Изобретение относится к покрытиям для поверхностей твердых тканей или поверхностей в области рта. Более конкретно настоящее изобретение относится к прочным покрытиям для поверхностей твердых тканей или поверхностей в области рта.
Настоящая заявка является частичным продолжением заявки США N 08/163028, поданной 6 декабря 1993 г., в настоящее время находящейся на рассмотрении.
Бляшка служит общей причиной кариеса, заболеваний десен и изменения цвета зубов и в значительной степени способствует их развитию. Бляшка появляется, когда к кутикуле, т. е. белковой пленке на поверхности зубов, пристают кариогенные бактерии. В свою очередь бляшка служит ядром для формирования зубного камня. По мере формирования и затвердевания зубного камня он имеет тенденцию к окрашиванию в связи с поглощением пищевых красителей. Кроме того, восстановительные материалы области рта могут быть по своей сущности подвержены накоплению налета из пищевых красителей. Желательно иметь средства, позволяющие избежать поглощения и приставания бактерий к твердой ткани и к поверхностям в области рта.
Для подавления процесса окрашивания предлагается включать в средства для чистки зубов силиконовые масла, для которых характерна гидрофобность. Однако они обычно обладают относительно низкой способностью прилипать и удерживаться на поверхности зубов.
В патенте США N 5078988, выданном Лину и др., описаны средства для чистки зубов, включающие модифицированные аминоалкилсиликоны. Утверждают, что модифицированные силиконы образуют на поверхности зуба гидрофобный слой, предупреждающий кариес и окрашивание. Заявка по договору о патентной кооперации WO 91/13608, поданная Роллой и др., описывает средства для чистки зубов, содержащие жидкое силиконовое масло и жирорастворимое антибактериальное средство, которое, как указывают, обеспечивает защиту зубов от формирования бляшек за счет медленного выделения антибактериального средства в слюну.
В патенте США N 4981903, выданном Гарбе и др., описаны адгезивные составы, склеивающие и несклеивающие при надавливании и состоящие из полихлорвиниловой основы с привитыми боковыми полисилоксанами. Показано, что эти составы могут быть полезны при местном использовании в качестве связующих материалов в косметических и лекарственных средствах, а также в качестве герметиков для таких пористых материалов как бумага и древесина. См. колонку 3, строки 25-31. В патенте США N 4972037, выданном Гарбе и др., описаны составы, которые могут служить адгезивом при комнатной температуре и которые содержат сополимер, включающий как боковые фторхимические группы, так и привитые боковые полисилоксаны. Эти составы также могут быть полезны при местном применении, включая применение косметических и лекарственных средств, и в качестве герметиков с пористыми материалами, такими как бумага и древесина (см. колонку 3, строки 25-33). В патенте США N 4981902, выданном Митре и др., описаны несклеивающие при надавливании адгезивные акрилатные или метакрилатные полимеры с боковыми привитыми полисилоксанами. Эти полимеры содержат мономеры с полярной функциональностью и описаны как полезные составные части в составах покрытий для тел животных.
В патенте США N 4693935, выданном Мазуреку, описаны склеивающие при надавливании адгезивные составы, состоящие из сополимера, включающего полихлорвиниловую основу с привитыми к ней полисилоксановыми частями. В патенте США N 4728571, выданном Клеменсу и др., описаны составы антиадгезионных покрытий, содержащие полисилоксановый привитой сополимер и его смеси на листовом материале.
Настоящее изобретение предлагает покрытия для поверхностей твердых тканей или поверхностей в области рта, и такие покрытия содержат полимер, состоящий из повторяющихся звеньев:
A) 1-80 мас.% полярной или поляризуемой группы;
B) 0-98 мас.% модулирующей группы;
C) 1-40 мас.% гидрофобной привитой полисилоксановой цепи с молекулярной массой, равной по меньшей мере 500.
Настоящее изобретение относится также к зубным составам, пригодным для нанесения покрытий на поверхности в области рта человека, которые содержат полимер, состоящий из повторяющихся звеньев:
A) 1-80 мас.% полярной или поляризуемой группы;
B) 0-98 мас.% модулирующей группы;
C) 1-40 мас.% гидрофобной привитой полисилоксановой цепи с молекулярной массой, равной по меньшей мере 500,
в которых указанный полимер дополнительно содержит по меньшей мере одну силановую часть, способную к реакции конденсации.
Эти составы в качестве необязательных добавок могут также содержать катализаторы, способствующие реакции конденсации силана, и дополнительное соединение, состоящее по меньшей мере из двух центров реакции конденсации силикона, в которых возможно возникновение реакции конденсации. Это дополнительное соединение действует как мостиковое соединение между описанными выше полимерами после завершения реакции конденсации.
Кроме того, неожиданно было обнаружено, что обработка поверхностей с указанным покрытием поверхностно-активным веществом способствует значительному улучшению сопротивления окрашиванию и адгезии бактерий.
Настоящее изобретение предлагает также другие примеры осуществления полимеров для покрытия поверхностей твердых тканей или поверхностей в области рта, которые могут сшиваться на поверхности.
И еще в одном примере осуществления изобретения предлагаются зубные приспособления, включающие описанную выше полимерную систему.
На фиг. 1 показана фотография осколка зуба с нанесенным на него покрытием по настоящему изобретению, на которой можно видеть, что краситель не удерживается на зубном материале.
На фиг. 2 показана фотография осколка зуба с нанесенным на него покрытием из сравнительного примера 1 по настоящему изобретению, на которой можно видеть, что краситель удерживается на зубном материале.
На фиг. 3 показана схема, демонстрирующая относительный уровень сцепления протеинов и бактерий с частицами эмали, промытыми фосфатным буферным солевым раствором.
На фиг. 4 показана схема, демонстрирующая относительный уровень сцепления протеинов и бактерий с частицами эмали, промытыми раствором NP-40, содержащим поверхностно-активное вещество.
Настоящее изобретение относится к покрытиям для твердых тканей, таких как дентин, эмаль, цементное вещество и кость. Кроме того, покрытие может быть нанесено на другие поверхности в области рта, включая поверхности зубных пломб и ортодонтических или ортопедических зубных изделий. Зубные пломбы включают пломбы, изготовленные из составов на основе смол, амальгамы, стеклянных иономеров, керамики и полученных из них разнообразных смешанных материалов. Ортодонтические изделия включают ортодонтические кронштейны, проволоку и т.п. Ортопедические изделия включают зубные мосты, коронки, зубные протезы и т.п.
Покрытие наносят в количестве, достаточном для того, чтобы обеспечить сопротивление поверхности с покрытием адгезии бактерий, образованию бляшек или окрашиванию пищей или красителями. Покрытие может быть нанесено в виде сплошного или наполовину сплошного слоя. Предпочтительно нанесение покрытия в количестве, достаточном для получения на покрытой поверхности одного, по существу сплошного слоя описанного выше полимера.
Покрытия в соответствии с настоящим изобретением в высокой степени субстантивны к перечисленным выше поверхностям. Эти покрытия обладают низким коэффициентом трения и высокой устойчивостью к бляшке, бактериям, окрашиванию пищей и т.п.
Кроме того, неожиданно было обнаружено, что обработка поверхности с описанным выше покрытием составом, содержащим поверхностно-активное вещество, способствует повышению сопротивления адгезии бактерий и белковых веществ. Операция обработки поверхностно-активным веществом обеспечивает этот необычный положительный эффект даже в тех случаях, когда поверхность с покрытием подвергается перед операцией обработки поверхностно-активным веществом воздействию бактерий и белковых веществ. Таким образом, описанное здесь покрытие, подвергнутое обработке содержащим поверхностно-активное вещество составом, очевидно физически отличается от покрытий, которые не были подвергнуты обработке составом, содержащим поверхностно-активное вещество. При отсутствии теоретических объяснений полагают, что обработка поверхностно-активным веществом ориентирует полисилоксановый компонент полимера покрытия, улучшая таким образом показатели сопротивления адгезии бактерий и окрашиванию покрытия.
Обработка поверхностно-активным веществом может быть выполнена (i) как часть первоначального нанесения покрытия, (ii) после первоначального нанесения покрытия, но до того, как поверхность с покрытием подвергнется воздействию нежелательных организмов и белковых веществ или (iii) после того как поверхность с покрытием подвергнется воздействию бактерий и т.п. В последнем случае обработка поверхностно-активным веществом может периодически повторяться.
Покрытие, являющееся предметом настоящего изобретения, содержит виниловый сополимер, включающий повторяющиеся звенья A, B и C, где A является производным этиленоненасыщенного мономера, содержащего по меньшей мере одну полярную или поляризуемую группу, B является производным этиленоненасыщенного мономера, которое может содержать модифицирующие группы, и C является производным этиленоненасыщенной органосилоксановой цепи. Предпочтительно, чтобы растворимость полимера в воде составляла менее 0,1%.
Более конкретно звено A является производным виниловых мономеров, таких как акрилаты, метакрилаты, кротонаты, итаконаты и т.п. Полярные группы могут быть кислотными, основными или солью. Эти группы также могут быть ионными или нейтральными.
В качестве примера полярных или поляризуемых групп можно назвать нейтральные группы, такие как окси-, тио-, замещенная и незамещенная амидогруппы, циклические эфиры (такие как оксаны, оксетаны, фураны и пураны), основные группы (такие как фосфины и амины, включая первичные, вторичные и третичные амины), кислотные группы (такие как кислородные кислоты, тиокислородные кислоты С, S, P, В) и ионные группы (такие как четвертичный аммоний, соль карбоновой кислоты, соль сульфокислоты и т.п.) и исходные вещества, и защищенные формы этих групп. Кроме того, А может быть макромономером. Ниже следуют более конкретные примеры этих групп.
Звенья А могут быть производными моно- или многофункциональной карбоксильной группы, содержащей молекулы, представленные следующей обобщенной формулой:
CH2=CR2G-(COOH)d
где R2 - H, метил, этил, циано, карбокси или карбоксиметил, d = 1-5 и G является связью или связующей группой радикала гидрокарбила, содержащей 1-12 атомов углерода с валентностью d+1, и может замещаться и/или прерываться замещенным или незамещенным гетероатомом (таким как О, S, N и P).
Возможно представление этого звена в форме соли. Предпочтительными мономерами этого класса являются акриловая кислота, метакриловая кислота, итаконовая кислота и N-акрилоилглицин.
Звенья a могут, например, быть производными моно- или многофункциональной карбоксильной группы, содержащей молекулы, представленные следующей обобщенной формулой:
CH2=CR2-CO-L-R3-(OH)d
где R2 - H, метил, этил, циано, карбокси или карбоксиалкил, L - O, NH, d = 1-5 и R3 является радикалом гидрокарбила, содержащим 1-12 атомов углерода с валентностью d+1. Предпочтительными мономерами этого класса являются оксиэтил (мет)акрилат, оксипропил (мет)акрилат, оксибутил (мет)акрилат, глицерин моно(мет)акрилат, три(оксиметил)этан моноакрилат, пентаэротритол моно(мет)акрилат, N-оксиметил (мет)акриламид, оксиэтил (мет)акриламид и оксипропил (мет)акриламид.
Звено A может, с другой стороны, быть производным моно- или многофункциональной карбоксильной группы, содержащей молекулы, представленные следующей обобщенной формулой:
CH2=CR2-CO-L-R3-(NR4R5)d,
где R2, L, d и R3 имеют значения, указанные выше, а R4 и R5 являются H или алкильными группами, содержащими 1-12 атомов углерода или же вместе они образуют карбоксильную или гетероксильную группу. Предпочтительными мономерами этого класса являются аминоэтил (мет)акрилат, аминопропил (мет)акрилат, N, N-диметиламиноэтил (мет)акрилат, N, N-диэтиламиноэтил (мет)акрилат, N,N-диметиламинопропил (мет)акриламид, N-изопропиламинопропил (мет)акриламид и 4-метил-1-акрилоил-пиперазин.
Звено A может также быть производным алкокси замещенных (мет)акрилатов или (мет)акриламидов, таких как метоксиэтил (мет)акрилат, 2(2-этоксиэтокси)этил (мет)акрилат, полиэтиленгликоль моно(мет)акрилат или полипропиленгликоль моно(мет)акрилат.
Звенья могут быть производными замещенных или незамещенных мономеров аммония, представленных следующей обобщенной формулой:
CH2CR2-CO-L-R3-(NR4R5R6)dQ,
где R2, R3, R4, R5, L и d имеют значения, указанные выше, R6 является H или алкилом, содержащим 1-12 атомов углерода, a Q является органическим или неорганическим анионом.
Предпочтительными примерами таких мономеров являются 2-N,N,N-триметиламмониевый этил(мет)акрилат, 2-N, N,N-триэтиламмониевый этил(мет)акрилат, 3-N, N, N-триметиламмониевый пропил(мет)акрилат, N(2-N',N',N'-триметиламмониевый) этил(мет)акриламид, N-(димeтилoкcиэтилaммoниeвый) пропил(мет)акриламид и т.п., где противоионом может быть фторид, хлорид, бромид, ацетат, пропионат, лаурат, пальмитат, стеарат и т.п. Мономер может также быть N,N-диметилдиалиламмониевой солью органического или неорганического противоиона.
Полимеры, содержащие группу аммония, могут также быть получены путем использования в качестве звена A любой из аминовых групп, содержащих описанный выше мономер, и подкисления полученных полимеров органической или неорганической кислотой до значения pH, при котором дополнительные аминовые группы по существу присоединяют протон. Полимеры, содержащие полностью замещенную группу аммония, могут быть получены путем алкилирования описанных выше аминовых полимеров алкилирующими группами, способом, общеизвестным как реакция Менчуткина.
Звено A по настоящему изобретению может также быть производным группы мономеров, содержащих сульфокислоту, таких как винилсульфокислота, стиролсульфокислота, 2-акриламидо-2-метил пропансульфокислота, аллилоксибензолсульфокислота и т.п. C другой стороны, звено A может быть производным группы мономеров, содержащих фосфорную кислоту или борную кислоту. Эти мономеры могут использоваться в качестве мономеров в форме кислоты с добавленным протоном, и полученные соответствующие полимеры могут быть нейтрализованы органическим или неорганическим основанием, чтобы перевести полимеры в форму солей.
Звено B является производным акрилата или метакрилата, или иных виниловых полимеризуемых исходных мономеров и может содержать функциональности, модулирующие такие характеристики как температура стеклования, растворимость в носителе, соотношение гидрофильности и гидрофобности и т.п.
В качестве примеров мономеров звена B можно назвать эфиры метакриловой кислоты от низших до промежуточных, являющиеся производными прямых, разветвленных или циклических спиртов с 1-12 атомами углерода. Другими примерами мономеров звена B являются стирол, виниловые эфиры, хлористый винил, 1,1-дихлорэтилен, мономеры акрилоила и т.п.
Еще одними примерами мономеров В являются сложные эфиры акриловой или метакриловой кислоты 1,1-дигидроперфторалканолов (1) и гомологов (2):
(1) CF3(CF2)xCH2OH,
(2) CF3(CF2)x(CH2)yOH,
где x составляет от нуля до 20 и у составляет по меньшей мере от 1 до 10,
(3) w-гидрофторалканолы 3, HCF2(CF2)x(CH2)yOH,
где x составляет от 0 до 20 и y составляет по меньшей мере от 1 до 10,
(4) фторалкилсульфонамидные спирты 4,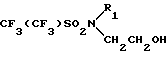
где x составляет от нуля до 20 и R1 является алкилом или алкиларилом с количеством атомов углерода до 20 или циклоалкилом с количеством кольцевых углеродных атомов до 6,
(5) циклические фторалкиловые спирты 5,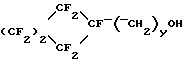
где z составляет от нуля до 7 и у составляет по меньшей мере от 1 до 10,
(6) CF3(CF2CF2--O)q(CF2O)x (CH2)yOH,
где q составляет от 2 до 20 и превышает x, x составляет от 0 до 20 и y составляет по меньшей мере от 1 до 10,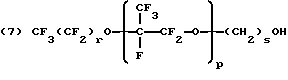
где p и s составляют по меньшей мере 1, a r равно от 1 до 6.
Предпочтительные полимеризованные основные составы мономера A включают полимеры фторакрилатов 8-13: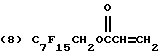
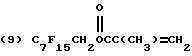
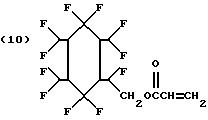
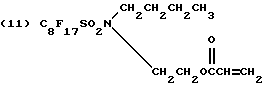
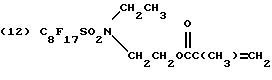
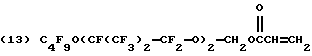
B может также быть производным макромономеров, полученных из стирола, альфа-метистирола, винилтолуола или метилметакрилата. Предпочтительно, чтобы такие макромономеры имели молекулярную массу 500-100000.
Звено C может быть производным этиленоненасыщенной предварительно сформированной органосилоксановой цепи. Молекулярная масса этого звена составляет в общем более 500.
Звено C настоящего изобретения может быть производным мономера, имеющего обобщенную формулу
X(Y)n-Si(R)3-mZm,
в которой X является виниловой группой, сополимеризуемой с мономерами A и B;
Y является двухвалентной связующей группой (например, алкилен, арилен, алкарилен и аралкилен с 1-30 атомами углерода), включающей гетероатомы, например О, N, S, P. Примерами являются группы сложных эфиров, амидов, уретанов и мочевины;
n равняется нулю или 1;
m - целое число от 1 до 3;
R - водород, низший алкил (т.е. от 1 до 4 атомов углерода, метил, этил или пропил), арил (т.е. от 6 до 20 атомов углерода, фенил или замещенный фенил) или алкокси (предпочтительно низший алкокси с количеством атомов углерода от 1 до 4);
Z - одновалентная силоксановая полимерная часть со средней молекулярной массой, превышающей приблизительно 500, которая по существу не вступает в реакции в условиях сополимеризации.
Предпочтительный мономер C может также быть описан как включающий группу X, имеющую обобщенную формулу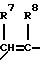
в которой R7 - атом водорода или группа COOH и R8 - атом водорода, метиловая группа или группа CH2COOH.
Группа Z мономера C имеет обобщенную формулу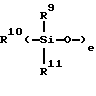
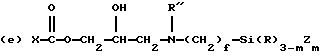
где R9 и R11 являются независимо низшим алкилом, арилом или фторалкилом, причем низший алкил и фторалкил оба относятся к алкильным группам, включающим от одного до трех атомов углерода и где арил относится к фенилу или замещенному фенилу (до 20 атомов углерода); R10 может быть алкилом (от 1 до 20 атомов углерода), алкокси (от 1 до 20 атомов углерода), алкиламино (от 1 до 20 атомов углерода), арилом (до 20 атомов углерода), гидроксилом или фторалкилом (от 1 до 20 атомов углерода), а e является целым числом от приблизительно 5 до приблизительно 700. Предпочтительно мономер C имеет обобщенную формулу, выбранную из группы, включающей следующие элементы, где m равно 1, 2 или 3, g равно нулю или 1, R'' может быть алкилом (от 1 до 10 атомов углерода) или водородом, f является целым числом от 2 до 6, h является целым числом от нуля до 2, и X, R и Z соответствуют приведенному выше определению: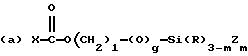
(b) X-Si(R)3-mZm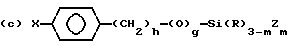
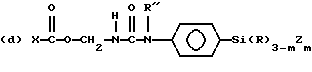
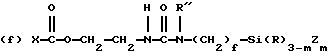
Наиболее предпочтительные для использования в соответствии с настоящим изобретением полимеры имеют состав, в котором
группа A является производной моно- или многофункциональной карбоксильной группы, содержащей молекулы, представленные следующей обобщенной формулой:
CH2=CR2G-(COOH)d,
где R2 - H или метил, d = 1, и
G является связью или связующей группой радикала гидрокарбила, содержащей 1-12 атомов углерода с валентностью d+1 или его солью, и
группа C является производной мономера, имеющего формулу
(a) 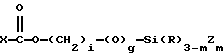
или (b) X-Si(R)3-mZm,
в которой X является виниловой группой, сополимеризуемой с мономерами A и B;
m - целое число от 1 до 3;
R - водород, низший алкил;
Z - одновалентная силоксановая полимерная часть со средней молекулярной массой, превышающей приблизительно 500, которая по существу не вступает в реакции в условиях сополимеризации.
Мономеры, используемые для получения звена C по настоящему изобретению являются предельно функциональными полимерами, включающими единственную функциональную группу (виниловую, этиленово-ненасыщенную, акрилоиловую или метакрилоиловую группу), которые иногда называют макромономерами или "макромерами". Такие мономеры известны и могут быть получены способом, описанным Милковичем и др. в патентах США NN 3786116 и 3842059. Получение макромономера полидиметилсилоксана и последующая сополимеризация с виниловым мономером описаны в нескольких статьях Й.Ямаситы и др. [Polymer J.14, 913 (1982); ACS Polymer Preprints 25 (1), 245 (1984); Makromol. Chem.l85,9 (1984)]. Этот способ получения макромономера включает анионную полимеризацию мономера гексаметилциклотрисилоксана (D3) для получения устойчивого полимера с контролируемой молекулярной массой, а обрыв цепи осуществляется с помощью хлорсилановых соединений, содержащих полимеризуемую виниловую группу. Сополимеризация свободного радикала монофункционального силоксанового макромономера с виниловым мономером или мономерами дает сополимер с привитым силоксаном четко обозначенной структуры, т.е. с контролируемой длиной и количеством привитых силоксановых ветвей.
