Изобретение относится к области омега-гидрофторалкиловых эфиров, их приготовлению и применению. Кроме того, настоящее изобретение относится к перфторовым (алкоксиалканоидным) кислотам, их производным и их приготовлению. Оно также относится к приготовлению перфторовых (алкоксиалканоидных) кислот путем прямого фторирования их аналогов в виде углеводородных алканоидных кислот или эфиров и приготовлению омега-гидрофторалкиловых эфиров, например, путем декарбоксилирования названных кислот или их алкиловых эфиров. Наконец, настоящее изобретение относится к использованию перфторовых (алкоксиалканоидных) кислот и их производных.
В связи с непрекращающимся потоком неблагоприятных новостей о повреждении озонового слоя, страны-участники Монреальского протокола о веществах, разрушающих озоновый слой, договорились о приближении срока введения запрета на производство промышленными странами хлор- и фторорганических веществ ("CFCs") и других химических веществ, повреждающих озоновый слой - см. Zurer, P. S. , "Looming Ban on Production of CFCs, Halons Spurs Switch to Substitutes", ноябрь 15, 1993, Chemical & Engineering News, 12.
Идет работа по замене хлор- и фторорганических веществ и галонов, таких как CCl2F2, CCl3F, СF3Вr и CCl2FCClF2, заменяющими или альтернативными соединениями и технологиями. Ряд фторуглеводородов ("HFCs"), например, CH2FCF3 ("HFC-134a"), применяется или предлагается в качестве замены CFC (и HFC-134a характеризовался как менее "вредный для озона" - см. патент США 5118494 (Schultz et al. , )). Хлорфторуглеводороды ("HCFCs"), такие как CH3CCl2F ("HCFC-141b), в соответствии с вышеупомянутой статьей в "Chemical & Engineering News", являются заменителями CFC, и хотя они не столь вредные, эти соединения содержат хлор, разрушающий стратосферный озон. Другая предлагаемая замена - это простой омега-гидродифторметил перфторметиловый эфир, CF3OCF2H - см. J. L. Adcock et al. , "Fluorinated Ethers - A New Family of Halons", 1991 CFC Conference Proceedings (1991). Другой гидрофторалкиловый эфир (или гидрид эфира), F[СF(СF3)СF2O] 4СFНСF3, приготовленный путем декарбоксилирования фторированной соли 2-алкоксипропионовой кислоты, испытывался в качестве эмульсии крови - см. Chem. Pharm. Bull. 33, 1221 (1985).
В патенте США 4173654 (Scherer) указывается, что фторуглероды в связи с их инертностью нашли применение в электронике в качестве охладителя или жидкостей для проверки протечек, а другие соединения, хорошо растворяющие кислород, исследовались как искусственные заменители крови. Настоящий патент описывает некоторые фторуглеродные "гибридные" материалы с метаболически активными углеводородными составляющими, такими как, например, помимо других, -СН2-(СН2)m-Н. Согласно патенту США 4686024 (Scherer et al. ), описывающего некоторые перфторциклические соединения эфира, различные перфторсоединения характеризуются в патентах как подходящие переносчики кислорода и двуокиси углерода. А международная заявка, опубликованная как WO 93/11868 (Kaufman et al. ) описывает некоторые хлорфтор-соединения и их эмульсии как соединения, применимые в различных практических целях для транспортировки кислорода, например, в качестве переносчиков кислорода или "искусственной крови".
Имеется ряд других патентов, описывающих различные фторуглеродные эфиры или полиэфиры. Патент США 3342875 (Selman et al. ) описывает некоторые "фторуглеродные эфиры с модифицированным водородом" (или "полиэфиры в водородном колпаке"), приготовленные, среди прочего, путем пиролиза водородсодержащего производного эфира, такого как фторуглеродная эфирная кислота или аммонийная соль, каковой эфир получен путем полимеризации фторуглеродных эпоксидов. Патент Великобритании 1194431 (Montecatini Edison S. P. A. ) описывает некоторые перфторированные эфиры и полиэфировые производные, имеющие общую формулу
СF3-O-(С3F6О)M-(СF2О)N-(СF(СF3)-O)L-СF2Х
где, помимо прочего, каждый индекс М, N и L является нулевым или целым числом от 1 до 99, а Х - это атом водорода или -СООМе, где металл эквивалентен щелочному или щелочноземельному металлу, примером чему является пентафтордиметиловый эфир, СF3-О-СF2Н.
Патент США 3597359 (Smith) описывает вещество, содержащее перфторалкиленовый эфир, представленное формулой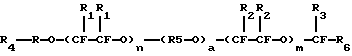
в которой, помимо прочего, R представляет собой алкилен, алкоксиалкилен или перфторалкилен, R1 - фтор или трифторметил, при условии, что не более одного R1 представлено трифторметилом, R2 - фтор или трифторметил, при условии, что не более одного R2 представлено трифторметилом, R3 - фтор или трифторметил, R4 - водород или галоген, при условии, что если R - алкилен или алкоксиалкилен, то R4 - водород, R5 - перфторалкилен по крайней мере с 2 атомами углерода, R6 представляет собой, помимо прочего, водород, трифторметил или перфторэтил, а - ноль или 1, n и m - целые числа от 0 до 50, а n+m составляет от 1 до 50.
Патент США 3962460 (Croix et al. ) описывает алифатические эфиры, включая таковые с формулами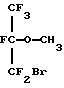
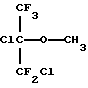
Международная патентная заявка WO 90/01901 (Long) описывает определенные гидриды перфторуглерода, такие как перфтороктилгидрид, применяемые в эмульсиях для переноса кислорода к тканям тела животных. Публикация о европейской патентной заявке 0482938 А1 (Chambers et al. ) описывает фторированные эфиры с формулой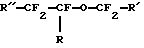
где R представляет собой водород, фтор или алкил, или фторалкил из 1-6 атомов углерода, R' - водород или алкил, или фторалкил из 1-6 атомов углерода и R'' - фтор или алкил, или фторалкил из 1-6 атомов углерода.
Другими патентами, описывающими одну или несколько различных фторалкоксиалканоидных кислот или эфиры, или другие их производные, а также их приготовление являются патенты США N 2713593 (Brice et al. ), 3214478 (Milian, Jr. ), 3393228 (Braun), 4118421 (Martini), 4357282 (Anderson et al. ), 4729856 (Bernonge), 4847427 (Nappa), 4940814 (Schwertfeger), 4973716 (Calini et al. ), 5053536 (Bierschenk et al. ), 5093432 (Bierschenk et al. ) и 5118494 (Schultz et al. ) и международные заявки РСТ N WO 90/03357 (Moore et al. ) и WO 90/06296 (Costello et al. ). Вышеупомянутый патент Brice et al. описывает фторуглеродные кислоты, приготовленные путем электрохимического фторирования, включая кислоту с точкой кипения 225oС, которой соответствует формула n-C8F17OC2F4CO2H. Вышеупомянутые публикации Nappa, Bierschenk et al. , Moore et al. и Costello et al. описывают приготовление фторированных соединений прямым фторированием исходных углеводородов-аналогов.
В одном аспекте данное изобретение обеспечивает получение обычно жидкого (то есть жидкого при определенной температуре и относительном давлении окружающей среды) фторалкил-эфирного соединения или обычно жидкого состава, состоящего или в основном состоящего из выбранной смеси таких соединений, причем указанное соединение имеет насыщенную перфторалифатическую цепь атомов углерода (например, от 4 до 30), прерванную одной или несколькими (например, от 2 до 8) группами эфира или заключенными в цепь (то есть находящимися внутри цепи) атомами кислорода. Атом углерода на одном конце цепи (далее именуемом проксимальным концом) связан с атомом водорода (то есть омега-гидрозаместителем, или первичным атомом водорода) и с двумя атомами фтора, причем указанный проксимальный атом углерода представляет собой атом углерода дифторметильной группы или части, -CF2H, который непосредственно связан с другим атомом углерода цепи, таким как перфторалкиленовый сегмент цепи, -CNF2N, или с указанным кислородом эфира. Атом углерода на другом конце цепи (дистальный конец) представляет собой часть дистальной группы, выбранной из группы, состоящей из дифторметила, дифторхлорометила, -CF2Cl, перфторалкила, замещенного насыщенной алициклической группой, например, с-С6F11-, перфторалкилом с прямой цепью и перфторалкилом с разветвленной цепью. В указанном соединении, где указанный проксимальный конец цепи оканчивается дифторметильной группой, связанной с атомом кислорода в эфире, указанный перфторалкил с прямой цепью имеет по крайней мере 6 атомов углерода цепи, например, от 6 до 16 атомов углерода, а указанный перфторалкил с разветвленной цепью имеет по крайней мере 4 атома углерода, например, от 4 до 16 атомов углерода. Примеры таких омега-гидрофторалкильных соединений следующие:
CF3(CF2)4-O-CF2CF2H
СF3(СF2)5-О-СF2Н
CF3(CF2)7-O-(CF2)5H
CF3(CF2)5-O-(CF2)2-O(CF2)2H
H(CF2)2-O-(CF2)2H
Cl(CF2)4-O-(CF2)4H
Если указанная "выбранная смесь", то есть предопределенная смесь выбранных омега-гидрофторалкилэфирных соединений нужна для определенного применения, указанный состав данного изобретения может быть приготовлен в составе или главным образом в составе смеси из двух или более указанных компонентов, причем каждый из них имеет желаемую отдельную, неслучайную молекулярную массу, причем избираемые составные части предпочтительно те, которые имеют дополнительные свойства, например, для придания повышенной стойкости эмульсиям, там где они включаются в качестве переносчиков кислорода при применении в медицине.
Термин "перфтор", как в случае "перфторалифатический", "перфторалкиленовый" или "перфторалкиловый", означает, что, если не указано иначе, нет атомов водорода, связанных с углеродом, замещаемых фтором, и нет никакого ненасыщения.
Омега-гидрофторалкильные эфиры данного изобретения гидрофобны и менее олеофобны, чем аналоги перфторалкильных эфиров, химически инертны, термостабильны, нерастворимы в воде. Они являются жидкими в нормальных условиях (например, при 20oС) и могут быть приготовлены согласно данному изобретению с большим выходом, высокой чистотой и при большом диапазоне молекулярных масс. Ковалентная связь между омега-водородом и терминальным углеродом, то есть связь С-Н, в целом разрушима при фотоокислении в атмосфере, и, таким образом, омега-гидрофторалкильные эфиры приемлемы в отношении природной среды, то есть совместимы с ней. Омега-гидрофторалкилэфирные соединения или составы жидкие в нормальных условиях, состоящие или в основном состоящие из них, могут применяться в случаях, когда применялись вышеупомянутые CFCs, HCFCs или галоны, например, в качестве растворителей в точной механике или для чистки металла электронных деталей, таких как диски или схемные платы, теплоносители, охладители в компрессорах холодильников или морозильников, или кондиционеров, вспучиватели или регуляторы размера пузырьков в приготовлении пенополиуретановых изоляционных материалов или в химических огнетушителях для струйного применения, полного затопления, подавления и гашения взрывов и в качестве растворителей для высокофторированных полиэфиров, применяемых как смазка в средствах магнитной записи. Другая область применения омега-гидрофторалкильных эфиров - это эмульсии, применяемые в различных областях медицины и для переноса кислорода, например, для искусственной или синтетической крови.
