Настоящее изобретение относится к новым химическим соединениям, их получению, фармацевтическим композициям, содержащим их, и использованию их в медицине, в частности профилактике и лечении мигрени.
Рецепторы, которые являются промежуточным звеном действий 5-окситриптамина (5-НТ), идентифицировали у млекопитающих как в периферии, так и в головном мозге. Согласно классификации и номенклатуре, предложенным в недавней статье (Bradley et al. Neuropharmac, 25, 563 (1986)), эти рецепторы могут быть классифицированы в трех основных типах, а именно, "5-НТ1- подобные", 5-НТ2 и 5-НТ3. Различные классы соединений предлагали в качестве 5-НТ агонистов (веществ, обладающих сродством к рецептору) или антагонистов для терапевтического использования, но они не всегда специфичны к определенному типу 5-НТ рецептора. Описание изобретения патента Европы 0313397 описывает класс 5-НТ агонистов, которые являются специфическими к определенному типу "5-НТ1-подобного" рецептора и являются эффективными лекарственными средствами для лечения клинических состояний, в которых селективный агонист для этого типа рецептора является показательным (требуется). Например, рассматриваемый рецептор является промежуточным звеном (посредничает в) вазоконстрикции (сужения кровеносных сосудов) в каротидном васкулярном ложе и тем самым изменяет кровоток в нем. Поэтому соединения, описанные в описании изобретения Европейского патента, являются благотворными при лечении или профилактике состояний, в которых вазоконстрикция в каротидном васкулярном ложе является показательной (требуется), например мигрени, состояния, связанного с избыточной дилатацией (расширением) каротидной сосудистой сети. Однако в пределах объема ранней заявки является то, что "ткань-мишень" может быть любой тканью, в которой действие связывается с посредничеством "5-НТ1-подобных" рецепторов названного выше типа.
Мы в настоящее время открыли дополнительный класс соединений, обладающих необычным агонизмом к "5-НТ1-подобным" рецепторам и превосходным поглощением (абсорбцией) после пероральной дозы. Эти свойства делают соединения особенно полезными для некоторых лекарственных применений, особенно профилактики и лечения мигрени, "гистаминовой" головной боли и головной боли, связанной с васкулярными нарушениями, в дальнейшем все вместе называемых "мигренью".
Соответственно согласно первому аспекту настоящего изобретения предлагается соединение формулы (I)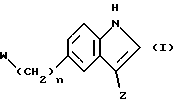
в которой n является целым числом от 0 до 3,
w является группой формулы (i), (ii) или (iii)

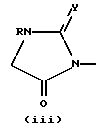
в которой R является водородом или C1-4-алкилом,
X является -O-, -S-, -NH- или -CH2-,
Y является кислородом или серой, а хиральный центр* в формуле (i) или (ii) находится в своей (S)- или (R)-форме или является их смесью в любых пропорциях; является группой формулы (iv), (v) или (vi)
-CH2CH2NR1R2 (iv)

в которой R1 и R2 независимо выбираются из водорода и C1-4-алкила, а
R3 является водородом или C1-4-алкилом;
и соли, сольваты и физиологически функциональные производные его, при условии, что в данный объем не входит соединение N,N-диметил-2-[5-(2-оксо-1,3-оксазолидин-4-илметил)-1H-индол-3-ил] этиламин.
Соединения формулы (I), имеющие особенно желательные свойства для лечения и профилактики мигрени, включают соединения, в которых является I, W является группой формулы (i), a Z является группой формулы (iv) или (vi). Из них особенно предпочтительными являются соединения формулы (I), в которой n является 1, W является группой формулы (i), в которой R является водородом, X является -O-, а Y является кислородом и Z является группой формулы (iv) или (vi), в которой R1 = R2 = водород или метил.
Двумя соединениями формулы (I), имеющими исключительные свойства для лечения и профилактики мигрени, являются N,N-диметил-2-[5-(2-оксо-1,3-оксазолидин-4-ил-метил)-1H-индол-3-ил] -этиламин и 3-(1-метил-4-пиперидил)-5-(2-оксо-1,3-оксазолидин-4-илметил)-1H-индол, либо в их (S)- или (R)-форме, либо в виде смеси их в любых соотношениях. Соли и сольваты этих соединений, например гидраты малеатов, являются особенно предпочтительными.
Физиологически приемлемые соли являются особенно пригодными для лекарственного применения из-за их более высокой растворимости в воде по сравнению с родственными, т.е. основными, соединениями. Такие соли определенно должны иметь физиологически приемлемый анион. Подходящие физиологически приемлемые соли соединений настоящего изобретения включают соли, получаемые из уксусной, соляной, бромистоводородной, фосфорной, яблочной, малеиновой, фумаровой, лимонной, серной, молочной или винной колоты. Сукцинатные или хлоридные соли являются особенно предпочтительными для лечебных целей. Соли, обладающие физиологически неприемлемым анионом, находятся в пределе объема изобретения в качестве полезных интермедиатов для получения физиологически приемлемых солей и/или для использования в нелечебных, например in vitro (в лабораторном сосуде), ситуациях.
По второй особенности настоящего изобретения, предлагается соединение формулы (I) или физиологически приемлемая соль, сольват или физиологически функциональное производное их для использования в качестве терапевтического средства, в частности в качестве агониста для "5-НТ1-подобного" рецептора, например в качестве каротидного вазоконстриктора при профилактике и лечении мигрени. Однако, как показывается, "органы-мишени" для представленных соединений, кроме каротидной сосудистой сети, находятся в пределах объема настоящего изобретения.
Количество соединения формулы (I)> или соли, или сольвата его, которое требуется для достижения соответствующего биологического действия, зависит от ряда факторов, таких как конкретное соединение, использование, для которого оно предполагается, способы применения и реципиент. Типичная суточная доза для лечения мигрени, как можно полагать, лежит в пределах от 0,01 до 5 мг на кг живого веса. Унифицированные дозы могут содержать от 1 до 100 мг соединения формулы (I), например ампулы для инъекции могут содержать от 1 до 10 мг, а перорально принимаемые унифицированно дозированные лекарственные формы, такие как таблетки или капсулы могут содержать от 1 до 100 мг. Такие унифицированные дозы могут применяться один или более раз в сутки, отдельно или в составах его. Внутривенная доза, как можно полагать, лежит в пределах от 0,01 до 0,15 мг/кг и обычно применяется в виде вливания от 0,0003 до 0,15 мг на кг в минуту. Растворы для вливания, пригодные для этой цели, могут содержать от 0,01 до 10 мг/мл.
Когда действующее соединение является солью или сольватом соединения формулы (I), доза основывается на катионе (для солей) или несольватированном соединении.
В дальнейшем ссылками на "соединение (я) формулы (I)" предполагают включать физиологически приемлемые соли и сольваты их.
По третьей особенности настоящего изобретения, поэтому, предлагаются фармацевтические композиции, включающие в качестве действующего ингредиента по меньшей мере одно соединение формулы (I) и/или фармакологически приемлемую соль или сольват его вместе с по меньшей мере одним фармацевтическим носителем или наполнителем. Эти фармацевтические композиции могут быть использованы при профилактике или лечении клинических состояний, для которых агонист для "5-НТ1-подобного" рецептора является показательным, например мигрени. Носитель должен быть фармацевтически приемлемым для реципиента и должен быть совместим с, т.е. не иметь вредного воздействия на, ингредиентами (ы) в композиции. Носитель может быть твердым или жидким и предпочтительно включается в состав вместе с по меньшей мере одним соединением формулы (I) в виде лекарственной формы с унифицированной дозой, например таблетки, которая может содержать от 0,05 до 95% вес. действующего ингредиента. Если желательно, в фармацевтические композиции изобретения могут быть введены также другие физиологически действующие ингредиенты.
Возможные лекарственные формы включают формы, пригодные для перорального, подъязычного, трансбуккального (внутриротового), парентерального (например, подкожного, внутримышечного или внутривенного), ректального, наружного и интраназального (внутриносового) применения. Наиболее подходящий способ применения для конкретного пациента зависит от природы и остроты состояния, подлежащего вылечиванию, и от природы действующего соединения, но, где это возможно, пероральное применение является предпочтительным.
Лекарственные формы, пригодные для перорального применения, могут быть выработаны в виде раздельных лекарственных форм, таких как таблетки, капсулы, крахмальные капсулы или лепешки, каждая из которых содержит заранее определенное количество действующего соединения; в виде порошков или гранул; в виде растворов или суспензий в водных или неводных жидкостях; или в виде эмульсий типа масло в воде или вода в масле.
Лекарственные формы, пригодные для подъязычного или трансбуккального применения, включают лепешки, содержащие действующее соединение и, обычно, вкусовую и ароматизирующую основу, такую как сахар и акация или трагакант, и пастилы, содержащие действующее соединение в инертной основе, такой как желатина и глицерин или сахароза и акация.
Лекарственные формы, пригодные для парентерального применения, обычно включают стерильные водные растворы, содержащие заранее определенную концентрацию действующего соединения; раствор предпочтительно является изотоническим с кровью предполагаемого реципиента. Хотя такие растворы предпочтительно применяются внутривенно, они могут быть применены также путем подкожной или внутримышечной инъекции.
Лекарственные формы, пригодные для ректального применения, предпочтительно вырабатываются в виде суппозиторий с унифицированной дозой, включающих действующий ингредиент и один или более твердых носителей, образующих основу для суппозиторий, например масло какао.
Лекарственные формы, пригодные для наружного или интраназального применения, включают мази, кремы, лосьоны, пасты, гели, аэрозоли и масла. Подходящими носителями для таких лекарственных форм являются нефтяное желе, ланолин, полиэтиленгликоли, спирты и их сочетания. Действующий ингредиент обычно содержится в таких лекарственных формах при концентрации от 0,1 до 15% вес.
Лекарственные формы изобретения могут быть приготовлены по любому подходящему способу, обычно путем равномерного и однородного смешивания действующего(их) соединения(й) с жидкими или тонко размолотыми твердыми носителями, или с обоими носителями, в требуемых пропорциях, а затем, если необходимо, формования получающейся смеси в целевую форму.
Например, таблетка может быть получена путем прессования однородной смеси, включающей порошок или гранулы действующего ингредиента и одного или более необязательных ингредиентов, таких как связующее, смазочное вещество, инертный разбавитель или поверхностно-активное диспергирующее вещество, или путем формования однородной смеси порошкообразного действующего ингредиента и инертного жидкого разбавителя.
Водные растворы для парентерального применения обычно получаются растворением действующего соединения в достаточном количестве воды, чтобы получить целевую концентрацию, а затем превращением полученного раствора в стерильный и изотонический раствор.
Таким образом, по четвертой особенности настоящего изобретения, предлагается использование соединения формулы (I) при получении лекарственного средства для профилактики или лечения клинического состояния, для которого показан агонист для "5-НТ1-подобного" рецептора, например мигрени.
В соответствии с пятым аспектом изобретения, предлагается способ профилактики и лечения клинического состояния у млекопитающих, например человека, для которых показан агонист для "5-НТ1- подобного" рецептора, например мигрени, который (способ) включает введение указанным млекопитающим терапевтически эффективного количества соединения формулы (I) или физиологически приемлемой соли, сольвата или физиологически функционального его производного.
Согласно шестому аспекту изобретения, соединения формулы (I), в которой Z является группой формулы (iv), могут быть получены реакцией взаимодействия соединения формулы (II) (выделенной или in situ (на месте))
в которой n и W имеют определенные выше значения, с соединением формулы (III)
или с карбонилзащищенной формой его, такой как диметил- или диэтилацеталем, в которой L является подходящей уходящей группой, такой как хлор, или защищенной аминогруппой, любая из которых может быть превращена in situ в аминогруппу, или является группой -NR1R2, где R1 и R2 имеют определенные выше значения. Реакция обычно проводится кипячением с обратным холодильником соединений в системе полярных растворителей, например этанол/вода, разбавленная уксусная кислота или вода в присутствии кислотной ионообменной смолы, например "Амберлита 15".
Стандартные методы N-алкилирования могут быть использованы, чтобы превратить соединения формулы (I), в которой Z является группой формулы (iv), а R2 и/или R2 являются водородом, в соответствующие соединения, в которых R1 и/или R2 являются C1-4-алкилом.
Соединения формулы (I), в которой Z = (iv), a R1 = R2 = C1-4-алкил, могут быть получены из соответствующего соединения, в котором R1 = R2 = H, с помощью методов N,N-диалкилирования, хорошо известным специалистам в данной области техники, например путем обработки с соответствующим альдегидом в присутствии восстанавливающей системы, например цианоборогидрид натрия/уксусная кислота, в полярном растворителе, таком как метанол.
Соединения формулы (I), в которой Z = (iv), а R1 или R2 = C1-4-алкил, могут быть получены из соответствующего соединения, в котором R1 = R2 = H, путем N-бензилирования, используя бензальдегид и подходящий восстановитель, например борогидрид натрия, в полярном растворителе, таком как этанол, за которым следует N-алкилирование, используя подходящее средство, такое как соответствующий диалкилсульфат, обычно в присутствии основания, например безводного карбоната калия, в полярном апротонном растворителе, таком как ДМФА, и, наконец, N-дебензилирование, обычно путем каталитического гидрирования, используя, например, Rd/C в молярном растворителе, таком как этанол.
