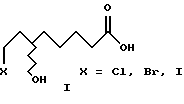
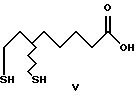
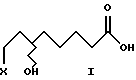
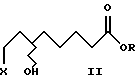
Изобретение относится к новым (+) и (-)- 8-галоген-6-гидроксиоктановым кислотам (галоген = хлор, бром и иод) формулы I, их алкильным эфирам формулы II и их солям с оптически активным α-метилбензиламином формулы III, которые в свою очередь используются в качестве промежуточных продуктов для получения α-липоевых кислот энантиомерной чистоты формулы IV, а также дигидролипоевых кислот энантиомерной чистоты формулы V. α-Липоевая кислота - 3-(карбоксибутил)-1,2-дитиолан (тиоктовая кислота).



Предшествующий уровень техники
R-энантиомер α-липоевой кислоты - природное вещество, присутствующее в незначительном количестве практически во всех тканях животных и растений. В качестве кофермента α-липоевая кислота принимает участие в реакции окислительного декарбоксилирования α-кето-кислот (например, пировиноградной кислоты). α-Липоевая кислота представляет интерес для фармакологии, поскольку оказывает противовоспалительное, обезболивающее и цитопротективное действие. Важным медицинским показанием является лечение α-липоевой кислотой диабетического полиневрита. Согласно последним данным (СА 116: 207360) α- липоевая кислота может оказаться полезной при лечении заболеваний, вызванных вирусами иммунодефицита HIV-1 и HTLV IIIB.
Что касается свойств оптически чистых изомеров α-липоевой кислоты (R- и S-энантиомеров, т. е. R-α-липоевой и S-α-липоевой кислот), то, в отличие от рацемата, R-энантиомер оказывает преимущественно противовоспалительное, а S-энантиомер - в основном обезболивающее действие (ЕР 0427247, 08.11.90). Вследствие этого синтез чистых энантиомеров имеет большое практическое значение.
Известные способы получения α-липоевой кислоты энантиомерной чистоты включают расщепление рацемата конечного продукта или его предшественников, асимметрические синтезы с использованием хиральных соединений и реагентов, энантиоспецифические синтезы на основе природных оптически активных предшественников, а также микробиологический синтез (обзор: J.S.Yadav et al., J. Sci. Ind.Res. 1990, 49, 400; а также: А.Г.Толстиков и др., Биоорг.хим. 1990, 16, 1670: L. Dasaradhi et al., J.Chem.Soc., Chem. Commun. 1990, 729; A.S. Gopalan et al., J.Chem.Perkin Trans. 1 1990, 1897: A.S.Gopalan et al., Tetrahedron Lett. 1989, 5705; EP 0487986 A2, 14.11.91).
Из перечисленных методов наиболее практичным до настоящего времени остается метод расщепления рацематов, основанный на образовании диастереомерных солей α-липоевой кислоты с оптически активным α-метилбензиламином (DE-OS 4137773.7, 16.11.91). К недостаткам метода относится то обстоятельство, что расщепление рацематов осуществляется лишь на последней стадии синтеза, когда побочный энантиомер уже невозможно рацемизовать или инвертировать. В других известных способах, когда расщепление рацематов проводят на предварительных этапах синтеза, в каждом случае лишь один энантиомер можно использовать для получения целевого продукта α- липоевой кислоты энантиомерной чистоты), и, следовательно, теоретически выход может достигать лишь 50% (E. Waltonet al., J. Am. Chem. Soc. 1955, 77, 5144; D.S. Acker und W.J. Wayne, J. Am. Chem. Soc. , 1957, 79, 6483; Л.Г. Чеботарева, А.М. Юркевич, Хим.-Фарм. Ж., 1980, 14, 92).
Сущность изобретения
Целью изобретения является синтез промежуточных продуктов для получения α-липоевой и дигидролипоевой кислот энантиомерной чистоты, на основе которых можно получить требуемый энантиомер α-липоевой кислоты или дигидролипоевой кислоты с выходом 100% от теории.
Исходные соединения для синтеза промежуточных продуктов - рацемические 8-хлор-6-гидроксиоктановую и 8-бром-6- гидроксиоктановую кислоты формулы I (X= Cl,Br) получают известным способом, гидролизуя их рацемические алкильные эфиры формулы II (X= Cl, Br) (Y.Deguchi und K.Nakanishi, Yakugaku Zasshi 1963, 83, 701). Рацемическую 8-иод-6-гидроксиоктановую кислоту формулы I (X= I) получают с высоким выходом путем обработки рацемической 8- хлор-6-гидроксиоктановой кислоты формулы I (X=Cl) йодистым натрием в ацетоне.
