Данное изобретение относится к группе новых соединений ряда пиперазина, имеющих полезные фармакологические свойства.
Было обнаружено, что соединения формулы (а):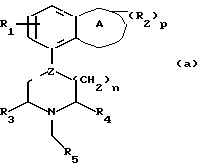
где А представляет гетероциклическую группу с 5-7 атомами в кольце, где присутствуют 1-3 гетероатома из группы О, N и S,
R1 представляет водород или фтор,
R2 представляет С1-4-алкил, С1-4-алкокси или оксогруппу и р равно 0, 1 или 2,
Z представляет углерод или азот, и пунктирная линия представляет одинарную связь, когда Z представляет азот, и одинарную или двойную связь, когда Z представляет углерод,
R3 и R4, независимо, представляют водород или С1-4-алкил,
n имеет величину 1 или 2,
R5 представляет 2-пиридил, 3-пиридил или 4-пиридил, замещенный в мета-положении относительно метиленового мостика группой Y и необязательно замещенный (R6)q,
Y представляет фенил, фуранил или тиенил, причем эти группы могут быть замещены 1-3 заместителями из группы, включающей гидрокси группу, галоген, CF3, С1-4-алкокси, С1-4-алкил, циано, аминокарбонил, моно- или ди-С1-4-алкиламинокарбонил,
R6 представляет галоген, гидрокси, С1-4-алкокси или С1-4алкил и q равно 0, 1, 2 или 3, и их соли, обладающие полезными фармакологическими свойствами.
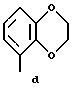
Предпочтительными соединениями по данному изобретению являются соединения формулы (а), где А вместе с фенильной группой представляет группу формулы а, b, с, d, e, f или g: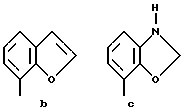
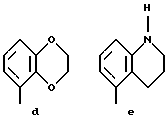
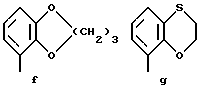
и R1, (R2)p, R3, R4, R5, (R6)q, Y и Z имеют указанные выше значения и n равно 1, и их соли.
Особенно предпочтительными соединениями формулы (а) являются соединения, где А вместе с фенильной группой представляет группу формулы (с) или (d), R5 имеет указанное выше значение, и Y представляет фенил, который может быть замещен, как указано выше, и где R2 имеет указанное выше значение, р = 0 или 1, n равно 1, R3 и R4 представляют водород, R6 представляет гидрокси, метокси или галоген, q равно 0 или 1, Z представляет азот, и их соли.
Определенным предпочтительным соединением является соединение, имеющее формулу (а), где А вместе с фенильной группой представляет группу формулы (d), R1, (R2)p, R3 и R4 представляют водород, n равно 1, Z представляет азот и R5 представляет группу 5-(4-фторфенил)-пирид-3-ил, и его соли.
Из Европейского патента 0650964 известно, что соединения формулы: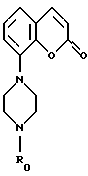
где R0 представляет С1-14-алкил, причем соединения могут быть замещены в фенильной группе и/или гетероциклической группе, и/или пиперазиновой группе, оказывают действие на центральную нервную систему путем связывания с 5-НТ-рецепторами (5-НТ обозначает 5-гидрокситриптамин, или серотонин). Особенно эти соединения связываются с подтипами 5-НТ-рецептора, т.е. 5-НТ1A и 5-НТ1D-рецепторами.
Теперь неожиданно было обнаружено, что соединения по данному изобретению проявляют сродство к D2-рецептору допамина (диапазона рКi 7-9,5) и D4-рецептору допамина (диапазон рКi 6,5-9,5) без значительного предпочтения к одному из указанных выше двух рецепторов. Кроме того, соединения по данному изобретению проявляют сродство к 5-НТ1A-рецепторам серотонина (диапазон рКi 7-9,5). Эту комбинацию сродства к рецепторам допамина и серотонина можно использовать для лечения шизофрении и других психотических нарушений и можно разрешить для более полного лечения всех симптомов таких болезней (например, положительных симптомов, отрицательных симптомов и дефицита познавательной способности).
Соединения проявляют переменную активность в качестве либо частичных агонистов, либо антагонистов в отношении D2-, D3- и -D4-рецепторов допамина. Некоторые соединения проявляют действия, подобные агонисту рецепторов допамина, однако, они оказывают сильное антагонистическое действие на апоморфин-индуцированное карабкающееся поведение мышей (величины ЕD50<1 мг/кг при пероральном введении). Соединения проявляют варьируемую активность в качестве агонистов 5-НТ1A-рецепторов и индуцируют аспекты серотонинового поведенческого синдрома до различных интенсивностей.
Эти соединения активны в терапевтических моделях, чувствительных к клинически уместным антипсихотическим средствам (например, обусловленная реакция избегания; Van der Heyden and Brad ford, Behav. Brain Res., 1988, 31: 61-67), антидепрессантам (например, дифференциальное усиление медленных ответных реакций, van Hest et al., Psychopharmacologe, 1992, 107: 474-479) и анксиолитикам (например, подавление индуцированной стрессом громкой звуковой реакции; van der Poel et al., Psychopharmacology, 1989, 97: 147-148).
