Область, к которой относится изобретение
Изобретение относится к соединениям N-[[2'-[[(4,5-диметил-3-изоксазолил)амино] сульфонил] -4-(2-оксазолил)[1,1'-бифенил] -2-ил]метил]-N,3,3-триметилбутанамид и N-(4,5-диметил-3-изоксазолил)-2'-[(3,3-диметил-2-оксо-1-пирролидинил)метил]-4'-(2-оксазолил)[1,1'-бифенил]-2-сульфонамид и их солям, которые применимы в качестве антагонистов эндотелина.
Краткое описание изобретения
Антагонисты эндотелина, которые представляют собой соединения, способные, между прочим, ингибировать связывание пептидов эндотелина с рецепторами эндотелина, применимы для лечения зависящих от эндотелина нарушений, таких как гипертензия и застойная сердечная недостаточность. Помимо увеличения способности антагонистов ингибировать (подавлять) эндотелин, в технике продолжается поиск путей улучшения параметров, связанных с общей in vivo функциональной активностью.
Настоящее изобретение предоставляет соединения N-[[2'-[[(4,5-диметил-3-изоксазолил)амино] сульфонил]-4-(2-оксазолил)[1,1'-бифенил]-2-ил]метил]-N, 3,3-триметилбутанамид и N-(4,5-диметил-3-изоксазолил)-2'-[(3,3-диметил-2-оксо-1-пирролидинил)метил]-4'-(2-оксазолил)[1,1'-бифенил]-2-сульфонамид и их соли, которые помогают достигнуть такого улучшения. В дополнение к своей высокой активности предлагаемые антагонисты эндотелина обладают исключительной биодоступностью при пероральном приеме, продолжительностью действия и предсистемной метаболической стабильностью внутри пищеварительного тракта и, следовательно, особенно пригодны при лечении зависящих от эндотелина нарушений.
Подробное описание изобретения
Соединение N-[[2'-[[(4,5-диметил-3-изоксазолил)амино]сульфонил]-4-(2-оксазолил)[1,1'-бифенил] -2-ил] метил] -N, 3,3-триметилбутанамид по данному изобретению имеет следующую структуру:
Соединение N-(4,5-диметил-3-изоксазолил)-2'-[(3,3-диметил-2-оксо-1-пирролидинил)метил] -4'-(2-оксазолил)[1,1'-бифенил]-2-сульфонамид по данному изобретению имеет следующую структуру: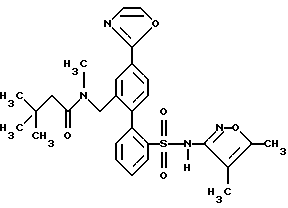
Любая из солей и все соли N-[[2'-[[(4,5-диметил-3-изоксазолил)амино] сульфонил] -4-(2-оксазолил)[1,1'-бифенил] -2-ил]метил]-N,3,3-триметилбутанамида и N-(4,5-диметил-3-изоксазолил)-2'-[(3,3-диметил-2-оксо-1-пирролидинил)метил] -4'-(2-оксазолил)[1,1'-бифенил] -2-сульфонамида входят в объем данного изобретения и, в частности, таковые, образованные присоединением неорганических или органических оснований. Предпочтительны фармацевтически приемлемые (т. е. нетоксические, физиологически приемлемые) соли, хотя применимы также другие соли, например, для выделения или очистки данных соединений.
Предпочтительны соли щелочных металлов, такие как натриевые, калиевые и литиевые соли, щелочноземельных металлов, такие как соли кальция и магния, а также соли с органическими основаниями (например, с органическими аминами), такими как дициклогексиламин, трет-бутиламин, бензатин, N-метил-D-глюкамид и гидрабамин, и с аминокислотами, например, такими как аргинин, лизин и тому подобное.
Соли согласно данному изобретению могут быть получены, например, реакцией N-[[2'-[[(4,5-диметил-3-изоксазолил)амино] сульфонил] -4-(2-оксазолил)[1,1'-бифенил] -2-ил] метил] -N, 3,3-триметилбутанамида или N-(4,5-диметил-3-изоксазолил)-2'-[(3,3-диметил-2-оксо-1-пирролидинил)метил]-4'-(2-оксазолил)[1,1'-бифенил] -2-сульфонамида с заданным ионом (например, при взаимодействии с эквивалентным количеством основания) в среде (например, в среде, из которой соль выпадает, или в водной среде с последующей лиофилизацией).
