В настоящей заявке испрашивается приоритет на основании предварительной заявки США рег. 60/011974, поданной 20, февраля 1996 и включенной в настоящее описание в виде ссылки.
Область изобретения
Настоящее изобретение относится к способам получения бифенилизоксазолсульфонамидов и их промежуточных соединений. Настоящее изобретение также относится к новым промежуточным соединениям, полученным этими способами. Бифенилизоксазолсульфонамиды, полученные способами по настоящему изобретению, являются антагонистами эндотелина, которые могут быть использованы помимо прочего для лечения гипертензии.
Краткое описание изобретения
Способы настоящего изобретения позволяют получить бифенилсульфонамиды следующей формулы I: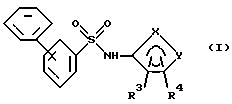
где фенильные кольца бифенильной группы могут быть независимо незамещенными или замещены одной или несколькими замещающими группами; а также их энантиомеры и диастереомеры, и соли, предпочтительно фармацевтически приемлемые соли. Предпочтительными замещающими группами для бифенильной группы являются группы R11-R14, описанные ниже, а особенно предпочтительно, если бифенильная группа является 2-бифенильной группой,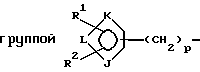
в 4'-положении. Предпочтительные способы настоящего изобретения позволяют получить соединения следующей формулы Iа: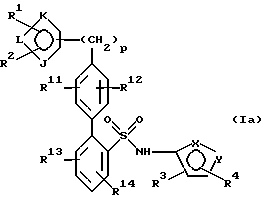
их энантиомеры и диастереомеры, и соли, предпочтительно фармацевтически приемлемые соли. В описании настоящего изобретения вышеуказанные символы имеют следующие значения:
один из Х и Y представляет N, а другой представляет О;
каждый из R1, R2, R3 и R4 непосредственно связан с углеродом кольца и каждый независимо представляет:
(a) водород;
(b) алкил, алкенил, алкинил, алкокси, циклоалкил, циклоалкилалкил, циклоалкенил, циклоалкенилалкил, арил, арилокси, аралкил или аралкокси, каждый из которых может быть замещен Z1, Z2 и Z3;
(c) галоген;
(d) гидроксил;
(e) циано;
(f) нитро;
(g) -С(О)Н или -C(О)R5;
(h) -CO2H или -CО2R5;
(i) -Z4-NR6R7;
(j) -Z4-N(R10)-Z5-NR8R9; или
(к) R3 и R4, взятые вместе, могут также представлять алкилен или алкенилен, каждый из которых может быть замещен Z1, Z2 и Z3, с образованием 4-8-членного насыщенного, ненасыщенного или ароматического кольца вместе с атомами углерода, к которым они присоединены;
R5 представляет алкил, алкенил, алкинил, циклоалкил, циклоалкилалкил, циклоалкенил, циклоалкенилалкил, арил или аралкил, каждый из которых может быть замещен Z1, Z2 и Z3;
каждый из R6, R7, R8, R9 и R10 независимо представляет:
(a) водород; или
(b) алкил, циклоалкил, циклоалкилалкил, циклоалкенилалкил, арил или аралкил, каждый из которых может быть замещен Z1, Z2 и Z3; или
R6 и R7, взятые вместе, могут представлять алкилен или алкенилен, каждый из которых может быть замещен Z1, Z2 и Z3, с образованием 3-8-членного насыщенного или ненасыщенного кольца вместе с атомом азота, к которому они присоединены; либо любые два из R8, R9 и R10, взятые вместе, представляют алкилен или алкенилен, каждый из которых может быть замещен Z1, Z2 и Z3, образуя 3-8-членное насыщенное или ненасыщенное кольцо вместе с атомами, к которым они присоединены;
каждый из R11, R12, R13 и R14 независимо представляет:
(a) водород;
(b) алкил, алкенил, алкинил, алкокси, циклоалкил, циклоалкилалкил, циклоалкенил, циклоалкенилалкил, арил, арилокси, аралкил или аралкокси, каждый из которых может быть замещен Z1, Z2 и Z3;
(c) гетероцикл, замещенный гетероцикл или гетероциклокси;
(d) галоген;
(e) гидроксил;
1(f) циано;
(g) нитро;
(h) -C(О)H или -C(О)R5;
(i) -CО2H или -CO2R5;
(j) -SH; -S(О)nR5, -S(О)m-OH; -S(О)m-OR5;
-O-S (O)m-OR5; -О-S(O)m-OH; или -О-S(О)m-OR5;
(k) -Z4-NR6R7; или
(1) -Z4-N(R10)-Z5-NR8R9;
каждый из Z1, Z2 и Z3, независимо, представляет:
(a) водород;
(b) галоген;
(c) гидрокси;
(d) алкил;
(e) алкенил;
(f) арил;
(g) аралкил;
(h) алкокси;
(i) арилокси;
(j) аралкокси;
(k) гетероцикл, замещенный гетероцикл или гетероциклоокси
(l) -SH; -S(О)nZ6, -S(О)m-ОН; -S(О)m-OZ6;
-О-S(O)m-OZ6; -О-S(О)m-ОН или -О-S(O)m-OZ6;
(m) оксо;
(n) нитро;
(о) циано;
(р) -С(О)Н или -C(О)Z6;
(q) -СО2Н или -CO2Z6;
(r) -Z4-NZ7Z8;
(s) -Z4-N(Z11)-Z5-H;
(t) -Z4-N(Z11)-Z5-Z6; или
(u) -Z4-N(Z11)-Z5-NZ7Z8;
каждый из Z4 и Z5 независимо представляет:
(a) простую связь;
(b) -Z9-S(O)n-Z10-;
(c) -Z9-C(O)-Z10-;
(d) -Z9-C(S)-Z10-;
(e) -Z9-O-Z10-;
(f) -Z9-S-Z10-;
(g) -Z9-O-C(O)-Z10-; или
(h) -Z9-C(O)-O-Z10-;
Z6 представляет алкил; алкил, замещенный одной-тремя группами, выбранными из галогена, арила, арилокси и алкокси; алкенил; алкинил; циклоалкил; циклоалкил, замещенный одной-тремя группами, выбранными из алкила, арила, алкенила и алкоксиарила; циклоалкил, который конденсирован с бензольным кольцом; арилокси, замещенный одним или двумя атомами галогена; циклоалкилалкил; циклоалкенил; циклоакенилалкил; арил; арил, замещенный метилендиокси или одной-четырьмя группами, выбранными из алкила, диалкиламино, циано, галогена, тригалогеналкила, алкокси и тригалогеналкокси; или гетероцикл или замещенный гетероцикл;
каждый из Z7 и Z8 независимо представляет водород, алкил, циклоалкил, циклоалкилалкил, циклоалкенилалкил, арил, или аралкил; или Z7 и Z8, взятые вместе, представляют алкилен или алкенилен с образованием 3-8-членного насыщенного или ненасыщенного кольца вместе с атомом азота, к которому они присоединены;
каждый из Z9 и Z10 независимо представляет простую связь, алкилен, алкенилен или алкинилен;
Z11 представляет:
(a) водород; или
(b) алкил; алкил, замещенный одним, двумя или тремя галогенами, циклоалкил, циклоалкилалкил, циклоалкенилалкил, арил, или аралкил;
или любые два из Z7, Z8 и Z11, взятые вместе, представляют алкилен или алкенилен с образованием 3-8-членного насыщенного или ненасыщенного кольца вместе с атомами, к которым они присоединены;
J представляет О, S, N или NR15;
К и L представляют N или С при условии, что, по крайней мере, один из К или L представляет С;
R15 представляет водород, алкил, гидроксиэтоксиметил или метоксиэтоксиметил;
каждый m независимо представляет 1 или 2;
каждый n независимо представляет 0, 1 или 2; и
р представляет 0 или целое число от 1 до 2.
В соответствии с указанным соединение формулы I или его соль могут быть получены способом, включающим следующие стадии:
(а) взаимодействие сложного эфира пинакола формулы II или его соли: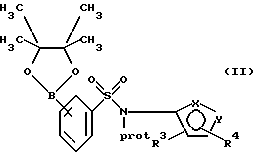
где фенильное кольцо указанной формулы II может быть, кроме того, замещенным одной или несколькими группами, описанными здесь для групп R11-R14, с производным галогенфенила формулы III или с его солью: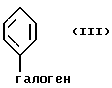
где фенильное кольцо указанной формулы III может быть, кроме того, замещено одной или несколькими группами, описанными здесь для групп R11-R14, и, в частности, если бифенильная группа указанного соединения формулы I или его соли является 2-бифенилом, то
находится в пара-положении по отношению к галогеновой группе, в присутствии палладиевого(0) катализатора и предпочтительно основания с образованием азотзащитного производного формулы IV или его соли:
где фенильные кольца бифенильной группы могут быть независимо незамещенными или замещены одной или несколькими замещающими группами заместителей; и
(b) удаление защиты у атома азота указанного соединения формулы IV или его соли с образованием указанного соединения формулы I или его соли.
Обозначение "Prot", используемое в формуле II и во всем описании, означает азотзащитную группу, которая может быть любой подходящей азотзащитной группой, такой как 2-этоксиэтил, 2-метоксипропил, метоксиэтоксиметил или группой, описанной в публикации Европейской патентной заявки 569193 (1993), включенной здесь в качестве ссылки и предпочтительно метоксиэтоксиметил ("MEM"). Группой галогена в формуле III предпочтительно является бром или йод, а наиболее предпочтительно йод.
В предпочтительном варианте настоящего изобретения, соединение формулы Iа или его соль могут быть получены способом, включающим следующие стадии:
(а) взаимодействие сложного эфира пинакола формулы IIа или его соли: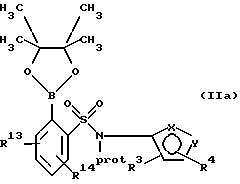
с галогенфенильным производным формулы IIIa или его солью: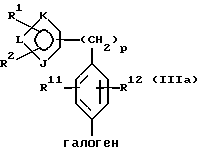
в присутствии палладиевого(0) катализатора и предпочтительно основания с образованием азотзащищенного производного формулы IVa или его соли:
и (b) удаление защиты у атома азота соединения указанной формулы IVa или его соли с образованием соединения формулы Iа или его соли.
Рассматриваемые способы получения соединения формулы I или его соли имеют то преимущество, что они позволяют получить высокие выходы нужного соединения с минимальными примесями или без них.
Кроме того, приложены новые промежуточные соединения по способам настоящего изобретения и новые способы получения этих промежуточных соединений.
Подробное описание изобретения
Далее представлено более подробное описание изобретения. Ниже приводятся определения терминов, используемых в настоящем описании. Если это не оговорено особо, то эти определения относятся к терминам, используемым во всем описании независимо от того, используются ли они отдельно или являются составной частью другой группы.
Термин "алкил" или "алк-" относится к прямым или разветвленным углеводородным группам, имеющим 1-10 атомов углерода, а предпочтительно 1-7 атомов углерода. Понятие "низший алкил" относится к алкильным группам, имеющим 1-4 атомов углерода.
Термин "алкокси" означает алкил-О-.
Термин "арил" или "ар-" относится к фенилу, нафтилу и бифенилу.
Термин "алкенил" означает прямые или разветвленные углеводородные группы с 2-10 атомами углерода, имеющие, по крайней мере, одну двойную связь. При этом предпочтительными являются группы с 2-4 атомами углерода.
Термин "алкинил" означает прямые или разветвленные углеводородные группы с 2-10 атомами углерода, имеющие, по крайней мере, одну тройную связь. При этом предпочтительными являются группы с 2-4 атомами углерода.
Термин "алкилен" означает мостик с прямой цепью и 1-5 атомами углерода, присоединенный посредством одинарных связей (например, (СН2)х-, где х=1-5), который может быть замещен 1-3 низшими алкильными группами.
Термин "алкенилен" означает мостик с прямой цепью и 2-5 атомами углерода, который имеет одну или две двойные связи, которые присоединены посредством простых связей, и может быть замещен 1-3 низшими алкильными группами. Примерами алкениленовых групп являются -СН=СН-СН=СН-, СН2-СН=СН-, -СН2-СН=СН-СН2-, -С(СН3)2СН=СН- и -СН(C2H5)-СН=СН-.
Термин "алкинилен" означает мостик с прямой цепью и 2-5 атомами углерода, который имеет тройную связь, присоединен посредством простых связей и может быть замещен 1-3 низшими алкильными группами. Примерами алкиниленовых групп являются -С≡С-, -СН2-С≡С-, -СН(СН3)-С≡С- и -C≡C-CH(C2H5)CH2-.
Термин "алканоил" означает группы формулы С(О)алкил.