Гидразины формулы (II) могут быть получены из соответствующего анилина формулы (IV)
в которой n и W имеют определенные выше значения,
путем диазотирования, за которым следует восстановление. Диазотирование обычно проводят путем использования нитрата натрия/конц. HCl и получающееся диазосоединение восстанавливают in situ, используя, например, хлорид олова (II)/конц. HCl. Получающийся гидразин может быть выделен или превращен в соединение формулы (I) in situ.
Анилины формулы (IV) могут быть получены путем восстановления соответствующего паранитросоединения формулы (V)
в которой n и W имеют определенные выше значения,
обычно путем каталитического гидрирования, используя, например, Rd/C в системе полярных растворителей, такой как подкисленная смесь этанола, воды и этилацетата.
Анилины формулы (IV), в которой W является группой формулы (i) или (ii), могут быть получены также путем циклизации соединения формулы (XXXIII)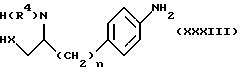
или (XXXIV)
в которых n и X имеют определенные выше значения, а R4 является группой -COOR5, где R5 является C1-4-алкилом,
обычно путем нагревания в присутствии основания, такого как метоксид натрия.
Соединения формулы (XXXIII), где X является кислородом, могут быть получены путем восстановления соответствующего C1-4-алкилового сложного эфира, используя, например, борогидрид натрия, в системе полярных растворителей, такой как этанол/вода, при 0oC. Сложный эфир может быть получен путем этерификации соответствующей карбоновой кислоты, используя, например, соответствующий спирт и HCl, или путем восстановления соответствующего паранитросоединения, например, путем каталитического гидрирования. Как кислота, так и паранитросоединение могут быть получены из соответствующей паранитроаминокислоты; кислота путем N-алкоксикарбонилирования, используя, например, R5OCOCl, где R5 имеет определенные выше значения, за которым следует восстановление нитрогруппы, например, путем каталитического гидрирования, или путем восстановления нитрогруппы, за которым следует N-алкоксикарбонилирование, а паранитросоединение путем N-алкоксикарбонилирования (что касается кислоты), за которым следует этерификация, используя, например, соответствующий спирт и HCl, или путем этерификации, за которой следует N-алкоксикарбонилирование. Паранитроаминокиcлота может быть получена коммерческим путем или получена из легко доступных исходных веществ по методам, известным специалистам в данной области техники или получаемым из химической литературы, например, путем паранитрования соответствующей аминокислоты, используя, например, конц. H2SO4/конц. HNO3 при 0oC.
Соединения формулы (XXXIV), в которой X является кислородом, могут быть получены путем восстановления соответствующего динитросоединения, обычно путем каталитического гидрирования, используя, например, Rd/C в полярном растворителе, таком как этанол. Динитросоединение может быть получено реакцией соответствующего альдегида с нитрометаном, обычно в присутствии основания, например, метоксида натрия, в полярном растворителе, таком как метанол, за которой следует паранитрование, используя, например, конц. H2SO4/конц. HNO3, или паранитрование соответствующего альдегида, за которым следует реакция с нитрометаном. Альдегид может быть получен коммерческим путем или получен из легко доступных исходных веществ по методам, известным специалистам в данной области техники, или получаемым из химической литературы.
Паранитросоединения формулы (V) могут быть получены:
а) в случае, когда W является группой формулы (i), в которой является кислородом или серой, реакцией соединения формулы (VI)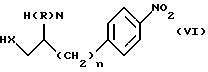
в которой n, R и X имеют определенные выше значения,
с соединением формулы (VII)
в которой Y имеет определенные выше значения, a L и L', которые могут быть одинаковыми или различными, являются уходящими группами, например хлором, этокси-, трихлорметилом, трихлорметокси-группой или имидазолилом, например, в случае, когда L=L'=хлор, в неполярном растворителе, таком как толуол, в присутствии основания, например, гидроокиси калия.
б) в случае, когда W является группой формулы (ii), в которой Y является кислородом или серой, реакцией соединения формулы (VIII)
в которой n, R и X имеют определенные выше значения,
с соединением формулы (VII), в которой Y, L и L' имеют определенные выше значения, используя обычно условия реакции, описанные а (а).
в) в случае, когда W является группой формулы (iii), реакцией соединения формулы (IX)

в которой n имеет определенные выше значения,
c соединением формулы (X)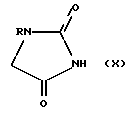
в которой R имеет определенные выше значения,
определенном апротонном растворителе, таком как ДМФА, в присутствии диэтилового эфира азодикарбоновой кислоты и Ph3P (ДЭАД/Ph3P).
Соединения формулы (VI) могут быть получены реакцией раскрытия цикла соединения формулы (V), в которой n имеет определенные выше значения, a W является группой формулы (i), в которой R, X и Y имеют определенные выше значения, например путем кипячения с обратным холодильником в 2N водной КОН.
Соединения формулы (VI), в которой X является кислородом, могут быть получены этерификацией соответствующей карбоновой кислоты, обычно обработкой с хлористым тионилом и соответствующим спиртом при -10oC, за которой следует восстановление сложного эфира, используя, например, борогидрид натрия, в системе полярных растворителей, такой как этанол/вода, при 0oC. Кислота может быть получена коммерческим путем или получена из легко доступных веществ по методам, известным специалистам в данной области техники или получаемым из химической литературы, например, путем паранитрования соответствующей аминокислоты, используя, например, конц. H2SO4/конец. HNO3 при 0oC.
Соединения формулы (VIII) могут получаться реакцией раскрытия цикла соединения формулы (V), в которой n имеет определенные выше значения, а W является группой формулы (ii), в которой R, X и Y имеют определенные выше значения, например, путем кипячения с обратным холодильником в 2N водной КОН.
Соединения формулы (III), (VII), (IX) и (X) могут быть получены коммерческим путем или получены из легко доступных исходных веществ по методам, известным специалистам в данной области техники или получаемым из химической литературы.
Паранитросоединения формулы (V), в которой W является группой формулы (i) или (ii), могут быть получены также путем паранитрования соединения формулы (XXXVI)
в которой n и W имеют определенные выше значения, используя, например, конц. H2SO4/конц. HNO3 при 0oC.
Соединения формулы (XXXVI) могут быть получены реакцией соединения формулы (XXXVII)
или (XXXVIII)
в которой n, R и X имеют определенные выше значения,
с соединением формулы (VII), в которой Y, L и L' имеют определенные выше значения,
обычно в присутствии основания, например, гидроокиси калия в неполярном растворителе, таком как толуол.
Соединения формулы (XXXVII) и (XXXVIII) могут быть получены путем восстановления соответствующих нитросоединений, обычно каталитическим гидрированием, используя, например, Rd/C, в полярном растворителе, таком как этанол. Нитросоединение, соответствующее соединению формулы (XXXVII), может быть получено реакцией соединения формулы (XXIV)
в которой n имеет определенные выше значения,
с параформальдегидом в полярном апротонном растворителе, таком как ДМФА, в присутствии основания, например, метоксида натрия, при 0oC, или этерификацией соответствующей карбоновой кислоты, обычно обработкой с хлористым тионилом и соответствующим спиртом при -10oC, за которой следует восстановление сложноэфирной группы, используя, например, борогидрид натрия, в системе полярных растворителей, такой как этанол/вода, при 0oC. Нитросоединение, соответствующее соединению формулы (XXXVIII), может быть получено реакцией соответствующего альдегида с нитрометаном, обычно в присутствии основания, например, метоксида натрия, в полярном растворителе, таком как метанол. Соединение формулы (XXIV), кислота и альдегид могут быть получены коммерческим путем или получены из легко доступных исходных веществ по методам, известным специалистам в данной области техники или получаемым из химической литературы.
Паранитросоединения формулы (V), в которой W является группой формулы (i), (ii) или (iii), в которой R является C1-4-алкилом, могут быть получены из соответствующего соединения формулы (V), в которой R является водородом, путем N-алкилирования, используя подходящий реагент, такой как соответствующий диалкилсульфат, обычно в присутствии основания, например, гидрида натрия, в неполярном растворителе, таком как ТГФ.
Соединения формулы (I), в которой W является группой формулы (i) или (ii), могут быть получены также реакцией соединения формулы (XV)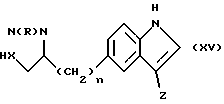
или (XXV)
в которой n, R, X и Z имеют определенные выше значения,
с соединением формулы (VII), в которой Y, L и L' имеют определенные выше значения, например, в случае, когда L = L'-этокси, путем нагревания в присутствии основания, например карбоната калия.
Соединения формулы (XV) могут быть получены реакцией раскрытия цикла соединения формулы (I), в которой n и Z имеют определенные выше значения, a W является группой формулы (i), в которой R , X и Y имеют определенные выше значения, например, путем кипячения с обратным холодильником в 2N водной КОН.
Соединения формулы (XV), в которой X является кислородом, могут быть получены этерификацией соответствующей карбоновой кислоты, обычно обработкой с хлористым тионилом и соответствующим спиртом при -10oC, за которой следует восстановление сложного эфира, используя, например, борогидрид натрия, в системе полярных растворителей, такой как этанол-вода, при 0oC. Кислота может быть получена реакцией раскрытия цикла соединения формулы (XVI)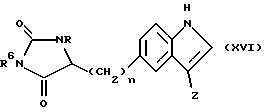
в которой n, R и Z имеют определенные выше значения, а
R6 является водородом или бензилом,
обычно путем кипячения с обратным холодильником в воде, в присутствии основания, например, гидроокиси бария.
Соединения формулы (XVI), в которой n≠0 , могут быть получены восстановлением соединения формулы (XVII)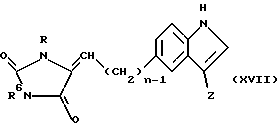
в которой n, R, R6 и Z имеют определенные выше значения,
обычно путем каталитического гидрирования, используя, например, Rd/C в системе полярных растворителей, таких как этанол/вода. Наоборот, чтобы восстановить двойную связь и тем самым ввести хиральный центр в положение 4 диоксоимидазольного цикла, может быть использован энантиоселективный восстановитель, такой как Rh (код) (дипамп)+BF4 - (JCS. Chem. Comm., 275 (1991)). Стадия восстановления может быть использована, чтобы превратить соединение формулы (XVII), в которой Z является группой формулы (v), в соединение формулы (XVI), в которой Z является группой формулы (vi).
Соединение формулы (XVII) могут быть получены реакцией соединения формулы (XVIIII)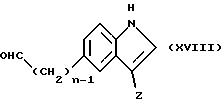
в которой n и Z имеют определенные выше значения, в случае, когда R6 должен быть водородом,
с соединением формулы (X), в которой R имеет определенные выше значения, обычно путем нагревания в ледяной уксусной кислоте в присутствии ацетата аммония.
Соединения формулы (XVIII) могут быть получены путем восстановительного гидролиза соответствующего нитрила, обычно используя никель Ренея и гипофосфит натрия в смеси воды, уксусной кислоты и пиридина. Нитрил может быть получен реакцией соединения формулы (XIX)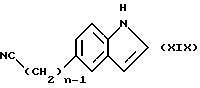
в которой n имеет определенные выше значения,
с соответствующим соединением формулы (XXVIII), в случае, когда Z должен быть группой формулы (v) или (vi),
в которой R3 имеет определенные выше значения,
обычно путем кипячения с обратным холодильником в полярном растворителе, таком как метанол, в присутствии основания, например гидроокиси калия.
Соединения формулы (XIX) и (XXVIII) могут быть получены коммерческим путем или получены из легко доступных исходных веществ по методам, известным специалистам в данной области техники или получаемым из химической литературы. Соединения формулы (XVI),
в которой n = 0, могут быть получены одинаковым способом.
Соединения формулы (XVI), в которой R6 является бензилом, a Z является группой формулы (iv), могут быть получены реакцией соединения формулы (XXXV)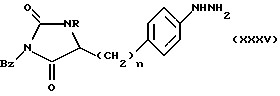
в которой n и R имеют определенные выше значения,
с соединением формулы (III), в которой Z имеет определенное выше значение, используя обычно условия реакции, описанные выше для реакции соединения (II) с соединением (III).
Гидразины формулы (XXXV) могут быть получены из соответствующего анилина, используя обычно условия реакции, описанные выше для превращения соединения (IV) в (II). Анилин может быть получен путем восстановления соответствующего паранитросоединения, используя обычно условия реакции, описанные выше для превращения соединения (V) в (IV). Паранитросоединение может быть получено реакцией соответствующей паранитроаминокислоты с бензил-изоцианатом в присутствии основания, например, гидроокиси калия, в полярном растворителе, таком как вода. Паранитроаминокислота может быть получена коммерческим путем или получена из легко доступных исходных веществ по методам, известным специалистам в данной области техники или получаемым из химической литературы, например, путем паранитрования соответствующей аминокислоты, используя, например, конц. H2SO4/конц. HNO3 при 0oC.
Соединения формулы (XV), в которой R является водородом, могут быть получены путем восстановления соединения формулы (XX)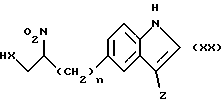
в которой n, X и Z имеют определенные выше значения,
обычно каталитическим гидрированием, используя, например, Rd/C в полярном растворителе, таком как этанол. Подобная стадия может быть использована, чтобы превратить соединение формулы (XX), в которой Z является группой формулы (v), в соединение формулы (XV) в которой Z является группой формулы (vi).
