Настоящее изобретение относится к усовершенствованному способу получения бутилакрилата. В частности, изобретение относится к новому способу перегонки, рекуперации и возврата в процесс нормального бутанола ("BuOH"), акриловой кислоты ("AK") и нормального бутилакрилата ("БА") из одного или нескольких технологических потоков во время катализируемого кислотой процесса этерификации БА. Настоящее изобретение включает две новых составляющих способа, одна из которых относится к гидролитическому выделению ценных реагентов из их более высококипящих продуктов присоединения, а вторая относится к усовершенствованной перегонке сырого продукта с получением БА, практически свободного от AK. Относящаяся к гидролитическому выделению составляющая изобретения может быть также использована в процессах с получением помимо БА определенных акриловых эфиров. Прежде всего изобретение относится к высокоэффективному способу непрерывного получения БА высокой чистоты с высоким выходом продукта.
Процесс прямой этерификации AK спиртом является равновесным. Константа равновесия определяет суммарную скорость и степень конверсии AK и спирта; для достижения постоянных высоких скоростей конверсии смесь не должна приближаться к равновесному состоянию. Таким образом, для поддержания высокой скорости конверсии AK используют избыток спирта относительно количества AK, а образующуюся при этерификации воду удаляют отгонкой в виде ее азеотропа со спиртом и эфиром. Этот азеотроп удаляют с помощью дистилляционной колонны, смонтированной непосредственно на реакторе для этерификации. В случае метилового или этилового эфира из головки дистилляционной колонны отводят воду этерификации, избыток спирта и полученный эфир и практически полностью освобождают от AK. Водной экстракцией удаляют спирт, который перегонкой концентрируют для возврата в реактор. Промытый эфир азеотропно обезвоживают и в заключение перегоняют с получением в качестве продукта чистого эфира. Однако при получении бутилакрилата отделение акриловой кислоты от реакционной воды, избытка спирта и полученного эфира оказывается более трудным, а дистиллят, получаемый из этерификационного реактора во время этого непрерывного процесса, обычно содержит 1-3% AK. Эту AK, как правило, экстрагируют водной щелочью. Хотя некоторое количество этой AK можно удалить из образовавшегося водного солевого раствора подкислением сильной кислотой с последующей экстракцией органическим растворителем, например, бутилакрилатом или смесью бутилакрилата с бутанолом, значительные потери с большими количествами сточных вод неизбежны. Затем бутилакрилат и избыток бутанола азеотропно обезвоживают, отделяя при этом избыток бутанола от полученного эфира в виде азеотропа бутанол/бутилакрилат, с последующим возвратом в реакцию этерификации. Заключительная перегонка дает чистый бутилакрилат. Во всех случаях из этерификационного реактора отводят небольшой отходящий поток недогона и для удаления из процесса высококипящих побочных продуктов и ингибиторных остатков на стадии заключительной перегонки продукта отбирают небольшой поток кубовых остатков. Эти потоки отпаривают для выделения свободной AK, спирта и ценных алкилакрилатных продуктов, однако при этом рекуперируют лишь незначительное количество или не рекуперируют вовсе ценных материалов, содержащихся в высококипящих побочных продуктах. Так, например, недостаток известных способов получения сложных C1- C4эфиров состоит в потерях выхода продукта с высококипящими побочными продуктами, а способ получения C4-продуктов дополнительно характеризуется таким недостатком, как прямые потери AK из-за затруднений технологического порядка при отделении AK от бутанола, воды и эфира.
На современном уровне техники при рекуперации реагентов из более высококипящих продуктов их присоединения, образующихся в процессе (так называемых "тяжелых фракций"; при получении БА они включают, например, бутил-β-бутоксипропионат и эфиры серной кислоты), и их возврате в процесс осуществляется лишь с ограниченной степенью успеха. Так, например, в относящемся к получению этилакрилата ("ЭА") из этилена и AK патенте США 4968834 (далее '834) описан способ выделения ЭА из потока "отработавшей черной кислоты", содержащего остатки серной кислоты, а также продукты присоединения, отводимые из куба дистилляционной колонны. В способе по патенту '834 предусмотрены использование спиртового растворителя с целью упростить выделение в виде головного дистиллята этилакрилата и стадии обработки остатков черной кислоты водной спиртовой смесью. Ни в реактор для получения ЭА, ни в дистилляционную колонну, из которой получают поток черной кислоты, непосредственно никаких материалов не возвращают. Таким образом, по способу из патента '834 частичная рекуперация этанола, ЭА и АК хотя и обеспечивается, но только путем водной обработки, которая от реактора для проведения этилен- АА-процесса отделена. В других способах частичного выделения свободных АК, БА и BuOH из отводимых потоков реакционной смеси применяют дистилляционные установки (часто называемые "отпарными секциями для отходящих потоков"), но в той степени, в которой тяжелые фракции выделяют при проведении этой операции, они химически остаются в форме более высококипящих продуктов (тяжелой фракции), и их не превращают в такие ценные целевые продукты, как АК, БА и BuOH.
При получении БА обычно используют перегонку. Так, например, в патенте США 4012439 (далее '439) описан способ непрерывного получения БА, в котором реакторную этерификационную смесь перегоняют в колонне для выделения АК с получением в качестве головного погона смеси БА с бутанолом и водой, а из куба колонны отводят поток концентрированной АК, который возвращают в реактор. Во время отделения смеси как головного погона от АК осуществление способа по патенту '439 позволяет очень большую долю (>97%) воднофазового дистиллята возвращать в головку колонны для отделения АК. Для столь большого количества возвращаемой в процесс водной фракции (т.е. коэффициент водного орошения равен приблизительно 32:1) требуется использование большой колонны и большой расход энергии при возврате в процесс больших объемов воды, что является существенным недостатком этого способа.
Таким образом, при катализируемом кислотой получении алкильных эфиров акриловой кислоты ("алкилакрилатов"), в частности БА, сохраняются значительный расход энергии и проблемы с рекуперацией реагентов. Существует необходимость в разработке способа, который позволил бы рекуперировать реагенты из более высококипящих, составляющих тяжелую фракцию продуктов присоединения, образующихся при получении акриловых эфиров, например, БА, и который позволял бы возвращать выделенные реагенты и сложный эфир в этерификационный реактор или на какую-либо другую стадию процесса для повторного использования. Существует, кроме того, необходимость в создании способов, которые обеспечили бы более эффективное использование воды реакции как для упрощения дистилляционного отделения акрилового эфира от АК, так и для более эффективного выделения и возврата в процесс непрореагировавшей АК, прежде всего если эти стадии осуществляют с уменьшенными энергетическими затратами. Решение одной или нескольких этих задач обеспечило бы повышение эффективности процесса и/или использования материала. Более того, если бы такие усовершенствованные способы привели бы к уменьшенному количеству образующегося в качестве побочного продукта дибутилового эфира (ДБЭ) в сравнении с обычным для известных способов количеством, результатом явилась бы еще большая эффективность процесса.
В соответствии с настоящим изобретением был разработан способ получения алкилакрилатов с достижением высокого выхода продукта, например, предпочтительно БА, который позволяет решить эти поставленные задачи. Согласно новому способу по изобретению обеспечивается возможность рекуперировать "ценные продукты", т. е. реагенты и алкилакрилатный продукт, из тяжелых фракций, получаемых в процессе. В новом способе по изобретению предусмотрено использование по меньшей мере одной из нижеследующих составляющих: 1. рекуперация ценных продуктов из гидролизной реакторной установки ("ГРУ"), питаемой от источника тяжелых фракций, в частности от этерификационного реактора; 2. рекуперация дополнительных количеств ценных продуктов из крекинг-реактора, предпочтительно применяемого в сочетании с гидролизным реактором, и 3. (непосредственно для процесса непрерывного получения БА) перегонка с использованием колонны для выделения акриловой кислоты эффективным, новым путем и обеспечение возможности выделения БА, который практически не содержит АК. Преимущество нового способа по изобретению состоит в возможности обеспечить очень низкое содержание ДБЭ в получаемом БА, поскольку этерификационный реактор работает в мягких условиях температуры и давления и при относительно низкой концентрации кислотного катализатора.
Таким образом, в соответствии с наиболее широким применением составляющей изобретения, относящейся к гидролитическому выделению, предлагается способ выделения АК, C1-C4алкилакрилата и C1-C4спирта из тяжелых фракций, образующихся при получении C1-C4 алкилакрилата, включающий стадии:
а) подачу общего сырьевого потока водной и тяжелых фракций, включающего тяжелые фракции, воду, остаточный кислотный катализатор и необязательно сильную кислоту, выбранную из минеральных кислот или сульфокислот, в гидролизный реактор, в котором поддерживают температуру 90-140oC и давление 50-1000 мм рт.ст., а продолжительность пребывания составляет 0,5-20 ч из расчета на общий поток водного и органического сырья;
б) отгонку потока головного погона, содержащего АК, C1-C4алкилакрилат, C1-C4спирт и воду, из гидролизного реактора при одновременном поддержании в жидкости гидролизного реактора концентрации 5-40 вес.% воды и по меньшей мере 1 вес.% кислоты, включающей остаточный кислотный катализатор и необязательно сильную кислоту;
в) конденсацию потока головного погона;
г) выделение из сконденсированного потока головного погона органической фазы, включающей C1-C4алкилакрилат, C1-C4спирт и АК, и водной фазы, включающей прежде всего воду, АК и C1-C4спирт;
д) удаление выделенной органической фазы;
е) возврат выделенной водной фазы в гидролизный реактор и
ж) отвод из гидролизного реактора 20-70 вес.% (в пересчете на общий сырьевой поток водной и тяжелых фракций) отходящего потока продуктов.
Непосредственно для случая получения БА предлагается способ выделения АК, н-бутилакрилата (БА) и н-бутанола (BuOH) из тяжелых фракций, образующихся во время катализируемой кислотой этерификации АК с использованием BuOH, включающий стадии:
а) подачу общего сырьевого потока водной и тяжелых фракций, включающего АК, БА, BuOH, воду, тяжелые фракции, остаточный кислотный катализатор и необязательно сильную кислоту, выбранную из минеральных кислот или сульфокислот, в гидролизный реактор, в котором поддерживают температуру 90-140oC и давление 50-1000 мм рт.ст., а продолжительность пребывания составляет 0,5-20,0 ч из расчета на общий сырьевой поток водной и тяжелых фракций;
б) отгонку потока головного погона, содержащего АК, БА, BuOH и воду из гидролизного реактора при одновременном поддержании в жидкости гидролизного реактора концентрации 5-40 вес.% воды и по меньшей мере 25 вес.% кислоты, включающей остаточный кислотный катализатор и необязательно сильную кислоту;
в) конденсацию потока головного погона;
г) выделение из сконденсированного потока головного погона органической фазы, включающей БА, BuOH и АК, и водной фазы, включающей прежде всего воду, АК и BuOH;
д) удаление выделенной органической фазы;
е) возврат выделенной водной фазы в гидролизный реактор и
ж) отвод из гидролизного реактора 20-70 вес.% (в пересчете на общий сырьевой поток водной и тяжелых фракций) отходящего потока продуктов.
По другому варианту выполнения изобретения предлагается способ непрерывного выделения АК, н-бутилакрилата (БА) и н-бутанола (BuOH) из тяжелых фракций, образующихся во время катализируемой кислотой этерификации АК с использованием BuOH, включающий стадии:
а) непрерывный отвод реакторного потока продуктов, отходящего из этерификационного реактора, содержащего этерификационную реакционную смесь, включающую АК, БА, BuOH, воду, тяжелые фракции и остаточный кислотный катализатор, при одновременной отгонке из этой этерификационной реакционной смеси АК, БА, BuOH и воды;
б) подачу общего потока водного и органического сырья, включающего отходящий из реактора поток материалов, воду, необязательно сильную кислоту, выбранную из минеральных кислот или сульфокислот, и необязательно дополнительные тяжелые фракции, в гидролизный реактор, в котором поддерживают температуру 90-140oC и давление 50-1000 мм рт.ст., а продолжительность пребывания составляет 0,5-20 ч из расчета на общий поток водного и органического сырья;
в) отгонку потока головного погона, содержащего АК, БА, BuOH и воду, из гидролизного реактора при одновременном поддержании в жидкости гидролизного реактора концентрации 5-40 вес. % воды и по меньшей мере 1 вес.% кислоты, включающей остаточный кислотный катализатор и необязательную сильную кислоту;
г) конденсацию потока головного погона;
д) выделение из сконденсированного потока головного погона органической фазы, включающей БА, BuOH и АК, и водную фазу, содержащую прежде всего воду, АК и BuOH;
е) удаление выделенной органической фазы;
ж) возврат выделенной водной фазы в гидролизный реактор и
з) отвод из гидролизного реактора 20-70 вес.% (в пересчете на общий поток водного и органического сырья) отходящего потока материалов.
Рекуперация дополнительных количеств ценных реагентов из тяжелых фракций достигается за счет применения крекинг-реактора в последовательном сочетании со способами гидролитического выделения, описанными выше. Такой способ осуществляют с использованием любого из вышеописанных способов гидролитического выделения, включая дополнительные стадии:
а) подачу до 100% отходящего из гидролизного реактора потока материалов в крекинг-реактор, в котором поддерживают температуру 90-140oC и давление 20-200 мм рт.ст., а продолжительность пребывания составляет 0,5-20 ч из расчета на подаваемый поток материалов, отходящий из реактора;
б) отгонку из крекинг-реактора потока головного погона, содержащего АК, С1-С4алкилакрилат, С1-С4спирт и воду, при одновременном поддержании в жидкости крекинг-реактора концентрации кислоты по меньшей мере 7,5 вес.%;
в) конденсацию отгоняемого из крекинг-реактора потока головного погона и
г) выделение из отгоняемого из крекинг-реактора потока головного погона АК, C1-C4алкилакрилата, C1-C4спирта и воды.
Предпочтительным алкилакрилатом является БА. Более предпочтительно описанный крекинг-реактор использовать в последовательном сочетании с гидролитическим реактором в процессе непрерывного катализируемого кислотой получения БА.
