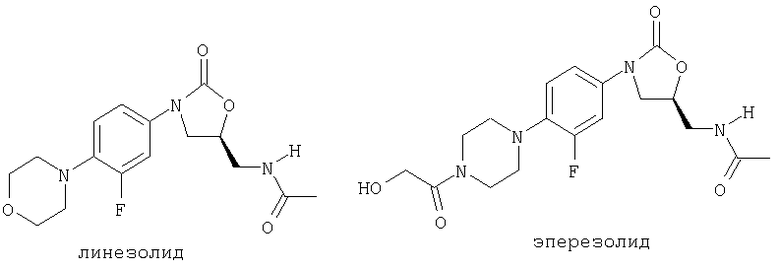
Настоящее изобретение относится к области соединений фенилоксазолидинона, обладающих антибактериальной активностью против грамположительных и грамотрицательных бактерий, содержащих их фармацевтические композиции, и к способам лечения бактериальных инфекций с помощью этих соединений.
В последние двадцать лет оксазолидиноны были определены как новый класс антибактериальных агентов, которые обладают активностью против различных грамположительных организмов, устойчивых ко множеству лекарственных препаратов. Особенно проблематичные патогены включают устойчивые к метициллину Staphylococcus aureus (MRSA), устойчивые к промежуточным гликопептидам Staphylococcus aureus (GISA), устойчивые к ванкомицину enterocci (VRE) и устойчивые к пенициллину и цефалоспорину Streptococcus pneumoniae. Как класс, оксазолидиноны демонстрируют уникальный механизм действия. Исследования показали, что эти соединения селективно связываются с 50S рибосомной субъединицей и ингибируют бактериальную трансляцию в начальной фазе синтеза белков. Членами класса оксазолидинонов являются линезолид (см. WO 95/07271) и эперезолид.
Патент США №5792765, выданный Riedl с сотр., раскрывает ряд замещенных оксазолидинонов (цианогуанидин, цианоамидины и амидины), которые можно использовать в качестве антибактериальных лекарственных препаратов.
В патенте США №5910504, выданном Hutchinson, раскрыт ряд фенилоксазолидинонов с замещенными гетероароматическими кольцами, включая индолилзамещенные соединения, которые можно использовать в качестве антибактериальных агентов.
В WO 98/54161 (Hester с сотр.) раскрыты амиды, тиоамиды, мочевины и тиомочевины, которые являются антибактериальными агентами.
В WO 95/07271 (Barbachyn с сотр.) раскрыты оксазиновые и тиазиноные производные оксазолидинона, такие как линезолид и его аналоги, которые можно использовать в качестве противомикробных агентов и которые эффективны против ряда патогенов человека и животных, включая грамположительные аэробные бактерии, такие как устойчивые ко многим лекарствам стафилококки, стрептококки и энтерококки, а также анаэробные организмы, такие как Bacteroldes spp. и Clostndia spp. виды, и кислотоустойчивые организмы, такие как Mycobactenum tuberculosis, Mycobacterium avium и Mycobacterium spp.
В WO 93/09103 (Barbachyn с сотр.) раскрыты замещенные арил- и гетероарилфенилоксазолидиноны, которые можно использовать в качестве антибактериальных агентов.
Настоящее изобретение относится к соединениям фенилоксазолидинона формулы I:
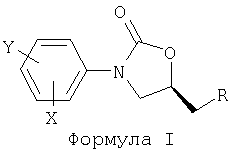
где:
R выбирают из группы, состоящей из ОН, N3, OR', O-арила, O-гетероарила, OSO2R'', -NR'''R'''', или
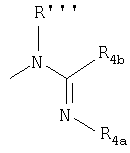
где:
(i) R' представляет неразветвленный или разветвленный ацил, содержащий вплоть до 6 атомов углерода, или бензил;
(ii) R'' представляет неразветвленный или разветвленный алкил, содержащий вплоть до 5 атомов углерода, фенил или толил; и
(iii) R''' и R'''' независимо выбирают из группы, состоящей из Н, циклоалкила, содержащего от 3 до 6 атомов углерода, фенила или трет-бутоксикарбонила, флуоренилоксикарбонила, бензилоксикарбонила, неразветвленного или разветвленного алкила, содержащего вплоть до 6 атомов углерода, который необязательно замещен циано- или алкоксикарбонилом, содержащим вплоть до 4 атомов углерода, -CO2-R1, -CO-R1, -CO-SR1, -CS-R1, Р(О)(OR2)(OR3) и -SO2-R4, где
R1 выбирают из группы, состоящей из Н, циклоалкила, содержащего от 3 до 6 атомов углерода, трифторметила или фенила бензила или ацила, содержащего вплоть до 5 атомов углерода, неразветвпенного или разветвленного алкила, содержащего вплоть до 6 атомов углерода, причем указанный алкил необязательно замещен неразветвленным или разветвленным алкоксикарбонилом, содержащим вплоть до 5 атомов углерода, ОН, циано, вплоть до 3 атомов галогенов и -NR5R6, где R5 и R6 одинаковы или различны, и их выбирают из Н, фенила или неразветвленного или разветвленного алкила, содержащего вплоть до 4 атомов углерода;
R2 и R3 одинаковы или различны и их выбирают из водорода или неразветвленного или разветвленного алкила, содержащего вплоть до 4 атомов углерода; и
R4 выбирают из неразветвленного или разветвленного алкила, содержащего вплоть до 4 атомов углерода или фенила; и
R4a представляет CN, COR4c, COOR4c, CONHR4c, CO-NR4cR4d, SO2R4с, SO2NHR4c, SO2-NR4c R4d или NO2;
R4b представляет Н, алкил, OR4c, SR4c, амино, NHR4c, NR4cR4d, (C1-8) алкиларил или моно-, ди-, три- и пергало(C1-8)-алкил;
R4с и R4d независимо выбирают из Н, алкила, арила или в случае любой NR4cR4d группы R4c и R4d, взятые вместе с атомом азота, к которому они присоединены, образуют незамещенную или замещенную группу пирролидинила, пиперидинила или морфолинила;
Х представляет от 0 до 4 членов, независимо выбранных из группы, состоящей из галогена, ОН, меркапто, нитро, гало-C1-8-алкила, C1-8 алкокси, тио-С1-8алкила, C1-8 алкиламино, ди(С1-8-алкил) амино, формила, карбокси, алкоксикарбонила, C1-8 алкил-СО-O-, C1-8 алкил-CO-NH-, карбоксамида, арила, замещенного арила, гетероарила, замещенного гетероарила, CN, амина, С3-6-циклоалкила, С1-8алкила, необязательно замещенного одним или более из членов, выбранных из группы, состоящей из F, Cl, ОН, С1-8алкокси и C1-8 ацилокси; и
Y представляет радикал формулы II или III:
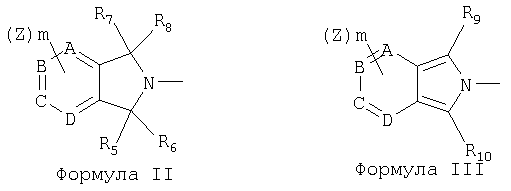
где
R5, R6, R7 и R8 независимо представляют Н, алкил, CN, нитро, С1-8алкил, гало-С1-8алкил, формил, карбокси, алкоксикарбонил, карбоксамид, арил, замещенный арил, гетероарил, замещенный гетероарил или R5 и R6 и/или R7 и R8 вместе образуют оксогруппу;
R9 и R10 независимо представляют Н, галоген, алкил, ОН, CN, меркапто, нитро, C1-8алкил, гало-С1-8алкил, C1-8алкокси, тио-С1-8алкил, амино, C1-8алкиламино, ди(C1-8алкил) амино, формил, карбокси, алкоксикарбонил, C1-8-алкил-СО-О-, C1-8-алкил-CO-NH-, карбоксамид, арил, замещенный арил, гетероарил, замещенный гетероарил или амин;
А, В, С и D выбирают из С, S, О и N с образованием любого 5-10 членного ароматического или гетероароматического кольца, причем указанное гетероароматическое кольцо содержит от одного до четырех членов, выбранных из группы, состоящей из S, О и N;
Z выбирают из галогенов, алкила, арила, замещенного арила, гетероарила, замещенного гетероарила, CN, CHO, СОалкила, амина (диалкиламино)алкила, где диалкиламино выбирают из диметиламина, диэтиламина, морфолинила, тиоморфолинила, пирролидинила или пиперидинила, или алкокси, или NHCO-(C1-8алкила); и
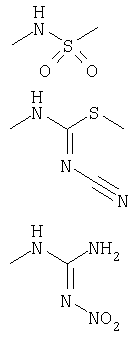
m представляет 0 или 1,
и его фармацевтически приемлемые соли и сложные эфиры.
Соединения приведенных выше формул полезны в качестве антибактериальных агентов для лечения бактериальных инфекций у человека и животных.
Настоящее изобретение относится также к способу лечения субъектов, состояния которых вызваны бактериальными инфекциями, или состояния, в которые вносят вклад бактериальные инфекции, который включает введение указанному млекопитающему терапевтически эффективное количество соединения формулы I.
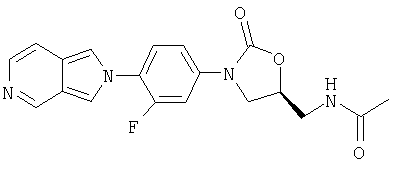
Настоящее изобретение относится также к способу профилактики у субъектов состояний, вызванных бактериальными инфекциями, или состояний, в которые вносят вклад бактериальные инфекции, который включает введение указанному млекопитающему профилактически эффективной дозы фармацевтической композиции соединения формулы I.
Другие цели и преимущества станут очевидны специалистам из последующего описания.
Подробное описание изобретения
В отношении приведенных выше описаний соединений фенилоксазолидинона настоящего изобретения использованы следующие определения.
Если нет других указаний, термины "алкил", "алкенил" и "алкинил" относятся к неразветвленным или разветвленным группам, содержащим от 1 до 8 атомов углерода.
Термин "ацил" относится к органическому радикалу, содержащему определенное число атомов углерода, полученному из органической кислоты в результате удаления гидроксильной группы формулы RCO, как в случае ацетила, где R представляет СН3.
Термин "арил" означает незамещенную карбоциклическую ароматическую группу, включая (но этим не ограничиваясь) фенил, 1-или 2-нафтил и т.п. Термин "гетероарил" относится к циклическому ароматическому радикалу, содержащему от пяти до десяти атомов в кольце; где от одного до трех кольцевых атомов независимо являются гетероатомами, такими как S, О и N, а остальными атомами кольца являются атомы углерода, например пиридинильный, пиразинильный, пиримидинильный, пирролильный, пиразолильный, имидазолильный, тиазолильный, оксазолильный, изоксазолильный, тиадиазолильный, оксадиазолильный, тиенильный, фуранильный, хинолинильный или изохинолинильный радикалы и т.п.
Термин "замещенный арил" или "замещенный гетероарил" относится к арилу или гетероарилу, у которых от 1 до 3 атомов водорода независимо замещены галогеном, ОН, CN, меркапто, нитро, С1-8алкилом, гало-С1-8алкилом, C1-8алкокси, тио-С1-8алкиламином, C1-8алкиламином, ди(C1-8алкил) амино, формилом, карбокси, алкоксикарбонилом, С1-8алкил-СО-О-, С1-8алкил-СО-NH- или карбоксамидом. Кроме того, замещенный гетероарил может быть замещен монооксо, образуя в результате, например, 4-оксо-1-Н-хинолин. Замещенный гетероарил может быть также замещен замещенным арилом или вторым замещенным гетероарилом, в результате чего образуется, например, 4-фенилимидазол-1-ил или 3-пиридинил-имидазол-1-ил и т.п.
Термин "гало" или "галоген" означает фтор, хлор, бром или йод, (моно-, ди-, три-, и пер-)галоалкил означает алкильный радикал, полученный в результате независимого замещения его атомов водорода галогеном. P обозначает фосфор.
Соединения настоящего изобретения являются асимметричными в оксазолидиноновом кольце в 5-положении и таким образом существуют в виде оптических антиподов. Как таковые, все возможные оптические антиподы, энантиомеры или диастереоизомеры, образующиеся в результате дополнительных асимметричных центров, которые могут существовать в оптических антиподах, рацематах и рацемических смесях, также являются частью настоящего изобретения. Антиподы можно разделить известными специалистам способами, такими как, например, фракционная перекристаллизация диастереоизомерных солей энантиомерно чистых кислот. В другом варианте антиподы можно разделить хроматографически на колонке Pirkle.
Фраза "фармацевтически приемлемые соли" относится к солям свободных оснований, которые обладают необходимой фармакологической активностью свободного основания и не являются нежелательными ни с биологической, ни с какой-либо иной точки зрения. Эти соли можно получить из неорганических или органических кислот. Примерами неорганических кислот являются хлористоводородная кислота, азотная кислота, бромистоводородная кислота, серная кислота или фосфорная кислота. Примерами органических кислот являются уксусная кислота, пропионовая кислота, гликолевая кислота, молочная кислота, пировиноградная кислота, малоновая кислота, янтарная кислота, яблочная кислота, малеиновая кислота, фумаровая кислота, винная кислота, лимонная кислота, бензойная кислота, коричная кислота, миндальная кислота, метансульфоновая кислота, этансульфоновая кислота, паратолуолсульфоновая кислота, метилсульфоновая кислота, салициловая кислота и т.п. Кроме того, подходящими солями являются соли органических и неорганических оснований, таких как КОН, NaOH, Са(ОН)2, Al(ОН)3, пиперидин, морфолин, этиламин, триэтиламин и т.п.
В объем настоящего изобретения включены также гидратные формы соединений, которые содержат различные количества воды, например гидратные, полугидратные и сесквигидратные формы.
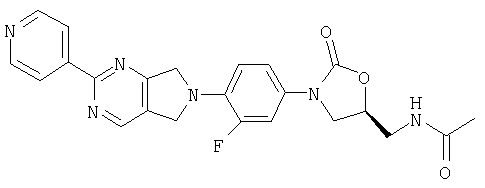
Термин "субъект" включает (без ограничений) любое животное или искусственно измененное животное. В предпочтительном варианте этот термин относится к человеку.
Термин "устойчивый к лекарству" или "устойчивость к лекарству" относится к таким характеристикам микробов, как способность выживать в присутствии доступного в настоящее время антимикробного агента в его обычной эффективной концентрации.