Для получения солей формулы III оптически чистых изомеров 8-галоген-6-гидроксиоктановых кислот с оптически чистыми изомерами α-метилбензиламина изомеры растворяют в подходящем растворителе при высоких температурах, например, от 30oC до 100oC, предпочтительно от 40oC до 60oC, и при 10oC - 30oC, предпочтительно при 20oC и с помощью кристаллизации выделяют чистые диастереомерные соли. В качестве растворителя наряду с водой используются алифатические углеводороды с длиной углеводородной цепи от 3 до 10 атомов углерода, жидкие ароматические углеводороды, эфиры алифатических и циклоалифатических карбоновых кислот с 2-6 атомами углерода и алифатических и циклоалифатических спиртов с 2-6 атомами углерода, алифатические и циклоалифатические спирты с 1-6 атомами углерода, простые эфиры и эфиры гликоля или гомогенные смеси всех перечисленных растворителей. Наиболее предпочтительными растворителями являются этиловый эфир уксусной кислоты, циклогексан, толуол и их гомогенные смеси.
Следует заметить, что диастереомерные соли указанных соединений проявляют значительное различие в растворимости, которое можно использовать при обработке рацемата 8-галоген-6- гидроксиоктановых кислот формулы I оптически чистым изомером α-метилбензиламина для селективного выделения одной диастереомерной соли формулы III. Причем для селективного получения определенного изомера необходимо добавить к раствору рацемата 8-галоген-6- гидроксиоктановой кислоты 0,3-0,8 мол-экв, предпочтительно 0,5-0,6 мол-экв чистого энантиомера α-метилбензиламина. Повысить содержание энантиомера соответствующей 8-галоген-6-гидроксиоктановой кислоты, находящегося в данный момент в маточном растворе в избытке, можно при добавлении другого энантиомера α-метилбензиламина. Этот метод применим для непрерывного способа получения как (+)-8-галоген-6-гидроксиоктановых кислот, так и (-)- 8-галоген-6-гидроксиоктановых кислот, причем оба чистых энантиомера могут быть получены с большим выходом без потерь. Для дополнительной очистки этих диастереомерных солей используют перекристаллизацию из чистых вышеперечисленных растворителей или их гомогенных смесей.
Разложение чистых солей формулы III (+)-8- галоген-6-гидроксиоктановых кислот и R-(+)-α-метилбензиламина и, соответственно, (-)-8-галоген-6-гидроксиоктановых кислот и S-(-)-α-метил-бензиламина, полученных вышеперечисленными способами, проводят либо кислотами, например, минеральными кислотами, либо основаниями, например, гидроксидами щелочных металлов, затем путем экстракции выделяют чистые (+)-8-галоген-6-гидроксиоктановые кислоты и (-)-8-галоген-6-гидроксиоктановые кислоты.
Согласно изобретению энантиомеры 8-галоген-6- гидроксиоктановых кислот формулы 1 можно стереоспецифично перевести с сохранением конфигурации в их алкильные эфиры формулы II, предпочтительно метиловые эфиры, в присутствии каталитических количеств HCl. Обработку проводят при температуре от 50oC до 100oC, предпочтительно при 60oC, при этом в качестве растворителя используют соответствующий спирт.
Оптическую чистоту изомеров и диастереомерных солей определяют по величине удельного оптического вращения. Относительное содержание оптических изомеров 8-галоген-6-гидроксиоктановых кислот формулы I и -α-липоевой кислоты формулы IV определяют методом ВЭЖХ на оптически активных сорбентах с точностью определения 0,5%. Кроме того, определение оптической чистоты алкильных эфиров 8-галоген-6- гидроксиоктановой кислоты формулы II проводят с помощью метода 1H-ЯМР, предварительно превратив их обработкой (5)-(+)-O- ацетилминдальной кислотой в смесь диастереомерных эфиров.
Данное изобретение позволяет получить (+)- и (-)-8-галоген-6- гидроксиоктановые кислоты (галоген = хлор, бром и иод) формулы I, их алкиловые эфиры формулы II и их соли с оптически активным α-метилбензиламином формулы III, являющиеся промежуточными продуктами при получении α-липоевых кислот формулы IV энантиомерной чистоты, а также дигидролипоевых кислот формулы V энантиомерной чистоты простым и эффективным способом, с высокими химическим и оптическим выходами.
