Область техники
Настоящее изобретение раскрывает способ получения гликозидов колхицина и тиоколхицина и их производных.
Уровень техники изобретения
Колхицин (I, R1=R2=OMe) представляет собой алкалоид, широко применяемый в течение длительного времени в терапии для лечения подагры. Кроме того, колхицин является очень сильнодействующим антибластическим агентом, который действует, блокируя образование митотического веретена в клеточном делении, это свойство полностью исследовано для любой антибластомной активности, и для этой цели получен большой ряд производных колхицина. Колхицин и тиоколхицин (I, R1=SMe, R2=OMe) являются исходными веществами, пригодными для получения серии активных препаратов.
Глюкозид 3-О-деметилтиоколхицина или тиоколхикозид (I, R1=SMe, R2=β-D-O-глюкозил) является активным ингредиентом в высшей степени важного применения в фармацевтической области, главным образом в терапии заболеваний костно-мышечной системы. Глюкозид 3-О-деметилколхицина или колхикозид (I, R1=OMe, R2=β-D-O-глюкозил) также является известным и наделен фармакологическими активностями.
Следовательно, были бы важными эффективные способы гликозидирования соединений типа колхицина для облегчения получения обоих соединений и новых производных для применения в фармакологическом исследовании.
FR 2112131 раскрывает способ глюкозидирования тиоколхицина, включающий взаимодействие 2,3,4,6-тетра-O-ацетил-α-D-глюкопиранозилбромида с деметилтиоколхицином. Этот способ является неудовлетворительным из-за низкого выхода и времени, требуемого для длительного и четкого процесса.
US 5777136 раскрывает процесс гликозидирования, который включает применение защищенных фторидов сахаров, в частности защищенных производных 1-фторглюкозы и 1-фторфукозы. Описанный процесс, в частности, ограничивается получением 3-О-глюкозильных или 3-О-фукозильных производных колхицина и тиоколхицина. Даже если описанные выходы являются удовлетворительными, этот способ требует получения подходящим образом активированных производных сахаров, т.е. фторидов сахара, что подразумевает дополнительную стадию при синтезе и получении и хранении реагентов относительной стабильности.
Сущность изобретения
Способ изобретения основан на применении защищенных производных 1-ацетилсахаров и, в частности, перацетилированных сахаров в альтернативе к 1-галогенсахарам в качестве гликозилирующих агентов для субстратов на основе колхицина и тиоколхицина. Эти соединения находятся в числе наипростейших, самых дешевых и определенно стабильных производных моносахаридов, и их применение является преимущественным для синтеза гликозилколхиноидов.
Следовательно, изобретение относится к способу получения соединений, имеющих формулу I:
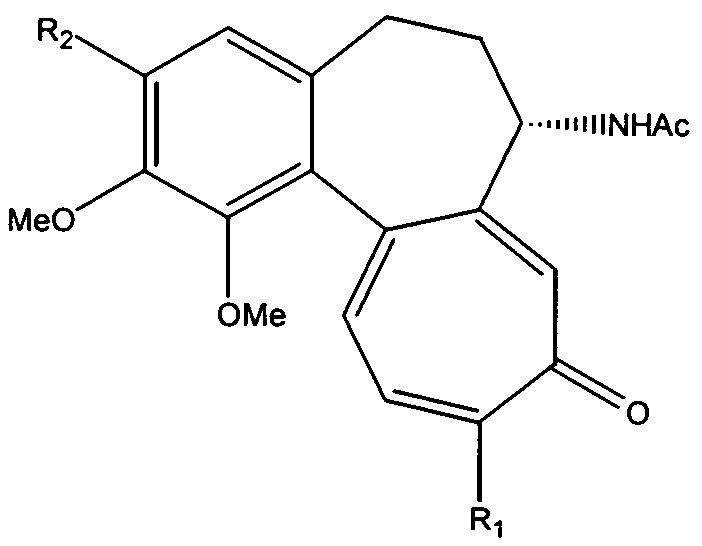
в которой:
- R1 является метокси или метилтиогруппой;
- R2 является О-гликозилокси остатком.
