Область техники
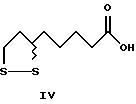
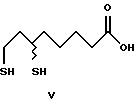
Изобретение относится к способу получения α-липоевых кислот формулы IV, а также дигидролипоевых кислот формулы V энантиомерной чистоты из 8-хлор- 6-гидроксиоктановых кислот формулы VI энантиомерной чистоты. Изобретение включает описание новых (+)- и (-)-8-хлор-6- сульфонилоксиоктановых кислот формулы I и их алкильных эфиров формулы II энантиомерной чистоты, новых эфиров (+)- и (-)-6,8- дихлороктановой кислоты формулы III и способов их получения, а также способов получения α -липоевых кислот и дигидролипоевых кислот энантиомерной чистоты.
α-Липоевая кислота - 3-(4-карбоксибутил)-1,2-дитиолан (тиоктовая кислота).

Уровень техники
R-энантиомер α -липоевой кислоты - природное вещество, присутствующее в незначительном количестве практически во всех тканях животных и растений. В качестве кофермента α -липоевая кислота принимает участие в реакции окислительного декарбоксилирования α -кето-кислот (например, пировиноградной кислоты). α -Липоевая кислота представляет интерес для фармакологии, поскольку оказывает противовоспалительное, обезболивающее и цитопротективное действие. Важным медицинским показанием является лечение α -липоевой кислотой диабетического полиневрита. Согласно последним данным (СА 116: 207360) α-липоевая кислота может оказаться полезной при лечении заболеваний, вызванных вирусами иммунодефицита HIV-1 и HTLV IIIВ.
Что касается свойств оптически чистых изомеров α -липоевой кислоты (R- и S-энантиомеров, т.е. R- α -липоевой и S- α -липоевой), то, в отличие от рацемата, R-энантиомер оказывает преимущественно противовоспалительное, а S-энантиомер - в основном обезболивающее действие (ЕР 0427247, 08.11.90). Вследствие этого синтез чистых энантиомеров, в особенности R-формы, имеет большое практическое значение.
Известные способы получения α -липоевой кислоты энантиомерной чистоты включают расщепление рацематов конечного продукта или его предшественников, асимметрические синтезы с использованием хиральных соединений и реагентов, энантиоспецифические синтезы на основе природных оптически активных предшественников, а также микробиологический синтез (обзор: J.S.Yadav et al., J. Sci. Ind. Res. 1990, 49, 400; а также: А.Г.Толстиков с соавт., Биоорг.хим., 1990, 16, 1670; L.Dasaradhi et al., J.Chem.Soc., Chem. Commun. 1990, 729: A. S. Gopalan et al. , J.Chem.Perkin Trans. 1 1990, 1897: A.S.Gopalan et al., Tetrahedron Lett. 1989, 5705; EP 0487986 A2, 14.11.91).
Из перечисленных методов наиболее практичным до настоящего времени остается метод расщепления рацематов, основанный на образовании диастереомерных солей α -липоевой кислоты с оптически активным α -метилбензиламином (DE-OS 4137773.7, 16.11.91). К недостаткам метода относится то, что расщепление рацемата осуществляется лишь на последней стадии синтеза, когда побочный энантиомер уже невозможно рацемизовать или инвертировать. В других известных способах, когда расщепление рацематов проводят на предварительных этапах синтеза, лишь один энантиомер может быть использован для получения целевого продукта (α-липоевой кислоты энантиомерной чистоты), и, следовательно, теоретически выход может достигать лишь 50% (E.Walton et al., J.Am. Chem.Soc. 1955, 77, 5144; D.S.Acker und W.J.Wayne, J.Am.Chem.Soc., 1957, 79, 6483; Л.Г.Чеботарева, А.М. Юркевич, Хим. - Фарм. ж., 1980, 14, 92).
Сущность изобретения
Целью настоящего изобретения является разработка метода получения α -липоевой и дигидролипоевой кислот энантиомерной чистоты, когда расщепление рацемата проводится на возможно более ранних стадиях синтеза, причем превращение обоих побочных энантиомеров 8-хлор-6-гидроксиоктановой кислоты в целевой энантиомер α-липоевой кислоты происходит с теоретическим выходом 100% без дополнительной рацемизации и инверсии.