Термины "циклоалкил" и "циклоалкенил" означают циклические углеводородные группы с 3-8 атомами углерода.
Термин "гидроксиалкил" означает алкильную группу, включающую один или несколько гидроксирадикалов, таких, как -СН2СН2ОН, -СН2СН2ОНСН2ОН, -CH(CH2OH)2, и т.п.
Термины "галоген" и "галогеновая группа" относятся к фтору, хлору, брому и йоду.
Термины "гетероцикл", "гетероциклический" и "гетеробицикло" относятся к необязательно замещенной, полностью насыщенной или ненасыщенной, ароматической или неароматической циклической группе, например, такой, которая представляет собой 4-7-членную моноциклическую, 7-11-членную бициклическую, или 10-15-членную трициклическую кольцевую систему, которая имеет, по крайней мере, один гетероатом, по крайней мере, в одном кольце, содержащем один атом углерода. Каждое кольцо гетероциклической группы, содержащее гетероатом, может иметь 1, 2 или 3 гетероатома, выбранные из атомов азота, кислорода и серы, где указанные гетероатомы азота и серы могут быть необязательно окислены, а гетероатомы азота могут быть необязательно кватернизованы. Гетероциклическая группа может быть связана с любым гетероатомом или атомом углерода.
Примерами моноциклических гетероциклических групп являются пирролидинил, пирролил, пиразолил, оксетанил, пиразолинил, имидазолил, имидазолинил, имидазолидинил, оксазолил, оксазолидинил, изоксазолинил, изоксазолил, тиазолил, тиадиазолил, тиазолидинил, изотиазолил, изотиазолидинил, фурил, тетрагидрофурил, тиенил, оксадиазолил, пиперидинил, пиразинил, 2-оксопиперазинил, 2-оксопиперидинил, 2-оксопирролидинил, 2-оксоазепинил, азепинил, 4-пиперидонил, пиридил, пиразинил, пиримидинил, пиридазинил, тетрагидропиранил, морфолинил, тиаморфолинил, тиаморфолинилсульфоксид, тиаморфолинилсульфон, 1,3-диоксолан, и тетрагидро-1,1-диоксотиенил, и т.п.
Примерами бициклических гетероциклических групп являются индолил, бензотиазолил, бензоксазолил, бензотиенил, хинуклидинил, хинолинил, тетрагидроизохинолинил, изохинолинил, бензимидазолил, бензопиранил, индолизинил, бензофурил, хромонил, кумаринил, бензопиранил, циннолинил, хиноксалинил, индазолил, пирролопиридил, фуропиридинил (такой как фуро[2,3-с]-пиридинил, фуро[3,2-b] пиридинил, или фуро[2,3-b]пиридинил), дигидроизоиндолил, дигидрохиназолинил (такой как 3,4-дигидро-4-оксохиназолинил), тетрагидрохинолинил, и т.п.
Примерами трициклических гетероциклических групп являются карбазолил, бензидолил, фенантролинил, акридинил, фенантридинил, ксантенил, и т.п.
Выражение "замещенный гетероцикл" означает гетероцикл, замещенный 1, 2 или 3 из следующих групп:
(a) алкил, особенно низший алкил;
(b) гидрокси (или защищенный гидрокси);
(c) галоген;
(d) оксо (то есть, = О);
(e) амино, алкиламино или диалкиламино;
(f) алкокси;
(g) карбоцикло, такой, как циклоалкил;
(h) карбокси;
(i) гетероциклоокси;
(j) алкоксикарбонил, такой как незамещенный низший алкоксикарбонил;
(k) карбамил, алкилкарбамил или диалкилкарбамил;
(l) меркапто;
(m) нитро;
(n) циано;
(о) карбоалкокси;
(р) сульфонамидо; сульфонамидоалкил; или сульфонамидодиалкил;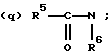

(s) арил;
(t) алкилкарбонилокси;
(u) арилкарбонилокси;
(v) арилтио;
(w) арилокси;
(х) алкилтио;
(y) формил;
(z) арилалкил или
(а') арил, замещенный алкилом, циклоалкилом, алкокси, гидрокси, амино, алкиламино, диалкиламино, галогеном или тригалогеналкилом.
Термин "гетероциклоокси" означает гетероциклическую группу, присоединенную посредством кислородного мостика.
Во всем описании группы и их заместители могут быть выбраны так, чтобы они обеспечивали получение стабильных радикалов и соединений.
Соединение формулы I и их промежуточные соединения могут образовывать соли, которые также входят в объем настоящего изобретения. При этом предпочтительными являются фармацевтически приемлемые соли (то есть нетоксичные физиологически приемлемые соли), хотя могут быть также использованы и другие соли, например, для выделения и очистки соединений настоящего изобретения.
Соединения формулы I и их промежуточные соединения могут образовывать соли со щелочными металлами, такими как натрий, калий и литий; с щелочноземельными металлами, такими как кальций и магний; с органическими основаниями, такими как дициклогексиламин, трет-бутиламино, бензатин, N-метил-D-глюкамид и гидрабамин; и с аминокислотами, такими как аргинин, лизин и т.п. Такие соли могут быть получены посредством реакции этих соединений с нужным ионом в среде, в которой соль осаждается, или в водной среде с последующей лиофилизацией.
Если группы, такие как заместители R1-R4 или R11-R14, содержат основную часть, такую как амино или замещенный амино, то соединения формулы I и их промежуточные соединения могут образовывать соли с рядом органических и неорганических кислот. Такими солями являются соли, образованные хлористоводородной кислотой, бромистым водородом, метансульфоновой кислотой, серной кислотой, уксусной кислотой, малеиновой кислотой, бензолсульфонатом, толуолсульфонатом и различными другими сульфонатами, нитратами, фосфатами, боратами, ацетатами, тартратами, малеатами, цитратами, сукцинатами, бензоатами, аскорбатами, салицилатами и т.п. Такие соли могут быть образованы посредством реакции указанных соединений в эквивалентном количестве кислоты в среде, в которой соль осаждается, или в водной среде с последующей лиофилизацией.
Кроме того, если группы, такие как заместители R1-R4 или R11-R14, содержат основную часть, такую как амино, то могут быть образованы цвиттерионы ("внутренние соли").
Некоторые группы, такие как заместители R1-R4 или R11-R14 соединений настоящего изобретения, могут содержать асимметрические атомы углерода. Поэтому соединения настоящего изобретения, такие как соединения формулы I и их соли, могут существовать в энантиомерных и диастереомерных формах и в виде их рацемических смесей. Все указанные формы входят в объем настоящего изобретения. Кроме того, такие соединения, как соединения формулы I и их соли, могут существовать в виде энантиомеров даже при отсутствии асимметрических атомов углерода. Все указанные энантиомеры входят в объем настоящего изобретения.
Заявка на патент США рег. 08/493331, поданная 24 июля 1995 (Attorney Docket НА662с) Murugesan et al., и являющаяся ее частичным продолжением заявка на патент США рег. 08/603975, поданная 20 февраля 1996 (Attorney Docket НА662с) Murugesan et al. под названием "Substituted Biphenyl Isoxazole Sulfonamides" ("Замещенные бифенилизоксазолсульфонамиды"), в которых описаны антагонисты эндотелина, исходные соединения и методы, вводятся в настоящее описание во всей своей полноте посредством ссылки.
Реакция сочетания соединений формул II и III и удаление защиты
Соединение формулы I или его соль могут быть получены путем реакции взаимодействия сложного эфира пинакола формулы II или его соли с галогенфениловым соединением формулы III или его солью с последующим удалением защиты у атома азота соединения IV или его соли, образованного (или образованной) с помощью вышеуказанной реакции.
Реакцию взаимодействия соединений формул II и III или их солей проводят в присутствии палладиевого (0) катализатора, предпочтительно ацетата палладия/трифенилфосфина или другой соли палладия (II)/трифенилфосфина, тетракисфенилфосфинпалладия или трис(дибензилиденацетон)дипалладия и предпочтительно основания, предпочтительно водного карбоната калия или карбоната натрия, в результате чего получают замещенное у атома азота соединение формулы IV или его соль. Предпочтительное мольное отношение соли палладия(II) к трифенилфосфину составляет от 1:1 до 1:3. Условия катализа описаны в работах Suzuki et al. . Pure & Applied Chemistry, 63, 419-422 (1991); А. Martin et al. , Acta. Chem. Scand., 47, 221 (1993); Н. Jendralla et al., Liebig Ann., 1253 (1995), которые вводятся в настоящее описание посредством ссылки.
Если галогенфенильным производным III является соединение IIIa, то в некоторых случаях для облегчения реакции взаимодействия может оказаться желательной защита гетероатомов J и К или L. Так, например, если J и К или L представляют N, то одна из этих групп может быть защищена подходящей защитной группой, такой как трет-бутоксикарбонил, и т.п. Конкретные группы R11-R14 могут быть выбраны в соответствии с реакционными условиями. Кроме того, конкретные группы R11-R14 могут быть преобразованы в альтернативные группы R11-R14 либо до, либо после реакции взаимодействия с использованием любых подходящих способов, известных специалистам.
Эту реакцию предпочтительно проводят при температуре от около 25oС до около 100oС (наиболее предпочтительно от около 45oС до около 75oС), под давлением около 1 атм, в атмосфере аргона или азота. Мольные отношения сложного эфира пинакола II или его соли к галогенфенильному соединению III или его соли составляют предпочтительно от около 1:1 до около 1:1,2. Количества палладиевого(0) катализатора и основания выбирают для катализа реакции сочетания, и эти количества предпочтительно составляют от около 2,5 мол. % до около 10 мол.%, и от около 2,5 эквивалентов до около 7 эквивалентов соответственно. Используемые в реакции растворители предпочтительно выбирают из водных или органических жидких растворителей, таких как ацетон, этанол, толуол, тетрагидрофуран, диметоксиэтан и вода, или их смесей, предпочтительно смеси толуола и этанола. Количества растворителя являются предпочтительно такими, при которых сложный эфир пинакола II или его соль составляет от около 4 до около 9 мас.% исходя из общей массы растворителя и сложного эфира пинакола II или его соли. Так, например, примерные количества растворителя сложного эфира пинакола II/основания составляют: тетрагидрофуран (30-70 мл), толуол (100-200 мл), этанол (80-160 мл)/ сложный эфир пинакола II (15-20 г)/водный 2М карбонат натрия (100-150 мл).
Остатки палладиевого катализатора предпочтительно удаляют либо перед, либо после удаления защиты у соединения формулы IV или его соли путем взаимодействия с хелатообразующим агентом, таким как тритиоциануровая кислота ("ТМТ"). В настоящем изобретении также предусматривается осуществление кристаллизации соединения формулы V или его соли после удаления защиты при условии, что кристаллическая форма соединения формулы I или его соли является подходящей для целей настоящего изобретения. Кристаллизацию предпочтительно осуществляют из перенасыщенного этанольного раствора в отсутствии или в присутствии сорастворителей, таких как гептан или вода, особенно с использованием затравки желаемой кристаллической формы. Наиболее предпочтительно, когда кристаллизацию осуществляют способами, описанными в нижеприведенных примерах.
Соединения формулы III и его соли могут быть получены методами, аналогичными методам, описанным в заявке на патент США, рег. 08/493331, и в вышеуказанной заявке, являющейся ее частичным продолжением. Предпочтительно оксазольные производные формулы IIIa или его соли получают новыми способами, разработанными для их получения и описанными в настоящей заявке. Соединения формулы II и его соли предпочтительно получают новыми способами, разработанными для их получения и описанными в настоящей заявке.
Удаление защиты соединения формулы IV или его соли, образованного (или образованной) путем взаимодействия, осуществляемого способом по настоящему изобретению, может быть проведено подходящим методом, таким как методы, аналогичные описанным в заявке на патент США, рег. 08/493331, и в вышеуказанной заявке, являющейся ее частичным продолжением. Причем, если "prot" обозначает MEM, то удаление защиты предпочтительно проводят путем нагревания в смеси водной НС1 и этанола.