Касательно непрерывного получения БА в соответствии со второй составляющей настоящего изобретения предлагается способ непрерывного выделения н-бутилакрилата (БА), практически свободного от АК, из этерификационной реакционной смеси, включающий стадии:
а) непрерывную подачу в этерификационный реактор АК и BuOH в молярном соотношении 1:1,1-1:1,7 и кислотного катализатора;
б) взаимодействие АК и BuOH с получением БА при конверсии по меньшей мере 60% в пересчете на АК и образованием этерификационной реакционной смеси, включающей АК, БА, BuOH, воду, тяжелые фракции и кислотный катализатор;
в) отгонку из этерификационного реактора испаренной смеси, содержащей АК, БА, BuOH и воду;
г) конденсацию испаренной смеси с получением первого конденсата, содержащего органическую фазу и водную фазу;
д) возврат 0-30% органической фазы в каплеотбойник, смонтированный на этерификационном реакторе;
е) подачу 70-100% органической фазы и 50-100% водной фазы в колонну для выделения акриловой кислоты;
ж) отгонку из колонны для выделения акриловой кислоты под давлением 35-800 мм рт. ст. в водном режиме и при коэффициенте водного орошения 8,5: 1-17: 1 смеси головного погона, представляющей собой азеотропную смесь бутанола, бутилакрилата и воды;
з) удаление из дистилляционной колонны потока богатых акриловой кислотой кубовых остатков;
и) возврат потока богатых акриловой кислотой кубовых остатков, отводимого из колонны для выделения акриловой кислоты, в этерификационный реактор;
к) конденсацию смеси головного погона с получением второго конденсата;
л) разделение второго конденсата на богатую бутилакрилатом органическую фазу и выделенную водную фазу и
м) выделение богатой бутилакрилатом органической фазы, практически свободной от АК.
Стадию выделения БА, практически не содержащего АК, можно также проводить путем подачи испаренной из реактора смеси прямо в колонну для выделения АК, минуя предшествующие стадии г), д) и е). Когда такую испаренную смесь направляют непосредственно в колонну, диапазон коэффициентов орошения водой сужают до 13: 1-17:1; все остальные стадии идентичны, за исключением того, что при этом, разумеется, отсутствует "первый конденсат".
В соответствии с кратким пояснением к чертежам способ, включающий обе составляющих настоящего изобретения, схематически показан на фиг. 1. На фиг. 2 представлен график зависимости количества остаточной АК в органическом дистилляте, полученном перегонкой в колонне для выделения кислоты, от расхода потока воды на орошение, что достигается в условиях, описанных ниже. На фиг. 1 показаны оборудование и технологические линии, включая этерификационный реактор 1, линию 3 для отходящего потока, направляемого в гидролизную реакторную установку (ГРУ) 5, а также связанные с ними технологические потоки и линии, в частности линия 8 возврата органической фазы в реактор 1 и линия 7, возвращающая водную фазу в ГРУ. Крекинг-реактор 10 также снабжен соответствующими линиями, например, дренажной и дистиллятной, обеспечивающими возврат сконденсированного потока головного погона, отводимого из сепаратора 31, в реактор 1 по линии 12. Касательно краткого пояснения к чертежам, относящегося к дистилляционной составляющей, на фиг. 1 показаны линия 2, подающая испаренную из этерификационного реактора смесь (в данном варианте) в конденсатор 62 и конденсат в фазовый сепаратор 14, а также соответствующие линии от фазового сепаратора к колонне 15 для выделения акриловой кислоты (одна или несколько линий 43, 53), для необязательной подачи некоторого количества водной фазы в ГРУ 5 (линия 42) и необязательной подачи по линии 41 доли органической фазы в каплеотбойник, смонтированный на реакторе 1, когда применяют сепаратор 14. Для необязательной подачи BuOH в колонну для выделения АК предусмотрена линия 54. Линии от колонны для выделения акриловой кислоты включают линию 17, возвращающую поток кубовых остатков, богатых АК, в реактор 1, и линию 16, подающую отогнанную смесь головного погона через конденсатор 63 в фазовый сепаратор 18, а также связанные с этим линии, а именно, линию 21 для возврата регулируемой доли водной фазы 20 в верхнюю часть колонны 15 для выделения акриловой кислоты, линию 22 для поступательного перемещения регулируемой доли выделенной водной фазы и линию 23, поступательно транспортирующую всю богатую БА органическую фазу 19. Линия 52 обеспечивает возврат БА/BuOH в реактор 1 во время последующих обычных обработки и выделения готового БА-продукта. Фиг. 1 и 2 более подробно описаны ниже.
Подробное описание изобретения
В соответствии с первой составляющей изобретения, относящейся к гидролитической рекуперации, которая позволяет рекуперировать из тяжелых фракций ценные материалы, предусмотрено использование преимущества известной возможности сильной кислоты, например, минеральной кислоты, такой, как серная кислота, катализировать индивидуальные используемые реакции прямой этерификации, сложноэфирного гидролиза, дегидратации и ретро-реакции Михаэля. Таким образом, каталитические способы, при которых в гидролизном реакторе происходит гидролиз сложных эфиров и тяжелых фракций, а в более широком смысле в крекинг-реакторе происходят дегидратация и ретро- присоединения Михаэля, являются новыми эффективными способами рекуперации, например, ценных БА, BuOH и АК, из компонентов тяжелых фракций, образующихся при уже известной реакции, в частности в этерификационном реакторе для получения БА. Такие тяжелые фракции подробно проиллюстрированы на примерах для случая получения БА с использованием серной кислоты в качестве катализатора в реакторе и ГРУ; из описания этих примеров для любого специалиста в данной области техники очевидны аналогичные "тяжелые фракции" -двойники, образующиеся при получении любых C1-C4алкилакрилатов. C1-C4алкильными группами могут являться метил, этил, пропил и изопропил, а также бутильные изомеры, предпочтительно н-бутил. Тяжелые фракции образуют продукты присоединения с более высокой температурой кипения, чем исходные реагенты, и применительно к используемому в данном описании в качестве примера случаю бутилакрилатного продукта они включают, в частности, акрилоксипропионовую кислоту ("АОПК") и ее бутилэфирное производное, β - гидроксипропионовую кислоту и ее бутилэфирное производное, β - бутоксипропионовую кислоту и ее бутилэфирное производное и другие неполимерные продукты присоединения исходных реагентов. Кроме того, в акриловой кислоте и сернокислотном катализаторе содержатся малеиновокислотные и бензойнокислотные примеси в виде монобутилового эфира малеиновой кислоты, бутилбензоата и монобутилсульфата. Более того, при проведении непрерывного процесса одновременное выделение БА, BuOH и АК путем перегонки потоков, отводимых как из гидролизного, так и крекинг-реактора, позволяет реакциям рекуперации протекать за пределами равновесных ограничений, свойственных периодическому процессу, и таким образом повышать выход продуктов процесса. Другое преимущество гидролитической рекуперации как составляющей настоящего изобретения состоит в том, что в технологическом потоке гидролитической рекуперации можно обрабатывать один или несколько дополнительных потоков тяжелых фракций, что позволяет, следовательно, выделять дополнительное количество ценных продуктов.
Ниже приведены примеры продуктов тяжелых фракций, содержащихся в общем сырьевом потоке водных и органических материалов (индивидуальные компоненты тяжелых фракций, смеси компонентов тяжелых фракций, реагенты и продукт), которые гидролизуют в гидролизном реакторе для рекуперации таких ценных продуктов, как АК и описанные выше алкилакрилаты и спирты. Сложные алкильные эфиры β -алкоксипропионатов являются обычными продуктами тяжелых фракций. Вместо алкилоксигруппы в β - положении алкильных эфиров может также содержаться гидроксильная группа. В состав тяжелых фракций могут также входить β - акрилоксикислотные производные C1-C4алкильных эфиров. Так, например, в продуктах тяжелых фракций в случае получения БА обычно содержится бутил ( β - акрилокси)пропионат вместе с его соответствующей кислотой. Содержатся также C1-C4эфиры сернокислотного катализатора, которые гидролизуют до серной кислоты и соответствующих C1-C4спиртов.
Реакции, которые протекают в ГРУ, в общем виде могут быть представлены следующими уравнениями 1 и 2: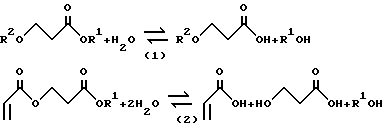
где R1 обозначает C1-C4алкильную группу, которая описана выше; R2 обозначает C1-C4алкильную группу или Н. Аналогичным образом дополнительно могут быть гидролизованы насыщенные и ненасыщенные эфиры, такие как C1-C4алкиловые эфиры бензойной кислоты и C1-C4алкиловые эфиры малеиновой кислоты, а также C1-C4алкилсульфат с получением эквивалентов C1-C4спирта. Более того, одновременное выделение БА, BuOH и АК путем перегонки потока, отводимого из ГРУ, позволяет рекуперационным реакциям протекать за пределами равновесных ограничений и повышать общий выход продуктов процесса. Однако в ГРУ исходную карбоновую кислоту, т.е. источник нескольких тяжелых продуктов, выделить невозможно, поэтому для этих продуктов необходима дополнительная схема рекуперации, которую осуществляют в крекинг-реакторе.
Реакции, которые протекают в крекинг-реакторе, в общем виде могут быть представлены следующими уравнениями 3 и 4, где значения R2 указаны выше: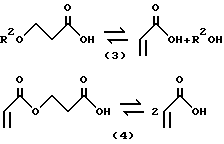
Конверсия C1-C4алкилового эфира β - гидроксипропионовой кислоты, β - алкоксипропионовой кислоты и β - акрилоксипропионовой кислоты в исходную кислоту в ГРУ (путем гидролиза сложных эфиров) оказывается достаточно эффективной, так как хорошо известно, что эти материалы подвергаются дегидратации и ретро-присоединению Михаэля в кислотной форме. Таким образом, в относительно сухих условиях в крекинг-реакторе соединения, такие как β-н-бутоксипропионовая кислота и β - гидроксипропионовая кислота могут подвергаться дегидратации с выделением в результате акриловой кислоты и бутанола. Хорошо известно, что димер акриловой кислоты (АОПК) подвергается крекингу с образованием 2 молей акриловой кислоты. В этом случае непрерывное выделение продуктов также позволяет реакции протекать за пределами равновесных ограничений и увеличивать общий выход продуктов процесса.
Как показано на фиг. 1, в соответствии с предпочтительным вариантом осуществления непрерывной гидролитической рекуперационной составляющей настоящего изобретения применительно к получению БА поток материалов 3, отходящий из этерификационного реактора 1, направляют в гидролизную реакторную установку ("ГРУ") 5. Способ гидролитической рекуперации по любому варианту выполнения изобретения можно осуществлять в многоходовой тарельчатой реакционно-дистилляционной колонне или другом многоступенчатом реакторе, причем его предпочтительно осуществляют в условиях непрерывного перемешивания, в частности в проточном реакторе с мешалкой ("ПРМ"). Под "отходящим потоком материалов" понимается любой технологический поток, который непрерывно отводят из одного сосуда и направляют в другой, в частности из одного реактора в другой реактор или дистилляционную колонну. В данном случае отходящий из этерификационного реактора поток 3 материалов содержит кислотный катализатор, воду, АК, БА, BuOH и тяжелые фракции; могут содержаться также ингибиторы полимеризации. Подаваемое по линии 4 дополнительное сырье может включать воду и может содержать также минеральную кислоту, например, серную кислоту, или сульфокислоту, такую, как метан-, бензол- или толуолсульфокислота. Эту минеральную или сульфоновую кислоту добавляют в соответствии с заданной минимально необходимой, рабочей концентрацией в ГРУ. Можно также вводить дополнительно один или несколько потоков материалов, содержащих тяжелые фракции, из источников, отличных от этерификационного реактора. Это сырье можно вводить по одной или нескольким питающим линиям, обозначенным позицией 4. Такие дополнительные тяжелые фракции могут в общей сложности составлять до 80 вес.% всех водных и органических исходных материалов. Серная кислота наиболее предпочтительна для использования в качестве как реакторного кислотного катализатора, так и минеральной кислоты во всех вариантах выполнения изобретения. В ГРУ смесь описанных потоков сырья в определенных условиях поддерживают в кипящем состоянии. Продолжительность пребывания 0,5-20 ч берется из расчета на общий поток водного и органического сырья (термин "общий" означает сумму водных и тяжелых фракций и/или отходящих из реактора потоков), направляемый в ГРУ. Предпочтительная продолжительность пребывания составляет 0,5-5 ч, более предпочтительно равна 0,5-3 ч. Из смеси в ГРУ отгоняют поток головного погона и по линии 6 после конденсации в конденсаторе 60 направляют в фазовый сепаратор 30. Этот сконденсированный поток головного погона разделяют на органическую фазу, богатую БА, BuOH и АК, и водную фазу, содержащую главным образом (т.е. > 50%) воду и некоторое количество BuOH и АК. Выделенную водную фазу возвращают в гидролизный реактор 5 по линии 7, а выделенную органическую фазу в виде потока 8 в данном варианте возвращают в этерификационный реактор 1, рекуперируя таким образом ценные БА, BuOH и АК для последующих реакции и выделения продукта. Эту выделенную органическую фазу можно также направлять в сепаратор 14 для удаления по линии 43 и перегонки в колонне 15. Неперегнанный остаток, т.е. 20-70 вес.%, общего потока водного и органического сырья отводят в виде отходящего из гидролизного реактора потока 9 продуктов и направляют для дальнейшей обработки (например, в качестве потока отходов по линии 51 или предпочтительно по линии 9 в качестве сырья в крекинг-реактор).
В предпочтительном варианте выполнения обеспечивается рекуперация дополнительных количеств АК, БА и BuOH. Как показано на фиг. 1, отходящий из гидролизного реактора поток 9 продуктов направляют в крекинг-реактор 10 и обрабатывают согласно описанному ниже. Конструкция такого крекинг-реактора может быть аналогичной конструкции ГРУ, а в предпочтительном варианте он представляет собой ПРМ. Концентрацию минеральной кислоты, предпочтительно серной, в жидкости крекинг-реактора поддерживают на уровне по меньшей мере 7,5 вес.%, и в нем содержатся также смесь акриловой кислоты, BuOH, БА, некоторое количество тяжелых фракций и остаточные ингибиторы полимеризации. В жидкость крекинг-реактора можно добавлять дополнительное количество минеральной или сульфоновой кислоты (по линии, которая не показана). Смесь в крекинг-реакторе поддерживают в кипящем состоянии в описанных выше условиях крекинга, одновременно с этим отгоняя из крекинг-реактора поток головного погона и направляя его по линии 11 с конденсацией в конденсаторе 61 в сепаратор 31. Конденсат включает поток органического дистиллята, содержащий АК, БА и BuOH, а также некоторое количество воды; весь этот сконденсированный поток головного погона возвращают в виде потока 12 в этерификационный реактор, обеспечивая тем самым рекуперацию дополнительных количеств ценных АК, БА и BuOH. Поток 13 крекинг- остатков сливают из крекинг-реактора для дальнейшей обработки, обычно в виде отходов. Предпочтительная и более предпочтительная продолжительность пребывания в крекинг-реакторе та же, что и указанная для ГРУ, а именно, соответственно составляет 0,5-5 ч и 0,5-3 ч.