Подходящими мономерами для использования в упомянутой выше анионной полимеризации являются, в целом, диорганоциклосилоксаны с формулой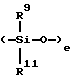
где R9 и R11 соответствуют определениям, приведенным выше, а e является целым числом от 3 до 7. Предпочтительными являются циклические силоксаны, в которых e равно 3 или 4, a R9 и R11 оба являются метилом, причем эти циклические силоксаны далее обозначены соответственно как D3 и D4. Особенно предпочтительным является D3 с деформированной кольцевой структурой.
Инициаторов анионной полимеризации выбирают таким образом, чтобы получался монофункциональный устойчивый полимер. К подходящим инициаторам относятся углеводороды щелочных металлов, такие как алкил или арил соединений лития, натрия или калия, содержащие в алкильном или арильном радикале до 20 или более атомов углерода, предпочтительно до 8 атомов углерода. Примерами таких соединений являются этилнатрий, пропилнатрий, фенилнатрий, бутилкалий, октилкалий, метиллитий, этиллитий, n-бутиллитий, сек- бутиллитий, трет-бутиллитий, фениллитий и 2-этилгексиллитий. Предпочтительными в качестве инициаторов являются соединения лития. В качестве инициаторов также подходят алкоксиды, гидроксиды и амиды щелочных металлов, а также триорганосиланолаты с формулой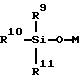
где M - катион щелочного металла, тетраалкиламмония или тетраалкилфосфония, и где R9, R10 и R11 соответствуют определениям, приведенным выше. Предпочтительным триоргансиланолатовым инициатором является триметилсиланолат лития (LTMS). В целом предпочтительно использование как деформированного циклического мономера, так и литиевого инициатора снижает вероятность реакций перераспределения и позволяет таким образом получить силоксановый макромономер с узким разбросом молекулярных масс, который в достаточной степени свободен от нежелательных циклических олигомеров.
Молекулярная масса определяется соотношением между инициатором и циклическим мономером, и таким образом количество инициатора может варьировать от приблизительно 0,004 до приблизительно 0,4 моль органометаллического инициатора в расчете на один моль мономера. Предпочтительно это количество составит от приблизительно 0,008 до приблизительно 0,04 моль инициатора в расчете на один моль мономера.
Для инициирования анионной полимеризации может использоваться инертный, предпочтительно полярный органический растворитель. Распространение анионной полимеризации с противоионом лития требует либо сильного полярного растворителя, такого как тетрагидрофуран, диметилсульфоксид или гексаметилфосфорный триамид, либо смеси такого полярного растворителя с неполярным алифатическим, циклоалифатическим или ароматическим углеводородным растворителем, таким как гексан, гептан, октан, циклогексан или толуол. Полярный растворитель служит для того, чтобы "активировать" ион силанолата, делая распространение возможным.
Обычно полимеризация может осуществляться при температурах в диапазоне от приблизительно -50oC до приблизительно 100oC, предпочтительно от приблизительно -20oC до приблизительно 30oC. Требуются отсутствие влаги и инертная атмосфера, такая как азот, гелий или аргон.
Прекращение анионной полимеризации обычно достигается путем непосредственной реакции действующего полимерного аниона с галогенсодержащими обрывателями цепи, т.е функционализированными хлорсиланами, с получением полимерных мономеров с виниловым завершением. Такие обрыватели цепи могут быть представлены обобщенной формулой X(Y)nSi(R)3-mClm, где m равно 1, 2 или 3 и где X, Y, n и R соответствуют приведенным выше определениям. Предпочтительным обрывателем цепи является метакрилоксипропилдиметилхлорсилан. Реакция обрыва цепи осуществляется путем добавления небольшого молярного избытка обрывателя цепи (относительно количества инициатора) к развивающемуся полимеру при температуре полимеризации. Согласно упомянутым выше статьям Й.Ямашиты и др. реакционную смесь после добавления обрывателя цепи можно подвергнуть обработке ультразвуком с целью улучшения функциональности макромономера. Очистка макромономера может быть осуществлена путем добавления метанола.
Сополимер, применяемый в настоящем изобретении, обычно получают путем сополимеризации исходных мономерных звеньев A, B и C с помощью обычной техники полимеризации.
Этот полимер может также содержать одну или несколько сшиваемых групп для последующей фиксации покрытия или поверхностного состава последующей реакцией сшивания после помещения полимера на предназначенную для этого подложку. Сополимеры, где группа В содержит сшиваемую группу, могут быть получены путем осуществления реакции электрофильной или нуклеофильной части сополимера с другим соединением, содержащим подходящую реакционноспособную группу и по меньшей мере одну сшиваемую группу, такую как этиленовая группа или эпоксидная группа. Электрофильная или нуклеофильная часть может в некоторых случаях быть такой же, как присутствующая в звене A сополимера.
Настоящее изобретение таким образом рассматривает также новые полимеры, содержащие повторяющиеся звенья:
A) 1-80 мас.% полярной или поляризуемой группы;
B) 0-98 мас.% модулирующей группы;
C) 1-40 мас.% гидрофобной привитой полисилоксановой цепи с молекулярной массой, равной по меньшей мере 500, в которых полимер дополнительно содержит боковые сшиваемые группы.
Сшиваемая группа может подвергнуться свободно радикальной или катионной реакции сшивания. Подходящие сшиваемые группы включают, не ограничиваясь ими, полимеризуемые этиленоненасыщенные группы и полимеризуемые эпоксидные группы. Этиленоненасыщенные группы являются предпочтительными, в особенности те из них, которые могут быть полимеризованы посредством механизма свободных радикалов, примерами которых являются замещенные и незамещенные акрилаты, метакрилаты, алкены и акриламиды. В водных системах полимеризуемые группы, которые полимеризуются катионным механизмом, т.е. полимеризуемые этиленоненасыщенные группы, такие как группы дивинилового эфира и полимеризуемые эпоксидные группы, являются менее предпочтительными, поскольку в таких системах механизм свободных радикалов применять легче, чем катионный механизм.
Сшиваемые полимеры могут быть получены разнообразными искусственными путями, включая, но не ограничиваясь им, осуществление реакции полимера, содержащего электрофильные или нуклеофильные группы с менее чем одним эквивалентом подходящего соединения для того, чтобы образовать боковые сшиваемые группы, оставляя таким образом электрофильные или нуклеофильные группы непрореагировавшими. С другой стороны, подходящие мономеры могут быть сополимеризованы с боковой сшиваемой группой, уже присутствующей в мономере. Реакция в этом процессе должна тщательно контролироваться, чтобы избежать осуществления полной реакции всех групп на стадии полимеризации, или же реакция, используемая для получения полимера, должна отличаться от реакции, используемой для образования поперечных связей между полимерами.
Первый искусственный путь из описанных выше для получения сшиваемого полимера может быть в настоящее время осуществлен путем использования "сочленяющего соединения", т.е. соединения, содержащего как боковую сшиваемую группу, так и реакционноспособную группу, которая может вступать в реакцию с полимером через функциональность, существующую на исходном материале полимера, чтобы образовать ковалентную связь между сочленяющим соединением и электрофильной или нуклеофильной группой, связывая таким образом боковую сшивающую группу с основной цепью полимера. Подходящими сочленяющими соединениями являются органические соединения, которые могут содержать неинтерферирующие заместители и/или неинтерферирующие соединительные группы между боковой сшивающей группой и реакционноспособной группой.
Сочленяющие соединения, пригодные для использования при получении полимеров согласно настоящему изобретению, включают соединения, которые содержат по меньшей мере одну группу, способную вступать в реакцию с полярной группой с целью формирования ковалентной связи, а также по меньшей мере одну полимеризуемую этиленово-ненасыщенную группу. Когда полярная группа является карбоксильной, с ней может вступать в реакцию довольно много групп, включая как электрофильные, так и нуклеофильные группы. Примерами таких групп являются следующие части и группы, содержащие эти части: -ОН, -NH2 -NCO, -COCl, и
Когда местом связи является спирт, несколько групп способны вступать в реакцию со спиртом. Примеры таких групп включают следующие части и группы, содержащие эти части: -NCO, - COCl,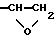
Примерами подходящих сочленяющих соединений для присоединения сшиваемых групп служат, например, акрилоилхлорид, метакрилоилхлорид, винилазалоктон, аллиловый изоцианат, 2- оксиэтилметакрилат, 2-аминоэтилметакрилат и 2- изоцианатоэтилметакрилат. Другие примеры подходящих сочленяющих соединений включают соединения, описанные в патенте США N 4035321. Примерами предпочтительных сочленяющих соединений являются, например, следующие метакрилатовые соединения и соответствующие им акрилаты: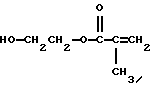
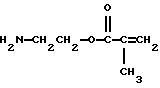
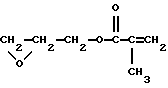
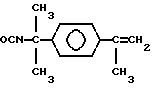
и следующее аллиловое соединение: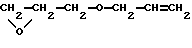
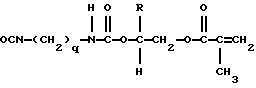
Особенно предпочтительными сочленяющими соединениями являются следующие метакрилатовые соединения и соответствующие им акрилаты, в которых R и q соответствуют приведенным выше определениям.
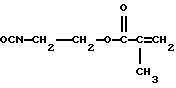
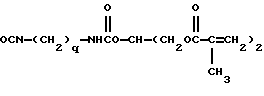
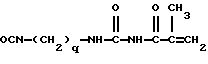
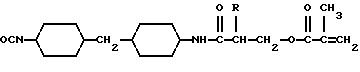
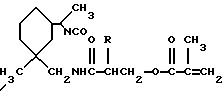
Полимер, являющийся предметом настоящего изобретения, может дополнительно содержать по меньшей мере одну силановую часть, которая может осуществлять реакцию конденсации. Реакция конденсации является реакцией объединения двух молекул, с исключением третьего соединения. Третьим соединением может быть вода или, в зависимости от структуры определенных реагентов, это третье соединение может быть спиртом, амином или любым другим подобным соединением, которое удаляется в ходе реакции. Эта силановая часть может быть, например, введена во время получения полимера посредством совместной реакции описанных выше звеньев A, B и C со звеном D, которое является производным этилен-ненасыщенного мономера, сополимеризуемого с мономерами для A, B и C. Это звено имеет обобщенную формулу
X(Y)n-Si(R12)iTj,
в которой - X является виниловой группой, сополимеризуемой с мономерами A и B;
Y является поливалентной связующей группой (например, алкилен, арилен, алкарилен и аралкилен с 1-30 атомами углерода), необязательно включающей гетероатомы, например. О, N, S, P. Примерами являются группы сложных эфиров, амидов, уретанов и мочевины.
n равняется нулю или 1;
R12 - H или низший алкил;
i - целое число из 0-2;
j - целое число из 1-3; и
i + j = 3.
T является окси или гидролизуемой группой, включающей атомы галогенов, алкокси, алкенокси, акилокси, карбокси, амино, амидо, диалкилиминокси, кетоксим, алдоксим и аналогичные группы. Предпочтительно гидролизуемую группу выбирают из группы, содержащей алкокси, алкенокси, акилокси, кетоксим и алдоксим. Более предпочтительно, чтобы гидролизуемые группы являлись алкокси группами, такими как метокси и этокси, что связано с их наличием на рынке, низкой стоимостью и низкой токсичностью. Примерами таких звеньев D являются, не ограничиваясь ими, акрилато- и метакрилато- алкилалкоксисиланы, представленные следующими ниже формулами.
где R13 является низшим алкилом.
В некоторых случаях также могут использоваться винилорганоалкоксисиланы, такие как винилтриметоксисилан, винилтриэтоксисилан и винил три(2-метоксиэтокси)силан.
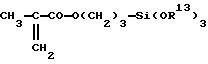
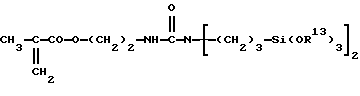
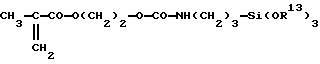
Эти соединения звена D могут использоваться в негидролизованной, частично гидролизованной или полностью гидролизованной форме. В последних двух формах, и в особенности в последней форме, необходимо принять меры предосторожности, чтобы свести к минимуму образование гелей, связанное с димеризацией и олигомеризацией силана через силоксановые связи. Для этого возможно применение любого способа, известного специалистам в данной области, такого как тщательный контроль значения pH или блокирование гидроксильных групп с целью замедления реакций силоксана.
Количество силанового соединения звена D, применяемого при синтезе описанного выше полимера, предпочтительно должно быть таким, чтобы силановая часть составляла 0,1-30 молярных процентов от полимера. Более предпочтительно, чтобы силановая часть составляла 0,1-20 молярных процентов полимера, а наиболее предпочтительно 0,1-10 молярных процентов полимера.
Описанные выше сополимеры, содержащие звено D, могут быть обычно получены путем сополимеризации исходных мономерных звеньев A, B, C и D с помощью стандартной техники полимеризации винила. С другой стороны, возможна модификация фракции полярных групп A полученного полимера с соединением, содержащим по меньшей мере одну силановую часть, которая способна осуществить реакцию конденсации, которая дополнительно содержит группу, способную вступать в реакцию с полярной группой A.
Предпочтительно, чтобы состав покрытия включал три компонента. Компонент I является сополимером, содержащим силановую часть, способную вступать в описанную выше реакцию конденсации. Компонент II является материалом, имеющим по меньшей мере два места реакции конденсации силикона, в которых возможно осуществление реакции конденсации. Компонент III является используемым в необязательном порядке катализатором, способствующим реакции конденсации между полимерами компонента I и/или между полимерами компонента I и соединениями компонента II. Компонент II является соединением, имеющим по меньшей мере два места реакции конденсации силикона, в которых возможно осуществление реакции конденсации, и поэтому действует как мостиковое соединение между полимерами компонента I в настоящей системе. Этот компонент может иметь сравнительно небольшую молекулу, или же может быть полимерным по природе. Предпочтительно, чтобы средняя молекулярная масса компонента II составляла приблизительно 64-3000.
Примерами компонента II являются тетраэтилортосиликат и его частично или полностью гидролизованные формы. Компонент II предпочтительно описывается следующей формулой:
Y-[Si(R12)iTj]k,
в которой Y является поливалентной связующей группой (например, алкилен, арилен, алкарилен и аралкилен с 1-30 атомами углерода), необязательно включающей гетероатомы, например О, N, S, P. Примерами являются группы сложных эфиров, амидов, уретанов и мочевины.
R12 - H или низший алкил;
i - целое число из 0-2;
j - целое число из 1-3;
i + j = 3;
k = 2 - 50.
T является окси или гидролизуемой группой, включающей атомы галогенов, алкокси-, алкенокси-, акилокси-, карбокси-, амино-, амидо-, диалкилиминоокси-, кетоксим-, алдоксим- и аналогичные группы. Предпочтительно гидролизуемую группу выбирают из группы, содержащей алкокси, алкенокси, акилокси, кетоксим и алдоксим. Более предпочтительно гидролизуемые группы являются алкокси группами, такими как метокси и этокси группы, что связано с их наличием на рынке, низкой стоимостью и низкой токсичностью.
Примерами компонента II являются следующие: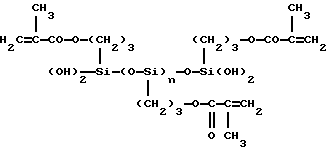
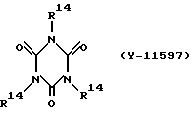
где R14 - -(CH2)3--Si(OCH3)3
Компонент III является катализатором, способствующим конденсации силановой части, которая способна вступать в реакцию конденсации. Обычно таким реакциям отверждения способствует влага. Для этой цели можно использовать любой катализатор конденсации силикона.
Предпочтительные отверждающие катализаторы для сшивания полимеров настоящего покрытия включают органометаллические катализаторы, содержащие металлы из группы III-A, IV-A, V-A, Vl-A, VIII-A, I-В, II-В, III-B, IV-B и V-B. Также предпочтительными для реакции конденсации силикона являются органические аминовые и органические кислотные катализаторы. Особенно предпочтительными катализаторами являются диоктоат олова, нафтенат олова, дилаурат дибутилолова, диацетат дибутилолова, диоксид дибутилолова, диоктоат дибутилолова, хелаты циркония, хелаты алюминия, титанаты алюминия, изопропоксид титана, диамин триэтилена, p-толуолсульфокислота, n-бутилфосфорная кислота и их смеси.
Одного лишь сочетания компонентов I и III, при отсутствии компонента II, может оказаться достаточно для получения достаточного сшивания для данного случая применения. С другой стороны, в некоторых случаях применения может оказаться достаточно соединить только компоненты I и II, в особенности когда компонент II представлен в частично гидролизованной форме. Скорость отверждения в нужной сфере применения так же, как фактическая молекулярная архитектура полимера компонента 1 и мостикового соединения компонента II будут определять выбор конкретного сочетания компонентов в данном изобретении. За счет обоснованного выбора растворителей и упаковочного материала, а также системы доставки можно получить систему из одной или нескольких частей. В последнем случае несколько частей можно смешать непосредственно перед применением или наносить последовательными слоями.
Полимеры, содержащие силановую часть, которая может вступить в реакцию конденсации, могут быть нанесены таким же образом, как и другие полимеры, описанные здесь. Так, например, эти полимеры могут быть нанесены на поверхность изделия перед его помещением в рот или же могут быть полимеризованы на месте применения на поверхности в области рта. Для достижения дополнительных преимуществ покрытия могут быть обработаны составом, содержащим поверхностно-активное вещество.
Особый интерес представляет покрытие поверхностей описанными здесь составами после размещения ортодонтических изделий. Защита поверхностей зубов, прилегающих к связанным скобам и т.п., очень важна из-за трудностей в обеспечении качественной гигиены рта и из-за того, что ортодонтические изделия сами образуют щели, в которых могут скапливаться бактерии и т.д. Важным способом использования материалов настоящего покрытия является их нанесение после сцепления ортодонтических изделий как на само изделие, так и на поверхность зуба, прилегающую к изделию.
В способе, являющемся предметом настоящего изобретения, желательно осуществлять предварительную обработку кислотой поверхности рта, на которую намечено нанести покрытие. Подходящими кислотами являются лимонная кислота, малеиновая кислота, азотная кислота, щавелевая кислота, кислоты фосфорная, серная, борная и т.п. Кроме того, в качестве состава предварительной обработки поверхности рта возможно использование дающих определенный эффект слабых кислотных растворов, подобных тем, что используются для обработки фтором.
При нанесении сополимера в качестве покрытия обычно полезно наносить его в сочетании с растворителем-носителем. Затем с помощью подходящего способа, например сушки, этот растворитель-носитель удаляют. Примерами растворителей-носителей являются вода, этанол, изопропанол, ацетоносиликоновые жидкости, такие как D4 и их смеси. Покрытие может также наноситься в виде эмульсии, например масла в воде или воды в масле.
Покрытия и обработка поверхностно-активными веществами, являющиеся предметом настоящего изобретения, могут применяться в форме полоскания рта или в форме профессионально нанесенного покрытия, которое может быть закреплено путем дальнейшей полимеризации или сшивания посредством этиленовых ненасыщенностей, присутствующих в модулирующей группе. Ингредиенты могут также быть включены в состав средств для чистки зубов, таких как зубная паста, зубной гель, зубной порошок, жевательная резинка, таблетки и т.п. Покрытия и обработка могут также быть частью профилактической пасты или полировочной пасты, которые наносятся в ходе отделочного или полировочного процесса с профилактической чашкой, углом, диском и т.п. Они могут также быть нанесены ниткой для чистки зубов с целью достижения межпроксимальных и других труднодоступных участков.
В отношении жидкостей для промывания и полоскания рта жидкой средой, являющейся носителем полимера или поверхностно-активного вещества, могут быть водные или водноспиртовые растворы, которые могут содержать другие неорганические растворители. В составах доставки полимеров могут присутствовать такие поверхностно-активные вещества, как моющие средства.
Зубные пасты, гели, жевательная резинка, таблетки и ротовые пластыри, используемые для доставки полимера или поверхностно-активного вещества, могут дополнительно содержать увлажнители (такие как глицерол, сорбитол и полиэтиленгликоль), полировальные составы (такие как кремнезем, карбонат кальция и трикальцийфосфат) и загустители (обычно природные или синтетические смолы, такие как каррагенан, оксиметиловая целлюлоза или синтетический сгуститель, такой как мелкодисперсный оксид кремния). Состав становится пастой, когда модуль неэластичности (иначе известный как "модуль потерь") становится меньше модуля упругости состава. Состав становится гелем, когда модуль неэластичности равен модулю упругости состава. Состав считается "краскоподобным", когда его можно нанести на выбранную подложку с помощью щеток, тампонов или иных подобных средств нанесения, обычно применяемых в зубоврачебном деле.