Ниже приведена схема получения R(+)-α-липоевой кислоты как пример получения -α-липоевой кислоты или дигидролипоевой кислоты энантиомерной чистоты из промежуточных продуктов в соответствии с данным изобретением. Отдельные стадии превращения соответствующих рацематов можно проводить известными способами (см. схему в конце описания).
Аналогичным способом получают S(-)-липоевую кислоту, причем в приведенной схеме слева используют(-)-8-хлор-6- гидроксиоктановую кислоту и справа - (+)-8-хлор-6- гидроксиоктановую кислоту. Оптически чистые дигидролипоевые кислоты получают восстановлением оптически чистых α-липоевых кислот известными способами.
Сведения, подтверждающие возможность осуществления изобретения
Сущность изобретения подтверждается следующими примерами.
Пример 1.
1,94 г (10 ммоль) (+)-8-хлор-6-гидроксиоктановой кислоты (+)-1 (X=Cl) растворили при 60oC в 30 мл смеси этилацетат-циклогексан (1:1). В течение 5 мин добавляли 1,21 г (10 ммоль) R-(+)-α-метилбензиламина. Через 1 час смесь охладили до 20oC. Осадок отфильтровали и промыли смесью этилацетат-циклогексан (1: 1) (2х3 мл). Соль высушили в вакууме при 40oC. Получили 3,12 г (99% от теории) соли (+)-8-хлор-6-гидроксиоктановой кислоты и R-(+)-α-метилбензиламина (+)/(+)-III (X=Cl), [α]
Пример 2.
1,94 г (10 ммоль) (-)-8-хлор-6-гидроксиоктановой кислоты (-)-I (X=Cl) растворили при 60oC в 30 мл смеси этилацетат-циклогексан (1:1). В течение 5 мин добавляли 1,21 г (10 ммоль) В-(+)-α-метилбензиламина. Через 1 час смесь охладили до 20oC. Осадок отфильтровали и промыли смесью этилацетат-циклогексан (1:1) (2х3 мл). Соль высушили в вакууме при 40oC.
Получили 3,05 г (97% от теории) соли (-)-8-хлор-6- гидроксиоктановой кислоты и R-(+)-α-метилбензиламина (-)/(+)-III (X=Cl), [α]
Пример 3.
1,94 г (10 ммоль) (-)-8-хлор-6-гидроксиоктановой кислоты (-)-I (X=Cl) растворили при 60oC в 30 мл смеси этилацетат-циклогексан (1:1). В течение 5 мин добавляли 1,21 г (10 ммоль) S-(-)-α-метилбензиламина. Далее провели обработку, как описано в примере 1. Получили 3,11 г (99% от теории) соли (-)-8-хлор-6-гидроксиоктановой кислоты и S-(-)-7а0-метилбензиламина (-)/(-)-III (X= Cl), [α]
Пример 4.
1,94 г (10 ммоль) (+)-8-хлор-6-гидроксиоктановой кислоты (+)-I (X=Cl) растворили при 60oC в 30 мл смеси этилацетат-циклогексан (1:1). В течение 5 мин добавляли 1,21 г(10 ммоль) S-(-)-α-метилбензиламина. Далее провели обработку, как описано в примере 1.
Получили 3,04 г (97% от теории) соли (+)-8-хлор-6- гидроксиоктановой кислоты и S-(-)-α-метилбензиламином (+)/(-)-III (X=Cl), [α]
Пример 5.
39,9 r (204 ммоль) рацемической 8-хлор-6-гидроксиоктановой кислоты (+)/(-)-I (X= Cl) растворили при 40oC в 155 мл смеси этилацетат-циклогексан (1:1). В течение 10 мин прибавляли порциями 13,5 г (112 ммоль) R-(+)-α-метилбензиламина. Затем через 2 ч охладили реакционную смесь до 20oC, осадок отделили фильтрованием, промыли 20 мл смеси этилацетат-циклогексан (1: 1) и 30 мл циклогексана. Соль дважды перекристаллизовали из 400 мл смеси этилацетат: циклогексан (3:1) и высушили в вакууме при 40oC. Получили 20,5 г (+)-(+)-диастереомерной соли, α = +22,7° (с=1; этанол).