Соединение формулы (XX), в которой X является кислородом, могут быть получены реакцией соединения формулы (XXI)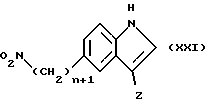
в которой n и Z имеют определенные выше значения,
с параформальдегидом в полярном апротонном растворителе, таком как ДМФА, в присутствии основания, например метоксида натрия, при 0oC.
Соединения формулы (XXI) могут быть получены реакцией соединения формулы (XXII)
в которой n имеет определенные выше значения, в случае, когда должен быть группой формулы (v) или (vi),
с соответствующим соединением формулы (XXVIII), в которой R3 имеет определенные выше значения, обычно путем нагревания в ледяной уксусной кислоте.
Соединения формулы (XXII), в которой n≠0 , могут быть получены путем восстановления соединения формулы (XXIII)
в которой n имеет определенные выше значения,
используя, например борогидрид натрия и 40% вес./об. водную NaOH в полярном апротонном растворителе, таком как ацетонитрил, при 0oC.
Соединения формулы (XXIII) могут быть получены путем нагревания соответствующего альдегида с нитрометаном в присутствии ацетата аммония. Альдегид может быть получен из соединения формулы (XIX), в которой n имеет определенные выше значения, используя условия реакции, описанной выше для получения соединения формулы (XVIII) из соответствующего нитрила.
Соединение формулы (XXII), в которой n = 0, могут быть получены коммерческим путем или получены из легко доступных исходных веществ по методам, известным специалистам в данной области техники или получаемым из химической литературы.
Соединения формулы (XXI), в которой n≠0, могут быть получены также из соединения формулы (XXXIX)
в которой n и Z имеют определенные выше значения,
используя условия реакции, аналогичные используемым при превращении (XXIII) в (XXII). Соединения формулы (XXXIX) могут быть получены из соединения формулы (XVIII), в которой n и Z имеют определенные выше значения, используя условия реакции, аналогичные используемым при получении (XXIII) из соответствующего альдегида и нитрометана.
Соединения формулы (XX), в которой X не является кислородом, могут быть получены коммерческим путем или получены из легко доступных исходных веществ по методам, известным специалистам в данной области техники или получаемым из химической литературы.
Соединения формулы (XXV) могут быть получены реакцией раскрытия цикла соединения формулы (I), в которой n и Z имеют определенные выше значения, а W является группой формулы (ii), в которой R, X и Y имеют определенные выше значения, например, путем кипячения с обратным холодильником в 2N водной КОН.
Соединения формулы (I), в которой W является группой формулы (i), где Y является серой, могут быть получены путем кипячения с обратным холодильником соединения формулы (XV), в которой n, R и X имеют определенные выше значения, с соединением формулы (VII), в которой Y является серой, a L и L' имеют определенные выше значения, например, N,N-тиокарбонилимидазолом, обычно в апротонном растворителе, таком как ТГФ.
Соединения формулы (I), в которой W является группой формулы (ii), в которой Y является серой, могут быть получены путем кипячения с обратным холодильником соединения формулы (XXV), в которой n, R и X имеют определенные выше значения, с соединением формулы (VII), в которой Y является серой, а L и L' имеют определенные выше значения, например, N,N-тиокарбонилимидазолом, обычно в апротонном растворителе, таком как ТГФ.
Соединения формулы (I), в которой W является группой формулы (iii), a Z является группой формулы (v) или (vi), могут быть получены также циклизацией соединения формулы (XXVI):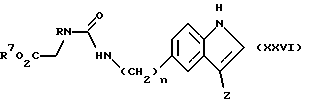
в которой n и R имеют определенные выше значения,
Z является группой формулы (v) или (vi),
R7 является C1-4-алкилом,
обычно путем нагревания в водной кислоте, например 2N HCl.
Соединения формулы (XXVI), в которой Z является группой формулы (v), могут быть получены реакцией соединения формулы (XXVII)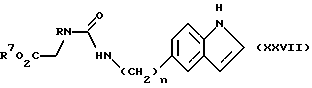
в которой n, R и R7 имеют определенные выше значения,
с соединением формулы (XXVIII), в которой R3 имеет определенные выше значения, обычно путем нагревания в неводной кислоте, например, ледяной уксусной кислоте.
Соединения формулы (XXVI), в которой Z является группой формулы (vi), могут быть получены путем восстановления соединения формулы (XXVI), в которой Z является группой формулы (v), обычно путем каталитического гидрирования, используя, например, Rd/C в системе полярных растворителей, такой как подкисленный метанол/вода.
Соединения формулы (XXVII) могут быть получены реакцией соединения формулы (XXIX)
в которой n имеет определенные выше значения,
с соединением формулы (XXX)
в которой R7 имеет определенные выше значения,
обычно в апротонном растворителе, таком как ДСМ (хлористый метилен).
Соединения формулы (XXIX) и (XXX) могут быть получены коммерческим путем или получены из легко доступных исходных веществ по методам, известным специалистам в данной области техники или получаемым из химической литературы.
Соединения формулы (I), в которой Z является группой формулы (iv), могут быть получены также из соединения формулы (XXXI)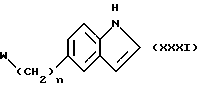
в которой n и W имеют определенные выше значения, по методам, известным специалистам в данной области техники или получаемым из химической литературы, например, обработкой с (COL)2, где L является подходящей уходящей группой, например, хлором, чтобы получить соответствующее соединение 3-COCOL, которое может быть обработано затем с HNR1R2, где R1 и R2 имеют определенные выше значения, и восстановлено, используя, например, литийалюминийгидрид. Или же соединение формулы (XXXI) может быть обработано смесью CH2O/KCN, чтобы получить соответствующее 3-цианометильное соединение, которое затем может быть подвергнуто каталитическому гидрированию над никелем Ренея в присутствии HNR1R2, который определен выше.
Упомянутое выше 3-цианометильное соединение может быть получено также путем циклизации соединения формулы (XXXX)
в которой n и W имеют определенные выше значения, обычно путем кипячения с обратным холодильником в апротонном растворителе, таком как хлороформ, в присутствии полифосфатного сложного эфира.
Соединения формулы (XXX) могут быть получены реакцией соединения формулы (II), в которой n и W имеют определенные выше значения, с 3-цианопропаналем или с карбонил-защищенной формой его, такой как диэтилацеталь, обычно в водной кислоте, например, разбавленной HCl.
Соединения формулы (I), в которой Z является группой формулы (v), могут быть получены также реакцией соединения формулы (XXXI), в которой n и W имеют определенные выше значения, с соединением формулы (XXVIII), в которой R3 имеет определенные выше значения, обычно путем нагревания в ледяной уксусной кислоте,
Соединения формулы (XXXI) могут быть получены путем восстановления соединения формулы (XXXII)
в которой n и W имеют определенные выше значения,
обычно путем нагревания с никелем Ренея в полярном растворителе, таком как IPA (изопропиловый спирт).
Соединения формулы (XXXII) могут быть получены реакцией гидразина формулы (II), в которой n и W имеют определенные выше значения, с фенилтиоацетальдегидом или с карбонил-защищенной формой его, например, диэтилацеталь, в полярном растворителе, таком как подкисленный этанол.
Соединения формулы (I), в которой Z является группой формулы (vi), могут быть получены также путем восстановления соединения формулы (I), в которой Z является группой формулы (v), обычно путем каталитического гидрирования, используя, например, Rd/C в системе полярных растворителей, такой как подкисленный метанол/вода.
Для лучшего понимания изобретения, в качестве пояснения представляются следующие примеры.
Синтетические примеры
Синтетический пример 1. Получение (S)-2-[5-(2-оксо-1,3-оксазолидин-4-ил-метил)-1H-индол-3-ил]-этиламина
(а) Хлоргидрат метилового эфира (S)-4-нитрофенилаланина
Метанол (110 мл) обрабатывают по каплям хлористым тионилом (26,3 г) при -10oC и к полученному раствору добавляют в виде твердого вещества L-4-нитрофенилаланин (Флюка, 21,7 г). Смесь перемешивают в течение ночи при комнатной температуре и метанол удаляют в вакууме, чтобы получить целевой продукт реакции в виде бледно-желтого твердого вещества (21,2 г).
(б) (S)-2-Амино-3-(4-нитрофенил)-пропанол
Продукт реакции из стадии (а) (21,2 г) растворяют в смеси этанола/воды (190 мл, 100/90 в объемном отношении) и раствор добавляют по каплям при 0oC к перемешиваемому раствору борогидрида натрия (13,0 г) в смеси этанол/вода (190 мл, 100/90 в объемном отношении). Получающуюся смесь кипятят с обратным холодильником в течение 2,5 часов, охлаждают и выпадающий осадок отфильтровывают. Этанол частично удаляют из фильтрата в вакууме и получающийся осадок отфильтровывают и высушивают, чтобы получить целевой продукт реакции в виде бледно-желтого твердого вещества (7,5 г).
(в) (5)-4-(4-Нитробензил)-1,3-оксазолидин-2-он
Продукт реакции из стадии (б) (4,9 г) суспендируют в толуоле, суспензию охлаждают до 0oC и раствор гидроокиси калия (7,0 г) в воде (56 мл) добавляют по каплям. К полученному раствору добавляют по каплям раствор фосгена (62,5 мл 12% вес. /объем раствора в толуоле) в течение 30 минут и перемешивании, продолжающемся в течение 1 часа. Смесь экстрагируют этилацетатом и экстракты промывают солевым раствором, высушивают и выпаривают в вакууме, чтобы получить желтое масло. Кристаллизация из этилацетата дает целевой продукт реакции в виде кристаллов бледно-желтого цвета (2,3 г).
(г) Хлоргидрат (S)-4-(4-аминобензил)-1,3-оксазолидин-2-она
Суспензию продукта реакции из стадии (в) (0,79 г) и 10% палладия на угле (0,26 г) в смеси этанола (15 мл), воды (11 мл), этилацетата (2,0 мл) и водной 2N HCl (2,3 мл) перемешивают при атмосферном давлении водорода до тех пор, пока не прекратится поглощение водорода. Смесь фильтруют через Hyflo и фильтрат выпаривают в вакууме, чтобы получить целевой продукт реакции в виде бледно-желтого пенообразного вещества (0,79 г).
(д) Хлоргидрат (S)-4-(4-гидразинобензил)-1,3-оксазолидин-2-она
Продукт реакции из стадии (г) (0,79 г) суспендируют в воде (4,8 мл) и по каплям добавляют конц. HCl (8,1 мл). Полученную смесь охлаждают до -5oC и раствор нитрита натрия (0,24 г) в воде (2,4 мл) добавляют по каплям к перемешиваемой смеси в течение 15 минут, за которым следует 30-минутное перемешивание при температуре от -5o до 0oC. Затем раствор добавляют при 0oC в течение 15 минут к перемешиваемому раствору хлористого олова (II) (3,8 г) в конц. HCl (6,9 мл), за которым следует 3-часовое перемешивание при комнатной температуре. Раствор выпаривают в вакууме и остаток растирают в порошок с помощью простого эфира, чтобы получить целевой продукт реакции в виде бледно-желтого твердого вещества (0,96 г).
(е) (S)-2-[5-(3-Оксо-1,3-оксазолидин-4-ил-метил)-1H- -индол-3-ил]-этиламина
Продукт реакции из стадии (д) (0,84 г) растворяют в смеси этанол/вода (125 мл, 5: 1) и раствор обрабатывают диметилацеталем 4-хлорбутанальдегида (JACS 1365 (1951), 0,52 г). Смесь кипятят с обратным холодильником в течение 2 часов, растворитель удаляют в вакууме и остаток элюируют через силикагелевую колонку, используя смесь хлористый метилен/EtOH/NH4OH (30:8:1) в качестве элюента. Целевой продукт реакции получают в виде бесцветного масла (0,21 г).
Соль синтетического примера 1
Малеат (соль малеиновой кислоты)
Этанольный раствор малеиновой кислоты (1,0 экв.) добавляют по каплям к свободному основанию (0,21 г) и этанол выпаривают в вакууме. Полученную смолу высушивают при температуре ниже 0oC, чтобы избавиться от воды и получить целевой продукт реакции в виде белого продукта лиофилизации (лиофилата) (0,22 г), [α]
1H-ЯМР (ДМСО-d6, δ): 2.7-3.5 (6H, мультиплет, CH2), 3.35 (2H, синглет, NH2), 4.05 (2H, мультиплет, CH2), 4.25 (1H, мультиплет, CH), 6.05 (2H, синглет, малеиновая кислота), 6.98 (1H, дублет, Ar), 7.2 (1H, синглет, Ar), 7.3 (1H, дублет, Ar), 7.4 (1H, синглет, Ar), 7.75 (1H, синглет, NH), и 10.9 (1H, синглет, NH).
Микроанализ: C 55,03 (54,96); H 5,54 (5,85); N 10,30 (10,68)
Синтетический пример 2. Получение 0,9 изопропанолата 0,5 гидрата (S)-N, N-диметил-2-[5-(2-оксо-1,3-оксазолидин-4-ил-метил)-1H-индол-3-ил]-этиламина
Раствор формальдегида (0,03 г) в метаноле (1,8 мл) добавляют к раствору свободного основания из стадии (е) Синтетического примера 1 (0,12 г) и цианоборогидрида натрия (0,04 г) в смеси метанола (5,5 мл) и ледяной уксусной кислоты (0,14 г) и полученную смесь перемешивают в течение ночи при комнатной температуре. pH доводят до 8,0, используя водный K2CO3 смесь экстрагируют этилацетатом. Объединенные экстракты промывают солевым раствором, высушивают и выпаривают, чтобы получить бесцветное масло (0,14 г), которое кристаллизуют из изопропилового спирта, чтобы получить целевой продукт реакции в виде белого кристаллического твердого вещества (0,10 г), т.пл. 139-141oC.
