Область техники
Изобретение относится к новым полициклическим алкалоидам, которые способны связывать или быть антагонистами NMDA (М-метил-(D)-аспарагиновая кислота) рецепторного комплекса, или иным образом защищать нейроны против дегенерации, индуцированной рецептором стимуляторных аминокислот. В другом аспекте изобретение относится к способу ингибирования активации NMDA-рецептора у млекопитающих, используя новые полициклические алкалоиды изобретения.
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
Стимуляторные аминокислоты, такие как L-глутамат (Glu) и L-аспартат (Asp), являются главными нейротрансмиттерами в центральной нервной системе млекопитающих. Для этих кислотных нейротрансмиттеров аминокислот существует множество подтипов кислотных рецепторов аминокислот. Например, они включают ионные канал-связанные рецепторы, опосредующие деполяризацию нейронов, названные по про-типичным антагонистам N-метил-D-аспартат (NMDA), альфа-амино-5-метил-4-изоксазолепроприониковая кислота (АМРА), каинат и мнимый пресинаптический стимулятор L-2-амино-4- фосфонобутират (L- АР4). Пятым рецептором стимуляторных аминокислот является метаботропический рецептор, связанный с фосфоинозитидным метаболизмом. (Farooqui и Horrocks, Brain Res. Rev. 16, 171, 1991).
NMDA-рецепторы играют специализированную роль вследствие уникальных особенностей связанных с ними ионных каналов и участвуют в разнообразных пластических нейронных событиях, включая инициацию длительной потенциации, которая является предположительным субстратом научения, памяти и установления синаптических контактов во время развития нейронов. NMDA-рецепторы также вовлечены в другие процессы, такие, как передача сенсорной информации (MacDermott и Dale, Trends Neurosci. 10, 280, 1987).
Кроме их важной физиологической роли стимуляторные аминокислоты, такие, как NMDA, также вовлечены в патофизиологические события в центральной нервной системе.
Ненормально низкие уровни глутаминовой кислоты (Glu) могут изменить нормальные уровни стимуляции и приводить, например, к дефициту научения и памяти. Избыточные уровни Glu могут привести к токсичным эффектам. Термин "стимулотоксичность" был создан Олни (Olney) (в Hyhan W.L. [изд.]: "Heritage Disorders of Amino Acids Metabolism" New York: Macmillan pp. 501-512, 1989) для описания процесса, посредством которого стимуляторные аминокислоты могут вызывать смерть нейронных клеток.
Имеются доказательства, что NMDA-рецепторы существуют в периферических тканях и что активация этих рецепторов может быть вовлечена в механизм повреждения легких и других органов (Said, S.I. et al., Letters to Neuroscience, 65, 943-946, 1995). Этот цитотоксический процесс, в основном, опосредован сверхстимуляцией NMDA-рецепторов и может иметь место в случаях церебрального паралича, церебральной ишемии, эпилепсии, болезни Алцгеймера, при слабоумии, связанном со СПИДом, при травматических повреждениях мозга и других нейродегенеративных расстройствах (Olney, Ann. Rev. Pharmacol. Toxicol. 30: 47- 71, 1990; Foster et al., в "Current and future Trends in Anticonvulsant, Anxiety and Stroke Therapy" Wiley-Liss, Inc. pp. 301-329, 1990; Rogawski and Porter, Pharmacol. Rev., 42: 223-286, 1990).
NMDA-рецепторы содержат несколько связывающих доменов, которые взаимодействуют друг с другом для должного функционирования и модулирования активности нервных клеток. Имеется теория о том, что NMDA-рецептор формирует комплекс, действующий как ионный канал, связанный с рецептором. Точнее, функцией рецептора является связывание NMDA или натуральных аминокислот, Глу или Асп, и раскрытие связанного ионного канала, что позволяет натрию (Na+) или кальцию (Ca2+) проникать в стимулированный нейрон, так же как калию (К+) покидать.
Тогда как ионные каналы рецепторов других стимуляторных аминокислот (АМРА, каинатный и L-AP4) проницаемы только для Na+ и К+, канал NMDA-рецептора также проницаем для Ca2+. Эта особенность может быть важной для предполагаемой роли этого рецептора для и кратковременной, и длительной пластичностей, таких, как научение, память и нейропатология.
Внутриклеточный Ca2+ отвечает за регуляцию большого разнообразия клеточных процессов (Farooqui и Horrocks, Brain Res. Rev. 16, 171; 1991). Сверхстимуляция NMDA-рецепторов мозга, наблюдаемая в случаях аноксии, ишемии и гипогликемии, приводит к накоплению концентрации Ca++ в стимулированных нейронах и к каскаду внутриклеточных событий (активации фосфолипаз [PLA2, PLC] , липаз, протеаз и эндонуклеаз), что приводит к смерти нейронных клеток (Farooqui и Horrocks, Brain Res. Rev. 16, 171; 1991).
Поэтому существует необходимость в соединениях, которые могут связывать или быть антагонистами комплекса NMDA-рецептора или другим образом защищать нейроны против дегенерации, индуцированной рецептором стимуляторной аминокислоты.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к новым полициклическим алкалоидам, которые действуют как антагонисты к ионотропному NMDA-рецептору (N-метил-(D)-аспарагиновая кислота), с общей структурой, представленной формулой I: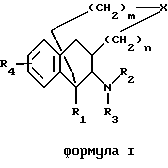
где R1 является H, C1-6 алкилом или С6-12 арилом, выборочно замененными на полярные группы;
R2 и R3 являются, независимо, H, ОН, С1-6 алкилом, -C(NH)-NH2, положительно заряженной группой или С7-13 аралкилом, выборочно замененными на NH2, ОН, C1-6 алкил или галоген; или R2 и R3 совместно формируют от 5 до 6-элементное кольцо, выборочно содержащее гетероатом;
R4 является H, С1-6 алкилом, OR6, SR6 или N(R6)2, отличающимися тем, что каждый R6 является, независимо, H или C1-6 алкилом;
X является О, S, SO, SO2, N-R5, или C-(R5)2, отличающимися тем, что каждый R5 является, независимо, H, C1-6 алкилом или C7-13 аралкилом, выборочно прерванными одним или более гетероатомами;
n является целым числом от 0 до 2;
m является целым числом от 0 до 3;
с условием, что когда X является CH2, тогда R1 не является CH3, R2 и R3 оба не являются H, R4 не является OH, m не является 3 и n не является 0.
Профессионалам в области будет понятно, что соединения формулы (I), в зависимости от заменителей, могут содержать один или более хиральных центров и, таким образом, существовать в форме многих различных изомеров, оптических изомеров (т. е. энантиомеров) и их смесей, включая рацемические смеси. Все такие изомеры, энантиомеры и их смеси, включая рацемические смеси, внесены в рамки изобретения.
В другом аспекте настоящего изобретения обеспечивается способ ингибирования активации NMDA-рецептора у млекопитающего, состоящий из введения вышеупомянутому млекопитающему соединения по пункту 1 в количестве, антагонистичном NMDA-рецептору.
В другом аспекте обеспечивается способ лечения или предотвращения повреждения или цитотоксичности клеток, опосредованных активацией NMDA-рецептора у млекопитающего, состоящий из введения вышеупомянутому млекопитающему фармацевтически эффективного количества соединения по пункту 1.
В другом аспекте изобретения обеспечиваются способы лечения или предотвращения нейродегенеративного заболевания, паралича, эпилептических припадков и конвульсий у млекопитающего, состоящий из введения вышеупомянутому млекопитающему фармацевтически эффективного количества соединения по пункту 1.
КРАТКОЕ ОПИСАНИЕ ФИГУР
Фигура 1 описывает эффект дозозависимой нейрозащитной активности декстрорфана после введения мышам NMDA.
Фигура 2 описывает эффект дозозависимой нейрозащитной активности соединения # 13 после введения мышам NMDA.
Фигура 3 описывает эффект дозозависимой нейрозащитной активности соединения #9 после введения мышам NMDA.
ДЕТАЛЬНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к новым полициклическим алкалоидам по формуле I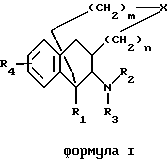
где R1-R4, X, m и n тип таковы, как определено ранее.
Использованы следующие общие сокращения во всем описании:
"EAA" относится к стимуляторной аминокислоте;
"NMDA" относится к N-метил-(D)-аспарагиновой кислоте;
"AMPA" относится к альфа-амино-5-метил-4-изоксазол-пропиониковой кислоте.
Термин CD50, как показано в Таблице I, определен как доза лекарства, которая индуцирует 50% понижение числа смертей, вызванных введением NMDA.
Будучи использован в этой заявке, термин "алкил" представляет насыщенный или ненасыщенный, замененный (одним или более галогеном, гидроксилом, амино или С6-20 арильной группой) или незамененный; прямая цепь, разветвленная цепь или циклическая гидроуглеродная доля может быть прервана одним или более гетероатомами (таким, как кислород, азот или сера).
Термин "арил" представляет карбоциклическую долю, которая выборочно заменена (например, одним или более C1-6 алкилом, галогеном, гидроксилом, аминогруппой), прервана одним или более гетероатомами (например, N, О или S) и содержит по меньшей мере одно кольцо бензоидного типа (например, фенил или нафтил).
Термин "аралкил" представляет арильную группу, присоединенную к смежному атому алкилом (например, бензил).
Соединения настоящего изобретения представлены формулой (I), как определено выше.
Предпочтительно, R1 является циклогексилом.
Предпочтительно, R1 является фенилом, выборочно замененным полярными группами.