Вышеуказанные соединения согласно данному изобретению, обладающие превосходной предсистемной метаболической стабильностью внутри желудочно-кишечного тракта, содержат 3-изоксазольную группу. Так как эта группа может придавать предсистемную метаболическую стабильность внутри желудочно-кишечного тракта, данное изобретение также представляет новые соединения, применимые в качестве антагонистов эндотелина и обладающие прекрасной метаболической стабильностью, следующей общей формулы:
Z*-SO2-NН-(3-изоксазольная группа),
где Z* означает органический радикал (остаток), например незамещенный или замещенный нафтил, фенил, бифенил, или гетероциклический остаток (например, тиофенил), и где ''3-изоксазольная группа'' представляет собой незамещенную или замещенную изоксазольную группу с NH-группой в положении 3. Предпочтительными ''3-изоксазольными группами'' являются незамещенные изоксазолы или таковые, имеющие в качестве заместителей алкил (особенно насыщенные с числом атомов углерода в цепи от 1 до 12, например метил) и/или галоид (т.е. фтор, хлор, иод и бром). Ряд антагонистов эндотелина описан в следующих документах, введенных в виде ссылок в данное описание во всей полноте, особенно, но не только, в связи с конкретными, описанными в них соединениями: патент США 5,378,715; патент США 5,514,696; патент США 5,420,123; заявка США 114,251, поданная 30 августа 1993 г.; Заявка США 08/728,238, поданная 8 октября 1996 г; европейская заявка 702,012; заявка США 08/754,715, поданная 21 ноября 1996 г.; заявка США 08/692,869, поданная 25 июля 1996 г.; заявка США 60/011,974, поданная 20 февраля 1996 г.; заявка США 60/013,491, поданная 12 марта 1996 г.; заявка США 60/015,072, поданная 2 апреля 1996 г.; Международная заявка 94/27979; патент США 5,464,853; патент США 5,514,691; ЕР 601386; ЕР 633259; патент США 5,292,740; ЕР 510526; ЕР 526708; ЕР 658548; патент США 5,541,186; WO 96/19454; WO 96/19455; ЕР 713875; WO 96/20177; ЕР 733626; японский патент JP 8059635; ЕР 682016; английский патент GB 2295616; WO 95/26957; патент США 5,571,821; ЕР 743307 и WO 96/31492; например, описаны, как указано, следующие соединения (введены в данное описание ссылками, как дано выше): босентан (Ro 47-0203, Clozel, M., et al., "Pharmacological Characterization of Bosentan, A New Potent Orally Active Nonpeptide Endothelin Receptor Antagonist", The Journal of Pharmacology and Experimental Therapeutics, vol. 270(1), pp.228-235 (1994)); и ТВС-11251, т.е.:
(IBC Международная конференция по ингибиторам эндотелина, Coronado, СА (февраль 1996 г. ) и 211-й Национальный конгресс Американского Химического Общества, New Orleans, LA (март 1996 г.). Эти соединения содержат сульфонамидную группу -SO2-NH-, в которой органический остаток связан с остальной частью молекулы через сульфонильную группу. Предпочтительными в качестве группы Z* в вышеприведенной общей формуле Z*-SO2-NН-(3-изоксазольная группа) по данному изобретению являются связанные через сульфонильную группу органические остатки соединений, описанных в вышеприведенных документах. Помимо представления таких метаболически стабильных соединений данное изобретение также обеспечивает способ применения этих соединений, отличающийся тем, что они вводятся для лечения эндотелинзависящего расстройства.
Соединения по данному изобретению представляют собой антагонисты ЕТ-1, ЕТ-2 и/или ЕТ-3 и применимы для лечения состояний, ассоциирующихся с повышенными уровнями ЕТ (например, диализ, травма или хирургическое вмешательство), и всех эндотелинзависимых расстройств. Они, следовательно, пригодны в качестве антигипертензивных веществ. При введении композиции, содержащей одно (или комбинацию) из соединений по данному изобретению, кровяное давление хозяина-млекопитающего (например, человека) снижается. Они также применимы при вызванной беременностью гипертензии и комой (преэклампсия и эклампсия), острой портальной гипертензией и гипертензией в результате побочного действия эритропоэтина.
Соединения по данному изобретению также применимы при терапии поражения клеток почек, почечных клубочков и мезангия, включая острую и хроническую почечную недостаточность, повреждение клубочка, нарушения деятельности почек как следствие возраста или в результате диализа, нефросклероз (особенно гипертензивный нефросклероз), нефротоксичность (включая нефротоксичность, связанную с визуализацией и контрастными веществами и с циклоспорином), почечную ишемию, первичный везикоуретеральный рефлюкс, гломерулосклероз и тому подобное. Соединения по данному изобретению могут также применяться при лечении расстройств, связанных с паракринной и эндокринной функцией.
Соединения согласно данному изобретению также применимы при терапии эндотоксемии (наличия в крови эндотоксинов) или эндотоксинового бактериально-токсического шока, а также геморрагического шока.
Соединения по данному изобретению также применимы при гипоксической болезни и в качестве антиишемического средства для лечения, например, ишемии сердца, почек или мозга, и реперфузии (например, после хирургической установки аппарата искусственного кровообращения), коронального или церебрального вазоспазма и тому подобное.
Кроме того, соединения по данному изобретению также могут применяться в качестве средств против аритмии; против стенокардии; против фибрилляции; как противоастматические средства; как средства против атеросклероза и артериосклероза; как добавки к растворам для кардиоплегии в аппаратах искусственного кровообращения; как вспомогательные средства при тромболитической терапии и в качестве средства против диареи. Соединения по данному изобретению можно применять при терапевтическом лечении инфаркта миокарда; при терапии периферического сосудистого заболевания (например, болезни Рейно или болезни Такаяши); для терапии сердечной гипертрофии (например, гипертрофической кардиомиопатии); для лечения первичной легочной гипертензии (например, плексогенной или эмболической) у взрослых и новорожденных и легочной гипертензии, вызванной сердечной недостаточностью, повреждениями вследствие облучения или химиотерапии или другими травмами; для лечения сосудистых расстройств центральной нервной системы, таких как удар (приступ), мигрень и субарахноидальное кровоизлияние; в терапии поведенческих нарушений центральной нервной системы; при терапии заболеваний желудочно-кишечного тракта, таких как язвенный колит, болезнь Крона, повреждение слизистой желудка, язвенная и ишемическая болезни пищеварительного тракта; при терапии заболеваний желчного пузыря и желчных протоков, таких как холангит; при лечении панкреатитов; для регуляции роста клеток; при терапии доброкачественной гипертрофии простаты; рестеноза после ангиопластики или любых процедур, включая трансплантацию; при терапии застойной сердечной недостаточности, включая ингибирование фиброза; уменьшение дилатации, изменения (коррекции) и дисфункции левого желудочка и при терапии гепатотоксичности и внезапной смерти. Соединения по данному изобретению можно применять при лечении болезни дрепаноцитов, включая стимуляцию и/или эволюцию болезненных кризисов этого заболевания; при лечении вредных последствий присутствия ЕТ-продуцирующих опухолей, таких как гипертензия, являющаяся следствием гемангиоперицитомы; при лечении раннего и запущенного заболевания и повреждения печени, включая сопутствующие осложнения (например, гепатотоксичность, фиброз и цирроз); при лечении спастических состояний мочевых путей и/или мочевого пузыря; при лечении гепаторенального синдрома; при лечении иммунологических, включая васкулит, заболеваний, таких, например, как волчанка, системная склеродермия, смешанная криоглобулинемия; и при лечении фиброза, связанного с почечной дисфункцией и гепатотоксичностью. Соединения по данному изобретению можно применять при терапии метаболических и неврологических расстройств; рака; инсулинзависимого и инсулиннезависимого сахарного диабета; невропатии; ретинопатии; материнского респираторного дистресс-синдрома; дисменореи; эпилепсии; кровотечения и приступа ишемии; костного ремоделирования (изменения); псориаза и хронических воспалительных заболеваний, таких как ревматоидный артрит, остеоартрит, саркоидоз и экзематоидный дерматит (все виды дерматита).