Получение соединений формулы II
Сложные эфиры пинакола формулы II и его соли могут быть сами получены новыми способами, описанными в настоящей заявке. В соответствии с этим сложный эфир пинакола формулы II или его соль могут быть получены способом, включающим следующие стадии:
(а) взаимодействие соединения формулы V или его соли: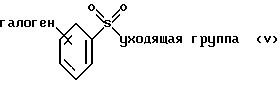
где фенильная группа указанной формулы V может быть, кроме того, замещена одной или несколькими группами, описанными выше для групп R11-R14, а галоген является предпочтительно бромом, хлором или йодом, и наиболее предпочтительно бромом, с амином формулы VI или его солью: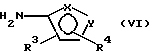
в присутствии органического основания и органического растворителя с образованием соединения формулы VII или его соли: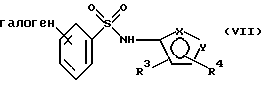
где фенильная группа в указанной формуле VII может быть, кроме того, замещена одной или несколькими группами, описанными выше для групп R11-R14,
(b) защиту атома азота указанного соединения формулы VII или его соли с образованием соединения формулы VIII или его соли: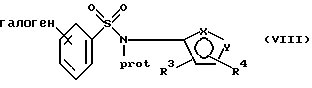
где фенильная группа в указанной формуле VIII может быть, кроме того, замещена одной или несколькими группами, описанными выше для групп R11-R14;
(с) получение литиевого производного указанного соединения формулы VIII или его соли с использованием алкил- или ариллитиевого соединения и взаимодействие образованного литиевого производного с триалкилборатом с последующим гидролизом с образованием бороновой кислоты формулы IX или ее соли:
где фенильная группа в указанной формуле IX может быть, кроме того, замещена одной или несколькими группами, описанными выше для групп R11-R14; и
(d) взаимодействие указанного соединения формулы IX или его соли с пинаколом (то есть 2,3-диметил-2,3-бутандиолом) с удалением воды и образованием в результате указанного соединения формулы II или его соли.
В предпочтительном варианте настоящего изобретения сложный эфир пинакола формулы IIа или его соль могут быть получены способом, включающим следующие стадии:
(а) взаимодействие соединения формулы Va или его соли: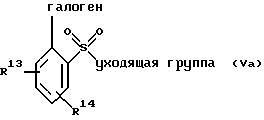
с амином формулы VIa или его солью: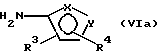
в присутствии органического основания или органического растворителя с образованием соединения формулы VIIa или его соли:
(b) защиту азота указанного соединения формулы VIIa или его соли с получением соединения формулы VIIIa или его соли: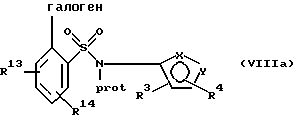
(с) получение литиевого производного указанного соединения формулы VIIIa или его соли с использованием алкил- или ариллитиевого производного и взаимодействие образованного литиевого производного с триалкилборатом с последующим гидролизом и образованием бороновой кислоты формулы IХа или ее соли: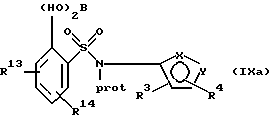
(d) взаимодействие указанного соединения формулы IХа или его соли с пинаколом с удалением воды и получением в результате указанного соединения формулы IIа или его соли.
Термин "уходящая группа", используемый в настоящем описании, означает любую подходящую уходящую группу, такую как галогеновая группа, предпочтительно хлор. На стадии (а) может быть использовано любое подходящее органическое основание. Предпочтительными органическими основаниями являются амины, такие как пиридин или триалкиламин. Органическим растворителем, используемым в стадии (а), является предпочтительно галогеналкан, такой как дихлорметан или 1,2-дихлорэтан либо в качестве растворителя может быть использовано органическое основание, такое как чистый пиридин.
Как описано выше, соединения формулы VIII и его соли могут быть получены путем взаимодействия соединения формулы V или его соли с производным амина формулы VI или его солью с последующей защитой у атома азота полученного соединения VII или его соли. Затем соединение формулы VIII или его соль подвергают преобразованию в литиевое производное с использованием алкил- или ариллитиевого соединения, предпочтительно н-бутиллития или фениллития, предпочтительно при температурах от около -40oС до около -105oС (особенно от около -70oС до около -100oС) с образованием соединения: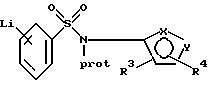
где фенильная группа в указанном соединении может быть, кроме того, замещена одной или несколькими группами, описанными выше для групп R11-R14, или его соли; а предпочтительно соединения:
или его соли. Обработка литиевого производного или его соли триалкилборатом, таким как триизопропилборат или предпочтительно триметилборат, предпочтительно при температурах от около -40oС до около -105oС (особенно от около -70oС до около -100oС) приводит к получению следующего боронатного эфира: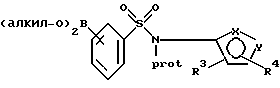
где фенильная группа в указанном соединении может быть, кроме того, замещена одной или несколькими группами, описанными выше для групп R11-R14, или его соли; предпочтительно боронатного эфира:
или его соли, который (или которая) может быть затем гидролизован(а) подходящей кислотой, предпочтительно водной кислотой, такой как водная соляная кислота; либо подходящим основанием с образованием бороновой кислоты IX или ее соли. Стадия гидролиза с образованием бороновой кислоты IX или ее соли является предпочтительной, поскольку указанная бороновая кислота обладает повышенной стабильностью по сравнению с боронатным сложным эфиром, из которого она была получена. Вышеуказанные стадии могут быть проведены методами, аналогичными методам, описанным в публикации европейской патентной заявки 569193 (1993), в заявке на патент США рег. 08/493331 и в вышеуказанной заявке, являющейся ее частичным продолжением, а также с использованием соединений, полученных методами, аналогичными методам, описанным в указанных работах.
Бороновая кислота IX или ее соль могут быть затем подвергнуты взаимодействию с пинаколом при удалении воды с образованием соответствующего сложного эфира пинакола II или его соли. Удаление воды может быть осуществлено, например, путем добавления осушителя, такого как сульфат магния, или путем азеотропного удаления воды при нагревании с растворителем, таким как толуол. Эту реакцию предпочтительно осуществляют при температуре от около 110oС до около 120oС (наиболее предпочтительно от около 112oС до около 115oС), под давлением около 1 атм, в атмосфере аргона или азота. Мольные отношения пинакола к бороновой кислоте IX или ее соли составляют предпочтительно от около 1:1 до около 1,1:1. Используемые растворители предпочтительно выбраны из органических жидких растворителей, таких как толуол. Количества растворителя являются предпочтительно такими, при которых бороновая кислота IX или ее соль составляет от около 4 до около 10 мас.%, исходя из общей массы растворителя и бороновой кислоты IX или ее соли.
Бороновая кислота IX или ее соль (предпочтительно бороновая кислота IХа или ее соль) могут быть подвергнуты непосредственному взаимодействию с галогенфенильным производным III или его солью с образованием соединения формулы IV или его соли. Этот метод, особенно когда галогенфенильное производное III или его соль является йодфенильным соединением III или его солью (предпочтительно йодфенильным соединением IIIa или его солью), также рассматривается в настоящем изобретении. Однако по сравнению с бороновой кислотой IX или ее солью сложный эфир пинакола II или его соль может иметь определенные преимущества, поскольку сложноэфирные соединения пинакола являются в высокой степени стабильными и в результате взаимодействия с галогенфенильным производным III или его солью могут образовываться меньшие количества примесей и могут быть получены более высокие выходы соединения формулы IV.
Получение соединений формулы III
Галогенфенильные производные формулы III или их соли могут быть получены методами, аналогичными описанным в заявке на патент США, рег. 08/493331, и в вышеуказанной заявке, являющейся ее частичным продолжением. Предпочтительные соединения формулы IIIa и их соли, несущие оксазольное кольцо, могут быть также получены новыми способами, описанными в настоящей заявке. В соответствии с этим оксазол формулы IIIа(1) или его соль могут быть получены способом, включающим следующие стадии:
(а) взаимодействие фенилгалогенангидрида Х или его соли: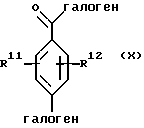
с аминоацеталем XI или его солью: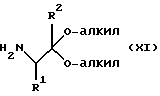
в присутствии основания и растворителя с образованием амидоацеталя формулы XII или его соли: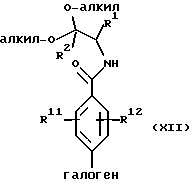
(b) циклизацию амидоацеталя формулы XII или его соли в присутствии циклизующего агента с образованием оксазолфенилгалогенида формулы IIIa(l) или его соли: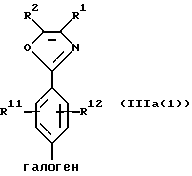
Исходный фенилгалогенангидрид Х или его соль является коммерчески доступным продуктом либо он может быть легко получен любым специалистом. Галогеновой группой галогенангидридной части является предпочтительно хлор; а галогеновой группой, находящейся в пара-положении по отношению к галогенангидридной части, является предпочтительно бром, хлор или йод и наиболее предпочтительно йод. Исходный аминоацеталь IX или его соль также является коммерчески доступным продуктом, либо он может быть легко получен каждым специалистом. Алкильными группами ацетальной части являются предпочтительно метил или этил и наиболее предпочтительно метил.
Основанием, используемым на стадии (а), может быть любое подходящее основание, предпочтительно карбонат, бикарбонат или гидроксид щелочного металла, и наиболее предпочтительно бикарбонат калия (в растворителе, таком как вода и/или ацетон), или карбонат калия (в растворителе, таком как метиленхлорид).
Циклизацию осуществляют путем взаимодействия амидоацеталя XII или его соли с циклизующим агентом, которым может быть любое соединение, осуществляющее реакцию циклизации, предпочтительно реактив Итона (Eaton) (то есть метансульфоновая кислота и оксид фосфора) или полифосфорная кислота (РРА) и наиболее предпочтительно реактив Итона. Циклизацию предпочтительно осуществляют при температуре от около 125oС до около 150oС (наиболее предпочтительно от около 130oС до около 135oС), под давлением около 1 атм, в атмосфере аргона или азота. Количества циклизующего агента выбирают так, чтобы осуществлялась реакция циклизации, и эти количества предпочтительно составляют от около (катализатор/субстрат) 8 мл/г до около 15 мл/г при использовании реактива Итона. Реактив Итона представляет собой раствор P2O5 в метансульфоновой кислоте и может быть также использован как предпочтительный растворитель для циклизации. Примерный состав реактива Итона содержит 7,5-15 мас.% P2O5 в метансульфоновой кислоте.
Соединения формулы IIIa и его соли могут быть также использованы в другом способе ("обратного взаимодействия") получения соединений формулы Iа или их солей, рассматриваемом в настоящем изобретении и включающем стадии:
(а) получение литиевого производного соединения формулы IIIa или его соли: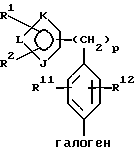
предпочтительно соединения формулы IIIа(1) или его соли с использованием алкил- или ариллитиевого соединения в присутствии триалкилбората с последующим гидролизом и получением бороновой кислоты формулы XIII или ее соли: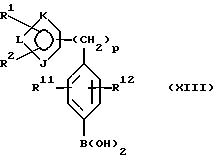
(b) взаимодействие бороновой кислоты формулы XIII или ее соли с соединением формулы VIIIa или его соли:
где галогеном является предпочтительно бром, йод или хлор и наиболее предпочтительно бром в присутствии палладиевого(0) катализатора и предпочтительно основания с образованием азотзащитного соединения формулы IVa или его соли: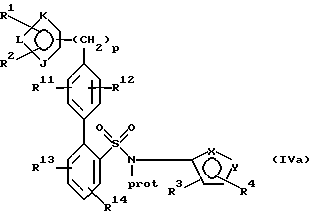
(с) удаление защитной группы у атома азота указанного соединения формулы IVa или его соли с образованием указанного соединения формулы Iа или его соли.
По этому способу получение литиевого производного проводят в присутствии триалкилбората с последующим гидролизом, который может быть осуществлен в условиях, описанных в настоящей заявке для получения бороновых кислот формулы IX и их солей. Реакция взаимодействия в присутствии палладиевого(0) катализатора и предпочтительно основания и удаление защиты у азотзащищенного продукта могут быть осуществлены в условиях, указанных в настоящем описании для проведения реакции взаимодействия соединений формул II и III и их солей, и удаление защиты у продукта этой реакции. После реакции введения лития получают соединение, имеющее следующую структуру, или его соль: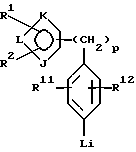
а после взаимодействия с триалкилборатом получают следующий сложный эфир бороновой кислоты или его соль: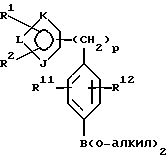
Предпочтительные соединения
Предпочтительно, чтобы соединения, используемые по способам настоящего изобретения или полученные способами настоящего изобретения, содержали один или несколько, предпочтительно все (если это необходимо) из нижеследующих заместителей:
Х представляет О, а У представляет N;
кольцо, несущее К, L и J, является 2-оксазолом;
р=0;
каждый из R1 и R2 независимо представляет водород, алкил, алкокси, арил, гидроксиалкил, -CO2R5 или Z4-NZ7Z8, а наиболее предпочтительно низший алкил или водород;
каждый из R3 и R4 независимо представляет алкил, а наиболее предпочтительно низший алкил, а особенно метил;
каждый из R11, R12, R13 и R14 независимо представляет водород, гидрокси, амино, гетероцикло, алкенил, алкокси, карбоксамид или замещенный низший алкил, а наиболее предпочтительно, если R12-R14 представляют водород, a R11 представляет водород, гидрокси, амино, гетероцикло, алкенил, алкокси, карбоксамид или замещенный низший алкил.
