Настоящее изобретение относится к биотехнологии, а именно к технологии получения лекарственных форм рекомбинантных белков.
Терапевтически значимые белки, полученные на основе технологии рекомбинантных ДНК, в последние годы получили широкое распространение для лечения широкого круга заболеваний. К наиболее значимым и распространенным препаратам данного класса следует отнести препараты, созданные на основе рекомбинантных эритропоэтина, интерферона-альфа, гранулоцитарного колониестимулирующего фактора и некоторых других цитокинов человека.
Общим для данного класса веществ является практическая невозможность и нецелесообразность выделения их из природных источников и, как следствие, получение их в искусственно созданных прокариотических (как правило, E.Coli) или эукариотических (обычно СНО) системах.
Белки данного класса обладают высокой удельной биологической активностью, вследствие чего их разовая терапевтическая доза находится в микрограммовом диапазоне. Некоторые из используемых в медицинской практике белков данного класса (как правило, получаемых в прокариотических системах) находятся в частично денатурированном состоянии и склонны к агрегации и выпадению в осадок. В первую очередь это относится к рекомбинантным интерферонам-альфа. В процессе приготовления лекарственных форм на основе белков данного класса наиболее распространенным в настоящее время подходом является введение в состав готового лекарственного средства в качестве стабилизатора сывороточного альбумина и/или сублимационная сушка. Однако оба этих подхода не свободны от недостатков. Введение в качестве стабилизатора сывороточного альбумина человека повышает риск вирусных контаминаций препарата и ограничивает его применение пациентами некоторых религиозных конфессий. Сублимационная сушка не только снижает активность препарата, но и вызывает частичную денатурацию альбумина, что в свою очередь иногда приводит к аллергическим реакциям у пациентов. Ранее было описано и реализовано приготовление лекарственной формы препарата рекомбинантного интерферона-альфа человека, свободного от сывороточного альбумина человека. Для этого в состав для сушки в качестве наполнителя вводили декстраны (РЕАЛЬДИРОН, БИОТЕХНА, Литва). Подобный подход позволяет произвести сушку препарата, но стабильность в растворе остается низкой за счет потерь на стенках сосуда и агрегации.
Другим подходом к решению проблемы стабильности рекомбинантных белков в готовых лекарственных формах является введение в композицию мочевины, комплекса амфотерных электролитов (аминокислот) и детергентов с последующей лиофильной сушкой (RU 2043118), где удалось получить стабильную лекарственную форму рекомбинантного эритропоэтина человека. В то же время имеются существенные ограничения на срок хранения препарата после его растворения.
Наиболее близким по совокупности существенных признаков к заявляемому способу является способ приготовления готовых форм рекомбинантных белков [US 5,656,730, 1996, МПК А 61 К 035/14; А 61 К 38/00; С 07 К 014/505; С 07 К 014/535] , согласно которому лекарственные формы рекомбинантных белков приготовляли с использованием сложных композиций, состоящих из комплекса аминокислот, хлорбутанола, бензилового спирта, бензалкониумхлорида и неорганических компонентов буферных растворов. Согласно прототипу для получения готовой лекарственной формы, содержащей 10-2000 мкг/мл рекомбинантного белка, в указанном растворе растворяют соответствующее количество субстанции и используют полученный раствор в равной степени для приготовления жидкой или лиофилизованной формы. Существенным недостатком данного подхода является необходимость использования крайне сложной композиции, содержащей такие компоненты, как 1,1,1-трихлор-2-метил-2-пропанол, бензиловый спирт и бензалкониум хлорид.
Задачей изобретения является разработка более простого и дешевого способа получения лекарственных форм рекомбинантных белков, обладающих стабильной биологической активностью при хранении.
Технический результат заключается в том, что в результате разработанного способа получают более дешевый и безопасный препарат, не включающий токсичных компонентов.