Из соединений по данному изобретению можно готовить препараты в сочетании с ингибиторами эндотелинпревращающего фермента (ЭПФ, ЕСЕ), такими как фосфорамидон; с антагонистами рецепторов тромбоксана; с веществами, способствующими открытию калиевых каналов; с ингибиторами тромбина (например, гирудином и тому подобное); ингибиторами факторов роста, например модуляторами PDGF-активности; антагонистами фактора активации тромбицитов (PAF); антагонистами рецепторов ангиотензина II (АII); ингибиторами ренина; ингибиторами ангиотензинпревращающего фермента (АПФ, АСЕ), такими как каптоприл, зофеноприл, фосиноприл, церанаприл, алацеприл, эналаприл, делаприл, пентоприл, хинаприл (quinapril), рамиприл, лизиноприл и соли этих соединений; ингибиторами нейтральной эндопептидазы (NEP); NEP-ACE-ингибиторами двойного действия; ингибиторами HMG СоА-редуктазы, такими как правастатин и мевакор; ингибиторами скваленсинтетазы; секвестрантами желчных кислот, такими как квестран; блокаторами кальциевых каналов; активаторами калиевых каналов; бета-адренергическими препаратами; антиаритмическими препаратами; диуретиками, такими как хлортиазид, дихлотиазид (гидрохлортиазид), флуметиазид, гидрофлуметиазид, бендрофлуметиазид, метилхлортиазид, трихлорметиазид, политиазид или бензотиазид, а также этакриновая кислота, трикринафен, оксодолин (хлорталидон), фуросемид, мусолимин, буфенокс (буметанид), триамтерен, амилорид и спиронолактон и соли этих соединений; сердечные гликозиды, например дигоксин; и с препаратами, влияющими на свертываемость крови, такими как активатор плазминогена тканей (tPA), рекомбинантный tPA, стрептокиназа, урокиназа, проурокиназа и комплекс анизоилированного плазминогена с активатором - стретокиназой (APSAC). Если нужно готовить в виде фиксированной дозы, то для таких комбинаций веществ используют соединения по данному изобретению в интервале доз, описанном ниже, и другой активный агент в лучшем для него (оптимальном) интервале доз. Соединения по данному изобретению могут готовиться в виде препарата с, или применяться совместно с, противогрибковыми средствами и иммунодепрессантами, такими как амфотерицин В, циклоспорины и тому подобное для того, чтобы предотвратить гломерулярное сокращение и нефротоксичность, вызываемые этими соединениями. Соединения по данному изобретению можно также применять в сочетании с гемодиализом.
Соединения по данному изобретению можно вводить любым подходящим способом, например перорально или парентерально, различным видам млекопитающих, о которых известно, что они могут подвергаться таким заболеваниям, например человеку, в эффективных количествах, таких как количества в интервале доз от примерно 0,1 до примерно 100 мг/кг, предпочтительно от примерно 0,2 до примерно 50 мг/кг и более, предпочтительно от примерно 0,5 до примерно 25 мг/кг (или от примерно 1 до 2500 мг, предпочтительно от примерно 5 до примерно 2000 мг) в виде однократной или от 2 до 4 разделенных суточных доз.
Активное вещество может применяться в композициях в виде таблетки, капсулы, раствора или суспензии, содержащих от примерно 5 до примерно 500 мг в расчете на стандартную дозу, соединения или смеси соединений по данному изобретению или в виде форм для местного применения при заживлении ран (например, от 0,01 до 5% весовых соединения по данному изобретению, от 1 до 5 обработок в день). Соединения по данному изобретению могут объединяться соответствующим образом с физиологически приемлемым наполнителем или носителем, эксципиентом, связующим, консервантом, стабилизатором, корригентом и т. д. , или с локальным носителем, таким как Plastibase (гель полиэтилена в минеральном масле), как принято в обычной фармацевтической практике.
Соединения по данному изобретению можно также применять местно для лечения периферических сосудистых заболеваний и в таком случае они могут быть в виде крема или мази.
Соединения по данному изобретению могут также быть в виде композиций, таких как стерильные растворы или суспензии для парентерального введения. Например, от около 0,1 до 500 миллиграмм соединения по данному изобретению может быть объединено с физиологически приемлемым наполнителем, носителем, эксципиентом, связующим, консервантом, стабилизатором и т.д., в виде стандартной дозы, как принято в обычной фармацевтической практике. Количество активного вещества в этих композициях или препаратах предпочтительно таково, что получается соответствующая доза в указанном интервале.