Из представляющих интерес соединений, являются помимо прочего соединения, где используется, по крайней мере, один из (i)-(iv): (i) по крайней мере, один из R11, R12, R13 или R14 представляет гетероцикл, замещенный гетероцикл или гетероциклоокси; (ii) по крайней мере, один из Z1, Z2 и Z3 представляет арил, гетероцикл, замещенный гетероцикл или гетероциклоокси; (iii) Z6 представляет алкил, который замещен 1-3 группами, выбранными из галогена, арила, арилокси и алкокси, где, по крайней мере, один заместитель не является арилом; алкил, замещенный двумя или тремя арильными группами; циклоалкил, замещенный 1-3 группами, выбранными из алкила, арила, алкенила и алкоксиарила; циклоалкил, с которым конденсировано бензольное кольцо; арилокси, замещенный одним или двумя атомами галогена; арил, который замещен метилендиокси; арил, который замещен 1-4 группами, выбранными из алкила, диалкиламино, циано, галогена, тригалогеналкила, алкокси и тригалогеналкокси; либо гетероцикл или замещенный гетероцикл; или (iv) Z11 представляет алкил, замещенный одним, двумя или тремя атомами галогена.
Использование соединений формулы I и их солей в качестве антагонистов эндотелина
Соединения формулы I и их соли являются антагонистами ЕТ-1, ЕТ-2 и/или ЕТ-3 и могут быть использованы для лечения состояний, ассоциированных с повышенными уровнями ЕТ (например, диализ, травма и хирургическая операция), и всех эндотелийзависимых расстройств. Эти соединения могут быть использованы в качестве гипотенсивных средств. Путем введения композиции, содержащей одно соединение (или комбинацию соединений) настоящего изобретения может быть снижено кровяное давление у млекопитающих (например, человека), страдающих гипертензией. Указанные соединения могут быть также использованы для лечения связанной с беременностью гипертензии и комы (преэклампсии и эклампсии), острой портальной гипертензии и вторичной гипертензии, вызываемой введением эритропоэтина.
Соединения настоящего изобретения могут быть также использованы для лечения заболеваний почек, связанных с нарушением функции гломерулярных и мезангиальных клеток, включая острую и хроническую почечную недостаточность, вторичные нарушения функции почек, связанные с возрастом или с диализом, нефросклероз (особенно гипертенсивный нефросклероз), нефротоксичность (включая нефротоксичность, связанную с введением визуализирующих и контрастных агентов, а также циклоспорина), ишемию почек, первичный везикоуретеральный рефлюкс, гломерулосклероз и т.п. Соединения настоящего изобретения могут быть также использованы для лечения расстройств, ассоциированных с паракринной и эндокринной функцией.
Соединения настоящего изобретения могут быть также использованы для лечения эндотоксемии или эндотоксинового шока, а также геморрагического шока.
Соединения настоящего изобретения могут быть также использованы при гипоксии и ишемии, а также в качестве противоишемических средств для лечения, например, ишемии сердца, почек и мозга, реперфузионных повреждений (которые имеют место после хирургической операции по созданию искусственного [экстракорпорального] кровообращения) коронарных и церебральных вазоспазмов, и т.п.
Кроме того, соединения настоящего изобретения могут быть также использованы в качестве средств против аритмии, средств против стенокардии; средств против фибрилляции, противоастматических средств; средств против атеросклероза и средств против артериосклероза; в качестве добавок к кардиоплегическим растворам для искусственного кровообращения; в качестве вспомогательных средств при тромболитической терапии; и средств против диареи. Соединения настоящего изобретения могут быть использованы для лечения инфаркта миокарда; лечения периферических васкулярных заболеваний (болезни Рейно и болезни Такаясу); лечения гипертрофии сердца (например, гипертрофической кардиомиопатии); лечения первичной легочной гипертензии (например, плексогенной и эмболической) у взрослых и у новорожденных, и легочной вторичной гипертензии, связанной с сердечной недостаточностью, радиацией и химиотерапевтическим воздействием, или других травм; лечения васкулярных нарушений центральной нервной системы, таких как инсульт, мигрень, и субарахноидальное кровоизлияние; лечения расстройств поведения, ассоциированных с нарушениями центральной нервной системы; лечения заболеваний желудочно-кишечного тракта, таких как язвенный колит, болезнь Крона, повреждения слизистой желудка, язва и ишемическая болезнь кишечника; лечения болезней мочевого пузыря или желчных протоков, таких как холангит; лечения панкреатита; регуляции клеточного роста; лечения доброкачественной гипертрофии предстательной железы; лечения повторного стеноза после ангиопластики или после любых процедур, включая трансплантацию; лечения застойной сердечной недостаточности, включая ингибирование фиброза; ингибирование дилатации, ремоделирования и дисфункции левого желудочка; и лечения гепатотоксичности и внезапной смерти.
Соединения настоящего изобретения могут быть использованы для лечения серповидно-клеточной болезни, включая инициацию и/или развитие болевых кризов при этом заболевании; лечения неблагоприятных последствий ЕТ-продуцирующих опухолей, таких как гипертензия, вызываемая гемангиоперицитомой; лечения ранней и продвинутой стадии болезней печени и нарушений, включая сопутствующие осложнения (например, гепатотоксичность, фиброз и цирроз); лечения спазмов мочевого тракта и/или мочевого пузыря; лечения гепаторенального синдрома; лечения иммунологических болезней, включая васкулиты, такие как красная системная волчанка, смешанная криоглобулинемия; и лечения фиброза, ассоциированного с дисфункцией почек и гепатотоксичностью. Соединения настоящего изобретения могут быть также использованы при лечении метаболических и неврологических расстройств; рака; инсулинзависимого и инсулиннезависимого сахарного диабета; нейропатии; ретинопатии; материнского респираторного дистресс-синдрома; дисменореи; эпилепсии; геморрагического и ишемического шока; ремоделирования костной ткани; псориаза; и хронических воспалительных заболеваний, таких как ревматоидный артрит; остеоартрит, саркоидоз и экземы (все типы дерматита).
Соединения формулы I и их соли могут быть также использованы в комбинации с ингибиторами эндотелин-конвертирующего фермента (ЕСЕ), такими как фосфорамидон; антагонистами рецептора тромбоксана; открывателями калиевых каналов; ингибиторами тромбина (например, гирудином и т.п.); ингибиторами фактора роста, такими как как модуляторы PDGF-активности (PDGF - тромбоцитарный фактор роста); антагонистами фактора активации тромбоцитов (PAF); антагонистами рецептора ангиотензина II (AII); ингибиторами ренина; ингибиторами ангиотензин-конвертирующего фермента (АСЕ), такими как каптоприл, зофеноприл, фозиноприл, керанаприл, алацеприл, эналаприл, делаприл, пентоприл, хинаприл, рамиприл, лизиноприл и соли указанных соединений; ингибиторами нейтральной эндопептидазы (NEP); двойными ингибиторами NEP-ACE; ингибиторами СоА-редуктазы HMG, такими как правастатин и мевакор; ингибиторами скваленсинтетазы; веществами, усиливающими секрецию желчных кислот, такими как квестран; блокаторами калиевых каналов; активаторами калиевых каналов; бета-адренергическими агентами; средствами против аритмии; диуретиками, такими как хлортиазид, гидрохлортиазид, флуметиазид, гидрофлуметиазид, бендрофлуметиазид, метилхлортиазид, трихлорметиазид, политиазид или бензотиазид, а также этакриновая кислота, трикринафен, хлорталидон, фуросемид, мусолимин, буметанид, триамтерен, амилорид и спиронолактон и соли указанных соединений; и тромболитическими агентами, такими как тканевый активатор плазминогена (tPA), рекомбинантный tPA, стрептокиназа, урокиназа, проурокиназа, и анизоилированный комплекс стрептокиназного активатора плазминогена (APSAC).
Если необходимо изготовить препарат с фиксированной дозой, то в такие комбинированные препараты включают соединения настоящего изобретения в дозах, описанных ниже, и другой фармацевтически активный агент в его апробированных дозах.
Соединения настоящего изобретения могут быть также изготовлены или использованы в сочетании с противогрибковыми и иммуносупрессорными агентами, такими как амфотерицин В, циклоспорины и т.п., для предотвращения гломерулярного сокращения и вторичной нефротоксичности, вызываемых такими соединениями. Соединения настоящего изобретения могут быть также использованы в сочетании с гемодиализом.
Соединения настоящего изобретения могут быть введены перорально или парентерально млекопитающим различных видов, которые, как известно, страдают указанными заболеваниями, например человеку, в эффективном количестве в интервалах доз, составляющих от около 0,1 до около 100 мг/кг, предпочтительно от около 0,2 до около 50 мг/кг, а более предпочтительно от около 0,5 до около 25 мг/кг (или от около 1 до около 2500 мг, предпочтительно от около 5 до около 2000 мг) в виде одноразовой дозы или в виде дробных доз, вводимых 2-4 раза в день.
Активное соединение может быть использовано в виде композиции, такой как таблетка, капсула, раствор или суспензия, содержащей от около 5 до около 500 мг соединения или смеси соединений формулы I на одну лекарственную форму либо в виде формы для местного применения при лечении ран (0,01-5 мас.% соединения формулы I, 1-5 обработок в день). Эти препараты могут быть изготовлены стандартными методами с использованием физиологически приемлемого носителя или наполнителя, разбавителя, связующего, консерванта, стабилизатора, отдушки, и т. п., или с использованием носителя для местного применения, такого как Plastibase (минеральное масло, желатинированное полиэтиленом), обычно используемого в фармацевтической практике.
Соединения настоящего изобретения могут быть также введены местно для лечения периферических васкулярных заболеваний и могут быть использованы как таковые в виде крема или мази.
Соединения формулы I могут быть также использованы в форме композиций, таких как стерильные растворы или суспензии для парентерального введения. Эти композиции, содержащие около 0,1-500 миллиграммов соединения формулы I в сочетании с физиологически приемлемым наполнителем, носителем, разбавителем, связующим, консервантом, стабилизатором и т.п., могут быть изготовлены в виде стандартной лекарственной формы, обычно используемой в фармацевтической практике. Количество активного вещества в этих композициях или препаратах должно быть таким, чтобы можно было получить нужную дозу в указанных интервалах.
Более подробно, настоящее изобретение описано в нижеследующих рабочих примерах, которые иллюстрируют предпочтительные варианты осуществления настоящего изобретения.
Пример 1
Получение сложного эфира пинакола II N-[(2-Метоксиэтокси)метил]-N-(3,4-диметил-5-изоксазолил)-2-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензолсульфонамид
А. 2-Бром-N-(3,4-диметил-5-изоксазолил)бензолсульфонамид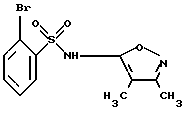
В 2-литровую трехгорлую колбу, снабженную механической мешалкой, 250 мл капельной воронкой и вводной трубкой для подачи аргона загружали 2-бромбензолсульфонилхлорид (150 г, 587 ммоль, коммерчески доступный) и безводный пиридин (150 мл). Полученный светло-желтый раствор охлаждали до -18oС (внутренняя температура) на ледяной/солевой бане. Затем через капельную воронку при перемешивании в течение одного часа по каплям добавляли раствор 5-амино-3,4-диметилизоксазола (69,1 г, 616 ммоль, коммерчески доступный) в безводном пиридине (195 мл). В процессе добавления внутренняя температура реакции не превышала -6oС. После завершения добавления ледяную/солевую баню удаляли и реакционную смесь нагревали до комнатной температуры, перемешивали в течение одного часа, а затем перемешивали в течение 21 часа при 40oС.
Реакционную смесь охлаждали до комнатной температуры и выливали в смесь ледяной воды (3 л) и целита (37,5 г). После перемешивания в течение 20 минут ее фильтровали и промывали водой (250 мл х 3). К фильтрату добавляли уголь (45 г). Смесь перемешивали при комнатной температуре в течение 40 минут и фильтровали через слой целита. Слой целита промывали водой (500 мл х 3). Фильтрат подкисляли путем добавления по каплям холодной HCl (6 н., 750 мл) при энергичном перемешивании в течение 2 часов. Продукт осаждался, и после добавления HCl, смесь перемешивали еще один час.