Соединения настоящего изобретения обладают антибактериальной активностью против грамположительных и некоторых грамотрицательных бактерий. Они полезны в качестве антибактериальных агентов для лечения бактериальных инфекций у человека и животных. В частности, эти соединения обладают антибактериальной активностью против S.aureus, S.epidermidls, S.pneumoniae, E.faecalis, E.faecium, Moraxella catarrhalis и H.influenzae. Более конкретно эти соединения можно использовать против устойчивых бактерий, таких как MRSA и GISA, и они мало подвержены механизмам приобретения устойчивости. Соединения формулы I, наиболее предпочтительные для этих целей, это те соединения, в которых R принимает любое из следующих значений:
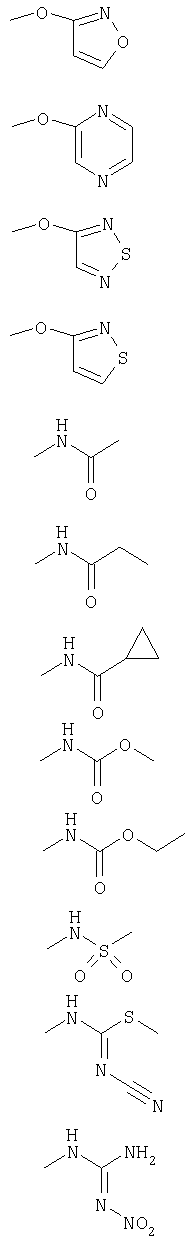
Кроме соединений формул I, которые наиболее предпочтительны для таких целей, предпочтительны соединения, в которых Y имеет следующие значения:
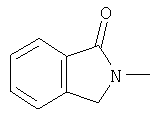 изоиндолон-;
изоиндолон-;
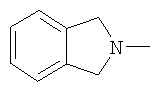 (1,3-дигидро-2Н-изоиндол-2-ил)-;
(1,3-дигидро-2Н-изоиндол-2-ил)-;
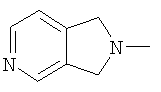 (1,3-дигидро-2Н-пирроло[3,4-с]пиридин-6-ил)-;
(1,3-дигидро-2Н-пирроло[3,4-с]пиридин-6-ил)-;
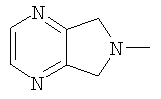 (5,7-дигидро-6Н-пирроло[3,4-b]пиразин-6-ил)-;
(5,7-дигидро-6Н-пирроло[3,4-b]пиразин-6-ил)-;
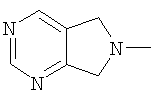 (5,7-дигидро-6Н-пирроло[3,4-d]пиримидин-6-ил)-;
(5,7-дигидро-6Н-пирроло[3,4-d]пиримидин-6-ил)-;
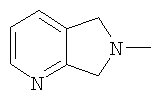 (5,7-дигидро-6Н-пирроло[3,4-b]пиридин-6-ил)-;
(5,7-дигидро-6Н-пирроло[3,4-b]пиридин-6-ил)-;
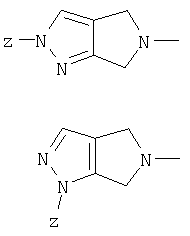
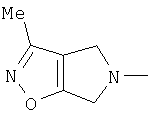 (4,6-дигидро-3-метил-5Н-пирроло[3,4-d]изоксазол-5-ил)-;
(4,6-дигидро-3-метил-5Н-пирроло[3,4-d]изоксазол-5-ил)-;
 (3,5-дигидро-5-метилпирроло[3,4-с]пиррол-2(1Н)-ил)-; и
(3,5-дигидро-5-метилпирроло[3,4-с]пиррол-2(1Н)-ил)-; и
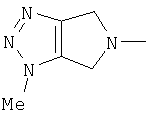 (4,6-дигидро-1-метилпирроло[3,4-d]-1,2,3-триазол-5(1Н)-ил)-
(4,6-дигидро-1-метилпирроло[3,4-d]-1,2,3-триазол-5(1Н)-ил)-
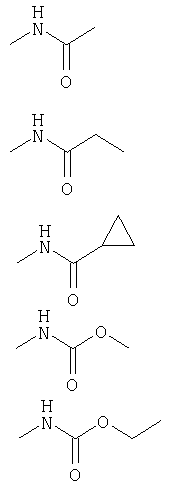
Конкретные примеры соединений настоящего изобретения включают следующие:
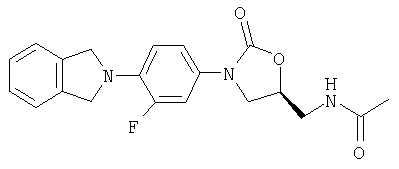
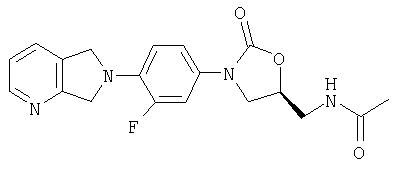
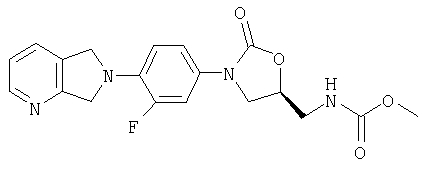
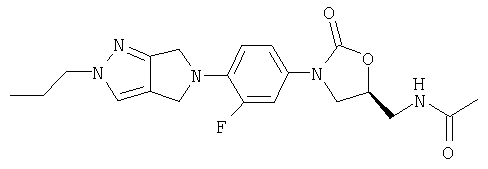
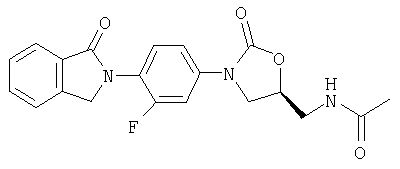
N-[[(5S)-3-[4-(1,3-дигидро-2Н-изоиндол-2-ил)-3-фторфенил]-2-оксо-5-оксазолидинил]метил]ацетамид;
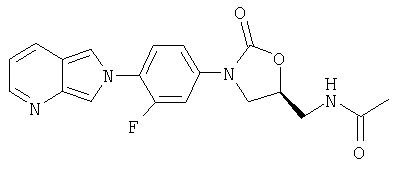
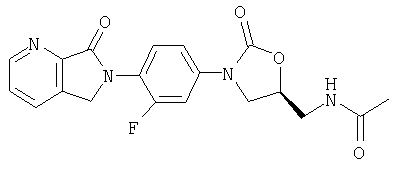
N-[[(5S)-3-[4-(1,3-дигидро-2Н-пирроло[3,4-с]пиридин-2-ил)-3-фторфенил]-2-оксо-5-оксазолидинил]метил]ацетамид;
N-[(5S)-3-[3-фтор-4-(5-оксидо-2Н-пирроло[3,4-с]пиридин-2-ил)фенил]-2-оксо-5-оксазолидинил]метил]ацетамид;
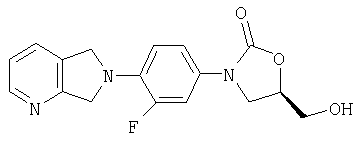
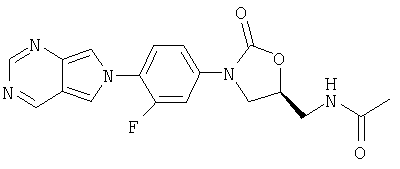
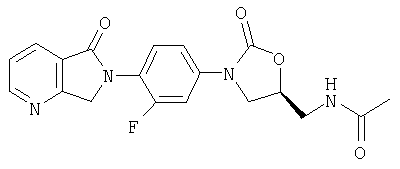
N-[[(5S)-3-[4-(5,7-дигидро-6Н-пирроло[3,4-b]пиридин-6-ил)-3-фторфенил]-2-оксо-5-оксазолидинил]метил]ацетамид;
N-[[(5S)-3-[4-(1,3-дигидро-1-оксо-2Н-изоиндол-2-ил)-3-фторфенил] -2-оксо-5-оксазолидинил]метил]ацетамид; и
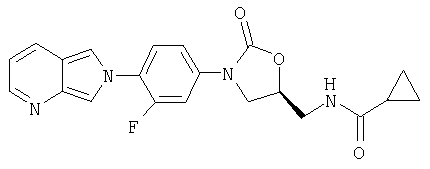
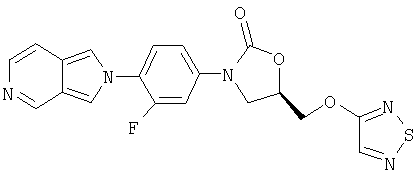
(5R)-3-[4-(5,7-дигидро-6Н-пирроло[3,4-b]пиридин-6-ил)-3-фторфенил]-5-(гидроксиметил)-2-оксазолидинон.
Соединения формулы I, которые являются предметом настоящего изобретения, можно получить из легко доступных исходных материалов, таких как индол (Gawley et al., J. Org. Chem., 1988, 53:5381), 6,7-дигидро-5Н-пирроло[3,4-с]пиридина и 6,7-дигидро-5Н-пирроло [3,4-b] пиридина (патент США №5371090 Petersen с сотр.) в соответствии со способами синтеза, хорошо известными специалистам. Примеры способов представлены на схемах I-V:
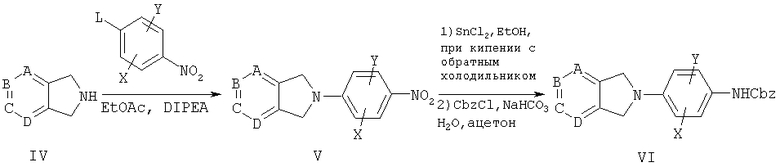
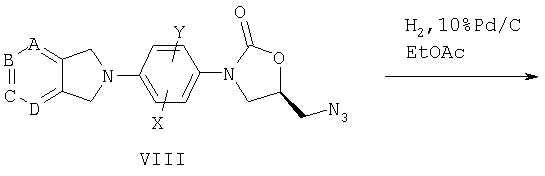
В соответствии со схемой I бициклические гетероциклы общей формулы IV обрабатывают замещенным производным нитробензола (L представляет соответствующую отщепляемую группу, такую как галоген или трифторметансульфонилокси) в подходящем основании и растворителе, таком как диизопропиламин и этилацетат, получая соединение замещенного нитрофенила V.
Затем производное нитробензола V восстанавливают до анилина в соответствующей реакции, например, обрабатывая SnCl2, или каталитическим гидрированием в присутствии соответствующего катализатора, такого как палладий-на-угле. Затем анилин обрабатывают бензил- или метилхлорформиатом и бикарбонатом натрия, получая соответствующее производное бензил- или метилкарбамата VI.
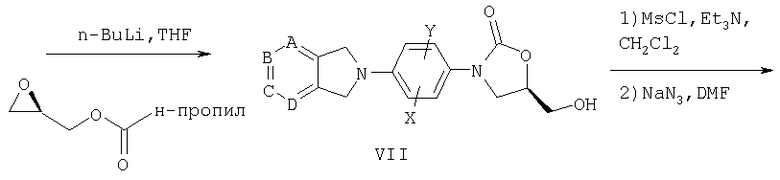
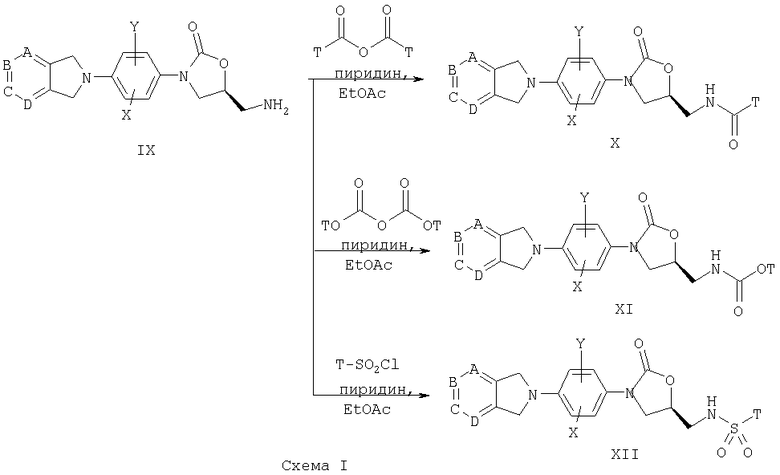
Затем Cbz анилин VI депротонируют, используя литиевое основание, такое как н-бутиллитий, и осуществляют взаимодействие с (R)-глицидилбутиратом, получая оксазолидинон VII. Затем гидроксиметильную группу превращают в амид, как представлено на схеме I, получая мезилат, превращая в азид VIII и восстанавливая до амина IX соответствующим способом, таким как гидрирование. В другом варианте замещение мезилата (схема II) или соответствующей отщепляемой группы, такой как тозилат или хлор, на фталимид калия и удаление фталоильной защитной группы гидразинолизом приводит к получению амина IX. Амин IX можно превратить в амид Х в реакции ацилирования, используя известные специалистам способы, такие как обработка уксусным ангидридом в присутствии основания, такого как пиридин. В другом варианте амин IX можно превратить в карбамат XI в результате обработки метилхлорформиатом и пиридином или в результате взаимодействия с сульфонилхлоридом в инертном растворителе в присутствии органического основания, такого как пиридин, получая сульфонамид XII
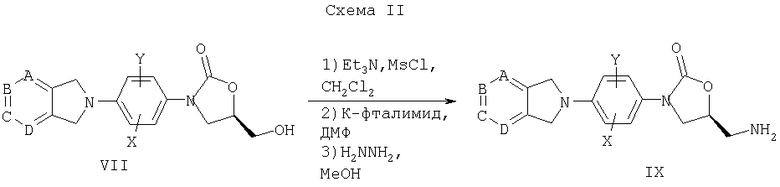

Для получения оксазолидинона, в котором R=О-гетероарил (XIII), оксазолидинонкарбинол VII можно превратить в соответствующий мезилат или другую подходящую отщепляемую группу и осуществить взаимодействие с HO-Het (подходящий гетероцикл, содержащий гидроксил), либо в присутствии основания, либо с HO-Het как полученного заранее алкоксида в соответствующем растворителе, например в ДМФ или в ацетонитриле (схема III)). В другом варианте можно использовать условия Митсунобу для присоединения VII к НО-гетероциклу в результате обработки трифенилфосфином или диизопропилазодикарбоксилатом (DIAD) в соответствующем растворителе, таком как ТГФ, при соответствующей температуре, предпочтительно при комнатной температуре. Условия реакции и соответствующие ссылки можно найти в WO 99/64416, Gravestock с сотр.
Креме того, обрабатывая VII подходящим ненуклеофильным основанием, например NaH, замену отщепляемой группы (LG), такой как хлор или бром, можно осуществить, используя соответствующий реакционноспособный аза-гетероцикл (LG-Het)(схема III).
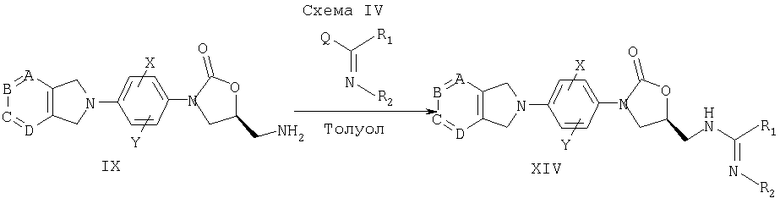
Соединения строения XIV можно получить по способу, представленному на схеме IV. Амин IX можно превратить в различные функционализированные амидины в реакции с активированными иминами, где Q представляет отщепляемую группу, такую как метилтио или метокси, в подходящем растворителе без катализатора или в присутствии катализатора (такого как AgNO3) при температурах в интервале 0-110°С.