Способ получения соединений формулы I включает взаимодействие защищенного 1-ацетилированного сахара с соединением, имеющим формулу II:
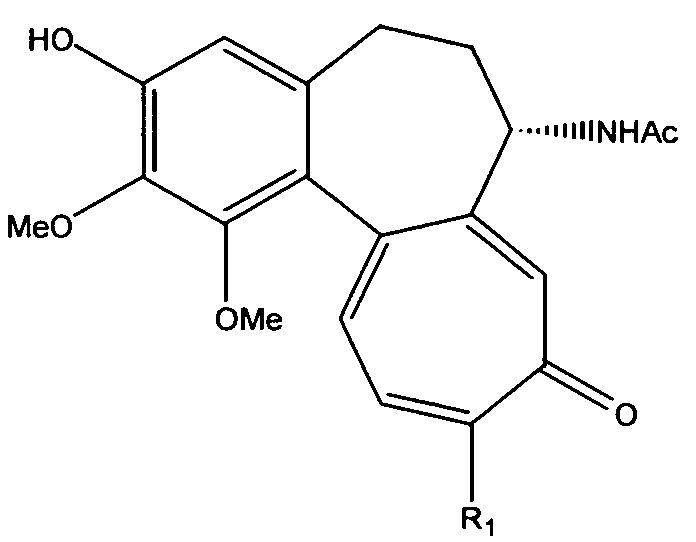
в которой R1 такой, как определено выше, с последующим гидролизом любых защитных групп, присутствующих в гликозильном остатке. 1-Ацетилгликозы обычно защищают группами сложных эфиров, типично ацетильными группами, таким образом, конечной стадией способа изобретения является удаление защитных ацетильных групп.
Подробное описание изобретения
Изобретение включает взаимодействие защищенного 1-ацетилсахара с соединением, имеющим формулу II. Затем полученный продукт подвергают гидролизу защитных групп, присутствующих в остатке глюкозы, с образованием гликозида колхицина или тиоколхицина, имеющего формулу I, в которой:
- R1 является метокси или метилтиогруппой;
- R2 является О-гликозилокси остатком.
Реакцию гликозидирования предпочтительно проводят в растворителе, выбираемом из ацетонитрила, нитрометана, галогенсодержащих углеводородов и их смесей. Особенно предпочтительным является применение ацетонитрила. Реакцию проводят в присутствии кислоты Льюиса, предпочтительно трифторида бора, в течение времени в интервале от 15 минут до 6 часов, предпочтительно от 30 минут до 2 часов, при температурах от 0°С до температуры флегмы растворителя, предпочтительно при комнатной температуре в среде инертной атмосферы. Для протекания реакции также требуется присутствие органического основания. Особенно предпочтительным является применение 1,1,3,3-тетраметилгуанидина. Способ изобретения имеет преимущество, заключающееся в стереоселективности, предоставленной только одним из двух возможных аномерных изомеров. В частности, в случае сахаров, принадлежащих к глюко-рядам (экваториальный С-2 заместитель, такой как в D-глюкозе, D-галактозе и D-ксилозе) селективно получают β-(1,2-транс) изомер. В случае сахаров, принадлежащих к манно-рядам (аксиальный С-2 заместитель, как в L-рамнозе) в качестве исходного материала, образуется исключительно α-(1,2-транс) изомер. Удаление защитных групп можно осуществить щелочным гидролизом в водной среде, когда не выделяется промежуточный перацетилгликозид, или нуклеофильным замещением, например, вторичными аминами, такими как диметиламин, диэтиламин, пирролидин, пиперидин или подобные.
Также конкретной целью настоящего изобретения является улучшенный способ синтеза широко известного тиоколхикозида или 3-О-β-D-глюкопиранозил-3-О-деметилтиоколхицина.
Способ изобретения предоставляет разнообразные преимущества по отношению к известным способам получения колхицина и тиоколхицина или их производных. Эти преимущества включают:
- обход получения подходящим образом активированного гликозильного реагента, такого как 1-галоген-сахар;
- применение стабильных и легко получаемых защищенных 1-ацетилом гликоз (например, перацетилгликоз);
- возможность кристаллизации конечного продукта непосредственно из исходной реакционной смеси;
- получение гликозилколхиноидов, с трудом получаемых с помощью способов предшествующего уровня техники, таких как галактозиды, рамнозиды и т.д.
Примеры
Следующие примеры иллюстрируют изобретение более подробно.
1) Синтез тиоколхикозида (3-О-β-D-глюкопиранозил-3-О-деметилтиоколхицина)
3-О-деметилтиоколхицин (2,0 г) суспендируют в ацетонитриле (20 мл) в атмосфере азота при комнатной температуре с последующим добавлением 1,1,3,3-тетраметилгуанидина (1,8 мл), раствора 1,2,3,4,6-пента-О-ацетил-β-D-глюкопиранозы (5,60 г) в ацетонитриле (10 мл) и в конце трифторида бора (7,2 мл).