Таким образом, настоящее изобретение предоставляет новые способы применения новых соединений, данных в настоящем описании, и фармацевтические композиции, содержащие их. В данное изобретение особо (главным образом) входят способы терапии эндотелинзависящих расстройств у млекопитающих, которые (способы) заключаются во введении млекопитающему эффективного для терапии эндотелинзависимого расстройства количества соединения по данному изобретению. Данное изобретение также особо (главным образом) охватывает фармацевтические композиции для терапии эндотелинзависящих расстройств, содержащие соединения по данному изобретению в количестве, эффективном для этой цели, и физиологически приемлемый наполнитель или носитель. Соединение по данному изобретению можно, например, применять в способах или фармацевтических композициях по данному изобретению одно (как таковое), в сочетании с одним или более других соединений по данному изобретению и/или в сочетании с, по меньшей мере, еще одним (с другим) активным агентом, например с антагонистом рецептора ангиотензина II (АII), ингибитором ренина, ингибитором ангиотензинпревращающего фермента (АПФ, АСЕ), ингибитором двойного действия нейтральной эндопептидазы NEP-ACE, диуретиком или сердечным гликозидом, или другим активным средством, перечисленным выше.
В способах по данному изобретению подобный(-е) другой(-ие) активный(-е) агент(ы) может(могут) вводиться до, одновременно с или после введения соединения(-й) по данному изобретению. В фармацевтические композиции по данному изобретению подобный(-е) другой(-ие) активный(-е) агент(ы) может(могут) входить вместе с соединением(-ями) по данному изобретению или вводиться отдельно, как описано выше для способов по данному изобретению.
В частности, предпочтительны такие способы и композиции для терапии гипертензии, особенно гипорениновой гипертензии (например, такой, как описана в заявке США 60/035,825, поданной 30 января 1997 г. J.E. Bird, озоглавленной "Способ предотвращения или лечения гипорениновой гипертензии путем введения антагониста эндотелина" или легочной гипертензии, в частности первичной легочной гипертензии; доброкачественной гипертрофии предстательной железы; мигрени; поражений клеток почек, почечных клубочков и мезангия; эндотоксемии; ишемии; атеросклероза; повторного стеноза; субарахноидального кровоизлияния и застойной сердечной недостаточности.
N-[[2'-[[(4,5-диметил-3-изоксазолил)амино] сульфонил] -4-(2-оксазолил)[1,1'-бифенил] -2-ил]метил]-N,3,3-триметилбутанамид и N-(4,5-диметил-3-изоксазолил)-2'-[(3,3-диметил-2-оксо-1-пирролидинил)метил] -4'-(2-оксазолил)[1,1'-бифенил] -2-сульфонамид и их соли могут быть получены такими методами, которые описаны в европейской заявке N 702,012 и заявке США N 08/754,715, поданной 21 ноября 1996 г., и/или методами, представленными в экспериментальной части данного описания. Данное изобретение дополнительно поясняется следующими рабочими примерами, которые представляют собой предпочтительные формы воплощения настоящего изобретения. Эти примеры призваны быть скорее иллюстрирующими, нежели ограничивающими изобретение.
ПРИМЕР 1
Получение N-[[2'-[[(4,5-диметил-3-изоксазолил)амино]сульфонил]-4-(2-оксазолил)[1,1'-бифенил]-2-ил]метил]-N,3,3-триметилбутанамила
А. Гидрохлорид 4,5-диметил-3-изоксазоламина
К 1,1-диметилэтиловому эфиру (4,5-диметил-3-изоксазолил)карбаминовой кислоты (25,0 г, 117,79 ммол, полученному как описано Konoike, Т. et al., Tet. Lett., 37, 3339-3342 (1996)), помещенному в колбу, добавляют 100 мл 4N НСl в диоксане. Смесь перемешивают при комнатной температуре в течение 5 час и концентрируют с получением обозначенного в заглавии этой стадии продукта в виде твердого вещества, которое используют в следующей стадии без дополнительной очистки.
В. 2-Бром-N-(4,5-диметил-3-изоксазолил)бензсульфонамид
К смеси твердого вещества, полученного на Стадии А, и 4-диметиламинопиридина (1,44 г, 11,78 ммол) в 79 мл пиридина при 0oС добавляют порциями в течение 10 мин 2-бромбензсульфонилхлорид (28,59 г, 111,90 ммол). Смесь перемешивают при 40oС в течение 6,5 час и концентрируют. Остаток растворяют в 300 мл метанола ("МеОН"), добавляют 1000 мл 3% водного раствора NaHCO3, и смесь концентрируют в вакууме для удаления большей части МеОН. Твердый остаток отфильтровывают и водный фильтрат подкисляют до рН 1 с помощью 6N НСl при 0oС, и экстрагируют этилацетатом ("ЕtOАс", 2•400 мл). Экстракт промывают 100 мл 1N HCl, 100 мл Н2О и 100 мл рассола, сушат и концентрируют с получением обозначенного в заглавии этой стадии продукта (34,32 г, ~95%-ной чистоты, выход 84% на две стадии). Rf=0,57, силикагель, 1:1 гексан/ЕtOАс.
С. 2-Бpoм-N-(4,5-диметил-3-изоксазолил)-N-[(2-метоксиэтокси)метил]бензсульфонамид
К продукту, полученному на Стадии В (32,60 г, 102,78 ммол), в 343 мл диметилформамида ("ДМФА") при 0oС добавляют порциями NaH (60%-ный в минеральном масле, 4,93 г, 123,34 ммол). После перемешивания при комнатной температуре в течение 30 минут смесь охлаждают в бане со смесью лед-соль (-15oС) и добавляют по каплям в течение 20 минут 2-метоксиэтоксиметилхлорид (16,0 г, 128,48 ммол). Реакционную смесь перемешивают в бане со смесью лед-соль в течение 20 минут и затем при комнатной температуре в течение 1,5 час. Добавляют к реакционной смеси 1400 мл смеси 1:1 гексан/ЕtOАс. Органический слой отделяют и промывают 2•800 мл воды, 400 мл рассола, сушат и концентрируют. Остаток хроматографируют на силикагеле с использованием смеси 2,5:1 гексан/ЕtOАс и получают обозначенный в заглавии этой стадии продукт (32,12 г, 75%) в виде масла.