Составы для подачи полимера или поверхностно-активного вещества могут также содержать другие адъюванты, такие как ароматизаторы (как природные, так и синтетические, такие как мятное масло, ментол и подсластители), красители, модификаторы вязкости, консерванты, антиоксиданты и противомикробные средства (такие как гидрохинон, ВНТ, аскорбиновая кислота, p-оксибензойная кислота, сложные алкиловые эфиры, сорбат натрия и тимол), другие антибляшные добавки (такие как органофосфонаты, триклозан и другие добавки, подобные описанным в патенте США N 3488419), пероральные терапевтические средства (такие как фтористые соли, хлорэксидин и аллантоин), пигменты и красители, и буферы для контроля ионной силы.
Составы для подачи полимера могут дополнительно включать этиленово-ненасыщенное соединение. Примерами предпочтительных этиленоненасыщенных соединений являются 2,2-бис[4-(2-окси-3- метакрилоксипропокси)фенил] пропан ("BIS-GMA") и 2- оксиэтилметакрилат ("НЕМА").
Описанные полимеры годятся не только для включения в состав зубных паст и тому подобного, но могут также использоваться в качестве составов для наружных покрытий посторонних изделий, которые на временной или постоянной основе помещаются в рот. Так, например, эти являющиеся покрытиями составы могут быть нанесены на зубные изделия, изготовленные вне рта и затем помещенные в рот, такие как ортодонтические скобы, проволока, мосты, коронки, пломбы и т. п. Эти составы могут быть нанесены либо до ввода в рот, либо после ввода стоматологом или пациентом. Когда эти покрывающие составы наносятся на существующую структуру или искусственные изделия во рту, покрывающий состав может быть нанесен в виде полимера или исходных веществ полимера, который в свою очередь полимеризуется вне рта или внутри рта посредством тепловой, фотоинициируемой или окислительно-восстановительной полимеризации. Низкие коэффициенты трения восстановительных материалов, покрытых составами, содержащими эти полимеры, способствуют повышению износостойкости этих восстановительных материалов по сравнению с аналогичными материалами, не содержащими этих полимеров.
Когда полимеры, входящие в настоящие составы, содержат боковые этиленоненасыщенные части, которые могут вступить в реакцию в ходе последующей операции после нанесения на выбранную подложку, составы содержат также катализатор полимеризации, способствующий реакции этиленоненасыщенной группы. Такой катализатор может содержать фотоинициирующий катализатор или сочетание окислителя и восстановителя.
Предпочтительно из соображений безопасности в теле человека использовать инициирующий реагент.
Фотоинициатор должен быть способен обеспечить сшивание свободного радикала этиленоненасыщенного компонента в результате облучения светом подходящей длины волны и интенсивности. Предпочтительно он является также в достаточной степени устойчивым при хранении и свободным от нежелательного окрашивания, что допускает его хранение и использование в типичных для зубов условиях. Предпочтительными являются фотоинициаторы, реагирующие на видимый свет. Фотоинициатор часто может использоваться отдельно, но обычно он используется в сочетании с подходящим донорским соединением или подходящим ускорителем (например, амины, перекиси, соединения фосфора, кетоны и соединения альфа-дикетонов).
К предпочтительным инициаторам, реагирующим на видимый свет, относятся камфорхинон (который обычно сочетается с подходящим донором водорода, таким как амин), диарилйодониевые простые или металлические сложные соли, хромофор-замещенные галометил-s-триазины или галометилоксидиазолы. Особенно предпочтительные фотоинициаторы, реагирующие на видимый свет, включают сочетания альфа-дикетона, например камфорхинона, и диарилйодониевой соли, например дифенилйодониевый хлорид, бромид, йодид или гексафторфосфат, с дополнительными донорами водорода или без них (такими как бензолсульфокислый натрий, амины и аминовые спирты). Предпочтительные инициаторы полимеризации, реагирующие на ультрафиолетовый свет, включают кетоны, такие как бензил и бензоин, и ацилоины, и ацилоиновые эфиры. Предпочтительные поставляемые промышленностью инициаторы полимеризации, реагирующие на ультрафиолетовый свет, включают 2,2-диметокси-2-фенилацетофенон ("IRGACURE 651") и бензоинметиловый эфир (2-метокси-2-фенилацетофенон), которые поставляются фирмой "Ciba-Geigy Corp".
Фотоинициатор должен присутствовать в количестве, достаточном для того, чтобы обеспечить достаточную интенсивность фотополимеризации. Это количество будет зависеть отчасти от источника света и коэффициента экстинкции фотоинициатора. Обычно компоненты фотоинициатора имеют суммарную массовую долю от общей массы (включая воду) неотвержденных компонентов покрытия, составляющую от приблизительно 0,01 до приблизительно 5%, более предпочтительно от приблизительно 0,1 до приблизительно 5%.
Альтернативные инициаторы полимеризации включают окислительно-восстановительные системы, представляющие собой сочетание восстановителя и окислителя. Эти реагенты должны реагировать или взаимодействовать иным образом друг с другом для образования свободных радикалов, способных инициировать полимеризацию этиленоненасыщенной части. Восстановитель и окислитель предпочтительно обладают достаточной устойчивостью к хранению и не подвергаются нежелательному окрашиванию, что допускает их хранение и использование в типичных для зубов условиях. Они должны обладать достаточной растворимостью или смешиваемостью со средой-носителем. Восстановитель и окислитель должны присутствовать в количествах, достаточных для того, чтобы обеспечить достаточную интенсивность реакции образования свободных радикалов. Подходящие пары восстановителей и окислителей приведены в публикации "Redox Polymerization", G.S.Misra and U.D.N. Bajpai, Prog. Polym. Sci.8, 61-131 (1982).
Предпочтительные восстановители включают аскорбиновую кислоту, хлорид кобальта (II), хлорид железа, сульфат железа, гидразин, ароматические и алифатические амины, гидроксиламин (в зависимости от выбора окислителя) щавелевой кислоты, тиомочевину, серную кислоту и ее соли, и соли с анионом дитионита или сульфита. Предпочтительные окислители включают хлорид кобальта (III), гидроперекись трет-бутила, хлорное железо, гидроксиламин (в зависимости от выбора восстановителя), надборную кислоту и ее соли, и соли с анионом перманганата или персульфата. Возможно использование также перекиси водорода, хотя обнаружено, что в некоторых случаях она мешает фотоинициатору.
Количество восстановителя и окислителя должно быть достаточным для того, чтобы обеспечить нужную степень полимеризации этиленоненасыщенного компонента. Предпочтительное количество восстановителя и окислителя должно составлять от приблизительно 0,01 до приблизительно 10% для каждого, более предпочтительно от приблизительно 0,02 до приблизительно 5% от суммарной массы (включая воду) компонентов. Производится обработка поверхностно-активным веществом, в особенности нейтральным и катионным.
Для сшиваемых полимеров, которые полимеризуются катионным механизмом, подходящими инициаторами являются соли, которые способны выделять катионы, такие как диарилйодиновые, триарилсульфониевые и арилдиазониевые соли.
Как упоминалось выше, описанная здесь дополнительная обработка покрытия составом, содержащим поверхностно-активное вещество, обеспечивает значительное уменьшение адгезии бактерий и белковых материалов. Поверхностно-активные вещества могут быть включены в очень небольших количествах в составы, получаемые после нанесения покрытий, и могут быть или неионными, или ионными поверхностно-активными веществами. В особенности предпочтительными для использования в процессе обработки после нанесения покрытия являются неионные поверхностно-активные вещества.
Предпочтительные ионные поверхностно-активные вещества включают соли алифатических кислот с длинными цепями, такие как додецилсульфат натрия или октадецилсульфат натрия. Полимер покрытия может также содержать ионную функциональность, которая действует в отношении поверхностно-активного вещества как противоион.
Предпочтительные неионные поверхностно-активные вещества основываются на полигидроксильных сложных эфирах жирных кислот с длинными цепями или полигидроксильных простых эфирах спиртового жирного ряда с длинными цепями. В особенности предпочтительными являются полиоксиэтилен, сорбированные эфиры жирных кислот с длинными цепями, например поверхностно-активные вещества TweenTM 20, 40, 60 или 80.
Описанные здесь покрытия могут, кроме того, использоваться для нанесения на медицинское оборудование и изделия, которые находят применение в медицине и для которых было бы полезным уменьшение адгезии к их поверхностям. Примеры такого медицинского оборудования включают изделия, которые на временной или постоянной основе имплантируются в тело, такие как кардиостимуляторы, сита кровяных сосудов, материалы для скрепления и замены костей и т.д. Изделия, которые входят в контакт с жидкостями тела, такие как катетеры и хирургические инструменты, также могут быть улучшены за счет применения покрытия, являющегося предметом настоящего изобретения. Кроме того, нанесение настоящих покрытий способствует улучшению изделий, используемых в целях недопущения переноса инфекции, таких как перчатки, маски, халаты, простыни и т.п.
Действенность покрытий, являющихся предметом настоящего изобретения, можно измерить рядом способов. Так, например, можно определить химическими средствами, сохраняется или нет покрытие после других видов воздействия на покрытие. Одним из таких аналитических средств является оценка продвигающегося угла контакта с помощью описанного ниже равновесия Вильгельми. Предпочтительно угол контакта, измеренный для воды, превышает 55o.
С другой стороны, сохранение эффективности покрытия можно оценить путем определения устойчивости подложки к окрашиванию или адгезии бактерий. Устойчивость покрытия можно определить путем применения физического воздействия или вымачивания покрытия. Физическое воздействие может осуществляться в виде чистки щеткой при заданной нагрузке в течение ограниченных периодов времени. С другой стороны, физическое воздействие может осуществляться в виде повторяющейся шлифовки или полирования зубов механическим средством, предназначенным для того, чтобы имитировать действие зубов во рту.
Равновесие Вильгельми
С целью определения гидрофобности и гидрофильности покрытий, являющихся предметом настоящего изобретения, для измерения углов контакта при продвижении и отступании воды соответственно использовали хорошо известный способ равновесия Вильгельми. Этот способ описан, например, в публикации "Wettability," John C. Berg, editor. Marcel Dekker, Inc., New York, 1993, pp 11-25. Измерение выполняется на образце со сплошным покрытием. Все измерения угла контакта выполняются с водой.
Предпочтительно, чтобы покрытия, являющиеся предметом настоящего изобретения, являлись в достаточной степени подходящими для данной подложки так, чтобы угол контакта при продвижении воды составлял по меньшей мере 55o при измерении его способом равновесия Вильгельми, как описано здесь для образца, в течение двух недель подвергавшегося вымачиванию в дистиллированной воде при температуре 37oC. Более предпочтительно, чтобы угол контакта при продвижении составлял по меньшей мере 55o для образца, вымачивавшегося таким образом в течение трех месяцев.
Следующие примеры приведены с целью проиллюстрировать настоящее изобретение, не ограничивая его рамки. Все части и процентные соотношения, если иное не оговорено особо, являются массовыми, а все молекулярные массы представляют собой среднюю молекулярную массу.
Пример 1
Изо-бутилметакрилат (14 г), акриловую кислоту (2 г), этиленоненасыщенный силиконовый макромер с молекулярной массой 10000, полученный согласно процедуре приготовления "мономера C 3b" в колонке 16 патента США N4693935 ("макромер PDMS") (4 г) и азобисизобутиронитрил ("AIBN", 0,1 г) были растворены в изопропаноле. После удаления кислорода раствору дали возможность полимеризоваться при температуре 55oC. Полимер обозначили как P1. Затем раствор независимо нанесли на протравленную эмаль и дентин. Углы контакта измерили способом равновесия Вильгельми с помощью анализатора угла контакта DCA 322, поставленного компанией ATI Instrument, Мэдисон, шт. Висконсин. Использовалось до трех циклов. Полученные результаты приведены в табл. 1 и 2.
Из приведенных примеров очевидно, что нанесение покрытия с полимером P1 на эмаль делает поверхность гидрофобной. Покрытие нелегко теряется, поскольку отмечено, что угол контакта при продвижении остается достаточно большим даже после активной обработки. Износ начинается только после механической обработки грубыми марками абразивных изделий.
Аналогичные исследования были проведены после нанесения полимера P1 на брусок дентина. Полученные результаты приведены в табл. 3.
Пример 2
Изо-бутилметакрилат (13 г), акриловую кислоту (3 г), макромер PDMS (4 г) и AIBN (0,1 г) были растворены в THF. После удаления кислорода раствору дали возможность полимеризоваться при температуре 55oС. Примерно одна третья часть карбоксилатных групп полученного полимера была соединена с изоцианатоэтиловым метакрилатом с целью получения боковых ненасыщенных групп. Затем был выполнен обмен THF с 60 г изопропанола. К 10 г этого раствора добавили 0,0123 г дифенилйодинового гексафторфосфата и 0,0031 г камфорхинона. Этот полимер обозначили как P2. Покрытие, полученное из полимера, было далее сшито путем облучения источником видимого света, VisiluxTM 2 компании 3М.
Результаты определения угла контакта способом равновесия Вильгельми показаны в табл. 4.
Результаты измерения угла контакта при продвижении показывают, что после обработки полимером P2 гидрофобность поверхностей как эмали, так и дентина значительно возрастает.
Бруски зубного состава Z100 покрыли P1 или P2. Покрытие P2 подвергли затем, как показано выше, сшиванию. После этого измерили углы контакта. Полученные результаты приведены в табл. 5.
Пример 3
Этилметакрилат (11,4 г), акриловую кислоту (2 г), макромер PDMS (4 г) и AIBN (0,1 г) растворили в 75 мл чистого этилового спирта. Реакционную смесь в течение 15 минут продували с азотом, после чего в течение 8 часов выдерживали при температуре 55oС. Затем ее охладили до комнатной температуры и разбавили равным объемом изопропанола. Раствор полимера обозначили как P3.
Брусок бычьей эмали отполировали наждачной бумагой, после чего определили ее угол контакта с водой по способу равновесия Вильгельми. Затем брусок высушили, покрыли раствором P3, высушили на воздухе и вновь определили угол контакта. Значения для контрольных образцов эмали без покрытия и образцов эмали с покрытием показаны в табл. 6. Эксперимент повторили с бруском бычьего дентина; его результаты показаны в табл. 7. Подготовили и подвергли аналогичной обработке брусок отвержденного стеклянного иономера VitremerTM. Результаты определения угла контакта этого материала до и после нанесения покрытия показаны в табл. 8.
Из указанных данных очевидно, что покрытие из раствора с полимером P3 повышает гидрофобность поверхностей и что покрытие является также устойчивым.
Пример 4
Синтез полимеров A-F
Реагенты, перечисленные в табл. 9, загрузили в 250-миллилитровую колбу с круглым дном и тремя горловинами, снабженную трубкой для подвода азота, конденсатором и термометром. В колбу ввели стержень магнитного перемешивания, после чего реагенты перемешивали в течение 15 минут с барботированием однородного раствора сухим азотом. Через 15 минут расход азота уменьшили и переключили с барботирования на режим покрытия слоем газа. Раствор выдерживали при температуре 60oC при помешивании, используя для этого масляную баню, снабженную электронным регулятором температуры. Нагрев продолжался в течение 8 часов. Затем реакционную смесь осадили в воде, используя 5 мл воды на каждый миллилитр раствора полимера. Белый полимерный осадок был затем собран осаждением, промыт холодной водой и высушивался в вакуумной печи при температуре 70o в течение нескольких суток.
Приготовление брусков из эмали и дентина
Брусок эмали
Лабиальную поверхность извлеченного бычьего зуба зачистили наждачной бумагой зернистостью 120 с тем, чтобы обнажить чистую поверхность дентина, на которой отсутствует эмаль. Затем для получения гладкой поверхности ее отшлифовали бумагой крупностью 600. Алмазной пилкой отрезали тонкую пластинку (приблизительно 1/2 мм), включающую полированную поверхность. Затем две такие пластинки склеили между собой зубной адгезивной системой ScotchbondTM Multipurpose (SBMP) компании 3M таким образом, чтобы отполированные поверхности были обращены наружу. Склеенную слоеную структуру обрезали, получив прямоугольник размерами приблизительно 8 мм х 6 мм. Затем к одной из коротких сторон эмалевой слоеной структуры адгезивом SBMP приклеили маленькую плоскую кнопку из зубного композиционного материала с тем, чтобы она служила ручкой.
Брусок дентина
Лабиальную поверхность извлеченного бычьего зуба зачистили наждачной бумагой зернистостью 120 с тем, чтобы обнажить большой участок дентина. Параллельно отшлифованной поверхности алмазной пилкой отрезали тонкий плоский брусок. Толщина бруска составила около 1 мм. При этом обращали внимание на то, что на отрезанной поверхности был только дентин (отсутствие видимых признаков полости коронки). Обе плоские поверхности бруска дентина отполировали бумагой крупностью 600. После этого алмазной пилкой вырезали прямоугольный участок размерами 6 мм х 8 мм. Затем к одному концу (короткая сторона) посредством SBMP приклеили маленькую плоскую кнопку.
Пример 5
Обнаженный брусок эмали сначала протравили гелем 35% фосфорной кислоты и затем отполировали шлифовальной шкуркой зернистостью 600. Брусок без покрытия проверили на контакт с водой. Затем на брусок нанесли раствор (7,5% полимера в изопропаноле) одного из полимеров A-F путем погружения на 5 минут и промокания с последующей сушкой на воздухе. Покрытый полимером брусок эмали испытали на угол контакта при продвижении воды согласно способу равновесия Вильгельми. Затем образец эмали с покрытием в течение двух суток выдерживали в деионизированной воде, после чего испытали снова. После этого образец выдерживали в течение 90 минут при постоянной температуре в емкости со слюной, промакнули досуха и испытали на угол контакта. Затем указанный образец выдержали в воде в течение 10 минут и испытали вновь. Промытый брусок после повторного испытания почистили щеткой со средним размером щетины и повторно испытали на угол контакта. Результаты измерения угла контакта при продвижении приведены в табл. 10. Значения угла контакта при продвижении в третьем цикле сообщаются по той причине, что в это время достигается равновесие.
Приведенные выше результаты показывают, что после нанесения покрытия на эмаль поверхность зуба становится гидрофобной. Выдерживание в воде не вызывает значительного уменьшения угла контакта, что указывает на действенность покрытия в этот момент. После выдерживания в слюне входящие в состав слюны протеины и другие компоненты в определенной степени пристают к покрытым поверхностям, полученным из полимеров A, B и E. Следовательно, поверхность этих полимеров, обработанная слюной, является гидрофильной, поскольку допускает прилипание протеинов и других компонентов слюны. При промывке обработанной слюной поверхности водой (операция 5) часть из приставших веществ из слюны смывается с поверхности, образованной полимером Е. При чистке зубов щеткой (операция 6) большая часть компонентов слюны удаляется и вновь открываются гидрофобные полимерные поверхности, что демонстрирует более высокие значения угла контакта (близкие к первоначальным значениям для образцов с покрытием). У зубов с покрытием из полимеров C и F не наблюдается заметного уменьшения угла контакта даже вскоре после выдерживания в слюне, что указывает, таким образом, на низкую адгезию к этим подложкам.
Образец 6
Чтобы продемонстрировать действенность покрытий, вырезали бруски из эмали и дентина (без какой-либо другой обработки), для которых сначала определили угол контакта. Затем на них независимо нанесли раствор полимеров B и C при концентрации 7,5 мас.%, как показано в табл. 11 и 12, после чего просушили.
Образцы с покрытием выдерживали в течение 18 часов в дистиллированной воде при температуре 37oC, промакнули досуха и определили углы контакта. Полученные результаты приведены в табл. 11 и 12.
Затем эти образцы погрузили в воду и выдерживали при температуре 37oC в течение одной недели, после чего определили углы контакта. Эти значения приведены в табл. 11. Углы контакта повторно определили через 2 недели выдерживания, и затем снова через 3 месяца выдерживания.
Образцы были изготовлены в нескольких экземплярах. Средние значения угла контакта при продвижении для двух полимеров во втором цикле приведены в табл. 11 и 12.
Пример 7
Эффект предварительной обработки эмали или дентина
Обрезанные и отполированные бруски эмали или дентина либо (a) протравливали с течение 15 секунд гелем фосфорной кислоты с последующей промывкой водой и сушкой перед нанесением полимерного покрытия, либо (b) протирали грубой полировальной пастой NuproTM (каждую сторону брусков эмали полировали в течение 10 минут, каждую сторону брусков дентина - в течение 4 минут), тщательно промыли водой и определили угол контакта.
Затем на бруски нанесли полимер С и определили углы контакта после выдерживания при температуре 37oC в дистиллированной воде в течение 18 часов, 1 недели и 2 недель.
Угол продвижения после второго цикла этих экспериментов приведен в табл. 13 и показывает, что действенность этих покрытий сохраняется даже после продолжительного хранения.
Сравнительный пример 1
Была проведена оценка на действенность и гидрофобность двух полидиметилсилоксановых полимеров, содержащих диметилпропиламиновые группы. Использовали два поставляемых промышленностью полимера такого типа: PS 510 (молекулярная масса 2500) и PS 513 (молекулярная масса 27000) компании Petrach, Huls.
Приготовили 7,5% раствор PS 510 в ацетоне. Затем этим раствором покрыли эмалевые бруски с последующей сушкой. После этого определили углы контакта (см. табл. 14).
Уменьшение угла контакта показывает, что покрытие стало недействительным из-за утери гидрофобности.