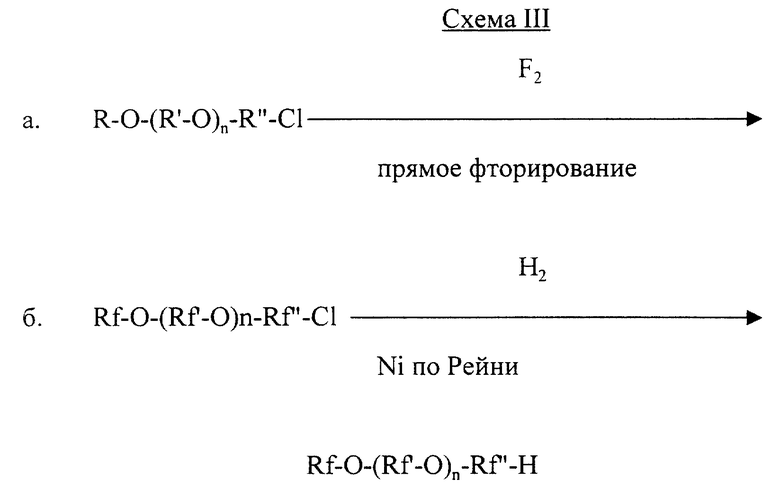
Описанные выше омега-гидрофторалкиловые эфиры данного изобретения могут быть приготовлены путем декарбоксилирования соответствующих исходных фторалкилэфирных карбоновых кислот или их солей, предпочтительно, омылением их алкиловых сложных эфиров. Кроме того, омега-гидрофторалкиловые эфиры могут быть приготовлены восстановлением соответствующих омега-хлорфторалкиловых эфиров (например, описанных в WO 93/11868, см выше). Сами перфторалкилэфирные карбоновые кислоты (и сложные эфиры) - некоторые из них считаются новыми соединениями, а их приготовление представляет другие аспекты данного изобретения - могут быть приготовлены прямым фторированием их соответствующих углеводородных аналогов. Омега-гидрофторалкильные эфиры - в основном чистые фторированные соединения, которые могут применяться в качестве таковых или в форме жидких в нормальных условиях композиций, состоящих или в основном состоящих из выбранной смеси таких соединений. Исходные соединения перфторалкилэфирной карбоновой кислоты и сложные эфиры, как и вышеописанные омега-гидрофторалкильные соединения данного изобретения, имеют насыщенную перфторалифатическую цепь из многих атомов углерода, указанная цепь подобным образом прерывается одним или многими атомами кислорода эфира, проксимальный конец цепи связан с карбоксильной группой или ее алкиловым сложным эфиром. Эта карбоксильная группа (или ее соли, или ее омыляемый алкиловый сложный эфир) может быть декарбоксилирована, как упомянуто выше, и замещена вышеупомянутым омега-гидрозаместителем получаемого омега-гидроалкильного эфира данного изобретения.
Вышеупомянутые новые перфторалкиловые эфирокислоты и сложные эфиры могут быть преобразованы в различные другие производные, такие как их аммонийные соли, которые находят применение в качестве поверхностно-активных агентов для изменения поверхностного натяжения или межфазного натяжения жидкостей. Эти соединения более растворимы в водных средах и в органических растворителях, чем соответствующие производные перфторалканоидной кислоты, что способствует их применению в качестве поверхностно-активных агентов. Удобно готовить эти соединения путем прямого фторирования соответствующих карбоновых эфирных кислот или производных, таких как сложный эфир, с высоким выходом вещества одного молекулярного состава.
Класс жидких в нормальных условиях, омега-гидрофторалкильных эфирных соединений данного изобретения может быть представлен общей формулой:
X-Rf-O-(Rf'-O)n-Rf''-H, I
где Н - первичный атом водорода;
Х - атом фтора, первичный атом водорода, или первичный атом хлора, связанный с дифторметиленом (или Rf);
n - целое число от 0 до 7, предпочтительно от 0 до 3;
Rf, Rf' и Rf'' - те же или другие перфторалкиленовые группы (линейные или разветвленные), например, -CF2CF2-, которые не замещены или замещены перфторорганогруппой, которая может содержать кислород эфира, например, Rf, может быть
-CF2CF(Rf''')CF2- или -Rf'''CF2-
где Rf''' - насыщенная перфторалициклическая группа с 4 до 6 атомами углерода в кольце, такая как перфторциклогексил или перфторциклогексилен;
при условии, что когда Х есть H или Cl, Rf имеет от 1 до 18, предпочтительно от 2 до 18 атомов в цепи углерода, Rf' имеет от 1 до 12, от 2 до 12 атомов в цепи углерода, a Rf'' имеет от 2 до 12 атомов в цепи углерода;
и при дальнейшем условии, что когда Х есть F, то Rf имеет по крайней мере 4, и предпочтительно от 4 до 18 атомов в цепи углерода, Rf' имеет 1 или более, предпочтительно от 1 до 12, а более предпочтительно от 2 до 12 атомов в цепи углерода, a Rf'' имеет 2 или более, и предпочтительно от 2 до 12 атомов в цепи углерода.
Подкласс полиэфирных соединений в пределах общей формулы I представлен общей формулой:
X-Rf-O-(CF2CF2-O)m-Rf''-H, II
где m - целое число от 0 до 7, а Н, Х и Rf'' соответствуют значениям, указанным для формулы I.
Другой подкласс соединений в пределах общей формулы I представлен общей формулой:
F-Rf-O-(Rf'-O)p-Rf''-H, III
где р - целое от 0 до 2 и Н, Rf, Rf' и Rf'' как указано для формулы I, за исключением того, что Rf имеет от 4 до 12 атомов углерода в цепи, Rf'' имеет от 1 до 12 атомов углерода в цепи, a Rf'' имеет от 2 до 12 атомов углерода в цепи.
Другой класс жидких, в нормальных условиях омега-гидрофторалкилэфирных соединений изобретения может быть представлен общей формулой:
X-Rf-O(Rf'-O)nRf''-H,
где Н - первичный атом водорода;
Х - атом фтора, первичный атом водорода или первичный атом хлора;
n - целое число от 0 до 7 и
Rf, Rf' и Rf'' независимо выбраны из группы, состоящей из линейных или разветвленных, незамещенных перфторалкиленовых групп; линейных или разветвленных перфторалкил- или замещенных перфторциклоалкилом перфторалкиленовых групп; и линейных или разветвленных перфторалкиленовых групп, замещенных эфирной кислородсодержащей частью;
при условии, что когда Х есть Н или С1, то Rf имеет от 1 до 18 атомов в цепи углерода и каждый из Rf' и Rf'' независимо имеет от 1 до 12 атомов в цепи углерода;
при дальнейшем условии, что когда Х есть F, то Rf имеет по крайней мере 4 атома в цепи углерода и каждый из Rf' и Rf'' имеет 1 и более атомов в цепи углерода;
и при дальнейшем условии, что когда n равен нулю, то Rf представляет собой замещенную перфторциклоалкилом перфторалкиленовую группу.
Список репрезентативных примеров омега-гидрофторалкиловых эфирных соединений настоящего изобретения следующий:
ТАБЛИЦА А
1. CF3(CF2)5-O-CF2H
2. CF3(CF2)-O-(CF2)2H
3. c-C6F11CF2-O-(CF2)2H
4. CF3(CF2)3-O-CF2C(CF3)2CF2H
5. (CF3)2CFCF2-O-CF2H
6. CF3(CF2)4-O-(CF2)5H
7. CF3(CF2)6-O-CF2H
8. CF3(CF2)5-O-(CF2)2H
9. CF3(CF2)5-O-(CF2)3H
10. CF3(CF2)6-O-(CF2)2H
11. CF3(CF2)7-O-CF2H
12. CF3(CF2)7-O-(CF2)5H
13. CF3(CF2)7-O-(CF2)6H
14. CF3(CF2)5-O-(CF2)2-O-CF2H
15. CF3(CF2)5-O-(CF2)2-O-(CF2)2H
16. H-(CF2)2-O-(CF2)2H
17. H-(CF2)4-O-(CF2)4H
18. H-(CF2)2-O-(CF2)2-O-(CF2)2H
19. H-CF2-O-CF2C(CF3)2CF2-O-CF2H
20. Cl(CF2)4-O-(CF2)4H
21. H(CF2)2OCF2C(CF3)2CF2O(CF2)2H
22. C8F17OCF2OC3F6H
23. (CF3)3COC2F4OCF2OC2F4OCF2H
Как упомянуто выше, омега-гидрофторалкилэфирные соединения или составы по данному изобретению могут быть приготовлены путем декарбоксилирования их соответствующих исходных перфторалкилэфирных карбоновых кислот, гидролизуемых производных карбоновых кислот, или также гидролизуемых исходных веществ (некоторые из которых считаются новыми). Класс таких исходных веществ может быть представлен следующей общей формулой:
Rfp-O-(Rf'-O)n-Rf''-Z, IV
где Rfp - ROC(O)Rf или F-Rf, причем Rf перфторалкиленовая группа соответствует определению для формулы I;
Rf' и Rf'' - также перфторалкиленовые группы по определению для формулы I;
n - число также по определению для формулы I; и
Z'' - одна из групп СO2Н, CO2R, COF, COCl, CONR1R2, или -CF2OC(O)Rf, где R выбран из группы, состоящей из водорода, алкила (такого как низшая алкильная группа с 1 до 6 атомами углерода), циклоалкила, фторалкила и арила, и где R1 и R2 - выбраны независимо из группы, состоящей из водорода, алкила, циклоалкила и циклоалкила с разными атомами в цикле.
При декарбоксилировании соединений формулы IV часть Z' замещается атомом водорода.
Подклассы названных эфирокислот и их производных, которые, кроме их использования в качестве исходных веществ для получения омега-гидроэфирных соединений данного изобретения, находят применение, например, в качестве поверхностно-активных веществ (сурфактантов), как упомянуто выше, и которые считают новыми, могут быть представлены общими формулами V, VI, VII, VIII и IX, приведенными ниже,
Rfo-O-Rfo'-Z, V
где Rfo - перфторалкиленовая группа (линейная или разветвленная), имеющая, например, от 1 до 18 атомов в цепи углерода, а предпочтительно от 1 до 12 атомов;
Rfo' - перфторалкильная группа (линейная или разветвленная), имеющая, например, от 2 до 11 атомов в цепи углерода, причем по крайней мере один из Rfo и Rfo' имеет 8 атомов в цепи углерода; и
Z - СООН, -СООМ1/v, -COONH4, -COOR, -СН2ОН, -COF, -COCl, -CR, -CONRR, -CH2NH2, -CH2NCO, -CN, -CH2OSO2R, -CH2OCOR, -СН2ОСОСR= СН2, -СОNН(СН2)mSi(ОR)3, или -СН2O(СН2)mSi(ОR)3,
где М - радикал аммония или атом металла с валентностью "v" от 1 до 4, такого как Na, К, Ti, или А1, и каждый R является независимо алкилом (например, имеющим от 1 до 14 атомов углерода в цепи) или циклоалкилом, причем эти группы могут быть частично или полностью фторированы, или арилом (например, имеющим от 6 до 10 атомов углерода в кольце), причем любая из этих групп может содержать гетероатом (гетероатомы) и m целое число от 1 до около 11.
Rfq-O-(CF2CF2)aOCF2-Z, VI
где Rfq - перфторалкильная группа (линейная или разветвленная), имеющая приблизительно от 6 до приблизительно 18 атомов углерода в цепи, предпочтительно от 6 до 12 атомов углерода, индекс а - целое число, равное по крайней мере 2, предпочтительно от 3 до 7, но когда а равно 2, тогда Rfq имеет по крайней мере 8 атомов углерода; и
Z - по определению для формулы V.
Rfr-O-CF2-O-Rfr'-Z, VII
где Rfr - перфторакильная группа (линейная или разветвленная), имеющая, например, от 2 до 18 атомов углерода, предпочтительно от 4 до 12 атомов углерода;
Rfr' - перфторалкиленовая группа (линейная или разветвленная), имеющая, например, от 1 до 11 атомов углерода и предпочтительно от 1 до 5 атомов углерода; и
Z - по определению для формулы V; сумма числа атомов углерода в группах Rfr и Rfr' составляет по крайней мере около 7.
Rfs-O-(CF2)b-Z, VIII
где Rfs - перфторалкильная группа (линейная или разветвленная), имеющая, например, от 1 до 18 атомов углерода, предпочтительно от 1 до 12 атомов углерода;
b - целое число, равное по крайней мере 3, предпочтительно от 3 до 11;
Z - по определению для формулы V.
Rft-(O-Rft')c-O-(CF2)d-Z, IX
где Rft - перфторалкильная группа (линейная или разветвленная), имеющая например, от 1 до 18 атомов углерода, предпочтительно от 1 до 12 атомов углерода;
Rft' - перфторалкиленовая группа (линейная или разветвленная), имеющая например, от 1 до 11 атомов углерода, предпочтительно от 2 до 4 атомов углерода;
с - целое число, равное по крайней мере 1, предпочтительно от 1 до 4;
d - целое число от 3 или более, предпочтительно от 3 до 9; и
Z - по определению для формулы V.
Карбоновые кислоты формул от V до IX представляют собой полезные промежуточные продукты для приготовления многих других производных формул V-IX. Эти производные включают нефункциональные или функциональные производные, такие как, например, карбоновые кислоты, соли, сложные эфиры, амиды, нитрилы, спирты, акрилаты и виниловые эфиры. Различные патенты описывают процессы приготовления ряда функциональных производных оксиперфторалкиленовых соединений, то есть перфторполиэфиров, например, см. патенты США N 3250808 (Mitsch et al. ) и 4094911 (Moore et al. ). Эти производные находят ряд применений в качестве поверхностно-активных веществ, такие как эластомеры, покрытия, смазки, вещества, применяемые в приготовлении материалов для жидких кристаллов, таких как указаны в патенте США 5262082 (Janulis et al. ), и для обработки волокнистых субстратов для придания им масло- и водоотталкивающих свойств. Аммонийные соли производных карбоновых кислот, в частности, находят практическое применение как поверхностно-активные вещества.