ГРУ может представлять собой многоходовую тарельчатую реакционно-дистилляционную колонну, единственным условием при этом является достаточное число тарелок, обеспечивающее заданную продолжительность пребывания. Когда в качестве ГРУ применяют реакционно- дистилляционную колонну, для достижения приемлемой рекуперации ценных продуктов отдельная крекинг-реакторная установка может не понадобиться. В большинстве рабочих условий крекинг-реактор предпочтительно применять в последовательном сочетании с гидролитической реакционно-дистилляционной колонной, аналогично его применению в случае, когда ГРУ представляет собой ПРМ. Одним из недостатков реакционно-дистилляционной колонны по сравнению с ПРМ является периодическое накопление твердых продуктов на тарелках колонны, который может обусловить нежелательный простой колонны при ее очистке.
Добавление одного или нескольких дополнительных потоков сырья в отходящий из этерификационного реактора поток или непосредственно в гидролизный реактор позволяет выделить дополнительные количества АК и, например, БА и BuOH, благодаря процессам, протекающим в гидролизном реакторе, и в крекинг-реакторе, когда его применяют. Для эффективности процесса жидкость в гидролизном реакторе включает по меньшей мере 5 вес.% воды, в предпочтительном варианте жидкость ГРУ содержит 9-18 вес.%, более предпочтительно 10-16 вес. % воды, что позволяет добиться эффективных скоростей гидролиза в номинальных условиях температуры и давления и компактности оборудования. Содержание воды поддерживают сочетанием возврата всего сконденсированного и выделенного водного потока по линии 7 в гидролизный реактор и добавлением дополнительных количеств воды из других источников, например, по линиям 4 и 42, для компенсации потерь воды при перегонке органических продуктов и в отходящем из ГРУ потоке продуктов. Предпочтительным источником воды в процессе непрерывного получения БА является вода, добавляемая из отогнанной из этерификационного реактора водной фазы по линии 42. С целью поддержать эффективные скорости дегидратации и ретро-реакции Михаэля в крекинг-реакторе содержание воды в крекинг-реакционной смеси должно быть более низким, чем в ГРУ-смеси. Обычно содержание воды менее 5 вес.%, предпочтительно менее 1 вес. %, достигается путем проведения процесса в крекинг-реакторе как в одностадийной установке, т.е. путем непрерывной отгонки из крекинг-реактора всей воды, поступившей с потоком материалов, отходящим из гидролизного реактора, и всей дополнительной воды, которая образуется во время реакций крекинга.
Дополнительные количества кислоты можно добавлять в рекуперационные установки по мере необходимости для достижения практических скоростей реакций; кислоту предпочтительно добавляют в виде одного или нескольких потоков сырья. "Остаточный кислотный катализатор" представляет собой кислотный катализатор, который остается в виде кислоты в потоке продуктов, отходящем из этерификационного реактора, и таким образом перемещается дальше, попадая в ГРУ. Предпочтительная концентрация кислоты в ГРУ составляет 3,5-15 вес.%, наиболее предпочтительно 5-8 вес.%. Концентрация кислоты в крекинг-реакторе обычно составляет 7,5-20 вес.%, но может быть и выше этого значения, например, составлять до 50%. Предпочтительная концентрация кислоты составляет 10-13 вес.%, в частности при получении БА. Количество тяжелых фракций в потоке продуктов, отходящем из этерификационного реактора, может варьироваться, но, как правило, лежит в пределах 10-50 вес.% от общего количества водного и органического сырья, содержащегося в потоке.
Для достижения эффективных скоростей гидролиза температура во время реакции гидролиза должна находиться в пределах 90-140oC, предпочтительно 105-125oC; температуры свыше 140oC могут привести к термически инициированной полимеризации алкилакрилатов и тяжелых фракций, компоненты которых содержат акрилоксигруппы, обусловливая потерю продукта. Предпочтительная продолжительность пребывания в ГРУ, необходимая для реакции гидролиза, составляет 0,5-5 ч, более предпочтительно 0,5-3 ч, причем более короткая продолжительность экономически выгоднее. Более низкая температура и присутствие воды способствуют также уменьшению количества образующегося ДБЭ. Температура в крекинг-реакторе составляет 90- 140oC, предпочтительно 110-125oC, а давление при крекинге, как правило, поддерживают в пределах 20-200 мм рт.ст., хотя можно создавать и более высокое давление, достигающее 800 мм рт.ст. В этих условиях предпочтительная продолжительность пребывания в крекинг-реакторе для дегидратации и других реакций составляет 0,5- 3 ч. В случае непрерывного получения БА рекуперацию ценных продуктов доводят до максимума с использованием двух последовательно установленных ПРМ, один из которых ГРУ, а другой крекинг-реактор.
Чтобы предотвратить полимеризацию на любой стадии при любой составляющей способа, можно добавлять эффективное количество одного или нескольких ингибиторов полимеризации. Технологический поток из этерификационного реактора, как правило, содержит достаточное количество ингибитора для предотвращения полимеризации в ГРУ и крекинг-реакторе. В случае необходимости добавления дополнительного количества ингибитора можно использовать любой из большого количества известных ингибиторов, например, гидрохинон, монометиловый эфир гидрохинона, бутилированный гидроксианизол, нафтахинон, антранил и их производные.
Вторая составляющая настоящего изобретения, а именно, дистилляционная составляющая, позволяет в еще большей степени усовершенствовать известные способы перегонки сырого БА и обеспечивает получение БА, практически свободного от АК, путем более эффективных обработки дистиллята и водного орошения. В частности новый способ перегонки обеспечивает получение БА в богатом БА потоке, содержащем менее 2000 част./млн АК, который далее поступает на последующую обычную стадию выделения. По такому способу обеспечивается также получение рециркуляционного потока с АК, содержащего ничтожные количества БА, и в частности рециркуляционного потока с АК (кубовый остаток, богатая АК фаза), содержащего менее 10 част./млн, предпочтительно менее 5 част./млн БА. При получении сырого БА для реализации новой, дистилляционной составляющей изобретения АК и BuOH вначале направляют по линии 70 вместе с кислотным катализатором в этерификационный реактор при молярном соотношении между АК и BuOH в интервале 1: 1,1-1: 1,7, предпочтительно 1:1,25-1:1,45, и взаимодействие проводят до конверсии в 60-95%, предпочтительно 75- 85% в пересчете на АК, используя вышеописанный кислотный катализатор типа минеральной или сульфоновой кислоты или сильнокислотную ионообменную смолу; предпочтительно использовать серную кислоту. Соотношение между реагентами и конверсия в БА обеспечивают создание потока сырого БА, который можно обрабатывать путем проведения в колонне для выделения акриловой кислоты стабильного процесса в "водном режиме" (подробно описанном ниже). Во время непрерывной перегонки испаренной смеси АК, БА, BuOH и воды содержимое реактора поддерживают в кипящем состоянии.
Как показано на фиг. 1, испаренную смесь по линии 2 из реактора 1 конденсируют в конденсаторе 62 и направляют в фазовый сепаратор 14 (в данном варианте) с получением первого конденсата. Альтернативно этому испаренную смесь можно направлять непосредственно в колонну 15 для дистилляции согласно описанному выше. С целью уменьшить или устранить захват кислотного катализатора испаренной смесью на реакторе можно смонтировать также каплеотбойник, который не показан, уменьшая таким образом дальше по ходу движения материала возможность коррозии. Фазовый сепаратор 14 наиболее целесообразно использовать в случае применения каплеотбойника, поскольку при этом обеспечивается возврат органического орошающего слоя в каплеотбойник, а также в качестве устройства создания необязательного водного потока 42 для ГРУ. Первый конденсат включает органическую фазу, состоящую главным образом (т.е. свыше 50%) из БА и BuOH и некоторого количества АК, и водную фазу, состоящую главным образом из воды и некоторого количества BuOH и АК. Обе эти фазы можно целиком направлять в колонну 15 для выделения акриловой кислоты по одной или нескольким линиям, например, по линиям 43, 53, или необязательно до 50 вес.% водной фазы можно отводить по линии 42 в гидролитическую рекуперационную установку 5 (когда ее использование предпочтительно). В эту колонну по линии 54 необязательно можно подавать дополнительное количество бутанола. Азеотропную смесь как головной погон отгоняют из колонны для выделения акриловой кислоты в вышеуказанных условиях давления, температуры и водного орошения и конденсируют подачей по линии 16 в конденсатор 63, после чего направляют в фазовый сепаратор 18, получая второй конденсат, включающий богатую БА органическую фазу 19 и водную фазу 20. Под "богатой БА" или "богатой АК" подразумевают то, что основным органическим компонентом (>50 вес.%) данной фазы является БА или АК. Параллельно из нижней части колонны для выделения акриловой кислоты отводят богатый АК поток кубовых остатков, содержащий ничтожное количества БА, и по линии 17 возвращают в этерификационный реактор 1. Количество возвращаемого водного потока 21 регулируют с тем, чтобы обеспечить в колонне 15 для выделения АК минимальный коэффициент водного орошения по меньшей мере 8,5:1 с целью поддержать работу колонны в имеющем решающее значение "водном режиме". При работе в водном режиме в колонне для выделения АК обеспечивается неожиданно эффективное выделение АК из содержащего БА исходного потока (т. е. потока первого конденсата или потока испаренной смеси, вводимого в колонну), обусловливающее небольшие потери АК в БА-дистилляте и, следовательно, повышенный выход БА, как более подробно описано ниже. Небольшую долю в 6-11 вес.% выделенной водной фазы 20, как правило, направляют далее в виде потока 22 совместно с поступательно подаваемой богатой БА органической фазой 19 в потоке 23 для последующего обычного выделения конечного БА-продукта. Как правило, коэффициент водного орошения определяют как соотношение между расходом потока возвращаемой воды и расходом потока воды, движущейся поступательно, в данном случае соотношение между расходом воды в потоках 21 и 22. Поддержание заданного соотношения имеет решающее значение для эффективной работы колонны для выделения акриловой кислоты по изобретению.
Колонна для выделения акриловой кислоты может включать 20-50, предпочтительно 30-40 тарелок и, как правило, оснащена контуром кубового кипятильника (не показан) и линией 16 подачи головного дистиллята через конденсатор 63 в фазовый сепаратор 18. Исходный первый конденсат, как правило, подают примерно на 10-ю тарелку 40-тарельчатой колонны, если исходить из нумерации тарелок от нижней части колонны. Если используют необязательную добавку BuOH, его обычно вводят на 8-ю или 9-ю тарелку. Эта колонна работает в вышеуказанных пределах, предпочтительно под давлением 90-135 мм рт.ст., что соответствует предпочтительной температуре в кубе 80-85oC. Во время отгонки смеси в виде головного погона предпочтительный коэффициент водного орошения составляет 8,5-12,5, наиболее предпочтительно 9,5-10,5. Расход потока, отводимого из нижней части колонны по линии 17, регулируют таким образом, чтобы он превышал количество АК в подаваемом в колонну сырье на 5-25 вес.%, так как это обеспечивает то, что вся АК остается в кубе колонны. Поток 17 обычно содержит 5-20 вес.% воды, а баланс составляет прежде всего АК и АОПК. В колонне для выделения акриловой кислоты, работающей аналогично описанному выше, получают БА, практически свободный от АК (< 2000 част./млн), и поток кубовых остатков с АК, содержащий ничтожное количество БА (<10 част./млн).
При моделировании и последующей демонстрации работы колонны для выделения акриловой кислоты неожиданным оказалось существование двух стационарных режимов в одних и тех же рабочих условиях (т.е. при тех же самых расходе сырья, составе сырья, расходе потока воды на орошение и расходе отводимых кубовых остатков). Один из стационарных режимов, выше названный "водным режимом", имеет решающее значение для достижения очень низкого содержания АК в богатой БА фазе и БА в богатом АК потоке кубовых остатков, как указано выше. В водном режиме колонна для выделения акриловой кислоты работает в относительно "холодном" состоянии, на всех тарелках содержатся существенные количества воды, вода содержится в потоке кубовых остатков и в этом потоке кубовых остатков содержится ничтожное количество БА. Однако, как неожиданный контраст, в тех же самых условиях (т.е. при тех же расходе сырья, составе сырья, расходе потока воды на орошение и расходе потока кубовых остатков) существует второй режим - "органический режим", - который нежелателен. При работе колонны для выделения акриловой кислоты в этом органическом режиме она приблизительно на 30-35oC горячее, чем в водном режиме, а в потоке кубовых остатков содержатся, как установлено, значительные количества (>10 вес. %) БА и концентрация АК в смеси БА головного погона по меньшей мере на порядок выше максимальной концентрации АК (2000 част./млн), достигаемой при проведении процесса в водном режиме. Кроме того, в нежелательном органическом режиме колонна не только горячее, чем в водном режиме, но вся вода концентрируется на нескольких верхних тарелках, а поток кубовых остатков оказывается практически сухим. Примеры 1-6 и описанные ниже модельные исследования предоставляют дополнительные подробности об этих неожиданно обнаруженных режимах и об основной скрытой причине такой работы колонны для выделения акриловой кислоты.