1H-ЯМР (ДМСО-d6, δ ): 2.2 (6H, синглет, NMe2), 2.5 (2H, мультиплет, CH2Ar), 2.7-3.0 (4H, мультиплет, CH2), 4.1 (2H, мультиплет, CH2O), 4.3 (1H, мультиплет, СH), 6.9 (1H, дублет, Ar), 7.1 (1H, синглет, Ar), 7.3 (1H, дублет, Ar), 7.4 (1H, синглет, Ar), 7.7 (1H синглет, NHCO) и 10.7 (1H, синглет, NH).
Микроанализ: C 64,26 (64,11); H 8,28 (8,34); N 12,02 (12,00).
[α]
Соли синтетического примера 2
Малеат
Раствор малеиновой кислоты (0,17 г) в этаноле (5 мл) добавляют к раствору свободного основания (0,5 г) в этаноле (5 мл). Смесь выпаривают в вакууме и полученное масло растирают в порошок с помощью простого эфира и метанола, чтобы получить малеатную соль в виде белого твердого вещества, которое кристаллизуют из этанола (0,45 г), т.пл. 151-152oC.
Хлоргидрат
Эфирный раствор HCl (1,1 экв.) добавляют по каплям к перемешиваемому раствору свободного основания (0,35 г) в метаноле (1 мл) при 0oC. Хлоргидратная соль выпадает в осадок в виде масла. Смесь выпаривают в вакууме и получающееся пенообразное вещество кристаллизуют из изопропилового спирта, чтобы получить целевой продукт реакции в виде белого твердого вещества (0,36 г), т.пл. 118-120oC.
[α]
Сукцинат (соль янтарной кислоты)
Раствор янтарной кислоты (0,36 г) в этаноле (10 мл) добавляют к раствору свободного основания (1,0 г) в этаноле (10 мл). Смесь выпаривают в вакууме и полученное пенообразное вещество растирают в порошок с помощью изопропилового спирта, чтобы получать сукцинатную соль в виде белого твердого вещества (1,0 г), т.пл. 122-123oC.
Бензоат
Раствор бензойной кислоты (0,37 г) в этаноле (10 мл) добавляют к раствору свободного основания (1,0 г) в этаноле (10 мл). Смесь выпаривают в вакууме и полученное пенообразное вещество кристаллизуют из этилацетата, чтобы получить бензоатную соль в виде белого твердого вещества (0,74 г), т.пл. 90-92oC.
Синтетический пример 3. Другой метод получения 0,9 изопропанолата 0,5 гидрата (S)-N,N-диметил-2-[5-(2-оксо-1,3-оксазолидинил-метил)- 1H-индол-3-ил]-этиламина
Диэтилацеталь 4-диметиламинобутанальдегида (Croatica Chemica Acta 36, 103, (1964), 3,9 г) добавляют к раствору продукта реакции из стадии (д) Синтетического примера 1 (10,4 г) в смеси уксусной кислоты (50 мл) и воды (150 мл) и полученную смесь кипятят с обратным холодильником в течение 4,5 часов. Смесь охлаждают, выпаривают в вакууме и остаток элюируют через колонну с кремнеземом, используя смесь хлористый метилен/EtOH/NH4OH (50:8:1) в качестве элюента, чтобы получить целевой продукт реакции в виде бледно-желтого масла, которое кристаллизуют из изопропилового спирта в виде белого кристаллического твердого вещества (3,5 г), т.пл. 138-140oC.
1H-ЯМР, микроанализ и [α]D такие же, что и у продукта реакции Синтетического примера 2.
Синтетический пример 4. Получение (±)-3-(1-метил-4-пиперидил)-5-(2-оксо-1,3-оксазолидин-4-ил-метил)- -1H-индола
(а) 3-(1-Метил-1,2,3,6-тетрагидро-4-пиридил)-1H-индол-5- карбонитрил
5-Цианоиндол (Альдрих, 20,0 г) добавляют к раствору КОН (22,4 г) в метаноле (200 мл). Затем добавляют N,N-метил-4-пиперидон (Альдрих, 40,4 г) по каплям и полученную смесь кипятят с обратным холодильником в течение 4 часов, затем охлаждают и выливают в воду. Получающийся осадок отфильтровывают и высушивают, чтобы получить целевой продукт реакции в виде бледно-розового кристаллического твердого вещества (32,6 г).
(б) 3-(1-Метил-1,2,3,6-тетрагидро-4-пиридил)-1H-индол- 5-карбальдегид
Никель Ренея (приблизительно 10 г) добавляют к раствору продукта реакции из стадии (а) (5,0 г) и гипофосфита натрия (6,0 г) в смеси воды (25 мл), ледяной уксусной кислоты (25 мл) и пиридина (25 мл) при 45oC. Полученную смесь перемешивают при 45oC в течение 1 часа, охлаждают и подщелачивают до pH 9 с помощью 0,88 NH4OH. Смесь фильтруют через Hyflo и фильтрат экстрагируют хлороформом. Объединенные экстракты высушивают и выпаривают в вакууме, чтобы получить целевой продукт реакции в виде не совсем белого твердого вещества, которое перекристаллизовывают из этанола (2,4 г).
(в) 5-[3-(1-Метил-1,2,3,6-тетрагидро-4-пиридил)-1H-индол- 5-ил-метилен] -2,4-имидазолидиндион
Смесь продукта реакции из стадии (б) (2,4 г), гидантоина (Альдрих, 0.98 г) и ацетата аммония (0,74 г) в ледяной уксусной кислоте (2,4 мл) нагревают при 120oC в течение 4 часов. Смесь охлаждают и получающийся осадок отфильтровывают и высушивают, чтобы получить целевой продукт реакции в виде желтого твердого вещества (2,4 г).
(г)(±)-5-(2,5-Диоксо-4-имидазолидинилметил)-3-(1-метил-4-пиперидил)-1H-индол
Продукт реакции из стадии (в) (2,4 г) суспендируют в смеси воды (100 мл) и этанола (200 мл) и добавляют 10% в весовом отношении Rd/C (0,25 г). Смесь перемешивают при 1-атмосферном давлении водорода в течение 17 часов, когда завершается поглощение. Смесь фильтруют через Hyflo и фильтрат выпаривают в вакууме, чтобы получить целевой продукт реакции в виде бесцветного твердого вещества (2,4 г).
(д) (±)-3-[3-(1-Метил-4-пиперидил)-1H-индол-5-ил]аланин
Раствор продукта реакции из стадии (г) (2,4 г) и гидрата гидроокиси бария (8,4 г) в воде (50 мл) кипятят с обратным холодильником в течение 72 часов, затем охлаждают и выпаривают в вакууме. Остаток переносят в горячий метанол и фильтруют, чтобы удалить соли бария. Фильтрат выпаривают в вакууме, остаток растворяют в воде и добавляют сухой лед, чтобы высадить в осадок карбонат бария. Последний отфильтровывают и фильтрат выпаривают в вакууме, чтобы получить целевой продукт реакции в виде желтого пенообразного вещества (1,3 г).
(е) Метиловый эфир (±)-3-[3-(1-метил-4-пиперидил)-1H-индол- 5-ил]-аланина
Раствор продукта реакции из стадии (д) (6,2 г) в метаноле (40 мл) добавляют по каплям к раствору хлористого тионила (2,9 мл) в метаноле (35 мл) при -10oC. Полученную смесь перемешивают в течение ночи при комнатной температуре, затем выпаривают в вакууме и остаток элюируют через колонку с кремнеземом, используя смесь хлористого метилена/EtOH/NH4OH (30:8:1) в качестве элюента. Элюат выпаривают в вакууме, чтобы получить целевой продукт реакции в виде желтого пенообразного вещества (4,8 г).
(ж) (±)-3-[3-(1-Метил-4-пиперидил)-1H-индол-5-ил]-2- амино-1-пропанол
Раствор продукта реакции из стадии (е) (4,8 г) в воде (20 мл) и этаноле (20 мл) добавляют по каплям к суспензии борогидрида натрия (0,61 г) в смеси воды (20 мл) и этанола (20 мл) при 0oC. Полученную смесь кипятят с обратным холодильником в течение 3 часов, затем выпаривают в вакууме и остаток элюируют через колонку с кремнеземом, используя смесь хлористого метилена/EtOH/NH4OH (30:8:1) в качестве элюента. Элюат выпаривают в вакууме и остаток перекристаллизовывают из смеси изопропилового спирта/простого эфира, чтобы получить целевой продукт реакции в виде бесцветного кристаллического твердого вещества (1,1 г), т.пл. 191-192oC.
1H-ЯМР (ДМСО-d6, δ ): 1.6-1.8 (2H, 2•CHNMe), 1.8-2.1 (4H, 2•CH2), 2.2 (3H, синглет, NMe), 2.6-3.0 (2H, 2•CHNMe; 1H, CH; 2H, CH2Ar), 3.9-4.1 (2H, мультиплет, CH2O), 4.2-4.4 (1H, мультиплет, CHII), 6.9 (1H, дублет, Ar), 7.1 (1H, дублет, Ar), 7.3 (1H, дублет, Ar ), 7.4 (1H, синглет, Ar), 7.8 (1H, синглет, NHCO) и 10.7 (1H, синглет, NH).
Соль синтетического примера 4
Хлоргидрат
К перемешиваемому раствору свободного основания (1,1 г) в этаноле (5 мл) добавляют по каплям при 5oC конц. HCl (1,0 экв.). Добавление простого эфира к полученной смеси вызывает выпадение в осадок целевого продукта реакции в виде белого твердого вещества (1,1 г), т.пл. 235-236oC (разл.).
Синтетический пример 5. Другой способ получения (±)-3-(1-метил-4-пиперидил)-5-(1,3-оксазолидин-4-ил-метил -1H-индола
(а) 1H-Индол-5-карбальдегид
Никель Ренея (6,7 г) добавляют к раствору 5-цианоиндола (Альдрих, 10,0 г) и гипофосфита натрия (20,0 г) в смеси воды (73 мл), ледяной уксусной кислоты (73 мл) и пиридина (145 мл) при 45oC. Полученную смесь перемешивают при 45oC в течение 2 часов, затем охлаждают и фильтруют через Hyflo. Фильтрат разбавляют водой и экстрагируют этилацетатом. Объединенные экстракты промывают водой, 10%-ной водной лимонной кислотой, 1N водной HCl, водой и солевым раствором; высушивают и выпаривают в вакууме, чтобы получить целевой продукт реакции в виде тускло-желтого твердого вещества, которое перекристаллизовывают из хлороформа (7,5 г).
(б) 5-(2-Нитроэтенил)-1H-индол
Смесь продукта реакции из стадии (а) (7,5 г), ацетата аммония (1,5 г) и нитрометана (77 мл) нагревают при 110oC в течение 2 часов, затем охлаждают и выпаривают в вакууме. Остаток растирают в порошок с помощью воды, чтобы получить целевой продукт реакции в виде желтого твердого вещества, которое отфильтровывают и высушивают (9,2 г).
(в) 5-(2-Нитроэтил)-1H-индол
Раствор борогидрида натрия (2,0 г) и 40% вес./об. водной NaOH добавляют по каплям к раствору продукта реакции из стадии (б) (1,9 г) в ацетонитриле (55 мл) при 0oC. pH поддерживают при 3-6 путем периодических добавлений 2N водной HCl. Полученный раствор перемешивают при 0oC в течение 2 часов, затем разбавляют водой и экстрагируют хлористым метиленом. Объединенные экстракты промывают солевым раствором, высушивают и выпаривают в вакууме, чтобы получить желтое масло, которое элюируют через колонку с кремнеземом, используя хлороформ в качестве элюента, чтобы получить целевой продукт реакции в виде бледно-желтого масла (0,78 г).
(г) 3-(1-Метил-1,2,3,6-тетрагидро-4-пиридил)-5- (2-нитроэтил)-1H-индол
N-Метил-4-пиперидон (Альдрих, 4,2 г) добавляют к раствору продукта реакции из стадии (в) (2,3 г) в ледяной уксусной кислоте (35 мл) при 100oC. Полученный раствор нагревают при 100oC в течение 1 часа, охлаждают и выливают в смесь 0,88 NH4OH (61 мл) и льда (61 г). Полученное твердое вещество отфильтровывают, высушивают и перекристаллизовывают из этанола, чтобы получить целевой продукт реакции в виде белого твердого вещества (1,6 г).
(д) (±)-3-[3-(1-Метил-1,2,3,6-тетрагидро-4-пиридил)-1H- индол-5-ил]-2-амино-1-пропанол
Метоксид натрия (0,30 г) добавляют к раствору продукта реакции из стадии (г) (1,5 г) в ДМФА (15 мл) при 0oC. К полученному раствору добавляют по каплям суспензию параформальдегида (0,19 г) в ДМФА (20 мл). Полученную смесь перемешивают при 0oC в течение 1,5 часов, затем выливают в воду и экстрагируют этилацетатом. Объединенные экстракты промывают водой и солевым раствором, высушивают и выпаривают в вакууме, чтобы получить желтое масло, которое элюируют через колонку с кремнеземом, используя смесь хлористого метилена/EtOH/NH4OH (50: 8: 1) в качестве элюента, чтобы получить целевой продукт реакции в виде не совсем белого твердого вещества (0,85 г), которое перекристаллизовывают из этанола.