Соединения карбоновых кислот формулы V тверды в нормальных условиях. Соединения карбоновых кислот формул VI, VII, VIII и IX обычно жидкие в нормальных условиях. Могут быть приготовлены композиции, жидкие в нормальных условиях, которые состоят или в основном состоят из выбранных смесей таких соединений.
Ниже приводится список репрезентативных примеров фторалкилэфирных кислот (или производных), которые могут быть использованы для приготовления омега-гидрофторалкиловых эфиров данного изобретения:
ТАБЛИЦА Б
1. CF3(CF2)7-O-CF2CO2H
2. CF3(CF2)11-O-CF2CO2H
3. CF3(CF2)6-O-C2F4CO2H
4. CF3(CF2)4-O-C2F4CO2H
5. CF3(CF2)5-O-C2F4CO2H
6. CF3(CF2)8-O-C2F4CO2H
7. CF3(CF2)7-O-C2F4CO2H
8. CF3(CF2)9-O-C2F4CO2H
9. CF3(CF2)11-O-C2F4CO2H
10. CF3(CF2)5-OC2F4O-C2F4CO2H
11. C8F17-O-(CF2)5CO2H
12. C10F21-O-(CF2)5CO2H
13. CF3-O-(CF2)7CO2H
14. C2F5-O-(CF2)7CO2H
15. C3F7-O-(CF2)7CO2H
16. CF3-O-(CF2)9CO2H
17. CF3-O-(CF2)10CO2H
18. CF3(CF2)5-O-C2F4-O-C2F4-O-C2F4-O-CF2CO2H
19. CF3(CF2)7-O-C2F4-O-C2F4-O-C2F4-O-CF2CO2H
20. CF3(CF2)9-O-C2F4-O-C2F4-O-C2F4-O-CF2CO2H
21. CF3(CF2)11-O-C2F4-O-C2F4-O-C2F4-O-CF2CO2H
22. CF3(CF2)11-(OC2F4)1-5-O-CF2CO2H из ацетата Brijtm30
23. C6F13OCF20(CF2)5CO2H
24. CF3(CF2)7-O-CF2CF2CO2H
25. CF3(CF2)7-O-CF2-C3F6CO2H
26. (CF3)3COC2F4OCF2OC2F4CO2H
27. C4F9-O-(CF2)3CO2H
28. C5F11-O-(CF2)3CO2H
29. C6F13-O-(CF2)3CO2H
30. C5F11-O-(CF2)4CO2H
31. CF3-O-(CF2)5CO2H
32. C4F9-O-(CF2)5CO2H
33. C5F11-O-(CF2)5CO2H
34. C4F9-O-C4F8-O(CF2)3CO2H
35. C6F13-O-C4F8-O(CF2)3CO2H
36. C4F9-O-C2F40-C2F40(CF2)3CO2H
37. CF3-O-(C2F40)3-(CF2)3CO2H
38. C8F17OCF2OC5F10CO2H
39. (CF3)3COC2F4OCF2OC2F4OCF2CO2H
40. (CF3)2CFCF2CF2O(CF2)5CO2H
41. CF3(CF2)7OC2F4OC2F4OCF2CO2H
42. CF3(CF2)11OC2F4OC2F4OCF2CO2H
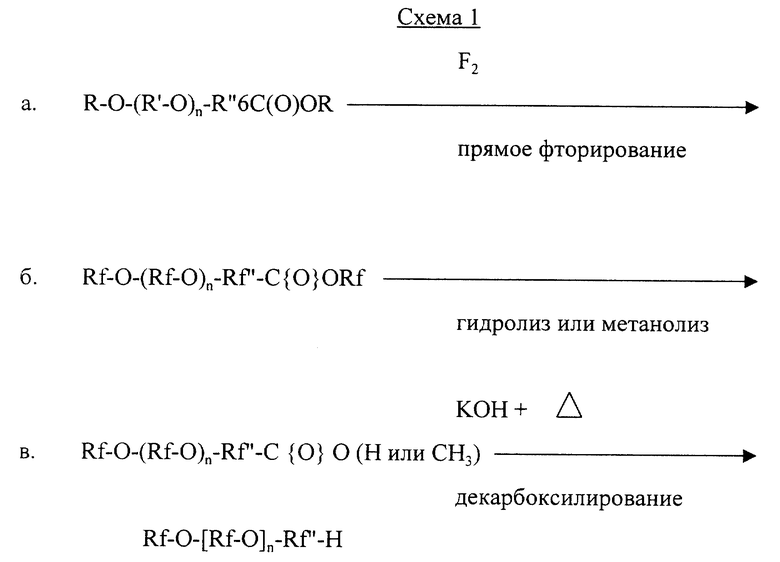
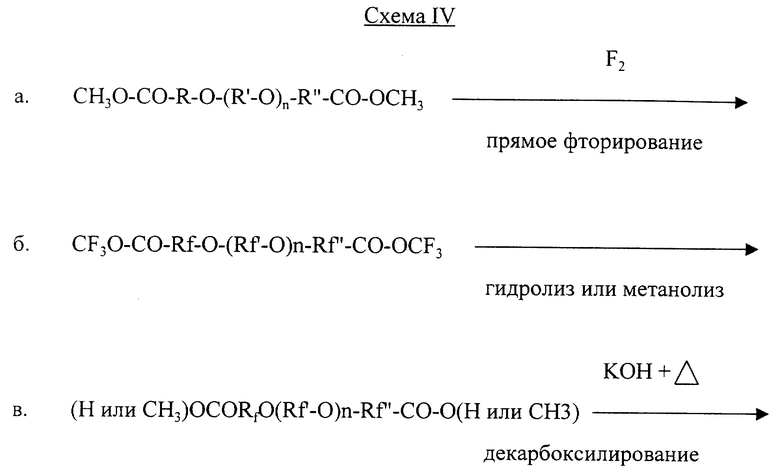
В конце текста приводятся общие схемы реакций, которые могут быть применены для приготовления омега-гидрофторалкилэфиров данного изобретения с применением общих формул приведенных выше. В данных схемах показанная реакция дает продукт, формула которого изображена на схеме I.
Альфа и омега - дигидриды эфира, в случае когда Х в формуле I есть Н, могут быть приготовлены по аналогичным схемам. Например, Схема IV аналогична Схеме I.
На Схеме I в процессе прямого фторирования, операция "а", фторируемый исходный сложный эфир эфирной карбоновой кислоты, например, С4Н9-O-(СН2)5СООСН3, непосредственно фторируется путем контакта с газообразным фтором. (Термин "фторируемый" означает, что исходное вещество содержит атомы водорода в связи с углеродом, которые могут замещаться фтором, и что исходное вещество может содержать ненасыщение, которое может быть насыщено фтором). Получаемое фторированное соединение в виде сложного эфира эфирной кислоты, изображенное на этапе операции "б", может быть приготовлено с существенно тем же числом и тем же пространственным расположением атомов углерода и кислорода, как у его исходного вещества. Если желательна композиция фторированной эфирокислоты, которая состоит или в основном состоит из выбранной смеси фторированных эфирных соединений, то фторированию может быть подвергнута выбранная смесь соответствующих исходных соединений, или, альтернативно, выбранные исходные соединения могут фторироваться раздельно и затем быть смешаны.
Прямое фторирование фторируемого эфирного исходного вещества может быть выполнено при температурах, обычно применяемых при прямом фторировании, например, умеренных, или температурах, близких к температуре окружающей среды, таких как от -20oС до +50oС, с применением стехиометрического избытка газообразного фтора, который предпочтительно разбавлять инертным газом, таким как азот, для минимизации или устранения опасностей, которые представляет чистый газообразный фтор, и для уменьшения избытка тепла, вырабатываемого при контакте исходного вещества с фтором. Фторирование предпочтительно проводить в бескислородной и безводной среде и может проводиться в присутствии твердого, измельченного поглотителя, например, фторида натрия, для побочно продуцируемого фтороводорода. Можно применять прямое фторирование в жидкой фазе с применением в качестве реакционной среды инертной жидкости, такой как жидкий фторуглерод или хлорфторуглерод. При желании можно применить и поглотитель, и реакционную среду из инертной жидкости. Предпочтительно проводить фторирование путем прямого фторирования в жидкой фазе в отсутствии поглотителя фтороводорода, применяя температуру и скорость инертного газа, достаточные для улетучивания побочно продуцируемого фтороводорода и удаления его из зоны фторирования по мере его образования.
В другом аспекте данное изобретение обеспечивает получение фторхимической композиции, содержащей фторированную эфирную кислоту или ее производные, описанные выше, в качестве единственного существенного компонента фторхимической композиции.
Хотя прямое фторирование представляет собой метод замещения, включающий замещение атомов водорода фтором, прямое фторирование дает высокие выходы и более чистые продукты по сравнению с другими методами замещения, такими как электрохимическое фторирование и методы с трифторидом кобальта см. , например, патент США 5093432 (Bierschenk et al. ). Чистота композиций из перфторированной эфирокислоты (или сложного эфира) данного изобретения далее повышается применением изолированных исходных соединений или их выбранных (а не случайных) смесей.
Предпочитаемый метод фторирования - это "методика прямого фторирования в жидкой фазе", предусматривающая приготовление очень разбавленной дисперсии, или, предпочтительно, раствора исходного вещества (веществ) в жидких реакционных средах, которые относительно инертны к фтору при применяемых температурах фторирования, причем концентрация фторируемого стартового материала, таким образом, относительно низка и облегчает управление температурой реакции. Реакционная смесь может также содержать или иметь в себе поглотитель фтороводорода, такой как фторид натрия, причем весовое отношение поглотитель: исходное вещество может быть, например, от около 0,5: 1 до 7: 1. Реакционная смесь может интенсивно перемешиваться при барботировании через нее газообразного фтора, причем фтор предпочтительно применять в смеси с инертным газом, например, азотом, при концентрации около 5 до 50 объемных процентов, предпочтительнее около 10 до 25 объемных процентов, при поддержании стехиометрического избытка в течение всего фторирования, например, до 15-40% или более, в зависимости от определенного исходного материала и эффективности применяемого оборудования (для перемешивания реактора). Выходы обычно бывают в пределах около 30-77 мол. %, а с опытом можно достигнуть 65 до 85 мол. % перфторированного продукта, применяя этот метод.
Подходящие жидкости в качестве реакционных сред для прямого фторирования в жидкой фазе - это хлорфторуглероды, такие как ФреонTM 11 фтортрихлорометан; хлорфторэфиры; ФлюоринертTM - электронные жидкости FC-75, FC-72 и FC-40; перфторалканы, такие как перфторпентан и перфтордекалин; перфторполиэфиры и перфторацетали. Можно применять смеси таких жидкостей, например, чтобы обеспечить хорошее диспергирование исходного вещества и промежуточных продуктов реакции. Реакционные среды удобно применять при атмосферном давлении. Члены вышеуказанных классов реакционных сред с более низкими молекулярными массами также могут применяться, но тогда нужно применять повышенные давления для поддержания жидкой фазы.
Реакция прямого фторирования в жидкой фазе обычно осуществляется при температуре между около -10oС до +50oС, предпочтительно между около -10oС до 0oС, если применяется поглотитель фтороводорода, а если такой поглотитель не применяется - между около 0oС до 150oС, предпочтительно от около 0oС до 50oС, еще предпочтительнее от около 10oС и до 30oС, температуре достаточной для улетучивания фтороводорода, образующегося в виде побочного продукта, и для удаления побочного продукта из реактора для фторирования по мере его образования, с помощью инертного газа, подаваемого с достаточной скоростью. При этих температурах жидкости, применяемые в качестве реакционных сред, незначительно реагируют с растворенным фтором и в основном инертны. Реакционная среда и другие органические вещества могут присутствовать в какой-то мере в газовых выбросах реактора, и может применяться конденсатор для конденсирования газовой реакционной среды и таких веществ в выбросах, позволяя возвратить конденсат в реактор. Конденсатор может действовать с таким режимом, чтобы минимизировать или предотвратить возврат в реактор побочного продукта в виде фтороводорода (который может отрицательно воздействовать на выход продукта, если допустить, что он оставался бы в реакторе в процессе фторирования). Возврат фтороводорода можно минимизировать или предотвратить путем избирательной конденсации органических материалов при пропускании фтороводорода через конденсатор или путем полной конденсации как фтороводорода, так и органических материалов в отдельной емкости и затем, если нужно, путем разделения фтороводорода в виде верхнего слоя жидкости и возврата нижней части жидкой фазы.