И, наконец, предлагается наиболее предпочтительный способ осуществления непрерывного процесса, в котором использовано сочетание всех составляющих настоящего изобретения, при получении БА, практически не содержащего акриловой кислоты (АК), и при рекуперации АК, БА, н-бутанола (BuOH) и воды из этерификационной реакторной смеси, содержащей АК, БА, BuOH, воду, тяжелые фракции и кислотный катализатор, который включает следующие стадии:
а) подачу в этерификационный реактор АК и BuOH в молярном соотношении от 1:1,1 до 1:1,7 и кислотного катализатора;
б) взаимодействие АК с BuOH с получением БА при конверсии по меньшей мере 60% в пересчете на АК и с образованием этерификационной реакционной смеси, содержащей АК, БА, BuOH, воду, тяжелые фракции и кислотный катализатор;
в) отвод потока реакционной смеси, отходящей из этерификационного реактора для непрерывной конверсии, с одновременной параллельной отгонкой из этерификационной реакционной смеси АК, БА, BuOH и воды;
г) подачу общего потока водного и органического сырья, включающего отходящий из реактора поток, воду, необязательно сильную кислоту, выбранную из минеральных кислот или сульфокислот, и необязательно дополнительные тяжелые фракции, в гидролизный реактор, в котором поддерживают температуру 90-140oC и давление 50-1000 мм рт. ст., а продолжительность пребывания составляет 0,5-20 ч из расчета на общий поток водного и органического сырья;
д) отгонку потока головного погона, содержащего АК, БА, BuOH и воду, из гидролизного реактора при одновременном поддержании в жидкости гидролизного реактора концентрации 5-40 вес. % воды и по меньшей мере 1 вес.% кислоты, включающей кислотный катализатор и необязательно сильную кислоту;
е) конденсацию потока головного погона;
ж) выделение из сконденсированного потока головного погона органической фазы, содержащей БА, BuOH и АК, и водной фазы, содержащей прежде всего воду, АК и BuOH;
з) подачу выделенной органической фазы в этерификационный реактор;
и) подачу выделенной водной фазы в гидролизный реактор;
к) отвод из гидролизного реактора 20-70 вес.% (в пересчете на общий поток водного и органического сырья) отходящего потока продуктов;
л) подачу до 100% отходящего из гидролизного реактора потока продуктов в крекинг-реактор, в котором поддерживают температуру 90-140oC и давление 20-200 мм рт.ст., а продолжительность пребывания составляет 0,5-20 ч из расчета на подаваемый поток материалов, отходящий из реактора;
м) отгонку из крекинг-реактора потока головного погона, содержащего АК, БА, BuOH и воду, при одновременном поддержании в жидкости крекинг-реактора концентрации кислоты по меньшей мере 7,5 вес.%;
н) конденсацию отгоняемого из крекинг-реактора потока головного погона;
о) возврат в этерификационный реактор сконденсированного потока головного погона из крекинг-реактора, содержащего АК, БА, BuOH и воду;
п) одновременно с осуществлением вышеприведенных стадий в)-о) отгонку из этерификационного реактора испаренной смеси, содержащей АК, БА, BuOH и воду;
р) конденсацию испаренной смеси с получением первого конденсата, содержащего органическую фазу и водную фазу;
с) возврат 0-30 процентов органической фазы в каплеотбойник, смонтированный на этерификационном реакторе;
т) подачу 70-100 процентов органической фазы и 50-100 процентов водной фазы в колонну для выделения акриловой кислоты;
у) отгонку из колонны для выделения акриловой кислоты под давлением 35-800 мм рт. ст. в водном режиме и при коэффициенте водного орошения 8,5: 1-17: 1 смеси головного погона, представляющей собой азеотропную смесь бутанола, бутилакрилата и воды;
ф) удаление из дистилляционной колонны потока богатых акриловой кислотой кубовых остатков;
х) возврат в этерификационный реактор потока богатых акриловой кислотой кубовых остатков, отводимого из колонны для выделения акриловой кислоты;
ц) конденсацию смеси головного погона с получением второго конденсата;
ч) разделение второго конденсата на богатую бутилакрилатом органическую фазу и выделенную водную фазу и
ш) выделение богатой бутилакрилатом органической фазы, практически свободной от АК.
Можно также осуществлять описанный выше способ, в котором отсутствуют стадии р), с) и т) и в котором 100 процентов испаренной смеси направляют непосредственно в колонну для выделения акриловой кислоты на стадии у) с последующей перегонкой согласно описанному выше. Когда испаренную смесь направляют непосредственно в эту колонну, диапазон коэффициентов водного орошения сужают до 13:1-17:1, а все остальные стадии идентичны, за исключением, разумеется, того, что при этом отсутствует "первый конденсат".
В описанных выше способах проведения непрерывных процессов кислотный катализатор можно выбирать из серной кислоты, сульфоновой кислоты, предпочтительно метан-, бензол- и толуолсульфоновой кислоты, или сильнокислотной ионообменной смолы. Предпочтительна серная кислота, которую используют и как кислотный катализатор, и как необязательно добавляемую минеральную кислоту. Предпочтительный интервал давлений для процесса перегонки в колонне для выделения АК составляет 90-135 мм рт.ст. Предпочтительный коэффициент водного орошения также равен 8,5-12,5. Общий поток водного и органического сырья можно направлять либо в гидролизный реактор, который представляет собой многоходовую реакционно-дистилляционную колонну, либо предпочтительно ПРМ, как описано выше, обеспечивая таким образом проведение гидролитической реакции в условиях непрерывного перемешивания. Дополнительные тяжелые фракции при этом могут также включать до 80 вес.% общего потока водного и органического сырья.
Возвращаясь к фиг. 1, необходимо отметить, что потоки 20 и 19 подают далее в линию 23 или раздельно по линиям 22 и 23, а БА-продукт далее выделяют обычными средствами. Таким образом, начиная с этого этапа способ можно далее осуществлять обычным путем, например, подачей потоков 22 и 23 в сепаратор, где общий поток нейтрализуют щелочью, а всю образующуюся соль АК экстрагируют водой. Затем свободную от АК органическую фазу обезвоживают пропусканием через дистилляционную колонну, удаляя последние следы воды. В следующей колонне непрореагировавший BuOH выделяют из головного погона в виде его азеотропа с БА для возврата в этерификационный реактор (поток 52), а поток кубовых остатков, содержащих практически чистый БА и ингибиторы, направляют в последнюю дистилляционную колонну для получения продукта. В этой последней колонне чистый БА отгоняют с головным погоном обычным путем, а отходящий поток материалов, содержащий технологические ингибиторы, удаляют из нижней части для повторного использования. Характерная чистота БА, получаемого по описанному выше способу, обычно превышает 99,8% БА.
Примеры
Общая часть
Материалы: Потоки АК, сырого и чистого н-бутилакрилата (БА), н-бутанола (BuOH) и тяжелых фракций получали из потоков, отводимых из производственной установки, где это указано, с обозначением качества/чистоты. Технические ингибиторы полимеризации использовали в том виде, в котором они поставляются на рынок, в указанных концентрациях; они включали гидрохинон (ГХ), метиловый эфир ГХ (МЭГХ) и фенотиазин (ФТЗ). Компоненты тяжелых фракций в этих примерах включали нижеследующие материалы: АОПК, бутил- β -бутоксипропионат ("БББП"), бутил- β - гидроксипропионат ("ББГП"), бутоксиАОПК ("БАОПК"), бутилмалеат и ДБЭ.
Аббревиатуры: Они включают, помимо уже указанных выше, следующие термины: дополнительный (доп.), водный (вдн.), сравнительный (сравн.), пример (прим.), фигура (фиг.), грамм (г), граммов в час (г/ч), килограмм (кг), часы (ч), тяжелые фракции или потоки тяжелых фракций (тяжелые), вес (в.), миллиметров ртутного столба (мм рт.ст.), миллимоли (ммоли), фунты (фнт.), испаренная смесь (исп.см.), круглодонная колба (крдон. колба), менее (<), более (>), точка (тч. ), стационарный режим (ст.реж.). На фиг. 2 соответствующие данным точки сокращенно представлены в следующем виде: незаштрихованные квадратики отражают водный ст.реж. (режим), треугольники отражают органический ст. реж., а кружочками обозначены точки, соответствующие данным экспериментов/примеров с указанием их номеров.
Анализы: Для определения воды прибегали к стандартным методам; содержание мономеров, BuOH, остаточных примесей и тяжелых фракций определяли газожидкостной хроматографией (ГЖХ) на хроматографе Varian модели 3700, пользуясь пламенным ионизационным определением. Серную кислоту определяли с применением pH-датчика Orion Research Ion analyzer и титрованного спиртового раствора гидроксида тетрабутиламмония. Во всех случаях, за исключением специально оговоренных, приведенные в примерах данные концентрации H2SO4 получены титрованием. Везде, кроме специально оговоренных случаев, содержание выражено в весовых процентах.
Рекуперация ценных продуктов: Рекуперированные "ценные продукты" рассчитывали и определяли в типичных тяжелых фракциях, образующихся, например, при получении БА, следующим образом. Поскольку все тяжелые фракции, связанные с получением БА, в конечном итоге образовывались на основе АК и BuOH, данные о рекуперированных ценных продуктах рассчитывали таким образом, чтобы они отражали утилизацию этих реагентов даже несмотря на то, что некоторое количество рекуперировали в форме БА-продукта. Так, например, 100 молей БББП содержат эквивалент 100 молей акриловой кислоты и 200 молей BuOH. Подобным же образом 100 молей БАОПК содержат эквивалент 100 молей BuOH и 200 молей АК. В целях расчета весовых количеств подразумевается, что "смесь тяжелых" (состав которой неясен) представляет собой смесь акриловой кислоты с BuOH в молярном соотношении 1: 1 и с молекулярной массой 146 г/моль. БА-мономер включает эквивалентные молярные количества АК и BuOH. Так называемые "свободные" ценные продукты представляют собой просто те же самые ценные продукты в "свободной" форме (не входящие в состав тяжелых фракций). Ниже в качестве типичного примера приведен список ценных BuOH- и АК-материалов, которыми характеризуются тяжелые фракции, отводимые из реакции этерификации с получением, например, БА.
Компонент - Ценные продукты
Бутил- β -бутоксипропионат (БББП) - 2 BuOH, 1 АК
Бутил- β-гидроксипропионат (ББГП) - 1 BuOH, 1 АК
Бутилакрилоксипропионат (БАОПК) - 1 BuOH, 2 АК
Акрилоксипропионовая кислота (АОПК) - 2 АК
н-Бутилмалеат - 1 BuOH
Кислый сульфат бутила - 1 BuOH
Смесь тяжелых - 1 BuOH, 1 АК
Выход продукта процесса: Технологический выход продукта рассчитывали следующим образом. С АК и BuOH, содержавшимися в любых вводимых потоках, направляемых в ГРУ, оперировали при расчете выхода продукта так, как если бы при подаче в этерификационный реактор они представляли собой свежие (т.е. исходные материалы) АК и BuOH. С БА-мономером, содержавшимся во вводимых потоках, направляемых в ГРУ (т.е. в рециркуляционных или дополнительных потоках), оперировали при расчете выхода продукта так, как если бы он представлял собой возвращаемый в процесс БА, получаемый при дальнейшем выделении, т. е. из-за всего возвращаемого в процесс БА никакого прироста выхода продукта не происходило. Следовательно, когда согласно приведенным данным ценные продукты (описанные выше) рекуперировали из потоков тяжелых фракций, отводимых из ГРУ и ГРУ/крекинг-реактора, выход продукта в пересчете на АК или БА мог превышать 100%. Таким образом, суммарный расчетный выход продукта составляет:
и
Оборудование: В нижеприведенных примерах ГРУ 5 представляла собой 1-литровую 4-горлую крдон. колбу, снабженную мешалкой, охлаждаемой водой дистилляционной насадкой, выходное отверстие которой сообщалось с 250-миллилитровым отборником фракций, фазовым сепаратором 30. Эта ГРУ дополнительно была оборудована питающими впускными отверстиями 3 и 4 для отходящего из реактора потока материалов, потока тяжелых фракций и других вводимых потоков; погружной трубкой из сплава "Хастеллой" с внешним диаметром 6 мм, которая посредством линии 9 сообщалась с крекинг-реактором 10 (когда его применяли) или посредством линии 52 с резервуаром для сбора отходящих потоков. Содержимое ГРУ нагревали с помощью греющей рубашки и перемешивали механической мешалкой. Различные сырьевые и направляемые на орошение и отходящие потоки закачивали в реактор из стеклянных питательных воронок, соответственно откачивали из него с помощью дозировочных насосов. Тепловой режим ГРУ регулировали электронным регулятором температуры, подключенным к калиброванной термопаре. Все линии для технологических потоков, которые испытывали воздействие потоков, содержавших серную кислоту, выполняли из сплава Hastelloy СTM или политетрафторэтилена (ПТФЭ).
В качестве крекинг-реактора 10 использовали 500-миллилитровую колбу, которая по конструкции была аналогичной ГРУ, если иметь в виду температурный контроль и линии технологических потоков. По линии 9 подачи отходящего из ГРУ потока питали крекинг-реактор через впускное отверстие и насосом. Ресивером 31 служил 125-миллилитровый отборник фракций.
Колонны для выделения акриловой кислоты описаны в конкретных примерах.
Во всех случаях, за исключением специально оговоренных, содержание выражено в весовых процентах от веса смеси, которая включала указанный компонент.
Модельные эксперименты с колонной для выделения акриловой кислоты
Модельные исследования проводили с помощью усовершенствованного имитатора технологической схемы "Aspen PlusTM" фирмы Aspen Technology, Inc. Все данные получали с использованием модели колонны "Aspen", которая обладала эффективностью в 13 теоретических тарелок, включала испаритель и декантатор и работала под давлением в головке 75 мм рт.ст. Питающей тарелкой служила 4-я теоретическая тарелка снизу. При работе в водном режиме параметры отводимого из колонны потока кубовых остатков соответствовали содержанию 90 вес.% АК и 10% воды. На фиг. 2 показаны два "стационарных режима" (т.е. требуемый "водный" и нежелательный "органический" режимы, которые описаны выше) работы колонны для выделения акриловой кислоты с использованием сырья, приведенного в таблице 1, что соответствовало 80%-ной конверсии АК в БА в реакторе при величине молярного соотношения между АК и BuOH 1-1,35. По данным строили представленный на фиг. 2 график зависимости концентрации АК в органическом дистилляте от расхода потока воды на орошение в колонне.
Работа на моделях показала, что минимальный расход потока воды на орошение, который необходим для работы колонны для выделения акриловой кислоты в требуемом водном стационарном режиме с использованием сырья из таблицы 1А, составлял приблизительно 15546 кг/ч (32000 фунтов/ч). Место перехода с водного режима в органический рассчитывали исходя из того, что разделение БА и АК в колонне для выделения АК достигается азеотропной отгонкой БА с использованием воды как азеотропообразующего компонента. В нижеприведенных таблицах 1А и 1Б показано, каким образом рассчитывали минимальное количество вводимой воды, необходимое для азеотропной отгонки всего БА в колонне для выделения акриловой кислоты. Первый азеотроп, который проявлял себя в колонне, представлял собой самый низкокипящий тройной азеотроп БА/бутанола/воды, который под давлением 100 мм рт.ст. кипел при 46,4oC и содержал 36,0% БА, 26,4% BuOH и 37,6% воды. Этот азеотроп обеднял сырье бутанолом и отбирал 10429 кг/ч (22944 фунта/ч) БА в виде головного погона из общего количества 20315 кг/ч (44692 фунта/ч), содержавшегося в сырье. Количество воды, которое требовалось для образования этого первого азетропа, превышало ее количество в сырье на 8198 кг/ч (18036 футов/ч). После расхода бутанола следующий наиболее низкокипящий азеотроп, который проявлял себя в колонне, представлял собой бинарный азеотроп БА/воды, который под давлением 100 мм рт.ст. кипел при 47,6oC и содержал 61,0% БА и 39,0% воды. Этот второй азеотроп отбирал оставшиеся 9885 кг/ч (21748 фунтов/ч) БА в виде головного погона, потребляя на образование азеотропа этого состава 6320 кг/ч (13904 фунта/ч) воды. Комбинированный анализ обоих азеотропов показывает, что общее количество воды, которое потреблялось на азеотропную отгонку всего БА, содержавшегося в сырье, превышало ее количество в водном сырье на 6320 кг/ч (31940 фунтов/ч). Это соответствует минимальному количеству воды, которое необходимо подавать посредством водного орошения для отгонки всего БА с головным погоном и, таким образом, проведения процесса в водном режиме. Было установлено, что эти расчетные данные превосходно согласуются с местом перехода с водного режима на органический, определенным по данным фиг. 2.