Реакционную смесь перемешивают при комнатной температуре в течение 2 часов, затем охлаждают до 5°С и охлаждают добавлением 2 М КОН до рН≈6 (приблизительно 20 мл). Водный слой отделяют и экстрагируют ацетонитрилом (10 мл). Комбинированные органические слои последовательно промывают 0,5 М NaHSO4 (20 мл), 6% NaHCO3 (20 мл) и насыщенным солевым раствором (20 мл).
Растворитель удаляют под вакуумом и заменяют 95% этанолом (30 мл). Добавляют 2 М NaOH (40 мл) и раствор перемешивают до завершения (приблизительно 2 часа).
1 М NaHSO4 добавляют до рН=7, затем под вакуумом выпаривают этанол.
Водный слой дважды экстрагируют дихлорметаном (2х20 мл) и органическую фазу обратимо экстрагируют водой (20 мл), затем отбрасывают. Комбинированные водные слои экстрагируют смесью дихлорметана и этанола (1:1) до полной экстракции тиоколхикозида. Органический слой промывают 20% раствором NaCl (30 мл), затем концентрируют до 20 мл и оставляют кристаллизоваться при перемешивании в течение 2 ч при комнатной температуре. Продукт собирают фильтрацией. Получают 2,00 г тиоколхикозида (выход 71%) с физическими и спектроскопическими данными, идентичными тем, которые описаны в литературе.
2) Синтез 3-О-β-D-галактопиранозил-3-О-деметилтиоколхицина
3-О-деметилтиоколхицин (1,0 г) суспендируют в ацетонитриле (10 мл) в атмосфере азота при комнатной температуре с последующим добавлением 1,1,3,3-тетраметилгуанидина (0,9 мл), раствора 1,2,3,4,6-пента-О-ацетил-β-D-галактопиранозы (2,80 г) в ацетонитриле (10 мл) и в конце трифторида бора (3,6 мл).
Реакционную смесь перемешивают при комнатной температуре в течение 2 часов, затем охлаждают до 5°С и охлаждают добавлением 2 М КОН до рН≈6 (приблизительно 10 мл). Водный слой отделяют и экстрагируют ацетонитрилом (10 мл). Комбинированные органические слои последовательно промывают 1 М NaHSO4 (10 мл), 6% NaHCO3 (10 мл) и солевым раствором (10 мл).
Растворитель удаляют под вакуумом и замещают 95% этанолом (20 мл). Добавляют 2 М NaOH (20 мл) и раствор перемешивают до завершения (приблизительно 2 часа).
0,5 М NaHSO4 добавляют до рН=7, затем под вакуумом выпаривают этанол.
Водный слой дважды экстрагируют дихлорметаном (2х20 мл) и органическую фазу обратимо экстрагируют водой (20 мл), затем отбрасывают. Комбинированные водные слои экстрагируют смесью дихлорметана и этанола (1:1) до полной экстракции тиоколхикозида. Органический слой промывают 20% раствором NaCl (20 мл), затем растворитель заменяют метанолом и концентрируют до 15 мл и оставляют кристаллизоваться при перемешивании в течение 2 ч при комнатной температуре. Продукт собирают фильтрацией. Получают 855 г продукта (выход 61%), mp=255-6°С.
ЯМР 1Н (ДМСО-d6) δ (м.д.): 1,86 (1Н, м); 1,87 (3Н, с); 2,06 (1Н, м); 2,24 (1Н, м); 2,43 (3Н, с); 2,55 (1Н, м); 3,45 (1Н, м); 3,57 (3Н, с); 3,60 (1Н, м); 3,62 (1Н, м); 3,67 (1н, м); 3,74 (1Н, м); 3,87 (3Н, с); 4,36 (1Н, м); 4,52 (1Н, д, J=4,4 Гц); 4,68 (1Н, т, J=5,7 Гц); 4,85 (1Н, д, J=5,7 Гц); 4,91 (1Н, д, J=7,8 Гц); 5,14 (1Н, д, J=5,6 Гц); 6,88 (1Н, с); 7,04 (1Н, с); 7,17 (1Н, д, J=10,7 Гц); 7,29 (1Н, д, J=10,7 Гц); 8,60 (1Н, д, J=7,3 Гц).