Реакция фторирования в жидкой фазе может проводиться в прерывистом режиме, причем исходное вещество добавляется в жидкость перед фторированием, чтобы обеспечить концентрацию исходного вещества примерно до 10% по весу, затем газ, содержащий фтор, барботируется через жидкость, содержащую исходное вещество. Реакцию также можно провести в полунепрерывном режиме, в котором исходное вещество непрерывно подкачивается или подается иным способом в реактор в чистом виде или в виде разбавленного раствора или дисперсии в подходящей жидкости типа той, что и реакционная среда, например, со скоростью около от 1 до 3 г в час в 400 мл жидкой реакционной смеси, по мере того как барботируется фтор, например, при скорости подачи фтора от приблизительно 40 до 120 мл/минуту и скорости подачи инертного газа около от 150 до 600 мл/минуту. Фторирование также может проводиться в непрерывном режиме, при котором исходное вещество (в чистом виде или в растворенном, или диспергированном в подходящей среде того же типа, что и реакционная среда) непрерывно закачивается или иначе подается в реактор с реакционной средой при подаче газа с фтором, как описано выше, и поток неотреагировавшего фтора, газообразного фтороводорода и инертного газа-носителя непрерывно удаляется из реактора, как и поток жидкости с перфторированным продуктом, неполностью фторированным исходным веществом и инертной жидкой реакционной средой, а затем производятся необходимые разделения для регенерации фторалкилэфирной композиции. При желании неотреагировавший фтор и неполностью фторированное исходное вещество могут быть снова пущены в оборот. Количество инертной реакционной жидкой среды в реакторе можно поддерживать на постоянном уровне путем добавления регенерированной или свежей жидкости.
В связи с чрезвычайно высокой экзотермичностью реакции фторирования обычно применяют охлажденную жидкость или ледяную баню для достижения приемлемых скоростей реакции. Когда реакция завершена, реактор очищают от фтора и удаляют содержимое реактора. Если фторирование выполняется методом фторирования в жидкой фазе в присутствии поглотителя фтороводорода, использованный поглотитель может быть отфильтрован и отделен с помощью декантации от жидкого содержимого реактора и последнее затем разделено путем перегонки на реакционную среду и полуфабрикат. Если фторирование выполнено по методу фторирования в жидкой среде без применения поглотителя, продукт может быть отделен от реакционной смеси с помощью перегонки.
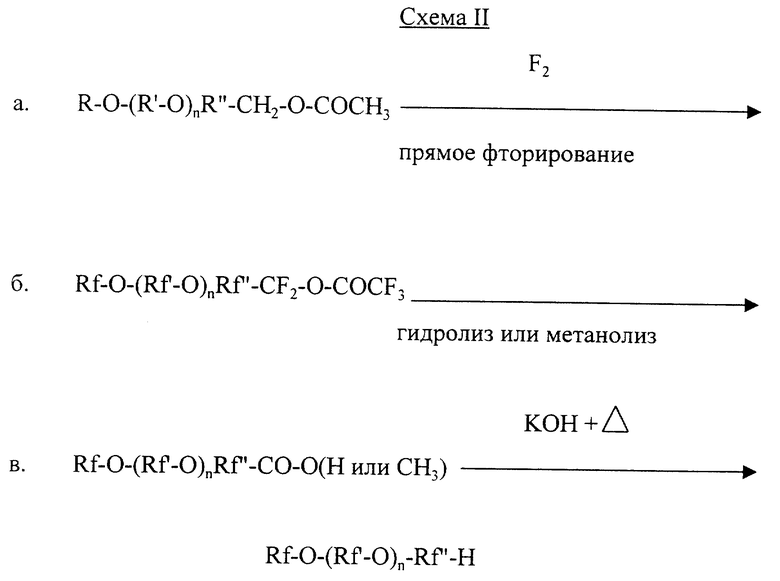
Полезные репрезентативные исходные фторируемые сложные эфиры эфирокислот, которые могут быть применены для приготовления омега-гидрофторалкильных эфиров данного изобретения - это углеводородные дубликаты структур, перечисленных в табл. А, исключая то, что вместо концевого атома водорода структуры сложных эфиров оканчиваются на -Z (где Z - как указано для формулы IV) или на -CН2OC(O)R (как на схеме II) и что исходные вещества могут содержать ненасыщенные части.
Репрезентативные примеры фторэфирокислот данного изобретения или применяемых в нем включают перфторированные (то есть практически со всеми водородами, замещенными фтором) дубликаты исходных фторируемых кислых эфиров, описанные выше. Когда исходные вещества имеют недонасыщение, соответствующие фторированные эфирокислоты насыщены.
Как указано выше, фторэфирокислоты и производные могут применяться в качестве исходных веществ в приготовлении омега-гидрофторалкильных эфиров и они также находят применение, например, в качестве поверхностно-активных веществ.
Вышеописанные фторэфирокислоты или их сложные эфиры, например, алкиловые сложные эфиры, такие как метиловый сложный эфир, могут конвертироваться путем процесса декарбоксилирования в омега-гидрофторалкилэфиры данного изобретения. В одном таком процессе готовят раствор КОН в этиленгликоле и к нему добавляют фторэфирную кислоту или исходный сложный эфир (чистый или в виде раствора в инертном растворителе, таком как перфторированная жидкость), предпочтительно по каплям при помешивании при температуре окружающей среды или комнатной температуре. Полученную смесь затем медленно нагревают, например, до 190oС, за это время отгоняются метанол (от омыления метилового эфира), вода (от нейтрализации кислоты) и декарбоксилированный продукт. Омега-гидрофторалкиловые эфиры данного изобретения удивительно стойки при таких жестких основных условиях. Инертный растворитель, если он применялся, может быть удален, например, при низкой температуре под
вакуумом после нейтрализации. Полученный дистиллят, содержащий продукт (омега-гидрофторалкиловый эфир), может быть промыт водой, высушен силикагелем или сульфатом магния и затем подвергнут перегонке для очистки продукта. Если нужно, продукт (гидрофторалкиловый эфир) может быть подвергнут обратной перегонке с раствором перманганата калия в ацетоне для удаления легко окисляемых примесей. Выходы эфирного продукта обычно высоки, продукт обычно совершенно чист и состоит или в основном состоит из желаемого омега-гидрофторалкилового эфира.
Композиции омега-гидрофторалкилового эфира нетоксичны и могут растворять и транспортировать кислород, поэтому они могут найти применение в качестве заменителей крови, которые могут применяться внутренне при лечении травм, сужении сосудов, в качестве адъювантов при лечении рака облучением или химиотерапии, и в качестве веществ, повышающих контрастность изображения. Для таких применений могут быть приготовлены эмульсии композиций с применением методов, описанных, например, в патентах США N 3911138 (Clark) и 5077036 (Long) и в Международной заявке РСТ, опубликованной как WO 93/11868 (Kaufman et al. ). Композиции омега-гидрофторалкилового эфира могут также применяться в качестве растворителей для чистки и сушки, как описано в патентах США N 5125089 (Flynn et al. ), 3903012 (Brandreth) и 4169807 (Zuber). Небольшие количества альтернативных компонентов, например, поверхностно-активных веществ, могут добавляться к фторэфирным композициям для придания им различных желаемых качеств для разных применений. Эфирные композиции также применяются как теплоносители или охладители в компрессорах холодильников или морозильников или в кондиционерах, как вспучиватели или регуляторы размера пузырьков при изготовлении изоляционных материалов из пенополиуретана, как химическое средство пожаротушения, применяемое как струйное, для полного затопления, для подавления и гашения взрывов, а также как растворитель для высокофторированных полиэфиров, применяемых в качестве смазки при магнитной записи.
При применении композиций омега-гидрофторалкиловых эфиров данного изобретения для сушки и для замещения воды на поверхности изделий, таких как схемные платы, могут применяться процессы сушки и замещения воды, описанные в патенте США N 5125978 (Flynn et al. ). В широком смысле эти процессы заключаются в том, что поверхность изделия покрывают эфирной композицией данного изобретения, предпочтительно в смеси с неионизированным фторалифатически поверхностно-активным агентом. Влажное изделие опускают в жидкую композицию и встряхивают в ней, отделяют замещенную воду от жидкой композиции и вынимают готовое, свободное от воды изделие из жидкой композиции. Дальнейшее описание процесса и изделий, которые могут обрабатываться таким образом, приведено в упомянутом патенте США N 5125978.
Применяя эфирную композицию данного изобретения в качестве теплопереносящей жидкости, можно применить процесс, описанный в патенте США N 5104034 (Hansen). Вкратце, такой процесс включает погружение объекта, подлежащего пайке, в пары, содержащие эфирную композицию данного изобретения, для растворения припоя. При выполнении такого процесса емкость с жидкой эфирной композицией данного изобретения может быть нагрета до кипения в баке для образования насыщенного пара в пространстве между кипящей жидкостью и конденсирующим средством, при этом деталь, подлежащая пайке, погружается в пары, пары конденсируются на поверхности детали, расплавляя и удаляя припой, подлежащая пайке деталь затем вынимается из пространства с парами.
Применяя эфирную композицию данного изобретения как пенообразователь при изготовлении пластмассовой пены, такой как пенополиуретан, могут применяться реагирующие вещества и условия реакции, описанные в патенте США N 5210106 (Dams et al. ). Выполняя этот процесс, органический полиизоцианат и высокомолекулярное вещество по крайней мере с 2 реактивными атомами водорода, такое как полиол, смешивают в присутствии пенообразующей смеси, включающей эфирную композицию данного изобретения, катализатор и поверхностно-активное вещество.
Данное изобретение далее иллюстрируется следующими примерами, причем конкретные материалы и их количества, указанные в этих примерах, как и другие условия и подробности, не должны рассматриваться в качестве ограничения данного изобретения.
Пример 1. Приготовление C8F17-O-C2F4H из С8F17-О-С2F4СО2СН3
Исходный органический материал C8F17-O-C2H4CO2CH3 был приготовлен путем присоединения по Микаэлю n-октанола при основном катализе к акрилонитрилу, с последующим метанолизом при кислотном катализе. Метиловый эфир подвергали прямому фторированию F2 для получения фторированного сложного эфира, С8F17-О-С2F2СО2CF3. Данное фторирование проводили в 2-литровом, изолированном рубашкой, реакционном сосуде из металла МонельTM, с магнитной мешалкой, устройствами для подачи газа, для подачи органического реагента и с обратным холодильником. Газ подавали по трубке диаметром 0,3 см, конец которой находился под нижним уровнем мешалки. Подающая трубка имела диаметр 0,15 см и была связана со шприцевым насосом. Обратный холодильник имел две спиральные концентрические трубки длиной около 6 метров, причем внутренняя трубка имела диаметр 1,27 см, а внешняя - диаметр 2,54 см. Газы из реактора охлаждались во внутренней трубке охладителем (этилен гликоль и вода), протекающим по зазору между двумя трубками. Реактор заряжали примерно 1,8 литрами хлорфторуглеводорода ФреонTM 113 и очищали 650 мл/мин азота в течение 20 минут. Поток газа затем заменяли смесью 310 мл/мин фтора и 1100 мл/мин азота. Через примерно 12 мин, 100 г С8F17-О-С2H4-CO2СН3, разбавленного 260 мл хлорфторуглеводорода ФреонTM 113, подавали в реактор со скоростью 13 мл/ч (5 г в час). В процессе фторирования у содержимого реактора поддерживали температуру 16-18oС. Температура конденсора была около -22oС. Поток фтора поддерживали в течение десяти минут после полного добавления органического материала. Реактор затем очищали азотом в течение одного часа. Раствор полуфабриката перфторированного эфира ФреонTM 113 обрабатывали, используя 150 мл 14% ВF3 в метаноле и интенсивно перемешивали в течение 24 ч. Смесь промывали водой, просушивали над MgSO4 и перегоняли (точка кипения 40oС и 0,2 Торр) и получали C8F17-O-C2F4-CO2CH3 (выход 47%). Для декарбоксилирования растворяли 39 г 85% КОН в примерно 300 мл этиленгликоля и добавляли по каплям вышеописанный фторированный метиловый сложный эфир при помешивании к раствору КОН при комнатной температуре. После всех добавок реакционная смесь имела рН от 8 до 9. Смесь медленно нагревали при помешивании и перегоняли продукт декарбоксилирования, C8F17-O-C2F4H, с метанолом от омыления метилового сложного эфира, водой от КОН и небольшим количеством этиленгликоля. По достижении реакционной смесью температуры 170oС нагревание прекращали. Нижнюю фторхимическую фазу дистиллята отделяли, промывали водой, сушили и перегоняли через трехслойную колонку Снайдера. Главная фракция, кипящая при 146-150oС, дала 122 г продукта. Газовая хроматография и масс-спектрометрия (ГХ/МС) пробы продукта показали, что проба имеет чистоту 94%, и подтвердили, что состав соответствует C8F17-O-C2F2H.