Соль суспендировали при 20oC в 220 мл диэтилового эфира. При охлаждении и перемешивании медленно подкисляли с помощью 3 н. соляной кислоты до pH 1, при этом соль перешла в раствор. Через 30 мин разделили фазы и органическую фазу однократно промыли 20 мл 2 н. соляной кислоты, двукратно - 20 мл воды и высушили сульфатом магния. После отгонки растворителя в вакууме получили 10,8 г (54% от теории) (+)-8-хлор-6-гидроксиоктановой кислоты (+)-I (X=Cl); [α]
Пример 6.
33,9 г (173 ммоль) рацемической 8-хлор-6-гидроксиоктановой кислоты (+)/(-)-I (X= Cl) растворили при 40oC в 130 мл смеси этилацетат-циклогексан (1: 1). В течение 10 мин прибавляли порциями 11,5 г (95 ммоль) S-(-)-α-метилбензиламина. Затем через 2 ч охладили реакционную смесь до 20oC, осадок отделили фильтрованием, промыли 17 мл смеси этилацетат-циклогексан (1: 1) и 25 мл циклогексана. Соль дважды перекристаллизовали из 340 мл смеси этилацетат: циклогексан (3:1) и высушили в вакууме при 40oC. Получили 17,2 г (-)-(-)-диастереомерной соли, α = -22,7° (с=1; этанол).
Соль суспендировали при 20oC в 190 мл диэтилового эфира. При охлаждении и перемешивании медленно подкислили с помощью 3 н. соляной кислоты до pH 1, при этом соль перешла в раствор. Через 30 мин разделили фазы и органическую фазу однократно промыли 17 мл 2 н. соляной кислоты, двукратно - 20 мл воды и высушили сульфатом магния. После отгонки растворителя в вакууме получили 9,1 г (53% от теории) [α]
Пример 7.
6,4 г (32,9 ммоль) (+)-8-хлор-6-гидроксиоктановой кислоты (+)-I (X=Cl) кипятили с обратным холодильником в 100 мл абсолютного метанола в присутствии 0,4 мл конц. соляной кислоты в течение 2 ч. Затем растворитель отогнали в вакууме. Получили 6,6 г (97% от теории) метилового эфира (+)-8-хлор-6-гидроксиоктановой кислоты (+)-II (X=Cl, R=Me), [α]
Пример 8.
7,7 г (39,5 ммоль) (-)-8-хлор-6-гидроксиоктановой кислоты (-)-I (X=Cl) кипятили с обратным холодильником в 120 мл абсолютного метанола в присутствии 0,5 мл конц. соляной кислоты в течение 2 ч. Затем растворитель отогнали в вакууме. Получили 7,9 г (97% от теории) метилового эфира (-)-8-хлор-6-гидроксиоктановой кислоты (-)-II (X=Cl, R=Me), [α]
Пример 9.
9,0 г (46,3 ммоль) рацемической 8-хлор-6- гидроксиоктановой кислоты I (X= Cl) и 10,6 г (70,0 ммоль) йодистого натрия растворили в 100 мл ацетона и кипятили с обратным холодильником в течение 12 ч. Осадок отфильтровали в вакууме, промыли 10 мл ацетона и полученный фильтрат сконцентрировали до 30 мл. Затем добавили 100 мл диэтилового эфира, промыли 10 мл воды и высушили сульфатом натрия. Растворитель отогнали в вакууме. Получили 12,1 г (91% от теории) рацемической 8-иод-6- гидроксиоктановой кислоты I (X=I).
Пример 10.
2,85 г (10 ммоль) (+)-8-иод-6-гидроксиоктановой кислоты (+)-I (X=I) растворили при 60oC в 30 мл смеси этилацетат-циклогексан (1:1). В течение 5 мин добавили 1,21 г (10 ммоль) R-(+)-α-метилбензиламина. Через 1 час смесь охладили до 20oC. Осадок отфильтровали и промыли смесью этилацетат-циклогексан (1:1) 2 раза по 3 мл. Соль высушили в вакууме при 40oC. Получили 4,01 г (99% от теории) соли - (+)-8- иод-6-гидроксиоктановой кислоты и R-(+)-α-метилбензиламина (+)/(+)-III (X=J), [α]
Пример 11.
2,85 г (10 ммоль) (-)-8-иод-6-гидроксиоктановой кислоты (-)-I (Х=I) растворили при 60oC в 30 мл смеси этилацетат-циклогексан (1:1). В течение 5 мин добавили 1,21 г (10 ммоль) R-(+)-α-метилбензиламина. Через 1 час смесь охладили до 20oC. Осадок отфильтровали и промыли смесью этилацетат-циклогексан (1: 1) (2х3 мл.) Соль высушили в вакууме при 40oC. Получили 3,93 г (97% от теории) соли (-)-8-иод-6- гидроксиоктановой кислоты и R-(+) -α- метилбензиламина (-)/(+)-III (X=I), [α]
Пример 12.
