Изобретение относится к медицине, точнее к радиофармпрепаратам для диагностических целей, и может найти применение в позитронно-эмиссионной томографии.
[2-18F] -2-дезоксиглюкоза (ФДГ) является наиболее употребляемым радиодиагностическим препаратом при ПЭТ исследованиях. Она предназначена для прижизненной оценки локальной скорости метаболизма глюкозы методом ПЭТ по накоплению фтора-18 в участках тела, в которых энергетический метаболизм включает потребление углеводов. Причем короткоживущий радионуклид фтор-18 (период полураспада 109.77 мин) по своим ядерно-физическим характеристикам является единственно пригодным изотопом фтора для целей радионуклидной диагностики методом позитронной эмиссионной томографии (ПЭТ). Его отличительной особенностью является относительно низкая радиационная нагрузка на кровь и организм в целом. ФДГ широко применяют для диагностики самых разнообразных патологий: первичных опухолей и метастазов, дифференциальной диагностики рецидивов опухолей и опухолевого некроза, локализации эпилептических очагов, дегенеративно-дистрофических заболеваний и др. Получение ФДГ осуществляют в день исследования и вводят ее в организм путем одномоментной внутривенной инъекции. Препарат описан в Фармакопеях США и Европейского Союза.
Синтез ФДГ основан на нуклеофильном радиофторировании 1,3,4,6-тетра-О-ацетил-2-О- трифторметансульфонил-бета-Д-маннозы (ТАТМ). Данный способ, предложенный Хамахером и сотр. [1], является наиболее распространенным и в настоящее время единственно перспективным методом получения ФДГ. Этот способ взят нами в качестве прототипа.
Поскольку известно, что ТАТМ - продукт нестабильный, а от качества ее зависит как качество, так и выход целевого продукта - ФДГ - в связи с этим синтез ФДГ начинают исходя из Д-маннозы. По данным различных авторов, радиохимический выход ФДГ в пересчете на радиоактивный распад фтора-18 в течение синтеза составляет максимально 69% [1], 58-73% [2]. Радиохимический выход ФДГ при получении нами с использованием ТАТМ производства UKE-Zyclotron (Германия) по методу Хамахера составил 46-71% в пересчете на распад фтора-18.
Получение ФДГ по способу-прототипу [2] осуществляют, начиная с 1,3,4,6-тетра-О-ацетил-2-О-трифторметансульфонил-бета-Д-маннопиранозы (ТАТМ), которую по Хамахеру [3] синтезируют из 1,3,4,6-тетра-О-ацетил-бета-Д-маннопиранозы, а ее - из Д-маннозы [4].
Полная схема синтеза ФДГ представлена на чертеже.
Получение 1,3,4,6-тетра-О-ацетил-бета-Д-маннопиранозы [4].
Несколько миллиграмм Д-маннозы добавляют к уксусному ангидриду (25 мл), затем прибавляют 2 капли 70% раствора хлорной кислоты и порциями 6.6 г Д-маннозы, перемешивают в течение 20 мин, температуру внутри реакционной смеси поддерживают в пределах 40-45oC. Смесь выдерживают при комнатной температуре в течение 60 мин, затем охлаждают до 15oC и прибавляют по каплям 4.3 мл трехбромистого фосфора, поддерживая внутреннюю температуру 20-25oC. Затем добавляют 2.3 мл воды и смесь выдерживают 90 мин при комнатной температуре. Медленно прибавляют раствор тригидрата ацетата натрия (20 г) в воде (25 мл) при 5oC, поддерживая внутреннюю температуру 35-40oC. Образовавшийся желтый раствор выдерживают при этой температуре 25 мин, после чего его выливают на лед и экстрагируют хлороформом (3х30 мл). Хлороформные вытяжки объединяют, промывают холодной водой, холодным раствором гидрокарбоната натрия и снова водой и сушат сульфатом магния. Раствор выпаривают досуха, остаток кристаллизуют из безводного диэтилового эфира. Получено 2.3 г, после кристаллизации - 1.8 г (29%) с Тпл. 164-165oC.
Получение 1,3,4,6-тетра-О-ацетил-2-О-трифторметансульфонил-бета- Д-маннопиранозы (ТАТМ) [3].
К раствору 1,3,4,6-тетра-О-ацетил-бета-Д-маннопиранозы (3.14 г, 9 ммоль) в сухом дихлорметане (80 мл) с добавлением пиридина (1.7 мл) прибавляют ангидрид трифторметансульфокислоты (3.4 мл, 2.1 ммоль) по каплям при -20oC. Желто-зеленую суспензию выдерживают при комнатной температуре 1 ч, промывают холодной водой, раствором гидрокарбоната натрия и снова водой, сушат сульфатом натрия, фильтруют и концентрируют досуха. Осадок дважды кристаллизуют из этанола. Получают 2.6 г (60%) с Тпл. 120oC.
Получение ФДГ [2,6].
1,3,4,6-тетра-О-ацетил-2-О-трифторсульфонил-бета-Д-маннопиранозу растворяют в ацетонитриле и при температуре 85oC в течение 5 мин добавляют фторид-18 и межфазные катализаторы, растворитель отгоняют, к остатку добавляют 1-2 М раствор соляной кислоты [2], выдерживая смесь в течение 6-10 мин при температуре 120-130oC, или 0.2-0.5 М раствор едкого натра [6], выдерживая в течение 1-2 мин при температуре 20-30oC, добавляют воду и полученную массу очищают посредством анионо-, катионообменных и обращеннофазных смол с получением целевого продукта с выходом 58-73% (с использованием смолы 900-PS-HCО3). Радиохимическая чистота, характеризуемая содержанием фторида-18 в препарате, колеблется в пределах 0.16-1.39%, в среднем 0.42+0.44%.