Предпочтительными полярными группами являются COOH, NH2 или гуанидино.
Более предпочтительно, R1 является Н.
Наиболее предпочтительно, R1 является CH3.
Предпочтительно, R2 -C(NH)-NH2.
Предпочтительно, R2 является положительно заряженной аминогруппой.
Более предпочтительно, R2 является H.
Предпочтительно, R3 является C1-6 алкилом.
Более предпочтительно, R3 является OCH3.
Наиболее предпочтительно, R3 является H.
Наиболее предпочтительно, R3 является OH.
Предпочтительно, R4 является ОС1-6 алкилом.
Предпочтительно, R4 является галогеном.
Более предпочтительно, R4 является ОН.
Наиболее предпочтительно, R4 является ОCH3.
Предпочтительно, X является SO.
Предпочтительно, X является SO2.
Предпочтительно, X является NR5.
Более предпочтительно, X является О.
Наиболее предпочтительно, X является S.
Предпочтительно, R5 С1-6 алкил.
Более предпочтительно, R5 является CH3.
Наиболее предпочтительно, R5 является Н.
Предпочтительно, n является 0.
Предпочтительно, m является 3.
Предпочтительное соединение изобретения включает:
соединение # 8а: 5,6,7,8,9,11,12-гептагидро-3- метокси-5-метил-10-тиа-5,11- метанобензоциклодесен-13- β гидроксиламин;
Предпочтительное соединение изобретения включает:
соединение #8б: 5,6,7,8,9,11,12-гептагидро-3-метокси-5-метил-10-тиа-5,11 метанобеноциклодесен-13-амин;
Предпочтительное соединение изобретения включает:
соединение #9: 5,6,7,8,9,11,12-гептагидро-3- гидрокси-5-метил- 10-тиа-5,11-метанобензоциклодесен-13-амин (сульфазоцин);
Предпочтительное соединение изобретения включает:
соединение # 9а: (-)-транс-5,6,7,8,9,11,12-гептагидро-10-тиа-3-гидрокси-5-метил-5,11- метанобензоциклодесен-13-амин;
Предпочтительное соединение изобретения включает:
соединение # 9б: (+)-транс-5,6,7,8,9,11,12-гептагидро-10-тиа-3-гидрокси-5- метил-5,11-метанобензоциклодесен-13 амин;
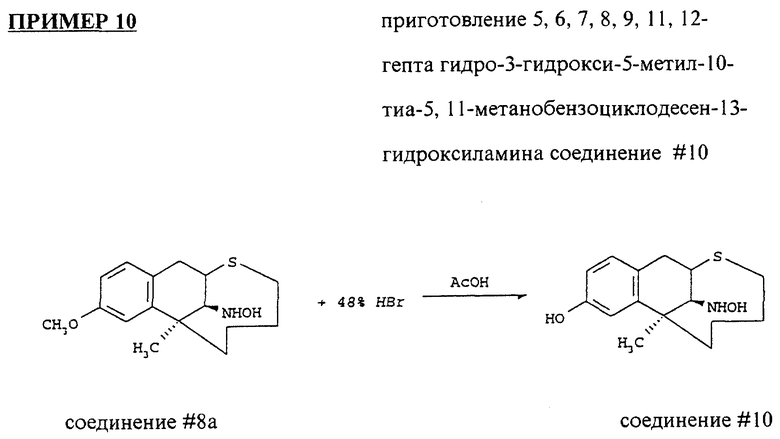
Предпочтительное соединение изобретения включает:
соединение #10: 5,6,7,8,9,11,12-гептагидро-3- гидрокси-5-метил-10-тиа-5,11 - метанобензоциклодесен-13- β гидроксиламин;
Предпочтительное соединение изобретения включает:
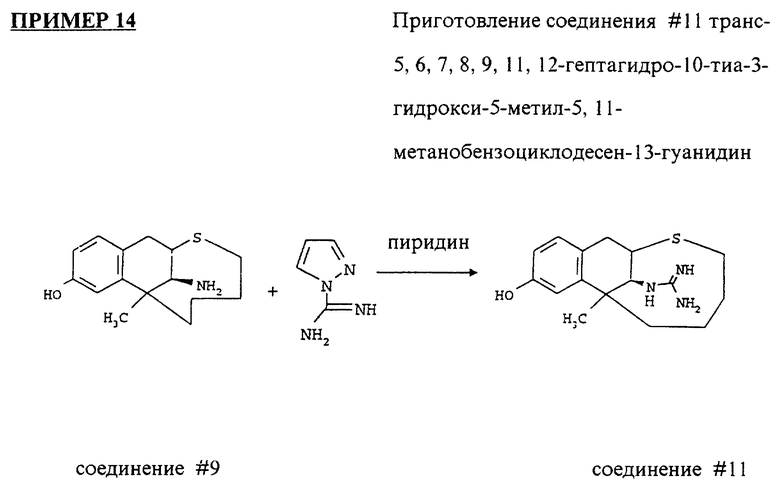
соединение #11 транс-5,6,7,8,9,11,12-гептагидро-10-тиа-3- гидрокси-5-метил-5,11-метанобензоциклодесен- 13-гуанидин.
Предпочтительное соединение изобретения включает:
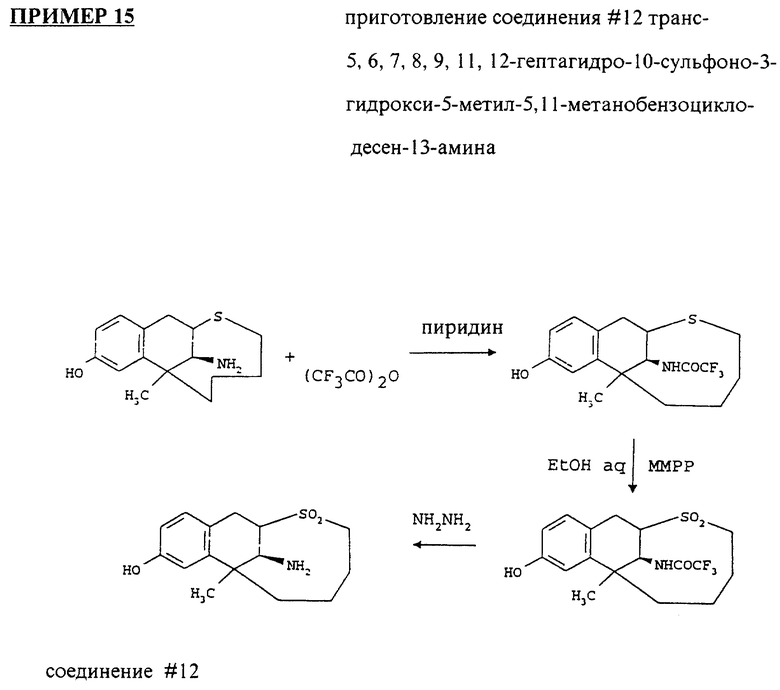
соединение # 12: транс-5,6,7,8,9,11,12-гептагидро-10-сульфоно-3-гидрокси-5- метил-5,11-метанобензоциклодесен-13-амин;
Предпочтительное соединение изобретения включает:
соединение # 13: 5,6,7,8,9,11, 12-гептагидро-3-гидрокси-5-метил-5,11-метанобензоциклодесен-13-амин.
Более предпочтительным соединением изобретения является:
соединение #11: транс-5,6,7,8,9,11,12-гептагидро-10-тиа-3- гидрокси-5-метил-5,11-метанобензоциклодесен-13-гуанидин.
Наиболее предпочтительным соединением изобретения является:
соединение # 9: 5,6,7,8,9,11,12-гептагидро-3-гидрокси-5- метил-10-тиа-5,11 метанобензоциклодесен-13-амин (сульфазоцин)
Наиболее предпочтительным соединением изобретения является:
соединение #10: 5,6,7,8,9,11,12-гептагидро-3-гидрокси-5- метил-10-тиа-5,11 метанобензоциклодесен-13-β-гидроксиламин;
Наиболее предпочтительным соединением изобретения является:
соединение #11: транс-5,6,7,8,9,11,12-гептагидро-10-тиа-3- гидрокси-5-метил-5,11-метанобензоциклодесен-13-гуанидин.
Соединения настоящего изобретения могут быть синтезированы с использованием традиционных препаративных шагов и способов получения, известным профессионалам в области органического и биоорганического синтезов, обеспечивая в то же время новые и уникальные комбинации для полного синтеза каждого соединения.