Сравнительный пример 2
Бруски дентина после обрезки и полировки покрыли раствором PS 510 (7,5% в ацетоне) или PS 513 (7,5% в метилэтилкетоне). У образцов с покрытием измерили угол контакта. Затем их подвергли старению при температуре 37oC, после чего вновь измерили угол контакта. Результаты измерений приведены в табл. 15.
Уменьшение угла контакта указывает на недействительность покрытий.
Для дальнейшей оценки эффективности настоящих покрытий был выполнен более детальный анализ с использованием биологических методик. Биологическая оценка касается в общем способов и материалов.
Частицы эмали
Частицы эмали были получены от приблизительно 100 бычьих зубов, с которых сначала удалили всю приставшую ткань путем обдирания скальпелем и зубной кюреткой. Корни отделили режущим диском в месте соединения цементного вещества и эмали, удалив пульпу. Затем бруски погрузили на 20 минут в жидкий азот, извлекли оттуда и немедленно раздробили молотком. С самых больших кусков дентин удалили шлифовальным диском. Затем бруски эмали поместили в аналитическую мельницу и обрабатывали в ней в течение приблизительно 5 минут при температуре 40oC. Полученный порошок просеяли для того, чтобы получить частицы крупностью приблизительно 80-120 мкм. Частицы промыли, очистили кислотой и хранили в сухом виде при температуре 40oC до момента использования.
Сбор и обработка слюны
Слюну получали у добровольцев, которым предложили жевать в течение 2-3 минут квадрат Parafilm размером 2 кв. дюйма, сплевывая в охлаждаемую пробирку емкостью 50 мл. Затем образцы собирали вместе и центрифугировали в течение 20 мин при температуре 4oC со скоростью 10000 об/мин. Осветленную слюну разлили по 10-миллилитровым мензуркам и хранили при температуре -70oC вплоть до момента использования.
БИОЛОГИЧЕСКАЯ ОЦЕНКА ПОКРЫТИЙ 1
Приготовили образцы измельченной эмали с покрытием из четырех полимеров, для чего использовали полимеры A, B, C и E.
Эти образцы были приготовлены для испытаний на биологическое прилипание. Сначала на частицы с покрытием воздействовали тремя материалами:
IgG - протеин иммуноглобулина PI 7.5,
OVA - протеин овальбумина PI 3.5,
P.gin - бактерии P.gingivalis.
Способы и материалы
Флуоресцентное мечение бактерий
Десять мл бактерий P.gingivalis в количестве 109/мл в фосфатном буферном солевом растворе (PBS) объединили с 0,1 мл изотиоцианата флуоресцеина (FITC) в количестве 1 мг/мл в PBS. Смесь в течение одного часа вращали при комнатной температуре, а затем промывали порциями PBS по 40 мл до тех пор, пока оптическая плотность всплывающего слоя при длине волны 495 нм не стала равной нулю. Окрашенный бактериальный осадок был затем повторно разведен в 10 мл PBS с 0,1% азида натрия и хранился до применения при температуре 4oC.
Нанесение покрытия на частицы эмали
Сто пятьдесят мг частиц (как с полимерным покрытием, так и без него) перемешали с 2,5 человеческой слюны и вращали при комнатной температуре в течение 1-2 часов в полипропиленовых пробирках. В то же время частицы без покрытия в качестве контрольного образца вращали в PBS. Затем частицы трижды промыли в PBS с целью удаления несвязанной слюны, разбавили до содержания 100 мг/мл в PBS и хранили до применения при температуре 4oC.
Анализ прилипания
Пять мг частиц (в трех экземплярах) пипетировали в лунки планшета микрофильтра с коническим основанием (NUNC) и трижды промыли 200 мкл PBS. В каждую лунку добавили сто мл FITC-меченых бактерий, IgG или овальбумина, а плашку выдерживали в течение часа при комнатной температуре при постоянном встряхивании. Затем лунки трижды промыли 250 мкл PBS и повторно развели в 250 мкл того же самого буферного раствора. Извлекли порции частиц по 20 мкл, которые поместили в лунки аналитического планшета IDEXX с дном в виде стекловолоконного фильтра. Затем планшеты поместили в отцеживающий аппарат IDEXX, жидкость удалили посредством вакуумного фильтрования и определили связанную флуоресценцию. После считывания первоначальных результатов планшеты дважды промыли PBS, содержащим 0,125% NP-40, после чего повторно определили в отцеживающем аппарате связанную флуоресценцию. Полученные результаты выражены в относительных единицах флуоресценции (RFU).
Результаты
Частицы испытывали как с полимерным покрытием, так и без него. Половина тех и других была подвергнута воздействию слюны. Эти обработанные слюной частицы были затем промыты фосфатным буферным соляным раствором (PBS) и подвергнуты проверке на прилипание не обработанных слюной частиц. Связанная флуоресценция была рассчитана как процентная доля от позитивного контроля, т.е. прилипания бактерий или протеина к частицам эмали, не имеющим покрытия. Частицы промыли также PBS, содержащим 0,125% NP-40, и измерили прилипание бактерий и протеина.
На фиг. 3 показаны результаты для частиц, промытых PBS, а на фиг. 4 - для частиц, промытых в PBS+NP-40. Эмаль без покрытия поглощала большие количества всех трех испытательных биологических материалов (что составляет 100% относительной шкалы); покрытие эмали слюной несколько уменьшает это прилипание для IgG, но не для OVA и P.gin. Нанесение на частицы полимерного покрытия значительно снижает прилипание, однако частицы с полимерным покрытием, на которых сверху нанесена слюна, имеют тенденцию к возвращению части поглотительной способности. Частицы с покрытием, обработанные слюной, а затем биологическими материалами, подвергнутые краткой промывке в PBS, содержащем неионный моющий состав NP-40, продемонстрировали значительное снижение прилипания IgG и OVA для всех покрытий и P.gin.
БИОЛОГИЧЕСКАЯ ОЦЕНКА ПОКРЫТИЙ 2
Рост штаммов бактерий
Штаммы Mutans streptococci были получены в Стоматологической школе университета Миннесоты (штаммы 43-2, 43-3 и RL19) в качестве свежих культур у лиц, обратившихся за зубоврачебной помощью. Дополнительные штаммы Mutans streptococci были получены из Американской коллекции типов культур (штаммы 10558, 12396, 27351, 27352, 27609 и 33399). Все штаммы выращивали в бульоне Тодд Хьюитт, в анаэробной камере при температуре 37oC. Клетки собрали в последней лог-фазе роста и трижды промыли в стерильном отфильтрованном солевом растворе. Затем клетки развели при оптической плотности ("OD"), равной 1,0 (600 нм), в стерильном отфильтрованном буферном растворе KCl (0,1 М NaCl, 0,05 М KCl, 0,1 М MgCl2, 0,1 mM фосфата калия и 1 мМ CaCl2, pH 7,0).
Получение цельногеномных зондов
ДНК изолировали от бактерий с использованием комплекта ASAPTM (Boeringer Manheim) согласно указаниям изготовителя. Около 109 клеток различных штаммов объединили с 2 мл буфера лизиса, дополненного 1 мг/мл мутанолизина (Sigma). Затем добавили и смешали плавной инверсией шестьдесят микролитров термообработанной рибонуклеазы и 160 мкл раствора лизоцима. Взвешенные клетки выдерживали затем в течение 30 минут при температуре 37oC с добавлением 100 мкл протеиназы K. После смешивания путем осторожного переворачивания взвешенные клетки в течение 60 минут выдерживали при температуре 60oC. Было добавлено 4 мл связующего буферного раствора, смешанного путем осторожного переворачивания, и затем весь образец добавлен к колонке связующей матрицы ДНК. Колонку дренировали гравитационным способом, добавили дополнительные 3 мл связующего буферного раствора и снова дренировали. Добавили половину миллилитра первичного элюирующего буферного раствора и колонку дренировали для элюирования РНК и протеина с последующими 2 мл элюирующего буфера ДНК, который был собран в пробирке 10 мм х 100 мм. Затем ДНК выделили изопропанолом, промыли этанолом и просушили. Полученный осадок ДНК развели затем в 50 мкл буфера Tris-EDTA ("TE"), а количество и чистоту каждого препарата определяли по оптической плотности при длине волны 260/280 нм и электрофорезом по 0,85% TE.
ДНК была помечена ник-трансляцией с использованием нерадиоактивного набора для помечивания ДНК GENIUSTM (компания Boeringer, Мангейм) согласно указаниям изготовителей. Если кратко, то 4,5 мкг ДНК из каждого штамма бактерий объединяли со сбалансированной смесью гексануклеотидов, дезоксинуклеозидтрифосфатов, помеченных дигоксигенином, ферментом Klenow и водой до 20 мкл. Пробирки центрифугировали в течение 10 с, после чего выдерживали при температуре 37oC в течение ночи.
Дот-блоттинг приставших бактерий
Частицы эмали с покрытием и без него независимо добавили в 1 мл слюны и вращали при комнатной температуре в течение 1 часа. Частицы промыли буферным раствором KCl, добавили 1 мл бактерий при оптической плотности 1,0 (длина волны 600 нм) и вращали полученную смесь при комнатной температуре еще час. Затем частицы промыли в одном из пяти буферных растворов (см. ниже). После этого частицы осадили, добавили 50 мкл буфера экстракции ДНК и взвешенные частицы прокипятили для извлечения и солюбилизации ДНК. Частицы быстро охладили до 40oC, центрифугировали и повторяющиеся образцы по 10 мкл из каждой пробирки точками нанесли на мембрану Zeta-ProbeTM (Bio Rad). Затем мембраны обработали раствором предгибридизации, промыли и добавили в составе 25 мл раствора гибридизации 100 нг ник-транслированного зонда ДНК. Гибридизацию выдержали в течение ночи при температуре 65oC перед тем, как трижды промыть ее для удаления несвязанного зонда. Затем мембраны выдержали в течение 1 часа в молочном блокирующем растворе и добавили козий антидигоксигенин (1: 1000 в блокирующем буфере). После дополнительного выдерживания в течение часа мембраны промыли с целью удаления несвязанных антител и погрузили на 1-2 часа в раствор ферментного субстрата. Реакцию прекратили путем промывки мембраны в растворе EDTA, а интенсивность пятен оценивал один наблюдатель, пользуясь для этого следующей шкалой:
0 = отсутствие окраски,
1 = едва видимое,
2 = ясно видимый круг,
3 = ясно видимый темный круг,
4 = очень темный, в высшей степени интенсивный круг.
Опыты по промывке с целью определения прочности прилипания
Для опытов по промывке были приготовлены пять разных буферных растворов: буферный KCl; буферный KCl + 1% Tween 20, буферный KCl + 1% Tween 80, буферный KCl + 1% растворимого экстракта зубной пасты и буферный KCl + 1% полоскания для рта. Каждый буфер использовали отдельно для заключительной промывки при опытах по определению прилипания микробов.
Эксперименты по определению прилипания микробов
Показатели прилипания для различных сочетаний бактерий, устойчивых к образованию бляшки покрытий и контрольных образцов приведены в табл. 16. В общем наблюдалось общее снижение прилипания приблизительно на 25% (в диапазоне 10-47%) в случае нанесения на эмаль всех экспериментальных материалов и в сравнении с контрольными образцами в буфере KCl. Кроме того, наблюдалось значительное уменьшение, на 81-29%, прилипания к частицам с покрытием в случае добавления к промывочному буферу различных поверхностно-активных веществ или материалов для рта по сравнению с необработанными образцами эмали и уменьшение прилипания на 86-13% по сравнению с контрольными образцами, обработанными слюной. Это уменьшение оказалось больше того, которое ожидалось в связи с добавлением одних лишь моющих растворов.
БИОЛОГИЧЕСКАЯ ОЦЕНКА РАСТВОРОВ ПОКРЫТИЯ 3
Полистироловые лунки для анализа, предварительно покрытые малеиновым ангидридом, были обработаны различными перечисленными ниже растворами:
i) контрольный, без покрытия;
ii) с покрытием из раствора полимера C (7,5% в изопропаноле);
iii) с покрытием из раствора полимера C (7,5% в изопропаноле) с последующей промывкой буферным раствором, содержащим 1 мас.% Tween 80;
iv) с покрытием из раствора полимера E (7,5% в изопропаноле); или
v) с покрытием из раствора полимера E (7,5% в изопропаноле) с последующей промывкой буферным раствором, содержащим 1 мас.% Tween 80.
Посевной материал клеток S.Challis посеяли в концентрации 1х107 клеток на лунку. После стандартных процедур промывания с помощью сцинтилляционного счетчика подсчитали количество клеток, прилипших к лункам, выразив его в процентной доле от первоначального посевного материала. Полученные результаты приведены в табл. 17 как относительная процентная доля от контрольного образца с подложкой без покрытия (100%).
Приведенные выше результаты показывают, что нанесение полимерного покрытия уменьшает прилипание бактерий области рта. Обработка покрытия промывочным раствором, содержащим поверхностно-активные вещества, перед попаданием на него бактерий способствует дополнительному снижению степени прилипания бактерий.
Подробное описание графических материалов
Для сравнения покрытия зубных брусков растворами полимера и PDMS. На бруски дентина наносили покрытие либо из 7,5% растворов полимера С в изопропаноле (пример 4), либо из 7,5% раствора полидиметилсилоксана PS 510 в ацетоне (сравнительный пример 1). Раствор был приготовлен путем растворения 0,01 г метиленовой сини в 50 мл дистиллированной воды. Бруски дентина с покрытием погрузили в этот раствор на 5 мин, затем извлекли и промыли дистиллированной водой. Бруски высушили на воздухе и с них фотоаппаратом "Полароид" были выполнены снимки с использованием микроскопа с увеличением в 7,5 раз.
Брусок дентина, представленный на фиг. 1, был чисто белым с обеих сторон, что показывает отсутствие красителей на бруске дентина, покрытом полимером C. В отличие от этого на фиг. 2 показано, что бруски дентина, покрытые силоксаном из сравнительного примера 1, сохраняли краситель на своей поверхности, на что указывают темные участки.
Как упоминалось при анализе прилипания в Биологической оценке 1, на фиг. 3 и 4 показано прилипание бактерий к частицам эмали, не имеющим покрытия или имеющим перечисленные ниже покрытия:
1. Эмаль без покрытия и без обработки слюной.
2. Эмаль без покрытия с обработкой слюной.
3. Эмаль с покрытием B без обработки слюной.
4. Эмаль с покрытием B с обработкой слюной.
5. Эмаль с покрытием A с обработкой слюной.
6. Эмаль с покрытием C с обработкой слюной.
7. Эмаль с покрытием E с обработкой слюной.
8. Отрицательный контрольный образец.
Частицы эмали, имеющие покрытие, демонстрируют значительное уменьшение прилипания IgG, OVA и/или P.gin.
Пример 8
В табл. 18 описаны реакционноспособные составы полимеров с различными звеньями A. Каждый из полимеров, указанных в табл. 18, был приготовлен путем непрерывного процесса питающей полимеризации, описанного ниже.
В 250-миллилитровую колбу с круглым дном и тремя горловинами добавили изопропанол (20 мл) вместе с магнитным стержнем для перемешивания. В изопропаноле (60 мл) развели AIBN (0,2 г) и этот раствор поместили в 60-миллилитровую капельную воронку и установили на реакционной колбе. Описанное в табл. 18 звено A наряду с метилметакрилатом (26 г) и макромером PDMS (4 г) развели в изопропаноле (28 мл), поместили во вторую 60-миллилитровую капельную воронку и установили на реакционной колбе. В третью горловину реакционной колбы поместили конденсатор. Реакционный сосуд деоксигенировали при комнатной температуре путем барботирования загруженных растворов газообразным азотом в течение 15 минут. Содержимое двух капельных воронок постепенно добавляли в реакционную колбу с постоянной скоростью (10 мл/ч) в течение шести часов, поддерживая температуру реакционной колбы на уровне 60oC под слоем азота и при умеренном помешивании. После завершения добавления растворов мономера и инициатора реакционную колбу выдерживали при температуре 60oC и под слоем азота в течение дополнительных трех часов.
После полимеризации полимеров G и H в каждую реакционную смесь независимо добавили количество ацетона, достаточное для разведения полимеров. Каждый полимер был очищен путем добавления по каплям с интенсивным перемешиванием каждой реакционной смеси к количеству воды, по объему в четыре раза превышающему общий объем реакционной смеси. Каждый осажденный полимер извлекали из водной смеси и затем в течение нескольких суток сушили в вакуумной печи при температуре приблизительно 60oC. Полученные твердые полимеры измельчили и хранили в виде порошка.
Полимер 1 использовали без дополнительной очистки. Массовое содержание полимера в реакционной смеси определяли гравиметрическими способами, а затем реакционную смесь развели до приблизительной концентрации полимера 10% в смеси равных долей изопропанола и ацетона.
Гидрофобность и гидрофильность полимерных покрытий описана в табл. 18, где она характеризуется соответственно углами контакта при продвижении и при отступлении воды. Углы контакта при продвижении и отступлении воды измеряли, используя пластинчатый способ Вильгельми. В качестве пластинок для измерений использовали покрытые полимером стеклянные пластинки (22 мм х 22 мм x 0,15 мм). Перед нанесением покрытия погружением в растворы полимеров стеклянные пластинки были обработаны силаном с гамма-метакрилоксипропилтриметоксисиланом ("A-174", компания OSi Specialties, Inc.). Углы контакта при продвижении и отступлении воды на пластинках с покрытием измеряли, используя скорость погружения пластинок 50 мкм/с. Угол контакта приведен в табл. 18 и представляет собой среднее для результатов двух или трех повторных измерений, показывая, что полимерные покрытия были более гидрофобными и менее гидрофильными, чем открытая, полированная эмаль (показано в табл. 6).
Сопротивление полимеров абразивному воздействию зубной щетки и зубной пасты, показанное в табл. 18 для эмали, было определено с помощью следующей процедуры, названной "Способ I". Бычьи резцы поместили в эпоксидную смолу. Затем щечные поверхности отполировали влажной и сухой наждачной бумагой из карбида кремния зернистостью 120 и 600 с тем, чтобы получить чистые, плоские поверхности эмали. Для дальнейшей очистки полированные поверхности эмали протравили 10% раствором лимонной кислоты в воде в течение 15 секунд, промыли и просушили. Полимеры (10%), описанные в табл. 18, развели в растворах, указанных в табл. 19, и нанесли на полированные поверхности эмали стержнем для нанесения покрытий N 75 компании RD Specialties, Inc. Толщина сухих полимерных пленок равнялась приблизительно 5 мкм.
Каждую поверхность эмали с покрытием чистили мягкой прямой зубной щеткой ORAL ВTM 35 под нагрузкой 2,7 H при частоте 3 цикла/с до тех пор, пока не осталось только 10% эмали, покрытой полимером. Поверхность эмали и зубную щетку во время процесса чистки погрузили в суспензию, состоящую из равных массовых долей зубной пасты CRESTTM Regular Flavor и дистиллированной воды.
В процессе чистки через регулярные интервалы с помощью процедуры окрашивания определяли долю полированной поверхности эмали, на которой сохранилось полимерное покрытие. Полированную поверхность протравили за одну секунду 37% фосфорной кислотой, промыли, погрузили в 0,2% водный раствор кислотного фиолетового N 17 (Aldrich Chemical Company, Inc., Милуоки, шт. Висконсин) приблизительно на 30 секунд, промыли и просушили. Устойчивые к бляшке полимеры оказались относительно незатронутыми травлением фосфорной кислотой и противостояли окрашиванию кислотным фиолетовым N 17, в то время как на тех участках, где не осталось покрытия, наблюдалось окрашивание. Доля полированной поверхности, сохранившей покрытие после чистки щеткой, определяли визуальным осмотром и обозначали как процентную долю поверхности, оставшейся неокрашенной.
В табл. 19 показано сопоставление сопротивления абразивному воздействию зубной щетки и зубной пасты со стороны полимеров G, H и I. Результаты, которые представляют среднее значение по пяти повторным измерениям, показывают, что полимеры G, H и I обладают хорошей устойчивостью к абразивному воздействию, возникающему при чистке зубов.
Пример 9
Ниже в табл. 20 описаны реакционные составы устойчивых к бляшке полимеров с различными звеньями В. Каждый из полимеров из табл. 20 приготовили путем помещения в 250-миллилитровую колбу с круглым дном акриловой кислоты (6 г), макромера PDMS (4 г), AIBN (0,2 г), описанного звена В (26 г) и растворителя (108 мл). Реакционную смесь деоксигенировали при комнатной температуре путем барботирования загруженных растворов газообразным азотом в течение 15 минут. Затем при слабом помешивании температуру реакционной смеси подняли до 60oC. Реакцию осуществляли в течение восьми часов под слоем азота.
Полимеры J и L были очищены путем независимого добавления по каплям с интенсивным перемешиванием каждой реакционной смеси к количеству воды, по объему в четыре раза превышающему общий объем реакционной смеси. Осажденные полимеры извлекали из водной смеси и затем в течение нескольких суток сушили в вакуумной печи при температуре приблизительно 60oC. Просушенные полимеры подвергли тонкому измельчению и хранили в форме порошка. Полимер K подвергли очистке с помощью сходной процедуры за исключением того, что к реакционной смеси добавили 36 г ацетона для того, чтобы развести полимер перед добавлением реакционной смеси по каплям к воде.