Что касается качества ФДГ, то известно, что фторид-18 является основной радиохимической примесью ее. Содержание его в неочищенном целевом продукте может достигать 50%. Известно, что фторид-18 при поступлении в организм накапливается в костях скелета и в отличие от ФДГ не выводится из организма. Поэтому наличие фторида-18 в готовом препарате приводит к увеличению дозных нагрузок на организм пациента и затрудняет интерпретацию данных медико-диагностического исследования. Это, в свою очередь, диктует необходимость тщательной очистки от фторида-18. Для этой цели используют микроколонки, наполненные сильными анионобменными смолами, Дауэкс IX-10, TIN-100 или 900-PS-HCО3 (Nuclear Interface), Германия, содержащими тетраалкиламмониевые группы. При использовании двух первых смол удается добиться экстракции из водных растворов 96-99,3% фторида-18. В настоящее время производителями модуля для синтеза ФДГ ("Нуклеар Интерфейс") для очистки от фторида-18 предложена третья смола (900-PS-HCО3). С ее использованием содержание фторида-18 в целевом продукте (по нашим данным) составляет от 0.16% до 1.39%, в среднем 0.42+0.44%.
Это побудило нас искать возможности повышения качества ФДГ.
Технический результат настоящего изобретения состоит в повышении качества и выхода ФДГ за счет усовершенствования режимов синтеза ТАТМ и очистки промежуточных и целевого продуктов.
Этот результат достигается тем, что в известном способе получения ФДГ, состоящем в том, что к уксусному ангидриду добавляют Д-маннозу в присутствии 70% раствора хлорной кислоты, выдерживают при комнатной температуре до получения 1,2,3,4,6-пентаацетата Д-маннозы, затем при температуре не выше 25oC добавляют трехбромистый фосфор и при охлаждении воду, смесь выдерживают при комнатной температуре до получения альфа-ацетобром-Д-маннозы, к ней добавляют раствор ацетата натрия при температуре не выше 5oC и выдерживают при комнатной температуре до получения смеси 2,3,4,6- и 1,3,4,6-тетраацетатов Д-маннопиранозы, реакционную массу выливают на лед и несколько раз экстрагируют органическим растворителем, экстракт сушат, упаривают, а остаток кристаллизуют из диэтилового эфира, полученную 1,3,4,6-тетра-О-ацетил-бета-Д-маннопиранозу растворяют в сухом хлористом метилене с добавлением к нему безводного пиридина и при температуре -20oC прибавляют ангидрид трифторметансульфокислоты, реакционную массу выдерживают при комнатной температуре, растворитель удаляют, полученную 1,3,4,6-тетра-О-ацетил-2-О-трифторметансульфонил-бета-Д-маннопиранозу кристаллизуют, затем ее растворяют в ацетонитриле и при температуре 85oC в течение 5 мин добавляют фторид-18 и межфазные катализаторы, растворитель отгоняют, к остатку добавляют 1-2 М раствор соляной кислоты, выдерживая смесь в течение 6-10 мин при температуре 120-130oC, или 0.2-0.5 М раствор едкого натра, выдерживая в течение 1-2 мин при температуре 20-30oC, добавляют воду и полученную массу очищают посредством анионо-, катионообменных и обращеннофазных смол с получением целевого продукта, согласно изобретению, при получении 1,2,3,4,6-пентаацетата Д-маннозы температуру реакционной массы поддерживают в пределах 35-40oC и оставляют на ночь при температуре 0-4oC, трехбромистый фосфор и воду добавляют к нему при мольном соотношении 1,2,3,4,6-пентаацетат Д-манноза: PBr3 : H2O - 1 : 2.5 : 13.6, причем при добавлении воды температуру реакционной массы поддерживают не выше 10oC, в качестве органического растворителя для экстракции смеси 2,3,4,6- и 1,3,4,6-тетраацетатов Д-маннопиранозы используют хлористый метилен, полученные экстракты выпаривают до объема около 125 мл и перед кристаллизацией из диэтилового эфира наносят на хроматографическую колонку с нейтральной окисью алюминия, элюируя смесью гексан - хлористый метилен с градиентом последнего от 10 до 50%, отбирают вторую фракцию, содержащую в основном 1,3,4,6-тетра-О-ацетил-бета-Д- маннопиранозу, растворители упаривают в вакууме, сиропообразный остаток выдерживают при 5-10 мм рт.ст. и температуре 35-40oC в течение 1.5-2 ч, после чего кристаллизуют из диэтилового эфира, после получения 1,3,4,6-тетра-О-ацетил-2-О-трифторметансульфонил-бета-Д- маннопиранозы реакционную смесь подвергают флэш-хроматографии на силикагеле, после чего растворитель удаляют, остаток кристаллизуют из диэтилового эфира, причем полноту превращения Д-маннозы в 1,2,3,4,6-пентаацетат Д-маннозы, последующее превращение его в альфа-ацетобром-Д-маннозу, последней - в 2,3,4,6- и 1,3,4,6-тетраацетаты Д-маннопиранозы, разделение этих изомеров, полноту перехода 1,3,4,6-тетра-О-ацетил-бета-Д-маннопиранозы в 1,3,4,6-тетра-О-ацетил-2-О-трифторметансульфонил-бета-Д-маннопиранозу и полноту выделения 1,3,4,6-тетраацетил-бета-Д-маннопиранозы при элюировании на хроматографической колонке контролируют посредством тонкослойной хроматографии в системе гексан:ацетон 2:1, для повышения выхода целевого продукта первую фракцию после колоночной хроматографии и маточный раствор после кристаллизации тетраацетатов Д-маннопиранозы выпаривают, объединяют, растворяя их в хлористом метилене, последний удаляют, а с остатком, содержащим преимущественно 2,3,4,6-тетраацетат Д-маннопиранозы, а также продукты неполного ацетилирования Д-маннозы, вновь проводят полный цикл вышеописанных химических реакций, повторяя такую процедуру не менее трех раз, полученную в трех циклах 1,3,4,6-тетра-О-ацетил-бета-Д- маннопиранозу объединяют и очищают кристаллизацией из абсолютного этилового спирта, а при очистке целевого продукта посредством анионообменной смолы используют смолу AB-17.