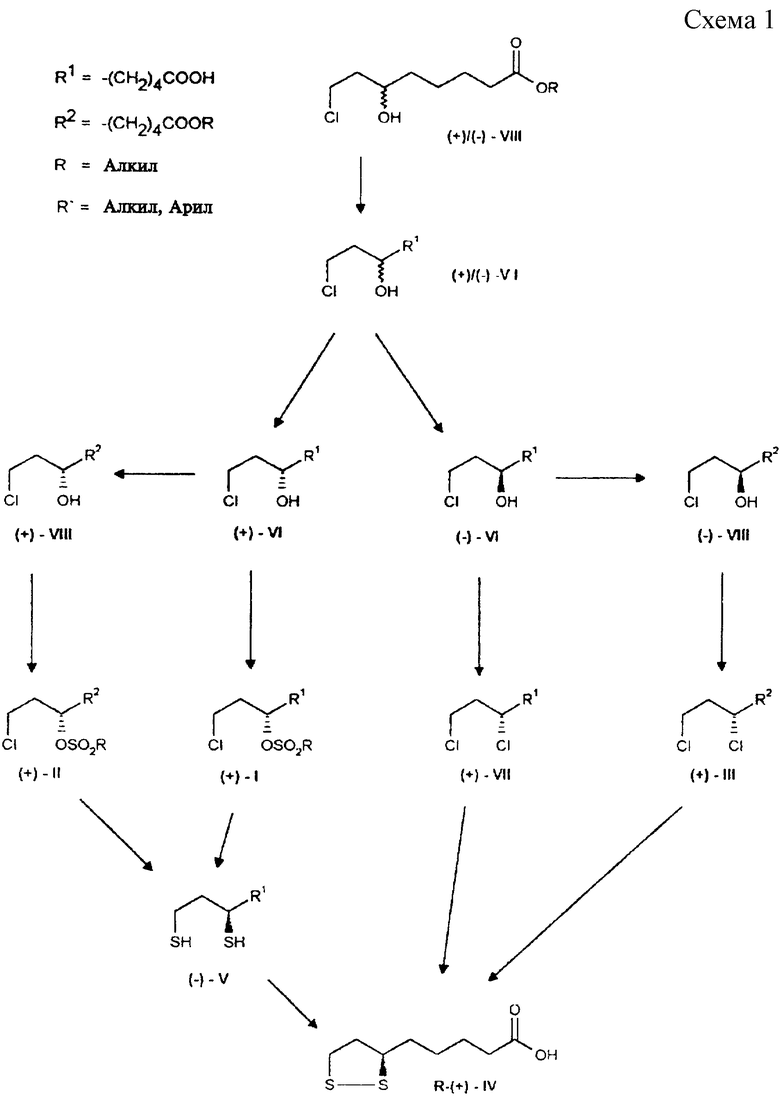
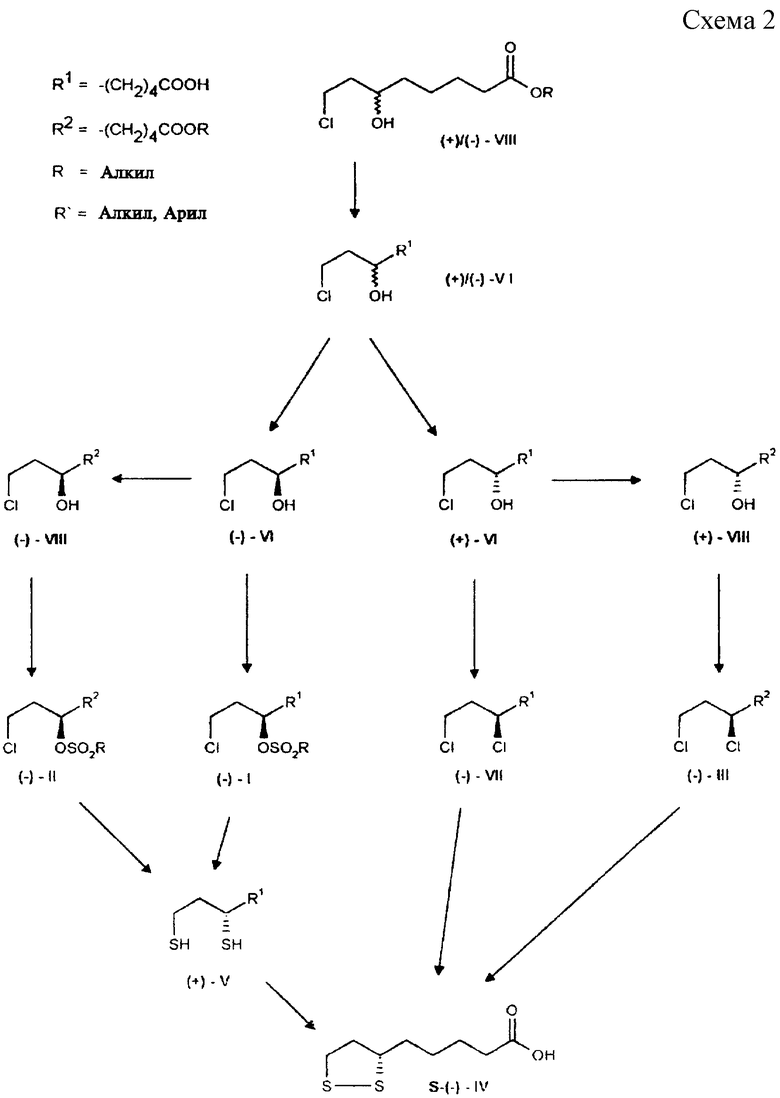
На схемах 1 и 2 представлена последовательность реакций, приводящих к получению R-(+)- и S-(-)- α -липоевых кислот. На схеме R - преимущественно линейные или разветвленные C1-C4-алкильные группы, в том числе метил-, этил-, н-пропил, изо-пропил, н-бутил-, изо-бутил, трет-бутил-, главным образом метил-; R'- преимущественно метил-, п-толил-группы.
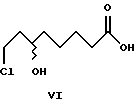
Исходное соединение, рацемическую 8-хлор-6-гидроксиоктановую кислоту формулы VI получали по известному методу путем гидролиза рацемических алкильных эфиров формулы VIII (Y.Deguchi und K.Nakanishi, Yakugaku Zasshi, 1963, 83, 701).
Оптически чистые (+)-8-хлор-6-гидроксиоктановую кислоту и (-)-8-хлор-6- гидроксиоктановую кислоту получали путем взаимодействия рацемической 8-хлор-6-гидроксиоктановой кислоты с оптическими антиподами α -метилбензиламина, образования диастеромерных солевых пар и выделения плохо растворимых солей, с последующим разложением чистых диастереомерных солей на (+)-8-хлор-6-гидроксиоктановую кислоту и R-(+)- α -метилбензиламин и, соответственно, на (-)-8-хлор-6-гидроксиоктановую кислоту и S-(-)- α -метилбензиламин. Разложение солей проводили кислотами, например, минеральными кислотами или основаниями, например, гидроксидами щелочных металлов.