Пример 2. Приготовление C8F17-O-C2F4H из C8F17-O-C2F4CO2H
C8F17-O-C2F4CO2H готовили путем присоединения по Микаэлю при основном катализе n-октанола к акрилонитрилу с последующим метанолизом при кислом катализе. Этот сложный эфир карбоновой кислоты подвергали прямому фторированию по процедуре фторирования в общем соответствующей той, что описана в Примере 1 для получения соответствующей эфирокислоты C8F17-O-C2F4COOH после гидролиза. Дифференциальная сканирующая калориметрия выявила множественные переходы, что характерно для полиморфизма.
Раствор 116 г 85% КОН в 800 мл этиленгликоля готовили в 3-литровой круглодонной колбе. 1000 г C8F17OC2F4-CO2H добавляли по каплям к раствору КОН при помешивании. После полного растворения, добавляли еще 10 г КОН и смесь нагревали. Фторхимический продукт декарбоксилирования перегоняли с небольшим количеством воды от нейтрализации кислоты. Отделяли нижнюю фторхимическую фазу дистиллята, промывали соленой водой, сушили над Na2SO4 и перегоняли как в Примере 1. Выход - 817 г C8F17-O-C2F4H.
Пример 3. Приготовление C7F15-O-C2F4H из C7F15-O-C2F4CO2CH3
C7Н15-O-C2F4CO2CH3 готовили путем присоединения на основе реакции Микаэля при основном катализе n-гептанола к акрилонитрилу, с последующим метанолизом при кислом катализе. 550 г соответствующего метилового сложного эфира, C7F15-O-C2F4COOCH3 (приготовленного практически по тем же процедурам фторирования и метанолиза, как в Примере 1), добавляли по каплям к раствору 166,6 г КОН в примерно 880 мл этиленгликоля. Фторхимический продукт выделяли по принципиально тем же процедурам, что и в Примере 1, с выходом 440 г, которые перегоняли через шестислойную колонку Снайдера и собирали фракцию, кипящую при температуре от 130 до 131oС (340 г). Соединяли эту фракцию с 8,5 г КМnO4 и с приблизительно 350 г ацетона и кипятили с целью обратной перегонки. Через четыре часа добавляли еще 5 г КМnO4 и нагревали полученную смесь еще 3 часа. Смесь фильтровали, осадок на фильтре промывали ацетоном и добавляли к фильтрату воду, причем образовывалась нижняя фторхимическая фаза, которую затем промывали водой, затем концентрированной H2SO4, снова водой и затем профильтровывали через кремнезем. 1Н ЯМР (ядерно-магнитный резонанс) и 19F ЯМР подтвердили, что продукт реакции имеет желаемое строение, С7F15-O-С2F2Н. Газожидкостная хроматография пробы показала чистоту 98,7%.
Пример 4. Приготовление C6F13-O-C2F4-O-CF2H из C6F13-O-C2F4-OCF2CO2CH3
Исходный материал C6Н13-O-C2H4-O-C2H4-O-COCH3 готовили путем ацетилирования гексилоксиэтокси этанола ацетил хлоридом. Ацетат затем превращали в C6F13-О-C2F4-OCF2CO2CH3 путем по существу тех же процедур фторирования и метанолиза, указанных в Примере 1, 548 г этого фторхимического продукта соединяли с 144,2 г КОН в 600 г этиленгликоля. Полученную жидкость нагревали, дистиллировали, а продукт C6F13-O-C2F4-OCF2H изолировали как в Примере 1. Общий выход составил 433 г. Продукт затем вновь перегоняли (точка кипения 131oС) через колонку с перфорированными пластинками длиной 30,5 см при атмосферном давлении. Структура продукта подтверждена 1Н и 19F ЯМР как C6F13-O-C2F4-OCF2H. ГХ/МС показали чистоту пробы 99,6%.
Пример 5. Приготовление C8F17-O-CF2H из C8F17-O-CF2-CO2CH3
C8H17-O-C2H4-O-(CO)CF3 готовили ацетилированием октилоксиэтанола трифторуксусным ангидридом. 100 г трифторацетата подвергали прямому фторированию по принципиально тем же процедурам, что указаны в Примере 1, и полученный продукт фторирования гасили раствором ВF3 в метаноле для получения полуфабриката C8F17-O-CF2-CO2CH3, который далее очищали перегонкой при кипении при температуре 92-97oС и 20 Торр.
Пробу в 58 г названного сложного метилового эфира декарбоксилировали с 10,8 г КОН в этиленгликоле, а продукт C8F17-O-CF2H изолировали так же как в Примере 1. Структура продукта подтверждена 19F ЯМР. Газожидкостная хроматография показала чистоту продукта 99,6%, кипение при температуре 134-136oС.
Пример 6. Приготовление С4F9-О-С2F4Н из С4F9-O-С2F4-СО2СН3
Метиловый сложный эфир С4Н9-O-С2H4-СО2СН3 готовили путем присоединения на основе реакции Микаэля при основном катализе n-бутанола к акрилонитрилу с последующим метанолизом при кислотном катализе. Метиловый сложный эфир затем преобразовывали в соответствующий фторированный метиловый сложный эфир С4F9-O-СF2CF2-СО2СН3, путем по существу тех же процедур фторирования и метанолиза, описанных в Примере 1.
1160 г названного метилового сложного эфира добавляли по каплям при помешивании к 3103 г этиленгликоля и 129,5 г NaOH. Продукт перегоняли (точка кипения 83oС), подвергали воздействию KMnO4 в ацетоне и обрабатывали так же, как описано в Примере 3. Структура очищенного соединения С4F9-O-СF2СF2Н подтверждена 1Н и 19F ЯМР и ГХ/МС.
Пробу данного соединения оценивали на пригодность в прецизионной очистке, измеряя растворимость избранных углеводородных растворителей в данной пробе. Считали, что высокая растворимость указывала бы на лучшее воздействие в качестве очищающего средства по отношению к перфторированным растворителям. Следующие углеводородные растворители оказались растворимыми в количествах до 50 весовых % с гидридом эфира: гексан, гептан, толуен, ацетон, 2-бутанон, 4-метил-2-пентанон, этилацетат, метанол, этанол, изопропанол, диметил формамид, транс-1,2-дихлороэтилен и изопропиловый эфир. о-Ксилол оказался растворимым до 19 весовых %. Хлороформ оказался растворимым до 45 весовых %, этиленгликоль - до менее 15 весовых %, легкое углеводородное масло - до менее 0,05 весовых %.
Проба данного соединения была также оценена в отношении ее применения для высушивания без пятен, как описано в патенте США N 5125978 (Flynn et al. ). Композиция, замещающая воду, была приготовлена путем растворения 0,2 весовых % C4F9OC2F4OCF2C2ОNHCH4ОH в С4F9-O-С2F4Н. Раствор нагревался до 45oС в ультразвуковой бане. Применяя процедуру, описанную в патенте США 5125978, испытуемые отрезки стекла и нержавеющей стали смачивали водой и затем погружали в этот раствор при ультразвуковом перемешивании. Вся вода была замещена за 60 секунд.
Проба этого соединения была также оценена в отношении применения в качестве очищающего агента при очистке с другими растворителями. (Применение для такой очистки указывает, например, Международная патентная публикация WO 92/22678 (Petroferm Inc. ). Органические сложные эфиры, такие как метилдеканоат, нашли применение в качестве сольватирующих агентов при двухфазной очистке с применением перфторгексана как жидкости-носителя и очищающего агента). Метилдеканоат и С4F9ОС2F4Н помещали в отдельные контейнеры и нагревали до 50oС в ультразвуковой бане. Алюминиевую пластинку размером 50 мм • 25 мм • 1,5 мм загрязняли в 0,0831 г легкого углеводородного масла. Загрязненную пластинку сначала погружали в метилдеканоат примерно на 60 секунд, а затем погружали в С4F9OС2F4Н примерно на 60 секунд. C4F9OC2F4H удаляла с пластинки 100% (как определялось по разнице весов) масла и метилдеканоата. При тех же условиях перфторгексан удалял лишь 98,5% масла и метилдеканоат, что указывает на большую эффективность C4F9OC2F4H в качестве жидкости-носителя и очищающего агента, чем перфторгексан.
Пример 7. Приготовление НСF2СF2-О-СF2СF2-О-СF2СF2Н из СН3ОC(O)C2F4-O-С2F4-O-С2F4C(O)OСН3
Исходный материал СН3ОC(O)C2Н4-O-С2Н4-O-С2Н4C(O)OСН3 готовили путем присоединения на основе реакции Микаэля этиленгликоля к акрилонитрилу при основном катализе с последующим метанолизом при кислотном катализе. Исходный материал затем фторировали и метанолизировали по преимущественно тем же процедурам, что описаны в Примере 1, для получения СН3ОC(O)C2Н4-O-С2Н4-O-С2F4C(O)OСН3.
1136 г фторированного сложного эфира добавляли к смеси 305,6 г КОН в 2665 г этиленгликоля. Декарбоксилирование проводили преимущественно так же, как описано в Примере 1, и полуфабрикат перегоняли после разделения фаз, но без промывания водой. Дистиллят все еще содержал метанол, который удаляли промыванием с концентрированной серной кислотой с последующими двумя промывками водой. Получено 695 г желаемого продукта в виде гидрида эфира, с диапазоном кипения при температуре 93-94oС.
Пример 8. Приготовление С4F9-O-(СF2)5Н из C4F9-O-(CF2)5-CO2H
Перемешивали с обратным потоком 118,2 г (1,0 моль) гексан-1,6-диола, 4,4 г четвертичной соли амония АдогенTM 464, 80,0 г (2,0 моля) NaOH и 250 мл тетрагидрофурана. Для лучшего перемешивания прибавляли 80 мл воды. Спустя 20 минут добавляли 137 г (1 моль) бутилбромида в течение 0,5 ч и перемешивали с обратным потоком до следующего утра. Реакционную смесь помещали в 1 л Н2О и соединяли верхний слой с эфирным экстрактом нижнего слоя, высушивали над MgSO4 и отгоняли легкие фракции на роторном испарителе. Обработка полученного освобожденного от легких фракций слоя (151 г) в 100 мл СНС13 150 миллилитрами ацетилхлорида, добавляемого по каплям, и последующее нагревание с обратным холодильником в течение 4 ч и удаление растворителя дало 225,4 г жидкости. Перегонка жидкости дала 176,0 г (кипение при 100-104oС и 0,9 Торр) дистиллята. Газожидкостная хроматография показала, что 56% из нее представляет собой нужный 6-бутоксигексил ацетат вместе с гександиол диацетатом и дибутоксигексаном. 100 г этой смеси фторировали, как указано в Примере 1. В результате обработки полученного фторированного продукта 30 миллилитрами 10% (по весу) раствором H2SO4 в Н2О, встряхивания при комнатной температуре в течение 2 ч, фильтрования твердой фторированной адипиновой кислоты, отделения слоя F-113, сушки над MgSО4 и перегонки получена основная фракция массой 73,4 г, кипение при температуре 116oС и 20 Торр, 96% чистоты С4F9-О-(СF2)5ООН. Последняя была прибавлена к раствору 10,0 (0,25 моля) NaOH и 100 мл этиленгликоля, смесь была нагрета до температуры 120oС, причем примесь C4F9-O-(CF2)6-O-C4F9 от фторирования собирали в ловушку Дина-Старка. После дальнейшего нагревания началось выделение газа, и жидкость C4F9-O-(CF2)5H (44,6 г) собирали в ловушку, закончив процесс при температуре 170oС. Собранную жидкость сушили над силикагелем и перегоняли через колонку Вигре длиной 10,2 см, до получения 38,8 г, точка кипения 131oС. Структура продукта F ЯМР показала, что продукт имеет желаемое строение C4F9-O-(CF2)5H и высокую чистоту.
Пример 9. Приготовление C5F11-O-(CF2)5H из C5F11-O-(CF2)5COOH
Способом, описанным в примере 8, гександиол алкилировали n-пентилбромидом, продукт ацетилировали, сырой ацетат C5F11-O-(CF2)6OC(O)CH3 перегоняли (температура кипения 125oС и 3 Торр), а дистиллят фторировали по существенно той же процедуре фторирования, как описано в Примере 1. Фторированный сложный эфир гидролизовали до соответствующей кислоты. Декарбоксилирование фторированной кислоты C5F11(CF2)5COOH с NaOH обеспечило выход 829 г продукта. Продукт промывали водой, сушили над MgSО4 и перегоняли, получив 555 г С5F11-O-(CF2)5H с кипением при температуре 145-149oС.