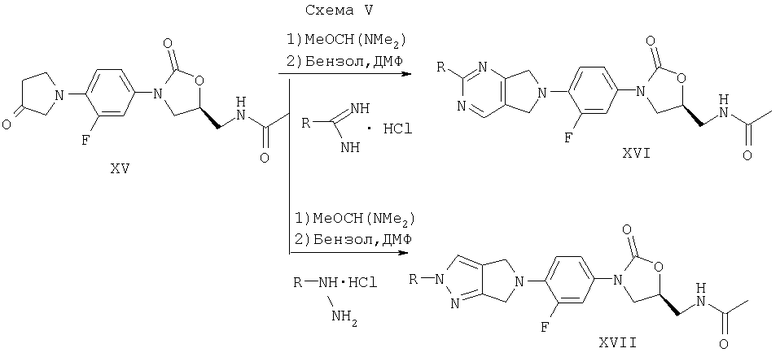
В соответствии со схемой V пирролидинон XV (полученный по способу WO96/13502) вначале подвергают взаимодействию с метокси-бис-(диметиламином) или другим активированным реагентом диметилформамида и затем нагревают в подходящем растворителе (например, в ДМФ или в бензоле), либо с замещенными амидинами с образованием пирролопиримидиноксазолидинонов, таких как XVI, либо с замещенными гидразинами с образованием пирролопиразолоксазолидинонов, таких как XVII. Получение енаминных, алкоксиметиленовых или алкоксикарбонильных производных пирролидинона XV по способу Brighty с сотр. патента США 5037834А также дает возможность доступа к таким системам.
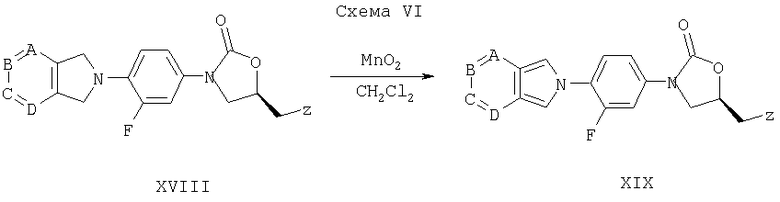
Как представлено на схеме VI, соединения строения XIX можно получить окислением различных соединений XVIII, используя соответствующий окисляющий агент (например, диоксид марганца, пероксиукусную кислоту, DDQ или воздух) в подходящем растворителе, таком как метиленхлорид.
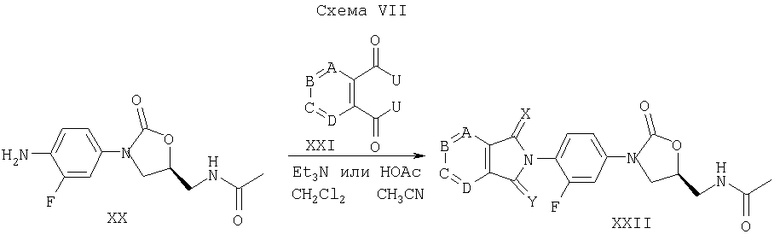
Оксопроизводное формулы XXII на схеме VII (X=0, Y=Н2 или Х=H2, Y=0) можно получить, осуществляя взаимодействие 1,2-арилдикарбоксальдегидов (где в XXI U=Н) с анилином XX (полученным по способу WO 96/23788) в присутствии кислот, таких как уксусная кислота, в подходящем растворителе, таком как метиленхлорид. Ди-оксопроизводные (формулы XXII, где Х=Y=0) получают в результате взаимодействия анилина XX с выбранными реагентами 1,2-арилдикарбонила с подходящими отщепляемыми группами (XXI, где U=Cl, Br и т.д.).
Определения
Все температуры даны в °С
Термин "рассол" относится к водному насыщенному раствору хлорида натрия
ДМФ обозначает N,N-диметилформамид
ТГФ обозначает тетрагидрофуран
Cbz обозначает карбобензилокси
n-BuLi обозначает н-бутиллитий
MS обозначает масс-спектр, выраженный как m/е или масса/единицу заряда
[М+Н] обозначает положительный ион соединения плюс атом водорода
Эфир обозначает диэтиловый эфир
кт обозначает комнатную температуру
Мр обозначает температуру плавления
CH2Cl2 обозначает метиленхлорид
NaOH обозначает гидроксид натрия
МеОН обозначает метанол
EtOAc обозначает этилацетат
ppt обозначает осадок
Эти соединения обладают антимикробной активностью против бактериальных патогенов, которые обладают или предполагается, что они обладают устойчивостью к лекарствам против таких патогенов, как S.aureus, S.epidermidis, S.pneumoniae, S.pyogenes, Enterococcus spp., Moraxella catarrhalis и Н.influenzae. Эти соединения особенно полезны против устойчивых к лекарствам грамположительных кокков, таких как устойчивые к метициллину S.aureus и устойчивые к ванкомицину энтерококки. Эти соединения полезны для лечения приобретенной в обществе пневмонии, респираторных инфекций верхних и нижних дыхательных путей, кожных инфекций и инфекций мягких тканей, приобретенных в больнице легочных инфекций, костных и суставных инфекций и других бактериальных инфекций.
Минимальная ингибирующая концентрация (МИК), которая является показателем антибактериальной активности in vitro, широко используется специалистами. In vitro антимикробную активность соединений настоящего изобретения определяют способом микроразбавлений бульона с последующим тестовым способом Национального комитета по лабораторным стандартам (National Committee for Laboratory Standards) (NCCLS). Этот способ раскрыт в NCCLS Document M7-A4, Vol.17, №2 "Methods for Dilution Antimicrobial Susceptibility Test for Bacteria that Grow Aerobically-Fourth Edition", который включен сюда для ссылки.
В этом способе двукратные сериальные разбавления лекарств в бульоне Mueller-Hinton с добавленным катионом добавляют в лунки планшетов для микроразбавлений. Тестовые организмы приготавливают, регулируя мутность культур в бульонах с активным ростом таким образом, чтобы конечная концентрация тестового организма после его добавления в лунки составляла примерно 5×104 культурообразующих единиц/лунку.
После инокулирования планшетов с микроразбавлениями планшеты инкубируют при 35°С в течение 16-20 час, а затем считывают показания. МИК представляет наинизшую концентрацию тестового соединения, которая полностью ингибирует рост тестового организма. Количество растущих организмов в лунках, содержащих испытываемое соединение, сравнивают с количеством организмов в лунках с контролируемым ростом (без испытываемого соединения), используемых для каждого планшета. Как видно из таблицы 1, некоторые соединения настоящего изобретения были протестированы в отношении различных патогенных бактерий, что дало интервал активностей от 1 до ≥128 мкг/мл в зависимости от тестированного организма. S.aureus OC2878 представляет MRSA и Е. faecium OC3312 представляют устойчивый к ванкомицину энтерококк.
Величины МИК для некоторых соединений формулы I
Далее в настоящем изобретении предложен способ лечения бактериальных инфекций или усиления или потенциирования активности других антибактериальных агентов у субъекта, состояние которого вызвано бактериальной инфекцией или в состояние которого вносит вклад бактериальная инфекция, который включает введение животным соединения настоящего изобретения отдельно или в смеси с другим антибактериальным агентом в форме лекарства в соответствии с настоящим изобретением. Термин "лечение" и "обработка" включает одновременное, раздельное или последовательное введение фармацевтически эффективного количества соединения, содержащего одно или более из раскрытых здесь соединений, субъекту, у которого необходимо ингибировать рост бактерий. Фармацевтически эффективное количество соединения, которое используют в практике настоящего изобретения для лечения, меняется в зависимости от способа введения, возраста, веса и общего состояния подлежащего лечению субъекта и, в конечном счете, определяется врачами или ветеринарами.
Соединения настоящего изобретения можно вводить субъекту, такому как человек, любым из способов, соответствующих подлежащему лечению состоянию, причем подходящие способы включают пероральный, ректальный, назальный, наружный (включая введение за щеку и под язык), вагинальный и парэнтеральный (включая подкожный, внутримышечный, внутривенный, чрезкожный, через трахею и эпидуральный). Предпочтительный способ может меняться, например, в зависимости от состояния реципиента, а также от типа препарата и удобства введения.
Если соединения используют для указанных выше целей, их можно объединять с одним или более из фармацевтически приемлемых носителей, то есть растворителей, разбавителей и т.п., и их можно вводить перорально в таких формах, как таблетки, капсулы, диспергируемые порошки, гранулы или суспензии, содержащие, например, от около 0,5% до 5% суспендирующего агента, сиропы, содержащие, например, от около 10% до 50% сахара, и эликсиры, содержащие, например, от около 20% до 50% этанола и т.п., или парэнтерально в форме стерильных растворов или суспензий для инъекций, содержащих от около 0,5% до 5% суспендирующего агента в изотонической среде. Эти фармацевтические препараты могут содержать, например, от около 0,5% вплоть до около 90% активного ингредиента в сочетании с носителем, обычно от 5% до 60%.
Композиции для наружного применения могут иметь форму жидкостей, кремов или желе, содержащих терапевтически эффективные концентрации соединения настоящего изобретения в смеси с дерматологически приемлемым носителем.
При получении композиций в дозовой форме для перорального введения можно использовать любую из фармацевтически приемлемых сред. Твердые носители включают крахмал, лактозу, дикальцийфосфат, микрокристаллическую целлюлозу, сахарозу и каолин, тогда как жидкие носители включают стерильную воду, полиэтиленгликоли, неионные поверхностно-активные агенты и пищевые масла, такие как кукурузное, арахисовое и кунжутное масла, сообразуясь с природой активного ингредиента и желательной конкретной формой введения. С успехом можно включать адъюванты, которые обычно используют при приготовлении фармацевтических композиций, такие как вкусовые агенты, красители, консерванты и антиоксиданты и т.д., например витамин Е, аскорбиновая кислота, ВНТ и ВНА.
Предпочтительными фармацевтическими композициями с точки зрения простоты приготовления и введения являются твердые композиции, особенно таблетки и капсулы с твердым или жидким содержанием. Предпочтительно пероральное введение соединений. Эти активные соединения можно также вводить парэнтерально или внутрибрюшинно. Растворы или суспензии этих активных соединений в виде свободных оснований или фармакологически приемлемых солей можно получить в воде, соответствующим образом смешанной с поверхностно-активным агентом, таким как гидроксипропилцеллюлоза. Дисперсии можно также приготовить в глицерине, в жидких полиэтиленгликолях и их смесях в маслах. В обычных условиях хранения и использования эти препараты могут содержать консерванты для предотвращения роста микроорганизмов.
Фармацевтические формы для использования в виде инъекций включают стерильные водные растворы или дисперсии и стерильные порошки для приготовленных для немедленного введения препаратов стерильных растворов или дисперсий для инъекций. Во всех случаях форма должна быть стерильной и должна быть жидкой до такой степени, чтобы ее можно было легко вводить с помощью шприца. Она должна быть стабильной в условиях изготовления и хранения и должны быть приняты меры, предотвращающие загрязнение микроорганизмами, такими как бактерии и грибки. Носителем может быть растворитель или диспергирующая среда, содержащая, например, воду, этанол, полиол (например, глицерин, пропиленгликоль и жидкий полиэтиленгликоль), их подходящие смеси и растительные масла.
Используемая эффективная доза активного ингредиента может меняться в зависимости от конкретного используемого соединения, способа введения и тяжести состояния, подлежащего лечению. Однако обычно удовлетворительные результаты достигаются, если соединения настоящего изобретения вводят в дневной дозе от около 0,1 мг/кг до около 400 мг/кг веса тела животного, предпочтительно вводимой в разделенных дозах от двух до четырех раз в день. Для большинства крупных млекопитающих полная дневная доза составляет от около 0,07 г до 7,0 г, предпочтительно от около 100 мг до 1000 мг. Дозовые формы, пригодные для внутреннего использования, включают от около 100 мг до 500 мг активного соединения, тщательно смешанного с твердым или жидким фармацевтически приемлемым носителем. Такой дозовый режим можно установить для обеспечения оптимальной терапевтической реакции. Например, несколько разделенных доз можно вводить ежедневно или дозу можно пропорционально уменьшить в соответствии с показаниями терапевтической ситуации.
Получение вышеуказанных фармацевтических композиций и лекарств можно осуществить любым из известных специалистам способов, например, смешивая активный ингредиент (ингредиенты) с разбавителем (разбавителями) для получения фармацевтической композиции (например, гранулята), а затем формируя композицию в виде лекарств (например, в виде таблеток).
Нижеприведенные примеры подробно раскрывают способы химического синтеза представительных соединений настоящего изобретения. Эти способы являются иллюстрациями и не следует рассматривать изобретение как ограниченное химическими реакциями и условиями, которые в них указаны. Не было предпринято попыток оптимизировать выходы, достигаемые в этих реакциях, и специалистам должно быть очевидно, что, варьируя времена реакций, температуры, растворители и/или реагенты, выходы можно увеличить.
Пример 1
(5R)-3-[4-(1,3-Дигидро-1-оксо-2Н-изоиндол-2-ил)-3-фторфенил]-5-(гидроксиметил)-2-оксазолидинон
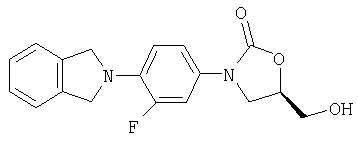
Изоиндолин синтезируют, используя способ R.Е.Gawley, S.R.Chemburkar, A.L.Smith, Т.V.Anklekar J. Org. Chem. 1983, 53, 5381.
Стадия 1:
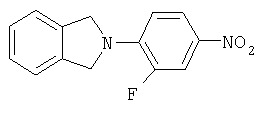
К 3,4-дифторнитробензолу (3,02 мл, 27,3 ммоль) в этилацетате при комнатной температуре добавляют диизопропилэтиламин (5,03 мл, 28,9 ммоль), затем изоиндолин (3,50 г, 29,4 ммоль) и перемешивают в течение ночи. Образующийся желтый осадок (ppt) собирают на фильтре, промывают водой и эфиром и сушат в вакуумном термостате (30°С), получая продукт в виде твердого вещества желтого цвета (6,69 г, 95% выход). Т. плавления = 200-202°С. Масс-спектр (М+1)=327 m/z.