Цель изобретения достигается тем, что оба энантиомера 8-хлор-6-гидроксиоктановой кислоты формулы VI непосредственно переводят в R- α -липоевую кислоту формулы (+)-IV по следующей последовательности реакций: (+)-8-хлор-6-гидроксиоктановую кислоту и, соответственно, ее алкильные эфиры, обрабатывают сульфохлоридами с сохранением конфигурации, а (-)-8-хлор-6-гидроксиоктановую кислоту, и, соответственно, ее алкильные эфиры, хлорируют с обращением конфигурации. При последующем введении в молекулу атомов серы все промежуточные соединения в качестве конечного продукта дают R- α -липоевую кислоту превосходной оптической чистоты (с учетом ошибки определения: > 99%, хиральная ВЭЖХ).
Из энантиомера 8-хлор-6-гидроксиоктановой кислоты формулы VI можно стереоспецифически с сохранением конфигурации в присутствии каталитического количества HCl получить алкильные эфиры формулы VIII, преимущественно метиловый эфир.
Затем из алкил-(+)-8-хлор-6-гидроксиоктаноата формулы (+)-VIII энантиомерной чистоты можно с сохранением конфигурации получить алкил-(+)-8-хлор-6-сульфонилоксиоктаноат формулы (+)-II, из которого синтезировать (-)-дигидролипоевую формулы (-)-V или (+)- дигидролипоевую формулы (+)-V кислоты.
Однако (+)-8-хлор-6- гидроксиоктановую кислоту формулы (+)-VI можно и непосредственно через промежуточное получение (+)-8-хлор-6-сульфонилоксиоктановой кислоты формулы (+)-I с высоким выходом стереоспецифически направленно перевести в (-)-дигидролипоевую формулы (-)-V или R- α -липоевую формулы (+)-IV кислоты. В этом случае синтез формулы (+)-I проводят в присутствии 2,0-2,2 мол. экв. сульфохлорида и 1,5-2,5, предпочтительно 2,0-2,1 мол. экв., третичного азотистого основания, предпочтительно триэтиламина.