D. N-(4,5-Диметил-3-изоксазолил)-2'-формил-N-[(2-метоксиэтокси)метил]-4'-(2-оксазолил)[1,1'-бифенил]-2-сульфонамид
К раствору продукта, полученному на Стадии С (22,16 г, 52,85 ммол), в 264 мл тетрагидрофурана ("ТГФ") при -95oС добавляют н-бутиллитий ("n-BuLi", 2M раствор в пентане, 29,07 мл, 58,14 ммол). Смесь перемешивают при -95oС в течение 10 минут, добавляют триметилборат (6,59 г, 63,42 ммол) и перемешивают при -78oС в течение 15 минут. Удаляют охлаждающую баню и смесь медленно доводят до комнатной температуры и перемешивают при комнатной температуре в течение 0,5 час. Затем смесь охлаждают до 0oС и добавляют по каплям 100 мл 3N НСl. После перемешивания в течение 30 минут смесь экстрагируют СН2Сl2 (300 мл, 100 мл). Объединенные органические экстракты промывают рассолом, сушат и концентрируют с получением 2-бороно-N-(4,5-диметил-3-изоксазолил)-N-[(2-метоксиэтокси)метил]бензсульфонамида в виде смолы.
К 2-бороно-N-(4,5-диметил-3-изоксазолил)-N-[(2-метоксиэтокси)-метил]бензсульфонамиду и 2-бром-5-(2-оксазолил)бензальдегиду (13,32 г, 58,14 ммол, полученному как описано в примере 21 европейской заявки N 702,012), в 264 мл толуола и 132 мл 95%-ного этанола ("EtOH") добавляют 106 мл 2М водного раствора карбоната натрия и тетракис(трифенилфосфин)-палладий(0) (6,11 г, 5,29 ммол), и реакционную смесь нагревают в атмосфере аргона при 85oС в течение 4 час, охлаждают и разбавляют 250 мл EtOAc. Органический слой отделяют и промывают 100 мл Н2О и 50 мл рассола, сушат и концентрируют. Остаток хроматографируют на силикагеле с использованием смеси 1:1 гексан/ЕtOАс и получают обозначенный в заглавии этой стадии продукт (16,95 г, 62,7% на две стадии) в виде бесцветной смолы.
1Н ЯМР (CDCl3) d 1,89 (с, 3Н), 2,28 (с, 3Н), 3,28 (с, 3Н), 3,43 (м, 2Н), 3,60-3,76 (м, 2Н), 4,40-4,59 (м, 2Н), 7,28-8,68 (м, 9Н), 9,76 (с, 1Н).
Е. N-(4,5-Диметил-3-изоксазолил)-2'-формил-4'-(2-оксазолил)[1,1'-бифенил]-2-сульфонамид
К продукту, полученному на Стадии D (16,95 г, 33,14 ммол), в 414 мл 95%-ного EtOH добавляют 414 мл 6N НСl. Смесь кипятят с обратным холодильником в течение 55 минут и выливают на 800 г льда. После выстаивания в течение 2 час, отделяют фильтрованием белый осадок с получением обозначенного в заглавии этой стадии продукта (13,17 г, выход 92%).
Rf(силикагель)=0,31 (5% метанол в СН2Сl2).
F. N-(4,5-Диметил-3-изоксазолил)-2'-[(метиламино)метил] -4'-(2-оксазолил)[1,1'-бифенил]-2-сульфонамид
К продукту, полученному на Стадии Е (12,91 г, 30,48 ммол), и молекулярным ситам 3А в 305 мл CH2Cl2 добавляют уксусную кислоту ("АсОН", 4,58 г, 76,20 ммол) и затем добавляют метиламин (8,03 М в EtOH, 13,29 мл, 106,68 ммол). Смесь перемешивают в течение 15 минут и добавляют триацетоксиборгидрид натрия (19,38 г, 91,44 ммол). Реакционную смесь перемешивают при комнатной температуре в течение 2 час, разбавляют 700 мл CH2Cl2 и 100 мл МеОН и фильтруют через целит. Фильтрат промывают 150 мл Н2О, сушат и концентрируют. Остаток растирают с этиловым эфиром (50 мл, 30 мл, 30 мл). Азеотропная отгонка со смесью СН2Сl2-гептан (несколько раз) приводит к обозначенному в заглавии этой стадии продукту в виде твердого вещества серого цвета, которое используют в следующей стадии без дополнительной очистки.
1Н ЯМР (СDСl3/СD3ОD 3: 1) d 1,83 (с, 3Н), 2,13 (с, 3Н), 2,71 (с, 3Н), 3,87-4,27 (м, 2Н), 7,11-8,09 (м,9Н).
G. N-[[2'-[[(4,5-диметил-3-изоксазолил)амино] сульфонил] -4-(2-оксазолил)-[1,1'-бифенил]-2-ил]метил]-N,3,3-триметилбутанамид
К продукту, полученному на Стадии F, в 300 мл СН2Сl2, при 0oС добавляют триэтиламин (6,17 г, 60,96 ммол) и перемешивают в течение 5 минут. К смеси добавляют по каплям в течение 10 минут трет-бутилацетилхлорид (398 г, 29,57 ммол). Реакционную смесь перемешивают при 0oС в течение 10 минут и при комнатной температуре в течение 1 час. Добавляют 100 мл 10%-ного водного раствора NaHSO4. Водный слой экстрагируют 100 мл СН2Сl2. Объединенные органические экстракты промывают 100 мл Н2О и 50 мл рассола, сушат и концентрируют. Остаток хроматографируют на силикагеле с использованием смеси 60:40:1 гексан/EtOAc/AcOH и получают обозначенный в заглавии этого примера продукт (13,10 г, 80% на две стадии) в виде твердого вещества белого цвета. Температура плавления=120-128oС (аморфный).