Представленные результаты моделирования соответствуют конкретному сырью колонны для выделения акриловой кислоты. Однако для расчета минимальной потребности колонны в водном орошении тот же самый анализ применим к любому подаваемому в колонну сырью, соответствующему любым конкретно заданным реакторным параметрам. Возможность точного предсказания минимальной потребности в воде колонны для выделения акриловой кислоты, основанная только на одном составе сырья, позволяет выбрать рабочий коэффициент орошения, который сводит к минимуму тепловую нагрузку и диаметр колонны, обеспечивая в то же самое время стабильную работу в требуемом водном режиме.
Моделирование давало возможность регулировать стационарный режим (водный или органический), в котором колонна работала в начале эксперимента в крайней точке (т.е. в водном стационарном режиме при очень высоком расходе потока на орошение или в органическом стационарном режиме при очень низком расходе потока на орошение), где существует только один целевой стационарный режим, с последующим продвижением вдоль линии, соответственно либо уменьшая, либо увеличивая расход потока на орошение, до тех пор, пока не достигали целевой точки процесса. Этого добивались проверкой чувствительности процесса к ключевым переменным; в данном исследовании изучали влияние расхода потока на водное орошение. Обе линии стационарного режима по фиг. 2 получали путем осуществления программы двух исследований чувствительности. В первом исследовании расход потока на водное орошение сначала был очень высоким и составлял 61364 кг/ч (135000 фунтов/ч) [коэффициент орошения примерно 40 при расходе направляемого дальше потока водного сырья 1591 кг/ч (3500 фунтов/ч)], и постепенно его уменьшали до очень низкого значения в 4545 кг/ч (10000 фунтов/ч) (коэффициент орошения приблизительно 3). Это исследование давало нижнюю, "водную линию" на фиг. 2, которая соответствовала целевому водному режиму, где содержание АК в органическом дистилляте оказывалось очень низким [в этой программе наименьшего содержания АК в дистилляте (27 част./млн) достигали с минимальным количеством жидкости при орошении, т.е. примерно 15454 кг/ч (32000 фунтов/ч), при коэффициенте орошения около 9, что указывало на работу колонны в целевом стационарном режиме]. Когда расход потока на орошение становился слишком низким, работа колонны в водном режиме становилась невозможной, а при расходе потока на орошение примерно 14090 кг/ч (31000 фунтов/ч) происходил внезапный и очень большой прирост содержания АК в дистилляте. При расходе менее 14090 кг/ч (31000 фунтов/ч) колонна работала только в органическом режиме, т.е. оба режима сходились в одной точке.
При втором исследовании чувствительности начинали с низкого расхода потока на водное орошение в 4545 кг/ч (10000 фунтов/ч) и постепенно увеличивали его до примерно 61364 кг/ч (135000 фунтов/ч). Это исследование давало верхнюю, "органическую линию" на фиг. 2, которая соответствовала нежелательному органическому режиму, где содержание АК в органическом дистилляте оказывалось намного выше, как указано. Последовательное перемещение вдоль этой линии (точки 4-7) до расхода выше примерно 54545 кг/ч (120000 фунтов/ч), когда воды оказывалось достаточно для принудительного перевода колонны в водный режим работы, обе линии сходились в единственном водном стационарном режиме. В пределах водной линии рабочий диапазон в этом имитированном исследовании, ведущий к получению БА, практически не содержавшего АК (целевое содержание АК 2000 част./млн, предпочтительно <1000 част./млн), является небольшим диапазоном на нижней, водной линии на фиг. 2. Программа позволяет прогнозировать также высокое содержание БА (например, 23-74 вес.%) в потоке кубовых остатков, когда колонна работает в органическом режиме. Возврат БА в этерификационный реактор нежелателен, так как он снижает скорость конверсии АК и BuOH.
При последующем моделировании двух стационарных режимов в колонне для выделения АК было установлено, что байпасирование реакторного конденсатора и фазового сепаратора 14 и подача испаренной смеси непосредственно в эту колонну давали преимущество, состоявшее в уменьшении потребности колонны в нагрузке водяным паром. Однако поскольку вода в сырье уже испарялась, она по существу оказывалась недоступной для образования с БА азеотропа, и для компенсации ее дефицита требовалось дополнительное количество воды на орошение. Для случая подачи пара в колонну точка перехода из водного режима в органический на фиг. 2 при увеличении количества воды в сырье сдвигается вправо, а диапазон коэффициентов водного орошения при водном режиме работы сужается до 13:1-17:1.
Моделирование показывало также, что возврат на орошение части органической фазы оказывает вредное влияние на работу колонны, поскольку весь БА и бутанол, возвращаемые в колонну при органическом орошении, будет просто необходимо вновь удалять азеотропной отгонкой с дополнительным расходом воды. Кроме того, АК при органическом орошении возвращают в самый верх колонны, не оставляя ни одной тарелки для ректификации этой доли АК с отводимыми сверху парами. Эти факторы увеличивают минимальное количество воды, необходимое для работы в водном режиме, уменьшают ширину окна водного режима и повышают минимальное содержание АК, которое может быть достигнуто при дистилляции.
Данные равновесия между паром и жидкостью (РПЖ) указывают на то, что бутанол проявляет эффект подавления летучести АК. Моделирование в соответствии с данными РПЖ показывает, что поступающие в колонну потоки сырья, которые богаты бутанолом, дают потоки дистиллятов с низким содержанием АК. Поэтому низкая конверсия и высокие величины соотношения бутанол/АК в реакторе, которые дают богатые бутанолом отходящие потоки, благоприятно сказываются на разделении БА и АК и обеспечивают, как описано выше, широкие окна (8,5: 1-17:1) рабочих коэффициентов водного орошения. В случае невозможности работы реактора в указанных выше условиях с целью обеспечить широкое окно в водном режиме работы независимо от условий в реакторе можно предусмотреть отдельный поток свежего бутанола, вводимый непосредственно в колонну для выделения АК. Свежий бутанол лучше всего вводить по месту ввода основного сырья или несколько ниже. Бутанол никогда не следует вводить над точкой ввода основного сырья, содержащего АК (ввод бутанола как легкокипящего компонента над местом ввода основного сырья ведет к его быстрой отпарке с головным погоном, из-за чего тарелки между уровнями ввода основного сырья и бутанола остаются с таким количеством бутанола, которого недостаточно для подавления летучести АК).
Лабораторное подтверждение водного и органического режимов
Существование двух стационарных режимов в колонне для выделения АК было подтверждено экспериментально при проведении многодневного непрерывного лабораторного испытания, из которого были взяты примеры 1-6 и сравнительные примеры 1 и 2. Материальный баланс технологических потоков в таблице 1 и в имитациях, в результате чего получили приведенную на фиг. 2 диаграмму, моделировали в масштабе установки; в приведенных ниже примерах с колонной для выделения акриловой кислоты в таком лабораторном эксперименте масштаб расхода потоков уменьшали так, что 250 кг/ч (550 фунтов/ч) на упомянутой модели в масштабе установки оказались эквивалентными 1 г/ч. Расширенный эксперимент, который проводили приблизительно следуя точкам, отмеченным на фиг. 2 кружочками, начинали демонстрацией непрерывного процесса в колонне в водном режиме при различных расходах направляемого на орошение потока (точки 1 и 2). За этой частью эксперимента следовало намеренное уменьшение расхода воды на орошение с целью перевести работу колонны в органический режим (точки 3 и 4). В дальнейшем для восстановления в колонне водного режима меняли условия парообразования в кипятильнике. На фиг. 2 точки 1 и 5, 2 и 4, 6 и 8 представляют собой пары согласующихся точек, т.е. точек одинакового расхода потока на орошение соответственно в водном и органическом режимах. После достижения водного режима его поддерживали с помощью коэффициента орошения 8,5:1-17:1, получая отогнанный БА с целевым содержанием АК (< 2000 част./млн), а также поток выделенной водной АК, практически не содержавший БА. Согласно данным измерений содержание АК в БА в точках 1 и 2 составляло соответственно 950 и 200 част./млн, а в АК БА не содержался (<1 част./млн).
Пример 1
Водный режим работы при коэффициенте водного орошения 16 (точка 1 на (фиг. 2)
В качестве колонны 15 для выделения акриловой кислоты использовали 30-тарельчатую ректификационную колонну Oldershaw диаметром в 1 дюйм, снабженную стеклянным холодильником на линии 16 и паровым испарителем из нержавеющей стали. Эта колонна работала под давлением в головке 75 мм рт.ст. В колонну для выделения акриловой кислоты в час подавали 10,8 г (0,15 моля) АК, 30,6 г (0,41 моля) бутанола, 82,4 г (0,64 моля) БА и 12,0 г (0,67 моля) воды. Состав этой смеси соответствовал реакторному конденсату, образовывавшемуся в системе, в которой реактор 1 работал при соотношении АК/BuOH 1: 1,35 и 80%-ной конверсии в пересчете на АК, при этом в час получали 0,18 г возвращаемого в процесс БА на грамм непрореагировавшего BuOH и 0,11 г возвращаемой в процесс воды на грамм непрореагировавшей АК. Сырье вводили на 10-ю тарелку снизу. Смесь в виде головного погона отгоняли при температуре 43,5oC, ее конденсировали и разделяли на две фазы в ресивере 18. Из богатой БА органической фазы 19 отбирали 117,3 г/ч (грамма в час) материала, который содержал по весу 70,3% БА, 26,0% бутанола, 3,6% воды и 0,1% АК. Из выделенной водной фазы 20 110,2 г/ч (94,3% фазы) по линии 21 возвращали в головку колонны, а 6,7 г/ч (5,7%) по линии 22 направляли дальше, достигая коэффициента водного орошения 16,4. Эта водная фаза содержала 96,6% воды, 3,2% бутанола, 0,2% БА и 354 част./млн АК. Из богатого АК остаточного продукта отбирали 12,0 г/ч потока 17, содержавшего 89,2% АК и 10,8% воды. Конечная температура в кубе составляла 60,0oC. Пример 1 соответствовал точке 1 на фиг. 2.
Пример 2
Водный режим работы при коэффициенте водного орошения 11 (точка 2 на фиг. 2)
Оборудование, расход сырья, состав сырья, место ввода сырья, давление в колонне и основные условия работы колонны были такими же, что и в примере 1. С целью уменьшить расход воды на орошение колонны уменьшали расход потоков водяного пара в кипятильник и возвращаемого водного конденсата. Головной продукт перегонки, отводимый по линии 16 и получаемый при температуре 42,6oC, конденсировали и в ресивере 18 разделяли на две фазы. Собирали 117,1 г/ч богатой БА органической фазы 19, которая содержала 70,4% БА, 26,0% бутанола, 3,6% воды и 218 част./млн АК. Из выделенной водной фазы 20 77,2 г/ч (91,8%) по линии 21 возвращали в головку колонны и 6,9 г/ч (8,2%) по линии 22 направляли дальше, достигая коэффициента водного орошения 11,2. Эта водная фаза содержала 96,6% воды, 3,2% бутанола, 0,2% БА и 81 част./млн АК. Отбирали 12,0 г/ч направляемого по линии 17 остаточного продукта, содержавшего 90,0% АК и 10,0% воды. Конечная температура в нижней части колонны составляла 60,4oC. Этот пример соответствовал точке 2 на фиг. 2.
Сравнительный пример 1
Органический режим работы (точка 3 на фиг. 2)
Оборудование, расход сырья, состав сырья, место ввода сырья, давление в колонне и основные условия работы колонны были такими же, что и в примере 1, и на начальном этапе колонна работала по принципу, аналогичному принципу работы в примере 2. Далее с целью еще больше уменьшить расход воды на орошение колонны уменьшали расход потоков водяного пара в кипятильник и возвращаемого водного конденсата. Головной продукт перегонки, отводимый по линии 16 и получаемый при температуре 50,1oC, конденсировали и в ресивере 18 разделяли на две фазы. Собирали 118,5 г/ч богатой БА органической фазы 19, которая содержала 62,6% БА, 25,6% бутанола, 6,1% АК и 5,7% воды. Из выделенной водной фазы 20 36,0 г/ч (86,2%) по линии 21 возвращали в головку колонны и 5,8 г/ч (13,8%) по линии 22 направляли дальше, достигая коэффициента водного орошения 6,3. Эта водная фаза содержала 94,4% воды, 3,1% бутанола, 2,2% АК и 0,3% БА. Отбирали 11,8 г/ч направляемого по линии 17 остаточного продукта, содержавшего 70,7% БА, 29,1% АК и 0,2% бутанола. Конечная температура в нижней части колонны составляла 88,3oC. Этот пример соответствовал точке 3 на фиг. 2 и показывал, что работа колонны при коэффициенте орошения ниже диапазона коэффициентов орошения по настоящему изобретению ведет к нежелательной работе в органическом режиме. Полученные результаты включали высокое содержание АК в богатой БА органической фазе 19, высокое содержание БА в потоке кубовых остатков 17 и высокую температуру в колонне в сравнении с условиями водного режима примеров 1 и 2.