(е) (±)-3-[3-(1-Метил-4-пиперидил)-1H-индол-5-ил]-2- амино-1-пропанол
Продукт реакции из стадии (д) (0,08 г) растворяют в этаноле (25 мл) и добавляют 10% в весовом отношении Rd/C (0,23 г). Смесь перемешивают при 1-атмосферном давлении водорода в течение 7 часов, когда завершается поглощение водорода. Смесь фильтруют через целит и фильтрат выпаривают в вакууме, чтобы получить целевой продукт реакции в виде бесцветного масла, которое элюируют через колонку с кремнеземом, используя смесь хлористого метилена/EtOH/NH4OH (50:8:1) в качестве элюента.
(ж) (±)-3-(1-Метил-4-пиперидил)-5-(1,3-оксазолидин-4- ил-метил)-1H-индол
Смесь продукта реакции из стадии (е) (1,6 г), диэтилкарбоната (0,71 г) и карбоната калия (0,08 г) нагревают при 130oC в течение 5 часов. Смесь охлаждают, переносят в метанол и нерастворимый карбонат калия отфильтровывают. Фильтрат выпаривают в вакууме и остаток элюируют через колонку с кремнеземом, используя смесь хлористого метила/EtOH/NH4OH (30:8:1) в качестве элюента, чтобы получить бесцветное пенообразное вещество, которое кристаллизуют из смеси изопропилового спирта/простого эфира, чтобы получить целевой продукт реакции в виде бесцветного кристаллического твердого вещества (1,1 г), т. пл. 191-192oC. 1H-ЯМР и микроанализ являются такими же, что и для продукта реакции из синтетического примера 4.
Синтетический пример 6. Получение (R)-2-[5-(2-оксо-1,3-оксазолидин- 4-ил-метил)-1H-индол-3-ил]-этиламина
(а) (R)-4-(4-Нитробензил)-1,3-оксазолидин-2-он
Раствор Д-4-нитрофенилаланина (Флюка, 53 г) в диметоксиэтане (250 мл) нагревают до 67oC и в течение 1 часа добавляют BF3•Et2O (Альдрих, 37 мл). Полученный раствор перемешивают при 67oC в течение 1 часа, затем нагревают до 80oC и добавляют в течение 1 часа при 80-85oC BH3•Me2S (Альдрих, 40 мл). Полученный раствор нагревают при 85oC в течение 4 часов, затем охлаждают и добавляют метанол (40 мл). Раствор нагревают до 80oC и растворители удаляют перегонкой до 1/3 первоначального объема. Добавляют 6N водную NaOH (136 мл) к горячему раствору, который затем нагревают при 85oC в течение 1/2 часа, охлаждают и добавляют хлористый метилен (100 мл). Раствор охлаждают до от -15o до -20oC и раствор трихлорметилового эфира хлормуравьиной кислоты (Альдрих, 18,2 мл) в хлористом метилене (23 мл) добавляют при температуре ниже -10oC. pH поддерживают при 9-11 путем периодических добавлений 6N водной NaOH. Полученный раствор перемешивают при комнатной температуре в течение 1 часа, затем разбавляют водой и экстрагируют хлористым метиленом. Объединенные экстракты промывают водой и солевым раствором, высушивают и выпаривают в вакууме, чтобы получить целевой продукт реакции в виде бледно-коричневого твердого вещества, которое перекристаллизовывают из этилацетата, чтобы получить бледно-желтое твердое вещество (35 г), т.пл. 113-115oC,
[α]
(б) Хлоргидрат (R)-4-(4-аминобензил)-1,3-оксазолидин-2-она
Продукт реакции из стадии (а) (10,0 г) суспендируют в смеси воды (120 мл), этанола (60 мл) и 2N водной HCl (22,5 мл) и добавляют 10% вес. Rd/C (1,0 г). Смесь перемешивают при 1-атмосферном давлении водорода в течение 8 часов, когда завершается поглощение водорода. Смесь фильтруют через Hyflo и фильтрат выпаривают в вакууме, чтобы получить целевой продукт реакции в виде бесцветного стеклообразного вещества (10,3 г).
(в) Хлоргидрат (R)-4-(4-гидразинобензил)-1,3-оксазолидин- 2-она
Продукт реакции из стадии (б) (10,3 г) суспендируют в воде (53 мл) и по каплям добавляют конц. HCl (106 мл). Полученную смесь охлаждают до -5oC и к перемешиваемой смеси в течение 15 минут добавляют по каплям раствор нитрата натрия (3,2 г) в воде (30 мл), за которым следует 30-минутное перемешивание при температуре от -5o до 0oC. Раствор добавляют затем при 0oC в течение 15 минут к перемешиваемому раствору хлористого олова (II) (51 г) в конц. HCl (91 мл), за которым следует 3-часовое перемешивание при комнатной температуре. Раствор выпаривают в вакууме и остаток растирают в порошок с помощью простого эфира, чтобы получить целевой продукт реакции в виде бледно-желтого твердого вещества (11 г).
(г) (R)-2-[5-(2-Оксо-1,3-оксазолидин-4-ил-метил)-1H- индол-3-ил]-этиламин
Продукт реакции из стадии (в) (8,8 г) растворяют в смеси этанол/вода (500 мл, 5: 1 в объемном отношении) и раствор обрабатывают диметилацеталем 4-хлорбутанальдегида (J.Amer. Chem. Soc., 1365 (1951), 5,5 г). Смесь кипятят с обратным холодильником в течение 2 часов, растворитель удаляют в вакууме и остаток элюируют через колонку с кремнеземом, используя смесь хлористого метилена/EtOH/NH4OH (30:8:1 в объемном отношении) в качестве элюента. Целевой продукт реакции получают в виде бледно-желтого масла (0,60 г).
Соль синтетического примера 6
Хлоргидрат
К перемешиваемому раствору свободного основания (0,16 г) в этаноле (2 мл) при 0oC добавляют по каплям конц. HCl (0,06 мл). Хлористоводородная соль выпадает в осадок в виде желтовато-коричневого твердого вещества, т.пл. 269-271oC.
[α]
Синтетический пример 7. Получение (R)-N,N-диметил-2-[5- (2-оксо-1,3-оксазолидин-4-ил-метил)-1H-индол-3-ил]этиламина
Раствор 35% вес./объемн. водного формальдегида (0,3 мл) в метаноле (2,0 мл) добавляют к раствору продукта реакции из стадии (г) синтетического примера 6 (0,44 г) и цианоборогидрида натрия (0,13 г) в смеси метанола (8,5 мл) и ледяной уксусной кислоты (0,51 г) при 10oC и полученную смесь перемешивают при комнатной температуре в течение 2,5 часов. Добавляют 2N водную NaOH (1,3 мл), затем борогидрид натрия (0,19 г), после чего 2N водную HCl (1,3 мл). Метанол выпаривают в вакууме и остающийся раствор разбавляют водой, доводят до pH 7 с помощью твердого карбоната калия и промывают этилацетатом. Добавляют дополнительное количество карбоната калия до pH 11 и раствор экстрагируют этилацетатом. Объединенные экстракты выпаривают в вакууме, чтобы получить целевой продукт реакции в виде белого пенообразного вещества (0,45 г).
Соль синтетического примера 7
Хлоргидрат
К перемешиваемому раствору свободного основания (0,45 г) в этаноле (4,5 мл) добавляют по каплям при 0oC конц. HCl (0,16 мл). Смесь выпаривают в вакууме и полученное пенообразное вещество растирают в порошок с помощью этилацетата, чтобы получить целевой продукт реакции в виде белого твердого вещества, т.пл. 130oC.
[α]
Синтетический пример 8. Получение хлоргидрата (S)-N,N-диметил- 2-[5-(2-тиа-1,3-оксазолидин-4-ил-метил)-1H-индол-3-ил]-этиламина
(а) (S)-N,N-Диметил-2-[5-(2-амино-1-пропанол)-1H- индол-3-ил]-этиламин
Раствор хлористоводородной соли продукта реакции из синтетического примера 2 (0,33 г) в 2N водной КОН (10 мл) кипятят с обратным холодильником в течение 4 часов, затем охлаждают и экстрагируют этилацетатом. Объединенные экстракты высушивают и выпаривают в вакууме, чтобы получить целевой продукт реакции в виде бесцветного масла (0,25 г).
(б) Хлоргидрат (S)-N, N-диметил-2-[5-(2-тиа-1,3-оксазолидин- 4-ил-метил)-1H-индол-3-ил]-этиламина
Раствор N,N'-тиокарбонилимидазола (Альдрих, 0,21 г) в ТГФ (4 мл) добавляют по каплям к перемешиваемому раствору продукта реакции из стадии (а) (0,31 г) в ТГФ (4 мл) и смесь кипятят с обратным холодильником в течение 23 часов, затем охлаждают и выпаривают в вакууме. Остаток хроматографируют через колонку с кремнеземом, используя смесь хлористого метилена/EtOH/NH4OH (20: 8: 1) в качестве элюента, чтобы получить целевой продукт реакции в виде бесцветного масла.
Соль синтетического примера 8
Хлоргидрат
К свободному основанию добавляют по каплям 1 М этанольный раствор HCl (1,0 экв. ) и этанол выпаривают в вакууме. Полученную смолу высушивают при температуре ниже 0oC, чтобы удалить воду и получить целевой продукт реакции в виде белого твердого вещества (0,71 г), т.пл. 133-136oC (размягчается при 128oC).
[α]
Синтетический пример 9. Получение бромгидрата (S)-2-[5-(3- метил-2-оксо-1,3-оксазолидин-4-ил-метил)-1H-индол-3-ил]-этиламина
(а) (S)-3-Метил-4-(4-нитробензил)-2-оксазолидинон
Гидрид натрия (0,80 г в виде 60%-ной в весовом отношении дисперсии в масле) добавляют при комнатной температуре к перемешиваемому раствору продукта реакции из стадии (в) синтетического примера 1 (4,4 г) в сухом ТГФ (150 мл). Смесь перемешивают в течение 1,5 часов, затем добавляют диметилсульфат (2,1 мл) и перемешивание продолжают в течение дополнительных 16 часов. Добавляют добавочно 0,40 г гидрида натрия и перемешивание продолжают в течение еще 2 часов. Смесь выпаривают в вакууме и остаток суспендируют в этилацетате и фильтруют. Фильтрат выпаривают в вакууме и остаток кристаллизуют из смеси этилацетата/гексана, чтобы получить целевой продукт реакции в виде желтых кристаллов (3,7 г), т.пл. 146-147oC.
[α]
(б) Хлоргидрат (S)-3-метил-4-(4-аминобензил)-2-оксазолидинона
Суспензию продукта реакции из стадии (а) (4,0 г) и 10% вес. Rd/C (0,20 г) в смеси этанола (70 мл) и разбавленной HCl (2N водной HCl (12 мл) + вода (55 мл)) гидрируют при давлении 45 psi (3,16 атм) в течение 1 часа. Смесь фильтруют через Hyflo и фильтрат выпаривают в вакууме, чтобы получить целевой продукт реакции в виде пенообразного вещества.
(в) Хлоргидрат (S)-3-метил-4-(4-гидразинобензил)-2-оксазолидинона
Раствор продукта реакции из стадии (б) (4,1 г) в воде (24 мл) охлаждают до -5oC и добавляют конц. HCl (40 мл). Раствор нитрита натрия (1,2 г) в воде (12 мл) добавляют, а затем продолжают перемешивание в течение 0,5 часа. Полученный раствор добавляют по каплям при -5oC к перемешиваемому раствору дигидрата хлористого олова (II) (18,8 г) в конц. HCl (34 мл). Полученную смесь перемешивают при 0oC в течение 2,5 часов, затем выпаривают в вакууме. Остаток переносят в воду, доводят до pH 2,5, используя 10N водную NaOH и фильтруют. Фильтрат выпаривают в вакууме и остаток растирают в порошок с помощью этанола и фильтруют. Фильтрат выпаривают в вакууме, чтобы получить целевой продукт реакции в виде пенообразного вещества.