Смесь фильтровали и твердое вещество промывали холодной водой (750 мл х 4) и сушили в вакууме в течение 3 дней. Целевое соединение этой стадии получали в виде желтовато-белого твердого вещества (171 г) с выходом 88% (площадь ВРЖХ = 97,4%).
Тонкослойная хроматография (ТСХ): Rf=0,47 (силикагель от Whatman; этилацетат (EtOAc): гексан = 1:1; визуализация САМ или УФ).
Альтернативное получение целевого соединения данной стадии
В 1-литровую трехгорлую колбу загружали 2-бромбензол-сульфонилхлорид (50 г, 196 ммоль) и безводный 1,2-дихлорэтан (125 мл) в атмосфере аргона. Полученный бесцветный раствор охлаждали до 0oС, добавляли безводный пиридин (40 мл, 396 ммоль), а затем добавляли твердый 5-амино-3,4-диметилизоксазол (24,1 г, 196 ммоль). После добавления ледяную баню удаляли и реакционную смесь нагревали до 55oС в течение 21 часа с получением неочищенной реакционной смеси, содержащей целевое соединение этой стадии.
В. 2-Бром-N-(3,4-диметил-5-изоксазолил)-N-[(2-метоксиэтокси)-метил]бензолсульфонамид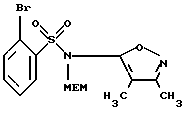
В 1-литровую трехгорлую колбу, снабженную механической мешалкой, загружали карбонат калия (130,5 г, 944 ммоль) и безводный диметилформамид (ДМФ, 286 мл) в атмосфере аргона. Гетерогенную смесь перемешивали в течение 15 минут при комнатной температуре. Затем добавляли целевое соединение стадии А (125 г, 378 ммоль) в виде твердого вещества. Полученную смесь снова перемешивали в течение 15 минут при комнатной температуре. Затем через капельную воронку по каплям в течение 40 минут добавляли метоксиэтоксиметилхлорид (MEMCl, 47,5 мл, 415,8 ммоль). После добавления, реакционную смесь перемешивали в течение 40 минут. Реакцию контролировали с помощью ВРЖХ.
Реакционную смесь разбавляли путем добавления этилацетата (400 мл), а затем перемешивали 5 минут и фильтровали. Твердое вещество промывали этилацетатом (200 мл х 2) и гексаном (250 мл х 2). Фильтрат обрабатывали углем (25 г), перемешивали при комнатной температуре в течение 1 часа и фильтровали через слой целита. Слой целита промывали этилацетатом (50 мл х 3). Этилацетатные слои объединяли и промывали Nа2СО3 (1М, 500 мл). В водном слое происходило осаждение. Это осаждение подавляли добавлением воды (750 мл). Водный слой выделяли и отбрасывали. Органический слой промывали водой (750 мл) и солевым раствором (500 мл х 2), а затем сушили Na2SO4, фильтровали и концентрировали с получением желтоватого полутвердого вещества (157,3 г, 99% массовый баланс).
Остаток растворяли в этаноле (125 мл) и помещали в холодильник (0oС) на 20 часов. В результате происходила кристаллизация. Твердое вещество фильтровали и сушили в вакууме. Целевое соединение этой стадии было получено в виде желтовато-белого твердого вещества (выход 75,65%, площадь ВРЖХ = 98,2%). ТСХ: Rf= 0,55 (силикагель от Whatman; EtOAc : гексан = 1/1; визуализация: САМ или УФ).
Альтернативный способ получения целевого соединения этой стадии
Реакционную смесь, полученную альтернативным способом получения целевого соединения стадии А, охлаждали до комнатной температуры и концентрировали при пониженном давлении на роторном испарителе с образованием темного густого маслянистого вещества (102 г) при 40oС. Это темное маслянистое вещество (98 г, 188 ммоль) растворяли в безводном дихлорметане (240 мл). После этого добавляли диизопропилэтиламин (97 мл, 4 эквивалента), а затем по каплям добавляли метоксиэтоксиметилхлорид (25,7 мл, 225,6 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 4 часов.
Реакционную смесь концентрировали при пониженном давлении на роторном испарителе с получением густого маслянистого вещества, которое растворяли в EtOAc (400 мл), и добавляли уголь (10 г). Угольную смесь перемешивали при комнатной температуре в течение 30 минут и фильтровали через слой целита. Слой целита промывали EtOAc (100 мл х 3) и гексаном (200 мл х 2). Фильтрат переносили в делительную воронку и промывали водой (100 мл х 2), HCl (0,5 н. , 100 мл х 1), водой (100 мл х 2) и солевым раствором (100 мл х 2), а затем сушили Na2SO4, фильтровали и концентрировали с получением густого маслянистого продукта (64,9 г, массовый баланс 83%).
Густой маслянистый продукт растворяли в этаноле (EtOH, 65 мл), охлаждали до 0oС в ледяной бане, вводили целевое соединение этой стадии в качестве затравки и перемешивали при 0oС в течение 6 часов. После этого происходила кристаллизация. Твердое вещество фильтровали и сушили в вакууме. Целевое соединение этой стадии было получено в виде желтоватого твердого вещества (полный выход 62%, площадь ВРЖХ = 98,2%). ТСХ: Rf=0,55 (силикагель от Whatman; EtOAc : гексан 1:1, визуализация: САМ или УФ).
С. 2-Бороно-N-(3,4-диметил-5-изоксазолил)-N-[(2-метоксиэтокси)-метил]бензолсульфонамид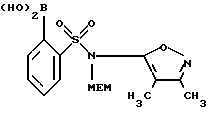
В сухую 1-литровую трехгорлую круглодонную колбу, снабженную верхней механической мешалкой, трубкой для подачи газа, термопарой и перегородкой, загружали целевое соединение стадии В (40,0 г, 95,4 ммоль), а затем тщательно дегазировали и помещали в атмосферу аргона. Затем шприцем добавляли тетрагидрофуран (ТГФ, 185 мл) и смесь охлаждали до около -100oС (внутренняя температура). После этого в течение 16 минут по каплям добавляли н-бутиллитий (n-BuLi, 42,5 мл, 101 ммоль, 2,38 М в гексане), поддерживая при этом внутреннюю температуру от -97 до -101oС. Светлый оранжево-желтый раствор перемешивали еще 16 минут при температуре от около -98 до -101oС.
В течение 14,5 минут по каплям добавляли триметилборат (16,0 мл, 140,9 ммоль) в ТГФ (24 мл) при поддержании внутренней температуры в пределах от -96 до -99oС. Полученную смесь перемешивали в течение около 44 минут при температуре от около -93 до -101oС, а затем еще 39 минут при температуре от около -93 до -72oС. HCl (3,0н., 120 мл, 360 ммоль) добавляли к реакционной смеси (экзотермический раствор до около -7oС), и смесь перемешивали в течение 20 минут (от -7oС до +6oС). Два слоя разделяли в делительной воронке и водную фазу промывали толуолом (120 мл х 3) и трет-бутилметиловым эфиром (ТВМЕ, 100 мл х 3). Объединенный органический слой промывали солевым раствором (100 мл х 4), сушили (Na2SO4) и концентрировали на роторном испарителе до объема около 100 мл вещества, содержащего целевое соединение этой стадии (площадь ВРЖХ = 97,7%).
Альтернативный способ получения целевого соединения этой стадии
В 500-миллилитровую трехгорлую колбу, снабженную стержнем для перемешивания, загружали целевое соединение стадии В (20,0 г, 47,7 ммоль) и продували аргоном в течение 0,5 часа. Затем шприцем добавляли безводный ТГФ (200 мл) и колбу охлаждали до -78oС в бане с ацетоном/сухим льдом. После этого через капельную воронку в течение 25 минут добавляли фениллитий (PhLi, 37,1 мл, 48,2 ммоль, 1,3 М в циклогексане-эфире, титрованный, как описано в J.Organomet. Chem., 186, 155 (1980) и определенный как 1,3М). PhLi добавляли так, чтобы внутренняя температура реакционной смеси поддерживалась ниже -75oС. Полученный раствор перемешивали в течение 15 минут при -78oС, а затем в реакционную смесь в течение 15 минут через канюлю по каплям вводили раствор триметилбората (10,8 мл, 95,4 ммоль) в ТГФ (5 мл). Перед добавлением раствор триметилбората в ТГФ охлаждали в бане со льдом и водой. Добавление проводили так, чтобы внутренняя температура реакционной смеси не превышала -73oС. Реакционную смесь перемешивали в течение получаса при температуре -78oС, а затем гасили путем добавления по каплям раствора уксусной кислоты (15 мл) в ТГФ (10 мл). Подкисленный раствор перемешивали в течение 10 минут при -78oС, а затем нагревали до 0oС. К этому раствору по каплям добавляли 1н. HCl (25 мл). (1н. HCl была получена путем разбавления 42 мл 12н. HCl в 500 мл воды. Избыток кислоты добавляли для полного прекращения реакции. Перед добавлением раствор HCl предварительно охлаждали в бане со льдом/водой). Реакционную смесь нагревали до комнатной температуры и экстрагировали трет-бутилметиловым эфиром (ТВМЕ, 250 мл х 4). Органические слои объединяли и экстрагировали 0,5н. водным NaOH (25 мл х 4). Водные слои объединяли и подвергали обратной экстракции ТВМЕ (100 мл х 1). Водный экстракт охлаждали до 0oС, а рН доводили до 2,0 (рН-метр) путем добавления по каплям 6н. HCl при быстром перемешивании. Подкисленный раствор экстрагировали ТВМЕ (250 мл х 4), а органические слои объединяли и сушили безводным МgSO4. Суспензию фильтровали и раствор концентрировали с получением целевого соединения бороновой кислоты этой стадии в виде светло-коричневого маслянистого продукта (17,1 г, 93%, площадь ВРЖХ=88%). ВРЖХ-условия: колонка - YMC ODS-A, 6х250 мм; мониторинг при 233 нм; скорость потока 1,5 мл/мин; растворитель А (%): Н2О/МеОН/Н3РO4, 90: 10: 0,2; растворитель В (%): Н2О/МеОН/Н3РО4, 90:10:0,2; Градиент: линейный градиент 40%В - 100%В, 10 минут, 100%В - 5 минут, 40%В - 4 минуты; время удерживания для целевого соединения этой стадии = 8,3 минуты.
D. N-[(2-Метоксиэтокси)метил] -N-(3,4-диметил-5-изоксазолил)-2-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)бензолсульфонамид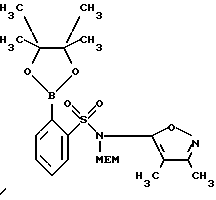
Смесь со стадии С разбавляли толуолом (170 мл) до полного объема около 270 мл и колбу снабжали ловушкой Дина-Старка и магнитным стержнем для перемешивания. Затем добавляли пинакол (11,6 г, 98,2 ммоль) и полученную смесь нагревали с обратным холодильником в течение около 1,25 часа. Воду сливали из ловушки Дина-Старка, а затем раствор использовали непосредственно в примере 3. (98%-ное превращение, площадь ВРЖХ=93,6%).
Обращенно-фазовая ВРЖХ-колонка: YMC-Pack ODS-A; 150х6 мм, S-5 мм, 120A и мониторинг при @233 нм; растворитель: А = 90% воды, 10% метанола, и 0,2% Н3РО4; В = 10% воды, 90% метанола, и 0,1% Н3РO4.
Скорость потока: 100 мл в минуту; градиент: 40%В-100%В в течение 10 минут. Время пребывания: 5 минут при 100%В. Понижение до 40%В и время пребывания = 5 минут. Время удерживания для целевого продукта этого примера = 11,5 минут.
Альтернативный способ получения целевого соединения этого примера
Бороновую кислоту, полученную альтернативным способом получения целевого соединения стадии С (17,1 г, 44,5 ммоль), растворяли в растворе безводного толуола (425 мл) и пинакола (5,51 г, 46,7 ммоль). Колбу помещали на масляную баню и нагревали до 120oС в течение 2 часов (примечание: реакция была завершена через первые 40 минут), а воду непрерывно удаляли с использованием ловушки Дина-Старка (колбу и ловушку заворачивали в фольгу; смесь быстро кипятили приблизительно в течение получаса) и холодильника. Анализ аликвоты (полученной путем повторной азеотропной перегонки с CDCl3) с помощью ВРЖХ указывал на полное превращение исходного соединения, а именно бороновой кислоты, в целевое соединение данного примера. Реакционную смесь охлаждали до комнатной температуры и концентрировали с получением целевого соединения этого примера в виде толуольного раствора, площадь ВРЖХ=86%. 100%-ный выход давал основание предположить, что бороновая кислота превратилась в сложный эфир пинакола. Часть неочищенного раствора целевого соединения этого примера была использована в примере 3. Время удерживания целевого соединения этого примера = 12,4 минуты (с использованием ВРЖХ-условий, описанных для исходного продукта бороновой кислоты).