Стадия 2:
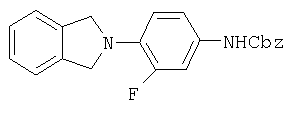
К вышеуказанному нитросоединению (2,62 г, 10,2 ммоль) в этаноле (100 мл) добавляют SnCl2 (9,84 г, 50,9 ммоль) и кипятят с обратным холодильником в течение 16 час. После охлаждения до комнатной температуры реакционную смесь добавляют к 10% водному раствору NaOH (300 мл) и экстрагируют СН2Cl2 (6×50 мл). Объединенные органические экстракты промывают рассолом (100 мл), сушат над Na2SO4 и концентрируют, получая 2,63 г оливково-зеленого твердого вещества (анилин), который используют без дальнейшей очистки. К этому анилину в ацетоне (150 мл) и воде (20 мл) добавляют NaHCO3 (1,84 г, 21,9 ммоль) и затем бензилхлорформиат (1,68 мл, 11,8 ммоль). После перемешивания в течение ночи смесь выливают в ледяную воду (100 мл) и полученный желто-коричневый осадок собирают на фильтре, промывают водой и сушат в вакууме, получая Cbz анилин в виде желто-коричневого твердого вещества (3,50 г, 95% выход). Т. плавления = 146-148°С. Масс-спектр (М+1)=363 m/z.
Стадия 3:
К вышеуказанному Cbz анилину (0,74 г, 2,04 ммоль) в ТГФ (10 мл) при -78°С по каплям добавляют н-BuLi (2,5 М, 0,82 мл, 2,05 ммоль). После перемешивания в течение 40 минут по каплям добавляют (R)-глицидилбутират (0,31 мл, 2,10 ммоль) в ТГФ (0,5 мл) и полученную смесь оставляют выстаиваться при комнатной температуре в течение ночи. Образовавшийся белый осадок собирают на фильтре и промывают водой и эфиром. В результате обработки на хроматографической колонке с силикагелем 25% смесью этилацетат/гексан в качестве элюента получают продукт в виде белого твердого вещества (0,58 г, 87% выход). Масс-спектр (М+1)=329 m/z.
Пример 2
(5R)-3-[4-(1,3-дигидро-1-оксо-2Н-изоиндол-2-ил)-3-фторфенил]-5-[[(метилсульфонил)окси]метил]-2-оксазолидинон
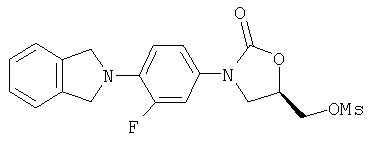
К оксазолидинонкарбинолу, полученному в примере 1 (0,58 г, 1,78 ммоль) в ДМФ (10 мл), и ацетонитрилу (10 мл) при 0°С добавляют триэтиламин (0,74 мл, 5,31 ммоль) и через 10 мин добавляют метансульфонилхлорид (0,28 мл, 3,62 ммоль). Реакционную смесь оставляют на 1 час при комнатной температуре и после этого все еще присутствует исходный материал, поэтому повторяют охлаждение и добавление триэтиламина (0,37 мл, 2,65 ммоль) и метансульфонилхлорида (0,14 мл, 1,81 ммоль). Эту смесь выливают в воду (50 мл) и экстрагируют CH2Cl2 (6×20 мл), промывают рассолом (4×10 мл), сушат над Na2SO4 и концентрируют, получая сырой продукт в виде масла коричневого цвета (0,95 g). Масс-спектр (М+1)=407 m/z.
Пример 3
(5R)-5-(азидометил)-3-[4-(1,3-дигидро-1-оксо-2Н-изоиндол-2-ил)-3-фторфенил]-2-оксазолидино
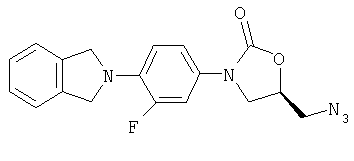
К мезилату, полученному в примере 2 (0,95 г, 1,78 ммоль) в ДМФ (25 мл), добавляют азид натрия (0,47 г, 7,23 ммоль) и нагревают при 70°С в течение 16 часов. После охлаждения до комнатной температуры добавляют воду, и полученную смесь экстрагируют этилацетатом (6×25 мл), промывают рассолом (4×10 мл), сушат над Na2SO4 и концентрируют, получая 0,48 г твердого вещества желто-коричневого цвета. Масс-спектр (М+1)=354 m/z.
Пример 4
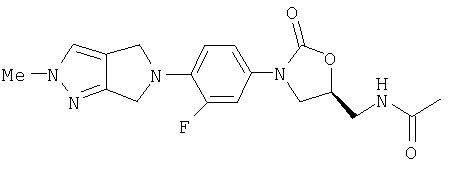
N-[[(5S)-3-[4-(1,3-дигидро-2Н-изоиндол-2-ил)-3-фторфенил]-2-оксо-5-оксазолидинил]метил]ацетамид
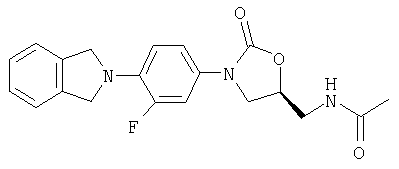
Соединение 1
Азид, полученный в примере 3, в этилацетате (25 мл) помещают в склянку Паара и через нее барботируют азот в течение 15 мин, после чего добавляют 10% Pd/С (0,15 г, 0,14 ммоль) для смеси создают давление 50 пси Н2 (г) и встряхивают в течение 16 час, после чего добавляют дополнительное количество 10% Pd/C (0,15 г, 1,4 ммоль) и полученную смесь встряхивают дополнительно в течение 6 час (в этот момент масс-спектр (М+1)=328 m/z). После того как смесь помещают в атмосферу азота, добавляют пиридин (0,22 мл, 2,72 ммоль) и затем Ас2О (0,51 мл, 5,30 ммоль), и полученную смесь перемешивают в течение 2 час. Смесь фильтруют через целит, промывают этилацетатом (100 мл), концентрируют и очищают на хроматографической колонке с силикагелем (градиентное элюирование 1%-5% МеОН/СН2Cl2), затем тщательно растирают с этилацетатом (3×3 мл), в результате чего получают 0,19 г твердого вещества белого цвета (Соединение 1, 29% выход за 4 стадии). Т. плавления = 240-242°С. Масс-спектр (М+1)=370 m/z.
Пример 5
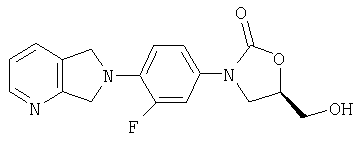
Соединение 2
Стадия 1:
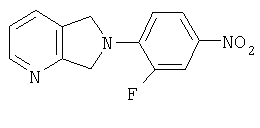
6,7-дигидро-6-(2-фтор-4-нитрофенил)-5Н-пирроло[3,4-b]пиридин: К дигидрохлоридной соли 6,7-дигидро-5Н-пирроло-[3,4-b]пиридина (как раскрыто у Petersen с сотр. (Bayer) ЕР0520277А2) (42,8 г, 222 ммоль) в ДМФ (1/2 л) добавляют 2,4-дифторнитробензол (25 мл, 224 ммоль). Смесь нагревают до 60°С и по каплям добавляют DIPEA (195 мл, 1,12 моль) через капельную воронку в течение 2 час. После нагревания в течение ночи реакционную смесь охлаждают до комнатной температуры, выливают в воду (3 л), фильтруют и сушат в вакуумном термостате (50°С), получая твердое вещество желто-зеленого цвета (53,8 г, 94% выход). Масс-спектр (М+1)=260 m/z.
Стадия 2:
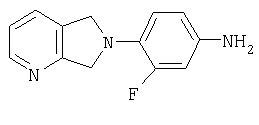
6,7-дигидро-6-(2-фтор-4-аминофенил)-5Н-пирроло [3,4-b]-пиридин
К полученному выше нитросоединению (53,8 г, 208 ммоль) в ТГФ (175 мл) и метанолу (600 мл) добавляют формиат аммония (59,0 г, 907 ммоль). Через реакционную смесь барботируют азот в течение примерно 30 минут, после чего добавляют 10% Pd/C (2/20 г, 21 ммоль). После перемешивания в течение ночи при комнатной температуре в атмосфере азота реакционную смесь фильтруют через слой целита, тщательно промывают метанолом (400 мл) и концентрируют до объема примерно 200 мл. Добавляют воду (300 мл) и смесь экстрагируют этилацетатом (5×200 мл). Обьединенные органические слои промывают рассолом, сушат (Na2SO4), фильтруют и используют непосредственно на следующей стадии без дополнительной очистки. Масс-спектр (М+1)=230 m/z.
Стадия 3:
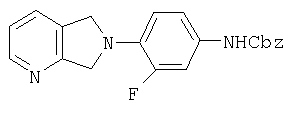
6,7-дигидро-6-(2-фтор-4-(аминокарбоксибензил)фенил)-5Н-пирроло-[3,4-b]пиридин
Полученный выше анилин (примерно 208 ммоль) в ацетоне (1 л) и воде (160 мл) охлаждают до 0°С, после чего добавляют бикарбонат натрия (37,4 г, 445 ммоль), а затем по каплям добавляют бензилхлорформиат (34,2 мл, 228 ммоль). Реакционную смесь оставляют нагреться до комнатной температуры и перемешивают в течение ночи, после чего образуется осадок. Реакционную смесь выливают в ледяную воду (2 л) и полученный осадок собирают фильтрованием. Полученное твердое вещество промывают водой и сушат в вакуумном термостате (50°С), получая Cbz производное (73,0 г, 97% выход) в виде порошка цвета само. Масс-спектр (М+1)=364 m/z.
Стадия 4:
(Соединение 2)
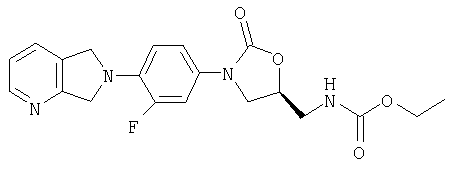
Полученное выше производное Cbz (40,8 г, 112 ммоль) в ТГФ (1 л) охлаждают до -78°С в атмосфере азота. К этой смеси по каплям через шприц добавляют n-BuLi (2,5 М, 45,8 мл, 114,5 ммоль) в течение 15 минут. Реакционную смесь нагревают до комнатной температуры и оставляют при перемешивании на 45 минут, прежде чем снова охлаждают до -78°С. В этот момент добавляют (R)-глицидил бутират (17,2 мл, 117 ммоль) и реакционную смесь оставляют нагреваться до комнатной температуры в течение ночи, при этом образуется осадок. Осадок собирают, промывают несколькими порциями эфира (5×100 мл) и сушат в вакуумном термостате (50°С), получая 40,6 г эфирного сольвата алкоксида лития в виде пушистого порошка желто-коричневого цвета. Этот материал промывают несколькими порциями воды (4×200 мл) и сушат в вакуумном термостате (50°С), получая оксазолидиноновый спирт (34,1 г, 92% выход) в виде гранулированного твердого вещества желто-коричневого цвета. Т. плавления = 208-212°С с разложением. Масс-спектр (М+1)=330 m/z.
Пример 6
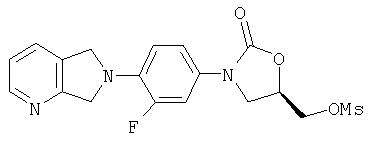
Оксазолидинон мезилат. Полученный выше оксазолидинонкарбинол (из примера 4) (33,8 г, 103 ммоль) суспендируют в ДМФ (1,25 л, предварительно дегазированный азотом) при комнатной температуре в атмосфере азота. Добавляют триэтиламин (50 мл, 360 ммоль), а затем по каплям добавляют метансульфонилхлорид (13,5 мл, 174 ммоль). После перемешивания в течение 3 часов реакционную смесь выливают в воду (200 мл) и добавляют метиленхлорид (1 л). Осадок отфильтровывают, промывают водой (3×200 мл) и сушат в вакуумном термостате (50°С), получая мезилат в виде твердого вещества желто-коричневого цвета (28,1 г, 67%). Органический слой сушат (Na2SO4), фильтруют и выпаривают, в результате чего также получают мезилат (11,7 г, 28% выход) в виде твердого вещества желто-коричневого цвета. Оба их характеризуют с помощью масс-спектроскопии. Масс-спектр (М+1)=408 m/z.
Пример 7
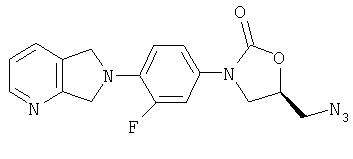
Оксазолидинон азид. Полученный выше мезилат (из примера 5) (27,8 г, 68,2 ммоль) и азид натрия (17,7 г, 271 ммоль) в безводном ДМФ (1 л), дегазированном ранее азотом, нагревают при 95°С в течение 6 часов в атмосфере азота. После охлаждения смесь выливают в перемешиваемую ледяную воду (2 л), в результате чего образуется пушистый белый осадок. Осадок собирают на фильтровальной бумаге, промывают водой (4×200 мл), сушат в вакуумном термостате (50°С), получая азид в виде твердого вещества светло-бежевого цвета (22,7 г, 94% выход). Т. плавления = 175-180°С с разложением. Масс-спектр (М+1)=355 m/z.
Пример 8
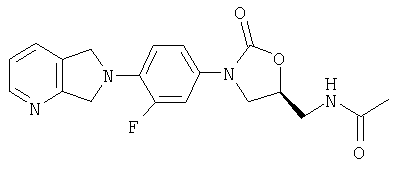
Соединение 3
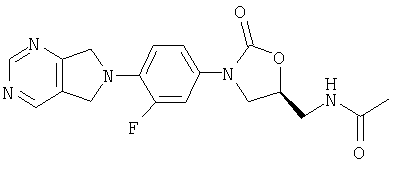
Оксазолидинонацетамид. Полученный выше азид (из примера 6) (21,67 г, 61,16 ммоль), растворенный в ДМФ (400 мл) и ТГФ (500 мл), дегазируют азотом в течение 30 минут, после чего добавляют 10% Pd/C (4,74 г, 4,4 ммоль) и реакционную смесь гидрируют в аппарате Парра (60 пси водорода) в течение 14 час. Реакционную смесь удаляют из аппарата Парра и помещают в атмосферу азота, после чего добавляют пиридин (5,44 мл, 67,3 ммоль) и уксусный ангидрид (6,35 мл, 67,3 ммоль). После перемешивания в течение 1 часа реакционную смесь фильтруют через слой целита, тщательно промывают метанолом и затем большими количествами 50% MeOH/CH2Cl2 (примерно 2 л). Полученный фильтрат выпаривают, получая неочищенный ацетамид в ДМФ. Смесь медленно добавляют к воде (2 л) и осадок собирают на фильтре, промывают водой (5×400 мл) и сушат в вакуумном термостате (50°С), получая ацетамид в виде аналитически чистого твердого вещества белого цвета (14,2 г, 63% выход). Объединенные фильтраты экстрагируют метиленхлоридом (5×200 мл), сушат над Na2SO4 и концентрируют. К остатку добавляют воду и полученный осадок отфильтровывают и сушат в вакуумном термостате (50°С), получая вторую порцию ацетамида в виде пушистого твердого вещества светло-желто-коричневого цвета (5,61 г, 25%). Для аналитически чистого материала Т. плавления = 229-230°С с разложением. Масс-спектр (М+1)=371 m/z.