MS+ (масса/заряд): 1149,0 [2M+Na]+, 1126,7 [2M+H]+, 586,3 [M+Na]+, 564,2 [M+Н]+, 402,2 [M-гал+Н]+.
3) Синтез 3-О-α-L-рамнопиранозил-3-О-деметилтиоколхицина
3-О-деметилтиоколхицин (2,0 г) суспендируют в ацетонитриле (20 мл) в атмосфере азота при комнатной температуре с последующим добавлением 1,1,3,3-тетраметилгуанидина (1,8 мл), раствора 1,2,3,4-тетра-О-ацетил-β-L-рамнопиранозы (4,77 г) в ацетонитриле (10 мл) и в конце трифторида бора (8,4 мл).
Реакционную смесь перемешивают при комнатной температуре в течение 3 часов, затем охлаждают до 5°С и охлаждают добавлением 2 М NaOH до рН≈6 (приблизительно 10 мл). Водный слой отделяют и экстрагируют ацетонитрилом. Комбинированные органические слои последовательно промывают 1 М NaHSO4, 6% NaHCO3 и солевым раствором.
Растворитель удаляют под вакуумом и заменяют 95% этанолом (20 мл). Добавляют 2 М NaOH (15 мл) и раствор перемешивают до завершения (приблизительно 2 часа).
1 М NaHSO4 добавляют до рН=7, затем под вакуумом выпаривают этанол.
Водный слой дважды экстрагируют дихлорметаном (2х20 мл) и органическую фазу обратимо экстрагируют водой (20 мл), затем отбрасывают. Комбинированные водные слои экстрагируют смесью дихлорметана и этанола (1:1) до полной экстракции тиоколхикозида. Органический слой промывают 20% раствором NaCl (30 мл), затем растворитель заменяют метанолом и концентрируют до 15 мл и оставляют кристаллизоваться при перемешивании в течение 2 ч при комнатной температуре. Продукт собирают фильтрацией. Получают 2,03 г 3-О-α-L-рамнопиранозил-3-О-деметилтиоколхицина (выход 78%), mp=254-5°С.
ЯМР 1Н (ДМСО-d6) δ (м.д.): 1,19 (3Н, д, J=6,6 Гц); 1,87 (1Н, м); 1,88 (3Н, с); 2,04 (1Н, м); 2,22 (1Н, м); 2,44 (3Н, с); 2,61 (1Н, м); 3,35 (1Н, м); 3,59 (3Н, с); 3,70 (1Н, м); 3,71 (1Н, м); 3,85 (3Н, с); 4,35 (1Н, м); 4,82 (1Н, д, J=5,7 Гц); 4,94 (1Н, д, J=5,7 Гц); 5,12 (1Н, д, J=4,3 Гц); 5,39 (1Н, д, J=1,8 Гц); 6,88 (1Н, с); 7,05 (1Н, с); 7,16 (1Н, д, J=10,7 Гц); 7,29 (1Н, д, J=10,7 Гц); 8,68 (1Н, д, J=7,5 Гц).
MS+ (масса/заряд): 1117,1 [2M+Na]+, 570,3 [M+Na]+, 548,2 [M+Н]+, 402,2 [M-rha+Н]+.
4) Синтез 3-О-β-D-ксилопиранозил-3-О-деметилтиоколхицина
15,0 г 3-О-деметилтиоколхицина суспендируют в 140 мл ацетонитрила при перемешивании в атмосфере азота.
Добавляют 13,5 мл 1,1,3,3-тетраметилгуанидина, превращая смесь в раствор кроваво-красного цвета. Добавляют 34,3 г тетраацетата D-ксилозы в 60 мл ацетонитрила и в конце добавляют по каплям 45 мл BF3·ET2O, сохраняя внутреннюю температуру при приблизительно 20°С. Раствор перемешивают в течение 2 часов до завершения, затем охлаждают до 5°С и регулируют до рН~7 с помощью 120 мл 2 М NaOH.
Фазы разделяют и водную фазу обратимо экстрагируют с помощью 20 мл ацетонитрила. Органический слой последовательно промывают 50 мл 1 М NaHSO4, 60 мл 5% NaHCO3 и в конце 50 мл соляного раствора. Органический слой концентрируют до объема, равного 150 мл, добавляют 50 мл простого трет-бутилметилового эфира и смесь оставляют кристаллизоваться при комнатной температуре в течение 1 ч. Твердое вещество собирают фильтрацией, промывают с помощью 40 мл смеси ацетонитрила и простого трет-бутилметилового эфира в соотношении 1:1 и высушивают, получая 19,8 г 3-О-β-D-(2',3',4'-триацетил)ксилопиранозил-3-О-деметилтиоколхицина.
