Предлагаемый способ относится к области получения закрепленных на носителе олигомерных хелатных комплексов металлов, которые могут быть использованы в качестве гетерогенизированных катализаторов жидкофазных или газофазных процессов окисления непредельных соединений.
Известно, что хелатные соединения металлов закрепляют на твердых носителях для их последующего использования как гетерогенных катализаторов [Ф. Хартли. Закрепленные металлокомплексы. Новое поколение катализаторов.- М.: Мир, 1989].
Известен способ нанесения (1-ацетил)фталоцианината железа (II) на цеолит NaA последовательной обработкой цеолита диэтилкетоном, гексаметилендиамином и хелатным соединением железа (II) в кипящем бензоле в анаэробных условиях [Can.J.Chem. 1997. Vol. 76. P. 955].
Олигомерные хелатные соединения меди (II) из арилгидразониминов никогда не наносили на щелочные формы цеолитов, и результаты закрепления на этих подложках неизвестны.
Данная задача решается предложенным способом, который состоит в том, что дегидратированный цеолит CaA последовательно обрабатывают дипропилкетоном (ДПК), бензидином в органическом растворителе при температуре кипения и смесью в хлороформе α,ω- полиметилендиамина (ПМДА) с 5-10%-ным избытком арилгидразонимината меди (II) при кипячении в течение 1-4 ч.
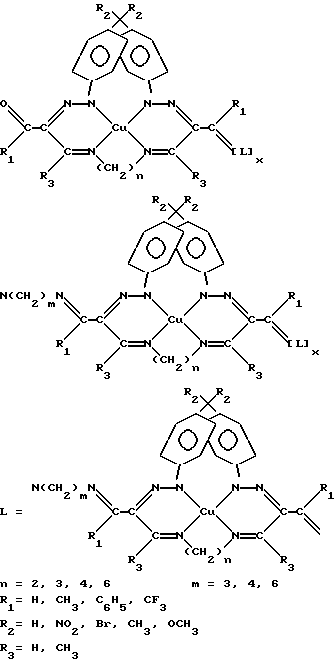
Олигомерные хелаты меди (II) являются гетерогенными катализаторами окисления в жидкофазных условиях, поскольку практически нерастворимы в органических растворителях и воде [Успехи химии. 1963. Т. 32. Вып. 12. С. 1488; Кинетика и катализ. 1962. Т. 3. Вып. 5. С. 680]. Однако использование этих соединений в собственной фазе неэффективно, т.к. в каталитической реакции участвуют только поверхностные центры.
В предлагаемом способе в качестве подложки выбран цеолит CaA со средним эффективным диаметром входных окон 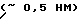 , соизмеримым с минимальным размером линейной молекулы ДПК, которая может проникать в пустоты носителя только при движении в направлении углеводородного скелета.
, соизмеримым с минимальным размером линейной молекулы ДПК, которая может проникать в пустоты носителя только при движении в направлении углеводородного скелета.
Для удаления молекул воды из больших полостей с целью размещения в них ДПК - предшественника якорного фрагмента - необходима стандартная предварительная термообработка цеолита CaA.
Выбор ДПК обусловлен требованиями, предъявляемыми к строению молекул предшественника, который должен иметь: псевдоцилиндрическую форму, поскольку цеолит CaA адсорбирует углеводороды и простейшие молекулы только нормального строения; линейный размер, превышающий эффективный диаметр входного окна в большую полость, и карбонильную группу в середине углеводородного скелета для последующего образования Т-образного фрагмента, выполняющего роль топологического якоря.
Молекула ДПК имеет линейное строение и эффективные размеры 0,49 нм х 1,15 нм, отвечающие перечисленным условиям.
После адсорбции ДПК добавляют бензидин в кипящем органическом растворителе, необходимый для образования жесткого Т-образного якорного фрагмента в соответствии с реакцией (1).