Сопротивление полимеров абразивному воздействию зубной щетки и зубной пасты, показанное в табл. 20 для эмали, было определено с помощью следующей процедуры, названной "Способ II". Бычьи резцы поместили в полиметилметакрилат так, что щечные поверхности выступали над обрамляющим их материалом. Затем щечные поверхности отполировали влажной и сухой наждачной бумагой из карбида кремния зернистостью 120 и 600 с тем, чтобы получить чистые, плоские поверхности эмали. Для дальнейшей очистки полированные поверхности эмали протравили 10%-ным раствором лимонной кислоты в воде в течение 15 секунд, промыли и просушили. Растворы полимеров (10%), описанных в табл. 20, в изопропаноле (полимеры J и L) или в составе из равных долей изопропанола и ацетона (полимер K) нанесли на полированные поверхности эмали небольшой кистью и дали просохнуть. Образцы эмали с покрытием хранили перед чисткой щеткой в течение 24 часов при температуре 37oC и относительной влажности 97%.
Каждую поверхность эмали с покрытием чистили мягкой прямой зубной щеткой ORAL ВTM 35 под нагрузкой 140 г при частоте 50 циклов/мин в течение пятнадцати минут. Поверхность эмали и зубную щетку во время процесса чистки погрузили в суспензию, состоящую из равных массовых долей зубной пасты CRECTTM Regular Flavor и дистилированной воды. После чистки с помощью процедуры окрашивания, приведенной в примере 8 способа I, определили долю в % полированной поверхности эмали, на которой сохранилось покрытие. В табл. 20 показано сопоставление сопротивления абразивному воздействию зубной щетки и зубной пасты со стороны полимеров с различными звеньями В. Результаты, представляющие собой средние значения по двум измерениям, показывают, что полимерные покрытия обладают хорошей устойчивостью к абразивному воздействию, возникающему при чистке зубов.
Пример 10
В табл. 21 описаны реакционные составы устойчивых к бляшке полимеров с разным относительным содержанием звеньев A, B и C. Каждый из полимеров из табл. 21 приготовили путем помещения в 250-миллилитровую колбу с тремя горловинами и круглым дном мономеров, AIBN (0,2 г) и изопропанола (108 мл). Реакционную смесь деоксигенировали при комнатной температуре путем барботирования смеси газообразным азотом в течение 15 минут. Затем при слабом помешивании температуру смеси повысили до 60oC. Реакцию осуществляли в течение восьми часов под слоем азота. После полимеризации мономеров, представленной в табл. 21, в каждую реакционную смесь добавили количество ацетона, достаточное для разведения полимеров.
Каждый полимер был очищен путем добавления по каплям с интенсивным перемешиванием каждой реакционной смеси к количеству воды, по объему в четыре раза превышающему общий объем реакционной смеси. Каждый осажденный полимер извлекали из водной смеси и затем в течение нескольких суток сушили в вакуумной печи при температуре приблизительно 60oC. Полученные твердые полимеры измельчили и хранили в виде порошка.
Гидрофобность и гидрофильность полимерных покрытий описана в табл. 21, где она характеризуется соответственно углами контакта при продвижении и при отступлении воды. Для измерений углов контакта при продвижении и отступлении воды использовали метод сидячей капли. Образцы для определения угла контакта по методу сидячей капли были приготовлены путем помещения сначала бычьих резцов в эпоксидную смолу. Затем щечные поверхности отполировали влажной и сухой наждачной бумагой из карбида кремния зернистостью 120 и 600, алмазными пастами 6 и 3 мкм и глиноземной суспензией 0,05 мкм, чтобы обнажить чистые, плоские поверхности эмали. Растворы устойчивых к бляшкам полимеров (10%) в табл. 21 в составе из равных долей изопропанола и ацетона были нанесены на отполированные поверхности эмали небольшой кисточкой. Угол контакта приведен в табл. 21 и представляет собой среднее значение для результатов двух повторных измерений, показывая, что полимерные покрытия были более гидрофобными и менее гидрофильными, чем открытая, полированная эмаль (показано в табл. 6).
Устойчивость полимеров в табл. 21 абразивному воздействию зубной щетки и зубной пасты измерили по способу I, за исключением того, что для нанесения растворов полимеров на подготовленную поверхность эмали использовали стержень для нанесения покрытий N 40 компании RD Specialties, Inc, получив сухую пленку толщиной приблизительно 3 мкм. Полимерные пленки были получены из полимеров (10%) в растворах, состоящих из равных частей изопропанола и ацетона. Зубы чистили щеткой до полного удаления полимеров. Приведенные в табл. 21 результаты сопротивления абразивному воздействию зубной щетки и зубной пасты, представляющие средние значения по трем или пяти последовательным измерениям, показывают, что полимерные покрытия обладают хорошей устойчивостью к абразивному воздействию, возникающему при чистке зубов щеткой.
Пример 11
Сцепление кариогенных бактерий с полимерами N и K из примера 10 определяли с помощью следующей процедуры. Чистые бычьи зубы протравили в течение 15 с 10%-ной лимонной кислотой, промыли и просушили. На коронки протравленных кислотой зубов нанесли покрытие методом погружения в 10%-ные растворы полимера в составе из равных долей изопропанола и ацетона с последующей сушкой на воздухе.
Бычьи зубы с полимерным покрытием поместили в пробирки из полипропилена размерами 10 мм х 30 мм и закрепили на месте, используя оттисковый материал 3MTM ImprintT (компании 3M). Зубы поместили таким образом, чтобы открытыми оставались только коронки зуба. Затем зубы поместили коронкой вниз в лунки 24-луночного культурального планшета (компании Costar, Inc., Кембридж, шт. Массачусетс), которые заполнили человеческой слюной (2,3 мл), полностью покрывшей коронки. Зубы выдерживали в течение часа при комнатной температуре со встряхиванием. В слюну добавили "Mutans streptococci" (Американская коллекция типов культур, Роквилл, шт. Мэриленд), промытые в буфере KCl (0,1 М NaCl, 0,05 М KCl, 1 мМ KH2PO4, 0,1 мМ MgCl2, 1 мМ CaCl2, pH 7,0) (109 на зуб), и при встряхивании выдерживали еще два часа при комнатной температуре. Затем зубы дважды промыли либо буфером KCl, либо буфером KCl, дополненным 0,3% TWEEN-80 (Sigma, Inc. , Сент-Луис, шт. Миннесота). Зубы извлекли из оттискового материала и поместили коронкой вниз на новые 24-луночные планшеты. В каждую лунку добавили буфер KCl либо буфер KCl, дополненный TWEEN-80 (2 мл), после чего зубы промыли еще два раза. В каждую лунку добавили буфер экстракции ДНК (0,4 М NaOH, 10 мМ этилендиаминтетрауксусной кислоты (EDTA)) (2,3 мл) и в течение 12 минут планшеты выдерживали при температуре до 95-100oC. Солюбилизированную ДНК извлекли из лунок планшета и разделили на три равные части. Каждую часть добавили в лунку слот-блоттерного устройства, где определялся уровень бактериальной ДНК в каждом образце. Затем зубы извлекли из лунок планшета и пронумеровали.
Уровень бактериальной ДНК в каждом образце определяли с помощью следующей процедуры слот-блоттинга ДНК. Лист мембраны гибридизации зета-зонда (BioRad Laboratories, Inc., Ричмонд, шт. Калифорния) приготовили путем погружения мембраны в дистиллированную воду. Затем влажную мембрану вставили в слот-блоттерное устройство (Minifold II, компания Schleicher & Schuell, Inc. ), создали вакуум и каждую лунку промыли 0,5 мл буфера TE (10 мМ трис(оксиметил)аминометангидрохлорида (гидрохлорид TRIZMATM, Sigma, Inc.), 1 мМ EDTA, pH 8,0). Солюбилизированные образцы ДНК были добавлены в каждую лунку и промыты один раз 0,4 М NaOH. После прокачивания всей жидкости сквозь мембрану источник вакуума отсоединили, устройство разобрали и ДНК зафиксировали на полусухой мембране, подвергнув в течение 3 минут воздействию ультрафиолетового света (StratLinker, Stratagene, Inc., Лайолла, шт. Калифорния). Мембраны подвергли короткой промывке в 0,3 М NaCl, 0,03 М цитрата натрия и просушили в термостате при температуре 37oС. Сухие мембраны поместили в стеклянную камеру гибридизации с добавлением 15 мл предгибридизационного раствора (компания Life Technologies, Inc, Грэнд Айленд, шт. Нью Йорк). Пробирки в течение одного часа вращали при температуре 65oС в гибридизационной печи (Hybridizer 700, компания Stratagene, Inc.). В предгибридизационный раствор (15 мл) были добавлены меченные дигоксигенином цельные геномные зонды испытываемого организма (150 нг). Разведенные зонды кипятили в течение 12 минут. Предгибридизационный раствор извлекли затем из стеклянных гибридизационных камер и заменили разведенным раствором зонда. Слот-блоттер выдержали с зондом в течение ночи при температуре 65oC.
Слот-блоттерные мембраны, выдержанные в течение ночи с меченными дигоксигенином зондами, извлекли из гибридизационной печи и дважды промывали в течение 5 минут в 0,3 М NaCl, 0,03 М цитрата натрия с 0,1% додецилсульфата натрия ("SDS") на стеклянном лотке, установленном на качалке, при комнатной температуре. Мембраны дважды промыли в 50 мл 15 мМ NaCl, 1,5 мМ цитрата натрия с 0,1% SDS в гибридизационной печи при температуре 65oC в течение 30 минут. Затем мембраны поместили на стеклянный лоток и промывали в течение 2 минут при комнатной температуре в буфере малеиновой кислоты (0,15 М NaCl, 0,1 М малеиновой кислоты, pH 7,5). Мембраны в течение 1 часа выдерживали в буфере малеиновой кислоты при комнатной температуре с добавлением 10% протеинов снятого молока (KPL Laboratories, Гэйтсбург, шт. Мэриленд) с целью блокирования неспецифических мест реакции на мембране. Антидигоксигениновое антитело, меченное щелочной фосфатазой (Boehringer Mannheim, Inc., Индианаполис, шт. Индиана), было разведено в соотношении 1:5000 в буфере малеиновой кислоты с 10% протеинов снятого молока и добавлено к мембранам на 1 час при комнатной температуре. Раствор антитела был удален и мембрану дважды промывали в течение 15 минут в буфере малеиновой кислоты с 0,3% TWEEN-20. После извлечения из промывочного буферного раствора мембрану уравновешивали в течение 5 минут в буфере ферментного субстрата (0,1 М гидрохлорида TRIZMA, pH 9,5). Добавили раствор субстрата фермента бромхлориндолфосфат/нитроголубой тетразол (KPL Laboratories, Inc.) и мембрану в течение 30 минут выдерживали в темноте при комнатной температуре. Развитие цвета было остановлено путем переноса мембраны на 5 минут в буфер TE с последующим вымачиванием в течение 5-10 минут в дистиллированной воде. После этого мембраны для сушки разместили на бумажных полотенцах.
Просушенные мембраны разместили на транспортном лотке плотномера (модель 325, Molecular Dynamics, Inc., Саннивейл, шт. Калифорния) и подвергли сканированию. Полученные данные были объединены в цифровой файл с использованием программного обеспечения Molecular Dynamics Image QuantTM. Определяли чистую оптическую плотность каждого паза и сравнивали ее со стандартной кривой по опытам с микробным ДНК на каждой мембране. Микробные эквиваленты каждого паза были рассчитаны и нормализованы по площади поверхности коронки зуба, определенной с помощью метода профилометрии.
В табл. 22 показаны результаты прилипания бактерий к полимерам N и K для четырех штаммов "mutans streptococci" из Американской коллекции типов культур (АТСС), Роквилл, шт. Мэриленд. В табл. 22 показано уменьшение в процентах количества связанных бактерий по сравнению с эмалью без покрытия. Четыре штамма бактерий оценивали с поверхностно-активным веществом TWEEN-80, включенным в промывочные составы KCl, и без него. Когда TWEEN-80 включали в промывочные составы KCl для зубов с полимерным покрытием, TWEEN-80 включали также в промывочные составы KCl для контрольных зубов без полимерного покрытия. Результаты, представляющие среднее значение по пяти измерениям, показывают, что когда на эмаль были нанесены покрытия, устойчивые к бляшке, было достигнуто значительное уменьшение прилипания кариогенных бактерий.
Пример 12
В табл. 23 описаны реакционные составы устойчивых к бляшке полимеров с различными звеньями D, включающими А-174 и алкоксисиланы, показанные ниже.
В табл. 23 сравнивается также полимер, приготовленный без звена D. Каждый из шести полимеров в табл. 23 был приготовлен путем загрузки в 250-миллилитровую колбу с круглым дном акриловой кислоты (6 г), изобутилметакрилата, макромера PDMS (4 г), описанного звена D, AIBN (0,2 г) и изопропанола (108 мл). Реакционную смесь подвергли деоксигенации при комнатной температуре путем барботирования смеси в течение пятнадцати минут азотом. Затем реакционную смесь нагрели до 60oC при осторожном помешивании и в течение восьми часов выдерживали под слоем азота.
Полимер B был очищен путем добавления по каплям с интенсивным перемешиванием каждой реакционной смеси к количеству воды, по объему в четыре раза превышающему общий объем реакционной смеси. Осажденный полимер извлекли из водной смеси и затем в течение нескольких суток сушили в вакуумной печи при температуре приблизительно 60oC.
Содержащие звено D полимеры, описанные в табл. 23 (полимеры Q-U), использовали без дополнительной очистки. Массовое содержание полимера в каждой реакционной смеси определяли гравиметрическими способами, а затем каждую реакционную смесь развели до концентрации полимера 10% в изопропаноле.
Гидрофобность и гидрофильность полимерных покрытий описана в табл. 23, где она характеризуется соответственно углами контакта при продвижении и при отступлении воды. Углы контакта при продвижении и отступлении воды определяли пластинчатым способом Вильгельми. Полученные результаты приведены в табл. 23. Результаты, представляющие собой среднее значение для результатов двух измерений, показывают, что полимерные покрытия были более гидрофобными и менее гидрофильными, чем открытая, полированная эмаль (показано в табл. 6).
Сопротивление полимеров абразивному воздействию зубной щетки и зубной пасты, показанное в табл. 23 для эмали, было определено с помощью способа II. Растворы полимеров (10%), описанных в табл. 23, в изопропаноле нанесли на полированные поверхности эмали небольшой кистью и просушили на воздухе. Образцы перед чисткой щеткой держали во влажностной печи при температуре 37oC в течение по меньшей мере 24 часов. Результаты, которые представляют собой среднее значение по меньшей мере двух измерений, показывают, что добавление звена D способствует повышению устойчивости полимерных покрытий к абразивному воздействию зубной щетки и зубной пасты.
Пример 13
Ниже в табл. 24 показаны углы контакта при продвижении и отступлении воды для полимеров Q-U из примера 12, когда в раствор покрытия был добавлен катализатор конденсации. Для определения углов контакта использовали пластинчатый способ Вильгельми. В качестве катализатора конденсации использовали октоат олова (5% от массы полимера). Приведенные в табл. 24 результаты, являющиеся средними значениями для двух измерений, показывают, что полимерные покрытия были более гидрофобными и менее гидрофильными, чем открытая, полированная эмаль (показано в табл. 6).
Кроме того, в табл. 24 показана устойчивость к абразивному воздействию зубной щетки и зубной пасты нанесенных на эмаль полимеров Q-U, определенная способом II при добавлении в покрывающий раствор катализатора конденсации (5% от массы полимера). Растворы устойчивых к бляшке полимеров (10%) в изопропаноле были приготовлены с использованием в качестве катализатора конденсации октоата олова, дибутилового дилаурата олова или триэтилендиамина. Растворы покрытия нанесли на полированные поверхности эмали небольшой кистью и просушили на воздухе. Образцы перед чисткой щеткой держали во влажностной печи при температуре 37oC в течение по меньшей мере 24 часов. Результаты, которые представляют собой среднее значение по меньшей мере двух измерений, показывают, что добавление сшиваемого алкоксисиланового звена D способствует повышению устойчивости полимерных покрытий к абразивному воздействию, возникающему при чистке зубов.
Пример 14
В табл. 25 показаны углы контакта при продвижении и отступлении воды для полимеров Q и R из примера 12, когда в раствор покрытия было добавлено мостиковое соединение для сшивания звеньев D. Мостиковое соединение под названием "PHS", указанное в табл. 25, было получено из А-174 после гидролиза и частичной конденсации. Концентрация PHS, показанная в табл. 25, выражена в мас. % от массы полимера в растворе. Для определения углов контакта использовали пластинчатый способ Вильгельми. Приведенные в табл. 25 результаты, являющиеся средними значениями для двух измерений, показывают, что полимерные покрытия были более гидрофобными и менее гидрофильными, чем открытая, полированная эмаль (показано в табл. 6).
В табл. 26 и 27 показаны соответственно углы контакта при продвижении и отступлении воды соответственно для полимеров Q и R из примера 12, когда к раствору покрытия был добавлен Y-11597 (три[N-(триметоксисилил)пропил]изоционурат, поставляемый в промышленных масштабах компанией OSi Specialties, Inc. ). Выраженное в молях количество этого мостикового соединения, добавленное к раствору покрытия, было рассчитано на основе одного моля единицы А-174 в полимере. В табл. 26 и 27 показано также воздействие катализатора конденсации в сочетании с мостиковым соединением на углы контакта. В качестве катализатора конденсации использовали октоат олова при концентрации 5% от массы полимера в растворе. Для определения угла контакта использовали пластинчатый способ Вильгельми. Приведенные в табл. 26 и 27 результаты, являющиеся средними значениями для двух измерений, показывают, что полимерные покрытия были более гидрофобными и менее гидрофильными, чем открытая, полированная эмаль (показано в табл. 6).
В табл. 25 и 28 соответственно показана устойчивость к абразивному воздействию зубной щетки и зубной пасты нанесенных на эмаль полимеров Q и R, определенная способом II при добавлении в раствор покрытия мостикового соединения PHS или Y-11597. В табл. 28 показано воздействие катализатора конденсации в сочетании с мостиковым соединением. Растворы полимеров (10%) в изопропаноле были приготовлены с использованием различных количеств мостикового соединения и, в качестве необязательной добавки, катализатора конденсации. В качестве катализатора конденсации использовали октоат олова при концентрации 5% от массы полимера в растворе. Растворы покрытия нанесли на полированные поверхности эмали небольшой кистью и просушили на воздухе. Образцы перед чисткой держали во влажностной печи при температуре 37oC в течение по меньшей мере 24 часов. Результаты, которые представляют собой среднее значение по меньшей мере для двух измерений, показывают, что добавление сшиваемого алкоксисиланового звена D способствует повышению устойчивости покрытий к абразивному воздействию.
Пример 15
Прилипание кариогенных бактерий к полимерам Q и S-U из примера 12 при использовании в качестве катализатора конденсации октоата олова и без него (5% от массы полимера в растворе) было измерено с использованием процесса, описанного в примере 11. В табл. 29 приведены результаты для штамма Streptococcus sobrinus (АТСС 27351) и для штамма Streptococcus gordonii (АТСС 10558). В табл. 29 в процентах показано уменьшение количества связанных бактерий по сравнению с эмалью без покрытия. Два вида бактерий оценивали при включении TWEEN-80 в промывочный состав KCl и без него. При включении Tween-80 в промывочный состав KCl для зубов с полимерным покрытием Tween-80 включали и в промывочный состав KCl для контрольных зубов с эмалью без покрытия. Результаты, представляющие собой среднее значение по пяти измерениям, показывают, что когда на эмаль были нанесены покрытия, устойчивые к бляшке, было достигнуто значительное уменьшение прилипания кариогенных бактерий.
Пример 16
В табл. 30 показаны реакционные составы полимера с метилметакрилатом звена В и А-174 звена D. Для сопоставления в табл. 30 приведен реакционный состав сходного полимера, приготовленного без соответствующего звена D. Оба полимера, приведенные в табл. 30, были приготовлены с помощью процесса полимеризации с непрерывной загрузкой, описанного в примере 8. Исходные материалы для мономера включали акриловую кислоту (6 г), метилметакрилат (26 г), макромер PDMS (4 г) и, в качестве необязательного компонента, А-174 звена D (2,6 г) в изопропаноле (28 мл). Исходный материал инициатора состоял из AIBN (0,2 г) в изопропаноле (60 мл). Исходные материалы мономера и инициатора были помещены в соответствующие 60-миллилитровые капельные воронки, а воронки были укреплены на 250-миллилитровой колбе с круглым дном и тремя горловинами. Первоначально в 250-миллилитровую колбу с круглым дном поместили небольшое количество изопропанола (20 мл) и магнитный стержень для перемешивания. В третью горловину реакционной колбы поместили конденсатор. Реакционную колбу деоксигенировали при комнатной температуре путем барботирования загруженных растворов газообразным азотом в течение 15 минут. Содержимое двух капельных воронок постепенно добавляли в реакционную колбу с постоянной скоростью (10 мл/ч) в течение шести часов, поддерживая температуру реакционной колбы на уровне 60oC под слоем азота при умеренном помешивании. После завершения добавления растворов мономера и инициатора реакционную колбу выдерживали при температуре 60oC и под слоем азота в течение дополнительных трех часов.
