 (3), 333-347].
Новые перспективы открывает комплекс, состоящий из адъюванта, связанного с полисахарид-белковой структурой. Технология химического синтеза биоконъюгатов разработана хорошо и позволяет сегодня осуществлять множество проектов, которые были немыслимыми несколько лет назад, используя многочисленные гомо- и гетерофункциональные реагенты и процедуры связывания, широко описанные в литературе [Hermanson, (1996), Bioconjugate Techniques, Academic Press, New York].
В качестве примера можно назвать метод восстановительного аминирования [Roy et al., (1984), Canad. J. Biochem. Cell. Biol. 62, 270-279; Hermanson, (1995), p. 472], позволяющий осуществить конъюгацию молекулы, имеющей альдегидную функцию, с первичным амином, содержащимся в пептиде или белке, с белок-полисахаридным конъюгатом или с каким-либо фармакофором. Эта реакция приводит к образованию очень стабильного диалкиламина.
ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Более конкретно, предметом изобретения являются псевдодипептиды, являющиеся функционализированными производными аминокислот, свободные аминные функции которых амидированы жирными кислотами, и одна из концевых групп которых имеет вспомогательное функционализированное ответвление.
Специфическим предметом изобретения являются N-ацилированные псевдодипептиды, имеющие кислотную группу в нейтральной или заряженной форме на одном из концов псевдодипептида, а на другом конце имеющие вспомогательное функционализированное ответвление, отвечающие общей формуле I:
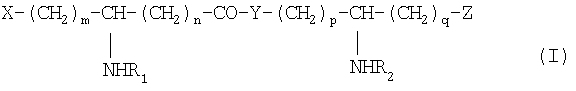
в которой R1 и R2 обозначают, каждый, ацильную группу насыщенной или ненасыщенной, линейной или разветвленной карбоновой кислоты, имеющей от 2 до 24 атомов углерода, незамещенной или имеющей заместитель или заместители, выбранные из групп: гидроксил, алкил, алкокси, ацилокси, амино, ациламино, ацилтио и С1-С24-алкилтио,
коэффициенты m и n принимают значения от 0 до 10,
коэффициенты р и q принимают значения от 1 до 10,
Y обозначает О или NH,
Х и Z обозначают вспомогательное функционализированное ответвление или кислотную группу в нейтральной или заряженной форме, выбранную из следующих групп:
- карбоксил
- карбокси-С1-С5-алкокси
- карбокси-С1-С5-алкилтио
- фосфоно-С1-С5-алкокси
- фосфоно-С1-С5-алкилтио
- дигидроксифосфорилокси-С1-С5-алкокси
- дигидроксифосфорилокси-С1-С5-алкилтио
- дигидроксифосфорилокси
- гидроксисульфонилокси
- гидроксисульфонил-С1-С5-алкокси
- гидроксисульфонил-С1-С5-алкилтио
- гидроксисульфонилокси-С1-С5-алкокси
- гидроксисульфонилокси-С1-С5-алкилтио
- (карбокси-С1-С5-алкил)аминокарбонил
- (дикарбокси-С1-С5-алкил)аминокарбонил
- (аммонио-С1-С5-алкил)аминокарбонил
- карбокси(амино-С1-С5-алкил)аминокарбонил
при условии, что по крайней мере один из заместителей Х и Z обозначает вспомогательное функционализированное ответвление.
Вспомогательная группа Х или Z отвечает формуле II:

где А обозначает О, S или NH,
коэффициент r имеет значение 0 или 1,
коэффициент s имеет значение от 1 до 10,
W выбирают из групп:
- формил
- ацетил
- циано
- галоген
- амино
- бром- или йодацетамидо
- ациламидо
- диацилимидо
- сульфгидрил
- алкилтио
- гидроксил
- 1,2-дигидроксиэтил
- алкокси
- ацилокси
- винил
- этинил
- свободный карбоксил
- карбоксил в форме сложного эфира, смешанного ангидрида, амида или гидразида
- азидо
- тиоциано.
Изобретение наиболее конкретно относится к новым N-ацилированным псевдодипептидам, имеющим кислотную группу в нейтральной или заряженной форме на одном из концов псевдодипептида, а на другом конце имеющим вспомогательное функционализированное ответвление, отвечающим общей формуле I:
в которой R1 и R2 обозначают, каждый, ацильную группу насыщенной или ненасыщенной, линейной или разветвленной карбоновой кислоты, имеющей от 2 до 24 атомов углерода, незамещенной или имеющей заместитель или заместителей, выбранные из групп: гидроксил, алкил, алкокси, ацилокси, амино, ациламино, ацилтио и С1-С24-алкилтио,
коэффициенты m и n принимают значения от 0 до 10,
коэффициенты р и q принимают значения от 1 до 10,
Х и Z обозначают кислотную группу в нейтральной или заряженной форме или вспомогательное функционализированное ответвление,
при условии, что по крайней мере один из заместителей Х и Z обозначает вспомогательное функционализированное ответвление,
Y обозначает О или NH,
кислотную группу Х или Z выбирают преимущественно из групп:
- карбоксил
- карбокси-С1-С5-алкокси
- карбокси-С1-С5-алкилтио
- фосфоно-С1-С5-алкокси
- фосфоно-С1-С5-алкилтио
- дигидроксифосфорилокси-С1-С5-алкокси
- дигидроксифосфорилокси
- гидроксисульфонилокси
- гидроксисульфонил-С1-С5-алкокси
- гидроксисульфонил-С1-С5-алкилтио
- гидроксисульфонилокси-С1-С5-алкокси
- гидроксисульфонилокси-С1-С5-алкилтио
и вспомогательная группа Х или Z отвечает общей формуле II за исключением дигидроксиэтильного радикала.
Когда заместители Х или Z обозначают кислотную группу в нейтральной форме, речь идет о свободной карбоксильной, сульфоновой, фосфоновой или фосфорнокислотной форме. Когда речь идет о кислотной группе в заряженной форме, это означает карбоксильную, сульфоновую, фосфоновую или фосфорнокислотную группу в форме соли, в частности, образованную добавлением минерального или органического основания, предпочтительно терапевтически совместимого. Когда основания терапевтически не совместимы, они могут служить в качестве средства для идентификации, очистки или расщепления.
Из терапевтически совместимых солеобразующих оснований можно, в частности, назвать щелочи, такие как гидроксиды натрия, калия, лития, соли аммония; щелочно-земельные основания, такие как гидроксиды кальция или стронция, соли магния, соли черных металлов и им подобных; органические основания, такие как производные первичных, вторичных или третичных аминов, аминокислоты с щелочной реакцией, такие как лизин или орнитин, или аминосахара.
Не пригодными для терапевтического использования основаниями являются, например, бруцин, стрихнин, N-метилглюкозамин или N-метилморфолин. Как было отмечено выше, образуемые ими соли могут служить в качестве средства для разделения, идентификации или очистки.
Когда m равно 1, а n равно 0, молекула происходит, в частности, от серина или аспарагиновой кислоты. Когда m равно 2, a n равно 0, молекула происходит от гомосерина или от глутаминовой кислоты.
Когда Y обозначает NH, р равно 3 и q равно 1, речь идет о производном от цитруллина, орнитина или аргинина.
Когда р равно 4, а q равно 1, речь в данном случае идет, в частности, о производном от гомоаргинина или лизина.
Когда заместитель Х или Z обозначает вспомогательную группу общей формулы III:
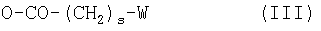
где коэффициент s принимает значения от 1 до 10, но особенно 4, 5 или 6,
W выбирают преимущественно из групп:
- формил
- амино
- гидроксил
- 1,2-дигидроксиэтил
- карбоксил.
Из составляющих предмет изобретения псевдодипептидов следует выделить в качестве особо предпочтительных соединения общей формулы IV:
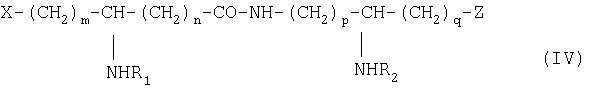
в которой R1 и R2 обозначают каждый ацильную группу насыщенной или ненасыщенной, линейной или разветвленной карбоновой кислоты, имеющей от 2 до 24 атомов углерода, незамещенной или имеющей один или несколько заместителей, выбранных из групп: гидроксил, алкил, алкокси, ацилокси, амино, ациламино, ацилтио и С1-С24-алкилтио,
коэффициенты m и n принимают значения от 0 до 10,
коэффициенты р и q принимают значения от 1 до 10,
и в которой один из заместителей Х и Z обозначает карбоксил, радикал гидроксифосфорилокси, группу карбокси-С1-С5-алкокси, карбокси-С1-С5-алкилтио, карбокси-С1-С5-алкиламинокарбонил, дикарбокси-С1-С5-алкиламинокарбонил или карбокси (амино-С1-С5-алкил)аминокарбонил и в которой другим заместителем является радикал ацилокси, выбранный из групп: 6-аминогексаноилокси, 6-оксогексаноилокси, 6-гидроксигексаноилокси, 6,7-дигидроксигептаноилокси и 3-карбоксипропаноилокси.
Определение R1 и R2 охватывает ацилированные производные с цепью переменной длины в пределах от 2 до 24 атомов углерода, одинаковые или разные, разветвленные или прямоцепочечные, насыщенные или ненасыщенные, которые могут иметь один или несколько заместителей, выбранных из группы, в которую входят алкил, амино, ациламино, гидроксил, алкокси, ацилокси, ацилтио и алкилтио. Из представляющих интерес ацильных групп предпочтительными для данного изобретения являются ацильные группы, образованные от лауриновой, 2-гидроксиоктановой, 3-гидроксимиристиновой, 2-деканоилоксиоктановой, 3-лаурилоксимиристиновой, 3-миристилоксимиристиновой, и 3-пальмитилоксимиристиновой кислот.
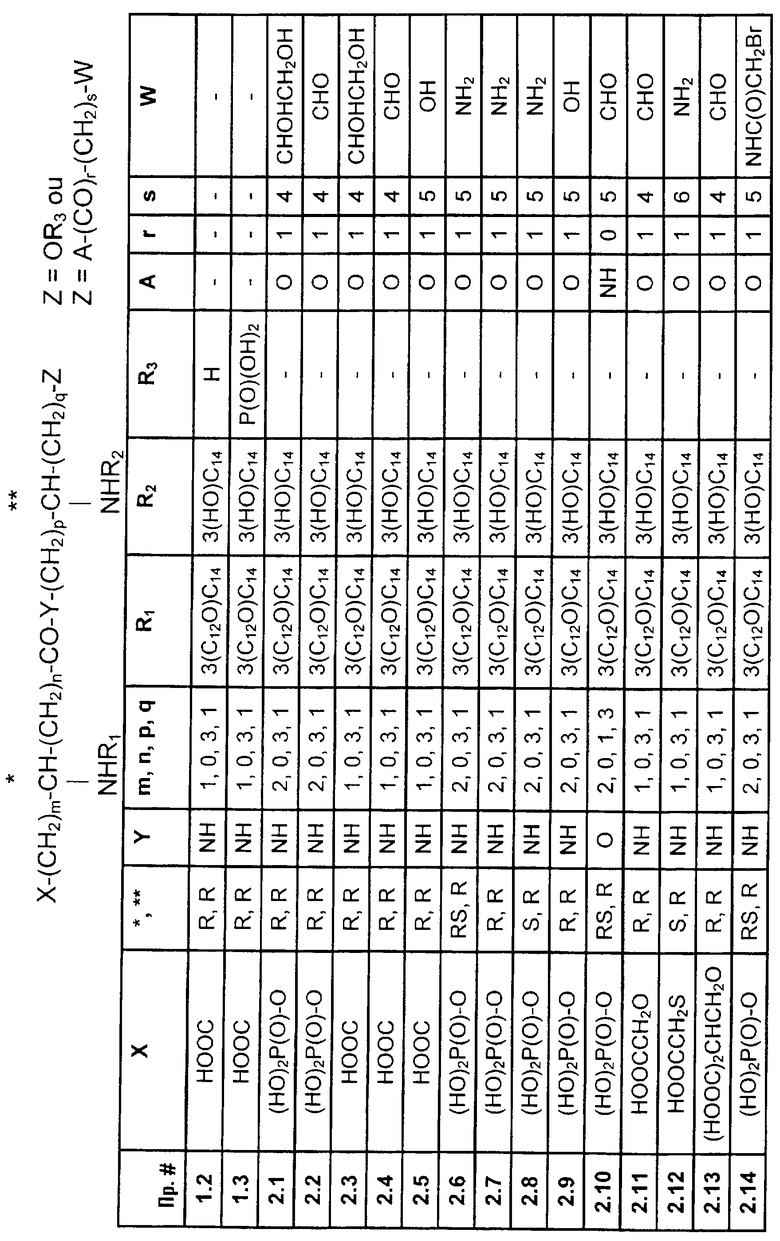
В качестве предпочтительных соединений, к которым относится, в частности, изобретение, являются псевдодипептиды, представленные в приведенной ниже таблице:

Изобретение включает, в частности, следующие особо предпочтительные соединения:
- α(-n{(4R)-5-гидрокси-4[(R)-3-гидрокситетрадеканоиламино]пентил}амид N[(R)-3-додеканоилокситетрадеканоиламино]-D-аспарагиновой кислоты и его солевые аддукты с неорганическим или органическим основанием (пример 1.2)
- 10(6,7-дигидроксигептаноат) 3[(R)-3-додеканолокситетрадеканоиламино]-4-оксо-5-аза-9-[(R)-3-гидрокситетрадеканоиламино]-декан-1,10-диол-1-дигидрофосфат и его солевые аддукты с неорганическим или органическим основанием (пример 2.1)
- 10-(6-оксогексаноат) 3-[(R)-3-додеканоилокситетрадеканоил-амино]-4-оксо-5-аза-9-[(R)-3-гидрокситетрадеканоиламино]-декан-1,10-диол-1-дигидрофосфата и его солевые аддукты с неорганическим или органическим основанием (пример 2.2)
- 5-O(6,7-дигидроксигептаноат) α-N{(4R)-5-гидрокси-4[(R)-3-гидрокситетрадеканоиламино]пентил}амид N-[(R)-3-додеканоилокситетрадеканоиламино]-D-аспарагиновой кислоты и его солевые аддукты с неорганическим или органическим основанием (пример 2.3)
- 5-O-(6-оксогексаноат) α-N{(4R)-5-гидрокси-4-[(R)-3-гидрокситетрадеканоиламино]пентил}амида N-[(R)-3-додеканоилокситетрадеканоиламино]-D-аспарагиновой кислоты и его солевые аддукты с неорганическим или органическим основанием (пример 2.4)
- 10-(6-аминогексаноат) (3RS, 9R), (3R, 9R) и (3S, 9R)-3-[(R)-3-додеканоилокситетрадеканоиламино]-4-оксо-5-аза-9[(R)- 3-гидрокситетрадеканоиламино]декан-1,10-диол-1-дигидрсфосфат и его солевые аддукты с неорганическим или органическим основанием (примеры 2.6, 2.7 и 2.8)
- 10-(6-гидроксигексаноат) 3-[(R)-3-додеканоилокситетрадеканоиламино]-4-оксо-5-аза-9-[(R)-3-гидрокситетрадеканоиламино]-декан-1,10-диол-1-дигидрофосфат и его солевые аддукты с неорганическим или органическим основанием (пример 2.9)
- {2-[(R)-3-гидрокситетрадеканоиламино]-5-(6-оксогексил)амино}пентиловый эфир {2-[(R)-3-додеканоилокситетрадеканоиламино]-4-(дигидроксифосфорилокси)бутановой кислоты и его солевые аддукты с неорганическим или органическим основанием (пример 2. 10)
- α-N{(4R)-5(6-аминогексаноилокси)-4[(R)-3-гидрокситетрадеканоиламино]-пентил}амид N[(R)-3-додеканоилокситетрадеканоил]-D-аспарагиновой кислоты и его солевые аддукты с неорганическим или органическим основанием (пример 2.16)
- α-N-{(4R)-5-сукцинилокси-4-[(R)-3-гидрокситетрадеканоиламино]-пентил}амид N-[(R)-3-додеканоилокситетрадеканоил]-D-аспарагиновой кислоты и его солевые аддукты с неорганическим или органическим основанием (пример 2.19)
- α-N-{(4R)-5-(6-аминогексаноилокси)-4-[(R)-3-гидрокситетрадеканоиламино]-пентил}амид N-[(R)-3-додеканоилокситетрадеканоил]-D-аспарагиновой кислоты β-N-((1S)-1-карбокси-5-аминопентил]амид и его солевые аддукты с неорганическим или органическим основанием (пример 2.22)
- α-N-{(4R)-5(6-аминогексаноилокси)-4[(R)-3-гидрокситетрадеканоиламино]- пентил}амид N[(R)-3-додеканоилокситетрадеканоил]-D-аспарагиновой кислоты β-N-((1S)-1,2-дикарбоксиэтил]амид и его солевые аддукты с неорганическим или органическим основанием (пример 2.24).
Эти соединения отличаются своими интересными и оригинальными фармакологическими свойствами, в частности иммуномодулирующими. Они представляют особый интерес при приготовлении вакцинных композиций путем их смешения или в качестве конъюгатов, ковалентно связанных с антигенами полипептидной или полисахаридной природы, или с соединениями, состоящими из полипептидов, конъюгированных с полисахаридами. Их можно использовать, в частности, для предупреждения инфекций вирусного, микробного и протозойного происхождения или при терапии некоторых аутоиммунных заболеваний.
Эти соединения находят также применение в качестве вектора молекулы, представляющей терапевтический интерес, благодаря их способности к нековалентной ассоциации в зависимости от большего или меньшего гидрофильного или гидрофобного характера их вспомогательного ответвления. Их химические свойства, которые позволяют сочетать их с молекулами, представляющими терапевтический интерес, а также их амфифильный характер благоприятствуют вводу в композицию и транспорту ассоциированных с ними молекул к мембранным рецепторам, а также к стенкам и цитоплазме клетки.
Эти соединения могут применяться, индивидуально или в виде ковалентой или нековалентной ассоциации с представляющей терапевтический интерес молекулой, перорально, парентерально, ректально, местно, через кожу или через слизистые ткани.
Они могут применяться, индивидуально или в виде ковалентной или нековалентной ассоциации, с представляющими терапевтический интерес молекулами, путем отдельно проводимой инкубации ex viva с кровяными клетками с целью придания этим клеткам иммунокомпетентности с последующей реинокуляцией клеток in vivo парентеральным путем.
Молекулы проявляют похожие свойства в качестве адъювантов к иммунной системе, используемых, например, для вакцинации, в ковалентной или нековалентной ассоциации с соответствующими антигенами против заболеваний вирусного, паразитарного, микробного и грибкового происхождения. Эти молекулы, конъюгированные или неконъюгированные, могут быть, кроме того, использованы при терапии некоторых аутоиммунных заболеваний.
Некоторые из соединений по изобретению, напротив, в результате ковалентной или нековалентной ассоциации проявляют различные свойства в отношении их способности индуцировать продукцию цитокинов или созревание иммунокомпетентных стволовых клеток, поступающих из гемопоэтических и лимфоидных органов.
Некоторые из соединений по изобретению могут способствовать созреванию и дифференцировке моноцитов в функциональные дендритные клетки в присутствии или в отсутствие соответствующего антигена и вносят, таким образом, вклад в усиление гуморального и клеточного иммунитета.
Некоторые из соединений способствуют дифференцировке клеток гемопоэтической системы, в частности костного мозга, и позволяют исправлять или корректировать некоторые нарушения иммунной системы. Они проявляют, в частности, антивирусные свойства.
Соединения по изобретению представляют особый интерес благодаря их низкой токсичности. Их используют в ковалентной или нековалентной ассоциации с антигенами при профилактике или терапии инфекционных заболеваний человека и животных в дозах, которые варьируют от 0,005 до 100 мг при разовом приеме и от 0,005 до 200 мг в сутки по показаниям и в зависимости от веса пациента.
Предметом настоящего изобретения является также способ получения N-ацилированных псевдодипептидов, имеющих кислотную группу в нейтральной или заряженной форме на одном из концов псевдодипептида, а на другом конце имеющих функционализированное вспомогательное ответвление и отвечающих общей формуле I, который состоит в блокировке аминных функций в положении (q+1) и YH в ω-положении ω-функционализированной аминокислоты ортогональными блокирующими реагентами, в действии на оставшуюся свободной карбоксильную функцию восстанавливающим агентом с образованием соответствующего спирта, в освобождении аминной функции в положении (q+1), которую затем ацилируют с помощью функционального производного карбоновой кислоты формулы R2OH, где R2 определен выше, после чего освобождают концевую функцию с получением функционализированного аминоспирта общей формулы V:
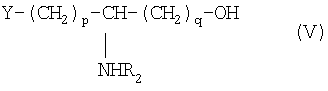
в которой Y обозначает НО или, предпочтительно, NH2;
R2 обозначает ацильную группу насыщенной или ненасыщенной карбоновой кислоты, имеющей от 2 до 24 атомов углерода, незамещенной или имеющей один или несколько определенных выше заместителей;
р и q обозначают, каждый, целое число от 1 до 10,
который конденсируют в присутствии агента пептидной конденсации в инертном растворителе с производным ω-функционализированной аминокислоты общей формулы VI:
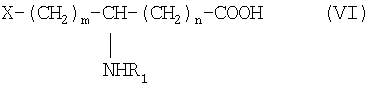
в которой R1 обозначает ацильную группу насыщенной или ненасыщенной карбоновой кислоты, имеющей от 2 до 24 атомов углерода, незамещенной или имеющей один или несколько определенных выше заместителей;
m и n являются целыми числами от 0 до 10 и
Х обозначает определенную выше кислотную группу, которая может находиться в эстерифицированной форме,
с образованием псевдодипептида общей формулы VII:
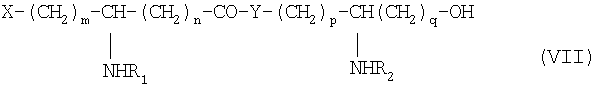
в которой заместители R1 и R2 и коэффициенты m, n, p и q такие, как определены выше,
свободная концевая спиртовая функция которого при желании может быть замещена, проалкилирована или проацилирована с помощью реагента замещения, алкилирования или ацилирования общей формулы VIII:
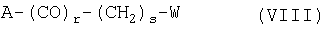
где А может быть уходящей группой, функцией OH, SH или NH2;
коэффициент r по преимуществу равен 1, но может также принимать значение O;
коэффициент s варьирует преимущественно от 2 до 6, но в общем случае может принимать значение от 1 до 10;
W выбирают преимущественно из групп: формил, ацетил, циано, галоген, амино, бром- или йодацетамидо, ациламидо, диацилимидо, сульфгидрил, алкилтио, гидроксил, 1,2-дигидроксиэтил, ацилокси, винил, этинил, карбоксил, свободный или в форме сложного эфира, смешанного ангидрида, амида или гидразида, азидо, тиоциано и их предшественники,
если необходимо, в присутствии сочетающего агента, после чего псевдодипептид подвергают каталитическому гидрированию или какому-либо другому процессу снятия защиты, получая производное общей формулы I:
в которой X, Y, Z, R1, R2, n, m, p и q имеют приведенные выше значения.
Изобретение относится также к способу получения фосфопсевдодипептидов общей формулы IV:
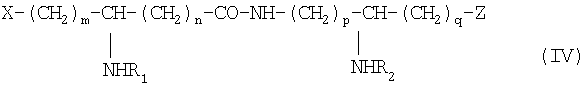
в которой R1 и R2 обозначают каждый ацильную группу насыщенной или ненасыщенной, линейной или разветвленной карбоновой кислоты, имеющей от 2 до 24 атомов углерода, незамещенной или имеющей один или несколько заместителей, выбранных из групп: гидроксил, алкил, алкокси, ацилокси, амино, ациламино, ацилтио и C1-C24-алкилтио,
коэффициенты n, p и q принимают значения от 1 до 10,
коэффициент n принимает значения от С до 10,
Х и Z обозначают, каждый, кислотную группу в нейтральной или заряженной форме или вспомогательное функционализированное ответвление,
отличающемуся тем, что осуществляют блокировку аминных функций в положениях (q+1) и ω диаминокислоты формулы H2N(СН2)pCHNH2(СН2)q-1COOH с помощью блокирующих реагентоз, лабильных, соответственно, к ацидолизу и гидрогенолизу, оставшуюся свободной карбоксильную функцию подвергают действию восстанавливающего агента с образованием соответствующего спирта, освобождают аминную функцию в положении (q+1), которую ацилируют с помощью функционального производного карбоновой кислоты формулы R2OH, в которой R2 определен выше, после чего освобождают гидрогенолизом концевую аминную функцию с получением аминоспирта общей формулы IX:
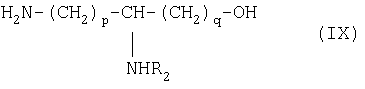
в которой R2 обозначает ацильную группу насыщенной или ненасыщенной карбоновой кислоты, имеющей от 2 до 24 атомов углерода, незамещенной или имеющей один или несколько определенных выше заместителей;
р и q обозначают целое число от 1 до 10,
который конденсируют в присутствии агента пептидной конденсации в инертном растворителе с производным ω-гидроксилированной аминокислоты общей формулы VI:
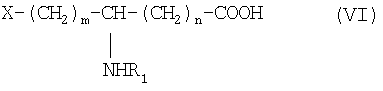
в которой R1 обозначает ацильную группу насыщенной или ненасыщенной карбоновой кислоты, имеющей от 2 до 24 атомов углерода, незамещенной или имеющей один или несколько заместителей;
m есть целое число от 1 до 10;
n есть целое число от 0 до 10; и
Х обозначает группу диалкилокси- или диарилокси-фосфорилокси формулы 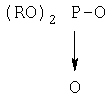 с образованием псевдодипептида общей формулы Х:
с образованием псевдодипептида общей формулы Х:
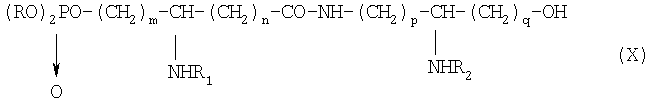
в которой заместители R1 и R2 и коэффициенты m, n, p и q являются такими, как определены выше, a R обозначает лабильный к гидрогенолизу радикал,
свободная концевая спиртовая функция которого при желании может быть замещена, проалкилирована или проацилирована с помощью реагента замещения, алкилирования или ацилирования общей формулы VIII:

где А может быть уходящей группой, функцией ОН, SH или NH2;
коэффициент r по преимуществу равен 1, но может также принимать значение 0;
коэффициент s варьирует преимущественно от 2 до 6, но в общем случае может принимать значение от 1 до 10;
W выбирают преимущественно из групп: формил, ацетил, циано, галоген, амино, бром- или йодацетамидо, ациламидо, диацилимидо, сульфгидрил, алкилтио, гидроксил, 1,2-дигидроксиэтил, ацилокси, винил, этинил, карбоксил, свободный или в форме сложного эфира, смешанного ангидрида, амида или гидразида, азидо, тиоциано и их предшественники,
если необходимо, в присутствии сочетающего агента, после чего образовавшийся продукт подвергают каталитическому гидрированию или какому-либо другому процессу снятия защиты, получая производное общей формулы XI:
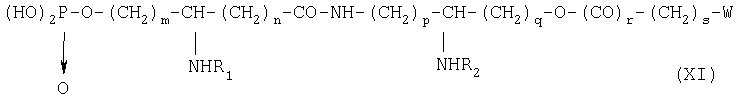
в которой заместители W, R1, R2, m, n, p, q, r, s имеют приведенные выше значения.
Изобретение относится также к способу получения фосфопсевдодипептидов общей формулы XII:
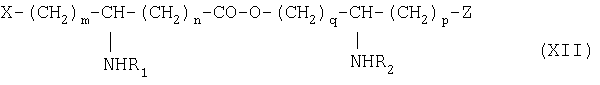
в которой R1 и R2 обозначают, каждый, ацильную группу насыщенной или ненасыщенной, линейной или разветвленной карбоновой кислоты, имеющей от 2 до 24 атомов углерода, незамещенной или имеющей один или несколько заместителей, выбранных из групп: гидроксил, алкил, алкокси, ацилокси, амино, ациламино, ацилтио и С1-С24-алкилтио,
коэффициенты m, p и q принимают значения от 1 до 10,
коэффициент n принимает значения от 0 до 10,
и в которой Х и Z обозначают, каждый, кислотную группу или вспомогательное функционализирсзанное ответвление,
состоящему в том, что осуществляют блокировку аминных функций в положениях (q+1) и ω диаминокислоты формулы H2N(CH2)pCHNH2(CH2) q-1-COOH с помощью блокирующих реагентов, лабильных, соответственно, к ацидолизу и гидрогенслизу, оставшуюся свободной карбоксильную функцию подвергают действию восстанавливающего агента с образованием соответствующего спирта, освобождают аминную функцию в положении (q+1), которую ацилируют с помощью функционального производного карбоновой кислоты формулы R2OH, в которой R2 определен выше, освобождают гидрогенолизом концевую аминную функцию и затем алкилируют ее с помощью сложноэфирного реагента с ω-функционализированным алкилом, таким как трифторметансульфонат, с получением аминоспирта обшей формулы XIII:
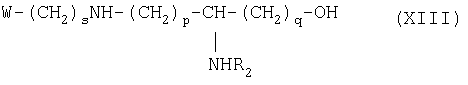
в которой R2 обозначает ацильную группу насыщенной или ненасыщенной карбоновой кислоты, имеющей от 2 до 24 атомов углерода, незамешенной или имеющей один или несколько определенных выше заместителей;
р и q обозначают целое число от 1 до 10,
в то время как коэффициент s преимущественно составляет от 2 до 7, но может принимать значение в пределах от 1 до 10,
который конденсируют в присутствии агента конденсации в инертном растворителе с производным ω-гидроксилированной аминокислоты общей формулы VI:
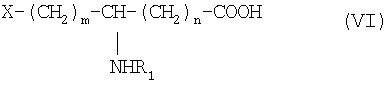
в которой R1 обозначает ацильную группу насыщенной или ненасыщенной карбоновой кислоты, имеющей от 2 до 24 атомов углерода, незамещенной или имеющей один или несколько заместителей;
m есть целое число от 1 до 10;
n есть целое число от 0 до 10; и
Х обозначает группу диалкилокси- или диарилокси-фосфорилокси формулы с образованием псевдодипептида общей формулы XIV:
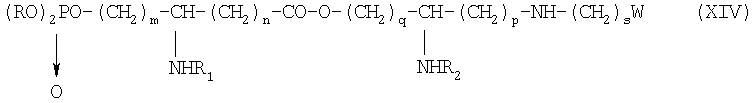
в которой заместители R1 и R2 и коэффициенты m, n, p, q и s являются такими, как определены выше, а R обозначает лабильный к гидрогенолизу радикал,
после чего псевдодипептид подвергают каталитическому гидрированию или какому-либо процессу снятия защиты с получением производного общей формулы XV:
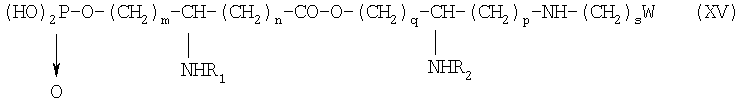
в которой заместители W, R1, R2, n, n, p, q, r и s имеют указанные выше значения,
причем W преимущественно выбирают групп: формил, ацетил, циано, галоген, бром- или йодацетамидо, ациламидо, диацилимидо, ацилокси, винил, этинил, карбоксил, свободный или в форме сложного эфира, смешанного ангидрида, амида или гидразида, азидо, тиоциано и их предшественники.
Изобретение относится также к способу получения карбоксипсевдодипептидов общей формулы IV:
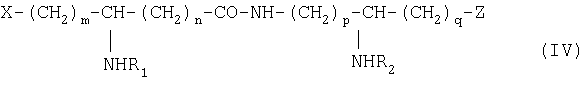
в которой R1 и R2 обозначают, каждый, ацильную группу насыщенной или ненасыщенной, линейной или разветвленной карбоновой кислоты, имеющей от 2 до 24 атомов углерода, незамещенной или имеющей один или несколько заместителей, выбранных из групп: гидроксил, алкил, алкокси, ацилокси, амино, ациламино, ацилтио и С1-С24-алкилтио,
коэффициенты m, р и q принимают значения от 1 до 10,
коэффициент n принимает значения от 0 до 10,
и в которой Х и Z обозначают, каждый, кислотную группу или вспомогательное функционализированное ответвление,
который состоит в том, что осуществляют блокировку аминных функций в положениях (q+1) и в ω диаминскислоты формулы H2N(CH2)pCHNH2(СН2)q-1COOH с помощью блокирующих реагентов, лабильных, соответственно, к ацидолизу и гидрогенолизу, оставшуюся свободной карбоксильную функцию подвергают действию восстанавливающего агента с образованием соответствующего спирта, освобождают аминную функцию в положении (q-1), которую ацилируют с помощью функционального производного карбоновой кислоты формулы R2ОН, в которой R2 определен выше, и освобождают гидрогенолизом концевую аминную функцию, получая аминоспирт обшей формулы IX:
в которой R2 обозначает ацильную группу насыщенной или ненасыщенной карбоновой кислоты, имеющей от 2 до 24 атомов углерода, незамещенной или имеющей один или несколько определенных выше заместителей;
р и q обозначают целое число от 1 до 10,
который конденсируют в присутствии агента пептидной конденсации в инертном растворителе с функциональным производным ω-аминокарбоновой кислоты общей формулы VI:
в которой R1 обозначает ацильную группу насыщенной или ненасыщенной карбоновой кислоты, имеющей от 2 до 24 атомов углерода, незамещенной или имеющей один или несколько определенных выше заместителей;
m есть целое число от 1 до 10,
n есть целое число от 0 до 10 и
Х обозначает радикал RO-CO-,
с образованием псевдодипептида общей формулы XVI:
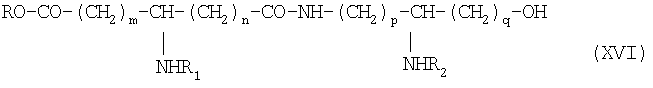
в которой заместители R1 и R2 и коэффициенты m, n, p и q являются такими, как определены выше, a R обозначает лабильную к гидрогенолизу группу, такую как, например, бензильная группа,
свободная концевая спиртовая функция которого при желании может быть замещена, проалкилирована или проацилирована с помощью реагента замещения, алкилирования или ацилирования общей формулы VIII:
где А обозначает уходящую группу, функцию ОН, SH или NH2;
коэффициент r по преимуществу равен 1, но может также принимать значение 0;
коэффициент s варьирует преимущественно от 2 до 6, но может принимать значение от 1 до 10;
W выбирают преимущественно из групп: формил, ацетил, циано, галоген, амино, бром- или йодацетамидо, ациламидо, диацилимидо, сульфгидрил, алкилтио, гидроксил, 1,2- дигидроксиэтил, ацилокси, винил, этинил, карбоксил, свободный или в форме сложного эфира, смешанного ангидрида, амида или гидразида, азидо, тиоциано и их предшественники,
если необходимо, в присутствии сочетающего агента, после чего соединение подвергают каталитическому гидрированию или какому-либо процессу снятия защиты, получая производное общей формулы XVII:
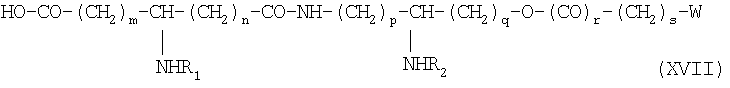
в которой заместители W, R1, R2, m, n, р, q, r, s имеют приведенные выше значения.
Изобретение относится также к способу получения фосфодипептидов общей формулы IV:
в которой R1 и R2 обозначают, каждый, ацильную группу насыщенной или ненасыщенной, линейной или разветвленной карбоновой кислоты, имеющей от 2 до 24 атомов углерода, незамещенной или имеющей один или несколько заместителей, выбранных из групп: гидроксил, алкил, алкокси, ацилокси, амино, ациламино, ацилтио и С1-С24-алкилтио,
коэффициенты m, р и q принимают значения от 1 до 10,
коэффициент п принимает значения от 0 до 10,
и в которой Х и Z обозначают, каждый, кислотную группу или вспомогательное функционализированное ответвление,
который состоит в том, что осуществляют блокировку свободной гидроксильной функции псевдодипептида общей формулы XVI:
в которой заместители R1 и R2 и коэффициенты m, n, p и q являются такими, как определены выше, a R обозначает лабильную к гидрогенолизу группу, такую как, например, бензильная группа,
с помощью лабильной к ацидолизу защитной группы, освобождают карбоксильную функцию, присутствующую в виде сложного эфира, подвергают эту карбоксильную функцию, возможно в активированной форме, действию восстанавливающего агента с образованием соответствующего первичного спирта, превращают спиртовую функцию в фосфорноэфирную действием фосфорилирующего агента, защищенную гидроксильную функцию освобождают действием кислоты и вводят в реакцию замещения, алкилирования или ацилирования с реагентом общей формулы VIII:
где А может быть уходящей группой, функцией ОН, SH или NH2;
коэффициент r по преимуществу равен 1, но может также принимать значение 0;
коэффициент s варьирует преимущественно от 2 до 6, но вообще может принимать значение от 1 до 10;
W выбирают преимущественно из групп: формил, ацетил, циано, галоген, амино, бром- или йодацетамидо, ациламидо, диацилимидо, сульфгидрил, алкилтио, гидроксил, 1,2-дигидроксиэтил, ацилокси, винил, этинил, карбоксил, свободный или в форме сложного эфира или какого-либо другого производного,
если необходимо, в присутствии сочетающего агента, после чего полученный продукт деблокируют, например, с помощью гидрогенолиза, получая продукт общей формулы XI:
в которой заместители W, R1, R2 m, n, p, q, r и s имеют приведенные выше значения.
Изобретение относится также к способу получения фосфодипептидов общей формулы IV:
в которой R1 и R2 обозначают, каждый, ацильную группу насыщенной или ненасыщенной, линейной или разветвленной карбоновой кислоты, имеющей от 2 до 24 атомов углерода, незамещенной или имеющей один или несколько заместителей, выбранных из групп: гидроксил, алкил, алкокси, ацилокси, амино, ациламино, ацилтио и С1-С24-алкилтио,
коэффициенты m, p и q принимают значения от 1 до 10,
коэффициент n принимает значения от 0 до 10,
и в которой Х и Z обозначают, каждый, кислотную группу или вспомогательное функционализированное ответвление,
который состоит в том, что осуществляют деблокирование карбоксильной функции, присутствующей в виде сложного эфира в псевдодипептиде общей формулы XVI:
в которой заместители R1 и R2 и коэффициенты m, n, p и q являются такими, как определены выше, a R обозначает лабильную к гидрогенолизу группу, такую как, например, бензильная группа,
после чего подвергают образовавшуюся таким образом кислоту реакции пептидного сочетания с частично защищенными диаминоалканом или аминокислотой, такими, например, как 3-бензилоксикарбониламино-1-аминопропан, дибензил-L-аспартат или бензиловый эфир ε-N-бензилоксикарбонил-L-лизина, в присутствии агента пептидного сочетания, после чего продукт сочетания подвергают при желании реакции замещения, алкилирования или ацилирования с использованием реагента общей формулы VIII:
где А может быть уходящей группой, функцией ОН, SH или NH2;
коэффициент r по преимуществу равен 1, но может также принимать значение 0;
коэффициент s варьирует преимущественно от 2 до 6, но вообще может принимать значение от 1 до 10;
W выбирают преимущественно из групп: формил, ацетил, циано, галоген, амино, бром- или йодацетамидо, ациламидо, диацилимидо, сульфгидрил, алкилтио, гидроксил, 1,2-дигидроксиэтил, ацилокси, винил, этинил, карбоксил, свободный или в форме сложного эфира или какого-либо другого производного,
если необходимо, в присутствии активирующего агента, после чего полученный продукт деблокируют, например, с помощью гидрогенолиза, получая продукт обшей формулы XVIII:
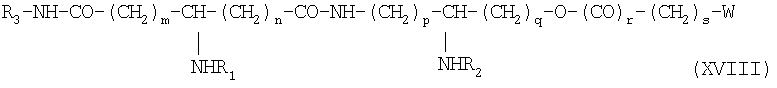
в которой заместители W, R1, R2, m, n, p, q, r и s имеют приведенные выше значения, а группа R3 обозначает аминоалкил, карбоксиалкил, дикарбоксиалкил или аминокарбоксиалкил.
Стереохимия центров, содержащих ациламиногруппы, определяется конфигурацией исходных аминокислот и конфигурацией ациламиногрупп, обусловленной конфигурацией исходных жирных кислот. Можно исходить из диаминокислот с конфигурацией L, D или рацемических. Можно исходить из гидроксилированных аминокислот с конфигурацией L, D или рацемических. Все энантиомеры или диастереоизомеры соединений общей формулы I, IV, XII или XVI составляют часть изобретения.
В общем случае соединения по изобретению получают сочетанием кислотной функции N-ацилированной ω-функционализированной аминокислоты и аминной функции аминоспирта, образующегося при восстановлении карбоксила моно-N-ацилированной диаминокислоты, с последующим О-ацилированием или -алкилированием оставшейся свободной спиртовой функции для введения вспомогательного функционализированного ответвления, которое может быть модифицировано после закрепления с целью освобождения реакционноспособной функции. Деблокирование конечного продукта освобождает кислотную функцию.
В предпочтительном для настоящего случая методе получения соединений по изобретению (фиг.1) ω-функционализированной аминокислотой является ω-гидрокси-α-аминокислота, такая как серин или гомосерин, которую подвергают последовательности реакций N-блокировки (например, в виде производного трет-бутоксикарбонила), образования бензилового эфира путем О-алкилирования карбоксилата и фосфорилирования функции ОН с целью введения защищенной фосфатной группы. Типичными защитными группами для фосфатов могут быть фенил, бензил или о-ксилил. Предпочтительным для настоящего случая является фенил. Аминную функцию после этого освобождают удалением защитной группы (например, обработкой трет-бутоксикарбонильного производного трифторуксусной кислотой) и затем ацилируют активированным производным жирной кислоты, преимущественно производным 3-гидрокситетрадекановой кислоты, таким как 3-додекансилокситетрадекановая кислота. Активированной формой может быть ацилхлорид, активированный сложный эфир, смешанный ангидрид или любая другая химическая форма, делающая возможным образование амидной связи. Возможно присутствующий бензиловый эфир удаляют затем селективным гидрогенолизом, получая карбоновую кислоту с ациламидной группой в положении α и с группой (RO)2P(O)O- в положении ω.
Партнер по реакции пептидного сочетания получают преимущественно исходя из α,ω-диаминокислоты, такой как орнитин или лизин, с использованием последовательности реакций селективной защиты аминной функции в положении и, например, в виде бензилоксикарбонильного производного, с помощью медного комплекса по методу, описанному в Organic Preparations [Organic Preparations and Procedures International (1992), 23, 191-194], элиминирования медного комплекса и защиты аминной функции в положении α, например, в виде трет-бутоксикарбонильного производного. Могут быть использованы и другие защитные группы. Свободную карбоксильную функцию восстанавливают до первичного спирта, используя, например, диметилсульфид-борановый комплекс, или действием смешанного ангидрида, предварительного полученного с использованием борогидрида натрия, по методу, описанному в Tetrahedron Letters (1991), 32, 923-92. Аминную функцию в положении α освобождают в кислой среде (например, действием трифторуксусной кислоты), затем проводят N-ацилирование активированным производным какой-либо жирной кислоты, преимущественно производным 3-гидрокситетрадекановой кислоты, таким как 3-бензилокситетрадекановая кислота. Свободную функцию ОН на этой стадии при необходимости защищают, например, в виде бензилоксиметилового эфира. ω-Аминную функцию освобождают действием реагента, инертного по отношению к другим имеющимся защитным группам, например, с помощью селективного гидрогенолиза в гидроксильном растворителе, содержащем триэтиламин в том случае, когда защитной группой является бензилоксикарбонил.
В предпочтительном для настоящего случая варианте синтеза полученный указанным способом амин сочетают с ω-фосфорилированной α-ациламинированной карбоновой кислотой, полученной, как описано выше, в присутствии 2-изобутокси-1-изобутоксикарбонил-1,2-дигидрохинолина (IID2) или какого-либо другого агента пептидного сочетания, получая защищенный фосфорилированный псевдодипептид. Этот продукт подвергают О-ацилированию по остающейся свободной гидроксильной функции с помощью ω-функционализированной кислоты, такой как 6-гептеновая кислота, в присутствии 1-(3-диметиламинопропил)-3-этилкарбодиимида (EDCI) или какого-либо другого агента эстерификации (фиг.5). Алкенильную функцию этого эфира подвергают реакции дигидроксилирования в присутствии тетроксида осмия в каталитическом или стехиометрическом количестве, после чего защитные группы фосфата и гидроксильную функцию, возможно в форме бензилового эфира, удаляют с помощью гидрогенолиза в присутствии подходящего катализатора (пример 2.1). Во второй стадии вицинальный диол подвергают окислению, например, с помощью йодной кислоты, с образованием реакционно-способной альдегидной функции (пример 2.2). Восстановление альдегидной функции в первично-спиртовую приводит к производному с ω-гидроксиацильной группой (пример 2.9).
В одном из вариантов способа продукт пептидного сочетания подвергают О-ацилированию с помощью производного ω-аминоалкановой кислоты, такой как 6-бензилоксикарбониламиногексановая кислота (фиг.11). Полученный в результате этого продукт подвергают реакции полного деблокирования с помощью гидрогенолиза в присутствии катализатора, что дает псевдодипептид, имеющий вспомогательное аминоалканоильное ответвление (пример 2.6).
В другом предпочтительном варианте кислотный партнер реакции пептидного сочетания представляет собой D- или L-аспарагиновую кислоту или, в некоторых случаях, D- или L-глутаминовую кислоту (фиг.2). Производное аспарагиновой кислоты получают N-ацилированием аминной функции β-бензилового эфира этой аминокислоты с помощью жирной кислоты, преимущественно производным 3-гидроксизамещенной жирной кислоты, таким как 3-додеканоилокситетрадекановая кислота, в присутствии агента ацилирования. Этот продукт сочетают с аминоспиртом описанного выше типа, после чего полученный таким образом псевдодипептид подвергают реакции гидрогенолиза в присутствии катализатора, такого как палладий на угле или платина на носителе, получая псевдодипептид с кислотной карбоксильной функцией (пример 1.2), или реакции фссфорилирования, например, методом с фосфорамидитом, с последующей реакцией гидрогенолиза, приводящей к псевдодипетиду с карбоксильной функцией и фосфорнокислой функцией (пример 1.3).
В одном из вариантов гидроксильную функцию полученного выше псевдодипетида (фиг.2) ацилируют ω-функционализированной кислотой. Такой кислотой преимущественно является ω-алкеновая или ω-аминоалкановая кислота. В случае гептеновой кислоты (фиг.8) псевдодипетид дает O-гептеноильное производное, которое последовательно подвергают реакциям дигидроксилирования в присутствии тетроксида осмия, затем деблокирования (пример 2.3) и перйодатного окисления (пример 2.4). Восстановление образовавшейся при этом альдегидной функции с помощью восстанавливающего агента, такого, например, как борогидрид натрия, дает псевдодипетид с ω-гидроксиалканоильной функцией (пример 2.5). В случае ω-аминоалкановой кислоты, например, N-бензилоксикарбонильного производного 6-аминогексановой кислоты или глицина (фиг.24) ацилирование псевдодипетида приводит к защищенным О-аминоацилированным производным, которые затем подвергают реакции деблокирования с помощью гидрогенолиза (примеры 2.16 и 2.18).
В другом варианте (фиг.13) описанный на фиг.2 промежуточный псевдодипетид подвергают реакции блокировки свободной гидроксильной функции, преимущественно в виде тетрагидропиранильного эфира, после чего присутствующую в виде сложного эфира карбоксильную функцию деблокируют с помощью гидрогенолиза или какого-либо другого метода деблокирования и затем восстанавливают (преимущественно после активации в форме смешанного ангидрида) восстанавливающим агентом, таким как борогидрид натрия. Образовавшуюся в результате этого гидроксильную функцию подвергают реакции фосфорилирования, преимущественно методом с фосфорамидитом, после чего тетрагидропиранильный эфир гидролизуют в кислой среде, и регенерированную таким образом гидроксильную функцию подвергают реакции ацилирования с использованием ω-функционализированной карбоновой кислоты. ω-Функционализированной карбоновой кислотой преимущественно является ω-алкеновая или ω-аминоалкановая кислота. В случае 6-бензилоксикарбониламиногексановой кислоты псевдодипетид приводит к защищенному О-аминоацилированному производному, которое затем подвергают реакции гидрогенолиза (примеры 2.7 и 2.8). Промежуточный монофосфорилированный продукт из предыдущего абзаца может быть также получен (фиг.15) пептидным сочетанием N-ацилированного производного β-бензилового эфира аспарагиновой кислоты (фиг.2) с какой-либо защищенной формой, например, в виде бензилоксиметилового, тетрагидропиранильного или силилового эфира аминоспирта, образованного от орнитина (фиг.1), с последующим освобождением кислотной функции, присутствующей в виде бензилового эфира, восстановления ее до первично-спиртовой, фосфорилирования этой функции и деблокирования спиртовой функции, защищенной в виде бензилоксиметилового, тетрагидропиранильного или силилового эфира.
В третьем варианте описанный на фиг.2 промежуточный псевдодипетид подвергают реакции О-ацилирования (фиг.27) с помощью производного дикарбоновой кислоты, преимущественно янтарного ангидрида, в присутствии основания или сочетающего агента, после чего образовавшийся таким образом новый сложный эфир подвергают реакции деблокирования гидрогенолизом (пример 2.19). Действие на деблокированные О-аминоацильные производные янтарным ангидридом дает псевдодипетиды с сукциниламиноацильной группой (пример 2.17).
В третьем предпочтительном методе полученный из орнитина или лизина аминоспирт, как это описано выше, алкилируют ω-алкенилтрифлатом, таким как гепт-6-енилтрифлат (фиг.17). Сочетание этого аминоспирта, имеющего вторичную аминогруппу, с описанной выше ω-фосфорилированной α-ациламинированной кислотой в присутствии EDCI или какого-либо другого агента ацилирования дает сложный эфир. Алкенильную группу после этого подвергают реакции дигидроксилирования в присутствии тетроксида осмия, и затем защитные группы удаляют гидрогенолизом в присутствии подходящих катализаторов, а вицинальную диоловую функцию окисляют перйодатом натрия с образованием реакционно-способной альдегидной функции (пример 2.10).
В четвертом предпочтительном методе (фиг.19) N-защищенное производное серина, например, п-метоксибензилоксикарбонильное производное, полученное по методу, описанному в Synthesis (1989), 36-37, подвергают О-алкилированию с помощью бензилбромацетата. Защитную группу аминной функции удаляют, например, действием трифторуксусной кислоты в дихлорметане, а аминную функцию ацилируют, преимущественно производным 3-гидрокси-жирной кислоты, таким как 3-додеканоилокситетрадекановая кислота, в присутствии агента ацилирования или с использованием хлорангидрида кислоты или какой-либо другой активированной формы жирной кислоты. Полученное таким образом N-ацилированное производное О-алкилированного серина сочетают с списанным выше (фиг.1) аминоспиртом в присутствии агента пептидного сочетания, такого как IIDQ, после чего свободную функцию ОН ацилируют ω-функционализированной алкановой кислотой в присутствии активирующего агента, такого как карбодиимид. Такой функционализированной кислотой преимущественно является ω-алкеновая кислота или какое-либо производное ω-аминоалкановой кислоты. Например, с гепт-6-еновой кислотой псевдодипетид приводит к О-(6-гептеноильному) производному, которое последовательно подвергают реакциям дигидроксилирования в присутствии тетроксида осмия и затем деблокированию гидоогенолизом и перйодатному окислению (пример 2.11).
В предпочтительном для настоящего случая пятом методе исходной аминокислотой является цистеин или гомоцистеин (фиг.20). Например, цистеин подвергают S-алкилированию п-метоксибезилбромацетатом и затем N-ацилируют, преимущественно производным 3-гидрокси-жирной кислоты, таким как 3-тетрадеканоилокситетрадекановая кислота, в присутствии агента анилирования или используя галогенангидрид кислоты или какую-либо другую активированную форму жирной кислоты. Полученное таким образом S-алкилированное и N-ацилированное производное цистеина сочетают с 5-амино-2[(R)-3-гидрокситетрадеканоиламино]пентан-1-олом в присутствии IIDQ или агента пептидного сочетания. Свободную первичную функцию ОН подвергают затем О-ацилированию ω-функционализированной алкановой кислотой, используемой в виде активированного сложного эфира, например О-бензотриазолилового эфира. Этой кислотой преимущественно является ω-аминоалкановая кислота, такая как 7-(п-метоксибензилоксикарбониламино)гептановая кислота. Полученный при этом продукт подвергают реакции полного деблокирования в кислой водной среде с образованием псевдодипетида, имеющего 7-аминогептаноильное вспомогательное ответвление (пример 2.12).
В другом предпочтительном методе (фиг.21) N-п-метоксибензилоксикарбонильное производное серина подвергают O-алкилированию с помощью дибензилового эфира метиленмалоновой кислоты в щелочной среде. Защитную группу амина удаляют действием кислоты и освобожденную таким образом аминную функцию ацилируют, преимущественно производным 3-гидрокси-жирной кислоты, таким как 3-додеканоилокситетрадекановая кислота, в присутствии агента ацилирования или с использованием функционального производного жирной кислоты, такого как хлорангидрид кислоты или какая-либо другая активированная форма жирной кислоты. Полученное таким образом N-ацилированное производное O-алкилированного серина сочетают с описанным выше (фиг.1) аминоспиртом в присутствии IICQ или любого другого агента пептидного сочетания. Псевдодипетид после этого подвергают O-ацилированию по свободной функции ОН с помощью ω-функционализированной алкановой кислоты. Такой кислотой преимущественно является ω-алкеновая кислота или какое-либо производное ω-аминоалкановой кислоты, например гепт-6-еновая кислота, и, таким образом, псевдодипетид приводит к О-(6-гептеноильному) производному, которое подвергают последовательно реакциям дигидроксилирования в присутствии тетроксида осмия и затем деблокированию гидрогенолизом и периодатному окислению, в результате чего получают производное, имеющее 6-оксогексаноильное вспомогательное ответвление и малонильную группу (пример 2.13).
В альтернативном и также предпочтительном методе (фиг.22) описанное выше (фиг.11) 6-аминогексаноильное производное подвергают N-ацилированию бромацетамидной группой, используя в качестве ацилирующего агента, например, O-сукцинимидиловый эфир бромуксуснсй кислоты. Эта реакция дает производное, имеющее 6-(бромацетамидо)гексаноильное вспомогательное ответвление (пример 2.14).
В восьмом предпочтительном методе (фиг.23) бензиловый эфир O-фосфорилированного гомомерина (см. фиг.1) подвергают N-ацилированию 3-бензилокситетрадекановой кислотой, преимущественно используя смешанный ангидрид, полученный исходя из 3-бензилокситетрадекановой кислоты и изобутилхлорформиата. Бензиловый эфир после этого подвергают селективному гидрогенолизу, как описано выше. ω-N-Бензилоксикарбонильное производное диаминоспирта, образованного от орнитина (см. фиг.1), подвергают Nα-ацилированию с помощью 3-додеканоилокситетрадекановой кислоты в присутствии сочетающего агента или с использованием активированного эфира или любой другой активированной формы жирной кислоты, а ω-аминную функцию освобождают селективным гидрогенолизом. Этот амин сочетают с N-(3-бензилокситетрадеканоил)-О-(дифенилоксифосфорильным) производным гомосерина в присутствии IIDQ или другого агента пептидного сочетания. Полученный таким образом псевдодипетид подвергают O-ацилированию ω-функционализированной алкановой кислотой, например, в присутствии карбодиимида. Этой кислотой преимущественно является ω-алкеновая кислота или какое-либо производное ω-аминоалкановой кислоты. Например, с гепт-6-еновой кислотой псевдодипетид приводит к О-(6-гептеноильному) производному, которое подвергают последовательно реакциям дигидроксилирования в присутствии тетроксида осмия и затем деблокированию гидрогенолизом в присутствии подходящих катализаторов и перйодатному окислению с образованием производного, имеющему 6-оксогексаноильное вспомогательное ответвление (пример 2.15).
В девятом предпочтительном методе (фиг.29, 31 и 34) описанный на фиг.2 промежуточный псевдодипетид подвергают реакции деблокирования карбоксильной функции, присутствующей в виде сложного эфира, после чего карбоксильную функцию сочетают с функционализированным аминоалканом в присутствии сочетающего агента. Функционализированным аминоалканом преимущественно является производное α,ω-диаминоалкана или аминокислота. В случае 1-амино-3-бензилоксикарбониламинопропана (фиг.3) продукт сочетания подвергают далее реакции гидрогенолиза (пример 2.20). В случае бензилового эфира Nε-Z-лизина (фиг.31) и α,β-дибензиласпартата (фиг.34) продукт сочетания подвергают далее реакции деблокирования, преимущественно гидрогенолизом (примеры 2.21 и. 2.23), либо реакции O-ацилирования ω-функционализированной алкановой кислотой, преимущественно N-бензилоксикарбонильным производным 6-аминогексановой кислоты. Образовавшийся при этом сложный эфир после этого деблокируют гидрогенолизом или с помощью какого-либо другого способа деблокирования (примеры 2.22 и 2.24).
Изобретение относится также к промежуточным продуктам общей формулы V, VI, IX, XIII, XVI в форме индивидуальных энантиомеров и/или стереоизомеров или в форме смеси стереоизомеров.
Изобретение относится также к фармацевтическим композициям, содержащим в качестве активного начала по меньшей мере одно соединение общей формулы I в нейтральной или заряженной форме, в ассоциации или смеси с нетоксичным и фармацевтически приемлемым инертным эксципиентом или носителем.
Изобретение относится более конкретно к фармацевтическим композициям, содержащим в качестве активного начала по меньшей мере одну соль соединения общей формулы I с терапевтически совместимым минеральным или органическим основанием.
Изобретение относится также к фармацевтическим композициям на основе соединения общей формулы I в форме индивидуального энантиомера или в форме смеси стереоизомеров в ассоциации или в смеси с фармацевтическим наполнителем или носителем.
Из предусматриваемых фармацевтических форм можно назвать те формы, которые пригодны для пищеварительного тракта, парентерального применения, для ингаляции, местного применения, введения через кожу или слизистые ткани, например, в форме таблеток, драже, желатиновых капсул, пригодных для инъекций растворов или суспензий, аэрозолей, гелей, пластырей или проникающих растворов.
Соединения по изобретению могут быть с успехом привиты к какому-либо антигену с целью модифицирования иммунного ответа или же привиты к какому-либо фармакофору для улучшения его действия или его позиционирования для преимущественного использования в виде инъекций в форме конъюгатов, растворов или водных суспензий, возможно нейтрализованных каким-либо амином или гидроксиламином. В качестве примера можно назвать антиген H1N1, нонапептидный антиген SYVPSAEQI плазмодия и фармакофоры, такие как AZT, d4T, а также антибиотики, такие как макролиды, и вещества, оказывающие воздействие на центральную нервную систему.
Приведенные ниже примеры, представленные на фиг.1-86, иллюстрируют изобретение, не ограничивая его объема.
ПРИМЕРЫ
1-ая серия примеров: получение синтетических полупродуктов
ПРИМЕР 1.1
1-(Дифенилоксифосфорилокси)-3-[(R)-3-додеканоилокситетрадеканоиламино]-4-оксо-5-аза-9-[(R)-3-бензилокситетрадеканоиламино]декан-10-ол (фиг.1)
1.1.1. 4-(Дифенилоксифосфорилокси)-2-[(R)-3-додеканоилокситетрадеканоиламино]бутановая кислота
а. N-Трет-бутидоксикарбонил-DL-гомосерин
К раствору гидробромида гомосерина (2 г; 16,78 ммоль) в Н2O (20 мл) добавляют 1 М раствор NaOH (16,78 мл), затем карбонат цезия (3,01 мг; 9,23 ммоль). После 5 мин перемешивания раствор охлаждают на водяной бане со льдом. Добавляют после этого диоксан (60 мл) и трет-бутилпирокарбонат. Реакционную смесь перемешивают на водяной бане со льдом в течение 1 часа и затем 5 час при комнатной температуре. Упаривают в вакууме растворитель и сухой остаток непосредственно используют для следующей стадии.
b. Бензиловый эфир N-трет-бутилоксикарбонил-DL-гомосерина
К полученному выше остатку добавляют диметилформамид (20 мл) и досуха упаривают. После этого к реакционной смеси добавляют диметилформамид (60 мл) и бензилбромид (4,5 мл; 20,13 ммоль) - образуется белый осадок. Реакционную смесь перемешивают 16 час, упаривают вакууме растворитель и остаток экстрагируют этилацетатом (2×20 мл). Органическую фазу промывают Н2O (20 мл) и затем солевым раствором (20 мл). После высушивания над MgSO4 упаривают растворитель, а остаток используют как таковой для следующей стадии.
с. Бензиловый эфир N-трет-бутилоксикарбонил-О-(дифенилоксифосфорил) -DL-гомосерина
К раствору сухого остатка предыдущей стадии в СН2Cl2 (60 мл) добавляют 4-(N,N-диметиламино)пиридин (DMAP) (4,11 г; 33,56 ммоль). Реакционную смесь перемешивают в течение 20 мин, добавляют пиридин (12 мл) и дифенилхлорфосфат (6,95 мл; 33,56 ммоль). Раствор перемешивают 18 час при комнатной температуре, затем промывают 1 н. HCl (5×20 мл), H2O (30 мл) и затем солевым раствором (30 мл). Органическую фазу высушивают над MgSO4 и упаривают в вакууме растворитель. Очистка флэш-хроматографией на силикагеле (элюент гексан/этилацетат 4/1) позволяет выделить фосфорилированный продукт (7,49 г; 82,4%) в виде кристаллического твердого вещества с т.пл. 63,5-64,0°С.
d. Трифторацетат бензилового эфира О-(дифенилоксифосфорил)-DL-гомосерина
Раствор фосфорилированного продукта предыдущей стадии (7,88 г; 15,4 ммоль) в трифторуксусной кислоте (15 мл) перемешивают при комнатной температуре в течение 2,5 час и упаривают в глубоком вакууме растворитель. Очистка флэш-хроматографией на силикагеле (элюент МеОН/СН2Cl2 10/1) позволяет выделить трифторацетатную соль деблокированного амина (7,17 г; 88,9%) в виде кристаллического твердого вещества с т.пл. 73,0-73,5°С.
е. Бензиловый эфир 2-[(R)-3-додеканоилокситетрадеканоиламино]-4-(дифенилоксифосфорилокси)бутановой кислоты
Приготовляют раствор (R)-3-додеканоилокситетрадекановой кислоты (4,284 г; 10,07 ммоль), полученной по методу, описанному в Bull. Chem. Soc. Jpn 60 (1987), 2205-2214, в ТГФ (30 мл) и охлаждают до -15°С. После этого добавляют N-метилморфолин (1,108 мл; 10,07 ммоль; 1 экв) и изобутилхлорформиат (1,31 мл; 10,07 ммоль; 1 экв). Реакционную смесь перемешивают в течение 30 мин при -15°С и добавляют раствор бензилового эфира О-(дифенилоксифосфорил)-DL-гомосерина (5,724 мл; 10,07 ммоль; 1 экв) в смеси ТГФ/Et3N (30 мл/5 мл). После перемешивания в течение 18 час при комнатной температуре упаривают в вакууме растворитель, остаток разбавляют H2O (20 мл) и экстрагируют этилацетатом (2 х 30 мл). Органические фазы объединяют, последовательно промывают водой (20 мл) и рассолом (20 мл), после чего высушивают над Mg3O4 и упаривают. Очистка флэш-хроматографией на силикагеле (элюент гексан/этилацетат 2/1) позволяет выделить желаемый бензиловый эфир (7,455 г; 87%) в виде кристаллического твердого вещества с т.пл. 31,0-32,1°С. 1Н-ЯМР (CDCl3, 250 МГц), δ в м.д.: 7,4-7,1 (м, 15Н); 6,90 (2д, 1Н, 3J=7,6 Гц, NH); 5,3-5,1 (м, 3Н); 4,7 (м, 1Н); 4,35 (м, 2Н); 2,45 (м, 2Н); 2,4-2,1 (м, 4Н); 1,6 (м, 4Н); 1,4-1,1 (м, 34H); 0,9 (т, 6Н). 13С-ЯМР (CDCl3, 63 МГц), δ в м.д.: 173,01; 171,08; 169,66; 150,18 (д, 2Jp,с = 7,1 Гц); 135,01; 129,60; 128,33; 128,14; 127,96; 125,21; 119,80 (д, 3Jp,c = 5,0 Гц); 70,69; 67,05; 65,19 (д, 2Jp,с = 5,6 Гц); 49,13; 40,97; 40,77 (2 диаст.); 34,20; 33,98; 33,82; 31,70; 29,42; 29,34; 29,14; 28,94; 25,01; 24,77; 22,47; 13,91.
f. 4-(Дифенилоксифосфорилокси)-2-[(R)-3-додеканоилокситетрадеканоиламино]бутановая кислота
Раствор полученного выше бензилового эфира (2,23 г; 2,6 ммоль) в метаноле для ЖХВР (300 мл) гидрируют в течение 1 часа в трехгорлой колбе в присутствии 10% Pd/C (1 г) при комнатной температуре и атмосферном давлении водорода. Удаляют фильтрацией катализатор и упаривают фильтрат с образованием бесцветного сиропа. Последний однороден при хроматографировании в тонком слое и в ЯМР и без дальнейшей очистки непосредственно используется для стадии сочетания. Rf = 0,75 (CH2Cl2/MeOH/Et3N 10/1/0,5). ЯМР (CDCl3, 250 МГц), δ в м.д.: 7,4-7,1 (м, 10Н); 6,85 (д, 1Н, NH); 5,15 (м, 1Н); 4,6 (м, 1Н); 4,35 (м, 2Н); 2,45 (м, 2Н); 2,4-2,15 (м, 4Н); 1,6 (м, 4Н); 1,4-1,1 (м, 34Н); 0,9 (т, 6H). 13С-ЯМР (CDCl3, 63 МГц), δ в м.д.: 173,35; 173,30 (2 диаст.); 172,75; 170,37; 150,0 (д, 2Jp,с = 7,5 Гц); 129,55; 125,28; 119,71 (д, 3Jp,с = 4,4 Гц); 70,78; 65,65 (д, 3Jp,с = 5,9 Гц); 49,00; 40,77; 40,63 (2 диаст.); 34,13; 33,86; 33,76; 31,59; 29,31; 29,25; 29,03; 28,82; 24,88; 24,68; 22,36; 13,76.
1.1.2. (2R)-5-Амино-2[(R-3-бензилокситетрадеканоиламино]пентан-1-ол
а. Медная соль D-орнитина
К раствору D-орнитина (5,25 г; 30 ммоль) в 1 н. NaOH (30 мл) добавляют раствор сульфата меди(II)-пентагидарата (3,814 г; 15,3 ммоль) в H2O (50 мл), перемешивают 2 часа при комнатной температуре и упаривают досуха растворитель. К остатку добавляют метанол (60 мл), в результате чего образуется твердое вещество пурпурного цвета, которое отделяют, промывают последовательно диоксаном и метанолом, после чего непосредственно используют в следующей стадии.
b. (2R)-2-Амино-5-(бензилоксикарбониламино)пентаноат меди
Твердое пурпурного цвета вещество растворяют в смеси 1 н. NaOH (40 мл) и диоксана (70 мл). Реакционную смесь охлаждают на водяной бане со льдом и после этого добавляют бензилхлорформиат (5,14 мл; 36 ммоль). После 3 час перемешивания на водяной бане со льдом и затем 15 час при комнатной температуре выделяют фильтрацией пурпурный осадок, который последовательно промывают 95%-ным EtOH (40 мл), Н2O (50 мл) и EtOH (60 мл). Осадок сушат в сушильном шкафу (T<45°C, вакуум). Выход в двух стадиях составляет 8,27 г, т.е. 93% от теории (ссылка: Organic Preparations and procedures international 23 (1992), 191-194).
с. (2R)-5-(Бензилоксикарбониламино)-2(трет-бутилоксикарбониламино)пентановая кислота
Полученную выше медную соль растворяют в 2 н. растворе HCl (400 мл) и добавляют этилендиаминтетрауксусную кислоту (ЭДТА) (8,15 г; 27,8 ммоль). После 2,5 час перемешивания смесь нейтрализуют до рН 7 добавлением 5 н. NaOH (приблизительно 160 мл) - образуется белый осадок. После этого смесь перемешивают в течение 2 час 30 мин на водяной бане со льдом, отфильтровывают осадок, промывают его холодной водой до бесцветного смыва и сушат в сушильном шкафу при температуре ниже 60°С. Полученное твердое вещество растворяют в 1 н. NaOH (156 мл) и охлаждают раствор на водяной бане со льдом. После этого к раствору добавляют трет-бутилпирокарбонат (7,7 г; 35,2 ммоль) в диоксане (160 мл). Реакционную смесь перемешивают при 0°С в течение 45 мин и затем 16 час при комнатной температуре. Органический растворитель упаривают и остаток растворяют в этилацетате (70 мл). После этого органическую фазу подкисляют, добавляя 2 н. HCl до рН ˜3 и промывают AcOEt (100 мл). Органические фазы объединяют и промывают Н2О (30 мл) и солевым раствором (30 мл), после чего досуха упаривают. Очистка флэш-хроматографией на скликагеле (элюент CH2Cl2/MeOH 20/1) позволяет выделить целевой продукт в виде бесцветного масла (выход 8,42 г в двух стадиях, т.е. 76,7% от теории) (Rf = 0,19, СН2Cl2/МеОН 20/1).
d. (2R)-5-(Бензилоксикарбониламино)-2-(трет-бутилоксикарбониламино) пентан-1-ол
К холодному (-15°С) раствору полученного выше производного диаминопентановой кислоты (5,45 г; 14,8 ммоль) в ТГФ (60 мл) добавляют N-метилморфолин (1,65 мл; 14,8 ммоль) и изобутилхлорформиат (9,6 мл; 14,8 ммоль). Реакционную смесь перемешивают при -15°С в течение 1 мин, после чего добавляют раствор борогидрида натрия (5,1 г; 44,6 ммоль) в 10 мл воды. После перемешивания при -15°С в течение 10 мин к смеси добавляют для остановки реакции Н2O (400 мл). Раствор промывают AcOEt (2×100 мл), органические фазы объединяют и промывают Н2O (50 мл) и солевым раствором (60 мл), после чего высушивают над MgSO4. Растворитель упаривают и остаток кристаллизуют в смеси AcOEt/гексан, получая целевой кристаллический продукт (4,94 г; 94,9%), т.пл. 47,5-48°С.
е. Трифторацетат (2R)-5-(бензилоксикарбониламино)-2-аминопентан-1-ола
Приготовляют раствор полученного выше (2R)-5-(бензилоксикарбониламино)-2-(трет-бутилоксикарбониламино)пентан-1-ола (6,32 г; 18 ммоль) в трифторуксусной кислоте (25 мл) и перемешивают его 2,5 часа при комнатной температуре и упаривают растворитель. Очистка флэш-хроматографией на силикагеле (элюент MeOH/CH2CI2 10/1) позволяет выделить трифторацетатную соль в виде масла (5,45 г; 82,7%). Гидрохлорид плавится при 133,0-134,3°С (перекристаллизация из метанола).
f. (2R)-5-(Бензилоксикарбониламино)-2-[(R)-3-бензилокситетрадеканоиламино)пентан-1-ол
К охлажденному до -15°С раствору (R)-3-бензилокситетрадекановой кислоты (5,27 г; 15,8 ммоль) [Bull. Chem. Soc. Jpn 60 (1987), 2197-2204] в ТГФ (30 мл) добавляют N-метилморфолин (1,89 мл; 15,8 ммоль) и изобутилхлорформиат (2,21 мл; 15,8 ммоль). После перемешивания при -15°С в течение 30 мин добавляют раствор полученной выше трифторацетатной соли (15,25 г; 14,4 ммоль) в смеси ТГФ/Et3N (30 мл/1,44 мл). Перемешивание продолжают при комнатной температуре в течение 16 час, после чего реакционную смесь разбавляют Н2O (30 мл) и AcOEt (60 мл). Органическую фазу отделяют и промывают водную фазу AcCEt (60 мл). Органические фазы объединяют и промывают Н2О (30 мл) и солевым раствором (30 мл), после чего высушивают над MgSO4 и упаривают в вакууме. Остаток перекристаллизовывают из смеси AcOEt/гексан, получая целевой продукт в кристаллической форме (5,8 г; 71,2%). Т.пл. 117,5-118°С. Rf = 0,32, AcOEt/петролейный эфир, 3/1. 1Н-ЯМР (CDCl3, 250 МГц), δ в м.д.: 7,4-7,2 (м, 10Н); 6,5 (д, 1Н, NH); 5,1 (с, 2Н); 4,9 (м, 1Н, NH); 4,5 (2д, АВ, 2Н); 3,8 (м, 2Н); 3,5 (м, 2Н); 3,1 (м, 2Н); 2,4 (м, 2Н); 1,6-1,4 (м, 6Н); 1,4-1,2 (м, 18Н); 0,9 (т, 3Н). 13С-ЯМР (CDCl3, 63 МГц), δ в м.д.: 172,24; 156,49; 138,06; 136,53; 128,46; 128,04; 127,87; 76,76; 71,39; 66,60; 65,44; 51,54; 41,43; 40,65; 33,76; 31,87; 29,61; 29,30; 28,01; 26,47; 25,05; 22,65; 14,09.
g. (2R)-5-Амино-2-[(R)-3-бензилокситетрадеканоиламино]пентан-1-ол
К раствору (2R)-5-(бензилоксикарбониламино)-2-[(R)-3-бензилокситетрадеканоиламино)пентан-1-ола (3,0 г; 5,27 ммоль) в смеси EtOH для ЖХВР/Et3N (300 мл/6 мл) в трехгорлой колбе добавляют катализатор 20% Pd/C (150 мг), удаляют воздух путем вакуумирования и заполняют колбу водородом. Реакционную смесь гидрируют в течение 2 час при комнатной температуре, удаляют катализатор фильтрацией и упаривают фильтрат, получая целевой продукт в виде белого твердого вещества. Rf = 0,2, CH2Cl2/MeOH/Et3N 5/1/0,5. Т.пл 47-48°С. 1Н-ЯМР (CDCl3, 250 МГц), δ в м.д.: 7,4-7,2 (м, 5Н); 6,75 (д, 1Н, NN); 4,5 (2д, АВ, 2Н); 3,9 (м, 2Н); 3,5 (м, 2Н); 2,3-2,6 (м, 7Н); 1,7-1,2 (м, 24H); 0,9 (т, 3Н). 13С-ЯМР (CDCl3, 63 МГц), δ в м.д.: 171,86; 138,13; 128,37; 127,87; 127,75; 76,81; 71,50; 64,57; 51,38; 41,51; 41,17; 33,89; 31,82; 29,26; 28,57; 28,03; 25,07; 22,60; 14,04.
1.1.3. 1-(Дифенилоксифосфорилокси)-3-[(R)-3-додеканоилокситетрадеканоиламино]-4-оксо-5-аза-9-[(R)-3-бензилокситетрадеканоиламино]-декан-10-ол
IIDQ (2-изобутокси-1-изобутоксикарбонил-1,2-дигидрохинолин) (364 мг; 1,2 ммоль; 1 экв) добавляют к раствору (2RS)-4-(дифенилоксифосфорилокси)-2[(R)-3-додеканоилокситетрадеканоиламино]бутановой кислоты (850 мг; 1,0 ммоль; 1 экв) в безводном СН2Cl2 (20 мл) под аргоном при комнатной температуре. Реакционную смесь перемешивают 15 мин и добавляют раствор (2R)-5-амино-2-[(R)-3-бензилокситетрадеканоиламино]пентан-1-ола (757 мг; 1,0 ммоль; 1 экв) в безводном СН2Cl2 (10 мл). После 4 час перемешивания раствор досуха упаривают. Очистка флэш-хроматографией на силикагеле (элюент СН2Cl2/ацетон 5/2) позволяет выделить фосфорилированный псевдодипептид (620 г; 53%) в виде аморфного твердого вещества (Rf = 0,49 в CH2Cl2/MeOH/Et3N 10/1/0,5). 1Н-ЯМР (CDCl3, 250 МГц) δ в м.д.: 7,40-7,15 (м, 15Н), 7,00 (м, 1Н), 6,90 и 6,80 (2д, 2 диаст. 1Н), 6,65 (д, 1Н) (3 × NH), 5,15 (м, 1Н), 4,50 (м, 3Н), 4,30 (м, 2Н), 3,85 (м, 2Н), 3,45 (м, 2H), 3,15 (м, 2Н), 2,41-2,14 (м, 8Н), 1,6-1,4 (м, 8Н), 1,4-1,1 (м, 54Н), 0,9 (т, 9Н, 3СН3). 13С-ЯМР (CDCl3, 63 МГц), δ в м.д.: 173,11, 171,68, 170,52 (2 диаст.), 169,94 (2 диаст.), 150,0 (д, 2Jp,с= 7,2 Гц), 138,20 (2 диаст.), 129,58, 127,99, 127,49, 127,26, 125,24, 119,73 (т, 3Jp,c = 5,0 Гц), 76,48, 71,12, 70,71, 65,86 (ушир.), 64,22, 50,96, 49,71 (ушир.), 41,46, 41,05, 39,07, 34,13, 34,00, 32,70, 31,61, 29,34, 29,06, 28,87, 27,98, 25,25, 24,92, 24,72, 22,38, 13,80.
ПРИМЕР 1.2
α-N-{(4R)-5-гидрокси-4-[(R)-3-гидрокситетрадеканоиламино]пентил}амид N-[(R)-3-додеканоилокситетрадеканоил]-D-аспарагиновой кислоты (= ОМ-197-МС) (фиг.2)
1.2.1. β-Бензиловый эфир N-[(R)-3-додеканоилокситетрадеканоил]-D-аспарагиновой кислоты
К раствору (R)-3-додеканоилокситетрадекановой кислоты (3,35 г; 7,85 ммоль) в безводном ТГФ (25 мл) при -15°С под аргоном последовательно добавляют N-метилморфолин (0,86 мл; 7,85 ммоль; 1 экв) и изобутилхлорформиат (1,02 мл; 7,85 ммоль; 1 экв) - наблюдается быстрое осаждение гидрохлорида N-метилморфолина. После 30 мин перемешивания при -15°С добавляют раствор коммерческого H-D-Asp(OBn)-ОН (Senn Chemicals AG, CH-Dielsdorf) (1/75 г; 7,85 ммоль; 1 экв) в смеси СН3CN/Н2О 3,5/1 (85 мл), содержащий Et3N (3,7 мл). Реакционную смесь перемешивают после этого в течение ночи при комнатной температуре. Упаривают органическую фазу, водную фазу охлаждают до 0°С, подкисляют водным 10%-ным раствором лимонной кислоты до рН 2 и дважды экстрагируют AcOEt. Органическую фазу высушивают над MgSO4, фильтруют и упаривают. Очистка флэш-хроматографией на силикагеле (элюент петролейный эфир/AcOEt 2/1, содержащий 2% уксусной кислоты) с последующим упариванием совместно с толуолом позволяет выделить β-бензиловый эфир N[(R)-3-додеканоилокситетрадеканоил-D-аспарагиновой кислоты (4,00 г; 81%) в виде белого кристаллического вещества (Rf =0,42 в петролейный эфир/AcOSt, 1/1, содержащий 2% уксусной кислоты; УФ и фосфомолибденовый проявители). Т.пл. 67-69°С.
1.2.2. β-Бензиловый эфир α-N{(4R)-5-гидрокси-4[(R)-3-бензилокситетрадеканоиламино]пентил}амида N-[(R)-3- додеканоилокситетрадеканоил]-D-аспарагиновой кислоты (ASM-1)
К раствору полученного выше бензилового эфира (363 мг; 0,57 ммоль) и (R)-5-амино-2[(R)-3-бензилокситетрадеканоиламино]пентан-1-ола (см. 1.1.2) (250 мг; 0,57 ммоль; 1,0 экв) в безводном CH2Cl2 (6 мл) при 0°C (водяная баня - лед) под аргоном последовательно добавляют коммерческий HOAt (1-гидрокси-7-азабензотриазол) (94 мг; 0,69 ммоль; 1,2 экв) и затем коммерческий N,N'-диизопропилкарбодиимид (109 μл; 0,69 ммоль; 1,2 экв). Реакционную смесь перемешивают 1 час при 0°С и затем в течение ночи при комнатной температуре, промывают водой, 1 н. раствором HCl и насыщенным раствором NaHCO3, после чего разделяют фазы. Органическую фазу высушивают над MgSO4, фильтруют и упаривают. Очистка флэш-хроматографией на силикагеле (элюент СН2Cl2/ацетон 3/1) позволяет выделить продукт сочетания (436 мг; 72%) в виде белого кристаллического вещества (Rf = 0,27 в СН2Cl2/ацетон, 5/1; УФ и фосфомолибденовый проявители). Т.пл. 106-106°С. 13С-ЯМR (62,89 МГц, CDCl3,), δ в м.д.: 173,66; 172,09; 171,73; 170,33; 170,12; 138,23; 135,28; 128,53; 128,37; 128,13; 127,81; 127,71; 125,81; 76,71; 71,40; 71,16; 66,77; 65,01; 51,36; 49,39; 41,66; 39,25; 34,40; 33,98; 31,85; 29,58; 29,47; 29,29; 29,11; 28,00; 25,57; 25,17; 25,08; 24,94; 22,62; 14,05.
1.2.3. α-N-{(4R)-5-гидрокси-4[(R)-3-гидрокситетрадеканоиламино]пентил}амид N-[(R)-3-додеканоилокситетрадеканоил]-β-аспарагиновой кислоты (= ОМ-197-МС)
Раствор β-бензилового эфира α-N{(4R)-5-гидрокси-4-(R)-3-бензилокситетрадеканоиламино]пентил}амида N-[(R)-3-додеканоилокситетрадеканоил]-D-аспарагиновой кислоты (417 мг; 0,40 ммоль) в смеси MeOH/AcOEt 1/1 (36 мл) гидрируют в течение 3 час в присутствии 10% Pd на угле (20 мг) при комнатной температуре и атмосферном давлении водорода. Катализатор удаляют фильтрацией, промывают смесью СН2Cl2/МеОН 4/1 (50 мл), упаривают досуха фильтрат и сушат остаток с помощью крыльчатого насоса, получая свободную кислоту в виде белого кристаллического вещества (Rf = 0,30 в СН2Cl2/МеОН 9/1, содержащий 0,5% уксусной кислоты; фосфомолибденовый проявитель). Т.пл. 135-137°C. ES/MS: m/z 868.7 [M+H]+, 890.7 [M+Na]+ 868.7 [М+K]+, 912.7 [M-H+2Na]+ (фиг.3).
1.2.4. α-N-{(4R)-5-гидрокси-4-[(R)-3-гидрокситетрадеканоиламино]пентил}амид N-[(R)-3-додеканоилокситетрадеканоил]-L-аспарагиновой кислоты
Та же последовательность реакций, выполненная с коммерческим H-L-Asp(Obn)-ОН (Fluka, Buchs, Швейцария), приводит к эпимерному продукту ряда L-аспарагиновой кислоты.
ПРИМЕР 1.3
α-N-{(4R)-5-дигидроксифосфорилокси-4-[(R)-3-гидрокситетрадеканоиламино]пентил}амид N-[(R)-3-додеканоилокситетрадеканоил]-D-аспарагиновой кислоты (= ОМ-197-МС-МР) (фиг.2)
1.3.1. β-Бензиловый эфир α-N-{(4R)-5- дибензилоксифосфорилокси-4-[(R)-3-бензилокситетрадеканоиламино]пентил}амида N-[(R)-3-додеканоилокситетрадеканоил]-D-аспарагиновой кислоты
К раствору полученного выше β-бензилового эфира α-N-{(4R)-5-гидрокси-4-[(R)-3-бензилокситетрадеканоиламино]пентил}амида N-[(R)-3-додеканоилокситетрадеканоил]-D-аспарагиновой кислоты (300 мг; 0,29 ммоль) и 1H-тетразола (60 мг; 0,86 ммоль) в безводном ТГФ (12 мл) под аргоном и при комнатной температуре добавляют 85%-ный дибензилдиэтилфосфорамидит (231 μл; 0,66 ммоль) - наблюдают быстрое образование в реакционной среде белых кристаллов. После 30 мин перемешивания реакционную смесь охлаждают до -20°С и добавляют раствор mCPBA (57-86%; 183 мг; 1,06 ммоль) в СН2Cl2 (8 мл) - наблюдают исчезновение кристаллов. После 45 мин перемешивания при комнатной температуре добавляют насыщенный раствор Na2S2O3, перемешивают смесь 10 мин и разбавляют эфиром. Органическую фазу отделяют и промывают насыщенным раствором Na2S2O3 (5 раз), дважды насыщенным раствором NaHCO3 и 1 М HCl. Органическую фазу высушивают над MgSO4, фильтруют и упаривают. Очистка флэш-хроматографией на силикагеле (элюент CH2Cl2/ацетон 5/1) позволяет выделить фссфотриэфир (294 мн; 79%) в виде белого твердого вещества (Rf =0,27 в СН2Cl2/ацетон 5/1; УФ и фосфомолибденовый проявители). 13С-ЯМР (62,89 МГц, CDCl3), δ в м.д.: 173,26; 171,37; 171,01; 170,60; 170,03; 138,22; 135,50; 135,40; 135,28; 128,40; 128,33; 128,16; 128,08; 127,94; 127,82; 127,76; 127,53; 127,41; 76,43; 71,03; 70,90; 69,28; 66,47; 49,09; 48,37; 41,62; 41,35; 41,24; 39,02; 38,88; 25,05; 24,94; 24,82; 22,48; 13,90.
1.3.2 α-N-{(4R)-5-дигидроксифосфорилокси-4-[(R)-3-гидрокситетрадеканоиламино] пентил}амид N-[(R)-3-додеканоилокситетрадеканоил]-D-аспарагиновой кислоты (= ОМ-197-МС-МР)
Раствор полученного выше дибензилфосфата (249 мг; 190 μмоль) в EtOH для ЖХВР (12 мл) гидрируют в течение 4 час в присутствии 10% Pd на угле (30 мг) при комнатной температуре и атмосферном давлении водорода. Катализатор удаляют фильтрацией на миллипоре, фильтрат досуха упаривают и остаток сушат с помощью крыльчатого насоса, получая сырой свободный фосфат (180 мг; 100%). ES/MS: m/z 850.7 [М+Н-O(О)(ОН)3]+, 948.5 [М+Н]+, 970,5 [M+Na]+ (фиг.4).
ПРИМЕР 1.4
Определение степени эпимеризации полупродуктов синтеза
Стереохимию центров, содержащих ациламиногруппы, определяют по конфигурации исходных аминокислот. Однако пептидное сочетание, осуществленное между производным аспарагиновой или глутаминовой кислоты и аминоспирта, полученного из орнитина или лизина, может при определенных условиях привести к явлению эпимеризации у α-углерода кислотной компоненты реакции сочетания. Чтобы показать степень эпимеризации этой реакции, для соединений, являющихся производными от аспарагиновой кислоты, был применен следующий способ.
Образец (30 μг) упаривают в микропузырьке и вновь растворяют в 40 μл 6 М HCl. В течение 24 час осуществляют гидролиз при 110°С под аргоном. После этого образец досуха упаривают и вновь растворяют в 0,1 М тетраборатном буфере (100 мл), рН 9,2. Перед хроматографированием получают производное с помощью реагента OPA-IBLC (о-фталевый диальдегид-N-изобутирил-L-цистеин) в следующих пропорциях:
5 μл 0,1 М натрий-тетраборатного буферного раствора, рН 9,2
2 μл метанольного раствора 170 мМ ОРА, 260 мМ IBLC
2 μл анализируемого раствора
Разделение с помощью ЖХВР на колонке Hypersil ODS (250 х 4,6 мм, 5 μм, Supelco) позволяет количественно определить оба производных, образованных от присутствующих в исходном образце L- и D-форм аспарагиновой кислоты (Brückner et al. 1995, J. Chromatogr. A 711, 201-215). При проведении ЖХБР использованы следующие условия:
В этих условиях хроматографии для производных L- и D-аспарагиновых кислот время удерживания составляло, соответственно, 16,1 и 17,2 мин.
2-ая серия примеров: Получение псевдодипептидов, имеющих вспомогательное функционализированное ответвление
ПРИМЕР 2.1
3-[(R)-3-Додеканоилокситетрадеканоиламино]-4-оксо-5-аза-9[(R)-3-гидрокситетрадеканоиламино]декан-1,10-диол-1-дигидрофосфат 10-(6,7-дигидроксигептаноат) (=OM-197-FV6) (фиг.5)
2.1.1. 1-(Дифенилоксифосфорилокси)-3-[(R)-3-додеканоил-окситетрадеканоиламино]-4-оксо-5-аза-9-[(R)-3-бензилокситетрадеканоиламино]декан-10-ол (6-гептеноат)
К раствору 1-(дифенилоксифосфорилокси)-3-[(R)-3-додекансилокситетрадеканоиламино]-4-оксо-5-аза-9-[(R)-3-бензилокситетрадеканоиламино]-10-ола (пример 1.1) (875 мл; 0,74 ммоль) в безводном СН2Cl2 (25 мл) добавляют 6-гептеновую кислоту (141 μл; 1,04 ммоль; 1,4 экв). Раствор охлаждают до 0°С и добавляют EDCI (64 мг; 0,33 ммоль; 1,4 экв) и DMAP (41 мг; 0,33 ммоль; 0,14 экв). Реакционную смесь перемешивают 30 мин при 0°C и затем 3 часа при комнатной температуре. После разбавления СН2Cl2 органическую фазу последовательно промывают Н2О, 1 н. раствором HCl и Н2О. После этого органическую фазу высушивают над MgSO4 и упаривают при 40°С в вакууме. Очистка флэш-хроматографией на силикагеле (элюент петролейный эфир/AcOEt 1/1) позволяет выделить 1-(дифенилоксифосфорилокси)-3[(R)-3-додеканоилокситетрадеканоиламино]-4-оксо-5-аза-9-[(R)-3-бензилокситетрадеканоиламино]-10-ол (6-гептеноат) (820 мг; 85%) в виде беловатого пенообразного вещества (Rf=0,18 в смеси петролейный эфир/AcOEt 1/1; проявитель фосфомолибденовая кислота). 13С-ЯМР (62,89 МГц, CDCl3), δ в м.д.: 173,30; 171,18; 170,42; 169,91; 169,73; 150,10; 138,21; 138,14; 129,77; 128,25; 127,49; 125,44; 119,88; 114,56; 76,48; 71,11; 70,90; 66,01; 65,52; 49,90; 47,80; 41,34; 39,07; 33,92; 33,76; 33,69; 33,61; 33,17; 31,75; 29,47; 29,19; 29,00; 28,46; 28,11; 25,34; 25,05; 24,85; 22,52; 13,96.
2.1.2. 1-(Дифенилоксифосфорилокси)-3-[(R)-3-додеканоил-окситетрадеканоиламино]-4-оксо-5-аза-9-[(R)-3-бензилокситетрадеканоиламино]-10-ол (6,7-дигидроксигептаноат)
К раствору, содержащему К3Fe(CN)6 (373 мг; 1,13 ммоль; 3 экв), К2СО3 (157 мг; 1,13 ммоль; 3 экв) и 1,4-диазабицикло[2.2.2]октан (DABCO) (10,7 мг; 0,095 ммоль; 0,25 экв) в смеси трет-бутанол/вода (5 мл/5 мл) добавляют полученное выше соединение (486 мг; 0,38 ммоль) и затем 2,5%-ный раствор тетроксида осмия в трет-бутаноле (48 рл; 4,75 μмоль; 0,0125 экв). Реакционную среду интенсивно перемешивают в течение 16 час при комнатной температуре (27°С), добавляют Na2S2O5 (60 мг) и продолжают перемешивание в течение приблизительно 1 часа до тех пор, пока реакционная среда не станет бурой, а затем зеленой или голубоватой. Реакционную смесь разбавляют затем эфиром и отделяют органическую фазу. Водную фазу обильно промывают эфиром, органические фазы объединяют, высушивают над MgSO4 и упаривают при 40°С в вакууме. Получают таким образом сырой ожидаемый диод в виде зеленого масла. Очистка флэш-хроматографией на силикагеле (элюент СН2Cl2/ацетон 5/2) позволяет выделить чистый 1-(дифенилоксифосфорилокси)-3-[(R)-3-додеканоилокситетрадеканоиламино]-4-оксо-5-аза-9-[(R)-3-бензилокситетрадеканоиламино]-10-ол 6,7-дигидроксигептаноат (198 мг; 40%) в виде аморфного твердого вещества (Rf =0,24 в СН2Cl2/ацетон 5/2; проявитель фосфомолибденовая кислота). 13С-ЯМР (62,89 МГц, CDCl3), δ в м.д.: 173,46; 173,33; 171,34; 170,58; 170,02; 150,13; 138,23; 129,80; 128,26; 127,87; 127,54; 125,50; 120,01; 119,80; 71,79; 71,23; 70,97; 66,63; 66,03; 65,54; 49,93; 47,90; 41,46; 39,17; 33,98; 33,79; 32,86; 32,45; 31,77; 29,49; 29,21; 29,03; 28,51; 25,50; 25,07; 24,87; 24,77; 24,66; 22,54; 13,99. ЖХВР (210 нм): Время удерж.= 32,535 мин. ES/MS: m/z 1343.0 (M+Na+), 1321.0 (М+Н+), 1071.0 (М+Н+ монофенилфосфат).
2.1.3. 1-(Дифенилоксифосфорилокси)-3[(R)-3-додеканоилокситетрадеканоиламино]-4-оксо-5-аза-9-[(R)-3-гидрокситетрадеканоиламино]-10-ол (6,7-дигидроксигептаноат)
Раствор полученного выше диола (198 мг; 0,15 ммоль) в смеси EtOH для ЖХВР/AcOEt (1,4 мл) гидрируют в течение 2,5 час в присутствии 10% Pd на угле (70 мг) при комнатной температуре и атмосферном давлении водорода. Катализатор удаляют фильтрацией, досуха упаривают фильтрат, остаток сушат с помощью крыльчатого насоса и получают сырой дебензилированный продукт (168 мг; 91%) в виде аморфного вещества. 13С-ЯМР (62,89 МГц, CDCl3), δ в м.д.: 173,15; 173,51; 172,82; 171,04; 170,49; 170,35; 150,05; 129,79; 129,42; 125,53; 119,91; 119,83; 119,72; 71,80; 70,93; 68,58; 66,47; 65,94; 65,52; 50,02; 48,12; 42,59; 41,26; 39,13; 36,92; 34,29; 33,85; 32,32; 31,74; 29,50; 29,18; 29,00; 28,15; 25,47; 25,07; 24,85; 24,73; 24,56; 22,51; 13/99. ЖХВР (210 нм): Время удерж.= 30,7 мин. ES/MS: m/z 1253.0 [M+Na]+, 1231.0 [M+H]+.
2.1.4. 3-[(R)-3-Додеканоилокситетрадеканоиламино]-4-оксо-5-аза-9-[(R)-3-гидрокситетрадеканоиламино]декан-1,10-диол-1-дигидрофосфат 10-(6, 7-дигидроксигентаноат) (=ОМ-197-FV6)
К суспензии оксида платины PtO2 (91 мг) в этаноле (для ЖХВР) (1 мл) (предварительно активированной до платиновой черни в атмосфере водорода в течение 10 мин) добавляют раствор полученного выше триола (168 мг; 0,14 ммоль) в смеси EtOH для ЖХВР (8 мл)/ AcOSt (8 мл). Раствор гидрируют в течение 24 час в при комнатной температуре (27°С) и атмосферном давлении водорода. Катализатор удаляют фильтрацией, досуха упаривают фильтрат, остаток сушат с помощью крыльчатого насоса и получают сырой фосфат (130 мг; 88%). ЖХВР (210 нм): Время удерж.= 23,6 мин. ES/MS: m/z 1078.9 [М+Н]+, 1100.8 [M+Na]+, 1116.8 [М+К]+ (фиг.6).
ПРИМЕР 2.2
3-[(R)-3-Додеканоилокситетрадеканоиламино]-4-оксо-5-аза-9-[(R)-3-гидрокситетрадеканоиламино]декан-1,10-диол-1-дигидрофосфат 10-(6-оксогексаноат) (=OM-197-FV7) (фиг.5)
2.2.1. 3-[(R)-3-Додеканоилокситетрадеканоиламино]-4-оксо-5-аза-9-[(R)-3-гидрокситетрадеканоиламино]декан-1,10-диол-1-дигидрофосфат 10-(6-оксогексаноат) (=OM-197-FV7)
Реакцию перйодатного окисления осуществляют добавлением 1,86 мл 0,1 М NaIO4 (39,62 мг, 20 экв) к 10 мг (9,28 μмоль, 1 экв) полученного выше деблокированного триода в смеси изопропиловый спирт/вода (1:1). За реакцией следят с помощью жидкостной хроматографии с УФ-детектором (LC/UV), обнаруживая через 2 часа количественное превращение диоловой функции в альдегидную. Молекулярный ион m/z 1046.8, наблюдаемый в спектре ES/MS после стадии перйодатного окисления (фиг.7), свидетельствует о протекании ожидаемой реакции. В спектре видны натриевые аддукты с m/z 1068.8 [M+Na]+ и калиевые с m/z 1084.8 [М+К]+, а также фрагмент, соответствующий потере фосфорильной группы с m/z 948.8. Реакцию останавливают добавлением 1-2 капель этиленгликоля. Продукт может быть очищен с помощью SPE на носителе С18 с выходом 90% (порядка 5 мг диола).
ПРИМЕР 2.3
α-N-{(4R)-5-(6-гидрокси)-4-[(R)-3-гидрокситетрадеканоиламино]пентил}амид N-[(R)-3-додеканоилокситетрадеканоиламино]-D-аспарагиновой кислоты 5-О-(6,7-дигидроксигептаноат) (=OM-197-MC-FV5) (фиг.8)
2.3.1. β-Бензиловый эфир α-N{(4R)-5-гидрокси-4[(R)-3-бензилокситетрадеканоиламино]пентил}амида N-[(R)-3-додеканоилокситетрадеканоиламино]-D-аспарагиновой кислоты 5-O-(6-гептеноат)
К раствору β-бензилового эфира α-N-{(4R)-5-гидрокси-4-[(R)-3-бензилокситетрадеканоиламино]пентил}амида N-[(R)-3-додеканоилокситетрадеканоиламино]-D-аспарагиновой кислоты (пример 1.2.2) (122 мг; 0,116 ммоль) в безводном дихлорметане (5 мл) добавляют 6-гептеновую кислоту (22 μл; 0,169 ммоль; 1,4 экв). Раствор охлаждают до 0°С и добавляют EDCI (49,3 мг; 0,25 ммоль; 2,1 экв) и DMAP (5,6 мг; 0,046 ммоль; 0,4 экв). Реакционную смесь перемешивают 30 мин при 0°С и затем 6 час при комнатной температуре. После разбавления CH2Cl2 органическую фазу последовательно промывают Н2O, дважды 1 н. HCl, дважды NaHCO3 и дважды Н2О, получая чистый ожидаемый гептеновый эфир (119 мг; 89%) в виде белого твердого вещества (Rf =0,62 в CH2Cl2/ацетон 5/1; проявитель фосфомолибденовая кислота). 13С-ЯМР (62/89 МГц, CDCl3), δ в м.д.: 173,63; 173,38; 171,72; 171,11; 170,08; 138,14; 135,28; 128,46; 128,34; 128,24; 128,05; 127,64; 127,53; 114,62; 76,49; 71,14; 71,02; 66,66; 65,49; 49,17; 47,79; 41,69; 41,35; 39,09; 35,46; 34,33; 33,80; 33,65; 33,21; 31,79; 29,52; 29,23; 29,06; 28,46; 28,16; 22,56; 14,00. ЖХВР (210 нм): время удерж.= 36,9 мин. ES/MS: m/z 1159.0 (M-H+), 1176.0 (M-NH4 +). Т.пл. 81,5-84°С. [α]D 20 +7,3 (CHCl3).
2.3.2. (β-Бензиловый эфир α-N-{(4R)-5-гидрокси-4[(R)-3-бензилокситетрадеканоиламино]пентил}амида N-[(R)-3-додеканоилокситетрадеканоиламино]-D-аспарагиновой кислоты 5-О-(6,7-дигидроксигептаноат)
К раствору полученного выше гептенового эфира (60 мг; 0,052 ммоль) в смеси Н2О/ацетон (5 мл/3 мл) добавляют оксид N-метилморфолина (9 мг; 0,076 ммоль; 1,5 экв) и затем прибавляют по каплям 2,5%-ный раствор OsC4 в трет-бутаноле (123 μл; 0,012 ммоль; 0,23 экв). Реакционную смесь перемешивают 24 часа при комнатной температуре, добавляют Na2S2O5 и перемешивают при комнатной температуре в течение от 1 до 2 час. После этого раствор несколько раз экстрагируют эфиром, органические фазы объединяют, высушивают над MgSO4 и упаривают. Очистка флэш-хроматографией на силикагеле (элюент CH2Cl2/ацетон 5/2) позволяет выделить чистый ожидаемый диол (35 мг; 57%) в виде белого твердого вещества (Rf =0,15 в СН2Cl2/ацетон 5/1; проявитель фосфомолибденовая кислота). 13С-ЯМР (62,89 МГц, CDCl3), δ в м.д.: 173,67; 173,39; 171,81; 171,34; 170,27; 170,01; 138,18; 135,27; 128,56; 128,42; 128,18; 127,50; 127,68; 76,68; 71,82; 71,37; 71,17; 66,85; 66,74; 65,66; 49,27; 47,99; 41,88; 41,51; 39,31; 35,60; 34,42; 33,95; 33,51; 31,80; 29,60; 29,49; 29,30; 28,55; 25,65; 25,60; 25,19; 25,12; 24,97; 24,77; 24,14; 22,65; 14,08. ЖХВР (210 нм): Время удерж.= 34,16 мин. ES/MS: m/z 1193.0 (М+Н+), 1212.0 (M+NH4)+.
2.3.3. α-N-{(4R)-5-гидрокси-4[(R)-3-гидрокситетрадеканоиламино]пентил}амид N-[(R)-3-додеканоилокситетрадеканоиламино]-D-аспарагиновой кислоты 5-O-(6,7-дигидроксигептаноат) (=OM-197-MC-FV5)
Раствор полученного выше диола (35 мг; 0,029 ммоль) в смеси МеОН для ЖХВР (2 мл)/AcOEt (3 мл) гидрируют в течение 2,5 час в присутствии 10% Pd на угле (10 мг) при комнатной температуре и атмосферном давлении водорода. Катализатор удаляют фильтрацией, фильтрат досуха упаривают и остаток сушат с помощью крыльчатого насоса, получая сырой α-N-{(4R)-5-гидрокси-4-[(R)-3-гидрокситетрадеканоиламино]пентил}амид N-[(R)-3-додеканоилокситетрадеканоиламино]-D-аспарагиновсй кислоты 5-О-(6,7-дигидроксигептаноат) (26 мг; 88%) в виде белого твердого вещества. ЖХВР (210 нм): Время удерж.= 26,90 мин. Т.пл 94-97°С. [α]D 20 +11,1 (CHCl3/MeOH, 1/0,1). ES/MS: m/z 1012.7 (M+H)+, 1034.7 (M+Na)+, 1050.7 [М+К]+ (фиг.9).
ПРИМЕР 2.4
α-N-{(4R)-5-Гидрокси-4-[(R)-3-гидрокситетрадеканоиламино]пентил}амид N-[(R)-3-додеканоилокситетрадеканоиламино]-D-аспарагиновой кислоты 5-О-(6-оксогексаноат) (=OM-197-MC-FV6) (фиг.8)
2.4.1. α-N-{(4R)-5-Гидрокси-4[(R)-3-гидрокситетрадеканоиламино]пентил}амид N-[(R)-3-додеканоилокситетрадеканоиламино]-D-аспарагиновой кислоты 5-О-(6-оксогексаноат) (=OM-197-MC-FV6)
1,98 мл 0,1 М NaIO4 (42,25 мг, 20 экв) добавляют к 10 мг (9,88 μмоль, 1 экв) полученного выше деблокированного триола (пример 2.3.3) в смеси изопропиловый спирт/вода (1:1). Раствор перемешивают два часа при комнатной температуре и останавливают реакцию добавлением 1-2 капель этиленгликоля. После этой стадии перйодатного окисления в спектре ES/MS (фиг.10) наблюдают молекулярный ион [М+Н]+ при m/z 980,6, свидетельствующий о присутствии альдегидной функции. Видны также соответствующие пики натриевых аддуктов при m/z 1002.8 ([M-Na]+) и калиевых при m/z 1018.6 ([М-К]+). Очистка с помощью SPE на носителе С18 позволяет выделить продукт OM-197-MC-FV6 с выходом 90%.
ПРИМЕР 2.5
α-N-{(4R)-5-Гидрокси-4-[(R)-3-гидрокситетрадеканоиламино]пентил}амид N-[(R)-3-додеканоилокситетрадеканоиламино]-D-аспарагиновой кислоты 5-О-(6-гидроксигексаноат) (=OM-197-MC-FV7) (фиг.8)
2.5.1. α-N-{(4R)-5-Гидрокси-4-[(R)-3-гидрокситетрадеканоиламино]пентил}амид N-[(R)-3-додеканоилокситетрадеканоиламино]-C-аспарагиновой кислоты 5-О-(6-гидроксигексаноат) (=OM-197-MC-FV7)
К раствору полученного выше 6-оксогексаноильного производного (4,5 мг, 0,0043 ммоль) в 90%-ном изопропиловом спирте (20 мл) добавляют раствор (0,2 мл) NaBH4 в метаноле (1 мг/мл). Смесь перемешивают 3 мин при 25°С и добавляют избыток уксусной кислоты (0,2 мл). Очистка с помощью препаративной ЖХВР на колонке с С18 позволяет выделить продукт OM-197-MC-FV7 (2,3 мг, 51%) в растворе 90%-ного изопропилового спирта.
ПРИМЕР 2.6.
(3RS,9R)-3-[(R)-3-Додеканоилокситетрадеканоиламино]-4-оксо-5-аза-9-[(R)-3-гидрокситетрадеканоиламино]декан-1,10-диол 1-дигидрофосфат 10-(6-аминогексаноат) (=ОМ-197-МР-АС-RS,R) (фиг.11)
2.6.1. 1-(Дифенилоксифосфорилокси)-3-[(R)-3-додеканоилокситетрадеканоиламино]-4-оксо-5-аза-9-[(R)-3-бензилокситетрадеканоиламино]декан-10-ол (6-бензилоксикарбониламиногексаноат)
К раствору 1-(дифенилоксифосфорилокси)-3-[(R)-3-додеканоилокситетрадеканоиламино]-4-оксо-5-аза-9-[(R)-3-бензилокситетрадеканоиламино]-10-декан-10-ола (фиг.1) (230 мг; 0,20 ммоль) в безводном СН2Cl2 (8 мл) добавляют 6-бензилоксикарбониламиногексановую кислоту (88 мг; 0,33 ммоль; 1,65 экв). Раствор охлаждают до 0°С и добавляют ECCI (200 мг; 1,04 ммоль; 5,2 экв) и DMAP (13 мг; 0,10 ммоль; 0,5 экв). Реакционную смесь перемешивают 30 мин при 0°С и затем 4 часа при комнатной температуре. После разбавления CH2Cl2 органическую фазу последовательно промывают Н2О, 1 н. HCl и Н2О. Органическую фазу высушивают над MgSO4 и упаривают при 40°С в вакууме. Очистка флэш-хроматографией на силикагеле (элюент СН2Cl2/ацетон 7/1,2) позволяет выделить чистый сложный эфир (115 мг; 41%) в виде аморфного вещества (Rf =0,77 в СН2Cl2/ацетон 5/2; проявитель фосфомолибденовая кислота). 13С-ЯМР (62,89 МГц, CDCl3), δ в м.д.: 173,27; 171,20; 170,34; 169,88; 156,36; 150,16; 150,13; 138,28; 136,56; 129,80; 128,35; 128,26; 127,90; 127,51; 125,45; 119,97; 119,83; 71,05; 66,37; 65,97; 65,61; 49,94; 47,81; 41,39; 40,66; 39,09; 34,32; 34,25; 33,95; 33,65; 31,78; 29,50; 29,38; 29,22; 29,0; 28,55; 28,44; 25,95; 25,06; 24,87; 24,26; 22,55; 14,00. ЖХВР (210 нм): Время удерж.= 33,497 мин. ES/MS: m/z 1424.0 [M+H]+, 1174.0 [M+H-(PhO)2OPOH]+.
2.6.2. 1-(Дифенилоксифосфорилокси)-3-[(R)-3-додеканоилокситетрадеканоиламино] -4-оксо-5-аза-9[(R)-3-гидрокситетрадеканоиламино]декан-10-ол 10-(6-аминогексаноат)
Раствор полученного выше продукта (115 мг; 81 μмоль) в смеси EtOH для ЖХВР (15 мл)/ледяная уксусная кислота (0,4 мл) гидрируют в течение 4 час в присутствии палладия (10% на угле) (80 мг) при комнатной температуре и атмосферном давлении водорода. Катализатор удаляют фильтрацией, упаривают досуха фильтрат и сушат остаток с помощью крыльчатого насоса, получая дебензилированный продукт (90 мг; 97%) в виде аморфного вещества. ЖХВР (210 нм): Время удерж.= 29,185 мин. ES/MS: m/z 1200.0 [М+Н]+, 952.0 [M+Н-(PhO)2ОРОН]+.
2.6.3. 3-[(R)-3-Додеканоилокситетрадеканоиламино]-4-оксо-5-аза-9-[(R)-3-гидрокситетрадеканоиламино]-1,10-диол-1-дигидрофосфат 10-(6-аминогексаноат) (=ОМ-197-МР-АС)
К раствору оксида платины PtO2 (30 мг) в этаноле (для ЖХВР) (1 мл) (предварительно активированной до платиновой черни в атмосфере водорода в течение 10 мин) добавляют раствор полученного выше аминоспирта (90 мг, 75 μмоль) в смеси EtOH для ЖХВР (5 мл)/1 н. HCl (0,1 мл). Раствор гидрируют в течение 24 час при комнатной температуре и атмосферном давлении водорода. Катализатор удаляют фильтрацией, досуха упаривают фильтрат, остаток сушат с помощью крыльчатого насоса и получают ожидаемый аминофосфат (60 мг; 79%). ЖХВР (210 нм): Время удерж.= 28,51 мин. ES/MS: m/z 1047.5 [М+Н]+, 1069.6 [M+Na]+, 949.6 [М+Н-(НО)2OPOH]+ (фиг.12).
ПРИМЕР 2.7
(3R,9R)-3-[(R)-3-Додеканоилокситетрадеканоиламино]-4-оксо-5-аза-9-[(R)-3-гидрокситетрадеканоиламино]декан-1,10-диол 1-дигидрофосфат 10-(6-аминогексаноат) (=OM-197-MP-AC-R,R) (фиг.13)
2.7.1 β-Бензилозый эфир α-N{(4R)-5-(2-тетрагидропиранил)окси-4[(R)-3-бензилокситетрадеканоиламино]пентил}амида N[(R)-3-додеканоилокситетрадеканоил]-D-аспарагиновой кислоты (ASM-1-O-ТНР)
К раствору β-бензилового эфира α-N{[(4R)-5-гидрокси-4-[(R)-3-бензилокситетрадеканоиламино]пентил}амида N-[(R)-3-додеканоилокситетрадеканоил]-D-аспарагиновой кислоты (1,5 г; 1,43 ммоль) в безводном CH2Cl2 (30 мл) при комнатной температуре под аргоном последовательно добавляют 3,4-дигидро-2Н-пиран (DHP) (327 μл, 3,58 μмоль) и затем п-толуолсульфонат пиридиния (PPTS) (108 μл, 429 μмоль). После 18 час перемешивания снова добавляют 3,4-дигидро-2Н-пиран (DHP) (130 μл, 1,43 μмоль). Раствор после этого перемешивают еще 9 час при комнатной температуре, разбавляют CH2Cl2, промывают 5%-ным раствором NaHCO3 и затем Н2O. Органическую фазу высушивают над MgSO4, фильтруют и упаривают. Очистка флэш-хроматографией на силикагеле (элюент СН2Cl2/ацетон 7/1, затем 6/1) позволяет выделить β-бензиловый эфир α-N{[(4R)-5-(2-тетрагидропиранил)окси-4-[(R)-3-бензилокситетрадеканоиламино]-пентил}амида N-[(R)-3-додеканоилокситетрадеканоил]-D-аспарагиновой кислоты (1,45 г; 90%) в виде белого кристаллического вещества (Rf =0,57 в СН2Cl2/ацетон 5/1; проявители УФ и фосфомолибденовая кислота).
2.7.2. α-N{(4R)-5(2-Тетрагидропиранил)окси-4[(R)-3-бензилокситетрадеканоиламино]пентил}амид N-[(R)-3-додеканоилокситетрадеканоил]-D-аспарагиновой кислоты
Раствор полученного выше соединения (250 мг; 0,22 ммоль) в смеси EtOH/AcOEt 1/1 (16 мл), содержащей Et3N (0,4 мл), гидрируют в течение 2 час в присутствии 10% Pd на угле (10 мг) при комнатной температуре и атмосферном давлении. Катализатор удаляют фильтрацией, фильтрат досуха упаривают и сушат с помощью крыльчатого насоса, получая кислоту в виде триэтиламмониевой соли (250 мг).
2.7.3. 3-[(R)-3-Додеканоилокситетрадеканоиламино]-4-оксо-5-аза-9-[(R)-3-бензилокситетрадеканоиламино]-10-(2-тетрагидропиранил)окси-декан-1-ол
К раствору полученной выше кислоты (˜250 мг) в смеси СН2Cl2/безводный ТГФ 2/1 (3 мл) при 0°С под аргоном добавляют N-метилморфолин (72 μл; 0,66 ммоль; 3 экв) и затем изобутилхлорформиат (86 μл; 0,66 ммоль; 3 экв) - наблюдают быстрое выпадение в осадок гидрохлорида N-метилморфолина. Осуществляется автоматическое слежение за реакцией (Rf =0,90 в СН2Cl2/ацетон 2/1). После 30 мин перемешивания при комнатной температуре реакционную смесь охлаждают до 0°С и быстро добавляют раствор NaBH4 (33 мг; 0,88 ммоль; 4 экв) в Н2О (1 мл) и ТГФ (1 мл) и перемешивают после этого 5 мин при комнатной температуре. Раствор частично упаривают и разделяют фазы. Органическую фазу высушивают над MgSO4, фильтруют и упаривают. Очистка флэш-хроматографией на силикагеле (элюент CH2Cl2/ацетон 2/1) позволяет выделить спирт (138 мг; 61% по двум стадиям) в виде белого кристаллического вещества (Rf =0,33 в СН2Cl2/ацетон 3/1; проявители УФ и фосфомолибденовая кислота).
2.7.4. Дибензилфосфат 3-[(R)-3-додеканоилокситетрадеканоиламино]-4-оксо-5-аза-9-[(R)-3-бензилокситетрадеканоиламино]-10-(2-тетрагидропиранил)окси-декан-1-ола
К раствору полученного выше спирта (120 мг; 0,12 ммоль) и 1H-тетразола (25 мг; 0,35 ммоль; 3 экв) в безводном ТГФ (5 мл) при комнатной температуре под аргоном добавляют 85%-ный дибензилдиэтилфосфорамидит (95 μл; 0,27 ммоль; 2,3 экв) - наблюдают быстрое образование в реакционной среде белых кристаллов. После 30 мин перемешивания реакционную смесь охлаждают до -20°С и добавляют раствор mCPBA (57-86%; 75 мг; 0,43 ммоль; 3,7 экв) в CH2Cl2 (3 мл) - наблюдают исчезновение кристаллов. После 45 мин перемешивания при комнатной температуре добавляют насыщенней раствор Na2S2O3 (3 мл) и перемешивают смесь 10 мин. Раствор разбавляют после этого эфиром, отделяют органическую фазу, промывают ее 5 раз насыщенным раствором Na2S2O3 и 2 раза насыщенным раствором NaHCO3, фильтруют и упаривают. Очистка флэш-хроматографией на силикагеле (элюент СН2Cl2/ацетон 4/1, затем 2/1) позволяет выделить дибензилфосфат (126 мг; 84%) в виде аморфного вещества (Rf =0,53 в CH2Cl2/ацетон 3/1; проявители УФ и фосфомолибденовая кислота).
2.7.5. 1-(Дибензилфосфат) 3-[(R)-3-додеканоилокситетрадеканоиламино]-4-оксо-5-аза-9-[(R)-3-бензилокситетрадеканоиламино]декан-1,10-диола
К 1%-ному раствору HCl в метаноле (25 мл) при 0°С добавляют раствор полученного выше соединения (700 мг, 0,54 ммоль) в CH2CI2 (2,5 мл). После 45 мин перемешивания при 0°С, реакционную смесь нейтрализуют 5%-ным раствором МаНСО3, разбавляют СН2Cl2 и отделяют органическую фазу. Оставшуюся водную фазу трижды экстрагируют СН2Cl2. Органические фазы объединяют, сушат над MgSO4, фильтруют и упаривают, получая сырой спирт (640 мг; 98%) (Rf =0,50 в СН2Cl2/ацетон 3/1; проявители УФ и фосфомолибденовая кислота).
2.7.6. 1-(Дибензилфосфат) 3-[(R)-3-додеканоилокситетрадеканоиламино]-4-оксо-5-аза-9-(R)-3-бензилокситетрадеканоиламино]декан-1,10-диола 10-(6-бензилоксикарбониламиногексаноат)
К раствору полученного выше соединения (640 мг, 0,53 ммоль) и 6-(бензилоксикарбониламино)гексановой кислоты (423 мг, 1,60 ммоль) в сухом СН2Cl2 (25 мл) при 0°С под аргоном последовательно добавляют коммерческий гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида (306 мг, 1,60 ммоль) и 4-диметиламинопиридин (20 мг, 160 ммоль). После этого реакционную смесь перемешивают 30 мин при 0°С и затем в течение ночи при комнаткой температуре, вслед за чем промывают водой, затем 1 н. раствором HCl и разделяют фазы. Органическую фазу высушивают над MgSO4, фильтруют и упаривают. Очистка флэш-хроматографией на силикагеле (элюент СН2Cl2/ацетон 4/1 и затем 2/1) позволяет выделить продукт сочетания (537 мг; 71%). 13С-ЯМР (62,89 МГц, CDCl3), δ в м.д.: 173,18; 171,16; 170,38; 169,60; 156,30; 138,23; 136,50; 135,38; 135,28; 128,42; 128,26; 128,17; 127,79; 127,74; 127,44; 76,48; 71,15; 70,84; 69,47; 69,39; 69,31; 66,25; 65,62; 64,43; 64,37; 49,78; 47,76; 41,41; 41,34; 40,57; 38,97; 34,22; 34,16; 33,96; 33,57; 32,95; 31,70; 29,43; 29,15; 28,95; 28,32; 25,87; 25,46; 25,02; 24,80; 24,18; 22,49; 13,94.
2.7.7. 1-Дигидрофосфат 3-[(R)-3-додеканоилокситетрадеканоиламино]-4-оксо-5-аза-9-[(R)-3-гидрокситетрадеканоиламино]декан-1,10-диола 10-(6-аминогексаноат) (=OM-197-MP-AC-R,R) (фиг.13)
Раствор полученного выше соединения (500 мг, 0,35 ммоль) в смеси СН2Cl2/этанол 5/2 (70 мл), содержащей уксусную кислоту (10 мл), гидрируют в течение 12-24 час в присутствии 10% Pd на угле при комнатной температуре и атмосферном давлении водорода. Катализатор удаляют фильтрацией, фильтрат досуха упаривают и остаток сушат с помощью крыльчатого насоса, получая 1-дигидрофосфат 3[(R)-3-додеканоилокситетрадеканоиламино]-4-оксо-5-аза-9-[(R)-3-гидрокситетрадеканоиламино]декан-1,10-диола 10-(6-аминогексаноат) (368 мг, количественный выход). ES/MS: m/z 1047.9 [М+Н]+, 1069.8 (М+Na]+ (фиг.14)
ПРИМЕР 2.8
(3S,9R)-3-[(R)-3-Додеканоилокситетрадеканоиламино]-4-оксо-5-аза-9-[(R)-3-гидрокситетрадеканоиламино]декан-1,10-диол 1-дигидрофосфат 10-(6-аминогексаноат) (=ОМ-197-МР-АС-S,R)
Та же последовательность реакций, выполненная с β-бензиловым эфиром α-N-{(4R)-5-гидрокси-4-[(R)-3-гидрокситетрадеканоиламино]пентил}амида N-[(R)-3-додеканоилокситетрадеканоил]-L-аспарагиновой кислоты (пример 1.2.4.), приводит к продукту примера 2.8.
ПРИМЕР 2.9
3-[(R)-3-Додеканоилокситетрадеканоиламино]-4-оксо-5-аза-9-[(R)-3-гидрокситетрадеканоиламино]декан-1,10-диол 1-дигидрофосфат 10-(6-гидроксигексаноат) (=OM-197-FV8) (фиг.15)
2.9.1. Бензилоксиметиловый эфир (2R)-5-(амино)-2-[(R)-3-бензилокситетрадеканоиламино]пентан-1-ола
К раствору (2R)-5-(бензилоксикарбониламино)-2-[(R)-3-бензилокситетрадеканоиламино)пентан-1-ола (пример 1.1.2) (2,05 г; 3,60 ммоль) в безводном СН2Cl2 (40 мл) при комнатной температуре под аргоном последовательно добавляют BOMCl (бензилхлорметиловый эфир) (технический, 60%, 1,25 мл; 5,41 ммоль; 1,5 экв) и диизопропилэтиламин (942 μл; 5,41 ммоль; 1,5 экв). Реакционную смесь перемешивают после этого в течение ночи при комнатной температуре и досуха упаривают. Очистка флэш-хроматографией на силикагеле (элюент петролейный эфир/AcOEt 2/1) позволяет выделить О-бензилоксиметильное производное (2.28 г; 92%) в виде белого кристаллического вещества (Rf=0,70 в смеси петролейный эфир/EtGAc 1/3; проявители УФ и фосфомолибденовая кислота). Т. пл. 97-100°С. Раствор этого продукта (2,00 г; 2,90 ммоль) в EtOH для ЖВХР (220 мл), содержащем Et3N (4 мл), гидрируют в течение 3 час в присутствии 20% Pd(OH)2 на угле (200 мг) при комнатной температуре и атмосферном давлении водорода. Катализатор удаляют фильтрацией, фильтрат досуха упаривают и остаток сушат с помощью крыльчатого насоса, получая свободный амин (1,58 г; 98%) в виде аморфного вещества. [α]D -1° (С = 1,20; CHCl3). 1Н-ЯМР (250 МГц, CDCl3), δ в м.д.: 7,45-7,21 (м, 10Н, Ar), 6,52 (д, 1Н, NH), 4,80-4,45 (м, 6Н, 2 х CH2-ph, O-CH2-O), 4,10 (м, 1Н, Н-3'), 3,83 (м, 1Н, Н-2), 3,62 (дд, 1Н, Н-1), 3,47 (дд, 1Н, Н-1), 2,65 (т, 2Н, 2 × Н-5), 2,40 (т, 2Н 2 х Н-2'), 1,80-1,40 (т, 8Н, 2 × Н-4, 2 × Н-3, 2 × Н-4', NH2), 1,40-1,20 (т, 18Н, 9 × CH2), 0,88 (т, 3Н, СН3); 13С-ЯМР (62,89 МГц, CDCl3), δ в м.д.: 170,78; 138,24; 137,63; 128,38; 128,32; 127,66; 127,62; 94,82; 76,70; 71,26; 69,63; 69,44; 48,48; 41,82; 41,47; 33,83; 31,84; 29,88; 29,58; 29,56; 29,51; 29,27; 29,04; 25,11; 22,62; 14,06.
2.9.2. β-Бензиловый эфир α-N{[(4R)-5(бензилоксиметокси)-4-[(R)-3-бензилокситетрадеканоиламино]пентил}амид N-[(R)-3-додеканоилокситетрадеканоиламино]-L-аспарагиновой кислоты
Полученный амин, как описано выше, сочетают с β-бензиловым эфиром N-[(R)-3-додеканоилокситетрадеканоиламино]-L-аспарагиновой кислоты (полученным из β-бензилового эфира L-аспарагиновой кислоты, см. пример 1.2.1) в присутствии IIDQ в тех же условиях, которые описаны в примере 1.2.2. Очистка продукта на силикагеле (петролейный эфир/AcOEt 2:1 и затем 1:1) позволяет выделить соответствующий амид с выходом 65%. ES/MS: m/z 1169.7 ([М+Н]+).
2.9.3. 1-дибензилфосфат (3S,9R)-3-[(R)-3-додеканоилокситетрадеканоиламино]-4-оксо-5-аза-9-(R)-3-бензилокситетрадеканоиламино]декан-1,10-диола
Полученный выше продукт сочетания (1,05 г; 0,90 ммоль) в смеси EtOH/AcOEt 1/1 (65 мл), содержащей Et3N (1,5 мл), гидрируют в течение 1 часа в присутствии 10% Pd на угле (50 мг) при комнатной температуре и атмосферном давлении водорода. Катализатор удаляют фильтрацией, фильтрат досуха упаривают и остаток сушат с помощью крыльчатого насоса. После этого остаток растворяют в смеси i-PrOH/ СН2Cl2 1/1 (50 мл) и перемешивают 10 мин при комнатной температуре со смолой Amberlite IR-120 (Н+) (3 мл). Смолу удаляют фильтрацией, а фильтрат досуха упаривают, получая свободную кислоту (956 мг; 99%) в виде белого кристаллического вещества.
К раствору полученной выше кислоты (855 мг; 0,79 ммоль) в безводном ТГФ (5 мл) при 0°С под аргоном добавляют N-метилморфолин (87 μл; 0,79 ммоль; 1 экв) и затем изобутилхлорформиат (103 μл; 0,79 ммоль; 1 экв) - наблюдают быстрое выпадение в осадок гидрохлорида N-метилморфолина. После 30 мин перемешивания при комнатной температуре реакционную смесь охлаждают до 0°С и затем быстро добавляют раствор NaBH4 (60 мг; 1,58 ммоль; 2 экв) в Н2О (2 мл). По прекращении выделения газа (5 мин) раствор разбавляют Н2О (2 мл) и ТГФ (2 мл), после чего перемешивают 5 мин при комнатной температуре. Раствор затем упаривают, разбавляют СН2Cl2 и Н2О, нейтрализуют 1 М раствором HCl и разделяют фазы. Органическую фазу высушивают над MgSO4 фильтруют и упаривают. Очистка флэш-хроматографией на силикагеле (элюент СН2Cl2/ацетон 4/1) позволяет выделить продукт восстановления (387 мг; 46%) в виде белого кристаллического вещества.
К раствору полученного спирта (313 мг; 0,29 ммоль) и 1H-тетразола (62 мг; 0,88 ммоль; 3 экв) в безводном ТГФ (12 мл) при комнатной температуре под аргоном добавляют 85%-ный дибензилдиэтилфосфорамидит (267 μл; 0,67 ммоль; 2,3 экв) - наблюдают быстрое образование в реакционной среде белых кристаллов. После 30 мин перемешивания реакционную смесь охлаждают до -20°С и добавляют раствор mCPBA (57-86%; 187 мг; 1,08 ммоль; 3,7 экв) в СН2Cl2 (8 мл) - наблюдают исчезновение кристаллов. После 45 мин перемешивания при комнатной температуре добавляют насыщенный раствор Na2S2O3 (5 мл), перемешивают реакционную смесь 10 мин и разбавляют эфиром. Органическую фазу отделяют, промывают 5 раз насыщенным раствором Na2S2O3, 2 раза насыщенным раствором NaHCO3 и 1 раз 1М раствором HCl. Органическую фазу сушат после этого над MgSO4 фильтруют и упаривают. Очистка флэш-хроматографией на силикагеле (элюент СН2Cl2/ацетон 8/1, затем 5/1) позволяет выделить фосфотриэфир (361 мг; 93%) в виде аморфного вещества.
Полученный продукт подвергают реакции гидролиза бензилоксиметилацеталя в водной среде с ТТФ-HCl, который дает 1-дибензилфосфат (3S,9R)-3-[(R)-3-додеканоилокситетрадеканоиламино]-4-оксо-5-аза-9-[(R)-3-бензилокситетрадеканоиламино] декан-1,10-диола.
2.9.4. 1-дигидрофосфат (3S,9R)-3-[(R)-3-додеканоилокситетрадеканоиламино]-4-оксо-5-аза-9-[(R)-3-гидрокситетрадеканоиламино]декан-1,10-диола 10-(6,7-дигидроксигептаноат)
Полученный выше продукт подвергают О-ацилированию в положении 10 гепт-6-еновой кислотой в присутствии EDCI в дихлорметане при 0°С в присутствии DMAP (см. пример 2.3.1). Полученный гептеноильный эфир подвергают реакции гидроксилирования в присутствии тетроксида осмия (каталитическое количество) и N-метилморфолиноксида (см. пример 2.3.2), что приводит к соответствующему диолу (6,7-дигидроксигептаноильный эфир). Этот продукт деблокируют гидрогенолизом в этаноле при атмосферном давлении водорода в присутствии палладия на угле (см. пример 2.3.3).
2.9.5. 1-дигидрофосфат (3S,9R)-3-[(R)-3-додеканоилокситетрадеканоиламино]-4-оксо-5-аза-9[(R)-3-гидрокситетрадеканоиламино]декан-1,10-диола 10-(6-оксогексаноат)
Полученный выше деблокированный диол подвергают реакции перйодатного окисления в среде изопропиловый спирт-вода (последовательность операций, как в 2.2.1).
2.9.6. 1-дигидрофосфат (3S,9R)-3-[(R)-3-додеканоилокситетрадеканоиламино}-4-оксо-5-аза-9[(R)-3-гидрокситетрадеканоиламино]декан-1,10-диола 10-(6-гидроксигексаноат) (=OM-197-FV8)
К раствору полученного выше 6-оксогексаноильного производного (4,5 мг, 0,0043 ммоль) в 90%-ном изопропиловом спирте (20 мл) добавляют раствор (0,2 мл) NaBH4 в метаноле (1 мг/мл). Смесь перемешивают 3 мин при 25°С и добавляют избыток уксусной кислоты (0,2 мл). Очистка с помощью препаративной ЖХВР на колонке с С18 позволяет выделить продукт OM-197-FV8 (2 мг, 44%) в виде раствора в 90%-ном изопропиловом спирте. ES/MS: m/z 1048.5 [М+Н]+ 950.5 [М+Н-(НО)2ОРОН]+ (фиг.16).
ПРИМЕР 2.10
{2-[(R)-3-гидрокситетрадеканоиламино]-5-(6-оксогексил)амино}пентиловый эфир 2-[(R)-3-додеканоилокситетрадеканоиламино]-4(дигидроксифосфорилокси)-бутановой кислоты (=OM-144-FP9) (фиг.17)
2.10.1. Гепт-6-енил-трифторметансульфонат
Трифлиновый ангидрид Tf2O ((CF3SO2)2O (11 мл; 6,76 ммоль) прибавляют по каплям при -15°С к раствору гепт-6-ен-1-ола (515 мг; 4,51 ммоль) и Et3N (627 μл; 4,51 ммоль) в СН2Cl2 (10 мл) и перемешивают смесь 30-45 мин при -15°С до исчезновения спирта. После возвращения к комнатной температуре среду разбавляют СН2Cl2 и последовательно промывают Н2О, насыщенным водным раствором NaHCO3 и насыщенным водным раствором NaCl. Полученную таким образом органическую фазу высушивают над MgSO4 и упаривают в вакууме. Остаток забирают смесью этилацетат/петролейный эфир (1/2) и фильтруют на силикагеле (удаление образовавшихся при реакции триэтиламиновых солей). После упаривания фильтрата получают ожидаемый трифлат с выходом 87% (956 мг) и используют в следующей стадии без дальнейшей очистки. Rf: 0,8 в смеси этилацетат/петролейный эфир, 1/2. 1H-ЯМР (250 МГц, CDCl3), δ в м.д.: 5,7; 5,0; 4,45; 2,0; 1,8; 1,4. 13С-ЯМР (62,89 МГц, CDCl3), δ в м.д.: 138,21; 114,95; 77,68; 33,40; 29,13; 28,10; 24,53.
2.10.2. (2R)-2-[(R)-3-бензилокситетрадеканоиламино]-5-(гепт-6-енил)амино-пентан-1-ол
Раствор свежеполученного выше трифлата (956 мг, 3,88 ммоль) в СН2Cl2 (10 мл) прибавляют по каплям к раствору (2R)-5-амино-2-[(R)-3-бензилокситетрадеканоиламино]пентан-1-ола (пример 1.1.2) (1,69 г; 3,88 ммоль) в СН2Cl2 (10 мл) и перемешивают смесь 4 часа под аргоном при комнатной температуре. После разбавления СН2Cl2 реакционную смесь промывают насыщенным водным раствором NaHCO3 и затем Н2О. Полученную таким образом органическую фазу высушивают над MgSO4 и упаривают в вакууме. Очистка флэш-хроматографией на силикагеле (элюент СН2Cl2/ацетон 15/1) позволяет выделить ожидаемый вторичный амин (862 мг; 43%). Rf: 0,3 (СН2Cl2/MeOH 8/1). ES/MS: m/z 532.0 [М+Н]+. 1Н-ЯМР (CDCl3, 250 МГц), δ в м.д.: 7,2-7,4; 7,1; 5,8; 5,0; 4,6; 3,95; 3,5; 2,9; 2,5; 2,1; 1,9-1,5; 1,5-1,2; 0,9. 13С-ЯМР (CDCl3, 62,89 МГц,), δ в м.д.: 172,17; 138,28; 138,21; 128,39; 127,96; 127,71; 114,82; 76,80; 71,40; 63,81; 50,87; 48,30; 47,75; 41,57; 34,10; 33,39; 31,89; 29,66; 29,62; 29,33; 28,19; 28,03; 25,21; 26,11; 25,78; 22,82; 22,66; 14,10. 1H-ЯМР (CDCl3, 250 МГц), δ в м.д.: 7,2-7,4; 6,75; 5,8; 5,0; 4,5; 3,9; 3,5; 2,5; 2,0; 2,1; 1,7-1,2; 0,9. 13С-ЯМР (CDCl3, 62,89 МГц,), δ в м.д.: 171,77; 138,68; 138,14; 128,23; 127,72; 127,56; 114,22; 76,64; 71,37; 64,58; 51,45; 49,79; 49,37; 41,53; 33,90; 33,53; 31,77; 29,65; 29,20; 28,64; 28,57; 25,00; 26,68; 25,93; 22,53; 13,98 (приведены спектры аммониевой соли и свободного основания, соответственно).
2.10.3. (2-[(R)-3-бензилокситетрадеканоиламино]-5-(гепт-6-енил)амино}пентиловый эфир 2-[(R)-3-додеканоилокситетрадеканоиламино]-4-(дифенилоксифосфорилокси)бутановой кислоты
К раствору полученного выше вторичного амина (163 мг; 0,307 ммоль; 1 экв) и 4-(дифенилоксифосфорилокси)-2-[(R)-3-додеканоилокситетрадеканоиламино]бутановой кислоты (пример. 1.1) (278 мг; 0,368 ммоль; 1,2 экв) в СН2Cl2 (25 мл) добавляют N, N-диизопропилэтиламин (DIEA) (54 μл; 1 экв). Среду охлаждают до 0°С и добавляют после этого EDCI (71 мг; 1,2 экв) и 1-гидрокси-7-азабензотриазол (HOAt) (41 мг; 1 экв). Реакционную смесь перемешивают 2 часа при 0°С и затем 90 час при комнатной температуре, после чего раствор промывают водой, а органическую фазу сушат над MgSO4 и упаривают в вакууме при 40°С. Очистка флэш-хроматографией на силикагеле (элюент СН2Cl2/МеОН 15/1) позволяет выделить продукт O-ацилирования (126 мг; 32%).
2.10.4. (2-[(R)-3-бензилокситетрадекаосиламино]-5-(6,7-дигидроксигептил)амино}пентиловый эфир 2-(R)-3-додеканоилокситетрадеканоиламино]-4-(дифенилоксифосфорилокси)бутановой кислоты
Свежеприготовленный 2,5%-ный раствор OsO4 в пиридине (1,1 мл; 1,9 экв) прибавляют по каплям при 25°С к раствору полученного выше продукта О-ацилирования (70 мг; 0,055 ммоль) в безводном пиридине (5 мл). Смесь перемешивают 24-48 час при комнатной температуре, обрабатывают добавкой Na2S2O5 и, наконец, разбавляют СН2Cl2, после чего последовательно промывают Н2О, 1 н. водным раствором HCl и вновь Н2O. Образовавшуюся органическую фазу высушивают над MgSO4, фильтруют и упаривают. Остаток подвергают счистке флэш-хроматографией на силикагеле (элюент СН2Cl2/МеОН от 15/1 до 10/1), которая позволяет выделить ожидаемый диол (27 мг; 38%). ЖХВР (210 нм): Время удерж.= 37,25 мин. ES/MS: m/z 1307.0 [М+Н]+.
2.10.5. {2-[(R)-3-гидрокситетрадеканоиламино]-5-(6,7-дигидроксигептил)амино}пентиловый эфир 2-[(R)-3- додеканоилокситетрадеканоиламино]-4(дигидроксифосфорилокси)бутановой кислоты (=ОМ-144-ЕР8)
а. Дебензилирование
К раствору полученного выше диола (50 мг, 0,038 ммоль) в смеси МеОН для ЖХВР/АсОН (5 мл/О,2 мл) добавляют катализатор 10% Pd/C (10 мг). Реакционную смесь перемешивают 4 часа в атмосфере Н2 (атмосферное давление) при комнатной температуре. Катализатор удаляют фильтрацией на мелкопористом фильтре и упаривают фильтрат, получая без дальнейшей очистки дебензилированный продукт (46 мг; 99%). ЖХВР (210 нм): Время удерж.= 35,13 и 35,51 мин (наблюдаются два стереоизомера). ES/MS: m/z 1217.0 [М+Н]+.
b. Дефенилирование
Катализатор (PtO2) (25 мг; 3,8 экв), суспендированный в EtOH для ЖХВР (0,5 мл), предварительно активируют в течение 10 мин в атмосфере Н2. Предварительно дегазированный под аргоном раствор полученного выше дебензилированного продукта (35 мг, 0,029 ммоль) в EtOH для ЖХВР (2 мл) добавляют к суспензии катализатора. Реакционную смесь после этого перемешивают в течение 2 час в атмосфере Н2 при комнатной температуре. Катализатор отфильтровывают на мелкопористом фильтре и упаривают фильтрат, получая без дальнейшей очистки ожидаемый фосфат (26 мг, 87%). ЖХВР (210 нм): Время удерж.= 29,3 и 30,9 мин (наблюдаются два стереоизомера). ES/MS: m/z 1064.8 [М+Н]+, 1086.8 [M+Na]+ 1108.8 [M-H+2Na]+ (фиг.18).
2.10.6. {2-[(R)-3-гидрокситетрадеканоиламино]-5-(6-оксогексил)амино}пентиловый эфир 2[(R)-3-додеканоилокситетрадеканоиламино]-4-(дигидроксифосфорилокси)бутановой кислоты (=OM-144-FP9)
1,87 мл 0,1 М NaIO4 (40,17 мг, 20 экв) добавляют к 10 мг (9,39 μмоль, 1 экв) полученного выше деблокированного продукта в смеси изопропиловый спирт-вода (1:1). Раствор перемешивают в течение 2 час при комнатной температуре и останавливают реакцию добавлением 1-2 капель этиленгликоля. Время удерж.= 30,0 и 31,5 мин (наблюдаются два стереоизомера). ES/MS: m/z 1032.8 [М+Н]+, 1054.8 [M+Na]+.
ПРИМЕР 2.11
1-О-Карбоксиметиловый эфир (2R,8R)-2-[(R)-3-додеканоилокситетрадеканоиламино]-3-оксо-4-аза-8-[(R)-3-гидрокситетрадеканоиламино]нонан-1,9-диола 9-О-(6-оксогексаноат) (=OM-112-FV7) (фиг.19)
D-Серин защищают в форме N(п-метоксибензилоксикарбонильного) производного [ссылка: Chen и Wang, Synthesis (1989), 36-37], после чего алкилируют функцию ОН бензилбромацетатом в присутствии NaH (2 экв). Аминную функцию освобождают действием трифторуксусной кислоты в дихлорметане, после чего ацилируют хлорангидридом (R)-3-додеканоилокситетрадекановой кислоты в присутствии триэтиламина. Полученное в результате этого производное серина сочетают с амином (2R)-5-амино-2-[(R)-3-бензилокситетрадеканоиламино]пентан-1-ола (см. 4.1.1) в присутствии IIDQ, получая 1-O-бензилоксикарбонилметиловый эфир (2R,8R)-2-[(R)-3-додеканоилокситетрадеканоиламино]-3-оксо-4-аза-8-[(R)-3-бензилокситетрадеканоиламино]нонан-1,9-диола. Этот продукт затем подвергают O-ацилированию гепт-6-еновсй кислотой в присутствии EDCl. Двойная связь вспомогательного сложного эфира гидроксилируется с помощью тетроксида осмия (каталитическое количество, в присутствии N-метилморфолин-N-оксида), после чего диол деблокируют гидрогенолизом в присутствии палладия на угле в этаноле. Продукт (ОМ-112-FV-7) получают действием перйодата натрия в смеси изопропиловый спирт-вода. С65Р103N3О12. ММ 1010.45.
ПРИМЕР 2.12
(2R,8R)-1-(Карбоксиметил)тио-2-[(R)-3-тетрадеканоил-окситетрадеканоиламино]-3-оксо-4-аза-8-[(R)-3-гидрокситетрадеканоиламино]нонан-9-ол 9-О-(7-аминогептаноат) (=ОМ-212-АН1) (фиг.20)
D-Цистеин подвергают S-алкилированию п-метоксибензилбромацетатом в присутствии карбоната натрия в среде ТГФ-вода. Полученный в результате этого S-бензилоксикарбонилметилцистеин подвергают N-ацилированию хлорангидридом (R)-тетрадеканоилокситетрадекановой кислоты, после чего S-алкилированный и N-ацилированный D-цистеин сочетают с амином (2R)-5-амино-2-[(R)-3-гидрокситетрадеканоиламино]пентан-1-олом [полученным гидрогенолизом (2R)-5-амино-2-[(R)-3-бензилокситетрадеканоиламино]пентан-1-ола, см. 4.1.1] в присутствии IIDQ. Полученный продукт подвергают селективному O-ацилированию в первичное положение с помощью сложного эфира, полученного из HOBt и 7-(п-метоксибензилокси-карбониламино)гептановой кислоты. После этого удаляют обе п-метоксибензильные группы действием на сложный эфир водной трифторуксусной кислотой. С59H112O10S. ММ 1069.64.
ПРИМЕР 2.13
1-О-(2,2-Дикарбоксиэтиловый) эфир (2R,8R)-2-[(R)-3-додеканоилокситетрадеканоиламино]-3-оксо-4-аза-8-[(R)-3-гидрокситетрадеканоиламино]нонан-1,9-диола 9-0-(6-оксогексаноат) (=OM-312-FV7) (фиг.21)
N-(п-метоксибензилоксикарбонильное) производное D-серина подвергают О-алкилированию дибензилметиленмалонатом в присутствии NaH. Аминную функцию освобождают действием трифторуксусной кислоты в дихлорметане, после чего ацилируют хлорангидридом (R)-3-додеканоилокситетрадекановой кислоты в присутствии триэтиламина. Полученное в результате этого производное серина сочетают с амином (2R)-5-амино-2-[(R)-3-бензилокситетрадеканоиламино]пентан-1-олом (см. 4.1.1) в присутствии IIDQ, получая 1-О-[2, 2-бис-(бензилоксикарбонил)этил]овый эфир (2R,SR)-2-[(R)-3-додеканоилокситетрадеканоиламино]-3-оксо-4-аза-8-[(R)-3-бензилокситетрадеканоиламино]нонан-1,9-диола. Этот продукт затем подвергают О-ацилированию гепт-6-еновой кислотой в присутствии EDCl, после чего гептеноильный эфир подвергают гидроксилированию с тетроксидом осмия. Бензильные группы удаляют гидрогенолизом в присутствии палладия на угле в этаноле. Продукт OM-312-FV-7 получают действием на дебензилированный продукт перйодатом натрия в смеси изопропиловый спирт-вода. C58H105N3O14. MM 1068.49.
ПРИМЕР 2.14
3-[(R)-3-Додеканоилокситетрадеканоиламино]-4-оксо-5-аза-9[(R)-3-гидрокситетрадеканоиламино]-1,10-диол-1-дигидрофосфат 10-(6-бромацетамидогексаноат) (=ОМ-412-ВА7) (фиг.22)
3-[(R)-3-Додеканоилокситетрадеканоиламино]-4-оксо-5-аза-9[(R)-3-гидрокситетрадеканоиламино]-1,10-диол-1-дигидрофосфат 10-(6-аминогексаноат) (см. 4.2.3.3) подвергают реакции бромацетилирования с помощью сукцинимидилокси-эфира бромуксусной кислоты в среде вода-ДХФ в присутствии триэтиламина. Конечный продукт счищают с помощью ЖХВР. С57Р108BrN4O13Р. ММ 1168.4.
ПРИМЕР 2.15
(3RS,9R)-3-[(R)-3-Гидрокситетрадеканоиламино]-4-оксо-5-аза-9-[(R)-3-додеканоилокситетрадеканоиламино]декан-1,10-диол 1-дигидрофосфат 10-О-(6-оксогексаноат) (=OM-512-FV7) (фиг.23)
2-[(R)-3-Бекзилокситетрадеканоиламино]-4-дифенилоксифосфорилокси-бутановую кислоту [полученную N-ацилированием трифторацетатной соли бензилового эфира О-(дифенилоксифосфорил)-DL-гомосерина хлорангидридом (R)-3-бензилокситетрадекановой кислоты с последующим расщеплением бензилового эфира гидрогенолизом в этаноле в присутствии триэтиламина и палладия на угле] сочетают с (2R)-5-амино-2-[(R)-3-додеканоилокситетрадеканоиламино]пентан-1-олом [полученным N-ацилированием трифторацетатной соли (2R)-5-(бензилоксикарбониламино)-2-аминопентан-1-ола хлорангидридом (R)-3-додеканоилокситетрадекановой кислоты с последующим деблокированием аминогруппы в положении С-5 гидрогенолизом в этаноле в присутствии триэтиламина и палладия на угле] в присутствии IIDQ. Образовавшийся в результате этого амид О-ацилируют по свободной гидроксильной группе при помощи 6-гептеновой кислоты в присутствии EDCI и получают соответствующий сложный эфир. Двойную связь вспомогательного сложного эфира подвергают реакции гидроксилирования с тетроксидом осмия. Бензильную группу затем удаляют гидрогенолизом на Pd/C, а освобожденный фосфат гидрогенолизом на платиновой черни. Диоловую функцию подвергают реакции перйодатного оксиления с образованием 6-оксогексаноильного производного OM-512-FV7. C55H104N3O13P. MM 1046.42.
ПРИМЕР 2.16
α-N{(4R)-5-(6-аминогексаноилокси)-4-[(R)-3-гидрокситетрадеканоиламино]пентил}амид N-[(R)-3-додеканоилокситетрадеканоил]-D-аспарагиновой кислоты (=ОМ-197-МС-АС) (фиг.24)
2.16.1. β-Бензиловый эфир α-N{(4R)-5-(6-бензилоксикарбониламино)гексаноилокси)-4-[(R)-3-бензилокситетрадеканоиламино]пентил}амида N-[(R)-3-додеканоилокситетрадеканоил]-D-аспарагиновой кислоты
К раствору β-бензилового эфира α-N{(4R)-5-гидрокси-4-[(R)-3-бензилокситетрадеканоиламино]пентил}амида N-[(R)-3-додеканоилокситетрадеканоил]-D-аспарагиновой кислоты (400 мг, 0,38 ммоль) и 6-(бензилоксикарбониламино)гексановой кислоты (220 мг, 0,83 ммоль) и сухого СН2Cl2 (15 мл) при 0°С под аргоном последовательно добавляют коммерческий гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида (162 мг, 0,85 ммоль) и 4-диметиламинопиридин (12 мг, 98 μмоль). После этого реакционную смесь перемешивают 30 мин при 0°С и затем в течение ночи при комнатной температуре, промывают водой, затем 1 н. раствором HCl, после чего фазы разделяют. Органическую фазу высушивают над MgSO4, фильтруют и упаривают. Очистка флэш-хроматографией на силикагеле (элюент СН2Cl2/ацетон 9/1 и затем 4/1) позволяет выделить продукт сочетания (395 мг; 81%) в виде белого твердого вещества. 13С-ЯМР (62,89 МГц, CDCl3), δ в м.д.: 173,46; 173,29; 171,83; 171,14; 170,12; 156,38; 138,19; 136,60; 133,30; 128,51; 128,42; 128,31; 127,99; 127,70; 127,58; 119,97; 76,49; 71,23; 71,06; 66,73; 66,47; 49,21; 47,83; 41,42; 40,73; 39,14; 35,46; 34,38; 33,86; 33,70; 31,84; 29,57; 29,46; 29,28; 29,10; 26,01; 25,54; 25,17; 25,08; 24,94; 24,31; 22,61; 14,05.
2.16.2. α-N{(4R)-5(6-аминогексаноилокси)-4[(R)-3-гидрокситетрадеканоиламино]пентил}амид N-[(R)-3-додеканоилокситетрадеканоил]-D-аспарагиновой кислоты
Раствор полученного выше соединения (340 мг, 0,26 ммоль) в смеси СН2Cl2/метанол 5/1 (24 мл), содержащей уксусную кислоту (2 мл), гидрируют в течение 12-24 час в присутствии 10% Pd на угле (40 мг) при комнатной температуре и атмосферном давлении водорода. Катализатор удаляют фильтрацией. Фильтрат досуха упаривают и сушат остаток с помощью крыльчатого насоса, получая α-N{(4R)-5-(6-аминогексаноилокси)-4-[(R)-3-гидрокситетрадеканоиламино]пентил}амид N-[(R)-3-додеканоилокситетрадеканоил]-D-аспарагиновой кислоты (238 мг, количественный выход). С55Н104N4О10. ES/MS: m/z 981.9 ([М+Н]+) (фиг.25).
ПРИМЕР 2.17
α-N{(4R)-5-(6-суициниламидогексаноилокси)-4-[(R)-3-гидрокситетрадеканоиламино]пентил}амид N-[(R)-3-додеканоилокситетрадеканоил]-D-аспарагиновой кислоты (=ОМ-197-MC-AC-Succ) (фиг.24)
К раствору продукта примера 2.16 (пункта 2.16.2) (50 мг; 0,051 ммоль) в пиридине (3 мл) последовательно добавляют янтарный ангидрид (11 мг, 0,11 ммоль) и 4-N,N-диметиламинопиридин (7 мг; 0,57 ммоль). После 6 час перемешивания при 50°С под аргоном добавляют метанол (2 мл) и перемешивают реакционную среду еще 15 мин при комнатной температуре. Растворитель упаривают и продукт очищают флэш-хроматографией на силикагеле (элюент СН2Cl2/MeOH 7/1 и затем 5/1), получая продукт в виде белого твердого вещества (42 мг; 74%). C59H108N4O13 ES/MS: m/z 1082 ([M+H]+). Rf 0,1 (СН2Cl2/MeOH 4/1).
ПРИМЕР 2.18
α-N{(4R)-5-глицинилокси-4-[(R)-3-гидрокситетрадеканоиламино]пентил}амид N-[(R)-3-додеканоилокситетрадеканоил]-D-аспарагиновой кислоты (=OM-197-MC-Gly) (фиг.24)
2.18.1. β-Бензиловый эфир α-N{(4R)-5-бензилоксикарбониламиноацетокси-4-[(R)-3-бензилокситетрадеканоиламино]пентил}амида N-[(R)-3-додеканоилокситетрадеканоил] D-аспарагиновой кислоты
К раствору α-N{(4R)-5-гидрокси-4-[(R)-3-бензилокситетрадеканоиламино]пентил}амида N[(R)-3-додеканоилокситетрадеканоил]-D-аспарагиновой кислоты (300 мг, 0,29 ммоль) и коммерческого N-бензилоксикарбонилглицина (101 мг, 0,49 ммоль) в сухом СН2Cl2 (10 мл) при 0°С под аргоном последовательно добавляют коммерческий гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида (93 мг, 0,48 ммоль) и 4-диметиламинопиридин (6 мг, 49 μмоль). После этого реакционную смесь перемешивают 30 мин при 0°С и затем в течение ночи при комнатной температуре, промывают водой, затем 1 н. раствором HCl и разделяют фазы. Органическую фазу высушивают над MgSO4, фильтруют и упаривают. Счистка флэш-хроматографией на силикагеле (элюент СН2Cl2/ацетон 8/1 и затем 6/1) позволяет выделить продукт сочетания (313 мг; 88%) в виде белого твердого вещества. 13С-ЯМР (62,89 МГц, CDCl3), δ в м.д.: 173,71; 173,49; 171,80; 171,68; 171,21; 170,21; 170,06; 169,94; 169,84; 156,41; 138,18; 136,16; 135,26; 128,46; 128,40; 128,33; 128,26; 128,04; 127,93; 127,65; 127,55; 76,49; 71,14; 71,07; 70,93; 66,90; 66,67; 66,38; 66,26; 49,21; 49,03; 47,72; 47,66; 42,58; 41,83; 41,68; 41,32; 39,02; 35,58; 35,39; 34,34; 33,84; 31,80; 29,52; 29,24; 29,06; 28,31; 28,13; 25,34; 25,12; 25,03; 24,89; 22,57; 14,01.
2.18.2. α-N{(4R)-5-Глицинилокси-4[(R)-3-гидрокситетрадеканоиламино]пентил}амид N-[(R)-3-додеканоилокситетрадеканоил]-D-аспарагиновой кислоты (=OM-197-MC-Gly) (фиг.24)
Раствор полученного выше соединения (286 мг, 0,22 ммоль) в смеси СН2Cl2/этанол 5/1 (12 мл), содержащей уксусную кислоту (2 мл), гидрируют в течение 12-24 час в присутствии 10% Pd на угле (30 мг) при комнатной температуре и атмосферном давлении водорода. Катализатор удаляют фильтрацией. Фильтрат досуха упаривают и сушат остаток с помощью крыльчатого насоса, получая α-N((4R)-5-глицинилокси-4[(R)-3-гидрокситетра-деканоиламино]пентил}амид N-[(R)-3-додеканоилокситетрадеканоил]-D-аспарагиновой кислоты (200 мг, количественный выход). C51H96N4O10. ES/MS: m/z 925.7 ([M+H]+), 947.9 ([M+Na]+). (фиг.26).
ПРИМЕР 2.19.
α-N{(4R)-5-сукцинилокси-4-[(R)-3-гидрокситетрадеканоиламино]пентил}амид N-[(R)-3-додеканоилокситетрадеканоил]-D-аспарагиновой кислоты (=OM-197-MC-Succ) (фиг.27)
2.19.1. β-Бензиловый эфир α-N{(4R)-5-сукцинилокси-4-[(R)-3-бензилокситетрадеканоиламино]пентил}амида N-[(R)-3-додеканоилокситетрадеканоил}-D-аспарагиновой кислоты
К раствору янтарного ангидрида (25 мг, 0,25 ммоль) в сухом СН2Cl2 (2 мл) в присутствии Et3N (40 μл, 0,29 ммоль) при 0°С добавляют раствор β-бензилового эфира α-N{(4R)-5-гидрокси-4-[(R)-3-бензилокситетрадеканоиламино]пентил}амида N-[(R)-3-додеканоилокситетрадеканоил]-D-аспарагиновой кислоты (пример 1.2.2) (150 мг, 0,14 ммоль) в сухом СН2Cl2 (5 мл). Реакционную смесь перемешивают 10 мин при 0°С и затем в течение ночи при комнатной температуре, разбавляют СН2Cl2, промывают 1 н. раствором HCl и разделяют фазы. Органическую фазу высушивают над MgSO4, фильтруют и упаривают. Очистка флэш-хроматографией на силикагеле (элюент СН2Cl2/ацетон 9/1, содержащий 2% уксусной кислоты) позволяет выделить кислоту (148 мг; 90%) в виде белого твердого вещества. 13С-ЯМР (62,89 МГц, CDCl3), δ в м.д.: 175,03; 173,55; 173,35; 171,92; 171,72; 171,36; 170,97; 170,60; 170,46; 170,41; 138,05; 135,24; 128,85; 128,76; 128,41; 128,27; 128,20; 128,03; 127,59; 76,48; 71,31; 70,77; 66,67; 65,08; 49,20; 48,11; 41,48;41,31; 39,02; 35,80; 34,26; 34,13; 33,84; 31,77; 29,50; 29,21; 29,03; 25,05; 24,99; 24,83; 22,54; 13,98.
2.19.2. α-N{(4R)-5-Сукцинилокси-4[(R)-3-гидрокситетрадеканоиламино]пентил}амид N-[(R)-3-додеканоилокситетрадеканоил]-D-аспарагиновой кислоты (= OM-197-MC-Succ) (фиг.27)
Раствор полученного выше соединения (124 мг, 0,11 ммоль) растворяют при нагревании в EtOH (для ЖХВР) (12 мл), содержащем уксусную кислоту (1 мл), после чего гидрируют в течение 10 час в присутствии 10% Pd на угле (15 мг) при комнатной температуре и атмосферном давлении водорода. Катализатор удаляют фильтрацией. Фильтрат досуха упаривают и сушат остаток с помощью крыльчатого насоса, получая дикислоту (102 мг; 97%). С53Н97N3О12. ES/MS: m/z 968.6 ([М+Н]+), 990.7 ([M+Na]+). (фиг.28).
ПРИМЕР 2.20
α-N{(4R)-5-гидрокси-4-[(R)-3-гидрокситетрадеканоиламино]пентил}амид N-[(R)-3-додеканоилокситетрадеканоил]-D-аспарагиновой кислоты β-N-(3-аминопропил)амид (=ОМ-197-АР) (фиг.29)
2.20.1. α-N{(4R)-5-гидрокси-4-[(R)-3-бензилокситетрадеканоиламино]пентил}амид N-[(R)-3-додеканоилокситетрадеканоил]-D-аспарагиновой кислоты
Раствор β-бензилового эфира α-N((4R)-5-гидрокси-4[(R)-3-бензилокситетрадеканоиламино]пентил}амида N-[(R)-3-додеканоилокситетрадеканоиламино]-D-аспарагиновой кислоты (пример 1.2.2) (2,53 г; 2,4 ммоль) в смеси EtOH/EtOAc 1/1 (150 мл), содержащей Et3N (4 мл), гидрируют в течение 2 час в присутствии 10% Pd на угле (120 мг) при комнатной температуре и атмосферном давлении водорода. Катализатор удаляют фильтрацией, фильтрат досуха упаривают и сушат остаток с помощью крыльчатого насоса. После этого остаток растворяют в смеси i-PrOH/ СН2Cl2 1/1 (100 мл) и перемешивают в течение 10 мин при комнатной температуре со смолой Amberlite IR-120 (Н+) (5 мл). Смолу удаляют фильтрацией и фильтрат досуха упаривают, получая свободную кислоту (2,25 г; 97%) в виде белого кристаллического вещества (Rf: 0,45 в смеси СН2Cl2/МеОН 9/1, содержащей 1% уксусной кислоты; проявители УФ и фосфомолибденовая кислота). Т.пл. 115-117°С.
2.20.2. α-N{(4R)-5-гидрокси-4-[(R)-3-бензилокситетрадеканоиламино]пентил}амид N-[(R)-3-додеканоилокситетрадеканоил]-D-аспарагиновой кислоты |3-N-(3-бензилоксикарбониламинопропил)амид
Раствор IIDQ (2-изобутокси-1-изобутоксикарбонил-1,2-дигидрохинолин) (98 мг, 0,32 ммоль) в безводном СН2Cl2 (3 мл) добавляют к раствору полученного выше соединения (250 мг, 0,26 ммоль) в безводном СН2Cl2 (15 мл) при 0°С под аргоном. Реакционную смесь перемешивают 15 мин при 0°С и затем добавляют раствор коммерческого гидрохлорида 3-бензилоксикарбониламинопропиламина (70 мг, 0,29 ммоль) в безводном СН2Cl2 (7 мл), содержащем триэтиламин (40 μл, 0,29 ммоль). После 18 час перемешивания раствор досуха упаривают. Очистка флэш-хроматографией на силикагеле (элюент СН2Cl2/MeOH 20/1) позволяет выделить продукт сочетания (217 мг; 78%) в виде белого твердого вещества.
2.20.3. α-N{(4R)-5-гидрокси-4-[(R)-3-гидрокситетрадеканоиламино]пентил}амид N-[(R)-3-додеканоилокситетрадеканоил]-D-аспарагиновой кислоты (3-N-(3-аминопропил)амид (= ОМ-197-АР)
Раствор полученного выше соединения (50 мг, 44 μмоль) в смеси СН2Cl2/изопропиловый спирт 1/1 (8 мл), содержащей уксусную кислоту (1 мл), гидрируют в течение 6-8 час в присутствии 10% Pd на угле (10 мг) при комнатной температуре и атмосферном давлении водорода. Катализатор удаляют фильтрацией. Фильтрат досуха упаривают и остаток сушат с помощью крыльчатого насоса, получая α-N{(4R)-5-гидрокси-4-[(R)-3-гидрокситетрадеканоиламино]пентил}амид N-[(R)-3-додеканоилокситетрадеканоил]-D-аспарагиновой кислоты β-N-(3-аминопропил)амид (32 мг, 80%). C52H101N5O8. ES/MS: m/z 924.8 ([М+Н]+, 1848.8 ([2M+H]+). (фиг.30).
ПРИМЕР 2.21
α-N{(4R)-5-гидрокси-4-[(R)-3-гидрокситетрадеканоиламино]пентил}амид N-[(R)-3-додеканоилокситетрадеканоил]-D-аспарагиновой кислоты β-N[(1S)-1-карбокси-5-аминопентил)амид (=OM-197-Lys) (фиг.31)
2.21.1. α-N{(4R)-5-гидрокси-4[(R)-3-бензилокситетрадеканоиламино]пентил}амид N-[(R)-3-додеканоилокситетрадеканоил]-D-аспарагиновой кислоты β-N-[(1S)-1-бензилоксикарбонил-5-бензилоксикарбонидаминопентил)амид
Раствор IIDQ (2-изобутокси-1-изобутоксикарбонил-1,2-дигидрохинолин) (114 мг, 0,38 ммоль) в безводном СН2Cl2 (5 мл) добавляют к раствору α-N{(4R)-5-гидрокси-4-[(R)-3-бензилокситетрадеканоиламино]пентил}амида N[(R)-3-додеканоилокситетрадеканоил]-D-аспарагиновой кислоты (пример 2.20.1) (300 мг, 0,31 ммоль) в безводном СН2Cl2 (20 мл) при комнатной температуре под аргоном. Реакционную смесь перемешивают 15 мин при комнатной температуре и затем добавляют раствор коммерческого гидрохлорида бензилового эфира ε-N-бензилоксикарбонил-L-лизина (140 мг, 0,34 ммоль) в безводном СН2Cl2 (5 мл), содержащем триэтиламин (48 μл, 0,34 ммоль). После 18 час перемешивания раствор досуха упаривают. Очистка флэш-хроматографией на силикагеле (элюент СН2Cl2/MeOH 30/1 и затем 20/1) позволяет выделить продукт сочетания (317 мг; 77%) в виде белого твердого вещества. 13С-ЯМР (62,89 МГц, CDCl3), δ в м.д.: 173,68; 172,35; 171,92; 170,74; 170,62; 170,54; 156,75; 156,52; 138,31; 136,54; 135,12; 128,54; 128,40; 128,28; 128,21; 127,96; 127,577; 127,59; 76,67; 71,40; 71,18; 67,20; 67,09; 66,48; 64,67; 52,30; 51,19; 41,64; 40,34; 39,24; 37,58; 34,40; 34,10; 31,83; 29,58; 29,27; 29,09; 25,16; 25,11; 24,93; 22,60; 14,04.
2.21.2. α-N{(4R)-5-гидрокси-4-[(R)-3-гидрокситетрадеканоиламино]пентил}амид N-[(R)-3-додеканоилокситетрадеканоил]-D-аспарагиновой кислоты (3-N[(1S)-1-карбокси-5-аминопентил)амид (=OM-197-Lys)
Раствор полученного выше соединения (93 мг, 71 μмоль) в смеси СН2Cl2/этанол 1/1 (20 мл), содержащей уксусную кислоту (0,5 мл), гидрируют в течение 12-24 час в присутствии 10% Pd на угле (17 мг) при комнатной температуре и атмосферном давлении водорода. Катализатор удаляют фильтрацией. Фильтрат досуха упаривают и остаток сушат с помощью крыльчатого насоса, получая α-N{(4R)-5-гидрокси-4[ (R)-3-гидрокситетрадеканоиламино]пентил}амид N-[(R)-3-додеканоилокситетрадеканоил]-D-аспарагиновой кислоты β-N-[(1S)-1-карбокси-5-аминопентил)амид (71 мг, выход количественный). C55H105N5O10. ES/MS: m/z 996.9 ([М+Н]+), 1018.9 ([2М+Н]+). (фиг.32).
ПРИМЕР 2.22
α-N{(4R)-5-(6-аминогексаноилокси-4-[(R)-3-гидрокситетрадеканоиламино]пентил}амид N-[(R)-3-додеканоилокситетрадеканоил]-D-аспарагиновой кислоты β-N-[(1S)-1-карбокси-5-аминопентил)амид (=OM-197-Lys-AC) (фиг.31)
2.22.1 α-N{(4R)-5-(6-бензилоксикарбониламиногексаноилокси-4-[(R)-3-бензилокситетрадеканоиламино]пентил}амид N-[(R)-3-додеканоилокситетрадеканоил]-D-аспарагиновой кислоты β-N-[(1S)-1-бензилоксикарбонил-5-бензилоксикарбониламинопентил)амид
К раствору α-N{(4R)-5-гидрокси-4-[(R)-3-бензилокситетрадеканоиламино]пентил}амида N-[(R)-3-додеканоилокситетрадеканоил]-D-аспарагиновой кислоты (3-N-[(1S)-1-бензилоксикарбонил-5-бензилоксикарбониламинопентил)амида (пример 2.21.1) (317 мг, 0,24 ммоль) и 6-(бензилоксикарбониламино)гексановой кислоты (128 мг, 0,48 ммоль) в сухом СН2Cl2 (15 мл) при О°C под аргоном последовательно добавляют коммерческий гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимид (93 мг, 0,48 ммоль) и 4-диметиламинопиридин (12 мг, 98 μмоль). После этого реакционную смесь перемешивают 30 мин при 0°C и затем в течение ночи при комнатной температуре, промывают водой, затем 1 н. раствором HCl. Органическую фазу высушивают над MgSO4, фильтруют и упаривают. Очистка флэш-хроматографией на силикагеле (элюент СН2Cl2/ацетон 5/1) позволяет выделить продукт сочетания (266 мг; 71%) в виде белого твердого вещества.
2.22.2. α-N{(4R)-5(6-аминогексаноилокси-4[(R)-3-гидрокситетрадеканоиламино]пентил}амид N-[(R)-3-додеканоилокситетрадеканоил]-D-аспарагиновой кислоты β-N-[(1S)-1-карбокси-5-аминопентил)амид (= OM-197-Lys-AC)
Раствор полученного выше соединения (236 мг, 151 μмоль) в смеси СН2Cl2/изопропиловый спирт 1/1 (20 мл), содержащей уксусную кислоту (1 мл), гидрируют в течение 12-24 час в присутствии 10% Pd на угле (100 мг) при комнатной температуре и атмосферном давлении водорода. Катализатор удаляют фильтрацией. Фильтрат досуха упаривают и остаток сушат с помощью крыльчатого насоса, получая α-N{(4R)-5(6-аминогексаноилокси-4-[(R)-3-гидрокситетрадеканоиламино]пентил}амид N-[(R)-3-додеканоилокситетрадеканоил]-D-аспарагиновой кислоты β-N-[(1S)-1-карбокси-5-аминопентил)амид (140 мг, 83%). C61H116N6O11. ES/MS: m/z 555.5 ([М+Н]+), 1110.0 ([М+Н]+). (фиг.33).
ПРИМЕР 2.23
α-N{(4R)-5-гидрокси-4-[(R)-3-гидрокситетрадеканоиламино]пентил}амид N-[(R)-3-додеканоилокситетрадеканоил]-D-аспарагиновой кислоты β-N-[(1S)-1,2-дикарбоксиэтил]амид (=OM-197-Asp) (фиг.34)
2.23.1. α-N((4R)-5-гидрокси-4[(R)-3-бензилокситетрадеканоиламино]пентил}амид N-[(R)-3-додеканоилокситетрадеканоил]- D-аспарагиновой кислоты β-N-[(1S)-1,2-бис(бензилоксикарбонил)этил]амид
Раствор IIDQ (2-изобутокси-1-изобутоксикарбонил-1,2-дигидрохинолин) (96 мг, 0,32 ммоль) в безводном СН2Cl2 (5 мл) добавляют к раствору α-N{(4R)-5-гидрокси-4[(R)-3-бензилокситетрадеканоиламино]пентил}амида N-[(R)-3-додеканоилокситетрадеканоил]-D-аспарагиновой кислоты (пример 2.20.1) (252 мг, 0,26 ммоль) в безводном СН2Cl2 (20 мл) при комнатной температуре под аргоном. Реакционную смесь перемешивают 15 мин при комнатной температуре и затем добавляют раствор коммерческой п-толуолсульфонатной соли дибензилового эфира L-аспарагиновой кислоты (141 мг, 0,29 ммоль) в безводном СН2Cl2 (5 мл), содержащем триэтиламин (40 ил, 0,29 ммоль). После 18 час перемешивания раствор досуха упаривают. Очистка флэш-хроматографией на силикагеле (элюент СН2Cl2/MeOH 20/1) позволяет выделить продукт сочетания (260 мг; 73%). 13С-ЯМР (62,89 МГц, CDCl3), δ в м.д.: 173,68; 171,92; 171,17; 170,60; 170,46; 170,37; 170,18; 170,06; 138,31; 138,19; 135,20; 135,12; 134,87; 128,48; 128,26; 128,18; 127,75; 127,63; 127,56; 76,66; 71,40; 71,19; 70,98; 68,79; 67,59; 66,85; 65,14; 64,73; 51,25; 50,17; 48,78; 48,67; 41,70; 39,37; 39,21; 37,50; 35,94; 34,48; 34,38; 34,10; 31,81; 29,54; 29,25; 29,08; 27,93; 25,56; 25,14; 25,09; 24,91; 22,58; 14,02.
2.23.2. α-N{(4R)-5-гидрокси-4-[(R)-3-гидрокситетрадеканоиламино]пентил}амид N-[(R)-3-додеканоилокситетрадеканоил]-D-аспарагиновой кислоты β-N- [(1S)-1,2-дикарбоксиэтил]амид (=OM-197-Asp)
Раствор полученного выше соединения (260 мг, 207 μмоль) в смеси СН2Cl2 /изопропиловый спирт 1/1 (20 мл) гидрируют в течение 4 час в присутствии 10% Pd на угле (50 мг) при комнатной температуре и атмосферном давлении водорода. Катализатор удаляют фильтрацией. Фильтрат досуха упаривают и сушат с помощью крыльчатого насоса, получая дикислоту (108 мг, 88%). C55H98N4О12. ES/MS: m/z 983.7 ([М+Н]+), 1005.8 ([M+Na]+). (фиг.35).
ПРИМЕР 2.24
α-N{(4R)-5-(6-аминогексаноилокси)-4-[(R)-3-гидрокситетрадеканоиламино]пентил}амид N-[(R)-3-додеканоилокситетрадеканоил]-D-аспарагиновой кислоты β-N-[(1S)-1,2-дикарбоксиэтил]амид (=OM-197-Asp-AC) (фиг.34)
2.24.1. α-N{(4R)-5-(6-бензилоксикарбониламиногексаноилокси)-4-[(R)-3-бензилокситетрадеканоиламино]пентил}амид N-[(R)-3-додеканоилокситетрадеканоил]-D-аспарагиновой кислоты β-N-[(1S)-1,2-бис(бензилоксикарбонил)этил]амид
К раствору α-N{(4R)-5-гидрокси-4-[(R)-3-бензилокситетрадеканоиламино]пентил}амида N-[(R)-3-додеканоилокситетрадеканоил]-D-аспарагиновой кислоты β-N-[(1S)-1,2-бис(бензилоксикарбонил)этил]амида (пример 2.23.1) (158 мг, 0,13 ммоль) и 6-(бензилоксикарбониламино)гексановой кислоты (84 мг, 0,32 ммоль) в сухом СН2Cl2 (6 мл) при 0°С под аргоном последовательно добавляют коммерческий гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида (61 мг, 0,32 ммоль) и 4-диметиламинопиридин (4 мг, 33 μмоль). После этого реакционную смесь перемешивают 30 мин при 0°С и затем в течение ночи при комнатной температуре, после чего промывают водой, затем 1 н. раствором HCl и разделяют фазы. Органическую фазу высушивают над MgSO4, фильтруют и упаривают. Очистка флэш-хроматографией на силикагеле (элюент СН2Cl2/ацетон 6/1) позволяет выделить продукт сочетания (83 мг; 44%). 13С-ЯМР (62,89 МГц, CDCl3), δ в м.д.: 173,69, 173,53, 173,26, 171,21, 171,11, 170,61, 170,38, 170,31, 170,22, 156,36, 138,26, 136,58, 135,20, 134,87, 128,48, 128,38, 128,31, 128,27, 128,19, 127,95, 127,55, 76,50, 71,24, 71,10, 67,58, 67,48, 66,79, 66,42, 65,69, 49,99, 48,77, 47,86, 41,70, 41/49, 40,70, 39,15, 37,50, 35,94, 34,44, 34,37, 33,97, 33,67, 31,80,29,54, 29,24, 29,07, 28,37, 25,98,25,55, 25,13, 25,08, 24,91, 24,28, 22,58, 14,02.
2.24.2. α-N{(4R)-5-(6-аминогексаноилокси)-4[(R)-3-гидрокситетрадеканоиламино]пентил}амид N-[(R)-3-додеканоилокситетрадеканоил]-D-аспарагиновой кислоты β-N-[(1S)-1,2-дикарбоксиэтил]амид (=OM-197-Asp-AC)
Раствор полученного выше соединения (80 мг, 0,053 ммоль) в смеси СН2Cl2/этанол 1/4 (10 мл), содержащий уксусную кислоту (1 мл), гидрируют в течение 12-24 час в присутствии 10% Pd на угле (50 мг) при комнатной температуре и атмосферном давлении водорода. Катализатор удаляют фильтрацией. Фильтрат досуха упаривают и затем остаток сушат с помощью крыльчатого насоса, получая дикислоту (61 мг, выход количественный); ММ: рассч. для C59H109N5O13 1095.8; найдено; m/z 1097.0 ([М+Н]+). (фиг.36).
ПРИМЕР 2.25
α-N{(4R)-5-(6-аминогексаноилокси)-4-[(R)-3-гидрокситетрадеканоиламино]пентил}амид N-ацетил-D-аспарагиновой кислоты (= OM-197-N'С2-АС) (фиг.37)
2.25.1 β-Бензиловый эфир N-ацетид-D-аспарагиновой кислоты
К раствору уксусного ангидрида (85 μл, 0,90 ммоль) в ацетонитриле (1 мл) добавляют раствор коммерческого H-D-Asp(OBn)-OH (Senn Chemicals, CH-Dielsdorf) (200 мг, 0,90 ммоль) в смеси CH3CN/H2O/Et3N (3,5/1.0/0,4 мл). Реакционную среду перемешивают 18 час при комнатной температуре, упаривают органический растворитель, охлаждают водную фазу до 0°С, подкисляют ее 10%-ным водным раствором лимонной кислоты до рН 3 и дважды экстрагируют этилацетатом. Органические фазы объединяют, высушивают над MgSO4, фильтруют и упаривают, получая β-бензиловый эфир N-ацетил-D-аспарагиновой кислоты в виде белого кристаллического вещества (192 мг, 81%), и используют в следующей стадии без дальнейшей очистки. (Rf 0,30 в смеси СН2Cl2/MeOH 20/1, содержащей 2% уксусной кислоты; проявители УФ и фосфомолибденовая кислота).
2.25.2. β-Бензидовый эфир α-N{(4R)-5-гидрокси-4[(R)-3-бензилокситетрадеканоиламино]пентил}амида N-ацетил-D-аспарагиновой кислоты
Раствор IIDQ (2-изобутокси-1-изобутоксикарбонил-1,2-дигидрохинолин) (254 мг, 0,84 ммоль; 1,2 экв.) в безводном СН2Cl2 (5 мл) добавляют к раствору приготовленного выше β-бензилового эфира N-ацетил-О-аспарагиновой кислоты (185 мг; 0,70 ммоль) в безводном СН2Cl2 (15 мл) при комнатной температуре под аргоном. Реакционную смесь перемешивают 15 мин при 0°С и затем добавляют раствор (2R)-5-амино-2[(R)-3-бензилокситетрадеканоиламино]пентан-1-ола (пример 1.1.2) (334 мг; 0,77 ммоль; 1,1 экв) в безводном СН2Cl2 (10 мл). После 3 час перемешивания раствор досуха упаривают. Очистка флэш-хроматографией на силикагеле (элюент СН2Cl2/ацетон 5/3 и затем ацетон) позволяет выделить продукт сочетания (369 мг; 78%). (Rf: 0,22 в СН2Cl2/ацетон 5/2; проявители УФ и фосфомолибденовая кислота). 13С-ЯМР (62,89 МГц, CD3OD), δ в м.д.: 171,92; 171,23; 171,07; 170,61; 170,35; 138,11; 13524; 128,40; 128,24; 128,02; 127,69; 127,59; 71,25; 67,57; 64,56; 51,03; 49,41; 41,51; 39,21; 35,90,33,88; 31,73; 29,46; 29,17; 28,32; 28,02; 25,35; 25,24; 24,97; 22,83; 22,51; 13,97; ММ: рассч. для С39Н59N3O7 681,4; найдено: m/z 682,5 ([M+H]+), 704,5 ([M+Na]+).
2.25.3. β-Бензиловый эфир α-N{(4R)-5-[6-(бензилоксикарбониламино)гексаноилокси]-4-[(R)-3-бензилокситетрадеканоиламино]пентил}амида N-ацетил-D-аспарагиновой кислоты
К раствору β-бензилового эфира α-N{(4R)-5-гидрокси-4-[(R)-3-бензилокситетрадеканоиламино]пентил} амида N-ацетил-D-аспарагиновой кислоты (200 мг, 0,29 ммоль) и 6-(бензилоксикарбониламино)гексановой кислоты (156 мг, 0,59 ммоль) в сухом СН2Cl2 (8 мл) при 0°C под аргоном последовательно добавляют коммерческий гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида (112 мг, 0,59 ммоль) и 4-диметиламинопиридин (7 мг, 59 имоль). После этого реакционную смесь перемешивают 30 мин при 0°С и затем в течение ночи при комнатной температуре, после чего промывают водой, затем 1 н. раствором HCl и разделяют фазы. Органическую фазу высушивают над MgSO4, фильтруют и упаривают. Очистка флэш-хроматографией на силикагеле (элюент СН2Cl2/ацетон 6/1) позволяет выделить продукт сочетания (200 мг; 73%). 13С-ЯМР (62,89 МГц, CDCI3), δ в м.д.: 173,65, 171,71, 171,26, 170,30, 156,41, 138,16, 136,60, 135,33, 128,57, 128,46, 128,38, 128,20, 128,04, 127,76, 127,62, 71,21, 66,80, 66,53, 65,72, 65,63, 49,36, 47,80, 41,36, 40,76, 39,22, 39,13, 35,79, 33,75, 31,88, 29,61, 29,31, 28,58, 28,52, 26,04, 25,48, 25,11, 24,36, 23,13, 22,64, 14, 09.
2.25.4. α-N{(4R)-5-(6-аминогексаноилокси)-4-[(R)-3-гидрокситетрадеканоиламино]пентил}амид N-ацетил-β-аспарагиновой кислоты (= ОМ-197-N'С2-АС)
Раствор полученного выше соединения (200 мг, 0,22 моль) в смеси СН2Cl2/этанол 1/1 (16 мл), содержащей уксусную кислоту (2 мл), гидрируют в течение 12-24 час в присутствии 10% Pd на угле (40 мг) при комнатной температуре и атмосферном давлении водорода. Катализатор удаляют фильтрацией, фильтрат досуха упаривают и остаток сушат с помощью крыльчатого насоса, получая α-N((4R)-5(6-аминогексаноилокси)-4[(R)-3- гидрокситетрадеканоиламино]пентил}амид N-ацетил-О-аспарагиновой кислоты (132 мг, выход количественный); ММ: рассч. для C31H58N4О8 614,43; найдено; m/z 615.5 ([М+Н]+), 629.5 ([M+NH4]+), 637.5 ([M+Na]+) (фиг.38).
ПРИМЕР 2.26
α-N{(4R)-5-(6-аминогексаноилокси)-4-[(R)-3-гидрокситетрадеканоиламино]пентил}амид N-(2-деканоилоксиоктаноил)-D-аспарагиновой кислоты (= OM-197-N'(С10-20С8)-АС) (фиг.39)
2.26.1. Бензил-2-гидроксиоктаноат
К раствору коммерческой рацемической 2-гидроксиоктановой кислоты (1,00 г, 6,2 ммоль) в этилацетате для ЖХВР (30 мл) последовательно добавляют бензилбромид (2,22 мл; 18,7 ммоль), триэтиламин (2,6 мл; 18,7 ммоль) и тетрабутиламмониййодид (1,15 г; 3,12 ммоль). Раствор перемешивают 18 час при комнатной температуре и упаривают растворитель. Остаток забирают эфиром и промывают органическую фазу насыщенным раствором NaHCO3 и затем дважды Н2О. Органическую фазу высушивают над MgSO4, фильтруют и упаривают, получая сырой бензил-2-гидроксиоктаноат (Rf=0,57 в смеси петролейный эфир/EtOAc 3/1; проявители УФ и фосфомолибденовая кислота).
2.26.2. Бензил-2-деканоилоксиоктаноат
К раствору полученного выше сложного эфира (780 мг; 3,12 ммоль) в сухом СН2Cl2 (15 мл) при 0°С добавляют пиридин (0,9 мл) и затем прибавляют по каплям деканоилхлорид (654 мг; 3,43 ммоль). Смесь перемешивают 20 час при комнатной температуре, после чего реакционную среду вливают в ледяную воду, содержащую NaHCO3 (5%). Органическую фазу отделяют, промывают 1 н. раствором HCl и затем дважды H2O. Органическую фазу высушивают над MgSO4, фильтруют и упаривают. Очистка флэш-хроматографией на силикагеле (элюент петролейный эфир/EtOAc 30/1) позволяет выделить чистый бензил-2-деканоилоксиоктаноат.
2.26.3. 2-Деканоилоктановая кислота
Полученный выше бензил-2-деканоилоксиоктаноат (515 мг; 1,27 ммоль) в EtOH для ЖХВР (40 мл) гидрируют в течение 2 час в присутствии 10% Pd на угле (100 мг) при комнатной температуре и атмосферном давлении водорода. Катализатор удаляют фильтрацией на мелкопористой мембране, фильтрат досуха упаривают и остаток сушат с помощью крыльчатого насоса, получая 2-деканоилоксиоктановую кислоту (выход количественный).
2.26.4. β-Бензиловый эфир N-(2-деканоилоксиоктаноил)-D-аспарагиновой кислоты
К раствору 2-деканоилоксиоктановой кислоты (317 мг; 1,01 ммоль) в безводном ТГФ (2 мл) при -15°С под аргоном последовательно добавляют N-метилморфолин (111 μл; 1,01 ммоль; 1 экв) и изобутилхлорформиат (131 μл; 1,01 ммоль; 1 экв) - наблюдают быстрое выпадение в осадок гидрохлорида N-метилморфолина. После 30 мин перемешивания при -15°С добавляют раствор коммерческого H-D-Asp(OBn)-ОН (Senn Chemicals AG, CH-Dielsdorf) (225 мг; 1,01 ммоль; 1 экв) в смеси СН3CN/Н2О 3,5/1 (9 мл), содержащий Et3N (0,4 мл). Реакционную смесь перемешивают после этого в течение ночи при комнатной температуре. Упаривают органическую фазу, водную фазу охлаждают до 0°С, подкисляют водным 10%-ным раствором лимонной кислоты до рН 3 и дважды экстрагируют AcOEt. Органическую фазу высушивают над MgSO4, фильтруют и упаривают. Очистка флэш-хроматографией на силикагеле (элюент петролейный эфир/AcOEt 2/1, содержащий 2% уксусной кислоты) с последующим упариванием совместно с толуолом позволяет выделить β-бензиловый эфир N-(2-деканоилоксиоктаноил)-D-аспарагиновой кислоты (140 мг; 27%).
2.26.5. β-Бензиловый эфир α-N-{(4R)-5-гидрокси-4[(R)-3-бензилокситетрадеканоиламино]пентил}амида N(2-деканоилоксиоктаноил)-D-аспарагиновой кислоты
IIDQ (98 мг; 0,32 ммоль; 1,2 экв) добавляют к раствору [3-бензилового эфира N-(2-деканоилоксиоктаноил)-D-аспарагиновой кислоты (140 мг; 0,27 ммоль) в безводном СН2Cl2 (15 мл) при комнатной температуре под аргоном. Реакционную смесь перемешивают 15 мин и затем добавляют раствор (2R)-5-амино-2-[(R)-3-бензилокситетрадеканоиламино]пентан-1-ола (пример 1.1.2) (129 мг; 0,30 ммоль; 1,1 экв) в безводном СН2Cl2 (5 мл). После 18 час перемешивания снова раствор досуха упаривают. Очистка флэш-хроматографией на силикагеле (элюент СН2Cl2/ацетон 5/1, затем СН2Cl2/ацетон 1/1) позволяет выделить продукт сочетания (197 мг; 78%). (Rf =0,33 в СН2Cl2/ацетон 5/1; проявители УФ и фосфомолибденовая кислота).
2.26.6. β-Бензиловый эфир α-N-{(4R)-5-[6-бензилоксикарбониламино)гексаноилокси]-4-[(R)-3-бензилокситетрадеканоиламино]пентил}амида N-(2-деканоилоксиоктаноил)-D-аспарагиновой кислоты
К раствору β-бензилового эфира α-N-{(4R)-5-гидрокси-4-[(R)-3-бензилокситетрадеканоиламино]пентил}амида N-[(R)-2-деканоилоксиоктаноил]-D-аспарагиновой кислоты (197 мг, 0,21 ммоль) и 6-(бензилоксикарбониламино)гексановой кислоты 112 мг, 0,42 ммоль) в сухом СН2Cl2 (8 мл) при 0°С под аргоном последовательно добавляют коммерческий гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида (81 мг, 0,42 ммоль) и 4-диметиламинопиридин (6 мг, 42 μмоль). После этого реакционную смесь перемешивают 30 мин при 0°С и затем в течение ночи при комнатной температуре, вслед за чем промывают водой, затем 1 н. раствором HCl и разделяют фазы. Органическую фазу высушивают над MgSO4, фильтруют и упаривают. Очистка флэш-хроматографией на силикагеле (элюент СН2Cl2/ацетон 6/1) позволяет выделить продукт сочетания (170 мг; 68%).
2.26.7. α-N-((4R)-5-[6-Аминогексаноилокси]-4-[(R)-3-гидрокситетрадеканоиламино]пентил}амид N(2-деканоилоксиоктаноил)-D-аспарагиновой кислоты
Раствор полученного выше соединения (150 мг, 0,13 ммоль) в смеси СН2Cl2/этанол 1/1 (16 мл), содержащей уксусную кислоту (10 мл), гидрируют в течение 12-24 час в присутствии 10% Pd на угле (40 мг) при комнатной температуре и атмосферном давлении водорода. Катализатор удаляют фильтрацией, фильтрат досуха упаривают и остаток сушат с помощью крыльчатого насоса, получая α-N-{(4R)-5-[6-аминогексаноилокси]-4-[(R)-3-гидрокситетрадеканоиламино]пентил}амид N-(2-деканоилоксиоктаноил)-D-аспарагиновой кислоты (110 мг, выход количественный). ММ: рассч. для C47H88N4O10 868.65; найдено: m/z 870.0 ([M+H]+), 884.0 ([M+NH4]+), 891.5 ([M+Na]+ (фиг.40).
ПРИМЕР 2.27
Очистка и анализ соединений по изобретению
Продукты синтеза и ацилированные псевдодипептиды, имеющие вспомогательное функционализированное ответвление, растворяют в смеси вода-изопропиловый спирт (1:1 по объему). После этого добавляют 2 М бикарбонат аммония в количестве, необходимом для достижения концентрации 50 мМ.
2.27.1. Очистка
Очистку осуществляют с помощью препаративной ЖХВР в обращенной фазе в следующих условиях:

В том случае, когда имеет место присутствие ароматических продуктов (неполная стадия деблокирования), должна быть произведена более тонкая очистка. Эта дополнительная очистка проводится в следующих условиях:

Фракции, содержащие целевые соединения в виде аммониевой соли, объединяют и упаривают адсорбцией на фазе C18 Bondapack, 15-20 цм, 300 Å, Waters. Противоион может быть затем заменен промывкой водным раствором соли щелочного металла (такого, например, как NaCI или KCl) в количестве 10 г на 1 л смеси вода-изопропиловый спирт (9:1 по объему). После удаления избытка соли пропусканием 5 объемов смеси вода-изопропиловый спирт (9:1 по объему) на колонке соединение элюируют чистым изопропиловым спиртом.
2.27.2. Контроль за очисткой
После каждой стадии фракции анализируют с помощью аналитической ЖХВР в обращенной фазе при следующих условиях:

2.27.3 Количественное определение u анализ чистоты конечных продуктов
Количественное определение и анализ чистоты конечных продуктов осуществляют с помощью ЖХВР/УФ в вышеописанных условиях хроматографии. Согласно этим анализам, чистота получаемых продуктов составляет от 97 до 100%. Для выявления примесей, неактивных в УФ, были осуществлены анализы LC/ES-MS (ионизация типа electrosprey, положительная мода). Чтобы удовлетворить условиям ионизации, были использованы следующие хроматографические условия:

2.27.4 Спектроскопические анализы
Масс-спектрометрия
Спектры ES/MS (положительная и отрицательная моды) регистрировали на масс-спектрометрах различных типов (Finnigan LCQ, ion trap; Micromass Quattro II, triple stage quadrupole; Micromass Z-Bio-Q, triple stage quadrupole). Проводились также дополнительные анализы типа MS/MS.
Ядерный магнитный резонанс
Спектры 1H-ЯМР и 13С-ЯМР регистрировали на приборах типа Bruker DPX при 250,13 и 62,89 МГц.