Новые промежуточные соединения, полученные на стадиях D, Е и F вышеприведенного примера 1, названия которых приведены в заглавии этих стадий, также заявляются в данном изобретении. Озоглавленные в стадиях Е и F продукты могут сами по себе найти применение в качестве антагонистов эндотелина для лечения эндотелинзависящих расстройств.
ПРИМЕР 2
In vivo функциональная активность N-[[2'-[[(4,5-диметил-3-изоксазолил)амино] сульфонил] -4-(2-оксазолил)[1,1'-бифенил] -2-ил]метил]-N,3,3-триметилбутанамида
Превосходная функциональная активность in vivo обозначенного в заглавии соединения (включая биодоступность, эффективность и продолжительность действия и метаболическую стабильность) была продемонстрирована следующим образом.
(A) Биодоступность
Мужским особям крыс (n= 3) натощак давали разовую дозу испытуемого (обозначенного в заглавии) соединения либо внутривенно (10 мкмол/кг в виде 10-минутного вливания), либо перорально через желудочный зонд (20 мкмол/кг). Носителями были пропиленгликоль в случае внутривенного введения и PEG-400 для перорального введения. После введения дозы в течение определенного времени определяли среднюю концентрацию обозначенного в заглавии соединения в плазме, и вычисленная площадь под кривой AUCг (мкМ х час) составляла 31,3±2,9 и 72,8±17,3 для внутривенного и перорального введения соответственно. Так как нормированные на дозу значения AUCг для внутривенного и перорального введения статистически не различались, величина биодоступности данного соединения при пероральном приеме была определена примерно 100%.
(B) Вазопрессорный тест
(i) Внутривенная инъекция big ET-1 находящимся в сознании крысам с нормальным артериальным давлением приводит к временному повышению среднего артериального давления, которое может быть ослаблено антагонистом рецептора ЕТА.
Крысам Sprague-Dawley перед и через 5 минут после внутривенной инъекции дозы испытуемого (обозначенного в заглавии) соединения (0,01 мкмол/кг (n= 10), 0,03 мкмол/кг (n=10) и 0,1 мкмол/кг (n=3)) быстро вводили большие объемы ("болюс-инъекция") человеческого big ET-1 (1 нмол/кг, внутривенно; носитель: 1% Tween 80 в 5% NаНСО3). Значения максимальной прессорной реакции (пики) сравнивали для того, чтобы определить степень ингибирования, вызываемую испытуемым соединением. Доза испытуемого соединения, вызывающая 50% ингибирования (подавления) сосудосуживающей (прессорной) реакции big ET-1 (ED50), составляла 0,03 мкмол/кг.
(ii) Используя пероральное введение испытуемого соединения, проводили аналогичный эксперимент, который продемонстрировал его действие, а также его эффективность. "Болюс-инъекция" человеческого big ET-1 (1 нмол/кг, внутривенно; носитель: 1% Tween 80 в 5% NаНСО3) крысам Sprague-Dawley (n=3) вводили до и через 15, 105 и 195 минут после введения дозы 3 мкмол/кг испытуемого (обозначенного в заглавии) соединения. Пики значений прессорной реакции сравнивали для того, чтобы определить степень ингибирования, вызываемую испытуемым соединением в эти временные интервалы. Полученные результаты, демонстрирующие как эффективность, так и продолжительность действия испытуемого соединения, представлены в табл.1.
(С) Предсистемная метаболическая стабильность внутри желудочно-кишечного тракта
In vitro
Содержимое слепой кишки крыс быстро помещали в холодный дегазированный калий-фосфатный буфер (50 мМ, рН 7,4, продували азотом в течение, по меньшей мере, 30 минут). Каждая емкость (плашка) для культивирования (инкубации) содержала около 0,1 г слепокишечного содержимого/мл. Испытуемое (обозначенное в заглавии) соединение добавляли в плашку для термостатирования в виде раствора в бикарбонатном буфере. Термостатирование проводили с 200 мкМ испытуемого соединения, смешивали с ацетонитрилом в соотношении 1:1 и центрифугировали перед анализом. Образцы анализировали методами ВЭЖХ-УФ и ЖХ-MS (масс-спектрометрия) на колонке YMC-ODS AQ (4,6•150 мм, 3 мк) с градиентным элюированием ацетатом аммония/ацетонитрилом и определяли при 270 нм. Процентное содержание испытуемого (обозначенного в заглавии) соединения, остающегося интактным (т.е. метаболиты не наблюдались) после термостатирования с гомогенатом содержимого слепой кишки крысы в течение 1 часа, составляло 100%.
In vivo
Крысы, которым в желчные протоки введены канюли, голодали в течение ночи. Испытуемое (обозначенное в заглавии) соединение вводили в виде раствора в 5%-ном бикарбонате натрия (примерно, 8 мг/мл, 29 мг/кг) перорально с помощью желудочного зонда. Гомогенат содержимого желудочно-кишечного тракта готовили в 3 объемах воды и добавляли равный объем ацетонитрила. Испытуемое соединение идентифицировали методом ЖХ-MS/MS и количественно определяли методом ЖХ-УФ на колонке YMC-ODS AQ (4,6•150 мм, 3 мк) с градиентным элюированием системой ацетат аммония/ацетонитрил и детектированием при 270 нм. Процентное содержание остающегося интактным испытуемого (обозначенного в заглавии) соединения (т.е. метаболиты не наблюдались) в желудочно-кишечном тракте крыс через 9 часов после перорального приема дозы составляло 100%.