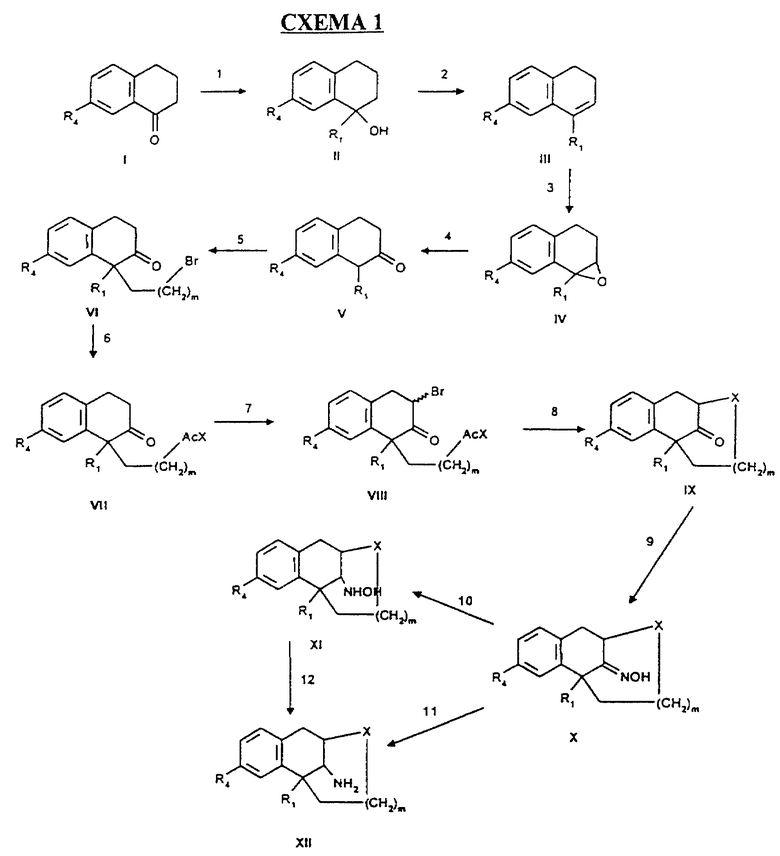
Прослеживаются пути синтеза для полупродуктов, вовлеченных в синтез, так же как и соединения-антагонисты NMDA-рецептора настоящего изобретения. Успешное получение этих соединений возможно путем нескольких способов синтеза, один из которых показан на Схеме 1.
Шаги, проиллюстрированные на Схеме 1, могут быть кратко описаны следующим образом:
Шаг 1: Соединение 1, алкил-1-тетралон, обрабатывают подходящим Реагентом Гринарда, таким, как метил магнезиум-бромид, в сухом неполярном растворителе, таком, как ТГФ, для накопления третичного спирта. Соединение II.
Шаг 2:
Спирт, Соединение II, дегидратируют при кислых условиях, таких, как водный насыщенный NH4Cl, для получения Соединения III.
Шаг 3:
Двойную связь в положении 1 на олефине эпоксидируют, используя стандартные реагенты и растворители, такие, как магниевая соль монопероксифталиковой кислоты в изо-пропаноле, для получения эпоксида, Соединение IV.
Шаг 4:
Эпоксид подвергают переаранжировке при кислых условиях, таких, как водный NaHCO3, используя стандартные методы для получения кетона. Соединение V.
Шаг 5:
Алкилирование бис-алкил-2-тетрона (Соединение V) проводят при щелочных условиях в неполярном растворителе, используя дигалоалкильный реагент, такой, как дибромобутан, для производства Соединения VI.
Шаг 6:
Нуклеофильное замещение бромида проводят с подходящим реагентом, таким, как триацетат калия, для получения Соединения VII.
Шаг 7:
Галогенируют 3 положение S-ацилированного тетралона (Соединение VII) в неполярном растворителе, таком, как смесь бензола и сухого ТГФ, используя подходящий реагент и неполярный растворитель, такой, как Бромин в сухом ТГФ, для получения Соединения VIII.
Шаг 8:
Циклизацию проводят при щелочных условиях с использованием стандартных реагентов и растворителей, таких, как бромид лития и сухой ТГФ под Аргоном, с добавлением щелочи, такой, как метоксид натрия, получая полициклическое соединение (Соединение IX).
Шаг 9:
Кетоновую группу соединения IX превращают в алкилоксимовую, используя стандартные процедуры, хорошо известные в области, получая соединение X.
Шаг 10 и 11:
Соединение Х восстанавливают, используя комплекс Боран-ТГФ. Если проводят в ТГФ, получают смесь 50:50 соединений XI и XII. Если реакцию проводят в диглиме (2- метоксиэтиловом эфире), селективно продуцируется амин (Соединение XII).
Шаг 12:
Соединение XI может быть использовано снова и восстановлено до амина, используя комплекс Боран-ТГФ, полученный в диглиме, для получения Соединения XII.
Соединения настоящего изобретения связываются и блокируют активацию ионотропного NMDA-рецептора и, таким образом, предотвращают избыточное проникновение Ca+2 в нейроны при событиях, опосредованных NMDA- рецепторами, которые типичны для гипоксии и/или ишемических состояний. Избыточное проникновение кальция в нейронные клетки является прелюдией к повреждению нейронов, что происходит после повреждения головы, паралича, и эпилептических припадков; связано с дегенеративными заболеваниями, такими, как болезнь Алцгеймера, болезнь Хантингтона, болезнь Паркинсона и амитрофический латеральный склероз (ALS); и периферической нейротоксичностью, включающей легкие и другие повреждения органов.
Соответственно, настоящее изобретение далее относится к использованию соединений по формуле (I) для ингибирования активации NMDA-рецептора, как и для лечения и предотвращения повреждения или цитотоксичности клеток, опосредованных активацией NMDA-рецептора у млекопитающих.
Настоящее изобретение обеспечивает соединения, которые блокируют EAA-индуцированные конвульсии на модели грызунов in vivo. Было обнаружено, что соединения настоящего изобретения блокируют смертность, индуцированную NMDA, AMPA или бикукуллином.
Принимается во внимание, что соединения настоящего изобретения могут быть модифицированы профессионалами в области таким образом, как присоединение меток, таких как радиоактивные метки, позволяющие определение соединения для использования в качестве радиопроводников. Соединения настоящего изобретения могут быть использованы как антагонисты NMDA in vitro или ex vivo, как в случае радиомеченых агентов, радиопроводников для использования в позитрон-эмиссионной томографии, парамагнитных агентов для использования в изображении магнитного резонанса и в случае антагонистов кальциевого канала, связанного с NMDA-рецептором.
Принимается во внимание, что соединения настоящего изобретения могут быть модифицированы профессионалами в области таким образом, как предотвращение доступа в центральную нервную систему, таким образом, что они могут функционировать как антагонисты NMDA-рецептора в периферических тканях для защиты против и/или снижения цитотоксичности (нейротоксичности), вовлеченных в события, опосредованные периферическим NMDA-рецептором.
Настоящее изобретение также обеспечивает фармацевтические составы, которые содержат фармацевтически эффективное количество соединения этого изобретения, или их фармацевтически приемлемые соли и, выборочно, фармацевтически приемлемые носитель или адъювант. Термин "фармацевтически эффективное количество" относится к количеству соединения, требуемому для введения млекопитающему для понижения или ингибирования активации NMDA-рецептора, повреждения клеток, конвульсий или симптомов, связанных с нейродегенеративными заболеваниями. Количество будет зависеть от таких факторов, как определенное показание к лечению, способ введения, размер больного, требующего лечения и т.д.
Терапевтические способы этого изобретения состоят из шага лечения пациентов фармацевтически приемлемым способом этими соединениями или составами. Такие составы могут быть в форме таблеток, капсул, таблеток в оболочке, порошков, гранул, леденцов, суппозиториев, общих порошков или жидких препаратов, таких как оральные или стерильные парентеральные растворы или суспензии.
Терапевтические агенты настоящего изобретения могут быть введены по отдельности или в комбинации с фармацевтически приемлемыми носителями. Пропорция каждого носителя определяется растворимостью и химической природой соединения, путем введения и стандартной фармацевтической практикой.
Для получения консистенции для введения предпочтительно, чтобы состав изобретения находился в форме единичной дозы. Формы, представляющие единичную дозу для орального введения, могут являться таблетками и капсулами и могут содержать традиционные экципиенты. Например, связывающие агенты, такие как акация, желатин, сорбитол или поливинилпиролидон; наполнители, такие как лактоза, сахар, кукурузный крахмал, фосфат кальция, сорбитол или глицин; таблетирующие смазки, такие как стеарат магния; дезинтеграторы, такие как крахмал, поливинилпирроликон, крахмал бриколлата натрия или микрокристаллическая целлюлоза; или фармацевтически приемлемые увлажняющие агенты, такие как лаурил сульфат натрия.
Соединения могут быть введены парентерально; это означает внутримышечно, внутривенно или подкожно. Для парентерального введения могут быть использованы соединения в форме стерильных растворов, содержащих другие растворы, например, солевой раствор или глюкозу, достаточных, чтобы сделать раствор изотоническим.
Соединения могут быть введены орально в форме таблеток, капсул или гранул, содержащих подходящие экципиенты, такие как крахмал, лактоза, белый сахар и подобное. Соединения могут быть введены орально в форме растворов, которые могут содержать агенты, придающие цвет и/или запах. Соединения могут быть введены подъязычно в виде таблеток или леденцов, в которых каждый активный ингредиент смешан с сахарным или кукурузным сиропами, агентами, придающими запах, и красителями, и затем достаточно дегидратированы, чтобы сделать смесь пригодной для прессирования в твердую форму.
Твердые оральные составы могут быть приготовлены традиционными способами смешивания, наполнения, таблетирования или подобными. Повторные операции смешивания могут быть использованы для распределения активного агента в тех композициях, где используются большие количества наполнителей. Такие процедуры являются, конечно, традиционными в области. Таблетки могут быть покрыты в соответствии с хорошо известными в нормальной фармацевтической практике способами, особенно, кишечными оболочками.