Поставленная задача решается посредством того, что при приготовлении жидкой лекарственной формы соответствующего рекомбинантного белка эффективное количество последнего растворяют в биосовместимом буферном растворе, содержащем неионный детергент и в некоторых случаях дополнительно стабилизатор. Для приготовления лекарственной формы берут буферный раствор со значением рН, отличающимся более чем на 1 единицу от изоэлектрической точки белка. Это обеспечивает стабильное нахождение белка при низких концентрациях (1-100 мкг/мл) в растворах без агрегации и олигомеризации.
В качестве детергента можно использовать Tween-20, Tween-80, Tween-60, Nonidet P-40 и др. В качестве биосовместимых буферных систем могут быть использованы изотонические буферные растворы на основе цитратов, ацетатов, фосфатов и др. В ряде случаев для корректировки осмолярности растворы могут содержать в качестве стабилизатора биосовместимые полимерные компоненты - многоатомные спирты, декстрины, поливинилпирролидоны и др.
Предложенный способ иллюстрируется следующими примерами.
Пример 1. Получение жидкой лекарственной формы рекомбинантного интерферона-альфа человека
Для получения жидкой лекарственной формы рекомбинантного интерферона-альфа использовали высокоочищенный препарат, выделенный из телец включения штамма E.Coli IF212S, не содержащий заметных примесей олигомерных форм, продуктов протеолиза и примесей белков клеток-продуцентов. Удельная биологическая активность препарата составляла 2•108 МЕ/мл, кажущаяся молекулярная масса 18,1 кD, изоэлектрическая точка pl=5,96.
Контроль биологической активности осуществляли по методу ингибирования цитопатогенного действия вируса везикулярного стоматита на клетках эмбрионального легкого человека линии Л-68. В лунки 96-ячеечных микропланшетов вносили по 0,1 мл суспензии клеток в питательной среде Игла DMEM с добавкой 80-160 мкг/мл гентамицина и 2% сыворотки плодов коров в концентрации 200-300 тыс. клеток/мл. Для определения активности интерферона готовят двукратные разведения (выше и ниже предполагаемого титра) исследуемых препаратов и стандарта активности интерферона в среде Игла DMEM, содержащей 2% сыворотки плодов коров и антибиотики (пенициллин 100 Ед./мл, гентамицин 80 мкг/мл). На каждое разведение препарата используют не менее 4 лунок с культурой клеток. В каждую лунку с клеточной культурой вносили по 0,1 мл приготовленных образцов интерферона. Четыре лунки с культурой в каждом микропланшете оставляли в качестве контрольных. Кроме того, 16 лунок оставляли для контроля дозы индикаторного вируса. В эти лунки вносили по 0,1 мл питательной среды. Инокулированные и контрольные культуры клеток инкубировали в течение 1 суток при (37,0±1,0)oС в атмосфере с (5,0±0,5)% СO2, после чего в каждую лунку с испытуемыми материалами вносили определенную заранее дозу вируса везикулярного стоматита, соответствующую 100 ТЦД50 в 0,1 мл. Одновременно осуществляли контроль взятой дозы вируса на предназначенных для этой операции 16 лунках с культурой. После внесения индикаторного вируса и титрования его дозы, культуру клеток инкубировали на протяжении 1-2 суток при температуре (37,0±1,0)oС в атмосфере с (5,0±0,5)% СO2 под контролем дозы вируса, после чего проводили учет активности интерферона. За титр интерферона принимали величину, обратную разведению препарата, при котором клеточная культура в 50% лунок оказалась полностью защищенной от цитопатического действия вируса.
Физическое содержание интерферона определяли методом иммуноферментного анализа с использованием тест-систем ProCon IF2 (Протеиновый контур, Россия). Для контроля олигомерных форм использовали твердофазный иммуноферментный тест с антителами PC/IF1 (Протеиновый контур, Россия) в качестве сорбирующего и открывающего реагентов.