(г) Гидробромид (S)-2-[5-(3-метил-2-оксо-1,3-оксазолидин- 4-ил-метил)-1H-индол-3-ил]этиламина
Диметилацеталь 4-хлорбутанальдегида (J.Amer. Chem. Soc., 1365, (1951), 2,3 г) добавляют к перемешиваемому раствору продукта реакции из стадии (в) (4,4 г) в смеси этанола/воды (150 мл/30 мл) и смесь кипятят с обратным холодильником в течение 2 часов. Охлажденную смесь выпаривают в вакууме и остаток элюируют через колонку с кремнеземом, используя смесь хлористого метилена/MeOH/NH4OH (60:8:1) в качестве элюента, чтобы получить коричневое масло (1,7 г). Часть масла этого (0,25 г) растворяют в этаноле и обрабатывают с избытком HBr в уксусной кислоте (приблизительно 45% вес./объем). Полученный раствор выпаривают в вакууме и остаток растирают в порошок с помощью эфира, затем кристаллизуют из смеси этанола/гексана, чтобы получить целевой продукт реакции в виде бледно-желтых кристаллов (0,14 г), т.пл. 203-205oC, [α]
Синтетический пример 10. Получение малеата 0,75 гидрата (S)- N/N-диметил-2-[5-(3-метил-2-оксо-1,3-окcазолидин-4-ил-метил)-1H- индол-3-ил]этиламина
Цианоборогидрид натрия (0,14 г), за которым следует ледяная уксусная кислота (0,54 мл), добавляют при комнатной температуре к перемешиваемому раствору свободного основания (0,52 г) из стадии (г) синтетического примера 9 в метаноле (9,0 мл). Когда завершается бурное выделение газа, добавляют раствор 37% вес. /об. водного формальдегида (0,16 г) в метаноле (2,0 мл) и смесь перемешивают в течение 1 часа, затем разбавляют водой, насыщают карбонатом калия и экстрагируют этилацетатом. Объединенные экстракты выпаривают в вакууме и остаток элюируют через колонку с кремнеземом, используя смесь хлористого метилена/MeOH/NH4OH (60:8:1) в качестве элюента, чтобы получить свободное основание целевого продукта реакции в виде бесцветного масла (0,25 г). Последнее растворяют в этаноле (10 мл), обрабатывают раствором малеиновой кислоты (0,09 г) в этаноле (1 мл) и полученный раствор выпаривают в вакууме, чтобы получить масло, которое растирают в порошок с помощью эфира, высушивают при температуре ниже 0oC, чтобы избавиться от воды и получить целевой продукт реакции в виде бесцветного стеклообразного вещества, [α]
Синтетический пример 11. Получение 0,75 гидрата малеата (S)- N-бензил-2-[5-(2-оксо-1,3-оксазолидин-4-ил-метил)-1H-индол-3-ил]-этиламина
Бензальдегид (0,70 г) добавляют при комнатной температуре к перемешиваемому раствору соединения синтетического примера 1 (1,7 г) в этаноле (20 мл). Раствор перемешивают в течение 36 часов, затем добавляют по частям борогидрид натрия (0,25 г) и перемешивание продолжают в течение еще 2 часов. Раствор выпаривают в вакууме и остаток охлаждают, подкисляют с помощью 2N водной HCl, подщелачивают бикарбонатом натрия, насыщают карбонатом калия и экстрагируют этилацетатом. Объединенные экстракты выпаривают в вакууме, чтобы получить масло, которое элюируют через колонку с кремнеземом, используя смесь хлористого метилена(EtOH/NH4OH (100: 8:1) в качестве элюента, чтобы получить свободное основание целевого продукта реакции в виде желтого пенообразного вещества (1,6 г). Часть этого вещества (0,13 г) растворяют в этаноле (10 мл), обрабатывают раствором малеиновой кислоты (43 мг) в этаноле (1 мл) и полученный раствор выпаривают в вакууме. Остаток высушивают при температуре ниже 0oC, чтобы избавиться от воды и получить целевой продукт реакции в виде бледно-желтого порошка (0,15 г), [α]
Элементарный анализ, 1H-ЯМР и масс-спектрометрия согласуются с предложенной структурой.
Синтетический пример 12. Получение гидрата малеата (S)-N-бензил-N-метил-2-[5-(2-оксо-1,3-оксазолидин-4-ил-метил)-1H-индол-3-ил]этиламина
К раствору свободного основания из синтетического примера II (0,45 г) в ДМФА (8,0 мл) добавляют при комнатной температуре безводный карбонат калия (0,34 г). Суспензию перемешивают в течение 0,5 часа, затем раствор диметилсульфата (0,17 г) в ДМФА (2,0 мл) добавляют и перемешивание продолжают в течение еще 3 часов. Добавляют воду (40 мл) и смесь экстрагируют этилацетатом. Объединенные экстракты выпаривают в вакууме, чтобы получить желтое масло, которое элюируют через колонку с кремнеземом, используя смесь хлористого метилена/EtOH/NH4OH (100: 8:1) в качестве элюента, чтобы получить свободное основание целевого продукта реакции в виде бесцветного масла (0,32 г). Часть этого масла (73 мг) растворяют в этаноле (10 мл), обрабатывают раствором малеиновой кислоты (23 г) в этаноле (1 мл) и полученный раствор выпаривают в вакууме. Остаток высушивают при температуре ниже 0oC, чтобы избавиться от воды и получить целевой продукт реакции в виде бледно-желтого порошка, [α]
Синтетический пример 13. Получение 0,5 гидрата малеата (S)- N-метил-2-[5-(2-оксо-1,3-оксазолидин-4-ил-метил)-1H-индол-3-ил]-этиламина
Суспензию свободного основания продукта реакции из синтетического примера 12 (0,25 г) и 10% вес, Rd/C (0,10 г) в этаноле (25 мл) гидрируют в течение 16 часов. Смесь фильтруют через Hyflo и фильтрат выпаривают в вакууме. Остаток элюируют через колонку с кремнеземом, используя смесь хлористого метилена/EtOH/NH4OH (30:8:1) в качестве элюента, чтобы получить свободное основание целевого продукта реакции (0,14 г). Последнее растворяют в этаноле (10 мл), обрабатывают раствором малеиновой кислоты (0,06 г) в этаноле (1 мл) и полученный раствор выпаривают в вакууме. Остаток высушивают при температуре ниже 0oC, чтобы избавиться от воды и получить целевой продукт реакции в виде гигроскопического твердого вещества, [α]
Синтетический пример 14. Получение 0,33 метанолата 0,75 гидрата (S)-3-(1-метил-1,2,3,6-тетрагидро-4-пиридил)-5-(2-оксо-1,3-оксазолидин- 4-ил-метил)-1H-индола
(а) (S)-3-Фенилтио-5-(2-оксо-1,3-оксазолидин-3-ил-метил)-1H-индол
Диэтилацеталь фенилтиоацетальдегида (J. C.S., Chem. Comm., 924 (1978), 9,1 г) добавляют при комнатной температуре к перемешиваемому раствору продукта реакции из стадии (д) синтетического примера 1 (9,8 г) в смеси этанола (150 мл) и воды (100 мл). Добавляют конц. HCl (5 капель) и смесь перемешивают при комнатной температуре в течение 2 суток, затем частично выпаривают в вакууме. Полученную водную суспензию экстрагируют этилацетатом и объединенные экстракты промывают водой и выпаривают в вакууме, чтобы получить коричневое масло. Последнее элюируют через колонку с кремнеземом, используя смесь хлористого метилена/EtOH/NH4OH (150:8:1) в качестве элюента, чтобы получить целевой продукт реакции в виде бледно-желтого масла (5,0 г).
(б) (S)-5-(2-оксо-1,3-оксазолидин-4-ил-метил)-1H-индол
Никель Ренея (3,0 г) добавляют к раствору продукта реакции из стадии (а) (3,1 г) в изопропиловом спирте (150 мл) и суспензию кипятят с обратным холодильником в течение 1 часа. Добавляют еще 2,0 г никеля Ренея и кипячение с обратным холодильником продолжают в течение дополнительных 2 часов. Суспензию фильтруют в горячем состоянии через Hyflo и фильтрат выпаривают в вакууме, чтобы получить масло. Последнее элюируют через колонку с кремнеземом, используя этилацетат в качестве элюента, чтобы получить целевой продукт реакции в виде пенообразного вещества (1,3 г). 1H-ЯМР и масс-спектрометрия согласуются с предложенной структурой.
(в) 0,33 Метанолат 0,75 гидрата (5)-3-(1-метил-1,2,3,6- -тетрагидро-4-пиридил)-5-(2-оксо-1,3-оксазолидин-4- -ил-метил)-1H-индола
1-Метил-4-пиперидон (0,74 г, Альдрих) добавляют к перемешиваемому раствору продукта реакции из стадии (б) (0,30 г) в ледяной уксусной кислоте (2,0 мл) и смесь перемешивают при 100oC в течение 2 часов. Охлажденную смесь выливают на смесь льда/NH4OH (20 мл) и полученное твердое вещество отфильтровывают. Последнее элюируют через колонку с кремнеземом, используя смесь хлористого метилена/EtOH/NH4OH (60: 8:1) в качестве элюента и кристаллизуют из этилацетата, чтобы получить целевой продукт реакции в виде бесцветного твердого вещества (0,11 г), т.пл. 225-227oC, [α]
Синтетический пример 15. Получение гидробромида (S)-3-(1- метил-4-пиперидил)-5- (2-оксо-1,3-оксазолидин-4-ил-метил)-1H-индола
Суспензию продукта реакции из синтетического примера 14 (0,35 г) и 10% вес. Rd/C (0,10 г) в смеси метанола (10 мл), воды (10 мл) и 1N водной HCl гидрируют в течение 5 часов. Смесь фильтруют через Hyflo и фильтрат выпаривают в вакууме. Остаток подщелачивают с помощью NH4OH, выпаривают в вакууме и элюируют через колонку с кремнеземом, используя смесь хлористого метилена/EtOH/ NH4OH (45:8:1) в качестве элюента, чтобы получить масло. Последнее растворяют в этаноле (5,0 мл) и обрабатывают избытком HBr в уксусной кислоте (приблизительно 45% вес./об.), чтобы получить целевой продукт реакции в виде бесцветных кристаллов (0,20 г), т.пл. 260-261oC, [α]
Синтетический пример 16. Получение гидрата (R)-3-(1-метил-1,2,3,6-тетрагидро-4-пиридил)-5-(2-оксо-1,3-оксазолидин-4-ил-метил)-1H-индола
(а) Хлоргидрат (R)-4-(4-гидразинобензил)-1,3-оксазолидин-2-она
По стадиям, одинаковым со стадиями от (а) до (в) из синтетического примера 6, превращают Д-4-нитрофенилаланин в хлоргидрат (R)-4-(4-гидразинобензил)-2-оксазолидинона.
(б) Гидрат (R)-3-(1-метил-1,2,3,6-тетрагидро-4-пиридил)-5- (2-оксо-1,3-оксазолидин-4-ил)-1H-индола
По стадиям, аналогичным стадиям от (а) до (в) из синтетического примера 14, превращают продукт реакции из стадии (а) в гидрат (R)-3-(1-метил-1,2,3,6-тетрагидро-4-пиридил)-5-(2-оксо-1,3- -оксазолидин-4-ил-метил)-1H-индола, т.пл. 229-231oC, [α]
Синтетический пример 17. Получение бромгидрата (R)-3-(1- метил-4-пиперидил)-5-(2-оксо-1,3-оксазолидин-4-ил-метил)-1H-индола
По методу, аналогичному из синтетического примера 15, превращают продукт реакции из синтетического примера 16 в бромгидрат (R)-3-(1-метил-4-пиперидил)-5-(2-оксо-1,3-оксазолидин-4-ил-метил)-1H-индола, т.пл. 260-261oC, [α]
Синтетический пример 18. Получение гидрата (R)-3-(1-бензил- -1,2,3,6-тетрагидро-4-пиридил)-5-(2-оксо-1,3-оксазолидин-4-ил-метил)-1H- индола
1-Бензил-4-пиперидон (Альдрих, 2,8 г) добавляют к перемешиваемой суспензии (R)-5-(2-оксо-1,3-оксазолидин-4-ил-метил)-1H- индола (1,0 г), промежуточного предшественника (исходного вещества) продукта реакции из синтетического примера 16, в ледяной уксусной кислоте (20 мл) и перемешивают при 100oC в течение 3 часов. Охлажденную смесь выпаривают в вакууме и остаток растворяют в метаноле, подщелачивают с помощью NH4OH и выпаривают в вакууме, чтобы получить темный деготь. Последний элюируют через колонку с кремнеземом, используя смесь хлористого метилена/EtOH/NH4OH (100:8:1) в качестве элюента, и обрабатывают хлористым метиленом. Полученный осадок отфильтровывают, чтобы получить целевой продукт реакции в виде желтых кристаллов (0,25 г), т.пл. 169-170,5oC. Элементарный анализ и 1H-ЯМР согласуются с предложенной структурой.
Синтетический пример 19. Получение бромгидрата (R)-3-(4-пиперидил)-5-(2-оксо-1,3-оксазолидин-4-ил-метил)-1H-индола
Суспензию продукта реакции из синтетического примера 18 (0,25 г) и 10% вес. Rd/C (0,10 г) в метаноле (25 мл) гидрируют при давлении 90 psi (6,32 атм) в течение 20 часов, когда прекращается поглощение водорода. Смесь фильтруют через Hyflo и фильтрат выпаривают в вакууме. Остаток элюируют через колонку с кремнеземом, используя смесь хлористого метилена/EtOH/NH4OH (30: 8: 1) в качестве элюента, чтобы получить масло. Последнее растворяют в изопропиловом спирте и обрабатывают избытком HBr в уксусной кислоте (приблизительно 45% вес./об.), чтобы получить гигроскопическое твердое вещество, которое высушивают при температуре ниже 0oC, чтобы удалить влагу и получить целевой продукт реакции в виде бледно-коричневого порошка. Элементарный анализ и 1H-ЯМР согласуются с предложенной структурой.