Пример 9
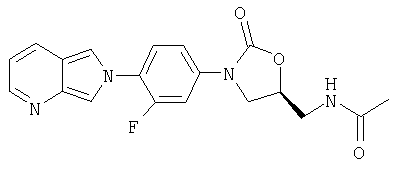
Соединение 4
Полученный выше ацетамид из примера 8 (2,51 г, 6,78 ммоль) помещают в СН2Cl2 и добавляют MnO2 (23,9 г, 234 ммоль). После перемешивания в течение ночи реакционную смесь фильтруют через целит, концентрируют и обрабатывают на хроматографической колонке с силикагелем, элюируя 10% MeOH/CH2Cl2 и получая продукт в виде твердого вещества светло-желтого цвета (0,48 г, 19% выход). Т. плавления = 220-225°С с разложением. Масс-спектр (М+1)=369 m/z.
Пример 10
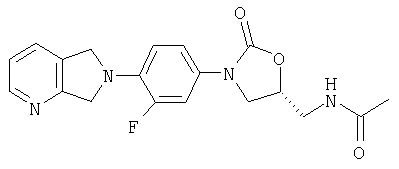
Соединение 5
Соединение 5 получают по способу примера 8 за исключением того, что при образовании оксазолидинона используют (S)-глицидилбутират. Продукт выделяют в виде твердого вещества светло-желто-коричневого цвета. Т. плавления = 227-230°С с разложением. Масс-спектр (М+1)=371 m/z.
Пример 11
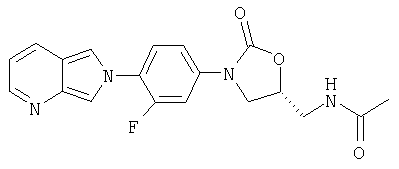
Соединение 6. Окисленный энантиомер
Соединение 6 получают по способу примера 9 и выделяют в виде твердого вещества светло-желтого цвета. Т. плавления = 181-185°С с разложением. Масс-спектр (М+1)=369 m/z.
Пример 12
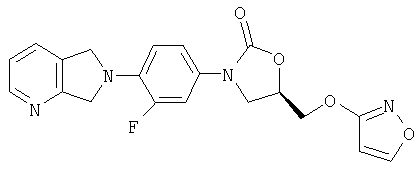
Соединение 7
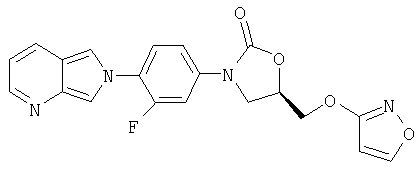
К 5-гидроксиизоксазолу (полученному по способу Chem Pharm Bull 1966, 14(11), 1277) (0,174 г, 2,04 ммоль) в ДМФ добавляют NaH (60% в масле) (0,105 г, 2,62 ммоль). После перемешивания в течение 30 минут одной порцией добавляют мезилат (из примера 6) (0,744 г, 1,82 ммоль) и полученную смесь перемешивают при 60°С в течение ночи. После охлаждения до комнатной температуры добавляют воду и осадок собирают на фильтре, сушат воздухом и обрабатывают на хроматографической колонке с силикагелем, элюируя 2,5% MeOH/CH2Cl2, в результате чего получают продукт в виде твердого вещества белого цвета (0,140 г, 19 % выход). Т. плавления = 182-185°С. Масс-спектр (М+1)=397 m/z.
Пример 13
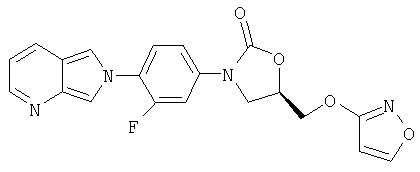
Соединение 8
К полученному выше оксазолидинону (из примера 12) (0,264 г, 6,66 ммоль), помещенному в CH2Cl2, добавляют MnO2 (1,66 г, 16,2 ммоль) двумя порциями в течение двух дней. После перемешивания в течение двух дней реакционную смесь фильтруют через целит, концентрируют и обрабатывают на хроматографической колонке с силикагелем, элюируя 10% MeOH/CH2Cl2 и получая продукт в виде твердого вещества светло-желтого цвета (0,086 г, 32% выход). Т. плавления = 133-135°С. Масс-спектр (М+1)=395 m/z.
Пример 14
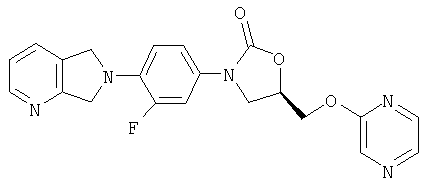
Соединение 9
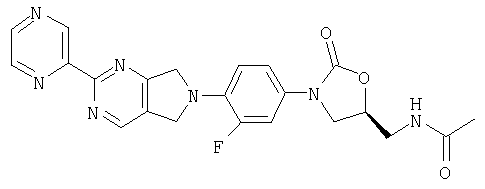
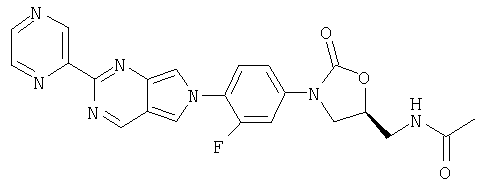
К NaH (60% по весу в масле) (0,03 г, 0,76 ммоль) в ДМФ (5 мл) добавляют оксазолидинонкарбинол (из примера 5) (0,23 г, 0,71 ммоль) четырьмя порциями. После перемешивания в течение 30 минут через шприц добавляют 2-хлорпиразин (0,065 мл, 0,71 ммоль) и перемешивают в течение ночи при комнатной температуре. Добавляют воду и осадок собирают на фильтре, сушат воздухом и обрабатывают на хроматографической колонке с силикагелем, элюируя 5% MeOH/CH2Cl2, получая продукт в виде твердого вещества белого цвета (0,067 г, 23 % выход). Т. плавления = 225-230°С. Масс-спектр (М+1)=408 m/z.
Пример 15

Соединение 10
К полученному выше оксазолидинону (из примера 14) (0,024 г, 0,058 ммоль) в CH2Cl2 (5 мл) добавляют MnO2 (0,07 г, 0,7 ммоль). После перемешивания в течение ночи реакционную смесь фильтруют через целит и концентрируют, получая продукт в виде твердого вещества очень светло-желтого цвета (0,015 г, 64% выход). Т. плавления = 192-194°С. Масс-спектр (М+1)=406 m/z.
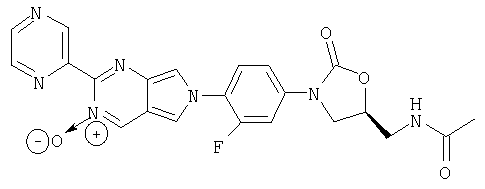
Пример 16
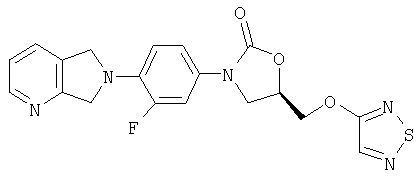
Соединение 11
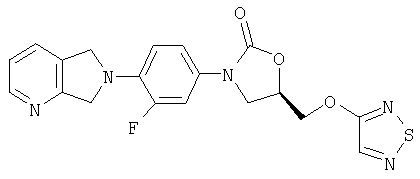
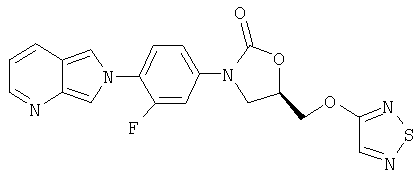
К суспензии оксазолидинонкарбинола (получен в примере 5) (330 мг, 1,0 ммоль), трифенилфосфина (260 мг, 1,1 ммоль) и 4-гидрокси-1,2,5-тиадиазола (100 мг, 1,0 ммоль) (получен по способу примера патента США 3391150 [7.2.68]) в ТГФ (8 мл) добавляют диизопропилазодикарбоксилат (0,20 мл, 1,1 ммоль). После перемешивания в течение ночи при комнатной температуре реакционную смесь фильтруют, промывают метанолом и сушат воздухом, получая кристаллическое твердое вещество желтого цвета (60 мг, 15% выход). Т. плавления = 185-187°С. Масс-спектр (М+1)=414 m/z.
Пример 17
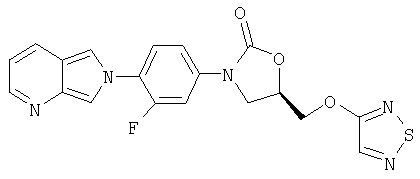
Соединение 12
К оксазолидинону (получен в примере 16) (160 мг, 0,39 ммоль), суспендированному в CH2Cl2 (1,0 мл), добавляют MnO2 (четырьмя порциями по 150 мг в течение четырех дней). Реакционную смесь фильтруют через слой целита, промывают СН2Cl2 (15 мл), концентрируют при пониженном давлении, получая продукт в виде твердого кристаллического вещества белого цвета (63 мг, 40% выход). Т. плавления = 185-188°С. Масс-спектр (М+1)=412 m/z.
Пример 18
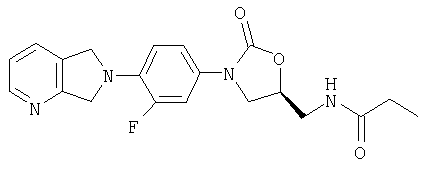
Соединение 13
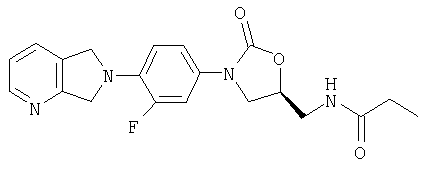
К амину (полученному в примере 8) (100 мг, 0,30 ммоль) и карбонату калия (100 мг, 0,72 ммоль), суспендированному в метаноле (1,0 мл), добавляют пропионилхлорид (50 мг, 0,54 ммоль). После перемешивания в течение ночи при 80°С реакционную смесь охлаждают и добавляют воду. Осадок отфильтровывают, промывают метанолом и сушат воздухом, получая продукт в виде кристаллического твердого вещества коричневого цвета (15 мг, 13 % выход). Т. плавления = 110-112°С. Масс-спектр (М+1)=385 m/z.
Пример 19
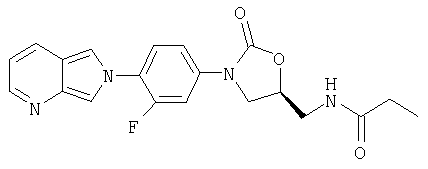
Соединение 14
К амиду (получен в примере 18) (15 мг, 0,04 ммоль), суспендированному в СН2Cl2 (1,0 мл), добавляют MnO2 (200 мг) при комнатной температуре. После перемешивания в течение ночи реакционную смесь фильтруют через слой целита, промывают CH2Cl2 (10 мл) и концентрируют при пониженном давлении, получая продукт в виде кристаллического твердого вещества светло-коричневого цвета (1,6 мг, 8 % выход). Масс-спектр (М+1)=383 m/z.
Пример 20
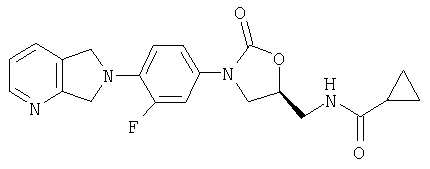
Соединение 15
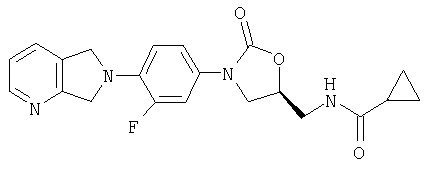
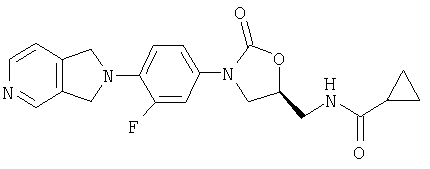
К амину (полученному по способу примера 8) (60 мг, 0,18 ммоль) и ацетату калия (60 мг, 0,61 ммоль), суспендированному в метаноле (1,0 мл), добавляют циклопропилкарбонилхлорид (120 мг, 1,15 ммоль). После перемешивания при комнатной температуре в течение ночи реакционную смесь фильтруют, промывают метанолом и затем концентрируют досуха при пониженном давлении. Полученный твердый остаток тщательно растирают с водой и фильтруют, получая продукт в виде кристаллического твердого вещества коричневого цвета (36 мг, 50% выход). Т. плавления = 235-240°С. Масс-спектр (М+1)=397 m/z.
Пример 21
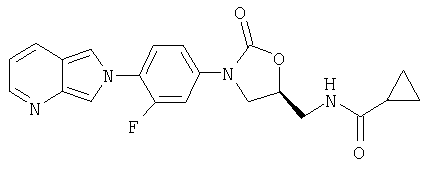
Соединение 16
К амиду (получен в примере 20) (36 мг, 0,09 ммоль), суспендированному в CH2Cl2 (1,0 мл), добавляют MnO2 (тремя порциями по 100 мг в течение трех дней) при комнатной температуре. Реакционную смесь фильтруют через слой целита, промывают CH2Cl2 (10 мл) и концентрируют при пониженном давлении, получая продукт в виде кристаллического твердого вещества грязно-белого цвета (3 мг, 8 % выход). Масс-спектр (М+1)=395 m/z.
Пример 22
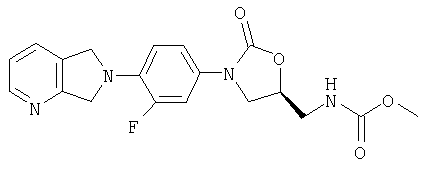
Соединение 17
К амину (получен в примере 8) (60 мг, 0,18 ммоль) и ацетату калия (60 мг, 0,61 ммоль), суспендированному в метаноле (1,0 мл), добавляют по каплям метилхлорформиат (120 мг, 1,27 ммоль). После перемешивания в течение четырех часов при комнатной температуре реакционную смесь фильтруют, разбавляют водой и концентрируют при пониженном давлении, удаляя метанол. Водный раствор экстрагируют этилацетатом (5×5 мл). Объединенные органические экстракты промывают водой, сушат над MgSO4, фильтруют и концентрируют, получая продукт в виде масла, которое тщательно растирают с эфиром, получая кристаллическое твердое вещество коричневого цвета (35 мг, 50% выход). Масс-спектр (М+1)=387 m/z.