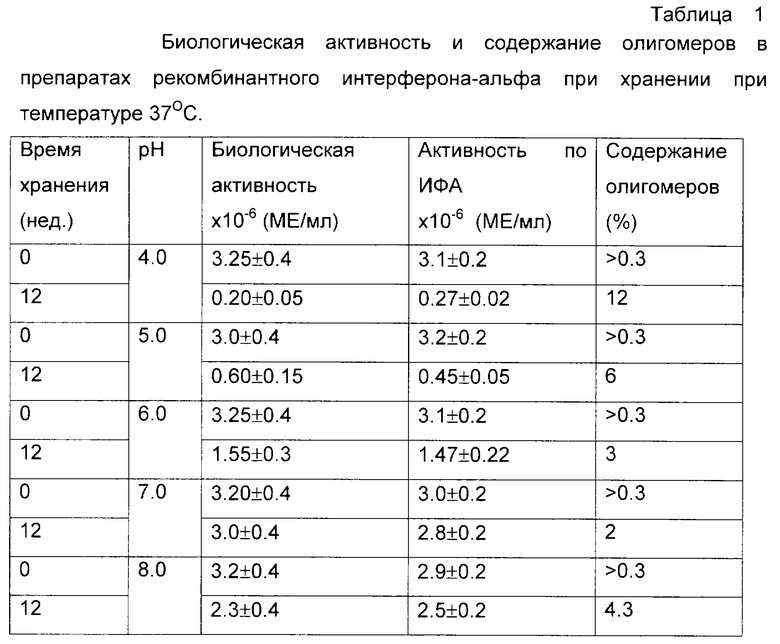
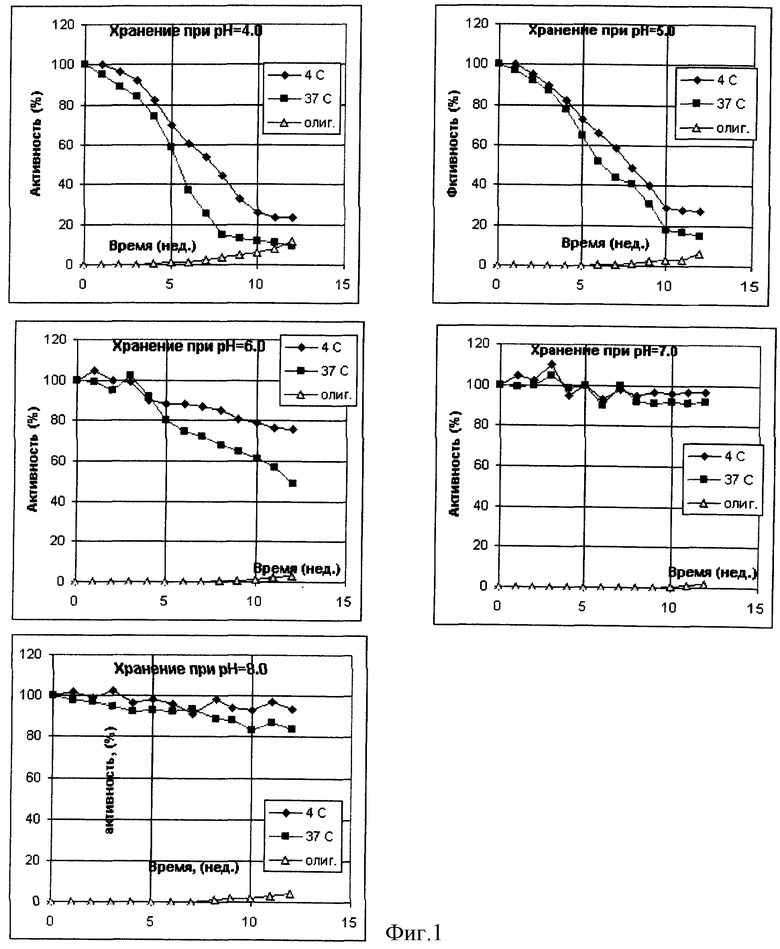
Была приготовлена серия 0,05 М аммонийацетатных буферных растворов, содержащих 0,1 М хлорида натрия и 0,02% Твин-20, в интервале рН от 4 до 8. В каждый из буферных растворов был внесен рекомбинантный интерферон-альфа из расчета 3•106 и 6•106 МЕ/мл, растворы подвергли стерилизующей фильтрации и стерильно аликвотировали по 1 мл в стеклянные ампулы. Ампулы были запаяны и в равных количествах помещены на хранение при температурах -70, +4 и +37oС. С периодичностью в 7 дней ампулы извлекали из термостатов и подвергали анализу на общее содержание интерферона и содержание олигомерных форм по отношению к замороженному препарату. В начале и в конце эксперимента были проведены дополнительно измерения биологической активности. Результирующие данные по хранению препарата с исходной активностью 3•106 МЕ/мл представлены на фиг. 1 и в таблице 1. Полностью аналогичные данные были получены и для препарата с активностью 6•106 МЕ/мл.