Сравнительный пример 2
Подтверждение существования двух стационарных режимов
Оборудование, расход сырья, состав сырья, место ввода сырья и давление в колонне были такими же, что и в примере 1, и на начальном этапе колонна работала в условиях, аналогичных условиям осуществления из сравнительного примера 1. Далее с целью увеличить расход воды на орошение колонны до такого же расхода, как в примере 2 (точка 2 на фиг. 2), увеличивали расход потоков водяного пара в кипятильник и возвращаемого водного конденсата. Головной продукт перегонки, отводимый по линии 16 и получаемый при температуре 43,9oC, конденсировали и в ресивере 18 разделяли на две фазы. Собирали 117,9 г/ч богатой БА органической фазы 19, которая содержала 63,9% БА, 25,8% бутанола, 5,2% воды и 5,1% АК. Из водной фазы 20 77,2 г/ч (92,5%) по линии 21 возвращали в головку колонны и 6,2 г/ч (7,5%) по линии 22 направляли дальше, достигая коэффициента водного орошения 12,4. Эта водная фаза содержала 94,7% воды, 3,1% бутанола, 1,9% АК и 0,3% БА. Отбирали 11,9 г/ч направляемого по линии 17 остаточного продукта, содержавшего 60,4% БА, 39,2% АК и 0,4% бутанола. Конечная температура в нижней части колонны составляла 88,3oC. Этот сравнительный пример соответствовал точке 4 на фиг. 2 и показывал, что даже при расходе потока на орошение в 77,2 г/ч, т.е. таком же, как и в примере 2, и коэффициенте водного орошения 12,4 колонна продолжала работать в нежелательном состоянии органического режима, что сопровождалось высоким содержанием АК в органическом дистилляте и БА в потоке кубовых остатков, а также высокой температурой в колонне по сравнению с результатами для условий водного режима из примеров 1 и 2.
Подтвердив существование режима точки 4 на фиг. 2, т.е. точки органического режима, аналогичной точке 2 водного режима, этот сравнительный пример показал, что в колонне действительно существуют два стационарных режима, как это было спрогнозировано вышеописанным моделированием. Этот сравнительный пример показал также, что линии двух таких стационарных режимов образуют "петлю гистерезиса" и что после установления в колонне нежелательного органического режима работы при поступлении достаточного количества тепла она остается в этом режиме работы даже после увеличения коэффициента водного орошения до уровня, эффективного для работы в водном режиме.
Пример 3
Восстановление водного режима работы после работы в органическом режиме
Оборудование, расход сырья, состав сырья, место ввода сырья и давление в колонне были такими же, что и в примере 1. При проведении эксперимента, идентичного описанному в сравнительном примере 1, на начальном этапе колонна работала в условиях точки 3 на фиг. 2. Затем на верхнюю тарелку колонны добавляли поток воды с расходом 41,2 г/ч. В совокупности с начальным расходом 36,0 г/ч на водное орошение этот дополнительный поток воды обеспечивал создание эффективного потока для орошения колонны с расходом 77,2 г/ч, т.е. с таким же расходом потока на орошение, как в примере 2 и сравнительном примере 2 (т.е. соответственно точки 2 и 4 на фиг. 2). Расход потока водяного пара, подаваемого в испаритель, поддерживали на том же уровне, что и в сравнительном примере 1 (точка 3 на фиг. 2). С помощью термопар, размещенных на чередующихся тарелках, наблюдали за перемещением вниз колонны холодного фронта, прежде всего воды в жидком состоянии, начинающегося на верхней тарелке и опускающегося с одной тарелки на другую до тех пор, пока он в конечном счете не достигал испарителя. Таким образом, без подачи дополнительного количества пара в испаритель для работы с повышенной нагрузкой дополнительное количество воды, подаваемой на верхнюю тарелку, проявляло предполагаемое поведение, оказывая охлаждающее воздействие на все тарелки. После достижения холодным фронтом испарителя, на что указывало резкое падение температуры с 88,3 до 57,0oC, подачу дополнительного потока свежей воды на верхнюю тарелку прерывали, расход потока подаваемого в испаритель водяного пара увеличивали для увеличения расхода потока на водное орошение с 36,0 до 77,2 г/ч и колонне давали достичь стационарного режима, в данном случае уже водного режима, при увеличенном расходе потока на орошение.
Смесь в виде головного погона, отводимого по линии 16, получаемую при температуре 42,6oC, конденсировали и в ресивере 18 разделяли на две фазы. Собирали 117,0 г/ч богатой БА органической фазы 19, которая содержала 70,5% БА, 26,0% бутанола, 3,5% воды и 263 част./млн АК. Из выделенной водной фазы 20 77,2 г/ч (91,8%) по линии 21 возвращали в головку колонны и 6,9 г/ч (8,2%) по линии 22 направляли дальше, достигая коэффициента водного орошения 11,2. Эта водная фаза содержала 96,6% воды, 3,2% бутанола, 0,2% БА и 75 част. /млн АК. Отбирали 12,1 г/ч богатого АК направляемого по линии 17 остаточного продукта, содержавшего 89,5 вес.% АК и 10,5 вес.% воды. Конечная температура в нижней части колонны составляла 60,2oC. Этот результат соответствовал точке 2 на фиг. 2 и был практически идентичным результату, достигнутому в примере 2. Таким образом, пример 3 показал кратчайший путь возврата работы колонны с точки на линии органического режима в желаемый водный режим работы. В водном режиме колонна для выделения акриловой кислоты работает с водой на всех тарелках и в потоке кубовых остатков 17, тогда как в нежелательном органическом режиме материал на нескольких верхних тарелках и в потоке кубовых остатков 17 лишен воды. Хотя в данном примере колонна для выделения акриловой кислоты начинала работать в органическом режиме, благодаря продемонстрированному техническому приему создавали возможность для работы колонны в целевом водном режиме. Этот результат особенно важен, принимая во внимание факты, обнаруженные в сравнительном примере 2, которые подтвердили, что оба стационарных режима в этой особой системе для получения БА образуют "петлю гистерезиса", как показано на фиг. 2.
Пример 4
Водный режим работы при коэффициенте водного орошения 9,6
С использованием оборудования, описанного в примере 1, работу колонны осуществляли под давлением в головке 75 мм рт.ст., подавая в час 5,6 г (0,08 моля) АК, 34,8 г (0,47 моля) бутанола, 96,4 г (0,75 моля) БА и 13,1 г (0,73 моля) воды. Состав этой смеси соответствовал испаренной из реактора смеси, которую получали при этерификации с образованием БА, где реактор 1 работал при соотношении между АК и бутанолом 1:1,5 и 90%-ной конверсии в пересчете на АК с возвратом 18% БА на весовую единицу непрореагировавшего бутанола в потоке 52 и 7% воды на весовую единицу непрореагировавшей АК в потоке 17. Сырье вводили на 10-ю тарелку снизу. Головной дистиллят 16 отгоняли при температуре 42,2oC, его конденсировали и разделяли на две фазы в ресивере 18. Отбирали 135,8 г/ч богатой БА органической фазы 19; она содержала 71,0 вес.% БА, 25,5 вес.% бутанола, 3,5 вес.% воды и 550 част./млн АК. Из водной фазы 20 78,9 г/ч (90,6%) в потоке по линии 21 возвращали в головку колонны, а 8,2 г/ч (9,4%) по линии 22 направляли дальше, достигая таким образом коэффициента водного орошения 9,6. Эта выделенная водная фаза содержала 96,7 вес.% воды, 3,1 вес.% бутанола, 0,2 вес.% БА и 209 част./млн АК. Собирали 6,0 г/ч потока богатого АК остаточного продукта, направляемого по линии 17, содержавшего 93,1 вес.% АК и 6,9 вес.% воды.
Пример 5
Водный режим работы при коэффициенте водного орошения 11,0
Оборудование, расход сырья, состав сырья, место ввода сырья и давление в колонне были такими же, что и в примере 4. Смесь в виде головного погона, получаемого при температуре 41,9oC и подаваемого по линии 16, конденсировали и разделяли на две фазы в ресивере 18. Отбирали 135,9 г/ч богатой БА органической фазы 19, которая содержала 70,9 вес.% БА, 25,4 вес.% бутанола, 3,5 вес.% воды и 779 част./млн АК. Из водной фазы 20 89,5 г/ч (91,6%) в виде потока 21 возвращали в головку колонны, а 8,2 г/ч (8,4%) в виде потока 22 направляли дальше, достигая коэффициента водного орошения 11,0. Эта водная фаза содержала 96,7 вес.% воды, 3,1 вес.% бутанола, 0,2 вес.% БА и 286 част. /млн АК. Собирали 6,0 г/ч потока 17 богатого АК остаточного продукта, содержавшего 92,8 вес.% АК и 7,2 вес.% воды. Этот пример продемонстрировал возрастание содержания АК в богатой БА органической фазе с 550 до 779 част./млн в таких условиях, когда расход потока воды на орошение возрастал по сравнению с расходом в примере 4 (коэффициент орошения 9,6).
Пример 6
Водный режим работы при коэффициенте водного орошения 9,7 в 35-тарельчатой колонне
К колонне, использованной в примере 1, добавляли 5- тарельчатую секцию, получая таким образом 35-тарельчатую ректификационную колонну Oldershaw диаметром в 1 дюйм, снабженную стеклянным холодильником и паровым испарителем из нержавеющей стали. Расход сырья, состав сырья, место ввода сырья и давление в колонне были такими же, что и в примере 4. Смесь в виде головного погона, получаемого при температуре 42,2oC и подаваемого по линии 16, конденсировали и разделяли на две фазы в ресивере 18. Отбирали 135,8 г/ч богатой БА органической фазы 19, которая содержала 71,0 вес.% БА, 25,5 вес.% бутанола, 3,5 вес.% воды и 193 част./млн АК. Из выделенной водной фазы 20 78,9 г/ч (90,7%) в виде потока 21 возвращали в головку колонны, а 8,1 г/ч (9,3%) в виде потока 22 направляли дальше, достигая коэффициента водного орошения 9,7. Эта водная фаза содержала 96,7 вес.% воды, 3,1 вес.% бутанола, 0,2 вес. % БА и 72 част./млн АК. Собирали 6,1 г/ч потока 17 богатых АК кубовых остатков, содержавших 92,5 вес.% АК и 7,5 вес.% воды. Конечная температура в кубе составляла 62,3oC. Этот пример показал, что добавление 5-тарельчатой ректификационной секции к колонне для выделения АК в еще большей степени снижал содержание АК в богатой БА органической фазе, а именно, с 550 в примере 4 до 193 част./млн.
Сравнительный пример 3
Обработка в крекинг-реакторе без применения ГРУ
Этот сравнительный пример проводили в вышеописанном 500-миллилитровом крекинг-реакторе, используя вышеописанные потоки сырья и не применяя ГРУ. Таким образом, 73,46 г/ч сырья, состав которого представлен в таблице 2, направляли в проточный ПРМ, в котором поддерживали температуру 130oC и давление 35 мм рт.ст. при продолжительности пребывания 60 мин и концентрации катализатора 8,07 вес.% (H2SO4). В целом выделяли 55,24 г/ч однофазного дистиллята, состав которого указан в таблице 3. 18,22 г/ч отходящего потока отводили из крекинг-реактора и сбрасывали в отход в виде отработавшего масла. Количество рекуперированных в качестве ценных продуктов АК и BuOH просуммировано в таблицах 10 и 11, данные которых показывают, что после рекуперации свободных ценных продуктов в виде ценных продуктов в тяжелых фракциях выделяли только 15,0% АК и 11,6% BuOH.
Пример 7
Оценка работы ГРУ в эффективных условиях
В общей сложности 73,46 г органического сырья, состав которого приведен в таблице 4, подавали в ГРУ, в которой поддерживали температуру 108oC и давление 760 мм рт.ст. при продолжительности пребывания 144 мин, 16 вес.% реакторной воды и концентрации катализатора 2,7 вес.% (H2SO4). Кроме того, для компенсации воды, отгоняемой и удаляемой совместно с органическим дистиллятом и отходящим из реактора потоком, и с целью имитации возврата водного дистиллята в ГРУ в эту последнюю подавали также 48,0 г/ч первого водного дистиллята из этерификационного реактора (включавшего 93,0% H2O, 6,0% АК и 1,0% BuOH). В общей сложности отбирали и анализировали 39,35 г/ч органического дистиллята и 38,27 г/ч водного дистиллята. Результаты анализа представлены в таблице 5. Органическую фазу выделяли и возвращали в этерификационный реактор, если его использовали. 43,24 г/ч отходящего потока отводили из ГРУ и использовали для создания всего потока сырья, вводимого в отпарную секцию ПРМ.
Пример 8
Оценка работы крекинг-реактора в эффективных условиях
В общей сложности 43,24 г/ч отходящего из ГРУ потока из примера 7 подавали в крекинг-реактор ПРМ, описанный выше, и выдерживали при температуре 130oC, давлении 100 мм рт.ст. и продолжительности пребывания 120 мин. Для возврата в этерификационный реактор получали в общей сложности 33,63 г/ч дистиллята в виде потока головного погона из крекинг-реактора. В общей сложности отбирали 9,61 г/ч потока остаточного продукта из крекинг-реактора и сбрасывали в отход в виде отработавшего масла. Все количество АК и BuOH, рекуперированных совместно из обеих установок (из ГРУ и крекинг-реактора), представлено в таблицах 10 и 11. Результаты показывают, что после выделения свободных ценных продуктов рекуперировали в виде ценных продуктов тяжелых фракций 68,7% АК и 59,5% BuOH.
Пример 9
Оценка работы ГРУ в более жестких условиях с последовательно установленным крекинг-реактором
В этом примере представлены более высокие показатели рекуперации АК и BuOH как ценных продуктов при работе ГРУ в более жестких условиях (повышенная концентрация кислоты), чем в примерах 7 и 8. Так, 73,46 г/ч потока сырья, состав которого представлен в таблице 7, подавали в ГРУ, в которой поддерживали температуру 114oC и давление 760 мм.рт.ст. при 5,1 вес.% H2SO4 [7,5 вес. % по массовому балансу (МБ)], продолжительности пребывания 144 мин и концентрации воды 15,5 вес.%. Кроме того, для имитации возврата в процесс водного дистиллята совместно с компенсацией потерь воды в органическом дистилляте и отходящем потоке кубовых остатков в ГРУ вводили 46,48 г водного сырья, содержавшего 93% воды, 6% АК и 1% бутанола. В общей сложности выделяли и анализировали 41,96 г/ч органического дистиллята из ГРУ (состав представлен в таблице 8), 38,80 г/ч водного дистиллята из ГРУ и 28,22 г/ч потока головного погона из крекинг- реактора (состав представлен в таблице 9). Поток головного погона из крекинг-реактора получали подачей отходящего из ГРУ потока (39,18 г/ч) в крекинг-реактор, в котором поддерживали следующие условия: 130oC, 100 мм рт.ст., продолжительность пребывания 120 мин, 26,8% H2SO4 (по МБ). Общие количества рекуперированных АК и BuOH для такого сочетания последовательных установок представлены в таблицах 10 и 11, данные которых показывают, что после выделения свободных ценных продуктов в виде ценных продуктов тяжелых фракций выделяли 81,7% АК и 65,2% BuOH.