Пример 23
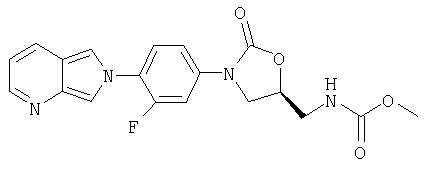
Соединение 18
К карбамату (получен в примере 22) (33 мг, 0,08 ммоль), суспендированному в СН2Cl2 (1,0 мл), добавляют MnO2 (150 мг). После перемешивания в течение ночи при комнатной температуре реакционную смесь фильтруют через слой целита, промывают CH2Cl2 (10 мл) и концентрируют при пониженном давлении, получая продукт в виде кристаллического твердого вещества желтого цвета (6,0 мг, 18% выход). Масс-спектр (М+1)=385 m/z.
Пример 24
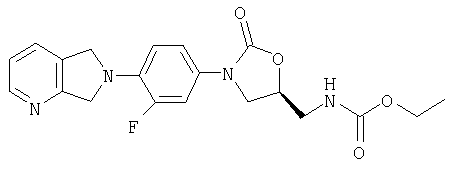
Соединение 19
К амину (получен в примере 8) (60 мг, 0,18 ммоль) и ацетату калия (60 мг, 0,61 ммоль), суспендированному в метаноле (1,0 мл), по каплям добавляют этилхлорформиат (0,1 мл, 1,04 ммоль). После перемешивания в течение ночи при комнатной температуре реакционную смесь фильтруют, разбавляют водой и концентрируют при пониженном давлении, удаляя метанол. Водный раствор экстрагируют этилацетатом (5×5 мл). Объединенные органические экстракты промывают водой, сушат над MgSO4, фильтруют и концентрируют. Полученное полутвердое вещество обрабатывают водой, фильтруют и сушат воздухом, получая кристаллическое твердое вещество коричневого цвета (18 мг, 30% выход). Масс-спектр (М+1)=401 m/z.
Пример 25
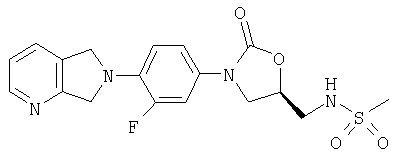
Соединение 20
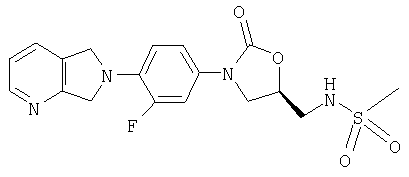
К амину (получен в примере 8) (95 мг, 0,29 ммоль), суспендированному в пиридине (0,5 мл), добавляют метансульфонилхлорид (0,08 мл, 1,0 ммоль). После перемешивания в течение ночи при комнатной температуре пиридин удаляют в атмосфере азота. Остаток обрабатывают водой, фильтруют и сушат воздухом, получая твердое вещество коричневого цвета (45 мг, 38% выход). Т. плавления = 172-176°С. Масс-спектр (М+1)=407 m/z.
Пример 26
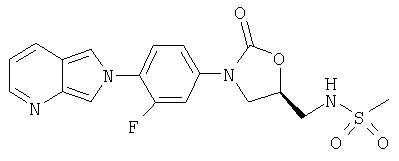
Соединение 21
К сульфонамиду (получен в примере 25) (10 мг, 0,02 ммоль), суспендированному в CH2Cl2 (1,0 мл), добавляют MnO2 (100 мг, 10 ммоль). После перемешивания в течение ночи реакционную смесь фильтруют через слой целита, промывают CH2Cl2 (10 мл) и концентрируют при пониженном давлении, получая продукт в виде кристаллического твердого вещества коричневого цвета (0,5 мг, 5% выход). Масс-спектр (М+1)=405 m/z.
Пример 27
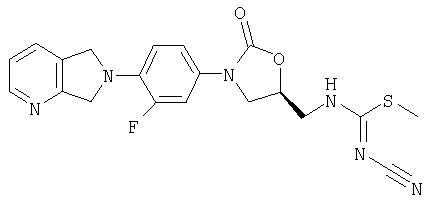
Соединение 22
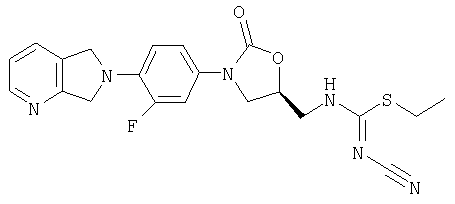
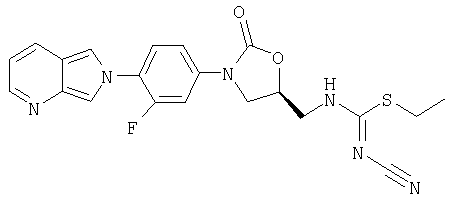
К амину (получен в примере 8) (200 мг, 0,61 ммоль), суспендированному в толуоле (8 мл), добавляют диметил-N-цианодитиоиминокарбонат (89 мг, 0,61 ммоль). После перемешивания в течение ночи при кипении с обратным холодильником толуол декантируют и маслянистый осадок обрабатывают метанолом, фильтруют и сушат воздухом, получая кристаллическое твердое вещество коричневого цвета (62 мг, 20% выход). Т. плавления = 204-207°С. Масс-спектр (М+1)=427 m/z.
Пример 28
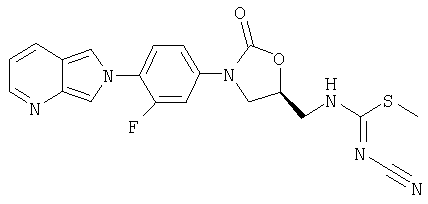
Соединение 23
Суспензию тиоимидата (из примера 27) (45 мг, 0,10 ммоль) и MnO2 (200 мг, 2,0 ммоль) в CH2Cl2 перемешивают при комнатной температуре в течение одного дня, после чего второй раз добавляют MnO2 (150 мг, 1,5 ммоль). После еще одного дня перемешивания смесь фильтруют через целит, промывают CH2Cl2 (10 мл) и концентрируют, получая кристаллическое твердое вещество желтого цвета (20 мг, 45% выход). Масс-спектр (М+1)=426 m/z.
Пример 29
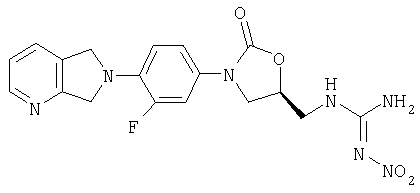
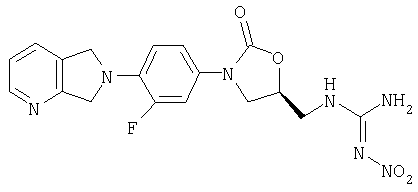
Соединение 24
Суспензию амина (получен в примере 8) (165 мг, 0,5 ммоль) и 2-метил-1-нитро-2-тиопсевдомочевины (94 мг, 0,70 ммоль) (получена по способу ЕР 0539204/1993) в метаноле (2 мл) кипятят с обратным холодильником в течение четырех часов. После охлаждения до комнатной температуры реакционную смесь фильтруют и сушат воздухом, получая кристаллическое твердое вещество желтого цвета (50 мг, 24% выход). Т. плавления = 202-206°С. Масс-спектр (М+1)=416 m/z.
Пример 30
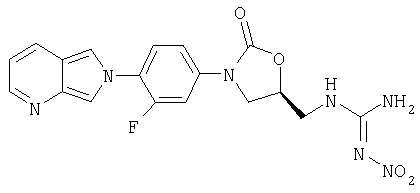
Соединение 25
К нитрогуанидину (получен в примере 29) (35 мг, 0,08 ммоль), суспендированному в CH2Cl2 (1,0 мл), добавляют MnO2 (тремя порциями по 100 мг в течение трех дней). Реакционную смесь фильтруют через слой целита, промывают CH2Cl2 (10 мл) и концентрируют при пониженном давлении, получая продукт в виде кристаллического твердого вещества желтого цвета (1,6 мг, 4% выход). Масс-спектр (М+1)=414 m/z.
Пример 31
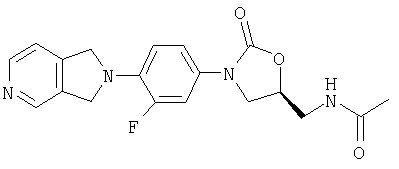
Соединение 26
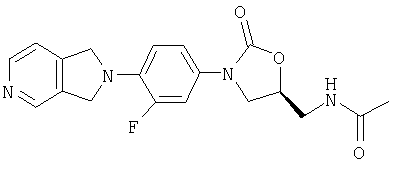
Исходный материал, 6,7-дигидро-5Н-пирроло[3,4-с]пиридин, получают по способу патента США № 5371090, выданного Petersen с сотр. Затем получают соединение 26 по способу примера 8 за исключением того, что ацетамид перекристаллизовывают из ацетонитрила, получая твердое вещество светло-желто-коричневого цвета. Т. плавления = 182-190°С с разложением. Масс-спектр (М+1)=371 m/z.
Пример 32
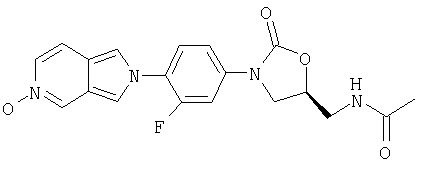
Соединение 27
Соединение 27 выделяют из конечной стадии примера 31 с помощью хроматографической обработки (5% MeOH/CH2Cl2 в качестве элюента) маточного раствора, полученного при перекристаллизации. Получают твердое вещество светло-желтого цвета, Т. плавления = 219-225°С с разложением. Масс-спектр (М+1)=385 m/z.
Пример 33
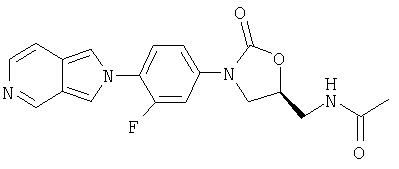
Соединение 28
Соединение 28 получают по способу примера 9 за исключением того, что в качестве элюента используют 10% CH2Cl2. Получают твердое вещество светло-желтого цвета. Т. плавления = 219-225°С с разложением. Масс-спектр (М+1)=369 m/z.
Пример 34
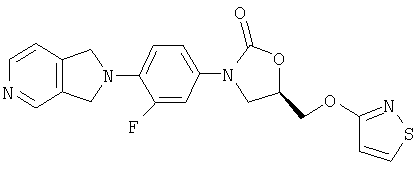
Соединение 29

Соединение 30
Изотиазол (0,088 г, 0,87 ммоль)(полученный по способу J. Heterocyclic Chem 1971, 8, 591) добавляют порциями при комнатной температуре к суспензии гидрида натрия (0,036 г, 0,91 ммоль, 60 в масле) в ДМФ (4 мл) в атмосфере азота. Смесь перемешивают в течение 30 минут, после чего добавляют сразу весь мезилат, полученный в примере 31 (0,31 г, 0,76 ммоль), в ДМФ (10 мл). После перемешивания в течение 6 час при 60°С реакционную смесь охлаждают до комнатной температуры, разбавляют водой (50 мл) и экстрагируют этилацетатом (3×50 мл). Объединенные органические экстракты промывают несколько раз водой, затем один раз рассолом, сушат над сульфатом натрия, концентрируют и обрабатывают на хроматографической колонке с силикагелем, элюируя 5% MeOH/EtOAc. В результате хроматографической обработки выделяют два продукта: 0,050 г соединения 29 и 0,022 г соединения 30. Полный выход 30%.
Соединение 29. Масс-спектр (М+1)=413,0.
Соединение 30. Масс-спектр (М+1)=411,1.
Пример 35
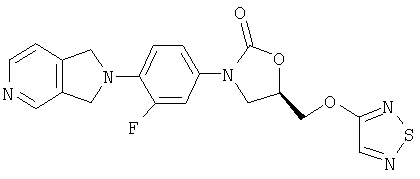
Соединение 31
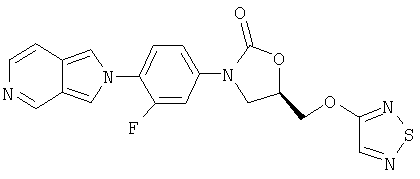
Соединение 32
К суспензии гидрида натрия (0,036 г, 0,91 ммоль, 60% в масле) в ДМФ (4 мл) при комнатной температуре в атмосфере азота добавляют порциями 4-гидрокси-1,2,5-тиадиазол (0,088 г, 0,87 ммоль) (получен по способу патента США 3391150 [7/2/68]). После перемешивания в течение 30 минут добавляют сразу весь мезилат, полученный в примере 31 (0,310 г, 0,76 ммоль) в ДМФ (10 мл). После перемешивания в течение 6 часов при 60°С реакционную смесь охлаждают до комнатной температуры, разбавляют водой (50 мл) и экстрагируют этилацетатом (3×50 мл). Объединенные органические экстракты промывают несколько раз водой, затем один раз рассолом, сушат над сульфатом натрия, концентрируют и обрабатывают на хроматографической колонке с силикагелем, элюируя 2% MeOH/EtOAc. В результате хроматографической обработки получают два продукта: 0,035 г Соединения 31 и 0,0093 г Соединения 32. Полный выход - 14%.
Соединение 31. Масс-спектр (М+1)=414,0.
Соединение 32. Масс-спектр (М+1)=412,1.
Пример 36
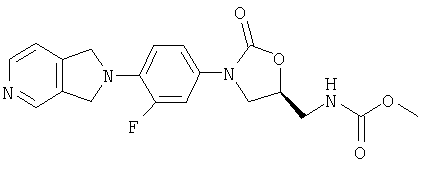
Соединение 33
Стадия 1:
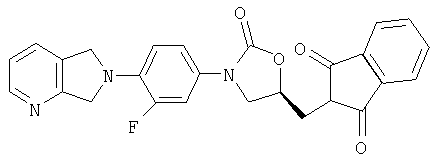
К мезилату, полученному в примере 31 (2,45 г, 6,01 ммоль), растворенному в дегазированном ДМФ (100 мл) в атмосфере азота, добавляют фталимид калия (2,23 г, 12,0 ммоль). После нагревания при 65°С в течение 3 часов реакционную смесь охлаждают, выливают в воду (300 мл) и экстрагируют метиленхлоридом (3×200 мл). Объединенные органические экстракты промывают водой (3×150 мл), сушат над сульфатом натрия и концентрируют, получая твердое вещество желто-коричневого цвета. Это твердое вещество промывают водой и сушат в термостате с высоким вакуумом при 50°С, получая 2,20 г ( 80%) фталимида оксазолидинона. Масс-спектр = 459,1 (М+1).
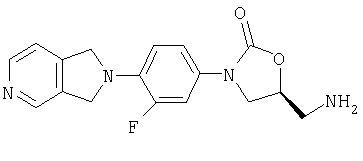
Стадия 2:
К полученному выше фталимиду (0,97 г, 2,1 ммоль) в дегазированном метаноле (30 мл) в атмосфере азота по каплям добавляют моногидрат гидразина (0,2 мл, 4,3 ммоль). После кипячения с обратным холодильником в течение 12 часов реакционную смесь охлаждают до комнатной температуры, концентрируют, суспендируют в CH2Cl2 и фильтруют. Сырой оксазолидинонамин концентрируют и используют без дальнейшей очистки.