В противоположность клинически релевантным антагонистам D2-рецептора допамина описанные здесь соединения имеют низкую склонность к индуцированию каталепсии, у грызунов и как таковые, вероятно, индуцируют более слабые экстрапирамидальные побочные действия, чем существующие антипсихотические средства.
Агонизм к 5-НТ1A-рецептору, присущий этим соединениям, может быть ответственен за пониженную тенденцию к индуцированию экстрапирамидальных действий и терапевтическое действие, наблюдаемое в поведенческих моделях, чувствительных либо к антидепрессантам, либо к анксиолитикам.
Данные соединения, по-видимому, ценны для лечения поражений или болезней центральной нервной системы, вызванных расстройством как в допаминергической, так и в серотининергической системах, например: болезни Паркинсона, агрессии, нарушений, проявляющихся в тревожных состояниях, аутизма, головокружения, депрессии, расстройства познавательной способности или памяти и, особенно, шизофрении и других психотических нарушений.
Подходящими кислотами, с которыми эти соединения могут образовать фармацевтически приемлемые кислотно-аддитивные соли, являются, например, хлористоводородная кислота, серная кислота, фосфорная кислота, азотная кислота, и органические кислоты, такие как лимонная кислота, фумаровая кислота, малеиновая кислота, винная кислота, уксусная кислота, бензойная кислота, п-толуолсульфоновая кислота, метансульфоновая кислота и нафталинсульфоновая кислота.
Соединения изобретения можно превратить в формы для введения посредством обычных способов с использованием вспомогательных веществ, например жидких или твердых материалов-носителей.
Соединения изобретения можно получить по способам (А и В), которые описываются ниже. Пиперазины, используемые в этих способах, обозначаются как от I-Н до III-Н, где I-III представляют следующие группы: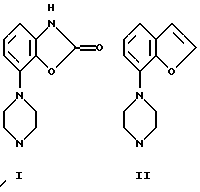
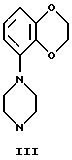
Синтез этих пиперазинов от I-Н до III-Н описывается в Европейском патенте 0189612.
Н-атом части N-Н соединений от 1-Н до III-Н может быть заменен на группу Q двумя различными химическими путями (А и В), в итоге приводящими к соединениям изобретения. Значения групп от Q1 до Q9 следующие:
Группы Q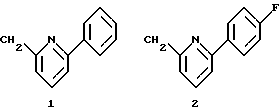
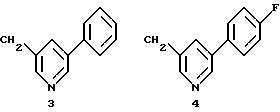
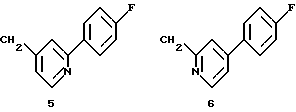
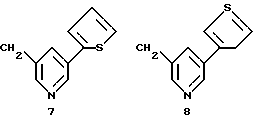
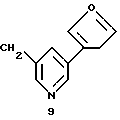
Синтетический путь А
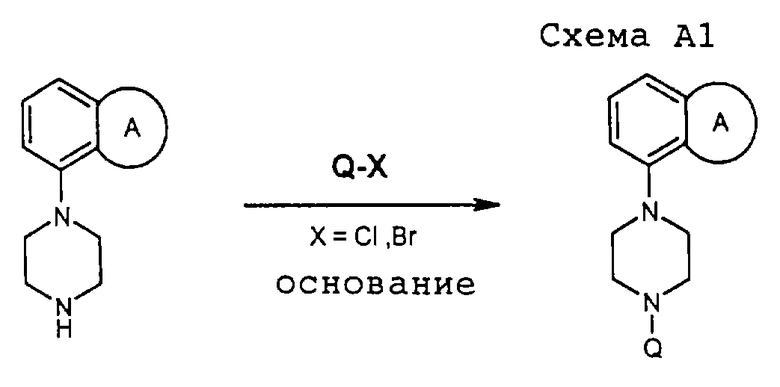
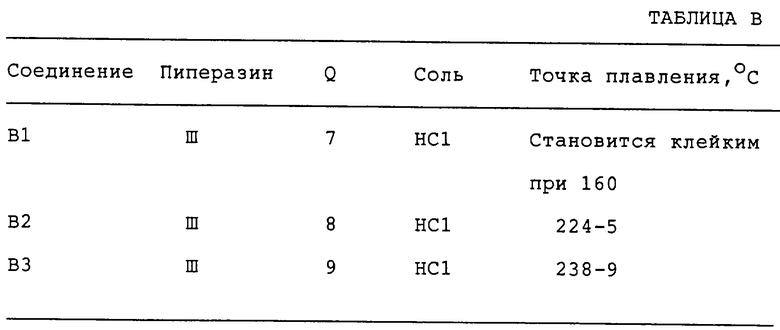
Соединения, перечисленные в таблице А, получали путем синтеза, изображенного на схеме А1: пиперазин подвергали реакции с соединением Q-Х (X = Сl, Вr), например в ацетонитриле с Et (i-Pr)2N, действующим в качестве основания; в некоторых случаях добавляли KI (или NaI). Вместо Еt(i-Pr)2N можно использовать Et3N.