ПРИМЕР 3
Получение N(4,5-диметил-3-изоксазолил)-2'-[(3,3-диметил-2-оксо-1-пирролидинил)метил]-4'-(2-оксазолил)[1,1'-бифенил]-2-сульфонамида
A. N-(4,5-Диметил-3-изоксазолил)-2'-(гидроксиметил)-N-[(2-метоксиэтокси)метил]-4'-(2-оксазолил)[1,1'-бифенил]-2-сульфонамид
К раствору, обозначенного в заглавии Стадии D примера 1 продукта (0,37 г, 0,76 ммол), в 10 мл МеОН при комнатной температуре добавляют боргидрид натрия (0,035 г, 0,93 ммол) и смесь перемешивают в течение 2 час. Прозрачный раствор затем концентрируют до 5 мл и разбавляют 100 мл воды, и водный раствор экстрагируют 3•100 мл EtOAc. Объединенный органический экстракт промывают затем один раз водой, сушат и упаривают с получением 0,36 г (95%) обозначенного в заглавии этой стадии продукта в виде бесцветной смолы.
Rf(силикагель)=0,32 (5% метанола в CH2Cl2).
В. 2'-(Бромметил)-N-(4,5-диметил-3-изоксазолил)-N-[(2-метоксиэтокси)метил]-4'-(2-оксазолил)[1,1'-бифенил]-2-сульфонамид
К раствору продукта, полученного на Стадии А (0,37 г, 0,72 ммол), в 5 мл ДМФА при 5oС добавляют трифенилфосфин (0,283 г, 1,08 ммол) и четырехбромистый углерод (0,358 г, 1,08 ммол), и смесь перемешивают в течение 5 час. Затем раствор разбавляют 100 мл воды, и водный раствор экстрагируют 3•100 мл EtOAc. Объединенный органический экстракт промывают затем один раз водой, сушат и упаривают. Полученный таким образом остаток хроматографируют на 20 г силикагеля с использованием смеси 2:1 гексан : ЕtOАс с получением 0,285 г (69%) обозначенного в заглавии этой стадии продукта.
Rf(силикагель)=0,34 (5% метанола в СН2Сl2).
С. 3,3-Диметил-2-пирролидон
В колбу, содержащую гидрохлорид 1,1-диметилэтилового эфира 3,3-диметил-2-оксо-1-пирролидинкарбоновой кислоты (0,5 г, 2,34 ммол, полученный как описано в J. Chem. Res. (Synopsis), 414-415 (1993)), добавляют lN HCl в эфире (15 мл) и смесь перемешивают в течение ночи. Затем раствор упаривают и остаток сушат в вакууме с получением 0,26 г (98%) обозначенного в заглавии этой стадии продукта в виде светло-желтой смолы, которая затвердевает при стоянии.
D. N-(4,5-диметил-3-изоксазолил)-2'-[(3,3-диметил-2-оксо-1-пирролидинил)метил]-N-[[2-метоксиэтокси)метил]-4'-(2-оксазолил)[1,1'-бифенил]-2-сульфонамид
К раствору продукта, полученного на Стадии С, (0,055 г, 0,49 ммол), в 3 мл ДМФА добавляют NaH (60% суспензия в минеральном масле, 0,019 г, 0,49 ммол) и смесь перемешивают при комнатной температуре в атмосфере аргона в течение 5 минут. Затем добавляют продукт, полученный на Стадии В (0,14 г, 0,24 ммол), и смесь перемешивают при комнатной температуре в течение ночи. В отдельной колбе перемешивают в течение 10 минут дополнительно 0,0275 г (0,25 ммол) продукта, полученного на Стадии С, и 0,01 г (0,25 ммол) гидрида натрия в 1 мл ДМФА и эту смесь затем добавляют к вышеполученному раствору, и реакционную смесь перемешивают дополнительно еще 3 часа. Затем смесь приливают к 100 мл воды и раствор экстрагируют 3•50 мл EtOAc. Объединенный органический экстракт промывают водой, сушат и упаривают с получением 0,16 г (100%) обозначенного в заглавии этой стадии продукта в виде бесцветной смолы.
Rf(силикагель)=0,24 (5% метанола в СН2Сl2).
Е. N-(4,5-диметил-3-изоксазолил)-2'-[(3,3-диметил-2-оксо-1-пирролидинил)метил]-4-(2-оксазолил)[1,1'-бифенил]-2-сульфонамид
К раствору продукта, полученного на Стадии D (0,157 г, 0,25 ммол), в 2 мл ацетонитрила добавляют триметилхлорсилан (0,157 г, 1,45 ммол) и иодистый натрий (0,15 г, 1,45 ммол) и смесь перемешивают при комнатной температуре в течение 1 часа. Добавляют дополнительные порции триметилхлорсилан (0,078 г, 0,726 ммол) и йодистый натрий (0,075 г, 0,726 ммол) и смесь перемешивают дополнительно в течение 1 часа. Смесь разбавляют 20 мл 1%-ного водного раствора тиосульфата натрия и экстрагируют 3•12 мл EtOAc. Объединенный органический экстракт промывают один раз водой, сушат и упаривают. Остаток очищают с помощью препаративной обращенно-фазовой ВЭЖХ на колонке 30x500 мм с ODS S10 с использованием 70% растворителя В (90% МеОН, 10% Н2О, 0,1% трифторуксусной кислоты ("TFA")) и 30% растворителя А (10% МеОН, 90% H2O, 0,1% TFA). Соответствующие фракции собирают и нейтрализуют водным раствором бикарбоната натрия до рН 7 и концентрируют до 10 мл. Затем раствор подкисляют до рН 4 с использованием водного раствора бисульфата натрия и белое твердое вещество отфильтровывают и сушат с получением 0,036 г (28%) обозначенного в заглавии этого примера продукта в виде твердого вещества белого цвета, т пл. 110-117oС (аморфное).