Пример 2
Получение галогенфенильного соединения III 2-(4-Иодфенил)оксазол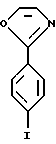
A. N-(2,2-Диметоксиэтил)-4-иодбензамид
К раствору диметилацеталя аминоацетальдегида (82,9 г, 86,0 мл, 0,79 моль, коммерчески доступного) в воде (900 мл) и ацетоне (400 мл) добавляли КНСО3 (80,0 г, 0,80 моль) и полученный раствор охлаждали на ледяной бане до 0oС. К раствору диметилацеталя аминоацетальдегида в течение 1,5 часа при механическом перемешивании через капельную воронку по каплям добавляли раствор 4-йодбензоилхлорида (200,0 г, 0,75 моль) в ацетоне (600 мл). Капельную воронку и стенки колбы промывали ацетоном (50 мл), а ледяную баню удаляли. Реакционную смесь перемешивали при комнатной температуре в течение 3 часов (реакция была завершена через 1,5 часа).
Реакционную смесь концентрировали путем удаления 1,0 л растворителя на роторном испарителе, а затем экстрагировали этилацетатом (350 мл х 4). Органические слои собирали, промывали насыщенным NаНСО3 (250 х 1 мл), а затем водой (250 мл х 1), сушили безводным MgSO4 (50,0 г), фильтровали и концентрировали при пониженном давлении с получением белого твердого вещества (во время упаривания растворителя твердое вещество время от времени соскабливали со стенок колбы). Твердое вещество сушили в вакууме, достигаемом в домашних условиях, в течение 12 часов (246,5 г, 98%, площадь ВРЖХ=99,7%). Температура плавления (т.пл.)=89-90oС.
Элементный анализ для C11H14NO3I (%):
Вычислено: С 39,42; Н 4,21; N 4,18;
Найдено: С 39,42; Н 4,22; N 4,07.
В. 2-(4-Иодфенил)оксазол
Реактив Итона получали путем добавления P2O5 (200 г) примерно 50 г - порциями в метансульфоновую кислоту (2000 мл) при 95oС в атмосфере аргона при интенсивном механическом перемешивании. Добавление P2О5 порциями предупреждает образование твердой массы на дне колбы. Таким образом был получен прозрачный раствор с очень бледным коричневым оттенком. Eaton et al., J. Org. Chem., 38, 4071-4073 (1973).
В 3-литровую круглодонную трехгорлую колбу, содержащую реактив Итона (2000 мл) и снабженную холодильником, загружали целевое соединение стадии А (200,0 г, 0,60 моль). Реакционную смесь помещали в масляную баню и нагревали, перемешивая, при повышенном давлении аргона. (Было обнаружено, что это лучше способствует нагреванию реакционной смеси до требуемой температуры, чем помещение ее в предварительно нагретую баню). Прослеживание реакции с помощью ТСХ указывало на то, что первой стадией реакции являлся гидролиз диметилацеталевой группы с образованием соответствующего альдегида (Rf= 0,11):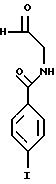
с последующей циклизацией. Температуру масляной бани корректировали так, чтобы поддерживать внутреннюю температуру реакционной смеси от 130 до 134oС. (Температуру масляной бани поддерживали в пределах 138-141oС). Через 7,5 часа анализ аликвоты (полученной путем добавления в H2O и экстракции EtOAc) с помощью ТСХ указывал на полное исчезновение исходного вещества и продуцирование одного нового пятна.
Реакционную смесь охлаждали до 30oС в атмосфере аргона, разделяли приблизительно на три равные порции и каждую порцию выливали в суспензию из льда/воды (приблизительно 6,0 л) при интенсивном перемешивании механической мешалкой и при наружном охлаждении. Это приводило к образованию коричневато-серого осадка. Полученную суспензию перемешивали в течение 2 часов и твердое вещество собирали путем вакуумной фильтрации через воронку из спеченного стекла со средним размером пор, а затем промывали водой со льдом (1 л) и осадок на фильтре измельчали. Твердое вещество сушили воздухом в течение 5 дней и получали серый порошок (151,7 г, 93%). Неочищенное вещество (151,7 г) растворяли в ацетонитриле (2 л) (путем мягкого нагревания бытовым феном; небольшое количество черного хлопьевидного твердого вещества оставалось нерастворенным в колбе), а затем добавляли активированный уголь (15,2 г) и смесь перемешивали при комнатной температуре в течение одного часа. Смесь фильтровали через слой целита (100,0 г). Затем слой целита промывали ацетонитрилом (100 мл х 2). Фильтрат концентрировали при пониженном давлении приблизительно до 1180 мл в 5-литровой круглодонной колбе. Колбу нагревали бытовым феном до полного растворения твердого вещества. К полученному горячему раствору двумя порциями добавляли кипящую воду (295 мл), а затем колбу нагревали до температуры, близкой к температуре кипения, и после каждого добавления мягко взбалтывали. Раствор снова один раз нагревали до температуры, близкой к температуре кипения, после чего колбу накрывали и оставляли на 48 часов при комнатной температуре (за 1,5 часа было обнаружено некоторое количество кристаллов), а затем хранили в течение 4 дней при 4oС. Образовавшееся кристаллы собирали путем фильтрации, промывали холодным ледяным раствором ацетонитрила и воды (4: 1, 150 мл), и сушили воздухом в течение 20 часов. Целевое соединение этого примера получали в виде очень бледных желтых кристаллов, которые затем сушили в условиях бытового вакуума (107,2 г, 66%, площадь ВРЖХ=99,7%). Второй сбор целевого соединения был получен после перекристаллизации желтого твердого вещества, полученного после концентрирования маточного раствора (27,4 г, 17%, площадь ВРЖХ=98,4%).
Т.пл.=107-109oС.
Элементный анализ для C9H6NOI
Вычислено: С 39,88, Н 2,23, N 5,17
Найдено: С 40,00, Н 2,09, N 5,14.
Пример 3
Получение соединения формулы I N-(3,4-Диметил-5-изоксазолил)-4'-(2-оксазолил)[1,1'-бифенил]-2-сульфонамид
Реакция взаимодействия пинаколового эфира II и галогенфенилового соединения III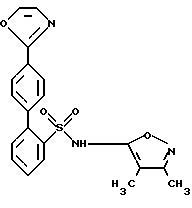
A. N-[(2-Метоксиэтокси)метил] -N-(3,4-диметил-5-изоксазолил)-4'-оксазол-2-ил-[1,1'-бифенил]-2-сульфонамид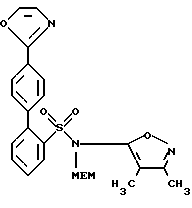
В осушенную 2-литровую трехгорлую круглодонную колбу, снабженную верхней механической мешалкой, трубкой для подачи газа, термопарой, обратным холодильником и перегородкой, загружали ацетат палладия (Pd(OAc)2, 0,54 г, 2,38 ммоль) и трифенилфосфин (Рh3Р, 1,7 г, 6,44 ммоль), а затем дегазировали и помещали в атмосферу аргона. После добавления ТГФ (85 мл), смесь нагревали при 65oС в течение 70 минут. Окраска раствора постепенно менялась с грязно-оранжевой на ярко-желтую. Затем шприцем добавляли дегазированный толуоловый (270 мл) раствор целевого соединения примера 2 (25,9 г, 95,6 ммоль) и неочищенное целевое соединение примера 1, полученное способом, описанным выше (см. стадию D примера 1, 44,45 г, 95,4 ммоль), после чего добавляли дегазированный 95% этанол (EtOH, 285 мл) и дегазированный 2,0 М раствор Nа2СО3 (306 мл, 612 ммоль). Полученный красный раствор нагревали приблизительно при 75oС (внутренняя температура) примерно в течение 1,75 часа. Баню для нагревания удаляли и смесь охлаждали до комнатной температуры. Слои выделяли в делительной воронке, после чего добавляли воду (250 мл) и водный слой промывали СН2Сl2 (400 мл х 6). Объединенные органические слои сушили МgSO4, обрабатывали нейтральным углем (Norit, 5,0 г), фильтровали через целит, концентрировали и выдерживали в динамическом высоком вакууме в течение ночи, в результате чего получали целевое соединение данной стадии (62,4 г, 140%) в виде красного маслянистого продукта (площадь ВРЖХ > 85%).
Альтернативный способ получения целевого соединения данной стадии
Получение катализатора:
В 2-литровую трехгорлую колбу, снабженную верхней механической мешалкой, обратным холодильником и устройством для впуска аргона, загружали Pd(OAc)2 и трифенилфосфин. Колбу продували медленным потоком аргона в течение 18 часов. Затем шприцем добавляли безводный дегазированный ТГФ (67 мл), а колбу погружали в масляную баню, предварительно нагретую до 75oС, и перемешивали. Через 20 минут внутренняя температура достигала 60oС. Смесь перемешивали один час, в течение которого появлялась бордовая окраска, после чего эту смесь охлаждали до комнатной температуры при слегка повышенном давлении аргона. См. J. Org. Chem. 1994, 59, 8151; Organometallics, 1992, II, 3009.
Реакция взаимодействия
Целевое соединение примера 2 (10,0 г) добавляли к порции толуолового раствора пинаколбороната, полученного альтернативным способом получения целевого соединения стадии D примера 1 (110 мл, 38,74 ммоль пинаколбороната, 87% полного объема неочищенного боронатного раствора). Смесь взбалтывали, добавляли безводный этанол (55 мл), колбу герметично закрывали мембраной и взбалтывали до получения раствора. Полученный раствор дегазировали путем барботирования аргона в раствор в течение одного часа при умеренной скорости. Затем раствор через канюлю вводили в реакционный сосуд, содержащий охлажденный раствор катализатора, при повышенном давлении аргона. Содержимое колбы промывали безводным дегазированным этанолом (55 мл) и через канюлю переносили в реакционный сосуд при повышенном давлении аргона. После этого через канюлю добавляли дегазированный 2М раствор Nа2СО3 (118 мл), начинали перемешивание, колбу погружали в предварительно нагретую масляную баню при 78oС и внутреннюю температуру доводили до 69-70oС. Через 1,75 часа, ВРЖХ- и ТСХ-анализ указывал на то, что реакция была завершена. После прохождения реакции в течение 2 часов смесь охлаждали до 27oС и при перемешивании добавляли воду (100 мл) и EtOAc (100 мл). Смесь переносили в делительную воронку, слои выделяли, а водный слой экстрагировали этилацетатом (100 мл х 3, 50 мл х 1). Объединенные органические экстракты промывали полусолевым раствором (60 мл х 1) и сушили безводным МgSO4 (40 г) при перемешивании. После фильтрации раствора, исходную колбу и воронку-фильтр промывали EtOAc (100 мл х 2). Таким образом был получен неочищенный раствор реакции сочетания с полным объемом 785 мл.
ВРЖХ-анализ давал площадь неочищенного ВРЖХ-продукта = 88,9%. Раствор делили на порции для оценки чистоты. Так, например, брали 5% по объему неочищенного реакционного раствора (39 мл) и концентрировали его на роторном испарителе. Остаток растворяли в толуоле (12 мл), добавляли продукт С (0,9 г, 100 мас.%) и смесь перемешивали в масляной бане при 88oС в течение 30 минут. После добавления тритиоциануровой кислоты (ТМТ, 407 мг, 2,3 ммоль, 50 молярных эквивалентов по отношению к предполагаемому содержанию Pd) смесь перемешивали в течение 30 минут в масляной бане, охлаждали до 0oС в ледяной бане, фильтровали через целит, а слой целита промывали толуолом (10 мл х 2), и фильтрат промывали 1н. NaOH (10 мл х 2). Для выделения фазы потребовалось ~3 минуты. Толуоловый слой сушили МgSO4, фильтровали и концентрировали с получением неочищенного целевого соединения этой стадии, 0,76 г, массовый баланс 87%. Было определено содержание палладия, которое составило <1 млн.д. Последующий эксперимент указывал на то, что 25 эквивалентов ТМТ являются достаточными для восстановления Pd до <10 млн.д.
10% по объему неочищенного реакционного раствора (78 мл), описанного выше, перемешивали с 50 мас.% соединения С (0,9 г) до около 60oС, оставляли для охлаждения на 30 минут, фильтровали через целит, промывали EtOAc и эту процедуру повторяли с использованием свежей порции угля. Раствор концентрировали и получали 2,29 г вещества (массовый баланс 131%). Остаток дважды подвергали азеотропной перегонке с безводным этиловым эфиром. Этот остаток растворяли в эфире (11 мл), раствор охлаждали до 0oС, а затем вводили целевое соединение в качестве затравки и перемешивали. Из раствора осаждалось твердое вещество. Через один час по каплям добавляли гексан (5 мл) и смесь перемешивали при 0oС в течение 2 часов, собирали и промывали холодным раствором эфира : гексана (2:1) со льдом, в результате чего получали целевое соединение этой стадии (1,07 г, 58% исходя из целевого соединения стадии В примера 1) в виде порошка с едва заметной желтой окраской, площадь ВРЖХ= 95,8% (в способе).