Синтетический путь В
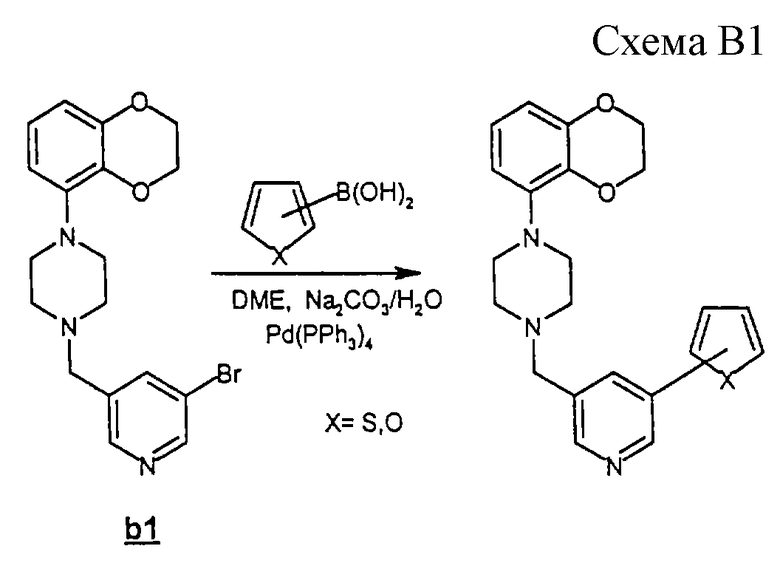
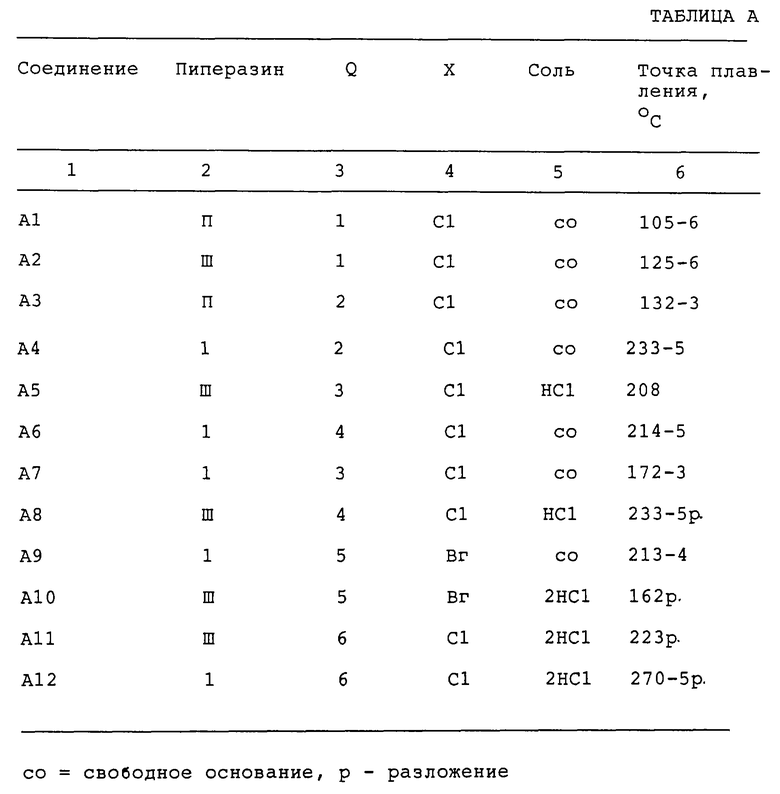
Соединения, перечисленные в таблице В, были получены посредством синтеза, изображенного на схеме В1 (смотри в конце описания); пиперазин подвергали реакции с 3-бром-5-хлорметилпиридином, получая промежуточный продукт b1 (схема В2), который сочетали с производным бороновой кислоты при помощи так называемой реакции перекрестного сочетания Сузуки.
Получение соединений формулы (а) и ряда промежуточных соединений будет теперь описываться подробно в следующих примерах, не ограничивающих, однако, настоящее изобретение.
Пример 1.
Методика А1 (схема А1):
К суспензии моногидрохлорида 1-(2,3-дигидро-1,4-бензодиоксин-5-ил)пиперазина, III-Н•НСl (1,1 г, 4,25 ммоль) в СН3СN (40 мл) добавляли Q4-C1 (1,0 г, 3,87 ммоль) и диизопропилэтиламин (2,45 г, 19 ммоль). Смесь перемешивали при кипячении с обратным холодильником в течение 3 час. После охлаждения и выпаривания растворителя в вакууме остаток растворяли в CH2Cl2, промывали 5% раствором NaHCO3, насыщенным NaCl, сушили (Na2SO4), фильтровали и выпаривали в вакууме. Получаемое темное масло очищали флэш-хроматографией на силикагеле (CH2Cl2/MeOH/NH4OH, 97,25/2,5/0,25), получая А8 (0,9 г, 58%) в виде масла. Продукт превращали в его моногидрохлоридную соль: остаток растворяли в Et2O и обрабатывали 1 экв. НСl в этаноле. Продукт осаждался в виде белого твердого вещества. Твердый А8•НСl собирали фильтрованием и сушили: т.пл. 233-5oС, разложение;
1Н NMR (400 MHz, DMSO/CDCl3, 4/1) δ (ppm) 3.1-3.6 (cluster, 8H), 4.24 (m, 4H), 4.58 (s, 2H), 6.49 (d, 1H, J = 8 Hz), 6.55 (d, 1H, J = 8 Hz), 6.74 (t, 1H, J = 8 Hz), 7.34 (m, 2H), 7.91 (m, 2H), 8.77 (m, 1H), 8.9 (m, 1H), 9.10 (m, 1H), 11.8 (brs, 1H, NH+).