Способ получения оптических изомеров α -липоевой кислоты формулы IV из дигидролипоевых кислот оптической чистоты путем окисления кислородом воздуха в присутствии каталитических количеств солей трехвалентного железа описан в литературе (E.Walton et al., J.Am.Chem.Soc. 1955, 77, 5144).
Из алкил-(-)-8-хлор-6-гидроксиоктаноата формулы (-)-VIII энантиомерной чистоты по реакции с тионилхлоридом в присутствии каталитических количеств пиридина с обращением конфигурации получают алкил-(+)-6,8-дихлороктаноаты формулы (+)-III. Последующее введение в молекулу атомов серы тионированием с помощью Na2S2 приводит к образованию R- α -липоевой кислоты формулы (+)-IV высокой оптической чистоты.
Аналогичным образом из (-)-8-хлор-6-гидроксиоктановой кислоты формулы (-)-VI можно с высоким выходом получить (+)-6,8- дихлороктановую кислоту формулы (+)-VII, если хлорирование проводить в присутствии 1,5-5, предпочтительно 2,0-2,5 мол.экв. тионилхлорида, а затем провести гидролиз реакционной смеси с помощью водных растворов оснований, предпочтительно едкого натра. Последующий перевод (+)-6,8-дихлороктановой кислоты с помощью Na2S2 в R- α -липоевую кислоту описан в литературе (D.S.Acker und W.J.Wayne, J.Am. Chem.Soc., 1957, 79, 6483).
(+)- и (-)-дигидролипоевые кислоты могут быть также получены путем гидрирования S(-)- α - и, соответственно, R(+)- α -липоевых кислот по известным методикам.
Все указанные реакции проводятся в подходящих органических растворителях. В качестве органического растворителя могут быть, например, использованы углеводороды, содержащие в цепи от 3 до 10 углеродных атомов, жидкие ароматические углеводороды, сложные эфиры алифатических и циклоалифатических карбоновых кислот, содержащие от 2 до 6 углеродных атомов, с алифатическими или циклоалифатическими спиртами, содержащими от 2 до 6 углеродных атомов, простые эфиры и гликоэфиры, или гомогенные смеси из перечисленных растворителей. Наиболее предпочтительными являются этилацетат, циклогексан, толуол, этанол и составленные из них гомогенные смеси.
Степень чистоты оптических изомеров и диастереомерных солей определяли по величине удельного оптического вращения. При этом относительное содержание оптического изомера 8-хлор-6- гидроксиоктановой кислоты формулы VI и α -липоевой кислоты формулы IV определяли методом ВЭЖХ на оптически активных сорбентах с точностью определения 0,5%. Оптическую чистоту алкил-(+)-8-хлор-6- гидроксиоктаноата формулы VIII дополнительно определяли методом 1H-ЯМР ее эфира (по окси-группе) с (S)-(+)-O-ацетилминдальной кислотой.
Предлагаемое изобретение позволяет получать энантиомеры α -липоевой кислоты простым и эффективным способом, с высоким химическим и оптическим выходом.
Сведения, подтверждающие возможность осуществления изобретения
Сущность изобретения подтверждается следующими примерами.