Оральные жидкие препараты могут быть в форме эмульсий, сиропов или эликсиров или могут быть представлены как сухие продукты для восстановления при помощи воды или других подходящих переносчиков перед использованием. Такие жидкие препараты могут содержать или не содержать традиционные добавки. Например, суспендирующие агенты, такие как сорбитол, сироп, метил целлюлозу, желатин, гидроксиэтилцеллюлозу, карбоксиметилцеллюлозу, гель стеарата алюминия или гидрогенированные съедобные жиры; эмульгирующие агенты, такие как моноолеат сорбитана или акацию; неводные переносчики (которые могут включать съедобные масла), такие как миндальное масло, фракционированное кокосовое масло, масляные эфиры, выбранные из группы, состоящей из глицерина, пропиленгликоля, этиленгликоля и этилового спирта; презерванты, например, метил парагидроксибензоат, этил парагидроксибензоат, n-пропил парагидроксибензоат или n-бутил парагидроксибензоат сорбитоловой кислоты; и, если требуется, традиционные агенты, придающие запах или цвет.
Для парентерального введения могут быть приготовлены формы жидкой единичной дозы путем использования соединения и стерильного переносчика, и, в зависимости от используемой концентрации, могут быть либо суспендированы, либо растворены в переносчике. Оказавшись в растворе, соединение может быть впрыснуто и простерилизовано на фильтре перед наполнением подходящей виалы или ампулы с последующим запечатыванием переносчика или упаковки для хранения. Адъюванты, такие как локальные анестетики, презервирующие или забуферивающие агенты, могут быть растворены в переносчике перед использованием. Стабильность фармацевтических композиций может быть усилена путем замораживания композиции после наполнения виалы и удаления воды под вакуумом (например, высушиванием с замораживанием состава). Парентеральные суспензии могут быть приготовлены, по существу, тем же образом за тем исключением, что соединение должно быть скорее суспендировано в переносчике, чем растворено, и, далее, стерилизация не достигается фильтрацией. Соединение может быть стерилизовано, однако, путем экспозиции его оксиду этилена перед суспендированием его в стерильном переносчике. Сурфактант или увлажняющий раствор могут быть выгодно внесены в состав для облегчения идентичного распределения соединения.
Фармацевтические составы этого изобретения состоят из фармацевтически эффективного количества соединения этого изобретения и фармацевтически приемлемого носителя. Обычно они содержат от примерно 0.1% до примерно 99% по весу, предпочтительно от примерно 10% до примерно 60% по весу соединения этого изобретения, в зависимости от того, какой способ введения используют.
Настоящее изобретение также обеспечивает способ для лечения опосредованных NMDA-рецептором цитотоксичности и/или заболевания у таких пациентов, как млекопитающие, включая человека, который состоит из шага введения пациенту фармацевтически эффективного количества соединения, его фармацевтически приемлемой соли или фармацевтического состава, как описано выше.
Врачи определят такую дозировку настоящих терапевтических агентов, которая будет наиболее подходящей. Дозировки могут варьировать в зависимости от способа введения и выбора определенного соединения. В дополнение, дозировка может варьировать для определенного пациента при лечении. Дозировка соединения, использованного в лечении будет варьировать в зависимости от степени расстройства, веса пациента, относительной эффективности соединения и выбора лечащего врача. Такая терапия может продлится в течение нескольких недель, с перерывами или без перерывов, до момента, когда симптомы пациента устранены.
Для дальнейшего помощи в понимании настоящего изобретения обеспечены следующие неограниченные примеры 1-17 (см. в конце описания).
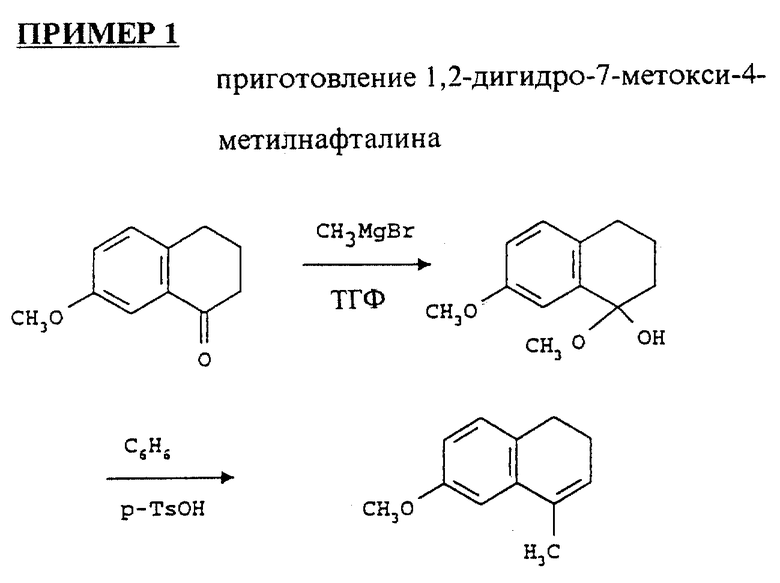
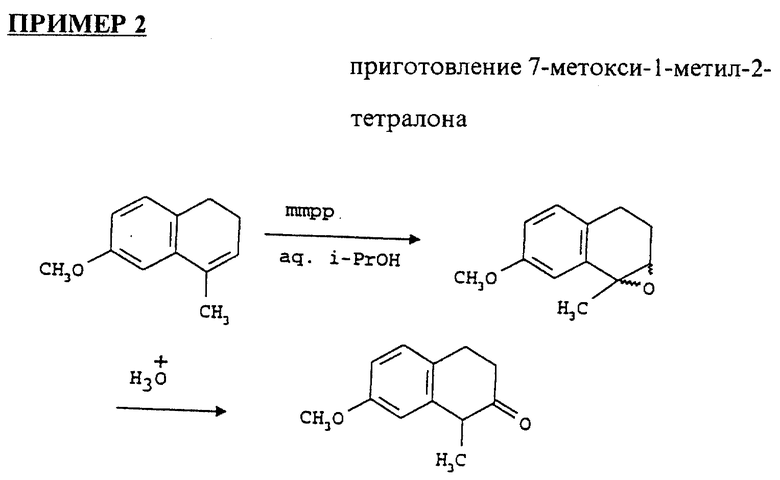
7-метокси-1-тетралон (25 г) высушивали азеотропной дистилляцией толуола и растворяли в сухом ТГФ (200 мл). Раствор охлаждали до -70oC (под аргоном) и добавляли хлорид метилмагния (1.4 м в смеси толуол/ТГФ, 187.5 мл). Объединенную реакционную смесь оставляли для перемешивания при комнатной температуре в течение ночи. Смесь осторожно обрабатывали водным насыщенным NN4Cl и экстрагировали этилацетатом. Последний раствор промывали соленой водой, высушивали над MgSO4 и выпаривали. Остаток растворяли в бензоле (150 мл), добавляли p-TSOH и нагревали смесь до обратного течения, используя конденсатор Dean-Stark до окончания реакции дегидратации. Этот раствор бензола разводили этилацетатом, промывали NaHCO3, высушивали над MgSO4 и выпаривали. Остаток экстрагировали гексасами, пропускали через колонку с силикагелем и элюировали смесью гексасов и этилацетата (1:0, 400:1, 200:1). Выход продукта был 20.87 г (84.42%).
1H ЯМР (300 МГц, CDCl3) δ: 2.05 (s, 3H); 2.23 (m, 2H); 2.69 (t, 2H); 3.81 (s, 3H) 5.88 (m, 1H); 6.68 - 7.06 (m, 3H).
Дигидро-7-метокси-4-метилнафталин (20.87 г) растворяли в изопропаноле (100 мл) и охлаждали в ледяной бане. Добавляли магниевую соль монопероксифталиевой кислоты (mmpp) (17 г), затем добавляли воду (50 мл) и перемешивали смесь при комнатной температуре в течение 2 часов. Когда окисление было закончено, смесь продукта гидролизовали с водным NaHCO3, частично выпаренным и проэкстрагированным этилацетатом. Последний экстракт промывали соляным раствором и выпаривали. Остаток растворяли в смеси этанола (156 мл), воды (121 мл) и конц. H2SO4 (24.3 мл), и нагревали до обратного течения под атмосферой N2 в течение 3 часов, охлаждали и нейтрализовали NaHCO3. После частичного выпаривания остаток экстрагировали этилацетатом, промывали соляным раствором, высушивали над MgSO4 и выпаривали. Продукт очищали на колонке с силикагелем, используя смесь гексасов и этилацетата (100:1, 50:1, 50:1.5). Выход продукта составил 16.2 г (71%).
1H ЯМР (300 МГц, CDCl3) δ: 1.47 (d, 3H); 2.55 (m, 2H); 3.02 (m, 2H); 3.5 (m, 1H); 3.81 (s, 3H); 6.75 - 6.77 (m, 3H) ppm.
IR (пленка) 1714 см-1
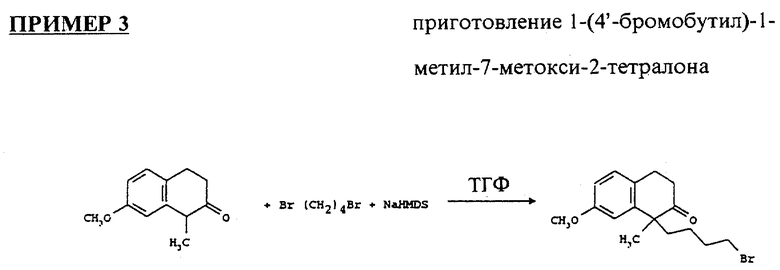
Высушивали 7-метокси-1-метил-2-тетралон (4 г) через азеотропную дистилляцию толуола, растворяли в сухом ТГФ (THF) (150 мл), охлаждали в ледяной бане под атмосферой аргона и добавляли раствор натрия-бис-(триметилсилил) амида (NaHMDS) (1 м в ТГФ, 23.13 мл), и перемешивали в течение 1/2 часа. Добавляли 1,4-дибромобутан (9.78 мл) оставляли реакционную смесь для нагревания до комнатной температуры на ночь, после чего ее гидролизовали солевым раствором, экстрагировали этилацетатом, высушивали над MgSO4 и выпаривали. Смесь продукта очищали на колонке с силикагелем, используя смесь гексанов и этилацетата(200:1, 150:1, 100:1, 75:1 и 50:1). Выход продукта составил 5.19г (76%).