Пример 2.
Методика Al (схема Al):
Суспензию 2-(п-хлорфенил)-4-бромметилпиридина, Q5-Br, (0,71 г, 2,67 ммоль) и 1-(2-бензоксазолинон-4-ил)пиперазин 1-Н•НСl (0,58 г, 2,27 ммоль) в ДМФ (20 мл) вместе с 2,1 эквивалента Et3N перемешивали при комнатной температуре в течение 2 час. Получаемый светлый раствор концентрировали, получая красное масло, которое очищали флэш-хроматографией (SiO2, элюирование CH2Cl2/MeOH/NH4OH, 92/7, 5/0,5), получая А9 (0,28 г, 26%) в виде желтого твердого вещества: т.пл. 213-4oС; 1H-NMR (400 MHz, DMSO/СDСl3, 4/1) δ (ppm) 2.62 (m, 4H), 3.24 (m, 4Н), 3.64 (s, 2H), 6.59 (d, 1H, J = 8 Hz), 6.63 (d, 1H, J = 8 Hz), 7.01 (t, 1H, J = 8 Hz), 7.27 (m, 2H), 7.32 (m, 1H), 7.85 (m, 1H), 8.13 (m, 2H), 8.6 (m, 1H), 11.5 (s, 1H).
Согласно приведенным выше синтезам, другие соединения А1-А12 получали аналогичным образом (см. таблицу A).
Пример 3.
Методика В1 (схема В1):
Раствор b1 (1,07 г, 2,75 ммоль) и Pd (PPh3)4 (0,1 г, 0,08 ммоль) в DME (5 мл) перемешивали при комнатной температуре в течение 10 минут в атмосфере N2. Затем последовательно добавляли 2-тиофенбороновую кислоту (0,39 г, 3,0 ммоль) и водный раствор Na2CO3 (2,75 мл 2 М раствора) и смеси давали реагировать при температуре флегмы в течение 1 часа. Раствор охлаждали, разбавляли Н2О и экстрагировали CH2Cl2. Органическую фазу выпаривали досуха в вакууме, получая сырой продукт В1, который очищали флэш-хроматографией (CH2Cl2/MeOH, 98/2) и затем превращали в моногидрохлоридную соль для получения В1•НСl (0,8 г, 74%) в виде белого твердого материала: т.пл. 160oС, разложение, материал становится клейким;
1H-NMR (400 MHz, СDСl3) δ (ppm) 3.0-3.8 (brb, 8Н, NH+, Н2О), 4.25 (m, 4Н), 4.63 (brs, 2H), 6.54 (d, 1H, J = 8 Hz), 6.64 (d, 1H, J = 8 Hz), 6.75 (t, 1H, J = 8 Hz), 7.14 (m, 1Н), 7.43 (d, 1H, J = 5 Hz), 7.74 (m, 1H).
Согласно приведенным выше синтезам, другие соединения В2 В3 получали аналогичным образом (см. таблицу B).
ПРОМЕЖУТОЧНЫЕ ПРОДУКТЫ, ИСПОЛЬЗОВАННЫЕ В ПУТИ А
Промежуточные продукты Q-X
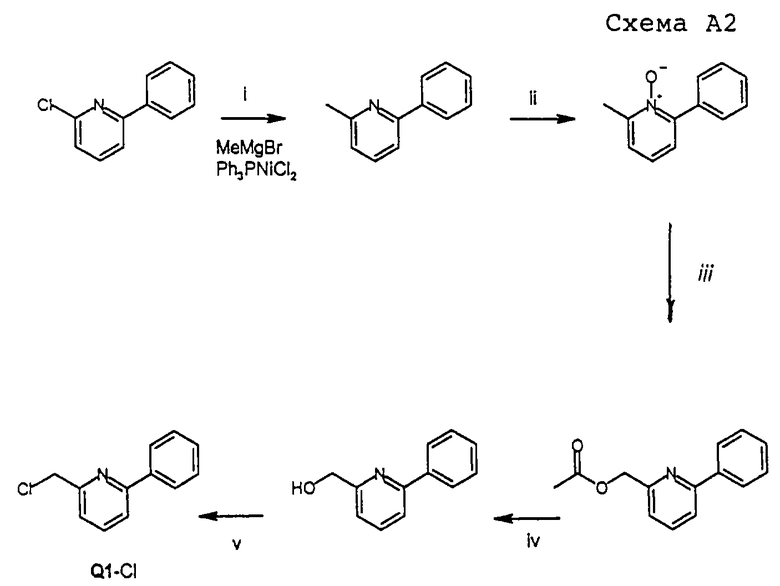
Q1-C1:
Этот промежуточный продукт синтезировали, как указано на схеме А2:
Стадия i (схема А2):
Эту стадию проводили по методике, аналогичной описанной в J.Het. Chem., 12, (1975), 443.