Повышение выхода ФДГ достигается авторами настоящего изобретения на основе оптимизации режимов (температурных, временных) проведения каждой стадии всего цикла получения ФДГ.
Так, поддержание температуры реакционной массы при ацетилировании Д-маннозы в пределах 35-40oC с отстаиванием ее в течение ночи при температуре 0-4oC способствует более полному превращению Д-маннозы в 1,2,3,4,6-пентаацетат Д-маннозы, что контролируется тонкослойной хроматографией (Rf 0.61 в системе гексан:ацетон 2:1).
Мольное соотношение 1,2,3,4,6-пентаацетат Д-маннозы : PBr3: H2O 1 : 2.5 : 13.6, найденное нами опытным путем, и последующее поддержание температуры в реакционной смеси при добавлении воды не выше 10oC обеспечивает по хроматографическому контролю полное превращение пентаацетата Д-маннозы в альфа-ацетобром-Д-маннозу (Rf 0.72).
Экстракция смеси 2,3,4,6-(Rf 0.41) и 1,3,4,6-(Rf 0.29) тетраацетатов Д-маннопиранозы хлористым метиленом позволяет, как показано нами, наиболее полно извлечь их из реакционной смеси, а последующее хроматографическое разделение изомеров - выделить искомую 1,3,4,6-тетра-О-ацетил-бета-Д-маннопиранозу с выходом 30% и одновременно удалить смолообразные побочные продукты, что позволяет в свою очередь использовать в дальнейшем и другие фракции.
Проведение двух дополнительных циклов получения 1,3,4,6-тетра-О-ацетил-бета-Д-маннопиранозы из "отходов" первого цикла позволяет получить дополнительное количество ее и довести выход до 52.3% в расчете на Д-маннозу против 29% в прототипе. Однако в способе-прототипе допущена ошибка при расчете выхода - он составляет не 29%, а 14%, исходя из приведенного выхода, в граммах. Таким образом, нам удалось повысить выход 1,3,4,6-тетра-О-ацетил- бета-Д-маннопиранозы в 3.5 раза по сравнению с прототипом.
Выполнение флэш-хроматографии на силикагеле для выделения 1,3,4,6-тетра-O-ацетил-2-O-трифторметансульфонил-бета-Д-маннопиранозы (ТАТМ) из реакционной смеси с последующей кристаллизацией ее из диэтилового эфира обеспечивает выход ТАТМ 94% (против 60% в прототипе) с получением стабильного продукта, сохраняющего постоянную точку плавления в течение года.
Точное соблюдение предлагаемых нами режимов обеспечивает выход целевого продукта (ФДГ) в пределах 65-94%, в среднем 76±12%, против 46-71%, в среднем 61±9% - в прототипе. Достоверность различия в сплошных выборках (7 и 7 синтезов) > 95%.
Использование для очистки целевого продукта от фторида-18 отечественной анионообменной смолы AB-17 в гидрокарбонатной форме с размерами частиц 100-150 меш и обменной емкостью 2.1 мг·экв/г обеспечивает высокую радиохимическую чистоту, а именно содержание в препарате фторида-18 колеблется от 0.06 до 0.21%, в среднем 0.12±0.06%, тогда как в прототипе оно составляет 0.16-1.39%, в среднем 0.42±0.44%.
Как видно из приведенных данных, достижение технического результата - повышение качества и выхода целевого продукта - в предлагаемом способе получения ФДГ обеспечено.
Способ осуществляется следующим образом.
Получение 1,3,4,6-тетра-О-ацетил-бета-Д-маннопиранозы.