1H ЯМР (300 МГц, DCDl3) δ:1.09 (m, 2H); 1.37 (s, 3H); 1.71 (m, 3H); 2.1 (m, 3H); 2.68 (m, 2H); 2.97 (m, 2H); 3.26 (t,2H); 3.8 (s, 3H), 6.72 - 7.09 (m, 3H) частей на миллион.
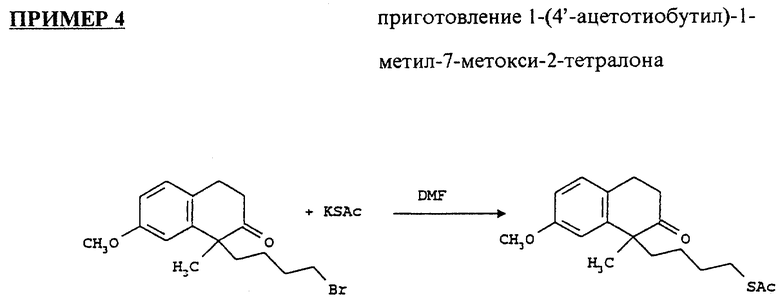
1-(4'-бромобутил)-1-метил-7-метокси-2-тетралон (4.48 г) высушивали азеотропной дистилляцией толуола и растворяли в сухом DMF (25 мл). Добавляли триацетат калия (5.86 г) и оставляли смесь для перемешивания под атмосферой аргона на ночь, ее экстрагировали этилацетатом, промывали солевым раствором, высушивали над MgSO4 и выпаривали. Остаток очищали на колонке с силикагелем, используя смесь гексанов и этилацетата (75:1, 50:1, 20:1). Выход продукта составил 3.9 г (88.3%).
1H ЯМР (300 МГц, CDCl3) δ: 0.99 (m, 2H); 1.5 (m, 6H); 2.07 (m, 1H); 2.25 (s, 3H); 2.65 (m, 4H); 2.95 (m, 2H); 3.79 (s, 3H); 6.71 - 7.08 (m, 3H) частей на миллион.
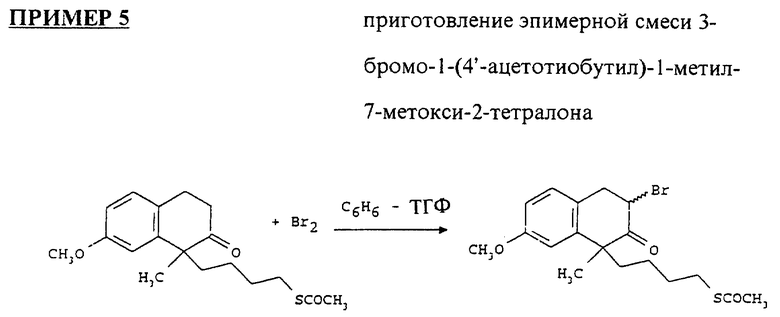
1-(4'-ацетотиобутил)-1-метил-7-метокси-2-тетралон (4 г) высушивали азеотропной дистилляцией толуола. Его растворяли в смеси бензола (244 мл) и сухого ТГФ (64 мл) и перемешивали при комнатной температуре под атмосферой аргона. Бромин (0.8 мл) растворяли в сухом ТГФ (26 мл) и постепенно добавляли к реакционной смеси под течением аргона. После 1 часа перемешивания смесь продукта гидролизовали водным NaHCO3, экстрагировали этилацетатом, промывали солевым раствором, высушивали над MgSO4 и выпаривали. Остаток высушивали азеотропной дистилляцией толуола и далее высушивали под высоким вакуумом.
Эпимерную смесь 3-бромо-1-(4'-ацетотиобутил)-1-метил- 7-метокси-2-тетралона (примерно 6.25 ммоль) высушивали азеотропной дистилляцией толуола и растворяли в сухом ТГФ (200 мл), добавляли бромид лития (сухой, 0.54 г), дегазировали раствор аргоном при комнатной температуре в течение одного часа и охлаждали в ледяной бане, хорошо перемешивая, с мягким течением аргона, проходящего через смесь. Растворяли метоксид натрия (0.5 м в метаноле, 13.75 мл) в сухом ТГФ (75 мл), дегазировали под аргоном при комнатной температуре в течение одного часа, после чего его добавляли к последнему раствору через шприц насосом в течение 4 часов. Объединенную реакционную смесь перемешивали в течение дополнительных 1/2 часа, разбавляли этилацетатом (100 мл), промывали солевым раствором, высушивали над MgSO4 и выпаривали. Остаток очищали на колонке с силикагелем, используя смесь гексанов и этилацетата (75:1, 50: 1). Выход продукта составил приблизительно 50-55%. Он кристаллизовался при стоянии.
1H ЯМР (300 МГц, CDCl3 δ:1.4- 1.95 (m, 4H); 2.85 (m, 2H); 2.7 - 3 (m, 2H); 3.4 (m, 1H); 3.82 (m, 4H); 6.7 - 7.1 (m, 3H) ppm.
IR (пленка) 1693, 1609 см-1
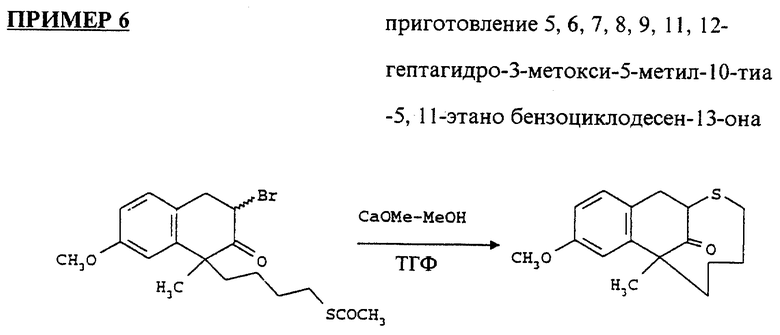
Высушивали 7,8,9,11,12-гептагидро-3-метокси-5-метил-10- тиа-5, 11-метанобензоциклодесен-13-он (1.32 г) азеотропной дистилляцией толуола, смешивали с гидрохлоридом гидроксиламина (2.64 г) и добавляли сухой пиридин (5.2 мл). Объединенную смесь нагревали до 80oC в течение двух дней. Ее охлаждали, разбавляли CH2CL2 и промывали солевым раствором. После высушивания над MgSO4, выпаривали растворитель очищали остаток на колонке с силикагелем, используя смесь гексанов и этилацетата (50:1, 25:1, 10:1,5:1,2:1). Выход продукта составил 1.22 г (92%).
1H ЯМР (300 МГц, CDCl3) δ: 1.2 - 1.9 (m, 9H); 2.4 (m, 2H); 2.85 (m,2H); 3.2 (dd, 1H); 3.8 (s,3H); 5.11 (t, 1H); 6.6 - 7.1 (m, 3H) частей на миллион.
IR (пленка) 1609, 2200, 3250 см-1
Масс спектрофотометрия: m/z 292.
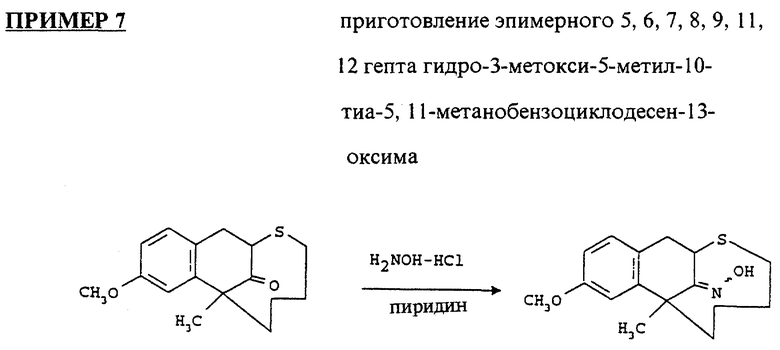
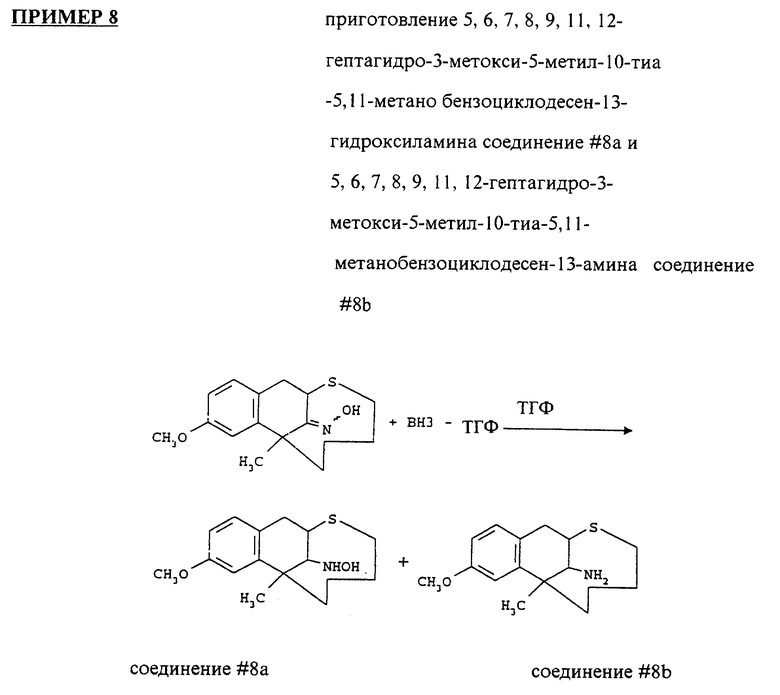
Высушивали 7,8,11,12-гептагидро-3-метокси-5-метил-10-тиа- 5, 11-метанобензоциклодесен-13-оксим (смесь изомеров, 0.3 г) с толуолом и растворяли в сухом ТГФ (30 мл). Его охлаждали в ледяной бане под атмосферой аргона. Добавляли комплекс боран-ТГФ (1 М раствор в ТГФ, 7.87 мл) и нагревали объединенную смесь до обратного течения в течение 30 часов. Охлаждали ее в ледяной бане. Осторожно добавляли воду (0.4 мл) и концентрированную HCl (0.6 мл) в соответствующем порядке. Нагревали смесь до обратного течения в течение 15 минут, охлаждали и высушивали. Остаток защелачивали концентрированным NH4OH до pH 12, экстрагировали CH2Cl2, промывали солевым раствором, высушивали над MgSO4 и выпаривали. Остаток очищали на колонке с силикагелем, используя смесь гексанов и этилацетата (50:1, 20:1, 10:1, 5:1, 5:1.5, 2:1, 1:1 и 1:2). Выход 5,6,7,8,9, 11, 12-гептагидро-3-метокси-5-метил-10-тиа-5,11-метанобензоциклодесен-13-гидроксиламина составил 0.073 г (23.1%). Он кристаллизовался из смеси этилацетата и гексанов.