Новые промежуточные соединения, полученные на стадиях А, В и D вышеприведенного примера, также заявляются в данном изобретении.
ПРИМЕР 4
Альтернативный способ получения N-(4,5-диметил-3-изоксазолил)-2'-[(3,3-диметил-2-оксо-1-пирролидинил)метил] -4'-(2-оксазолил)[1,1'-бифенил]-2-сульфонамида
А. 4-[[[2'-[[(4,5-Диметил-3-изоксазолил)амино] сульфонил]-4-(2-оксазолил)[1,1'-бифенил]-2-ил]метил]амино]-2,2-диметилбутановая кислота
К продукту, полученному на стадии Е из примера 1 (2,00 г, 4,72 ммол), 3-карбокси-3-метилбутиламмоний хлориду (1,58 г, 9,45 ммол, полученному как описано в J. Chem. Res. (S)., 414-415 (1993)) и молекулярным ситам 3А в 47 мл CH2Cl2 добавляют АсОН (0,85 г, 14,17 ммол) и затем добавляют ацетат натрия (0,775 г, 9,45 ммол).
Смесь перемешивают в течение 10 минут и добавляют триацетоксиборгидрид натрия (3,00 г, 14,17 ммол). Реакционную смесь перемешивают при комнатной температуре в течение 1 часа и 40 минут, разбавляют 100 мл CH2Cl2 и фильтруют через целит. Фильтрат промывают 2•30 мл Н2О, 30 мл рассола, сушат и концентрируют с получением остатка, содержащего обозначенный в заглавии продукт.
Rf(силикагель)=0,06 (СН2Сl2: метанол 10:1).
В. N-(4,5-диметил-3-изоксазолил)-2'-[(3,3-диметил-2-оксо-1-пирролидинил)метил]-4'-(2-оксазолил)[1,1'-бифенил]-2-сульфонамид
Весь остаток, полученный на стадии А, растворяют в 50 мл СН2Сl2 и добавляют 1,3-диизопропилкарбодиимид (775 мг, 6,14 ммол). Реакционную смесь перемешивают при комнатной температуре в течение 1 часа и разбавляют 50 мл СН2Сl2, промывают 30 мл Н2О, 30 мл рассола, сушат и концентрируют. Остаток хроматографируют на силикагеле с использованием смеси 50:50:1 гексан/ЕtOАс/АсОН с получением обозначенного в заглавии этого примера продукта (1,47 г, 60% на две стадии) в виде твердого вещества белого цвета, п. пл. 206-208oС (ЕtOН/Н2О).
Новое промежуточное соединение, полученное на стадии А вышеприведенного примера, также заявляется в данном изобретении. Это промежуточное соединение само по себе может найти применение в качестве антагониста эндотелина для лечения эндотелинзависящих расстройств.
ПРИМЕР 5
In vivo функциональная активность N-(4,5-диметил-3-изоксазолил)-2'-[(3,3-диметил-2-оксо-1-пирролидинил)метил] -4'-(2-оксазолил)[1,1'-бифенил]-2-сульфонамида
Превосходная in vivo функциональная активность испытуемого (обозначенного в заглавии) соединения (включая биодоступность, эффективность и продолжительность действия и метаболическую устойчивость) продемонстрирована следующим образом.
(A) Биодоступность
Применяя метод, приведенный в примере 2(А), определили, что биодоступность испытуемого соединения составляет примерно 78%.
(B) Вазопрессорный тест
Применяя метод, приведенный в Примере 2(B)(i), определили, что доза испытуемого (обозначенного в заглавии) соединения, вызывающая 50% ингибирования (подавления) сосудосуживающей (прессорной) реакции big ET-1 (ED50), составляет, примерно, 0,01 мкмол/кг.
Эффективность и продолжительность действия испытуемого соединения демонстрируют результаты, приведенные в табл. 2, которые получены при применении метода, описанного в примере 2(B)(ii).
(С) Предсистемная метаболическая стабильность внутри желудочно-кишечного тракта
Используя методы, описанные в примере 2(С), получили следующие данные стабильности испытуемого (обозначенного в заглавии) соединения.
In vitro
Количество испытуемого (обозначенного в заглавии) соединения, остающегося интактным после термостатирования с гомогенатом содержимого слепой кишки крысы в течение 1 часа, составляло 100%.
In vivo
Количество испытуемого (обозначенного в заглавии) соединения, остающегося интактным в желудочно-кишечном тракте крыс через 9 часов после перорального приема дозы вещества, составляло 100%.


Изобретение относится к соединениям: N-[[2'-[[(4,5-диметил-3-изоксазолил)амино] сульфонил] -4-(2-оксазолил)[1,1'-бифенил] -2-ил] метил]-N,3,3-триметилбутанамид и N-(4,5-диметил-3-изоксазолил)-2'-[(3,3-диметил-2-оксо-1 -пирролидинил)метил] -4'-(2-оксазолил)[1,1'-бифенил] -2-сульфонамид и их фармацевтически приемлемым солям, таким как литиевая, натриевая или калиевая соль или соль с основанием, являющимся органическим амином. Данные соединения могут использоваться в качестве антагонистов эндотелина. Технический результат - получение новых соединений, являющихся антагонистами эндотелина. 9 с. и 6 з.п. ф-лы, 2 табл.
| Перхлораты -арил-2,4,6-п-триметоксифенилпиридиния,обладающие люминесцентными свойствами | 1977 |
|
SU702012A1 |
| ЕР 0569193 А1, 10.11.1993 | |||
| Способ получения 3-амино-5-трет.бутилизоксазола | 1979 |
|
SU1003757A3 |