К 200 мл свежеперегнанного уксусного ангидрида прибавляют 0.3 мл 70% раствора хлорной кислоты и затем при перемешивании небольшими порциями присыпают 54 г (0.1 моль) сухой Д-маннозы в течение 1 ч, поддерживая температуру реакционной массы в пределах 35-40oC. Смесь перемешивают в течение 1.5-2 ч при 20-25oC и оставляют на ночь при 0-4oC. Полноту превращения Д-маннозы в 1,2,3,4,6-пентаацетат Д-маннозы контролируют тонкослойной хроматографией (ТСХ) (для пентаацетата Rf 0.61 в системе гексан:ацетон 2:1). По окончании ацетилирования к реакционной массе при охлаждении ледяной водой прикапывают при перемешивании 45.9 мл (0.25 моль) свежеперегнанного трехбромистого фосфора при мольном соотношении 1,2,3,4,6-пентаацетат Д-маннозы: PBr3:H2O - 1 : 2.5 : 13.6 и температуре не выше 25oC. Массу охлаждают льдом с солью и при интенсивном перемешивании медленно прикапывают 25 мл воды, не допуская подъема температуры реакционной массы выше 10oC, и выдерживают образующийся раствор в течение 2-2.5 ч до полного превращения пентаацетата Д-маннозы в альфа-ацетобром-Д-маннозу (Rf 0.72). Затем медленно прикапывают охлажденный раствор ацетата натрия (98.7 г, 0.59 моль) в 220 мл воды, при этом температура реакционной массы не должна превышать 5-10oC. Снимают охлаждение и раствор перемешивают при комнатной температуре в течение 0.5-1.0 ч, фиксируя ТСХ полное превращение альфа-ацетобром-Д-маннозы в основном в 2,3,4,6- и 1,3,4,6-тетраацетаты Д-маннопиранозы (соответственно Rf 0.41 и 0.29). Массу выливают в 600 г льда и перемешивают до полного разложения уксусного ангидрида. Продукт тщательно экстрагируют хлористым метиленом (4 х 200 мл), органический слой промывают холодным раствором гидрокарбоната натрия, ледяной водой, экстракт сушат сульфатом магния. После отделения осушителя раствор обрабатывают активированным углем, фильтруют, хлористый метилен выпаривают на роторном испарителе до объема около 125 мл. Концентрированный раствор продукта наносят на колонку (4 х 70 см) с нейтральной окисью алюминия и элюируют смесью гексан:хлористый метилен с градиентом последнего от 10 до 50% до полного вымывания веществ под контролем ТСХ. Отбирают 2 фракции: первая содержит в основном 2,3,4,6-тетра-О-ацетил-бета-Д-маннопиранозу с небольшими примесями других продуктов дезацетилирования пентаацетата Д-маннопиранозы; вторая - в основном целевой продукт 1,3,4,6-тетраацетил-бета-Д-маннопиранозу. Вторую фракцию упаривают на роторном испарителе в вакууме, сиропообразный остаток выдерживают при 5-10 мм рт.ст. и температуре 35-40oC в течение 1-1.5 ч. Затем к продукту приливают 200 мл абсолютного диэтилового эфира, нагревают до кипения и после полного растворения медленно охлаждают до начала кристаллизации. После выдержки в течение ночи при 0-4oC бесцветный осадок отфильтровывают, промывают небольшим количеством сухого эфира. Получают 31.3 г (0.09 моль, 30%) белого кристаллического вещества - 1,3,4,6-тетра-О-ацетил-бета-Д-маннопиранозы с температурой плавления 162-163oC.
Для более полного использования исходной Д-маннозы первую фракцию после колоночной хроматографии и маточный раствор от кристаллизации тетраацетатов Д-маннозы выпаривают, затем объединяют путем растворения в минимальном количестве хлористого метилена, упаривают в вакууме до постоянного веса. Остаток - сырец, содержащий в основном 2,3,4,6-тетра-О-ацетил-бета-Д-маннопиранозу в количестве около 0.2 моль, вновь используют для получения 1,3,4,6-тетра-О-ацетил-бета-Д-маннопиранозы при точном соблюдении описанных выше условий реакции: в стадии ацетилирования сырец обрабатывают 140 мл уксусного ангидрида в присутствии 0.2 мл хлорной кислоты; при получении альфа-ацетобром-Д-маннозы используют 30.8 мл трехбромистого фосфора и 16.4 мл воды, а при гидролизе его - раствор 66 г ацетата натрия в 150 мл воды. Продукт выделяют и подвергают хроматографическому разделению на той же колонке с окисью алюминия. После кристаллизации из эфира получают 14.6 г (0.042 моль) 1,3,4,6-тетра-О-ацетил-бета-Д- маннопиранозы. В конце из остатков сырца тетраацетатов маннозы из хроматографических и маточных растворов (около 14 моль) получают 8.7 г (0.025 моль) с соблюдением тех же условий реакции и выделения продукта. Общий выход 1,3,4,6-тетра-О-ацетил-бета- маннопиранозы составляет 52.3% (54.6 г, 0.16 моль) в расчете на исходную Д-маннозу. После кристаллизации из абсолютного этанола и сушки в вакууме получают 52.4 г (0.15 моль, 50%) 1,3,4,6-тетра-O- ацетил-бета-Д-маннопиранозы в виде бесцветных кристаллов с точкой плавления 166-167oC.
Получение 1,3,4,6-тетра-O-ацетил-2-O- тифторметансульфонил-Д-маннопиранозы (ТАТМ).