Пример 2. Приготовление жидкой лекарственной формы рекомбинантного эритропоэтина человека
Для получения жидкой лекарственной формы рекомбинантного эритропоэтина человека использовали высокоочищенный препарат, выделенный из кондиционированной культуральной жидкости клеток СНО SP/M pZip NeoEPO SV(x) DFR в соответствии с методом, описанным в патенте [RU 2145610, М.кл6 C 12 N 11/00] и не содержащий заметных примесей олигомерных форм, продуктов протеолиза, и примесей белков клеток-продуцентов. Удельная биологическая активность препарата составляла 1,4•105 МЕ/мл, кажущаяся молекулярная масса - 35,2 кD, pl= 3,5-5,0.
Контроль биологической активности осуществляли in vitro по методу [Krystal G. , 1983] , основанном на оценке стимуляции пролиферации клеток крови полицитемических мышей в присутствии эритропоэтина. Анемическое состояние у мышей-гибридов F1 CDF•C57Bl вызывали двукратным, через 30 часов, внутрибрюшинным введением фенилгидразина в дозе 60 мкг/кг. Спленоциты выделяли через 3 суток после последнего введения фенилгидразина и культивировали в ячейках 96-луночных планшетов в среде DMEM с 5% телячьей эмбриональной сыворотки, 2 mM L-глутамина и 80 мкг/мл гентамицина и присутствии раститрованных образцов стандартного и исследуемого препаратов в СО2-инкубаторе в течение 24 часов. За два часа до окончания культивирования в ячейки вносили 40 мкБк 3H-тимидина. Клеточные культуры переносили на фильтры, отмывали и производили учет включения 3H-тимидина при помощи сцинциляционного счетчика RackBeta 1217, Фармация. По графику уровня включения 3H-тимидина для стандартного образца определяли биологическую активность исследуемых препаратов.
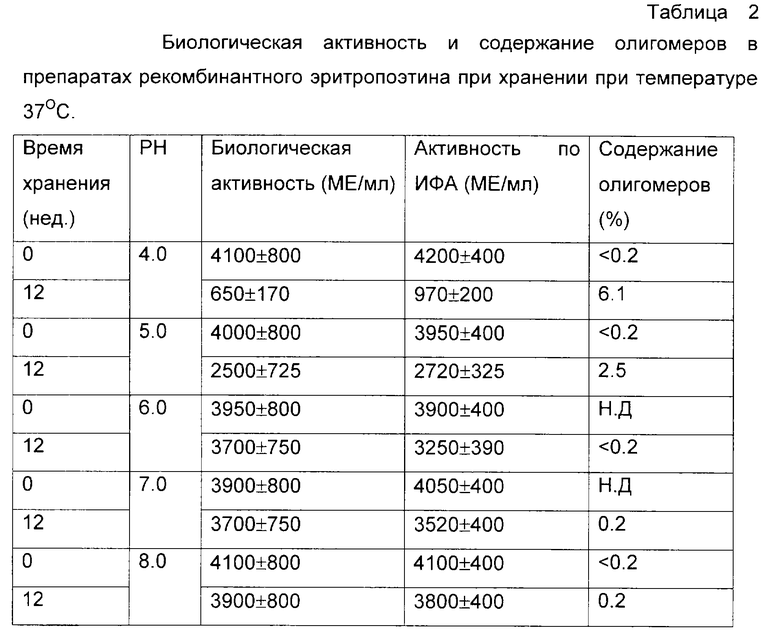
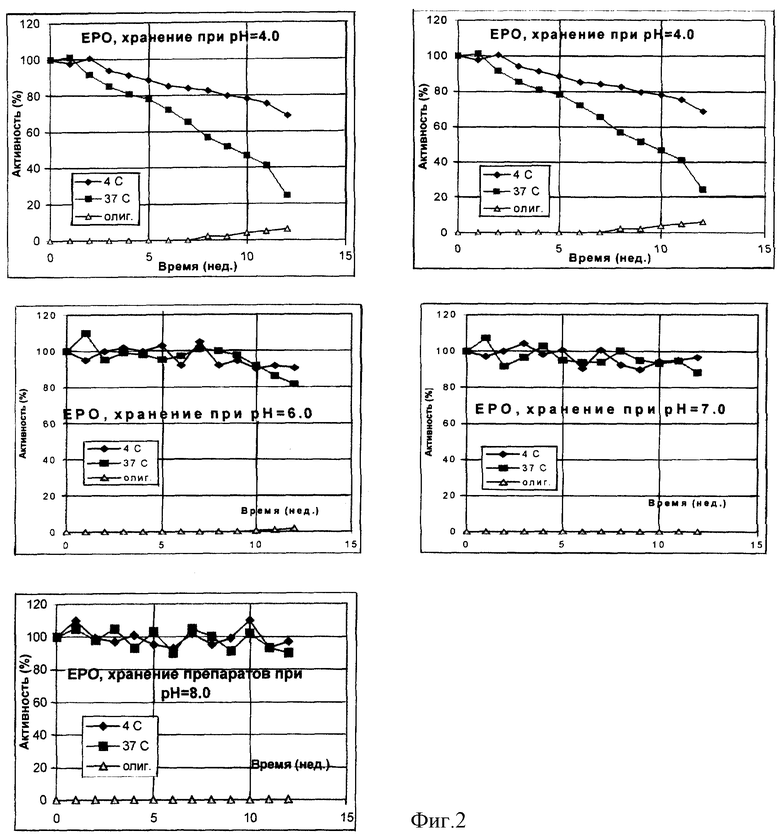