19,4 г этого промежуточного продукта суспендируют в 300 мл метанола, добавляют 16 мл диэтиламина и смесь нагревают до 40°С в течение 2 часов до завершения.
Полученный раствор концентрируют до объема, равного 110 мл, и дают возможность кристаллизоваться в течение 1 ч при комнатной температуре. Твердое вещество собирают фильтрацией, промывают с помощью 25 мл метанола и высушивают, получая 13,7 г продукта (общий выход: 70%), mp=233-4°С.
ИК см-1: 3295, 2940, 2867, 1636, 1601, 1558, 1507, 1480, 1424, 1348, 1317, 1074, 1029, 870, 594.
ЯМР 1Н (ДМСО-d6) δ (м.д.): 1,86 (1Н, м); 1,87 (3Н, с); 2,04 (1Н, м); 2,24 (1Н, м); 2,43 (3Н, с); 2,61 (1Н, м); 3,28 (1Н, м); 3,31 (1Н, м); 3,33 (1Н, м); 3,43 (1Н, м); 3,57 (3Н, с); 3,82 (1Н, дд, J=11,2 Гц, 5,1 Гц); 3,86 (3Н, с); 4,36 (1Н, м); 4,98 (1Н, д, J=7,2 Гц); 5,08 (1Н, д, J=4,8 Гц); 5,11 (1Н, д, J=4,8 Гц); 5,34 (1Н, д, J=5,7 Гц); 6,86 (1Н, с); 7,05 (1Н, с); 7,16 (1Н, д, J=10,7 Гц); 7,28 (1Н, д, J=10,7 Гц); 8,60 (1Н, д, J=7,3 Гц).
MS+ (масса/заряд): 1067,7 [2M+Na]+, 1066,5 [2M+H]+, 556,2 [M+Na]+, 534,2 [M+Н]+, 402,2 [M-ксил+Н]+.
5) Синтез 3-О-β-D-ксилопиранозил-3-О-деметилколхицина
2,0 г 3-О-деметилколхицина суспендируют в 18 мл ацетонитрила при перемешивании в атмосфере азота.
Добавляют 1,9 мл 1,1,3,3-тетраметилгуанидина, превращая смесь в раствор кроваво-красного цвета. Добавляют раствор 4,9 г тетраацетата D-ксилозы в 10 мл ацетонитрила и в конце добавляют по каплям 5,2 мл BF3·ET2O, сохраняя внутреннюю температуру при приблизительно 20°С. Раствор перемешивают в течение 2 часов до завершения, затем охлаждают до 5°С и регулируют до рН~7 с помощью 2 М NaOH.
Фазы разделяют и водную фазу обратимо экстрагируют с помощью 10 мл ацетонитрила. Органический слой последовательно промывают 1 М NaHSO4, 5% NaHCO3 и в конце соляным раствором. Растворитель заменяют метанолом (30 мл), добавляют 6,4 мл диэтиламина и смесь нагревают до 40°С в течение 2 ч до завершения.
Растворитель выпаривают и осадок очищают колоночной хроматографией с помощью DCM:MeOH 85:15. Собирают фракции, содержащие продукт, и удаляют растворитель, получая 2,29 г аморфного продукта (общий выход: 86%).
ЯМР 1Н (ДМСО-d6) δ (м.д.): 1,86 (1Н, м); 1,87 (3Н, с); 2,03 (1Н, м); 2,23 (1Н, м); 2,59 (1Н, м); 3,28 (1Н, м); 3,31 (1Н, м); 3,33 (1Н, м); 3,42 (1Н, м); 3,55 (3Н, с); 3,81 (1Н, дд, J=10,8 Гц, 5,0 Гц); 3,86 (3Н, с); 3,90 (3Н, с); 4,35 (1Н, м); 4,97 (1Н, д, J=7,2 Гц); 5,06 (1Н, д, J=4,6 Гц); 5,10 (1Н, д, J=4,8 Гц); 5,33 (1Н, д, J=5,3 Гц); 6,84 (1Н, с); 7,04 (1Н, д, J=10,7 Гц); 7,12 (1Н, д, J=10,7 Гц); 7,15 (1Н, с); 8,60 (1Н, д, J=7,3 Гц). MS+ (масса/заряд): 1056,8 [2M+Na]+, 540,3 [M+Na]+, 518,2 [M+Н]+, 386,2 [M-ксил+Н]+.