К раствору 52.4 г (0.15 моль) 1,3,4,6-тетра-О-ацетил-бета-Д-маннопиранозы в 1200 мл сухого хлористого метилена приливают 31.7 мл безводного пиридина. Раствор охлаждают до -20oC и в течение 1-1.5 ч прикапывают раствор 87.7 г (0.31 моль, d 1.677) ангидрида трифторметансульфокислоты в 300 мл хлористого метилена, поддерживая температуру реакционной массы в пределах (-10)-(-15)oC. Охлаждение убирают и реакционную массу перемешивают при комнатной температуре в течение 1.5-2 ч до полного исчезновения на хроматограмме исходного тетраацетата Д-маннозы. Реакционную смесь промывают ледяной водой, холодным раствором гидрокарбоната натрия и еще раз ледяной водой. Водные слои дополнительно экстрагируют хлористым метиленом, объединенный органический экстракт сушат сульфатом натрия. После отделения осушителя фильтрат обрабатывают осветляющим активированным углем, подвергают флэш-хроматографии на силикагеле и затем выпаривают на роторном растворителе при пониженном давлении. Продукт выдерживают при 30-35oC и 10-15 мм рт.ст. до полной кристаллизации и перекристаллизовывают из абсолютного диэтилового эфира (1г/15 мг). После сушки в вакууме получают 69.17 г (0.144 моль) 1,3,4,6-тетра-O-ацетил-2-O- трифторсульфонил-бета-Д-маннопиранозы (выход в расчете на 1,3,4,6-тетра-О-ацетил-бета-Д-маннопиранозу 96%, на исходную маннозу - 48%). Это белое кристаллическое вещество с Тпл.122-123oC. Спектральный и хроматографический анализы показывают, что продукт устойчив при длительном хранении (до 1 года) в холодильнике с защитой от влаги воздуха.
Получение ФДГ
Синтез [2-18F] ФДГ основан на нуклеофильном радиофторировании 1,3,4,6-тетра-О-ацетил-2-О- трифторметансульфонил-бета-Д-маннопиранозы
(ТАТМ) в ацетонитриле при каталитическом участии межфазных катализаторов и последующим гидролизом образующейся 1,3,4,6-тетра-О-ацетил-2-18F-бета-Д- глюкопиранозы.
Радиоактивным сырьем является фторид, фтор-18, который получают по ядерной реакции 18O(p, n)18F в мишени циклотрона при облучении протонами с энергией до 15 МэВ. Образующийся радионуклид стабилизируется в химической форме фторида, фтор-18. В качестве облучаемого вещества используют воду, обогащенную кислородом-18 (H2O-18), в качестве материала мишени - титан, радиохимический выход ядерной реакции составляет 120 мКи/ μ A · ч.
Предварительная стадия синтеза препарата - извлечение целевого радионуклида из облученного материала технологически объединена с регенерацией обогащенной воды и отделением аниона фторид, фтор-18 от соединений, содержащих радионуклидную примесь азота-13.
Далее следует обезвоживание реакционной массы. Оно достигается путем азеотропной отгонки воды с ацетонитрилом (20% ацетонитрила : 80% воды). Отгонку ацетонитрила проводят током инертного газа (гелий) при одновременном вакуумировании реакционного сосуда до практически полного обезвоживания реакционной среды.
Основной химической стадией синтеза является нуклеофильное замещение трифлатной группы в ТАТМ на фтор-18. Реакция проводится в растворе ацетонитрила при температуре 85oC при участии межфазных катализаторов: четвертичных аммониевых оснований. Она протекает по SN2 механизму, т.е. с обращением конфигурации, поэтому ее радиоактивным продуктом является производное Д-глюкозы - 1,3,4,6-тетра-O-ацетил-2-18F-бета-D- глюкопираноза. После окончания реакции фторирования проводят полную отгонку растворителя в токе гелия.
Гидролиз реакционной массы, содержащей 1,3,4,6-тетра-О-ацетил-2-18F-бета-D- глюкопиранозу, и непрореагировавшую ТАТМ, приводит к образованию целевого продукта - [2-18F] ФДГ и глюкозы. Гидролиз осуществляют при кислотном или щелочном катализе. Кислотный гидролиз проводят действием 2 М раствором соляной кислоты при температуре 125oC в течение 6-10 мин, а щелочной - 0.3 М раствором едкого натра при 25oC за 1-2 мин.
После завершения гидролиза в реакционный сосуд добавляют воду для полного смывания реакционной массы со стенок реакционного сосуда, после чего реакционную массу передают на очистительные колонки, в качестве наполнителей для которых используют катионообменную смолу сильного типа в H+ форме - KУ-2, анионообменную смолу сильного типа в гидрокарбонатной форме AB-17, нейтральный оксид алюминия Alox-N и обращеннофазную смолу HR-P типа C18. Выход очищенного целевого продукта составляет 65-94%, в среднем 76±12% (в прототипе 58-73%, при получении нами по методу Хамахера - 46-71%, в среднем 61±9%). Содержание фторида-18 колеблется от 0.06% до 0.21%, в среднем 0.12±0.06%.
Предлагаемый способ по сравнению с известными имеет целый ряд значительных преимуществ:
1. Обеспечивает выход целевого продукта (ФДГ) в среднем 76+12%, тогда как по прототипу он равен 61±9%.
2. Обеспечивает высокую радиохимическую чистоту целевого продукта - содержание фторида-18 составляет в среднем 0.12±0.06%, тогда как в прототипе - 0.42±0.44%.
3. Способ высокоэкономичен, если учесть стоимость 1 дозы ФДГ, которая на международном рынке составляет 500-1000 $, то повышение выхода ее за один цикл дает немалую экономию.
4. Способ пригоден для крупномасштабного производства ТАТМ, так как обеспечивает высокий выход (96% вместо 60% по прототипу) стойкого продукта, сохраняющегося без изменения свойств в течение года.