Физическое содержание эритропоэтина определяли методом иммуноферментного анализа с использованием тест-систем ProCon EPO (Протеиновый контур, Россия). Для контроля олигомерных форм использовали твердофазный иммуноферментный тест с антителами PC/ED7 (Протеиновый контур, Россия) в качестве сорбирующего и открывающего реагентов. Была приготовлена серия 0,05 М натрий-цитратных буферных растворов, содержащих 0,12 М хлорида натрия и 0,02% Твин-20, в интервале рН от 4 до 8. В каждый из буферных растворов был внесен рекомбинантный эритропоэтин человека из расчета 2•103 и 4•103 МЕ/мл, растворы подвергли стерилизующей фильтрации и стерильно аликвотировали по 1 мл в стеклянные ампулы. Ампулы были запаяны и в равных количествах помещены на хранение при температурах -70, +4 и +37oС. С периодичностью в 7 дней ампулы извлекали из термостатов и подвергали анализу на общее содержание эритропоэтина и содержание олигомерных форм по отношению к замороженному препарату. В начале и в конце эксперимента были проведены дополнительно измерения биологической активности. Результирующие данные по хранению препарата с исходной активностью 4•103 МЕ/мл представлены на фиг. 4 и в таблице 2. Полностью аналогичные данные были получены и для препарата с активностью 2•103 МЕ/мл.
Пример 3. Получение жидкой лекарственной формы рекомбинантного гранулоцитарного колониестимулирующего фактора (Г-КСФ) человека
Для получения жидкой лекарственной формы рекомбинантного Г-КСФ человека использовали высокоочищенный препарат, выделенный из кондиционированной культуральной жидкости клеток СНО pZip NeoGCSF CMV(x) DFR, не содержащий заметных примесей олигомерных форм, продуктов протеолиза, и примесей белков клеток-продуцентов (фиг. 5). Удельная биологическая активность препарата составляла 1,1•108 МЕ/мл, кажущаяся молекулярная масса 20 кD, pl=5,7-6,3.