Пример 10. Приготовление C8F17-O-(CF2)5H из C8F17-O-(CF2)5СООН
С помощью такой же процедуры, которая описана в Примере 8, гександиол алкилировали с n-октил бромидом, продукт ацетилировали и полученный продукт С8Н17-О-(СН2)6-О-СОСН3 подвергали прямому фторированию и гидролизации, как описано в Примере 8, до C8F17-O-(CF2)5COOH, которая рекристаллизовалась из перфторгексана. Рекристаллизованную кислоту (37,5 г) смешивали с 4,0 г NaOH и 100 г этиленгликоля и нагревали до 185oС. Продукт промывали водой, а остаток 27,9 г перегоняли, получая чистый C8F17-O-(CF2)5H при микроточке кипения 195oС.
Пример 11. Приготовление С4F9-O-СF2С(СF3)2СF2Н из С4F9-O-СF2С(СF3)2CF2Cl
Проводили алкилирование преимущественно так же, как описано в Примере 8, 2,2-диметил-1,3-пропандиола n-бутилбромидом с получением моноалкилированного полуфабриката, который обрабатывали SOCl2 с получением С4Н9-O-СН2С(СН3)2СН2С1, кипение при температуре 80-90oС и 20-30 Торр. Это соединение затем фторировали, как описано в Примере 1, для получения С4F9-O-СF2С(СF3)2СF2С1. 20,0 г хлорида последнего вещества смешивали с 5,3 г смоченного водой Ni по Рейни и 50 мл метанола, насыщенного NH3. Смесь встряхивали на гидрогенизационном аппарате Парра в течение 3 дней при температуре около 25oС, причем большая часть падения давления водорода величиной 21 кПа (3 psig) происходила в первый день. Отфильтровывали продукт, гасили его водой, получая 7,9 г при некоторой механической потере. 19F ЯМР подтвердила, что продукт представляет собой С4F9-O-СF2С(СF3)2СF2H. Масштабный переход к 100 г дал 47 г (перегонка при точке кипения 135oС).
Пример 12. Приготовление H(CF2)4-O-(CF2)4H из Cl(CF2)4-O-(CF2)4Cl
Фторировали Сl-(СН2)4-O-(СН2)4-Сl, как указано в Примере 1, для получения Cl(CF2)4-O-(CF2)4Cl. Смесь 30,3 г Cl(CF2)4-O-(CF2)4Cl, 11,3 г свежего смоченного водой Ni по Рейни и 200 г метанола очищали в течение нескольких минут с использованием NН3 и подвергали давлению 310 кПа водорода в гидрогенизационном аппарате Парра при температуре около 25oС. Через 17 ч давление снижали до 255 кПа, смесь становилась кислой, замечали травление стекла. Добавляли еще аммиака и продолжали восстановление, снижая давление еще на 62 кПа. Отфильтровывали продукт реакции, гасили в воде, получая 15,4 г продукта нижней фазы, газожидкостная хроматография подтвердила, что H(CF2)4-O-(CF2)4H составлял 68% чистого продукта. Перегонка дала 27,0 г, кипение при температуре 121-124oС, чистота 87%.
Пример 13. Приготовление H(CF2)4-O-(CF2)4H и Cl(CF2)4-O-(CF2)4H из Cl(CF2)4-O-(CF2)4Cl
Смесь 50,0 г Cl(CF2)4-O-(CF2)4Cl и 30 г Zn в бутаноле перемешивали при температуре 110oС в течение 2 дней. Газожидкостная хроматография полученного продукта реакции показала частичное превращение. Добавляли еще 21 г Zn и нагревали смесь в течение еще одного дня. Отфильтровывание и гашение получаемого материала в воде дало 27,0 г бесцветной жидкости. Продукт оказался на 35% Н(СF2)4-O-(СF2)4Н, на 42% моногидридом и на 16% невосстановленным дихлоридом.
Пример 14. Приготовление C6F13-O-CF2CF2H из C6F13-O-C2F4CO2HY
Исходный материал С6Н13-О-С2Н4-СО2СН3 готовили путем присоединения на основе реакции Микаэля гексанола к акрилонитрилу с последующей эстерификацией метанолом при кислом катализе. Полученный сложный эфир затем фторировали и гидролизовали с получением С6F13-O-С2F4СO2Н.
Медленно добавляли 500 г кислоты C6F13-O-C2F4CO2H к раствору 68,7 г КОН в 700 г этиленгликоля. По окончании добавления прибавляли еще 5 г КОН к гомогенному раствору, чтобы довести рН до 9. Проводили декарбоксилирование, как показано в Примере 1, затем перегоняли с получением 327 г продукта (кипение при температуре 104-107oС). Продукт обрабатывали перманганатом калия преимущественно так же, как описано в Примере 3. Газожидкостная хроматография, масс-спектрометрия, 19F ЯМР, 1Н ЯМР и ИК подтвердили структуру продукта как C6F13-O-CF2CF2H.
Пример 15. Приготовление C6F13-O-CF2H из C6F13-O-CF2CO2CH3
Исходный материал C6H13-O-C2H4OC(O)CH3, приготовленный ацетилированием этиленгликоль-моногексилового эфира, подвергали фторированию и декарбоксилированию с помощью существенно тех же процедур, которые указаны в Примере 1, с получением 146 г C6F13-O-CF2H (кипение при температуре 92-96oС).
Пример 16. Приготовление СF3СF(СF3)СF2-O-СF2 из СF3СF(СF3)СF2-O-СF2СO2СН3
Исходный материал СН3СН(СН3)СН2-O-СН2СН2-ОC(O)CН3 готовили ацетилированием этиленгликоль-моноизобутилового эфира и конверсией с помощью по существу тех же процедур фторирования и метанолиза, которые указаны в Примере 1, чтобы получить метиловый сложный эфир СF3СF(СF3)СF2-O-СF2СO2СН3 кипение при температуре 118-120oС.
Быстро по каплям добавляли 149 г метилового сложного эфира к 28,6 г КОН в 700 г этиленгликоля. Проводили декарбоксилирование, чтобы получить, после перегонки фракцию продукта массой 70 г, кипение при температуре 45-47oС, с чистотой 99% по газожидкостной хроматографии. Структура подтверждена газожидкостной хроматографией, масс-спектрометрией, 1Н-ЯМР и 19F-ЯМР как СF3-СF(СF3)СF2-O-СF2Н.
Пример 17. Приготовление C4F9-O-(CF2)4-O-(CF2)3H из С4F9-O-(СF2)4-O-(СF2)3СООСН3
Исходный материал С4Н9-O-С4Н8-O-(СН2)3СН2OСОСН3 подвергали прямому фторированию и метанолизу с помощью по существу тех же процедур, которые указаны в Примере 1, чтобы получить С4F9-O-С4F8-O-(СF2)3СO2СН3. 56 г последнего вещества быстро добавляли к раствору 5,6 г КОН в 250 мл этиленгликоля. Проводили декарбоксилирование и разделяли фазы продукта, промывали один раз рассолом и перегоняли с получением 36,6 г продукта (точка кипения 155-158oС), чистота по газожидкостной хроматографии 100%. Газожидкостная хроматография, масс-спектрометрия, 1Н и 19F-ЯМР подтвердили, что продукт представляет собой С4F9-O-(СF2)4-O-(СF2)3Н.
Пример 18. Приготовление (C2F5)2CFCF2-O-C2F4H из (C2F5)2CFCF2-O-CF2CF2-C(O)OCH3
Исходный материал (С2Н5)2СНСН2-O-СН2СН2C(O)OСН3, приготовленный присоединением на основе реакции Микаэля 2-этилбутанола к акрилонитрилу с последующей эстерификацией с метанолом при кислом катализе, фторировали и метанолизировали с помощью по существу тех же процедур, которые указаны в Примере 1, чтобы получить (С2F5)2СFСF2-O-СF2СF2-C(O)OСН3, точка кипения 159oС, выход прямого фторирования, основанного на исходном материале в виде метилового сложного эфира - 88%.
Декарбоксилирование проводили по существу так же, как указано в Примере 1, продукт перегоняли при температуре 108-110oC с получением 145 г. ИК-анализ подтвердил, что продукт имеет структуру (C2F5)2CFCF2-O-CF2CF2H.
Пример 19. Приготовление с-С6F11СF2-O-С2F4Н из с-С6F11СF2-O-С2F4C(O)OСН3
Исходный материал с-С6Н11СН2-О-С2H4С(O)ОСН3, приготовленный на основе реакции циклогексилметанола с акрилонитрилом с последующей эстерификацией с метанолом при кислом катализе, затем фторировали и метанолизировали с ВF3 в метаноле, используя по существу процедуры, указанные в Примере 1, для получения выхода 65% (основанного на фторировании) с-С6F11СF2-O-С2F4C(O)OСН3. Добавляли 224 г фторированного сложного эфира последнего вещества к раствору 28,2 г 85% КОН и 466 г этиленгликоля при поддержании температуры 60oС. Затем нагревали полученную жидкость до 100oС и доводили ее рН до величины более 7 добавлением 5 г водного 45% (по весу) раствора КОН. Проводили декарбоксилирование путем перегонки полученной жидкости. Отделяли нижнюю фторхимическую фазу полученного дистиллята, промывали равным объемом воды и перегоняли при температуре 123-126oС с получением 155 г продукта (чистота 99,7%). Обрабатывали продукт KMnO4 в ацетоне и получали c-C6F11CF2-O-C2F4H.
Пример 20. Приготовление С4F9-O-С2F4-O-С3F6Н из С4F9-O-С2F4-O-С3F6C(O)ОСН3
Фторировали и метанолизировали С4Н9-O-С2Н4-O-С4Н8ОC(O)CН3 с помощью по существу тех же процедур, которые описаны в Примере 1. Полученный продукт С4F9-O-С2F4-O-С3F6C(O)OСН3 в количестве 419 г быстро добавляли по каплям к смеси 49,4 г КОН в 800 г этиленгликоля. Полученную жидкость затем медленно нагревали до температуры колбы 190oС. В процессе этого нагревания метанол от омыления сложного эфира, воду и C4F9-O-C2F4-O-C3F6H отгоняли от реакционной смеси. Затем к дистилляту добавляли воду, отделяли и перегоняли (кипение при температуре 120-122oС) нижнюю фторхимическую фазу (355 г) с получением 308 г С4F9-O-С2F4-O-С3F6Н (выход 82%).
Пример 21. Приготовление C6F13-O-C4F8-H из C6F13-O-C4F8-CO2CH3
Исходный материал С6Н13-О-С5Н10-ОС(O)СН3 готовили моноалкилированием 1,5-пентандиола с гексилбромидом с последующим ацетилированием ацетилхлоридом. Фторировали и метанолизировали это соединение с помощью по существу той же процедуры, что описана в Примере 1, чтобы получить C6F13-O-C4F8-CO2CH3, точка кипения 100oС при давлении 13 Торр. Этот сложный эфир декарбоксилировали нагреванием раствора 200 г сложного эфира в 250 мл этиленгликоля с 30 г КОН до отгонки гидридного продукта. Эту жидкость промывали водой, сушили над MgSО4 с получением 128 г C6F13-O-C4F8-H чистотой 82%. Далее ее очищали перегонкой, применяя насадочную стеклянную колонку с 12 пластинками, при точке кипения 146oС. Структура подтверждена 19F-ЯМР.
Пример 22. Приготовление C6F13-O-C3F6-H из C6F13-O-C3F6-CO2 -K+
Исходный материал C6F13-O-C4F8-OC(O)CH3 готовили моноалкилированием 1,4-бутандиола с гексилбромидом с последующим ацетилированием уксусным ангидридом. Это соединение фторировали и метанолизировали с использованием по существу той же процедуры, которая указана в Примере 1, чтобы получить C6F13-O-C3F6-CO2CH3. Метиловый сложный эфир подвергали омылению с избытком КОН и затем просушивали в вакуумном шкафу для получения калийной соли. 575 г соли нагревали при помешивании в 250 мл этиленгликоля и полученный гидрид Y изолировали из дистиллята, точка кипения 129oС. Структура подтверждена 19F-ЯМР.