Синтетический пример 20. Получение ацетата (±)-N,N-диметил- 2-[5-(1-тио-2-тиа-3-оксазолидин-4-ил-метил)-1H-индол-3-ил]-этиламина
Сероуглерод (90 мкл) добавляют к перемешиваемому раствору продукта реакции из стадии (а) синтетического примера 8 (0,31 г) и гидроокиси калия (0,08 г) в этаноле (3,8 мл) и смесь кипятят с обратным холодильником, затем выпаривают в вакууме. Остаток экстрагируют эфиром, подкисляют и хроматографируют, используя обратимую фазовую ЖХВД-колонку с кремнеземом и элюируя смесью 10 ---> 90% об. воды/ацетонитрила вместе с 0,1 М водным аммоний-ацетатным буфером при pH 4.0 в течение 20 минут, чтобы получить целевой продукт реакции (0,01 г) и, после обработки с HCl, продукт реакции из синтетического примера 8 (0,11 г). Оба продукта высушивают при температуре ниже 0oC, чтобы удалить воду и получить 1H-ЯМР и масc-спектрометрию, которые согласуются с предложенной структурой.
Синтетический пример 21. Получение хлоргидрата (±)-N,N-диметил-2-[5-(2-оксо-1,3-оксазолидин-5-ил-метил)-1H-индол- 3-ил]-этиламина
(а) (±)-1-Нитрометил-2-фенилэтанол
Метоксид натрия (1,1 г) добавляют к перемешиваемому раствору нитрометана (Альдрих, 12,2 г) в метаноле (100 мл) при 0oC и смесь перемешивают в течение 10 минут. Раствор фенилацетальдегида (Альдрих, 24,0 г) в метаноле (50 мл) добавляют по каплям в течение 15 минут и смесь перемешивают в течение 45 минут при 0oC, затем доводят до комнатной температуры в течение 1 часа и перемешивают в течение ночи. Смесь выпаривают в вакууме и остаток растворяют в воде и экстрагируют эфиром. Объединенные экстракты промывают водой и солевым раствором и выпаривают в вакууме, чтобы получить целевой продукт реакции в виде желтого масла (29,0 г).
(б) Хлоргидрат (±)-1-аминометил-2-фенилэтанола
Суспензию продукта реакции из стадии (а) (10,0 г) и 10% вес. Rd/C (1,0 г) в этаноле (250 мл) гидрируют до тех пор, пока поглощение водорода не прекратится. Смесь фильтруют через Hyflo и фильтрат выпаривают в вакууме. Остаток растворяют в этилацетате и экстрагируют 2N водной HCl. Объединенные экстракты промывают этилацетатом, затем выпаривают в вакууме, чтобы получить целевой продукт реакции в виде розовато-белого твердого вещества (6,8 г).
(в) (±)-5-Бензил-1,3-оксазолидин-2-он
Раствор КОН (9,4 г) в воде (85 мл) добавляют к перемешиваемому раствору продукта реакции из стадии (б) (5,1 г) в толуоле (150 мл) при 0oC. Раствор фосгена (9,8 г) в толуоле (78,4 мл = 12,5% вес./об.) добавляют по каплям в течение 15 минут и смесь доводят до комнатной температуры, затем перемешивают в течение ночи. Водную фазу отделяют и экстрагируют этилацетатом. Объединенные экстракты выпаривают в вакууме, чтобы получить целевой продукт реакции в виде белого твердого вещества (2,2 г), т.пл. 106-108oC. Элементарный анализ согласуется с предложенной структурой.
(г) (±)-5-(4-Нитробензил)-1,3-оксазолидин-2-он
К продукту реакции из стадии (в) при 0oC добавляют конц. H2SO4 (1,6 мл), а затем конц. HNO3 (0,33 мл, приблизительно 0,05 мл за 5 минут) также при 0oC. Смесь перемешивают в течение 0,5 часа при 0oC, а затем в течение 0,5 часа при комнатной температуре. Добавляют смесь воды/льда (100 мл) и реакционную смесь экстрагируют этилацетатом. Объединенные экстракты выпаривают в вакууме, чтобы получить желтое масло, которое перекристаллизовывают из этилацетата, чтобы получить целевой продукт реакции в виде белого порошка (0,4 г), т.пл. 143-146oC.
(д) Хлоргидрат (±)-5-(4-аминобензил)-1,3-оксазолидин-2-она
Суспензию продукта реакции из стадии (г) (1,4 г) и 10% вес.
Rd/C (0,14 г) в смеси воды (21 мл), этанола (28 мл) и 2N водной HCl (3,2 мл) гидрируют в течение 2 часов, когда прекращается поглощение водорода. Смесь фильтруют через Hyflo и фильтрат выпаривают в вакууме, чтобы получить целевой продукт реакции в виде бледно-желтого пенообразного вещества (1,4 г).
(е) Хлоргидрат (±)-N, N-диметил-2-5-(2-оксо-1,3-оксазолидин-5-ил-метил)-1H-индол-3-ил-этиламина
К перемешиваемому раствору продукта реакции из стадии (д) (1,4 г) в воде (8,5 мл) при 0oC добавляют конц. HCl (14,5 мл). Раствор нитрита натрия (0,43 г) в воде (4,3 мл) добавляют по каплям в течение 15 минут при 0oC и смесь перемешивают в течение 0,5 часа при 0oC. Смесь добавляют затем по каплям к перемешиваемому раствору хлористого олова (II) (6,8 г) в конц. HCl (12,4 мл) при 0oC в течение 15 минут. Смесь доводят до комнатной температуры в течение 1 часа, затем выпаривают в вакууме. Остаток растворяют в воде (30 мл), доводят до pH 2,5, используя 10N водную NaOH и выпадающие в осадок соли отфильтровывают. Диэтилацеталь 4-диметил- аминобутанальдегида (Croatica Chemica Acta, 36, 103 (1964), 1,1 г), за которым следует ионообменная смола типа "Амберлит 15" (Альдрих, 3,0 г), добавляют к фильтрату и смесь нагревают в течение 3 часов при 100oC, фильтруют и фильтрат выпаривают в вакууме. Остаток обрабатывают горячим этанолом, фильтруют и фильтрат выпаривают в вакууме. Остаток растирают в порошок с помощью этилацетата, фильтруют, фильтрат выпаривают в вакууме. Остаток перекристаллизовывают из этанола, чтобы получить целевой продукт реакции в виде бледно-желтого твердого вещества (0,75 г), т.пл. 280-281oC.
1H-ЯМР и масс-спектрометрия согласуются с предложенной структурой.
Синтетический пример 22. Получение (S )-N,N-диметил-2-[5- (2-оксо-1,3-оксазолидин-4-ил-метил)-1H-индол-3-ил]-этиламина
(а) (S)-5-(4-Нитробензил)-1,3-имидазолидин-2,4-дион
Бензилизоцианат (Альдрих, 3,2 г) добавляют к раствору L-4- нитрофенилаланина (Альдрих, 4,2 г) и гидроокиси калия (1,3 г) в воде (40 мл) при 0oC. Смесь нагревают при 60-70oC в течение 2 часов, фильтруют и фильтрат подкисляют с помощью конц. HCl, чтобы получить не совсем белое твердое вещество, которое отфильтровывают, суспендируют в 2N водной HCl (20 мл) и кипятят с обратным холодильником в течение 2 часов. Охлажденную смесь разбавляют водой и фильтруют, чтобы получить целевой продукт реакции в виде белого твердого вещества (5,6 г).
(б) (S)-N, N-Диметил-2-[5-(2-оксо-1,3-оксазолидин-4-ил-метил)- 1H-индол-3-ил]-этиламин
По стадиям, аналогичным стадиям от (г) до (е) синтетического примера 1 и синтетического примера 2, или по стадиям (г) и (д) синтетического примера 1 и синтетического примера 3, и по стадиям от (д) до (з) синтетического примера 4, превращают продукт реакции из стадии (а) в (S)-N,N-диметил-2-[5-(2-оксо-1,3-оксазолидин- 4-ил-метил)-1H-индол-3-ил]-этиламин.
Синтетический пример 23. Получение (S)-N,N-диметил-2-[5- (2-оксо-1,3-оксазолидин-4-ил-метил)-1H-индол-3-ил]-этиламина
(а) Хлоргидрат (S)-4-(4-гидразинобензил)-1,3-оксазолидин-2-она
По стадиям, аналогичным стадиям от (а) до (в) из синтетического примера 6, превращают 1-4-нитрофенилаланин в хлоргидрат (S)-4-(4-гидразинобензил)-1,3-оксазолидин-2-она.
(б) (S)-4-[4-2-(3-цианопропилиден)-гидразинобензил] - 1,3-оксазолидин-2-он
1 М водную HCl (4,0 мл) добавляют к раствору продукта реакции из стадии (а) (2,4 г) в воде (35 мл). Диэтилацеталь 3-цианопропанальдегида (Альдрих, 1,7 г) добавляют при комнатной температуре и смесь перемешивают в течение 2 часов. Добавляют еще 0,20 г ацеталя и смесь перемешивают в течение дополнительных 20 минут. Водную фазу декантируют от получающейся смолы и экстрагируют этилацетатом. Экстракты объединяют со смолой и выпаривают в вакууме, чтобы получить целевой продукт реакции (2,5 г).
(в) (S)-3-Цианометил-5-(2-оксо-1,3-оксазолидин-4-ил- метил)-1H-индол
Раствор продукта реакции из стадии (б) (2,5 г) и полифосфатный сложный эфир (20,0 г) в хлороформе (40 мл) кипятят с обратным холодильником в течение 20 минут. К охлажденной смеси добавляют лед, и хлороформ выпаривают в вакууме. Оставшуюся водную фазу экстрагируют этилацетатом и объединенные экстракты выпаривают в вакууме, чтобы получить целевой продукт реакции в виде бледно-желтого масла (1,8 г).
(г) (S)-N, N-Диметил-2-[5-(2-оксо-1,3-оксазолидин-4-ил- метил)-1H-индол-3-ил]-этиламин
Суспензию продукта реакции из стадии (в) (1,3 г) и 10% вес. Rd/C (1,0 г) в 30% вес. этанольном диметиламине (25 мл) гидрируют в течение 24 часов и фильтруют через Hyflo. К фильтрату добавляют свежеприготовленный Rd/C (0,7 г) и этанольный диметиламин (5 мл) и гидрирование продолжают в течение еще 16 часов. Смесь фильтруют через колонку с кремнеземом, используя смесь хлористого метилена/EtOH/NH4OH (40:8:1) в качестве элюента, чтобы получить целевой продукт реакции в виде бесцветного пенообразного вещества (0,3 г). Элементарный анализ и 1H-ЯМР согласуются с предложенной структурой.
Синтетические примеры от 24 до 31.
По методам, аналогичным описанным в синтетических примерах от 1 до 23, получают следующие соединения формулы (I). ЯМР и микроанализ для каждого соединения согласуются с предложенной структурой.
24) 0,75 гидрат малеата 2-[5-(3-метил-2-оксоимидазолидин-4-ил-метил)-1H-индол-3-ил]-этиламина, т.пл. 94-98oC.
25) 0,95 гидрат малеата 2-[5-(3-метил-2-оксоимидазолидин-4- ил-метил)-1H-индол-3-ил]-N,N-диметилэтиламина (белый продукт лиофильной сушки),
26) гидрат хлоргидрата 2-[5-[2-(2,5-диоксоимидазолидинил)- этил]-1H-индол-3-ил]-этиламина, т.пл. 83-85oC,
27) гидрат малеата 2-[5-[2-(2,5-диоксоимидазолидинил)-этил]-1H-индол-3-ил]-N,N-диметилэтиламина (бледно-желтый продукт лиофильной сушки),
28) хлоргидрат 5-[2-(2,5-диоксоимидазолидинил)-этил]-3-(1- метил-4-пиперидинил)-1H-индола, т.пл. 320-322oC (разл.),
29) гидрат малеата 2-[5-(5-метил-2-оксоимидазолидин-4-ил-этил)- 1H-индол-3-ил]-этиламина, т.пл. 99oC (размягчается при 88oC),
30) 1,4 гидрат ацетата 5-[3-(4-пиперидил)-1H-индол-5-ил-метил]- 2,4-имидазолидиндиона, т.пл. 92-93oC (размягчается при 86oC) и
31) 2,75 гидрат диацетата 2-[5-(1-метил-2-оксо-4-имидазолидинил- метил)-1H-индол-3-ил]этиламина (бледно-желтый продукт лиофильной сушки).
Примеры приготовления лекарственных средств
В следующих примерах "действующим ингредиентом" может быть любое соединение формулы (I) и/или физиологически приемлемая соль, сольват или физиологически функциональное производное его.
(1) Приготовление лекарственных форм в таблетках
(i) пероральная форма (см. табл. 1).
(ii) подъязычная форма (см. табл. 2).
Лекарственные формы от A до E могут быть приготовлены путем влажного гранулирования первых шести ингредиентов с повидоном, за которым следует добавление стеарата магния и прессование в таблетку.
(iii) трансбуккальная форма (см. табл. 3).
Лекарственная форма может быть приготовлена непосредственным прессованием смешанных ингредиентов.
(2) лекарственные формы в капсулах
(i) порошок (см. табл. 4).
Лекарственные формы F и G могут быть приготовлены смешиванием ингредиентов и наполнением двусторонних жестких желатиновых капсул полученной смесью.
(ii) жидкое наполнение (см. табл. 5).