Контроль биологической активности осуществляли по методу оценки стимуляции пролиферации клеток костного мозга мышей [Okabe M., et al., 1990]. Костномозговые клетки мышей-гибридов F1 СВА•C57Bl выделяли стандартным методом и культивировали в концентрации 5•105 клеток/мл в ячейках 96-луночных планшетов в среде DMEM с 10% телячьей эмбриональной сыворотки, 2 mM L-глутамина и 80 мкг/мл гентамицина в присутствии раститрованных образцов стандартного и исследуемого препаратов в СO2-инкубаторе в течение 72 часов. За 20 часов до окончания культивирования в ячейки вносили по 40 мкБк 3Н-тимидина. Клеточные культуры переносили на фильтры, отмывали и производили учет включения 3Н-тимидина при помощи сцинциляционного счетчика RackBeta 1217, Фармация. По графику уровня включения 3Н-тимидина для стандартного образца определяли биологическую активность исследуемых препаратов.
Физическое содержание Г-КСФ определяли методом иммуноферментного анализа с использованием тест-систем ProCon G-CSF (Протеиновый контур, Россия). Для контроля олигомерных форм использовали твердофазный иммуноферментный тест с антителами PC/G36F11 (Протеиновый контур, Россия) в качестве сорбирующего и открывающего реагентов.
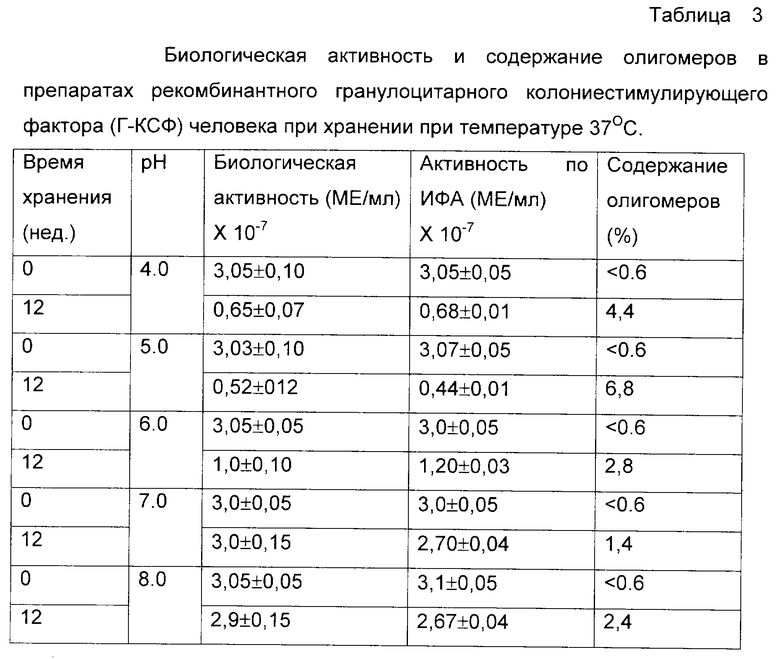
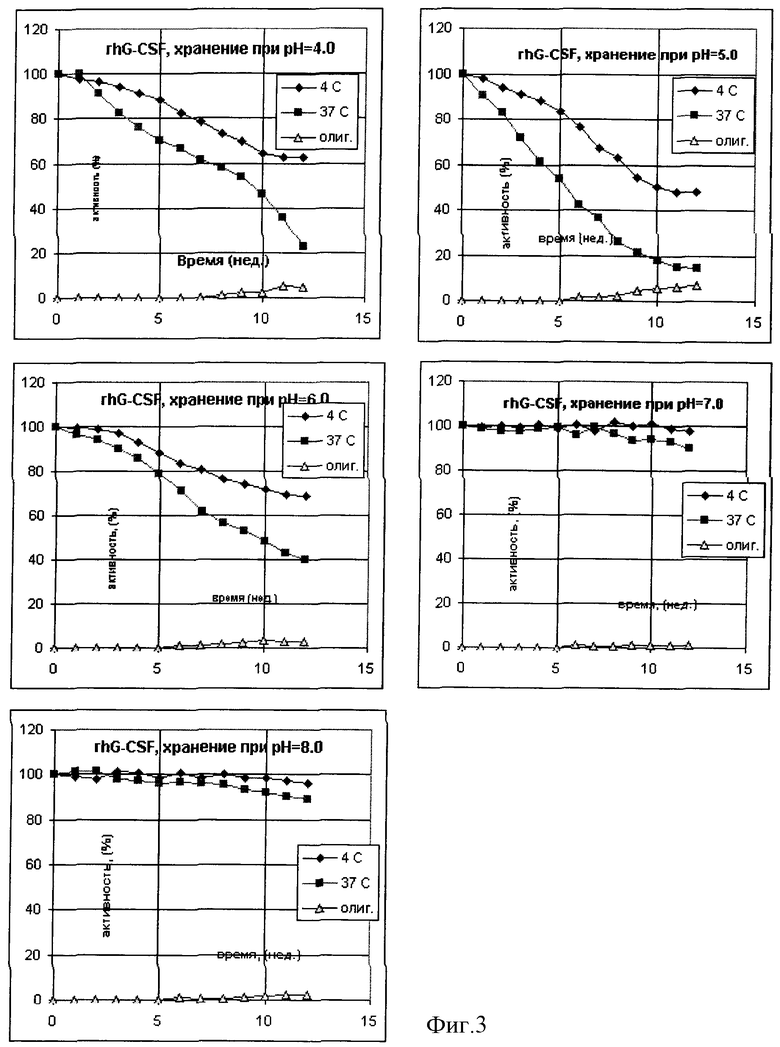
Была приготовлена серия 0,02 М натрий-фосфатных буферных растворов, содержащих 0,15 М хлорида натрия и 0,02% Твин-80 в интервале рН от 4 до 8. В каждый из буферных растворов был внесен рекомбинантный Г-КСФ человека из расчета 3•107 МЕ/мл, растворы подвергли