После полимеризации полимера K в реакционную смесь добавили ацетон (50 мл) для разведения полимеров. Затем полимер был очищен путем добавления по каплям с интенсивным перемешиванием каждой реакционной смеси к количеству воды, по объему в четыре раза превышающему общий объем реакционной смеси. Осажденный полимер извлекли из водной смеси и затем в течение нескольких суток сушили в вакуумной печи при температуре приблизительно 60oC. Полученный твердый полимер измельчили и хранили в виде порошка.
Полимер V использовали без дополнительной очистки. Процентный выход полимера в реакционной смеси, определенный гравиметрическим способом, составил 76,4%. Затем реакционную смесь развели до приблизительной концентрации полимера 10% в смеси равных долей изопропанола и ацетона.
В табл. 30 показаны также углы контакта при продвижении и при отступлении воды для полимера К и полимера V с мостиковым соединением PHS и без него, а также с катализатором конденсации октоатом олова и без него. Углы контакта определяли пластинчатым способом Вильгельми. Результаты, представляющие собой среднее значение для результатов двух измерений, показывают, что полимерные покрытия были более гидрофобными и менее гидрофильными, чем открытая, полированная эмаль (показано в табл. 6).
Сопротивление полимера K и полимера V абразивному воздействию зубной щетки и зубной пасты с мостиковым соединением PHS и без него, а также с катализатором конденсации октоатом олова и без него на эмали было определено с помощью способа I. Растворы полимеров (10%), описанные в табл. 30 и разведенные в составе из равных долей изопропанола и ацетона, нанесли на полированные поверхности эмали стержнем для нанесения покрытий N 75. Толщина сухих полимерных пленок равнялась приблизительно 5 мкм. Зубы с покрытием перед чисткой хранили во влажностной печи при температуре 37oC в течение 70 часов при относительной влажности 95%. Зубы с покрытием чистили до тех пор, пока всего 10% эмали оставались покрытыми полимером. Результаты, показанные в табл. 30, которые представляют собой среднее значение по пяти повторным измерениям, показывают, что включение сшиваемого алкоксисиланового звена D значительно повышает устойчивость покрытия к абразивному воздействию.
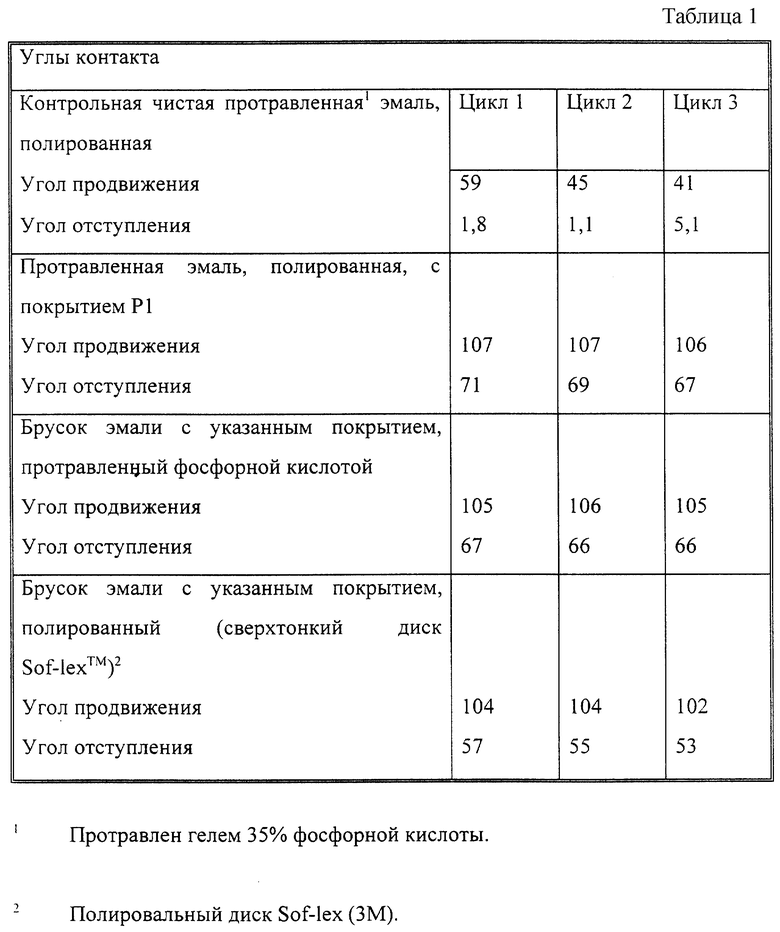
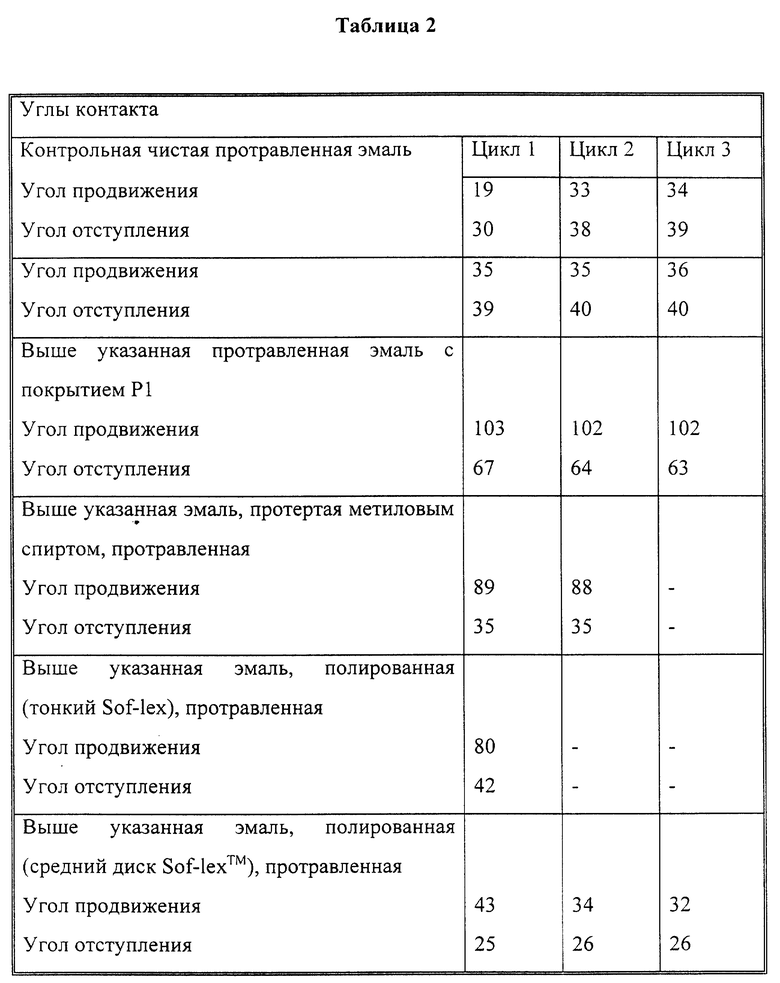
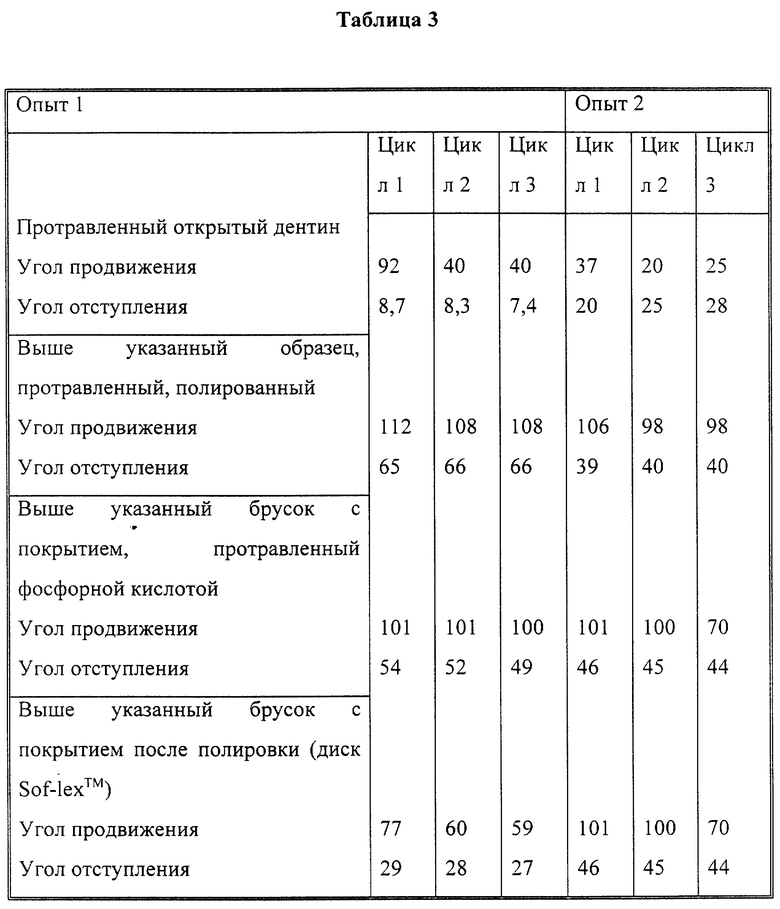
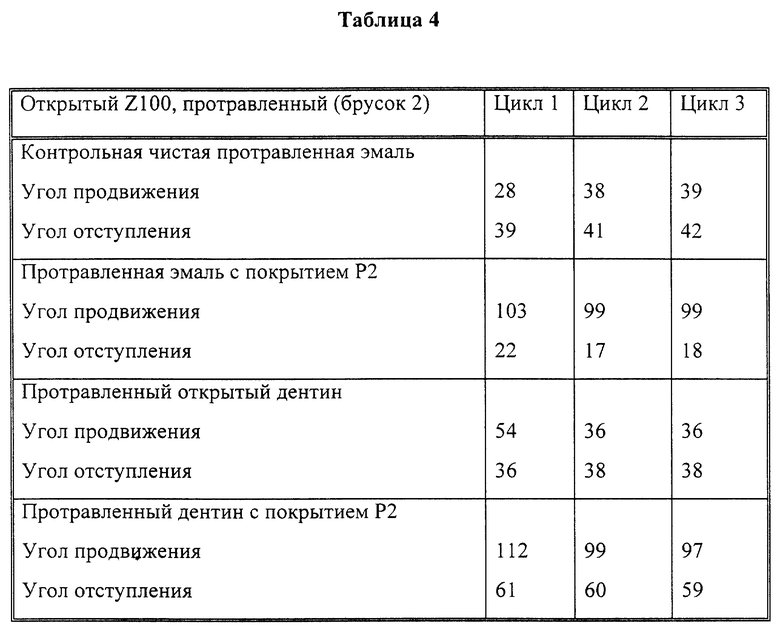
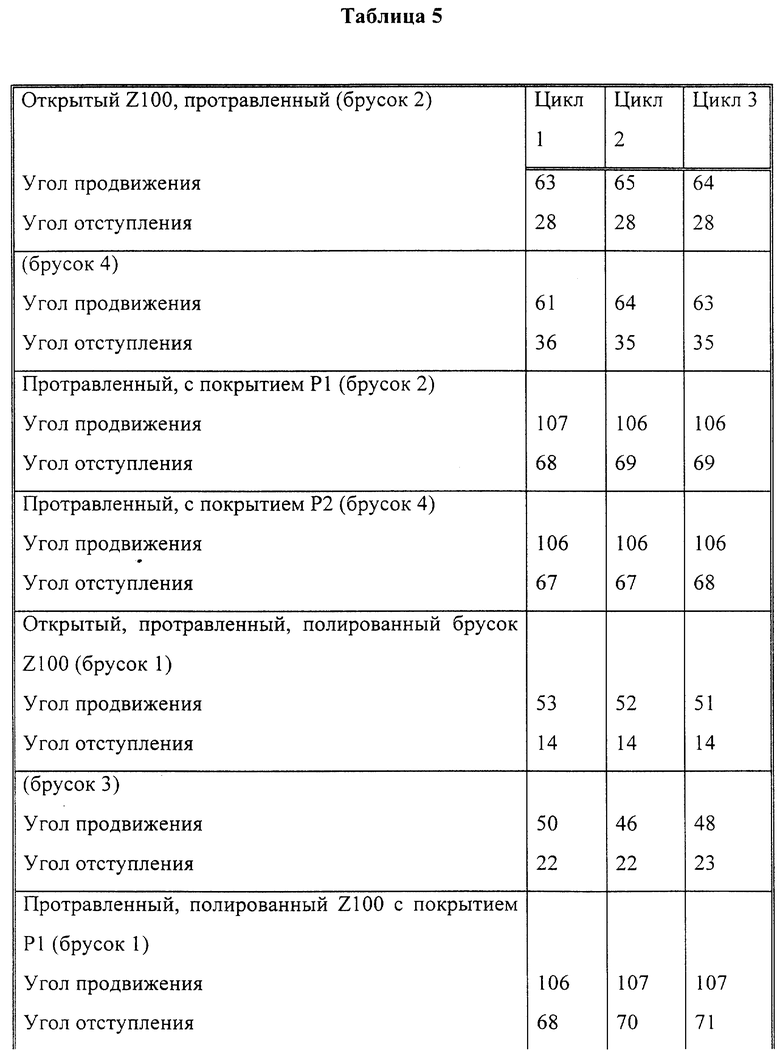

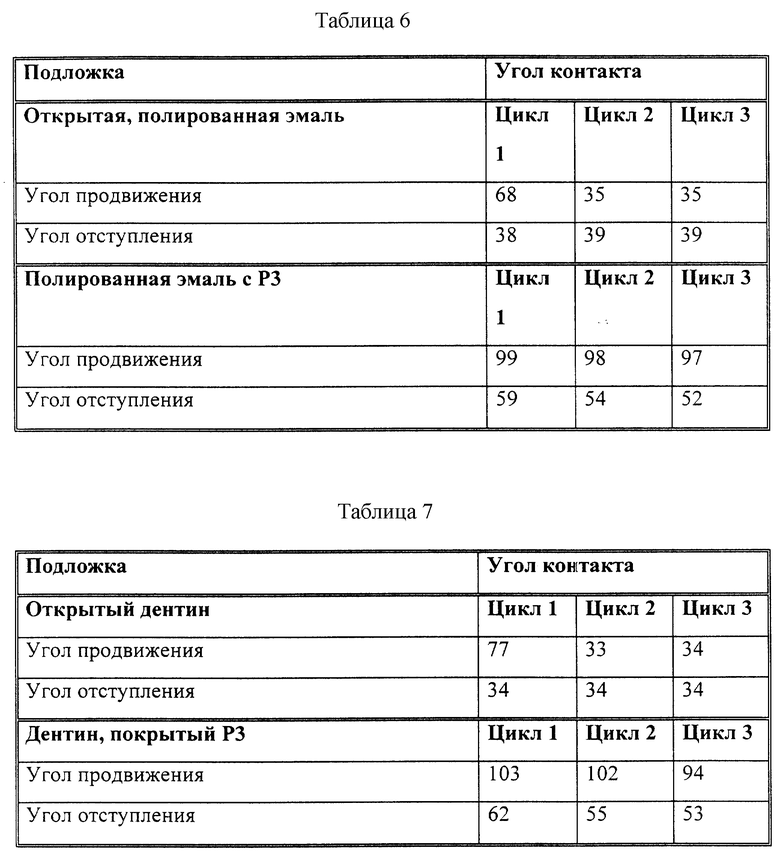
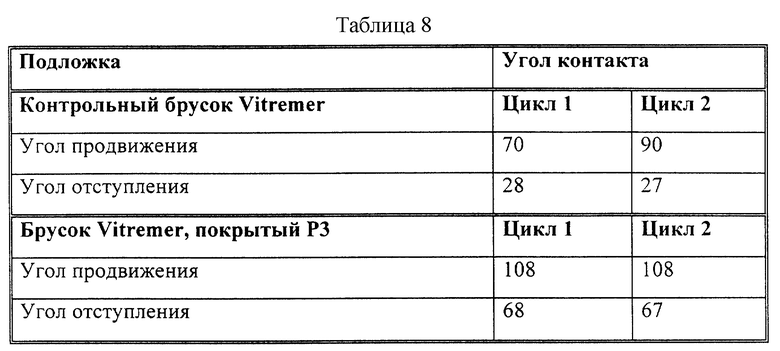
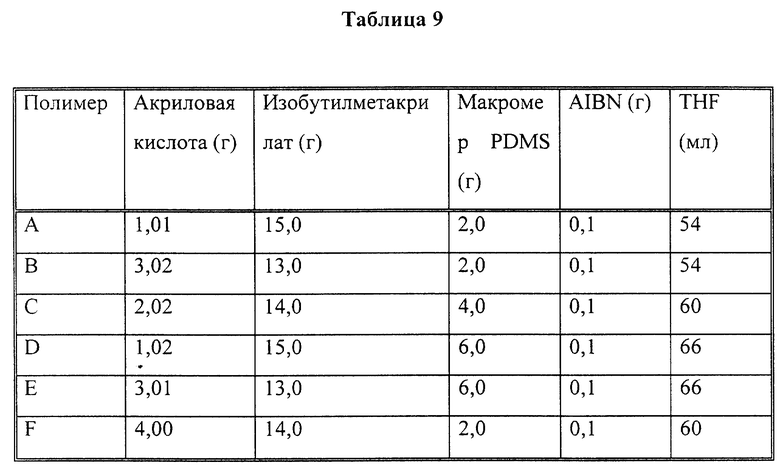
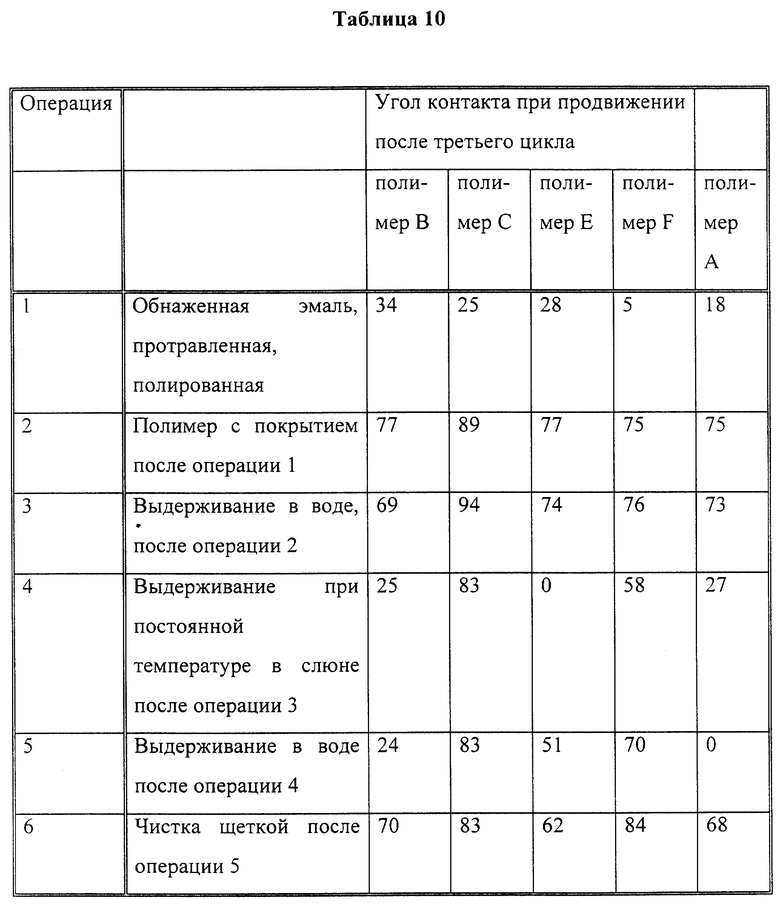
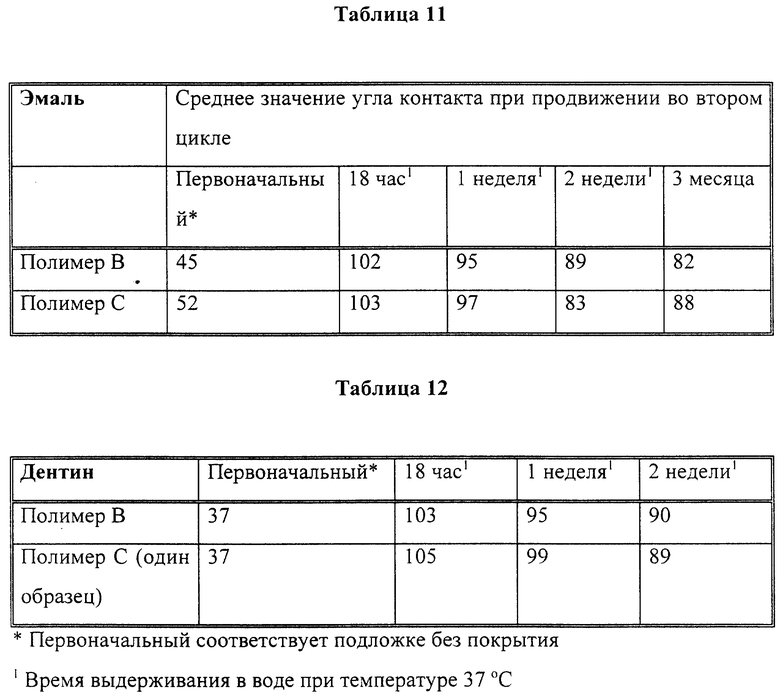
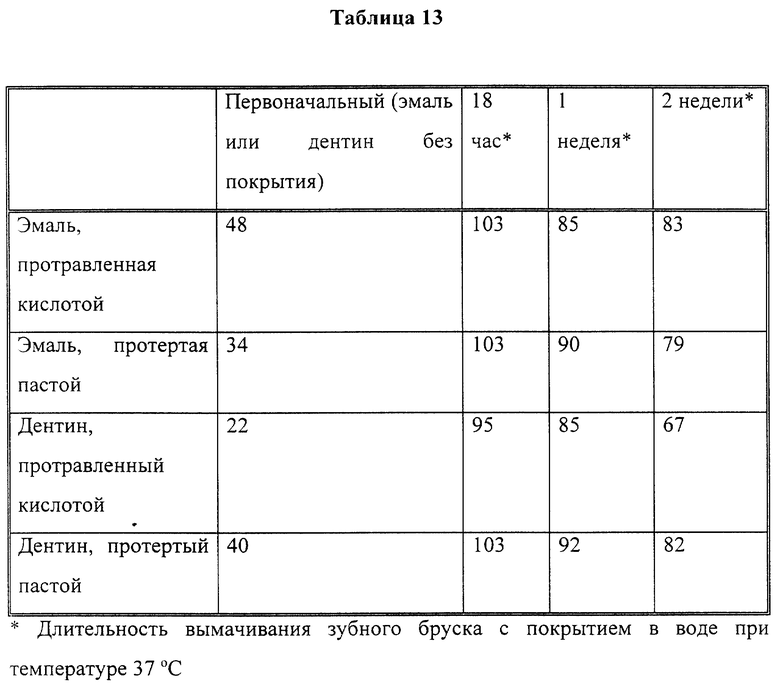
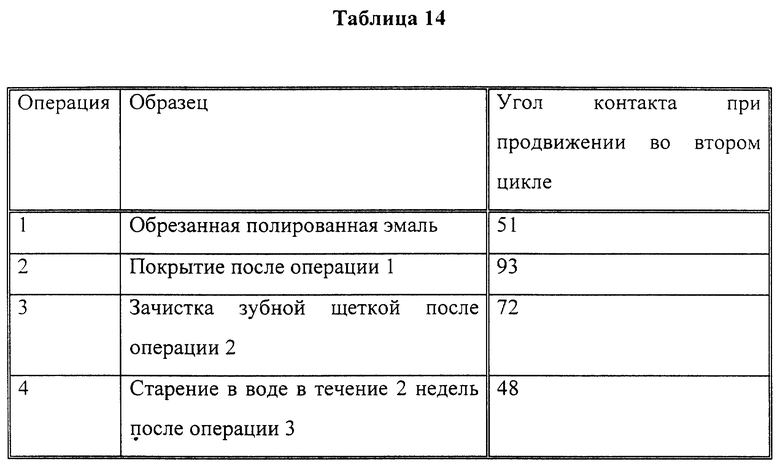

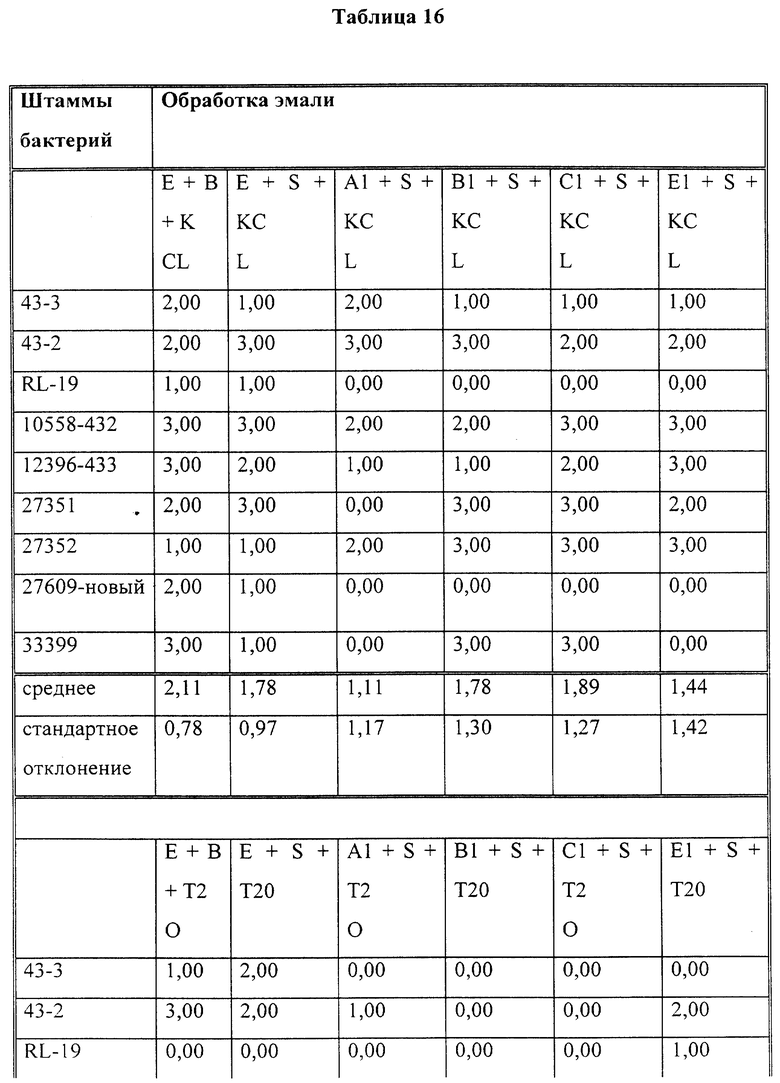
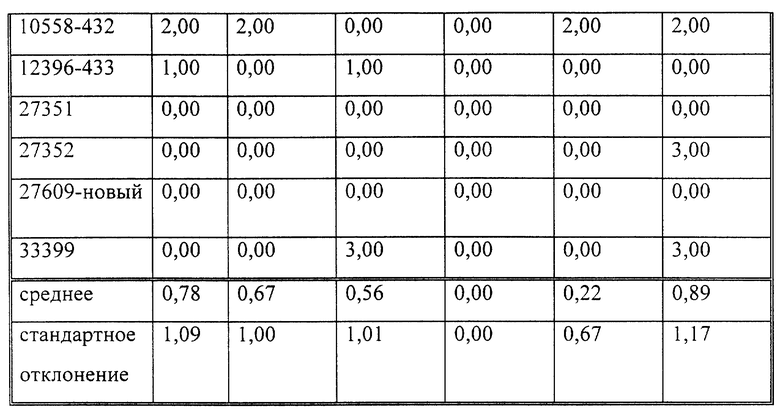
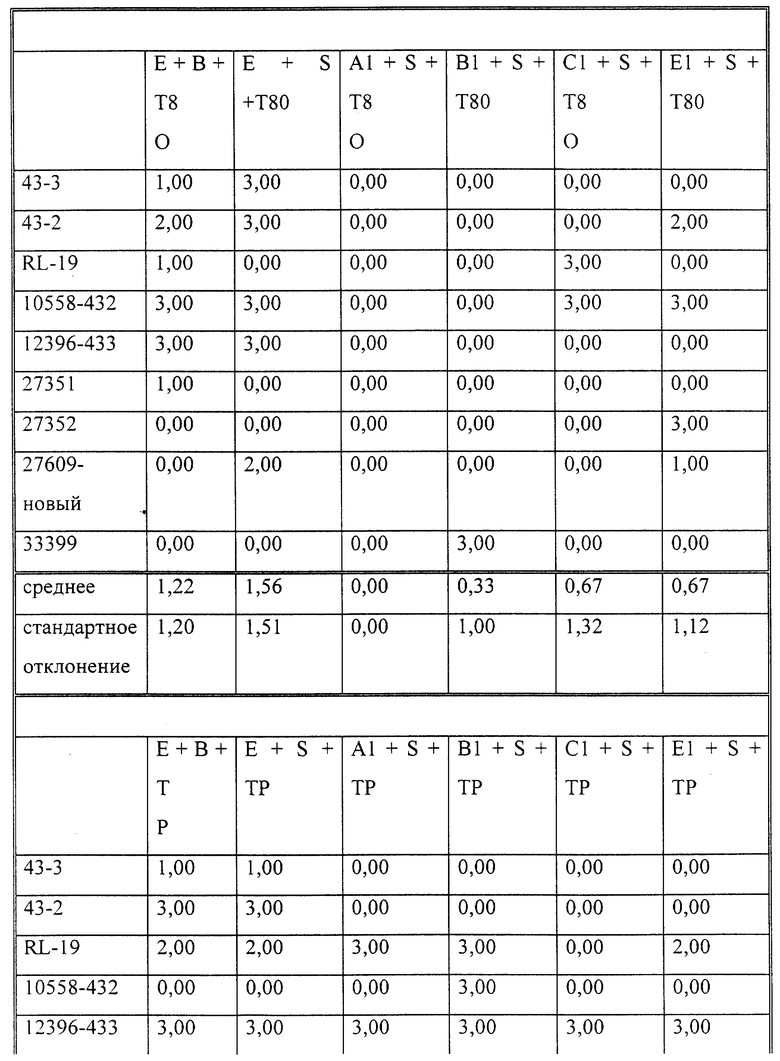
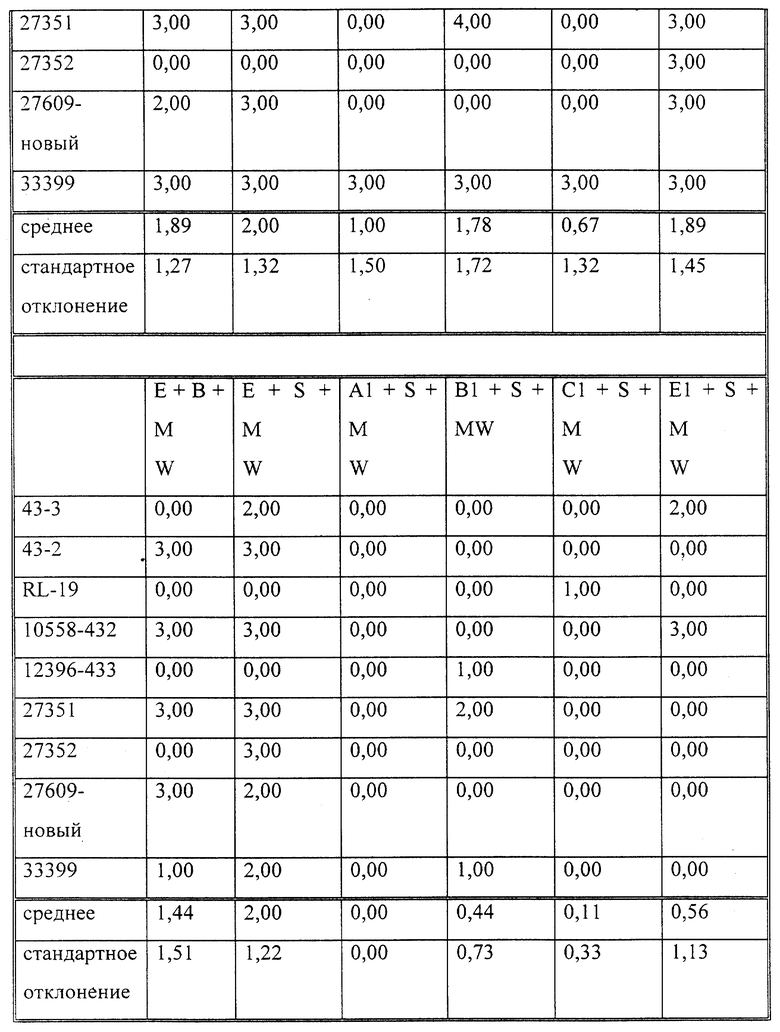

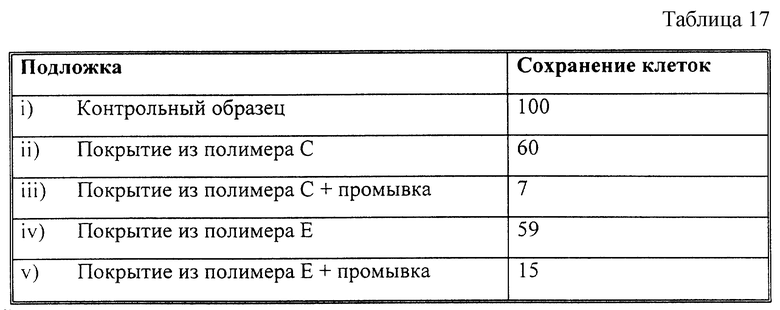
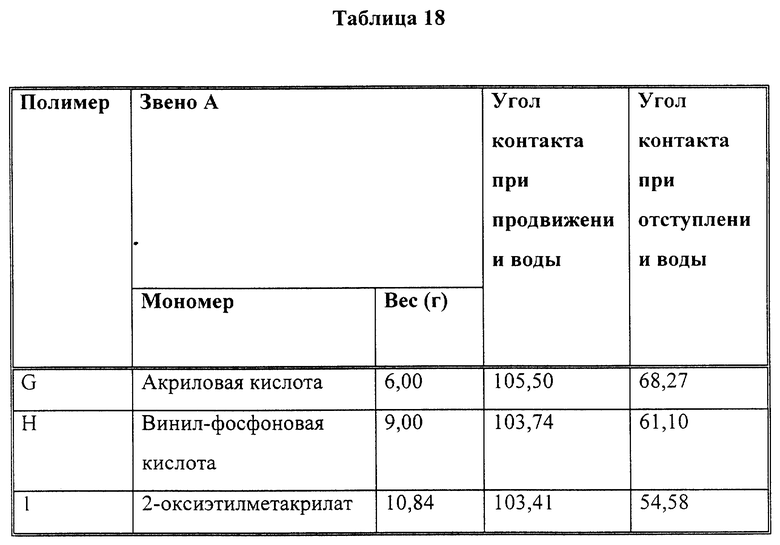
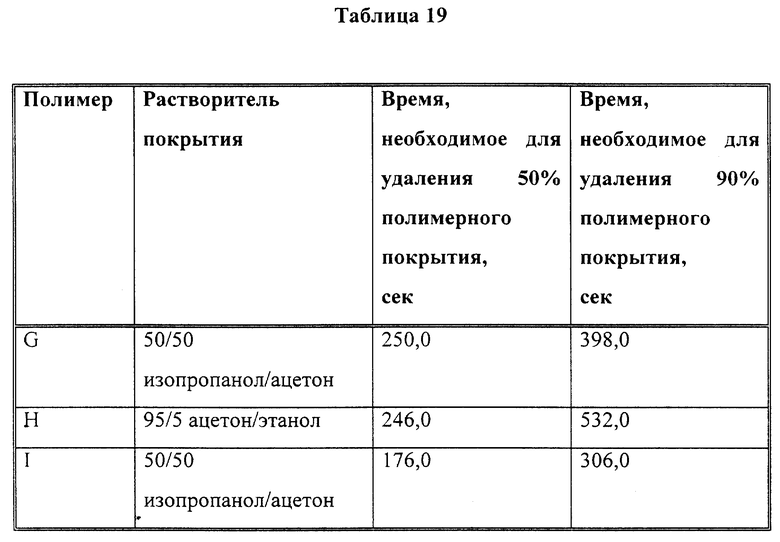

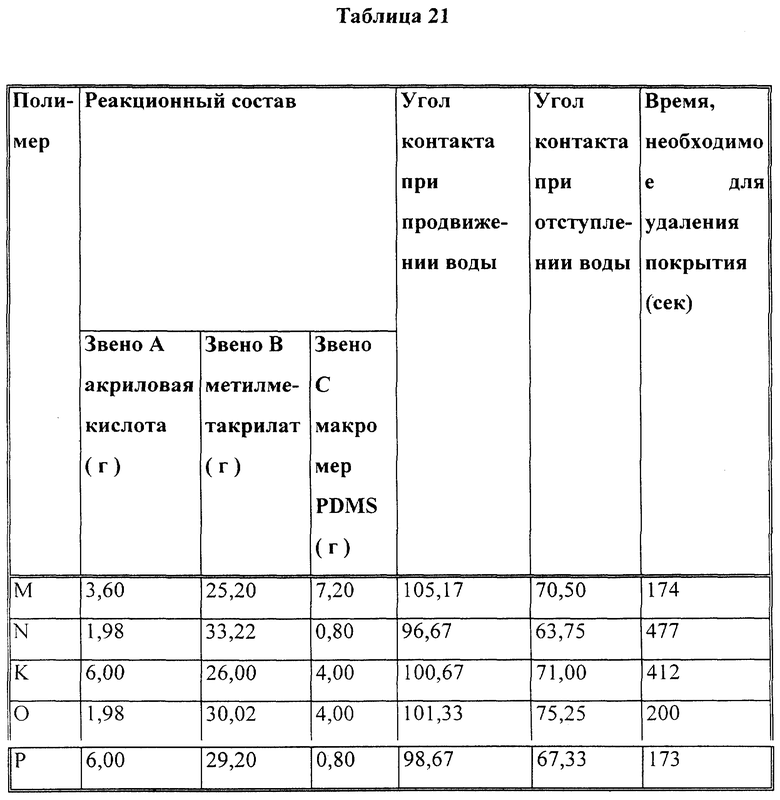
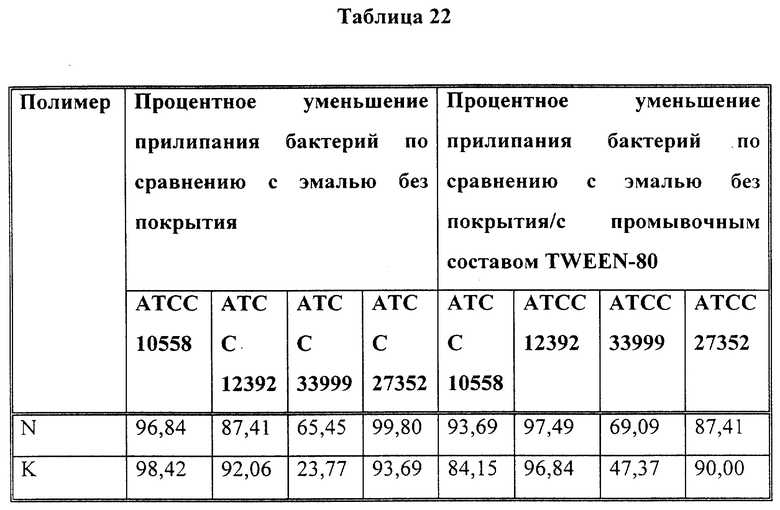
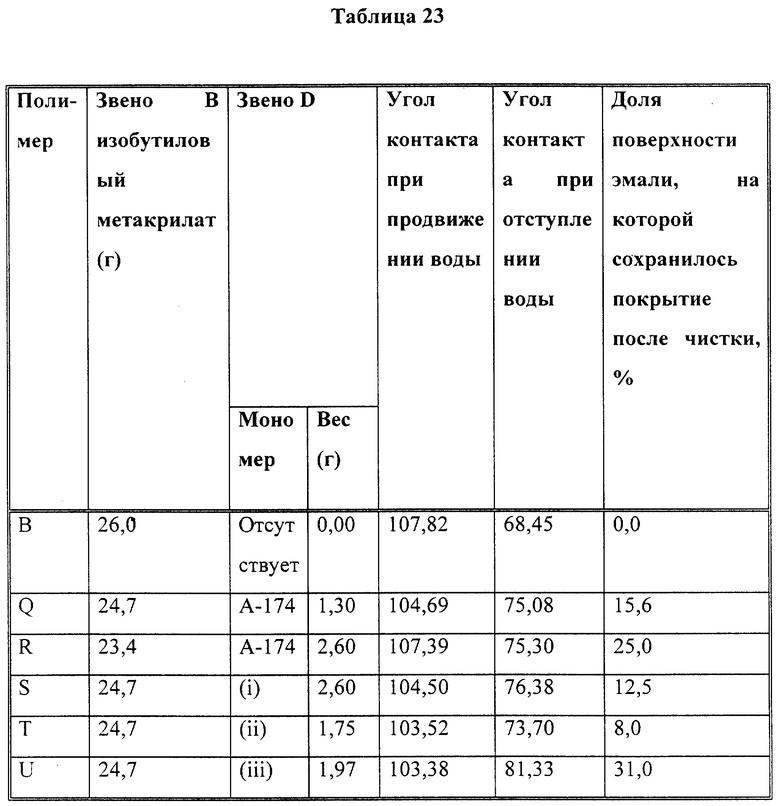
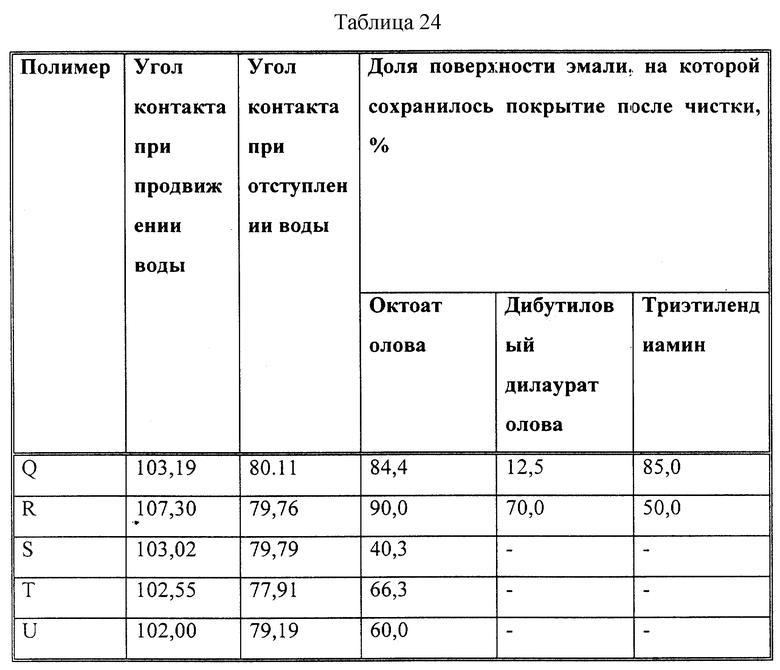