ПРИМЕР 2.28
(3S,9R)-3-[(R)-3-Додеканоилокситетрадеканоиламино]-4-оксо-5-аза-9-[(R)-3-гидрокситетрадеканоиламино]декан-1,10-диол 10-(6-аминогексаноат) 10-дигидрофосфат (=ОМ-197-АС МР) (фиг.87)
2,28.1 (3S,9R)-3[(R)-3-Додеканоилокситетрадеканоиламино]-4-оксо-5-аза-9-[(R)-3-бензилокситетрадеканоиламино]декан-1,10-диол 10-(2-тетрагидропиранил)-1-(6-бензилоксикарбониламиногексаноат)
К раствору (3S,9R)-3-[(R)-3-додеканоилокситетрадеканоиламино]-4-оксо-5-аза-9-[(R)-3-бензилокситетрадеканоиламино]-10-(тетрагидропиранил)оксидекан-1-ола (пример 2.7) (700 мг, 0,68 ммоль) и 6-(бензилоксикарбониламино)гексановой кислоты (541 мг, 2,04 ммоль) в сухом дихлорметане (30 мл) при 0°С под аргоном последовательно добавляют коммерческий гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида (393 мг, 2,04 ммоль, 3 экв) и 4-диметиламинопиридин (26 мг, 0,21 ммоль, 0,3 экв). После этого реакционную смесь перемешивают 30 мин при 0°С и затем в течение ночи при комнатной температуре. Реакционную среду разбавляют далее дихлорметаном (приблизительно 40 мл), промывают водой, затем 0,1 н. HCl и снова водой до нейтрального рН водной фазы. Органическую фазу отделяют, высушивают над MgSO4, фильтруют и упаривают. Очистка флэш-хроматографией на силикагеле (элюент СН2Cl2/ацетон 7/1 и затем 5/1) позволяет выделить продукт сочетания (711 мг; 81%). (Rf =0,75 в смеси СН2Cl2/ацетон 7/1; проявители УФ и фосфомолибденовая кислота). 13С-ЯМР (62,89 МГц, CDCl3), δ в м.д.: 173,7, 173,3, 171,1, 170,7, 169,7, 156,4, 138,2, 136,6, 127,5-128,3, 100,1, 98,7, 77,0, 71,2, 70,9, 69,8, 68,5, 66,3, 63,2, 62,3, 60,5, 50,1, 48,6, 48,3, 41,8, 41,5, 40,6, 39,2, 34,2, 33,9, 31,7, 30,5, 30,3, 29,0-29,5, 26,0, 25,1, 24,9, 24,4, 22,5, 14,0.
2.28.2. (3S,9R)-3-[(R)-3-Додеканоилокситетрадеканоиламино]-4-оксо-5-аза-9-[(R)-3-бензилокситетрадеканоиламино]декан-1,10-диол 1-(6-бензилоксикарбониламиногексаноат)
К раствору 1% HCl в метаноле (21 мл) добавляют при 0°С раствор полученного выше соединения (700 мг, 0,55 ммоль) в СН2Cl2 (7 мл). После 45 мин перемешивания при 0°С реакционную смесь нейтрализуют 20%-ным раствором NaHCO3, разбавляют СН2Cl2 (50 мл) и отделяют органическую фазу. Оставшуюся водную фазу трижды экстрагируют СН2Cl2 и объединяют органические фазы. Органическую фазу высушивают над MgSO4. фильтруют и упаривают, получая сырой спирт (651 мг, 99%). (Rf =0/55 в смеси СН2Cl2/ ацетон 4/1; проявители УФ и фосфомолибденовая кислота).
2.28.3. (3S,9R)-3-[(R)-3-Додеканоилокситетрадеканоиламино]-4-оксо-5-аза-9-[(R)-3-бензидокситетрадеканоиламино]декан-1,10-диол 1-(6-бензилоксикарбониламиногексаноат) 10-(дибензилфосфат)
К раствору полученного выше спирта (650 мг; 0,55 ммоль) и 1Н-тетразола (115 мг; 1,64 ммоль; 3 экв) в безводном ТГФ при комнатной температуре под аргоном добавляют 85%-ный дибензилдиэтилфосфорамидат (453 μл; 1,25 ммоль; 2,3 экв) - в реакционной среде наблюдается быстрое образование белых кристаллов. После 30 мин перемешивания реакционную смесь охлаждают до -18°С и добавляют раствор mCPBA (57-86%; 335 мг; 2,0 ммоль; 3,7 экв) в СН2Cl2 (20 мл) - происходит растворение кристаллов. После 45 мин перемешивания при комнатной температуре добавляют насыщенный раствор Na2S2O3 (20 мл), перемешивают реакционную смесь 10 мин и разбавляют эфиром (40 мл). Органическую фазу после этого отделяют, промывают 5 раз насыщенным раствором Na2S2O3 и 2 раза насыщенным раствором NaHCO3. Органическую фазу далее сушат над MgSO4, фильтруют и упаривают. Очистка флэш-хроматографией на силикагеле (элюент СН2Cl2/ацетон 5/1, 4/1, 2/1 и затем 1/1) позволяет выделить дибензилфосфат (645 мг; 82%) в виде аморфного вещества. (Rf=0,40 в смеси СН2Cl2/ацетон 1/3; проявители УФ и фосфомолибденовая кислота). 13С-ЯМР (62,89 МГц, CDCl3), δ в м.д.: 173,6, 173,4, 171,0, 170,7, 169,7, 156,4, 138,2, 136,7, 135,5, 127,6-128,6, -77,0, 71,1, 71,0, 69,5, 68,6, 66,4, 60,6, 50,0, 48,4, 41,6, 41,3, 40,7, 38,8, 34,3, 33,9, 31,8, 31,6, 29,0-29,5, 27,7, 26,1, 25,0, 24,4, 22,6, 14,1. C84H131N4O14P. IS/MS: m/z 1453.0 ([М+Н]+), 1469.0 ([M+NH]+), 1475.0 ([М+Na]+).
2.28.4. (3S,9R)-3-[(R)-3-Додеканоилокситетрадеканоиламино]-4-оксо-5-аза-9-[(R)-3-гидрокситетрадеканоиламино]декан-1,10-диол 1-(6-аминогексаноат) 10-дигидрофосфат
Раствор полученного выше соединения (540 мг, 0,37 ммоль) в смеси СН2Cl2/этанол 5/2 (70 мл), содержащий уксусную кислоту (10 мл), гидрируют в течение 48 час в присутствии 10% Pd на угле (270 мг) при комнатной температуре и атмосферном давлении водорода. Катализатор удаляют фильтрацией. Фильтрат досуха упаривают, остаток 2 раза упаривают вместе с толуолом и затем сушат с помощью крыльчатого насоса, получая (3S,9R)-3-[(R)-3-додеканоилокситетрадеканоиламино]-4-оксо-5-аза-9-[(Р)-3-гидрокситетрадеканоиламино]декан-1,10-диол 10-(6-аминогексаноат) 10-дигидрофосфат (380 мг, 98%). C55H107N4O12P. ES/MS: m/z 1047.9 ([М+Н]+) (фиг.88)
ПРИМЕР 2.29.
α-N-{(4R)-5-(6-аминогексаноилокси)-4-[(R)-3-гидрокситетрадеканоиламино]пентил}амид N-[(R)-3-додеканоилокситетрадеканоил]-L-аспарагиновой кислоты β-N-(6-аминогексил)амид (= ОМ-197-АН АС)(фиг.89)
Та же последовательность реакций, которая была использована в примерах 1.2.1, 1.2.2, 2.7.1 и 2.7.2, будучи примененной в отношении коммерческого H-L-Asp(Obn)-ОН (Fluka, Buchs, Швейцария), приводит к триэтиламмониевой соли α-N{(4R)-5(2-тетрагидропиранил)окси-4-[(R)-3-бензилокситетрадеканоиламино]пентил}амида N-[(R)-3-додеканоилокситетрадеканоил]-L-аспарагиновой кислоты.
2.29.1. α-N{(4R)-5-(2-тетрагидропиранил)окси-4[(R)-3-бензилокситетрадеканоиламино]пентил)амид N-[(R)-3-додеканоилокситетрадеканоил]-L-аспарагиновой кислоты β-N-(6-бензилоксикарбониламиногексил)амид IIDQ (2-изобутокси-1-изобутоксикарбонил-1,2-дигидрохинолеин) (101 мг, 0,33 ммоль) добавляют к раствору триэтиламмониевой соли α-N{(4R)-5-(2-тетрагидропиранил)окси-4-[(R)-3-бензилокситетрадеканоиламино]пентил}амида N-[(R)-3-додеканоилокситетрадеканоил]-L-аспарагиновой кислоты (250 мг, 0,22 ммоль) и коммерческого гидрохлорида 6-бензилоксикарбониламиногексиламина (70 мг, 0,24 ммоль) в безводном СН2Cl2 (15 мл) при 0°С под аргоном. Реакционную смесь перемешивают 5 час при 0°С, после чего раствор досуха упаривают. Очистка флэш-хроматографией на силикагеле (элюент СН2Cl2/ацетон 3/1) позволяет выделить продукт сочетания (128 мг; 46% по двум стадиям) в виде аморфного вещества. (Rf=0,48 в СН2Cl2/метанол 1/3; проявители УФ и фосфомолибденовая кислота). C75H127N5O11. IS/MS: m/z 1275.0 ([M+Н]+), 1289.0 ([M+NH4]+), 1297.0 ([M+Na]+).
2.29.2. α-N((4R)-5-гидрокси-4[(R)-3-бензидокситетрадеканоиламино]пентил}амид N-[(R)-3-додеканоилокситетрадеканоил]-L-аспарагиновой кислоты (3-N(6-бензилоксикарбониламиногексил)-амид
К раствору 1% HCl в метаноле (30 мл) при 0°С добавляют раствор полученного выше соединения (900 мг, 0,70 ммоль) в СН2Cl2 (3 мл). После 45 мин перемешивания при 0°С реакционную смесь нейтрализуют 10%-ным раствором NaHCO3, разбавляют СН2Cl2 (100 мл) и отделяют органическую фазу. Оставшуюся водную фазу трижды экстрагируют СН2Cl2 и объединяют органические фазы. Органическую фазу высушивают над MgSO4. фильтруют и упаривают, получая сырой спирт (826 мг; 98%). (Rf =0,15 в СН2Cl2/ацетон 3/1; проявители УФ и фосфомолибденовая кислота). C70H119N5O10. IS/MS: m/z 1191.0 ([M+H]+), 1213.0 ([M+Na]+). Т.пл. 148-149°С.
2.29.3. α-N{(4R)-5-[6(бензилоксикарбониламино)гексано-илокси]-4-[(R)-3-бензилокситетрадеканоиламино]пентил}амид N-[(R)-3-додеканоилокситетрадеканоил]-L-аспарагиновой кислоты (5-М-(6-бензилоксикарбониламиногексил)амид
К раствору α-N{(4R)-5-гидрокси-4-[(R)-3-бензилокситетрадеканоиламино]пентил}амида N-[(R)-3-додеканоилокситетрадеканоил]-L-аспарагиновой кислоты β-N-(6-бензилоксикарбонил-аминогексил)амида (641 мг, 0,54 ммоль) и 6-(бензилоксикарбониламино)гексановой кислоты (428 мг, 1,62 ммоль) в сухом СН2Cl2 (30 мл) при комнатной температуре под аргоном последовательно добавляют коммерческий гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида (309 мг, 1,62 ммоль) и 4-диметиламинопиридин (20 мг, 0,17 ммоль). После этого реакционную смесь перемешивают в течение ночи при комнатной температуре, промывают водой, затем 1 н. раствором HCl и разделяют фазы. Органическую фазу высушивают над MgSO4, фильтруют и упаривают. Очистка перекристаллизацией (этилацетат) позволяет выделить продукт сочетания (717 мг; 100%). (Rf =0,66 в СН2Cl2/ацетон 3/1; проявители УФ и фосфомолибденовая кислота). Т.пл. 150-152°С. 13С-ЯМР (62,89 МГц, CDCl3), δ в м.д.: 173,64, 173,26, 171,32, 171,11, 170,60, 170,00, 156,42, 138,20, 136,59 (d), 128,38, 128,33, 127,95, 127,61, 127,52, 71,24, 70,99, 66,42, 65,71, 49,89, 47,80, 41,97, 41,45, 40,72, 40,67, 39,23, 39,11, 37,21, 34,49, 34,37, 33,90, 31,81, 29,54, 29,43, 29,25, 29,07, 28,40, 26,11, 26,00, 25,74, 25,15, 25,05, 24,90, 24,31, 22,59, 14,02.
2.29.4. α-N{(4R)-5(6-аминогексаноилокси)-4[(R)-3-гидрокситетрадеканоиламино]пентил}амид N-[(R)-3-додеканоилокситетрадеканоил]-L-аспарагиновой кислоты [3-N-(6-аминогексил)амид
Раствор полученного выше соединения (690 мг, 0,48 ммоль) в смеси СН2Cl2/этанол 7/3 (100 мл), содержащей уксусную кислоту (15 мл), гидрируют в течение 3-8 дней в присутствии 10% Pd на угле при комнатной температуре и атмосферном давлении водорода. Катализатор удаляют фильтрацией, фильтрат досуха упаривают, остаток упаривают совместно с толуолом и затем сушат с помощью крыльчатого насоса, получая α-N{(4R)-5-(6-аминогексаноилокси)-4-[(R)-3-гидрокситетрадеканоиламино]пентил}амид N-[(R)-3-додеканоилокситетрадеканоил]-L-аспарагиновой кислоты β-N-(6-аминогексил)амид (420 мг, 75%). C61H118N6O9. ES/MS: m/z 1080.0 ([М+Н]+) (фиг.90).
ПРИМЕР 2.30.
α-N{(4R)-5-(6-дигидроксифосфорилокси)-4-[(R)-3-гидрокситетрадеканоиламино]пентил}амид N-[(R)-3-додеканоилокситетрадеканоил]-L-аспарагиновой кислоты β-N-(6-аминогексил)амид (= ОМ-197-АН МР) (фиг.91)
2.30.1. α-N{(4R)-5-дибензилоксифосфорилокси-4[(R)-3-бензилокситетрадеканоиламино]пентил}амид N-[(R)-3-додеканоилокситетрадеканоил]-L-аспарагиновой кислоты β-N-(6-бензилоксикарбониламиногексил)амид
К раствору α-N{(4R)-5-гидрокси-4-[(R)-3-бензилокситетрадеканоиламино]пентил}амида N-[(R)-3-додеканоилокситетрадеканоил]-L-аспарагиновой кислоты β-N-(6-бензилоксикарбониламиногексил)амида (пример 2.29.2) (174 мг; 0,15 ммоль) и 1Н-тетразола (31 мг; 0,44 ммоль) в безводном ТГФ (8 мл) добавляют при комнатной температуре под аргоном 85%-ный дибензилдиэтилфосфорамидит (120 μл; 0,34 ммоль) - в реакционной среде наблюдается быстрое образование белых кристаллов. После перемешивания в течение 1 часа 15 мин реакционную смесь охлаждают до 0°С и добавляют раствор тСРВА (57-86%; 93 мг; 0,54 ммоль) в СН2Cl2 (4 мл) - происходит исчезновение кристаллов. После перемешивания в течение 1 часа 15 мин при комнатной температуре добавляют насыщенный раствор Na2S2O3 (6 мл) и перемешивают смесь 10 мин. После этого раствор разбавляют эфиром, отделяют органическую фазу, промывают ее 5 раз насыщенным раствором Na2S2O3 и 2 раза насыщенным раствором NaHCO3, высушивают над MgSO4, фильтруют и упаривают. Очистка флэш-хроматографией на силикагеле (элюент СН2Cl2/ацетон 2,5/1, затем 1/1) позволяет выделить α-N{(4R)-5-дибензилоксифосфорилокси-4-[(R)-3-бензилокситетрадеканоиламино]пентил}амид N-[(R)-3-додеканоилокситетрадеканоил]-L-аспарагиновой кислоты β-N-(6-бензилоксикарбониламиногексил)амид (168 мг; 79%) в виде аморфного вещества (Rf = 0,44 в СН2Cl2/ацетон 3/1; проявители УФ и фосфомолибденовая кислота). C84H132N5O13P. IS/MS: m/z 1451.0 ([М+Н]+), 1474.0 ([M+Na]+). Т.пл.109-112°С. 13С-ЯМР (62,89 МГц, CDCl3), δ в м.д.: 173,67, 171,37, 171,16, 170,73, 170,04, 156,48, 138,38, 136,69, 135,69, 135,59, 128,60, 128,48, 128,36, 128,02, 127,98, 127,73, 127,61, 71,22, 71,05, 69,47, 69,12, 66,51, 53,40, 50,05, 48,66, 47,55, 48,46, 41,99, 41,44, 40,71, 39,34, 39,02, 37,52, 34,52, 34,46, 34,00, 31,90, 29,61, 29,51, 29,34, 29,14, 27,53, 26,20, 26,08, 25,72, 25,23, 24,97, 22,66, 14,10.
2.30.2. α-N((4R)-5-дигидроксифосфорилокси)-4[(R)-3-гидрокситетрадеканоиламино]пентил}амид N-[(R)-3-додеканоилокситетрадеканоил]-L-аспарагиновой кислоты β-N-(6-аминогексил)амид
Раствор полученного выше соединения (140 мг, 96 μмоль) в смеси СН2Cl2 (14 мл) и этанола (6 мл), содержащей уксусную кислоту (3 мл), гидрируют в течение 3-8 дней в присутствии 10% Pd на угле при комнатной температуре и атмосферном давлении водорода. Катализатор удаляют фильтрацией, фильтрат досуха упаривают, остаток упаривают совместно с толуолом и затем сушат с помощью крыльчатого насоса, получая α-N{(4R)-5-(дигидроксифосфорилокси)-4-[(R)-3-гидрокситетрадеканоиламино]пентил}амид N-[(R)-3-додеканоилокситетрадеканоил]-L-аспарагиновой кислоты β-N-(6-аминогексил)амид (98 мг, 97%). C55H108N5011P. IS/MS: m/z 1046.7 ([М+Н]+) (фиг.93)
ПРИМЕР 2.31
α-N{(4R)-5-(6-аминогексаноилокси)-4-[(R)-3-додеканоилокситетрадеканоиламино]пентил}амид N-[(R)-3-гидрокситетрадеканоил]-L-аспарагиновой кислоты (= ОМ-197-МС-АС inv) (фиг.94)
2.31.1. Гидрохдорид (2R)-5-(бензилоксикарбониламино)-2-аминопентан-1-ола
К раствору соединения (2R)-5-(бензилоксикарбониламино)-2-(трет-бутилоксикарбониламино)пентан-1-ола (пример 1.1.2d) (2,5 г, 7,09 ммоль) в этилацетате для ЖХВР (25 мл) при 0°C добавляют 1% (мас./об.) HCl в метаноле (200 мл). После 14 час перемешивания при комнатной температуре растворитель упаривают, получая гидрохлорид (2R)-5-(бензилоксикарбониламино)-2-аминопентан-1-ола (1,98 г, 97%) в виде желтоватого сиропа.
2.31.2. (2R)-5-(Бензилоксикарбониламино)-2-[(R)-3-додеканоилокситетрадеканоиламино]пентан-1-ол
К охлажденному до -15°С раствору (R)-3-додеканоилокситетрадекановой кислоты (3,03 г, 7,09 ммоль) в безводном ТГФ (40 мл) добавляют N-метилморфолин (0,78 мл; 7,09 ммоль) и изобутилхлорформиат (0,92 мл; 7,09 ммоль). После перемешивания в течение 1 часа при -10°С добавляют раствор гидрохлорида (2R)-5-(бензилоксикарбониламино)-2-аминопентан-1-ола (пример 2.31.1) (1,98 г; 7,09 ммоль) в безводном ТГФ (1 мл) и NaHCO3 (1,19 г, 14,2 ммоль). Продолжают перемешивание реакционной смеси при комнатной температуре в течение 16 час и разбавляют водой и эфиром. Органическую фазу отделяют, дважды промывают водой, высушивают над MgSO4 и упаривают в вакууме. Получают в результате этого без дальнейшей очистки (2R)-5-(бензилоксикарбониламино)-2-[(R)-3-додеканоилокситетрадеканоиламино]пентан-1-ол (4,54 г; 97%) в виде аморфного вещества. (Rf =0,22 в СН2С12/ацетон 6/1; проявители УФ и фосфомолибденовая кислота). 1H-ЯМР (250 МГц, CDCl3), δ в м.д.: 7,4 (м, 1Н); 6,1 (м, 1Н, NH); 5,2 (м, 1Н); 5,1 (с, 2Н); 4,9 (м, 1Н, NH); 4,0-3,8 (м, 1Н); 3,8-3,5 (м, 2Н); 3,2 (м, 3Н); 2,5 (д, 2Н); 2,3 (т, 2Н); 1,7-1,5 (м, 8Н); 1,4-1,2 (м, 34Н); 0,9 (м, 6Н). C39H68N2O6. IS/MS: m/z 661.5 ([М+Н]+), 683.5 ([М+Na]+), 699.5 ([М+К]+).
2.31.3. (2R)-5-амино-2-[(R)-3-додеканоилокситетрадеканоиламино]пентан-1-ол
Раствор (2R)-5-(бензилоксикарбониламино)-2-[(R)-3-додеканоил-окситетрадеканоиламино]пентан-1-ола (189 мг; 0,29 ммоль) в этаноле (17 мл) гидрируют в течение 2 час 30 мин в присутствии 10% Pd на угле при комнатной температуре и атмосферном давлении водорода. Катализатор удаляют фильтрацией, промывают этанолом, фильтрат досуха упаривают и остаток сушат с помощью крыльчатого насоса, получая свободный амин (123 мг; 81%) в виде полупрозрачного аморфного вещества. 1H-ЯМР (250 МГц, CDCl3), δ в м.д.: 5,2 (м, 1Н); 3,9 (м, 1Н); 3,6 (м, 2Н); 3,1 (м, 2Н); 2,5 (м, 2Н); 2,3 (т, 2Н); 1,9-1,5 (м, 8Н); 1,3 (м, 34Н); 0,9 (м, 6Н).
2.31.4. β-Бензиловый эфир N-[(R)-3-бензилокситетрадеканоиламино]-L-аспарагиновой кислоты
К раствору (R)-3-бензилокситетрадекановой кислоты (750 мг; 2,2 ммоль) [Bull. Chem, Soc, Jpn. 60 (1987), 2197-2204] в безводном ТГФ (7,2 мл) при -15°С под аргоном последовательно добавляют N-метилморфолин (250 μл; 2,2 ммоль; 1 экв) и изобутилхлорформиат (291 μл; 2,2 ммоль; 1 экв). После 45 мин перемешивания при -15°С добавляют раствор коммерческого H-L-Asp(OBn)-OH (Fluka, Buchs, Швейцария) (500 мг; 2,2 ммоль) в смеси CH3CN/H2O 3,5/1 (24 мл), содержащей Et3N (1 мл). После этого реакционную смесь перемешивают в течение ночи при комнатной температуре, упаривают органический растворитель, водную фазу охлаждают до 0°С, подкисляют 10%-ным водным раствором лимонной кислоты до рН 3 и трижды экстрагируют AcOEt. Органическую фазу высушивают над MgSO4, фильтруют и упаривают. Очистка флэш-хроматографией на силикагеле (элюент петролейный эфир/AcOEt 2/1 с 2% уксусной кислоты) с последующим совместным упариванием с толуолом позволяет выделить β-бензиловый эфир N-[(R)-3-бензилокситетрадеканоиламино]-L-аспарагиновой кислоты (1,19 г; 99%) в виде белого кристаллического вещества. (Rf=0,27 в смеси петролейный эфир/AcOEt 1/1 с 2% уксусной кислоты; проявители УФ и фосфомолибденовая кислота). 1H-ЯМР (CDCl3, 250 МГц,), δ в м.д.: 7,3 (м, 10Н); 5,0 (с, 2Н); 4,9 (м, 1Н); 4,5 (кв, 2H); 3,8 (м, 1Н); 3,1-2,8 (дд, 2H); 2,5 (м, 2H); 1,6 (м, 2H); 1,3 (м, 18Н); 0,9 (м, 3Н).
2.31.5. β-Бензиловый эфир α-N{(4R)-5-гидрокси-4-[(R)-3-додеканоилокситетрадеканоиламино]пентил}амида N-[(R)-3-бензилокситетрадеканоил]-L-аспарагиновой кислоты
К раствору полученного выше бензилового эфира (61 мг; 114 ммоль) и (2R)-5-амино-2[(R)-3-додеканоилокситетрадеканоиламино]пентан-1-ола (пример 2.31.3) (60 мг; 114 μмоль; 1,0 экв) в безводном СН2Cl2 (1/4 мл) при комнатной температуре под аргоном последовательно добавляют коммерческий HOOBt (3-гидрокси-1,2,3-бензотриазин-4(3Н)-он) (ACROS, Basel, Швейцария) (19 мг; 114 μмоль; 1,0 экв) и затем коммерческий N,N'-диизопропилкарбодиимид (23 μл; 114 μмоль; 1 экв). После этого реакционную среду разбавляют СН2Cl2, промывают водой и 1 н. раствором НС1, насыщенным раствором NaHCO3, после чего разделяют фазы. Органическую фазу высушивают над MgSO4, фильтруют и упаривают. Остаток забирают СН2Cl2, охлаждают раствор в течение 2 час при 0°С, удаляют осадок диизопропилмочевины фильтрацией, промывают его холодным СН2Cl2 и упаривают растворитель. Очистка флэш-хроматографией на силикагеле (элюент СН2Cl2/ацетон 3/1) позволяет продукт сочетания (58 мг; 49%) в виде белого кристаллического вещества (Rf = 0,50 в СН2Cl2/ацетон 3/1; проявители УФ и фосфомолибденовая кислота). 13С-ЯМР (62,89 МГц, CDCl3), δ в м.д.: 174,21; 171,91; 170,78; 170,62; 138,25; 135,67; 128,9; 128,8; 128,7; 128,5; 128,21; 76,88; 71,78; 71,66; 67,1; 64,89; 51,71; 49,79; 42,31; 41,62; 39,34; 35,78; 34,86; 34,7; 32,22; 29,95; 29,66; 29,49; 28,02; 26,24; 25,6; 25,54; 25,33; 23,75; 22,99; 14,43.
2.31.6. β-Бензиловый эфир α-N{(4R)-5-(6-(бензилоксикарбониламино)гексаноилокси)-4-[(R)-3-додеканоилокситетрадеканоиламино]пентил}амида N-[(R)-3-бензилокситетрадеканоил]-L-аспарагиновой кислоты
К раствору полученного выше бензилового эфира (58 мг, 56 μмоль) и 6-(бензилоксикарбониламино)гексановой кислоты (45 мг, 168 μмоль) в сухом СН2Cl2 (2,6 мл) при 0°С под аргоном последовательно добавляют коммерческий гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида (33 мг, 168 μмоль) и 4-диметиламинопиридин (3 мг, 17 μмоль). После этого реакционную смесь перемешивают 30 мин при 0°С и затем в течение ночи при комнатной температуре, разбавляют СН2Cl2, промывают водой с 1 н. раствором HCl и разделяют фазы. Органическую фазу высушивают над MgSO4, фильтруют и упаривают. Очистка флэш-хроматографией на силикагеле (элюент СН2Cl2/ацетон 5/1) позволяет выделить продукт сочетания (57 мг; 79%) в виде белого кристаллического вещества. (P.f =0,80 в СН2Cl2/ацетон 3/1; проявители УФ и фосфомолибденовая кислота). 13С-ЯМР (62,89 МГц, CDCl3), δ в м.д.: 173,83; 173,77; 171,9; 171,72; 170,79; 169,94; 156,79; 138,32; 136,96; 135,71; 128,89; 128,81; 128,68; 128,49; 128,37; 128,20; 76,89; 71,81; 71,56; 67,04; 66,87; 66,03; 49,62; 48,52; 42,06; 41,65; 41,1; 39,25; 35,83; 34,86; 34,53; 34,19; 34,08; 32,22; 29,95; 29,72; 29,66; 29,50; 28,43; 26,42; 26,23; 26,0; 25,58; 25,36; 22,99; 14,43.
2.31.7. α-N{(4R)-5-(6-аминогексаноилокси)-4[(R)-3-додеканоилокситетрадеканоиламино]пентил}амид N-[(R)-3-гидрокситетрадеканоил]-L-аспарагиновой кислоты. Раствор полученного выше соединения (47 мг, 36 μмоль) в смеси СН2Cl2/метанол 5/1 (3,3 мл), содержащей уксусную кислоту (280 рл), гидрируют в течение 72 час в присутствии 10% Pd на угле при комнатной температуре и атмосферном давлении водорода. Катализатор удаляют фильтрацией, фильтрат досуха упаривают и остаток сушат с помощью крыльчатого насоса, получая α-N{(4R)-5-(6-аминогексаноилокси)-4-[(R)-3-додеканоилокситетрадеканоиламино]пентил}амид N-[(R)-3-гидрокситетрадеканоил]-L-аспарагиновой кислоты (35 мг, выход количественный). C55H104N4O10. ES/MS: m/z 981.9 ([М+Н]+) (фиг.95).
ПРИМЕР 2.32
α-N{(4R)-5-(6-аминогексаноилокси)-4-[(R)-3-додеканоил-окситетрадеканоиламино]пентил}амид N-[(R)-3-додеканоилокситетрадеканоил]-L-аспарагиновой кислоты (= ОМ-197-МС-АС tétracyle (фиг.96)
Та же реакция, которая была использована в примере 1.2.1, примененная к коммерческому H-L-Asp(ОВп)-ОН (Fluka, Buchs, Швейцария), приводит к β-бензиловому эфиру N-[(R)-3-додеканоилокситетрадеканоил]-L-аспарагиновой кислоты.
2.32.1. β-Бензиловый эфир α-N{(4R)-5-гидрокси-4-[(R)-3-додеканоилокситетрадеканоиламино]пентил}амида N-[(R)-3-додеканоилокситетрадеканоил]-L-аспарагиновой кислоты
К раствору β-бензилового эфира N-[(R)-3-додеканоилокситетрадеканоил]-L-аспарагиновой кислоты (131 мг; 207 μмоль) и (2R)-5-амино-2-[(R)-3-додеканоилокситетрадеканоиламино]пентан-1-ола (пример 2.31.3) (109 мг; 207 μмоль; 1,0 экв) в безводном СН2Cl2 (2,6 мл) при комнатной температуре под аргоном последовательно добавляют коммерческий HOOBt (3-гидрокси-1,2,3-бензотриазин-4(3Н)-он) (ACROS, Basel, Швейцария) (349 мг; 207 μмоль; 1,0 экв) и затем коммерческий N,N'-диизопропилкарбодиимид (41 μл; 207 μмоль; 1 экв). После этого реакционную среду разбавляют СН2Cl2, промывают водой, 1 н. раствором HCl, насыщенным раствором NaHCO3 и разделяют фазы. Органическую фазу высушивают над MgSO4, фильтруют и упаривают. Очистка флэш-хроматографией на силикагеле (элюент СН2Cl2/ацетон 3/1 и затем 2/1) позволяет выделить позволяет выделить продукт сочетания (122 мг; 52%) в виде белого кристаллического вещества (Rf = 0,55 в СН2Cl2/ацетон 3/1; проявители УФ и фосфомолибденовая кислота). 13С-ЯМР (62,89 МГц, CDCl3), δ в м.д.: 174,18; 172,02; 170,72; 170,60; 157,53; 135,67; 128,87; 128,65; 128,43; 71,69; 71,36; 67,07; 64,81; 51,68; 49,63; 42,33; 42,15; 39,58; 36,08; 34,87; 34,77; 32,21; 29,94; 29,83; 29,72; 29,65; 29,49; 26,09; 25,6; 25,54; 25,34; 25,29; 22,98; 14,41.
2.32.2. β-Бензиловый эфир α-N{(4R)-5-(6-бензилоксикарбониламино)гексаноилокси)-4-[(R)-3-додеканоилокситетрадеканоиламино]пентил}амида N-[(R)-3-додеканоилокситетрадеканоил]-L-аспарагиновой кислоты
К раствору полученного выше β-бензилового эфира (122 мг, 107 μмоль) и 6-(бензилоксикарбониламино)гексановой кислоты (86 мг, 107 μмоль) в сухом СН2Cl2 (5 мл) при 0°C под аргоном последовательно добавляют коммерческий гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида (85 мг, 321 μмоль) и 4-диметиламинопиридин (4 мг, 32 μмоль). После этого реакционную смесь перемешивают 30 мин при 0°С и затем в течение ночи при комнатной температуре, разбавляют СН2Cl2, промывают водой, 1 н. раствором HCl и разделяют фазы. Органическую фазу высушивают над MgSO4, фильтруют и упаривают. Очистка флэш-хроматографией на силикагеле (элюент СН2Cl2/ацетон 5/1) позволяет выделить продукт сочетания (97 мг; 65%) в виде белого кристаллического вещества. (Rf=0,77 в СН2Cl2/ацетон 3/1; проявители УФ и фосфомолибденовая кислота). 13С-ЯМР (62,89 МГц, CDCl3), δ в м.д.: 172,13; 170,7; 170,43; 169,93; 156,76; 136,93; 135,68; 128,88; 128,78; 128,64; 128,4; 128,34; 71,60; 71,34; 67,03; 66,83; 65,97; 49,46; 48,55; 42,20; 42,10; 41,08; 39,35; 36,00; 34,86; 34,74; 34,51; 34,17; 32,20; 29,93; 29,81; 29,70; 29,63; 29,48; 29,44; 28,57; 26,39; 26,10; 25,60; 25,56; 25,35; 25,27; 24,71; 22,97; 14,40.
2.32.3. α-N{(4R)-5(6-аминогексаноилокси)-4[(R)-3-додеканоилокситетрадеканоиламино]пентил}амид N-[(R)-3-додеканоилокситетрадеканоил]-L-аспарагиновой кислоты
Раствор полученного выше соединения (90 мг, 65 μмоль) в смеси СН2Cl2/метанол 5/1 (6 мл), содержащей уксусную кислоту (500 ил), гидрируют в течение 72 час в присутствии 10% Pd на угле при комнатной температуре и атмосферном давлении водорода. Катализатор удаляют фильтрацией, фильтрат досуха упаривают и остаток сушат с помощью крыльчатого насоса, получая α-N{(4R)-5-(6-аминогексаноилокси)-4-[(R)-3-додекано-илокситетрадеканоиламино]пентил}амид N-[(R)-3-додеканоилокситетрадеканоил]-L-аспарагиновой кислоты (76 мг, выход количественный). С67Н126N4О11. ES/MS: m/z 1163.9 ([M+H]+) (фиг.97).
ПРИМЕР 2.33
(3S,9R)-3-[(R)-3-Додеканоилокситетрадеканоиламино]-4-оксо-5-аза-9-[(R)-3-гидрокситетрадеканоиламино]декан-1,10-диол 1-дигидрофосфат 10-(3-аминопропаноат) (= ОМ-197-МР-АР) (фиг.98)
2.33.1.(2R)-5-(Бензилоксикарбониламино)-2-[(R)-3-бензилокситетрадеканоиламино]-1-(2-тетрагидропиранилокси)пентан
К раствору (2R)-5-(бензилсксикарбониламино)-2-[(R)-3-бензилокситетрадеканоиламино]пентан-1-ола (пример 1.1.2f) (2,5 г; 4,39 ммоль) в безводном СН2Cl2 (83 мл) при комнатной температуре под аргоном последовательно добавляют 3,4-дигидро-2Н-пиран (DHP) (1,4 мл, 15,38 ммоль) и затем п-толуол-сульфонат пиридиния (PPTS) (441 мг, 1,75 ммоль). Раствор перемешивают 18 час при комнатной температуре, разбавляют СН2Cl2 (100 мл), промывают 5%-ным раствором NaHCO3 и затем водой. Органическую фазу высушивают над MgSO4, фильтруют и упаривают. Очистка флэш-хроматографией на силикагеле (элюент AcOEt/петролейный эфир 4/1) позволяет выделить (2R)-5-(бензилоксикарбониламино)-2-[(R)-3-бензилокситетрадеканоиламино]-1-(2-тетрагидропиранилокси)пентан (2,86 г; 100%) в виде белого кристаллического вещества. (Rf =0,66 в смеси AcOEt/петролейный эфир 4/1; проявители УФ и фосфомолибденовая кислота). C39H60N2O6. IS/MS: m/z 653.5 ([M+H]+), 675.5 ([M+Na]+). Т.пл.84-86°С.
2.33.2.(2R)-5-Амино-2-[(R)-3-бензилокситетрадеканоиламино]-1-(2-тетрагидропиранилокси)пентан
Раствор (2R)-5-(Бензилоксикарбониламино)-2-[(R)-3-бензилокситетрадеканоиламино]-1-(2-тетрагидропиранилокси)пентана (2,5 г; 4,4 ммоль) в этаноле (150 мл), содержащем триэтиламин (4 мл), гидрируют в течение 3 час 30 мин в присутствии 10% Pd на угле при комнатной температуре и атмосферном давлении водорода. Катализатор удаляют фильтрацией, фильтрат досуха упаривают и остаток сушат с помощью крыльчатого насоса, получая свободный амин (2,15 г; 96%) в виде белого аморфного вещества. C31H54N2O4. IS/MS: m/z 519.5 ([М+Н]+).
2.33.3. (S)-(α)-[(R)-3-Додеканоилокситетрадеканоиламино]-γ-бутиролактон
К раствору (R)-3-додеканоилокситетрадекановой кислоты (2,16 г; 5,1 ммоль) в безводном ТГФ (28 мл) при -15°С под аргоном последовательно добавляют N-метилморфолин (0,56 мл; 5,1 ммоль; 1 экв) и изобутилхлорформиат (657 μл; 5,1 ммоль; 1 экв). После 1 часа перемешивания при -15°С добавляют коммерческий гидробромид лактона L-гомосерина (916 мг; 5,1 ммоль; 1 экв), растворенный в 0,72 М растворе NaHCO3 (14 мл, 2 экв). После этого реакционную смесь перемешивают 20 час при комнатной температуре, разбавляют диэтиловым эфиром (130 мл), отделяют органическую фазу, промывают ее водой, высушивают над MgSO4, фильтруют и упаривают при пониженном давлении. Очистка кристаллизацией (минимум СН2Cl2 и избыток пентана при 0°С) позволяет выделить продукт сочетания (2,17 г; 85%) в виде белого твердого вещества. (Rf =0,41 в смеси петролейный эфир/AcOEt 1/1; проявитель фосфомолибденовая кислота). C30H55NO5. IS/MS: m/z 510.5 ([М+Н]+), 532.5 ([M+Na]+). Т.пл. 79-80°С.
2.33.4. (3S,9R)-3-[(R)-3-Додеканоилокситетрадеканоиламино]-4-оксо-5-аза-9-[(R)-3-бензилокситетрадеканоиламино]-10-(2-тетрагидропиранил)окси-декан-1-ол
К раствору(2R)-5-Амино-2-[(R)-3-бензилокситетрадеканоиламино]-1-(2-тетрагидропиранилокси)пентана (пример 2.33.2) (638 мг, 1,23 ммоль, 1,3 экв) в безводном СН2Cl2 (1,5 мл) при 20-21°С добавляют (S)-(α)-[(R)-3-додеканоилокситетрадеканоиламино]-γ-бутиролактон (пример 2.33.3) (483 мг, 0,95 ммоль). Раствор перемешивают 3 дня при 20-21°С, после чего растворитель упаривают при пониженном давлении. Очистка флэш-хроматографией на силикагеле (элюент СН2С12/ацетон 5/1 и затем 1/1) позволяет выделить спирт (829 мг, 85%) в виде белого твердого вещества. (Rf =0,37 в СН2С12/ацетон 2/1; проявители УФ и фосфомолибденовая кислота). С61Н109N3O9. IS/MS: m/z 1029.0 ([M+H]+), 1051.0 ([M+Na]+). Т.пл. 81-82°С.
2.33.5. (3S,9R)-3-[(R)-3-Додеканоилокситетрадеканоиламино]-4-оксо-5-аза-9-[(R)-3-бензилокситетрадеканоиламино]декан-1,10-диол 1-(дибензилфосфат) 10-(3-бензилоксикарбониламинопропаноат). Та же последовательность реакций, которая была использована в примерах 2.7.4 и 2.7.5, будучи примененной к полученному выше продукту: (3S,9R)-3-[(R)-3-додеканоилокситетрадеканоиламино]-4-оксо-5-аза-9-[(R)-3-бензилокситетрадеканоиламино]-10-(2-тетрагидропиранил)оксидекан-1-олу, приводит к (3S,9R)-3-[(R)-3-додеканоилокситетрадеканоиламино] -4-оксо-5-аза-9[(R)-3-бензилокситетрадеканоиламино]декан-1,10-диолу 1-(дибензилфосфату).
К раствору этого соединения (500 мг, 0,41 ммоль) и коммерческого N-CBz-β-аланина (278 мг, 1,24 ммоль) в сухом СН2Cl2 (20 мл) при 0°С под аргоном последовательно добавляют коммерческий гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида (239 мг, 1,25 ммоль) и 4-диметиламинопиридин (15 мг, 125 μмоль). После этого реакционную смесь перемешивают 30 мин при 0°С и затем в течение ночи при комнатной температуре, промывают водой, затем 1 н. раствором HCl и разделяют фазы. Органическую фазу высушивают над MgSO4, фильтруют и упаривают. Очистка флэш-хроматографией на силикагеле (элюент СН2Cl2/ацетон 5/1 и затем 1/2) позволяет выделить продукт сочетания (562 мг; 96%). (Rf =0,68 в СН2Cl2/ацетон 2/1; проявители УФ и фосфомолибденовая кислота). C84H125N4014P. IS/MS: m/z 1410.0 ([М+Н]+), 1432.0 ([M+Na]+).
2.33.6. (3S,9R)-3-[(R)-3-Додеканоилокситетрадеканоиламино]-4-оксо-5-аза-9-[(R)-3-гидрокситетрадеканоиламино]декан-1,10-диол 1-дигидрофосфат 10-(3-аминопропаноат).
Раствор полученного выше соединения (540 мг, 0,38 ммоль) в смеси СН2Cl2 (54 мл) и этанола (20 мл), содержащей уксусную кислоту (10 мл), гидрируют в течение 36 час в присутствии 10% Pd на угле при комнатной температуре и атмосферном давлении водорода. Катализатор удаляют фильтрацией, фильтрат досуха упаривают и остаток сушат с помощью крыльчатого насоса, получая (33,9R)-3-[(R)-3-додеканоилокситетрадеканоиламино]-4-оксо-5-аза-9-[(R)-3-гидрокситетрадеканоиламино]декан-1,10-диол 1-дигидрофосфат 10-(3-аминопропаноат) (358 мг, 93%). C52H101N4012P. ES/MS: m/z 1005.6 ([М+Н]+) (фиг.99).
ПРИМЕР 2.34
(3S,9R)-3-[(R)-3-Додеканоилокситетрадеканоиламино]-4-оксо-5-аза-9-[(R)-3-гидрокситетрадеканоиламино]декан-1,10-диол 1-дигидрофосфат 10-(12-аминододеканоат) (= OM-197-MP-AD) (фиг.100)
2.34.1. (3S,9R)-3-[(R)-3-Додеканоилокситетрадеканоиламино]-4-оксо-5-аза-9-[(R)-3-бензилокситетрадеканоиламино]декан-1,10-диол 1-дибензилфосфат 10-(12-бензилоксикарбониламинододеканоат)
К раствору (3S,9R)-3-[(R)-3-додеканоилокситетрадеканоиламино]-4-оксо-5-аза-9-[(R)-3-бензилокситетрадеканоиламино]-декан-1,10-диола 1-дибензилфосфата (пример 2.33.5) (659 мг, 0,55 ммоль) и коммерческой 12-(бензилоксикарбониламино)додекановой кислоты (287 мг, 0,82 ммоль) в сухом СН2Cl2 (25 мл) при 0°С под аргоном последовательно добавляют гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида (157 мг, 0,82 ммоль) и 4-диметиламинопиридин (10 мг, 82 μмоль). После этого реакционную смесь перемешивают 30 мин при 0°С и затем в течение ночи при комнатной температуре, промывают водой, 1 н. раствором HCl и разделяют фазы. Органическую фазу высушивают над MgSO4, фильтруют и упаривают. Очистка флэш-хроматографией на силикагеле (элюент СН2Cl2/ацетон 4/1 и затем 2/1) позволяет выделить продукт сочетания (840 мг; 89%). (Rf =0,70 в СН2Cl2/ацетон 3/1; проявители УФ и фосфомолибденовая кислота). 13C-ЯМР (62,89 МГц, CDCl3), δ в м.д.: 173,35, 171,18, 170,49, 169,40, 156,31, 138,28, 136,61, 135,53, 135,43, 135,36, 128,54, 128,41, 128,33, 127,97, 127,88, 127,57, 76,49, 71,25, 71,01, 69,74, 69,63, 69,52, 66,46, 65,64, 64,67, 64,58, 53,36, 49,85, 47,89, 41,62, 41,52, 41,03, 39,15, 34,41, 34,33, 34,00, 33,91, 33,57, 33,49, 31,82, 30,84, 29,87, 29,56, 29,42, 29,27, 29,17, 29,09, 29,04, 28,59, 26,64, 25,39, 25,15, 25,09, 24,94, 24,72, 22,61, 14,05.
2.34.2. (3S,9R)-3-[(R)-3-Додеканоилокситетрадеканоиламино]-4-оксо-5-аза-9-[(R)-3-гидрокситетрадеканоиламино]декан-1,10-диол 1-дигидрофосфат 10-(12-аминододеканоат) Раствор полученного выше соединения (607 мг, 0,40 ммоль) в смеси СН2Cl2/этанол 5/2 (70 мл), содержащей уксусную кислоту (12 мл), гидрируют в течение 12-24 час в присутствии 10% Pd на угле при комнатной температуре и атмосферном давлении водорода. Катализатор удаляют фильтрацией, фильтрат досуха упаривают и остаток сушат с помощью крыльчатого насоса, получая (3S, 9R)-3-[(R)-3-додеканоилокситетрадеканоиламино]-4-оксо-5-аза-9-[(R)-3-гидрокситетрадеканоиламино]декан-1,10- диол 1-дигидрофосфат 10-(12-аминододеканоат) (429 мг, 96%). C61H119N4O12P. ES/MS: m/z 1131.8 ([M+H]+), 1153.8 ([M+Na]+) (фиг.101).
ПРИМЕР 2.35.
α-N{(4R)-5-гидрокси-4-[(R)-3-гидрокситетрадеканоиламино]-пентил}амид N-[(R)-3-додеканоилокситетрадеканоил]-L-аспарагиновой кислоты β-N-(6-аминогексил)амид (=ОМ-197-АН ОН) (фиг.91)
2.35.1. α-N{(4R)-5-гидрокси-4[(R)-3-гидрокситетрадеканоиламино]пентил}амид N-[(R)-3-додеканоилокситетрадеканоид]-L-аспарагиновой кислоты β-N-(6-аминогексил)амид
Раствор α-N{(4R)-5-гидрокси-4[(R)-3-бензилокситетрадеканоил-амино]пентил}амида N-[(R)-3-додеканоилокситетрадеканоил]-L-аспарагиновой кислоты β-N-(6-бензилоксикарбониламиногексил)амида (пример 2.29.2) (80 мг, 0,67 ммоль) в смеси СН2Cl2/этанол 1/1 (10 мл), содержащей уксусную кислоту (1 мл), гидрируют в течение 18 час в присутствии 10% Pd на угле при комнатной температуре и атмосферном давлении водорода. Катализатор удаляют фильтрацией, фильтрат досуха упаривают, остаток упаривают совместно с толуолом и сушат с помощью крыльчатого насоса, получая α-N{(4R)-5-гидрокси-4-[(R)-3-гидрокситетрадеканоиламино]пентил}амид N-[(R)-3-додеканоилокситетрадеканоил]-L-аспарагиновой кислоты β-N-(6-аминогексил)амид (65 мг, 100%). C55H107N5O8. ES/MS: m/z 967.0 ([M+H]+), 989.0 ([М+Na]+) (фиг.92).
ПРИМЕР 2.36.
(3S,9R)-3-[(R)-3-Гидрокситетрадеканоиламино]-4-оксо-5-аза-9-[(R)-3-додеканоилокситетрадеканоиламино]декан-1,10-диол 1-дигидрофосфат 10-(6-аминогексаноат) (=ОМ-197МР AC-inv) (фиг.102)
2.36.1.(2R)-5-(Бензилоксикарбониламино)-2-[(R)-3-додеканоилокситетрадеканоиламино]-1-(2-тетрагидропиранилокси)-пентан
К раствору (2R)-5-(бензилоксикарбониламино)-2-[(R)-3-додеканоилокситетрадеканоиламино]пентан-1-ола (пример 2.31.2) (4,54 г, 6,87 ммоль) в безводном CH2CI2 (130 мл) при комнатной температуре под аргоном последовательно добавляют 3,4-дигидро-2Н-пиран (DHP) (2,19 мл, 0,024 моль) и затем п-толуолсульфонат пиридиния (PPTS) (961 мг, 2,75 ммоль). Раствор перемешивают 16 час при комнатной температуре, разбавляют СН2Cl2 (100 мл), промывают 5%-ным раствором NaHCO3 и затем водой. Органическую фазу высушивают над MgSO4, фильтруют и упаривают. Очистка флэш-хроматографией на силикагеле (элюент СН2Cl2/ацетон 9/1) позволяет выделить (2R)-5(Бензилоксикарбониламино)-2[(R)-3-додеканоилокситетрадеканоиламино]-1-(2-тетрагидропиранилокси)пентан (3,88 г; 76%) в виде аморфного твердого вещества (Rf =0,74 в СН2С12/ацетон 5/1; проявители УФ и фосфомолибденовая кислота). С44Н76N207. IS/MS: m/z 745.5 ([M+H]+), 768.0 ([M+Na]+), 783.5 ([М+К]+).
2.36.2.(2R)-5-Амино-2-[(R)-3-додеканоилокситетрадеканоиламино]-1 -(2-тетрагидропиранилокси)пентан
Раствор (2R)-5-(бензилоксикарбониламино)-2-[(R)-3-додеканоил- окситетрадеканоиламино]-1-(2-тетрагидропиранилокси)пентана (3,79 г, 5,21 ммоль) в этаноле (150 мл), содержащем триэтиламин (4,6 мл), гидрируют в течение 3 час в присутствии 10% Pd на угле при комнатной температуре и атмосферном давлении водорода. Катализатор удаляют фильтрацией и промывают этанолом. Фильтрат досуха упаривают и остаток сушат с помощью крыльчатого насоса, получая свободный амин (3,2 г; 100%) в виде бледно-желтого твердого вещества. С36Н70N2O5. IS/MS: m/z 611.5 ([М+Н]+), 633.5 ([M+Na]+).
2.36.3. (S)-(α)-[(R)-3-Бензилокситетрадеканоиламино]-γ-бутиролактон
К раствору (R)-3-бензилокситетрадекановой кислоты (1,5 г; 4,48 ммоль) [Bull. Chem. Soc. Jpn. 60 (1987) 2197-2204] в безводном ТГФ (25 мл) при -15°С под аргоном последовательно добавляют N-метилморфолин (497 μл; 4,48 ммоль; 1 экв) и изобутилхлорформиат (585 μл; 4,48 ммоль; 1 экв). После перемешивания в течение 1 час 15 мин при -15°С добавляют коммерческий гидробромид лактона L-гомосерина (815 мг, 4,48 ммоль) и затем раствор NaHCO3 (757 мг) в H2O (12 мл). После этого реакционную смесь перемешивают 20 час при комнатной температуре, разбавляют Et2O (50 мл), отделяют органическую фазу, промывают ее Н2O, высушивают над MgSO4, фильтруют и упаривают при пониженном давлении. Очистка кристаллизацией (минимум СН2Cl2 и избыток пентана при 0°С) позволяет выделить продукт сочетания (1,59 г; 85%) в виде белого твердого вещества. (Rf = 0,80 в смеси СН2Cl2/ацетон 2/1; проявитель фосфомолибденовая кислота). C25H39NO4. IS/MS: m/z 418.5 ([М+Н]+), 440.5 ([M+Na]+).
2.36.4. (3S,9R)-3-[(R)-3-Бензилокситетрадеканоиламино]-4-оксо-5-аза-9-[(R)-3-додеканоилокситетрадеканоиламино]-10-(2-тетрагидропиранил)оксидекан-1-ол
К раствору (2R)-5-амино-2-[(R)-3-додеканоилокситетрадеканоил-амино]-1-(2-тетрагидропиранилокси)пентана (пример 2.36.2) (800 мг, 1,3 ммоль, 1,3 экв) в безводном СН2С12 (2 мл) добавляют при 25°С (S)-(α)-[(R)-3-бензилокситетрадеканоиламино]-y-бутиролактон (пример 2.36.3) (421 мг, 1 ммоль). Раствор перемешивают 12 часа при 25°С и добавляют триэтиламин (140 ил, 1 ммоль, 1 экв). Реакционную смесь перемешивают при этой температуре в течение 10 дней и упаривают растворитель при пониженном давлении. Очистка флэш-хроматографией на силикагеле (элюент СН2С12/ацетон 4/3 и затем ацетон) позволяет выделить спирт (587 мг, 57%) в виде аморфного вещества. (Rf =0,52 в СН2Cl2/ацетон 2/1; проявители УФ и фосфомолибденовая кислота). C61H109N309. IS/MS: m/z 1029.0 ([М+Н]+), 1151.0 ([M+Na]+ ).
2.36.5. (3S,9R)-3-[(R)-3-Бензилокситетрадеканоиламино] -4-оксо-5-аза-9-[(R)-3-додеканоилокситетрадеканоиламино]-10-(2-тетрагидропиранил)оксидекан-1-ол 1-дибензилфосфат
К раствору полученного выше спирта (585 мг; 0,57 ммоль) и 1Н-тетразола (120 мг; 1,71 ммоль; 3 экв) в безводном ТГФ (23 мл) добавляют при комнатной температуре под аргоном 85%-ный дибензилдиэтилфосфорамидит (392 μл; 1,31 ммоль; 2,3 экв) - в реакционной среде наблюдается быстрое образование белых кристаллов. После 45 мин перемешивания при комнатной температуре добавляют раствор mCPBA (57-86%; 360 мг, 2,10 ммоль; 3,7 экв) в СН2Cl2 (15 мл) - наблюдается исчезновение кристаллов. После 45 мин перемешивания при комнатной температуре добавляют насыщенный раствор Na2S2O3 (20 мл), после чего реакционную смесь перемешивают 10 мин. Раствор разбавляют эфиром, отделяют органическую фазу, промывают ее 5 раз насыщенным раствором Na2S2O3 и 2 раза насыщенным раствором NaHCO3, высушивают над MgSO4, фильтруют и упаривают. Очистка флэш-хроматографией на силикагеле (элюент СН2Cl2/ацетон 6/1, 5/1 и затем 1/1) позволяет выделить дибензилфосфат (644 мг; 88%) в виде аморфного вещества (Rf = 0,25 в СН2Cl2/ацетон 1/1; проявители УФ и фосфомолибденовая кислота). C75H122N3O12P, IS/MS: m/z 1289.0 ([М+Н]+), 1312.0 ([M+Na]+).
2.36.6. (3S,9R)-3-[(R)-3-Бензилокситетрадеканоиламино]-4-оксо-5-аза-9-[(R)-3-додеканоидокситетрадеканоиламино]-декан-1,10-диол 1-дибензилфосфат
К 1%-ному раствору HCl в метаноле (23 мл) при 0°С добавляют раствор полученного выше соединения (634 мг, 0,49 ммоль) в СН2Cl2 (2 мл). После перемешивания в течение 1 часа при 0°С реакционную среду нейтрализуют 10%-ным раствором NaHCO3, разбавляют СН2Cl2 и отделяют органическую фазу. Оставшуюся водную фазу трижды экстрагируют СН2Cl2. Органические фазы объединяют, сушат над MgSO4, фильтруют и упаривают, получая сырой спирт (579 мг; 98%) в виде аморфного вещества. (Rf =0,29 в СН2Cl2/ацетон 3/1; проявители УФ и фосфомолибденовая кислота). C70H114N3O11P, IS/MS: m/z 1204.5 ([М+Н]+).
2.36.7. (3S,9R)-3-[(R)-3-Бензилокситетрадеканоиламино]-4-оксо-5-аза-9-[(R)-3-додеканоилокситетрадеканоиламино]-декан-1,10-диол 1-дибензилфосфат 10-(6-бензилоксикарбониламиногексаноат)
К раствору полученного выше соединения (542 мг, 0,45 ммоль) и коммерческой 6-(бензилоксикарбониламино)гексановой кислоты (238 мг, 0,90 ммоль) в сухом СН2Cl2 (15 мл) при 0°С под аргоном последовательно добавляют коммерческий гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида (173 мг, 0,90 ммоль) и 4-диметиламинопиридин (11 мг, 90 рмоль). После этого реакционную смесь перемешивают 30 мин при 0°С и затем в течение ночи при комнатной температуре, промывают водой, затем 1 н. раствором HCl и разделяют фазы. Органическую фазу высушивают над MgSO4, фильтруют и упаривают. Очистка флэш-хроматографией на силикагеле (элюент петролейный эфир/EtOAc 1/2) позволяет выделить продукт сочетания (537 мг; 82%) в виде аморфного вещества. (Rf =0,69 в СН2Cl2/ацетон 3/1; проявители УФ и фосфомолибденовая кислота). C84H131N4O14P. IS/MS: m/z 1451.8 ([M+H]+), 1475.0 ([M+Na]+), 1174.0 ([М-(BnO)2OPOH]+).
2.36.8. (3S,9R)-3-[(R)-3-Гидрокситетрадеканоиламино]-4-оксо-5-аза-9-[(R)-3-додеканоилокситетрадеканоиламино]декан-1,10-диол 1-дигидрофосфат 10-(6-аминогексаноат)
Раствор полученного выше соединения (500 мг, 0,34 ммоль) в смеси СН2Cl2/этанол 5/2 (70 мл), содержащей уксусную кислоту (10 мл), гидрируют в течение 36 час в присутствии 10% Pd на угле при комнатной температуре и атмосферном давлении водорода. Катализатор удаляют фильтрацией, фильтрат досуха упаривают и остаток сушат с помощью крыльчатого насоса, получая (3S,9R)-3-[(R)-3-гидрокситетрадеканоиламино]-4-оксо-5-аза-9-[(R)-3-додеканоилокситетрадеканоиламино]декан-1,10-диол 1-дигидрофосфат 10-(6-аминогексаноат) (301 мг, 84%) в виде аморфного вещества. C55H106N4O12P. ES/MS: m/z 1047.8 ([М+Н]+), 1069.8 ([M+Na]+) (фиг.103).
ПРИМЕР 2.37.
(3S,9R)-3-[(R)-3-Додеканоилокситетрадеканоиламино]-4-оксо-5-аза-9-[(R)-3-додеканоилокситетрадеканоиламино]декан-1,10-диол 1-дигидрофосфат 10-(6-аминогексаноат) (=ОМ-197-МР AC-tétracyle) (фиг.104)
2.37.1. (3S,9R)-3-[(R)-3-Додеканоидокситетрадеканоиламино]-4-оксо-5-аза-9-[(R)-3-додеканоилокситетрадеканоиламино]-10-(2-тетрагидропиранил)окси-декан-1-ол
К раствору (S)-(α)-[(R)-3-додеканоилокситетрадеканоиламино]-γ-бутиролактона (пример 2.33.3) (514 мг, 1,00 ммоль) в безводном СН2Cl2 (2 мл) добавляют при 25°С (2R)-5-амино-2-[(R)-3-додеканоилокситетрадеканоиламино]-1-(2-тетрагидропиранилокси)пентан (пример 2.36.2) (800 мг, 1,3 ммоль, 1,3 экв). Раствор перемешивают в течение 6 дней при 25°С (не обнаруживается никакого протекания реакции), после чего добавляют изопропиловый спирт (2 мл) и триэтиламин (140 μл, 1 ммоль, 1 экв). Раствор перемешивают еще 10 дней при 25°С и упаривают при пониженном давлении растворитель. Очистка флэш-хроматографией на силикагеле (элюент СН2С12/ацетон 3,5/1, затем 1/1) позволяет выделить спирт (394 мг, 35%) в виде аморфного вещества. (Rf = 0,34 в СН2Cl2/ацетон 3/1; проявители УФ и фосфомолибденовая кислота). С66Н125N3О10. IS/MS: m/z 1121.0 ([M+H]+).
2.37.2. (3S,9R)-3-[(R)-3-Додеканоилокситетрадеканоиламино]-4-оксо-5-аза-9-[(R)-3-додеканоилокситетрадеканоиламино]-10-(2-тетрагидропиранил)окси-декан-1-ол 1-дибензилфосфат
К раствору полученного выше спирта (393 мг; 0,35 ммоль) и 1Н-тетразола (74 мг; 1,05 ммоль; 3 экв) в безводном ТГФ (23 мл) добавляют при комнатной температуре под аргоном 85%-ный дибензилдиэтилфосфорамидит (242 μл; 0,807 ммоль; 2,3 экв) - в реакционной среде наблюдается быстрое образование белых кристаллов. После перемешивания в течение 1 часа 45 мин при комнатной температуре добавляют раствор mCPBA (57-86%; 221 мг, 1,3 ммоль; 3,7 экв) в СН2Cl2 (10 мл) - наблюдается исчезновение кристаллов. После 1 часа перемешивания при комнатной температуре добавляют насыщенный раствор Na2S2O3 (20 мл), после чего реакционную смесь перемешивают 10 мин. Раствор разбавляют эфиром, отделяют органическую фазу, промывают ее 5 раз насыщенным раствором Na2S2O3 и 2 раза насыщенным раствором NaHCO3, высушивают над MgSO4, фильтруют и упаривают. Очистка флэш-хроматографией на силикагеле (элюент СН2Cl2/ацетон 6/1, 5/1 и затем 1/1) позволяет выделить дибензилфосфат (450 мг; 92%) в виде аморфного вещества (Rf=0,28 в СН2Cl2/ацетон 4/1; проявители УФ и фосфомолибденовая кислота). C80H138N3O13P/ IS/MS: m/z 1381.0 ([М+Н]+), 1403.0 ([M+Na]+).
2.37.3. (3S,9R)-3-[(R)-3-Додеканоилокситетрадеканоиламино]-4-оксо-5-аза-9-[(R)-3-додеканоилокситетрадеканоиламино]декан-1,10-диол 1-дибензилфосфат
К 1% раствору HCl в метаноле (14 мл) при 0°С добавляют раствор полученного выше соединения (429 мг, 0,31 ммоль) в СН2Cl2 (15 мл). После 2 час перемешивания при комнатной температуре реакционную среду нейтрализуют 10%-ным раствором NaHCO3, разбавляют СН2Cl2 и отделяют органическую фазу. Оставшуюся водную фазу трижды экстрагируют СН2Cl2. Органические фазы объединяют, сушат над MgSO4, фильтруют и упаривают. Очистка флэш-хроматографией на силикагеле (элюент СН2Cl2/ацетон 4/1, 2/1 и затем 1/1) позволяет выделить дибензилфосфат (186 мг; 46%) в виде аморфного вещества. (Rf=0,39 в СН2Cl2/ацетон 3/1; проявители УФ и фосфомолибденовая кислота). С75Н130N3O12Р, IS/MS: m/z 1297.0 ([M+H]+), 1319.0 ([M+Na]+), 1019.0 ([М-(BnO)2ОРОН]+).
2.37.4. (3S,9R)-3-[(R)-3-Додеканоилокситетрадеканоиламино]-4-оксо-5-аза-9-[(R)-3-додеканоилокситетрадеканоиламино]декан-1,10-диол 1-дибензилфосфат 10-(6-бензилоксикарбониламиногексаноат)
К раствору полученного выше соединения (154 мг, 0,12 ммоль) и коммерческой 6-(бензилоксикарбониламино)гексановой кислоты (63 мг, 0,24 ммоль, 2 экв) в сухом СН2Cl2 (5 мл) при 0°С под аргоном последовательно добавляют коммерческий гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида (46 мг, 0,24 ммоль, 2 экв) и 4-диметиламинопиридин (3 мг, 0,02 ммоль, 0,2 экв). После этого реакционную смесь перемешивают 30 мин при 0°С и затем в течение ночи при комнатной температуре, разбавляют СН2Cl2, промывают водой, затем 1 н. раствором HCl и разделяют фазы. Органическую фазу высушивают над MgSO4, фильтруют и упаривают. Очистка флэш-хроматографией на силикагеле (элюент СН2Cl2/ацетон 3/1) позволяет выделить продукт сочетания (149 мг; 81%) в виде аморфного вещества. (Rf=0,75 в СН2Cl2/ацетон 3/1; проявители УФ и фосфомолибденовая кислота). C89H147N4O15P. IS/MS: m/z 1545.5 ([М+Н]+).
2.37.5. (3S,9R)-3-[(R)-3-Додеканоилокситетрадеканоиламино]-4-оксо-5-аза-9-[(R)-3-додеканоилокситетрадеканоиламино]декан-1,10-диол 1-дигидрофосфат 10-(6-аминогексаноат)
Раствор полученного выше соединения (140 мг, 0,091 ммоль) в смеси СН2Cl2/этанол 3/1 (20 мл) гидрируют в течение 24 час в присутствии 10% Pd на угле при комнатной температуре и атмосферном давлении водорода. Катализатор удаляют фильтрацией, фильтрат досуха упаривают и остаток сушат с помощью крыльчатого насоса, получая (3S,9R)-3-[(R)-3-додеканоилокситетрадеканоиламино]-4-оксо-5-аза-9-[(R)-3-додеканоилокситетрадеканоиламино]декан-1,10-диол 1-дигидрофосфат 10-(6-аминогексаноат) (92 мг, 83%). C67H129N4O13P. ES/MS: m/z 1229.9 [М+Н]+) (фиг.105).
ПРИМЕР 2.38.
(3S,9R)-3-Октадеканоиламино-4-оксо-5-аза-9-[(R)-3-додеканоилокситетрадеканоиламино]декан-1,10-диол 1-дигидрофосфат 10-(6-аминогексаноат) (= ОМ-197-МР AC-inv-Cis) (фиг.106)
2.38.1. (S)-(α)-Октадеканоиламино-γ-бутиролактон
К раствору коммерческой октадекановой кислоты (1,56 г; 5,49 ммоль) в безводном ТГФ (30 мл) при -15°С под аргоном последовательно добавляют N-метилморфолин (604 μл, 5,49 ммоль; 1 экв) и изобутилхлорформиат (713 ил; 5,49 ммоль; 1 экв). После перемешивания в течение 1 часа 15 мин при -10°С добавляют коммерческий гидробромид лактона L-гомосерина (1 г; 5,49 ммоль) и затем раствор NaHCO3 (923 мг, 11 ммоль, 2 экв) в H2O (14 мл). После этого реакционную смесь перемешивают 16 час при комнатной температуре, разбавляют Et2O (50 мл), отделяют органическую фазу, промывают ее водой, высушивают над MgSO4, фильтруют и упаривают при пониженном давлении. Очистка кристаллизацией (минимум СН2Cl2 и избыток пентана при 0°С) позволяет выделить продукт сочетания (1,38 г; 68%) в виде белого твердого вещества. (Rf =0,31 в СН2Cl2/ацетон 2/1; проявитель фосфомолибденовая кислота). C22H41NO3. IS/MS: m/z 368.5 ([М+Н]+), 390.5 ([M+Na]+).
2.38.2. (3S,9R)-3-Октадеканоиламино-4-оксо-5-аза-9-[(R) -3-додеканоилокситетрадеканоиламино]-10-(2-тетрагидропиранил)-окси-декан-1-ол
К раствору полученного выше (S)-(α)-октадеканоиламино-γ-бутиролактона (370 мг, 1 ммоль) в безводном СН2Cl2 (2 мл) добавляют при 25°С (2R)-5-амино-2-[(R)-3-додеканоилокситетра-деканоиламино]-1-(2-тетрагидропиранилокси)пентан (пример 2.36.2) (800 мг, 1,3 ммоль, 1,3 экв). Раствор перемешивают 72 часа при 25°С (не обнаруживается никакого протекания реакции), после чего добавляют триэтиламин (140 μл, 1 ммоль, 1 экв). Раствор перемешивают еще 40 дней при 25°С и упаривают при пониженном давлении растворитель. Очистка флэш-хроматографией на силикагеле (элюент СН2Cl2/ацетон от 4/3 до 1/1) позволяет выделить спирт (306 мг, 31%) в виде белого твердого вещества. (Rf = 0,17 в СН2Cl2/ацетон 3/1; проявители УФ и фосфомолибденовая кислота). С58Н111N3O8. IS/MS: m/z 979.0 ([М+H]+), 1001.0 ([M+Na]+).
2.38.3. (3S,9R)-3-Октадеканоиламино-4-оксо-5-аза-9-[(R)-3-додеканоилокситетрадеканоиламино]-10-(2-тетрагидропиранил)-окси-декан-1-ол 1-дибензидфосфат
К раствору полученного выше спирта (296 мг; 0,303 ммоль) и 1H-тетразола (296 мг; 0,303 ммоль; 3 экв) в безводном ТГФ (12 мл) при комнатной температуре под аргоном добавляют 85%-ный дибензилдиэтилфосфорамидит (209 μл; 0,7 ммоль; 2,3 экв) - в реакционной среде наблюдается быстрое образование белых кристаллов. После 45 мин перемешивания при комнатной температуре добавляют раствор mCPBA (57-86%; 191 мг; 1,12 ммоль; 3,7 экв) в СН2Cl2 (17 мл) - происходит исчезновение кристаллов. После 45 мин перемешивания при комнатной температуре добавляют насыщенный раствор Na2S2O3 (20 мл) и перемешивают реакционную смесь в течение 10 мин. Раствор разбавляют после этого эфиром, отделяют органическую фазу, промывают ее 5 раз насыщенным раствором Na2S2O3 и 2 раза насыщенным раствором NaHCO3, высушивают над MgSO4, фильтруют и упаривают. Очистка флэш-хроматографией на силикагеле (элюент СН2Cl2/ацетон 3/1) позволяет выделить дибензилфосфат (330 мг; 89%) в виде аморфного вещества (Rf =0,40 в СН2Cl2/ацетон 2/1; проявители УФ и фосфомолибденовая кислота). С72Н124N3О11Р. IS/MS: m/z 1239.0 ([М+Н]+), 1261.0 ([M+Na]+).
2.38.4. (3S,9R)-3-Октадеканоиламино-4-оксо-5-аза-9[(R)-3-додеканоилокситетрадеканоиламино]декан-1,10-диол 1-дибензилфосфат
К 1%-ному раствору НС1 в метаноле (11 мл) при 0°С добавляют раствор полученного выше соединения (320 мг, 0,258 ммоль) в СН2Cl2 (2 мл). После перемешивания в течение 1 часа при 0°С реакционную среду нейтрализуют 10%-ным раствором NaHCO3, разбавляют СН2Cl2 и отделяют органическую фазу. Оставшуюся водную фазу трижды экстрагируют СН2Cl2. Органические фазы объединяют, сушат над MgSO4, фильтруют и упаривают, получая спирт (254 мг; 85%) в виде аморфного вещества. (Rf =0,20 в СН2Cl2/ацетон 3/1; проявители УФ и фосфомолибденовая кислота).
2.38.5. (3S,9R)-3-Октадеканоиламино-4-оксо-5-аза-9-[(R)-3-додеканоилокситетрадеканоиламино]декан-1,10-диол 1-дибензилфосфат 10-(6-бензилоксикарбониламиногексаноат)
К раствору полученного выше соединения (243 мг, 0,211 ммоль) и коммерческой 6-(бензилоксикарбониламино)гексановой кислоты (101 мг, 0,379 ммоль, 1,8 экв) в сухом СН2Cl2 (9,5 мл) при 0°С под аргоном последовательно добавляют коммерческий гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида (73 мг, 0,379 ммоль, 1,8 экв) и 4-диметиламинопиридин (1 мг, 0,038 ммоль, 0,18 экв). После этого реакционную смесь перемешивают 30 мин при 0°С и затем в течение ночи при комнатной температуре, разбавляют СН2Cl2, промывают водой, затем 1 н. раствором HCl и разделяют фазы. Органическую фазу высушивают над MgSO4, фильтруют и упаривают. Очистка флэш-хроматографией на силикагеле (элюент EtOAc/петролейный эфир 2/1) позволяет выделить продукт сочетания (226 мг; 77%) в виде аморфного вещества. (Rf =0,53 в СН2Cl2/ацетон 3/1; проявители УФ и фосфомолибденовая кислота). C81H133N4O13P. IS/MS: m/z 1402.8 ([М+Н]+), 1425.0 ([M+Na]+).
2.38.6. (3S,9R)-3-Октадеканоиламино-4-оксо-5-аза-9-[(R)-3-додеканоилокситетрадеканоиламино]декан-1,10-диол 1-дигидрофосфат 10-(6-аминогексаноат)
Раствор полученного выше соединения (220 мг, 0,35 ммоль) в смеси СН2Cl2 (20 мл), этанол (9 мл), содержащей уксусную кислоту (10 мл), гидрируют в течение 24 час в присутствии 10% Pd на угле при комнатной температуре и атмосферном давлении водорода. Катализатор удаляют фильтрацией, фильтрат досуха упаривают и остаток сушат с помощью крыльчатого насоса, получая (3S,9R)-3-октадеканоиламино-4-оксо-5-аза-9-[(R)-3-додеканоилокситетрадеканоиламино]декан-1,10-диол 1-дигидрофосфат 10-(6-аминогексаноат) (134 мг; 81%). С59Н115N4O11Р. ES/MS: m/z 1087.7 ([М+Н]+) (фиг.107).
ПРИМЕР 2.39.
(3S,9R)-3-[(R)-3-Додеканоилокситетрадеканоиламино]-4-оксо-5-аза-9-[(R)-3-гидрокситетрадеканоиламино]декан-1,10-диол 1-дигидрофосфат 10-сукцинат (= OM-197-MP-Succ) (фиг.108)
2.39.1. (3S,9R)-3-[(R)-3-Додеканоилокситетрадеканоиламино]-4-оксо-5-аза-9-[(R)3-безилокситетрадеканоиламино]декан-1,10-диол 1-дибензилфосфат 10-сукцинат
Та же последовательность реакций, которая была использована в примерах 2.7.4 и 2.7.5, будучи примененной в отношении (3S,9R)-3-[(R)-3-додеканоилокситетрадеканоиламино]-4-оксо-5-аза-9-[(R)-3-бензилокситетрадеканоиламино]-10-(2-тетрагидропиранил)окси-декан-1-ола (пример 2.33.4), приводит к (3S,9R)-3-[(R)-3-додеканоилокситетрадеканоиламино]-4-оксо-5-аза-9-[(R)3-безилокситетрадеканоиламино]декан-1,10-диолу 1-дибензилфосфату.
К раствору (3S,9R)-3-[(R)-3-додеканоилокситетрадеканоиламино]-4-оксо-5-аза-9-[(R)3-безилокситетрадеканоиламино]декан-1,10-диола 1-дибензилфосфата (176 мг, 0,15 ммоль) в сухом СН2Cl2 (5 мл) в присутствии Et3N (41 μл, 0,29 ммоль) добавляют при комнатной температуре янтарный ангидрид (22 мг, 0,22 ммоль). Реакционную среду перемешивают в течение ночи при комнатной температуре, разбавляют СН2Cl2, промывают 1 н. раствором HCl и разделяют фазы. Органическую фазу высушивают над MgSO4, фильтруют и упаривают. Очистка флэш-хроматографией на силикагеле (элюент СН2Cl2/ацетон 4/1 с 1% уксусной кислоты и затем СН2С12/ацетон 2/1 с 1% уксусной кислоты) позволяет выделить кислоту (186 мг, 97%) в виде белого твердого вещества. (Rf =0,65 в СН2С12/ацетон 2/1 с 1% уксусной кислоты/проявители УФ и фосфомолибденовая кислота). C74H118N3O14P. ES/MS: m/z 1327.0 ([М+Н]+), 1027 [М+(BnO)2P(О)ОН+Н]+.
2.39.2. (3S,9R)-3-[(R)-3-Додеканоилокситетрадеканоиламино]-4-оксо-5-аза-9-[(R)-3-гидрокситетрадеканоиламино]декан-1,10-диол 1-дигидрофосфат 10-сукцинат
Раствор полученного выше соединения (54 мг; 0,041 ммоль) в этаноле (6 мл) гидрируют в течение 4 час 30 мин в присутствии 10% Pd на угле при комнатной температуре и атмосферном давлении водорода. Катализатор удаляют фильтрацией, фильтрат досуха упаривают и остаток сушат с помощью крыльчатого насоса, получая (3S,9R)-3-[(R)-3-додеканоилокситетрадеканоиламино]-4-оксо-5-аза-9-[(R)-3-гидрокситетрадеканоиламино]декан-1,10-диол 1-дигидрофосфат 10-сукцинат (33 мг, 77%) в виде белого твердого вещества. С53Н100N3О14Р. ES/MS: m/z 1034.5 ([М+Н]+) (фиг.109).
ПРИМЕР 2.40.
О-{(2R)-2-[(R)-3-гидрокситетрадеканоиламино]-5-(6-аминогексаноиламино)пентиловый} эфир N-[(R)-3-додеканоилокситетрадеканоил]-О-(фосфоно)-DL-гомосерина (= ОМ-197-МР-АС-ester) (фиг.110)
2.40.1.(2R)-5-[6-(Бензилоксикарбониламино]-гексаноиламино]-2-[(R)-3-бензилокситетрадеканоиламино]пентан-1-ол
К раствору (2R)-5-амино-2-[(2R)-3-бензилокситетрадеканоиламино]пентан-1-ола (пример 1.1.2g) (200 мг, 0,46 ммоль) в безводном СН2Cl2 (15 мл) под аргоном добавляют раствор IIDQ (181 мг, 0,598 ммоль, 1,3 экв) в безводном СН2Cl2 (5 мл). Реакционную смесь перемешивают 15 мин при комнатной температуре и добавляют под аргоном раствор 6-(бензилоксикарбониламино)гексановой кислоты (122 мг, 0,46 ммоль, 1 экв) в безводном СН2Cl2 (5 мл). После этого реакционную среду перемешивают 48 час при комнатной температуре и упаривают растворитель. Очистка остатка флэш-хроматографией на силикагеле (элюент СН2Cl2/ацетон 3/2) позволяет выделить продукт сочетания (216 мг; 69%).
2.40.2. О-{(2R)-2-[(R)-3-бензилокситетрадеканоиламино]-5-(6-бензилоксикарбониламино)гексаноиламино]пентиловый} эфир N-[(R)-3-додеканоилокситетрадеканоил]-О-(дифенилоксифосфорил)-DL-гомосерина
К раствору полученного выше соединения (110 мг, 0,161 ммоль) и 4-(дифенилоксифосфорилокси)-2-[(R)-3-додеканоилокситетрадеканоиламино]бутановой кислоты (пример 1.1.If) (122 мг, 0,161 ммоль) в безводном СН2Cl2 (5 ммоль) последовательно добавляют дициклогексилкарбодиимид (34 мг, 0,164 ммоль, 1,02 экв) и 4-диметиламинопиридин (3 мг, 0,024 ммоль, 0,15 экв). Реакционную смесь перемешивают затем 24 часа при комнатной температуре, разбавляют эфиром, охлаждают до 0°С, фильтруют на мелкопористом фильтре для удаления осадка дициклогексилмочевины и упаривают растворитель. Очистка флэш-хроматографией на силикагеле (элюент СН2Cl2/ацетон 3/1) позволяет выделить ожидаемый продукт сочетания (70 мг; 30%).
2.40.3. О-{(2R)-2-[(R)-3-гидрокситетрадеканоиламино]-5-(6-аминогексаноиламино)пентиловый} эфир N-[(R)-3-додеканоилокситетрадеканоил]-О-(дифенилоксифосфорил)-DL-гомосерина
Раствор полученного выше соединения (100 мг, 0,070 моль) в этаноле для ЖХВР (10 мл), содержащем уксусную кислоту (8 μл, 2 экв), гидрируют в течение 18 час в присутствии избытка 10% палладия на угле при комнатной температуре и атмосферном давлении водорода. Катализатор удаляют фильтрацией, фильтрат досуха упаривают и остаток сушат с помощью насоса, получая О-{(2R)-2-[(R)-3-гидрокситетрадеканоиламино]-5-(6-амино)гексаноиламино)пентиловый} эфир N-[(R)-3-додеканоилокситетрадеканоил]-О-(дифенилоксифосфорил)-DL-гомосерина (65 мг, 77%). C67H115N4O12P. IS/MS: m/z 1222.5 ([М-Na]+), 1200.0 ([М+Н]+).
2.40.4. О-{(2R)-2-[(R)-3-гидрокситетрадеканоиламино]-5-(6-аминогексаноиламино)пентиловый} эфир N-[(R)-3-додеканоилокситетрадеканоил]-О-(фосфоно)-DL-гомосерина
PtO2 предварительно активируют в платиновую чернь в суспензии в этаноле для ЖХВР (1 мл), действуя в течение 15 мин водородом при комнатной температуре и атмосферном давлении. Эту суспензию активированной платины переносят в раствор полученного выше соединения (60 мг) в метаноле для ЖХВР (10 мл), содержащего уксусную кислоту (10 мл). Реакционную смесь дегазируют в вакууме и затем выдерживают в течение 48 час в нормальной атмосфере водорода при комнатной температуре. Затем водород в реакционной колбе заменяют инертным газом, таким как аргон, для устранения опасности воспламенения платины, отфильтровывают катализатор на мелкопористом фильтре и упаривают растворитель, получая О-{(2R)-2-[(R)-3-гидрокситетрадеканоиламино]-5-(6-аминогексаноиламино)-пентиловый} эфир N-[(R)-3-додеканоилокситетрадеканоил]-О-(фосфоно)-DL-гомосерина (40 мг, 76%).
3-Я СЕРИЯ ПРИМЕРОВ: ПОЛУЧЕНИЕ КОНЪЮГАТОВ
ПРИМЕР 3.1
Конъюгаты с пептидом FGFG
Как показано на схемах синтезов, представленных на фиг.5, 8, 15, 17, 19, 21 и 23, реакцию вицинального гидроксилирования проводят на ацилированных псевдодипептидах, имеющих вспомогательное ответвление олефинового типа с целью образования устойчивой диоловой функции. После этого проводят реакцию перйодатного окисления с целью создания на вспомогательном ответвлении альдегидной функции. Последняя, благодаря реакции восстановительного аминирирования, помогает сочетать, например, пептид малого размера, такой как FGFG (фенилаланин-глицин-фенилаланин-глицин, Sigma). Однако для удаления присутствующих в растворе солей необходима стадия очистки альдегида. Очистку альдегида проводят на фазе Bondapack C18 (Waters), 15-20 μм, 300 Å при следующих условиях:
- адсорбция после разбавления в смеси изопропиловый спирт-вода (1:3);
- элюирование солей смесью изопропиловый спирт-вода (1:9);
- элюирование альдегида минимальным объемом изопропилового спирта.
Реакцию сочетания восстановительным аминированием проводят в водном растворе, соблюдая равную стехиометрию между альдегидной функцией (вспомогательное ответвление ацилированного псевдодипептида) и аминными функциями, содержащимися в сочетающемся пептиде или белке.
В примере пептида FGFG 2 мг очищенного OM-197-FV7 (1,86 μмоль, 1 экв) были добавлены к раствору 0,8 мг FGFG (1,86 μмоль, 1 экв), растворенного в 1 мл H2O-MeCN (1:1). После этого раствор перемешивают при комнатной температуре до образования имина и затем проводят стадию восстановления, добавляя 92 μл 1 М NaBH3CN (5,77 мг, 50 экв), в результате чего образуется особо устойчивая связь углерод-азот (фиг.41). Конъюгат очищают вначале на колонке Vydac C4 с целью удаления непрореагировавшего пептида и затем на фазе Bondapack C18 (Waters) для получения натриевой соли (см. параграф: очистка и анализ соединений изобретения). Конъюгат после этого повторно растворяют в стерильной Н2O с 0,01% триэтаноламина для соответствия условиям тестов на биологическую активность. Зарегистрированный после очистки масс-спектр ES/MS представлен на фиг.43 и свидетельствует о наличии молекулярного иона [М+Н]+ при m/z 1457.4, являющегося показателем ожидаемого сочетания. Наблюдаемые между m/z 400 и 900 ионы были идентифицированы с помощью анализов MS/MS в виде фрагментов, происходящих от молекулярного пика конъюгированного пептида. Ионы, соответствующие исходному пептиду FGFG (m/z 427.1 [М+Н]+ m/z 853.0 [2М+Н]+, фиг.42), не были детектированы.
ПРИМЕР 3.2
Конъюгаты с пептидом (NANP)6Р2Р30
Соединение OM-197-FV7, полученное по схеме синтеза, представленной на фиг.5, может сочетаться, например, с каким-либо пептидом, представляющим фармацевтический интерес, таким как (NANP)6P2P30 [Valmori et al., 1992, J. Immunol. 149, 717-721], имеющего следующую последовательность:

Пептид (NANP)6Р2Р30 обладает четырьмя центрами для возможного сочетания (концевой амин + три лизина). 5 мг очищенного OM-197-FV7 (4,64 μмоль, 4 экв) были добавлены к раствору 7,49 мг (NANP)6Р2Р30 (1,16 μмоль, 1 экв), растворенных в 1 мл смеси Н2О-изопропиловый спирт (1:1). Раствор перемешивают 30 мин при комнатной температуре, после чего осуществляют стадию восстановления, добавляя 230 μл 1 М NaBH3CN (14,43 мг, 50 экв). Раствор перемешивают два часа при комнатной температуре и затем реакционную смесь диализуют в течение 24 час против Н2O (диализная кассета 3,5 кД, Slide-A-Lyzer, Pierce). Масс-хроматографический анализ реакционной среды указывает на присутствие трех типов молекул, как это следует из хроматограмм, представленных на фиг.44: четко видны пики, соответствующие свободному пептиду (пик А), моноконъюгированному пептиду (пик В) и диконъюгированному пептиду (пик С). Зарегистрированные для каждого из них ионные оболочки представлены на фиг.45, 46 и 47 и свидетельствуют о ковалентном конъюгировании с одной или с двумя молекулами ОМ-197-FV7 соответственно для моноконъюгата и биконъюгата.
Фиг.45: Пептид (NANP)6Р2Р30 (свободный пептид). ММ: 6451 ионы с m/z 1076.2 (А6), 1291.2 (А5), 1613.7 (А4).
Фиг.46: Моноконъюгат OM-197-FV-(NANP)6Р2Р30. ММ: 7481 ионы с m/z 1247.8 (В6), 1497.4 (В5), 1871.2 (В4).
Фиг.47: Биконъюгат (OM-197-FV)2 - (NANP)6Р2Р30. ММ: 8511 ионы с m/z 1419.6 (С6), 1703.3 (С5), 2129.1 (С4).
Анализ конъюгатов OM-197-FV-(NANP)6Р2Р30 методом SDS-PAGE (электрофорез на полиакриламидном геле с додецилсульфатом натрия) позволил обнаружить различные индивидуальные полосы, соответствующие непрореагировавшему пептиду, моноконъюгату и биконъюгату. В использованных электрофоретических условиях (градиент 10-20% полиакриламида, Трис-трициновый буфер, Bio Rad, #161-1108) присутствующие стандарты позволили оценить с точностью до приблизительно одной тысячи а.е.м. (атомных единиц массы) разницу между изучаемыми полосами, подтвердив сочетание одной или двух молекул OM-197-FV с пептидом.
ПРИМЕР 3.3
Конъюгаты с пептидом Р2Р30
В реакции сочетания с помощью восстановительного аминирования с соединениями, имеющими вспомогательное ответвление альдегидного типа, могут участвовать также и другие пептиды. Ниже в качестве примера приведено сочетание OM-197-FV7 с пептидом Р2Р30.Последний соответствует детерминантной части токсина Tenanus toxoid и имеет следующую последовательность:

Пептид Р2Р30 обладает четырьмя центрами для возможного сочетания (концевой амин + четыре лизина) и массой 4200 а.е.м. Таким образом, были взяты пять эквивалентов OM-197-FV7. Были использованы описанные выше условия реакции. После диализа против Н2O (диализная кассета 3,5 кД, Slide-A-Lyzer, Pierce) масс-спектры полученных конъюгатов были зарегистрированы с помощью LC/ES-MS (жидкостная хроматография + масс-спектрометрия) и сравнены со спектрами, полученными для свободного пептида Р2Р30, включающего множественно заряженные ионы m/z 840.9 (А5), 1050.9 (А4) и 1401.0 (A3), которые образуют ионную оболочку пептида с молекулярной массой 4200 а.е.м. Спектр ES/MS моноконъюгата ОМ-197-FV-Р2Р30 включает множественно заряженные ионы m/z 872.8 (А6), 1047.1 (А5), 1308.6 (А4) и 1744.4 (A3) (фиг.49), которые образуют ионную оболочку конъюгата с молекулярной массой 5230 а.е.м., что соответствует прививке одной молекулы OM-197-FV на молекуле пептида P2Р30 с помощью восстановительного аминирования.
Аналогичным образом, масс-спектр, полученный для биоконъюгата (OM-197-FV)2P2Р30, не оставляет никакого сомнения в отношении присутствия двух молекул OM-197-FV7 на пептидную единицу. Ионная оболочка образована ионами m/z 1044.5 (А6), 1253.1 (А5), 1566.3 (А4) и 2088.4 (A3) (фиг.50), которые после преобразования свидетельствуют об ожидаемой молекулярной массе биоконъюгата (6261 а.е.м.).
ПРИМЕР 3.4
Конъюгаты с пептидом (NANP)3CS.T3
Аналогичное сочетание может быть проведено с пептидом (NANP)3CS.T3 (TNO, молекулярная масса 3527 а.е.м.), имеющим следующую последовательность:
После диализа против Н2О наблюдаемые в спектрах ES/MS ионы свидетельствуют о молекулярных массах от 4557 до 5587 а.е.м., что соответствует ожидаемым значениям для монои биконъюгированных соединений.
ПРИМЕР 3.5
Конъюгаты с пептидом MR99B
Соединение OM-197-MC-FV6, полученное по схеме синтеза, приведенной на фиг.8, может быть введено в сочетание, например, с представляющим фармацевтический интерес пептидом, таким как PyCS 245-253. Этот пептид соответствует детерминантной части Т у Plasmodium yoelil [Franke E.D. et al., 1997, J. Immunol. 159, 3424-3433], имеющим следующую последовательность:
SYVPSAEQI (PyCS 245-253)
Чтобы не изменить детерминантную часть Т в процессе конъюгирования, были добавлены аминокислоты SER с целью образования пептида, имеющего название MR99B и последовательность:
SERSYVPSAEQI (MR99B)
Поскольку эта последовательность не содержит лизина, пептид MR99B обладает единственным центром для конъюгирования восстановительным аминированием (по концевому амину). В частности, 2 мг очищенного OM-197-FV6 (2,04 μмоль, 1,4 экв) добавляют к раствору 2 мг MR99B (1,47 μмоль, 1 экв), растворенного в смеси Н2O-изопропиловый спирт (1:1). Раствор перемешивают в течение 30 мин при комнатной температуре, после чего осуществляется стадия восстановления путем добавления 29,4 μл 1 М NaBH3CN (29,4 μмоль, 20 экв). Раствор перемешивают в течение двух час при комнатной температуре. Анализ реакционной смеси с помощью LC/ES-MS позволяет показать образование конъюгата OM-197-MC-FV-MR99B с ожидаемой молекулярной массой (2330.4 а.е.м., фиг.51), о чем свидетельствуют спектры, приведенные на фиг.52 (ионная оболочка) и 53 (преобразованный спектр).
Очистку конъюгата OM-197-MC-FV-MR99B осуществляют с помощью полупрепаративной жидкостной хроматографии на фазе С4 при следующих условиях:

Для того, чтобы показать, что антигенная часть пептида не затронута конъюгированием, проводится расщепление конъюгата OM-197-MC-FV-MR99B трипсином. 0,5 мг конъюгата инкубируют 24 часа при 25°С с 83 μг трипсина (Roche, #109819) (отношение субстрат-фермент 6:1)) в 1 мл 50 мМ буфера Трис-HCl, 2 мМ CaCl2, pH 8.
Анализ LC/ES-MS после реакции представлен на фиг.54 и позволяет установить присутствие двух фрагментов, спектры ES-MS которых приведены на фиг.55 и 56. Первый фрагмент, представленный ионами m/z 993.5 ([М+Н]+) и 1015.6 ([M+Na]+, соответствует пептиду CSPy 245-253 (фиг.55). Второй фрагмент дает ионы m/z 678.1 ([М+Н]+2) и 1355.0 ([М+Н]+), что указывает на присутствие структуры типа OM-197-MC-FV-SER (фиг.56). Наблюдается также третий пик, который соответствует двум фрагментам SYVPS и AEQI (совместно элюируемым в хроматографических условиях). Такая реакция наблюдается и при расщеплении одного MR99B.
Полученные результаты четко показывают, что конъюгирование не изменяет детерминантную часть Т пептида MR99B, что иллюстрируется схемой на фиг.51.
ПРИМЕР 3.6
Конъюгаты с пептидом MR99A
С целью увеличения степени конъюгирования OM-197-MC-FV пептида и увеличения отношения адъювант/антиген без изменения детерминантной части Т к предназначенному для конъюгирования пептиду могут быть привиты последовательности типа KGG. Так, например, последовательность KGGKGGK может быть привита к пептиду MR99B с образованием следующего пептида MR99A:
KGGKGGKSERSYVPSAEQMR99A
Последний обладает, таким образом, четырьмя возможными центрами для реакции восстановительного аминирования (три лизина и концевой амин). В качестве примера: 12 мг очищенного OM-197-MC-FV6 (12,2 μмоль, 12 экв) были добавлены к раствору 2 мг MR95A (1,01 μмоль, 1 экв) в 1 мл смеси Н2О-изопропиловый спирт (1:1). Раствор перемешивают в течение 30 мин при комнатной температуре, после чего осуществляется стадия восстановления путем добавления 242 μл 1 н. NaBH3CN (244 μмоль, 242 экв). Раствор перемешивают в течение 2 час при комнатной температуре. Анализ реакционной смеси с помощью LC/ES-MS позволяет установить присутствие различных пиков, соответствующих разным степеням конъюгирования молекул OM-197-MC-FV с пептидом MR99A. Масс-спектры, соответствующие триконъюгату (OM-197-MC-FV)3-MR99A (4870 а.е.м.) показаны на фиг.57 (ионная оболочка) и 58 (преобразованный спектр), в то время как спектры, приведенные на фиг.59 (ионная оболочка) и 60 (преобразованный спектр), свидетельствуют об образовании тетраконъюгата (OM-197-MC-FV)4-MR99A (5834 а.е.м.). Наблюдаются также ионы, соответствующие пентаконъюгированной форме. Этот результат показывает, что в определенных условиях реакции (избыток OM-197-MC-FV) лизин не является единственной аминокислотой, способной участвовать в реакции восстановительного аминирования.
Очистку три- и тетраконъюгатов (OM-197-MC-FV)3-4-МR99А осуществляют с помощью полупрепаративной жидкостной хроматографии на фазе С4 при следующих условиях:

Так же как и в случае моноконъюгированного OM-197-MC-FV-MR99B, чтобы установить, что антигенная часть (CSPy 245-253) не затронута конъюгированием, проводится расщепление поликонъюгатов (OM-197-MC-FV)n-MR99A трипсином. 1 мг конъюгата инкубируют 24 часа при 25°С со 166 иг трипсина (Roche, #109819) (отношение субстрат/фермент 6:1) в 1 мл 50 мМ буфера Трис-HCl, 2 мМ CaCl2, pH 8.
Анализ LC/ES-MS после реакции позволяет установить присутствие многочисленных фрагментов, соответствующих расщеплению пептида MR99A в разных местах. В числе этих фрагментов выявляется фрагмент, представленный ионами m/z 993.5 ([М+Н]+) и 1015.6 ([M+Na]+), что соответствует пептиду CSPy 245-253. Присутствие последнего свидетельствует о целостности детерминантной части Т пептида даже после многократного конъюгирования. Два фрагмента, наблюдаемые в виде их трижды заряженных ионов m/z 1621.4 ([М+Н]3+) и 1942.8 ([М+Н]3+), представляют особый интерес. Действительно, они соответствуют фрагментам (OM-197-MC-FV)4-KGGKGGKSER и (ОМ-197-MC-FV)5-KGGKGGKSER, соответственно. Присутствие последних показывает, что различные молекулы OM-197-MC-FV эффективно прививаются к цепи, добавляемой к CSPy 245-253, даже в том случае, когда на стадии восстановительного аминирования в реакции участвовали аминокислоты, отличные от лизина. Следует также подчеркнуть, что стерически значимое присутствие ряда молекул OM-197-MC-FV на пептиде не влияет на активность протеазы, такой как трипсин. Этот результат приобретает особую важность в случае конъюгирования, например, с предшественником лекарства, когда активное начало высвобождается после селективного расщепления какой-либо лабильной связи.
ПРИМЕР 3.7
Конъюгаты с пептидом Аg85с
Структуры, имеющие вспомогательное ответвление формилвалерильного типа, могут быть конъюгированы с многочисленными пептидами, представляющими фармацевтический интерес. Назовем, например, прививку с помощью восстановительного аминирования молекул OM-197-MC-FV на концевую аминогруппу синтетических пептидов, последовательности которых восходят к антигенному белку Mycobacterium tuberculosis. Исходными пептидами являются следующие:
После реакции с OM-197-MC-FV с помощью анализа LC/ES-MS наблюдались следующие конъюгаты: в качестве примера, ОМ-197-MC-FV-MR100, имеющий молекулярную массу 2171,0 а.е.м. (фиг.61 и 62). Одновременно были получены также OM-197-MC-FV-MR101, 3140,0 а.е.м. и OM-197-MC-FV-LR72, 3643,0 а.е.м.
ПРИМЕР 3.8
Димер со структурами типа ОМ-197
Техника восстановительного аминирования может быть использована также и для синтеза димера со структурами типа ОМ-197.
Димер может быть образован, например, из двух соединений, имеющих вспомогательные функционализированные ответвления, таких как OM-197-MC-FV и ОМ-197-МС-АС. Например, 1 мг очищенного OM-197-MC-FV6 (0,98 μмоль, 1 экв) добавляют к раствору 1 мг ОМ-197-МС-АС (0,98 имоль, 1 экв), растворенного в 1 мл смеси Н2О-изопропиловый спирт (1:9). Раствор перемешивают в течение 1 часа при комнатной температуре, после чего осуществляется стадия восстановления путем добавления 19,6 μл 1 н. NaBH3CN (19,6 μмоль, 20 экв). Раствор перемешивают в течение 12 час при комнатной температуре. Анализ реакционной смеси с помощью LC/ES-MS позволяет установить образование димера OM-197-MC-FV-OM-197-MC-AC с ожидаемой молекулярной массой (1945,5 а.е.м.), о чем свидетельствует спектр, приведенный на фиг.63. Полученная молекула имеет, таким образом, две кислотные карбоксильные функции на концах скелета, несущего ацильные цепи.
В представленной на фиг.63 структуре одна из карбоксильных групп может быть заменена, например, фосфорильной группой, если проводить синтез исходя из соединения типа ОМ-197-МР-АС или OM-197-MP-FV. С помощью реакции восстановительного аминирования, аналогичной описанной выше, между двумя соединениями, имеющими каждое по одному функционализированному вспомогательному ответвлению, (OM-197-MP-FV и ОМ-197-МС-АС), может быть получен димер ОМ-197-MP-FV-OM-197-MC-AC. Приведенный на фиг.64 спектр ES/MS свидетельствует об ожидаемой молекулярной массе для этого соединения (2011,9 а.е.м.).
ПРИМЕР 3.9
Конъюгирование соединений, имеющих одно вспомогательное ответвление аминного типа
Соединения, имеющие одно вспомогательное ответвление аминного типа, также могут быть конъюгированы с пептидными антигенами с помощью реакции восстановительного аминирования. Возможно, в частности, прибегнуть к наличию в последовательности конъюгируемых пептидов, например, концевого серина. Вначале пептид подвергается перйодатному окислению, приводящему к образованию высокореакционноспособной глиоксилильной группы (-СО-СНО). Последняя может участвовать в реакции восстановительного аминирования с первичной аминогруппой, находящейся на функционализированном вспомогательном ответвлении соединений типа ОМ-197.
Синтетический пептид MR99B может быть, например, конъюгирован с соединением ОМ-197-МС-АС, как это показано на схеме синтеза на фиг.65. С этой целью 147 μл 0,1 М NaIO4 (14,6 μмоль, 20 экв) добавляют к раствору 1 мг пептида MR99B (0,73 μмоль, 1 экв) в 1 мл воды. Раствор перемешивают в течение двух час при комнатной температуре. Полученный таким образом альдегид очищают на фазе С18, собирая его с помощью 1 мл смеси изопропиловый спирт-вода (1:1). Добавляют затем 0,72 мг соединения ОМ-197-МС-АС (0,73 μмоль, 1 экв) вместе с 1 мл 0,2 М боратного буфера, рН 9,2. После перемешивания при комнатной температуре в течение 30 мин проводят восстановление, добавляя 14,6 μл 1 М NaBH3CN (14,6 μмоль, 20 экв). Раствор перемешивают в течение 24 час при комнатной температуре.
Анализ реакционной смеси с помощью LC/ES-MS позволяет установить образование конъюгата OM-197-MC-AC-MR99B с ожидаемой молекулярной массой (2299,1 а.е.м., фиг.65), о чем свидетельствуют спектры, приведенные на фиг.66 (ионная оболочка) и 67 (преобразованный спектр).
Аналогичным образом могут быть конъюгированы и другие соединения, имеющие вспомогательное ответвление аминного типа. Назовем в качестве примера ОМ-197-МС-АР, ОМ-197-МР-АС, ОМ-212-АН1, OM-197-MC-Gly, OM-197-MC-Lys, OM-197-Lys-AC или OM-197-Asp-AC.
ПРИМЕР 3.10
Конъюгаты с овальбумином
Белки, такие, например, как овальбумин, особенно хорошо подходят для сочетания с помощью восстановительного аминирования. Множество имеющихся в цепи лизинов (20 лизинов в овальбумине) являются возможными центрами конъюгации для псевдопептидолипидов, имеющих вспомогательное функционализированное ответвление с альдегидной группой. Меняя стехиометрию реакции восстановительного аминирования (отношение ацилированный псевдодипептид/белок), можно получать разные степени конъюгирования.
1 мг очищенного OM-197-FV7 (0,928 μмоль, 10 экв) был добавлен к раствору 4 мг овальбумина (0,09 μмоль, 1 экв) в 5 мл Н2O. Раствор перемешивают в течение 30 мин при комнатной температуре, после чего осуществляется стадия восстановления путем добавления 46 μл 1 М NaBH3CN (2,89 мг, 50 экв). Раствор перемешивают в течение двух час при комнатной температуре. После этого реакционную смесь диализуют в течение 12 час против Н2О (диализная кассета 3,5 кД, Slide-A-Lyzer, Pierce).
Из-за своего размера и собственной гетерогенности овальбумин создает множество аналитических проблем. Вследствие этого охарактеризовывание конъюгата OM-197-FV-овальбумин с помощью масс-спектрометрии оказалось чрезвычайно деликатным. Чтобы показать протекание конъюгирования, были проведены анализы с помощью SDS-PAGE на различных группах конъюгатов ОМ-197-FV-овальбумин. Как об этом свидетельствует гель, представленный на фиг.68 (В) (градиент 4-20% полиакриламид), наблюдается масса конъюгатов OM-197-FV-овальбумин (линии 3, 4 и 5) приблизительно на 3000 а.е.м. более высокая, чем у исходного овальбумина (линия 2). Эта разница в массе наводит на мысль о присутствии нескольких молекул OM-197-FV на одну молекулу овальбумина. Этот результат подтверждают анализы с помощью жидкостной хроматографии с УФ-детектором, выполненные на фазе С4. Действительно, в этих условиях исходный овальбумин четко появляется при времени удерживания 11,3 мин, в то время как после реакции конъюгирования пик, соответствующий исходному овальбумину, полностью исчезает, доказывая количественное превращение овальбумина. Конъюгат ОМ-197-ГУ-овальбумин, напротив, не элюируется. Такое хроматографическое поведение указывает также на присутствие на овальбумине нескольких молекул OM-197-FV, что препятствует элюции конъюгата в этих условиях.
ПРИМЕР 3.11
Конъюгаты с гемагглютинином H1N1
Белок гемагглютинина H1N1 также представляет собой пример, который был выбран для иллюстрации сочетания между молекулами типа ОМ-197 и антигенами белкового типа.
1 мг очищенного OM-197-FV7 (0,928 μмоль, 15 экв) был добавлен к раствору 5 мг H1N1 (гемагглютинина А / Beijing 262/95, Solvay Duphar, Weesp, Нидерланды) (0,06 μмоль, 1 экв) в 5 мл Н2O. Раствор перемешивают в течение 30 мин при комнатной температуре, после чего осуществляется стадия восстановления путем добавления 46 μл 1 н. NaBH3CN (2,89 мг, 50 экв). Раствор перемешивают в течение двух час при комнатной температуре. После этого реакционную смесь диализуют в течение 24 час против Н2O (диализная кассета 3,5 кД).
Конъюгат OM-197-FV-H1N1 анализируют с помощью SDS-PAGE в условиях, использованных для конъюгата ОМ-197-FV-овальбумин (градиент 4-20% полиакриламид). Электрофоретические профили, полученные для исходного белка (линия 6) и конъюгата до и после диализа (линии 8 и 7, соответственно), приведены на фиг.68 (С). Белок H1N1 появляется в виде трех полос, две из которых соответствуют субъединицам НА1 и НА2, которые описаны в литературе (Swiss-Prot, Swiss Institute of Bioinformatics, Geneva). В случае конъюгатов OM-197-FV-H1N1 для каждой полосы наблюдается разница в массе, что свидетельствует о сочетании молекул OM-197-FV с белком H1N1.
ПРИМЕР 3.12
Конъюгат с 3'-азидо-3'-деокситимидином (AZT) (фиг.69)
К раствору AZT (200 мг; 0,749 ммоль) в пиридине (4 мл) последовательно добавляют янтарный ангидрид (150 мг; 0,5 ммоль) и 4-диметиламинопиридин (50 мг; 0,41 ммоль). После проведения реакции в течение 12 час при 50°С добавляют метанол (2 мл) и перемешивают реакционную смесь еще 15 мин при комнатной температуре. Затем реакционную смесь упаривают и очищают Флэш-хроматографией на силикагеле с элюентом СН2Cl2/МеОН (5/1). Получают продукт в виде белого твердого вещества (266 мг; 97%). C14H17N5O7 (M = 367 г/моль); Rf = 0,25 (CH2Cl2/MeOH 4/1).
К раствору полученного таким образом сукцинил-AZT (60 мг; 0,163 ммоль) в ТГФ (3 мл) при 0°С под аргоном последовательно добавляют триэтиламин (24 μл; 0,172 ммоль) и этилхлорформиат (14 μл; 0,172 ммоль) - происходит быстрое выпадение в осадок гидрохлорида триэтиламина. После 40 мин перемешивания добавляют при 0°С раствор аминокапроильного производного (пример 2.16) (160 мг; 0,163 ммоль) в смеси ДМФ-вода 9/1 (10 мл), содержащей триэтиламин (24 μл; 0,172 ммоль). Реакционную смесь перемешивают 15 мин при 0°С и затем в течение ночи при комнатной температуре, после чего досуха упаривают при пониженном давлении. Сырой продукт забирают дихлорметаном (15 мл) и водой (5 мл) и отделяют органическую фазу. Водную фазу подкисляют 10%-ным водным раствором лимонной кислоты и дважды экстрагируют дихлорметаном. Органические фазы объединяют, высушивают над MgSO4, фильтруют и упаривают. Очистка флэш-хроматографией на силикагеле с элюентом CH2Cl2/MeOH (9/1 и затем 7/1) позволяет выделить конъюгированное с AZT соединение (123 мг; 57%) в виде белого твердого вещества. Rf = 0,2 (CH2Cl2/MeOH 4/1); C69H119N9O16 (M = 1329 г/моль); найдено: m/z 1330,9 ([М+Н]+). Аналитическая ЖХВР: время удерж.= 24,437 мин, колонка с С18 Supelcosil (15 см х 4,6 мм, 3 цм, 100 Å); растворитель А = 50% MeCN, 5 мМ ТВАР; растворитель В = 90% 2-пропанол, 5 мМ ТВАР. Градиент: 25-100% В за 37,5 мин; детекция УФ при 210 нм.
Препаративная ЖХВР: на колонке с С18 Kromasil (25 см х 21 мм, 5 μм, 100 Å); растворитель А = 50% MeCN, 5 мМ ТВАР; растворитель В = 90% 2-пропанол, 5 мМ ТВАР. Продукт элюируют при 80% В.
Конечная фаза: переход в Na+ соль с помощью SPE, фаза С18 Bondapack 15 μм, элюирование 90%-ным 2-пропанолом.
Конъюгат с d4T (фиг.69)
К раствору d4T (200 мг; 0,89 ммоль) в пиридине (4 мл) последовательно добавляют янтарный ангидрид (179 мг; 0,786 ммоль) и 4-диметиламинопиридин (109 мг; 0,89 ммоль). После 12 час перемешивания при комнатной температуре добавляют метанол (2 мл), перемешивают реакционную смесь еще 15 мин при комнатной температуре и упаривают. Сырой продукт очищают флэш-хроматографией на силикагеле с элюентом CH2Cl2/MeOH (5/1). Получают продукт в виде белого твердого вещества (280 мг; 97%). С14Н16N2O7 (M = 324 г/моль); Rf = 0,25 (CH2Cl2/MeOH 4/1).
К раствору полученного таким образом сукцинильного производного d4T (60 мг; 0,185 ммоль) в ТГФ (3 мл) при 0°С под аргоном последовательно добавляют триэтиламин (27 μл; 0,194 ммоль) и этилхлорформиат (16 μл; 0,194 ммоль) - происходит быстрое выпадение в осадок гидрохлорида триэтиламина. После 40 мин перемешивания добавляют при 0°С раствор аминокапроильного производного (пример 2.16) (181 мг; 0,185 ммоль) в смеси ДМФ-вода 9/1 (10 мл), содержащей триэтиламин (27 μл; 0,194 ммоль). Реакционную смесь перемешивают 15 мин при 0°С и затем в течение ночи при комнатной температуре, после чего досуха упаривают при пониженном давлении. Сырой продукт забирают дихлорметаном (15 мл) и водой (5 мл) и отделяют органическую фазу. Водную фазу подкисляют 10%-ным водным раствором лимонной кислоты и дважды экстрагируют дихлорметаном. Органические фазы объединяют, высушивают над MgSO4, фильтруют и упаривают. Очистка флэш-хроматографией на силикагеле с элюентом CH2Cl2/MeOH (9/1 и затем 7/1) позволяет выделить конъюгированное с d4T соединение (107 мг; 45%) в виде белого твердого вещества. Rf =0,2 (CH2Cl2/MeOH 4/1); С69Н118N6О16 (M = 1287 г/моль); найдено: m/z 1288 (МН+), 1310 (Na). Аналитическая ЖХВР: время удерж.= 24,455 мин, колонка с С18 Supelcosil (15 см × 4,6 мм, 3 μм, 100 Å); растворитель А = 50% MeCN, 5 мМ ТВАР; растворитель В = 90% 2-пропанол, 5 мМ ТВАР. Градиент: 25-100% В за 37,5 мин; детекция УФ при 210 нм.
Препаративная ЖХВР: на колонке с С18 Kromasil (25 см × 21 мм, 5 цм, 100 Å); растворитель А = 50% MeCN, 5 мМ ТВАР; растворитель В = 90% 2-пропанол, 5 мМ ТВАР. Продукт элюируют при 80% В.
Конечная фаза: переход в Na+ соль с помощью SPE, фаза С18 Bondapack 15 μм, элюирование 90%-ным 2-пропанолом.
4-ая серия примеров: фармакологическое исследования соединений изобретения (in vitro)
ПРИМЕР 4.1
Тест на определение эндотоксичности
Эндотоксичность определяли с помощью хромогенного кинетического теста Limulus Amoebocyte Lysate (продукт R1708K партия ЕК2251Е фирмы Charles River Endosafe). Этот тест (LAL) был использован для доказательства слабой эндотоксичности соединений по изобретению.
Биологический принцип этого теста основан на инициации с помощью бактериальных эндотоксинов профермента, содержащегося в лизате амебоцитов Limulus (LAL), который активирует цепь серин-протеаз. В присутствии бесцветного субстрата (продукт сочетания пептида S-2834 с п-нитроанилином) расщепление хромофора приводит к освобождению п-нитроанилина (pNA) и развитию желтой окраски, за которым можно следить спектрофотометрически при 405 нм.
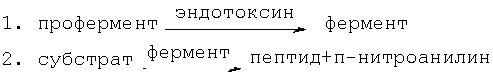
Кинетический хромогенный тест основан на том, что количество эндотоксина обратно пропорционально времени, необходимому для достижения оптической плотности (D) 0,2. Концентрацию определяют по отношению к эталонной кривой от 0,005 до 50 эн.ед./мл.
Продукты тестируют, начиная с разведения (5 х, 10 х, 50 х, 100 х, 500 х или 100 х) раствора продукта в концентрации 0,1 мг/мл. Результат LAL соответствует первому разведению, которое уже не оказывает ингибирующего действия для перекрытия перегрузки по липополисахариду (LPS).
Результаты выражены в эндотоксиновых единицах (эн.ед.) по отношению к соответствующему международному стандарту эталонному раствору (ЕС-6). Для данной серии измерений 1 эн.ед. составляет приблизительно 0,08 нг LPS Е. coli 055:B5.
Таблица результатов теста LAL для различных ацилированных псевдодипептидов, имеющих вспомогательное функционализированное ответвление, отвечающих общей формуле 1
Все протестированные псевдодипептиды обладают низкой эндотоксичностыо. Для многих соединений эндотоксичность даже ниже порога чувствительности теста. Количественное определение эндотоксичности ограничено необходимостью разбавлять образцы для нахождения перегрузки. Даже наиболее активные соединения в тесте LAL имеют эндотоксинную активность, в 5000 раз более низкую по сравнению с LPS.
Псевдодипептиды по изобретению представляют интерес благодаря их низкой эндотоксичности и их биологической активности.
Таблица результатов теста LAL для различных продуктов в эксперименте по иммунизации с использованием продуктов, конъюгированных с овальбумином
Есть основание полагать, что сочетание OM-197-FV с овальбумином повышает детектируемую активность LAL. Это повышение может быть обусловлено изменением состояния OM-197-FV. Все другие продукты из серии продуктов с овальбумином обладают эквивалентной активностью в тесте LAL. Результаты теста LAL очень различаются, в то время как различия между группами (3 х) и (4 х) не существенны. Максимальная активность LAL равна 2,4 нг-эквивалента LPS на одну инъекцию (25 μг/инъекция) для продуктов сочетания, в то время как для других групп максимальная активность LAL равна 0,012 нг-эквивалента LPS на одну инъекцию.
Таблица результатов теста LAL для различных продуктов в эксперименте по иммунизации с использованием продуктов, конъюгированных с гемагглютинином H1N1
* LPS - липополисахарид
Все продукты из серии с Н1N1 обладают эквивалентной активностью в тесте LAL. Результаты теста LAL очень различаются, в то время как различия между группами (3 х) и (4 х) не существенны. Сочетание не влияет на активность LAL. Максимальная активность LAL равна 0,008 нг-эквивалента LPS на одну инъекцию (5 μг/инъекция).
Таблица результатов теста LAL для различных продуктов в эксперименте по иммунизации с использованием продуктов, конъюгированных с пептидом (NANP)6P2P30:
Все продукты из серии с (NANP)6P2P30 обладают эквивалентной активностью в тесте LAL. Результаты теста LAL очень различаются, в то время как различия между группами 3 х и 4 х не существенны. Сочетание не влияет на активность LAL. Максимальная активность LAL равна 0,03 нг-эквивалента LPS на одну инъекцию (20 μг/инъекция).
ПРИМЕР 4.2.
Определение пролиферации стволовых клеток костного мозга мыши, стимулированной продуктами по изобретению
4.2.1. Эксперимент с пролиферацией
6-Недельные мыши-самцы C57/BL6 усыпляются путем ингаляции CO2. Отбираются кости задних конечностей, тазобедренный сустав, большеберцовая кость и бедренная кость. Костный мозг извлекается из просвета кости с помощью инъекции модифицированной Dulbecco среды Игла (среда DH) через предварительно рассеченные эпифизы. Стволовые клетки промывают и суспендируют в среде DH с добавкой 20% зародышевой телячьей сыворотки. Концентрацию клеток доводят до 500 000 клеток в 1 мл.
Продукты, растворенные в среде с добавками 20% зародышевой телячьей сыворотки, аминокислот и антибиотиков, последовательно разбавляют непосредственно на микроплашке. Продукты тестируют по три раза, причем на каждой микроплашке имеется отрицательный контроль, состоящий только из одной среды. Конечный объем в каждой лунке равен 100 μл.
100 μл клеточной суспензии добавляют к растворам продуктов и инкубируют клетки в течение 7 дней в термостате при 37°С с 8% CO2 при насыщающей влажности. Пролиферацию определяют измерением окисления хромогенного субстрата ХТТ (2,3-бис[2-метокси-4-нитро-5-сульфофенил]-2Н-тетразолий-5-карбоксанилид) в митохондриях живых клеток.
В конце инкубации в каждую лунку добавляют по 50 μл раствора субстрата ХТТ (1 мг/мл ХТТ + 0,008 мг/мл феназинметосульфата). После 8 час инкубации при 37°С с 8% CO2 в насыщенном влагой термостате микроплашки спектрофотометрируют при 492 нм, сравнивая с контрольной средой при 690 нм.
Результаты выражают как среднее измерений ± среднеквадратичное отклонение в виде кривой зависимости доза-ответ. Значения отрицательного контроля, состоящего из среды DH (среднее измерений ± среднеквадратичное отклонение) также приведены в графической форме.
На основании кривой доза-ответ рассчитываются 3 особые точки кривой:
Мах: максимальная амплитуда кривой и соответствующая ей концентрация;
ЕС50: концентрация, соответствующая 50% максимальной амплитуды;
Min: первая концентрация, индуцирующая значимую пролиферацию, соответствующую контролю + утроенное стандартное отклонение.
Концентрации, соответствующие ЕС50 и min, определяют по сегментам прямой, соединяющим разные точки кривой.
Если кривая не достигает своего максимума и продолжает подъем при наибольшей из тестируемых концентраций, перед значениями «Мах» и «ЕС50» ставится знак «>».
Если кривая не опускается ниже границы «контроль + утроенное стандартное отклонение», минимальная концентрация дается как "<" самой низкой из тестируемых концентраций.
4.2.2. Представление результатов
Пролиферация стволовых клеток костного мозга, индуцируемая ацилированными псевдодипептидами, имеющими вспомогательное функционализированное ответвление
Фиг.70 представляет активность различных ацилированных псевдодипептидов и демонстрирует влияние вспомогательного функционализированного ответвления на способность индуцировать пролиферацию стволовых клеток костного мозга мыши.
Некоторые из ацилированных псевдодипептидов способны индуцировать более сильную пролиферацию по сравнению с пролиферацией, индуцируемой положительным контролем, состоящим из LPS Е. coli. Минимальная концентрация продукта, способная индуцировать значимую пролиферацию, больше минимальной концентрации положительного контроля. На эту минимальную концентрацию продукта сильно влияет функциональность вспомогательного ответвления.
На фиг.71 приведена активность ацилированных псевдодипептидов, связанных с лекарственным средством. Эта активность доказывает то, что, несмотря на сочетание, соединения частично сохраняют свою активность. Было невозможно провести различие между изначальной активностью и активностью, возникающей в результате взаимодействия сложноэфирной связи с внутриклеточными эстеразами. Антивирусная активность этих продуктов предположительно доказывает, что по крайней мере часть сложноэфирных связей подверглась расщеплению. Какой бы ни был механизм, сохранение биологической активности доказывает большую значимость комбинации антивирусной активности с активностью продукта. Таблица результатов, демонстрирующая индуцирование пролиферации стволовых клеток костного мозга разными ацилированными псевдодипептидами, имеющими вспомогательное функционализированное ответвление и отвечающими общей формуле I:
0/μг/мл
D/μг/мл
nd - не определяется
Все ацилированные псевдодипептиды по изобретению, за исключением соединений с укороченными цепями, способны индуцировать значимую пролиферацию стволовых клеток костного мозга.
ПРИМЕР 4.3.
Определение продукции моноксида азота макрофагами мыши, стимулированными продуктами по изобретению
4.3.1. Эксперимент с продукцией моноксида азота:
6-Недельные мыши-самцы C57/BL6 усыпляются путем ингаляции CO2. Отбираются кости задних конечностей, тазобедренный сустав, большеберцовая кость и бедренная кость. Костный мозг извлекается из просвета кости с помощью инъекции модифицированной Dulbecco среды Игла (среда DH) через предварительно рассеченные эпифизы. Стволовые клетки промывают и суспендируют (до концентрации приблизительно 40000 клеток в 1 мл) в среде DH с добавкой 20% лошадиной сыворотки и 30% супернатанта L929. L929 представляют собой линию фибробластов мыши, супернатант которых обогащен фактором роста макрофагов (M-CSF). Клеточную суспензию распределяют по чашкам Петри, которые инкубируют в течение 8 дней при 37°С с 8% СО2 в насыщенном влагой термостате.
Через 8 дней стволовые клетки дифференцируются в зрелые макрофаги. Макрофаги отделяют действием холода, промывают и повторно суспендируют в среде DH с добавкой 20% зародышевой телячьей сыворотки, аминокислот и антибиотиков. Концентрацию клеток доводят до 700 000 клеток в 1 мл.
Продукты, растворенные в среде DH с добавками 5% зародышевой телячьей сыворотки, аминокислот и антибиотиков, последовательно разбавляют непосредственно на микроплашке. Продукты тестируют по три раза, причем на каждой микроплашке имеется отрицательный контроль, состоящий только из одной среды. Конечный объем в каждой лунке равен 100 μл.
По 100 μл клеточной суспензии добавляют к растворам продуктов и инкубируют клетки в течение 22 час в термостате при 37°С с 8% CO2 при насыщающей влажности. В конце инкубации с продуктами отбирают по 100 μл супернатанта и определяют концентрацию нитрита с помощью реакции Грисса.
В каждую лунку добавляют по 100 μл реактива Грисса (5 мг/мл сульфаниламида + 0,5 мг/мл гидрохлорида N-(1-нафтилэтилендиамина)) в 2,5%-ной водной фосфорной кислоте. Микроплашки спектрофотометрируют при 562 нм в сравненнии с контрольной средой при 690 нм. Концентрация нитрита пропорциональна концентрации продуцированного моноксида азота. Концентрацию нитрита определяют в сравнении с эталонной кривой.
Результаты выражают как среднее измерений ± стандартное отклонение в виде кривой зависимости доза-ответ.
На основании кривой доза-ответ рассчитываются 3 особые точки кривой:
Мах: максимальная амплитуда кривой и соответствующая ей концентрация;
ЕС50: концентрация, соответствующая 50% максимальной амплитуды;
Min: первая концентрация, индуцирующая значимую пролиферацию, соответствующую контролю + утроенное стандартное отклонение.
Концентрации, соответствующие ЕС50 и min, определяют по сегментам прямой, соединяющим разные точки кривой.
Если кривая не достигает своего максимума и продолжает подъем при наибольшей из тестируемых концентраций, перед значениями «Мах» и «ЕС50» ставится знак «>».
Если кривая не опускается ниже границы контроля + утроенное среднеквадратичное отклонение», минимальная концентрация дается как "<" самой низкой из тестируемых концентраций.
4.3.2. Представление результатов
Продукция моноксида азота макрофагами мыши, индуцируемая ацилированными псевдодипептидами, имеющими вспомогательное функционализированное ответвление
Фиг.72 представляет активность различных ацилированных псевдодипептидов и демонстрирует влияние вспомогательного функционализированного ответвления на способность индуцировать пролиферацию стволовых клеток костного мозга мыши.
ОМ-197-МР-АС (R/S/R) способен индуцировать более сильную пролиферацию по сравнению с пролиферацией, индуцируемой положительным контролем, состоящим из LPS Е. coli. Тем не менее, минимальная концентрация продукта, способная индуцировать значимую пролиферацию, значительно больше минимальной концентрации положительного контроля. На продукцию NO макрофагами мыши, стимулируемыми ацилированными псевдодипептидами, сильно влияет функциональность вспомогательного ответвления.
На фиг.73 приведена активность ацилированных псевдодипептидов, связанных с лекарственным средством. Эта активность доказывает то, что, несмотря на сочетание, соединения частично сохраняют свою активность. Было невозможно провести различие между изначальной активностью и активностью, возникающей в результате взаимодействия сложноэфирной связи с внутриклеточными эстеразами. Антивирусная активность этих продуктов предположительно доказывает, что по крайней мере часть сложноэфирных связей подверглась расщеплению. Какой бы ни был механизм, сохранение биологической активности доказывает большую значимость комбинации антивирусной активности с активностью продукта.
Таблица результатов, демонстрирующая индуцирование продукции моноксида азота макрофагами мыши разными ацилированными псевдодипептидами, имеющими вспомогательное функционализированное ответвление и отвечающими общей формуле I:
Все ацилированные псевдодипептиды по изобретению за исключением соединений с укороченными цепями (выпадение значения продукции в случае OM-197-N'(С10-2OС8)-МС-АС) способны индуцировать значительную продукцию моноксида азота макрофагами мыши.
ПРИМЕР 4.4.
Тест на дифференцировку дендритных клеток
Была оценена способность продуктов по изобретению индуцировать созревание предендритных клеток в дендритные клетки. Измерялись следующие параметры: инкорпорация Dextran-FITC и экспрессия поверхностных молекул CD83, CD86.
4.4.1. Пропись эксперимента
Одноядерные клетки периферической крови выделяют из "buffy coats" (светлых слоев кровяного сгустка) здоровых доноров. Очищенные адгезионным способом моноциты суспендируют в среде RPMI-1640, содержащей 10% зародышевой телячьей сыворотки и по 10 нг/мл GM-CSF и IL-4, в расчете на 1 × 106 клеток в 1 мл. Клетки распределяют по чашкам Петри в расчете на 10 × 106 клеток в чашке и культивируют в течение 6 дней со сменой среды после 3 дней. Полученные таким образом клетки называют предендритными (DC-6). Созревание предендритных клеток в зрелые дендритные клетки осуществляют инкубацией с растворами продуктов или LPS (положительный контроль) в течение дополнительных 3 дней (см. побочные продукты). На 9-ый день (DC-9) клетки собирают и оценивают различные параметры, свидетельствующие о созревании дентритных клеток: экспрессия поверхностных молекул CD83, CD86, а также способность инкорпорировать Dextran-FITC. Все параметры анализируют с помощью FACS EPICS-XL-MCL (Coulter Immunology, Hialeah, Finland).
Экспрессия поверхностных молекул выражается в среднем значении % флуоресценции клеток, стимулируемых LPS (положительный контроль), а инкорпорацию декстрана рассчитывают по отношению к инкорпорации удерживающихся в среде клеток и выражают в %.
Продукты: маточные растворы ОМ-197-МС, OM-197-FV6 и ОМ-197-МР-АС (R/S,R) приготовляют в концентрации 0,5 мг/мл в 0,9%-ном NaCl/вода с добавкой О,1%; триэтаноламина. Растворы инкубируют при 37°С в течение 20 мин, интенсивно перемешивают в течение 3 мин и разбавляют до 100 μг/мл в культуральной среде RPMI 1640, после чего используют, разбавляя от 10 μг/мл до 0,03 μг/мл.
Контрольный продукт: LPS E. coli (LPS, DIFCO, Detroit, MI, США), исходный раствор 5 мг/мл в забуференном фосфатом соляном растворе. Приготовляют промежуточный раствор с концентрацией 100 μг/мл в культуральной среде RPMI 1640. Тестируемыми концентрациями являются разбавления, исходя из 1 μг/мл и до 0,03 μг/мл.
В другой серии экспериментов все ацилирозанные псевдодипептиды были лиофилизированы и повторно растворены непосредственно в апирогенной воде. Разные продукты, а также контрольный LPS тестируют при концентрации 10 μг/мл. Результаты выражены в% дендритных клеток и в средней величине флуоресценции, обусловленной фагоцитозом декстрана, или экспрессией поверхностного маркера CD86. Эти результаты представляют собой среднее из 2-6 (в зависимости от продукта) опытов.
4.4.2. Оценка результатов
Незрелые дендритные клетки (DC-6), возникшие при дифференцировке моноцитов, на самом деле конъюгированные с GM-CSF и IL-4, способны инкорпорировать Dextran-FITC. В процессе созревания клетки теряют способность инкорпорировать Dextran-FITC. Анализы проводятся на стадии дифференцировки DC-9.
Результаты (фиг.74) выражены в% инкорпорации Dextran-FITC, наблюдаемого в нестимулированных клетках, инкубируемых в присутствии только среды. Клетки, обработанные липополисахаридом или ОМ-197-МР-АС (R/S,R), сохраняют фагоцитоз, соответственно, только на 15 и 22%. Необходим по меньшей мере 1 μг/мл ОМ-197-МР-АС (R/S,R) для индуцирования максимального уменьшения инкорпорации Dextran-FITC, в то время как эквивалентное уменьшение индуцируется 0,1 цг/мл липополисахарида. 10 μг/мл ОМ-197-МС необходимы для индуцирования уменьшения фагоцитоза на 22%. При этом, концентрации ОМ-197-МС ниже 3 μг/мл не оказывают никакого эффекта на фагоцитоз. По-видимому, OM-197-FV6 не оказывает никакого эффекта на созревание дендритных клеток.
Экспрессия одновременно стимулирующих поверхностных молекул является еще одним критерием созревания дендритных клеток. Было протестировано увеличение экспрессии CD83 и CD86 (фиг.75 и 76, соответственно). Результаты выражены в % средней величины флуоресценции по отношению к экспрессии этих маркеров, индуцируемой LPS. Эти результаты прекрасно подтверждают результаты, полученные на основании фагоцитоза. ОМ-197-МР-АС (R/S,R) индуцирует увеличение экспрессии поверхностных молекул начиная с 0,1 μг/мл, в то время как необходим по меньшей мере 1 μг/мл для того, чтобы ОМ-197-МС начал повышать экспрессию CD83 и CD86. OM-197-FV6 не оказывает никакого влияния на экспрессию поверхностных молекул, характеристических для дендритных клеток.
Эти результаты свидетельствуют об очень различном поведении ацилированных псевдодипептидов с разными вспомогательными ответвлениями. ОМ-197-МР-АС (R/S,R) проявляет исключительную способность индуцировать дифференцировку клеток DC-6 и DC-9 начиная уже с 0,1 μг/мл, в то время как необходимы концентрации выше 1 цг/мл для того, чтобы эту дифференцировку начал индуцировать ОМ-197-МС, а OM-197-FV6 полностью лишен такого типа активности.
Фагоцитоз декстрана, меченного флуоресцеином, и экспрессия CD86 после стимуляции предендритных клеток (DC6) ацилированными псевдодипептидами, имеющими вспомогательное функционализированное ответвление и отвечающими общей формуле I. Дифференцировка предендритных клеток в зрелые дендритные клетки.
% CD / флуоресценция
% CD / флуоресценция
При концентрации 10 μг/мл многие ацилированные псевдодипептиды способны индуцировать дифференцировку предендритных клеток в дендритные клетки. Содержащиеся в вспомогательном ответвлении функции модулируют эту способность активировать предендритные клетки.
Ацилированный псевдодипептид OM-197-MC-AC-Succ-AZT, подвергнутый сочетанию с лекарством, обладает значительной способностью индуцировать дифференцировку в дендритные клетки. Влияние составления композиций ацилированных псевдодипептидов имеет особую важность для способности индуцировать дифференцировку предендритных клеток в дендритные клетки. Некоторые продукты обладают очень различающимися активностями в зависимости от того, были ли они растворены в присутствии или в отсутствие 0,1% ТЕОА.
ПРИМЕР 4.5.
Анализ индуцирования белка МхА в дендритных клетках
Была оценена способность продуктов по изобретению индуцировать продукцию антивирусного белка МхА предендритными клетками. Продукцию белка МхА клетками измеряли после электрофоретического разделения методом SDS-PAGE с использованием western blotting.
4.5.1.Пропись эксперимента
Полученные, как описано выше, дендритные клетки на стадии DC-6 стимулировали в течение 48 час или не стимулировали IFN-α (500 ед./мл), LPS (μг/мл), TRANCE (100 μг/мл), CD40L (1 μг/мл), ОМ-197-МР-АС (1 μг/мл) или OM-197-FV6 (1 μг/мл). Экстракцию и анализ белков проводят следующим образом: отбирают клетки, промывают их 3 раза забуференным фосфатом соляным раствором и лизируют в 100 мМ буфере Трис-HCl, 150 мМ NaCI, 5 мМ EDTA, 1% Triton-100, содержащем 1 мМ фенилметилсульфонилфторида и 1 μг/мл пепстатина в качестве ингибиторов протеаз. Лизис осуществляют при соотношении 106 клеток на 100 μл буфера. Образцы нагревают до 95°С в течение 5 мин в герметичных пробирках Эпендорфа. Электрофорез проводят на минигеле 10 см х 10 см с 12% акриламида, используя количество образца, эквивалентное одному и тому же количеству клеток. Миграцию осуществляют в следующем буфере: 25 мМ Трис, 192 мМ глицина, 0,1% додецилсульфата натрия, рН 8,3.
Перенос на мембрану: белки переносят с геля SDS-PAGE на нитроцеллюлозную мембрану (буфер для переноса: 25 мМ Трис, 192 мМ глицина, 20% метанола, рН 8,3).
Инкубация с антителами: перенос проверяют по окрашиванию мембраны в присутствии Ponceau красного. Неспецифические центры на мембране блокируют 5%-ным обезжиренным молоком в забуференном Трис солевом растворе (10 мМ Трис-HCl, 150 мМ NaCI, pH 7,4) в течение 1 часа при комнатной температуре. Мембрану инкубируют в растворе моноклонального антитела анти-МхА (клон 143), разбавленном в 100 раз по объему смесью забуференного Трис солевого раствора с молоком и 3 раза промывают забуференным Трис солевым раствором с 0,1% Tween 20. Мембрану инкубируют со вторым антителом анти-мышь, связанным с пероксидазой (HRP, Sigma), разбавленным в 1000 раз по объему. Мембрану промывают 3 раза забуференным Трис солевым раствором с Tween. Полосы, содержащие МхА, выявляют после инкубации мембраны с хемилюминесцентными реагентами (ECL, Amersham Pharmacia). В этой системе HRP катализирует реакцию хемилюминесценции на основе люминола, который можно детектировать на пленке.
4.5.2. Результаты
DC-6 стимулируют в течение 0, 2, 4, 24 (фиг.7 7) или 48 час (фиг.78), как следует ниже.
Western blots, приведенные на фиг.77 и 78, демонстрируют продукцию МхА в течение времени: значительное увеличение белка МхА индуцируется продуктами ОМ-197-АС5 и OM-197-FV6 через 24 часа (фиг.77) и соответственно через 48 час (фиг.78). Эта продукция МхА эквивалентна продукции, индуцируемой положительным контролем: LPS или IFNα. Эта продукция выше продукции, индуцируемой, соответственно, отрицательным контролем, TRANCE (цитокин из семейства TNF-α, который индуцирует также созревание дендритных клеток) или CD40L (другой агент созревания дендритных клеток).
Соединения по изобретению ОМ-197-МР-АС и OM-197-FV6 проявляют антивирусные свойства благодаря их способности индуцировать белок МхА.
ПРИМЕР 4.6.
Определение способности соединений по изобретению активировать продукцию TNF-α одноядерными клетками периферической крови человека
4.6.1. Пропись
Одноядерные клетки периферической крови выделяют из "buffy coats" здоровых доноров. Клетки "buffy coats" переводят в суспензию и отделяют центрифугированием одноядерные клетки в градиенте Ficoll-Paque (Amersham Pharmacia). Очищенные одноядерные клетки повторно суспендируют в среде RPMI 1640 с добавкой 10% зародышевой телячьей сыворотки, антибиотиков, меркаптоэтанола и L-глутамина. Концентрацию клеток доводят до 2 × 106 живых клеток в 1 мл.
100 μл каждого из растворов ацилированных псевдодипептидов, имеющих вспомогательное функционализированное ответвление, разбавленных до 100, 10 и 1 μг/мл средой RPMI 1640, помещают на микроплашку. Образцы тестируют по три раза, причем на каждой микроплашке имеется отрицательный контроль, состоящий только из одной среды. Растворы LPS Е. coli (от 1 до 0,00001 цг/мл) служат в качестве положительного контроля.
По 100 μл клеточной суспензии добавляют в каждую лунку, содержащую растворы продуктов или контрольные растворы. Клетки инкубируют с продуктами в течение 18 час при 37°С в термостате, насыщенном влагой с 5% CO2.
В конце инкубации микроплашки центрифугируют, объединяют супернатанты и замораживают их при -80°С в виде образца до определения методом ELISA.
Концентрацию TNF-α, продуцированного одноядерными клетками, определяют с помощью хемилюминесцентного метода ELISA (kit R&D QuantiGlo # QTA00). Разбавленные в 4 раза супернатанты помещают в каждую лунку микроплашки, где находится в связанном состоянии антитело анти-TNF-α человека. Микроплашку инкубируют 4 часа при комнатной температуре. После промывки добавляют антитело анти-TNF-α человека, конъюгированное с пероксидазой. После 2 час инкубации и интенсивной промывки добавляют хемилюминесцентный субстрат и через 29 мин измеряют хемилюминесценцию. Концентрацию содержащегося в супернатанте TNF-α определяют по эталонной кривой в диапазоне от 7000 до 0,7 пг/мл.
Результаты выражены в пг/мл продуцированного TNF-α. Представленные результаты взяты из одного репрезентативного эксперимента на активность ацилированных псевдодипептидов.
4.6.2. Результаты
Продукция TNF-α одноядерными клетками периферической крови, индуцированная ацилированными псевдодипептидами, имеющими вспомогательное функционализированное ответвление и отвечающими общей формуле I
Отрицательный контроль, состоящий из среды RPMI, индуцирует базовую продукцию TNF-α ниже порога чувствительности, равного 0,7 пг/мл.
Продукция TNF-α одноядерными клетками периферической крови, индуцируемая липополисахаридом Е. coli
Положительный контроль, состоящий из LPS Е. coli, индуцирует сильную продукцию TNF-α даже при самых низких испытанных концентрациях.
Большая часть ацилированных псевдодипептидов индуцирует значительную продукцию TNF-α, даже если эта продукция ниже продукции, индуцируемой положительным контролем. При этом, чтобы индуцировать значимую продукцию TNF-α, нужны более высокие концентрации, чем концентрация LPS.
Некоторые соединения, ОМ-197-МС и ОМ-197-МС-МР, индуцируют значимую продукцию TNF-a лишь при концентрации 100 μг/мл.
Аминная функция представляет особый интерес для индуцирования продукции TNF-a, поскольку, с одной стороны, она увеличивет амплитуду и, с другой стороны, уменьшает минимальную концентрацию продукта, необходимую для индуцирования значимого ответа. Ацилированные псевдодипептиды с укороченными цепями: OM-197-N'С2-МС-АС и OM-197-N'(С10-2OС8)-МС-АС вообще не индуцируют продукцию TNF-a даже при концентрации 100 μг/мл.
ПРИМЕР 4.7.
Определение способности соединений по изобретению ингибировать продукции TNF-α одноядерными клетками периферической крови человека, индуцированное липополисахаридами Е. coli
4.7.1. Пропись
Одноядерные клетки периферической крови выделяют из "buffy coats" здоровых доноров. Клетки "buffy coats" переводят в суспензию и отделяют центрифугированием одноядерные клетки в градиенте Ficoll-Paque (Amersham Pharmacia). Очищенные одноядерные клетки повторно суспендируют в среде RPMI 1640, дополненной 10% зародышевой телячьей сывороткой, антибиотиками, меркаптоэтанолом и L-глутамином. Концентрацию клеток доводят до 2 х 106 живых клеток в 1 мл.
Ингибирование продуктами определяют путем преинкубации продуктов с одноядерными клетками и добавления через 1 час последовательных разведении LPS, индуцирующего продукцию TNF-α. Ингибирующая концентрация различных ацилированных псевдодипептидов, имеющих вспомогательное функционализированное ответвление, равно 10 μг/мл. LPS E. coli последовательно разбавляют от 1 до 0,00001 μг/мл (множитель 10 х).
50 μл продуктов, разбавленных до 10 μг/мл средой RPMI 1640, помещают на микроплашку. Каждый образец (10 μг/мл продукта + разбавленный LPS E. coli) трижды тестируют, причем на каждой микроплашке имеется отрицательный контроль, состоящий только из одной среды. Определение продукции TNF-α, индуцируемой только LPS (без ингибирования), производится добавлением к среде RPMI разведении LPS E. Coli.
По 100 μл клеточной суспензии добавляют в каждую лунку, содержащую разведения продуктов и контролей. Клетки инкубируют с продуктами в течение 1 часа в насыщенном влагой с 5% CO2 термостате при 37°С.
В конце преинкубации добавляют по 50 μл последовательных разведений LPS E. Coli. Инкубацию одноядерных клеток с продуктом и LPS проводят в течение 18 час. В конце стимуляции клеток микроплашки центрифугируют, а супернатанты объединяют, замораживают при -80°С в виде образца до определения методом ELISA.
Концентрацию TNF-α, продуцированного одноядерными клетками, определяют с помощью хемилюминесцентного метода ELISA (kit R&D QuantiGlo # QTA00). Разбавленные в 4 раза супернатанты помещают в каждую лунку микроплашки, где находится в связанном состоянии антитело анти-TNF-α человека. Микроплашку инкубируют 4 часа при комнатной температуре. После промывки добавляют антитело анти- TNF-α человека, конъюгированное с пероксидазой. После 2 час инкубации и интенсивной промывки добавляют хемилюминесцентный субстрат и измеряют хемилюминесценцию по истечении 20 мин. Концентрацию содержащегося в супернатанте TNF-α определяют по эталонной кривой в диапазоне от 7000 до 0,7 пг/мл.
Ингибирование характеризуется концентрацией LPS Е. Coli, который при совместной инкубации с продуктами индуцирует продукцию 50% TNF-α в сравнении с контролем, инкубируемым с одной RPMI. С повышением концентрации увеличивается ингибирующая активность продукта. Эту концентрацию рассчитывают из значений ингибирования продукции TNF-α, равной 100 - (продукция TNF-α, индуцированная продуктом и LPS) / (продукция TNF-α, индуцированная одним LPS) × 100. На основании этих данных по ингибированию концентрацию экстраполируют на отрезок прямой, соединяющей два разведения LPS с обеих сторон от ингибирования 50%.
Способность продуктов индуцировать ингибирование продукции TNF-α липополисахаридом характеризуется также максимальным ингибированием. Концентрация LPS, соответствующая этому максимальному ингибированию (ингибирование более 95%), характеризует также ингибирующую способность продуктов. Результаты рассчитаны из репрезентативного эксперимента, включающего все продукты.
4.7.2. Результаты
Ингибирование ацилированными псевдодипетидами, имеющими вспомогательное функционализированное ответвление и отвечающими общей формуле I, продукции TNF-α, индуцируемой липополисахаридом у одноядерных клеток человека
Отрицательный контроль, состоящий из одной среды RPMI, индуцирует базовую продукцию TNF-α ниже порога чувствительности, равного 0,7 пг/мл.
Все ацилированные псевдодипетиды способны индуцировать ингибирование продукции TNF-α, индуцируемой LPS Е. Coli. Концентрация продукта, необходимая для этого ингибирования, зависит от функций в вспомогательном ответвлении и от длины цепей жирных кислот. Даже ацилированные псевдодипетиды, связанные с лекарственными средствами сложноэфирной связью, способны индуцировать это ингибирование.
ПРИМЕР 4.8.
Определение способности соединений по изобретению активировать цитотоксичность клеток NK в отношении клеток-мишеней линии К562
4.8.1. Пропись
Одноядерные клетки периферической крови (РВМС) выделяют из "buffy coats" согласно прописи, приведенной выше в примере 4.6. При этом, после центрифугирования на Ficoll-Paque и тройной промывки, клетки были распределены на 6-лунковой плашке в расчете на 3 х 106 клеток/мл в 3 мл среды RPMI, содержащей 10% FCS, и инкубировались либо только с одной средой, либо будучи стимулированными IFN-γ (1000 ед/мл), LPS (1 μг/мл), ОМ-197-АС5 (1 μг/мл) или OM-197-FV6 (1 μг/мл). Клетки инкубировали в этих условиях в течение 24 час. Клетки К562 (клеточная линия больного лейкемией человека, АТСС #CCL 243, 20110-2209 Manassas (VA), USA) культивировали в среде RPMI, содержащей 10% FCS, с двумя пассажами в неделю.
Для измерения токсичности РВМС и клетки К562 дважды промывают HBSS и повторно суспендируют в среде RPMI без фенолового красного, содержащей 5% FCS. РВМС распределяют на плашке с 96 круглодонными лунками таким образом, чтобы получить отношения РВМС/мишени 20:1, 10:1 и 5:1 в объеме 50 μл. После этого в каждую лунку добавляют по 5000 клеток К562 (мишени) в 50 μл RPMI без фенолового красного. Конечный объем равен 100 μл. Плашку центрифугируют для улучшения контакта и затем инкубируют в течение 4 час при 37°С.
Цитотоксичность определяют с помощью нерадиоактивного набора CytoTox 96, основанного на измерении высвободившейся при клеточном лизисе лактатдегидрогеназы (LDH) (kit #G1780, lot #124100, Promega, Madison, WI), в соответствии с прописью поставщика.
Степень цитотоксичности рассчитывают по приведенной ниже формуле:
4.8.2. Результаты
Соединения ОМ-197-АС5 и OM-197-FV6 индуцируют активность NK такую же или несколько ниже активности, индуцируемой LPS, тестируемым в той же концентрации (1 μг/мл).
ПРИМЕР 4.9.
Определение способности соединений по изобретению индуцировать пролиферацию лимфоцитов мыши
4.9.1. Описание эксперимента
Объединяют клетки селезенки группы из 4 интактных мышей СВА, культивируют 4 раза в отсутствие или в присутствии адъюванта ОМ-197-МР-АС (от 0,1 до 10 μг/мл). Пролиферативный ответ этих клеток оценивают, измеряя инкорпорацию тритированного тимидина (3Н-TdR) после 1 или 2 дней культивирования. Приведенные в таблице значения представляют собой среднее арифметическое ± стандартное отклонение четырех культивирований.
Адъювант: приготовляют маточный раствор ОМ-197-МР-АС с концентрацией 0,9 мг/мл в воде для инъекций с добавкой 0,1% триэтаноламина.
Результаты
Продукт ОМ-197-МР-АС стимулирует пролиферацию клеток селезенки in vitro. Этот эффект, достигающий максимума после 1 дня культивирования, увеличивает инкорпорацию тимидина в 5 раз при концентрации 1 μг/мл и в 12 раз при концентрации 10 μг/мл. После 1 дня культивирования продукт ОМ-197-МР-АС в концентрации 1 μг/мл проявляет митогенетический эффект в такой же степени сильный, как Конканавалин А в концентрации 5 μг/мл (см. таблицу 1).
Таблица 1. Измерение пролиферативного ответа клеток селезенки мыши с адъювантом ОМ-197-МР-АС
Приведенные в таблице значения представляют собой среднее арифметическое ± стандартное отклонение измерений инкорпорации четырех культивирований. Конканавалин А (5 μг/мл), использованный в качестве положительного контроля, дает значение инкорпорации тимидина, равное 37,5±6,2×103 счетов/мин после 1 дня культивирования.
ПРИМЕР 4.10.
Определение антивирусной активности конъюгатов соединений по изобретению с 3'-азидо-3'-деокситимидином (AZT) и d4T в качестве предшественника лекарственного средства
4.10.1. Пропись
Лимфоцитарная линия Т МТ-4, иммортализованная инфекцией HTLV вируса, вызывающего одну из форм лейкемии, была использована для тестирования антивирусной (анти-ВИЧ) активности соединений по изобретению, конъюгированных с AZT и, соответственно, с d4T. Эти клетки обладают свойством не выживать при ВИЧ-инфицировании. Тест основан на защите с помощью продукта (такого как AZT) от уничтожающего эффекта вируса.
Продукты тестируют при последовательном разбавлении продукта на 96-лунковой плашке. В верхней части микроплашки продукты тестируют при разных концентрациях с клетками и вирусом (по три раза), что позволяет определить концентрацию продукта, которая защищает клетки от уничтожающего эффекта вируса. Концентрация, при которой наблюдают уменьшение на 50% числа живых клеток, есть IC50 продукта. Подсчет живых клеток производится с помощью МТТ - желтого тетразолиевого соединения, которое ферментативно восстанавливается (в митохондриях живых клеток) до формазана, имеющего пурпурный цвет. Лунки, которые содержат клетки в хорошем состоянии, имеют пурпурный цвет, а если клетки мертвы, сохраняется желтая окраска. Измерение оптической плотности (D) позволяет количественно оценить этот эффект.
Цитотоксичность продуктов оценивают таким же способом в нижней части плашки, где тест осуществляется путем разведения продуктов, инкубируемых на клетках без вируса, что позволяет определить токсический эффект продуктов на клетки. Он дается в виде СС50 - концентрации, при которой 50% клеток погибают в результате токсического действия продукта. Селективность (показатель селективности, Sl) продукта определяется как CC50/IC50. Этот параметр является важным: хороший продукт имеет повышенный показатель селективности.
4.10.2. Результаты
Продукты по изобретению, конъюгированные с AZT (ОМ-197-MC-succ-AZT) или с d4T, обладают антивирусными свойствами в отношении ВИЧ 1 IIIB и ВИЧ 2 ROD. Результаты двух экспериментов (по 3 раза) на клетках МТ-4:
Соединения по изобретению, конъюгированные с AZT или с d4T, демонстрируют антивирусную активность в модели с клетками МТ- 4. Одно из преимуществ этих предшественников лекарств состоит в липидной природе конъюгатов, которая делает возможным, с одной стороны, направлять соединения к инфицируемым клеткам, высвобождая антивирусные соединения в резервуарах вируса, и, с другой стороны, модулировать иммунологическую активность клеток-мишеней.
ПРИМЕР 4.11.
Определение способности соединений по изобретению активировать созревание предендритных клеток мыши в дендритные клетки
4.11.1. Пропись
Получение дендритных клеток: из бедренных и больших берцовых костей мышей (C57BI/6, возраст 8-10 недель) был извлечен костный мозг. Суспензия костного мозга была пропущена через клеточное сито 70 μм таким образом, чтобы получить одноклеточную суспензию. Клетки костного мозга были повторно суспендированы в среде Dulbecco, модифицированной Iscove (IMDM) с добавкой 10% зародышевой телячьей сыворотки, инактивированной нагреванием и антибиотиками. Клетки инкубировали в течение ночи при 37°С в атмосфере CO2 в насыщенном влагой термостате на микроплашках с 6 лунками. На следующий день (день 0) нелипкие клетки были собраны и промыты. Живые клетки были подсчитаны методом исключения трипановой сини. Концентрация клеток была доведена до 2×103 клеток/мл в среде IMDM, содержащей 20 нг/мл рекомбинантного GM-CSF мыши (rmGM-CSF) и 20 нг/мл рекомбинантного IL-4 мыши (rmIL-4), после чего инкубация была продолжена. На третий день были добавлены свежие rmGM-CSF и rmIL-4 в концентрации 10 нг/мл. На шестой день нелипкие клетки были собраны промыты и подсчитаны методом исключения трипановой сини. Концентрация клеток была доведена до 2×105 клеток/мл в среде IMDM, содержащей по 10 нг/мл rmGM-CSF и rmIL-4, после чего инкубация была продолжена. На восьмой день были собраны нелипкие клетки, состоящие из недоразвившихся и зрелых дендритных клеток, и использованы для инкубации с продуктами.
Инкубация с продуктами по изобретению: недоразвившиеся и зрелые дендритные клетки инкубировали в концентрации 5 х 105 клеток/мл на микроплашках с 24 лунками в присутствии следующих активаторов: положительный контроль LPS Е. Coli 0111:В4, 100 нг/мл; отрицательный контроль IMDM; а также с 2 соединениями по изобретению: OM-197-MC-FV5 и ОМ-197-МР-АС в концентрациях 0,1, 1 и 10 μг/мл. После 48 час инкубации и клетки были собраны и сохранены для анализов методом поточной цитометрии.
Анализ методом поточной цитометрии: недоразвившиеся и зрелые дендритные клетки были промыты забуференным фосфатом соляным раствором, содержащим 2% зародышевой телячьей сыворотки и 0,01% азида натрия (буфер FACS). После этого клетки инкубировали на льду в течение 30 мин с оптимальными концентрациями анти-CD86, анти-CD40, анти-МНС11, конъюгированного с FITC и анти-CD80, конъюгированного с пероксидазой. Далее клетки были трижды промыты, после чего была произведена маркировка анти-CD86 и анти-CD40 путем инкубации клеток на льду в течение 30 мин с антителами осла анти-Ig крысы, конъюгированного с пероксидазой. После инкубации клетки были промыты и повторно суспендированы в буфере FACS. На поточном цитометре FACScan™ была измерена флуоресценция. Проанализированы 15000 событий на образец. Клетки, которые инкубировали с одним конъюгатом, служили в качестве отрицательного контроля.
4.11.2. Результаты
Экспрессия клеток
MFI: средняя интенсивность флуоресценции
Продукты по изобретению способны индуцировать созревание предендритных клеток в дендритные клетки. Эти полученные in vitro результаты находятся в согласии с наблюдениями, проведенными на мышах in vivo.
in vivo: внутривенное или подкожное введение ОМ-197-МР-АС соответственно 20 μг и 5 μг на мышь индуцируют созревание и миграцию дендритных клеток в селезенке маргинальной зоны (расположенной между красной и белой пульпой) в направлении к белой пульпе, где локализованы клетки Т. Иммуноокрашивание гистологических срезов селезенок животных, получивших дозу продуктов по изобретению, выявляет совместную локализацию зрелых дендритных клеток и клеток Т в белой пульпе, так же как и при действии LPS, в то время как у животных, не получивших дозы, ни миграция, ни созревание не наблюдались.
ПРИМЕР 4.12.
Определение пролиферации стволовых клеток костного мозга мыши, стимулированной продуктами по изобретению
4.12.1. Эксперимент с пролиферацией
6-Недельные мыши-самцы C57/BL6 усыпляются путем ингаляции CO2. Отбираются кости задних конечностей, тазобедренный сустав, большеберцовая кость и бедренная кость. Костный мозг извлекается из просвета кости с помощью инъекции модифицированной Dulbecco среды Игла (среда DH) через предварительно рассеченные эпифизы. Стволовые клетки промывают и суспендируют в среде DH с добавкой 20% зародышевой телячьей сыворотки. Концентрацию клеток доводят до 500 000 клеток в 1 мл.
Продукты, растворенные в среде с добавками 20% зародышевой телячьей сыворотки, аминокислот и антибиотиков, последовательно разводят непосредственно на микроплашке. Продукты тестируют по три раза, причем на каждой микроплашке имеется отрицательный контроль, состоящий только из одной среды. Конечный объем в каждой лунке равен 100 μл.
По 100 μл клеточной суспензии добавляют к разведениям продуктов и инкубируют клетки в течение 7 дней в термостате при 37°С с 8% СО2 при насыщающей влажности. Пролиферацию определяют измерением окисления хромогенного субстрата ХТТ (2,3-бис[2-метокси-4-нитро-5-сульфофенил]-2Н-тетразолий-5-карбоксанилид) в митохондриях живых клеток.
В конце инкубации в каждую лунку добавляют по 50 μл раствора субстрата ХТТ (1 мг/мл ХТТ + 0,008 мг/мл феназинметосульфата). Инкубацию продолжают еще 8 час и затем спектрофотометрируют микроплашки при 490 нм, сравнивая с контрольной средой при 690 нм.
Результаты выражают как среднее измерений ± стандартное отклонение в виде кривой зависимости доза-ответ. Значения отрицательного контроля, состоящего из среды DH (среднее измерений ± стандартное отклонение) также приведены в графической форме.
На основании кривой доза-ответ рассчитываются 3 особые точки кривой:
Мах: максимальная амплитуда кривой и соответствующая ей концентрация;
ЕС50: концентрация, соответствующая 50% максимальной амплитуды;
Min: наинизшая концентрация, индуцирующая значимую пролиферацию, соответствующую контролю + утроенное стандартное отклонение.
Концентрации, соответствующие ЕС50 и min, определяют по сегментам прямой, соединяющим разные точки кривой.
Если кривая не достигает своего максимума и продолжает подъем при наибольшей из тестируемых концентраций, перед значениями «Мах» и «ЕС50» ставится знак «>».
4.12.2. Представление результатов
Пролиферация стволовых клеток костного мозга, индуцируемая ацилированными псевдодипептидами, имеющими вспомогательное функционализированное ответвление
Фиг.111 представляет активность различных ацилированных псевдодипептидов и доказывает влияние вспомогательных функционализированных ответвлений и распределения ацильных цепей на способность индуцировать пролиферацию стволовых клеток костного мозга мыши.
Изменения в ацилированных цепях существенным образом влияют на способность псевдодипептидов индуцировать пролиферацию стволовых клеток. Тетраацильные соединения, так же как и обращенные соединения, обладают в этой модели маргинальной активностью. Тип связи функционализированной группы с псевдодипептидом также меняет кривую доза-ответ для продуктов.
Минимальная концентрация продукта, способная индуцировать значимую пролиферацию стволовых клеток, выше минимальной концентрации положительного контроля, и, напротив, некоторые псевдодипептиды способны индуцировать такой же сильный ответ, какой индуцирует липополисахарид Е. coli.
Таблица, представляющая индуцирование пролиферации стволовых клеток костного мозга разными ацилированными псевдодипептидами, имеющими вспомогательное функционализированное ответвление и отвечающими общей формуле I:
D/μг/мл
D/μг/мл
D = оптическая плотность
Когда индуцируемая активность низка, невозможно определить активность ЕС50, поскольку она ниже минимальной активности.
Ацилированные псевдодипептиды по изобретению характеризуются большой разницей в способности индуцировать пролиферацию стволовых клеток костного мозга. Некоторые соединения обладают даже маргинальной активностью. В отличие от продуктов, образованных от липида А, увеличение числа ацильных цепей не коррелирует с активностью продуктов.
ПРИМЕР 4.13.
Определение продукции моноксида азота макрофагами мыши, стимулированными продуктами по изобретению
4.13.1. Эксперимент с продукцией моноксида азота:
6-Недельные мыши-самцы C57/BL6 усыпляются путем ингаляции СО2. Отбираются кости задних конечностей, тазобедренный сустав, большеберцовая кость и бедренная кость. Костный мозг извлекается из просвета кости с помощью инъекции модифицированной Dulbecco среды Игла (среда DH) через предварительно рассеченные эпифизы. Стволовые клетки промывают и суспендируют (до концентрации приблизительно 40000 клеток в 1 мл) в среде DH с добавкой 20% лошадиной сыворотки и 30% супернатанта L929. L929 представляют собой линию фибробластов мыши, супернатант которых обогащен фактором роста макрофагов (M-CSF). Клеточную суспензию распределяют по чашкам Петри, которые инкубируют в течение 8 дней при 37°С с 8% CO2 в насыщенном влагой термостате.
Через 8 дней стволовые клетки дифференцируются в зрелые макрофаги. Макрофаги отделяют действием холода, промывают и повторно суспендируют в среде DH с добавкой 5% зародышевой телячьей сыворотки, аминокислот и антибиотиков (среда DHE). Концентрацию клеток доводят до 700 000 клеток в 1 мл.
Продукты последовательно разводят средой DHE непосредственно на микроплашке. Продукты тестируют по три раза, причем на каждой микроплашке имеется отрицательный контроль, состоящий только из одной среды. Конечный объем в каждой лунке равен 100 μл.
По 100 μл клеточной суспензии добавляют к разведениям продуктов и инкубируют клетки в течение 22 час в термостате при 37°С с 8% СО2 при насыщающей влажности. В конце инкубации с продуктами отбирают по 100 μл супернатанта и определяют концентрацию нитрита с помощью реакции Грисса.
В каждую лунку добавляют по 100 μл реактива Грисса (5 мг/мл сульфаниламида + 0,5 мг/мл гидрохлорида N-(1-нафтил-этилендиамина)) в 2,5%-ной водной фосфорной кислоте. Микроплашки спектрофотометрируют при 562 нм в сравненнии с контрольной средой при 690 нм. Концентрация нитрита пропорциональна концентрации продуцированного моноксида азота. Концентрацию нитрита определяют в сравнении с эталонной кривой.
Результаты выражают как среднее измерений ± стандартное отклонение в виде кривой зависимости доза-ответ.
На основании кривой доза-ответ рассчитываются 3 особые точки кривой:
Мах: максимальная амплитуда кривой и соответствующая ей концентрация;
ЕС50: концентрация, соответствующая 50% максимальной амплитуды;
Min: наинизшая концентрация, индуцирующая значимую пролиферацию, соответствующую контролю + утроенное стандартное отклонение.
Концентрации, соответствующие ЕС50 и min, определяют по сегментам прямой, соединяющим разные точки кривой.
Если кривая не достигает своего максимума и продолжает подъем при наибольшей из тестируемых концентраций, перед значениями «Мах» и «ЕС50» ставится знак «>».
Если кривая не опускается ниже границы зоны «контроль + утроенное стандартное отклонение», минимальная концентрация дается как «<» самой низкой из тестируемых концентраций.
4.13.2.Представление результатов
Продукция моноксида азота макрофагами мыши, индуцируемая ацилированными псевдодипептидами, имеющими вспомогательное функционализированное ответвление
Фиг.112 представляет активность различных ацилированных псевдодипептидов и доказывает влияние вспомогательного функционализированного ответвления и модулирования ацильных цепей на способность индуцировать продукцию NO мышиными макрофагами.
Изменения в ацильных цепях существенным образом влияют на способность псевдодипептидов индуцировать продуцирование NO мышиными макрофагами как по амплитуде, так и по минимальной концентрации, способной индуцировать ответ. Тип связи функционализированной группы с псевдодипептидами также изменяет кривую доза-ответ для продуктов.
Минимальная концентрация продукта, способная индуцировать значимую продукцию NO мышиными макрофагами, значительно выше минимальной концентрации положительного контроля. Эта пороговая концентрация может существенным образом варьировать среди соединений по изобретению. Псевдодипептиды идуцируют более низкий по амплитуде ответ по сравнению с липополисахаридом Е. coli.
Таблица результатов, демонстрирующая индуцирование продукции моноксида азота мышиными макрофагами разными ацилированными псевдодипептидами, имеющими вспомогательное функционализированное ответвление и отвечающими общей формуле I:
μг/мл
На продукцию моноксида азота мышиными макрофагами сильно влияют как вспомогательные функционализированные ответвления, так и модулирование ацильных цепей, содержащихся в псевдодипептидах. Разнообразие функций, имеющихся в функционализированных ответвлениях, позволяет модулировать активность соединений даже тогда, когда имеются затруднения при выводе точных заключений, касающихся взаимоотношения структуры и активности. Некоторые соединения из-за их низкой активности могли бы служить в качестве антагонистов, в то время как более активные могли бы быть мощными агонистами.
ПРИМЕР 4.14
Тест на дифференцировку дендритных клеток
Была оценена способность продуктов по изобретению индуцировать созревание предендритных клеток в дендритные клетки. Методом цитометрии в потоке измерялись следующие параметры: потерю способности к фагоцитозу меченого флуоресцеином декстрана и экспрессию поверхностного маркера CD86.
4.14.1. Пропись эксперимента
Одноядерные клетки периферической крови выделяют из "buffy coats" (светлых слоев кровяного сгустка) здоровых доноров. Очищенные адгезионным способом моноциты суспендируют в среде RPMI-1640, содержащей 10% зародышевой телячьей сыворотки и по 10 нг/мл GM-CSF и IL-4, в расчете на 1×106 клеток в 1 мл. Клетки распределяют по чашкам Петри в расчете на 10×106 клеток в чашке и культивируют в течение 6 дней со сменой среды после 3 дней. Полученные таким образом клетки называют предендритными (DC-6). Созревание предендритных клеток в зрелые дендритные клетки осуществляют инкубацией с продуктами в концентрации 10 цг/мл или LPS (положительный контроль, 1 или 0,1 μг/мл) в течение дополнительных 3 дней.
На 9-ый день (DC-9) клетки собирают и параметры, свидетельствующие о созревании дентритных клеток (характеризующихся их большим размером и сложной внутренней архитектурой дифракция), измеряют на проточном цитометре (FACSCalibur BD Bioscience). Оценивают долю клеток, экспрессирующих поверхностный маркер CD86, или клеток, не инкорпорирующих Dextran-FITC.
4.14.2. Оценка результатов
Незрелые дендритные клетки (DC-6), возникшие при дифференцировке моноцитов, на самом деле конъюгированные с GM-CSF и IL-4, способны инкорпорировать Dextran-FITC. В процессе созревания клетки теряют способность инкорпорировать Dextran-FITC. Анализы проводятся на стадии дифференцировки DC-9.
Фагоцитоз декстрана, меченого флуоресцеином, и экспрессия CD86 после стимуляции предендритных клеток (DC6) ацилированными псевдодипептидами, имеющими вспомогательное функционализированное ответвление и отвечающими общей формуле I. Дифференцировка предендритных клеток в зрелые дендритные клетки.
% CD / флуоресценция
% CD / флуоресценция
Разница в усредненных величинах флуоресценции, обнаруженная, в частности, в случае маркировки CD86, обусловлена качеством клеток (доноров), а представленные результаты взяты из нескольких экспериментов.
При концентрации 10 μг/мл некоторые псевдодипептиды способны индуцировать дифференцировку предендритных клеток в дендритные клетки. Функции, имеющиеся во вспомогательном ответвлении, модулируют эту способность активировать предендритные клетки. Инверсия между Х и Y при одних и тех же функциях также может модулировать способность индуцировать созревание дендритных клеток. Соединение OM-197-AC-MP является практически неактивным в этой модели, в то время как соединение ОМ-197-МР-АС (прежний пример) было очень активным.
Еще более значимо изменение числа ацильных цепей, причем тетраацильные соединения обладают в настоящей модели маргинальной активностью.
Влияние составления композиции ацилированных псевдодипептидов особенно существенно для способности индуцировать дифференцировку предендритных клеток в дендритные клетки, причем некоторые тетраацильные соединения или соединения со слишком длинными функционализированными ответвлениями, например OM-197-MP-AD, очень плохо растворимы и очень мало активны.
5-ая серия примеров: фармакологическое исследование соединений по изобретению (in vivo)
ПРИМЕР 5.1.
Оценка свойств ацилированных псевдодипетидов, имеющих вспомогательное функционализированное средство, смешанных или сочлененных с (NANP)6Р2Р30 в иммунизационной модели мыши
5.1.1. Пропись эксперимента
Антиген: пептид (NANP)6P2P30 содержит повторяющуюся последовательность (NANP) из Plasmodium falciparum и две последовательности столбнячного токсина (P2: 830-843 и Р30: 947-967). Этот пептид известен как эпитоп "T-helper" и показал свою способность индуцировать повышенный и долговременный иммунный ответ в присутствии традиционных адъювантов. Пептид был получен синтетически по методу, описанному в литературе (Valmori et al., 1992, J. Immunol. 149, 717-721). Приготовлен маточный раствор антигена с концентрацией 0,4 мг/мл в 0,9%-ном водном растворе NaCI с рН 8,0.
Адъюванты: маточные растворы ацилированных псевдодипетидов (ОМ-197-МР, OM-197-FV6 и ОМ-197-МС) готовят с концентрацией 1 мг/мл в 0,9%-ном водном растворе NaCI с добавкой 0,1% триэтаноламина. Положительный контроль состоит из неполного адъюванта Фройнда (IFA de Difco, Detroit, MI, USA), а отрицательный контроль представляет собой 0,9%-ный водный раствор NaCI.
В рамках этой прописи эксперимента адъювант и антиген образуют в виде смеси или описанных выше конъюгатов. Были протестированы два типа конъюгатов (OM-197-FV)n(NANP)6Р2Р30. Они соответствуют разным степеням конъюгации, либо 1, 2 или 3 молекулы OM-197-FV на молекулу пептида (смесь моно-, би- и триконъюгата, n=l, 2 и 3, соответственно), либо 1 молекула OM-197-FV на молекулу пептида (моноконъюгат, n=l).
Иммунизация: 6-недельных мышей-самок (по 6 мышей в группе) иммунизируют в два приема с помощью 0,1-мл подкожной инъекции в основание хвоста. Применяемыми количествами являются 20 μг антигена и 50 цг адъюванта при первой инъекции и 10 μг антигена и 50 μг адъюванта при второй инъекции. В случае конъюгатов антигенная часть является определяющей для расчета инъецируемой дозы.
Таблицы экспериментальных групп:
Схема иммунизации и отбор проб
Отбор крови
Получение сыворотки: отбор крови производят в моменты 0 и 5 недель. Кровь оставляют на 60 мин при 37°С, затем выдерживают в течение ночи при 4°С. После этого сыворотку замораживают при -80°С до определения антител.
5.1.2. Определение антител IgG анти-(NANP)6P2P30
Количественное определение антител IgG, специфичным образом направленных к антигену (NANP)6P2P30, производится с помощью метода ELISA. Фиксацию антигена производят на микроплашке с 96 лунками (Maxisorp F 96, Nunc, DK) путем инкубации в течение ночи во влажном помещении при 4°С с 0,1 мл PBS (забуференного фосфатом соляного раствора), содержащего 0,001 мг/мл антигена (NANP)6P2P30. Насыщение микроплашки осуществляют с PBS, содержащим 1% бычьего сывороточного альбумина (BSA, Fluka, СН). Плашки промывают PBS, содержащим 0,05% Tween 20 (Sigma, Saint-Louis, МО, USA). Сыворотки, взятые за 5 недель, последовательно разводят буфером для разведения(PBS, содержащим 2,5% обезжиренного порошкового молока и 0,05% Tween 20), переносят на микроплашку и оставляют на 1 час при комнатной температуре. После этого плашки промывают PBS. Добавляют на плашки раствор для разведении, содержащий поликлональное антитело анти-IgG мыши, конъюгированное с щелочной фосфатазой (Sigma, Saint-Louis, МО, USA), и инкубируют 1 час при комнатной температуре. Плашки промывают PBS и выявляют специфичные антитела колориметрической реакцией с субстратом щелочной фосфатазы-п-нитрофенилфосфатом (Sigma, Saint-Louis, МО, USA). Измеряют поглощение при 405 нм с помощью считывающего устройства микроплашки (Dynatech 25000 ELISA reader, Ashford, Middlesex, UK), проводя каждую операцию дважды. Приведенные результаты представляют собой среднее измерений плотности при 405 нм, полученных для мышей из каждой группы.
5.1.3. Результаты: специфический ответ
Специфическая продукция антител Ig анти-(NANP)6P2P30, определенная методом ELISA, представлена графически для мышей, подвергшихся двойной иммунизации (фиг.79).
Две инъекции смеси антиген + адъювант (OM-197-FV6 или ОМ-197-МС) индуцируют серологический ответ несколько более высокий по сравнению с тем, который наблюдают при инъекции только одного антигена. Иммунизация моноконъюгатом OM-197-FV-(NANP)6P2P30 индуцирует ответ в виде антитела IgG несколько более высокий по сравнению с тем, который получают при использовании смеси антиген + OM-197-FV6. Более высокая степень конъюгации в (OM-197-FV)1,2,3-(NANP)6P2P30 не улучшает серологического ответа против этого антигена. Смесь адъюванта ОМ-197-МР и моноконъюгата OM-197-FV-(NANP)6Р2Р30 значительно увеличивает серологический ответ на этот антиген и превосходит значения, получаемые с положительным контролем, состоящим из смеси (NANP)6P2P30 + IFA.
Подобные же результаты получены, когда захват антител осуществляется по методу ELISA с использованием в качестве антигена пептида (NANP)6NA (фиг.80).
ПРИМЕР 5.2.
Группа иммунизации с помощью овальбумина: группа специфичных антител, генерируемых у мыши после 1 и 2 подкожных введений
5.2.1. Описание эксперимента
Цель этого исследования состоит в сравнении серологического ответа, индуцированного инъекцией смеси адъювант + антиген (ОМ-197-МР + овальбумин), с ответом, вызываемым конъюгатом с тем же антигеном (OM-197-FV-овальбумин). Для этой цели 30 мышей BALB/c (самки в возрасте 8 недель в начале обработки) были распределены по трем следующим группам:

Растворы:
Группа А: маточный раствор одного антигена (овальбумин, Fluka AG, Buchs, Швейцария) приготовлен в концентрации 125 μг/мл в Н2O+0,01% тиомерсала.
Группа В: маточный раствор смеси антиген + адъювант (овальбумин + ОМ-197-МР) содержит 125 μг/мл овальбумина и 250 μг/мл ОМ-197-МР в Н2O+0,01% тиомерсала.
Группа С: маточный раствор конъюгата OM-197-FV-овальбумин приготовлен в концентрации 125 μг/мл белка в Н20+0,01% тиомерсала.
Приготовление растворов для инъекции: каждый маточный раствор инкубируют 15 мин при 37°С, после чего во всех случаях к раствору добавляют адекватный объем 14%-ного NaCI для получения изотонического (0,9%-ный NaCI) раствора. Полученный материал встряхивают в течение 3 мин.
Иммунизация: инъекции производят в дни 0 и 14. Растворы вводят подкожно (100 μл в две разные точки, в сумме 200 μл на животное). Отбор крови производят в дни 14 и 28 (ретро-орбитальная пункция).
Количественное определение анти-овальбуминовых иммуноглобулинов: методом ELISA были по два раза определены следующие овальбумино-специфичные иммуноглобулины: IgGI, IgG2a и IgM. Для этого плашки с микролунками (NUNC Immunoplate, Roskilde, DK) инкубировали (coating overnight) при 4°С со 100 μл овальбумина (0,5 μг) в бикарбонатном буфере (рН 9,6). После промывки 0,5%-ным Tween-20 (Merck, Hohenbrunn, Германия) сыворотки разводили в 50, 200 и 800 раз (раствор для разведений: забуференный фосфатом соляной раствор (PBS) + 1% бычьего сывороточного альбумина (BSA, Sigma, St. Louis, Mo, USA) + 0,02% Tween-20)). По 100 μл каждой из разведенных сывороток были помещены в лунки. Инкубация длилась 45 мин при 37°С.
После второй промывки овальбумин-специфичные IgGI, IgG2a и IgM инкубируют 30 мин при 37°С со 100 μл антител (крыса анти-мышь) анти-IgGl, конъюгированных с пероксидазой (Serotec, Oxford, UK), IgG2a, конъюгированных с пероксидазой (Pharmingen, San Diego, CA, USA), предварительно разбавленными буфером PBS/BSA/Tween (разведения в 250, 1000 и 500 раз, соответственно). В случае IgM, после дополнительной промывки была необходима третья инкубация (30 мин при 37°С) с раствором 1/100 стрептавидина, конъюгированного с пероксидазой (Dako, Glostrup, DK).
После промывки добавляют 100 μл п-фенилендиамина (OPD, Merck, Darmstadt, Германия) для детекции пероксидазы, конъюгированной с вторичными антителами анти-IgGl, в то время как в случае IgG2a и IgM используемым реактивом является 3',3',5',5'-тетраметилбензидин (TMB, Sigma, St. Louis, Mo, USA). После 20-минутной инкубации при комнатной температуре реакцию останавливают добавлением 100 μл 2 н. H2SO4. Измеряют оптическое поглощение при 490 нм с помощью считывающего устройства для плашек Bio Rad 3550.
5.2.2. Результаты
Результаты каждого измерения при 490 нм выражены в произвольных единицах (пр.ед.) на мл. Эту возможность обеспечило сравнение каждого образца с эталоном, приготовленным путем разведении ряда образцов на 28-ой день из группы А (животные, инъецированные одним овальбумином). По определению, ряд образцов, разбавленных в 50 раз, обладают концентрацией 1000 пр.ед./мл. После этого отдельные результаты были скорректированы на соответствующий коэффициент разведения (50, 200 или 800 раз) и приведены на фиг.81, 82 и 83: полые кружки соответствуют среднему значению за 28 дней (сокращение Ova означает овальбумин).
Полученные результаты указывают на то, что конъюгат ОМ-197-ГУ-овальбумин является особенно иммуногенным в исследуемой модели, поскольку после второй инъекции он значительно увеличивает специфичные анти-овальбуминовые антитела, продуцируемые мышью - речь идет об исследуемом подклассе иммуноглобулинов (IgGI, IgG2a и IgM). В частности, нужно отметить весьма значительное увеличение анти-овальбуминовых IgG2a (фиг.84), что наводит на мысль о преимущественно ориентированном иммунитете ТН1. Нековалентная смесь овальбумин + ОМ-197-МР дает более низкий ответ по IgG2a по сравнению с конъюгатом.
ПРИМЕР 5.3.
Группа иммунизации с помощью гемагглютинина H1N1: определение специфичных антител, генерируемых мышью после 1 и 2 подкожных введений
5.3.1. Описание эксперимента
Цель этого исследования состоит в сравнении серологического ответа, индуцированного инъекцией смеси адъювант + антиген (ОМ-197-МР + H1N1), с ответом, вызываемым конъюгатом с тем же антигеном (OM-197-FV-H1N1). Для этой цели 30 мышей BALB/c (самки в возрасте 8 недель в начале обработки) были распределены по трем следующим группам:
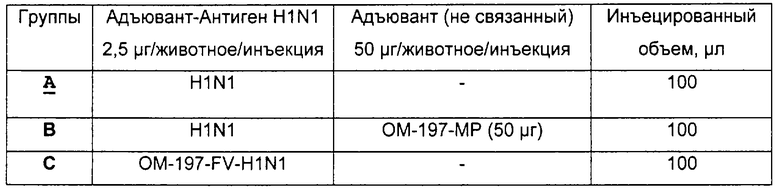
5.3.2. Приготовление растворов для инъекций
Группа А: маточный раствор H1N1 (гемагглютинин A/Beijing 262/95, Solvay Duphar, Weesp, Нидерланды) приготовлен в концентрации 25 μг/мл в H2O.
Группа В: маточный раствор смеси H1N1 + ОМ-197-МР содержит 25 μг/мл H1N1 и 500 μг/мл ОМ-197-МР в Н2О.
Группа С: маточный раствор OM-197-FV-H1N1 (H1N1 ковалентно связан с OM-197-FV) с концентрацией 25 μг/мл в Н2O.
Каждый маточный раствор инкубируют 15 мин при 37°С, после чего к 2 мл каждого раствора добавляют по 135 μл 14%-ного NaCI. Полученный материал встряхивают в течение 3 мин.
5.3.3. Инъекции и количественное определение анти-HlNl
Инъекции производят в дни 0 и 14. Растворы вводят подкожно (50 μл в две разные точки, в сумме 100 μл на животное). Отбор крови производят на 14-ый день (ретро-орбитальная пункция).
HlNl-специфичные IgM определяли по два раза методом ELISA. Для этого плашки с микролунками (NINC Immunoplate, Roskilde, DK) инкубировали (coating overnight) при 4°С со 100 μл H1N1 (0,5 μг) в бикарбонатном буфере (рН 9,6). После промывки 0,5%-ным Tween-20 (Merck, Hohenbrunn, Германия) сыворотки разбавляли в 50, 200 и 800 раз (раствор для разведений: забуференный фосфатом соляной раствор (PBS) + 1% бычьего сывороточного альбумина (BSA, Sigma, St. Louis, Mo, USA) + 0,02% Tween-20)). По 100 μл каждой из разбавленных сывороток были помещены в лунки. Инкубация длилась 45 мин при 37°С.
После второй промывки HlNl-специфичные IgM инкубируют 30 мин при 37°С со 100 μл антител (крыса анти-мышь) анти-IgGl, конъюгированных с биотином ((Pharmingen, San Diego, CA, USA), предварительно разбавленными буфером Tween (разбавление в 500 раз). После дополнительной промывки была проведена третья инкубация (30 мин при 37°С) с раствором 1/100 стрептавидина, конъюгированного с пероксидазой (Dako, Glostrup, DK).
После промывки добавляют 100 μл раствора 3',3',5',5'-тетраметилбензидина (ТМВ, Sigma, St. Louis, Мо, USA) для детектирования пероксидазы, конъюгированной с вторичными антителами анти-IgM. После 20-минутной инкубации при комнатной температуре реакцию останавливают добавлением 100 μл 2 н. H2SO4). Измеряют оптическое поглощение при 490 нм с помощью считывающего устройства для плашек Bio Rad 3550.
5.3.4. Результаты
Результаты каждого измерения при 490 нм выражены в произвольных единицах (пр.ед.) на мл. Эту возможность обеспечило сравнение каждого образца с эталоном, приготовленным путем разведении ряда образцов на 28-ой день из группы А (животные инъецированные одним H1N1). По определению, ряд образцов, разбавленных в 50 раз, обладают концентрацией 1000 пр.ед./мл. После этого отдельные результаты были скорректированы на соответствующий коэффициент разведения (50, 200 или 800 раз) и приведены на фиг.84: заполненные кружки соответствуют среднему значению за 28 дней.
Мыши, иммунизированные конъюгатом OM-197-FV-H1N1, имеют титр антител IgM анти-HlNl, более высокий по сравнению с контролем, состоящим из одного антигена H1N1, а также контролем, состоящим из смеси ОМ-197-МР + H1N1.
ПРИМЕР 5.4.
Оценка свойств адъюванта ОМ-197-МР-АС в модели мыши, иммунизованной подкожно антигеном Leishmania LmCPb
5.4.1. Описание эксперимента
Мышам СВА осуществляют в хвост одну подкожную инъекцию (100 μл) антигена паразита Leishmania LmCPb (3 μг на мышь) ± адъювант. Образованы две группы по 6 мышей, которые были обработаны следующим образом: один антиген (контрольная группа) и, соответственно, антиген + 50 иг ОМ-197-МР-АС.
Через 11 дней после инъекции клетки паховых и периаортальных лимфатических ганглиев (2 группы по 3 мыши) 3 раза культивирют в присутствии очищенного антигена LmCPb или цельного экстракта паразита (амастиготной формы). На 4-ый день отбирают супернатант и хранят его при -20°С для количественного определения IL-4 и ifn-y. Наконец, для измерения пролиферации к культурам добавляют тритированный тимидин (3Н-TdR).
Количественное определение цитокинов осуществляют методом sandwich ELISA (наборы OptEIA™ IL-4 и IFN-y, BD, PharMingen, Basel, Швейцария) и оценивают пролиферативный ответ, измеряя инкорпорацию тимидина (3Н-TdR). Измерение инкорпорации 3Н-TdR, выраженное в счетах/мин в виде среднеарифметического ± стандартное отклонение (2 группы по 3 мыши), и концентрация IL-4 даются индивидуально для каждой группы из 3 мышей в виде среднеарифметического из двух измерений (определение цитокинов).
Антиген: приготовляют маточный раствор LmCPb в концентрации 150 μг/мл в PBS.
Адъюванты: приготовляют маточный раствор ОМ-197-МР-АС в концентрации 0,9 мг/мл в воде для инъекций с добавкой 0,1% триэтаноламина.
Смесь антиген-адъювант: препарат адъюванта (4 объема), рассчитанный на конечную концентрацию в стерильном PBS, смешивают с маточным раствором антигена (1 объем) непосредственно перед инъекцией.
5.4.2. Результаты
Для мышей СВА, иммунизированных антигеном Leishmania LmCPb, достаточно одной дозы 50 μг адъюванта ОМ-197-МР-АС для индуцирования увеличения (от 2 до 6 раз) пролиферации ганглионарных клеток in vitro в качестве ответа на очищенный антиген (от 0,6 до 15 цг/мл) или на цельный экстракт амастиготной формы паразита (от 2 до 50 х 10-6/мл). Эти результаты приведены в таблице 1. Обработка мышей ОМ-197-МР-АС приводит также к появлению в супернатанте ганглионарных культур, стимулированных паразитарным антигеном, значительных количеств IL-4 (см. таблицу 2), в то время как продукция IFN-γ не детектируется (менее 100 пг/мл).
Таблица 1. Пролиферативный ответ in vitro ганглионарных лимфоцитов мышей СВА, иммунизированных LmCPb: влияние адъюванта ОМ-197-МР-АС, одного или в комбинации с CpG-ДНК
(СРМ)
Приведенные в таблице 1 значения представляют среднеарифметическое ± стандартное отклонение измерений инкорпорации, проведенное в двух группах лимфатических ганглионов (по 3 мыши на группу; каждую группу культивировали по 3 раза).
Таблица 2.Продукция IIL-4 in vitro лимфоцитами мышей СВА, иммунизированных LmCPb: влияние адъюванта ОМ-197-МР-АС, одного или в комбинации с CpG-ДНК
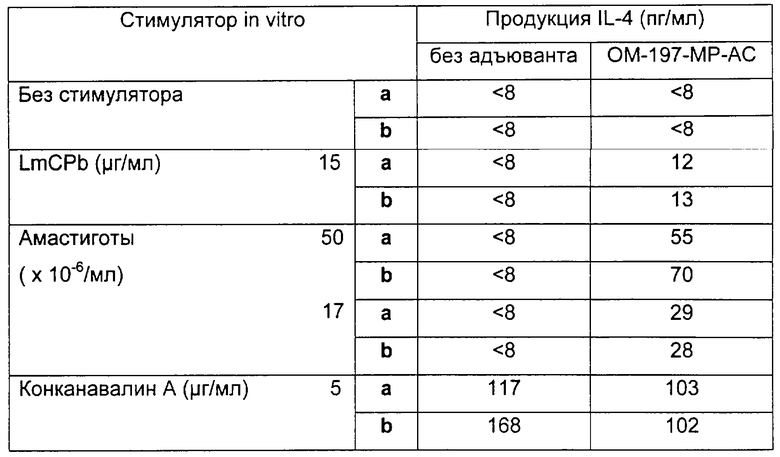
Приведенные в таблице значения представляют среднеарифметическое двух измерений цитокинов, проведенных в двух супернатантах двух групп (а и b; 3 мыши на группу) ганглионарных клеток, культивируемых по 3 раза.
Заключение
Продукт ОМ-197-МР-АС усиливает у мыши СВА, иммунизированной LmCPb (протеаза, изобилующая в амастиготной фазе паразита Leishmania), специфичный ответ Т, оцениваемый in vitro путем измерения лимфоцитарной пролиферации. Тот же адъювант способствует развитию лимфоцитов анти-LmCPb, секретирующих значительные количества IL-4.
ПРИМЕР 5.5.
Оценка на иммунизационной модели мыши свойств ацилированных псевдодипептидов, имеющих вспомогательное функционадизированное средство, связанных или смешанных с синтетическими пептидами белка циркумспорозоита Plasmodium yoelii (PyCS 252-260)
Целью этого эксперимента является оценка способности ацилированного псевдодипептида, имеющего вспомогательное функционализированное ответвление, привитого к или смешанного с синтетическим пептидом PyCS 252-260 белка циркумспорозоита Plasmodium yoelii индуцировать клеточный и гуморальный иммунный ответ специфичный по отношению к пептиду.
5.5.1. Пропись эксперимента
Комбинирование антигенов с адъювантами: синтетический пептид PyCS 252-260, соответствующий эпитопу Т белка циркумспорозоита Plasmodium yoelii последовательности SYVPSAEQI (таблица 1). Пептид 2 MR99B (SERSYVPSAEQI) был получен добавлением аминокислот SER к N-концу, а пептид 1 MR99A (KGGKGGKSERSYVPSAEQI) был получен добавлением аминокислот лизин-глицин-глицин, соединенных с М-концом описанного выше пептида MR99B, при этом пептиды были получены синтезом на твердой фазе (F-moc no Merrifield и Atherton (Atherton et al., Bioorg. Chem. 8: 350-351. 1979) с использованием реактивов и растворителей от фирм Bachem Feinchemikallien (Buddendorf, Швейцария), Novabiochem (Laufelfingen, Швейцария) и Fluka (Buchs, Швейцария). Пептиды очищали с помощью ЖХВР с обращенной фазой.
Моно- и поликонъюгаты пептидов с ацилированным псевдодипептидом, имеющим вспомогательное функционализированное ответвление (OM-197-MC-FV) были получены как описано в примере 3.6 ([OM-197-MC-FV]n-пептид 1) и примере 3.5 (ОМ-197-MC-FV-пептид 2). Стерильные конечные растворы конъюгированных пептидов были лиофилизованы.
Иммунизация мышей: доза антигена соответствует 20 цг пептида 2 на мышь в одной инъекции (таблица 2). Перед инъекцией лиофилизированные препараты растворяют в 0,9%-ном NaCI, встряхиваются в течение 3 мин и подвергаются 10-минутной обработке ультразвуком при 50°С. Составы с IFA готовят смешением одного объема антигена (14,5 цг пептида PyCS 252-260 + 50 цг РЗО (универсальный эпитоп Т) в PBS) и одного объема адъюванта.
4-Недельным мышам BALB/c (Harlan, Zeist, Нидерланды) проводят подкожную инъекцию в основание хвоста. Инъекция составов производится с помощью игл 23'' с конечным объемом 50 μл. Животные получают вторую дозу антигена в начале 5-ой недели и третью инъекцию в течение 9-ой недели.
Антигены
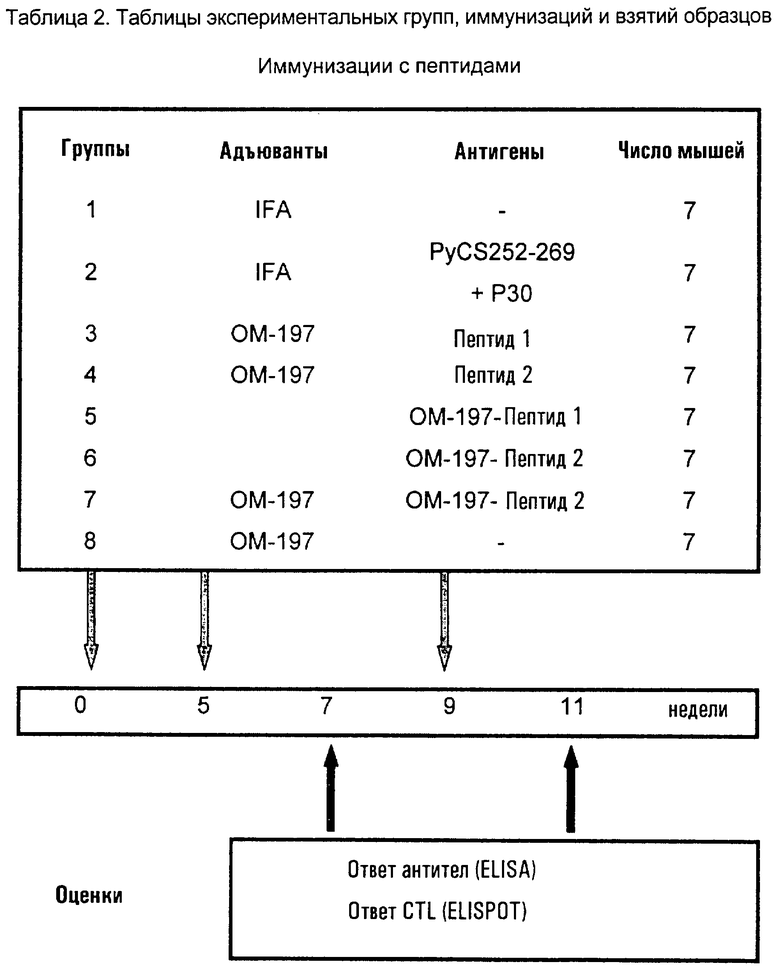
Отбор крови и лимфоидных органов:
Получение сыворотки: отбор крови проводят у всех мышей после 7 и 11 недель. Кровь оставляют на 60 мин при 37°С, после чего оставляют на ночь при 4°С. После этого сыворотку замораживают при -80°С для определения антител Отбор паховых ганглиев и селезенки: 2 животных из каждой группы забивают, соответственно, после 7 и 11 недель. Паховые ганглии и селезенку забирают хирургически и культивируют клетки для развития активности CTL методом ELISPOT (ответ интерферон-γ)
Определение титра антител анти-пептид-1:
Количественное определение антител, специфичным образом направленных к пептиду 1 (KGGKGGKSERSYVPSAEQI), производится с помощью метода ELISA. Фиксацию антигена производят на микроплашке с 96 лунками (Maxisorp F 96, Nunc, DK) путем инкубации в течение ночи во влажном помещении при 4°С с 0,05 мл PBS (забуференного фосфатом соляного раствора), содержащего 0,001 мг/мл пептида 1. Плашки промывают PBS, содержащим 0,05% Tween 20 (Sigma, Saint-Louis, МО, USA) (PBS-T) и производят в течение 1 часа насыщение микроплашки 5% снятого молока в PBS-T при комнатной температуре. Отдельные сыворотки (А, В, С, D, Е, F, G) мышей, взятые за 9 и 11 недель, последовательно разбавляют буфером для разведения (PBS, содержащим 2,5% обезжиренного порошкового молока и 0,05% Tween 20), после чего переносят на микроплашку и оставляют на 1 час при комнатной температуре. После этого плашки промывают PBS и добавляют на плашки раствор для разведении, содержащий поликлональное антитело козы анти-IgG мыши, конъюгированное с щелочной фосфатазой (Sigma, Saint-Louis, МО, USA)? и инкубируют 1 час при комнатной температуре. Плашки промывают PBS и выявляют специфичные антитела колориметрической реакцией с субстратом щелочной фосфатазы -п-нитрофенилфосфатом (Sigma, Saint-Louis, МО, USA). Измеряют поглощение при 405 нм с помощью считывающего устройства для микроплашки (Dynatech 25000 ELISA reader, Ashford, Middlesex, UK), проводя каждую операцию дважды. Приведенные результаты представляют собой среднее измерений плотности при 405 нм, полученных для мышей из каждой группы. Титр антитела определяют по последнему разведению, индуцирующему значимый положительный ответ, т.е. при значении оптической плотности выше значения фонового шума плюс 3 стандартных отклонения.
Количественные определения методом ELIPSOT-интерферон-γ:
Специфичные антитела анти-интерферон-γ мыши (01Е703В2) фиксируют инкубацией в течение ночи при 4°С во влажном помещении раствора антитела с концентрацией 50 иг/мл на микроплашке ELIPSOT, дно лунок которой покрыто нитроцеллюлозой (Millipore, Molsheim, F). Стадию насыщения проводят путем добавления DMEM (Life Technologies, Grand Island, NY, USA), содержащего 10% зародышевой телячьей сыворотки (FCS, Fakola, CH), в течение 2 час при 37°С. Клетки, полученные из лимфоидных органов (паховых ганглиев и селезенки) культивируют на микроплашках в количестве 200 000 или 400 000 клеток в лунке и затем культивируют в течение 24 час при 37°С совместно со 100 000 клеток, содержащих антиген (предварительно сенсибилизированных в течение 1 часа с пептидом PyCS 252-260 при 37°С и облученных дозой 10 крад с последующей 3-кратной промывкой) или в присутствии Конканавалина А в качестве контроля. После инкубации клетки удаляют и после промывки добавляют в течение 2 час второе антитело - биотинилированный анти-IFN-y мыши (ANI, 2 иг/мл в PBS с 1% бычьей сывороточного альбумина). Далее добавляют стрептавидин, конъюгированный с щелочной фосфатазой (Boehringer Mannheim, Mannheim, Германия), разбавленный 1:1000, инкубируют 1 час при 37°С и трижды промывают PBS, содержащим 0,05% Tween 20, и затем 3 раза PBS. Присутствие иммунных комплексов анти-IFN-y выявляется путем добавления субстрата BCIP/NBT (Sigma, St-Louis, МО, USA). Реакцию останавливают промывкой проточной водой. После этого подсчитывают IFN-y-положительные бляшки с помощью автоматического считывающего устройства Biosys GmbH (Karben, Германия). Специфические бляшки соответствуют разнице между числом бляшек, подсчитываемых в присутствии клеток, сенсибилизированных с пептидом, и числом бляшек, подсчитываемых в отсутствие пептида. Представленные результаты являются средним из значений, полученных для мышей из каждой группы. Они выражаются числом бляшек на миллион взятых для культивирования клеток.
Результаты
Гуморальный ответ представлен на фиг.85 (2 недели после второй инъекции) и на фиг.86 (2 недели после третьей инъекции). После двух иммунизации положительные ответы наблюдались у 5 из 7 животных из группы 5 (животные, иммунизированные пептидом 1, конъюгированным с помощью вспомогательного ответвления с 4 единицами ацилированного псевдодипептида). В этой группе доля мышей, у которых наблюдается ответ, увеличивается в случае третьей инъекции, поскольку мыши В и D обладают значимым, но низким титром антител. Группа 3, иммунизированная тем же пептидом в смеси с тем же адъювантом, не образует значимого титра антител. Особенно заметный ответ наблюдается после 3 иммунизации в группе 7 (моноконъюгат с пептидом 2, дополненный свободным адъювантом ОМ-197). Другие группы, получившие короткий пептид или никакого пептида не проявляют ответа антителами.
Таким образом, тетраконъюгат ОМ-197-пептид 2 способен индуцировать ответ антителами у всех иммунизированных мышей с титром от среднего до низкого.
Оценку клеточной активности производят по частоте CTL, секретирующих интерферон-γ, специфичных эпитопу PyCS 252-260. Результаты приведены в таблице 3. Положительный контроль представлен группой 2, а отрицательный контроль одними адъювантами (группы 1 и 8). Контроль стимуляции клеток Конканавалином А является положительным для всех мышей. Группы 5 (тетраконъюгат) и 7 (моноконъюгат + адъювант) дают положительные CTL-ответы.
В заключение следует сказать, что пептид, конъюгированный с 4 молекулами ацилированного псевдодипептида (ОМ-197-пептид 1), является эффективным, в то время как моноконъюгат (ОМ-197-пептид 2) не индуцирует CTL. Напротив, моноконъюгат, дополненный свободным ОМ-197, индуцирует CTL и в то же время он не индуцирует продукцию антител, что, по-видимому, обусловлено малым размером пептида 2 (12 минокислот).
Таблица 3: ответ CTL на пептид PyCS 252-260
ПРИМЕР 5.6.
Оценка пирогенности ацилированных псевдодипептидов в модели кролика
Целью этого эксперимента является определение наиболее высокой концентрации ацилированного псевдодипептида, имеющего привитое вспомогательное функционализированное ответвление, которая не вызывает повышения температуры у кролика при внутривенном введении
5.6.1. Пропись:
15 кроликов (New Zealand White Rabbits) были распределены по следующим группам:
Группа 1: 1,0 мг/кг
Группа 2: 0,1 мг/кг
Группа 3: 0,01 мг/кг
Группа 4: 0,001 мг/кг
Группа 5: вода для инъекций
В день тестирования кроликов взвешивают и обездвиживают в предусмотренных для этой цели ящиках. Датчики температуры вводят в их задний проход.
Через 1 час после ввода датчиков температуры (прединъекция) в течение 90 мин регистрируют их показания и определяется среднее значение.
После этого каждый кролик получает внутривенно продукт или контроль (стерильная вода с 0,9% NaCI) через латеральную ушную вену. Температуру замеряют каждые 30 мин в течение 3 час. Рассчитывают и регистрируют разницу между средней температурой прединъекции и максимальной температурой.
Для 3 кроликов на группу тест рассматривается как непирогенный, если сумма трех ответов не превышает 1,15°С, и пирогенным, если сумма превышает 2,65°С. Если полученное значение находится между двумя указанными выше, тест проводят заново с 3 дополнительными кроликами.
Результат
Результаты теста на пирогенность с соединениями ОМ-197-МС-МР и OM-197-MC-Asp приведены ниже:
Соединения по изобретению не являются пирогенными в дозах ниже 0,1 мг/кг.
5.6.2. Пропись:
15 кроликов (New Zealand White Rabbits) были распределены по следующим группам:
Группа 1: 0,9 мг/кг
Группа 2: 0,09 мг/кг
Группа 3: 0,009 мг/кг
Группа 4: 0,0009 мг/кг
Группа 5: вода для инъекций
В день тестирования кроликов взвешивают и обездвиживают в предусмотренных для этой цели ящиках. Датчики температуры вводят в прямую кишку.
Через 1 час после ввода датчиков температуры (прединъекция) в течение 90 мин регистрируют их показания и определяется среднее значение.
После этого каждый кролик получает внутривенно инъекцию продукта или контроля (стерильная вода с 0,9% NaCI) через латеральную ушную вену. Температуру замеряют каждые 30 мин в течение 3 час. Рассчитывают и регистрируют разницу между средней температурой прединъекции и максимальной температурой. Для 3 кроликов на группу тест рассматривается как Непирогенный, если сумма трех ответов не превышает 1,15°С, и пирогенным, если сумма превышает 2,65°С. Если полученное значение находится между двумя указанными выше, тест проводят заново с 3 дополнительными кроликами.
Результат
Результаты теста на пирогенность с соединением ОМ-197-МР-АС:
Соединение по изобретению не пирогенно при наиболее высокой из тестируемых доз - 0,9 мг/кг.
Тест с промежуточной дозой 0,009 мг/кг не был повторен, поскольку 2 более высокие дозы были непирогенными. Низкая пирогенность соединений по изобретению подтверждает данные по эндотоксичности in vitro, определенные в тесте LAL.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИММУНОМОДУЛЯТОРНЫЕ СОЕДИНЕНИЯ И ЛЕЧЕНИЕ ЗАБОЛЕВАНИЙ, СВЯЗАННЫХ СО СВЕРХПРОДУКЦИЕЙ ВОСПАЛИТЕЛЬНЫХ ЦИТОКИНОВ | 2007 |
|
RU2498813C2 |
| ФУНКЦИОНАЛИЗИРОВАННЫЕ БЕТА 1,6 ГЛЮКОЗАМИН-ДИСАХАРИДЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2007 |
|
RU2481352C2 |
| НОВЫЕ АЦИЛИРОВАННЫЕ ПСЕВДОДИПЕПТИДЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 1999 |
|
RU2223262C2 |
| ИНГИБИТОРЫ ДИПЕПТИДИЛПЕПТИДАЗЫ IV НА ОСНОВЕ КОНДЕНСИРОВАННЫХ ЦИКЛОПРОПИЛПИРРОЛИДИНОВ И СПОСОБ ИХ ПРИМЕНЕНИЯ | 2001 |
|
RU2286986C2 |
| ПРОФИЛАКТИЧЕСКОЕ И ТЕРАПЕВТИЧЕСКОЕ ЛЕЧЕНИЕ ИНФЕКЦИОННЫХ И ДРУГИХ ЗАБОЛЕВАНИЙ С ПОМОЩЬЮ ИММУНОЭФФЕКТИВНЫХ СОЕДИНЕНИЙ | 2002 |
|
RU2288723C2 |
| ПРОИЗВОДНЫЕ 2-ОКСО-1-ПИРРОЛИДИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2001 |
|
RU2291860C2 |
| ЗАМЕЩЕННЫЕ 4-ФЕНИЛТЕТРАГИДРОИЗОХИНОЛИНЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ЛЕКАРСТВЕННЫХ СРЕДСТВ, А ТАКЖЕ СОДЕРЖАЩИЕ ИХ ЛЕКАРСТВЕННЫЕ СРЕДСТВА | 2004 |
|
RU2343147C2 |
| ПОЛУЧЕНИЕ ПЕПТИДОВ СОМАТОСТАТИНА | 2004 |
|
RU2360921C2 |
| ИНГИБИТОРЫ КАСПАЗ | 1999 |
|
RU2274642C2 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛА ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ | 1991 |
|
RU2076100C1 |
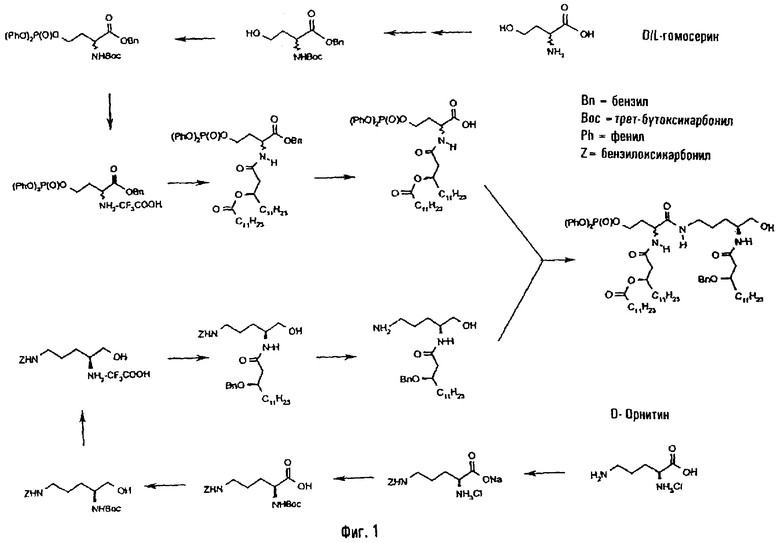
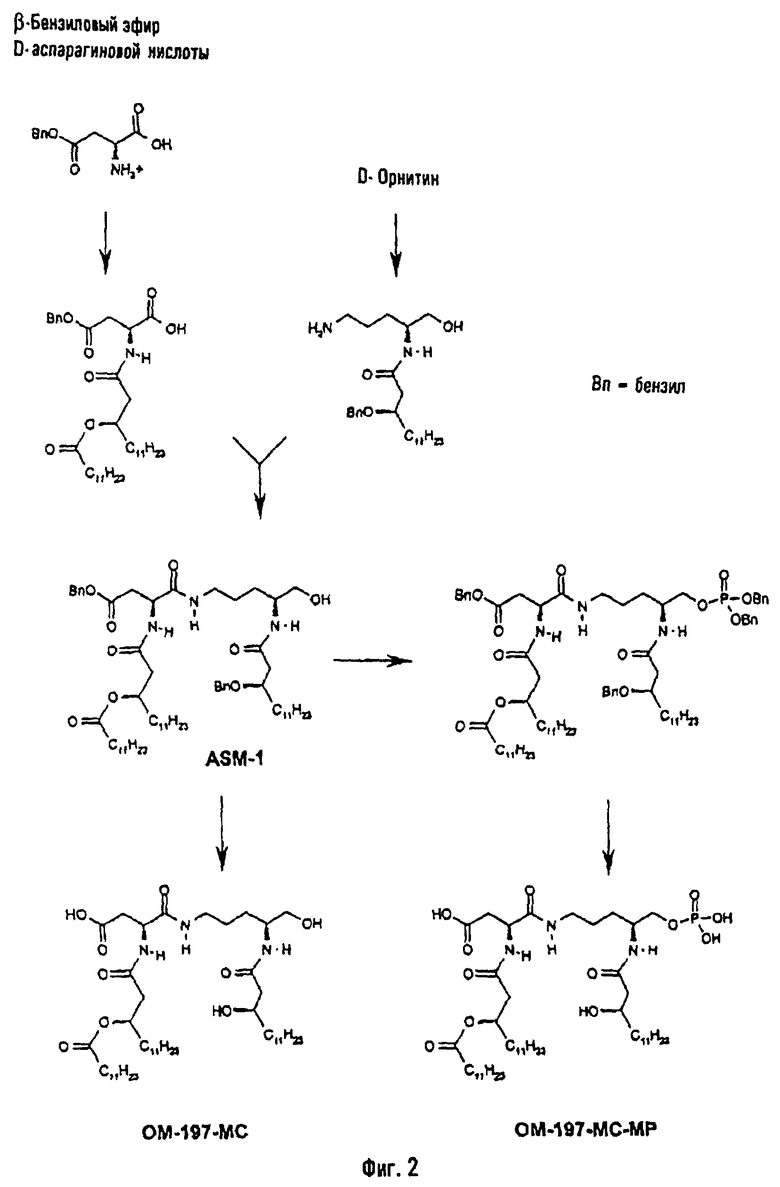
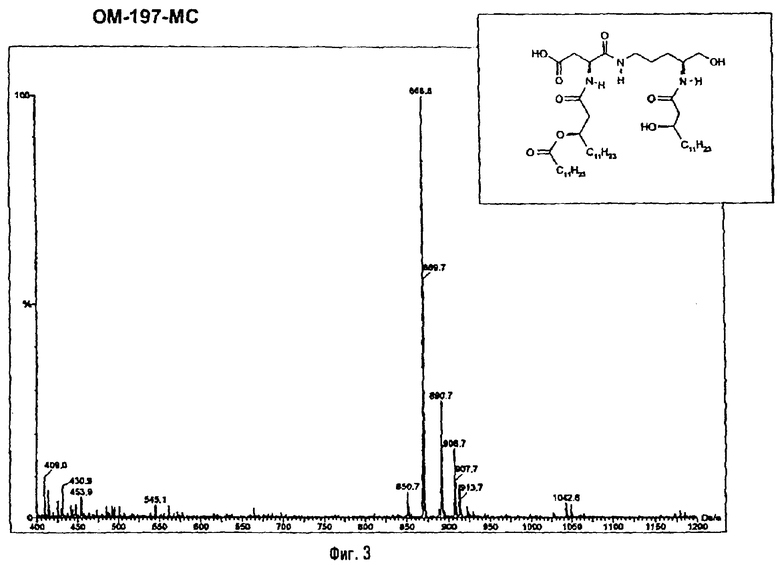
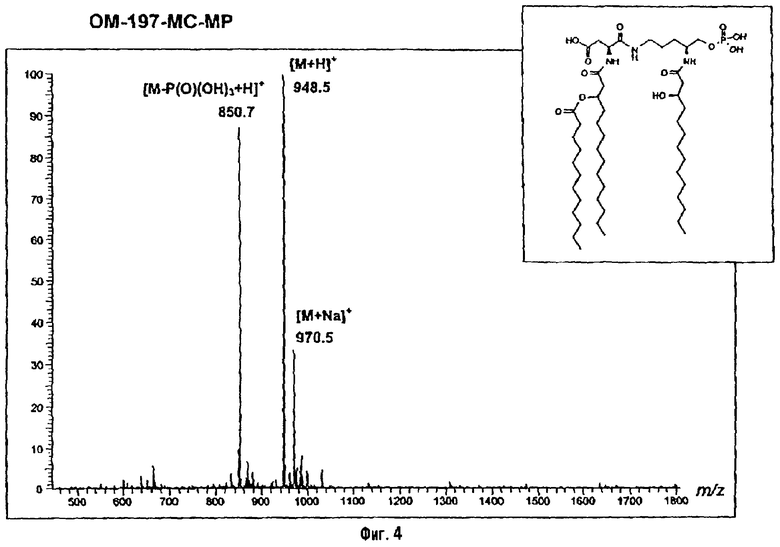

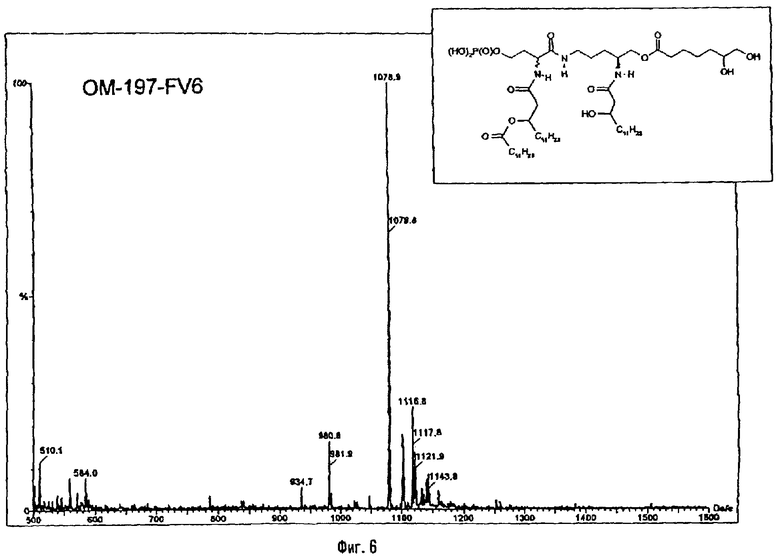
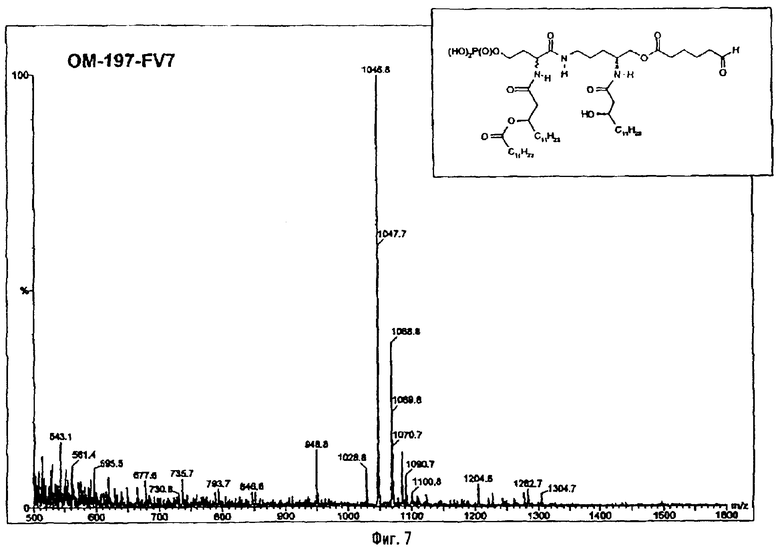
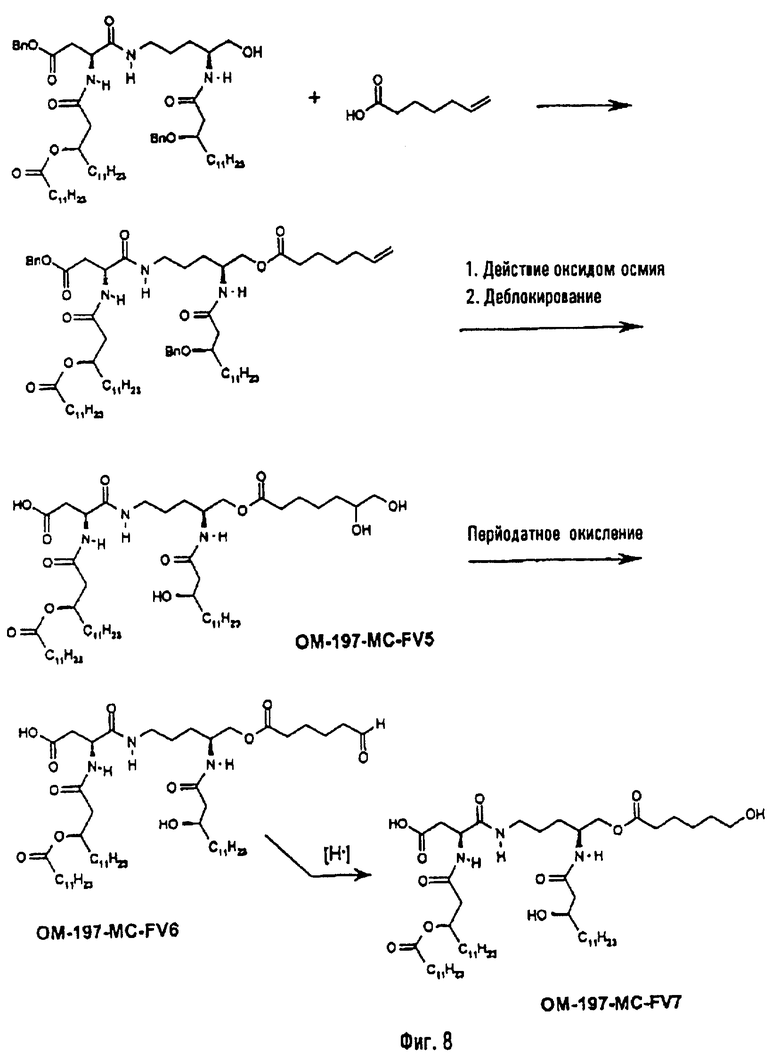
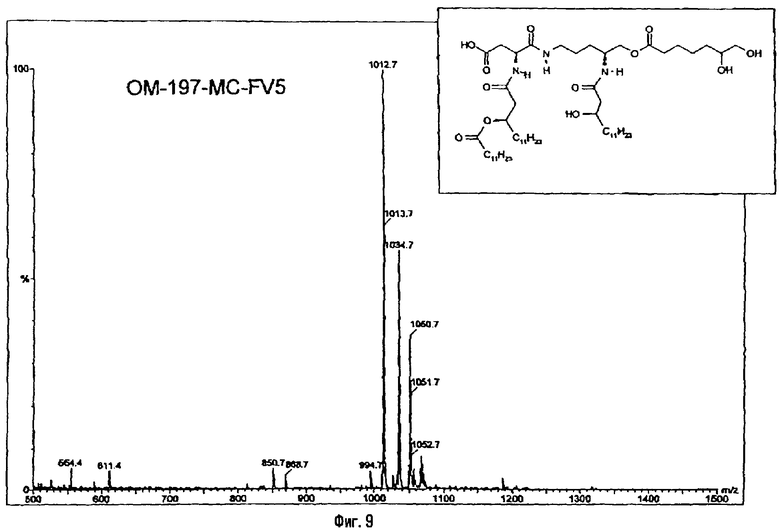
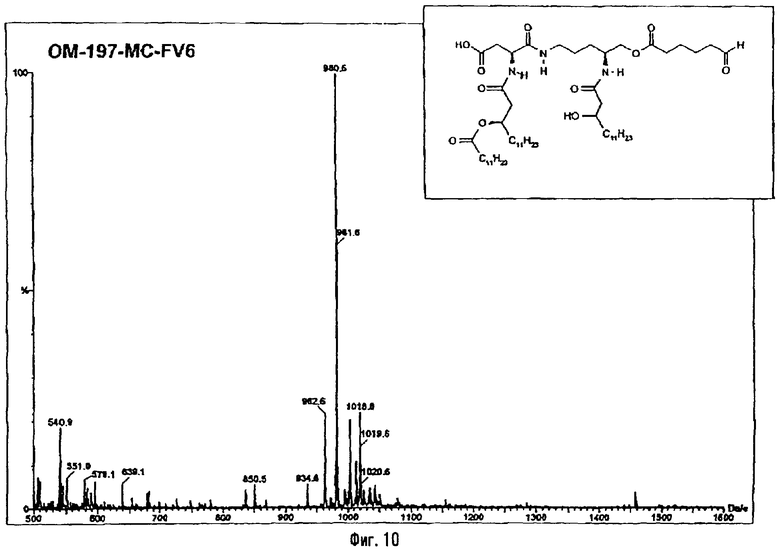
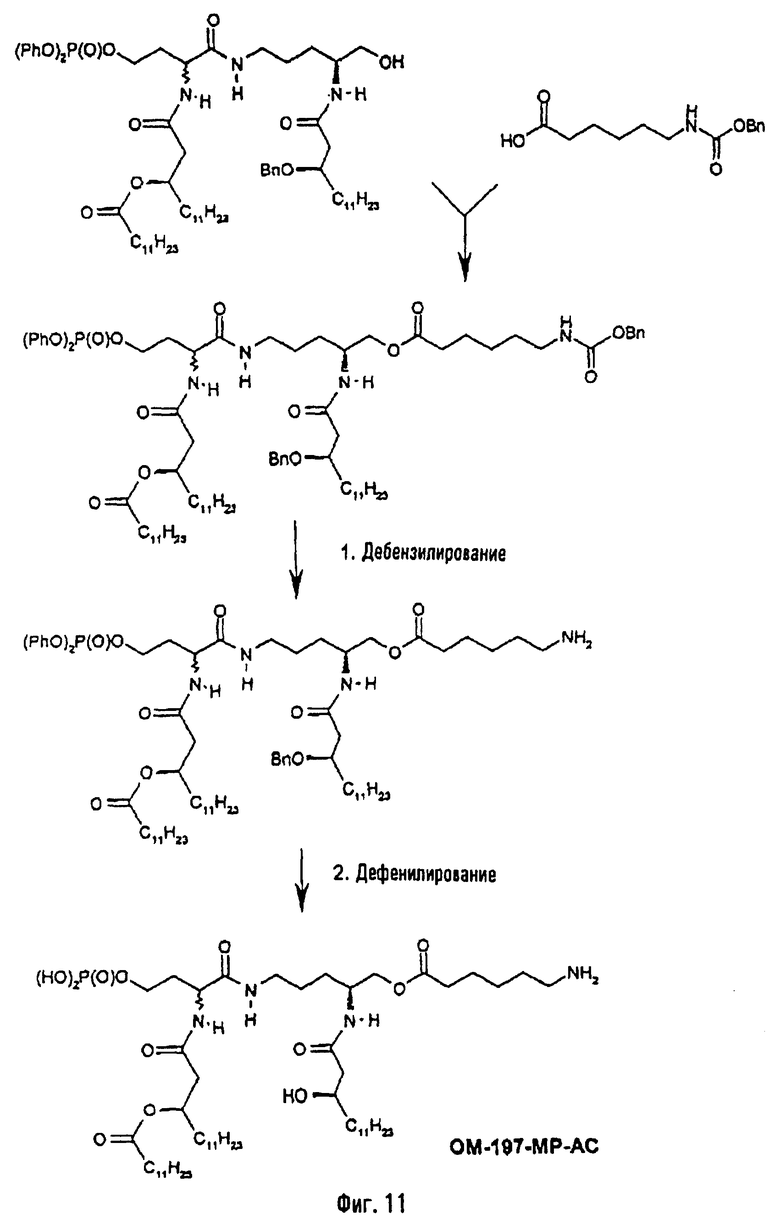
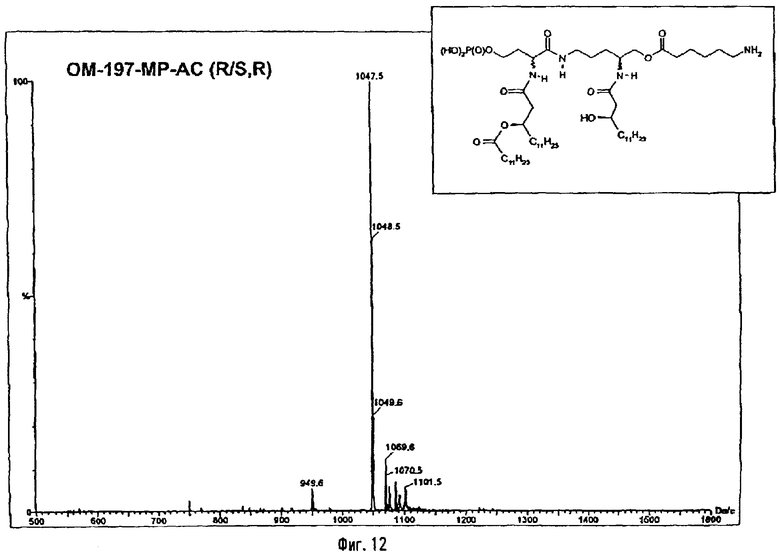
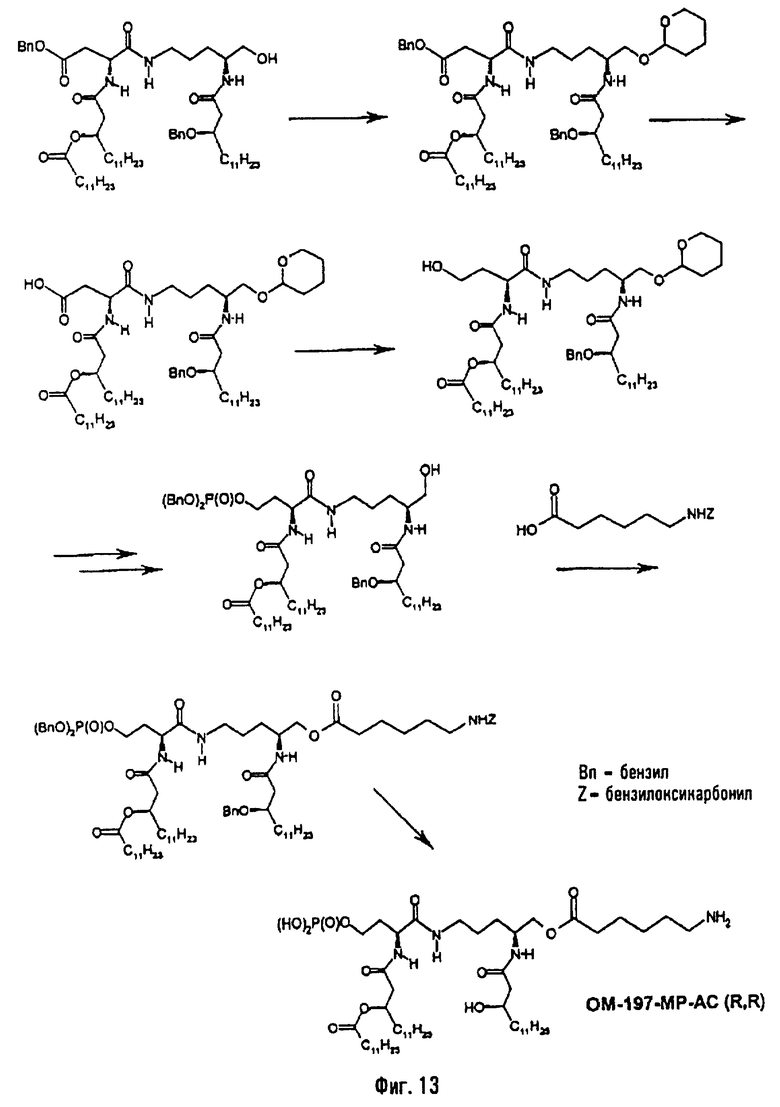
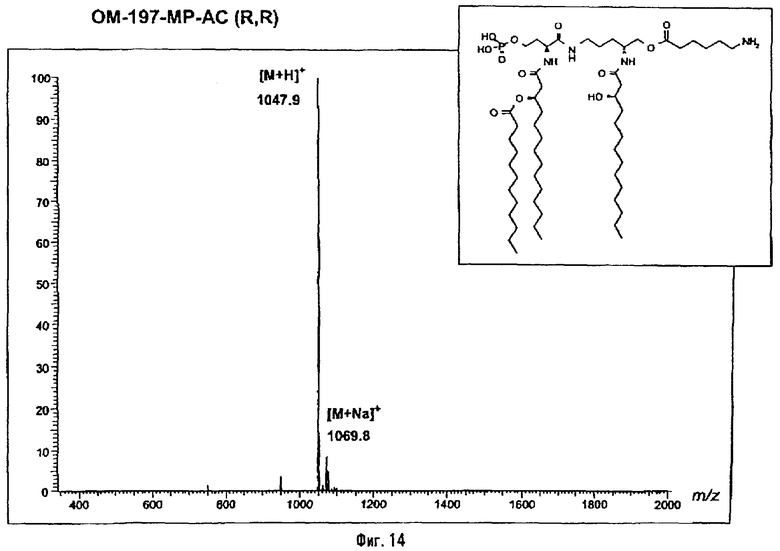
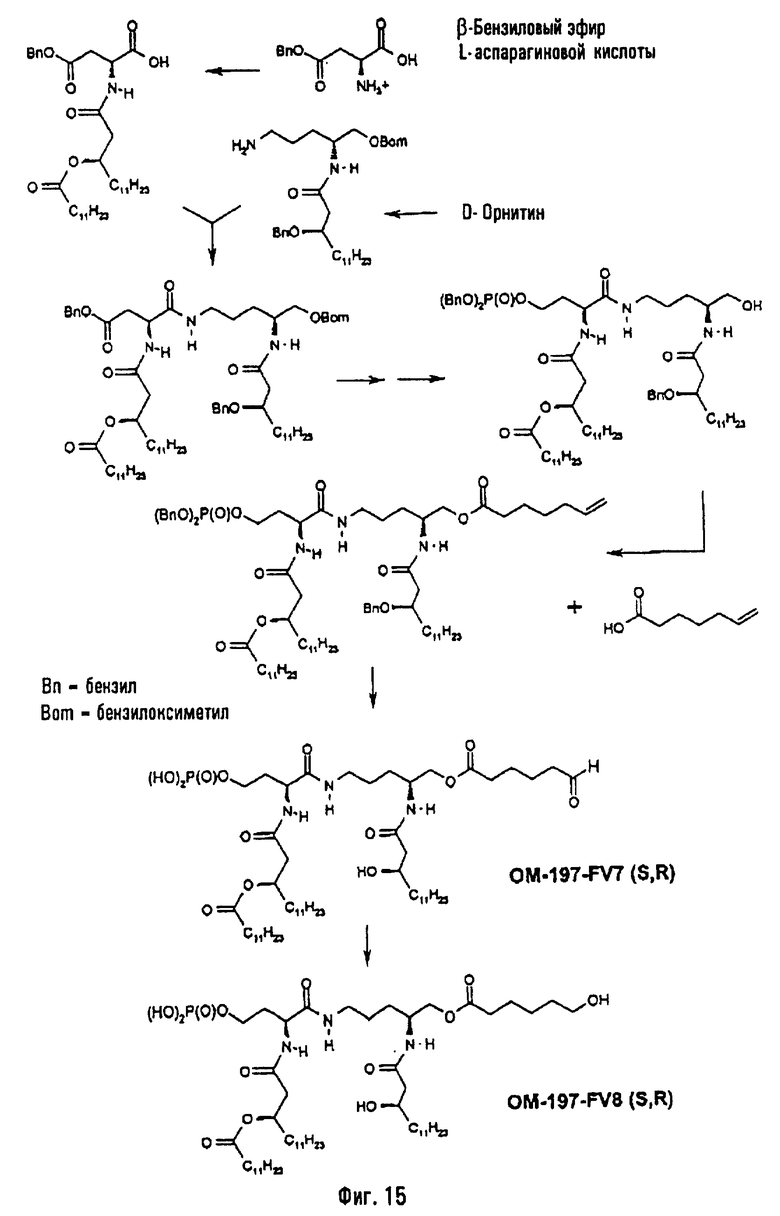
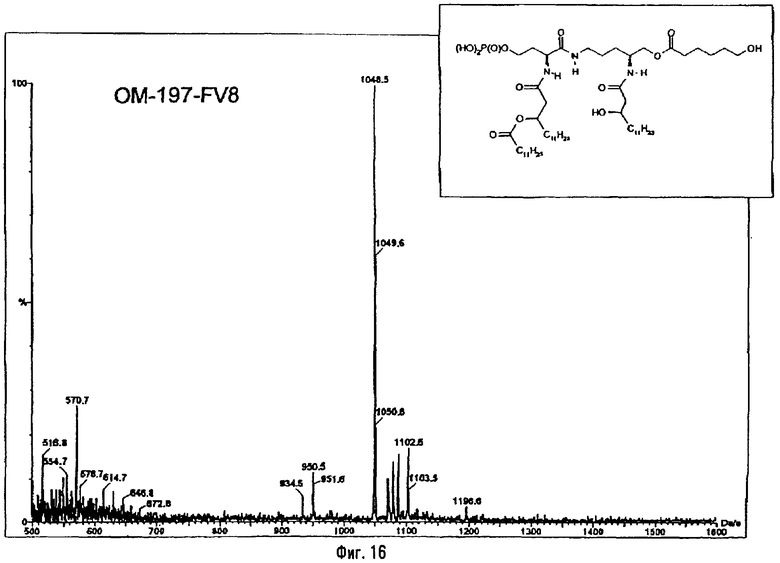

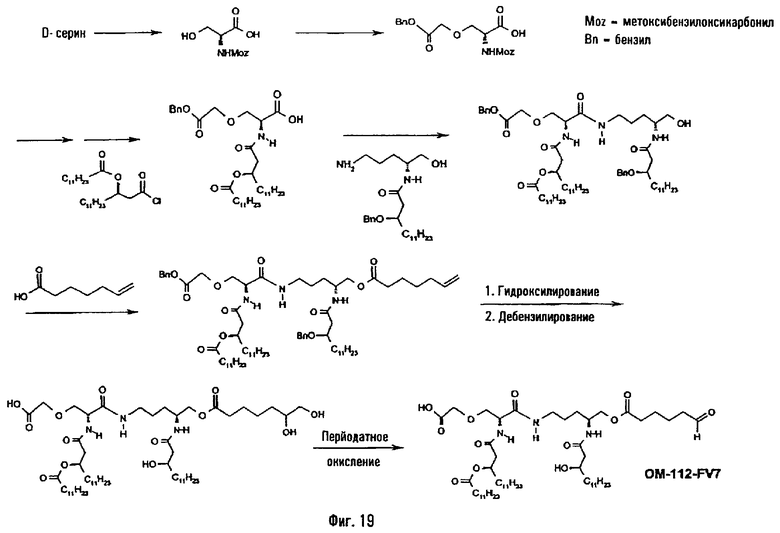
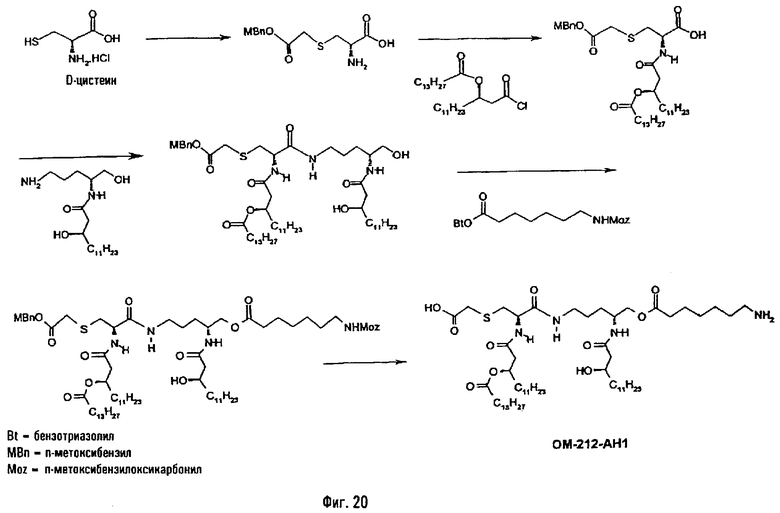
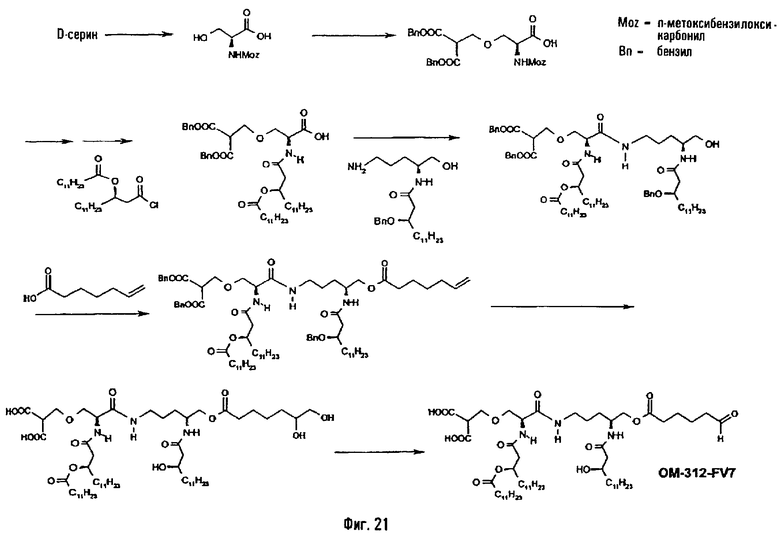
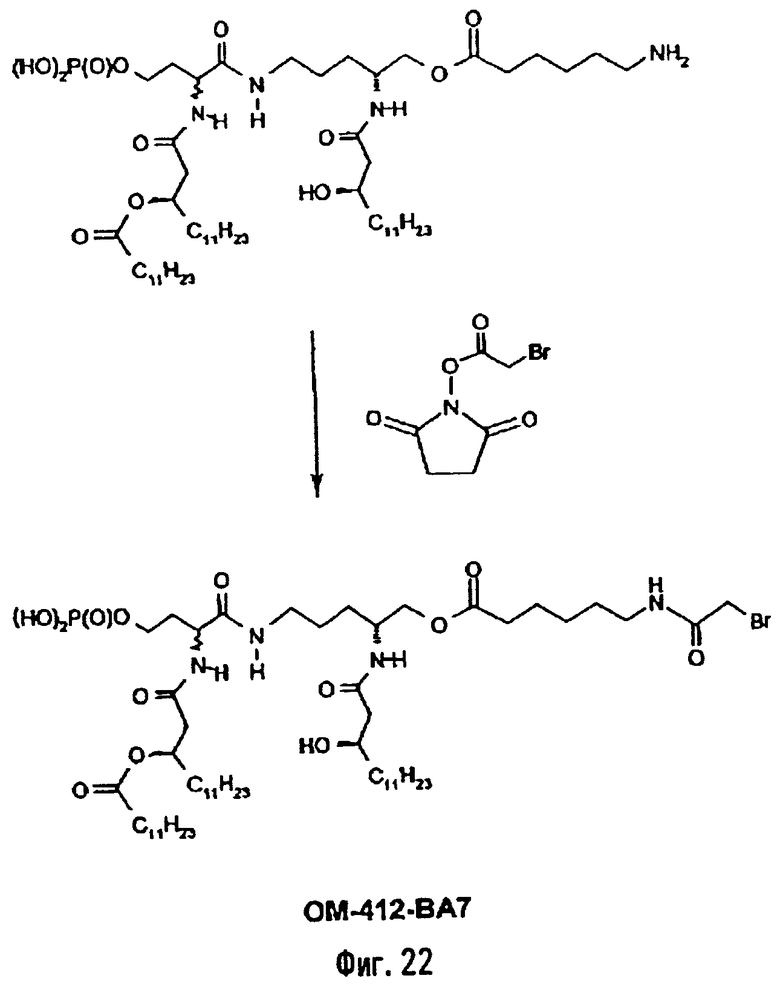
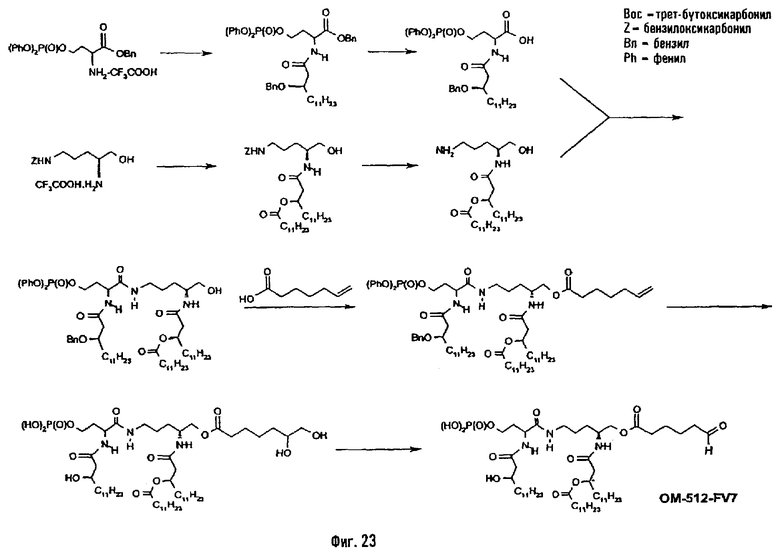

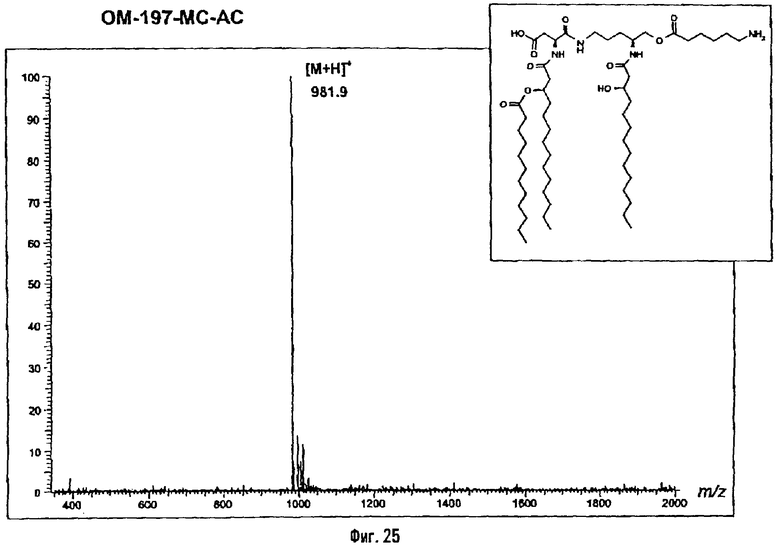
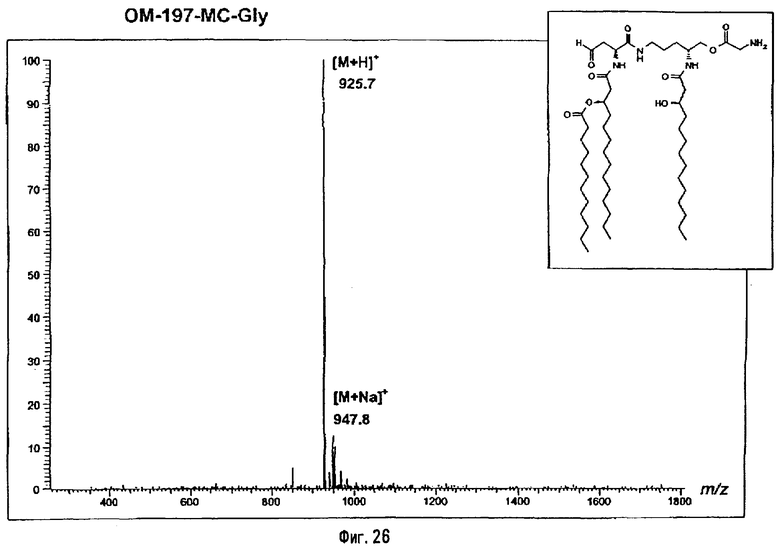
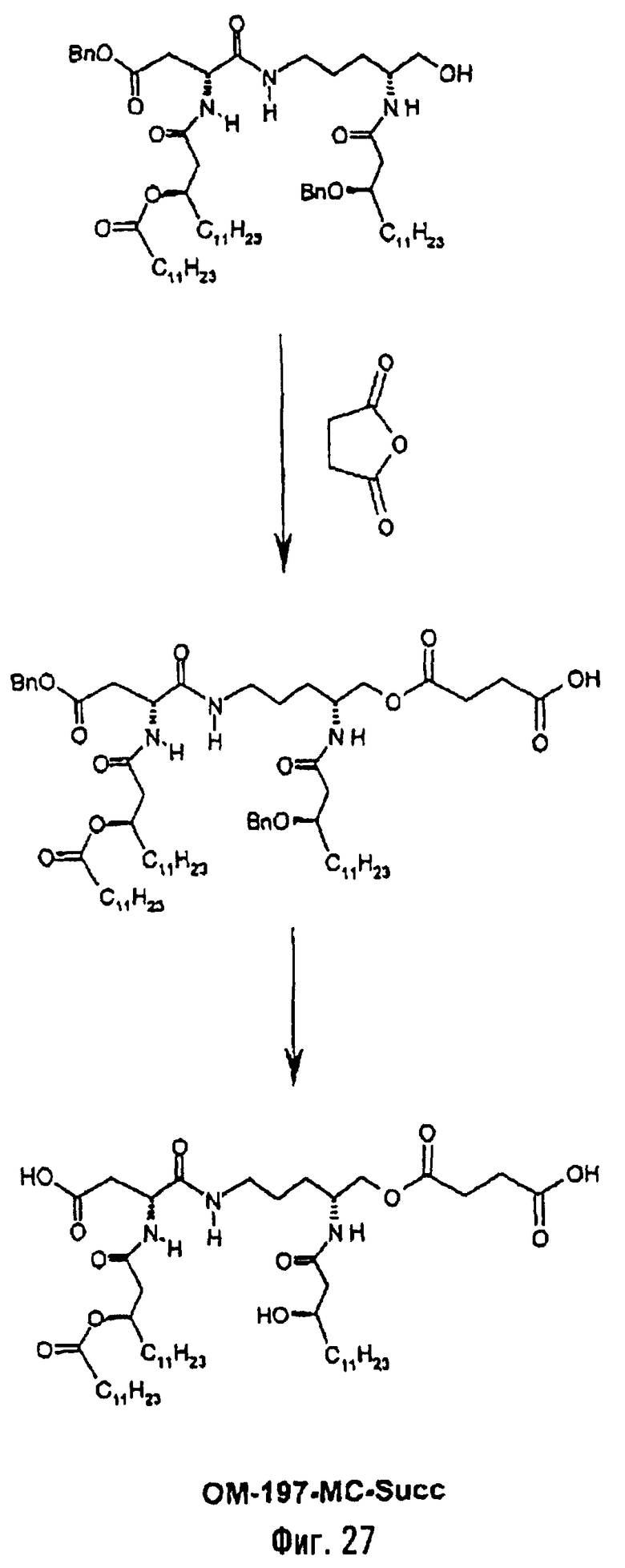
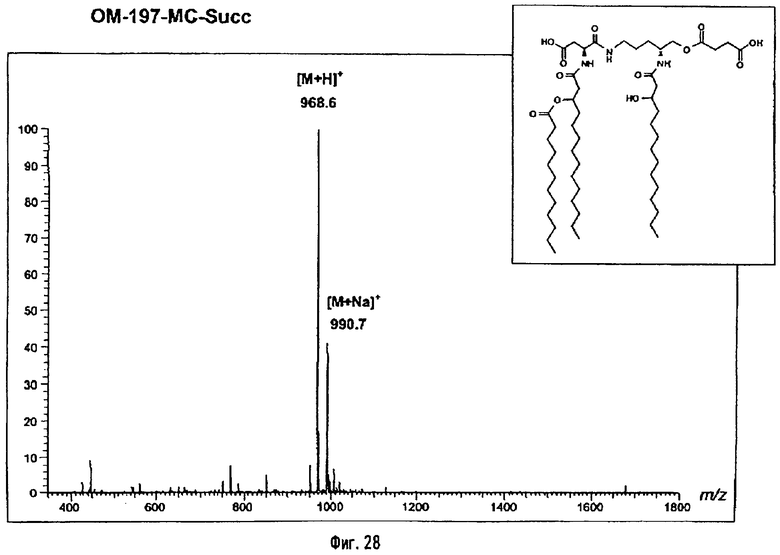