2,85 г (10 ммоль) (-)-8-иод-6-гидроксиоктановой кислоты (-)-I (X=I) растворили при 60oC в 30 мл смеси этилацетат-циклогексан (1:1). В течение 5 мин добавили 1,21 г (10 ммоль) S-(-)-α-метилбензиламина. Далее провели обработку, как описано в примере 10. Получили 4,02 г (99% от теории) соли (-)-8-иод-6-гидроксиоктановой кислоты и S-(-)-α-метилбензиламина (-)/(-)-III (X= I), [α]
Пример 13.
2,85 г (10 ммоль) (+)-8-иод-6-гидроксиоктановой кислоты (+)-l (X=I) растворили при 60oC в 30 мл смеси этилацетат-циклогексан (1:1). В течение 5 мин добавили 1,21 г (10 ммоль) S-(-)-α-метилбензиламина. Далее провели обработку, как описано в примере 10. Получили 3,90 г (96% от теории) соли (+)-8-иод-6-гидроксиоктановой кислоты и S-(-)-α-метилбензиламина (+)/(-)-III (X= I), [α]
Пример 14.
8,1 г соли (+)-8-иод-6-гидроксиоктановой кислоты и (R)-(+)-α-метилбензиламина (+)/(+)-III (X= I) суспендировали при 20oC в 90 мл диэтилового эфира. При охлаждении и перемешивании медленно подкислили с помощью 3 н. соляной кислоты до pH 1, при этом соль перешла в раствор. Через 30 мин разделили фазы и органическую фазу однократно промыли 10 мл 2 н. соляной кислоты, двукратно - 10 мл воды и высушили сульфатом магния. После отгонки растворителя в вакууме получили 5,2 г (90% от теории) (+)-8-иод-6- гидроксиоктановой кислоты (+)-I (X=I); [α]
Пример 15.
8,1 г соли (-)-8-иод-6-гидроксиоктановой кислоты и (S)-(-)-α-метилбензиламина (-)/(-)-III (X= J) суспендируют при 20oC в 90 мл диэтилового эфира. При охлаждении и перемешивании медленно подкисляют с помощью 3 н. соляной кислоты до pH 1, при этом соль переходит в раствор. Через 30 мин разделяют фазы и органическую фазу однократно промывают 10 мл 2 н. соляной кислоты, двукратно - 10 мл воды и высушивают сульфатом магния. После отгонки растворителя в вакууме получают 5,1 г (89% от теории) (-)- 8-иод-6-гидроксиоктановой кислоты (-)-I (X=I); [α]
Пример 16.
2,1 г (10 ммоль) метил-(-)-8-хлор-6- гидроксиоктаноата (-)-VIII (R=Me) и 1,0 г (10 ммоль) триэтиламина смешивают в 40 мл толуола. При охлаждении (10-15oC) медленно прибавляют 1,4 г (12 ммоль) метансульфохлорида. Перемешивают в течение 30 мин, добавляют 25 мл воды и вновь перемешивают в течение 30 мин, отделяют органическую фазу и высушивают сульфатом магния. Растворитель упаривают в вакууме. Получают 2,5 г (86% от теор.) метил-(-)-8-хлор-6-мезилоксиоктаноата (-)-II (R=R'=Me), [α]
Пример 17.
Смесь 3,1 г (13 ммоль) нонагидрата сульфида натрия и 0,41 г (13 ммоль) серы в 25 мл этанола кипятят с обратным холодильником в течение 15 мин. Спустя 2 ч при температуре 20oC добавляют раствор 3,3 г (11 ммоль) метил-(-)-8-хлор-6-мезилоксиоктаноата (-)-ll (R=R'= Me) в 5 мл этанола и перемешивают в течение 3 ч. Затем добавляют 15 мл 10% NaOH и перемешивают в течение 2 ч при 25oC. После упаривания этанола в вакууме к реакционной смеси в течение 10 мин при 25oC добавляют раствор 0,25 г (6,6 ммоль) натрийборгидрида в 10 мл 1% NaOH, при перемешивании медленно нагревают до 100oC, перемешивают при этой температуре в течение 1 ч. После охлаждения реакционную смесь подкисляют конц. HCl до pH 1 и экстрагируют диэтиловым эфиром (2х15 мл). Органическую фазу сушат сульфатом натрия, растворитель упаривают в вакууме. Получают 1,9 г (83% от теор.) (+)-дигидролипоевой кислоты (+)-V, [α]
Пример 18.