5. Способ обеспечивает также высокий выход 1,3,4,6-тетра-О-ацетил- бета-Д-маннопиранозы (50% против 29%, как сообщают авторы прототипа, на самом деле он составляет лишь 14%, а по сообщениям других авторов, от 6 до 9%).
Все вышеизложенные достоинства предлагаемого способа получения ФДГ выгодно отличают его от известных в настоящее время, способным заменить любой из них.
Способ разработан в отделении циклотрона ЦНИРРИ и используется для получения ФДГ для ПЭТ-исследований.
ЛИТЕРАТУРА:
[1] Hamacher К., Coenen H.H., Stoecklin G. J. Nucl. Med. 1986. V. 27. P. 235.
[2] Hamacher К., Blessing G., Nebeling B. Appl. Radiat. Isot. 1990. V. 41. P. 49/
[3] Hamacher K, Carbohydrate Res. Vol. 128, P. 291-295, 1984.
[4] Deferrari, Carbohydr. Res. 1967, Vol. 4 P. 432-434.
[5] Бовин Н.В. и др. Изв. АН СССР, Сер. Хим. 1981, С. 1638-1641.
[6] Fuechtner P., Steinbach J., Maeding P., Johannsen B. 1996. Appl. Radiat. Isot. V. 47. P. 61-66.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ФТОРИРОВАНИЯ ДЛЯ СИНТЕЗА 2-[F]-ФТОР-2-ДЕЗОКСИ-D-ГЛЮКОЗЫ | 2005 |
|
RU2394040C2 |
| СТАБИЛИЗАЦИЯ РАДИОФАРМАЦЕВТИЧЕСКИХ ПРЕДШЕСТВЕННИКОВ | 2005 |
|
RU2389510C2 |
| ТЕТРАВАЛЕНТНЫЕ НЕОГЛИКОКОНЪЮГАТЫ С УГЛЕВОДНЫМ РАЗВЕТВЛЯЮЩИМ ЯДРОМ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2014 |
|
RU2575925C1 |
| СПОСОБ ПОЛУЧЕНИЯ 6-O-[β-D-(2,3,4,6-ТЕТРА-O-АЦЕТИЛ)ГЛИКОПИРАНОЗИЛ]-d,l-α-ТОКОФЕРОЛОВ | 2007 |
|
RU2350620C1 |
| Сополимер 3-0-[4-0-( @ -D-маннопиранозил)- @ -L-рамнопиранозил- @ -АЛЛИЛ-D-ГАЛАКТОПИРАНОЗИДА С АКРИЛАМИДОМ,ОБЛАДАЮЩИЙ СЕРОЛОГИЧЕСКОЙ СПЕЦИФИЧНОСТЬЮ 0-ФАКТОРА 3 БАКТЕРИЙ РОДА САЛЬМОНЕЛЛА,ОТНОСЯЩИХСЯ К СЕРОЛОГИЧЕСКОЙ ГРУППЕ Е | 1979 |
|
SU879970A1 |
| Способ получения оптически активных асимметрично замещенных производных миониозита | 1974 |
|
SU505620A1 |
| Способ получения 1,2-0-(1-циан)-алкилиденовых производных сахаров | 1978 |
|
SU727656A1 |
| СПОСОБ ПОЛУЧЕНИЯ ГЛИКОЗИДА КУРКУЛИГОЗИДА А | 2024 |
|
RU2828122C1 |
| ДИ- И ТРИВАЛЕНТНЫЕ НЕБОЛЬШИЕ МОЛЕКУЛЫ ИНГИБИТОРОВ СЕЛЕКТИНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ИНГИБИРОВАНИЯ СЕЛЕКТИНА | 1996 |
|
RU2165931C2 |
| ЧУВСТВИТЕЛЬНЫЕ К ГЛЮКОЗЕ КОНЪЮГАТЫ ИНСУЛИНА | 2014 |
|
RU2676307C2 |
Изобретение относится к медицине, точнее к радиофармпрепаратам для диагностических целей, и может найти применение в позитронной эмиссионной томографии. К уксусному ангидриду добавляют Д-маннозу в присутствии 70%-ного раствора хлорной кислоты. Выдерживают при комнатной температуре до получения 1,2,3,4,6-пентаацетата Д-маннозы. Затем при температуре не выше 25°С добавляют трехбромистый фосфор и при охлаждении воду. Смесь выдерживают при комнатной температуре до получения альфа-ацетобром-Д-маннозы. К ней добавляют раствор ацетата натрия при температуре не выше 5°С и выдерживают при комнатной температуре до получения смеси 2,3,4,6- и 1,3,4,6-тетраацетатов Д-маннопиранозы. Реакционную массу выливают на лед и несколько раз экстрагируют органическим растворителем. Экстракт сушат, упаривают, а остаток кристаллизуют из диэтилового эфира. Полученную 1,3,4,6-тетра-О-ацетил-бета-Д-маннопиранозу растворяют в сухом хлористом метилене с добавлением к нему безводного пиридина и при температуре - 20°С прибавляют ангидрид трифторметансульфокислоты. Реакционную массу выдерживают при комнатной температуре, растворитель удаляют. Полученную 1,3,4,6-тетра-О-ацетил-2-О-трифторметансульфонил-бета-Д-маннопиранозу