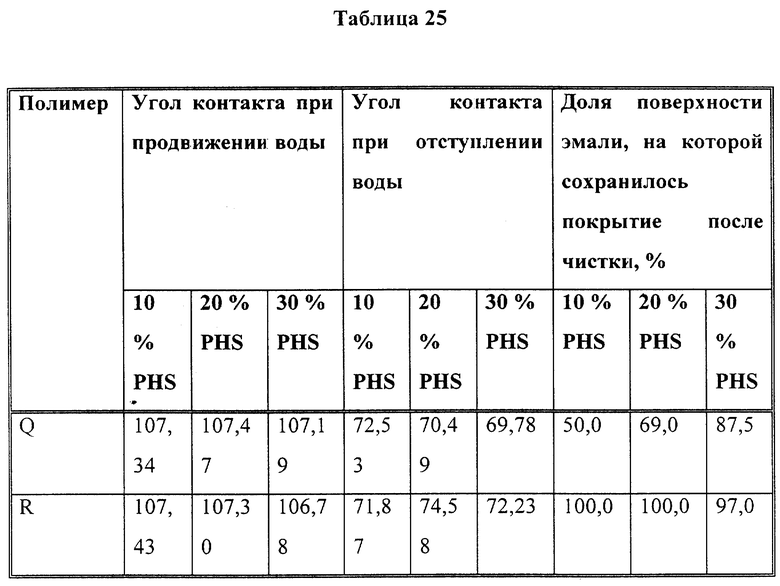
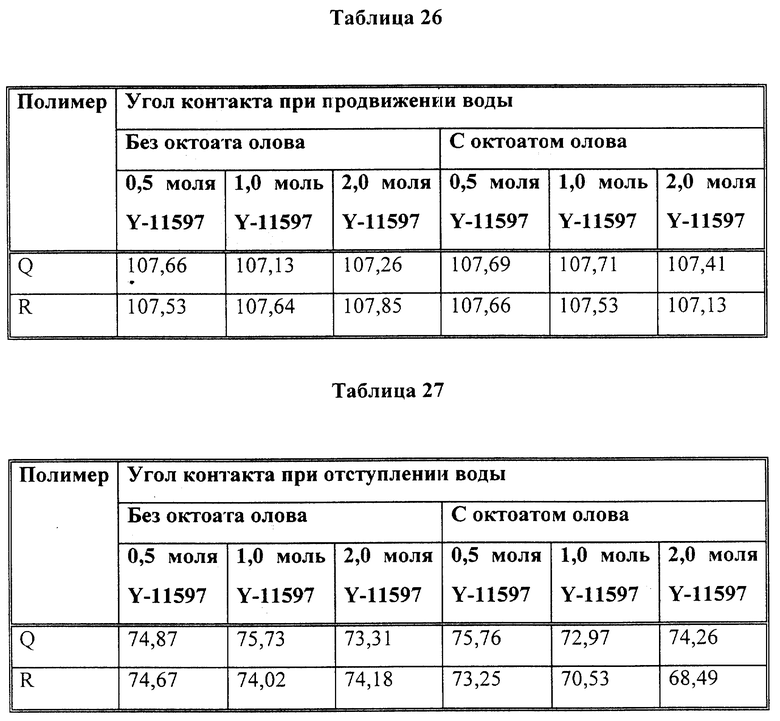
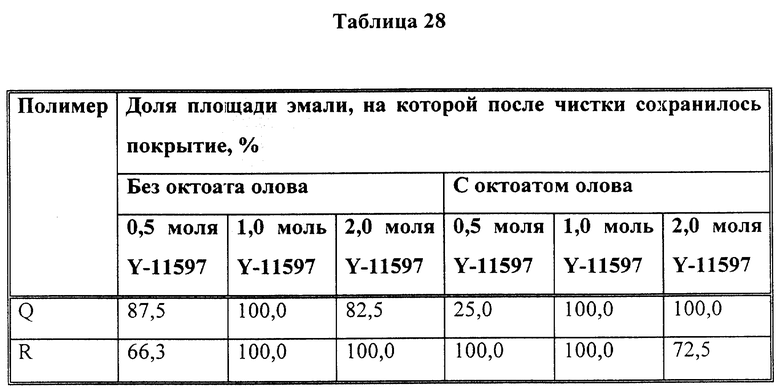
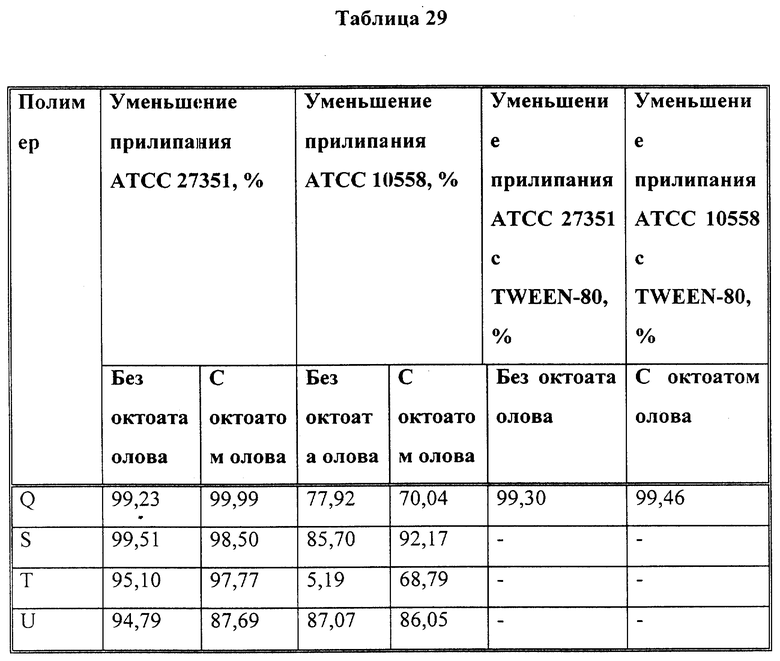
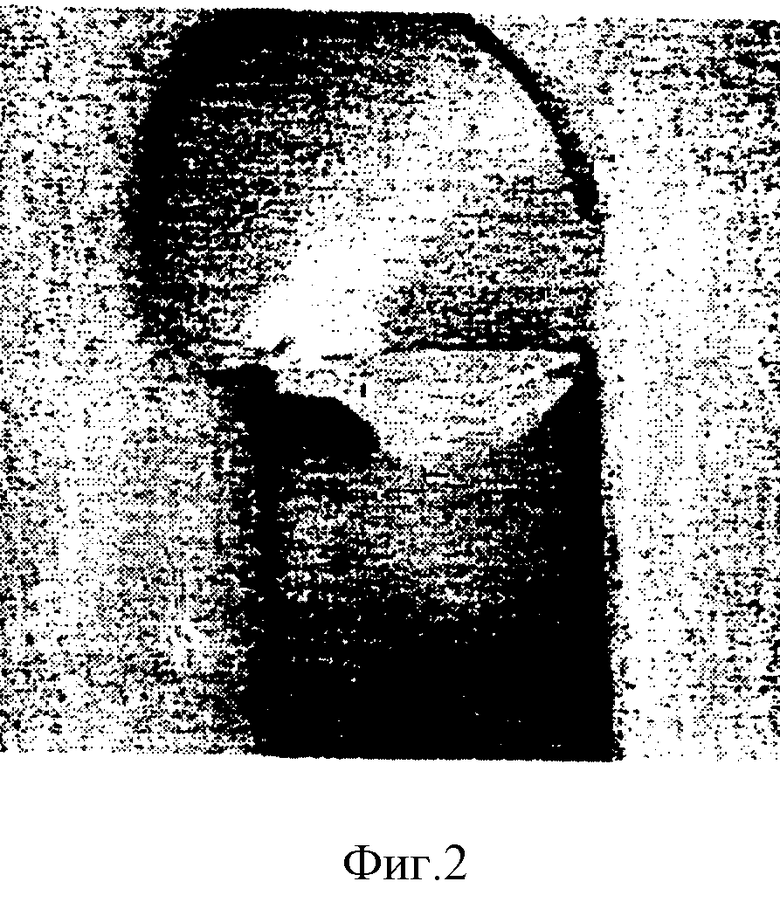
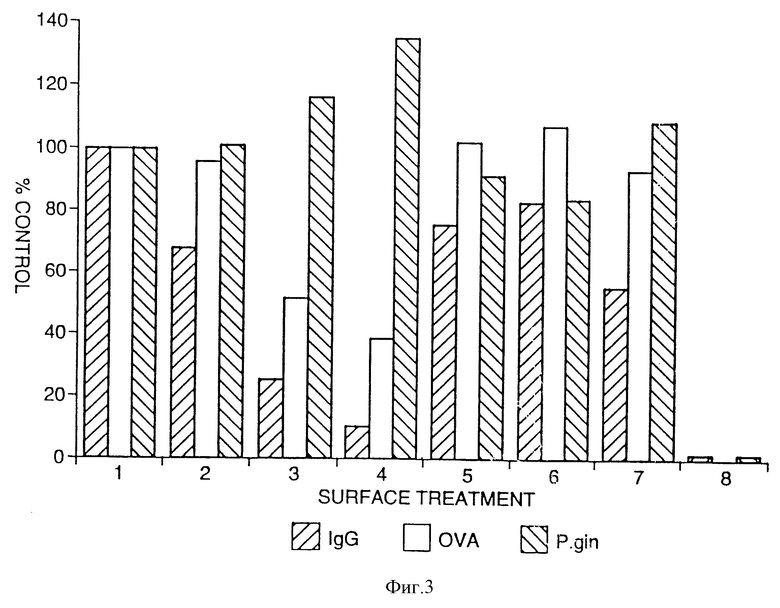
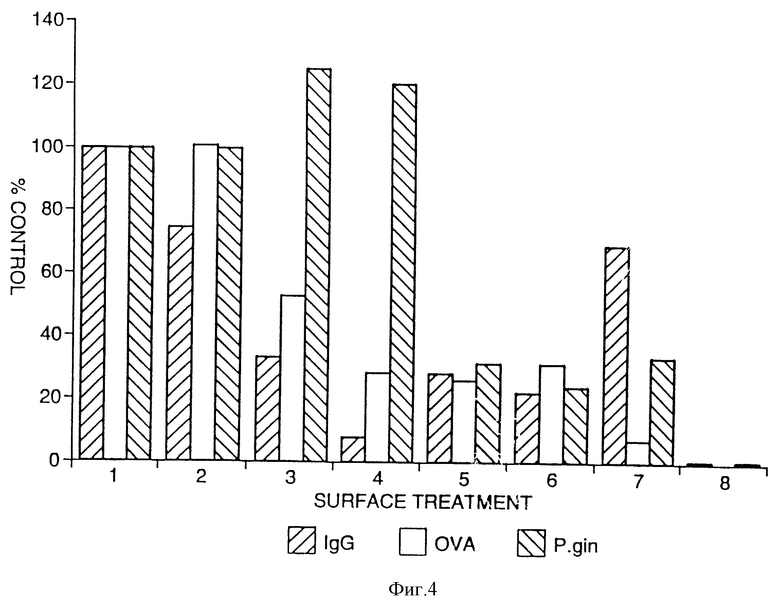
Изобретение относится к покрытиям для твердых тканей, таких как дентин, эмаль, цементное вещество и кость. Кроме того, покрытие может быть нанесено на другие поверхности в области рта, включая поверхности зубных пломб и ортодонтических или ортопедических зубных изделий. Состав, предназначенный для покрытия поверхностей твердых тканей или других поверхностей в области рта, включающих поверхности зубных пломб и ортодонтических или ортопедических зубных изделий, содержит полимер, содержащий повторяющиеся звенья: А) 1-80 вес. % полярной или поляризуемой группы; В) 0-98 вес.% модулирующей группы; С) 1-40 вес.% гидрофобной привитой полисилоксановой цепи с молекулярным весом, равным по меньшей мере 500. Эти покрытия обладают низким коэффициентом трения и высокой устойчивостью к бляшке, бактериям, окрашиванию пищей и т.п. Кроме того, неожиданно было обнаружено, что обработка поверхности с описанным выше покрытием составом, содержащим поверхностно-активное вещество, способствует повышению сопротивления адгезии бактерий и белковых веществ. Операция обработки поверхностно-активным веществом обеспечивает этот необычный положительный эффект даже в тех случаях, когда поверхность с покрытием подвергается перед операцией обработки поверхностно-активным веществом воздействию бактерий и белковых веществ. 22 с. и 49 з.п.ф-лы, 30 табл., 4 ил.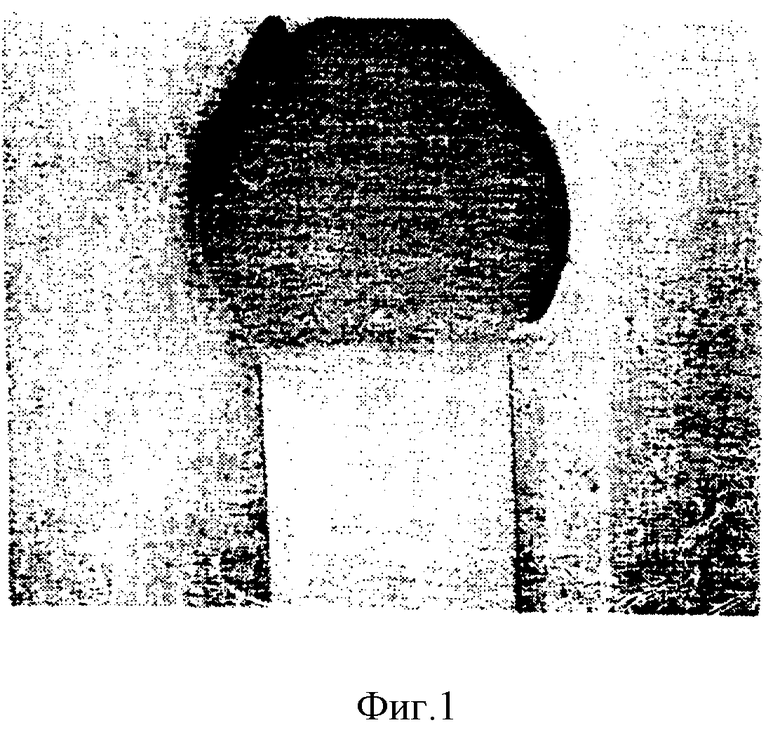
CH2 = CR2G-(COOH)d,
где R2 = H, метил, этил, циано, карбокси или карбоксиметил;
d = 1 - 5;
G является связью или связующей группой радикала гидрокарбила, содержащей 1 - 12 атомов углерода с валентностью d + l, и в необязательном порядке замещенной и/или прерванной замещенным или незамещенным гетероатомом,
и его солями.
CH2 = CR2-CO-L-R3-(OH)d,
где R2 = H, метил, этил, циано, карбокси или карбоксиалкил;
L = O, NH;
d = 1 - 5,
R3 - радикал гидрокарбила с валентностью d + l, содержащий 1 - 12 атомов углерода.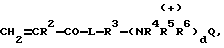
где R2 = H, метил, этил, циано, карбокси или карбоксиалкил;
L = O, NH;
d = 1 - 5;
R3 - радикал гидрокарбила с валентностью d + l, содержащий 1 - 12 атомов углерода;
R4 и R5 являются H или алкильными группами, содержащими 1 - 12 атомов углерода, или вместе они образуют карбоциклическую или гетероциклическую группу;
R6 является H или алкилом с 1 - 30 атомами углерода;
Q является органическим или неорганическим анионом.
X(Y)n-Si(R)3-mZm,
где X является виниловой группой, сополимеризуемой с мономерами А и В;
Y является двухвалентной связующей группой;
n = 0 или 1;
m - целое число от 1 до 3;
R - водород, низший алкил;
Z - одновалентная силоксановая полимерная часть со среднемассовой молекулярной массой, превышающей приблизительно 500, которая по существу не вступает в реакции в условиях сополимеризации.
X(Y)n-Si(R)3-mZm,
где Х имеет общую формулу
где R7 - атом водорода или группа СООН, a R8 - атом водорода, метиловая группа или группа СН2СООН;