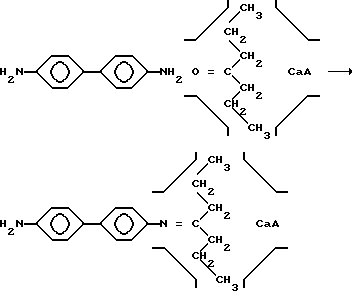
Реакция (1) между бензидином и адсорбированным в большой полости кетоном приводит к захвату цеолитом Т-образного якорного интермедиата в том случае, если она происходит на входе в большую полость, которая непроницаема для диамина с эффективными размерами 0,6 нм х 1,4 нм.
Выбор среды обусловлен необходимостью растворения бензидина, а кипячение смеси требуется для ускорения конденсации амина с адсорбированным в большой полости кетоном. При недостаточно высокой температуре кетон десорбируется в раствор, и реакция (1) происходит преимущественно в объеме жидкой фазы, что нежелательно, поскольку не приводит к закреплению якорного интермедиата.
Предлагаемая последовательность обработки цеолита реагентами вызвана тем, что стерически затрудненный фрагмент енамина образуется в большой полости из предварительно адсорбированного в ней кетона. Закрепление енамина происходит только тогда, когда реакция между ДПК и бензидином осуществляется на границе между раствором и большой полостью, открывающейся на внешнюю поверхность кристаллита. При этом енамин захватывается цеолитом согласно схеме. Все остальные случаи взаимодействия ДПК и кетона не приводят к продуктивному закреплению. В силу стерических затруднений перемещение якорного интермедиата возможно только в пределах полости.
Таким образом подготовленный цеолит CaA содержит Т-образные фрагменты с аминогруппами, обращенными в сторону внешней поверхности. Полученный образец обрабатывают смесью ПМДА и арилгидразонимината меди (II) в органическом растворителе в молярном соотношении 1:(1,05-1,10) соответственно и кипятят в течение 1-4 ч. Использование смеси ПМДА и арилгидразонимината меди (II) связано с необходимостью темплатного in situ синтеза олигомерного полихелата. Избыток арилгидразонимината меди (II) обеспечивает сохранение свободной карбонильной группы по крайней мере на одном конце олигомерных полихелатных цепей. Величина избытка арилгидразонимината меди (II) зависит от длины полиметиленового остова диамина. При использовании 1,3- пропилендиамина продукт олигомеризации обогащен макромолекулами с одной концевой карбонильной группой, начиная с 5%-ного избытка мономерного хелата. Дальнейшее увеличение соотношения мономер: диамин нецелесообразно. При использовании 1,4- тетраметилендиамина и 1,6-гексаметилендиамина минимальное соотношение мономер: диамин увеличивается до 1:1,08 и 1:1,10 соответственно.
При конденсации "поверхностных" аминогрупп якорных фрагментов с карбонильной функцией хелата меди (II) образуется топологическая связь комплекса с носителем, а конденсация карбонильных групп закрепленного мономера с аминогруппами ПМДА приводит к олигомеризации в растворе в соответствии со схемой.
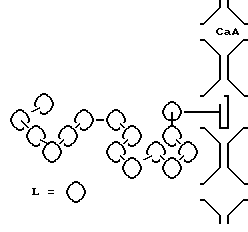
Полученные таким образом образцы охарактеризованы данными элементного анализа, йодометрии и электронных спектров диффузного отражения, которые сопоставлены с электронными спектрами индивидуальных олигомерных арилгидразониминатов меди (II) и их механических смесей с цеолитом CaA.
Предложенный способ получения нанесенных олигомерных арилгидразониминатов меди (II) позволяет проводить в их присутствии жидкофазное окисление циклогексена молекулярным кислородом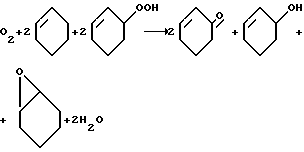
с более высокой эффективностью, поскольку количество работающих каталитических центров возрастает пропорционально средней степени олигомеризации.
По сравнению с использованием индивидуальных олигомерных арилгидразониминатов меди (II) в присутствии гетерогенизированных олигомерных арилгидразониминатов меди (II) наблюдаемая скорость окисления циклогексена, рассчитанная на единицу массы твердой фазы, в сопоставимых условиях увеличивается в среднем в 400-500 раз в зависимости от химической природы и средней степени олигомеризации хелатных соединений.