Пример 1
39,9 г (204 ммоль) рацемической 8-хлор-6- гидроксиоктановой кислоты (+)/(-)-VI растворили при 4oC в 155 мл смеси этилацетат/циклогексан (1:1). В течение 10 мин порциями прибавляли 13,5 г (112 ммоль) R-(+)- α -метилбензиламина. Спустя 2 ч охладили до 20oC, осадок отделили фильтрованием, промыли 20 мл смеси этилацетат/циклогексан (1:1) и 30 мл циклогексана. Соль дважды перекристаллизовали из 400 мл смеси этилацетат/циклогексан (3:1) и высушили в вакууме при 40oC. Получили 20,5 г (+)(+)- диастереомерной соли, [α]
Соль суспендировали при 20oC в 220 мл диэтилового эфира. При охлаждении и перемешивании медленно подкисляли с помощью 3 н. соляной кислоты до pH 1, при этом соль переходила в раствор. Спустя 30 мин отделили органическую фазу, однократно промыли 20 мл 2 н. HCl, двукратно 20 мл воды и высушили сульфатом магния. После отгонки растворителя в вакууме получили 10,8 г (54% от теор.) (+)-8-хлор- 6-гидроксиоктановой кислоты (+)-VI; [α]
Пример 2
33,9 г (173 ммоль) рацемической 8-хлор-6- гидроксиоктановой кислоты (+)/(-)-VI растворили при 40oC в 130 мл смеси этилацетат/циклогексан (1:1). В течение 10 мин порциями прибавляли 11,5 г (95 ммоль) S-(-)- α -метилбензиламина. Через 2 ч охладили до 20oC, осадок отделили фильтрованием, промыли 17 мл смеси этилацетат/циклогексан (1:1) и 25 мл циклогексана. Соль дважды перекристаллизовали из 340 мл смеси этилацетат/циклогексан (3:1) и высушили в вакууме при 40oC. Получили 17,2 г (-)(-)-диастереомерной соли, [α]
Соль суспендировали при 20oC в 190 мл диэтилового эфира. При охлаждении и перемешивании медленно подкисляли с помощью 3 н. соляной кислоты до pH 1, при этом соль перешла в раствор. Спустя 30 мин отделили органическую фазу, однократно промыли 17 мл 2 н. HCl, двукратно 20 мл воды и высушили сульфатом магния. После отгонки растворителя в вакууме получили 9,1 г (53% от теор.) (-)-8- хлор-6-гидроксиоктановой кислоты (-)-VI; [α]
Пример 3
6,4 г (32,9 ммоль) (+)-8-хлор-6-гидроксиоктановой кислоты (+)-VI кипятили с обратным холодильником в 100 мл абсолютного метанола в присутствии 0,4 мл конц. соляной кислоты в течение 2 ч. Затем растворитель отогнали в вакууме. Получили 6,6 г (97% от теор.) метил-(+)-8-хлор-6-гидроксиоктаноата (+)-VIII (R=Me), [α]
Пример 4
7,7 г (39,5 ммоль) (-)-8-хлор-6-гидроксиоктановой кислоты (-)-VI кипятили с обратным холодильником в 120 мл абсолютного метанола в присутствии 0,5 мл конц. соляной кислоты в течение 2 ч. Затем растворитель отогнали в вакууме. Получили 7,9 г (97% от теор.) метил-(-)-8-хлор-6-гидроксиоктаноата (-)-VIII (R=Me), [α]
Пример 5
3,9 г (20 ммоль) (+)-8-хлор-6-гидроксиоктановой кислоты (+)- VI и 4,1 г (40 ммоль) триэтиламина смешали в 80 мл толуола. При охлаждении (10-15oC) медленно прибавляли 3,5 г (30,6 ммоль) метансульфохлорида. Перемешивали в течение 30 мин, добавили 25 мл воды и вновь перемешивали в течение 30 мин, отделили органическую фазу и высушили сульфатом магния. Растворитель упаривали в вакууме. Получили 3,8 г (69% от теор.) (+)-8-хлор-6- мезилоксиоктановой кислоты (+)-I (R'=Me), [α]
Пример 6
6,6 г (34 ммоль) (-)-8-хлор-6-гидроксиоктановой кислоты (-)-VI и 7,0 г (68 ммоль) триэтиламина смешали в 140 мл толуола. При охлаждении (10-15oC) медленно прибавляли 6,0 г (52,6 ммоль) метансульфохлорида. Перемешивали в течение 30 мин, добавили 40 мл воды и вновь перемешивали в течение 30 мин, отделили органическую фазу и высушили сульфатом магния. Растворитель упаривали в вакууме. Получили 6,5 г (70% от теор.) (-)-8-хлор-6- мезилоксиоктановой кислоты (-)-I (R'=Me), [α]
Пример 7