1H ЯМР (300 МГц, CDCl3) δ: 1.1 - 1.91 (m, 9H); 2.3 (m, 2H); 3.30 (d, 1H); 3.37 (m, 2H); 3.7 (m, 1H); 3.78 (s, 3H); 6.6 - 7.1 (m, 3H) частей на миллион.
IR (пленка): 1612, 3300 см-1
Масс спектрофотометрия: 293.8, 275.8, 260.8.
Структура # 8а была подтверждена однокристаллической рентгеновской кристаллографией.
Выход 5,6,7,8,9,11,12-гептагидро-3-метокси-5-метил- 10-тиа-5,11-метанобензоциклодесен-13-амина составил 0.0968 г (32%). Свободное основание было растворено в гексанах.
1H ЯМР (300 МГц, CDCl3) δ: 0.8 - 2.5 (m, 11H); 3.18 (m, 3H); 3.6 (q, 1H); 3.8 (s, 3H); 6.6 - 7.1 (m, 3H) частей на миллион.
Этот продукт растворяли в эфире (40 мл) и закисляли смесью метанол-HCl.
Позволяли суспензии осаждаться и фильтровали. Осадок промывали эфиром и высушивали, получая 0.090 г продукта.
-1H ЯМР (300 МГц, CDCl3) δ: 10.8 - 1.7 (m, 6H); 1.8 (m, 2H); 2.0 - 2.5 (m, 3H); 3.45 (m, 2H); 3.5 (m, 2H); 3.8 (S, 3H) 6.7 - 7.1 (m, 3H) ppm.
Масс спектрометрия: m/z 278.
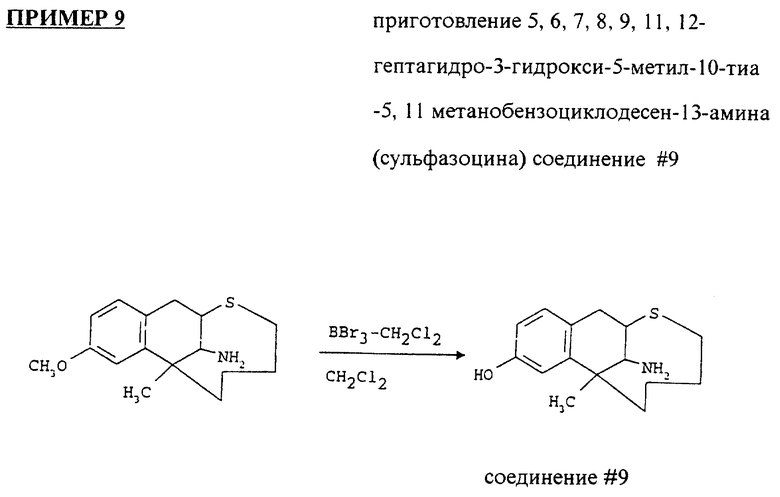
Высушивали 7,8,9,11,12-гептагидро-3-метокси-5-метил-10- тиа-5,11-метанобензоциклодесен-13-амин (0.260 г) азеотропной дистилляцией толуола и растворяли в сухом CH2Cl2 (40 мл). Охлаждали до -70oC под атмосферой аргона. Добавляли раствор трибромида борона (1М раствор в CH2Cl2, 187 мл) и оставляли объединенную смесь для перемешивания при окружающей температуре на ночь. Реакционную смесь гидролизовали NaHCO3, понижали pH при помощи NH4OH до 12 и экстрагировали CH2Cl2. Последний раствор высушивали над MgSO4 и выпаривали. Остаток очищали на колонке с силикагелем, используя смесь толуола и этилацетата. (10:1, 5:1, 2:1, 1:1, 1:2).
Выход 5,6,7,8,9,11,12-гептагидро-3-гидрокси-5-метил-10-тиа- 5,11 метанобензоциклодесен-13-амина составил 0.162 г (65%).
1H ЯМР (350 МГц, DMSO-D6 δ: 1.02 (m, 1H); 1.25 (m, 5H); 1.55 (m 2H); 2.01 (m, 2H); 2.55 (m, 1H); 2.97 ( d, 1H) 3.08 (m, 1H), 3.14 (m, 2H); 6.4 - 6.9 (m, 3H) частей на миллион.
Вышеупомянутый продукт был преобразован в его гидрохлоридную соль и очищен через HPLC (жидкостной хроматографией под высоким давлением).
Растворяли 8, 9, 11, 12-гептагидро-3-метокси-5-метил-10-тиа-5,11-метанобензоциклодесен-13-гидроксил амин (0.166 г) в смеси уксусной кислоты (4.5 мл) и 48% Hbr (4.5 мл). Ее охлаждали, осторожно нейтрализовали NaHCO3 и экстрагировали CH2Cl2. Последний раствор промывали солевым раствором, высушивали над MgSO4 и выпаривали. Остаток очищали на колонке с силикагелем, используя смесь гексанов и этилацетатов (10: 1, 10: 1.5, 5:1). Выход 5,6,7, 8,9,11,12- гептагидро-3-гидрокси-5-метил-10-тиа-5,11-метанобензоциклодесен-13-гидроксиламина составил 0.035 г (22%).
1H ЯМР (300 МГц, CDCl3) δ: 1.1-1.91 (m, 9H); 2.30 (m, 2H); 3.28 (m, 1H); 3.34 (m, 2H); 3.7 (m, 1H); 6.6 - 7.0 (m, 3H) частей на миллион.
Вышеупомянутый продукт был преобразован в его гидрохлоридную соль и очищен HPLC (жидкостной хроматографией под высоким давлением).
Соединение #9 (2.96 г) смешивали с D-тартаровой кислотой (2.01 г), растворенной в кипящем этаноле (95%, 50 мл), и фильтровали. Нерастворимую массу промывали горячим этанолом (25 мл). Объединенные фильтраты выпаривали досуха и остаток вновь растворяли осадок в горячем этаноле (20 мл). Собирали пушистую твердую массу и вновь растворяли в горячем этаноле (15 мл). Кристаллизацию оставляли происходить без движения при комнатной температуре в течение 2 дней. Полукристаллизованную массу далее еще дважды подвергали сходному процессу фракционной кристаллизации. Было обнаружено, что проба на этой стадии имеет диастерометрическую чистоту 98% по хиральной дериватизации с реагентом Marfey. (0.267 г).
Тартратовую соль названного соединения (0.1 г) растворяли в горячем метаноле (20 мл) и переносили на колонку, наполненную ион-обменной смолой Amberlite IRA-400 (С1-форма) (5 г, последовательно промытую метанолом, водой, 0.1 М HCl, водой и метанолом). Колонку последовательно промывали метанолом (100 мл) и водой(100 мл). Объединенные элюенты выпаривали и лиофилизовали.
Остаток (0.076 г).
Соединение #9-D-тартратовую соль (высокообогащенную декстровращающимися диастереомерами 1.5 г) смешивали с NH4OH (10 мл), насыщенным хлоридом натрия, и экстрагировали метилен хлоридом. Последний промывали солевым раствором, высушивали над MgSO4 и выпаривали. Остаток (1.27 г) смешивали с L-тартаровой кислотой (0.87 г) и кипятили с этанолом (95%, 100 мл), фильтровали, затем фильтрат оставляли кристаллизоваться при комнатной температуре в течение 2 дней. Осажденную массу оставляли медленно кристаллизоваться из горячего изопропанола. Было обнаружено, что проба имеет диастереометрическую чистоту 97% по хиральной дериватизации с реагентом Marfey, с выходом 0.1515 г. -
Тартратовую соль названного соединения (0.076 г) растворяли в горячем метаноле (25 мл) и переносили на колонку, набитую активированным Amberlite IRA-400 (С1-форма, 5 г), которую затем промывали метанолом (100 мл) и водой (100 мл) последовательно. Объединенные фильтраты выпаривали и лиофилизировали остаток (0.058 г).