Лекарственная форма H может быть приготовлена путем плавления Макрогола 4000 ВР, диспергирования действующего ингредиента в расплаве и наполнения двусторонних жестких желатиновых капсул смесью расплава. Лекарственная форма I может быть приготовлена путем диспергирования действующего ингредиента в лецитине и арахисовом масле и заполнения мягких, эластичных желатиновых капсул полученной дисперсией.
(iii) контролируемое выделение (см. табл. 6).
Лекарственная форма может быть приготовлена путем смешивания и экструдирования первых четырех ингредиентов и превращения в сферическую форму и высушивания экструдата. Высушенные шарики покрываются этилцеллюлозой в качестве мембраны, контролирующей выделение, и заполняются в двусторонние, жесткие желатиновые капсулы,
(3) Лекарственная форма для внутривенной инъекции (см. табл. 7).
Действующий ингредиент смешивается в цитратном буфере и добавляется достаточное количество соляной кислоты, чтобы повлиять на растворение и довести pH до 7. Полученный раствор доводится до объема и фильтруется через микропористый фильтр в стерильные стеклянные ампулы, которые укупориваются и запаиваются.
(4) Интраназальная лекарственная форма (см. табл. 8).
Действующий ингредиент поглощается в смеси оксибензоатов и цитратного буфера и добавляется достаточное количество соляной кислоты, чтобы повлиять на растворение и довести pH до 7. Полученный раствор доводится до объема и фильтруется через микропористый фильтр в стерилизованные стеклянные ампулы, которые укупориваются и запаиваются.
(5) Лекарственная форма для внутримышечной инъекции (см. табл. 9).
Действующий ингредиент растворяется в гликофуроле. Добавляется бензиловый спирт, который растворяется, и добавляется вода до 3 мл. Смесь фильтруется через микропористый фильтр в стерилизованные стеклянные ампулы, которые укупориваются и запаиваются.
(6) Сиропообразная лекарственная форма (см. табл. 10).
Бензоат натрия растворяется в части очищенной воды и добавляется раствор сорбита. Добавляется и растворяется действующий ингредиент. Полученный раствор смешивается с глицерином и доводится до требуемого объема с помощью очищенной воды.
(7) Лекарственная форма в виде суппозиторий (см. табл. 11).
Одна пятая часть Вайтепсола Н15 расплавляется в сосуде с паровой рубашкой при температуре максимум 45oC. Действующий ингредиент просеивается через сито с размером отверстий 200 мкм и смешивается с расплавленной основой, используя миксер Сильверсона, снабженный резательной головкой, до тех пор, пока не будет достигнута однородная дисперсия. Поддерживая смесь при 45oC, остальную часть Вайтепсола Н15 добавляют к суспензии, которая перемешивается, чтобы обеспечить гомогенную смесь. Вся суспензия пропускается затем через сито из нержавеющей стали и размером отверстий 250 мкм и при непрерывном перемешивании оставляют охлаждаться до 40oC. При температуре 30o-40oC, 2 г аликвоты смеси заполняются в подходящие пластмассовые формы и суппозитории оставляют охлаждаться до комнатной температуры.
(8) Лекарственная форма в виде пессарии (вагинальных суппозиторий) (см. табл. 12)
Вышеуказанные ингредиенты непосредственно смешиваются, и пессарии получаются прессованием полученной смеси.
Биологическое испытание
Соединения формулы (I), полученные в синтетических примерах от 1 до 17, каждое испытывают на их активность в качестве агонистов для "5-НТ1-подобного" рецептора. являющегося посредником сокращения гладкой мышцы, по следующему способу,
Подкожные вены правой и левой ног получают от новозеландских белых кроликов-самцов (2,4-2,7 кг), которых забили путем внутривенной инъекции пентобарбитона натрия (60 мг/кг). Кольцевые сегменты (шириной 3-5 мм), полученные из каждого сосуда (вены), подвешивают между двумя проволочными крючками и погружают в ванночки для органов, объемом 20 мл, содержащие раствор Кребса (pH 7.4) следующего состава (мМ): NaCl 118,41; NaHCO3 25,00; KCl 4,75; K2HPO4 1,19; MgSO4 1,19; глюкоза 11,10; и CaCl2 2,50. Кокаин (30 мкМ) содержится в растворе Кребса на всем протяжении эксперимента, чтобы предотвратить поглощение аминов симпатическими нейронами (нервными клетками). Раствор поддерживают при 37oC и непрерывно продувают (газируют) смесью 95% кислорода/5% углекислого газа. Повышения в изометрической силе ткани измеряют, используя датчики силового смещения типа Грасс FTOЗC и регистрируют на самопишущем приборе типа Гоулд ВД-212.
Силу в 1,0 г применяют к каждому препарату и повторно устанавливают дважды в течение последующего 30-минутного периода времени. Во время этого периода времени ткани подвергают действию паргилина (500 мкМ), чтобы необратимо ингибировать моноамин-оксидазу, и действию феноксибензамина (0,1 мкМ), чтобы инактивировать α1 -адреноцепторы. К концу 30 минут ингибиторы удаляют путем нескольких изменений раствора Кребса в ванночках для органов.
Активность агонистов оценивают путем совокупных добавлений испытуемого соединения, причем его концентрация увеличивается в 0,5 log10-единицах возрастаний до тех пор, пока дополнительные добавления не будут вызывать дальнейшего изменения в силе (смещения) ткани. В каждом эксперименте активность испытуемого соединения сравнивают с активностью 5-НТ. Активность выражают в виде p[A50](-log10[M], где M является молярной концентрацией агониста, требуемого для создания половины максимального действия). Результаты, полученные для соединений синтетических примеров 2/3 и 4/5, представляются в таблице 13.
Данные по токсичности
Хлористоводородную соль соединения из синтетических примеров 2/3 перорально путем кормления через желудочный зонд вводят крысам вида Вистар в виде раствора в дистиллированной воде при дозах 25, 100 и 200 мг/кг в основной форме и собакам Бигля (ищейкам) при дозах 0.25, 0.50, 1.0 и 2.0 мг/кг в основной форме один раз в сутки в течение 14 суток. В отдельном исследовании собаки в течение суток дозировка свободного основания была повышена от 2 мг/кг на первые сутки до 100 мг/кг на 30-е сутки. Свободное основание перорально вводят также собаковидным? (cynomolgus?) обезьянам при дозе 50 мг/кг один раз в сутки в течение 15 суток.
Наблюдают отсутствие доказательства о токсичности в любых из вышеупомянутых исследований при любых использованных дозировках.
Сравнительные данные по биодоступности для соединений EP-0313397
Данные показывают концентрацию в плазме крыс через 30 минут и 120 минут после ввода р.о. в дозе 10 мг на кг веса тела;
Соединение A
Биодоступность - менее 5%
Соединение B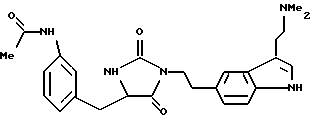
Биодоступность (30, 120 минут) - 0,00/0,01.
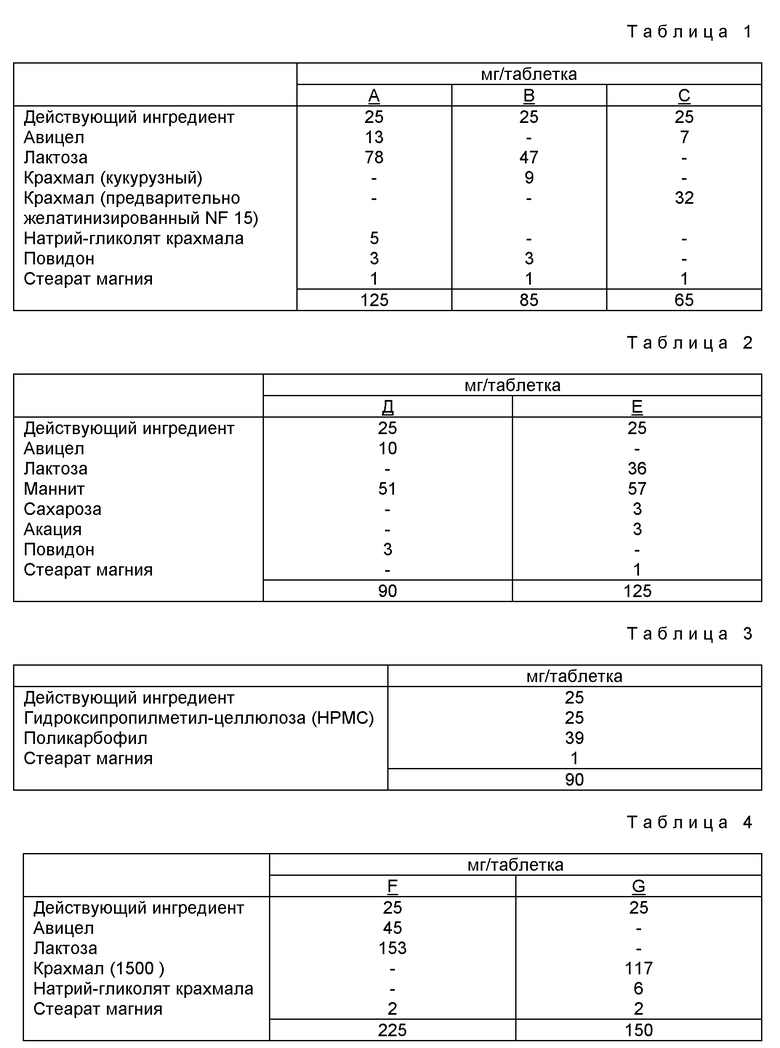


Предложены производные индола формулы I, где n равно от 1 до 3, W является группами формулы (i), (ii) или (iii), где R является водородом или C1-4-алкилом; X является -O-, -NH-; Y является O или S; хиральный центр * в формулах (i) или (ii) находится в (S)- или (R)-форме или является смесью их в любых соотношениях; Z является группами формул - -CH2CH2NR1R2, (V) или (VI), R1, R2 и R3 являются водородом или C1-4-алкилом; и их физиологически приемлемые соли или сольваты, при условии, что исключается соединение: N, N-диметил-2-[5-(2-оксо-1,3-оксазолидин-4-ил-метил)-1H-индол-3-ил] -этиламин. Производные индола формулы I могут быть использованы при профилактике клинических состояний, для которых показан прием агониста "5-HT1-подобного" рецептора. Предложены также способы их получения, лекарственное средство на основе производных индола формулы I и способ получения лекарственного средства. 7 с. и 10 з. п. ф-лы, 13 табл. 


в которой n является целым числом от 1 до 3;
W является группами формул (i), (ii) или (iii):


где R является водородом или C1-4-алкилом;
X является -O-, -NH-;
Y является кислородом или серой;
хиральный центр * в формулах (i) или (ii) находятся в своей (S)- или (R)-форме или является смесью их в любых соотношениях;
Z является группами формул (iv), (v) или (vi)
-CH2 CH2NR1R2 (iv)
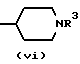
где R1 и R2 независимо выбирают из водорода или C1-4-алкила;
R3 является водородом или C1-4-алкилом,
и их физиологически приемлемые соли или сольваты, при условии, что исключается соединение N, N-диметил-2-[5-(2-оксо-1,3-оксазолидин-4-ил-метил)-1H-индол-3-ил]-этиламин.
где n и W имеют определенные выше значения,
с соединением формулы III
или с его карбонилзащищенной формой,
где L является подходящей удаляемой группой или защищенной аминогруппой, которая может быть превращена in situ в аминогруппу, или является группой - NR1R2, где R1 и R2 имеют определенные выше значения,
и необязательно превращают соединение формулы I, полученное таким образом, в соответствующую физиологически приемлемую соль или сольват.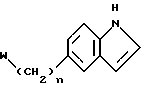
где n и W имеют определенные выше значения,
с соединением формулы XXVIII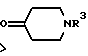
где R3 имеет определенные выше значения,
и необязательно превращают соединение формулы I, полученное таким образом, в соответствующую физиологически приемлемую соль или сольват.
или XXV
где n, R и X имеют определенные выше значения;
Z является группой формулы (vi),
с соединением формулы VII
где Y имеет определенные выше значения;
L и L', которые могут быть одинаковыми или различными, являются подходящими уходящими группами,
и необязательно превращают соединение формулы I, полученное таким образом, в соответствующую физиологически приемлемую соль или сольват.
Приоритет по пунктам:
07.06.90 по пп.1 - 4, 5 - 17, за исключением, когда Y представляет S в формулах (ii) и (iii);
01.02.91 по п.4;
06.02.92 по пп.1 - 4, 5 - 17, когда Y представляет S в формулах (ii) и (iii).
| ЭЛЕКТРОДУГОВОЙ ИСПАРИТЕЛЬ МЕТАЛЛОВ | 1999 |
|
RU2186874C2 |
| EP 0313397 А, 26.04.1989 | |||
| ЦИЛИНДРИЧЕСКАЯ ФРЕЗА ДЛЯ ОБРАБОТКИ ИЗВЕСТНЯКА | 0 |
|
SU237678A1 |
| 0 |
|
SU303506A1 | |
| УСТРОЙСТВО для ОПРЕДЕЛЕНИЯ ПОЛОЖЕНИЯ ПОВЕРХНОСТЕЙ | 0 |
|
SU303507A1 |
| Способ получения 3-/1-имидазолилалкил/индолов или их фармацевтически приемлемых кислотно-аддитивных солей | 1980 |
|
SU1277894A3 |