К раствору 2,9 г (13,2 ммоль) метил-(-)-8-хлор-6- гидроксиоктаноата (-)-VIII (R=Me) и 0,05 г (0,6 ммоль) пиридина в 10 мл толуола медленно добавляют раствор 1,9 г (16,2 ммоль) тионилхлорида в 6 мл толуола. Кипятят с обратным холодильником в течение 1 ч. После охлаждения до комнатной температуры добавляют к реакционной смеси 25 мл ледяной воды, отделяют органическую фазу, промывают 10 мл воды и высушивают сульфатом магния. Растворитель упаривают в вакууме. Получают 2,4 г (81% от теор.) метил-(+)-6,8- дихлороктаноата (+)-III (R=Me), [α]
Пример 19.
Смесь 0,62 г (2,6 ммоль) нонагидрата сульфида натрия и 0,08 г (2,6 ммоль) серы в 10 мл этанола кипятят с обратным холодильником в течение 15 мин. Спустя 1 ч при слабом кипении добавляют раствор 0,55 г (2,4 ммоль) метил-(+)-6,8-дихлороктаноата (+)-III (R= Me) в 5 мл этанола. Перемешивают в течение 15 мин, отгоняют 8 мл этанола. Затем добавляют 10 мл 0,5 н. NaOH и перемешивают в течение 12 ч при 25oC. После подкисления реакционной смеси конц. HCl до pH 1 экстрагируют диэтиловым эфиром (2х20 мл), органическую фазу сушат сульфатом натрия, растворитель упаривают в вакууме. После перекристаллизации из циклогексана получают 0,28 г (57% от теор.) R-(+)-α-липоевой кислоты (+)-IV, т.пл. 44-46oC, с учетом ошибки определения: > 99% (ВЭЖХ).

Изобретение относится к новым (+) или (-)-8-галоген-6-гидроксиоктановым кислотам формулы I, где Х обозначает Cl, Вr, I, их алкильным эфирам формулы II и их солям с α-метилбензиламином формулы III. Способ получения изомеров 8-галоген-6-гидроксиоктановой кислоты формулы I путем разложения чистых диастереомерных солей формулы III путем обработки неорганическими или органическими кислотами или основаниями. Способ получения алкиловых эфиров 8-галоген-6-гидроксиоктановой кислоты формулы II путем перевода 8-галоген-6-гидроксиоктановой кислоты формулы I в соединение формулы II в присутствии каталитических количеств НСl при температуре 50 - 100oС, предпочтительно при 60oС, в качестве растворителя используют соответствующий спирт, причем реакция проходит стереоспецифично с сохранением конфигурации. Способ получения солей формулы III путем обработки рацемической смеси (+)-8-галоген-6-гидроксиоктановой кислоты и (-)-8-галоген-6-гидроксиоктановой кислоты или любой другой смеси изомеров (+) и (-)-8-галоген-6-гидроксиоктановой кислоты энантиомерами α-метилбензиламина в растворе и затем проводят перекристаллизацию диастереомерных соединений. Технический результат - получение новых промежуточных соединений для получения α-липоевой и дигидролипоевой кислот. 7 с. и 3 з.п. ф-лы.


где Х обозначает Cl, Вr, I.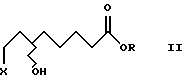
где Х обозначает Сl, Вr, I,
a R - линейные или разветвленные С1-С4-алкильные группы.
выбранные из солей (+)-8-галоген-6-гидроксиоктановой кислоты и R-(+)-α-метилбензиламина, (+)-8-галоген-6-гидроксиоктановой кислоты и S-(-)-α-метилбензиламина, (-)-8-галоген-6-гидроксиоктановой кислоты и R-(+)-α-метилбензиламина, (-)-8-галоген-6-гидроксиоктановой кислоты и S-(-)-α-метилбензиламина, где галоген обозначает хлор, бром или иод.
| Способ получения алифатических @ -оксикарбоновых кислот | 1988 |
|
SU1512964A1 |
| ЕР 0 487 986 А2, 03.06.1992 | |||
| US 5202467 А, 13.04.1993 | |||
| Способ уравновешивания движущихся масс поршневых машин | 1925 |
|
SU427A1 |