Y - двухвалентная связующая группа;
Z имеет обобщенную формулу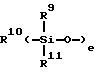
где R9 и R11 являются независимо низшим алкилом, арилом или фторалкилом;
R10 может быть алкилом, алкокси, алкиламино, арилом, гидроксилом или фторалкилом;
e является целым числом приблизительно от 5 до 700,
m = 1, 2 или 3,
n = 0 или 1.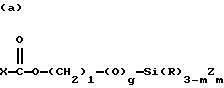
(b) X-Si(R)3-mZm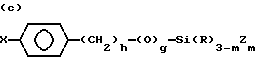
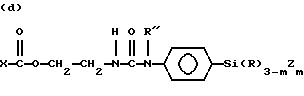
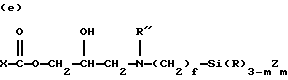
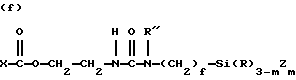
где Х является виниловой группой, сополимеризуемой с мономерами А и В;
m - целое число от 1 до 3;
R и R'' - водород, низший алкил;
Z - одновалентная силоксановая полимерная часть со среднемассовой молекулярной массой, превышающей приблизительно 500, которая по существу не вступает в реакции в условиях сополимеризации,
f - целое число от 2 до 6;
g = 0 или 1;
h - целое число от 0 до 2.
СН2 = CR2G-(COOH)d,
где R2 = H или метил;
d = 1;
G является связью или связующей группой радикала гидрокарбила, содержащей 1 - 12 атомов углерода с валентностью d + 1, или его солью;
звено С является производным мономера с формулой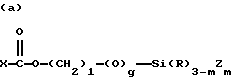
или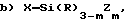
где Х является виниловой группой, сополимеризуемой с мономерами А и В;
m - целое число от 1 до 3;
R - водород, низший алкил;
Z - одновалентная силоксановая полимерная часть со среднемассовой молекулярной массой, превышающей приблизительно 500, которая по существу не вступает в реакции в условиях сополимеризации;
f - целое число от 2 до 6;
g - 0 или 1.
-Si(R12)iTj,
где X является виниловой группой, сополимеризуемой с мономерами А и В;
Y является поливалентной связующей группой;
n = 0 или 1;
R12 - H или низший алкил;
i - целое число из 0 - 2;
j - целое число из 1 - 3;
i + j = 3;
Т является гидрокси- или гидролизуемой группой, выбранной из группы, состоящей из атомов галогенов, алкокси, алкенокси, ацилокси, карбокси, амино, амидо, диалкилиминоокси, кетоксима и алдоксима.
X(Y)n-Si(R12)iТj,
в которой Х является виниловой группой, сополимеризуемой с мономерами А и В;
Y является поливалентной связующей группой;
n = 0 или 1;
R12 - H или низший алкил;
i - целое число из 0 - 2;
j - целое число из 1 - 3;
i + j = 3;
Т является гидрокси- или гидролизуемой группой, выбранной из группы, состоящей из атомов галогенов, алкокси, алкенокси, ацилокси, карбокси, амино, амидо, диалкилиминоокси, кетоксима и алдоксима.
Y-[Si(R12)iTj]k,
где Y является поливалентной связующей группой;
R12 - H или низший алкил;
i - целое число из 0 - 2;
j - целое число из 1 - 3;
i + J = 3;
k = 2 - 50;
T является гидрокси- или гидролизуемой группой, выбранной из группы, состоящей из атомов галогенов, алкокси, алкенокси, ацилокси, карбокси, амино, амидо, диалкилиминоокси, кетоксима и алдоксима.
где R14 = -(СН2)3-Si(ОСН3)3.
| WO 9323009 A, 25.11.1993 | |||
| EP 0412770 A, 13.02.1991 | |||
| Стоматологическое покрытие для профилактики кариеса | 1989 |
|
SU1804831A1 |
| Стоматологический материал | 1991 |
|
SU1792699A1 |
| Способ получения привитых сополимеров и способ получения композиции для базисов зубных протезов | 1981 |
|
SU994472A1 |