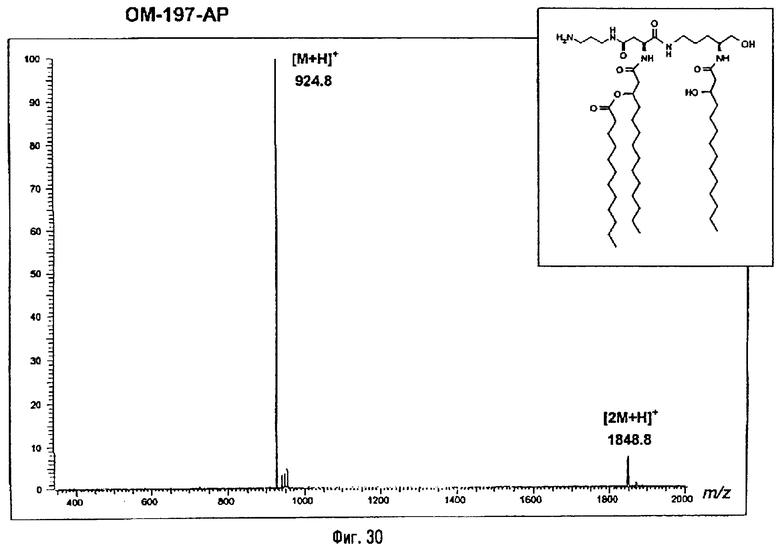

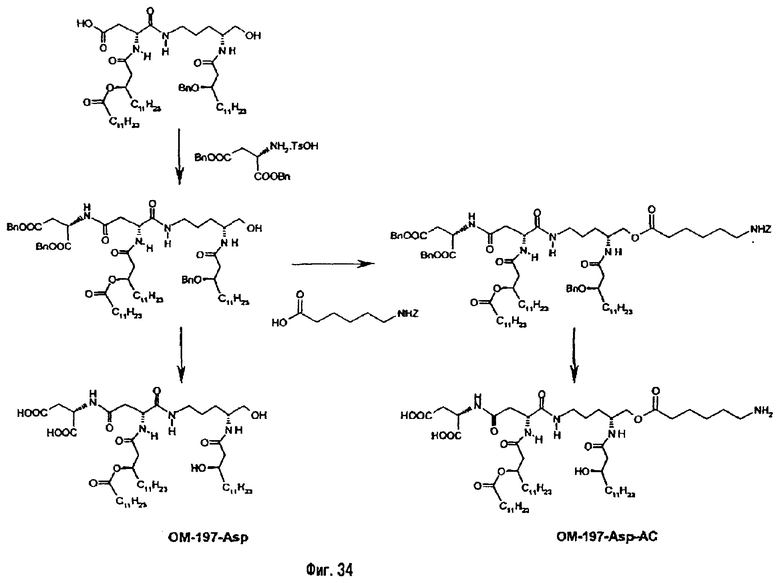
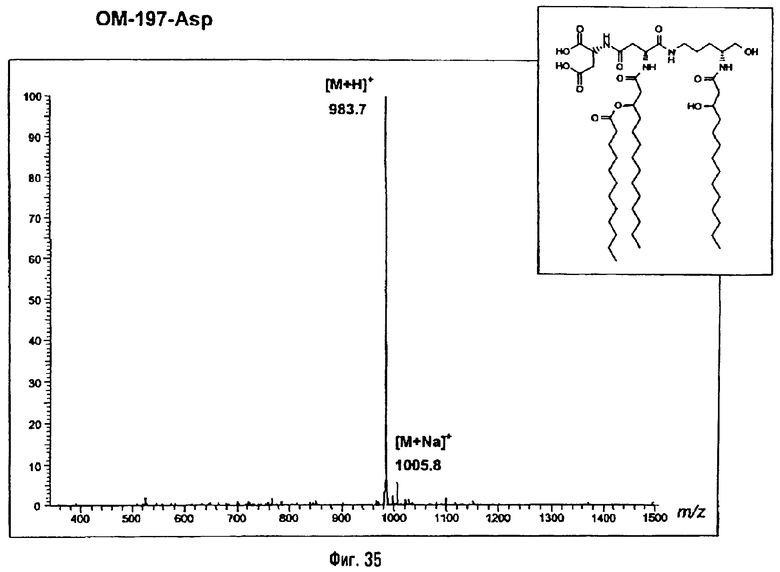
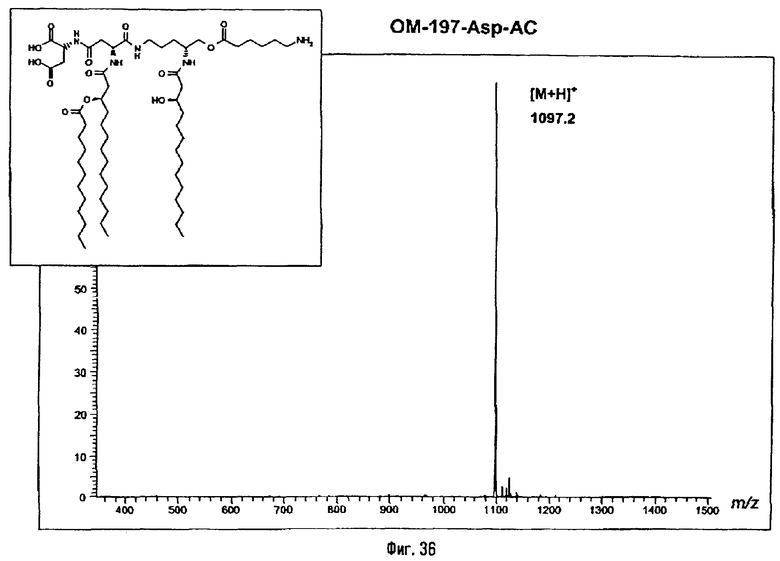
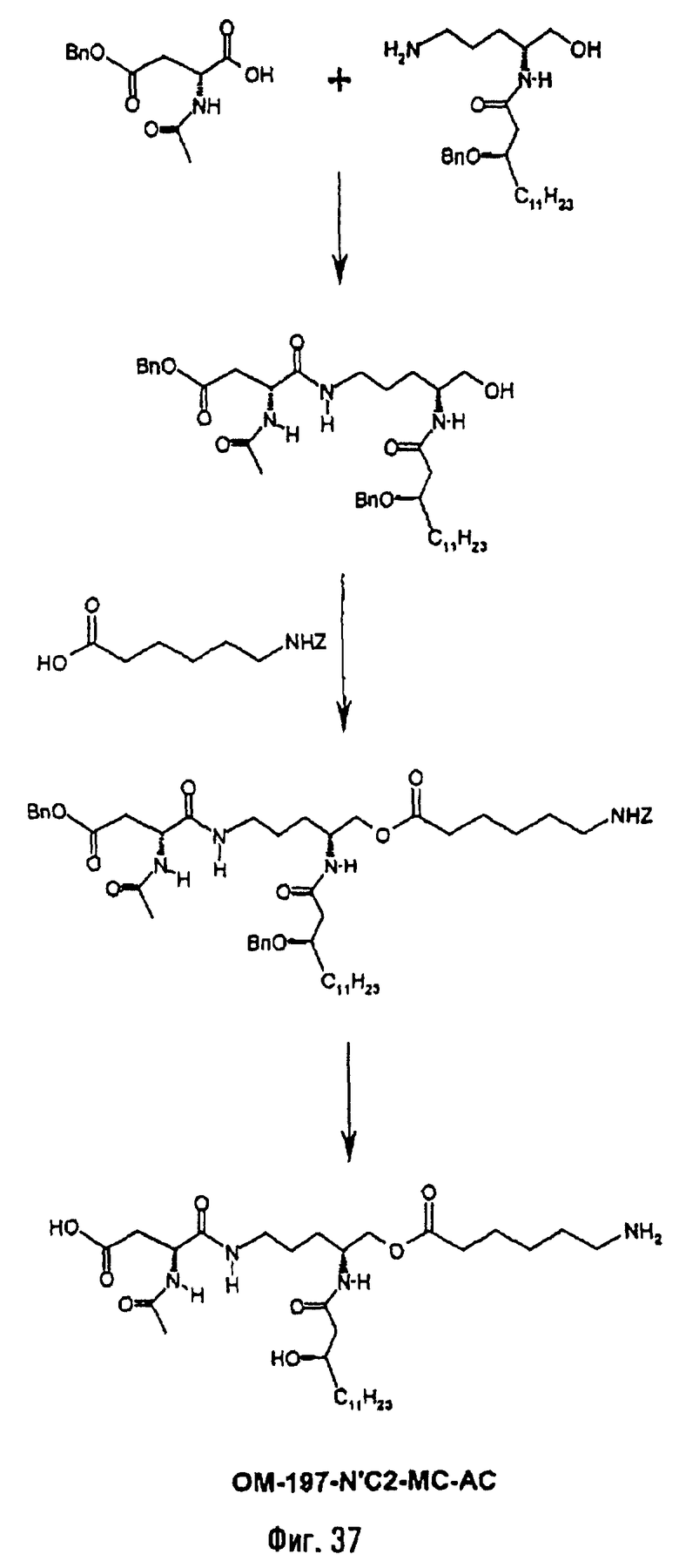

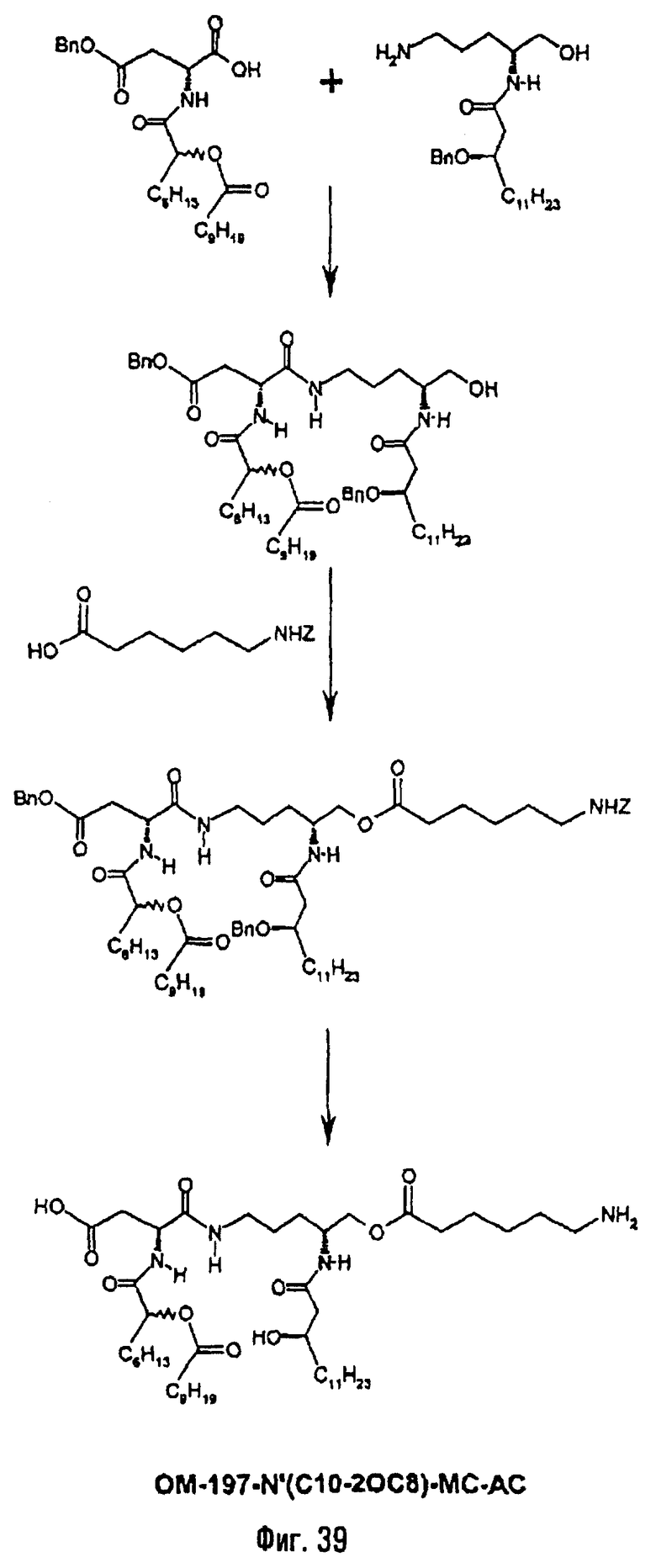
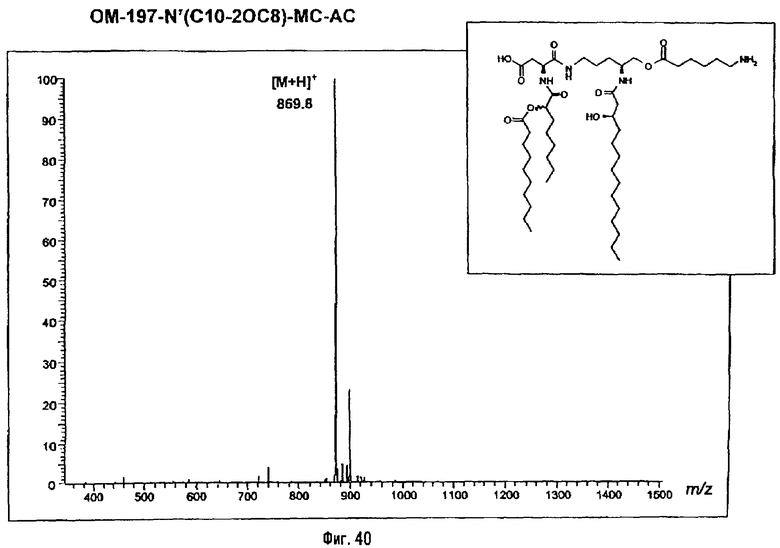
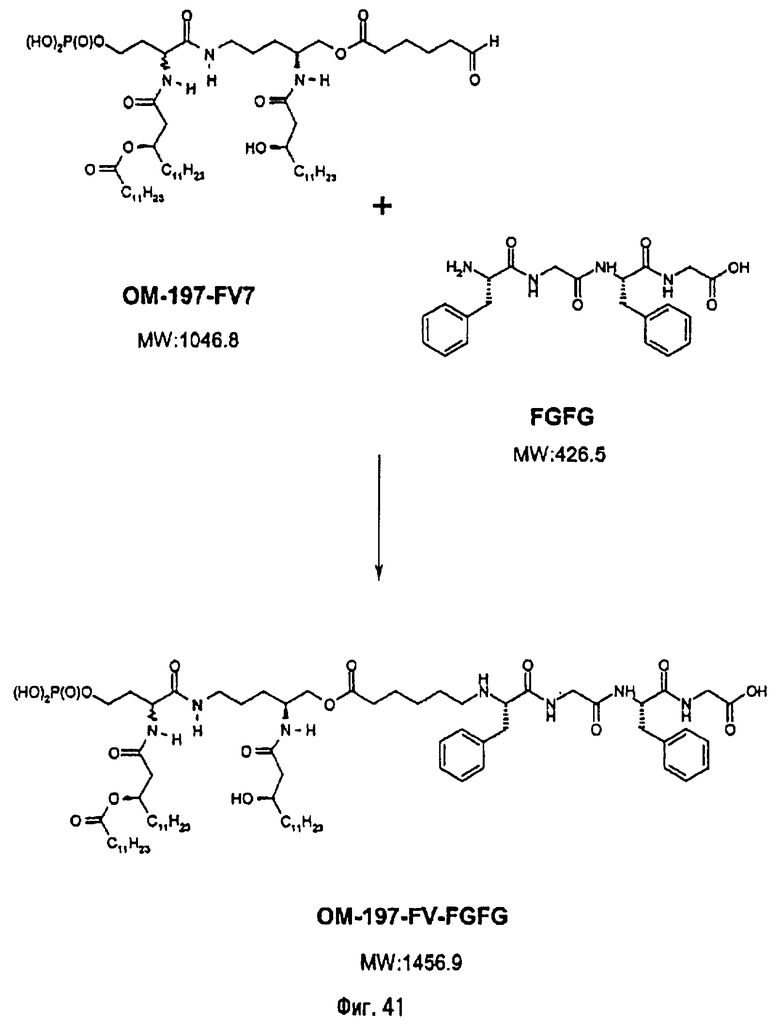
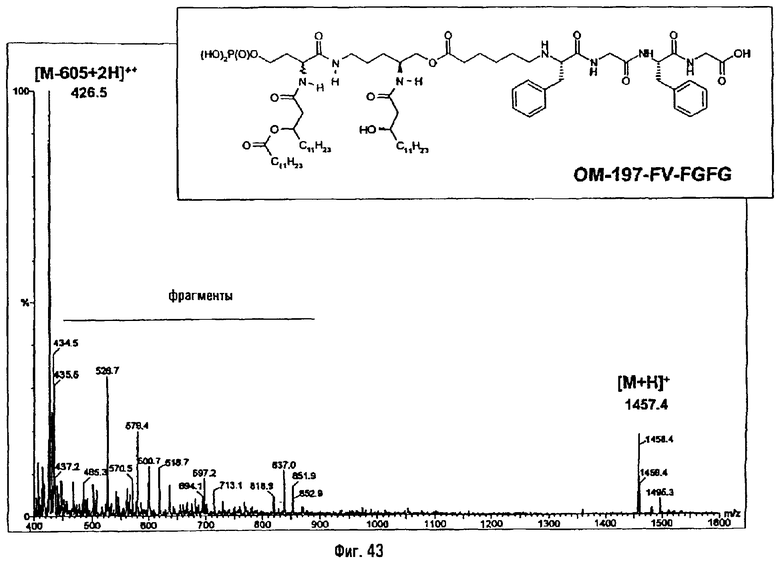
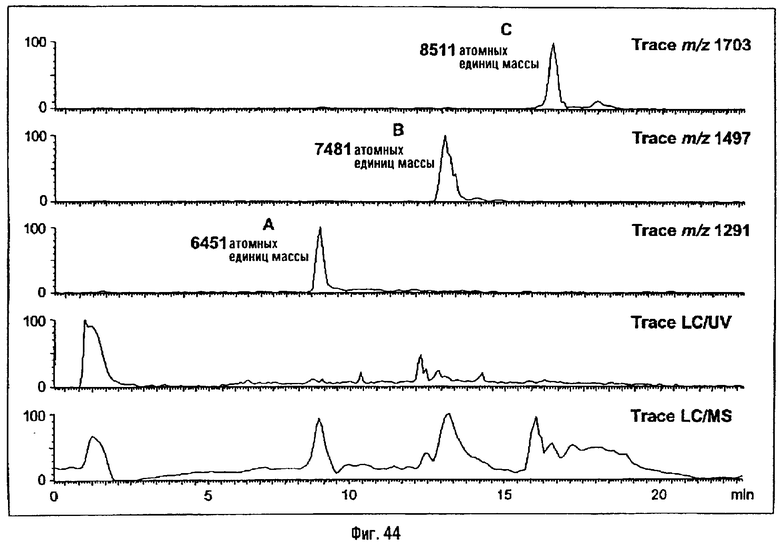
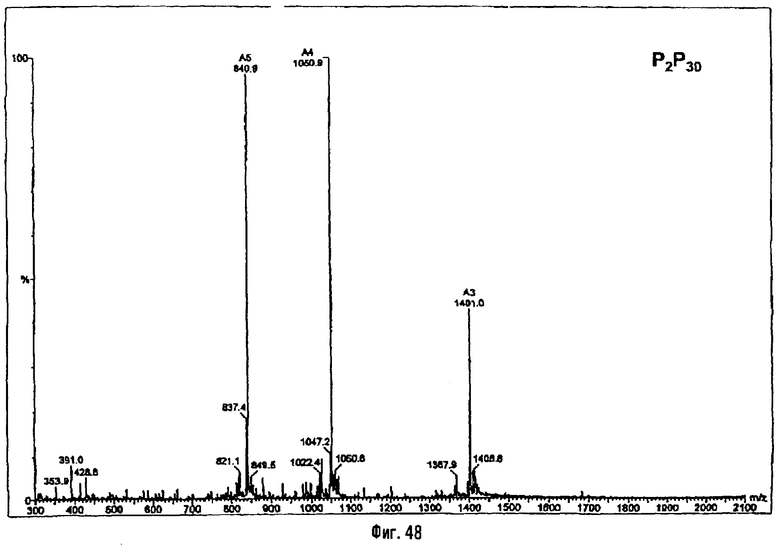
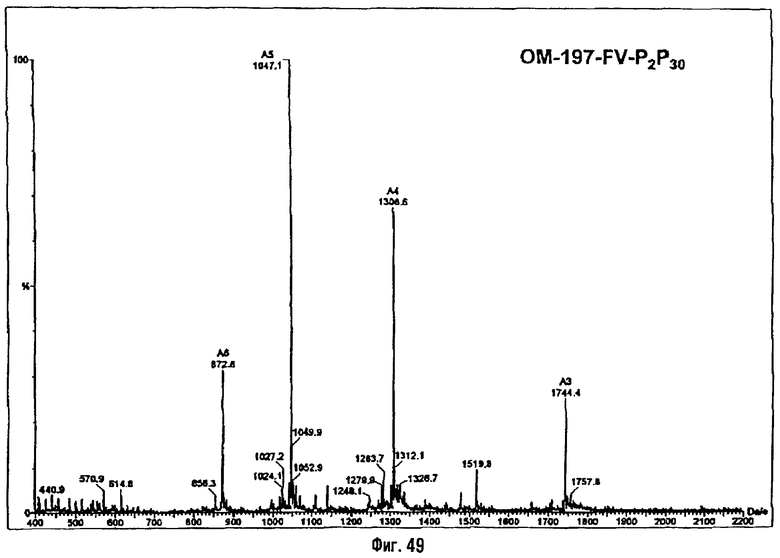
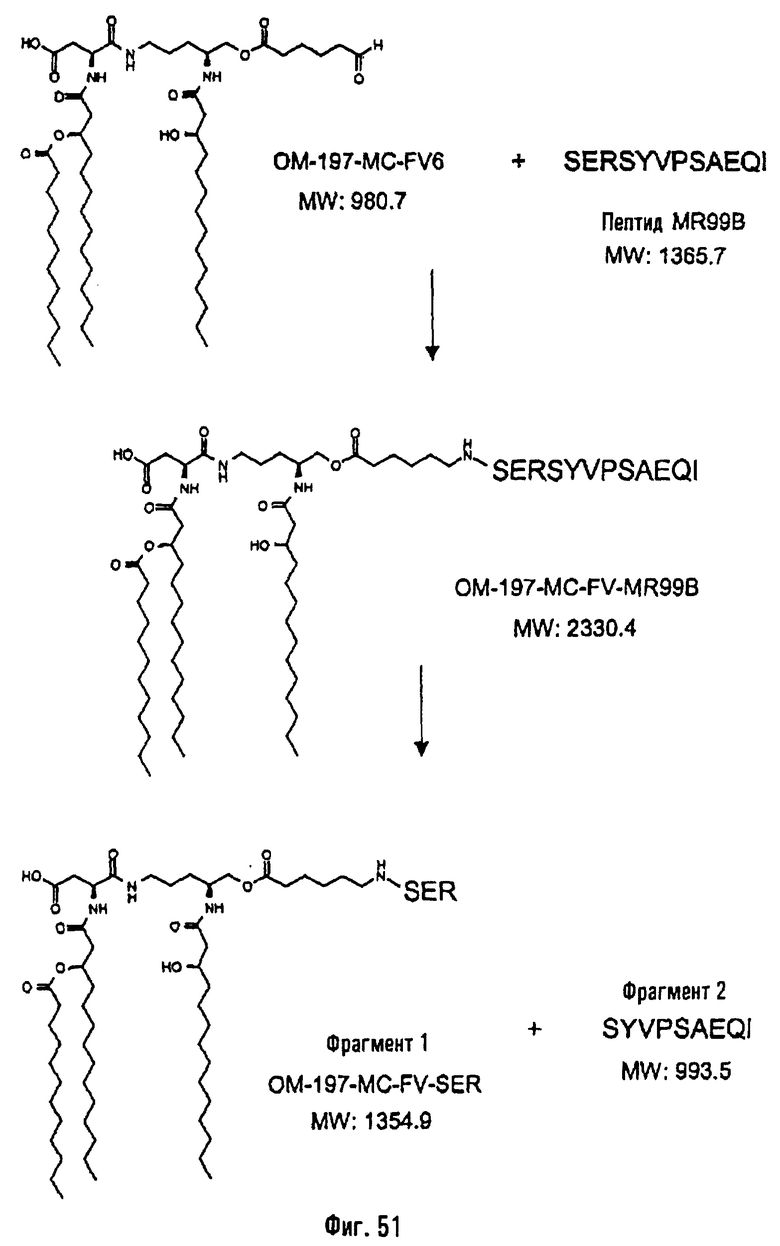
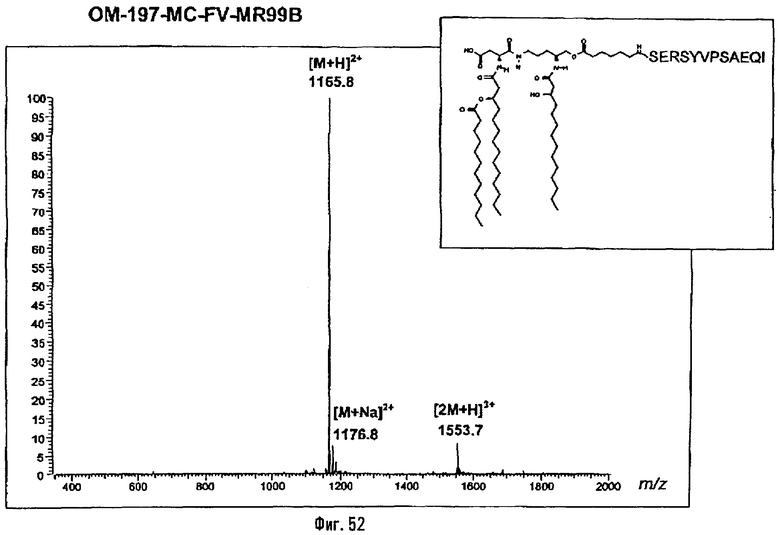


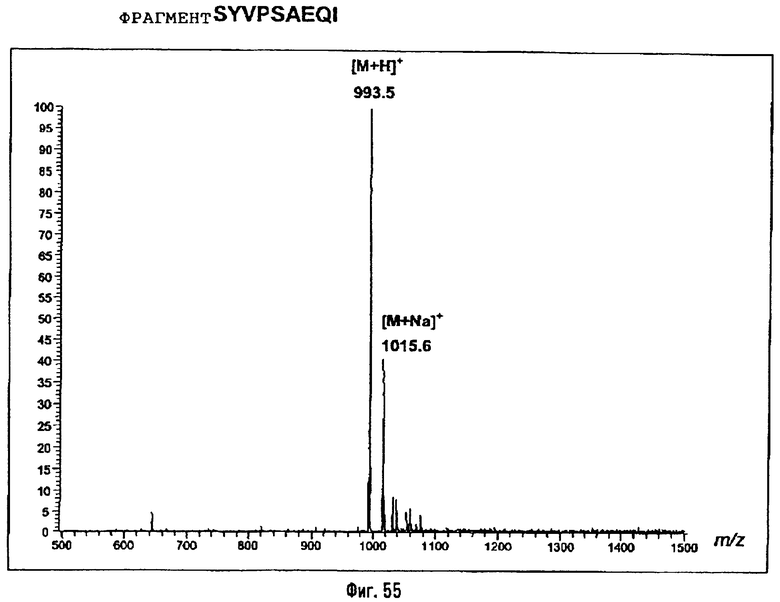
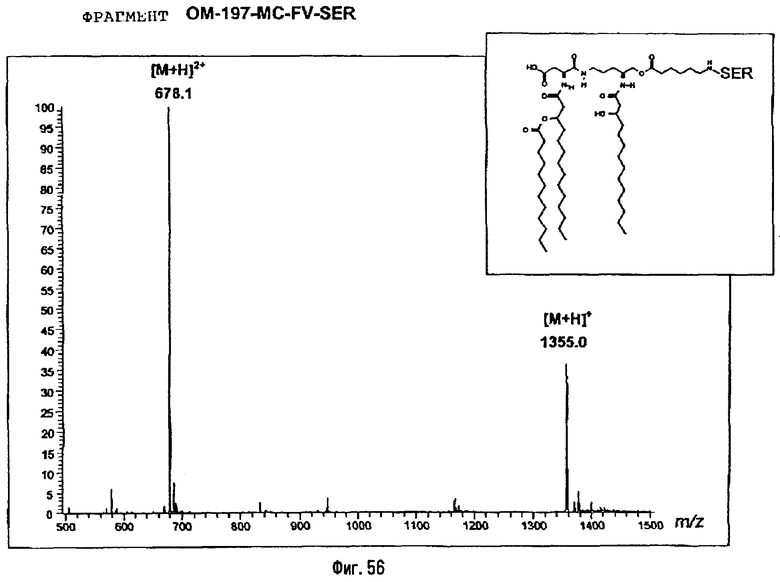
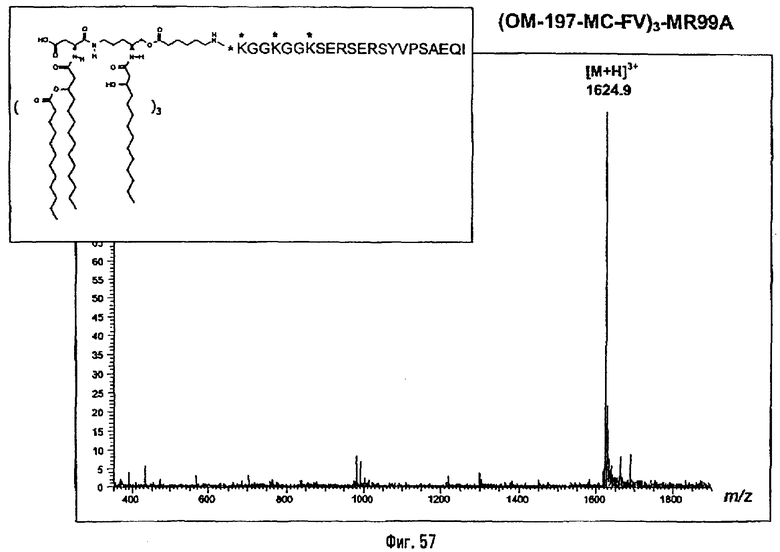
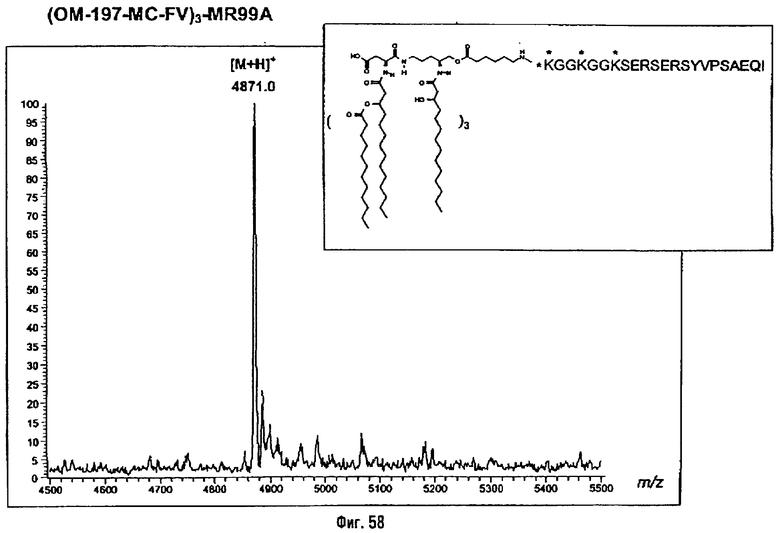
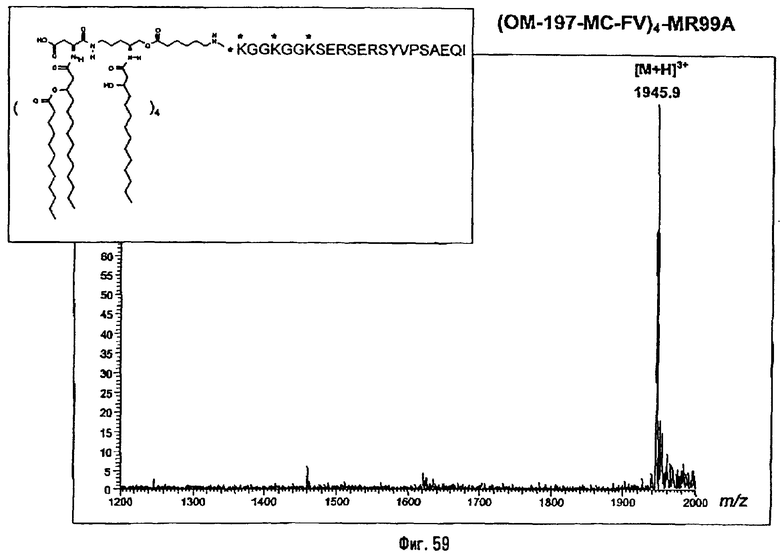
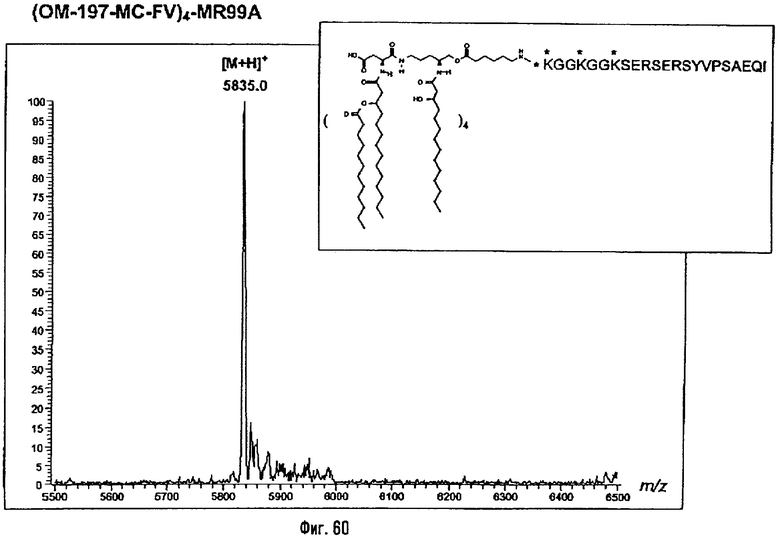

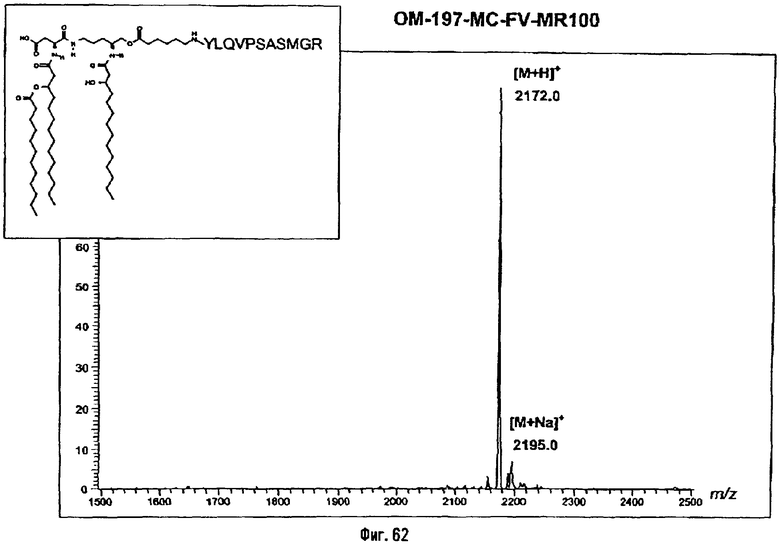
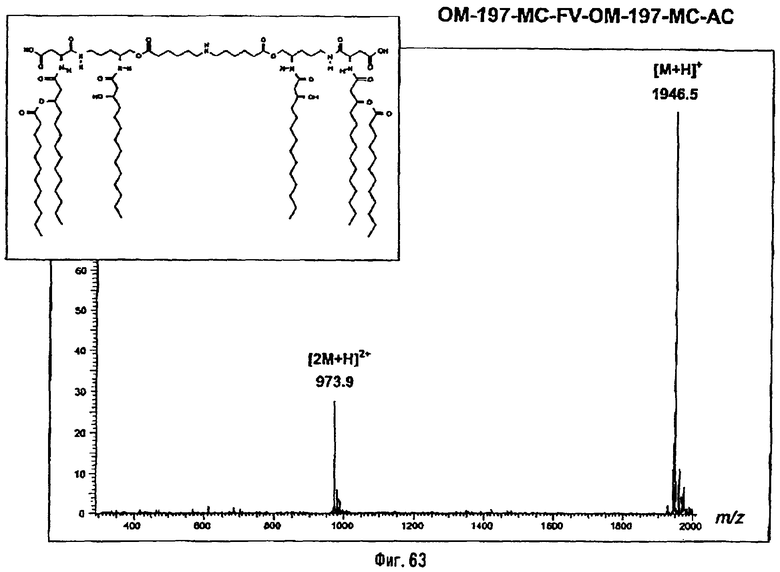
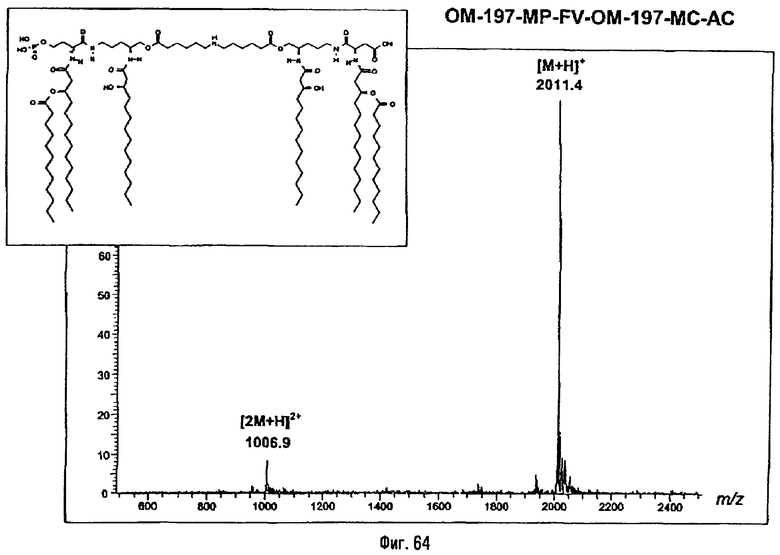
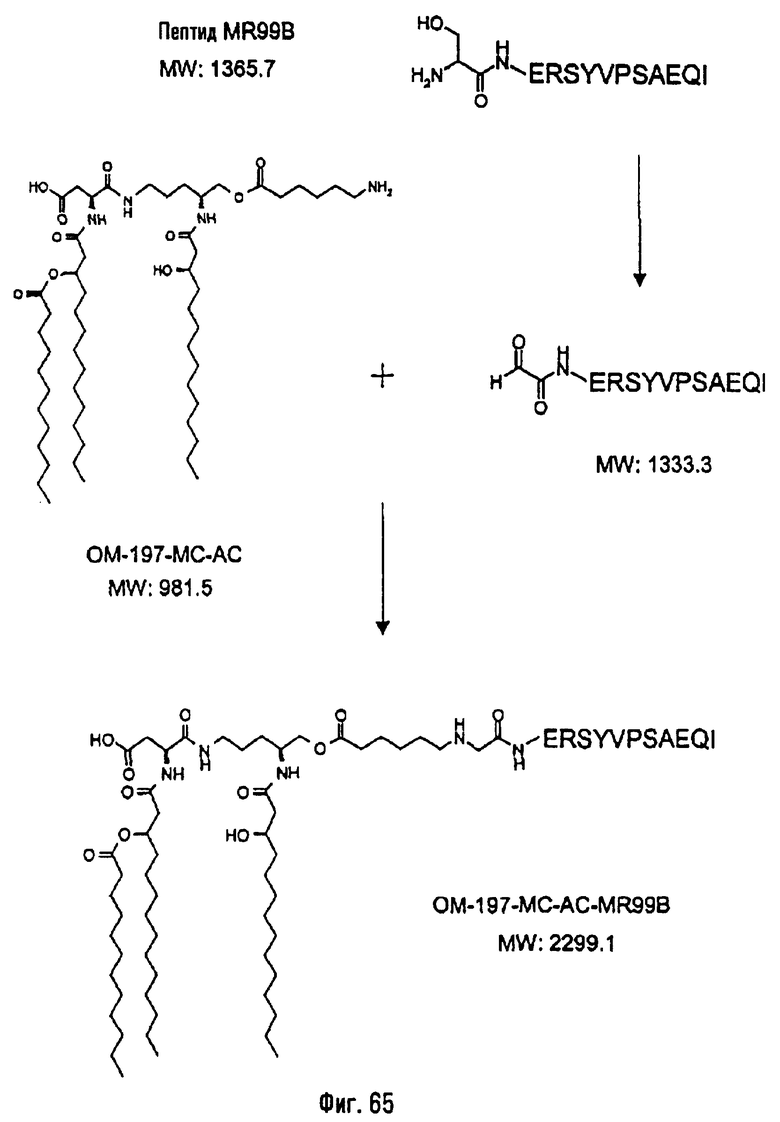
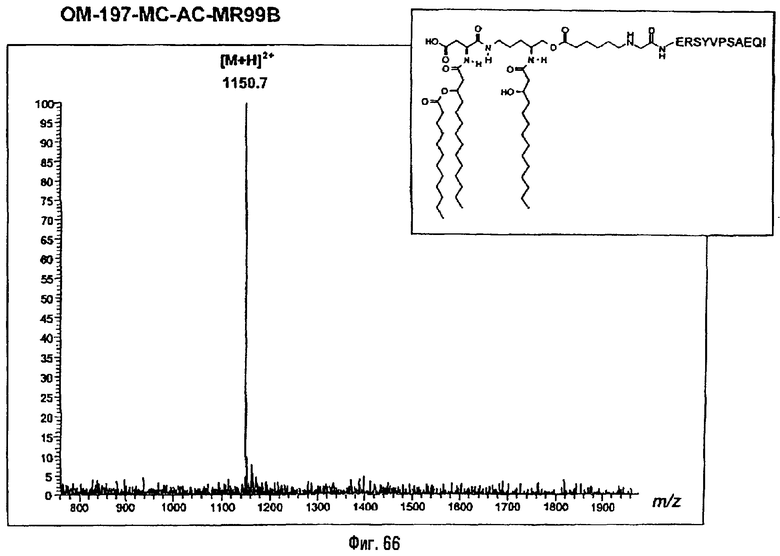
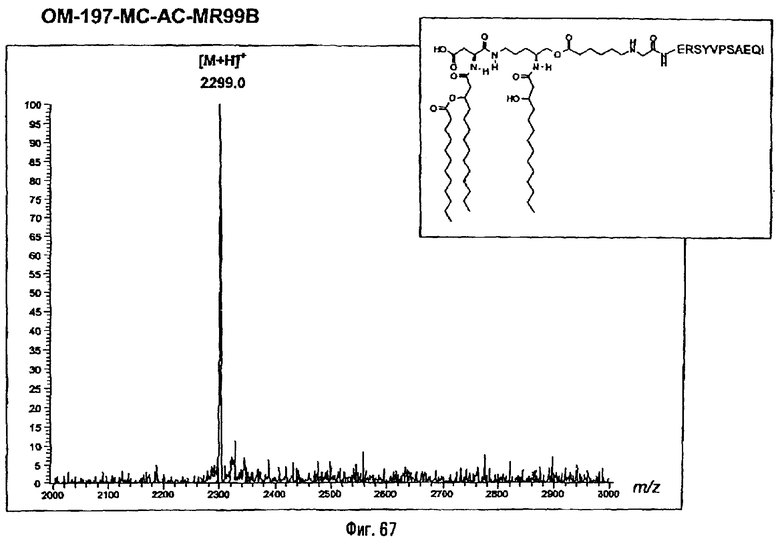

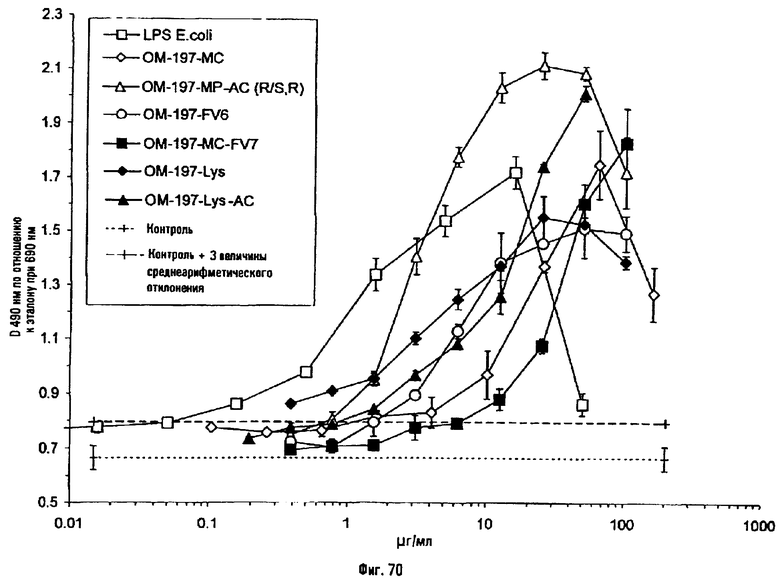
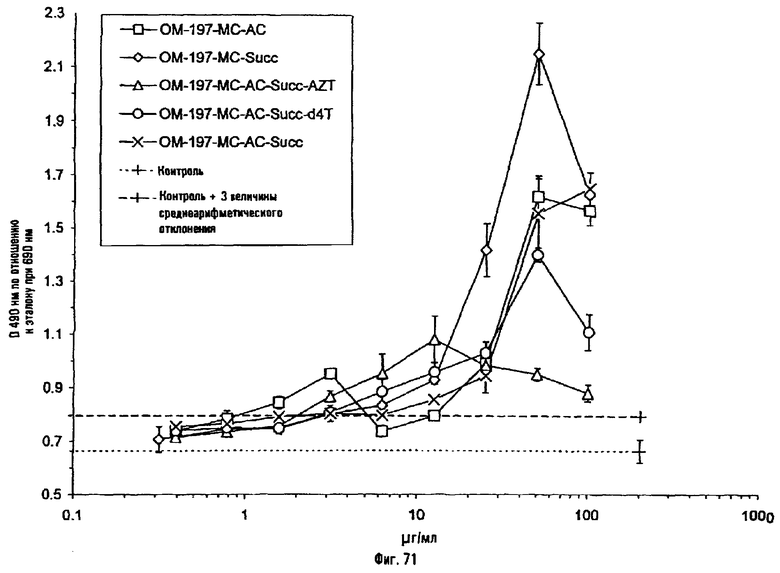
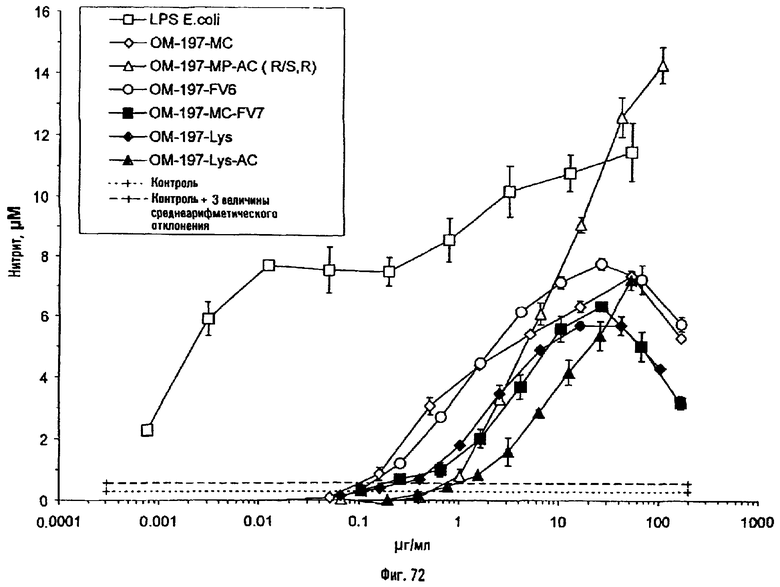
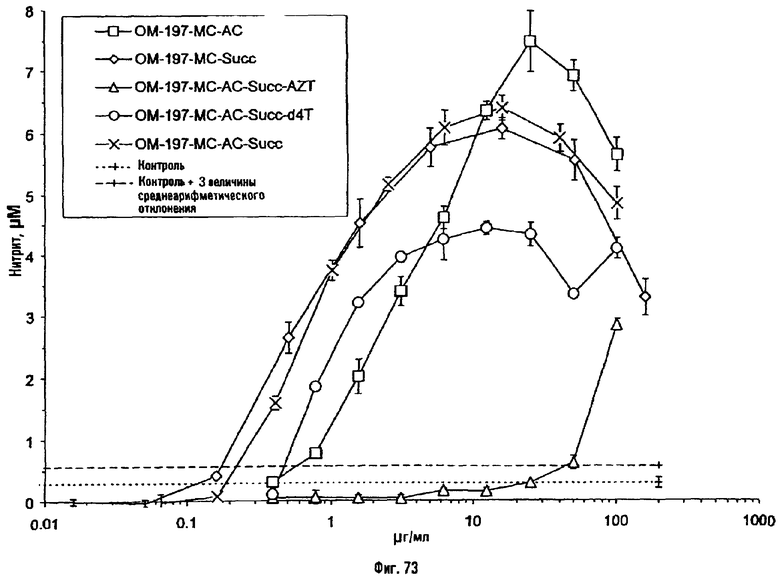
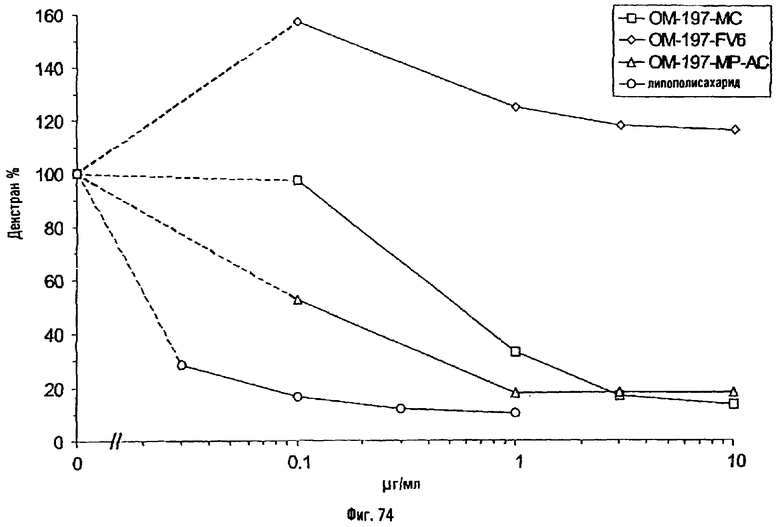



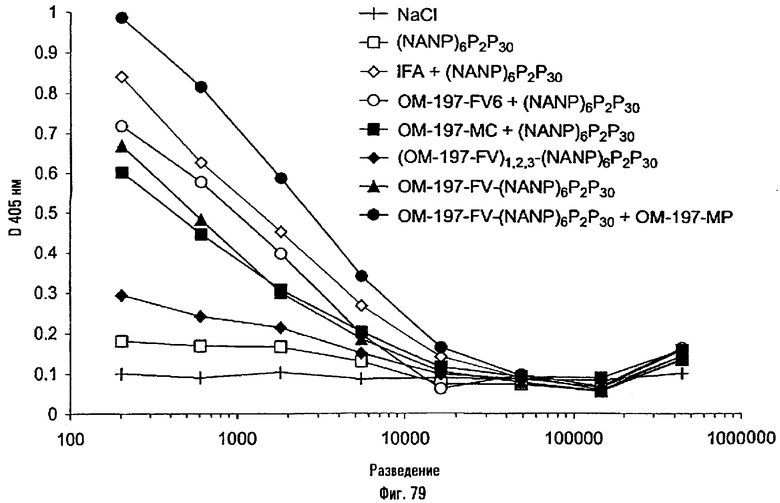
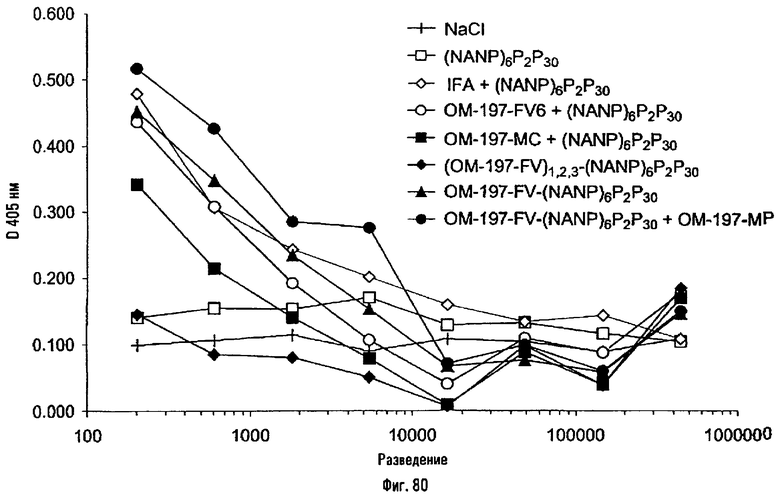
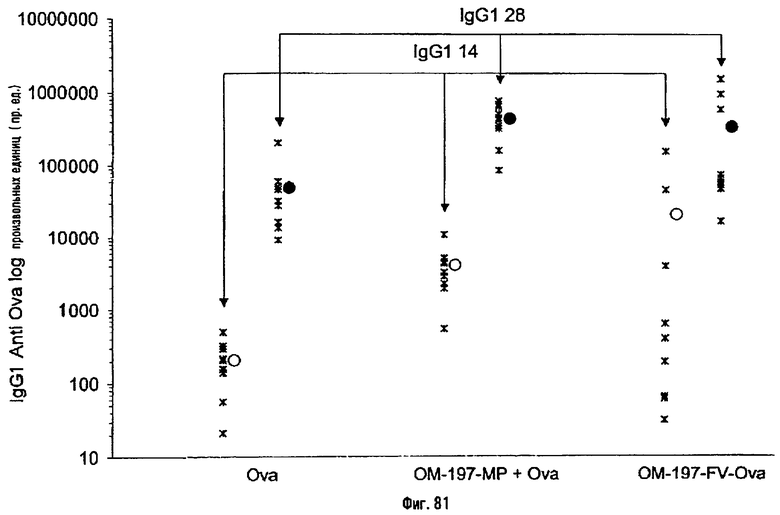
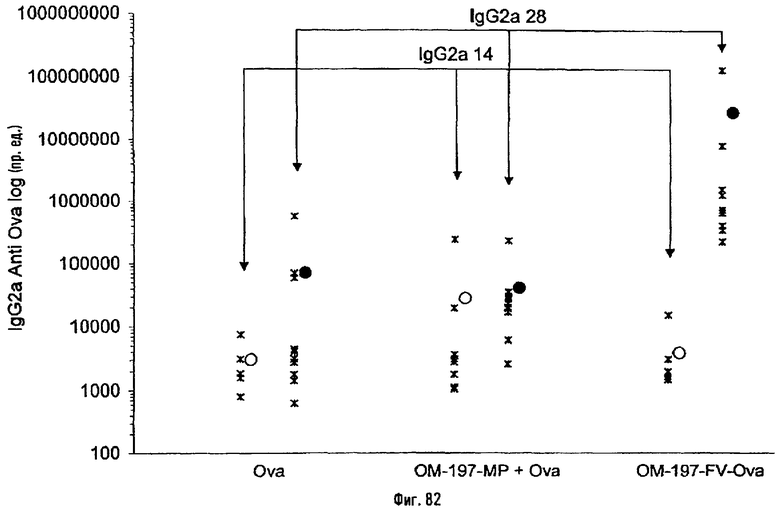
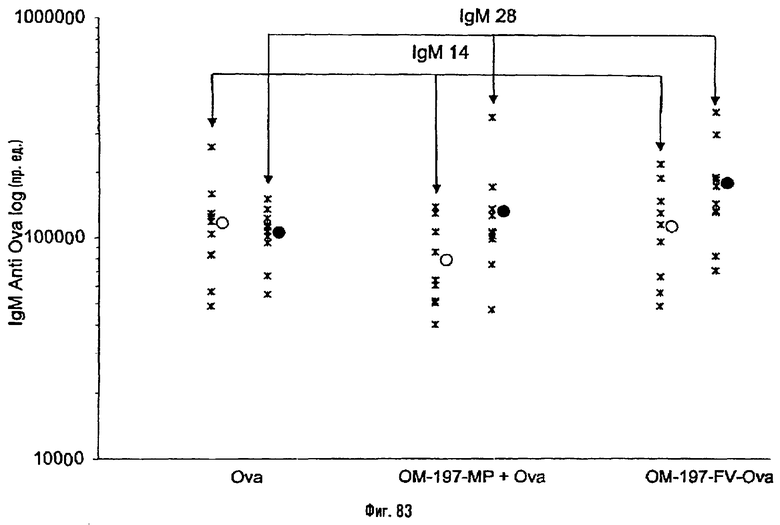
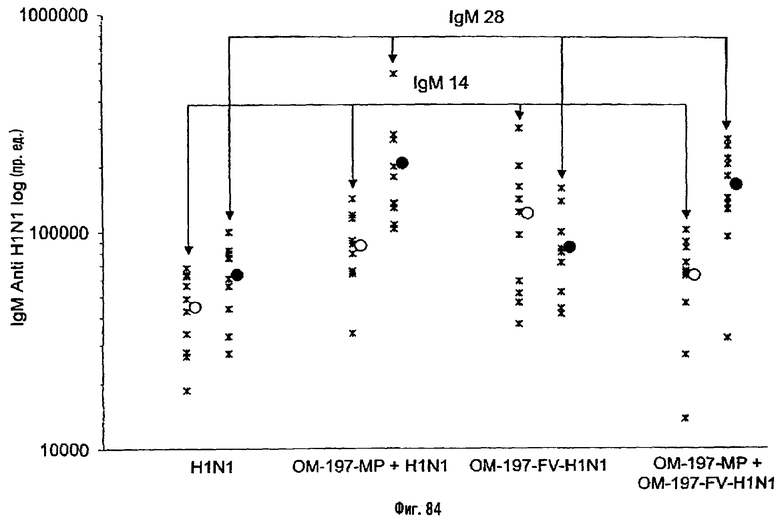
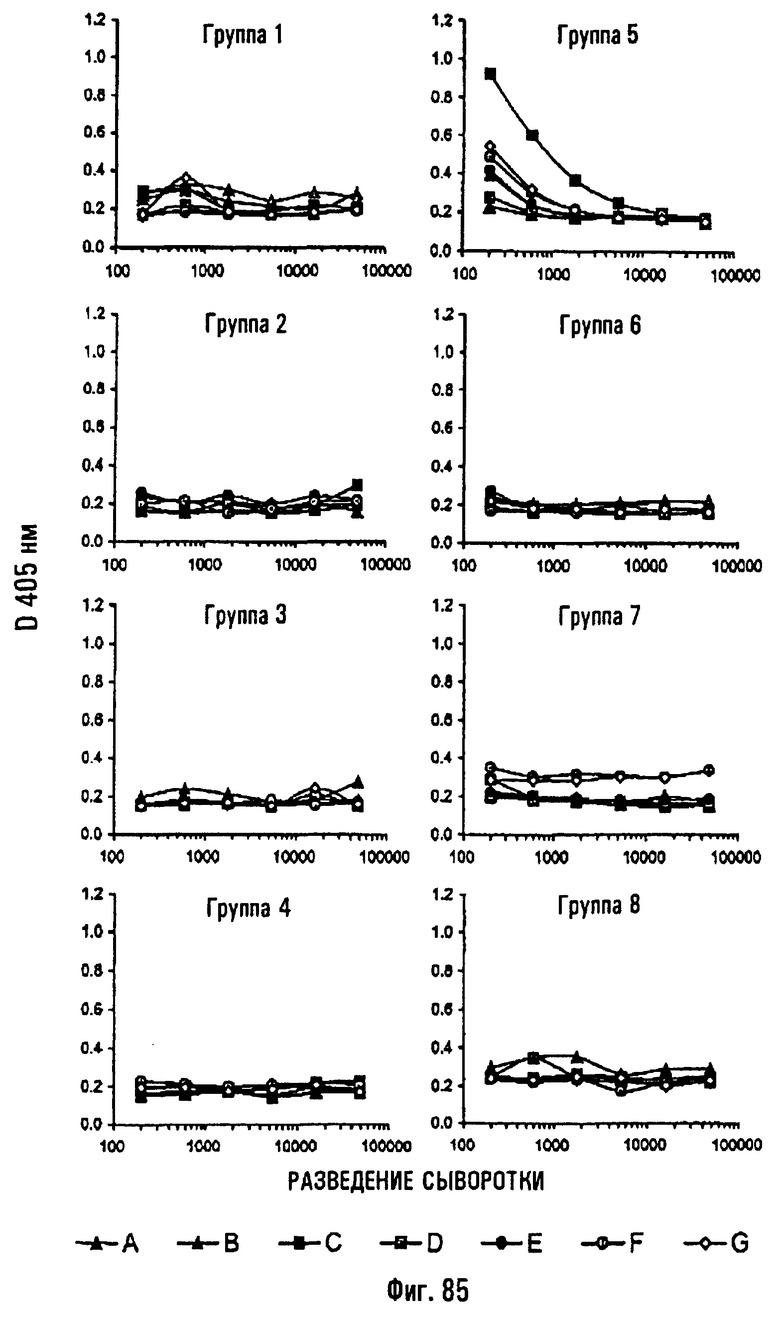
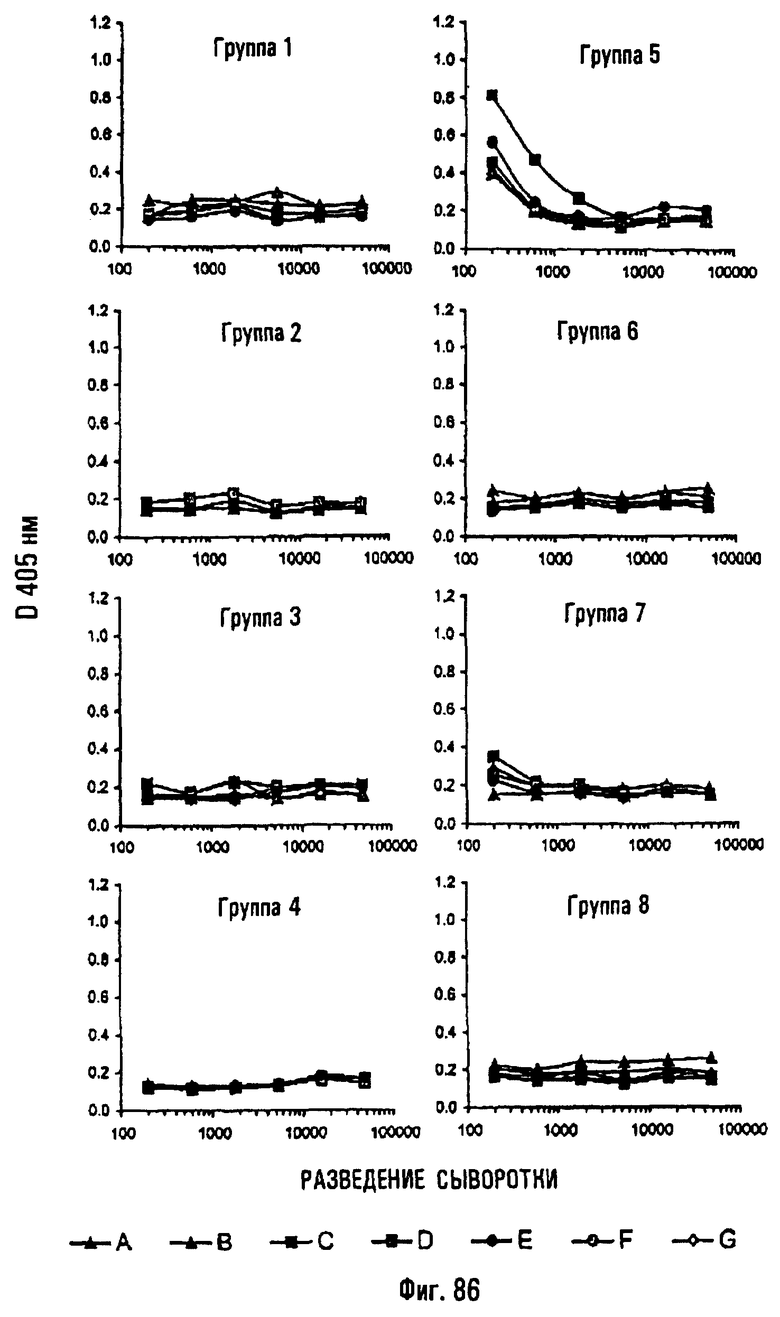
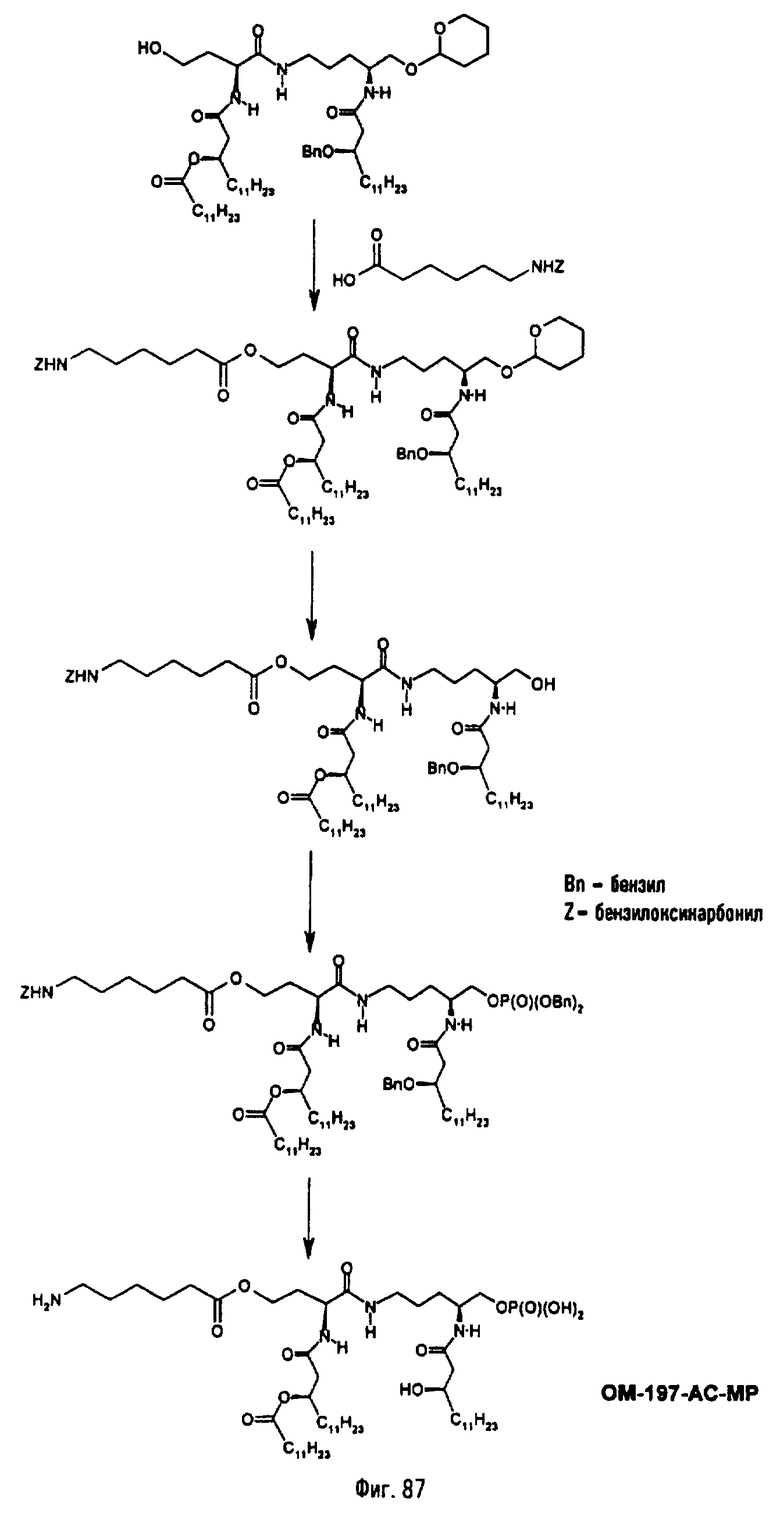
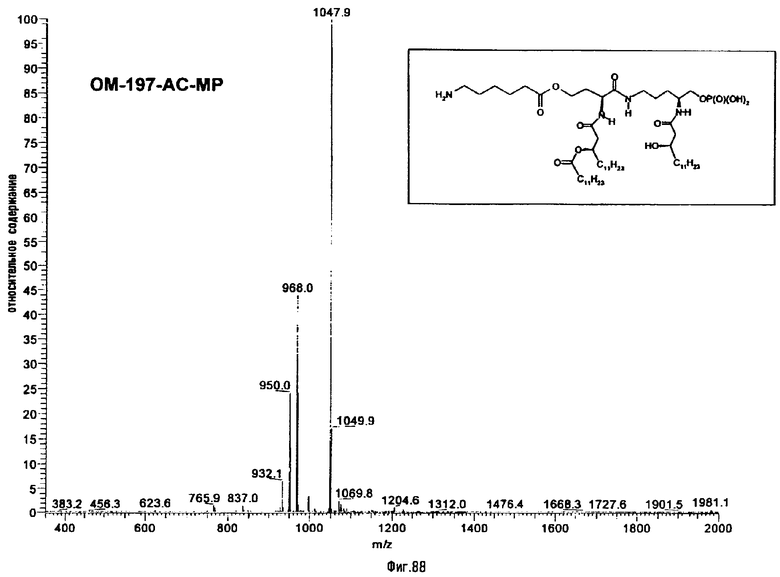


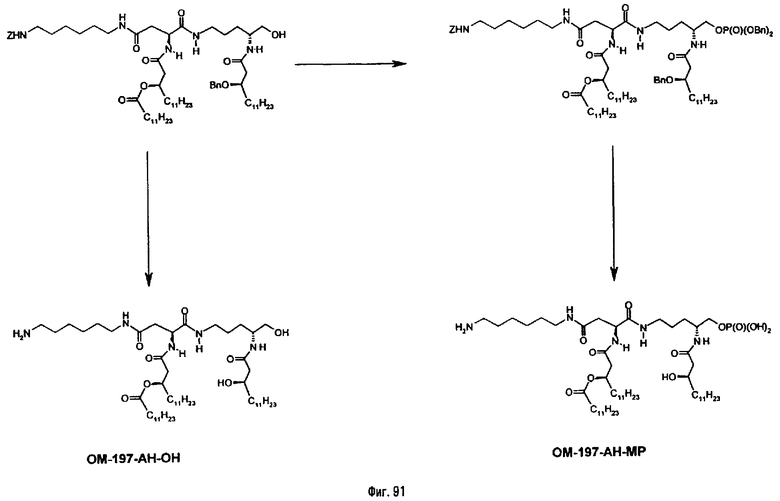
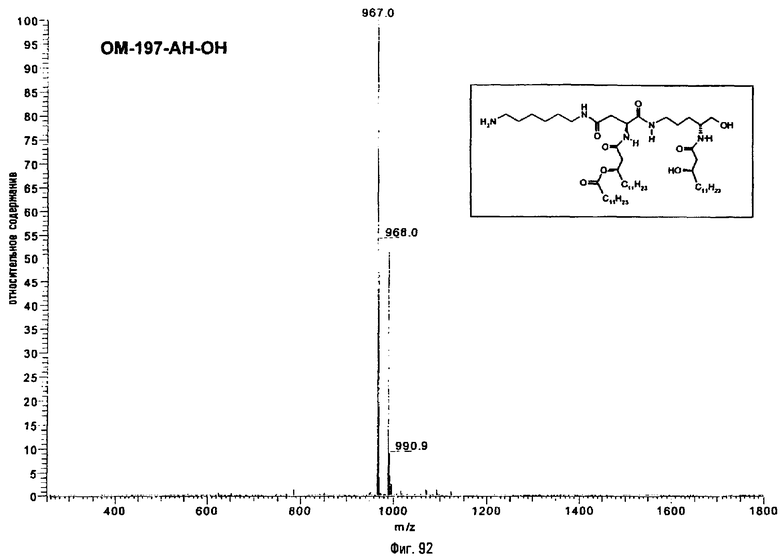
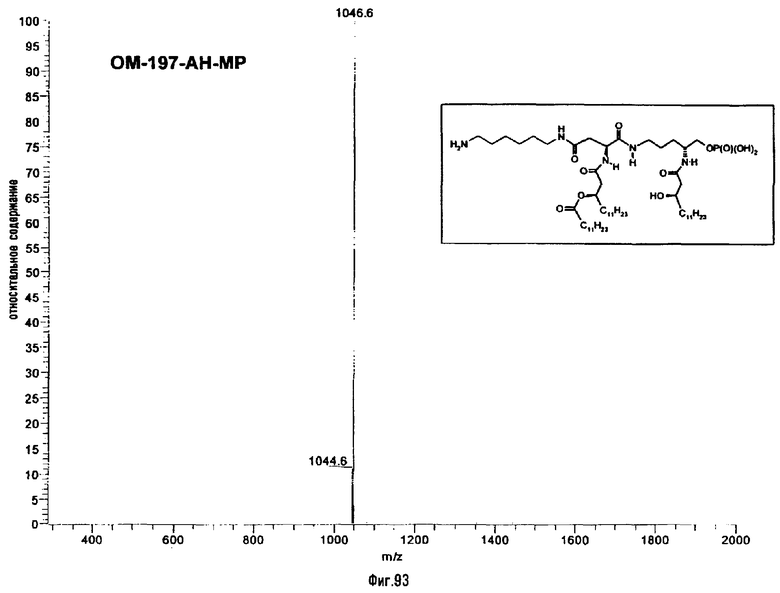