Соединение #9 (0.35 г) высушивали азеотропной дистилляцией с толуолом и растворяли в сухом пиридине (5 мл). Добавляли 1H-пиразол-1-карбоксамидин гидрохлорид (1.29 г) и диизо-пропил этиламин (1.74 мл). Объединенную смесь нагревали до 80oC под атмосферой азота в течение 4 дней. Растворитель выпаривали и очищали остаток на колонке с силикагелем, используя смесь метилен хлорида и метанола. Продукт (0.41 г) растворяли в метаноле, насыщенном хлоридом азота (5 мл) и выпаривали растворитель. Остаток очищали HPLC (жидкостной хроматографией под высоким давлением), с выходом 0.045 г конечного продукта.
Соединение 9 (0.1 г) высушивали азеотропной дистилляцией толуола, растворяли в сухом метилен хлориде (20 мл) и охлаждали в ледяной бане под атмосферой аргона. Добавляли трифторуксусный ангидрид (0.54 мл) и пиридин (0.5 мл). После перемешивания при комнатной температуре в течение ночи реакционную смесь гидролизовали водным раствором бикарбоната натрия, экстрагировали метилен хлоридом, промывали солевым раствором, высушивали над MgSO4 и выпаривали. Остаток очищали на колонке с силикагелем, используя смесь гексанов и метилен хлорида с выходом 0.068 г транс-5,6,7,8,9,11,12-гептагидро-10-тиа-3-гидрокси- 5-метил- 5,11-метанобензоциклодесен-13-трифлуороацетамида.
Транс-5,6,7,8,9,11,12-гептагидро-10-тиа-3-гидрокси-5-метил- 5,11-метанобензоциклодесен-13-трифлуороацетамида растворяли в смеси этанола (2 мл) и воды (1 мл), и затем охлаждали в ледяной бане. Добавляли монопероксифталевую кислоту, гексагидратную соль магния (0.21 г). После 1 часа добавляли водный насыщенный бикарбонат натрия (5 мл). Объединенную смесь перемешивали при комнатной температуре в течение ночи и выпаривали, и остаток экстрагировали метилен хлоридом. Последний раствор промывали солевым раствором и выпаривали, получая 0.123 г транс-5,6,7,8,9,11,12-гептагидро-10-сульфоно-3- гидрокси-5-метил-5,11-метано-бензоциклодесен-13- трифлуороацетамида.
Транс-5,6,7,8,9, 11, 12-гептагидро-10-сульфоно-3-гидрокси-5- метил-5,11-метанобензоциклодесен-13-трифлуороацетамид (0.123 г) высушивали азеотропной дистилляцией с толуолом и добавляли ангидрированный гидразин (2 мл). Смесь перемешивали при комнатной температуре в течение 2 дней, затем выпаривали досуха под вакуумом. Смесь продукта растворяли в метаноле (2 мл) и оставляли при комнатной температуре. Кристаллизованный материал (0.037 г) отфильтровывали и растворяли в насыщенном растворе хлорида азота в метаноле (2 мл), и затем выпаривали. Остаток лиофилизовали, получая 0.031 г названного соединения.
Для целей сравнения соединения настоящего изобретения и соответствующие стандарты вводили мышам и определяли во времени нейрозащитную активность непосредственно после введения NMDA. Соединения вводили i.c.v. или внутрибрюшинно в различное время и в разных концентрациях перед инъекцией NMDA (1 нмоль/мышь, I.C.V.). Уровень смертности (CD50) ТАБЛИЦА I и кривые ответа.
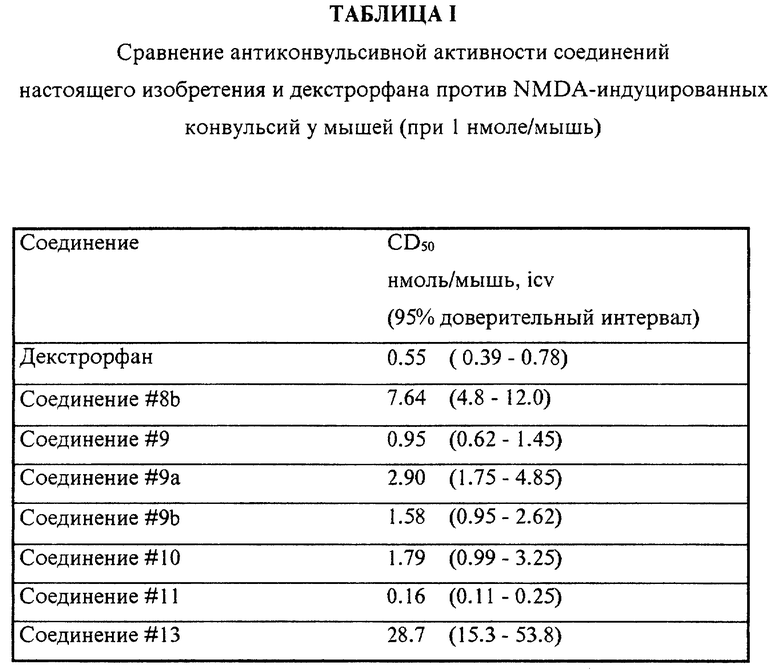
Результаты представлены на Фигурах 1, 2 и 3. Фигура 1 описывает влияние дозозависимой нейрозащитной активности декстрорфана, последовавший после введения мышам NMDA. Результаты демонстрируют зависимые по времени понижения защитных эффектов лекарства в различных дозах. На Фигуре 1 предварительно вводили декстрорфан i. c. v. в концентрациях 1.0 (замкнутое кольцо); 5.0 (открытый квадрат): и 10 (замкнутый квадрат) нмолей/мышь. Открытое кольцо представляет предварительно введенный физиологический раствор в концентрации 10 μл /мышь.
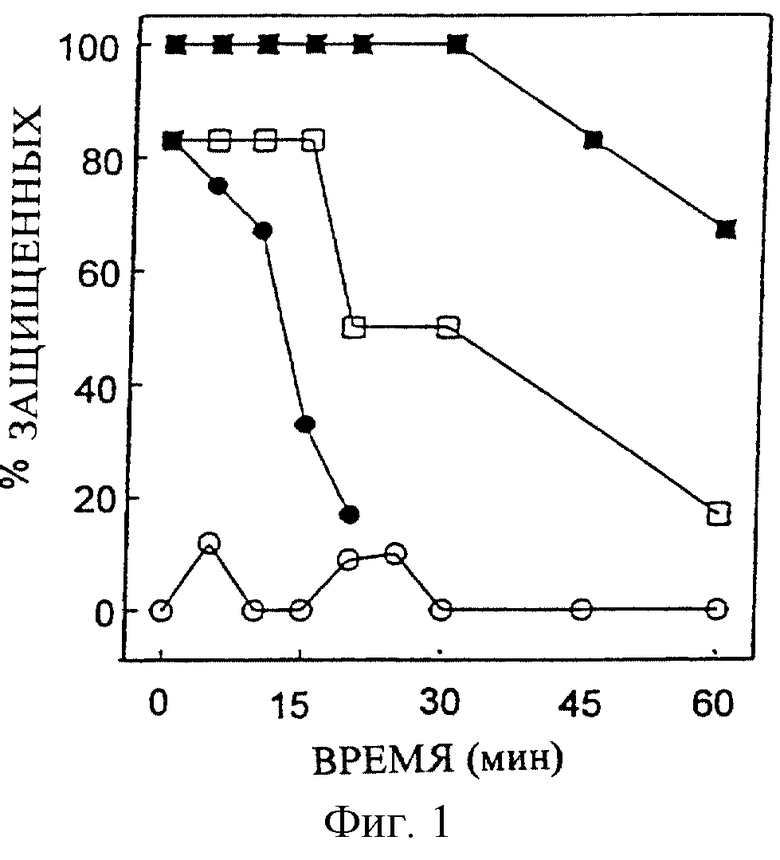
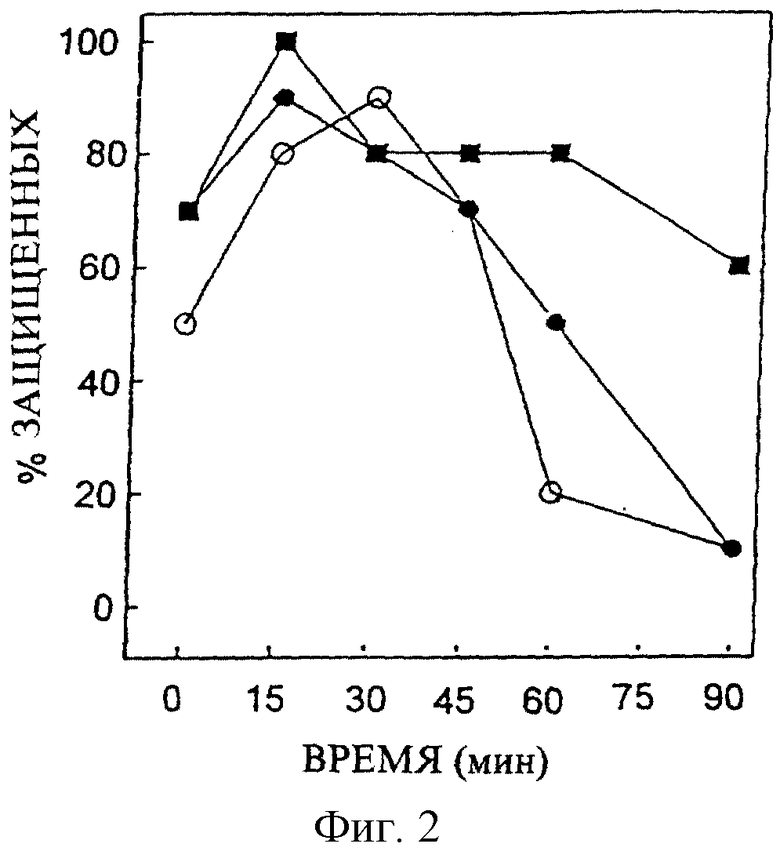
Профиль зависимости во времени нейрозащитной активности соединения #13 у мышей представлен на Фигуре 2. Фигура иллюстрирует зависимые во времени понижения защитных эффектов предварительного введения i.c.v. соединения #13 в различных дозах против NMDA. Дозозависимый временной профиль нейрозащитной активности соединения #9 против NMDA-индуцированных конвульсий у мышей показан на Фигуре 3. График иллюстрирует зависимые во времени понижения защитных эффектов Соединения #9 в различных дозах. На Фигурах 2 и 3 соответствующее соединение было предварительно введено в концентрациях 25 (открытое кольцо); 50 (замкнутое кольцо); и 100 (замкнутый квадрат) нмоль/мышь.