Пример 10
Способ непрерывного получения бутилакрилата
Этерификационный реактор 1 представлял собой 2-литровую круглодонную колбу из пирекса (высокотермостойкое стекло), снабженную двухтарельчатой дистилляционной колонной Oldershaw (диаметром 5,0 см; использовали в качестве сепараторного каплеотбойника для кислотного катализатора), холодильником, термопарой, патрубками для ввода сырья, присоединенными к соответствующим дозировочным насосам для текучей среды, и линиями, ведущими к гидролитической реакторной установке (ГРУ 5) и крекинг-реактору 10, которые более подробно описаны ниже. Рабочая емкость реактора соответствовала 750 мл реакционной смеси, содержавшей 2,50 вес.% сернокислотного катализатора. Реакционная температура была равной 89oC, а давление составляло 127 мм рт.ст. В реактор 1 подавали 182,90 г/ч свежей сырой АК (количественный анализ: 96 вес. % АК, 2435 ммолей/ч), 182,48 г/ч свежего н-бутанола (BuOH, 2466 ммолей/ч) и 1,71 г/ч свежей H2SO4 (95,5 вес.% кислоты). В общей сложности в реактор вводили 655,3 г/ч материала, состоявшего из 223,85 г/ч АК (3105 ммолей/ч), 316,90 г/ч BuOH (4282 ммолей/ч), сконденсированного и разделенного органического потока головного погона ГРУ, конденсата головного погона крекинг-реактора, остаточного продукта колонны для выделения АК и смеси БА/BuOH/H2, представлявшей собой потоки из операций рекуперации и возврата в процесс следующих потоков: (а) непрореагировавшего BuOH по ходу процесса после колонны для азеотропной отгонки BuOH/БА; (б) рекуперационного потока BuOH/БА из отпарки сбрасываемых в отход водных потоков перед подачей такого потока на очистку сточных вод и (в) части кубовых остатков дистилляционной колонны для конечного продукта [эти потоки представляли собой типичные сырьевые и дополнительные (например, возвращаемые в процесс) потоки, используемые в типичном непрерывном процессе в установке]. Таким образом, общее количество БА, направляемое в этерификационный реактор из таких источников, составляло 88,07 г/ч (688 ммолей/ч), из которых 50,85 г/ч составлял возврат в процесс из последующих по ходу процесса разделительных колонок. Следовательно, АК и BuOH использовали в молярном соотношении 1:1,38.
Продолжительность пребывания в реакторе поддерживали равной приблизительно 60 мин, в результате чего в общем в виде реакторного головного дистиллята через колонну Oldershaw отгоняли 749,8 г/ч материала, который конденсировали и разделяли на две фазы. Часть органической фазы (160 г/ч) возвращали в головку дистилляционной колонны на орошение. Оставшиеся 563,8 г/ч органического дистиллята, содержавшего 38,56 г/ч АК, 124,59 г/ч BuOH и 371,24 г/ч БА, подавали в колонну 15 для выделения акриловой кислоты. Реакторную водную сконденсированную испаренную смесь (26,0 г/ч), содержавшую 2,12 г/ч BuOH и 0,619 г/ч АК, разделяли и использовали следующим образом: 22,4 г/ч по линии 53 подавали в колонну для выделения АК и 3,6 г/ч по линии 42 направляли в ГРУ.
ГРУ 5 и крекинг-реактор 10 были идентичными таковым, описанным в примерах 7, 8 и 9. Соответственно 65,5 г/ч отходящего из этерификационного реактора потока, содержавшего 4,33 г/ч BuOH, 6,19 г/ч АК, 34,23 г/ч БА и другие относящиеся к процессу высококипящие материалы и ингибиторы, по линии 3 направляли в ГРУ 5 и выдерживали при 122oС, 760 мм рт.ст., продолжительности пребывания 317 мин, 6,26 вес.% H2O и концентрации кислотного катализатора 7,58 вес. % (H2SO4). Дополнительно в ГРУ вводили 3,6 г/ч водного сконденсированного дистиллята из реактора. Из ГРУ в виде потока головного погона отгоняли, конденсировали и разделяли в целом 80,5 г/ч продукта. Всю выделенную водную фазу (38,3 г/ч), содержавшую 2,47 г/ч BuOH и 1,01 г/ч АК, по линии 7 возвращали в ГРУ на орошение. Выделенную органическую фазу (42,1 г/ч), содержавшую 7,07 г/ч BuOH, 3,19 г/ч АК и 29,72 г/ч БА, по линии 8 возвращали в этерификационный реактор в виде потока рекуперированного рециркуляционного материала. Отходящий поток (27,0 г/ч) удаляли из ГРУ по линии 9 и направляли в крекинг-реактор 10, в котором поддерживали температуру 120oC, давление 35 мм рт.ст., продолжительность пребывания 815 мин и 20,5 вес.% H2SO4. В общей сложности отгоняли 17 г/ч материала, содержавшего 0,397 г/ч BuOH, 8,44 г/ч АК и 4,78 г/ч БА, и конденсировали в сепараторе 31. Этот объединенный конденсат по линии 12 в виде рециркуляционного потока возвращали в этерификационный реактор. Отходящий из крекинг-реактора поток по линии 13 сбрасывали в отход в виде отработавшего масла.
Колонна 15 для выделения акриловой кислоты диаметром 5,0 см включала 35 тарелок. Она представляла собой дистилляционную колонну Oldershaw, снабженную рубашкой для водяного пара, испарителем из нержавеющей стали и охлаждаемой водой конденсаторной системой. Соответственно в колонну 15, которая работала при давлении в головке 260 мм рт.ст., температуре в нижней части 82oC и коэффициенте водяного орошения 9,61, подавали 563,8 г/ч конденсированного дистиллята органического слоя этерификационного реактора и 22,4 г/ч конденсированного дистиллята водного слоя этерификационного реактора (вышеописанного состава). Отгонкой через колонну получали в общей сложности 907,3 г/ч смеси в виде головного погона, которую конденсировали и разделяли в сепараторе 18. Всего отбирали 400,7 г/ч выделенной водной фазы, 363,4 г/ч которой по линии 21 возвращали в головку колонны на орошение. Богатая БА органическая фаза (506,60 г/ч), включавшая БА-продукт, практически не содержала АК (1450 част./млн). Из колонны отводили 42,3 г/ч богатого АК потока кубовых остатков (35,35 г/ч) и по линии 17 возвращали в этерификационный реактор. Данные о рекуперации представлены в таблицах 10 и 11.
Во время проведения таким образом всего процесса получения БА достигали количественного выхода БА в пересчете на BuOH и выхода БА 102,7% в пересчете на АК (выход 104,8% от теоретического в пересчете на содержание АК и АОПК в свежей сырой АК).
Пример 11
Процесс непрерывного получения БА, включая возврат в процесс дополнительных потоков
Используемые в данном примере этерификационный реактор и связанное с ним технологическое оборудование были идентичными описанным в примере 10. Рабочая емкость реактора соответствовала 1000 мл реакционной смеси, содержавшей 2,25 вес.% сернокислотного катализатора. Питание всех рабочих установок осуществлялось аналогично описанному выше. В реактор 1 подавали 183,90 г/ч свежей сырой АК (количественный анализ: 96 вес.% АК, 2449 ммолей/ч), 207,03 г/ч свежего н-бутанола (BuOH, 2798 ммолей/ч) и 2,05 г/ч свежей H2SO4 (95,5 вес.% кислоты). В виде дополнительных потоков в ГРУ подавали 30,58 г/ч (424 ммоля/ч) АК и 4,11 г/ч (55,5 ммолей/ч) BuOH, доводя общее количество свежей АК, введенной в систему, до 207,12 г/ч (2873 ммолей/ч) и общее количество введенного свежего BuOH до 211,14 г/ч (2853 ммолей/ч). Таким образом, в реактор 1 подавали в общей сложности 866,9 г/ч материала, состоявшего из 260,10 г/ч АК (3607 ммолей/ч), 406,6 г/ч BuOH (5495 ммолей/ч), сконденсированного головного погона органического слоя ГРУ, потока головного погона крекинг-реактора, остаточного продукта колонны для выделения АК и смеси БА/BuOH/H2O, представлявшей собой потоки со стадий рекуперации и возврата в процесс следующих потоков: (а) непрореагировавшего BuOH по ходу процесса после колонны для азеотропной отгонки BuOH/БА; (б) рекуперационного потока BuOH/БА из отпарки сбрасываемых в отход водных потоков перед подачей такого потока на очистку сточных вод и (в) части кубовых остатков дистилляционной колонны для конечного продукта (эти потоки представляли собой типичные сырьевые и возвращаемые в процесс потоки, используемые в непрерывном процессе в установке с полным циклом). Общее количество БА, направляемое в этерификационный реактор из таких источников, составляло 144,6 г/ч, из которых 84,14 г/ч составлял БА, возвращаемый в процесс из последующих по ходу процесса разделительных колонок и дополнительных направляемых в отход потоков. Следовательно, АК и BuOH использовали в реакторе 1 в молярном соотношении 1:1,52.
Продолжительность пребывания в реакторе поддерживали равной приблизительно 60 мин, в результате чего в общем в виде реакторного головного дистиллята через дистилляционную колонну Oldershaw отгоняли 995,9 г/ч продукта, который конденсировали и разделяли на две фазы. Часть органической фазы (216,1 г/ч) возвращали в головку дистилляционной колонны на орошение. Оставшиеся 719,8 г/ч органического дистиллята, содержавшего 45,20 г/ч АК, 183,10 г/ч BuOH и 451,0 г/ч БА, подавали в колонну 15 для выделения акриловой кислоты, которая описана ниже. Реакторный водный дистиллят (60,10 г/ч), содержавший 4,78 г/ч BuOH и 1,06 г/ч АК, разделяли следующим образом: 36,5 г/ч подавали в колонну для выделения АК и 23,6 г/ч направляли в ГРУ.
ГРУ 5 и крекинг-реактор 10 были идентичными таковым, которые описаны в примерах 7, 8 и 9. Соответственно 87,1 г/ч отходящего из этерификационного реактора по линии 3 потока и 128,7 г/ч дополнительных потоков, содержавших 11,3 г/ч BuOH, 38,3 г/ч АК, 37,1 г/ч БА и другие относящиеся к процессу высококипящие материалы и ингибиторы, по линии 4 направляли в ГРУ, в котором поддерживали температуру 122oC, давление 760 мм рт.ст., продолжительность пребывания 150 мин, 12,8 вес.% H2O и концентрацию кислотного катализатора 9,3 вес.% (H2SO4). Дополнительно в ГРУ вводили 23,60 г/ч водного дистиллята из реактора. Из ГРУ в целом отгоняли, конденсировали и разделяли 198,7 г/ч продукта. Весь выделенный водный дистиллят (114,6 г/ч), содержавший 4,35 г/ч BuOH и 6,25 г/ч АК, по линии 7 возвращали в ГРУ на орошение. Органический дистиллят (84,1 г/ч), содержавший 12,2 г/ч BuOH, 9,94 г/ч АК и 56,3 г/ч БА, по линии 8 возвращали в этерификационный реактор в виде рециркуляционного материала. Отходящий из ГРУ поток (155,2 г/ч) удаляли из нижней части ГРУ и направляли в крекинг-реактор 10, в котором поддерживали температуру 120oC, давление 35 мм рт. ст., продолжительность пребывания 180 мин и 24,0 вес.% H2SO4. В общей сложности в виде потока головного погона из крекинг-реактора отгоняли и конденсировали 70,6 г/ч продукта, содержавшего 2,00 г/ч BuOH, 34,3 г/ч АК и 9,29 г/ч БА. Этот конденсат выделяли и в виде рециркуляционного потока возвращали в главный этерификационный реактор, а отходящий из крекинг-реактора поток кубовых остатков сбрасывали в отход в виде отработавшего масла.
Колонна 15 для выделения акриловой кислоты диаметром 5,0 см включала 35 тарелок. Она представляла собой дистилляционную колонну Oldershaw, снабженную рубашкой для водяного пара, испарителем из нержавеющей стали и охлаждаемой водой конденсаторной системой. Соответственно в колонну для выделения акриловой кислоты, которая работала при давлении в головке 260 мм рт. ст. , температуре в нижней части 82oC и коэффициенте водяного орошения 12,5, подавали 719,8 г/ч конденсированного органического дистиллята из этерификационного реактора и 36,5 г/ч конденсированного водного дистиллята из реактора (состав описан выше). Отгонкой через колонну для выделения АК получали в общей сложности 1193,8 г/ч смеси в виде головного погона, которую конденсировали и разделяли. В общей сложности отбирали 526,4 г/ч выделенной водной фазы, 487,9 г/ч которой по линии 21 возвращали в головку колонны для выделения АК на орошение, а остальную водную фазу направляли дальше совместно с остальной частью конденсата. Богатую БА органическую фазу (667,4 г/ч), включавшую БА-продукт, также направляли дальше для дополнительного выделения; она была практически свободной от АК (содержание АК составляло 1500 част. /млн). Из колонны для выделения АК отводили 50,4 г/ч богатого АК потока кубовых остатков (43,56 г/ч АК) и по линии 17 возвращали в этерификационный реактор. Данные о рекуперации представлены в таблицах 10 и 11.
Во время проведения всего процесса получения БА согласно методике, описанной в данном примере, достигали выхода БА в пересчете на BuOH 100,5% и выхода БА 99,8% в пересчете на АК.