4,0 г (19,2 ммоль) метил-(+)-8-хлор-6- гидроксиоктаноата (+)-VIII (R=Me) и 1,97 г (19,2 ммоль) триэтиламина смешали в 90 мл толуола. При охлаждении (10-15oC) медленно прибавляли 2,63 г (23,0 ммоль) метансульфохлорида. Перемешивали в течение 30 мин, добавили 30 мл воды и вновь перемешивали в течение 30 мин, отделили органическую фазу и высушили сульфатом магния. Растворитель упаривали в вакууме. Получили 4,8 г (88% от теор.) метил-(+)-8-хлор-6- мезилоксиоктаноата (+)-II (R=R'=Me), [α]
Пример 8
2,1 г (10 ммоль) метил-(-)-8-хлор-6- гидроксиоктаноата (-)-VIII (R=Me) и 1,0 г (10 ммоль) триэтиламина смешали в 40 мл толуола. При охлаждении (10-15oC) медленно прибавляли 1,4 г (12 ммоль) метансульфохлорида. Перемешивали в течение 30 мин, добавили 25 мл воды и вновь перемешивали в течение 30 мин, отделили органическую фазу и высушили сульфатом магния. Растворитель упаривали в вакууме. Получили 2,5 г (86% от теор.) метил-(-)-8-хлор-6-мезилоксиоктаноата (-)-II (R=R'=Me), [α]
Пример 9
К раствору 2,4 г (11,0 ммоль) метил-(+)-8-хлор-6- гидроксиоктаноата (+)-VIII (R= Me) и 0,04 г (0,5 ммоль) пиридина в 8 мл толуола медленно добавляли раствор 1,6 г (13,5 ммоль) тионилхлорида в 5 мл толуола. Кипятили с обратным холодильником в течение 1 ч. После охлаждения до комнатной температуры добавили к реакционной смеси 20 мл ледяной воды, отделили органическую фазу, промыли 10 мл воды и высушили сульфатом магния. Растворитель упаривали в вакууме. Получили 2,0 г (81% от теор.) метил-(-)-6,8-дихлороктаноата (-)-III (R=Me), [α]
Пример 10
К раствору 2,9 г (13,2 ммоль) метил-(-)-8-хлор-6- гидроксиоктаноата (-)-VIII (R= Me) и 0,05 г (0,6 ммоль) пиридина в 10 мл толуола медленно добавляли раствор 1,9 г (16,2 ммоль) тионилхлорида в 6 мл толуола. Кипятили с обратным холодильником в течение 1 ч. После охлаждения до комнатной температуры добавили к реакционной смеси 25 мл ледяной воды, отделили органическую фазу, промыли 10 мл воды и высушили сульфатом магния. Растворитель упаривали в вакууме. Получили 2,4 г (81% от теор.) метил-(+)-6,8- дихлороктаноата (+)-III (R=Me), [α]
Пример 11
К раствору 2,4 г (12,3 ммоль) (+)-8-хлор-6- гидроксиоктановой кислоты (+)-VI и 0,05 г (0,6 ммоль) пиридина в 30 мл толуола медленно добавляли 3,3 г (27,7 ммоль) тионилхлорида. Кипятили с обратным холодильником в течение 1 ч. После охлаждения до комнатной температуры добавили к реакционной смеси 50 мл ледяной воды, отделили органическую фазу, промыли 20 мл воды и перемешивали 4 ч с 30 мл 2 н. NaOH. Отделили водную фазу, подкислили 3 н. HCl до pH 1, экстрагировали диэтиловым эфиром (2х20 мл). Объединенный экстракт высушили сульфатом магния. Растворитель упаривали в вакууме. Получили 2,1 г (80% от теор.) (-)-6,8- дихлороктановой кислоты (-)-VII, [α]
Пример 12
К раствору 3,0 г (15,4 ммоль) (-)-8-хлор-6-гидроксиоктановой кислоты (-)-VI и 0,06 г (0,8 ммоль) пиридина в 40 мл толуола медленно добавляли 4,1 г (34,4 ммоль) тионилхлорида. Кипятили с обратным холодильником в течение 1 ч. После охлаждения до комнатной температуры добавили к реакционной смеси 60 мл ледяной воды, отделили органическую фазу, промыли 20 мл воды и перемешивали 4 ч с 35 мл 2 н. NaOH. Отделили водную фазу, подкислили 3 н. HCl до pH 1, экстрагировали диэтиловым эфиром (2х20 мл). Объединенный экстракт высушили сульфатом магния. Растворитель упаривали в вакууме. Получили 2,6 г (80% от теор.) (+)-6,8- дихлороктановой кислоты (+)-VII, [α]
Пример 13
Смесь 4,6 г (19 ммоль) нонагидрата сульфида натрия (Na2S•9H2O) и 0,61 г (19 ммоль) серы в 40 мл этанола кипятили с обратным холодильником в течение 15 мин. Спустя 2 ч при температуре 20oC добавили раствор 4,9 г (17 ммоль) метил-(+)-8-хлор-6-мезилоксиоктаноата (+)-II (R=R'=Me) в 5 мл этанола и перемешивали в течение 3 ч. Затем добавили 24 мл 10% NaOH и перемешивали в течение 2 ч при 25oC. После упаривания этанола в вакууме к реакционной смеси в течение 10 мин при 25oC добавляли раствор 0,37 г (9,7 ммоль) натрийборгидрида в 10 мл 1% NaOH, при перемешивании медленно нагревали до 100oC, перемешивали при этой температуре в течение 1 ч. После охлаждения реакционную смесь подкислили конц. HCl до pH 1 и экстрагировали диэтиловым эфиром (2х20 мл). Органическую фазу высушили сульфатом натрия, растворитель упаривали в вакууме. Получили 2,8 г (79% от теор.) (-)-дигидролипоевой кислоты (-)-V, [α]