Использование цеолита в качестве носителя способствует сдвигу равновесия (1) вправо за счет связывания воды, выделяющейся в процессе окисления циклогексена. Адсорбированная в цеолите вода удаляется вакуумной сушкой образца при 50-60oC в течение 5-6 ч без разрушения гетерогенизированных олигомерных арилгидразониминатов меди (II).
Примеры
Пример 1.
Фракционированный цеолит CaA (0,5-1 мм) в количестве 3 г прокаливают на воздухе при 550oC в течение 6 ч. К полученному дегидратированному образцу добавляют 0,8 см3 ДПК, смешивают с 0,3 мг бензидина в 50 см3 бензола, нагревают до кипения и кипятят в течение 30 мин. Полученную суспензию декантируют, осадок экстрагируют бензолом в аппарате Сокслета до полного удаления избытка бензидина. К образцу добавляют смесь, содержащую 6,7 мг (0,09 ммоль) 1,3-пропилендиамина и 45,4 мг (0,095 ммоль) N,N'-пропиленбис(1-ацетилглиоксаль-1-фенилгидразониминато)медь (II) в 75 см3 хлороформа, и кипятят в течение 2 ч. Твердую фазу отделяют от раствора, переносят в аппарат Сокслета и экстрагируют хлороформом.
При использовании 0,1 г образца, содержащего 0,003 ммоль олигомерного N, N'-пропиленбис(1-ацетилглиоксаль-1- фенилгидразониминато) медь (II), в качестве гетерогенного катализатора в жидкофазном окислении циклогексена молекулярным кислородом скорость реакции в сопоставимых условиях при 50oC возрастает в 431 раз по сравнению со скоростью той же реакции в присутствии 0,003 ммоль индивидуального олигомерного хелатного соединения.
Пример 2.
Цеолит CaA (фракция 0,5-1 мм) в количестве 3 г прокаливают на воздухе при 550oC в течение 6 ч. К дегидратированному образцу добавляют 0,8 см3 ДПК, смешивают с 0,3 мг бензидина в 50 см3 толуола, нагревают до кипения и кипятят в течение 30 мин. Полученную суспензию декантируют и осадок экстрагируют толуолом в аппарате Сокслета до полного удаления избытка бензидина. К образцу добавляют смесь 8,79 мг (0,076 ммоль) 1,6-гексаметилендиамина и 48,3 мг (0,083 ммоль) N,N'-гексаметиленбис(2,3,4-пентантрион-1 - n-нитрофенилгидразониминато)медь (II) в 100 см3 хлороформа и нагревают при кипении в течение 4 ч. Образец отделяют от раствора, переносят в аппарат Сокслета и экстрагируют хлороформом.
При использовании 0,2 г нанесенного образца, содержащего 0,0055 ммоль олигомерного N, N'-гексаметиленбис(2,3,4-пентантрион-2-n- нитрофенилгидразониминато)медь (II) в качестве гетерогенного катализатора в жидкофазном окислении циклогексена, скорость реакции возрастает при 50oC в 486 раз по сравнению со скоростью той же реакции в присутствии 0,0055 ммоль индивидуального олигомерного хелата меди (II) в сопоставимых условиях.
Примеры с остальными олигомерными арилгидразониминатами меди (II) протекают в условиях, аналогичных примеру 1.
Таким образом, предложенный способ позволяет закреплять олигомерные арилгидразониминаты меди (II) на цеолите CaA и при использовании конечного продукта как гетерогенного катализатора повышать скорость жидкофазного окисления циклогексена молекулярным кислородом в 400-500 раз по сравнению со скоростью того же процесса в присутствии индивидуальных олигомерных арилгидразониминатов меди (II).