Стадия ii (схема А2):
При перемешивании при комнатной температуре 4,8 г (28,5 ммоль) 2-фенил-6-метилпиридина растворяли в 50 мл хлороформа, после чего по каплям добавляли раствор 7,8 г 75% m-СРВА (33,9 ммоль) в 75 мл хлороформа. Реакционная смесь проявляла только слабое повышение температуры. После перемешивания в течение 1,5 часа реакционную смесь встряхивали два раза с 5% водным раствором NaHCO3 и два раза с водным раствором Na2S2O3 для удаления избытка mСРВА, после чего реакционная смесь давала отрицательную реакцию на влажную бумагу KI/крахмал. Органический слой сушили над MgSO4. Удаление осушителя фильтрованием и растворителя выпариванием в вакууме давало масло, которое кристаллизовалось при царапании стенок сосуда, давая 5,5 г (105%) сырого N-оксида 2-фенил-6-метилпиридина, которое использовали в следующей стадии без дальнейшей очистки.
Стадия iii (схема А2):
Перемешиваемый раствор сырого N-оксида 2-фенил-6-метил-пиридина (5,2 г, 28,5 ммоль) в Ас2О (25 мл) нагревали при температуре флегмы в течение 2 час. Ас2О удаляли при помощи масляного насоса (10 мм) при 40oС, получая красное масло, которое очищали флэш-хроматографией на силикагеле с использованием в качестве элюента смеси Et2O/петролейный эфир = 1:1, получая 2-фенил-6-(ацетоксиметил)пиридин (4,6 г, 70%) в виде масла.
Стадия iv (схема А2):
4,5 г 2-фенил-6-(ацетоксиметил)пиридина (20 ммоль) обрабатывали водным раствором НСl (15%, 10 мл) и смесь нагревали при температуре флегмы при перемешивании. Через 30 минут реакционную смесь концентрировали при помощи масляного насоса (10 мм) при 40oС, добавляли CH3CN и смесь выпаривали досуха в вакууме, получая 2-фенил-6-(гидроксиметил)пиридин (3,0 г, 80%) в виде масла.
Стадия v (схема А2):
К перемешиваемому раствору 2-фенил-6-(гидроксиметил)пиридина (1,0 г, 5,4 ммоль) в СНСl3 (7 мл) при комнатной температуре по каплям добавляли SOCl2 (1,22 г, 10,2 ммоль) и смесь нагревали при 60oC в течение 20 минут. После выпаривания растворителя в вакууме остаток очищали растиранием с Et2O. Получаемый осадок собирали фильтрованием и сушили, получая хлорид 2-фенил-6-(хлорметил)пиридиния, Q1-C1 (1,2 г, 92%) в виде белого твердого продукта.
Q2-C1:
Q2-C1 получали аналогично синтезу Q1-C1.
Q3-C1:
Q3-C1 получали аналогично синтезу Q4-C1 (см. ниже).
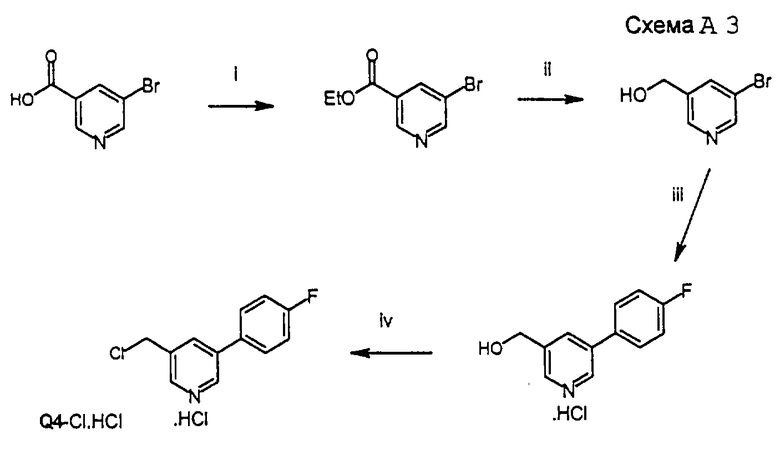
Q4-C1:
Этот промежуточный продукт синтезировали, как приведено на схеме A3.
Стадия i (схема A3):
Перемешиваемую смесь 3-бром-5-пиридинкарбоновой кислоты (10,1 г, 50 ммоль) и H2SO4 (1,5 мл) в EtОН (150 мл) кипятили с обратным холодильником в течение 6 часов. После охлаждения растворитель удаляли выпариванием в вакууме. Остаток разбавляли Н2О (100 мл), подщелачивали 5% раствором (водным) NaHCO3 и экстрагировали эфиром (4 х 100 мл). Объединенные органические экстракты промывали насыщенным NaCl и сушили над Na2SO4. Фильтрование и концентрирование в вакууме фильтрата давало этиловый эфир 3-бром-5-пиридинкарбоновой кислоты в виде масла, которое отверждалось при стоянии (9,8 г, 85%).