Пример 23. Приготовление C5F11-O-C4F8-H из C5F11-O-C4F8-CO2 -Na+
Исходный материал С5Н11-О-С5Н10-О-С(О)СН3 готовили моноалкилированием 1,5-пентандиола с пентилбромидом с последующим ацетилированием ацетилхлоридом. Это соединение фторировали и метанолизировали по существу по той же процедуре, которая указана в Примере 1, чтобы получить С5F11-О-C4F8-СО2СН3. Метиловый сложный эфир подвергали омылению с избытком NaOH, декарбоксилировали и перегоняли по существу так же, как это описано в Примере 22. Перегонка через насадочную стеклянную колонку с 12 пластинками дала чистую C5F11-O-C4F8-H, точка кипения 125oС. Структура подтверждена 19F-ЯМР.
Пример 24. Приготовление С4F9-O-С3F6-Н из C4F9-O-C3F6-CO2 -Na+
Исходный материал С4Н9-O-С4Н8-ОC(O)CН3 готовили моноалкилированием 1,4-бутандиола бутилбромидом с последующим ацетилированием ацетилхлоридом. Это соединение фторировали и метанолизировали по существу по той же процедуре, которая указана в Примере 1, чтобы получить C4F9-O-C3F6-CO2CH3. Этот метиловый сложный эфир подвергали омылению, декарбоксилировали, отделяли сырой гидрид, как указано в Примере 23, и затем перегоняли далее для получения чистой С4F9-O-С3F6-Н, точка кипения 90oС. Структура подтверждена 19F-ЯМР.
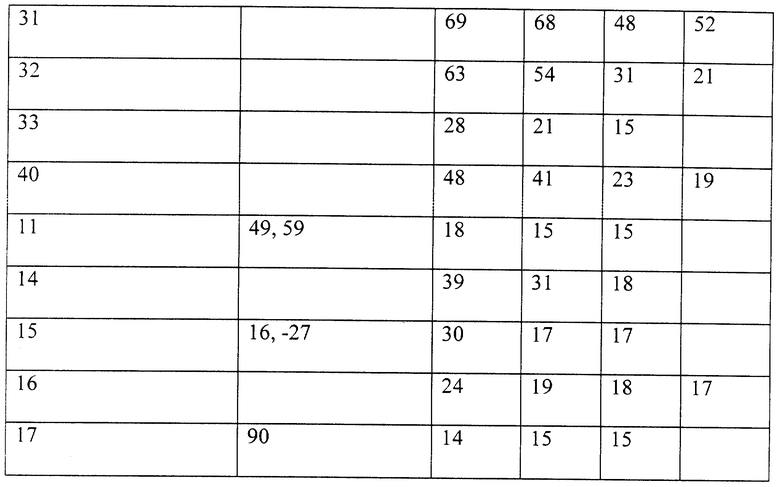
Пример 25. Оценка поверхностной активности перфтороэфирных карбоновых кислот
Поверхностную активность новых перфтороэфирных карбоновых кислот данного изобретения измеряли тензиометром ДеНуой после превращения кислот в соответствующие аммонийные соли. Кислоты готовили прямым фторированием их углеводородных предшественников с последующим гидролизом. Аммонийные соли готовили обработкой кислоты избытком водного раствора аммиака с последующей сушкой вымораживанием. Результаты указываются в динах/см в табл. В для кислот (номера как в табл. Б), соответствующих аммонийным солям.
Пример 26. Оценка эфиров как средств пожаротушения
Омега-гидрофторалкилэфирные соединения как средства пожаротушения оценивали по Стандарту пожарной защиты 2001 Национальной Ассоциации пожарной защиты, с чашечной горелкой, видоизмененной для жидких соединений. Результаты, показанные в табл. Г, свидетельствуют, что эти соединения могут быть эффективными средствами пожаротушения, подавления взрывов и агентами, нейтрализующими горючую атмосферу.
Пример 27. Приготовление пенополиуретана
Выполнили оценку омега-гидрофторалкилэфирных соединений данного изобретения в качестве вспучивателей с применением процедур, указанных в патенте США N 5210106 (Dams et al. ). Компонент А содержал 15,0 весовых частей метилендифенилдиизоцианата PAPITM 27 с изоцианатным эквивалентом 134,0, произведенным компанией Dow Chemical. Компонент Б пены содержал 10,5 весовых частей, полиэфирного полиола ВоранолTM 360 с гидроксильным числом 360, произведенного компанией Dow Chemical, 0,26 весовых частей воды, 0,26 весовых частей олигомерного фторхимического сурфактанта по описанию в Примере 1 патента США N 3787351, а также 0,13 весовых частей N, N-диметилциклогексиламинового катализатора ПоликатTM 8, произведенного компанией Air Products и 1,87 весовых частей C4F9OCF2CF2H как вспучивателя.
Составные части Компонента Б смешивали для получения эмульсии, которая затем смешивалась с Компонентом А и перемешивалась при 2500 оборотах в минуту в течение 10 с. Время до появления пены было около 10 с. Время поднятия и время отверждения "до отлипа" были примерно 2 и 3 мин, соответственно. Полученная полиуретановая пена была жесткой с равномерным распределением очень мелких, замкнутых ячеек.
Пример 28. Приготовление (СF3)3СОС2F4OCF2OCF2СO2СН3
Исходное вещество (t-C4H9OC2H4O)2CH2, приготовленное алкилированием метиленхлорида с t-бутоксиэтанолом, фторировали и метанолизировали по существу так же, как указано в Примере 1, чтобы получить (СF3)3СОС2F4OCF2OCF2СO2СН3,
имеющее диапазон кипения 80-82oС при 18 Торр и структуру, подтвержденную 19F ЯМР.
Пример 29. Приготовление C8F17OCF2OC3F6H из С8F17ОСF2ОС3F6СО2СН3
Исходное вещество C8H17OCH2OC4H8OH готовили моноалкилированием бутандиола с октилхлорометиловым эфиром. Исходное вещество сначала ацетилировали ацетилхлоридом в метиленхлориде, содержащем триэтиламин, затем фторировали, а порцию перфторированного полуфабриката гидролизовали путем обработки водным раствором серной кислоты, затем перегоняли с получением карбоновой кислоты C8F17OCF2OC3F6CO2H с диапазоном кипения 100-106oС при 1,1 Торр. С помощью дифференциальной сканирующей калориметрии определили, что кислота имеет Tg -97,0oС и несколько кристаллических экзотерм -77,4, -61,5 и -37,7oС, и широкую точку плавления при -9,0oС.
Другую порцию перфторированного полуфабриката метанолизировали по существу так же, как указано в Примере 1, чтобы получить C8F17OCF2OC3F6CO2CH3, имеющий пределы кипения 124-130oС при 25 Торр. Полученный метиловый эфир затем декарбоксилировали по процедуре, указанной в Примере 1, чтобы получить C8F17OCF2OC3F6H, имеющий пределы кипения 178-183oС. Структуры этого гидрида и исходного фторированного сложного эфира подтверждены 19F ЯМР.
Пример 30. Приготовление C8F17O-(C2F4O)2CF2CO2H
Исходное вещество готовили моноалкилированием триэтиленгликоля октилбромидом с последующим ацетилированием. Исходное вещество фторировали так же, как указано в Примере 1, гидролизовали обработкой водным раствором серной кислоты и отгоняли продукт C8F17O-(C2F4O)2CF2CO2H, имеющий пределы кипения 105-110oС при 1,4 Торр и точку плавления 24oС.
Пример 31. Приготовление НС3F6ОС3F6Н из СН3О(СО)С3F6ОС3F6СООСН3
Исходный диацетат CH3C(O)OC4H8O-(C4H8O)nC4H8OC(O)CH3 готовили ацетилированием политетраметиленгликоля (со средней молекулярной массой 250) с ацетилхлоридом. Диацетат затем превращали в СН3ОC(O)C3F6О-(С4F8O)nС3F6СООСН3,
используя по существу процедуру фторирования и метанолиза, описанную в Примере 1. 1400 г полученной смеси сложных диэфиров перегоняли с помощью насадочной стеклянной колонки с десятью пластинками и выделяли СН3ОC(O)C3F6ОС3F6СООСН3.
278 г изолированного фторхимического соединения соединяли с 72 г КОН в 250 мл этиленгликоля. Полученную смесь нагревали, перегоняли и выделяли продукт HC3F6OC3F6H, по существу тем же путем, который указан в Примере 1 (точка кипения 84oС). Структура продукта подтверждена 19F ЯМР.
Пример 32. Приготовление n-C12F25OC2F4OC2F4OCF2СO2H
Исходное вещество n-C12H25O(C2H4O)3H готовили моноалкилированием триэтиленгликоля с n-додецилбромидом. После ацетилирования фторировали полученный продукт по существу так же, как описано в Примере 1, концентрировали и обрабатывали фторированный продукт 55,0 г NaOH в 300 мл воды. После нагревания в течение 5 ч на водяной бане продукт подкисляли избытком 50% (по весу) раствора Н2SO4 в воде и затем экстрагировали ФлуоринертомTM FC-75 перфторированную жидкость (смесь C8 перфторсоединений, точка кипения 103oС), нагретую до примерно 60oС на паровой бане. Перегонка дала чистую n-C12F25OC2F4OC2F4OCF2CO2H (Tg = -62,7oС и Тm = 69,2oС по дифференциально сканирующей калориметрии).
Пример 33. Приготовление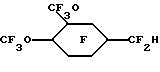
Исходный материал метил 2-(3,4-диметоксифенил)ацетат фторировали по существу так же, как в Примере 1, для получения перфторо-2-(3,4-диметоксициклогексил)уксусной кислоты после гидролиза. Ее затем декарбоксилировали, по существу так же, как описано в Примере 1, до гидрида перфторированного эфира.
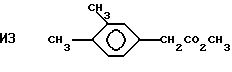
Пример 34. Приготовление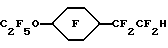
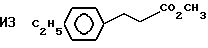
Исходный материал метил 3-(4-этоксифенил)-транс-2-пропеноат готовили конденсацией 4-этоксибензальдегида с малоновой кислотой с последующей эстерификацией. Этот метиловый сложный эфир фторировали, метанолизировали и декарбоксилировали по существу так же, как описано в Примере 1, для получения гидрида перфторированного эфира.
Пример 35. Приготовление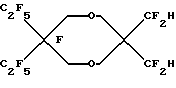
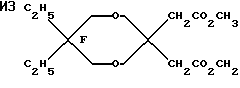
Исходный материал готовили конденсацией 2,2-диэтилпропандиола с диметил-3-оксоглютаратом. Этот диметиловый сложный эфир фторировали, метанолизировали до ди-сложного эфира и декарбоксилировали по существу так же, как описано в Примере 1, для получения дигидрида перфторированного эфира.
Пример 36. Приготовление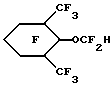
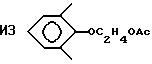
Исходный материал готовили на основе реакции 2,6-диметилфенола с карбонатом этилена с последующим ацетилированием ацетилхлоридом. Этот ацетат фторировали, метанолизировали и декарбоксилировали по существу так же, как описано в Примере 1, для получения гидрида перфторированного эфира (точка кипения 132oС).
Пример 37. Приготовление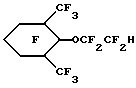
Исходный материал готовили обработкой 2-(2,6-диметилфенилокси) этанола (из Примера 36) тионил хлоридом. Он подвергался фторированию по существу так же, как указано в Примере 1, с последующим восстановлением хлорида Ni по Рейни, по существу так же, как описано в Примере 12, для получения гидрида перфторированного эфира.
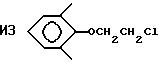
Пример 38. Приготовление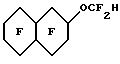
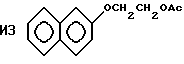
Исходный материал готовили на основе добавления $-нафтола к карбонату этилена с последующим ацетилированием ацетилхлоридом. Этот ацетат фторировали, метанолизировали и декарбоксилировали по существу так же, как описано в Примере 1, для получения гидрида перфторированного эфира (точка кипения 171oС).
Пример 39. Приготовление C5F11OCF2C(CF3)2CF2H из С5Н11ОCH2C(CH3)2CH2Cl
Исходный материал готовили по существу так же, как описано выше в Примере 11. Хлорид эфира фторировали по существу так же как в Примере 1 с последующим восстановлением хлорида Ni по Рейни по существу так же, как описано в Примере 11, для получения гидрида перфторированного эфира (точка кипения 148oС).
Пример 40. Приготовление (C4F9O)2CFCF2H из (C4H9O)2CHCH2Cl
Исходный материал готовили добавлением n-бутанола к 2-хлорацетальдегиду и фторировали по существу так же, как описано в Примере 1, с последующим восстановлением хлорида Ni по Рейни по существу так же, как описано в Примере 11, для получения гидрида перфторированного эфира.