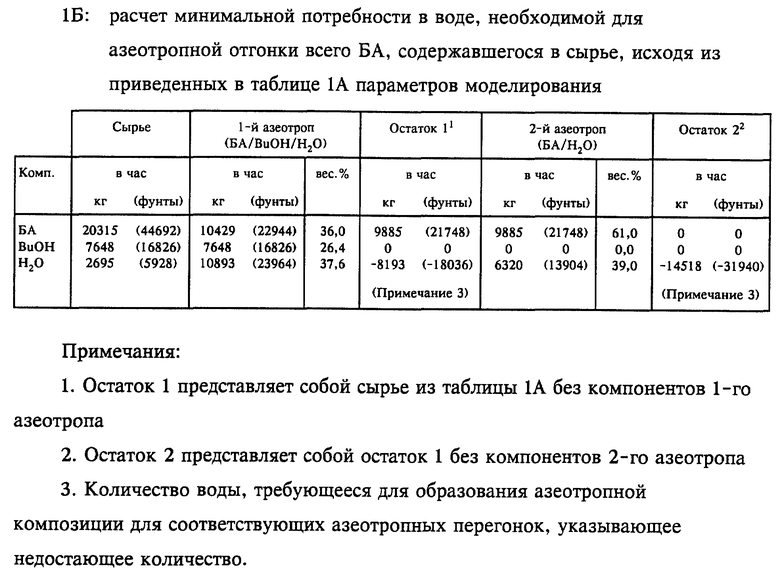
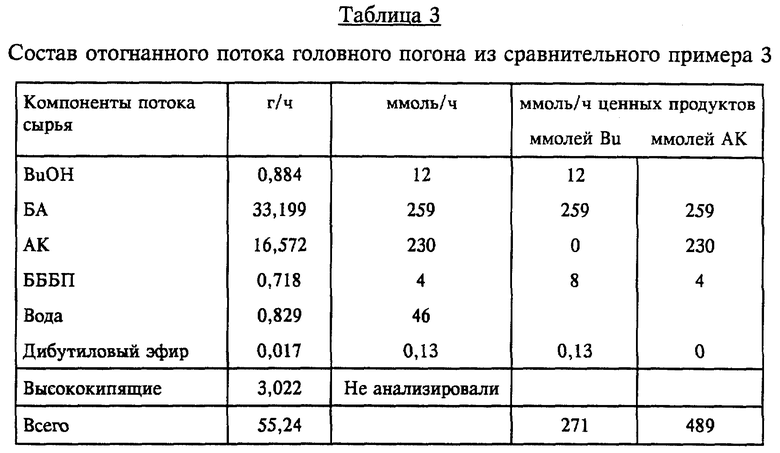
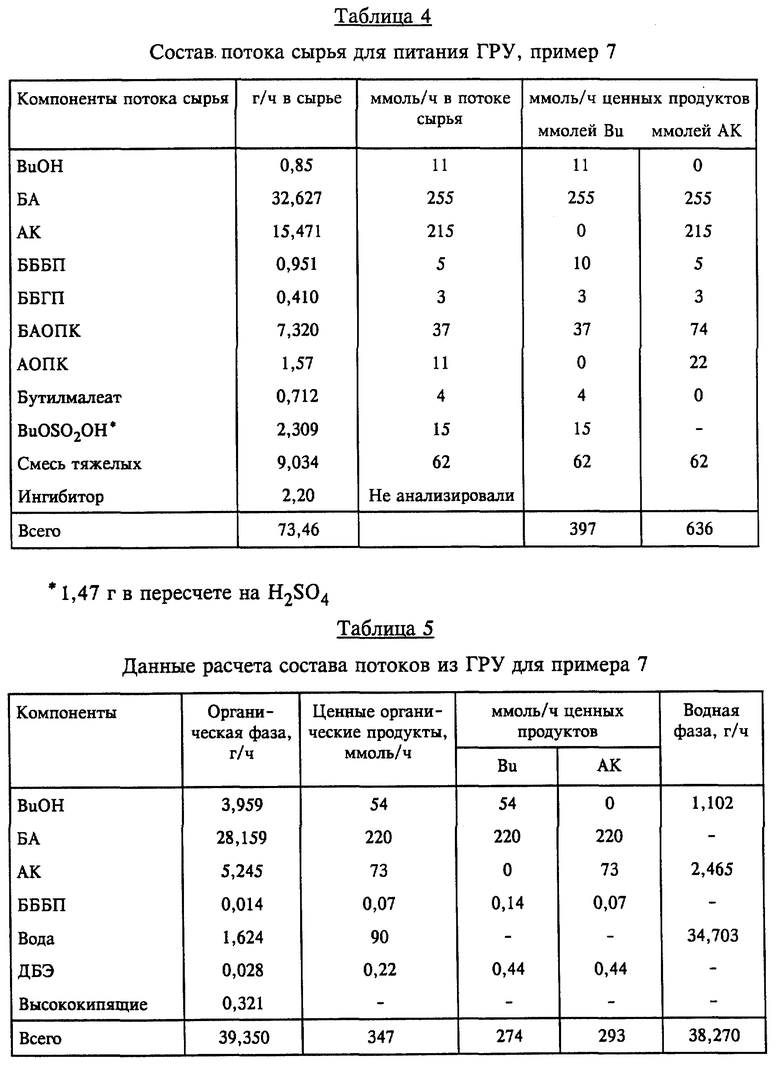
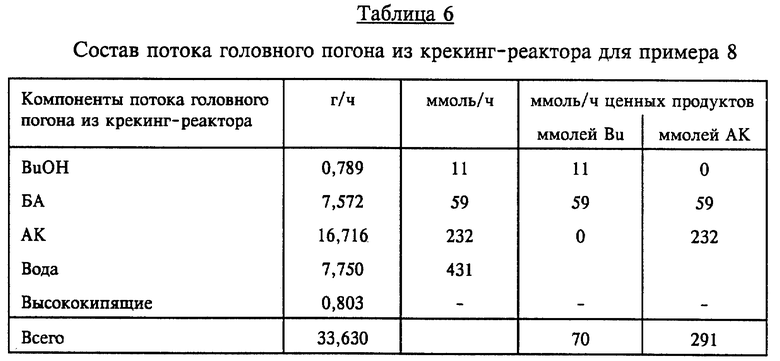
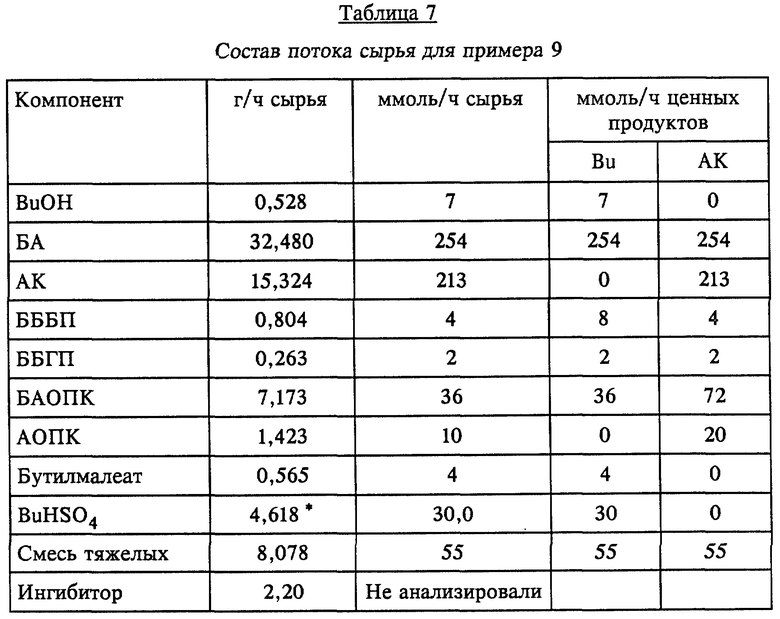
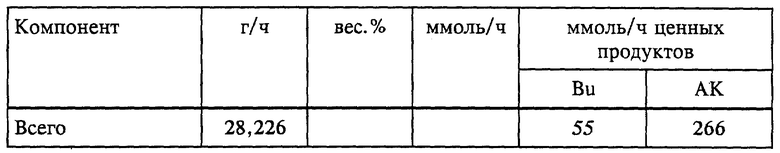
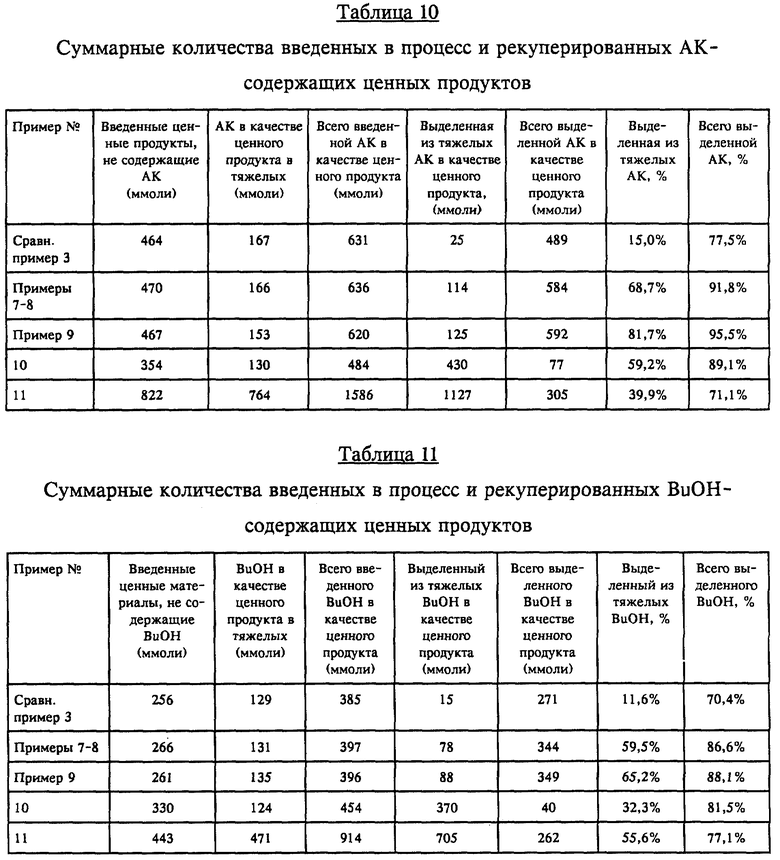
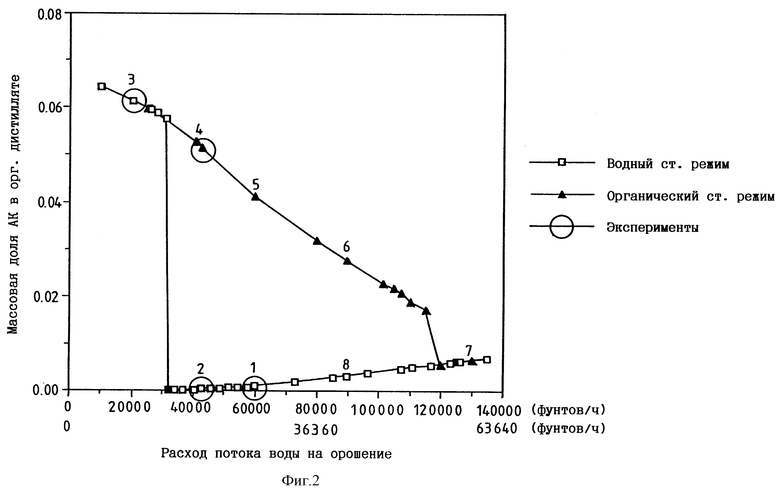
Изобретение относится к усовершенствованному способу получения алкилакрилата с высоким выходом продукта и высокой чистотой. Способ включает одну или несколько новых составляющих для проведения катализируемого кислотой процесса этерификации при получении алкилакрилата, в частности, н-бутилакрилата: 1) составляющую, связанную с гидролитической рекуперацией, согласно которой продукты присоединения в тяжелых фракциях, образующиеся во время катилизируемой кислотой этерификации, гидролизуют, выделяют и возвращают в процесс в виде ценных реагентов из гидролитической рекуперационной установки (ГРУ); 2) составляющую, связанную с крекинг-реактором, предпочтительно используемым совместно с ГРУ, согласно которой рекуперируют и после обработки в крекинг-реакторе возвращают в процесс дополнительное количество ценных реагентов; 3) новую дистилляционную составляющую, согласно которой поток сырого н-бутилакрилата эффективно перегоняют в водном режиме в колонне для выделения акриловой кислоты, в результате чего получают н-бутилакрилат, практически не содержащий акриловую кислоту, с высоким выходом продукта. Первые две составляющие применимы также к катализируемому кислотой процессу получения С1-С4 алкилакрилатов. Описан также способ непрерывного получения н-бутилакрилата, включающий все новые составляющие способа. Изобретение также включает выделение акриловой кислоты, С1-С4 алкилакрилата и С1-С4 спирта из тяжелых фракций, образующихся при получении С1-С4 алкилакрилата с использованием указанных составляющих. Преимуществом способа является получение алкилакрилата с низким содержанием акриловой кислоты и снижение потерь акриловой кислоты при отделении ее от этанола, воды и эфира. 5 с. и 12 з.п. ф-лы, 11 табл., 2 ил.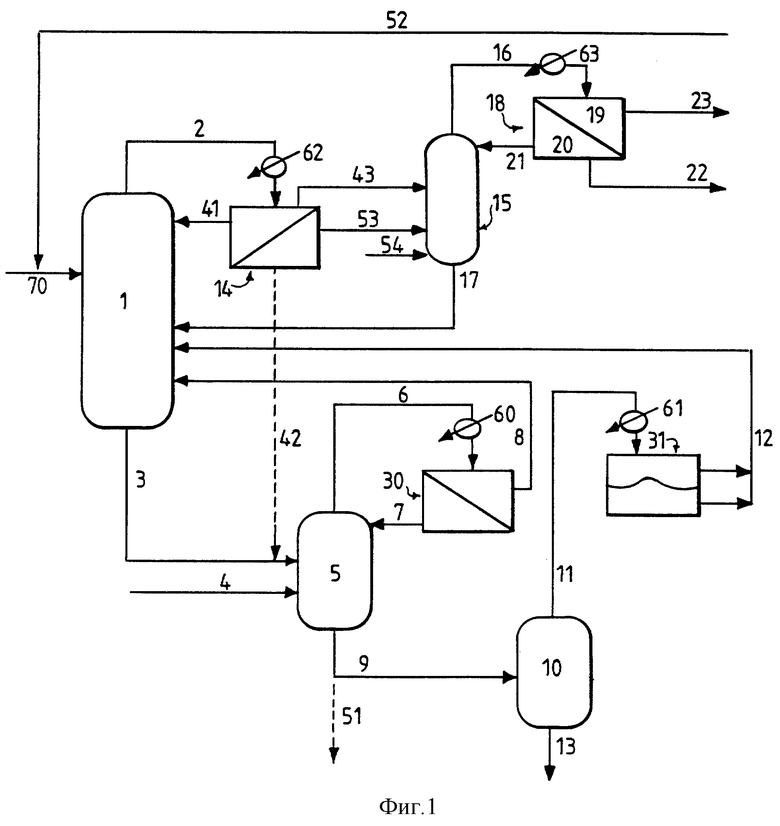
| Способ получения бутилакрилата | 1975 |
|
SU706397A1 |
| Контейнер для транспортирования и хранения жидких материалов | 1972 |
|
SU455906A1 |
| US 4012439 A, 19.03.1977 | |||
| US 4968834 A, 07.11.1990 | |||
| GB 1471800 A, 27.04.1977 | |||
| GB 1515620 A, 28.06.1978 | |||
| Способ определения качества плавленого сыра | 1977 |
|
SU1288596A1 |