Стадия ii (схема A3):
К перемешиваемому раствору этилового эфира 3-бром-5-пиридинкарбоновой кислоты (9,5 г, 41,3 ммоль) в EtOH (96%, 220 мл) медленно добавляли NaBH4 (14,4 г, 380 ммоль) при 25oС. Реакция была слабо эндотермической. Смесь перемешивали в атмосфере азота при комнатной температуре в течение 6 час. Получаемую молочную смесь разбавляли Н2O (150 мл), EtOH выпаривали в вакууме и остаток экстрагировали СН2Сl2 (3 раза). Объединенные органические слои сушили над Na2SO4. После фильтрования фильтрат концентрировали в вакууме, получая 9 г сырого масла, которое очищали флэш-хроматографией на силикагеле (элюент: Еt2O), получая 3-бром-5-гидроксиметилпиридин (3,5 г, 45%).
Стадия iii (схема A3):
К раствору 3-бром-5-гидроксиметилпиридина (3,3 г, 17,5 ммоль) в толуоле (35 мл) добавляли Pd(PPh3)4 (0,6 г, 0,52 ммоль), водный раствор Na2CO3 (17,5 мл 2 М раствора) и п-фторфенилбороновую кислоту (2,65 г, 19 ммоль, растворенная в 8,5 мл EtOH). Смесь нагревали при 80-90oС в течение 1 часа и энергично перемешивали. После завершения реакции двухфазную реакционную смесь охлаждали, органический слой собирали и промывали насыщенными NaCl. Водный слой промывали EtOAc и объединенные органические слои сушили над Na2SO4. Осушитель удаляли фильтрованием и растворитель выпаривали в вакууме, получая темное масло, которое очищали флэш-хроматографией на силикагеле (элюент: CH2Cl2/MeOH/NH4OH, 95/4, 5/0,5), получая 3-(п-фторфенил)-5-гидроксиметилпиридин (3,0 г, 84%). Продукт превращали в его моногидрохлоридную соль: остаток растворяли в Et2O и обрабатывали 16,5 экв. НСl в этаноле. Продукт, гидрохлорид 3-(п-фторфенил)-5-гидроксиметилпиридиния, Q4-OH•HCl, осаждался в виде белого твердого вещества, которое собирали фильтрованием и затем сушили.
Стадия iv (схема A3)
Гидрохлорид 3-(п-фторфенил)-5-гидроксиметилпиридиния Q4-ОН•НСl (3,5 г, 14,7 ммоль) добавляли к избытку SOCl2 (20 мл) и смесь нагревали при 60oС для начала реакции (происходит генерация НСl). После завершения превращения исходного материала (45 мин) реакционную смесь охлаждали и избыток SOCl2 удаляли в вакууме, оставляя сухой остаток. Кристаллизация из Et2O давала гидрохлорид 3-(п-фторфенил)-5-хлорметилпиридиния Q4-С1•НСl (2,5 г, 66%).
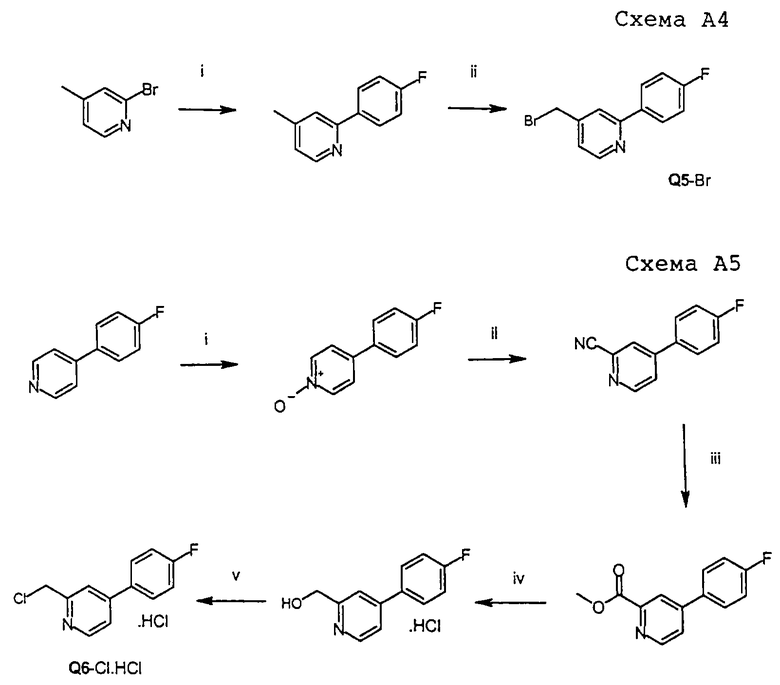
Q5-Вr:
Синтез Q5-Br изображен на схеме А4.
Стадия i (схема А4):
Раствор 2-бром-4-метилпиридина (10 г, 58 ммоль) и Pd(PPh3)4 (1,5 г, 1,3 ммоль) в толуоле (110 мл) перемешивали при комнатной температуре в атмосфере азота. Последовательно добавляли водный раствор Na2CO3 (58 мл 2 М раствора) и п-фторфенилбороновую кислоту (8,93 г, 63,8 ммоль) и получаемой смеси давали реагировать при 90-100oС в течение 4 час. Смесь охлаждали, водный слой отделяли и экстрагировали EtOAc (2 раза). Объединенные фракции EtOAc и толуола сушили над MgSO4. Отделение осушителя фильтрованием и удаление растворителя в вакууме давало розовое масло (28 г). Перегонка давала чистый 2-(п-фторфенил)-4-метилпиридин (6,10 г, 56%); т.кип. 110-116oС (6-7 мбар) в виде бесцветного масла.