ВРЖХ-условия:
Колонка = YMC ODS-A, 6х250 мм
Мониторинг при 233 нм;
Скорость потока = 1,5 мл/мин.
Растворитель А (%): Н2О/МеОН/Н3РО4, 90:10:0,2
Растворитель В (%): Н2О/МеОН/Н3РО4, 10:90:0,2,
Градиент: 40%В - 100%В; линейный градиент; 10 минут, 100%В 5 минут, 40%В 4 минуты.
Время удерживания для целевого соединения этой стадии: 12,8 минут.
Второе альтернативное получение целевого соединения этой стадии; метод обратной реакции сочетания
В 500-литровую круглодонную колбу, осушенную в печи, загружали целевое соединение примера 2 (10 г, 37,3 ммоль), безводный ТГФ (250 мл), толуол (50 мл) и триизопропилборат (В(ОiРr)3, 50 мл) в атмосфере аргона. Полученный бесцветный раствор охлаждали до -75oС и в течение одного часа по каплям добавляли н-бутиллитий (36 мл, 1,43 М в гексане, 51,5 ммоль). Во время добавления внутреннюю температуру поддерживали ниже -73oС. Реакцию гасили путем добавления уксусной кислоты (4 мл) в ТГФ (25 мл) и концентрировали на роторном испарителе. После добавления толуола (100 мл) и метанола (100 мл), растворитель удаляли на роторном испарителе при 40oС. Эту процедуру повторяли еще один раз.
Полученный остаток растворяли в NaOH (100 мл, 100 ммоль, 1н.) и дважды экстрагировали гексаном (25 мл и ТВМЕ (25 мл). Было получено следующее соединение: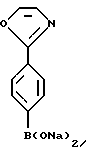
Водный слой добавляли к смеси NaHCO3 (8,4 г, 100 ммоль) в EtOH (100 мл), а затем добавляли раствор целевого соединения стадии В примера 1 (14,4 г, 34,4 ммоль) в толуоле (100 мл). Полученную смесь барботировали аргоном в течение 15 минут.
Во время прохождения вышеуказанной реакции в круглодонную 50 мл колбу загружали трифенилфосфин (676,8 мг, 2,58 ммоль) и ТГФ (30 мл) и барботировали аргоном в течение 15 минут. После добавления Pd(OAc)2 (193,4 мг, 0,86 ммоль) смесь нагревали до 65oС в течение 1 часа в атмосфере аргона. Смесь охлаждали до комнатной температуры и добавляли к вышеуказанной смеси этанола/толуола/воды. Полученную гетерогенную смесь нагревали до 75oС в течение 6 часов. ВРЖХ указывала на израсходование всего исходного продукта.
Реакционную смесь охлаждали до комнатной температуры и концентрировали на роторном испарителе. Затем добавляли этилацетат (250 мл) и уголь (10 г). Смесь перемешивали при комнатной температуре в течение 30 минут и фильтровали через слой целита. Слой целита промывали этилацетатом (25 мл х 4). Фильтрат переносили в делительную 500 мл воронку и промывали водой (100 мл) и солевым раствором (100 мл х 2). Этилацетатные слои объединяли, смешивали с тритиоциануровой кислотой (ТМТ, 4 г) и нагревали при 45oС в течение 45 минут. Затем слои фильтровали через слой целита. Слой целита промывали этилацетатом (25 мл х 4). Органические слои объединяли и промывали NaOH (200 мл х 2; ТВМЕ (300 мл) добавляли для минимизации эмульсии), водой (200 мл) и солевым раствором (200 мл), а затем сушили Na2SO4, фильтровали и концентрировали с получением 17,5 г вещества (98% массовый баланс).
Неочищенный продукт растворяли в этаноле (17,5 мл). Половину этого раствора (около 8,8 мл) помещали в холодильник при 0oС на 2 дня. После этого происходила кристаллизация. Твердое вещество фильтровали и сушили в вакууме. Продукт был получен в виде желтоватого твердого вещества (6 г, полный выход 67% исходя из целевого соединения примера 2, площадь ВРЖХ=99,1%).
ВРЖХ-условия:
Колонка = YMC ODS-A, 6х250 мм;
Мониторинг при 233 нм;
Скорость потока = 1,5 мл/мин.
Растворитель А (%): Н2О/МеОН/Н3РО4, 90:10:0,2
Растворитель В (%): Н2О/МеОН/Н3РO4, 10:90:0,2;
Градиент: 40%В --> 100%В; линейный градиент - 10 минут, 100%В - 5 минут, 40%В - 4 минуты.
Время удерживания целевого соединения этой стадии: 12,8 минут.
В. N-(3,4-Диметил-5-изоксазолил)-4'-(2-оксазолил)[1,1'-бифенил] -2-сульфонамид
В 3-литровую трехгорлую круглодонную колбу, снабженную верхней механической мешалкой, термопарой и обратным холодильником, загружали ярко-желтый раствор неочищенного целевого соединения стадии А (62,45 г) в 95% этанола (620 мл). К этому раствору при перемешивании быстро добавляли 6н. HCl (620 мл) и смесь нагревали с обратным холодильником при 87oС в течение 100 минут, в результате чего образовывался светло-оранжевый раствор. Реакционную смесь охлаждали до комнатной температуры, после чего охлаждали примерно до 0-4oС с использованием ледяной бани, а затем подщелачивали до рН ~13 путем добавления 5н. NaOH (~850 мл). Затем медленно в течение 10 минут добавляли раствор гидроксида натрия без повышения температуры до 30oС. Полученную смесь снова охлаждали до 0oС, добавляли 250 мл воды и полученный осадок фильтровали и промывали водой (200 мл). Фильтрат концентрировали в вакууме на роторном испарителе примерно при 40oС для удаления большого количества этанола. Во время концентрирования происходило осаждение натриевой соли целевого соединения этого примера. Суспензию охлаждали до ~10oС, фильтровали и промывали солевым раствором (150 мл х 2). Твердое вещество сушили воздухом в течение 15 минут, переносили в 2-литровую колбу и растворяли в 1 л горячей воды. Раствор тонко отфильтровывали, промывали водой (150 мл) и переносили в 2-литровую делительную воронку. Водный слой промывали этилацетатом : гексаном (1:1) (1 л х 2) и гексаном (500 мл). Затем желтый водный слой охлаждали до ~ 7oС и при интенсивном перемешивании, очень медленно подкисляли до рН ~ 9,0-2,6 для осаждения целевого соединения этого примера. После перемешивания в течение около 10 минут суспензию фильтровали, промывали избыточным количеством воды для удаления всех остатков кислоты и сушили воздухом, в результате чего получали 31,5 г (84%) продукта в виде беловатого твердого вещества.
5,0 г - часть вышеуказанного твердого вещества растворяли в этилацетате (25 мл) и обрабатывали тритиоциануровой кислотой (ТМТ, 0,23 г). ТМТ является хелатообразующим агентом, добавленным для облегчения удаления остатка палладия из продукта. Смесь подвергали мягкому кипячению, а затем нагревали в масляной бане в течение 15 минут при ~65oС. После добавления к смеси угля Norit (5,1 г), нагревание продолжали в течение еще 30 минут. Затем смесь охлаждали в ледяной бане, фильтровали через слой целита и промывали этилацетатом (25 мл). Фильтрат переносили в делительную воронку и промывали 1,0н. НCl (75 мл х 1) и водой (75 х 1 мл). HCl-промывку использовали для удаления остатка ТМТ. Затем органический слой обрабатывали углем Norit (0,5 г), нагревали до слабого кипения и фильтровали через слой целита. Фильтрат концентрировали, а остаток растворяли в абсолютном этаноле (20 мл), кипятили, а затем добавляли воду (10 мл). В раствор вводили затравочный кристалл, оставляли на 16 часов при перемешивании для охлаждения и кристаллизовали. Полученные кристаллы фильтровали, промывали этанолом : водой (1:1, 10 мл) и сушили в вакууме, в результате чего получали 4,0 г чистого целевого соединения этого примера (полный выход 66% исходя из целевого соединения стадии А, площадь ВРЖХ= 99,02%). Т.пл.=145,1oС (полиморф с высокой температурой плавления).
Пример 4
Получение соединения формулы I
N-(3,4-Диметил-5-изоксазолил)-4'-(2-оксазолил)[1,1'-бифенил] -2-сульфонамид
Реакция взаимодействия соли бороновой кислоты IX с иодфенильным соединением III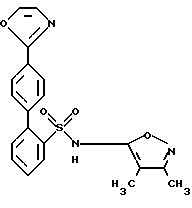
А. Динатриевая соль 2-бороно-N-(3,4-диметил-5-изоксазолил)-N-[(2-метоксиэтокси)метил]бензолсульфонамида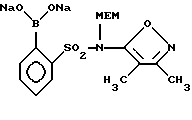
В осушенную трехгорлую круглодонную 100 мл колбу, снабженную магнитным стержнем для перемешивания, трубкой для подачи газа, термопарой и перегородкой, загружали целевое соединение стадии В примера 1 (5,0 г, 11,9 ммоль), а затем тщательно дегазировали и помещали в атмосферу аргона. Затем шприцем добавляли ТГФ (23 мл) и смесь охлаждали до -100oС (внутренняя температура) и энергично перемешивали. К этой смеси при интенсивном перемешивании в течение 19 минут по каплям вводили n-BuLi (5,30 мл, 12,6 ммоль), поддерживая при этом внутреннюю температуру в пределах от -95 до -100oС. Светлый оранжево-желтый раствор перемешивали в течение еще 12 минут приблизительно при -100oС. К раствору в течение 12,0 минут по каплям добавляли триметилборат (2,0 мл, 17,6 ммоль), растворенный в ТГФ (3,0 мл), поддерживая при этом температуру в пределах от -95 до -100oС. Смесь перемешивали примерно 50 минут при температуре от около -95 до -100oС, а затем еще 40 минут при температуре от около -78 до -72oС. Боратный сложный эфир гидролизовали путем добавления 1,0н. HCl (2,0 мл, 2,0 ммоль) при температуре около -72oС и раствор нагревали до комнатной температуры и перемешивали примерно 60 минут. После добавления раствора Nа2СО3 (55 мл, 2,0 М, 110 ммоль) смесь тщательно дегазировали и помещали в атмосферу аргона. Затем этот продукт был непосредственно использован в последующих стадиях (чистота = 95,9%, ВРЖХ). Колонка: YMC ODS-A, 150 мм х 6,0 мм, S-3 мкм, 120А; Скорость потока: 1,0 мл/мин. При 0,0 мин система растворителей состояла из растворителя А (40%) (А=90% МеОН, 10% Н2О, 0,1% Н3РО4) и растворителя В (60%) (В=90% H2О, 10% MeOH, 0,2% Н3РO4). При 15,0 мин состав системы растворителей = 100% А. При 15,1 - 20,0 мин состав системы растворителей = А (40%) и В (60%). Время удерживания (Rt) для целевого соединения этой стадии составляло 7,5 минут. Измерение на детекторе проводили при 233 нм.
В. N-[(2-Метоксиэтокси)метил] -N-(3,4-диметил-5-изоксазолил)-4'-оксазол-2-ил- [1,1'-бифенил]-2-сульфонамид
В сухую трехгорлую круглодонную 300 мл колбу, снабженную механической мешалкой, трубкой для подачи газа, термопарой, обратным холодильником и перегородкой, загружали Pd(OAc)2 (0,16 г, 0,71 ммоль) и Рh3Р (0,5 г, 1,93 ммоль), а затем дегазировали и помещали в атмосферу аргона. После добавления ТГФ (30 мл), смесь нагревали при ~65oС в течение около 70 минут. Окраска постепенно менялась с грязно-оранжевой на желтую. Затем шприцем добавляли дегазированный раствор толуола (85 мл) и этанольного (64 мл) раствора целевого соединения примера 2 (3,23 г, 11,9 ммоль), а затем добавляли дегазированный раствор целевого соединения стадии А. Раствор, окрашенный в красный цвет, нагревали при ~75oС (внутренняя температура) в течение 1,5 часа. Баню для нагревания удаляли и смесь охлаждали до комнатной температуры. Слои разделяли в делительной воронке, к водному слою добавляли воду (50 мл) и экстрагировали EtOAc (200 мл х 3). Объединенные органические слои промывали солевым раствором (75 мл х 1), сушили MgSO4, обрабатывали нейтральным углем Norit (5,0 г), фильтровали через целит, концентрировали, и выдерживали в вакууме (около 0,1 мм рт.ст.) в течение ночи, в результате чего получали целевое соединение этой стадии (6,17 г, 111%) в виде красного маслянистого вещества (чистота > 80%, ВРЖХ).