Пример 14
Смесь 3,1 г (13 ммоль) нонагидрата сульфида натрия и 0,41 г (13 ммоль) серы в 25 мл этанола кипятили с обратным холодильником в течение 15 мин. Спустя 2 ч при температуре 20oC добавили раствор 3,3 г (11 ммоль) метил-(-)-8-хлор-6- мезилоксиоктаноата (-)-II (R=R'=Me) в 5 мл этанола и перемешивали в течение 3 ч. Затем добавили 15 мл 10% NaOH и перемешивали в течение 2 ч при 25oC. После упаривания этанола в вакууме к реакционной смеси в течение 10 мин при 25oC добавляли раствор 0,25 г (6,6 ммоль) натрийборгидрида в 10 мл 1% NaOH, при перемешивании медленно нагревали до 100oC, перемешивали при этой температуре в течение 1 ч. После охлаждения реакционную смесь подкислили конц. HCl до pH 1 и экстрагировали диэтиловым эфиром (2х15 мл). Органическую фазу высушили сульфатом натрия, растворитель упаривали в вакууме. Получили 1,9 г (83% от теор.) (+)- дигидролипоевой кислоты (+)-V, [α]
Пример 15
Смесь 1,5 г (6,3 ммоль) нонагидрата сульфида натрия и 0,2 г (6,3 ммоль) серы в 15 мл этанола кипятили с обратным холодильником в течение 15 мин. Спустя 2 ч при температуре 20oC добавили раствор 1,5 г (5,5 ммоль) (+)-8-хлор-6-мезилоксиоктановой кислоты (+)-I в 4 мл этанола и перемешивали в течение 3 ч. Затем добавили 15 мл 10% NaOH и перемешивали в течение 2 ч при 25oC. После упаривания этанола в вакууме к реакционной смеси в течение 10 мин при 25oC добавляли раствор 0,12 г (3,2 ммоль) натрийборгидрида в 4 мл 1% NaOH, при перемешивании медленно нагревали до 100oC, перемешивали при этой температуре в течение 1 ч. После охлаждения реакционную смесь подкислили конц. HCl до pH 1 и экстрагировали диэтиловым эфиром (2х10 мл). Органическую фазу высушили сульфатом натрия, растворитель упаривали в вакууме. Получили 0,8 г (70% от теор.) (-)-дигидролипоевой кислоты (-)-V, [α]
Пример 16
Смесь 1,8 г (7,6 ммоль) нонагидрата сульфида натрия и 0,24 г (7,6 ммоль) серы в 18 мл этанола кипятили с обратным холодильником в течение 15 мин. Спустя 2 ч при температуре 20oC добавили раствор 1,8 г (6,6 ммоль) (-)-8-хлор-6-мезилоксиоктановой кислоты (-)-I в 5 мл этанола и перемешивали в течение 3 ч. Затем добавили 18 мл 10% NaOH и перемешивали в течение 2 ч при 25oC. После упаривания этанола в вакууме к реакционной смеси в течение 10 мин при 25oC добавляли раствор 0,14 г (3,8 ммоль) натрийборгидрида в 5 мл 1% NaOH, при перемешивании медленно нагрели до 100oC, перемешивали при этой температуре в течение 1 ч. После охлаждения реакционную смесь подкислили конц. HCl до pH 1 и экстрагировали диэтиловым эфиром (2х10 мл). Органическую фазу высушили сульфатом натрия, растворитель упаривали в вакууме. Получили 1,0 г (73% от теор.) (+)-дигидролипоевой кислоты (+)-V, [α]
Пример 17
Смесь 0,62 г (2,6 ммоль) нонагидрата сульфида натрия и 0,08 г (2,6 ммоль) серы в 10 мл этанола кипятили с обратным холодильником в течение 15 мин. Спустя 1 ч при слабом кипении добавили раствор 0,55 г (2,4 ммоль) метил-(+)-6,8- дихлороктаноата (+)-III (R=Me) в 5 мл этанола. Перемешивали в течение 15 мин, отогнали 8 мл этанола. Затем добавили 10 мл 0,5 н. NaOH и перемешивали в течение 12 ч при 25oC. После подкисления реакционной смеси конц. HCl до pH 1 экстрагировали диэтиловым эфиром (2х20 мл), органическую фазу высушили сульфатом натрия, растворитель упаривали в вакууме. После перекристаллизации из циклогексана получили 0,28 г (57% от теор.) R-(+)- α -липоевой кислоты (+)-IV, т.пл. 44-46oC, с учетом ошибки определения: > 99% (ВЭЖХ).