Стадия ii (схема А4):
Смесь 2-(п-фторфенил)-4-метилпиридина (0,5 г, 2,67 ммоль), N-бромсукцинимида (0,48 г, 2,69 ммоль) и каталитического количества бензоилпероксида в ССl4 (50 мл) перемешивали при температуре флегмы и облучали при помощи обычной УФ-лампы на 250 Вт в течение 4 часов. Потом реакционную смесь охлаждали и затем растирали со смесью Еt2О/петролейный эфир. Осадок удаляли фильтрованием, фильтрат концентрировали в вакууме, получая 2-(п-фторфенил)-4-бромметилпиридин (0,63 г, 88%, нестабильный) в виде темно-желтого масла.
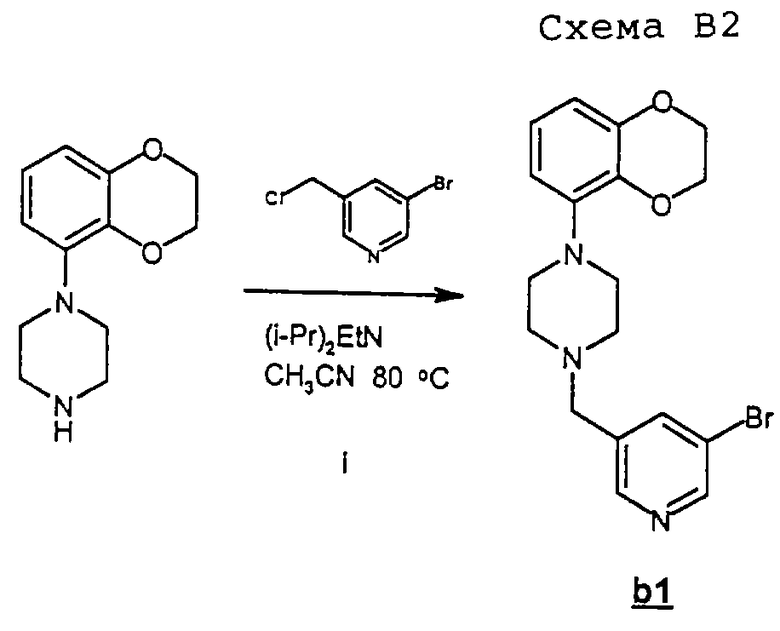
Q6-С1:
Промежуточный продукт Q6-C1 синтезировали по приведенной ниже схеме (схема А5):
Стадия i (схема А5):
4-(п-фторфенил)пиридин (13 г, 75 ммоль) растворяли в ледяной уксусной кислоте (100%; 50 мл) при 70-80oС. Затем при перемешивании добавляли Н2О2 (35%; 8 мл). Через 4 часа добавляли дополнительную порцию H2O2 (35%, 5 мл). Реакционной смеси давали охладиться, после чего ее выпаривали досуха в вакууме, оставляя желтое твердое вещество, которое разбавляли Н2О (150 мл), подщелачивали водным раствором NaOH (150 мл 2 М раствора) и экстрагировали CH2Cl2 (100 мл). Органический слой отделяли и сушили над Na2SO4. После удаления осушителя фильтрованием и удаления растворителя в вакууме было выделено 13 г (91%) целевого продукта, N-оксида 4-(п-фторфенил)пиридина.
Стадия ii (схема А5):
К 13 г N-оксида 4-(п-фторфенил)пиридина (68,7 ммоль) в атмосфере N2 при 80oС добавляли Me2SO4 (8,6 г, 68 ммоль), после чего смесь перемешивали при 100-110oС в течение 2 час. Смесь охлаждали и в реакционную смесь выливали 70% раствор диоксан/вода. Полученный темно-коричневый раствор по каплям добавляли к перемешиваемому раствору NaCN (10 г, 0,20 моль) в Н2О (85 мл) при 15-20oС. Смесь перемешивали при комнатной температуре в течение 3 час. Реакционную смесь фильтровали, остаток промывали СН2Сl2, который добавляли к двухфазному фильтрату. Органический слой фильтрата сушили над Na2SO4. Удаление осушителя фильтрованием и выпаривание растворителя в вакууме давало целевое соединение в виде светло-коричневого твердого вещества, которое очищали кристаллизацией из EtОН (300 мл), чтобы получить 2-циано-4-(п-фторфенил)пиридин (8,6 г, 68%): т.пл. 194-195oС.