Соединения изобретения продемонстрировали антиконвульсивную активность против индуцированных NMDA конвульсий у мышей. Соединение #11 демонстрировало наивысшую активность, обладая примерно в три раза большей силой, чем Декстрорфан.
ПРИМЕР 17 ПОВРЕЖДЕНИЕ ДВИЖЕНИЯ
А. Локомоторика и нарушение поведения
Мыши были расположены индивидуально в клетках для наблюдений на 60 минут для периода привыкания. Им инъецировали I.С.V. тестовые соединения и наблюдали в течение от 15 до 30 минут. Локомоторику и нарушения поведения оценивали в соответствии с процедурами Koek Colpaert (J. Pharm. Exp. Therm. 252 349-357, 1990). Для каждого животного записывали присутствие локомоторной активности и нарушения поведения. Статистическую значимость изменений, индуцированных лекарством, в соответствии с определенным поведением тестировали посредством методов Fray et al. (Psycopharmacol 69 253-259, 1980). MK-801, известный как имеющий существенную нейротоксичность, индуцировал нарушение поведения в таких низких дозах, как 1.5 нмоль/мышь.
Б. Оценка двигательной координации Ротародом.
Влияние на движение, усиленное различными антагонистами, оценивали с использованием топчака ротарода (модель 7600, EGO Basile, Италия). Использованный способ был схож с процедурой, описанной Dunhamand Miya (J. Am. Pharmac. Assoc. 46 208-209- 1957). Аппарат состоял из рейки диаметром 2.5 см, которая подвешена горизонтально на высоте 50 см над рабочей поверхностью. Рейку поворачивали при скорости 8 об/мин. Круглые разделители были помещены на интервалах вдоль рейки таким образом, чтобы можно было тестировать одновременно пять животных. У всех животных было выработано привыкание к рейке в течение двух последовательных дней перед экспериментированием.
Результаты исследования, в котором сравнительное повреждение движения было вызвано Соединением #13, сравнивали с декстрорфаном, при оценке ротародом. Процент животных, демонстрирующих влияние на движение, является выше для животных, обработанных декстрорфаном, чем для животных, обработанных Соединением #13.
| название | год | авторы | номер документа |
|---|---|---|---|
| СОЕДИНЕНИЕ СУЛЬФОНАМИДА ИЛИ ЕГО СОЛЬ | 2007 |
|
RU2425029C2 |
| СОЕДИНЕНИЯ КОНДЕНСИРОВАННОГО ИНДАНА | 2008 |
|
RU2451671C2 |
| АМИНОИНДАНОВОЕ ПРОИЗВОДНОЕ ИЛИ ЕГО СОЛЬ | 2007 |
|
RU2429222C2 |
| ПРОИЗВОДНОЕ ЦИКЛИЧЕСКОГО АМИНА ИЛИ ЕГО СОЛЬ | 2005 |
|
RU2347776C2 |
| СУЛЬФОНАМИДНЫЕ СОЕДИНЕНИЯ ИЛИ ИХ СОЛИ | 2009 |
|
RU2481329C2 |
| СИНЕРГИЧЕСКОЕ ЛЕЧЕНИЕ ПАРКИНСОНИЗМА | 1995 |
|
RU2176145C2 |
| ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛА В КАЧЕСТВЕ СЕЛЕКТИВНЫХ ИНГИБИТОРОВ КИСЛОТНОЙ ПОМПЫ | 2007 |
|
RU2412188C2 |
| ПРОИЗВОДНЫЕ ПИПЕРАЗИНА ИЛИ ПИПЕРИДИНА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1996 |
|
RU2194702C2 |
| ЗАМЕЩЕННЫЕ СОЕДИНЕНИЯ АМИДА | 2010 |
|
RU2609021C2 |
| АЛЬФА-ФЕНИЛ ИЛИ ПИРИДИЛ-ЭТАНОЛАМИНЫ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ АГОНИСТОВ β АДРЕНЕРГИЧЕСКИХ РЕЦЕПТОРОВ | 2003 |
|
RU2319698C2 |



Изобретение относится к новым полициклическим алкалоидам, которые способны связывать или быть антагонистами NMDA (N-метил-(D)-аспарагиновая кислота) рецепторного комплекса или иным образом защищать нейроны против дегенерации, индуцированной рецептором стимуляторных аминокислот. В другом аспекте изобретение относится к способу ангибирования активации NMDA-рецептора у млекопитающих, используя новые полициклические алкалоиды изобретения. 3 с.п. ф-лы, 1 табл., 3 ил.
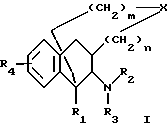
где R1 является H, C1-6 алкилом или C6-12 арилом, которые могут быть замещены полярными группами;
R2 и R3 являются, независимо, H, ОН, C1-6 алкилом, -C(NH)-NH2, положительно заряженной группой или C7-13 аралкилом, которые могут быть замещены NH2, ОН, C1-6 алкилом или галогеном, или
R2 и R3 совместно образуют 5- - 6-членное кольцо, которое может содержать гетероатом;
R4 является H, C1-6 алкилом, ОR6, SR6 или N(R6)2, где каждый R6 является, независимо, H, С1-3 алкилом;
Х является О, S, SO, SO2, N-R5 или C-(R5)2, где каждый R5 является, независимо, H, C1-6 алкилом или C7-13 аралкилом, который может содержать один или более гетероатомов;
n является целым числом от 0 до 2;
m является целым числом от 0 до 3,
с условием, что когда Х является СН2, тогда R1 не является СН3; R2 и R3 оба не являются H; R4 не является ОН; m не равно 3 и n не равно 0.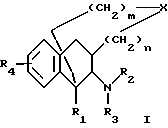
где R1 является H, C1-6 алкилом или C6-12 арилом, которые могут быть замещены полярными группами:
R2 и R3 являются, независимо, H, ОН, C1-6 алкилом, -C(NH)-NH2, положительно заряженной группой или C7-13 аралкилом, которые могут быть замещены NH2ОН, C1-6 алкилом или галогеном, или
R2 и R3 совместно образуют 5- - 6-членное кольцо, которое может содержать гетероатом;
R4 является Н, C1-6 алкилом, ОR6, SR6 или N(R6)2, где каждый R6 является, независимо, H, С1-3 алкилом;
Х является О, S, SO, SO2, N-R5 или C-(R5)2, где каждый R5 является, независимо, H, C1-6 алкилом или C7-13 аралкилом, который может содержать один или более гетероатомов;
n является целым числом от 0 до 2;
m является целым числом от 0 до 3,
с условием, что когда Х является СН2, тогда R1 не является СН3; R2 и R3 оба не являются H; R4 не является ОН; m не равно 3 и n не равно 0.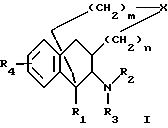
где R1 является H, C1-6 алкилом или C6-12 арилом, которые могут быть замещены полярными группами:
R2 и R3 являются, независимо, H, ОН, C1-6 алкилом, -C(NH)-NH2, положительно заряженной группой или C7-13 аралкилом, которые могут быть замещены NH2ОН, C1-6 алкилом или галогеном, или
R2 и R3 совместно образуют 5- - 6-членное кольцо, которое может содержать гетероатом;
R4 является H, C1-6 алкилом, OR6, SR6 или N(R6)2, где каждый R6 является, независимо, H, C1-3 алкилом;
Х является О, S, SO, SO2, N-R5 или C-(R5)2, где каждый R5 является, независимо, H, C1-6 алкилом или C7-13 аралкилом, который может содержать один или более гетероатом;
n является целым числом от 0 до 2;
m является целым числом от 0 до 3,
с условием, что когда Х является СН2, тогда R1 не является СН3; R2 и R3 оба не являются H; R4 не является ОН; m не равно 3 и n не равно 0.
| Farooqui and Horrocks, Brain Rev | |||
| Устройство для электрической сигнализации | 1918 |
|
SU16A1 |
| Способ выделения фактора роста нервной ткани из змеиного яда | 1982 |
|
SU1055732A1 |
| КОМПЕНСАЦИОННЫЙ СТАБИЛИЗАТОР НАПРЯЖЕНИЯ ПОСТОЯННОГО ТОКА | 0 |
|
SU408294A1 |