Пример 41. Приготовление CF3O(CF2)9H из СН3О(СН2)10ОАс
Исходный материал готовили моноалкилированием 1,10-декандиола с диметилсульфатом с последующим ацетилированием ацетилхлоридом. Этот ацетат фторировали, гидролизовали и декарбоксилировали по существу как в Примере 1 для получения гидрида перфторированного эфира.
Пример 42. Приготовление C9F19OCF2H из С9Н19ОС2Н4OАс
Исходный материал готовили моноалкилированием этиленгликоля с n-нонилбромидом с последующим ацетилированием ацетилхлоридом. Этот ацетат фторировали, гидролизовали и декарбоксилировали по существу так же, как описано в Примере 1, для получения гидрида перфторированного эфира (точка кипения 155oС).
Пример 43. Приготовление (изо-С3F7)2СFОС2F4Н из (изо-С3Н7)2СНOC2H4CO2CH3
Исходный материал готовили с помощью присоединения на основе реакции Микаэля 2,4-диметил-3-пентанола к акрилонитрилу с последующим метанолизом до метилового сложного эфира. Этот сложный эфир фторировали, гидролизовали и декарбоксилировали по существу так же, как описано в Примере 1, для получения гидрида перфторированного эфира.
Пример 44. Приготовление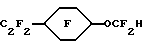
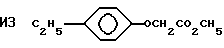
Исходный материал готовили алкилированием 4-этилфенола с метилхлороацетатом. Этот сложный эфир фторировали, гидролизовали и декарбоксилировали по существу так же, как описано в Примере 1, для получения гидрида перфторированного эфира (точка кипения 131oС).
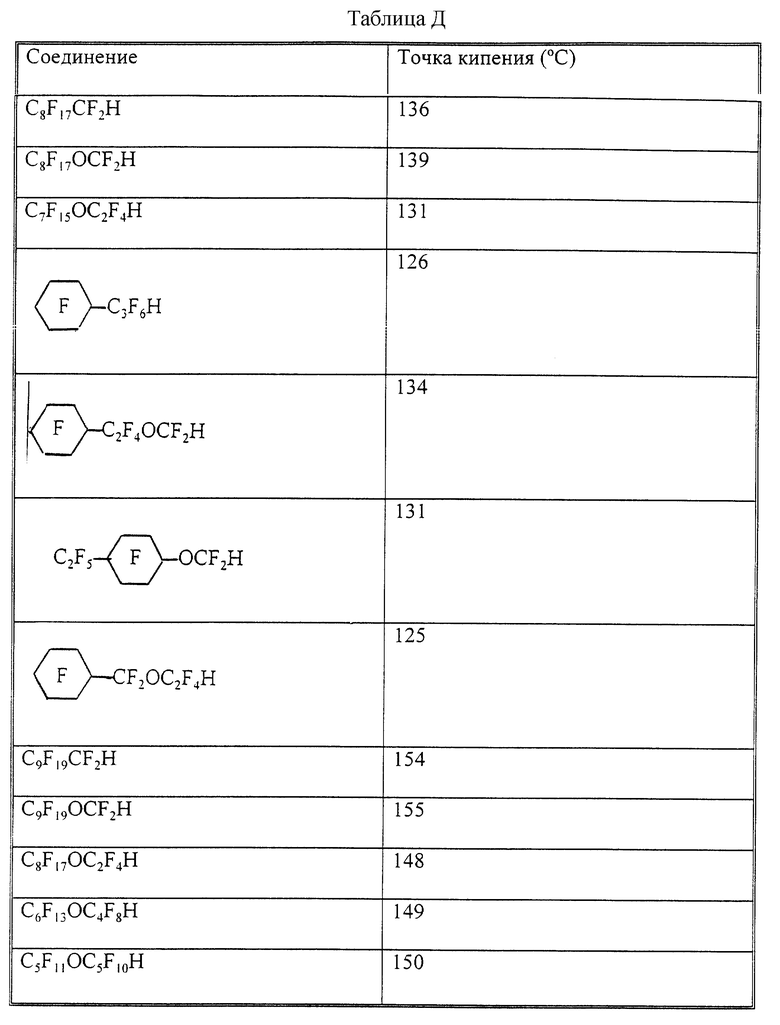
Пример 45. Сравнительная длительность существования в атмосфере и точки кипения.
Время существования в атмосфере различных компонентов пробы рассчитывали по методике, описанной в работе Y. Tang, Atomospheric Fate of Various Fluorocarbons, M. S. Thesis, Massachusetts Institute of Technology (1993). Как показано ниже в таблице, время существования в атмосфере эфирного гидридного соединения с двумя или более атомами углерода между атомом кислорода эфира и терминальным атомом водорода значительно короче, чем время существования в атмосфере эфирных гидридных соединений с одним только атомом углерода между атомом кислорода эфира и терминальным атомом водорода. В связи с более короткой длительностью существования соединений данного изобретения эти соединения более приемлемы в экологическом плане.
Соединение - Время существования в атмосфере (лет)
C6F13OC2F4OCF2H - >170
C4F9OC2F4OCF2H - >170
C8F17OCF2CF2H - 80
Кроме того, как показано в таблице Д, эфирные гидридные соединения, имеющие два или более атомов углерода между атомом кислорода эфира и терминальным атомом водорода, имеют более низкие точки кипения, чем аналогичные неэфирные соединения, и значительно более низкие точки кипения, чем аналогичные эфирные гидридные соединения, имеющие только один атом углерода между атомом кислорода эфира и терминальным атомом водорода. Низкие точки кипения соединения данного изобретения позволяют использовать эти соединения в процессах с участием субстратов, чувствительных к температуре, таких как пластмассы. (Например, при очистке в парах субстрат очищается в конденсирующихся парах кипящей жидкости, а при конденсационном нагревании субстрат нагревается при погружении в кипящую жидкость). При таких применениях предпочтительны жидкости с более низкой точкой кипения, чтобы избежать повреждения субстрата. Поскольку известно, что точки кипения можно понизить, выбирая соединения с меньшим числом атомов углерода, таким способом можно добиться снижения точки кипения на 25oС или более, что однако отрицательно воздействует на другие свойства, такие как растворяющая способность.
Специалистам в данной области очевидны различные модификации и изменения настоящего изобретения, не выходящие за пределы его существа и объема.

Изобретение относится к новым жидким в нормальных условиях омега-гидрофторалкиловым эфирам, которые обладают свойствами поверхностно-активных веществ и могут быть использованы для вытеснения воды с поверхности, в композициях для удаления загрязнителя с изделия, в композициях для пожаротушения, для изготовления пенопласта, при пайке в паровой фазе. Эфиры соответствуют общей формуле I
F-Rf-O(Rf'-O)nRf''-H,
где Н - атом водорода в концевой группе СF3, Rf, Rf' и Rf'' - независимо выбраны из группы, состоящей из линейных незамещенных или замещенных перфторалкилом перфторалкиленовых групп, причем Rf имеет 4-9 атомов углерода в цепи, Rf' имеет 1-6 атомов углерода в цепи, Rf'' имеет 2-6 атомов углерода в цепи, n - целое число от 0 до 7, или при n, равном 0, Rf означает перфторциклоалкилзамещенную перфторалкиленовую группу с 6-10 атомами углерода, в которой один или более атомов углерода в Rf могут быть замещены атомами кислорода, Rf', Rf'' независимо выбраны из группы линейных незамещенных перфторалкиленовых групп, имеющих в цепи 1-4 атома углерода. Способ получения эфиров включает декарбоксилирование соответствующей перфторкарбоновой кислоты или ее сложного эфира путем смешения с раствором неорганического основания в протонном растворителе с последующим нагреванием. Изобретение включает также новые перфторкарбоновые кислоты или их сложные эфиры в качестве исходных соединений для получения эфиров формулы I. Новые эфиры позволяют заменить вещества, способствующие разрушению озонового слоя земли. 9 с. п. ф-лы, 5 табл.
F-Rf-O(R'f-O)nR''f-H,
где Н - атом водорода в концевой группе CF2;
Rf, R'f и R''f - независимо выбраны из группы, состоящей из линейных незамещенных или замещенных перфторалкилом перфторалкиленовых групп, причем Rf имеет 4-9 атомов углерода в цепи, R'f имеет 1-6 атомов углерода в цепи, R''f имеет 2-6 атомов углерода в цепи, n - целое число от 0 до 7 или при n, равном O, Rf означает перфторциклоалкилзамещенную перфторалкиленовую группу с 6-10 атомами углерода, в которой один или более атомов углерода в Rf могут быть замещены атомами кислорода, R'f, R''f независимо выбраны из группы линейных незамещенных перфторалкиленовых групп, имеющих в цепи 1-4 атома углерода.
FRf-O-R''fH,
где Н - атом водорода в концевой группе CF2,
Rf означает перфторциклоалкилзамещенную перфторалкиленовую группу с 6-10 атомами углерода, в которой один или более атомов углерода в Rf могут быть замещены атомами кислорода;
R''f независимо выбраны из группы линейных незамещенных перфторалкиленовых групп, имеющих в цепи 1-4 атома углерода.
Rft-(O-R'ft)c-O-(CF2)d-Z,
где Rft - линейная или разветвленная перфторалкильная группа, имеющая от 1 до 18 атомов углерода;
R'ft - линейная или разветвленная перфторалкиленовая группа, имеющая от 1 до 11 атомов углерода;
с - целое число, равное по крайней мере 1;
d - целое число, равное по крайней мере 3;
Z - выбрано из группы, состоящей из -СООН, -COOM1/v, -COONH4, -COOR, где М - радикал аммония или атом металла с валентностью "v" от 1 до 4, при этом каждый R независимо выбран из группы, состоящей из алкильных групп, имеющих от 1 до 14 атомов углерода, фторалкильных групп, имеющих от 1 до 14 атомов углерода.
Приоритет по пунктам:
20.05.1994 и 12.05.1995 - по пп. 1-8;
20.05.1994 - по п. 9.
| СПОСОБ ИЗГОТОВЛЕНИЯ БИОПОЛЯРНЫХ ЭЛЕКТРОДОВ ХИМИЧЕСКИХ ИСТОЧНИКОВ ТОКАS ; | 0 |
|
SU261501A1 |
| Страховочное устройство | 1981 |
|
SU971369A1 |
| Способ выплавки высокохромистых сплавов и лигатур и шихта для его осуществления | 1980 |
|
SU1038365A1 |
| US 3706773 A, 19.12.1972 | |||
| US 3674800 A, 04.07.1972 | |||
| US 3766274 A, 16.10.1973 | |||
| Пенообразователь для тушения пожаров | 1980 |
|
SU874077A1 |
| SU 1180016 A, 23.04.1993 | |||
| D.D.DENSON ET AL | |||
| "Synthesis of some perfluoroalkylether compounds" Journal of fluorine chemistry, LAUSANNE, CH, 1977, v | |||
| Печь-кухня, могущая работать, как самостоятельно, так и в комбинации с разного рода нагревательными приборами | 1921 |
|
SU10A1 |
| Фальцовая черепица | 0 |
|
SU75A1 |
| RU 2060306 C1, 20.05.1996 | |||
| Т.S.CROFT ET AL "Fluoroalkyl-s-triazines" Journal of heterocyclic chemistry, Provo, US, 1973, v | |||
| Печь-кухня, могущая работать, как самостоятельно, так и в комбинации с разного рода нагревательными приборами | 1921 |
|
SU10A1 |
| Четырехсторонний сигнальный фонарь для городских дорог | 1924 |
|
SU943A1 |
| CHANG-MING HU ET AL "Mechanistic study on the photooxidation of perhalofluoralkyl sulfinates" Tetrahedron Letters, Oxford, GB, 1989, v | |||
| Способ обработки медных солей нафтеновых кислот | 1923 |
|
SU30A1 |
| СВЕРЛО ДЛЯ ОТБИРАНИЯ ПРОБЫ ИЗ ТОРФЯНЫХ КИРПИЧЕЙ | 1925 |
|
SU6717A1 |
| СПОСОБ ДУГОВОЙ СВАРКИ НЕПЛАВЯЩИМСЯ ЭЛЕКТРОДОМ В СРЕДЕ ИНЕРТНЫХ ГАЗОВ | 1991 |
|
RU2027564C1 |
| US 5210106 A, 11.05.1993 | |||
| DE 4221555 A1, 05.01.1994 | |||
| DE 4006491 А1, 05.09.1991 | |||
| СПОСОБ ПОВЫШЕНИЯ ЗАПАСЕННОЙ ЭНЕРГИИ В НАНОПОРОШКАХ МЕТАЛЛОВ | 2011 |
|
RU2461445C1 |
| DE 4231444 A1, 24.03.1994. | |||