стерилизующей фильтрации и стерильно аликвотировали по 1 мл в стеклянные ампулы. Ампулы были запаяны и в равных количествах помещены на хранение при температурах -70, +4 и +37oС. С периодичностью в 7 дней ампулы извлекали из термостатов и подвергали анализу на общее содержание Г-КСФ и содержание олигомерных форм по отношению к замороженному препарату. В начале и в конце эксперимента были проведены дополнительно измерения биологической активности. Результирующие данные по хранению препарата на фиг. 6 и в таблице 3.
Из данных таблиц 1-3 и фиг. 1-3 видно, что наиболее стабильные композиции растворов белков находятся в областях, удаленных от изоэлектрических точек рассматриваемых объектов более чем на 1 единицу. Пониженная стабильность интерферона-альфа в кислой области определяется, по-видимому, одновременно комплексом факторов - гидролитическим расщеплением белка при повышенной температуре, олигомеризацией за счет тиол-дисульфидного обмена с участием свободных сульфгидрильных групп не полностью ренатурированного белка и повышенной сорбции положительно заряженного белка на поверхностных силанольных группах стекла. В то же время переход в основную область за изоэлектрическую точку приводит к стабилизации данного белка. Хорошо видна явная зависимость стабильности препаратов эритропоэтина от рН. В области, близкой от изоэлектрической точки, стабильность понижена в связи с образованием агрегатов, а по мере отдаления от рl электростатические взаимодействия подавляются. Та же тенденция может быть отмечена и в случае Г-КСФ.