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ГЕТЕРОГЕНИЗИРОВАННЫХ АРИЛГИДРАЗОНИМИНАТОВ МЕДИ (II) | 1999 |
|
RU2159677C1 |
| СПОСОБ ЗАКРЕПЛЕНИЯ КОМПЛЕКСОВ МЕТАЛЛОВ НА ЦЕОЛИТЕ ТИПА А | 1999 |
|
RU2163508C1 |
| СПОСОБ БИФУНКЦИОНАЛЬНОЙ МОДИФИКАЦИИ ЦЕОЛИТА | 2001 |
|
RU2203853C1 |
| ЧУВСТВИТЕЛЬНЫЙ ЭЛЕМЕНТ ВОЛНОВОГО ТВЕРДОТЕЛЬНОГО ГИРОСКОПА | 2000 |
|
RU2166734C1 |
| СПОСОБ ПОЛУЧЕНИЯ КАПСУЛИРОВАННЫХ В ПОЛИМЕРНОЙ ПЛЕНКЕ ЖИДКОКРИСТАЛЛИЧЕСКИХ КОМПОЗИЦИЙ | 2002 |
|
RU2215770C1 |
| РЕЗИНОВАЯ КОМПОЗИЦИЯ | 2001 |
|
RU2215756C2 |
| СПОСОБ ОЧИСТКИ УГЛЕВОДОРОДНЫХ КОМПОЗИЦИЙ ОТ МЕРКАПТАНОВ | 2009 |
|
RU2404225C2 |
| СПОСОБ ПОЛУЧЕНИЯ ВЫСОКОТЕМПЕРАТУРНЫХ ПРОТОНСОДЕРЖАЩИХ ДВОЙНЫХ ФОСФАТОВ КАЛЬЦИЯ С ЖЕЛЕЗОМ CaFeH(PO) И МЕДЬЮ СаCuH(PO) | 2001 |
|
RU2229436C2 |
| КАТАЛИЗАТОР ДЛЯ ПОЛУЧЕНИЯ ЛИНЕЙНЫХ МОНОАЛКИЛБЕНЗОЛОВ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2009 |
|
RU2383387C1 |
| СПОСОБ СОВМЕСТНОГО ПОЛУЧЕНИЯ ХЛОРАЛКАНОВ И ХЛОРОФОРМА НА ТВЕРДОМ КАТАЛИЗАТОРЕ | 2010 |
|
RU2434838C1 |
Изобретение относится к области получения закрепленных на носителе олигомерных хелатных катализаторов, которые могут быть использованы для жидкофазных или газофазных процессов окисления непредельных соединений. Способ заключается в последовательной обработке дегидратированного цеолита СаА дипропилкетоном, бензидином в кипящем органическом растворителе и добавлением смеси в хлороформе α,ω-полиметилендиамина и арилгидразонимината меди (II) с последующим выдерживанием кипящей смеси в течение 1-4 ч. Способ позволяет закреплять олигомерные арилгидразониминаты меди (II) на цеолите СаА и при использовании конечного продукта как гетерогенного катализатора повышать скорость жидкофазного окисления циклогексена молекулярным кислородом в 400-500 раз по сравнению со скоростью того же процесса в присутствии индивидуальных олигомерных арилгидразониминатов меди (II).
Способ получения гетерогенизированных олигомерных арилгидразониминатов меди (II), заключающийся в том, что дегидратированный цеолит СаА последовательно обрабатывают дипропилкетоном, бензидином в кипящем органическом растворителе и смесью в хлороформе α,ω- полиметилендиамина и 5 - 10%-ного молярного избытка арилгидразонимината меди (II) при кипячении в течение 1 - 4 ч.
| ZEFIROF N.T | |||
| и др | |||
| "Catalysist by topologically anchored metal complexes", Can | |||
| J | |||
| Chem, 1997, v.76, p.955 | |||
| ХАРТЛИ Ф | |||
| Закрепленные металлокомплексы | |||
| Новое поколение катализаторов | |||
| - М.: Мир, 1989 | |||
| СПОСОБ ПРИГОТОВЛЕНИЯ КАТАЛИЗАТОРА | 0 |
|
SU389828A1 |
| Катализатор для жидкофазного окисления углеводородов | 1983 |
|
SU1126318A1 |