кристаллизуют. Затем ее растворяют в ацетонитриле и при температуре 85°С в течение 5 мин добавляют фторид-18 и межфазные катализаторы. Растворитель отгоняют. К остатку добавляют 1 -2 М раствор соляной кислоты и выдерживают смесь в течение 6-10 мин при температуре 120-130°С, или 0,2-0,5 М раствор едкого натра и выдерживают в течение 1-2 мин, при температуре 20-30°С. Добавляют воду и полученную массу очищают посредством анионо-, катионообменных и обращеннофазных смол с получением целевого продукта. При этом при получении 1,2,3,4,6-пентаацетата Д-маннозы температуру реакционной массы поддерживают в пределах 35-40°С и оставляют на ночь при температуре 0-4°С. Трехбромистый фосфор и воду добавляют при мольном соотношении 1,2,3,4,6-пентаацетат Д-манноза : PBr3 : H2O - 1 : 2.5:13.6. При добавлении воды температуру реакционной массы поддерживают не выше 10°С. В качестве органического растворителя для экстракции смеси 2,3,4,6- и 1,3,4,6-тетраацетатов Д-маннопиранозы используют хлористый метилен. Полученные экстракты выпаривают до объема 125 мл. Перед кристаллизацией из диэтилового эфира наносят на хроматографическую колонку с нейтральной окисью алюминия. Элюируют смесью гексан - хлористый метилен с градиентом последнего от 10 до 50%. Отбирают вторую фракцию, содержащую 1,3,4,6-тетра-О-ацетил-бета-Д-маннопиранозу. Растворители упаривают в вакууме. Сиропообразный остаток выдерживают при 5-10 мм рт. ст. и температуре 35-40°С в течение 1,5-2 ч, после чего кристаллизуют из диэтилового эфира. После получения 1,3,4,6-тетра-О-ацетил-2-О-трифторметансульфонил-бета-Д-маннопиранозы реакционную смесь подвергают флэш-хроматографии на силикагеле. Затем растворитель удаляют. Остаток кристаллизуют из диэтилового эфира. Первую фракцию после колоночной хроматографии и маточный раствор после кристаллизации тетраацетатов Д-маннопиранозы выпаривают, объединяют, растворяют их в хлористом метилене. Последний удаляют. С остатком проводят полный цикл вышеописанных химических реакций. Повторяют процедуру не менее трех раз. Полученную в трех циклах 1,3,4,6-тетра-О-ацетил-бета-Д-маннопиранозу объединяют и очищают кристаллизацией из абсолютного этилового спирта. При очистке целевого продукта посредством анионообменной смолы используют смолу АВ-17. Изобретение позволяет повысить качество и выход продукта. 1 ил.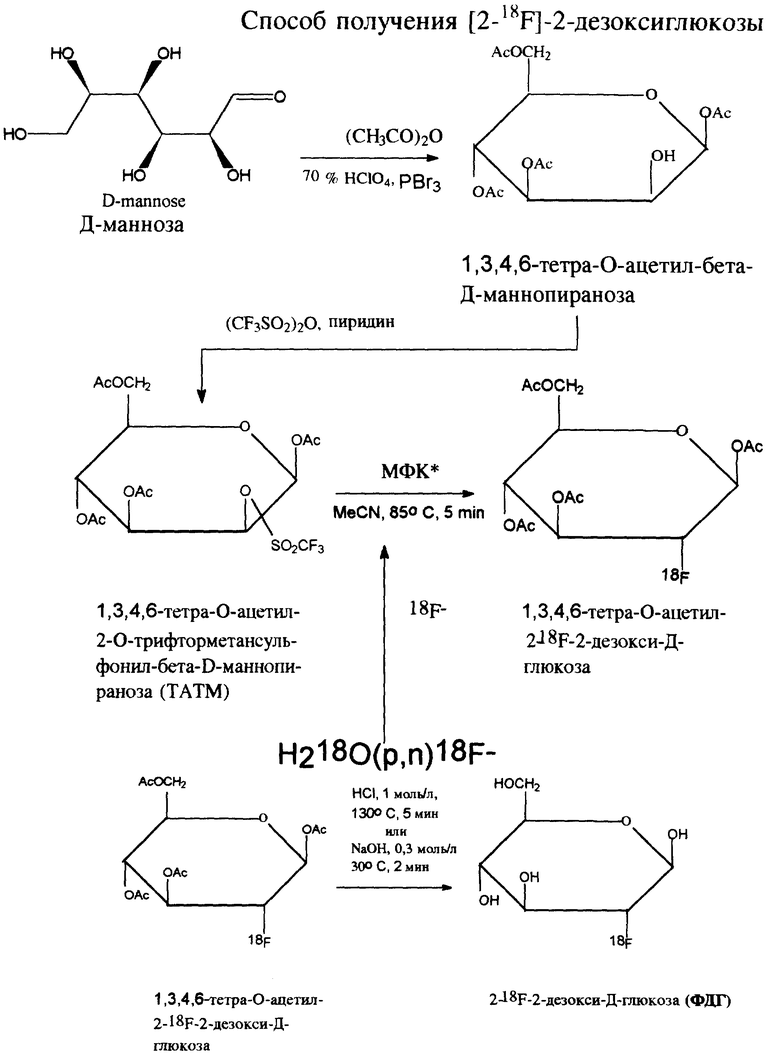
Способ получения [2 - 18F]-2-дезокси-глюкозы, состоящий в том, что к уксусному ангидриду добавляют Д-маннозу в присутствии 70%-ного раствора хлорной кислоты, выдерживают до получения 1,2,3,4,6-пентаацетата Д-маннозы, затем при температуре не выше 25oС добавляют трехбромистый фосфор и при охлаждении воду, смесь выдерживают при комнатной температуре до получения альфа-ацетобром-Д-маннозы, к ней добавляют раствор ацетата натрия при температуре не выше 5oС и выдерживают при комнатной температуре до получения смеси 2,3,4,6- и 1,3,4,6-тетраацетатов Д-маннопиранозы, реакционную массу выливают на лед и экстрагируют органическим растворителем, экстракт сушат, упаривают, а остаток кристаллизуют из диэтилового эфира, полученную 1,3,4,6-тетра-О-ацетил-бета-Д-маннопиранозу растворяют в сухом хлористом метилене с добавлением к нему безводного пиридина и при температуре -20oС прибавляют ангидрид трифторметансульфокислоты, реакционную массу выдерживают при комнатной температуре, растворитель удаляют, полученную 