С. N-(3,4-Диметил-5-изоксазолил)-4'-(2-оксазолил) [1,1'-бифенил] -2-сульфонамид
В осушенную круглодонную 250 мл колбу, снабженную обратным холодильником и магнитным стержнем для перемешивания, загружали неочищенный продукт стадии В (6,17 г, теоретич. 5,56 г, 11,9 ммоль), 95% EtOH (60 мл), 6,0н. HCl (60 мл) и нагревали с обратным холодильником в течение двух часов. Смесь охлаждали приблизительно до 5oС, добавляли водный раствор NaOH до рН ~13 при умеренном повышении температуры до приблизительно менее чем 25oС, а затем подвергали тонкой фильтрации и добавляли воду (50 мл). Раствор концентрировали при 40oС для удаления большого количества EtOH (~55-60 мл), после чего образовывалось белое твердое вещество. Смесь охлаждали до 0oС в течение 20 минут, фильтровали и полученное белое твердое вещество промывали солевым раствором (50 мл х 2), а затем сушили в вакууме (~0,1 мм рт.ст.) в течение 12 часов. Белое твердое вещество растворяли в горячей воде (80 мл) и подвергали тонкой фильтрации. Водный раствор помещали в делительную воронку и промывали EtOAc/гексаном (1/1) (50 мл х 2) и гексаном (75 мл). Раствор охлаждали до 0oС, интенсивно перемешивали, медленно добавляли 1,0н. HCl до рН 1,0, а затем смесь выдерживали при 0oС в течение 15 минут. После фильтрации и промывки водой (50 мл х 3) получали беловатое твердое вещество, которое сушили в вакууме в течение 3 часов. Затем белое твердое вещество растворяли в этилацетате (65 мл), добавляли тритиоциануровую кислоту (0,20 г, 1,13 ммоль) и смесь перемешивали при 65oС в течение 30 минут. После добавления угля (нейтральный уголь Norit, 5,0 г) смесь перемешивали еще 30 минут при 65oС. Смесь охлаждали до 0oС, фильтровали через слой целита, промывали EtOAc (50 мл х 3) и концентрировали до объема ~60 мл. Смесь промывали 1,0н. NaOH (50 мл х 3) и к водному слою добавляли NaCl (3,0 г, 637 ммоль), а также солевой раствор (50 мл). Водную смесь охлаждали до 0oС, фильтровали и промывали солевым раствором (50 мл х 2). Полученное белое твердое вещество растворяли в горячей воде (200 мл), охлаждали до 0oС, интенсивно перемешивали, и медленно добавляли 1,0н. НCl до рН 1,0, а затем смесь оставляли на 15 минут при 0oС. После фильтрации и промывки водой (50 мл х 3) получали беловатое твердое вещество, которое сушили в вакууме (0,1 мм рт.ст.) при ~45oС в течение около 16 часов, и получали 2,4 г (50% выход) беловатого твердого вещества. Это твердое вещество (2,25 г) растворяли в 95% ЕtOН/воде (9,0 мл/5,0 мл) при 70oС. Раствор медленно охлаждали до комнатной температуры (1,5 часа), а затем охлаждали до 5oС в течение ночи, в результате чего образовывалось белое кристаллическое вещество, которое фильтровали и промывали холодной смесью 95% ЕtОН/воды (3,0 мл/5,0 мл), а затем сушили в вакууме (~0,1 мм рт. ст. ) в течение 48 часов и получали 2,1 г целевого соединения данного примера в виде белого твердого продукта (46,9% полный выход исходя из целевого соединения стадии В). ТСХ (гексан/-ЕtОАс, 2/1, краситель КМnО4) указывала на то, что реакция была полностью завершена через 2 часа. Т.пл.= 143,07oС.
Изобретение относится к новому способу получения бифенилизоксазолсульфонамида формулы (I) его энантиомеров и диастереомеров и его солей, где Х представляет кислород, Y представляет азот, каждый из R3 и R4 непосредственно связан с углеродом кольца и независимо представляет алкил, каждый из R1 и R2 непосредственно связан с углеродом кольца и независимо представляет водород или алкил, каждый из R11, R12, R13 и R14 независимо представляет водород, J представляет кислород, К представляет азот, L представляет углерод, р представляет 0 или целое число от 1 до 2, который включает (а) взаимодействие сложного эфира пинакола формулы II с галогенфенильным соединением формулы III в присутствии палладиевого (0) катализатора и, необязательно, основания, с образованием азотзащищенного производного формулы IV и (b) удаление защиты у атома азота в соединении формулы IV с образованием соединения формулы I. Сложный эфир пинакола общей формулы II, где Х представляет кислород, Y представляет азот, каждый из R3 и R4 непосредственно связан с углеродом кольца и независимо представляет алкил, каждый из R13 и R14 непосредственно связан с углеродом кольца и независимо представляет водород, "prot" означает азотзащитную группу. Способ получения соединения формулы II, включающий (а) взаимодействие соединения формулы V с амином формулы VI в присутствии органического основания и органического растворителя с образованием соединения формулы VII, (b) защиту азота указанного соединения формулы VII с получением соединения формулы VIII, (с) получением литиевого производного указанного соединения формулы VIII с использованием алкил- или ариллитиевого соединения с последующей реакцией образованного литиевого производного с триалкилборатом и последующим гидролизом с образованием бороновой кислоты формулы IX, (d) взаимодействие указанного соединения формулы IX с пинаколом с удалением воды и получением соединения формулы II. Технический результат - новый способ получения бифенилизоксазолсульфонамидов. 5 с. и 29 з.п. ф-лы.

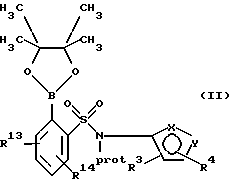
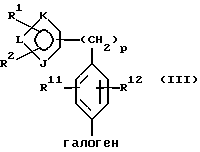




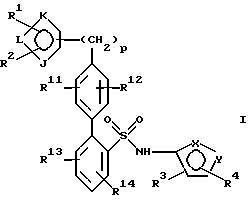
его энантиомеров и диастереомеров и его солей,
где Х представляет кислород;
Y представляет азот;
каждый из R3 и R4 непосредственно связан с углеродом кольца и независимо представляет алкил;
каждый из R1 и R2 непосредственно связан с углеродом кольца и независимо представляет водород или алкил;
каждый из R11, R12, R13 и R14 независимо представляет водород;
J представляет кислород;
К представляет азот;
L представляет углерод;
р представляет 0 или целое число от 1 до 2;
где указанный способ включает
(а) взаимодействие сложного эфира пинакола формулы II или его соли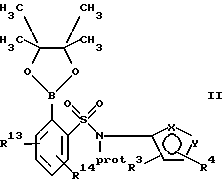
где указанные значения X, Y, R3, R4, R13, R14 указаны выше и "prot" означает азотзащитную группу, с галогенфенильным соединением формулы III или его солью
где указанные значения R1, R2, R11, R12, К, L, J и р указаны выше, в присутствии палладиевого (0) катализатора и, необязательно, основания, с образованием азот защищенного производного формулы IV или его соли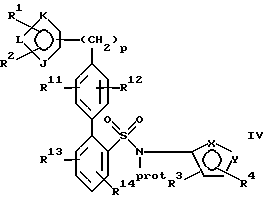
где указанные значения R1, R2, R3, R4, R11-R14, К, L, J, X, Y, "prot" и р указаны выше, и
(b) удаление защиты у атома азота в соединении указанной формулы IV или его соли с образованием указанного соединения формулы I или его соли.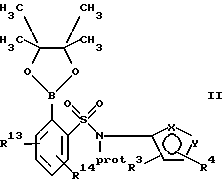
где Х представляет кислород;
Y представляет азот;
каждый из R3 и R4 непосредственно связан с углеродом кольца и независимо представляет алкил, каждый из R13 и R14 независимо представляет водород, "prot" означает азотзащитную группу.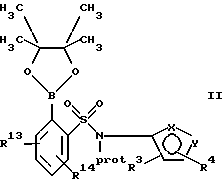
где фенильное кольцо указанной формулы II может быть, кроме того, замещено, где "prot" означает азотзащитную группу; и где X, Y, R3 и R4 имеют значения, указанные в п. 10,
где указанный способ включает следующие стадии:
а) взаимодействие соединения формулы V или его соли;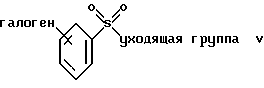
где фенильная группа указанной формулы V может быть, кроме того, замещена амином формулы VI или его солью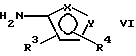
где X, Y, R3 и R4 имеют значения, указанные в п. 10, в присутствии органического основания и органического растворителя с образованием соединения формулы VII или его соли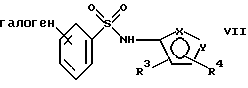
где X, Y, R3 и R4 имеют значения, указанные в п. 10, и где фенильная группа в указанной формуле VII может быть, кроме того, замещена;
(b) защиту азота указанного соединения формулы VII или его соли с получением соединения формулы VIII или его соли
где X, Y, R3 и R4, "prot" имеют значения, указанные в п. 10, и где фенильная группа в указанной формуле VIII может быть, кроме того, замещена;
(с) получение литиевого производного указанного соединения формулы VIII или его соли с использованием алкил- или ариллитиевого соединения с последующей реакцией образованного литиевого производного с триалкилборатом, и последующим гидролизом с образованием бороновой кислоты формулы IX или его соли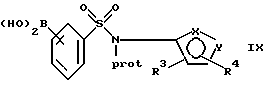
где X, Y, R3, R4 и "prot" имеют значения, указанные в п. 10, где фенильная группа в указанной формуле IX может быть, кроме того, замещенной; и
(d) взаимодействие указанного соединения формулы IX или его соли с пинаколом, с удалением воды, и получением, в результате, указанного соединения формулы II или его соли.
его энантиомеров или диастереомеров и его солей,
где Х представляет кислород;
Y представляет азот;
каждый из R3 и R4 непосредственно связан с углеродом кольца и независимо представляет алкил;
каждый из R1 и R2 непосредственно связан с углеродом кольца и независимо представляет водород или алкил;
каждый из R11, R12, R13 и R14 независимо представляет водород;
J представляет кислород;
К представляет азот;
L представляет углерод;
р представляет 0 или целое число от 1 до 2;
где указанный способ включает
(а) взаимодействие сложного эфира пинакола формулы IX или его соли
где указанные значения X, Y, R3, R4 указаны выше, и "prot" означает азотзащитную группу, с галогенфенильным производным формулы III или его солью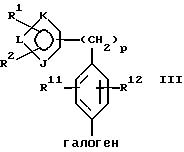
где указанные значения R1, R2, R11, R12, К, L, J и р указаны выше, в присутствии палладиевого (0) катализатора и, необязательно, основания, с получением азотзащищенного производного формулы IV или его соли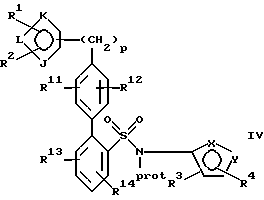
где указанные значения R1, R2, R3, R4, R13-R14, К, L, J, X, Y, "prot" и р указаны выше,
(b) удаление защиты у атома азота в соединении указанной формулы IV или его соли с образованием указанного соединения формулы I или его соли.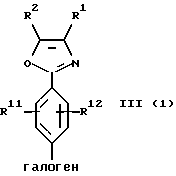
где R1, R2, R11, R12 имеют значения, указанные выше, взаимодействием фенилгалогенангидрида формулы Х или его соли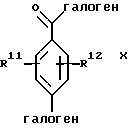
где R11 и R12 указаны выше, с аминоацеталем формулы XI или его солью
где значения R1, R2 указаны выше, в присутствии основания и растворителя с образованием амидоацеталя формулы XII или его соли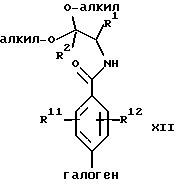
где R1, R2, R11, R12 имеют значения, указанные выше, и
(b) циклизацию амидоацеталя формулы XII или его соли в присутствии циклизующего агента с получением указанного оксазолфенилгалогенида формулы III (1) или его соли.
| Электронные часы, корректируемые радиосигналами точного времени | 1975 |
|
SU558258A1 |
| US 5086184 А, 04.02.1992 | |||
| SU 1375111 A3, 15.02.1988. | |||