Стадия 3:
Соединение 33
К сырому амину (0,14 г, 0,44 ммоль) в CH2Cl2 (5 мл) добавляют пиридин (0,14 мл, 18 ммоль), а затем пропионилхлорид (0,76 мл, 0,88 ммоль). После перемешивания в течение 5 часов при комнатной температуре раствор выливают в воду (20 мл) и экстрагируют метиленхлоридом (3×10 мл). Объединенные экстракты промывают водой (10 мл) и 1 М NaOH (водн.) (10 мл), сушат над сульфатом натрия, концентрируют и обрабатывают на хроматографической колонке с силикагелем, используя в качестве элюента чистый EtOAc и получая пропиониламид в виде золотистого масла (0,020 г, 12% выход). Масс-спектр = 385,2 (М+1).
Пример 37
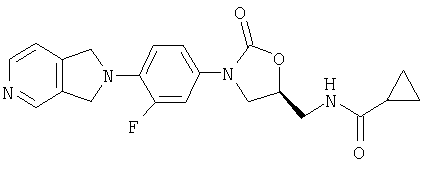
Соединение 34
К сырому амину (получен по способу примера 36) (0,144 г, 0,437 ммоль) в метиленхлориде (5 мл) добавляют пиридин (0,14 мл, 1,7 ммоль), а затем циклопропанкарбонилхлорид (0,08 мл, 0,88 ммоль). После перемешивания в течение 5 часов при комнатной температуре раствор выливают в воду (20 мл) и экстрагируют метиленхлоридом (3×10 мл). Объединенные экстракты промывают водой (10 мл) и 1 М NaOH (водн.) (10 мл), сушат над сульфатом натрия, концентрируют и обрабатывают на хроматографической колонке с силикагелем, используя градиентное элюирование от 1% до 5% до 10% МеОН/ EtOAc. Нужный продукт элюируют 5% МеОН/-EtOAc и концентрируют, получая продукт в виде порошка белого цвета (0,012 г, 7% выход). Масс-спектр = 397,2 (М+1).
Пример 38
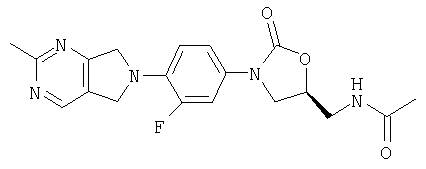
Соединение 35
Стадия 1
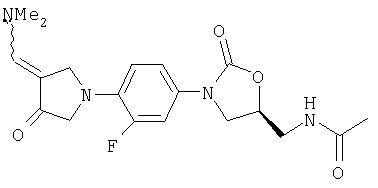
К N-[(3-пирролидинон-3-фторфенил) 5-оксазолидинил]метилацетамиду (полученному по способу WO 96/13502)(0,150 г, 0/447 ммоль) добавляют метокси-бис(диметиламино)метан (1 мл). После нагревания при 50°С в течение 15 минут реакционную смесь концентрируют, получая сырой β-кетоенамин, который используют без дополнительной очистки.
Стадия 2
Соединение 35
К этанольному раствору NaOEt (получен из 0,027 г Na в 3 мл EtOH) добавляют ацетамидингидрохлорид (0,113 г, 1,19 ммоль) и полученный выше β-кетоенаминоксазолидинон ацетамид. После кипячения с обратным холодильником в течение 3 часов реакционную смесь охлаждают до комнатной температуры, концентрируют, помещают в хлороформ и промывают водой (3×8 мл). После сушки над сульфатом натрия сырой продукт концентрируют, растворяют в 5% MeOH/EtOAc и фильтруют, получая продукт в виде грязно-белого твердого вещества (0,052 г, 45% выход). Т. плавления = 234°С с разложением. Масс-спектр = 385,9 (М+1).
Пример 39
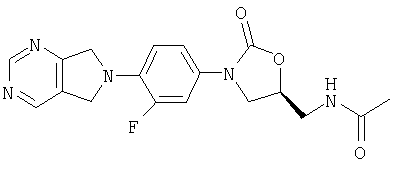
Соединение 36
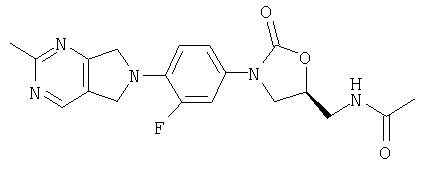
К N-[(3-пирролидинон-3-фторфенил) 5-оксазолидинил]метилацетамиду (полученному по способу WO 96/13502) (0,099 г, 0,29 ммоль) добавляют метокси-бис(диметиламино)метан (1,0 мл). После нагревания при 50°С в течение 2 часов реакционную смесь концентрируют, получая сырой β-кетоенамин. К этой смеси добавляют бензол (5 мл), ДМФ (1 мл) и ацетат формамидина (0,55 г, 5,3 ммоль). После нагревания в течение ночи при 95°С реакционную смесь охлаждают до комнатной температуры и добавляют воду (8 мл). Образовавшийся осадок собирают фильтрованием, сушат в вакуумном термостате (50°С) и обрабатывают на хроматографической колонке с силикагелем, используя в качестве элюента 5% MeOH/CH2Cl2, в результате чего получают продукт в виде порошка белого цвета (0,037 г, 34% выход). Т. плавления = 230-232°С. Масс-спектр (М+1)=372 m/z.
Пример 40
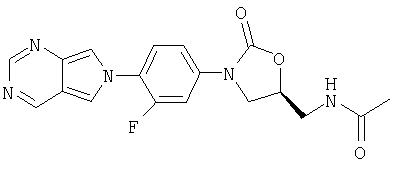
Соединение 37
Полученный выше ацетамид из примера 39 (0,020 мг, 0,054 ммоль) помещают в СН2Cl2 (5 мл) и добавляют MnO2 (0,10 г, 0,98 ммоль). После перемешивания в течение ночи при комнатной температуре реакционную смесь фильтруют через целит и концентрируют, получая продукт в виде твердого вещества светло-желтого цвета (0,016 г, 80% выход). Т. плавления = 164-166°С. Масс-спектр (М+1)=370 m/z.
Пример 41
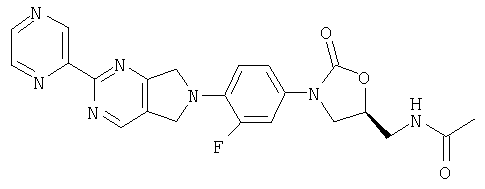
Соединение 38
К β-кетоенамину (полученному по способу примера 39) добавляют бензол (5 мл), ДМФ (1 мл) и пиразин-2-карбоксамидингидрохлорид (0,62 г, 3,9 ммоль). После нагревания в течение ночи при 95°С реакционную смесь охлаждают до комнатной температуры и добавляют воду (8 мл). Образовавшийся осадок собирают фильтрованием, сушат в вакуумном термостате (50°С) и обрабатывают на хроматографической колонке с силикагелем, используя в качестве элюента 5% MeOH/CH2Cl2, в результате чего получают продукт в виде твердого вещества светло-желтого цвета (0,0026 г, 2% выход). Т. плавления = 212-214°С.
Масс-спектр (М+1)=450 m/z.
Пример 42
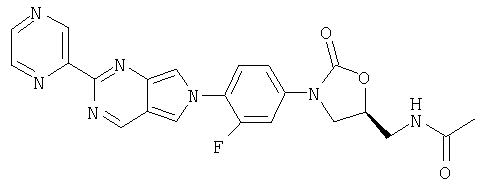
Соединение 39
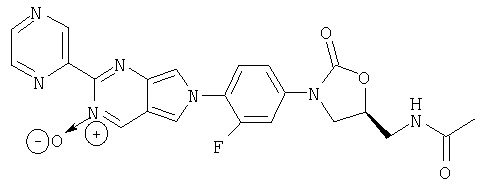
Соединение 40
Полученный выше ацетамид из примера 39 (0,040 г, 0,088 ммоль) помещают в СН2Cl2 (10 мл) и добавляют MnO2 (0,36 г, 3,5 ммоль) тремя порциями в течение трех дней. После перемешивания в течение трех дней реакционную смесь фильтруют через целит, концентрируют и обрабатывают на хроматографической колонке с силикагелем, используя в качестве элюента 7% МеОН/СН2Cl2. В результате хроматографической обработки выделяют два продукта: 0,001 г Соединения 39 в виде твердого вещества светло-желтого цвета (4% выход) и 0,002 г Соединения 40 в виде твердого вещества желтого цвета (4% выход).
Соединение 39: Масс-спектр (М+1)=448 m/z.
Соединение 40: Масс-спектр (М+1)=464 m/z.
Пример 43
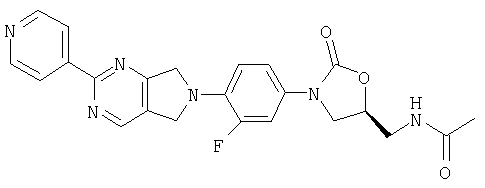
Соединение 41
К β-кетоенамину (полученному по способу примера 39) добавляют бензол (5 мл), ДМФ (1 мл) и 4-амидинопиридингидрохлорид (0,81 г, 5,2 ммоль). После нагревания в течение ночи при 95°С реакционную смесь охлаждают до комнатной температуры и добавляют воду (8 мл). Образовавшийся осадок собирают фильтрованием, сушат в вакуумном термостате (50°С) и обрабатывают на хроматографической колонке с силикагелем, используя в качестве элюента 5% МеОН/СН2Cl2, в результате чего получают продукт в виде твердого вещества светло-желтого цвета (0,072 г, 55% выход). Т. плавления = 245-250°С с разложением. Масс-спектр (М+1)=449 m/z.
Пример 44
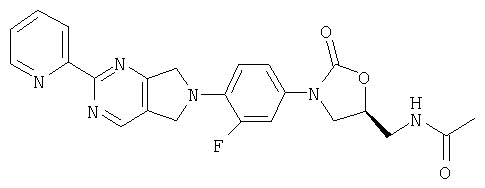
Соединение 42
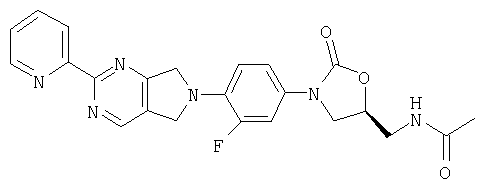
К β-кетоенамину (полученному по способу примера 39) добавляют бензол (5 мл), ДМФ (1 мл) и 2-амидинопиридингидрохлорид (0,61 г, 3,9 ммоль). После нагревания в течение ночи при 95°С реакционную смесь охлаждают до комнатной температуры и добавляют воду (8 мл). Образовавшийся осадок собирают фильтрованием, сушат в вакуумном термостате (50°С) и обрабатывают на хроматографической колонке с силикагелем, используя в качестве элюента 5% MeOH/CH2Cl2, в результате чего получают продукт в виде порошка желтого цвета (0/054 г, 40% выход). Т. плавления = 216-220°С. Масс-спектр (М+1)=449 m/z.
Пример 45
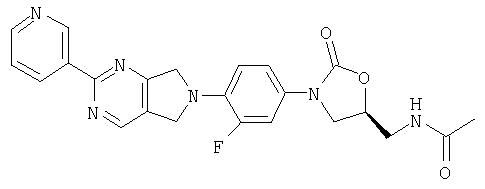
Соединение 43
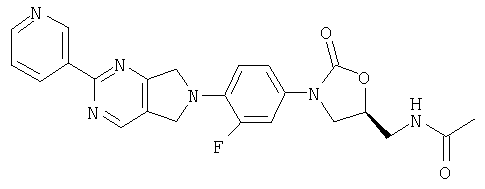
К β-кетоенамину (полученному по способу примера 39) добавляют бензол (5 мл), ДМФ (2 мл) и 3-амидинопиридингидрохлорид (0,49 г, 3,1 ммоль). После нагревания в течение ночи при 95°С реакционную смесь охлаждают до комнатной температуры и добавляют воду (8 мл). Образовавшийся осадок собирают фильтрованием, сушат в вакуумном термостате (50°С) и обрабатывают на хроматографической колонке с силикагелем, используя в качестве элюента 5% MeOH/CH2Cl2, в результате чего получают продукт в виде кристаллического твердого вещества светло-пурпурного цвета (0,044 г, 33% выход). Т плавления = 265-270°С. Масс-спектр (М+1)=449 m/z.
Пример 46
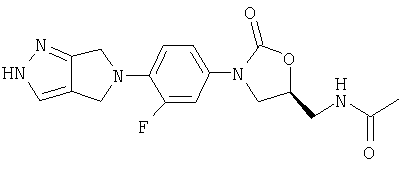
Соединение 44
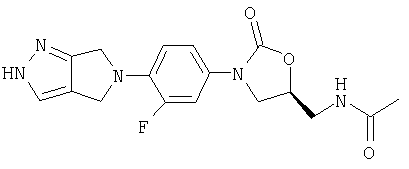
К β-кетоенамину (полученному по способу примера 39) добавляют бензол (5 мл), ДМФ (2 мл) и гидразингидрохлорид (0,22 г, 3,2 ммоль). После нагревания в течение ночи при 95°С реакционную смесь охлаждают до комнатной температуры и добавляют воду (8 мл). Образовавшийся осадок собирают фильтрованием, сушат в вакуумном термостате (50°С) и обрабатывают на хроматографической колонке с силикагелем, используя в качестве элюента 5% MeOH/CH2Cl2, в результате чего получают продукт в виде порошка грязно-белого цвета (0,022 г, 21% выход). Т. плавления = 244-247°С. Масс-спектр (М+1)=360 m/z.
Пример 47
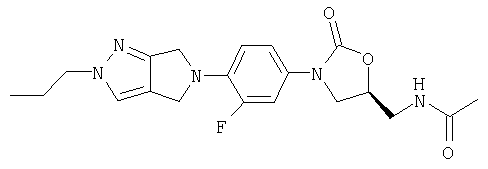
Соединение 45
К β-кетоенамину (полученному по способу примера 39) добавляют бензол (5 мл), ДМФ (2 мл) и н-пропилгидразиноксалат (0,87 г, 5,3 ммоль). После нагревания в течение ночи при 95°С реакционную смесь охлаждают до комнатной температуры и добавляют воду (8 мл). Образовавшийся осадок собирают фильтрованием, сушат в вакуумном термостате (50°С) и обрабатывают на хроматографической колонке с силикагелем, используя в качестве элюента 5% MeOH/CH2Cl2, в результате чего получают продукт в виде твердого вещества светло-желтого цвета (0,081 г, 55% выход). Т. плавления = 204-208°С. Масс-спектр (М+1)=402 m/z.