1,3,4,6-тетра-О-ацетил-2-О-трифторметансульфонил-бета-Д-маннопиранозу кристаллизуют, затем ее растворяют в ацетонитриле и при температуре 85oС в течение 5 мин добавляют фторид-18 и межфазные катализаторы, растворитель отгоняют, к остатку добавляют 1 - 2 М раствор соляной кислоты, выдерживая смесь в течение 6 - 10 мин при температуре 120 - 130oС, или 0,2 - 0,5 М раствор едкого натра, выдерживая в течение 1 - 2 мин при температуре 20 - 30oС, добавляют воду и полученную массу очищают посредством анионо-, катионообменных и обращеннофазных смол с получением целевого продукта, отличающийся тем, что при получении 1,2,3,4,6-пентаацетата Д-маннозы температуру реакционной массы поддерживают в пределах 35 - 40oС и оставляют на ночь при температуре 0 - 4oС, трехбромистый фосфор и воду добавляют к нему при мольном соотношении 1,2,3,4,6-пентаацетат Д-манноза : PBr3 : H2O 1 : 2.5 : 13.6, причем при добавлении воды температуру реакционной массы поддерживают не выше 10oС, в качестве органического растворителя для экстракции смеси 2,3,4,6- и 1,3,4,6-тетраацетатов Д-маннопиранозы используют хлористый метилен, полученные экстракты выпаривают до объема около 125 мл и перед кристаллизацией из диэтилового эфира наносят на хроматографическую колонку с нейтральной окисью алюминия, элюируя смесью гексан - хлористый метилен с градиентом последнего от 10 до 50%, отбирают вторую фракцию, содержащую в основном 1,3,4,6-тетра-О-ацетил-бета-Д-маннопиранозу, растворители упаривают в вакууме, сиропообразный остаток выдерживают при 5 - 10 мм рт. ст. и температуре 35 - 40oС в течение 1,5 - 2 ч, после чего кристаллизуют из диэтилового эфира, после получения 1,3,4,6-тетра-О-ацетил-2-О-трифторметансульфонил-бета-Д-маннопиранозы реакционную смесь подвергают флэш-хроматографии на силикагеле, после чего растворитель удаляют, остаток кристаллизуют из диэтилового эфира, причем полноту превращения Д-маннозы в 1,2,3,4,6-пентаацетат маннозы, последующее превращение его в альфа-ацетобромманнозу, последней - в 2,3,4,6- и 1,3,4,6-тетраацетаты Д-маннопиранозы, разделение этих изомеров, полноту перехода 1,3,4,6-тетра-О-ацетил-бета-Д-маннопиранозы в 1,3,4,6-тетра-О-ацетил-2-О-трифтор-метансульфонил-бета-Д-маннопиранозу и полноту выделения 1,3,4,6-тетраацетил-бета-Д-маннопиранозы при элюировании на хроматографической колонке контролируют посредством тонкослойной хроматографии в системе гексан : ацетон 2 : 1, первую фракцию после колоночной хроматографии и маточный раствор после кристаллизации тетраацетатов маннопиранозы выпаривают, объединяют, растворяя их в хлористом метилене, последний удаляют, а с остатком, содержащим преимущественно 2,3,4,6-тетраацетат Д-маннопиранозы, а также продукты неполного ацетилирования Д-маннозы, вновь проводят полный цикл вышеописанных химических реакций, повторяя такую процедуру не менее трех раз, полученную в трех циклах 1,3,4,6-тетра-О-ацетил-бета-Д-маннопиранозу объединяют и очищают кристаллизацией из абсолютного этилового спирта, а при очистке целевого продукта посредством анионообменной смолы используют смолу АВ-17.
| HAMACHER | |||
| K | |||
| И ДР | |||
| APPL | |||
| RADIAT | |||
| ISOT, 1990, v.41, p.49 | |||
| ПРЕПАРАТ, СОДЕРЖАЩИЙ ОРГАНИЧЕСКОЕ СОЕДИНЕНИЕ, МЕЧЕННОЕ БЕТА-РАДИОНУКЛИДОМ | 1995 |
|
RU2104035C1 |
| US 5425935 A, 20.06.1995 | |||
| Бесколесный шариковый ход для железнодорожных вагонов | 1917 |
|
SU97A1 |