| название | год | авторы | номер документа |
|---|---|---|---|
| АНАЛОГИ КОЛХИКОЗИДА | 2004 |
|
RU2343157C2 |
| ПРОИЗВОДНЫЕ КОЛХИЦИНА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1996 |
|
RU2163241C2 |
| СПОСОБ БИОТРАНСФОРМАЦИИ СОЕДИНЕНИЙ КОЛХИЦИНОИДОВ В СООТВЕТСТВУЮЩИЕ 3-ГЛИКОЗИЛПРОИЗВОДНЫЕ | 1997 |
|
RU2196826C2 |
| ПРОИЗВОДНЫЕ ТИОКОЛХИЦИНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, СОЕДИНЕНИЕ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1998 |
|
RU2190598C2 |
| БИОТРАНСФОРМАЦИЯ КОЛХИЦИНОИДНЫХ СОЕДИНЕНИЙ | 2004 |
|
RU2346050C2 |
| ПРОИЗВОДНЫЕ ТАКСАНА, ФУНКЦИОНАЛИЗИРОВАННЫЕ ПО 14-ПОЛОЖЕНИЮ, И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2003 |
|
RU2320652C2 |
| СПОСОБ ПОЛУЧЕНИЯ 2-(1-АДАМАНТИЛКАРБОНИЛ)-1,2-ДИГИДРОНАФТО[2,1-b]ФУРАНОВ | 2011 |
|
RU2495033C2 |
| Нуклеозидные производные 1,3-диаза-2-оксофеноксазина в качестве ингибиторов репликации герпесвирусов. | 2019 |
|
RU2731381C1 |
| ПРОИЗВОДНЫЕ КОЛХИЦИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1995 |
|
RU2145598C1 |
| ПРОИЗВОДНОЕ 23-ИН-ВИТАМИНА D | 2011 |
|
RU2558362C2 |
В настоящем изобретении раскрыт способ получения соединений, имеющих формулу I, в которой R1 является метокси или метилтиогруппой; R2 является О-гликозилокси остатком. Соединения формулы I получают посредством взаимодействия перацетилированной гликозы с соединением, имеющим формулу II, с последующим удалением защитных групп из гликозильного фрагмента. 8 з.п. ф-лы, 4 пр.


1. Способ получения соединений, имеющих формулу I:
где
- R1 является метокси или метилтиогруппой;
- R2 является О-гликозилокси остатком;
включающий взаимодействие перацетилированной гликозы с соединением, имеющим формулу II:
где R1 такой, как определено выше, с последующим удалением защитных групп из гликозильного фрагмента.
2. Способ по п.1, в котором указанную перацетилированную гликозу или D- или L-рядов защищают группами сложных эфиров.
3. Способ по пп.1, 2, в котором указанные защитные группы расщепляют гидролизом в щелочной среде.
4. Способ по пп.1, 2, в котором указанные защитные группы расщепляют через нуклеофильное вытеснение посредством взаимодействия с амином.
5. Способ по п.1, в котором взаимодействие проводят в растворителе, выбираемом из группы, состоящей из ацетонитрила, нитрометана, галогенсодержащих углеводородов и их смесей.
6. Способ по п.1, в котором взаимодействие проводят в присутствии кислоты Льюиса.
7. Способ по п.6, в котором кислотой Льюиса является трифторид бора.
8. Способ по п.1, в котором взаимодействие проводят в присутствии органического основания.
9. Способ по п.8, в котором указанным основанием является 1,1,3,3-тетраметилгуанидин.
| АЛМАЗНАЯ БУРОВАЯ КОРОНКА | 1996 |
|
RU2112131C1 |
| US 5777136 A, 07.07.1998 | |||
| Способ получения 3-глюкоз-2-ил3-деметилтиоколхицина или 2-глюкоз2-ил-2-деметилтиоколхицина | 1972 |
|
SU508204A3 |
| Приспособление в пере для письма с целью увеличения на нем запаса чернил и уменьшения скорости их высыхания | 1917 |
|
SU96A1 |
| EP 0789028 A2, 13.08.1997. | |||