Стадия iii (схема А5):
Перемешиваемому раствору 2-циано-4-(п-фторфенил)пиридина (8,6 г, 46,7 ммоль) в насыщенном растворе HCl-MeOH (200 мл) давали возможность реагировать при температуре кипения с обратным холодильником, в течение 6 час. Получаемый розовый раствор концентрировали в вакууме до объема приблизительно 50 мл, после чего его разбавляли 250 мл воды. Последний раствор подщелачивали водным раствором NH4OH (25%) и экстрагировали СН2Сl2. Органический слой сушили над Na2SO4. Удаление осуществляли фильтрованием и выпаривание растворителя в вакууме давало целевой продукт, метиловый эфир 4-(п-фторфенил)пиридин-2-карбоновой кислоты, в виде розового твердого вещества (5,0 г, 46%): т.пл. 97-8oС.
Стадия iv (схема А5):
NaBH4 (8,2 г, 0,2 моль) добавляли по частям к перемешиваемому раствору метилового эфира 4-(п-фторфенил)пиридин-2-карбоновой кислоты (5,0 г, 21,6 ммоль) в EtOH (96%, 100 мл) и смесь перемешивали при комнатной температуре в течение 6 час. Растворитель удаляли при пониженном давлении, после чего добавляли воду. Затем проводили экстракцию с помощью EtOAc. Органический слой сушили над MgSO4. Удаление осушителя и выпаривание растворителя в вакууме давало масло, которое растворяли в МеОН и обрабатывали 1,1 экв. HCl/EtOH, получая гидрохлорид 2-гидроксиметил-4-(п-фторфенил)пиридиния, Q6-OH•HCl, в виде желтой пены (4,47 г, 87%).
Стадия v (схема А5):
Эту реакцию проводили аналогично стадии iv в схеме A3:
Промежуточные продукты, использованные в пути В
Промежуточный продукт b1:
Этот промежуточный продукт синтезировали, как изображено на схеме В2.
Стадия i (схема Е2):
К суспензии моногидрохлорида 1-(2,3-дигидро-1,4-бензодиоксин-5-ил)пиперазина (5,4 г, 21 ммоль) в CH2CN (125 ммоль) добавляли 3-бром-5-хлорметилпиридин (4,6 г, 19 ммоль) и диизопропилэтиламин (12,3 г, 95 ммоль). Смесь перемешивали при температуре флегмы в течение 30 мин. После охлаждения смеси и выпаривания растворителя в вакууме остаток растворяли в CH2Cl2, промывали 5% водным раствором NaHCO3, насыщенным водным раствором NaCl, после чего органическую часть сушили над Na2SO4. После удаления осушителя фильтрованием и растворителя выпариванием в вакууме остаток очищали флэш-хроматографией на силикагеле (CH2Cl2/MeOH/NH4OH, 97, 25/2, 5/0, 25), получая b1 (7,2 г, 97%) в виде масла.
Настоящее изобретение относится к производным пиперазина формулы (а), где А представляет гетероциклическую группу с 5-6 атомами в кольце, где присутствуют 1-2 гетероатома, из группы О и N, R1 представляет водород, R2 представляет С1-4-алкил, С1-4-алкокси или оксогруппу и р = 0,1 или 2, Z представляет азот и пунктирная линия представляет одинарную связь, R3 и R4 представляют водород, n = 1, R5 представляет 2-пиридил, 3-пиридил или 4-пиридил. Соединения могут быть использованы для лечения шизофрении и других психотических нарушений. Описаны также промежуточные соединения, которые используются в синтезе. 2 с. и 3 з.п. ф-лы, 2 табл.
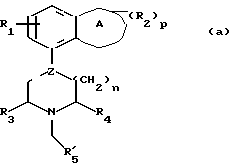
где А представляет гетероциклическую группу с 5-6 атомами в кольце, где присутствуют 1-2 гетероатома, из группы О и N;
R1 представляет водород;
R2 представляет С1-4-алкил, С1-4-алкокси или оксогруппу;
р = 0, 1 или 2;
Z представляет азот и пунктирная линия представляет одинарную связь;
R3 и R4 представляют водород;
n = 1;
R5 представляет 2-пиридил, 3-пиридил или 4-пиридил, замещенный в метаположении относительно метиленового мостика группой Y и необязательно замещенный заместителем (R6)q;
Y представляет фенил, фуранил или тиенил, причем фенил может быть замещен 1-3 заместителями из группы, включающей галоген;
R6 представляет С1-4алкил;
q = 0 или 1,
и их соли.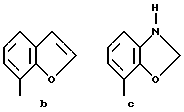
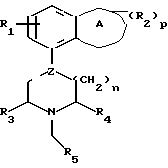
n = 1;
R1 и (R2)p, R3, R4, R5, (R6)q, Y и Z имеют значения, данные в п. 1,
и его соли.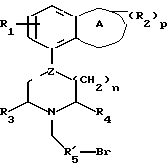
где R1, (R2)р, Z, n, R3 и R4 имеют значение, данное в п. 1;
R'5 представляет 2-пиридил, 3-пиридил или 4-пиридил, необязательно замещенный группой (R6)q, где R6 и q имеют значения, указанные в п. 1, и атом брома находится в метаположении относительно метиленовой группы.
| ЕР 0561319 А1, 22.09.1993 | |||
| Механизм подпора к ножницам длярезки проката | 1974 |
|
SU508347A1 |
| Способ получения производных пиперазина или их солей | 1974 |
|
SU549084A3 |