Пример 48
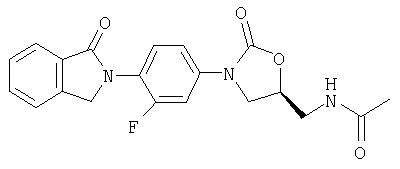
Соединение 46
Исходный материал - анилин (N-[[(5S)-3-(4-амино-3-фторфенил)-2-оксо-5-оксазолидинил]метил]ацетамид) получают по способу WO 96/23788. К фталевому дикарбоксальдегиду (0,0522 г, 0,378 ммоль) в ацетонитриле (1 мл) добавляют ледяную уксусную кислоту (0,05 мл, 0,87 ммоль) и затем вышеуказанный анилин (0,0955 г, 0,357 ммоль) в ацетонитриле (5 мл) по каплям. После 4 часов добавляют воду (10 мл) и осадок собирают на фильтре, промывают водой и эфиром, получая соединение 46 в виде твердого вещества светло-зеленого цвета (0,0655 г, 48%). Т. плавления = 211-214°С. Масс-спектр (М+1)=384 m/z.
Пример 49
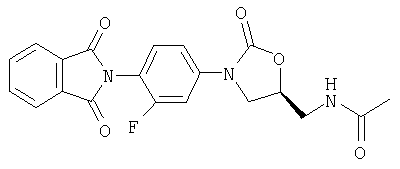
Соединение 47
К исходному материалу - анилин (N-[[(5S)-3-(4-амино-3-фторфенил)-2-оксо-5-оксазолидинил]метил]ацетамиду) (0,095 г, 0,36 ммоль) (полученному по способу патента WO 96/23788) в CH2Cl2 (5 мл) добавляют триэтиламин (0,15 мл, 1,1 ммоль) и фталоилдихлорид (0,056 мл, 0,39 ммоль). После перемешивания в течение ночи твердое вещество собирают на фильтре, промывают водой (10 мл) и сушат в вакуумном термостате (50°С), получая продукт в виде твердого вещества грязно-белого цвета (0,060, 42%). Т. плавления = 240-242°С. Масс-спектр (М+1)=398 m/z.
Пример 50
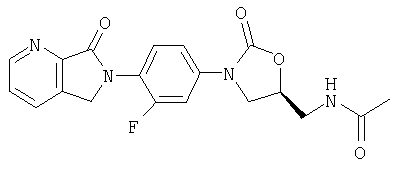
Соединение 48
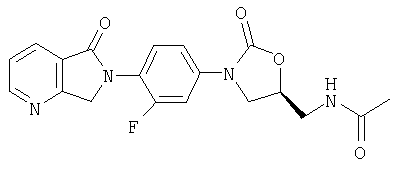
Соединение 49
К исходному материалу - анилин (N-[[(5S)-3-(4-амино-3-фторфенил)-2-оксо-5-оксазолидинил]метил]ацетамиду) (0,20 г, 0,75 ммоль) (полученному по способу патента WO 96/23788) в ацетонитриле (5 мл) добавляют 2,3-пиридиндикарбоксальдегид (0,10 г, 6,6 ммоль) и ледяную уксусную кислоту (0,050 мл, 0,87 ммоль). После перемешивания в течение 5 часов реакционную смесь концентрируют и обрабатывают на хроматографической колонке с силикагелем, используя в качестве элюента 2,5% MeOH/CH2Cl2, в результате чего получают два продукта: 0,035 г Соединения 52 (12%) в виде твердого вещества желтого цвета; и 0,011 г Соединения 53 (4%) в виде твердого вещества желтого цвета. Соединение 48: Т. плавления=230-232°С. Масс-спектр (М+1)=385 m/z. Соединение 49: Т. плавления=207-209°С. Масс-спектр (М+1)=385 m/z.
ПРИМЕР 51
Соединение 56
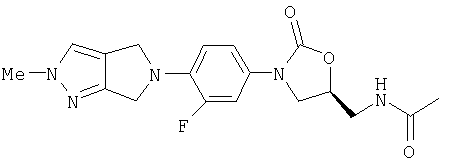
К соединению 44 (0.1098 г, 0.306 ммоль) в диметилформамиде (4 мл) при комнатной температуре добавляют гидрид натрия (60% в масле) (0.018 г, 0.45 ммоль) и смесь перемешивают в течение 30 мин с последующим добавлением MeI (23.0 мкл, 0.369 ммоль). Смесь региоизомеров (как показано Н1 ЯМР) перемешивают в течение 2 час и затем выливают в ледяную воду. Образующийся осадок собирают на фильтре, высушивают в вакуумном сушильном шкафу (50°C) и хроматографируют на кремнеземе, используя в качестве элюента 2.5% MeOH/CH2Cl2, получая целевой продукт в виде белого твердого вещества (0.0215 г, 19%). Точка плавления составляет 234-238°С. MS (M+1)=374 m/z.
Настоящее изобретение было раскрыто более подробно с конкретными ссылками на представленные выше его конкретные варианты. Приведенные выше варианты и примеры представлены лишь для иллюстрации объема и сути настоящего изобретения. На основании этих вариантов и примеров специалистам будут очевидны и другие варианты и примеры. Эти другие варианты и примеры включены в рассмотрение настоящего изобретения. Следует учитывать, что, не выходя за рамки настоящего изобретения, можно осуществить его различные вариации и модификации, поэтому настоящее изобретение ограничено только прилагаемой формулой изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ КАРБОНОВЫХ КИСЛОТ (ВАРИАНТЫ), ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ СЕЛЕКТИВНОГО ИНГИБИРОВАНИЯ СВЯЗЫВАНИЯ αβ ИНТЕГРИНА У МЛЕКОПИТАЮЩЕГО | 2000 |
|
RU2263109C2 |
| ПРОИЗВОДНЫЕ ИМИДАЗОЛА КАК ИНГИБИТОРЫ ПРЕНИЛТРАНСФЕРАЗЫ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБЫ ЛЕЧЕНИЯ НА ИХ ОСНОВЕ | 1999 |
|
RU2241712C9 |
| СИММЕТРИЧНЫЕ И НЕСИММЕТРИЧНЫЕ ПРОИЗВОДНЫЕ ДИФЕНИЛМОЧЕВИНЫ (ВАРИАНТЫ), ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ПОДАВЛЕНИЯ РОСТА ОПУХОЛЕВЫХ КЛЕТОК, ОПОСРЕДОВАННОГО КИНАЗОЙ RAF | 1998 |
|
RU2247109C9 |
| СПИРОПИПЕРИДИНЫ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ) | 1993 |
|
RU2168512C2 |
| ИНДОЛОПИРРОЛОКАРБАЗОЛЬНЫЕ ПРОИЗВОДНЫЕ САХАРОВ, СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ИНГИБИРОВАНИЯ РОСТА ОПУХОЛЕЙ | 1997 |
|
RU2167880C2 |
| ПРОИЗВОДНЫЕ ПРОПИОНОВОЙ КИСЛОТЫ (ВАРИАНТЫ), ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ СЕЛЕКТИВНОГО ИНГИБИРОВАНИЯ СВЯЗЫВАНИЯ αβ ИНТЕГРИНА | 2000 |
|
RU2255933C9 |
| БИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ АМИДИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ИНГИБИРОВАНИЯ СИНТЕТАЗЫ ОКИСИ АЗОТА | 1995 |
|
RU2155761C2 |
| 7-ОКСАБИЦИКЛОГЕПТИЛЗАМЕЩЕННЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ АМИДЫ ИЛИ ИХ СТЕРЕОИЗОМЕРЫ В КАЧЕСТВЕ АНТАГОНИСТОВ РЕЦЕПТОРОВ ТРОМБОКСАНА | 1991 |
|
RU2015980C1 |
| ПРОИЗВОДНЫЕ АНТРАНИЛОВОЙ КИСЛОТЫ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1997 |
|
RU2195454C2 |
| РАЦЕМИЧЕСКИЕ И ОПТИЧЕСКИЕ АКТИВНЫЕ ПРОИЗВОДНЫЕ ТЕТРАЛИНА И 7-ГИДРОКСИТЕТРАЛИНА | 1992 |
|
RU2073673C1 |
Изобретение относится к бициклическим гетероциклическим замещенным фенилоксазолидинонам, которые представляют собой соединения формулы I:
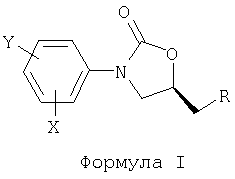
где: R выбирают из группы, состоящей из ОН, O-гетероарила, N3, OSO2R'', -NR'''R'''', или
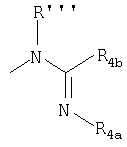
где: (ii) R'' представляет неразветвленный или разветвленный алкил, содержащий вплоть до 5 атомов углерода; и (iii) R''' и R'''' независимо выбирают из группы, состоящей из Н, -CO2-R1, -CO-R1, -CS-R1 и -SO2-R4, где R1 выбирают из группы, состоящей из циклоалкила, содержащего от 3 до 6 атомов углерода и неразветвленного или разветвленного алкила, содержащего вплоть до 6 атомов углерода; R4 выбирают из неразветвленного или разветвленного алкила, содержащего вплоть до 4 атомов углерода; и R4a представляет CN или NO2; R4b представляет SR4c, амино, NHR4c или NR4cR4d; R4c и R4d независимо выбирают из Н или алкила; Х представляет от 0 до 4 членов, независимо выбранных из группы, состоящей из галогена; и Y представляет радикал формулы II или III:

где R5, R6, R7 и R8 независимо представляют Н или R5 и R6 и/или R7 и R8 вместе образуют оксогруппу; R9 и R10 независимо представляют Н; А, В, С и D выбирают из С и N с образованием фенильного кольца или 5-6 членного гетероароматического кольца, причем указанное гетероароматическое кольцо содержит от одного до четырех членов, выбранных из группы, состоящей из N; Z выбирают из алкила, гетероарила, содержащего N; и m представляет 0 или 1. Эти соединения полезны в качестве антибактериальных агентов и могут быть использованы для лечения субъекта, состояние которого вызвано бактериальной инфекцией или вклад в которое вносит бактериальная инфекция, вызываемая S.aureus и Е.Faecium. 4 н. и 41 з.п. ф-лы, 1 табл.
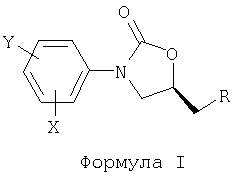
где R выбирают из группы, состоящей из ОН, O-гетероарила, N3, OSO2R'', NR'''R'''', или
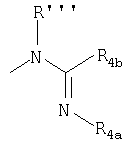
где (ii) R'' представляет неразветвленный или разветвленный алкил, содержащий вплоть до 5 атомов углерода, и
(iii) R''' и R'''' независимо выбирают из группы, состоящей из Н, -CO2-R1, -CO-R1, -CS-R1 и -SO2-R4, где
R1 выбирают из группы, состоящей из циклоалкила, содержащего от 3 до 6 атомов углерода, неразветвленного или разветвленного алкила, содержащего вплоть до 6 атомов углерода;
R4 выбирают из неразветвленного или разветвленного алкила, содержащего вплоть до 4 атомов углерода;
R4a представляет CN или NO2;
R4b представляет SR4c, амино, NHR4c или NR4cR4d;
R4c и R4d независимо выбирают из Н или алкила;
Х представляет от 0 до 4 членов, независимо выбранных из группы, состоящей из галогена, и
Y представляет радикал формулы II или III
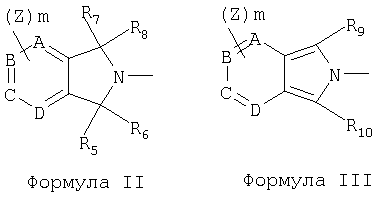
где
R5, R6, R7 и R8 независимо представляют Н или R5 и R6 и/или R7 и R8 вместе образуют оксогруппу;
R9 и R10 независимо представляют Н;
А, В, С и D выбирают из С и N с образованием фенильного кольца или 5-6 членного гетероароматического кольца, причем указанное гетероароматическое кольцо содержит от одного до четырех членов, выбранных из группы, состоящей из N;
Z выбирают из алкила, гетероарила, содержащего N, и
m представляет 0 или 1.
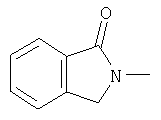 изоиндолон-;
изоиндолон-;
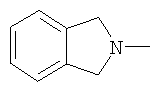 (1,3-дигидро-2Н-изоиндол-2-ил)-;
(1,3-дигидро-2Н-изоиндол-2-ил)-;
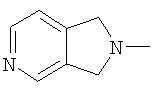 (1,3-Дигидро-2Н-пирроло[3,4-с]пиридин-6-ил)-;
(1,3-Дигидро-2Н-пирроло[3,4-с]пиридин-6-ил)-;
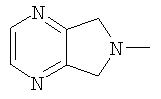 (5,7-дигидро-6Н-пирроло[3,4-b]пиразин-6-ил)-;
(5,7-дигидро-6Н-пирроло[3,4-b]пиразин-6-ил)-;
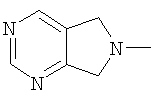 (5,7-дигидро-6H-пирроло[3,4-d]пиримидин-6-ил;
(5,7-дигидро-6H-пирроло[3,4-d]пиримидин-6-ил;
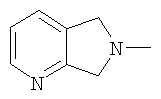 (5,7-дигидро-6H-пирроло[3,4-b]пиридин-6-ил;
(5,7-дигидро-6H-пирроло[3,4-b]пиридин-6-ил;
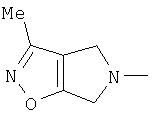 (4,6-дигидро-3-метил-5Н-пирроло[3,4-d]изоксазол-5-ил)-;
(4,6-дигидро-3-метил-5Н-пирроло[3,4-d]изоксазол-5-ил)-;
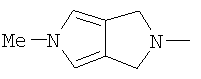 3,5-дигидро-5-метилпирроло[3,4-с]пиррол-2(1Н)-ил)- и
3,5-дигидро-5-метилпирроло[3,4-с]пиррол-2(1Н)-ил)- и
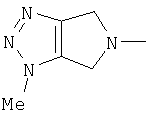 (4,6-дигидро-1-метилпирроло[3,4-d]-1,2,3-триазол-5(1Н)-ил)-.
(4,6-дигидро-1-метилпирроло[3,4-d]-1,2,3-триазол-5(1Н)-ил)-.

| Приспособление в пере для письма с целью увеличения на нем запаса чернил и уменьшения скорости их высыхания | 1917 |
|
SU96A1 |
| Походная разборная печь для варки пищи и печения хлеба | 1920 |
|
SU11A1 |
| ПРОИЗВОДНОЕ ОКСАЗОЛИДИНОНА ИЛИ ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМАЯ СОЛЬ, СПОСОБ ЛЕЧЕНИЯ МИКРОБНЫХ ИНФЕКЦИЙ У ТЕПЛОКРОВНЫХ ЖИВОТНЫХ | 1993 |
|
RU2105003C1 |