Пример 4. Получение жидкой лекарственной формы рекомбинантного гранулоцитарного колониестимулирующего фактора (Г-КСФ) человека, содержащей лактозу
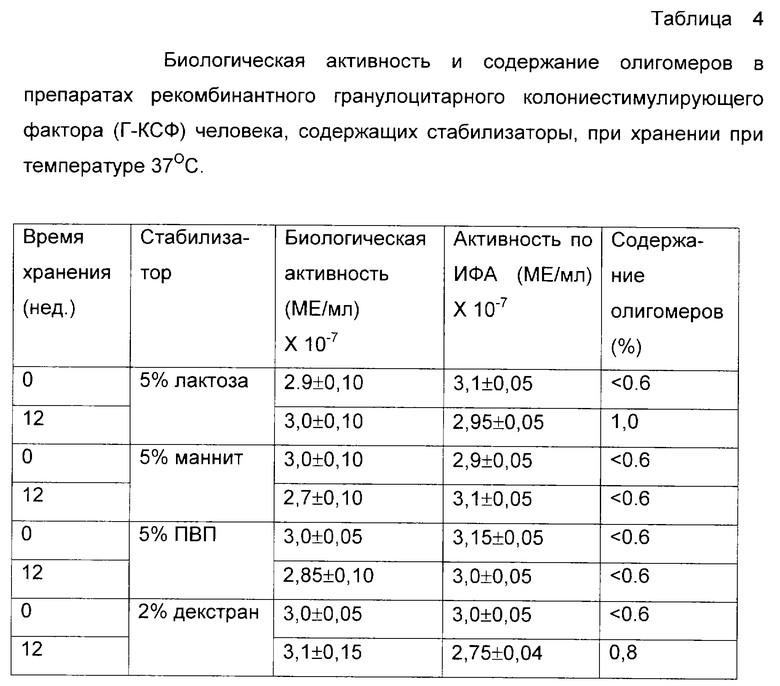
Для исследования влияния стабилизирующих свойств некоторых компонентов небелковой природы была приготовлена серия растворов лактозы, маннита, поливинилпирролидона 8000 (ПВП) и декстрана 10000 в 0,02 М натрий-фосфатном буферном растворе, содержащем 0,15 М хлорида натрия, 0,02% Твин-20, рН=7,0. Рекомбинантный Г-КСФ человека, как в примере 3, был внесен из расчета 3•107 МЕ/мл в каждый из растворов. Растворы аликвотировали и хранили так же, как в примере 3, подвергая еженедельному анализу на активность и содержание олигомерных форм. Результаты представлены в таблице 4.
В процессах получения готовых форм препаратов генно-инженерного происхождения в качестве стабилизирующих наполнителей часто используют компоненты мономерной и полимерной углеводной природы, многоатомные спирты и некоторые биосовместимые полимеры. Из таблицы 4 видно, что введение подобных компонентов в готовые формы препаратов белков генно-инженерного происхождения, находящихся при рН, существенно отличающихся от рl данного белка, не оказывает значительного эффекта на стабильность композиции.
Изобретение относится к области биотехнологии, а именно к технологии получения лекарственных форм рекомбинантных белков. Сущность изобретения состоит в том, что при приготовлении жидкой лекарственной формы соответствующего рекомбинантного белка эффективное количество последнего растворяют в биосовместимом буферном растворе, содержащем неионный детергент и в некоторых случаях дополнительно стабилизатор. Для приготовления лекарственной формы берут буферный раствор со значением рН, отличающийся более чем на 1 единицу от изоэлектрической точки белка. Это обеспечивает стабильное нахождение белка при низких концентрациях (1-100 мкг/мл) в растворах без агрегации и олигомеризации. В качестве детергента можно использовать Tween-20, Tween-80, Tween-60, Nonidet P-40 и др. В качестве биосовместимых буферных систем могут быть использованы изотонические буферные растворы на основе цитратов, ацетатов, фосфатов и др. В ряде случаев для корректировки осмолярности растворы могут содержать в качестве стабилизатора биосовместимые полимерные компоненты - многоатомные спирты, декстрины, поливинилпирролидоны и др. Технический результат заключается в том, что в результате разработанного способа получают более дешевый и безопасный препарат. 3 з.п. ф-лы, 4 табл., 3 ил.
| US A 6072040, 06.06.2000 | |||
| US A 5917021, 29.06.1999 | |||
| US A 5691312, 25.11.1997 | |||
| US A 5656730, 12.08.1997 | |||
| US A 5358708, 25.10.1994 | |||
| СТАБИЛИЗИРОВАННЫЙ ВОДНЫЙ РАСТВОР ЭРИТРОПОЭТИНА | 1998 |
|
RU2128517C1 |