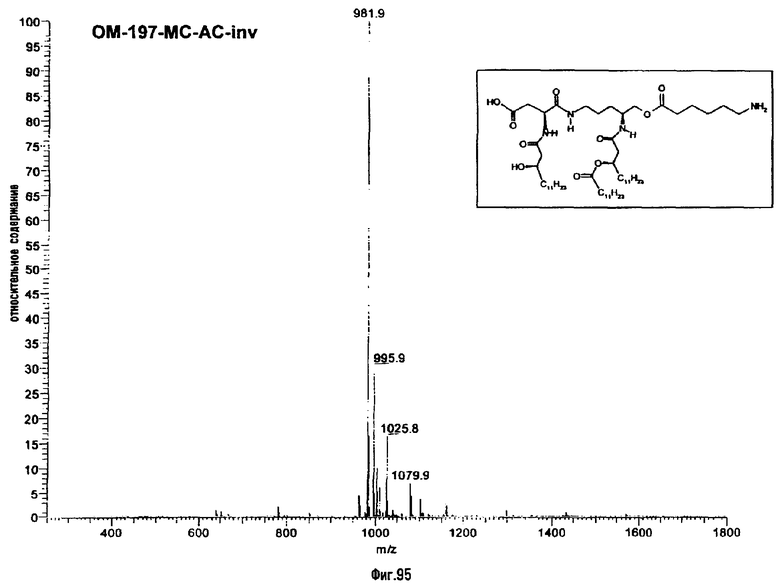
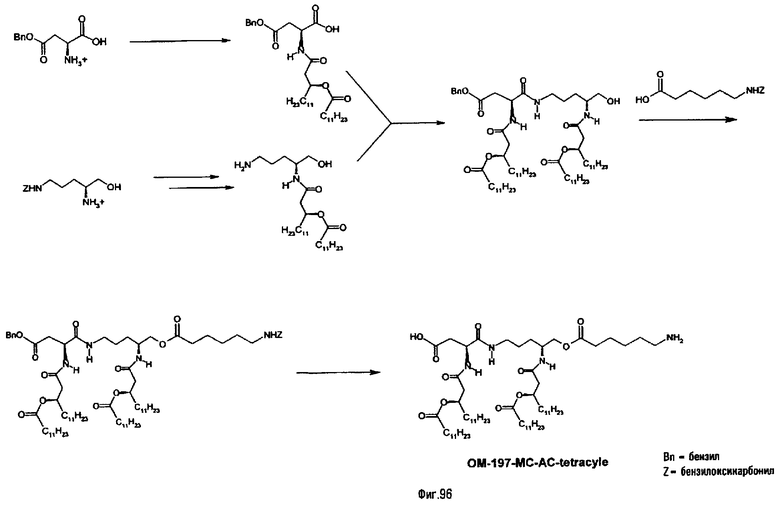
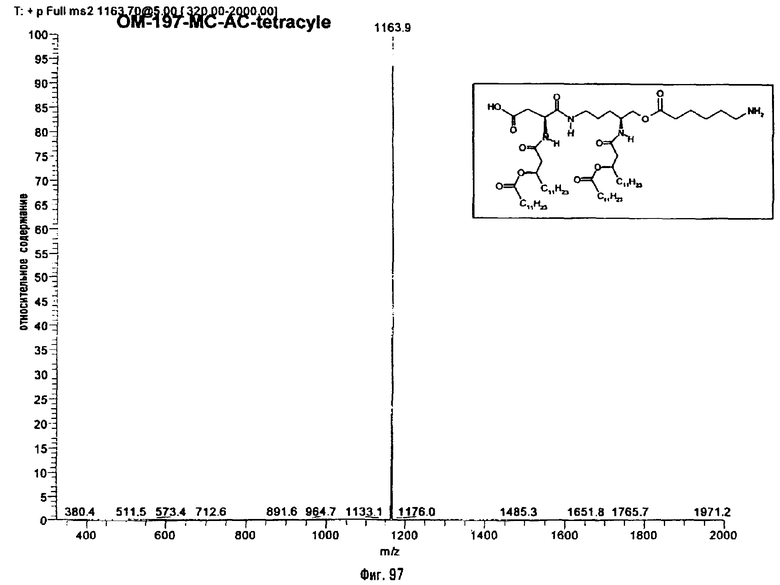
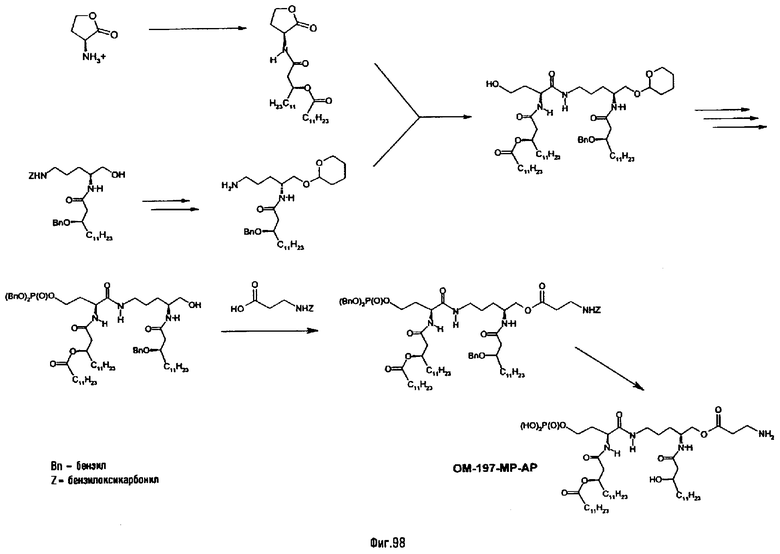
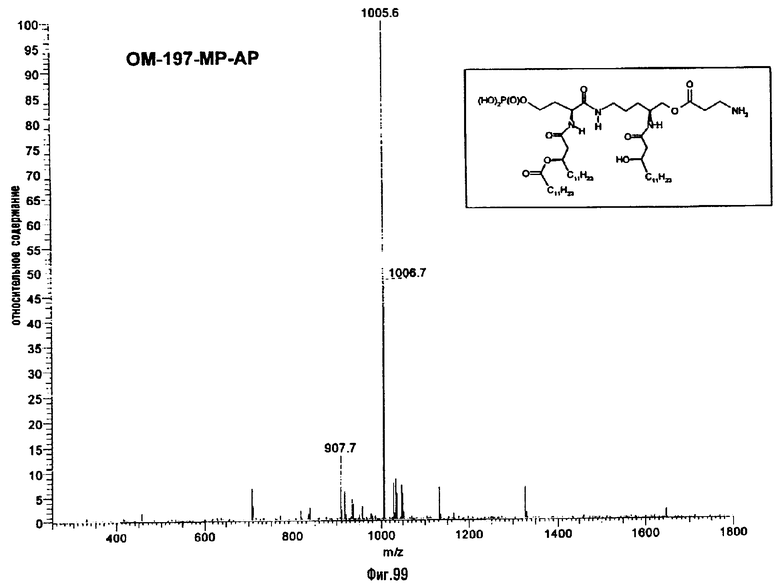
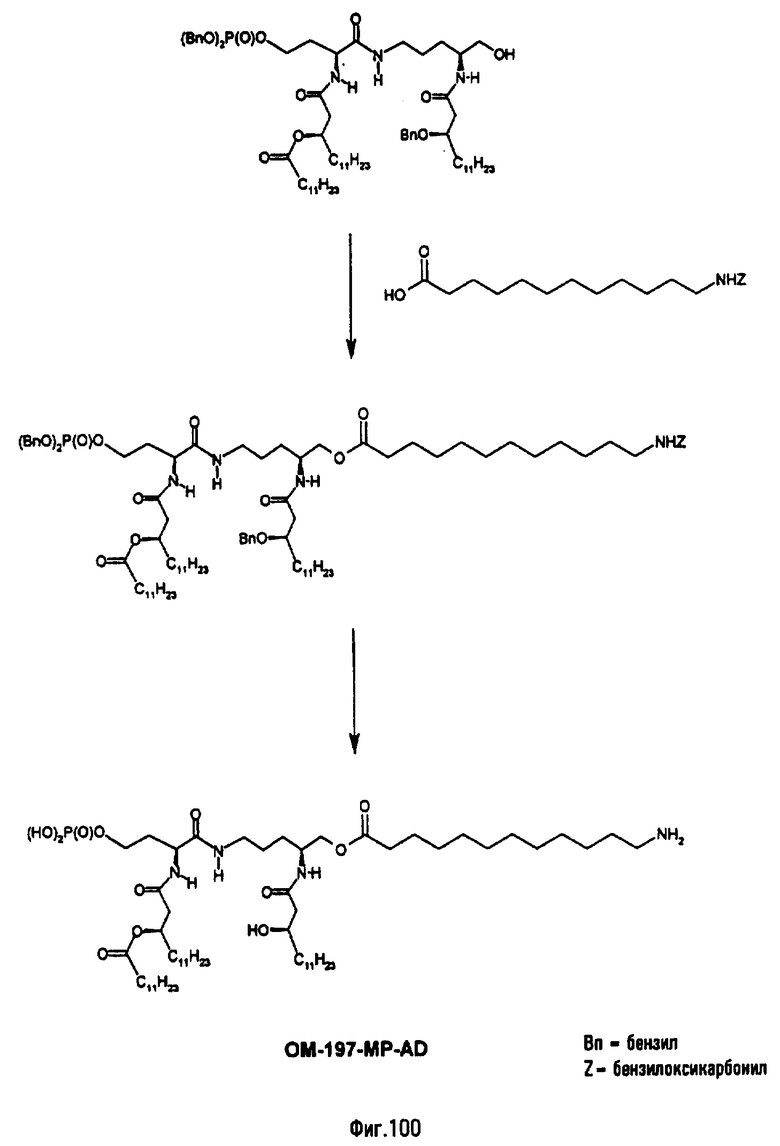
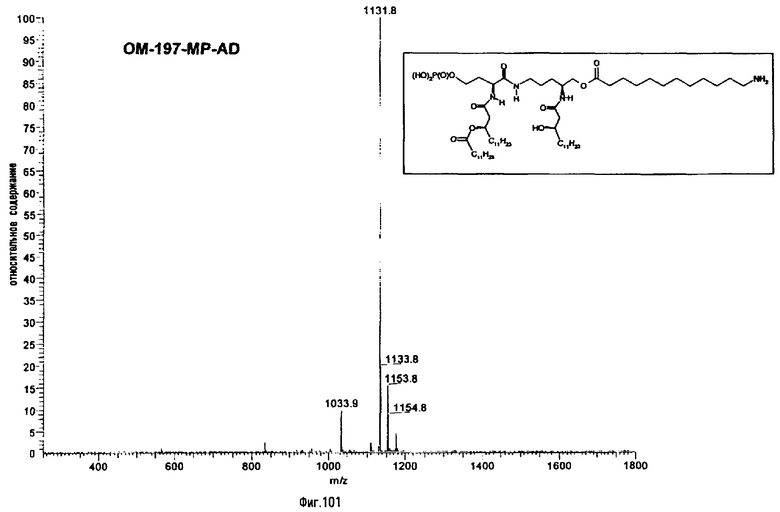
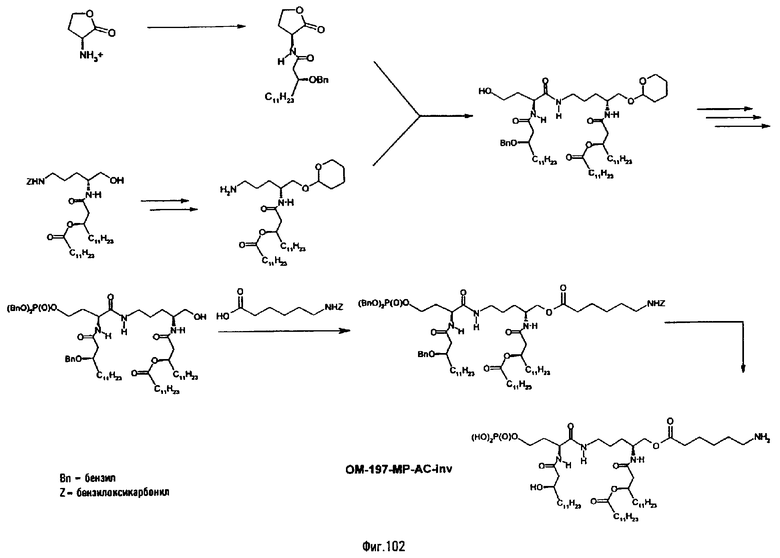
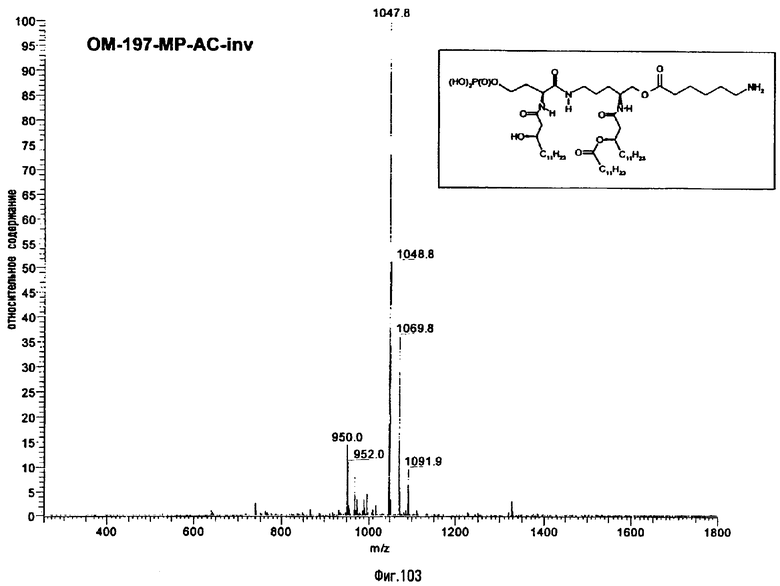
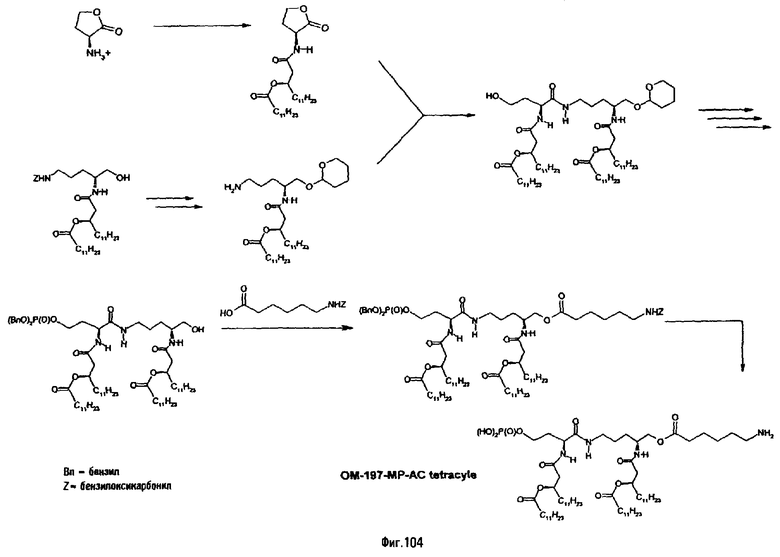
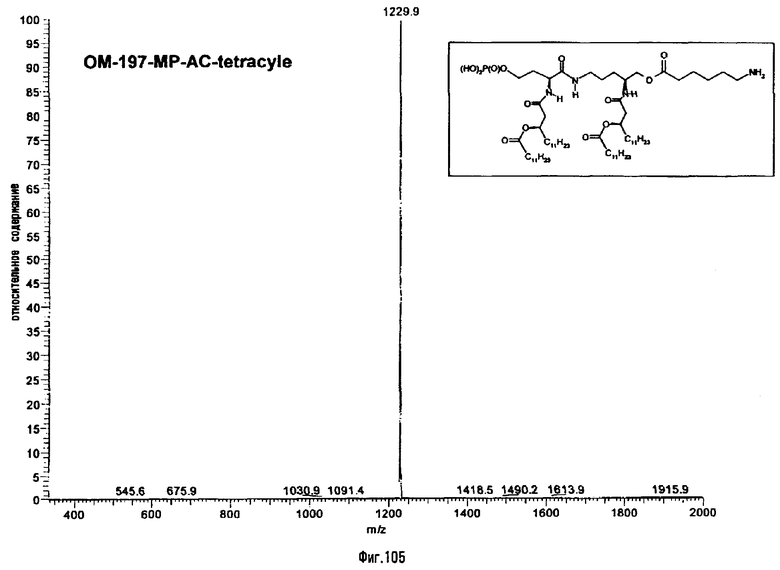
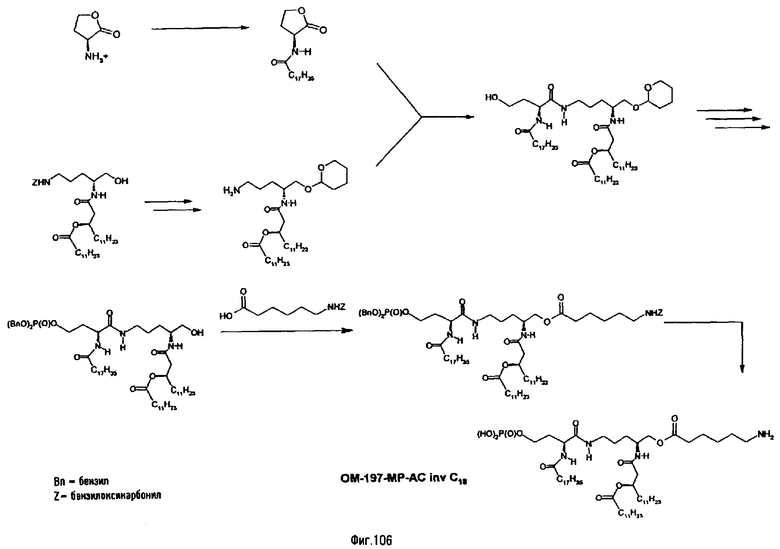
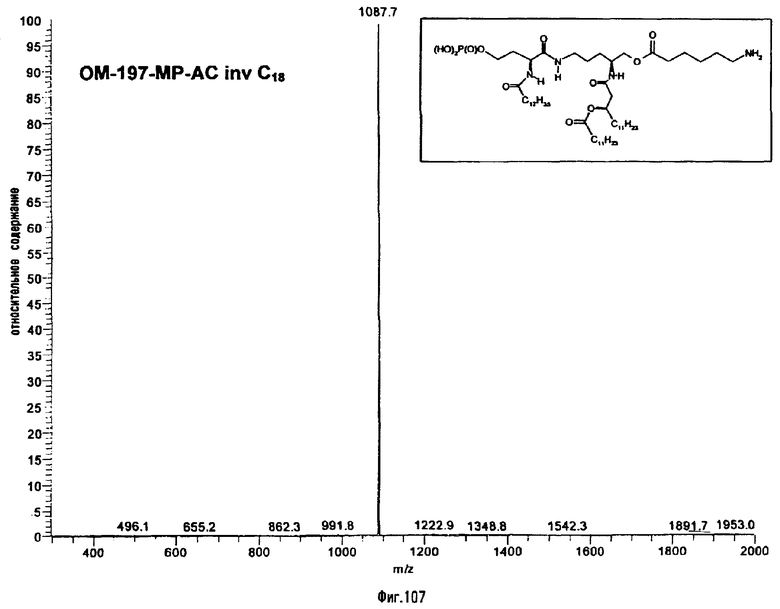
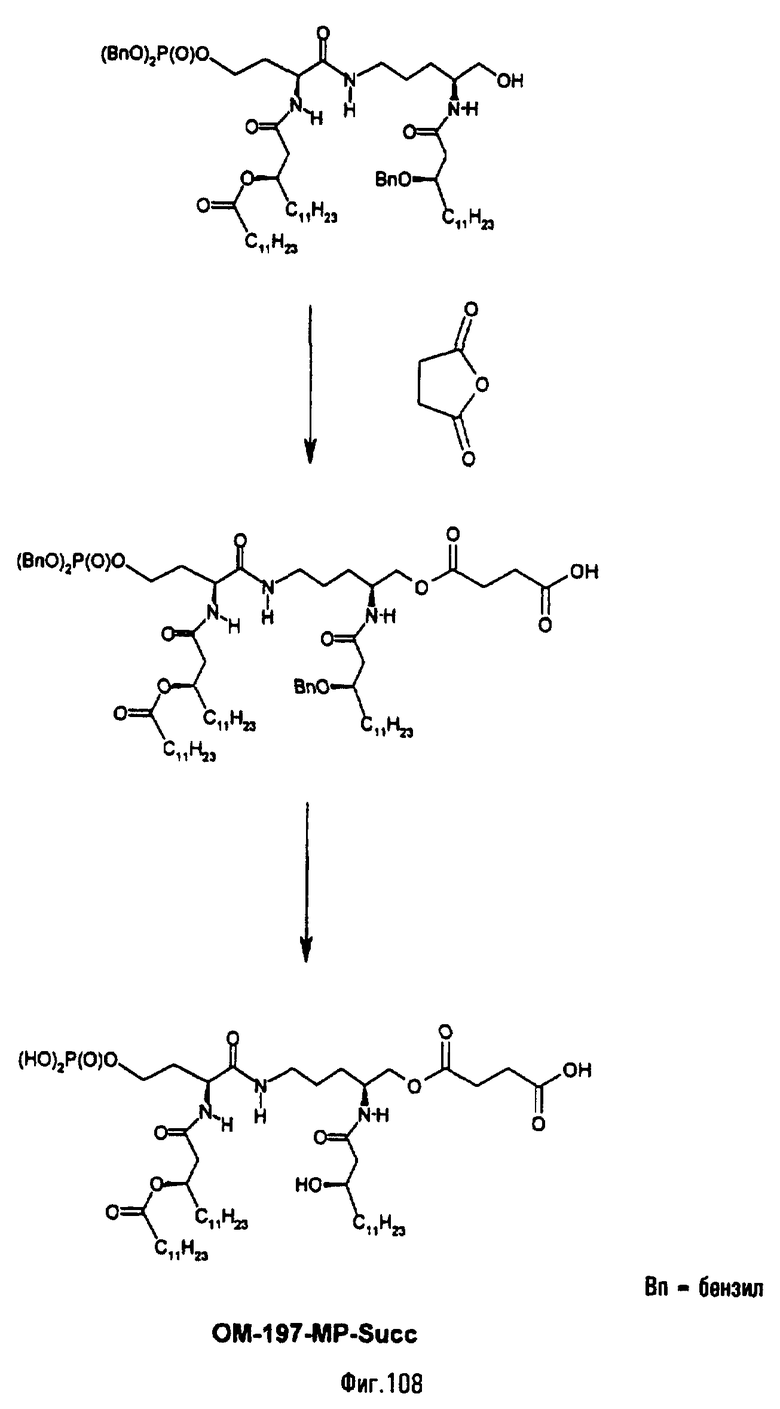
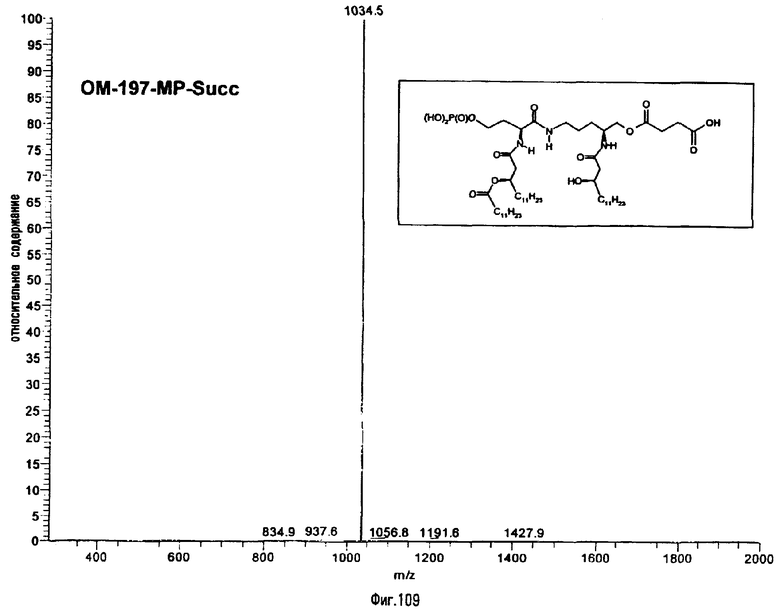
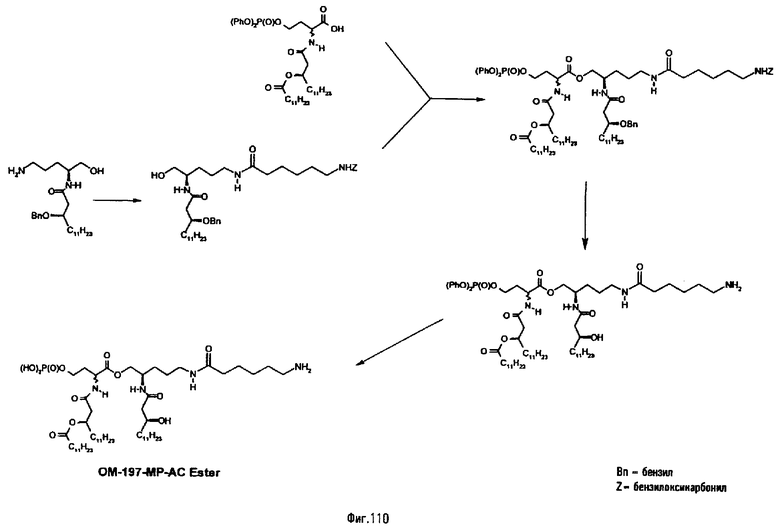
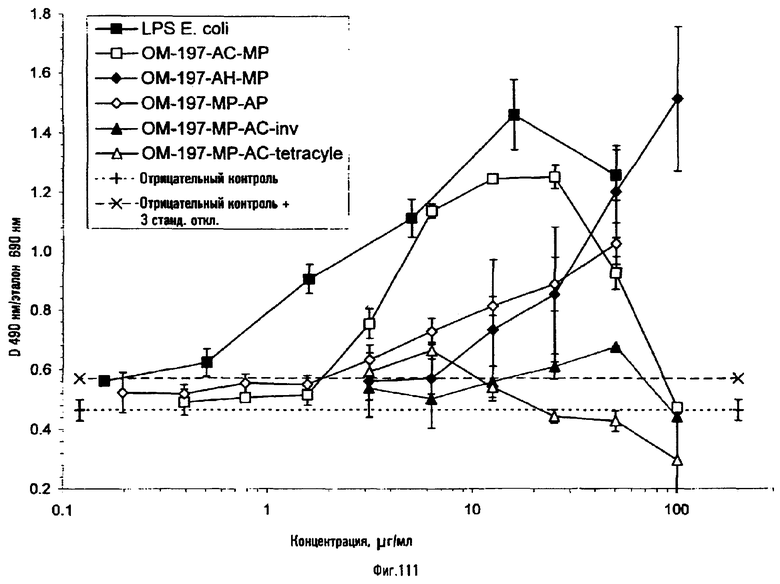
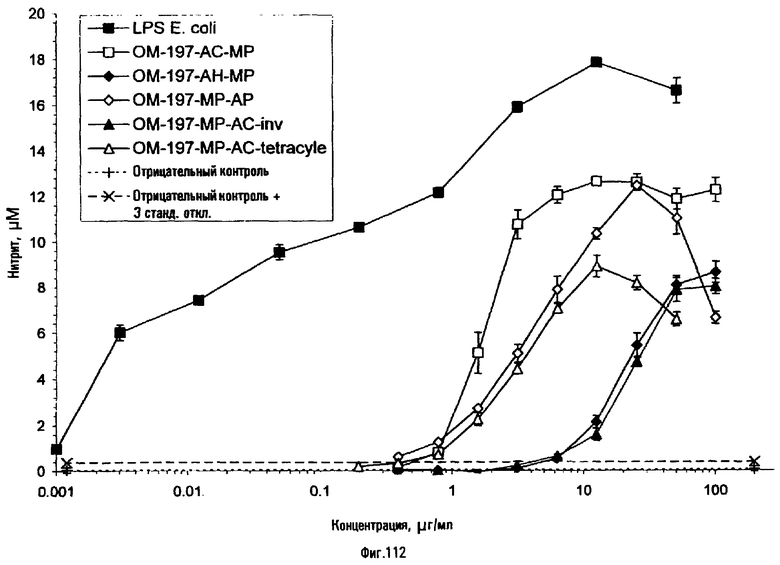
Изобретение относится к новым N-ацилированным псевдодипептидам, имеющим кислотную группу в нейтральной или заряженной форме на одном из концов псевдодипептида, а на другом конце имеющим вспомогательное функционализированное ответвление, отвечающие общей формуле I:
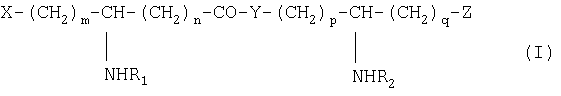
в которой R1 и R2 обозначают, каждый, ацильную группу насыщенной или ненасыщенной, линейной или разветвленной карбоновой кислоты, имеющей от 2 до 24 атомов углерода, незамещенной или имеющей заместитель или заместители, выбранные из групп: гидроксил, алкокси и ацилокси, коэффициент n принимает значения от 0 до 3, коэффициенты р и q принимают значения от 1 до 3, и коэффициент m принимает значения от 1 до 3, кроме случая, когда X является карбоксилом или одним из его производных; в этом случае он принимает значения от 0 до 3; Y обозначает О или NH, X и Z обозначают вспомогательное функционализированное ответвление или кислотную группу в нейтральной или заряженной форме, выбранную из следующих групп: карбоксил, карбокси-С1-С5-алкокси, карбокси-С1-С5-алкилтио, фосфоно-С1-С5-алкокси, дигидроксифосфорилокси, гидроксисульфонилокси, (карбокси-С1-С5-алкил)аминокарбонил, (дикарбокси-С1-С5-алкил)аминокарбонил, (аммонио-С1-С5-алкил)аминокарбонил, карбокси(амино-С1-С5-алкил)аминокарбонил, при условии, что по крайней мере один из заместителей X и Z обозначает вспомогательное функционализированное ответвление, и их энантиомеры и диастереоизомеры. Соединения обладают иммуномодулирующими свойствами как адъюванты, а также соединения могут быть привиты к антигену с целью модулирования иммунного ответа или могут быть привитыми на фармакофор для улучшения терапевтического действия или его направленной доставки. 7 н. и 41 з.п. ф-лы, 32 табл., 112 ил.
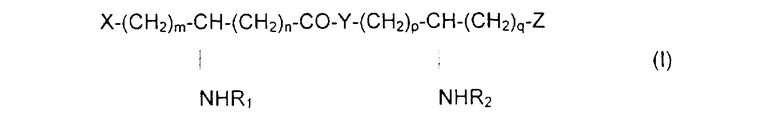
в которой R1 и R2 обозначают, каждый, ацильную группу насыщенной или ненасыщенной, линейной или разветвленной карбоновой кислоты, имеющей от 2 до 24 атомов углерода, незамещенной или имеющей заместитель или заместители, выбранные из групп: гидроксил, алкокси и ацилокси;
n принимают значения от 0 до 3;
р и q принимают значения от 1 до 3;
m принимает значения от 1 до 3, кроме случая, когда X является карбоксилом или одним из его производных; в этом случае он принимает значения от 0 до 3;
Y обозначает О или NH;
X и Z обозначают вспомогательное функционализированное ответвление или кислотную группу в нейтральной или заряженной форме, выбранную из следующих групп: карбоксил, карбокси-С1-С5-алкокси, карбокси-С1-С5-алкилтио, фосфоно-С1-С5-алкокси, дигидроксифосфорилокси, гидроксисульфонилокси, (карбокси-С1-С5-алкил)аминокарбонил, (дикарбокси-С1-С5-алкил) аминокарбонил, (аммонио-С1-С5-алкил) аминокарбонил, карбокси (амино-С1-С5-алкил) аминокарбонил, при условии, что по крайней мере один из заместителей X и Z обозначает вспомогательное функционализированное ответвление,
и их энантиомеры и диастереоизомеры.
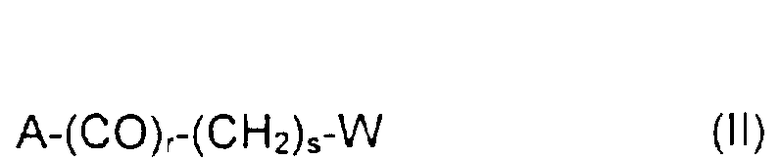
где А обозначает О, S или NH;
r имеет значение 0 или 1;
s имеет значение от 1 до 10;
W выбирают из групп: формил, амино, бром- или йодацетамидо, гидроксил, 1,2-дигидроксиэтил, свободный карбоксил, карбоксил в форме сложного эфира, амида или гидразида.
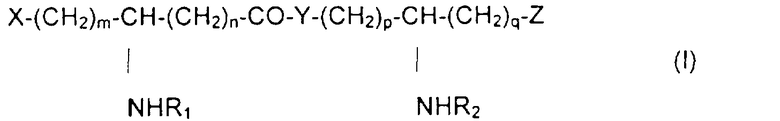
в которой R1 и R2 обозначают, каждый, ацильную группу насыщенной или ненасыщенной, линейной или разветвленной карбоновой кислоты, имеющей от 2 до 24 атомов углерода, незамещенной или имеющей заместитель или заместители, выбранные из групп: гидроксил, алкокси и ацилокси;
n принимают значения от 0 до 3;
р и q принимают значения от 1 до 3;
m принимает значения от 1 до 3, кроме случая, когда X является карбоксилом или одним из его производных; в этом случае он принимает значения от 0 до 3;
X и Z обозначают кислотную группу в нейтральной или заряженной форме или вспомогательное функционализированное ответвление, при условии, что по крайней мере один из заместителей X и Z обозначает вспомогательное функционализированное ответвление,
Y обозначает О или NH,
и вспомогательное функциональное ответвление X или Z имеет указанное выше значение за исключением 1,2-дигидроксиэтильного радикала.

s принимает значения от 1 до 10, в частности, 4, 5 или 6;
W выбирают преимущественно из групп: формил, амино, гидроксил, 1,2-дигидроксиэтил и карбоксил.
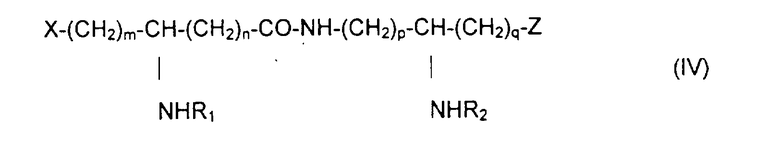
в которой R1 и R2 обозначают, каждый, ацильную группу насыщенной или ненасыщенной, линейной или разветвленной карбоновой кислоты, имеющей от 2 до 24 атомов углерода, незамещенной или имеющей один или несколько заместителей, выбранных из групп: гидроксил, алкокси, ацилокси;
n принимают значения от 0 до 3;
р и q принимают значения от 1 до 3;
m принимает значения от 1 до 3, кроме случая, когда X является карбоксилом или одним из его производных; в этом случае он принимает значения от 0 до 3;
один из заместителей X и Z обозначает кислотную функцию, выбранную из групп: карбоксил, дигидроксифосфорилокси, карбокси-С1-С5-алкокси, карбокси-С1-С5-алкилтио, карбокси-С1-С5-алкиламинокарбонил, дикарбокси-С1-С5-алкиламинокарбонил или карбокси(амино-С1-С5-алкил)аминокарбонил, и в которой другим заместителем является ацилокси, выбранный из групп: 6-аминогексаноилокси, 6-оксогексаноилокси, 6-гидроксигексакоилокси, 6,7-дигидроксигептаноилокси и 3-карбоксипропаноилокси,
а также их энантиомеры и диастереоизомеры.
α-N{[(R)-5-гидрокси-4-[(R)-3-гидрокситетрадеканоиламино]-пентил}амид N-[(R)-3-додеканоилокситетрадеканоиламино]-D-аспарагиновой кислоты и его солевые аддукты с неорганическим или органическим основанием;
10-(6,7-дигидроксигептаноат) 3-[(R)-3-додеканоилокситетра-деканоиламино]-4-оксо-5-аза-9-[(R)-3-гидрокситетрадеканоиламино]-декан-1,10-диол-1-дигидрофосфат и его солевые аддукты с неорганическим или органическим основанием;
10-(6-оксогексаноат) 3-[(R)-3-додеканоилокситетрадеканоиламино]-4-оксо-5-аза-9-[(R)-3-гидрокситетрадеканоиламино]-декан-1,10-диол-1-дигидрофосфат и его солевые аддукты с неорганическим или органическим основанием;
5-O-(6,7-дигидроксигептаноат) α-N{[(4R)-5-гидрокси-4-[(R)-3-гидрокситетрадеканоиламино]пентил}амида N-[(R)-3-додеканоилокситетрадеканоиламино]-D-аспарагиновой кислоты и его солевые аддукты с неорганическим или органическим основанием;
5-O-(6-оксогексаноат) α-N{[(4R)-5-гидрокси-4-[(R)-3-гидрокситетрадеканоиламино]пентил}амида N-[(R)-3-додеканоилокситетрадеканоиламино]-D-аспарагиновой кислоты и его солевые аддукты с неорганическим или органическим основанием;
5-O-(6-гидроксигексаноат) α-N{[(4R)-5-гидрокси-4-[(R)-3-гидрокситетрадеканоиламино]пентил}амида N-[(R)-3-додеканоилокситетрадеканоиламино]-D-аспарагиновой кислоты и его солевые аддукты с неорганическим или органическим основанием;
10-(6-аминогексаноат) (3RS,9R)-3-[(R)-3-додеканоилокситетрадеканоиламино]-4-оксо-5-аза-9-[(R)-3-гидрокситетрадеканоиламино]декан-1,10-диол-1-дигидрофосфат и его солевые аддукты с неорганическим или органическим основанием;
10-(6-аминогексаноат) (3R,9R)-3-[(R)-3-додеканоилокситетра-деканоиламино]-4-оксо-5-аза-9-[(R)-3-гидрокситетрадеканоил-амино]декан-1,10-диол 1-дигидрофосфат и его солевые аддукты с неорганическим или органическим основанием;
10-(6-аминогексаноат) (3S,9R)-3-[(R)-3-додеканоилокситетра-деканоиламино]-4-оксо-5-аза-9[(R)-3-гидрокситетрадеканоиламино]декан-1,10-диол-1-дигидрофосфат и его солевые аддукты с неорганическим или органическим основанием;
10-(6-гидроксигексаноат) 3-[(R)-3-додеканоилокситетрадеканоиламино]-4-оксо-5-аза-9-[(R)-3-гидрокситетрадеканоиламино]-декан-1,10-диол-1-дигидрофосфат и его солевые аддукты с неорганическим или органическим основанием;
{2-[(R)-3-гидрокситетрадодеканоиламино]-5-(6-оксогексил)-амино}пентиловый эфир 2-[(R)-3-додеканоилокситетрадеканоиламино]-4-(дигидроксифосфорилокси)бутановой кислоты и его солевые аддукты с неорганическим или органическим основанием;
9-O-(6-оксогексаноат) 1-O-карбоксиметилового эфира (2R,8R)-2-[(R)-3-додеканоилокситетрадеканоиламино]-3-оксо-4-аза-8-[(R)-3-гидрокситетрадеканоиламино]нонан-1,9-диола и его солевые аддукты с неорганическим или органическим основанием;
9-O-(7-аминогептаноат) (2S,8R)-1-(карбоксиметил)тио-2-[(R)-3-тетрадеканоилокситетрадеканоиламино]-3-оксо-4-аза-8-[(R)-3-гидрокситетрадеканоиламино]нонан-9-ола и его солевые аддукты с неорганическим или органическим основанием;
9-O-(6-оксагексаноат) 1-O-(2, 2-дикарбоксиэтилового) эфира (2R,8R)-2-[(R)-3-додеканоилокситетрадеканоиламино]-3-оксо-4-аза-8-[(R)-3-гидрокситетрадеканоиламино]нонан-1,9-диола и его солевые аддукты с неорганическим или органическим основанием;
10-(6-бромацетамидогексаноат) 3[(R)-3-додеканоилокситетра-деканоиламино]-4-оксо-5-аза-9-[(R)-3-гидрокситетрадеканоиламино]-1, 10-диол-1-дигидрофосфата и его солевые аддукты с неорганическим или органическим основанием;
10-O-(6-оксогексаноат) (3RS, 9R)-3-[(R)-3-гидрокситетрадеканоиламино]-4-оксо-5-аза-9-[(R)-3-додеканоилокситетра-деканоиламино]декан-1,10-диол-1-дигидрофосфата и его солевые аддукты с неорганическим или органическим основанием;
α-N-{(4R)-5-(6-аминогексаноилокси)-4-[(R)-3-гидрокситетрадеканоиламино] пентил}амид N-[(R)-3-додеканоилокситетрадеканоил]-D-аспарагиновой кислоты и его солевые аддукты с неорганическим или органическим основанием;
α-N-{(4R)-5-(6-сукциниламидогексаноилокси)-4-[(R)-3-гидрокси-тетрадеканоиламино]пентил}амид N-[(R)-3-додеканоилокситетрадеканоил]-D-аспарагиновой кислоты и его солевые аддукты с неорганическим или органическим основанием;
α-N-{(4R)-5-глицинилокси-4-[(R)-3-гидрокситетрадеканоиламино]-пентил}амид N-[(R)-3-додеканоилокситетрадеканоил]-D-аспарагиновой кислоты и его солевые аддукты с неорганическим или органическим основанием;
α-N-{(4R)-5-сукцинилокси-4-[(R)-3-гидрокситетрадеканоиламино]-пентил}амид N-[(R)-3-додеканоилокситетрадеканоил]-D-аспарагиновой кислоты и его солевые аддукты с неорганическим или органическим основанием;
α-N-{(4R)-5-гидрокси-4-[(R)-3-гидрокситетрадеканоиламино]пентил}амид N-[(R)-3-додеканоилокситетрадеканоил]-D-аспарагиновой кислоты β-N-(3-аминопропил) амид и его солевые аддукты с неорганическим или органическим основанием;
α-N-{(4R)-5-гидрокси-4-[(R)-3-гидрокситетрадеканоиламино]-пентил}амид N-[(R)-3-додеканоилокситетрадеканоил]-D-аспарагиновой кислоты β-N[(1S)-1-карбокси-5-аминопентил] амид и его солевые аддукты с неорганическим или органическим основанием;
α-N-{(4R)-5-(6-аминогексаноилокси)-4-[(R)-3-гидрокситетрадеканоиламино]-пентил}амид N-[(R)-3-додеканоилокситетрадеканоил]-D-аспарагиновой кислоты β-N((1S)-1-карбокси-5-аминопентил] амид и его солевые аддукты с неорганическим или органическим основанием;
α-N-{(4R)-5-гидрокси-4-[(R)-3-гидрокситетрадеканоиламино]-пентил}амид N-[(R)-3-додеканоилокситетрадеканоил]-D-аспарагиновой кислоты β-N-((1S)-1,2-дикарбоксиэтил] амид и его солевые аддукты с неорганическим или органическим основанием;
α-N-{(4R)-5-(6-аминогексаноилокси)-4-[(R)-3-гидрокситетрадеканоиламино]-пентил}амид N-[(R)-3-додеканоилокситетрадеканоил]-D-аспарагиновой кислоты β-N-((1S)-1, 2-дикарбоксиэтил] амид и его солевые аддукты с неорганическим или органическим основанием;
α-N-{(4R)-5-(6-аминогексаноилокси)-4-[(R)-3-гидрокситетра-деканоиламино]пентил}амид N-ацетил-D-аспарагиновой кислоты и его солевые аддукты с неорганическим или органическим основанием;
α-N-{(4R)-5-(6-аминогексаноилокси)-4-[(R)-3-гидрокситетрадеканоиламино]пентил}амид N-(2-деканоилоксиоктаноил)-D-аспарагиновой кислоты и его солевые аддукты с неорганическим или органическим основанием,
а также их энантиомеры и диастереоизомеры.
α-N{(4R)-5-гидрокси-4[(R)-3-гидрокситетрадеканоиламино] пентил}амид N-[(R)-3-додеканоилокситетрадеканоиламино]-D-аспарагиновой кислоты и его солевые аддукты с неорганическим или органическим основанием;
10-(6,7-дигидроксигептаноат) 3-[(R)-3-додеканоилокситетра-деканоиламино]-4-оксо-5-аза-9-[(R)-3-гидрокситетрадеканоиламино]-декан-1,10-диол-1-дигидрофосфат и его солевые аддукты с неорганическим или органическим основанием;
10-(6-оксогексаноат) 3-[(R)-3-додеканоилокситетрадеканоиламино]-4-оксо-5-аза-9-[(R)-3-гидрокситетрадеканоиламино]декан -1,10-диол-1-дигидрофосфат и его солевые аддукты с неорганическим или органическим основанием;
5-O-(6,7-дигидроксигептаноат) α-N [(4R)-5-гидрокси-4-[(R)-3-гидрокситетрадеканоиламино]пентил}амид N-[(R)-3-додеканоилокситетрадеканоиламино]-D-аспарагиновой кислоты и его солевые аддукты с неорганическим или органическим основанием;
5-O-(6-оксогексаноат) α-N[(4R)-5-гидрокси-4-[(R)-3-гидрокситетрадеканоиламино]пентил}амид N-[(R)-3-додеканоилокситетрадеканоиламино]-D-аспарагиновой кислоты и его солевые аддукты с неорганическим или органическим основанием;
10-(6-аминогексаноат) (3RS, 9R), (3R, 9R) и (3S, 9R)-3-[(R) -3-додеканоилокситетрадеканоиламино]-4-оксо-5-аза-9[(R)-3-гидрокситетрадеканоиламино]декан-1,10-диол 1-дигидрофосфат и их солевые аддукты с неорганическим или органическим основанием;
10-(6-гидроксигексаноат) 3-[(R)-3-додеканоилокситетрадеканоиламино]-4-оксо-5-аза-9-[(R)-3-гидрокситетрадеканоиламино]-декан-1,10-диол 1-дигидрофосфат и его солевые аддукты с неорганическим или органическим основанием;
{2-[(R)-3-гидрокситетрадеканоиламино]-5-(6-оксогексил)амино}-пентиловый эфир {2-[(R)-3-додеканоилокситетрадеканоиламино]-4-(дигидроксифосфорилокси)бутановой кислоты и его солевые аддукты с неорганическим или органическим основанием;
α-N-{(4R)-5-(6-аминогексаноилокси)-4-[(R)-3-гидрокситетрадеканоиламино]пентил}амид N-[(R)-3-додеканоилокситетрадеканоил]-D-аспарагиновой кислоты и его солевые аддукты с неорганическим или органическим основанием;
α-N-{(4R)-5-сукцинилокси-4-[(R)-3-гидрокситетрадеканоиламино] пентил}амид N-[(R)-3-додеканоилокситетрадеканоил]-D-аспарагиновой кислоты и его солевые аддукты с неорганическим или органическим основанием;
α-N-{(4R)-5-(6-аминогексаноилокси)-4-[(R)-3-гидрокситетра-деканоиламино]пентил}амид N-[(R)-3-додеканоилокситетрадеканоил]-D-аспарагиновой кислоты β-N-[(1S)-1-карбокси-5-аминопентил]амид и его солевые аддукты с неорганическим или органическим основанием;
α-N-{(4R)-5-(6-аминогексаноилокси)-4-[(R)-3-гидрокситетрадеканоиламино]пентил}амид N-[(R)-3-додеканоилокситетрадеканоил]-D-аспарагиновой кислоты β-N((1S)-1,2-дикарбоксиэтил] амид и его солевые аддукты с неорганическим или органическим основанием,
а также их диастереоизомеры и энантиомеры.
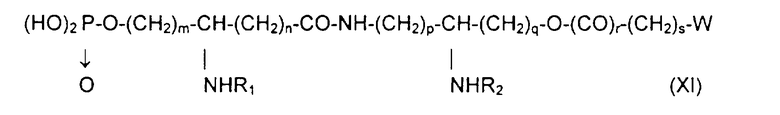
в которой заместители W, R1, R2, m, n, p, q, r, s имеют приведенные выше значения,
а также их энантиомеры и диастереоизомеры.
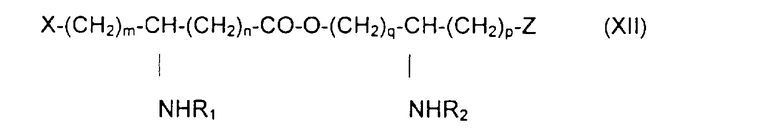
в которой R1 и R2 обозначают, каждый, ацильную группу насыщенной или ненасыщенной, линейной или разветвленной карбоновой кислоты, имеющей от 2 до 24 атомов углерода, незамещенной или имеющей один или несколько заместителей, выбранных из групп: гидроксил, алкокси и ацилокси;
р и q принимают значения от 1 до 3;
n принимают значения от 0 до 3;
m принимает значения от 1 до 3, кроме случая, когда X является карбоксилом или одним из его производных, в этом случае он принимает значения от 0 до 3;
Х и Z обозначают, каждый, кислотную группу или вспомогательное функционализированное ответвление,
а также их энантиомеры и диастереоизомеры.
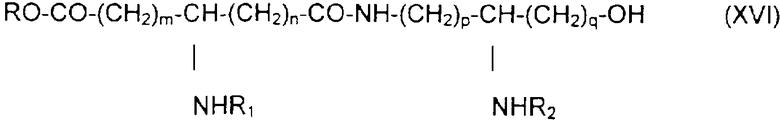
в которой заместители R1 и R2 и коэффициенты m, n, р и q являются такими, как определены выше, а R обозначает лабильную к гидрогенолизу группу,
а также их энантиомеры и диастереоизомеры.
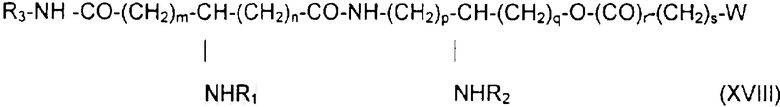
в которой заместители W, R1, R2, m, n, p, q, r и s имеют приведенные выше значения, а группа R3 обозначает аминоалкил, карбоксиалкил, дикарбоксиалкил или аминокарбоксиалкил,
а также их энантиомеры и диастереоизомеры.
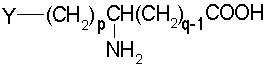
ортогональными блокирующими реагентами, в действии на оставшуюся свободной карбоксильную функцию восстанавливающим агентом с образованием соответствующего спирта, в освобождении аминной функции в положении (q+1), которую ацилируют с помощью функционального производного карбоновой кислоты формулы R2OH, где R2 определен выше, после чего освобождают спиртовую или аминную концевую функцию с получением функционализированного аминоспирта общей формулы V

в которой Y обозначает НО или NH2;
R2 обозначает ацильную группу насыщенной или ненасыщенной карбоновой кислоты, имеющей от 2 до 24 атомов углерода, незамещенной или имеющей один или несколько определенных выше заместителей;
р и q обозначают, каждый, целое число от 1 до 3,
который конденсируют в присутствии агента пептидной конденсации в инертном растворителе с производным ω-функционализированной аминокислоты общей формулы VI
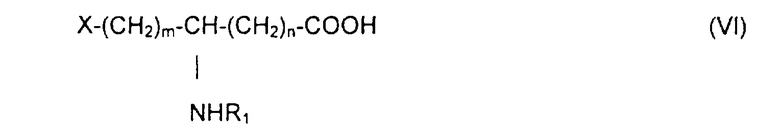
в которой R1 обозначает ацильную группу насыщенной или ненасыщенной карбоновой кислоты, имеющей от 2 до 24 атомов углерода, незамещенной или имеющей один или несколько определенных выше заместителей;
m и n имеют определенные выше значения;
Х обозначает определенную выше кислотную группу, которая может быть в свободной или этерифицированной форме, с образованием псевдодипептида общей формулы VII
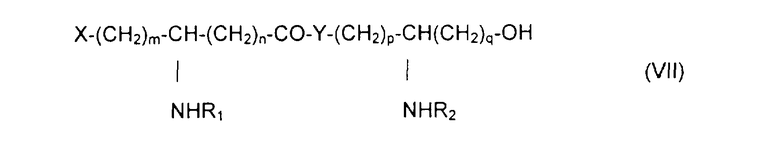
в которой заместители R1, и R2 и коэффициенты m, n, p и q такие, как определены выше,
свободная концевая спиртовая функция которого при желании может быть замещена, проалкилирована или проацилирована с помощью реагента замещения, алкилирования или ацилирования общей формулы VIII
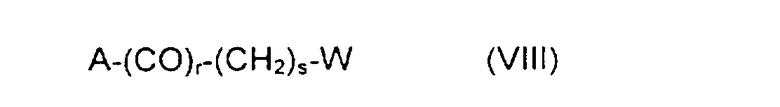
где А может быть уходящей группой, функцией ОН, SH или NH2;
r равен 1 или 0;
s варьирует от 1 до 10, преимущественно от 2 до 6;
W выбирают из групп: формил, амино, бром- или йодацетамидо, гидроксил, 1,2-дигидроксиэтил, карбоксил, свободный или в форме сложного эфира, амида или гидразида, и их предшественников,
если необходимо, в присутствии сочетающего агента, после чего псевдодипептид подвергают каталитическому гидрированию или какому-либо процессу снятия защиты, получая производное общей формулы I
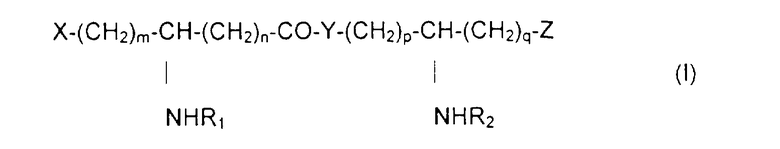
в которой X, Y, Z, R1, R2, n, m, p и q имеют приведенные выше значения.

в которой R1 и R2 обозначают, каждый, ацильную группу насыщенной или ненасыщенной, линейной или разветвленной карбоновой кислоты, имеющей от 2 до 24 атомов углерода, незамещенной или имеющей один или несколько заместителей, выбранных из групп: гидроксил, алкокси или ацилокси;
р и q принимают значения от 1 до 3;
n принимает значения от 0 до 3;
m принимает значения от 1 до 3, кроме случая, когда X является карбоксилом или одним из его производных; в этом случае он принимает значения от 0 до 3;
Х и Z обозначают, каждый, кислотную группу в нейтральной или заряженной форме или вспомогательное функционализированное ответвление,
отличающийся тем, что осуществляют блокировку аминных функций в положениях (q+1)- и ω диаминокислоты формулы H2N(CH2)pCHNH2(CH2)q-1COOH с помощью блокирующих реагентов, лабильных, соответственно, к ацидолизу и гидрогенолизу, оставшуюся свободной карбоксильную функцию подвергают действию восстанавливающего агента с образованием соответствующего спирта, освобождают аминную функцию в положении (q+1), которую ацилируют с помощью функционального производного карбоновой кислоты формулы R2OH, в которой R2 определен выше, после чего освобождают гидрогенолизом концевую аминную функцию с получением аминоспирта общей формулы IX
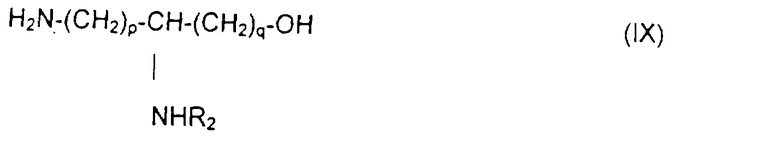
в которой R2 обозначает ацильную группу насыщенной или ненасыщенной карбоновой кислоты, имеющей от 2 до 24 атомов углерода, незамещенной или имеющей один или несколько определенных выше заместителей;
р и q обозначают целое число от 1 до 3,
который конденсируют в присутствии агента пептидной конденсации в инертном растворителе с производным ω-гидроксилированной аминокислоты общей формулы VI
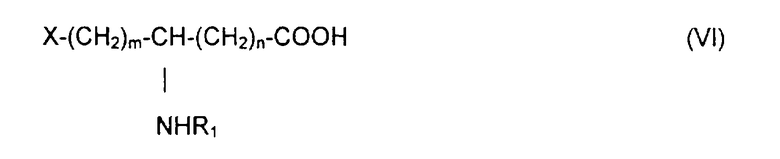
в которой R1 обозначает ацильную группу насыщенной или ненасыщенной карбоновой кислоты, имеющей от 2 до 24 атомов углерода, незамещенной или имеющей один или несколько определенных выше заместителей;
m есть целое число от 1 до 3;
n есть целое число от 0 до 3;
Х обозначает группу диалкилокси- или диарилокси-фосфорилокси формулы

с образованием фосфорилированного псевдодипептида общей формулы X
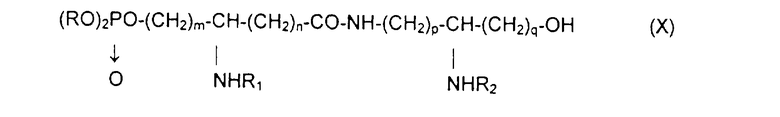
в которой заместители R1 и R2 и коэффициенты m, n, p и q являются такими, как определены выше, a R обозначает лабильный к гидрогенолизу радикал,
свободная концевая спиртовая функция которого при желании может быть замещена, проалкилирована или проацилирована с помощью реагента замещения, алкилирования или ацилирования общей формулы VIII

где А может быть уходящей группой, функцией ОН, SH или NH2;
r равен 0 или 1,
s варьирует от 1 до 10, предпочтительно от 2 до 6;
W выбирают из групп: формил, амино, бром- или йодацетамидо, гидроксил, 1,2-дигидроксиэтил, карбоксил, свободный или в форме сложного эфира, амида или гидразида и их предшественников,
если необходимо, в присутствии сочетающего агента, после чего псевдодипептид подвергают каталитическому гидрированию или какому-либо другому процессу снятия защиты, получая производное общей формулы XI
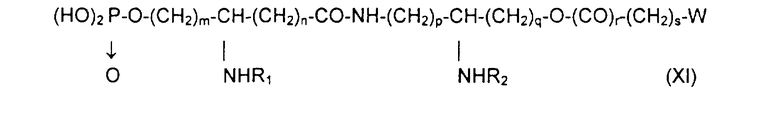
в которой заместители W, R1, R2, m, n, p, q, r, s имеют приведенные выше значения.
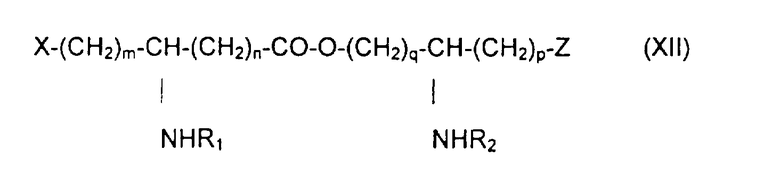
в которой R1 и R2 обозначают, каждый, ацильную группу насыщенной или ненасыщенной, линейной или разветвленной карбоновой кислоты, имеющей от 2 до 24 атомов углерода, незамещенной или имеющей один или несколько заместителей, выбранных из групп: гидроксил, алкокси и ацилокси;
р и q принимают значения от 1 до 3;
n принимает значения от 0 до 3;
m принимает значения от 1 до 3, кроме случая, когда X является карбоксилом или одним из его производных, в этом случае он принимает значения от 0 до 3;
Х и Z обозначают, каждый, кислотную группу или вспомогательное функционализированное ответвление,
который состоит в том, что блокируют аминные функции в положениях (q+1) и ω диаминокислоты формулы H2N(CH2)pCHNH2 (CH2)q-1COOH с помощью блокирующих реагентов, лабильных, соответственно, к ацидолизу и гидрогенолизу, оставшуюся свободной карбоксильную функцию подвергают действию восстанавливающего агента с образованием соответствующего спирта, освобождают аминную функцию в положении (q+1), которую ацилируют с помощью функционального производного карбоновой кислоты формулы R2OH, в которой R2 определен выше, освобождают гидрогенолизом концевую аминную функцию и затем алкилируют ее с помощью алкилирующего агента, получая аминоспирт общей формулы XIII
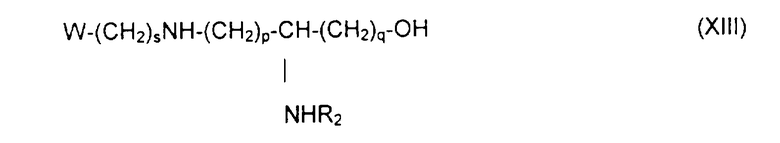
в которой R2 обозначает ацильную группу насыщенной или ненасыщенной карбоновой кислоты, имеющей от 2 до 24 атомов углерода, незамещенной или имеющей один или несколько определенных выше заместителей;
р и q обозначают целое число от 1 до 3;
s варьирует в пределах от 1 до 10, преимущественно от 2 до 7,
который конденсируют в присутствии агента конденсации в инертном растворителе с производным ω-гидроксилированной аминокислоты общей формулы VI

в которой R1 обозначает ацильную группу насыщенной или ненасыщенной карбоновой кислоты, имеющей от 2 до 24 атомов углерода, незамещенной или имеющей один или несколько заместителей;
m есть целое число от 1 до 3;
n есть целое число от 0 до 3;
Х обозначает группу диалкилокси- или диарилокси-фосфорилокси формулы
с образованием псевдодипептида общей формулы XIV
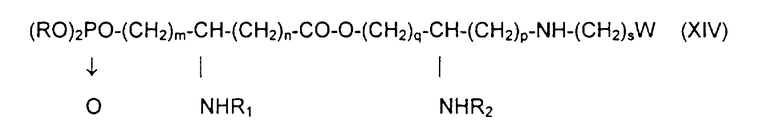
в которой заместители R1, R2 и W коэффициенты m, n, p, q и s являются такими, как определены выше, a R обозначает лабильный к гидрогенолизу радикал,
после чего псевдодипептид подвергают каталитическому гидрированию или какому-либо процессу снятия защиты с получением производного общей формулы XV
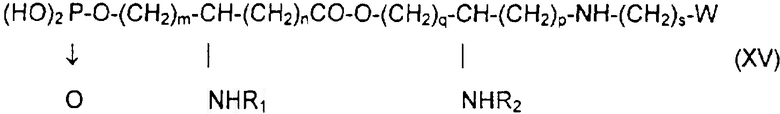
в которой заместители R1, R2, m, n, p, q, r и s имеют указанные выше значения,
причем W преимущественно выбирают из групп: формил, бром- или йодацетамидо, карбоксил, свободный или в форме сложного эфира, амида или гидразида и их предшественников.
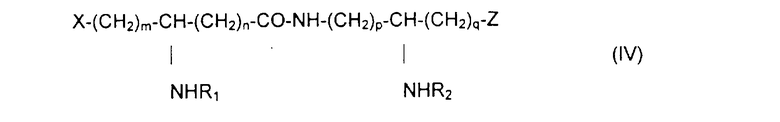
в которой R1 и R2 обозначают, каждый, ацильную группу насыщенной или ненасыщенной, линейной или разветвленной карбоновой кислоты, имеющей от 2 до 24 атомов углерода, незамещенной или имеющей один или несколько заместителей, выбранных из групп: гидроксил, алкокси и ацилокси;
р и q принимают значения от 1 до 3;
n принимает значения от 0 до 3;
m принимает значения от 1 до 3, кроме случая, когда X является карбоксилом или одним из его производных; в этом случае он принимает значения от 0 до 3;
Х и Z обозначают, каждый, кислотную группу или вспомогательное функционализированное ответвление,
который состоит в том, что осуществляют блокировку аминных функций в положениях (q+1) и в ω диаминокислоты формулы H2N(CH2)pCHNH2(CH2)q-1COOH с помощью блокирующих реагентов, лабильных, соответственно, к ацидолизу и гидрогенолизу, оставшуюся свободной карбоксильную функцию подвергают действию восстанавливающего агента с образованием соответствующего спирта, освобождают аминную функцию в положении (q+1), которую ацилируют с помощью функционального производного карбоновой кислоты формулы R2OH, в которой R2 определен выше, и освобождают гидрогенолизом концевую аминную функцию, получая аминоспирт общей формулы IX
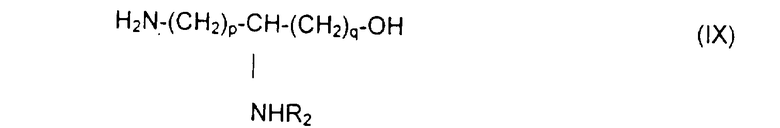
в которой R2 обозначает ацильную группу насыщенной или ненасыщенной карбоновой кислоты, имеющей от 2 до 24 атомов углерода, незамещенной или имеющей один или несколько определенных выше заместителей;
р и q обозначают целое число от 1 до 3,
который конденсируют в присутствии агента пептидной конденсации в инертном растворителе с функциональным производным ω-аминокарбоновой кислоты общей формулы VI

в которой R1 обозначает ацильную группу насыщенной или ненасыщенной карбоновой кислоты, имеющей от 2 до 24 атомов углерода, незамещенной или имеющей один или несколько определенных выше заместителей;
m обозначает целое число от 1 до 3, кроме случая, когда X является карбоксилом или одним из его производных, в этом случае он принимает значения от 0 до 3;
n есть целое число от 0 до 3;
Х обозначает радикал RO-CO-, в котором R обозначает лабильную алкильную группу,
с образованием псевдодипептида общей формулы XVI
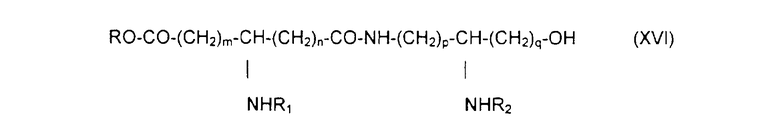
в которой заместители R1 и R2 и коэффициенты m, n, p и q являются такими, как определены выше, а R обозначает лабильную к гидрогенолизу группу,
свободная концевая спиртовая функция которого при желании может быть замещена, проалкилирована или проацилирована с помощью реагента замещения, алкилирования или ацилирования общей формулы VIII
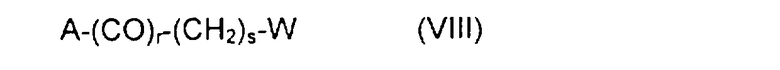
где А обозначает уходящую группу, функцию ОН, SH или NH2;
r равен 1 или 0;
s варьирует от 1 до 10, преимущественно от 2 до 6;
W выбирают из групп: формил, амино, бром- или йодацетамидо, ациламидо, 1,2-дигидроксиэтил, карбоксил, свободный или в форме сложного эфира, амида или гидразида и их предшественников,
если необходимо, в присутствии сочетающего агента, после чего соединение подвергают процессу деблокирования, в частности каталитическому гидрированию, получая производное общей формулы XVII
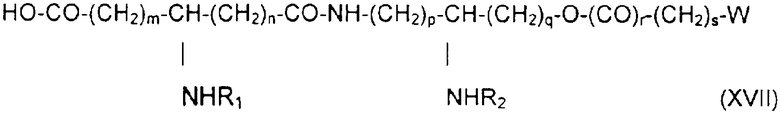
в которой заместители W, R1, R2, m, n, p, q, r и s имеют приведенные выше значения.
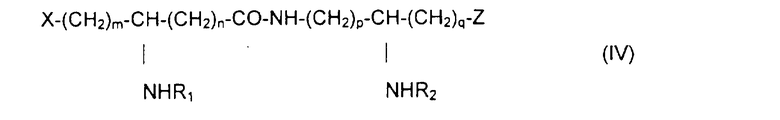
в которой R1 и R2 обозначают, каждый, ацильную группу насыщенной или ненасыщенной, линейной или разветвленной карбоновой кислоты, имеющей от 2 до 24 атомов углерода, незамещенной или имеющей один или несколько заместителей, выбранных из групп: гидроксил, алкокси и ацилокси;
p и q принимают значения от 1 до 3;
n принимает значения от 0 до 3;
m принимает значение от 1 до 3, кроме случая, когда X является карбоксилом или одним из его производных; в этом случае он принимает значения от 0 до 3;
Х и Z обозначают, каждый, кислотную группу или вспомогательное функционализированное ответвление,
который состоит в том, что блокируют свободную гидроксильную функцию псевдодипептида общей формулы XVI
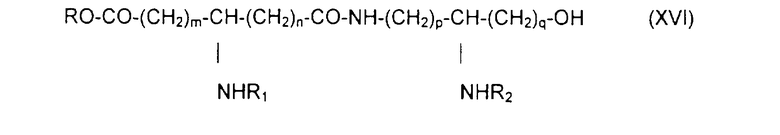
в которой заместители R1 и R2 и коэффициенты m, n, p и q являются такими, как определены выше, a R обозначает лабильную к гидрогенолизу группу,
с помощью лабильной к ацидолизу защитной группы, освобождают карбоксильную функцию, присутствующую в виде сложного эфира, подвергают эту карбоксильную функцию, возможно в активированной форме, действию восстанавливающего агента с образованием соответствующего первичного спирта, превращают спиртовую функцию в фосфорноэфирную действием фосфорилирующего агента, защищенную гидроксильную функцию освобождают действием кислоты и вводят в реакцию замещения, алкилирования или ацилирования с реагентом общей формулы VIII
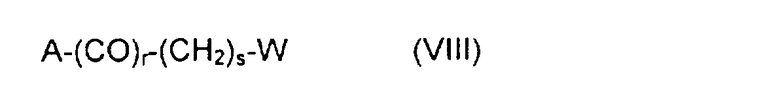
где А может быть уходящей группой, функцией ОН, SH или NH2;
r равен 1 или 0;
s варьирует от 1 до 10, преимущественно от 2 до 6;
W выбирают из групп: формил, амино, бром- или йодацетамидо, ациламидо, гидроксил, 1,2-дигидроксиэтил, карбоксил, свободный или в форме сложного эфира или другого производного;
если необходимо, в присутствии сочетающего агента, после чего полученный продукт деблокируют с помощью гидрогенолиза, получая продукт общей формулы XI

в которой заместители W, R1, R2, m, n, p, q, r и s имеют приведенные выше значения.
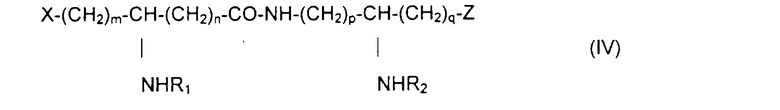
в которой R1 и R2 обозначают, каждый, ацильную группу насыщенной или ненасыщенной, линейной или разветвленной карбоновой кислоты, имеющей от 2 до 24 атомов углерода, незамещенной или имеющей один или несколько заместителей, выбранных из групп: гидроксил, алкокси и ацилокси;
p и q принимают значения от 1 до 3;
n принимает значения от 0 до 3;
m коэффициент принимает значение от 1 до 3, кроме случая, когда X является карбоксилом или одним из его производных, в этом случае он принимает значения от 0 до 3;
Х и Z обозначают, каждый, кислотную группу или вспомогательное функционализированное ответвление,
который состоит в том, что деблокируют карбоксильную функцию, присутствующую в виде сложного эфира в псевдодипептиде общей формулы XVI
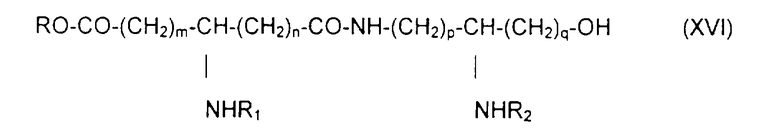
в которой заместители R1 и R2 и коэффициенты m, n, p и q являются такими, как определены выше, a R обозначает лабильную к гидрогенолизу группу,
после чего подвергают образовавшуюся таким образом кислоту реакции пептидного сочетания с частично защищенными аминокислотой или диаминоалканом в присутствии агента пептидного сочетания, после чего продукт сочетания подвергают при желании реакции замещения, ацилирования или алкилирования с использованием реагента общей формулы VIII

где А может быть уходящей группой, функцией ОН, SH или NH2;
r равен 0 или 1;
s варьирует от 1 до 10, преимущественно от 2 до 6;
W выбирают из групп: формил, амино, бром- или йодацетамидо, ациламидо, 1,2-дигидроксиэтил, карбоксил, свободный или в форме сложного эфира или какого-либо другого производного,
если необходимо, в присутствии активирующего агента, после чего полученный продукт деблокируют, например, с помощью гидрогенолиза, получая продукт общей формулы XVIII:
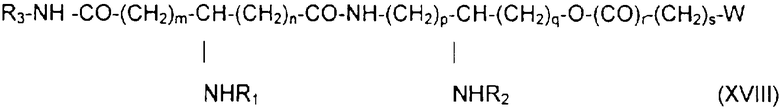
в которой заместители W, R1, R2, m, n, p, q, r и s имеют приведенные выше значения, а группа R3 обозначает аминоалкил, карбоксиалкил, дикарбоксиалкил или аминокарбоксиалкил.
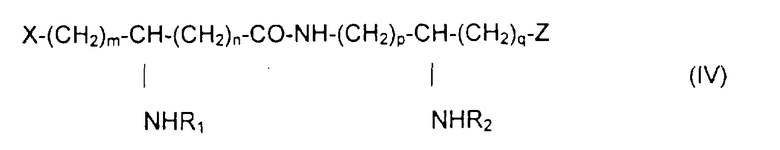
в которой R1 и R2 обозначают, каждый, ацильную группу насыщенной или ненасыщенной, линейной или разветвленной карбоновой кислоты, имеющей от 2 до 24 атомов углерода, незамещенной или имеющей один или несколько заместителей, выбранных из групп: гидроксил, алкокси и ацилокси;
p и q принимают значения от 1 до 3;
n принимает значение от 0 до 3;
m принимает значение от 1 до 3, кроме случая, когда X является карбоксилом или одним из его производных; в этом случае он принимает значение от 0 до 3;
X и Z обозначает, каждый, кислотную группу или функционализированное дистанцирующее ответвление,
в чистой энантиомерной форме или в виде смеси стереоизомеров.
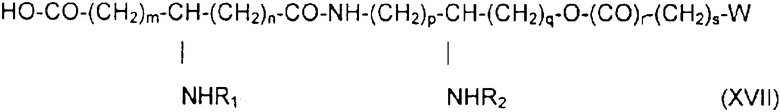
в которой заместители W, R1, R2, m, n, p, q, r, s имеют приведенные выше значения,
в чистой энантиомерной форме или в виде смеси диастереоизомеров.
O CO(CH2)sW ,
в котором s обозначает число от 1 до 10 и W имеет значение, определенное выше, соединено посредством аминогруппы, альдегидной группы, карбоксильной группы, свободной или комбинированной, или посредством атома галогена с антигеном или фармакофором.
| ПРОИЗВОДНЫЕ ПЕПТИДОВ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ПРИМЕНЕНИЕ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1997 |
|
RU2141483C1 |