Пример 18
Смесь 0,87 г (3,6 ммоль) нонагидрата сульфида натрия и 0,11 г (3,6 ммоль) серы в 15 мл этанола кипятили с обратным холодильником в течение 15 мин. Спустя 1 ч при слабом кипении добавили раствор 0,77 г (3,4 ммоль) метил-(-)-6,8- дихлороктаноата (-)-III (R=Me) в 7 мл этанола. Перемешивали в течение 15 мин, отогнали 12 мл этанола. Затем добавили 14 мл 0,5 н. NaOH и перемешивали в течение 12 ч при 25oC. После подкисления реакционной смеси конц. HCl до pH 1 экстрагировали диэтиловым эфиром (2х20 мл), органическую фазу высушили сульфатом натрия, растворитель упаривали в вакууме. После перекристаллизации из циклогексана получили 0,38 г (54% от теор.) S-(-)- α-α -липоевой кислоты (-)-IV, т.пл. 44-46oC, с учетом ошибки определения: > 99% (ВЭЖХ).
Настоящее изобретение относится к способу получения 8-хлор-6-сульфонилоксиоктановых кислот формулы I и, соответственно, их алкильных эфиров формулы II, а также 6,8-дихлороктановой кислоты и, соответственно, ее алкильных эфиров формулы III как промежуточных продуктов при синтезе энантиомеров α-липоевой кислоты, а также способы превращения энантиомеров 8-хлор-6-гидроксиоктановой кислоты в энантиомеры α-липоевой кислоты. Способ получения энантиомеров α-липоевой кислоты состоит в том, что 8-хлор-гидроксиоктановую кислоту этерифицируют с образованием алкиловых эфиров, которые затем подвергают реакции с сульфонилхлоридом и третичным азотистым основанием и получают алкиловые эфиры 8-хлор-6-сульфонилоксиоктановой кислоты, которые взаимодействием с дисульфидами щелочных металлов, серой переводят в дигидролипоевую кислоту, окислением которой получают α-липоевую кислоту. Настоящий способ с использованием новых промежуточных продуктов позволяет получить энантиомер α-липоевой кислоты с теоретическим выходом 100%. 15 с. и 3 з.п. ф-лы.
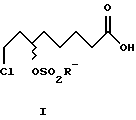
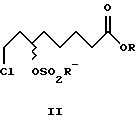
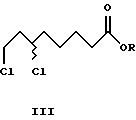
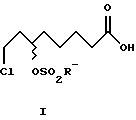
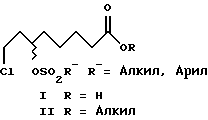
где R' - линейные и разветвленные С1-С4-алкильные группы.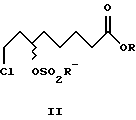
где R и R' обозначают линейные и разветвленные С1-С4-алкильные группы.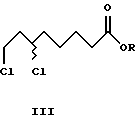
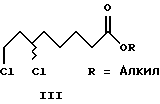
где R - линейные и разветвленные С1-С4-алкильные группы.
| ЕР, 0261336, А2, 30.03.1988 | |||
| 2-Полиоксиалкил-1,3-дитиан-4-валерьяновые кислоты или их натриевые соли, как исходные соединения для синтеза оптически активных липоевых кислот | 1976 |
|
SU667556A1 |
| ЕР, 0586987, А1, 16.03.1994 | |||
| US, 2792406, А, 14.05.1957. | |||

